遺傳性免疫缺乏疾病 (Genetic Immunodeficiency Diseases)
PART 21 代謝性、遺傳性與全身性疾病 (METABOLIC, GENETIC, AND SYSTEMIC DISEASES)
遺傳性免疫缺乏疾病總覽 (OVERVIEW)
原發性免疫缺乏疾病(primary immunodeficiency diseases)是免疫系統的遺傳性疾患,會導致對感染的易感性增加,以及罹病率與死亡率升高。¹ 許多此類遺傳性免疫缺乏疾病都與各式各樣的皮膚異常相關聯,辨識出這些臨床特徵,可使原發性免疫缺乏得以早期診斷。皮膚異常可能包括皮膚感染、異位樣 (atopic-like) 或脂漏樣 (seborrheic-like) 皮膚炎、斑狀紅斑 (macular erythemas)、禿髮 (alopecia)、傷口癒合不良、紫斑 (purpura)、瘀點 (petechiae)、毛細血管擴張 (telangiectasias)、色素稀釋 (pigmentary dilution)、皮膚肉芽腫 (cutaneous granulomas)、廣泛性疣 (extensive warts)、血管性水腫 (angioedema) 以及狼瘡樣 (lupus-like) 變化(表 132-1)。其他臨床特徵常包括生長遲滯 (failure to thrive)、內臟感染、自體免疫疾患、結締組織/風濕病、過敏反應與腫瘤。
臨床特徵 (CLINICAL FEATURES)
當病人出現持續時間延長或嚴重度增加的反覆感染,尤其是由不尋常微生物引起的感染時,應懷疑免疫缺乏。可能伴隨感染清除不完全、感染出現意料之外或嚴重的併發症,或對抗生素反應不佳。² 受影響的嬰兒常生長不良(生長遲滯)。最常見的非皮膚性異常為感染、腹瀉、嘔吐、肝脾腫大、關節炎、淋巴結腫大或淋巴結/扁桃腺稀少,以及血液學異常。遺傳性免疫缺乏疾患的分類包括:(a) 抗體缺乏 (antibody deficiencies)、(b) 細胞性缺乏 (cellular deficiencies)、(c) 合併抗體與細胞性缺乏 (combined antibody and cellular deficiencies)、(d) 吞噬作用與細胞殺傷障礙 (disorders of phagocytosis and cell killing),以及 (e) 補體缺陷 (complement defects)。各組別的特徵性臨床表徵提示適當的分類與實驗室檢查可用以確認診斷。各組免疫缺乏疾患相關的實驗室檢查與臨床疾病型態(可供其鑑別診斷)概述於表 132-2 與圖 132-1。在所有遺傳性免疫缺乏疾患的鑑別診斷中,最重要的疾病為 HIV 感染。除了遺傳性免疫缺乏病人經聚合酶連鎖反應 (polymerase chain reaction) 偵測不到 HIV 抗原之外,其他特徵也有助於區分這些疾患。HIV 感染病人傾向呈現倒置的 CD4 對 CD8 比值 (inverted CD4-to-CD8 ratio) 與高γ球蛋白血症 (hypergammaglobulinemia),與許多遺傳性免疫缺乏病人的低γ球蛋白血症 (hypogammaglobulinemia) 形成對比。
抗體缺乏疾患 (ANTIBODY DEFICIENCY DISORDERS)
無γ球蛋白血症 (Agammaglobulinemia)
重點一覽 (AT-A-GLANCE)
■ 同義詞:Bruton 病 (Bruton disease)。
■ 早發性、反覆性細菌感染。
■ 扁桃腺與頸部淋巴結組織缺如或幾乎無法偵測到。
■ 嚴重的低γ球蛋白血症,周邊 B 細胞減少或缺如。
臨床特徵
無γ球蛋白血症的特徵為反覆性化膿性感染 (pyogenic infections),通常在 6 個月大之後開始發作,與母源免疫球蛋白逐漸消退同時發生。這些病人扁桃腺與頸部淋巴結缺如或幾乎無法偵測到。³ 皮膚感染(尤其是癤病 furunculosis 與膿痂疹 impetigo)很常見,且常圍繞身體開口處。許多受影響的兒童曾被描述出現一種異位樣濕疹性疹 (atopic-like eczematous eruption),且對免疫球蛋白 (Ig) 治療無改善反應。亦曾有壞疽性膿皮症 (pyoderma gangrenosum) 與非感染性皮膚肉芽腫的報告。兒童期出疹性疾患可被妥善處理,但由於無法產生特異性抗體,感染可能復發。反覆性中耳炎、鼻竇炎、支氣管炎與肺炎是最早出現的感染表現,通常由肺炎球菌 (pneumococci)、葡萄球菌 (staphylococci) 或嗜血桿菌 (Haemophilus) 引起。未經治療的肺部感染可能導致進行性支氣管擴張 (bronchiectasis) 與慢性肺部疾病。⁴ 病人也可能罹患慢性腸病毒感染,以及因反覆中耳炎與鼻竇炎而導致聽力喪失。其他常見的細菌感染包括結膜炎、骨髓炎、化膿性關節炎與腦膜炎。遷延性腹瀉可能由感染引起,特別是梨形鞭毛蟲 (Giardia)、沙門氏菌 (Salmonella)、曲狀桿菌 (Campylobacter) 或隱孢子蟲 (Cryptosporidium) 等。有三類病毒會造成問題:腸病毒 (enterovirus)、B 型肝炎病毒 (hepatitis B virus) 與輪狀病毒 (rotavirus)。病人在接種活性小兒麻痺疫苗 (live polio vaccine) 後曾發生麻痺。一種類風濕性關節炎 (rheumatoid-like arthritis),特徵為大關節的慢性發炎與腫脹,可能發生於多達三分之一至二分之一的 X 連鎖無γ球蛋白血症 (X-linked agammaglobulinemia, XLA) 男孩,且常由黴漿菌感染 (Ureaplasma urealyticum) 引起。瀰漫性伊科病毒 (echovirus) 感染曾導致腦膜腦炎,以及一種類皮肌炎疾患 (dermatomyositis-like disorder),伴隨硬實水腫 (brawny edema)、肌肉硬化伴隨無力、肌肉攣縮與異色症 (poikiloderma)。
病因與發病機轉
無γ球蛋白血症肇因於 B 細胞發育失敗,最常見的原因是基因缺陷阻止了完整 B 細胞抗原受體 (B-cell antigen receptor) 的組裝。XLA 是無γ球蛋白血症最常見的病因,源於一種細胞質酪胺酸激酶 (cytoplasmic tyrosine kinase)——Bruton 酪胺酸激酶 (Bruton tyrosine kinase) 的缺陷。XLA 以 X 連鎖方式遺傳,約 50% 的受影響男孩有本病家族史。⁵ 體染色體隱性無γ球蛋白血症 (autosomal recessive agammaglobulinemia) 男女受影響機率相同,源於編碼前 B 細胞與 B 細胞受體組件的基因缺陷,或 BLNK(B-cell linker,一種組裝與前 B 細胞及 B 細胞受體相關之訊息分子的支架蛋白)的缺陷。
診斷
無γ球蛋白血症的診斷依據為血清 IgG、IgA、IgM 濃度遠低於適當對照組的 95% 信賴區間下限(通常總 Ig 少於 100 mg/dL),以及周邊循環中 B 細胞幾乎完全缺如(<正常值的 1%)。XLA 病人體內 Bruton 酪胺酸激酶蛋白的缺如,可透過流式細胞儀 (flow cytometry) 偵測。鑑定出無γ球蛋白血症已知遺傳病因之一的缺陷可確認診斷,並可用於遺傳諮詢與產前診斷。
臨床病程、預後與處置
早期經靜脈或皮下進行 Ig 替代治療,並使用抗生素,可顯著降低感染風險,雖然對於減少慢性肺病或慢性腸病毒感染的風險與罹病率可能無助益。
常見變異型免疫缺乏 (Common Variable Immunodeficiency)
重點一覽 (AT-A-GLANCE)
■ 一群異質性的疾患,其中可同時發現抗體缺乏與 T 細胞異常。
■ 最常見的潛在基因缺陷位於跨膜活化子與鈣調節親環素配體交互作用因子 (transmembrane activator and calcium-modulating cyclophilin ligand interactor, TACI) 基因。
■ 可能在兒童期出現,發病高峰在生命的第二與第三個十年。
■ 診斷需具備:血清免疫球蛋白 (Ig) G 低下,並伴隨 IgM 與/或 IgA 低下、對免疫接種(尤其是肺炎球菌抗原)的抗體反應缺陷,且有反覆感染與/或典型的自體免疫併發症。
■ 自體免疫與感染併發症的嚴重度多變。
臨床特徵
常見變異型免疫缺乏 (common variable immunodeficiency, CVID) 通常於年輕成人時表現,但約 20% 的病例在 21 歲之前被診斷。⁶
病人的感染與 XLA 病人相似,特別是鼻竇肺部感染 (sinopulmonary infections),但對腸病毒感染較不易感,而對梨形鞭毛蟲 (Giardia) 感染較易感。許多 CVID 病人有肝臟疾病與胃腸道疾病,造成吸收不良症候群 (malabsorption syndromes)。曾有皮膚(圖 132-2)、肺、肝、脾的非乾酪性肉芽腫 (noncaseating granulomas) 報告。皮膚與內臟的乾酪性肉芽腫 (caseating granulomas) 雖罕見,亦曾被描述。⁷,⁸
廣泛性疣可能是 CVID 患者的一大問題(圖 132-3)。一項比較 CVID 病人與 IgA 缺乏病人皮膚表現的研究發現,IgA 缺乏病人異位性皮膚炎 (atopic dermatitis) 的發生率增加但 IgE 並未升高,而 CVID 病人則乾癬 (psoriasis)、皮膚感染、痤瘡 (acne)、禿髮、白斑 (vitiligo) 與口瘡性潰瘍 (aphthous ulcers) 的盛行率增加。⁹ 淋巴組織常腫大,亦可見脾腫大伴脾功能亢進 (hypersplenism),有 8.2% 的 CVID 病人接受脾切除術。⁶ 自體免疫疾患特別常見(28.6%),尤其是自體免疫性血小板減少症、自體免疫性溶血性貧血、類風濕性關節炎、乾燥症候群 (sicca syndrome) 與惡性貧血 (pernicious anemia)。亦曾描述圓禿 (alopecia areata) 與狼瘡。在 10% 至 20% 的 CVID 病人中,至少有 1 名家庭成員亦有免疫缺乏,特別是 CVID 或 IgA 缺乏。¹⁰ 淋巴網狀惡性腫瘤 (lymphoreticular malignancy) 與胃癌的發生率顯著增加,特別是在生命的第五與第六個十年。
選擇性免疫球蛋白疾患 (Selective Immunoglobulin Disorders)
重點一覽 (AT-A-GLANCE)
■ 免疫球蛋白 A 缺乏 (Immunoglobulin A deficiency) 通常無症狀;只有 10% 至 15% 的受影響者會出現臨床表現,包括細菌性鼻竇肺部感染與自體免疫疾患。
細胞性缺乏 (CELLULAR DEFICIENCIES)
慢性皮膚黏膜念珠菌病 (Chronic Mucocutaneous Candidiasis)
重點一覽 (AT-A-GLANCE)
■ 一群異質性疾患,具有選擇性針對念珠菌 (Candida) 的免疫反應改變。
■ 皮膚、指甲與黏膜的反覆性、進行性念珠菌感染。
■ 可能與後續發生的內分泌病變相關(APECED〔自體免疫多內分泌腺病變、念珠菌病與外胚層失養症,autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy〕)。
慢性皮膚黏膜念珠菌病 (chronic mucocutaneous candidiasis, CMC) 已被定義出數種臨床亞型(表 132-3)。它們具有不同的臨床表現、不同程度的免疫缺乏,以及不同的遺傳方式。CMC 病人可能於兒童期或成熟期發病,並可能呈家族性或散發性發生。此外,CMC 可能伴或不伴內分泌病變。患有自體免疫多內分泌腺病變、念珠菌病與外胚層失養症(APECED,或稱第 1 型自體免疫多內分泌腺症候群 autoimmune polyendocrine syndrome, Type 1)的病人常有受影響的手足。APECED 與其他家族型 CMC 為體染色體隱性遺傳。在合併角膜炎 (keratitis) 的病人中可見體染色體顯性遺傳。
臨床特徵
CMC 病人有皮膚、指甲與黏膜的反覆性、進行性感染,最常由白色念珠菌 (Candida albicans) 引起。依亞型而定,臨床表現範圍從反覆、難治的鵝口瘡 (thrush)(圖 132-4),到伴有少數失養性指甲的輕度紅斑性鱗屑斑塊 (erythematous scaling plaques)(圖 132-5),乃至嚴重的全身性、結痂性肉芽腫斑塊 (crusted granulomatous plaques)(圖 132-6)。皮膚斑塊最常發生於間擦部位 (intertriginous areas)、開口周圍部位 (periorificial sites) 與頭皮,但也可能呈全身性。指甲變厚、脆裂、變色,甲溝周圍區域常呈紅斑、腫脹與壓痛。頭皮感染可能導致瘢痕與禿髮。雖然口腔黏膜是最常見的黏膜變化部位,食道、生殖器與喉部黏膜也可能受累。念珠菌感染在這些黏膜部位可能形成狹窄 (strictures)。來自皮膚或黏膜病灶的刮取物與培養可顯示念珠菌微生物。CMC 病人很少發展為全身性念珠菌病,但 50% 可能發展出由其他微生物引起的反覆或嚴重感染。在一項研究中,81% 的早發型 CMC 病人也合併有細菌、真菌與寄生蟲感染,包括細菌性敗血症。¹¹ 可能同時發生皮癬菌 (dermatophyte) 感染。在 APECED 病人中,念珠菌感染傾向在 5 歲前開始,雖然內分泌功能障礙可能要到 12 至 13 歲才明顯(見圖 132-6)。最常相關的內分泌病變為副甲狀腺低能症 (hypoparathyroidism)(88%)與腎上腺皮質低能症 (hypoadrenocorticism)(60%)。三分之一的病人有念珠菌病、副甲狀腺低能症與腎上腺功能缺陷。其他相關的內分泌病變或自體免疫疾患包括性腺功能不全 (gonadal insufficiency)(45%)、圓禿(20%)、惡性貧血(16%)、甲狀腺異常(12%)、慢性活動性肝炎或幼年型肝硬化(9%)、白斑、糖尿病與腦下垂體功能低下 (hypopituitarism)。慢性腹瀉與吸收不良已在 25% 的病人中報告,通常與副甲狀腺低能症相關。某些受影響的病人也有肺纖維化、牙釉質發育不全 (dental enamel hypoplasia) 與角結膜炎 (keratoconjunctivitis)。其「外胚層失養症 (ectodermal dysplasia)」特徵可能是繼發於念珠菌感染或自體免疫。APECED 病人常有自體免疫抗體,包括抗甲狀腺球蛋白 (antithyroglobulin)、抗微粒體 (antimicrosomal)、抗腎上腺 (antiadrenal) 與抗黑色素細胞 (antimelanocyte) 抗體,以及類風濕因子 (rheumatoid factor)。在沒有臨床內分泌疾病的 CMC 病人中也曾發現自體抗體。
