外胚層發育不良 (Ectodermal Dysplasias)
總論
- 定義:一群遺傳性疾病,特徵為 2 個或更多外胚層結構(毛髮、牙齒、指甲、皮脂腺與汗腺)出現發育異常;僅侵犯單一結構者不歸類為 ED。也可能合併非外胚層結構異常。
- 近 200 種疾病被歸為 EDs,臨床表現多樣且重疊,精確診斷困難。
- 異質性:臨床不同的疾病可源自同一基因不同突變(對偶基因異質性, allelic heterogeneity);臨床相似者可源自不同基因突變(基因座異質性, locus heterogeneity)。
- 診斷起點:先判定有無出汗(汗性 hidrotic vs 低汗/無汗 hypohidrotic/anhidrotic),再依其他外胚層與非外胚層結構侵犯與遺傳模式分支。