病因與發病機轉
T 輔助型 17 (Th17) 細胞在對抗念珠菌的免疫防禦中扮演關鍵角色,而其反應性在 APECED 與非 APECED 的 CMC 病人中皆受損。¹² 活化訊息傳遞與轉錄活化因子 1 基因 (signal transducer and activator of transcription 1 gene, STAT1) 的雜合性活化突變 (heterozygous mutations) 是最常見的病因,¹³,¹⁴ 會抑制介白素 (interleukin, IL)-17 的表現,同時促進干擾素訊息傳遞(見表 132-3)。¹⁵⁻¹⁸ IL-17 異型體、IL-17 受體,或調控 IL-17 反應的 TRAF3IP2 的突變,也曾被描述。APECED 是一種體染色體隱性型,源於自體免疫調節基因 (autoimmune regulator gene, AIRE) 的功能喪失突變。AIRE 調控胸腺髓質上皮細胞中自體抗原的表現,透過自體反應性 T 細胞的負向選擇 (negative selection) 與抗原特異性調節性 T 細胞 (regulatory T cells) 的生成,促進免疫耐受性。¹⁹ APECED 中周邊組織特異性自體反應性 T 細胞的保留,導致中和性自體抗體的產生,包括針對 Th17 相關細胞激素的抗體,從而引起念珠菌感染與自體免疫疾病。²⁰,²¹ 對念珠菌與皮癬菌感染的易感性也是 caspase 招募結構域家族成員 9 基因 (caspase recruitment domain family member-9 gene, CARD9)²² 與編碼 dectin-1 之基因突變的特徵。²³
診斷
來自病灶的刮取物與培養可顯示念珠菌微生物。若進行切片,可見白色念珠菌僅存於角質層 (stratum corneum)。在 75% 的 CMC 病人中可發現免疫缺陷的證據,包括皮膚試驗無反應性 (anergy) 以及對念珠菌抗原的體外淋巴增生 (lymphoproliferation) 或細胞激素釋放缺陷。然而,此變異性反映了 CMC 潛在的異質性。針對第 I 型干擾素 (Type I interferons) 的自體抗體是 APECED 敏感且特異的標記。²⁴
鑑別診斷
嬰兒頻繁的念珠菌感染(尤其是鵝口瘡)常伴隨反覆使用抗生素(如治療中耳炎時)出現;若無反覆念珠菌感染的明確原因,則應考慮 HIV 感染與 CMC。自體免疫疾患與內分泌病變是 IPEX(免疫失調、多內分泌腺病變、腸病變、X 連鎖,immune dysregulation, polyendocrinopathy, enteropathy, X-linked)症候群的特徵,²⁵ 但其感染傾向為細菌性。然而,在 IL-2 受體 α 鏈 (CD25) 缺乏症中,真菌、細菌與病毒感染可能伴隨自體免疫內分泌病變。²⁶
臨床病程、預後與治療
CMC 病人的念珠菌病灶一般對長期全身性投予的唑類 (azole) 抗真菌劑(itraconazole、fluconazole)或 terbinafine 有反應。²⁷ 抗藥性病人通常對 voriconazole(具光毒性 phototoxicity 風險)、posaconazole、棘白菌素類 (echinocandins) 與/或合併或不合併 flucytosine 的 amphotericin B 有反應。皮膚肉芽腫即使感染清除後仍常反應較差。其他療法包括顆粒球菌落刺激因子 (granulocyte colony-stimulating factor)(可能增加 IL-17 產生)²⁸、拔甲、膿瘍引流,以及厚痂性皮膚斑塊的清創。所有 CMC 病人都應每年接受內分泌評估,已記錄有內分泌病變或有 APECED 家族史者應接受更密切的監測。曾有念珠菌以外感染病史者,應進一步評估其免疫狀態。
軟骨毛髮發育不全症候群 (Cartilage–Hair Hypoplasia Syndrome)
重點一覽 (AT-A-GLANCE)
■ 同義詞:McKusick 型幹骺端軟骨發育不良 (metaphyseal chondrodysplasia McKusick type)。
■ 體染色體隱性疾患,常見於 Amish 與芬蘭族群。
■ 一種核糖核蛋白內切核糖核酸酶 (ribonucleoprotein endoribonuclease) 之 RNA 組件的突變,導致細胞介導性與體液性免疫缺陷。
■ 特徵為細、稀疏、色素減退的毛髮,以及短肢侏儒症 (short-limbed dwarfism)。
■ 支持性治療,適當使用抗生素;骨髓移植可矯正免疫缺陷,但無法矯正真皮或軟骨。
合併抗體與 T 細胞缺乏 (COMBINED ANTIBODY AND T-CELL DEFICIENCY)
高免疫球蛋白 M 症候群 (Hyperimmunoglobulin M Syndrome)
重點一覽 (AT-A-GLANCE)
■ 大多數高免疫球蛋白 M 症候群病例為 X 連鎖隱性型,伴有 CD40 配體 (CD40 ligand) 缺乏;常見的臨床特徵為反覆性鼻竇肺部與胃腸道感染、口腔潰瘍與疣 (verrucae)。
■ 體染色體隱性高免疫球蛋白 M 症候群(CD40 缺乏除外)不具對伺機性感染的易感性與淋巴增生。
威斯科特-奧德里奇症候群 (Wiskott-Aldrich Syndrome)
重點一覽 (AT-A-GLANCE)
■ X 連鎖隱性。
■ WASP (Wiskott-Aldrich syndrome protein) 基因突變。
■ 難治性皮膚炎。
■ 反覆性化膿性感染。
■ 因血小板減少與血小板功能障礙引起的出血。
■ 治療:骨髓幹細胞移植。
臨床特徵
威斯科特-奧德里奇症候群 (Wiskott-Aldrich syndrome, WAS) 是一種 X 連鎖隱性疾患,發生率約為每 100 萬名男嬰中 4 例。²⁹,³⁰ WAS 的典型三聯徵為:(a) 因血小板減少與血小板功能障礙引起的出血、(b) 反覆性化膿性感染,以及 (c) 難治性皮膚炎,但此三聯徵僅出現於 25% 的病人。出血傾向 (bleeding diathesis) 是 WAS 突變最常見的表現,存在於 84% 的病人,常於出生後最初數週或數月以血便初次表現。亦可能發生鼻出血、嘔血、血尿、皮膚黏膜瘀點與顱內出血。隨著經胎盤傳遞的母源抗體減少,反覆性細菌感染自嬰兒期開始。這些感染包括癤病、結膜炎、中耳炎與外耳炎、全鼻竇炎 (pansinusitis)、肺炎、腦膜炎與敗血症。以莢膜細菌如肺炎球菌 (Pneumococcus)、流感嗜血桿菌 (Haemophilus influenzae) 與腦膜炎雙球菌 (Neisseria meningitidis) 的感染為主。病人也易感染疱疹及其他病毒,以及肺囊蟲 (Pneumocystis jiroveci)。WAS 相關的異位性皮膚炎發生於約 80% 的病人,通常在出生後最初幾個月內出現,且可能相當嚴重。顏面、頭皮與屈側區域受累最嚴重,但病人常有伴隨進行性苔癬化的廣泛侵犯。其疹子可能比無 WAS 個體的異位性皮膚炎更具剝脫性 (exfoliative),且抓破處常有血清血性結痂 (serosanguineous crusts)(圖 132-7)。濕疹病灶繼發細菌感染很常見,疱疹性濕疹 (eczema herpeticum)(圖 132-8)與傳染性軟疣 (molluscum contagiosum) 也常見。除異位性皮膚炎外,亦可見 IgE 介導的過敏問題,如蕁麻疹、食物過敏與氣喘。多達 40% 的 WAS 病人會發展出自體免疫疾患。³¹ 最常見者為血管炎 (vasculitis)(特別侵犯皮膚、胃腸道、腦與心臟)佔 20% 的病人,自體免疫性溶血性貧血佔 14% 的病人,以及 IgA 腎病變 (IgA nephropathy) 佔多達 10% 的病人。³² 其他免疫介導的皮膚表現有血管性水腫、皮肌炎 (dermatomyositis)、壞疽性膿皮症與結節性紅斑 (erythema nodosum)。肝脾腫大常見,偶爾會出現淋巴結腫大、暫時性關節炎與關節積液。
病因與發病機轉
缺陷基因為 WASP,定位於 Xp11.22-11.23,編碼 WASP——一種造血特異性 (hematopoietic specific) 細胞質蛋白,功能在於訊息傳遞與細胞骨架組織。WASP 將細胞膜上產生的訊號與細胞骨架的重組偶聯,導致細胞活化並促進細胞運動。³³ WASP 突變會影響免疫突觸 (immunologic synapse) 的組織與 T 細胞活化、T 淋巴球與 B 淋巴球的遷移,以及初級抗體反應的啟動。本病存在強烈的表現型-基因型相關性;當 WASP 缺如或截短時發生典型 WAS,而當突變的 WASP 仍有表現時則發生 X 連鎖血小板減少症 (X-linked thrombocytopenia)。異位性皮膚炎可能與所觀察到的 CD4+ T 細胞分化偏向 Th2 細胞、同時 Th1 與調節性 T 細胞分化受抑制有關。³⁴ 周邊 B 細胞恆定的改變與調節性 T 細胞活化的減少,被認為會促進 WAS 自體免疫表現的發展。³⁵⁻³⁷
由於 WASP 在表皮蘭格漢細胞 (epidermal Langerhans cells) 上有表現,蘭格漢細胞與 T 細胞的異常交互作用,以及蘭格漢細胞在抗原刺激後遷移至淋巴結的能力,可能也參與其中。WAS 病人自生命最初幾年起即有 T 與 B 淋巴球的功能與數量減少。WAS 病人的淋巴球缺乏由肌動蛋白束 (actin bundles) 構成的微絨毛 (microvilli),導致趨化作用 (chemotaxis) 缺陷,且在某些病人中,淋巴球與血小板上唾液醣蛋白 (sialoglycoproteins)(如 CD43 等)的表現減少。體液免疫的缺陷包括血清 Ig 異常與對多醣抗原 (polysaccharide antigens) 的抗體反應減少。WAS 病人也有自然殺手 (natural killer, NK) 細胞細胞毒性、樹突細胞遷移與活化的缺陷,以及巨噬細胞趨化作用受損。
診斷
診斷根據血小板異常與相關的異位性皮膚炎而懷疑,並由實驗室檢查確認,特別是基因分型 (genotyping) 以鑑定 WASP 突變。WAS 的血小板減少是持續性的,血小板數可能介於 1000 至 80,000 顆血小板/µL。正式診斷標準要求血小板數 <70,000/µL。血小板體積小,且血小板凝集功能缺陷。IgM 與有時 IgG 的濃度低下,且缺乏同種血球凝集素 (isohemagglutinins)。IgA、IgE 與 IgD 濃度通常升高。亦可見嗜伊紅性白血球增多 (eosinophilia)、白血球減少 (leukopenia) 與淋巴球減少 (lymphopenia)。延遲性過敏 (delayed hypersensitivity) 皮膚試驗反應性減弱,且病人對多醣抗原無反應。WASP 可使用抗 WASP 抗體進行細胞內染色,以流式細胞儀偵測,而 WASP 基因的定序可確認 WAS 或 X 連鎖血小板減少症的診斷——後者也源於不影響免疫功能的 WASP 基因突變。突變分析可用於產前診斷。WAS 的女性帶因者可透過其淋巴球與血小板中異常 X 染色體的選擇性去活化 (selective inactivation) 而被偵測出來。³⁸
鑑別診斷
其他免疫缺乏疾病也以濕疹性皮膚炎與對感染易感性增加為特徵,但 WAS 通常可藉由出血傾向與微血小板減少 (microthrombocytopenia) 的實驗室證據來區分。
臨床病程、預後與治療
治療性介入使某些 WAS 病人得以存活至成年;然而,相當比例的病人在 10 歲前因繼發於出血的感染、惡性腫瘤或移植併發症而死亡。²⁹,³⁹
百分之十三的 WAS 病人會發展出淋巴網狀惡性腫瘤,尤其是非何杰金氏淋巴瘤 (non-Hodgkin lymphoma)(特別是瀰漫性大 B 細胞淋巴瘤),以結外 (extranodal) 與腦部侵犯為主。自體免疫性溶血性貧血的發生是不良預後因子,且與淋巴惡性腫瘤的發展相關;整體而言,25% 有自體免疫的病人會發展出惡性腫瘤。³² 10% 的病人死於這些惡性腫瘤,通常是在青春期或年輕成人時期。適當的抗生素、免疫接種,以及血小板與血漿的輸注,可降低致命性感染與出血的風險。免疫球蛋白替代治療對某些病人有用。脾切除術曾被提倡用於緩解反覆嚴重出血病人的出血異常,但此手術會增加莢膜細菌微生物感染的風險。局部糖皮質類固醇製劑與 Ig 替代可能改善皮膚炎,而對反覆性疱疹性濕疹的病人,長期口服投予 acyclovir 是適當的。骨髓或幹細胞移植是反覆出現問題(尤其是顯著自體免疫)病人的首選治療。²⁹,³⁹ 完全植入 (full engraftment) 可使血小板數量與功能正常、免疫狀態正常,並清除皮膚炎(T 淋巴球植入)。對於有人類白血球抗原 (human leukocyte antigen, HLA) 配對手足捐贈者的 5 歲以下兒童,5 年存活率為 87%;年齡較大的病人與不相配捐贈者的病人,存活率約為 50%。在基因治療試驗中,曾投予經慢病毒轉導 (lentivirally transduced)、WAS 重建、自體 CD34+ 細胞,使大多數受治療病人獲得持久的臨床益處;皮膚炎獲得改善或清除,感染與自體免疫表現的風險亦降低。³⁷,⁴⁰,⁴¹ 為避免病毒載體插入性致癌 (insertional oncogenesis) 的風險,正在進行的試驗使用自我去活化 (self-inactivating) 慢病毒載體,以及更近期作為矯正 WAS 細胞之非病毒方法的鋅指核酸酶 (zinc finger nucleases)。⁴²
嚴重複合型免疫缺乏 (Severe Combined Immunodeficiency)
重點一覽 (AT-A-GLANCE)
■ 一群異質性的 X 連鎖與體染色體隱性疾患,具有細胞介導性與體液性免疫缺乏。
■ 嬰兒早期生長遲滯;腹瀉;反覆性皮膚黏膜念珠菌病、細菌與病毒感染;移植物對抗宿主疾病 (graft-versus-host disease) 風險。
■ 造血幹細胞移植可能提供長期治癒。
臨床特徵
嚴重複合型免疫缺乏 (severe combined immunodeficiency, SCID) 的嬰兒,通常在反覆性鼻竇肺部與皮膚感染發作後,於 3 至 6 個月大時無法增加體重。⁴³,⁴⁴ 診斷時常有持續性皮膚黏膜念珠菌病,偶爾發生全身性念珠菌感染。SCID 病人也可能因病毒感染而有慢性腹瀉與吸收不良。肺囊蟲 (P. jiroveci [carinii]) 肺炎常為初發表現。雖然細菌感染通常對全身性抗生素有反應,病毒感染傾向致命。SCID 嬰兒儘管有反覆感染,仍缺乏可觸及的淋巴組織。除皮膚細菌與念珠菌感染外,最常見的皮膚疹為麻疹樣 (morbilliform) 或類似脂漏性皮膚炎。在某些 SCID 嬰兒中,切片切面顯示移植物對抗宿主疾病 (graft-versus-host disease, GVHD)(見第 129 章)。GVHD 可能源於子宮內暴露於母源淋巴球(通常非致命)、輸注未經照射的血液製品(通常致命),或可能繼發於幹細胞移植。GVHD 病人最常急性表現為麻疹樣紅斑、丘疹性皮膚炎或瀰漫性紅皮症 (erythroderma)。顏面、頸部、手掌與足底通常最先受累,之後疹子才變為全身性。嚴重病例會因瀰漫性水疱 (bullae) 或毒性表皮壞死溶解 (toxic epidermal necrolysis) 而複雜化。源於母源植入(無移植)之 GVHD 的臨床表現與源於移植之 GVHD 的臨床表現無法區分,但組織病理特徵不同。⁴⁵ 繼發於母源植入之 GVHD 的切片切面顯示乾癬樣增生 (psoriasiform hyperplasia),伴有角化不全 (parakeratosis) 與不等程度的海綿水腫 (spongiosis),與傳統 GVHD 所見的空泡性界面型態 (vacuolar interface pattern) 形成對比。整體而言,83% 有母源植入的 SCID 病人會發展出皮膚表現。SCID 也可見口腔與生殖器潰瘍,特別是在 Artemis 基因有缺陷的 Athabascan 語系美洲原住民兒童中。⁴⁶
患有 Omenn 症候群 (Omenn syndrome) 的病人呈現紅皮症、肝脾腫大與淋巴結腫大,伴有嗜伊紅性白血球增多與 IgE 產生增加。⁴⁷ B 細胞傾向無法偵測到,而 T 細胞雖然數量常增加,卻無功能。Omenn 症候群已被證實為具有數種潛在遺傳基礎的表現型。大多數病人有 RAG1/RAG2 突變,但 Omenn 症候群的表現型也曾在 Artemis 與 IL-7R 突變的病人中描述。兩名軟骨毛髮發育不全的個體與另一名完全型 DiGeorge 症候群的個體,也顯示 Omenn 症候群的特徵。隨著 SCID 與其他 T 細胞淋巴球減少症新生兒篩檢的引入,SCID 的自然病程正在改變。⁴⁸ 除了少數出生時可能不以 T 細胞淋巴球減少表現的 SCID 病因外,病人在出生後不久即由新生兒篩檢辨識出來,並在嚴重感染發生前確立診斷。早期幹細胞移植可提供極佳的結果,無論幹細胞來源或預處理方案為何。⁴⁹
外胚層失養症合併免疫缺乏 (Ectodermal Dysplasia with Immunodeficiency)
重點一覽 (AT-A-GLANCE)
■ 核因子 κB (nuclear factor κB, NF-κB) 必要調節因子 (essential modulator) 的突變,導致 NF-κB 訊息傳遞異常。
■ 無汗性外胚層失養症 (hypohidrotic ectodermal dysplasia) 的特徵性面容。
■ 常為嚴重的免疫缺乏;細菌、非典型分枝桿菌 (atypical mycobacterial) 與病毒感染常見。
■ 治療:幹細胞移植可矯正免疫缺乏,但無法矯正本病的其他特徵。
流行病學
X 連鎖隱性遺傳最常見,估計發生率為每 250,000 名活產男嬰中 1 例。⁵⁰
亦曾描述一種體染色體顯性型的外胚層失養症合併免疫缺乏。⁵¹
病因與發病機轉
數種遺傳疾患源於核因子 κB (NF-κB) 必要調節因子 (NEMO) 的基因突變,並導致 NF-κB 訊息傳遞異常(見第 75 與 131 章)。其中之一,無汗性外胚層失養症合併免疫缺乏,源於 NEMO 基因的弱效突變 (hypomorphic mutations)(亦稱為無汗性外胚層失養症合併免疫缺乏,或高免疫球蛋白 M、免疫缺乏、X 連鎖合併外胚層失養症)。NF-κB 是一種 DNA 結合轉錄因子,其轉錄基因的能力受一種細胞質抑制因子 IKB(NF-κB 抑制因子,inhibitor of nuclear factor κB)調控。NF-κB 訊息傳遞影響發炎、細胞凋亡、發育與免疫。NF-κB 活化需要 IKB 激酶 (IKB kinase, IKK) 對 IKB 進行磷酸化。IKK 由 2 個催化組件(IKKα 與 IKKβ)與一個調節次單元 IKKγ 或 NEMO 構成。NEMO 沒有催化功能,但是支撐 IKK 複合體的結構支架。當 NEMO 缺如或有缺陷時,無法形成具功能的 IKK 複合體,因此 NF-κB 無法轉位至細胞核並活化基因轉錄。導致無汗性外胚層失養症合併免疫缺乏的弱效突變允許男性早期存活,與導致女性帶因者出現色素失調症 (incontinentia pigmenti)、受影響男性胎兒死亡的 NEMO 大片段缺失(通常為第 4 至 10 外顯子)形成對比(見第 75 章)。弱效 NEMO 突變的女性帶因者常顯示色素失調症的輕微特徵,但也可能表現出伴隨暫時性免疫缺乏的色素失調症表現型。⁵² 在弱效 NEMO 突變的男孩中,外胚層失養症表現型源於 NF-κB 訊息傳遞受損。⁵³ 免疫缺乏源於 NF-κB 對經由抗原受體、類鐸受體 (Toll-like receptors)、IL-1 受體與腫瘤壞死因子 (tumor necrosis factor, TNF) 受體家族訊息傳遞之反應活化受損。⁵⁴ IKBα(NF-κB 抑制因子中被 IKK 磷酸化的組件)的功能增益突變 (gain-of-function mutations),會導致一種具有獨特免疫表現型的體染色體顯性型外胚層失養症,特徵為嚴重的 T 細胞缺乏,但 NK 細胞毒性與對分枝桿菌的反應正常。亦曾報告導致免疫缺乏但無外胚層失養症表現型的 NEMO 突變。
臨床特徵
受影響的病人呈現無汗性外胚層失養症的典型特徵(圖 132-9)。這些包括特徵性面容、毛髮稀少 (hypotrichosis) 或無毛 (atrichia)、少汗 (hypohidrosis)(導致耐熱不良 heat intolerance)、缺牙 (hypodontia) 或無牙 (anodontia) 伴錐狀門齒,以及相關的皮膚炎(見第 131 章)。一部分病人伴有骨硬化症 (osteopetrosis) 與淋巴水腫 (lymphedema),並伴隨嚴重免疫缺乏。亦曾描述源於 NEMO 突變但無無汗性外胚層失養症特徵的免疫缺乏男孩。在一項對 72 名 NEMO 突變病人的分析中,僅 77% 有外胚層失養症。⁵⁰ 嬰兒期早期的細菌感染常見,特別是敗血症、肺炎、中耳炎、鼻竇炎與淋巴結炎。反覆性肺炎可能導致支氣管擴張。常見病原體包括肺炎鏈球菌 (Streptococcus pneumoniae)、流感嗜血桿菌 (H. influenzae)、克雷伯氏菌 (Klebsiella)、沙門氏菌 (Salmonella) 與假單胞菌 (Pseudomonas)。亦曾報告非典型分枝桿菌與病毒感染,包括巨細胞病毒 (cytomegalovirus)(全身性與胃腸道侵犯)、單純疱疹病毒 (herpes simplex virus)、傳染性軟疣、人類乳突病毒 (human papillomavirus) 與肺囊蟲 (Pneumocystis carinii)。免疫缺乏常為嚴重,但其特性多變,具有明顯的基因型-表現型相關性,可進一步透過突變的體外重建 (in vitro reconstitution) 加以鑑定。免疫功能障礙包括 B 細胞 Ig 類別轉換 (class switching) 受損伴低γ球蛋白血症(且常伴 IgM 與/或 IgA 濃度增加)、特異性抗體產生受損(特別是對多醣抗原)、NK 細胞毒性缺陷、對 CD40 訊息傳遞反應之細胞激素產生不良,以及自體發炎 (autoinflammation),尤其是腸道的自體發炎。皮膚切片可能顯示角質細胞凋亡 (keratinocyte apoptosis) 的證據,反映 NF-κB 訊息傳遞的功能障礙,且必須與 GVHD 區分。
治療與預後
死亡率增加,36% 的病人於平均 6.4 歲死亡。⁵⁰ 治療依病人的臨床與免疫表現型而定。外胚層失養症採支持性治療(見第 131 章)。抗體產生受損的病人可能受益於 Ig 替代。所有感染(細菌與病毒)都應以適當的抗生素/抗病毒藥物積極治療。應考慮針對肺囊蟲 (P. jiroveci) 與鳥型分枝桿菌複合體 (Mycobacterium avium-intracellulare) 進行預防性投藥。幹細胞移植可能導致免疫重建,但可能無法矯正本病的其他表現。⁵⁵ 應為這些病人的母親與姊妹提供 NEMO 突變的基因檢測與諮詢。
運動失調-毛細血管擴張症 (Ataxia-Telangiectasia)
重點一覽 (AT-A-GLANCE)
■ 體染色體隱性疾患,具有運動失調-毛細血管擴張症突變基因 (ataxia-telangiectasia mutated, ATM) 突變。
■ 運動失調早發伴進行性神經退化;大多數病人在學齡前出現結膜毛細血管擴張 (conjunctival telangiectasia)。
■ 鼻竇肺部感染、淋巴網狀腫瘤 (lymphoreticular neoplasia)。
■ IgA、IgE、IgG2、IgG4 缺乏;T 細胞缺乏的多變表現;高濃度 α 胎兒蛋白 (α-fetoprotein);染色體斷裂;對放射線敏感。
流行病學
運動失調-毛細血管擴張症 (Ataxia-telangiectasia, AT;線上人類孟德爾遺傳 [Online Mendelian Inheritance in Man, OMIM] #208900),又稱 Louis-Bar 症候群,是一種體染色體隱性疾患,估計發生率可達 1:40,000,帶因率可達 1%。帶因者罹患乳癌、血液惡性腫瘤⁵⁶,⁵⁷ 與缺血性心臟病的風險增加,預期壽命約縮短 8 年。⁵⁸ 這些雜合子在體外暴露於放射線後染色體斷裂的風險增加,顯示已知 AT 帶因者的乳房 X 光攝影 (mammograms) 為禁忌。
臨床特徵
特徵性的眼皮膚毛細血管擴張 (oculocutaneous telangiectasias) 始於眼眥 (ocular canthi) 附近,並橫越球結膜 (bulbar conjunctivae) 進展(圖 132-10)。這些毛細血管擴張通常在病人 3 至 6 歲時出現;很少在更早年齡被描述。皮膚毛細血管擴張隨後可能發展於顴骨隆突、耳朵、眼瞼、前胸、膕窩與肘前窩,以及手足背側(圖 132-11)。毛細血管擴張可能不明顯且類似細小瘀點,尤其在屈側區域。毛細血管擴張的發展可能與陽光暴露有關,因為在受影響的深膚色兒童中會發展出眼部(但非皮膚)毛細血管擴張。毛細血管擴張的發展可能至少部分與某些 AT 株系對紫外線的敏感性有關。⁵⁹
皮膚的早老性 (progeric) 變化,包括乾燥症 (xerosis) 與灰髮,發生於 90% 的病人。⁶⁰ 在青春期,顏面皮膚可能逐漸變得更萎縮與硬化,造成面具樣 (mask-like) 外觀。偶爾,耳朵、手臂與手部也會變得硬皮病樣 (sclerodermatous)。毛髮可能在青春期前瀰漫性變灰,且皮下脂肪通常在兒童期流失。反覆性嚴重膿痂疹常發展。許多病人發生脂漏性皮膚炎,相關的眼瞼炎 (blepharitis) 可能導致被診斷為眼瞼結膜炎 (blepharoconjunctivitis) 而非眼部毛細血管擴張。斑駁的過度色素沉著與色素減退常見,與毛細血管擴張及萎縮一起,可類似放射性皮膚炎、光損傷或硬皮病的異色症 (poikiloderma)。其他色素變化包括可能呈皮節分布 (dermatomal distribution) 的咖啡牛奶斑 (café-au-lait spots)、多發性雀斑 (ephelides) 與白斑。手臂與腿部多毛症 (hypertrichosis)、圓禿、多發性疣、異位性皮膚炎、毛囊角化症 (keratosis pilaris)、錢幣狀濕疹 (nummular eczema) 與黑色棘皮症 (acanthosis nigricans) 也曾被描述與 AT 相關。最常見的皮膚表現之一為非感染性皮膚肉芽腫(圖 132-12)。⁶¹ 這些持續性、萎縮性且常潰瘍的病灶,常被誤認為其他肉芽腫性病程,包括類肉瘤病 (sarcoidosis)、脂質類壞死 (necrobiosis lipoidica diabeticorum)、環狀肉芽腫 (granuloma annulare) 與肉芽腫性皮膚炎 (granulomatous dermatitis)。
進行性小腦運動失調 (cerebellar ataxia) 通常在嬰兒期首次顯現(中位年齡:1.2 歲),表現為頭部與軀幹搖晃及眼球運動失用症 (apraxia of eye movements),常比皮膚或結膜異常發展早數年。在兒童期,構音障礙語言 (dysarthric speech)、流涎、舞蹈手足徐動症 (choreoathetosis) 與肌陣攣性抽動 (myoclonic jerks) 變得明顯。AT 的診斷通常在皮膚黏膜毛細血管擴張出現後,於中位年齡 7 歲時作出。病人通常在青少年期前即需要輪椅。多達 80% 的病人發生反覆性細菌與病毒性鼻竇肺部感染;這些是最常見的死因,通常源於支氣管擴張與呼吸衰竭。約 75% 的 AT 病人可能有生長遲滯與內分泌疾患,尤其是卵巢發育不全 (ovarian agenesis) 或睾丸發育不全 (testicular hypoplasia) 與胰島素阻抗型糖尿病。腫瘤發生於 40% 存活至青少年或年輕成人的病人,雖然淋巴惡性腫瘤也曾被描述為嬰兒期的初發徵象。最常見者為淋巴瘤(尤其是 B 細胞淋巴瘤;風險增加 200 倍)與白血病(尤其是 T 細胞慢性淋巴球性白血病;風險增加 70 倍)。年輕成人曾報告基底細胞癌。AT 病人傾向同時具有體液性與細胞性免疫異常。血清 IgA 與 IgE 分別在 70% 與多達 80% 的病人中缺如或缺乏。在有 IgA 缺乏的 AT 病人中,循環性抗 IgA 抗體常見。約 60% 的病人有選擇性 IgG2 與 IgG4 缺乏。70% 的病人發現細胞介導性免疫缺陷,特別是淋巴球減少與對抗原及有絲分裂原 (mitogens) 的體外反應缺陷;帶有 γ/δ 受體的 T 細胞數量增加,而 CD4+ T 細胞傾向減少。幾乎所有病人都有 α 胎兒蛋白濃度升高(在 2 歲後尤具意義),許多病人有可偵測到的癌胚抗原 (carcinoembryonic antigen)。AT 病人常有肝臟轉胺酶升高(40% 至 50%)與葡萄糖耐受不良。DNA 對 X 射線極為敏感,病人也顯示端粒縮短率增加。自發性染色體異常(碎片、斷裂、間隙與易位)在 AT 病人中的發生頻率比正常個體高 2 至 18 倍,主要涉及第 2、7、14 號染色體。第 7 與 14 號染色體的重排,尤其是 14:14 易位,似乎能預測淋巴網狀惡性腫瘤(包括白血病)的發展。胸腺缺如或發育不全,脾臟可能縮小。確認診斷的技術包括:8 個月以上兒童的血清 α 胎兒蛋白濃度升高、抗放射線 DNA 合成 (radioresistant DNA synthesis) 分析(顯示異常的 S 期檢查點)、以菌落存活分析 (colony survival assay) 進行放射敏感性檢測、ATM(運動失調-毛細血管擴張症突變)蛋白的免疫墨點法 (immunoblotting)、ATM 激酶活性評估,以及分子遺傳檢測。
預後、臨床病程與治療
AT 的治療為支持性,包括投予抗生素治療感染、針對肺部支氣管擴張的物理治療、預防神經功能障礙病人攣縮的物理治療,以及防曬與避免日曬以減少類光損傷變化。應積極篩檢病人的惡性腫瘤,尤其是在第一個十年之後。triamcinolone 病灶內注射有助於促進疼痛性相關潰瘍的癒合,雖然病灶經治療後不會完全清除。屍檢結果顯示約 50% 的病人死於肺部疾病,此為最常見的死因。淋巴網狀惡性腫瘤(包括淋巴球性白血病)是第二常見的死因(佔 AT 病人的 15%)。其餘病人傾向同時死於肺部疾病與惡性腫瘤。治療性放射線與類放射性化療藥物(尤其是 bleomycin)可能導致廣泛的組織壞死。投予小劑量的其他化療藥物與低劑量、分次放射線是處理這些惡性腫瘤傷害最小的方法。在一小群較輕度 AT 的病人中,以胺基醣苷類 (aminoglycosides) 治療可增加 ATM 基因功能。⁶² 死亡通常發生於兒童晚期或青春期早期;存活最久的病人於 50 歲死亡。目前產前診斷最好透過 DNA 分析達成。
吞噬作用與細胞殺傷障礙 (DISORDERS OF PHAGOCYTOSIS AND CELL KILLING)
吞噬細胞功能或細胞殺傷有缺陷的病人,典型於嬰兒期或兒童期出現反覆性、不尋常與/或難以清除的細菌感染。常見的感染包括皮膚或黏膜、肺、淋巴結、深部組織膿瘍,或兒童期牙周炎 (periodontitis) 的感染。吞噬作用與細胞殺傷最具特徵性的疾患為慢性肉芽腫病 (chronic granulomatous disease, CGD)、白血球黏附缺乏症 (leukocyte adhesion deficiencies)、高免疫球蛋白 E 症候群 (Hyperimmunoglobulinemia E syndrome, HIES),以及伴免疫缺乏的銀髮症候群 (silvery hair syndromes)。⁶³ 然而,數種伴有嗜中性白血球減少 (neutropenia)、嗜中性白血球功能障礙或細胞激素功能障礙的疾患,也會導致吞噬與/或殺傷微生物的能力下降。這些疾患回顧於表 132-5。
慢性肉芽腫病 (Chronic Granulomatous Disease)
重點一覽 (AT-A-GLANCE)
■ 一群疾患,其中活性氧中間產物 (reactive oxygen intermediates) 產生缺陷,損害對微生物的細胞內殺傷。
■ X 連鎖或體染色體隱性。
■ 編碼菸鹼醯胺腺嘌呤二核苷酸磷酸氧化酶系統 (nicotinamide adenine dinucleotide phosphate oxidase system) 組件之基因的突變。
■ 肺炎、淋巴結腫大、肝脾腫大與皮膚感染。
■ 肉芽腫,最常見於肺與肝。
■ 帶因者(X 連鎖)與病人的狼瘡樣發炎。
CGD 是一群異質性的 X 連鎖與體染色體隱性疾患。⁶⁴,⁶⁵ 90% 的本病病人為男性,整體發生率為每 200,000 至 250,000 人中 1 例。伴有 gp91phox 缺乏的 X 連鎖疾病發生於 70% 的受影響個體,徵象與症狀於生命第一年顯現。患有體染色體隱性疾病的個體較可能保有部分菸鹼醯胺腺嘌呤二核苷酸磷酸 (nicotinamide adenine dinucleotide phosphate, NADPH) 氧化酶活性,傾向較晚發病(平均發病年齡:8 歲)、有較輕微的徵象與症狀,且存活較長。⁶⁶,⁶⁷
臨床特徵
伴有區域性淋巴結腫大/淋巴結炎與皮膚炎的膿皮症 (pyodermas),傾向影響暴露於微生物的身體部位,特別是皮膚與肺。葡萄球菌膿瘍見於 40% 的病人,特別是肛門周圍、鼻孔與耳朵。化膿性發炎反應可能發生於淋巴結引流部位或輕微皮膚創傷處,並緩慢癒合伴瘢痕。壞疽性深膿瘡 (Ecthyma gangrenosum) 可能是新生兒的初發表現。⁶⁸ 葡萄球菌感染最常見,但其他經活性氧物種 (reactive oxygen species) 生成而被殺死的細菌與真菌微生物也可能引起感染(麴菌屬 Aspergillus spp.、洋蔥伯克氏菌 Burkholderia cepacia、念珠菌屬 Candida spp.、克雷伯氏菌屬 Klebsiella spp.、分枝桿菌〔包括嚴重/瀰漫性卡介苗感染〕⁶⁹、諾卡氏菌屬 Nocardia spp.,以及黏質沙雷氏菌 Serratia marcescens〔尤其是伴潰瘍與骨髓炎的膿瘍〕)。⁷⁰
CGD 病人常發展出慢性發炎性肉芽腫,最常見於肺與肝。皮膚肉芽腫呈結節狀且常壞死。肉芽腫可能阻塞重要結構,尤其是胃腸道與泌尿生殖系統。許多病人也曾被描述出現類似口瘡性口炎 (aphthous stomatitis) 的口內潰瘍、慢性牙齦炎、口周潰瘍、頭皮毛囊炎與脂漏性皮膚炎。X 連鎖 CGD 的女性帶因者沒有感染風險增加,但可能有圓盤狀或全身性紅斑性狼瘡的皮膚病灶、Jessner 淋巴球皮膚浸潤 (Jessner lymphocytic infiltration)、口瘡性口炎、肉芽腫性唇炎 (granulomatous cheilitis)、光敏感性,與/或雷諾現象 (Raynaud phenomenon)。⁷¹,⁷²
淋巴結、肺、肝、脾與胃腸道是最常見的非皮膚侵犯部位。伴有膿瘍與廔管形成的化膿性淋巴結炎通常影響頸部淋巴結。肺炎發生於幾乎所有受影響的兒童,並可能導致膿瘍形成、空洞化 (cavitation) 與膿胸 (empyema)。肝脾腫大已在 80% 至 90% 的病人中報告;超過 30% 的病人發展出肝膿瘍,而葡萄球菌肝膿瘍幾乎是 CGD 的特異性表現。其他過度發炎反應可包括噬血細胞性淋巴組織球增生症 (hemophagocytic lymphohistiocytosis)⁷³ 或類似發炎性腸道疾病的特徵(肛門廔管、腹瀉)、IgA 腎病變、類肉瘤病與類風濕性關節炎。⁷⁴,⁷⁵
患有細菌與諾卡氏菌屬感染的病人傾向有症狀,且常有白血球增多 (leukocytosis)、貧血與血沉升高。相對地,無發燒、紅血球沉降率正常與少有症狀,在麴菌屬感染中較常見。因此,實驗室數值在正常範圍內並不能排除 CGD 病人的感染。病人顯示紅血球沉降率增加、高γ球蛋白血症、白血球增多與輕度貧血;其他免疫功能檢查通常正常。
病因與發病機轉
CGD 由還原型 NADPH 氧化酶(負責生成超氧化物 superoxide 的酵素複合體)缺陷引起。吞噬作用後的正常殺菌活性需要 NADPH 氧化酶系統,該系統由 NADPH、一種不尋常的吞噬細胞細胞色素 b (cytochrome b, b558) 與細胞質蛋白構成。在 CGD 病人中,此膜結合 NADPH 氧化酶系統無法產生超氧化物與其他毒性氧代謝產物。這些氧化分子具有直接的殺微生物活性,但也作為細胞內訊息分子,活化吞噬泡 (phagocytic vacuole) 內初級顆粒蛋白嗜中性白血球彈性蛋白酶 (neutrophil elastase) 與組織蛋白酶 G (cathepsin G) 的釋放,而這些蛋白依次是殺死微生物所必需。CGD 的小鼠模型顯示調節性 T 細胞活性降低、γ/δ T 細胞活性不受抑制,以及 IL-1β、IL-8 與 IL-17 產生增加。⁷⁶ NADPH 氧化酶也調控 B 細胞的主要組織相容性複合體第 II 類抗原呈現。⁷⁷
在 X 連鎖家族中(約 70%),編碼細胞色素 b558 之 gp91phox(吞噬細胞氧化酶 phagocyte oxidase)次單元的 CYBB 基因發生突變。患有體染色體隱性 CGD 的病人缺乏 NADPH 氧化酶的細胞質因子(p47phox——由 NCF1 編碼——約佔 20% 的病人;p67phox——由 NCF2 編碼——≤5%;以及 p40phox——由 NCF4 編碼——非常罕見);偶爾,含有 p47phox 對接位點的 p22phox(細胞色素 b558 的 γ 次單元)缺乏(CYBA 突變;≤5% 的病人)。一般認為細胞色素 b558 是這些細胞質因子的膜附著位點,這些因子從細胞質轉位至質膜,組裝氧化酶組件以允許 NADPH 氧化酶的活化。在 CGD 病人中引起感染的微生物類型需要細胞內殺傷,且通常為觸酶陽性 (catalase-positive)。在北美與歐洲,僅 5 種微生物負責 CGD 絕大多數的感染:金黃色葡萄球菌 (Staphylococcus aureus)、黏質沙雷氏菌 (S. marcescens)、洋蔥伯克氏菌 (B. cepacia)、諾卡氏菌屬 (Nocardia spp.) 與麴菌屬 (Aspergillus spp.)。CGD 強烈的體液性與肉芽腫性反應,被認為是在對微生物強力但無效的反應中的代償性反應。
診斷
CGD 的診斷依據為仰賴超氧化物產生的分析。目前較受青睞的是基於二氫羅丹明流式細胞儀的檢測 (dihydrorhodamine flow cytometry-based test, DHR 123 assay),可輕易辨識 X 連鎖疾病的病人或帶因者⁷⁸;亞鐵細胞色素 c 還原分析 (ferricytochrome c reduction assay) 是另一種呼吸爆發 (respiratory burst) 的定量測量。CGD 的篩檢試驗為四唑氮藍 (nitroblue tetrazolium, NBT) 還原分析,其中黃色 NBT 在正常氧化代謝作用下沉澱時,會變成不溶性的氧化型(藍色甲䐀 formazan)。也可進行定量 NBT 試驗與化學發光分析 (chemiluminescence assays)。免疫墨點分析確認醣蛋白 91phox (gp91phox) 組件的缺如;由於細胞色素 b558 一個組件的缺乏會導致另一組件的缺如,若免疫墨點分析發現缺如,則需要進行 gp91phox 或 p22phox 基因的定序。顯示 p47phox 或 p67phox 缺如的免疫墨點可具診斷性。皮膚肉芽腫的切片顯示組織球浸潤,伴有異物巨細胞 (foreign-body giant cells) 與嗜中性白血球聚積伴壞死。雖然 CGD 病人與帶因者紅斑性狼瘡樣皮膚病灶的組織病理特徵可能類似狼瘡病人,但病灶皮膚的免疫螢光檢查通常為陰性。
鑑別診斷
雖然測量呼吸爆發的試驗是簡便的篩檢試驗,其他吞噬作用障礙可能與 CGD 混淆。這些包括白血球黏附缺陷(見「白血球黏附缺乏症」一節)、Shwachman-Diamond 症候群、髓過氧化酶缺乏 (myeloperoxidase deficiency),以及類鐸受體、干擾素或 IL-12 訊息傳遞的缺陷。
臨床病程、預後與治療
患有 X 連鎖 CGD、p22phox CGD 與 p67phox CGD 的病人,相較於 p47phox CGD 病人,傾向有較嚴重的臨床病程。X 連鎖 CGD 的平均診斷年齡為 3 歲,而體染色體隱性型為 8 歲。超過 90% 的非 p47phox CGD 病人有無法偵測到的超氧化物產生濃度。某些病人早在嬰兒期即發展出嚴重感染,而其他病人則在兒童期後期出乎意料地發展出典型 CGD 的嚴重感染。局部發炎的小病灶可能不伴發燒,且若未透過例行篩檢 X 光、掃描或超音波對肺、肝與骨骼進行積極調查,可能難以偵測。應進行培養以鑑定感染源,且可能需要侵入性程序以取得足夠的組織樣本。有感染證據的病人應以涵蓋金黃色葡萄球菌與革蘭氏陰性微生物的廣效非經腸道抗生素經驗性治療。⁷⁹ 靜脈治療應持續至少 10 至 14 天,隨後接續數週的口服抗生素療程。較深部的感染可能需要外科介入(引流、清創)(表 132-6)。Trimethoprim-sulfamethoxazole 治療可降低細菌感染的發生率,而不增加真菌感染的發生率。Itraconazole 是預防真菌感染的有效藥劑。⁸⁰ 預防性干擾素 (interferon, IFN)-γ 可降低 X 連鎖與體染色體隱性 CGD 感染的數量與嚴重度,而不增加慢性發炎併發症的發生率。⁸¹ 使用 IFN-γ 並不伴隨任何可測量的 NADPH 氧化酶活性改善;其臨床益處與透過非氧化機制增強的吞噬細胞功能與殺傷有關。顆粒球輸注 (Granulocyte transfusions) 曾用於快速進展、危及生命的感染。預防性投予抗生素與 IFN-γ 已將 CGD 的死亡率降至體染色體 CGD 約每病人年 2%,X 連鎖 CGD 每年約 5%。⁶⁵ 最常見的死因為由麴菌或洋蔥伯克氏菌引起的肺炎與/或敗血症。全身性糖皮質類固醇對有阻塞性內臟肉芽腫的病人有幫助。
對發炎表現可能有益的其他療法包括 anakinra、azathioprine、hydroxychloroquine、pioglitazone⁸²、sirolimus 與 thalidomide⁸³;TNF 抑制劑可改善結腸炎,但增加感染風險。幹細胞移植可能治癒 CGD,其使用已增加。⁸⁴ 移植時無感染的年輕病人預後最佳(存活率 >95%),但對於較年長個體(包括成人)與有難治性感染或發炎的病人,曾使用降低強度的調理方案 (reduced-intensity conditioning regimens)⁸⁵,且若在不可逆器官損傷發生前出現反覆嚴重感染或類固醇依賴性發炎疾病,應加以考慮。⁸⁶
基因治療已用於 p47phox 缺乏型與 X 連鎖型的 CGD。然而,反轉錄病毒的使用可能導致插入性致癌與骨髓增生異常 (myelodysplasia),因此需要更安全的方法,如自我去活化慢病毒載體或非病毒技術。⁸⁶⁻⁹⁰
白血球黏附缺乏症 (Leukocyte Adhesion Deficiencies)
重點一覽 (AT-A-GLANCE)
■ 一群 4 種疾患:3 種體染色體隱性(ITGB2、SLC35C1 或 FERMT3 突變)與 1 種體染色體顯性(Rac2 突變)。
■ 牙齦炎與牙周炎。
■ 傷口癒合不良;臍帶殘端分離延遲,以及受傷後發展出壞疽性膿皮症樣壞死性潰瘍 (pyoderma gangrenosum-like necrotic ulcerations)。
■ 危及生命的細菌與真菌感染。
高免疫球蛋白 E 症候群 (Hyperimmunoglobulinemia E Syndrome)
重點一覽 (AT-A-GLANCE)
■ 同義詞:Job 症候群 (Job syndrome)、Buckley 症候群 (Buckley syndrome)、高免疫球蛋白 E 反覆感染症候群。
■ 大多數病例為體染色體顯性,但也曾描述具有某些不同特徵的隱性型。
■ 典型三聯徵:(a) 反覆性葡萄球菌皮膚膿瘍、(b) 伴有肺氣囊腫 (pneumatocele) 形成的肺炎,以及 (c) 高血清 IgE 濃度。
■ 異位性皮膚炎是常見的表現。
■ 脊柱側彎 (scoliosis)、骨折與牙齒異常是較盛行的體染色體顯性型所獨有的特徵。
■ 體染色體隱性病例有嚴重病毒感染,並可發展出嚴重神經併發症。
HIES 罕見(發生率為 10⁶ 分之 1)。⁹¹ 男女比例相當。大多數病例為散發性或符合體染色體顯性遺傳。也曾描述體染色體隱性遺傳,但病人顯示不同的相關特徵。
臨床特徵
個別 HIES 病人的臨床表現可能差異甚大。⁹¹⁻⁹³ 表 132-7 摘要了散發型或體染色體顯性 HIES 病人最常見的臨床特徵與實驗室數值。HIES 的新生兒或嬰兒期皮疹常為丘疹膿疱性疹 (papulopustular eruption),伴有明顯結痂,分布於頭皮、顏面、頸部、腋窩與尿布區。⁹⁴,⁹⁵ 皮膚切片最一致的發現為嗜伊紅性海綿水腫性皮膚炎 (eosinophilic spongiotic dermatitis),有時以真皮毛囊為中心。⁹⁴ 皮膚、黏膜與指甲的早發性念珠菌感染與/或嬰兒期異位性皮膚炎是其他表現;在有嗜伊紅性丘疹膿疱的病人中,異位性皮膚炎常在嬰兒期後期才出現。皮膚炎合併金黃色葡萄球菌的二次感染非常常見⁹⁵,病人顯示高濃度的抗葡萄球菌 IgE 抗體。葡萄球菌皮膚感染包括膿痂疹、癤病、甲溝炎、蜂窩性組織炎,以及特徵性的「冷」膿瘍 (cold abscesses)(圖 132-13),其不顯現預期程度的紅斑、溫熱與化膿。膿瘍最常發生於頭頸部與間擦區域。也可能發展出反覆性鏈球菌膿皮症。肺部細菌性肺炎、膿瘍與膿胸是最常見的全身性感染,並可能導致肺氣囊腫,成為細菌(常為綠膿桿菌 Pseudomonas aeruginosa)或真菌(尤其是麴菌 Aspergillus)感染的病灶溫床。最常見的感染微生物為金黃色葡萄球菌與流感嗜血桿菌。除肺炎外,深部感染、菌血症與敗血症罕見。⁹⁶
顏面與骨骼異常常見。病人發展出進行性的面部特徵粗糙化(見圖 132-13),可能反映骨骼缺陷,但反覆的面部膿疱與皮膚炎引起的苔癬化可能也有貢獻。獨特的面部特徵,包括突出的前額、寬鼻樑與寬鼻尖,在 16 歲前普遍存在。牙齒異常包括乳牙滯留與恆牙未萌發。⁹⁷ 骨質減少 (Osteopenia) 常見,57% 的病人曾有至少 3 次病理性骨折,尤其是長骨、肋骨與骨盆。⁹⁸,⁹⁹ 脊柱側彎發生於 63% 的成年病人,關節過度伸展 (hyperextensibility) 發生於 68% 的病人。⁹⁶ 較輕的表現型可能反映鑲嵌現象 (mosaicism)。¹⁰⁰
體染色體隱性疾病與體染色體隱性 HIES 共有高血清 IgE 濃度、周邊嗜伊紅性白血球增多、慢性濕疹性皮膚炎,以及反覆性皮膚(包括冷膿瘍)與呼吸道葡萄球菌感染。然而,體染色體隱性 HIES 病人沒有形成肺氣囊腫的傾向,也沒有骨骼或牙齒異常。¹⁰¹ 取而代之的是,體染色體隱性 HIES 的特徵為反覆性病毒感染(如傳染性軟疣、疣、單純疱疹與水痘-帶狀疱疹)、伺機性感染、自體免疫,以及常於兒童期致命的毀滅性神經併發症(尤其是血管炎)(圖 132-14)。¹⁰²⁻¹⁰⁷ 在體染色體隱性 HIES 病人中,多達 24% 可能有新生兒期的丘疹膿疱性疹,且食物過敏/氣喘比體染色體顯性 HIES 更常見。體染色體隱性 HIES 病人罹患皮膚黏膜鱗狀細胞癌與淋巴瘤的風險增加。
病因與發病機轉
HIES 已鑑定出三種不同的基因缺陷。最常見者為訊息傳遞與轉錄活化因子 3 (signal transducer and activator of transcription 3, STAT3) 的顯性負向突變 (dominant negative mutation)¹⁰⁸,¹⁰⁹,並與 Th17 細胞的嚴重減少相關。¹¹⁰ STAT3 在 IL-6 的下游進行訊息傳遞,而 IL-6 與轉化生長因子-β (transforming growth factor–β) 一起,對 Th17 細胞的發育很重要。Th17 細胞缺乏解釋了對念珠菌感染易感性增加的原因,且由於 Th17 活化是關鍵,也解釋了透過抗微生物胜肽 (antimicrobial peptides) 發展而對上皮表面之先天免疫的重要性。細胞激素訊息傳遞減少可能解釋了「冷」膿瘍,以及因其他促發炎細胞激素表現增加而導致的破壞性發炎。HIES 的骨質減少被認為源於 STAT3 依賴性的破骨細胞 (osteoclast) 分化下調的喪失。體染色體隱性 HIES 最常源於胞質分裂作用 8 調節因子 (dedicator of cytokinesis 8, DOCK8) 的雙等位基因突變,該基因編碼一種與肌動蛋白細胞骨架調控相關的蛋白,而此調控對 T 細胞擴增與抗體反應至關重要。¹⁰²,¹⁰³
TYK2 突變曾在一名 HIES 病人中發現¹¹¹,雖然其他 TYK2 缺乏的病人有增加的病毒與分枝桿菌感染,但無 HIES,顯示 HIES 可能不是其重要特徵。磷酸葡萄糖變位酶 3 (phosphoglucomutase 3,一種醣基化酵素) 的雙等位基因突變,也被發現是一種類 HIES 體染色體隱性疾患的基礎,伴有 IgE 濃度升高、異位性 (atopy)、自體免疫與神經認知異常。¹¹²⁻¹¹⁴
診斷
根據定義,血清 IgE 濃度必須升高,但正常 IgE 濃度在兒童期會顯著上升。體染色體顯性 HIES 的診斷標準已建立,包括 IgE 濃度大於 1000 IU/mL 與 5 項臨床特徵的加權分數(典型新生兒皮疹、反覆性肺炎、病理性骨折、特徵性面容與高顎弓)¹¹⁵;這些特徵與 Th17 細胞缺乏可導致「可能」的診斷,但合併雜合性 STAT3 突變則為確定診斷。然而,應認識到正常 IgE 濃度是年齡依賴性的。青少年與成人可見大於 2000 IU/mL 的濃度;嬰兒期則見到相當低的濃度,因為 1 歲以下嬰兒正常上限為 12.7 IU/mL,5 歲時為 47.1 IU/mL;診斷需要 IgE 濃度為該年齡第 95 百分位的 10 倍以上。嗜伊紅性白血球增多至少高於正常值 2 個標準差(通常高於 700 cells/µL)也是一種臨床表現。總白血球計數通常正常,且在急性感染情況下常無法升高。在體染色體隱性型中,嗜伊紅性白血球增多傾向更嚴重(如 17,500/µL)。¹¹⁶ 這些病人有 T 細胞活化的整體缺陷(CD4>CD8 細胞),這可能解釋了他們除細菌與真菌感染外,對病毒感染(包括疱疹與軟疣)的易感性。與體染色體顯性 HIES 相反,體染色體隱性 HIES 與 Th2 細胞偏向 (Th2 cell skewing) 相關。DOCK8 表現的缺乏可透過流式細胞儀偵測。¹¹⁷
鑑別診斷
由於高 IgE 濃度、嗜伊紅性白血球增多、皮膚炎與反覆性葡萄球菌感染,HIES 常在患有異位性皮膚炎或 WAS 的年幼兒科病人中被考慮。體染色體顯性 HIES 的粗糙面部特徵、骨質減少、反覆性肺炎與冷膿瘍有助於區分它;血小板異常的存在也有助於區分 WAS。在一項對 70 名血清 IgE 濃度高於 2000 IU/mL 的兒科病人的研究中,54 名(77%)有異位性皮膚炎,僅 6 名(8%)有 HIES;IgE 濃度與 HIES 的診斷無相關性。¹¹⁸ 其他免疫缺乏症候群可能與 HIES 混淆(如 Omenn、DiGeorge、IPEX 與 IRAK-4〔IL-1R 相關激酶 4,IL-1R–associated kinase 4〕缺乏),以及 Netherton 症候群與 GVHD。早發性念珠菌感染可能導致考慮皮膚黏膜念珠菌病(而在鑲嵌型 STAT3 突變中,念珠菌感染可能存在而無可偵測到的 Th17 細胞缺乏)。¹⁰⁰
臨床病程、預後與治療
由於血清 IgE 濃度、嗜伊紅性白血球增多與對嚴重感染易感性之間缺乏相關性,臨床病程難以預測。體染色體隱性 HIES 病人因神經併發症而有較嚴重的病程。抗葡萄球菌抗生素對 HIES 病人大多數皮膚感染有效,口服三唑類 (triazole) 抗真菌劑可治療皮膚黏膜念珠菌病。預防性使用抗葡萄球菌抗生素可顯著降低皮膚膿瘍與肺炎的發生率。皮膚與肺部膿瘍常需要切開引流,並可能需要部分肺切除。靜脈注射免疫球蛋白治療已成功使用。靜脈注射免疫球蛋白可能因 Ig 分解代謝增加或 IgE 中和而影響 IgE 濃度。抗壞血酸 (Ascorbic acid) 與 cimetidine 在某些病人中降低了感染數量與趨化缺陷。Isotretinoin 曾被報告在一名孤立病人中消除反覆性葡萄球菌膿瘍,而免疫狀態無任何改變。Cyclosporine 也曾使用並有良好的臨床與實驗室反應。IFN-γ 對 IgE 濃度與感染易感性顯示不一致的效果。Alendronate 已用於骨質減少。⁹⁹ 有零星報告指出,使用 omalizumab(一種針對 IgE 的單株抗體)可改善 HIES 的濕疹性皮膚炎。¹¹⁹ IFN-α 已有效治療 DOCK8 缺乏型 HIES 的多發性疣¹²⁰ 與嚴重疱疹感染。¹²¹,¹²² 幹細胞移植一般保留給 DOCK8 缺乏型 HIES¹²³⁻¹²⁵,但在 STAT3 缺乏型 HIES 的嚴重病例中也曾成功。¹²⁶
WHIM 症候群 (WHIM Syndrome)
WHIM(疣、低γ球蛋白血症、感染與骨髓滯留,warts, hypogammaglobulinemia, infections, and myelokathexis)症候群是一種免疫缺乏,特徵為嗜中性白血球減少、低γ球蛋白血症,以及對人類乳突病毒感染的易感性。本疾患源於趨化激素受體基因 (chemokine receptor gene) CXCR4 的缺陷¹²⁷,¹²⁸,因此成熟的嗜中性白血球無法離開骨髓(骨髓滯留 myelokathexis)。對人類乳突病毒感染的易感性顯示 CXCR4 在對此病毒的免疫中扮演重要角色。Plerixafor(一種 CXCR4 拮抗劑)可增加循環中的白血球、減少感染,且與 imiquimod 合併使用時,可顯著減少先前對治療有抗藥性的相關疣。¹²⁹
銀髮症候群:Chédiak-Higashi 與 Griscelli 症候群 (Silvery Hair Syndromes: Chédiak-Higashi and Griscelli Syndromes)
銀髮症候群包含一群體染色體隱性疾患,其中可見毛髮(且常包括皮膚)有一種特殊的金屬光澤(另見第 75 章)。其中,Chédiak-Higashi 與 Griscelli(第 2 型)症候群伴有相關的免疫缺乏(表 132-8)。另一種伴有銀髮的症候群——Elejalde 症候群——不伴免疫缺乏,且可能與 Griscelli 症候群 (Griscelli syndrome, GS) 之肌凝蛋白 5a 缺乏型 (myosin 5a-deficient form) 為等位基因,伴有嚴重的相關中樞神經系統功能障礙。
Chédiak-Higashi 症候群 (Chédiak-Higashi Syndrome)
重點一覽 (AT-A-GLANCE)
■ 一種囊泡運輸 (vesicle trafficking) 的體染色體隱性疾患,導致巨大胞器,包括黑色素小體 (melanosomes)、白血球顆粒與血小板緻密顆粒。
■ LYST 基因突變。
■ 銀髮;鼻與耳的色素斑點,且常有過度色素沉著。
■ 不等程度的畏光 (photophobia) 與眼球震顫 (nystagmus)。
■ 診斷可藉由毛幹中的色素聚集與血液抹片中的白血球顆粒確認。
■ 反覆性化膿性感染與輕度出血傾向。
■ 危及生命的「加速期」(accelerated phase),因淋巴組織球浸潤而出現全血球減少 (pancytopenia) 與器官腫大。
■ 進行性神經退化。
■ 治療:移植。
流行病學
Chédiak-Higashi 症候群 (Chédiak-Higashi syndrome, CHS) 是一種罕見的體染色體隱性疾患。¹³⁰,¹³¹ 常報告父母近親聯姻,全球約有 300 例報告病例。
臨床發現
病人通常於嬰兒期或兒童早期首次顯現表現。色素異常發生於 75% 的病人,特別是毛髮的銀色光澤(圖 132-15A)。眼部色素減退可能引起畏光,斜視 (strabismus) 與眼球震顫常見。視力傾向不會降低。皮膚相較於其他家庭成員通常較白皙,但在深膚色個體的日曬皮膚中可能見到深色、板岩色 (slate-colored) 的色素沉著區域與瀰漫性、斑點狀的色素減退。¹³²⁻¹³⁴
感染可在新生兒期觀察到,並持續病人的終生。感染最常涉及皮膚、肺與呼吸道,這些部位金黃色葡萄球菌 (S. aureus)、化膿性鏈球菌 (Streptococcus pyogenes) 與肺炎球菌 (Pneumococcus) 等微生物盛行。曾描述類似壞疽性膿皮症的深部潰瘍。神經表現包括肌肉無力、顱神經與周邊神經病變,以及進行性神經退化。CHS 病人也表現出輕度凝血缺陷。病人容易瘀青、表現瘀點,並有一些黏膜出血,雖然血小板數量維持正常。
病因與發病機轉
CHS 由位於染色體 lq42 的 LYST 基因突變引起,該基因編碼一種囊泡運輸與分泌最終步驟所需的蛋白。¹³⁵ LYST 基因在溶酶體 (lysosomes) 與其他分泌性胞器(黑色素小體、細胞溶解顆粒與血小板緻密顆粒)中表現。一般認為,特徵性的巨大顆粒源於顆粒成熟與融合的改變。增大的黑色素小體無法轉移至角質細胞。NK 細胞與細胞毒性 T 淋巴球不釋放標靶細胞殺傷所需的蛋白水解酵素。細胞毒性 T 淋巴球相關抗原 (Cytotoxic T-lymphocyte–associated antigen) 被困在異常大的囊泡中而非細胞表面,因此可能無法調控 T 細胞活化,增加淋巴增生性疾病的風險。血小板緻密顆粒在分泌正常凝血所需的儲存池時延遲。常伴有嗜中性白血球與單核球趨化作用減少,以及細胞內微生物殺傷延遲。
診斷
由於毛髮的銀色光澤,診斷通常在臨床上即被懷疑。在血液白血球中發現大型細胞質顆粒極具診斷性。毛幹有均勻分布的小型色素顆粒(見圖 132-15B),與 GS 不規則、大型的色素聚集形成對比(見圖 132-15C)。雖然不為診斷目的而執行,皮膚切片顯示黑色素細胞與角質細胞中皆有色素稀釋,電子顯微鏡可見巨大黑色素小體。
鑑別診斷
銀髮光澤在 GS 中也可見,但 Hermansky-Pudlak 症候群也以不尋常的金屬髮澤與皮膚色素減退為組成部分(見第 75 章)。一名因先天性胎兒水腫 (congenital hydrops fetalis) 而有低蛋白血症的新生兒也曾被描述有銀髮;該毛髮隨臨床改善而自發性再色素化。¹³⁶ 在某些急性白血病病例的抹片上可見假性 CHS 顆粒 (Pseudo-CHS granules)。¹³⁷
與銀髮症候群淋巴組織球增生相關的噬血細胞症候群 (hemophagocytic syndrome),需要與其他免疫缺乏疾患中的大量淋巴增生區分,特別是 X 連鎖淋巴增生性疾患(見上文「抗體缺乏疾患」)、家族性噬血細胞性淋巴組織球增生症 (familial hemophagocytic lymphohistiocytosis)、自體免疫淋巴增生症候群 (autoimmune lymphoproliferative syndrome),以及由 IL-2R 突變引起的免疫缺乏。家族性噬血細胞性淋巴組織球增生症是一群體染色體隱性疾患,伴有噬血細胞作用 (hemophagocytosis) 與 NK 細胞毒性缺如,若未早期幹細胞移植則傾向致命。¹³⁸,¹³⁹ 此病況可源於 PRF1(編碼穿孔素 perforin)、UNC13D(編碼 Munc 13-4)¹⁴⁰、STX11(編碼 syntaxin 11)或 STXBP2(編碼 Munc 18-2)的突變。所有這些蛋白都參與 NK 細胞與細胞毒性 T 淋巴球之細胞毒性細胞內顆粒的胞吐作用 (exocytosis) 與功能。數個基因的突變可導致體染色體隱性或顯性自體免疫淋巴增生症候群,又稱 Canale-Smith 症候群。¹⁴¹,¹⁴² 這些包括編碼 Fas 或 CD95 (TNFRSF6)(細胞表面凋亡受體)及其配體 (TNFSF6) 的基因,以及 caspase 蛋白 8 與 10(導致凋亡之級聯反應中的蛋白酶)。這群疾患的特徵為自體免疫疾患(血球減少、白血球破碎性血管炎 leukocytoclastic vasculitis、紅斑性狼瘡)與淋巴瘤風險增加,並與循環中 CD4−/CD8− α/β T 細胞數量增加相關。伴有內臟廣泛淋巴球浸潤的免疫缺乏,也是 IL-2 受體 α 鏈體染色體隱性突變的一種表現。¹⁴³
由於發育中胸腺 T 淋巴球的早期凋亡,病人表現出細菌、病毒與真菌感染。
預後、臨床病程與治療
噬血細胞性淋巴組織球增生症的「加速期」發生於 85% 的 CHS 病人,通常由 Epstein-Barr 病毒感染觸發。內臟被淋巴與組織球細胞浸潤,這些細胞有時外觀非典型。伴有肝脾腫大與黃疸、淋巴結腫大、全血球減少,以及一種伴頰黏膜偽膜性剝脫的白血病樣牙齦炎。在此期,常見瘀點、瘀青與牙齦出血,源於血小板減少與凝血因子肝臟合成減少。加速期間的壓倒性感染或出血常導致死亡。CHS 可能以加速期表現。¹⁴⁴
未移植 CHS 病人的平均死亡年齡為 6 歲,通常死於淋巴瘤樣加速期間的壓倒性感染或出血。產前診斷已可透過檢查胎兒頭皮切片的毛髮與胎兒血液樣本的白血球達成。CHS 病人的首選治療為早期移植¹⁴⁵⁻¹⁴⁸,可矯正免疫狀態,但不影響色素異常,也不抑制隨年齡增長而日益惡化之神經疾患的發展。降低強度的調理已成功使用,特別是在加速期發生前進行移植的病人。細胞毒性 T 細胞功能的缺如已被宣稱可預測噬血細胞性淋巴組織球增生症的後續發生,並可作為早期移植的生物標記。CHS 的處置在其他方面大致為支持性,使用預防性抗生素以避免反覆感染。Acyclovir、高劑量靜脈注射 γ 球蛋白、vincristine、cyclosporine 與 prednisone 曾用於控制加速期,但加速期通常致命。
Griscelli 症候群 (Griscelli Syndrome)
重點一覽 (AT-A-GLANCE)
■ 體染色體隱性的一群疾患。
■ MYO5A(較偏神經性)、RAB27A(噬血細胞作用)或 Slac-2a 突變。
■ 銀髮是其標誌。
■ 診斷可藉由毛幹中的色素聚集確認,但抹片不顯示白血球顆粒。
■ 噬血細胞作用常由 Epstein-Barr 病毒感染促發。
■ 治療:移植。
流行病學
患有此體染色體隱性疾患的病人大多數為地中海或中東血統,且出生於近親聯姻的家庭。
臨床發現
GS 的色素稀釋常限於毛髮,特徵為帶有銀灰色光澤,少數病例有皮膚侵犯。毛幹顯示大型不均勻的色素叢集(見圖 132-15C),皮膚顯示角質細胞中色素稀釋,但黑色素細胞中黑色素小體聚積。電子顯微鏡可見表皮黑色素細胞中有眾多第 IV 期黑色素小體 (stage IV melanosomes)。與 CHS 相反,白血球抹片不顯示巨大顆粒。各亞型所具有的額外臨床特徵取決於缺陷蛋白的功能與組織表現。GS1 病人有原發性神經疾患,於嬰兒期以肌張力低下 (hypotonia) 與發育遲緩表現。以噬血細胞症候群或「疾病加速期」為特徵的嚴重免疫疾患見於 GS2。噬血細胞症候群的特徵為活化的淋巴球與巨噬細胞浸潤各器官系統。它於平均 36 個月齡時發展,且常由病毒(特別是 Epstein-Barr 病毒)促發。病人有發燒、肝脾腫大、神經損害、凝血障礙與全血球減少。GS3 病人僅以色素異常表現。¹⁴⁹,¹⁵⁰
病因與發病機轉
3 個基因的突變可能構成 GS 的基礎。GS1 由 MYO5A 基因(編碼肌凝蛋白 5a,一種負責細胞內胞器運輸的馬達蛋白)突變引起。MYO5A 在腦組織中大量表現,對神經傳遞物質分泌很重要。它不在細胞毒性細胞中表現。GS2 由 RAB27A 基因突變引起,該基因編碼 Rab27a,一種參與細胞內調控性分泌途徑功能的鳥苷三磷酸酶 (guanosine triphosphatase) 蛋白。它對 T 淋巴球與 NK 淋巴球的細胞毒性顆粒釋放與細胞死亡至關重要。此缺陷的細胞毒性活性最可能導致噬血細胞症候群中淋巴球與巨噬細胞的失控活化。MYO5A 與 RAB27A 皆定位於染色體 15q21.1。第三型 GS3 是 Slac2-a/melanophilin 的同型合子錯義突變 (homozygous missense mutation),或 MYO5A F 外顯子缺失 (F-exon deletion) 的結果。¹⁵¹ Melanophilin(也不在細胞毒性細胞中表現)編碼 Rab 效應子家族的成員,並與肌凝蛋白 5a(透過 F 外顯子)及 RAB27A 交互作用,形成黑色素小體捕捉與局部運輸至黑色素細胞所必需的三蛋白複合體(圖 132-16)。全部 3 種遺傳亞型都共有無法建構此黑色素小體運輸複合體的缺陷,因而導致 GS 特徵性的色素異常。
預後、臨床病程與治療
GS 過去一律致命,但現在可透過成功的造血幹細胞移植加以逆轉。在 GS2 病人中,85% 有導致蛋白早期截短的 RAB27A 基因突變。他們有最嚴重型的疾病,早期發展噬血細胞症候群且快速進展。這些病人從加速期發作至死亡的中位存活時間為 5 個月。大多數病人於 5 歲前因進行性中樞神經系統疾病或反覆感染而死亡。RAB27A 基因的錯義突變顯示較輕的表現型,噬血細胞症候群發作年齡較晚,且對化療治療反應良好。GS1 病人不發展噬血細胞症候群。產前診斷已可透過檢查胎兒頭皮切片的毛髮與 DNA 分析達成。
補體缺乏疾患 (COMPLEMENT DEFICIENCY DISORDERS)
重點一覽 (AT-A-GLANCE)
■ 早期補體組件的缺乏或功能障礙與自體免疫疾患相關,特別是全身性紅斑性狼瘡 (systemic lupus erythematosus)。
■ 早期補體組件的缺乏或功能障礙導致由莢膜細菌引起的感染風險。
■ 晚期補體組件的缺乏顯著增加對奈瑟氏菌 (neisserial) 感染的易感性。
遺傳性血管性水腫 (Hereditary Angioedema)
遺傳性血管性水腫 (Hereditary angioedema, HAE) 幾乎總是以體染色體顯性方式遺傳,自發突變率為 25%(臨床特徵與處置見第 41 章)。¹⁵² 它發生於每 150,000 人中 1 例,無種族或族裔偏好。75% 的病人報告有陽性家族史。HAE 有 3 種類型。第 I 型的特徵為 C1 抑制因子 (C1 inhibitor, C1 INH) 抗原濃度低,影響 85% 的病人,其餘病人為第 II 型,功能濃度低但抗原濃度正常/高。第三型 HAE 不顯示 C1 INH 的缺乏。此型 HAE 先前被認為僅影響女性,雖然最終曾描述一個有三名男性家庭成員受影響的家族。¹⁵³ 已鑑定出一個家族,其中本病因基因啟動子區域 (promoter region) 的突變而以體染色體隱性性狀傳遞。¹⁵⁴
HAE 造成顯著的罹病率,尤其當發作頻繁且喉部水腫 (laryngeal edema) 是死亡的重大風險時。有數種可用的治療模式,包括使用衰減型雄激素 (attenuated androgens)、抗纖溶劑 (antifibrinolytic agents)、血漿來源 C1 INH、一種激肽釋放酶抑制劑 (kallikrein inhibitor) 與一種緩激肽受體拮抗劑 (bradykinin receptor antagonist)。¹⁵² 血漿來源 C1 INH 可急性用於治療血管性水腫發作,或在發作頻繁時或已知程序之前預防性使用。
圖表 (Figures and Tables)
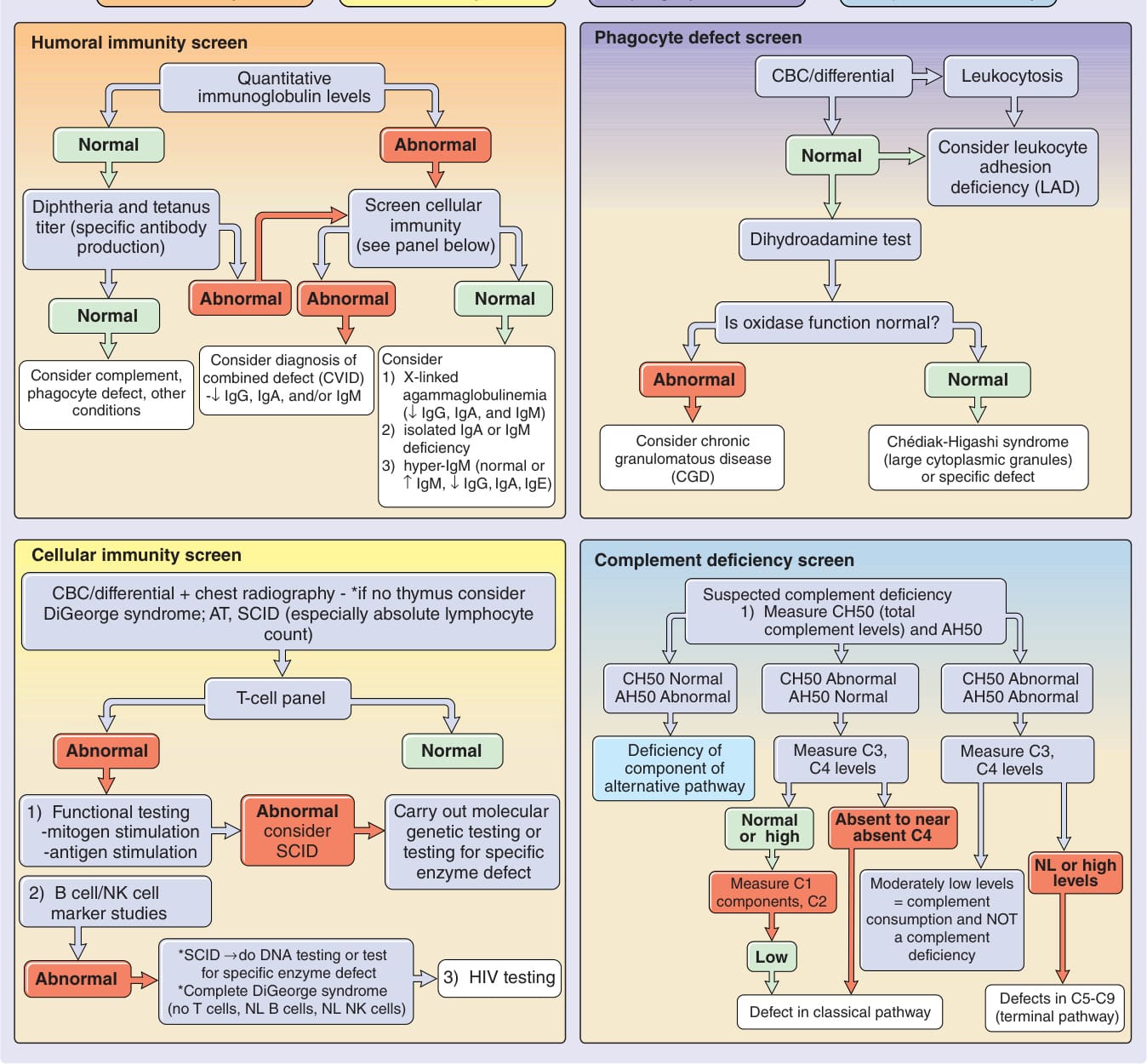
圖 132-1:新生兒與嬰兒免疫缺乏疾患的診斷流程。若臨床表現令人擔心為嚴重複合型免疫缺乏 (severe combined immunodeficiency, SCID),此為醫療緊急狀況,病人需立即轉診至免疫學專家。AH50,替代途徑 50% 溶血活性 (alternative pathway 50% hemolytic activity);AT,運動失調-毛細血管擴張症 (ataxia-telangiectasia);CBC,全血球計數 (complete blood cell count);CH50,50% 溶血補體 (50% hemolytic complement);CVID,常見變異型免疫缺乏 (common variable immunodeficiency);Ig,免疫球蛋白 (immunoglobulin);NK cell,自然殺手細胞 (natural killer cell);NL,正常 (normal);↓,減少;↑,增加。
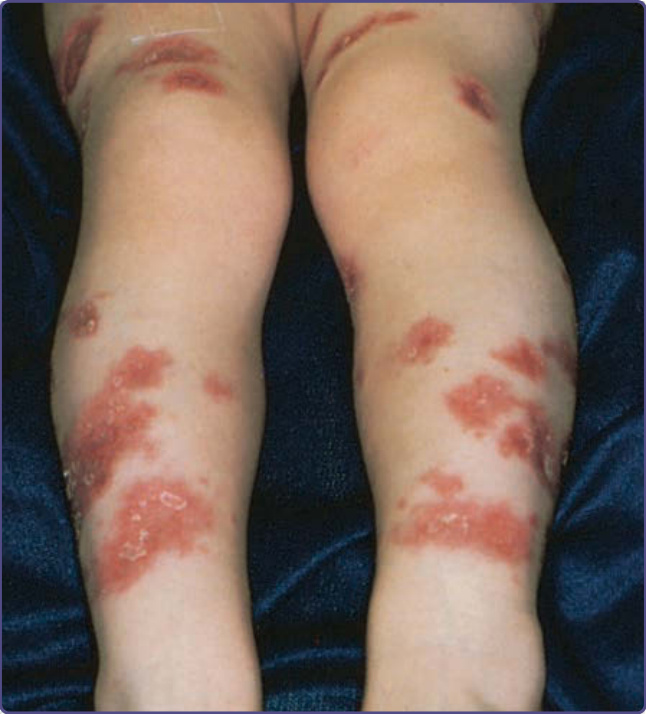
圖 132-2:一名常見變異型免疫缺乏 (common variable immunodeficiency) 兒童腿部的非乾酪性肉芽腫 (noncaseating granulomas)。培養與特殊染色未顯示微生物。
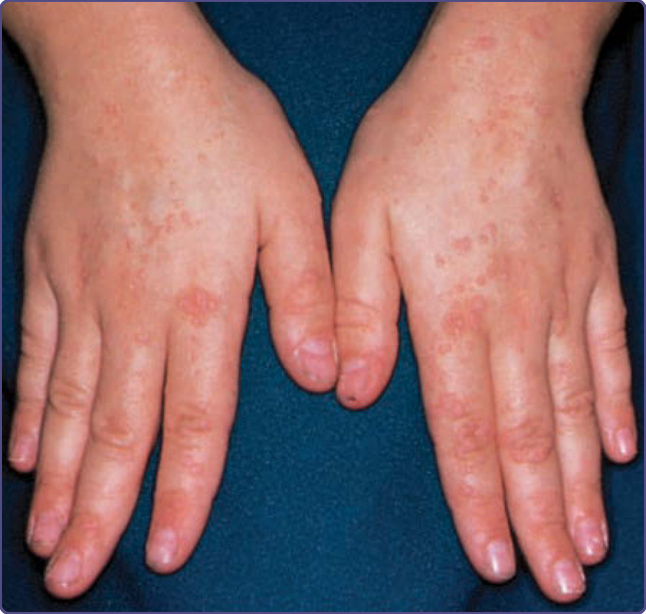
圖 132-3:這名患有常見變異型免疫缺乏 (common variable immunodeficiency) 的女孩,手背覆蓋著難治性尋常疣 (verrucae vulgaris)。
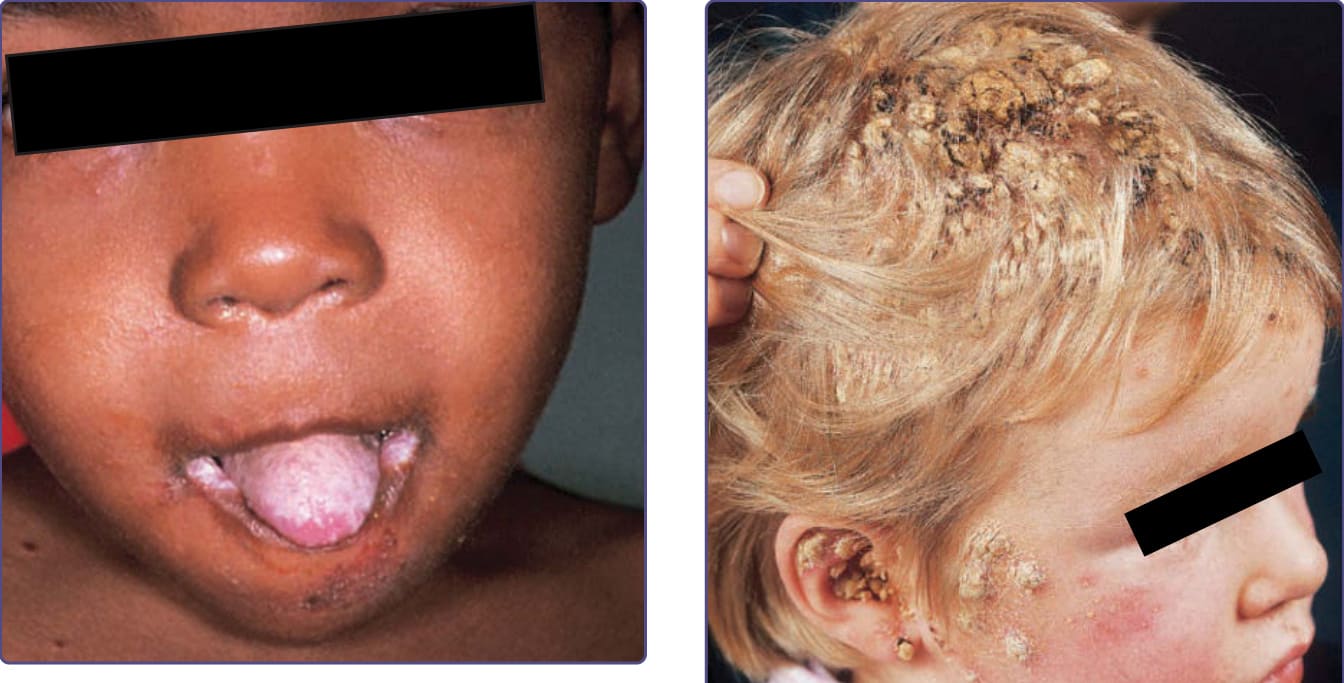
圖 132-4:一名患有皮膚黏膜念珠菌病 (mucocutaneous candidiasis) 男孩的反覆性鵝口瘡 (thrush) 與念珠菌性唇炎 (candidal cheilitis)。
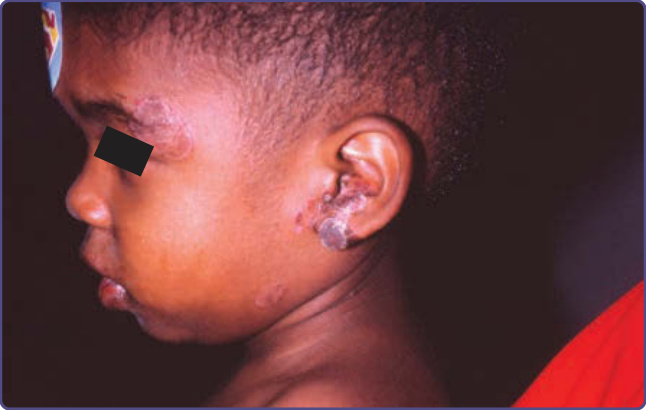
圖 132-5:伴有過度角化性念珠菌肉芽腫 (hyperkeratotic candidal granulomas) 的皮膚念珠菌感染。該兒童對口服唑類抗真菌藥物有反應,但對外用藥物無反應。
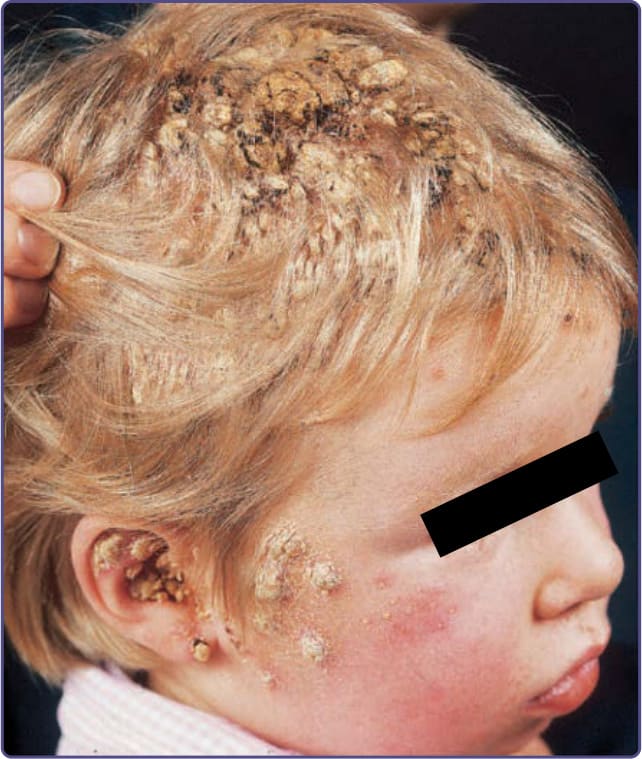
圖 132-6:這名 3 歲、患有甲狀腺低能症 (hypothyroidism) 的兒童有口腔鵝口瘡、間擦部位念珠菌病 (intertriginous candidiasis)、頭皮與顏面的疣狀結痂,以及念珠菌性甲癬 (candidal onychomycosis)。照片中所示的疣狀增生由乾燥的膿與血清組成,且僅培養出白色念珠菌 (Candida albicans)。
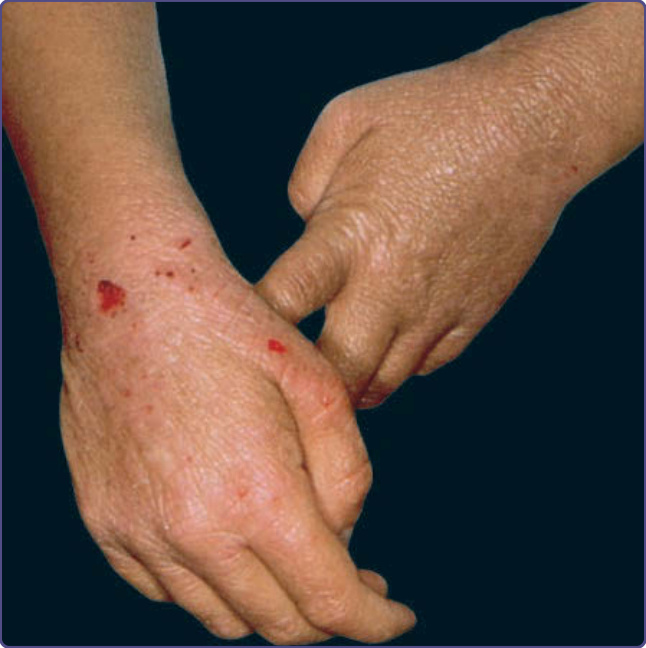
圖 132-7:一名患有威斯科特-奧德里奇症候群 (Wiskott-Aldrich syndrome) 男孩的嚴重異位性皮膚炎。注意血清血性結痂 (serosanguineous crusting)。
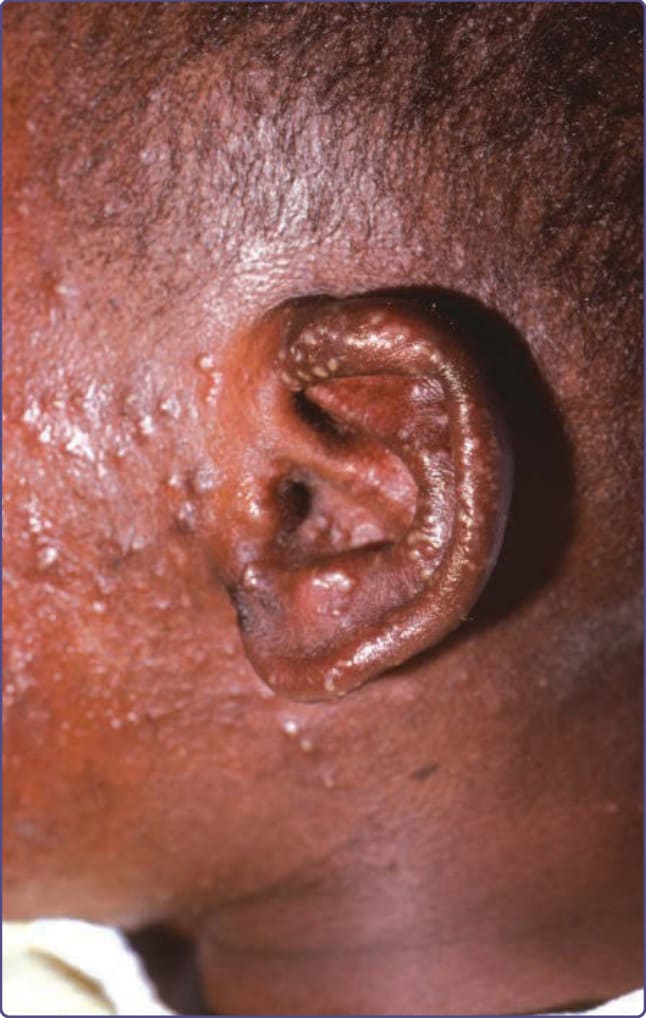
圖 132-8:一名患有威斯科特-奧德里奇症候群 (Wiskott-Aldrich syndrome) 青少年耳部伴有膿疱的疱疹感染。該病人因先前的眼部感染而左眼失明。在圖示的感染之後,病人接受預防性 acyclovir,此後十年無後續疱疹感染。最終,他對 acyclovir 產生抗藥性,並死於其免疫缺乏。
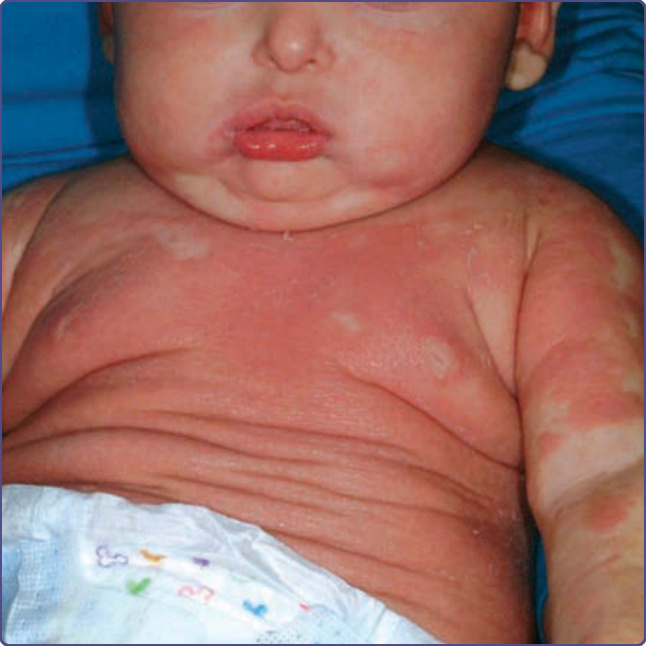
圖 132-9:外胚層失養症合併免疫缺乏基因突變 (Ectodermal dysplasia with immunodeficiency gene mutation)。這名 NEMO(核因子 κB 必要調節因子,nuclear factor κB essential modulator)有弱效突變的男孩,顯示生長遲滯、輕度剝脫性皮膚炎,以及無汗性外胚層失養症 (hypohidrotic ectodermal dysplasia) 的典型面部特徵。注意小下巴、「噘嘴」的下唇、薄上唇,以及伴顴骨發育不全 (malar hypoplasia) 的小而緊縮的鼻子。他的母親有沿 Blaschko 線分布的色素條紋,為色素失調症 (incontinentia pigmenti) 所見的典型表現。(Used with permission from Dr. Anthony Mancini.)
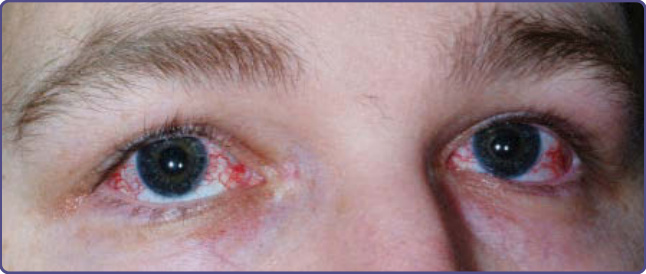
圖 132-10:一名運動失調-毛細血管擴張症 (ataxia-telangiectasia) 病人的球結膜毛細血管擴張 (bulbar conjunctival telangiectasias)。
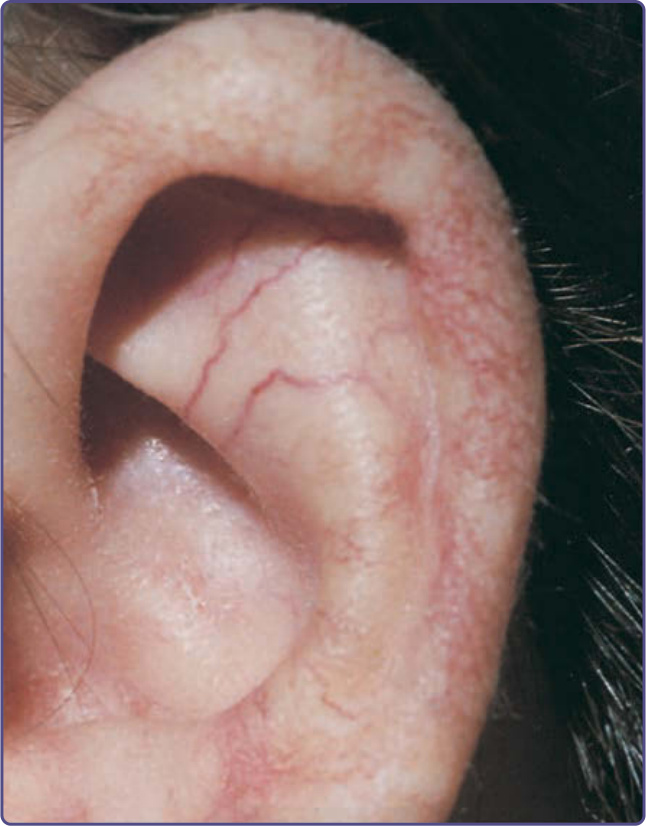
圖 132-11:運動失調-毛細血管擴張症 (Ataxia-telangiectasia)。耳輪 (helix) 內與耳輪上的毛細血管擴張。
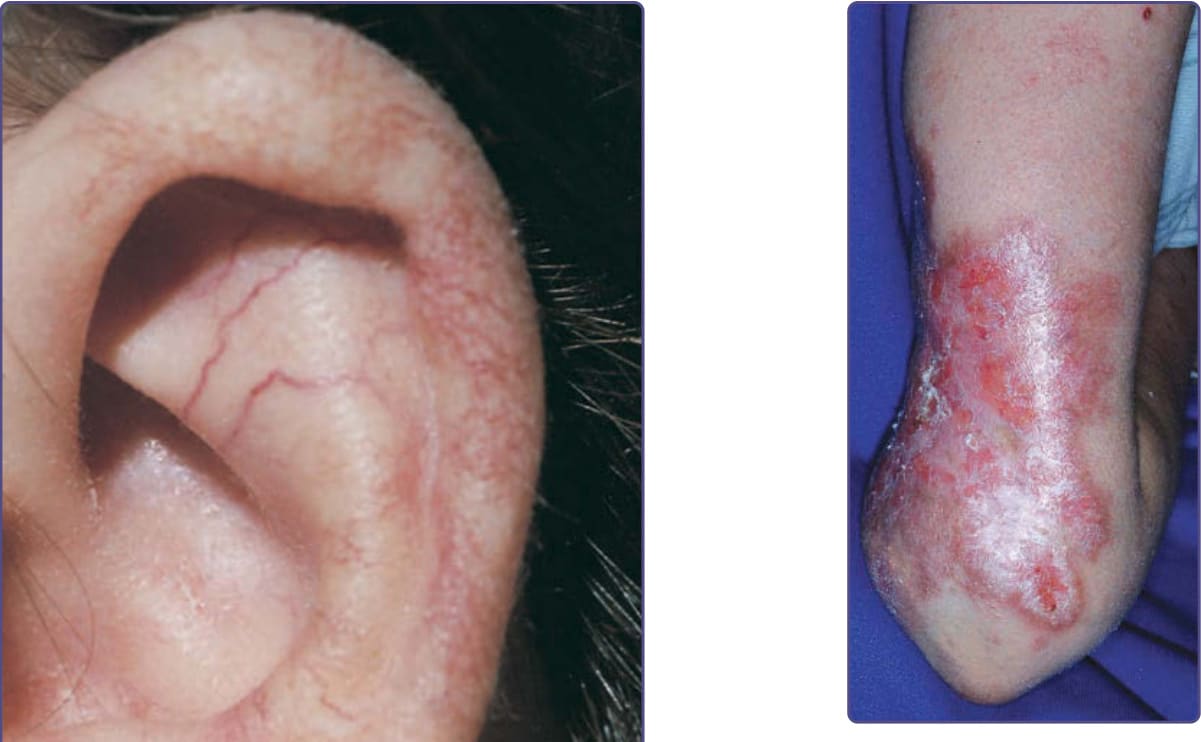
圖 132-12:一名運動失調-毛細血管擴張症 (ataxia-telangiectasia) 病人的非感染性肉芽腫性皮膚炎 (noninfectious granulomatous dermatitis)。這些持續性病灶傾向潰瘍,但常對 triamcinolone acetonide 注射有反應。
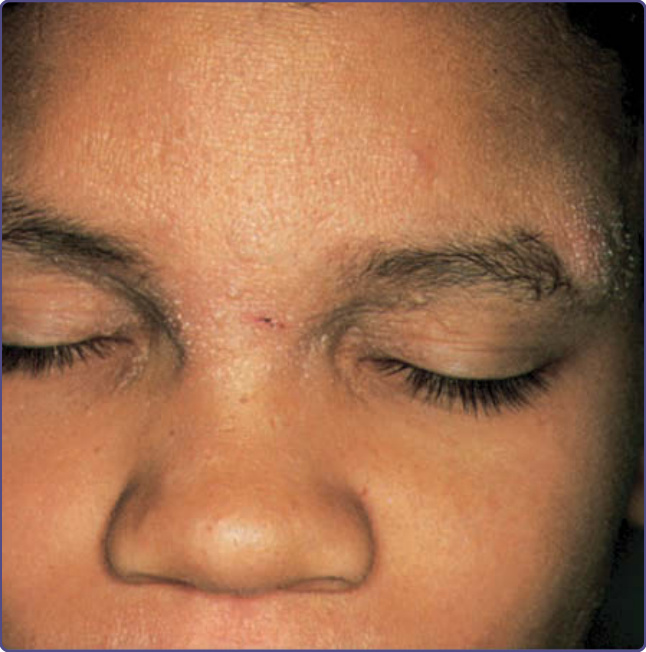
圖 132-13:一名患有高免疫球蛋白 E 症候群 (Hyperimmunoglobulinemia E syndrome) 男孩的粗糙面部特徵、異位性皮膚炎癤腫,以及眉間 (glabella) 的冷膿瘍 (cold abscess)。
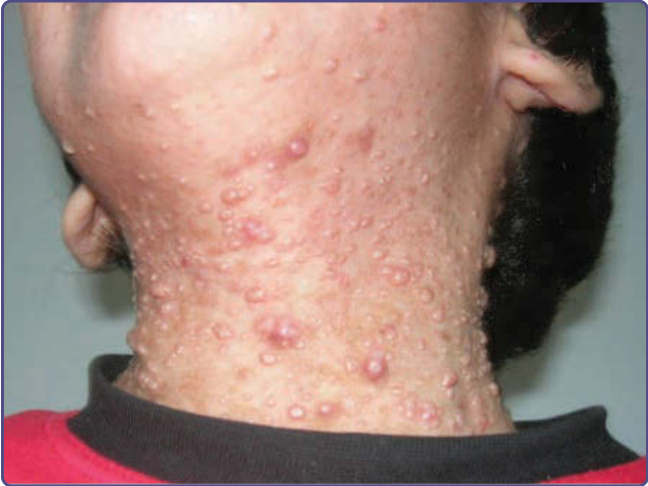
圖 132-14:這名因 DOCK8(胞質分裂作用 8 調節因子,dedicator of cytokinesis 8)突變引起的體染色體隱性 HIES 男孩,頸部顯示廣泛的傳染性軟疣 (molluscum contagiosum) 感染。(Used with permission from Dr. I. Barlan.)
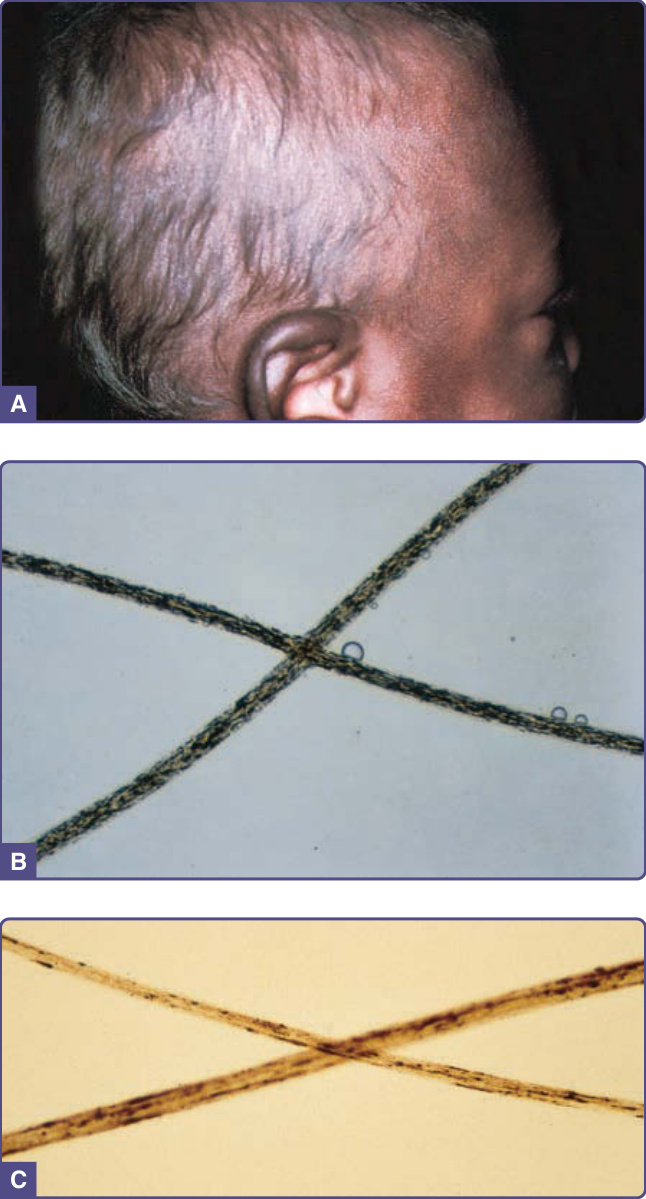
圖 132-15:銀髮症候群 (Silvery hair syndromes)。A,一名黑人嬰兒毛髮的銀色光澤;該病人有深色素的眼睛與皮膚。注意耳輪上色素的強化。B,Chédiak-Higashi 症候群毛髮中均勻聚集的小型色素顆粒。C,一名 Griscelli 症候群病人毛髮中不規則間隔的較大型色素聚集物。
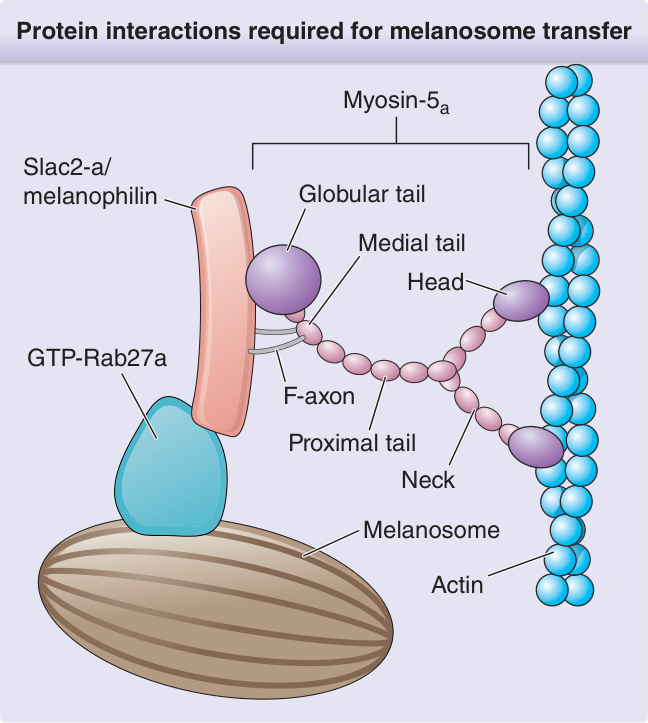
圖 132-16:黑色素小體轉移所需的蛋白交互作用 (Protein interactions required for melanosome transfer)。GTP,鳥苷三磷酸 (guanosine triphosphate)。(Adapted with permission of ASCI: Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456. Permission conveyed through Copyright Clearance Center, Inc.)
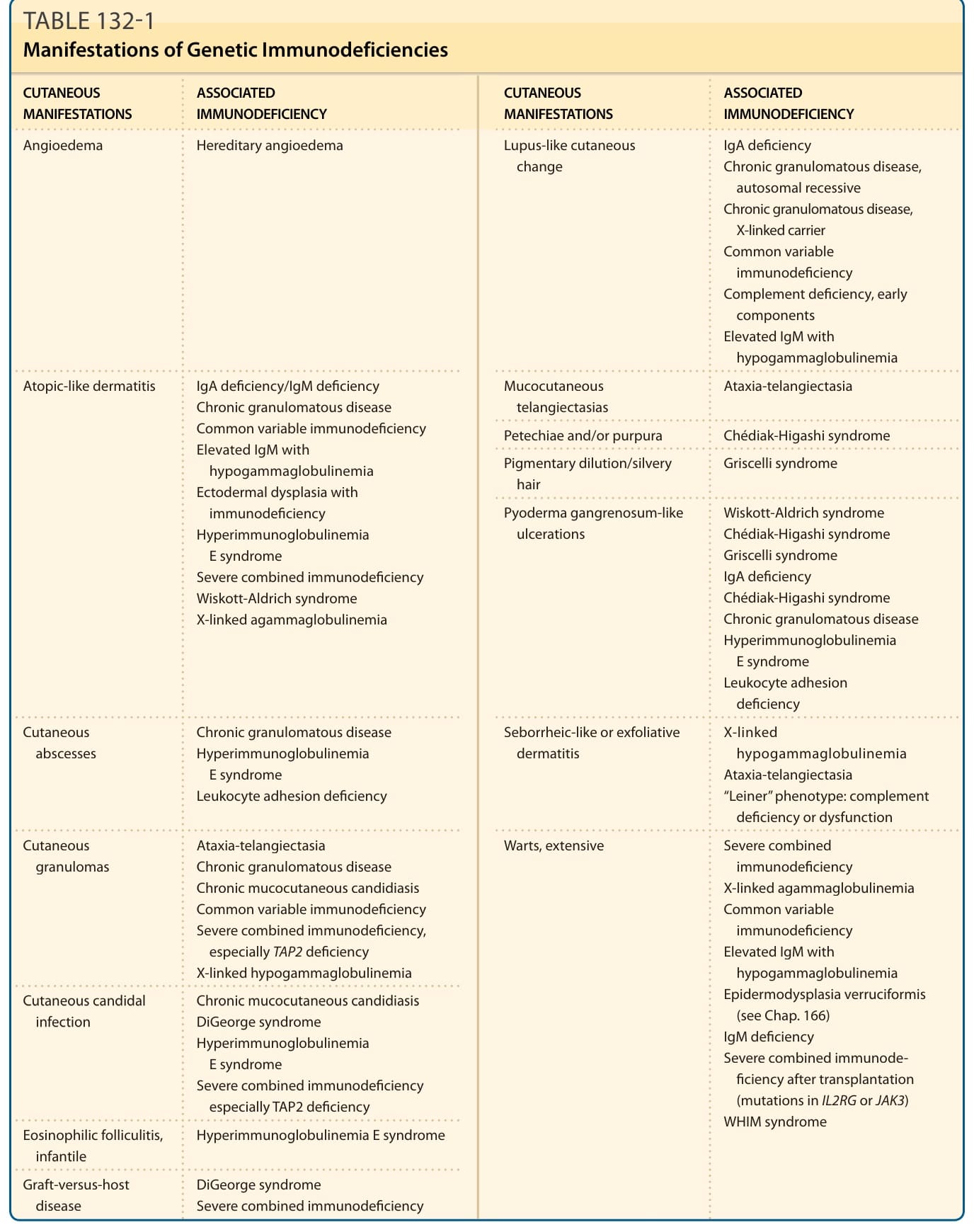
表 132-1:遺傳性免疫缺乏的表現 (Manifestations of Genetic Immunodeficiencies)
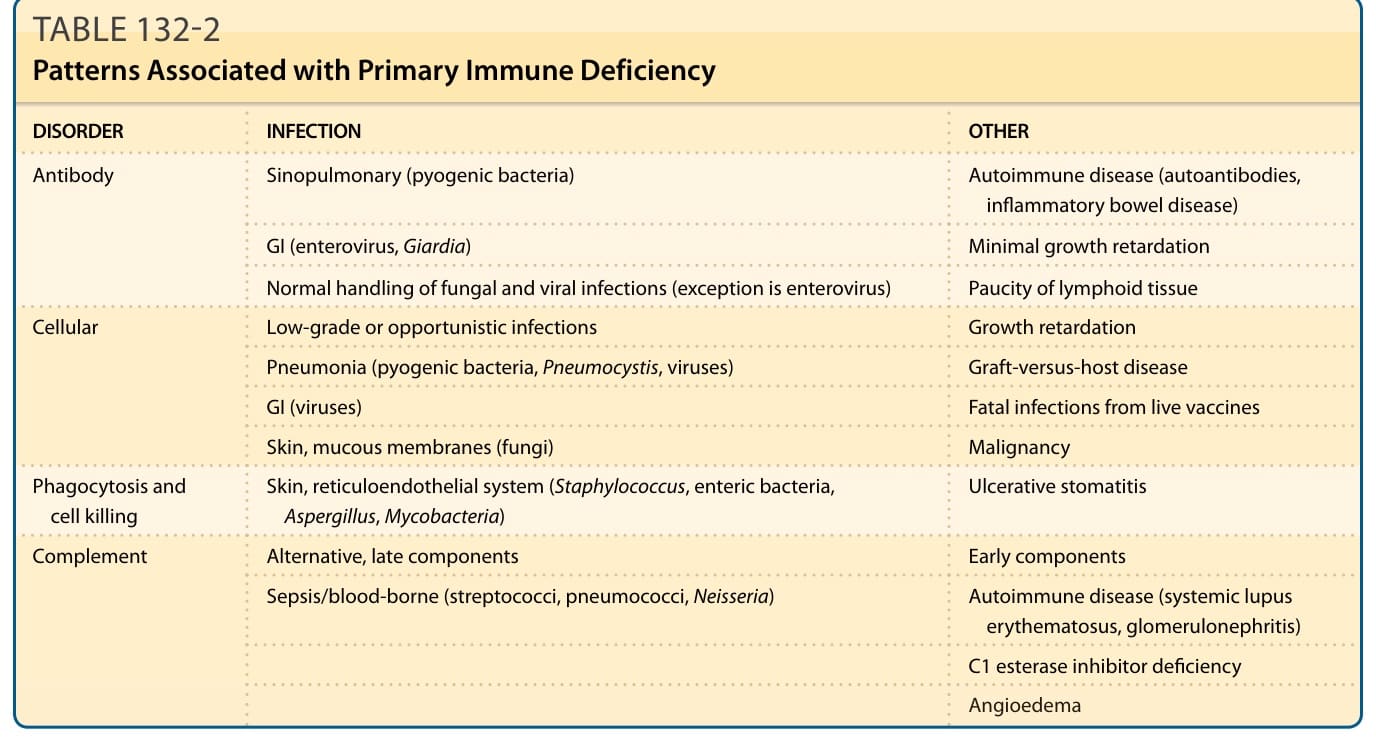
表 132-2:與原發性免疫缺乏相關的型態 (Patterns Associated with Primary Immune Deficiency)
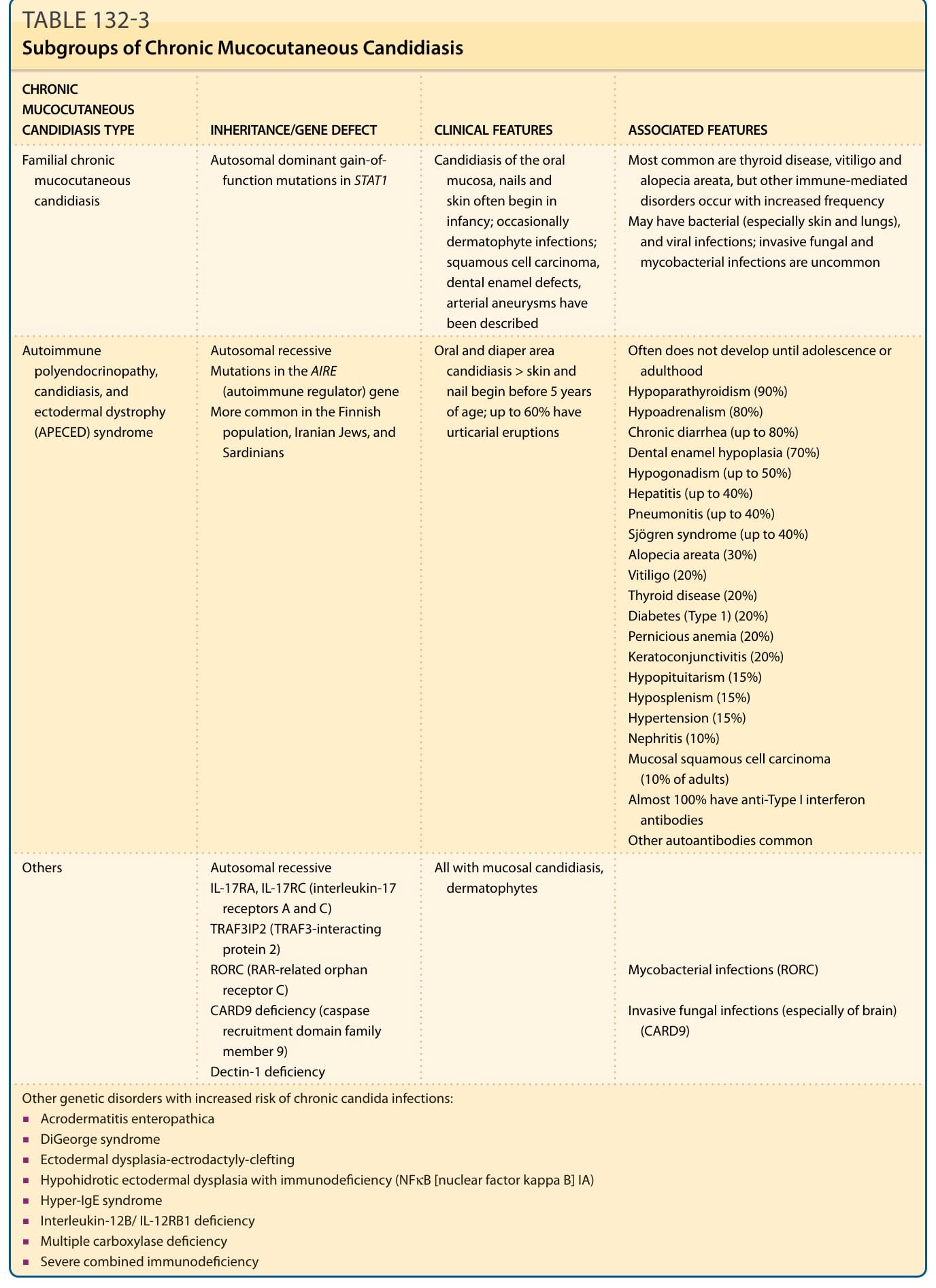
表 132-3:慢性皮膚黏膜念珠菌病的亞群 (Subgroups of Chronic Mucocutaneous Candidiasis)
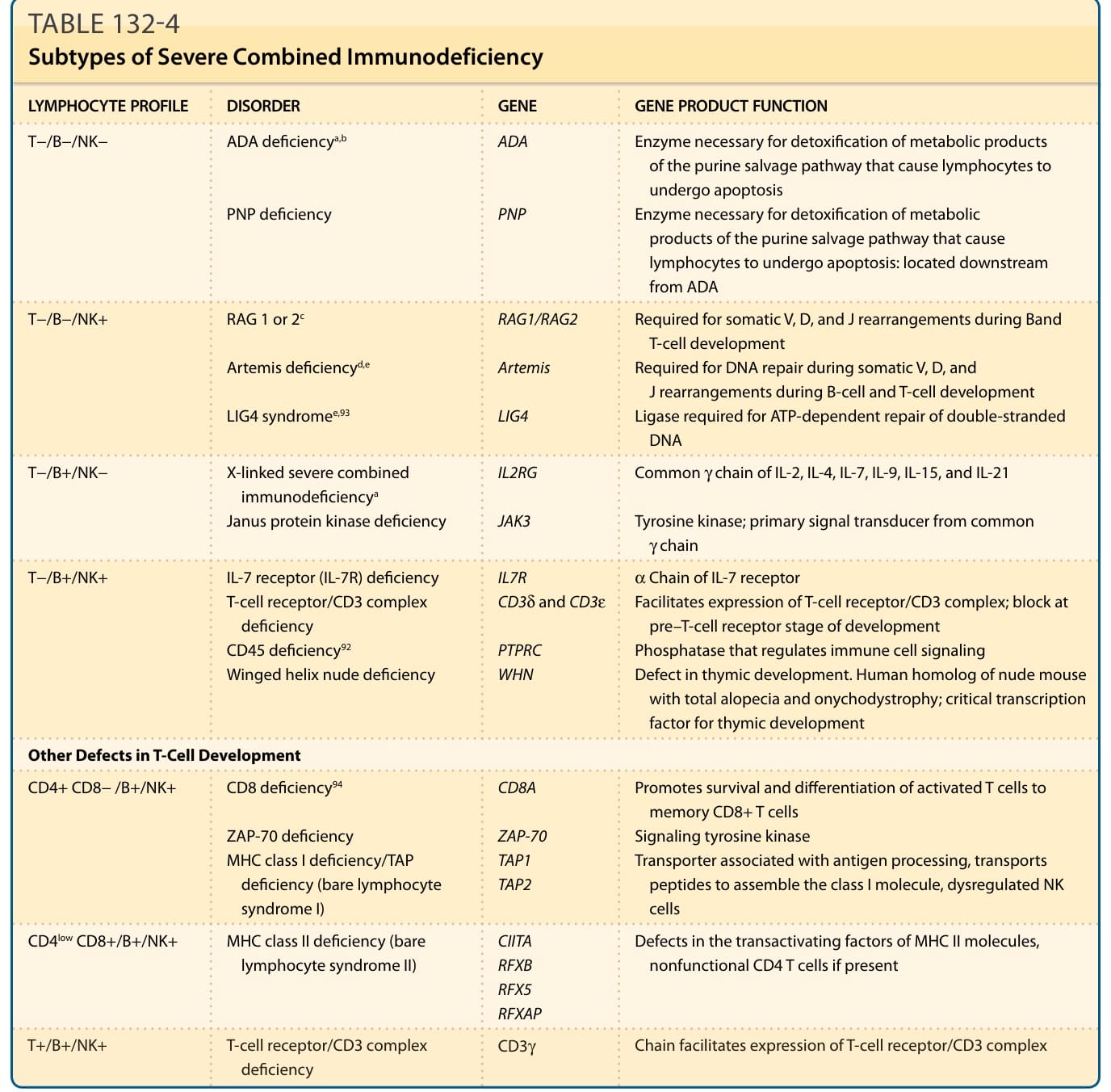
表 132-4:嚴重複合型免疫缺乏的亞型 (Subtypes of Severe Combined Immunodeficiency)。
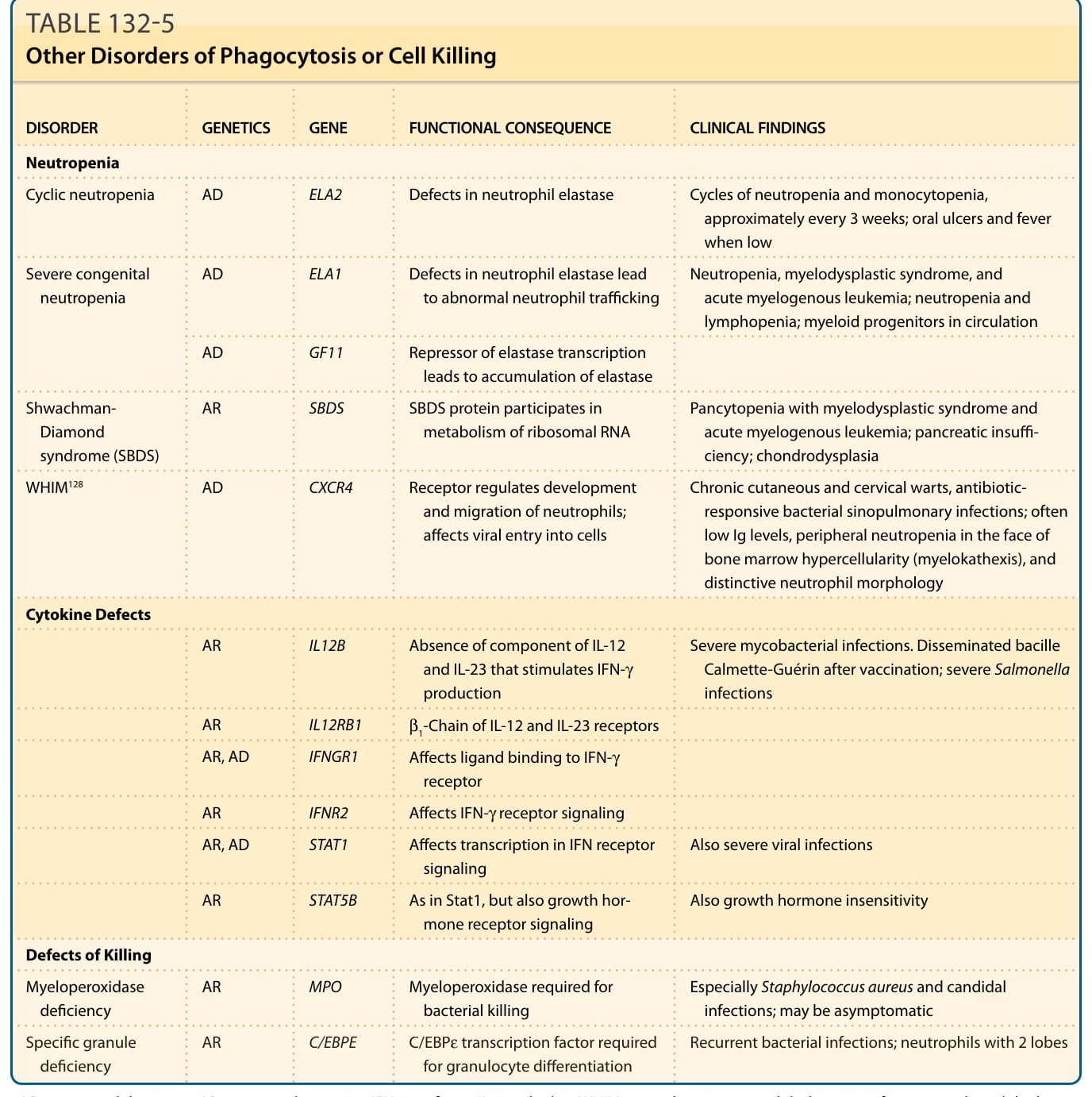
表 132-5:其他吞噬作用或細胞殺傷障礙 (Other Disorders of Phagocytosis or Cell Killing)
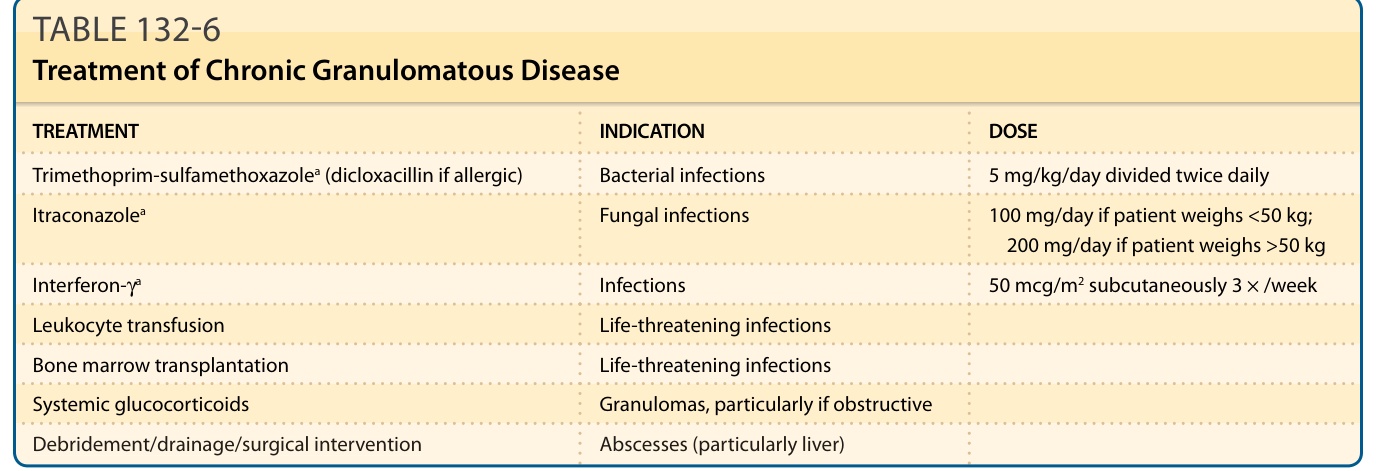
表 132-6:慢性肉芽腫病的治療 (Treatment of Chronic Granulomatous Disease)
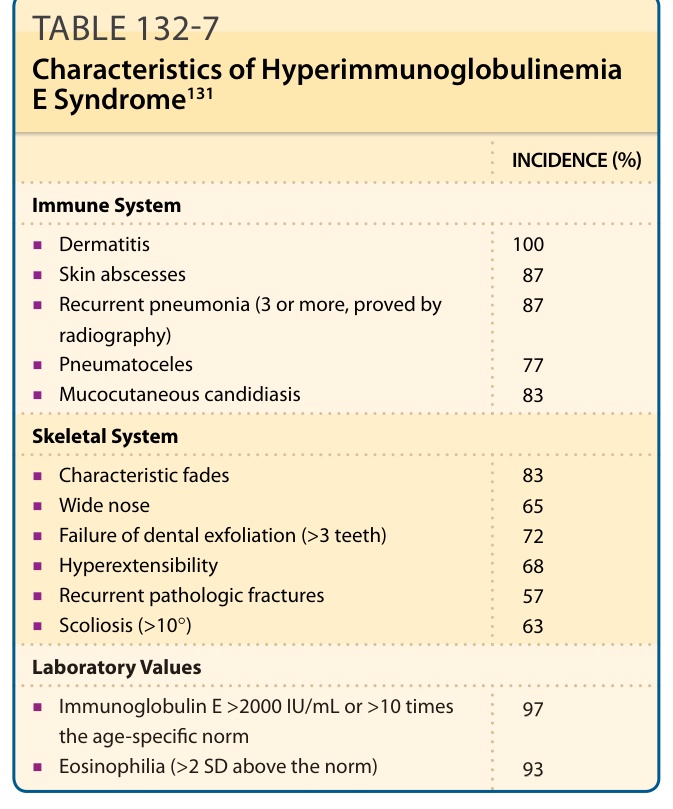
表 132-7:高免疫球蛋白 E 症候群的特徵 (Characteristics of Hyperimmunoglobulinemia E Syndrome)¹³¹
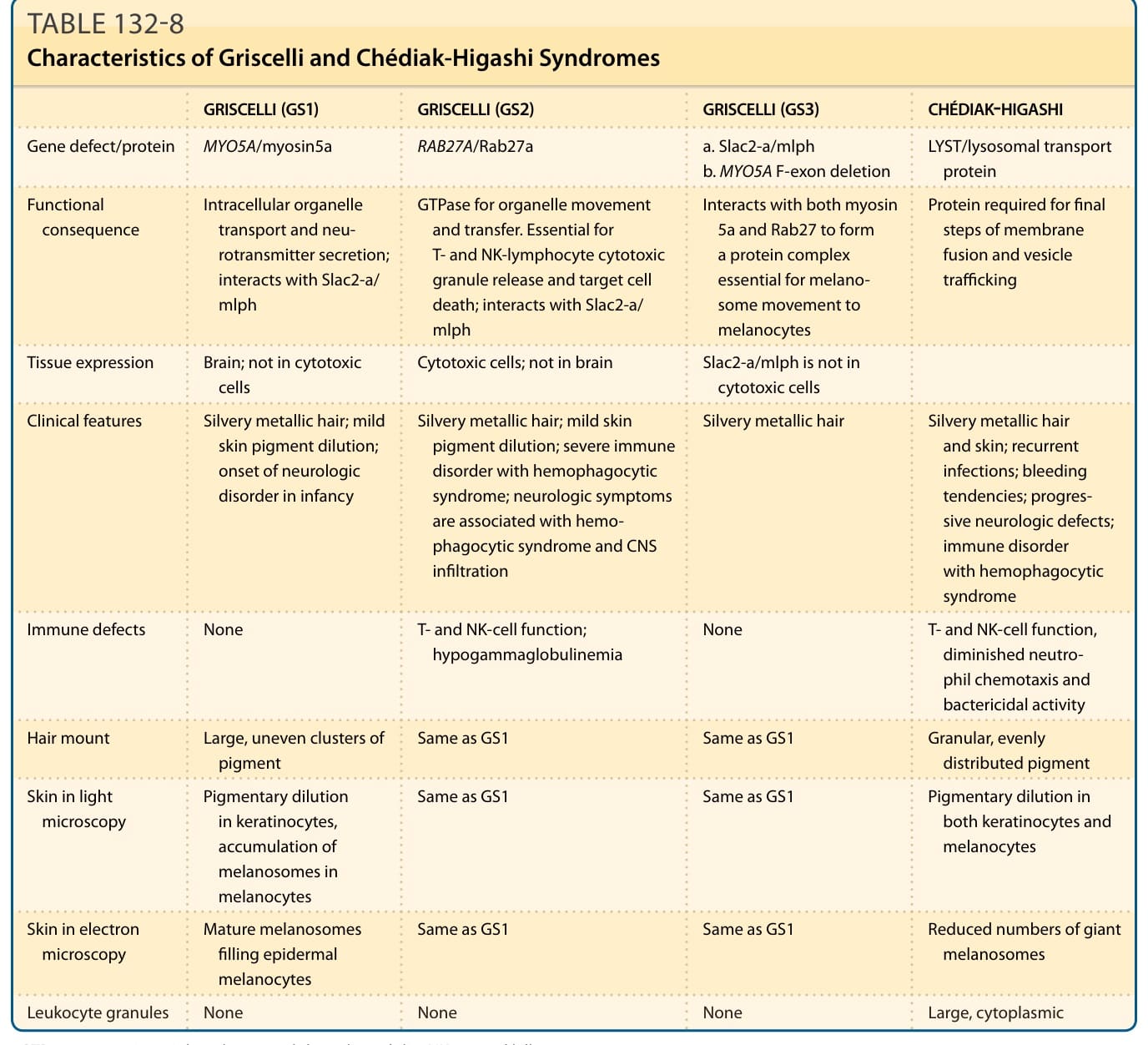
表 132-8:Griscelli 與 Chédiak-Higashi 症候群的特徵 (Characteristics of Griscelli and Chédiak-Higashi Syndromes)
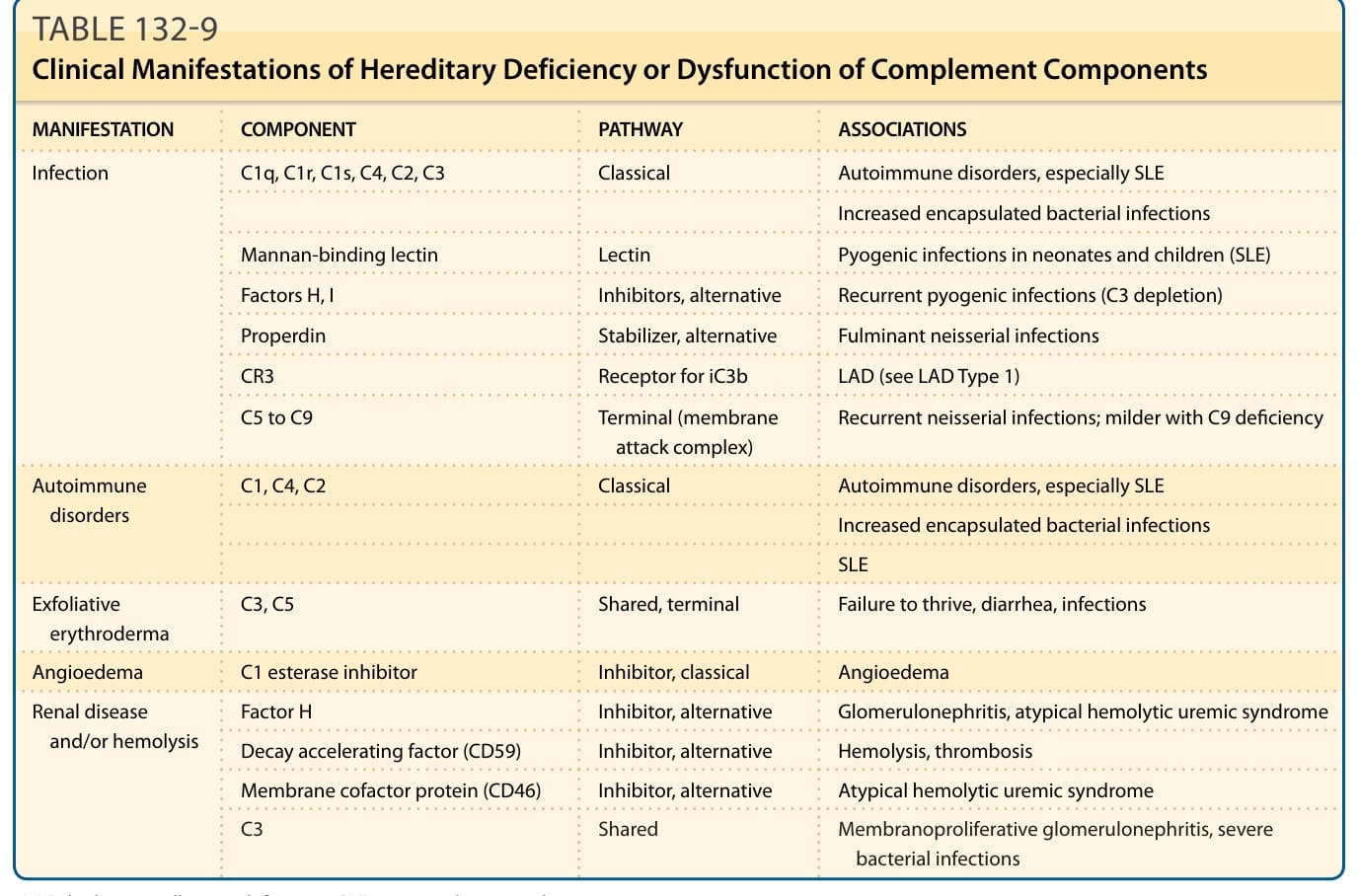
表 132-9:遺傳性補體組件缺乏或功能障礙的臨床表現 (Clinical manifestations of hereditary deficiency or dysfunction of complement components)。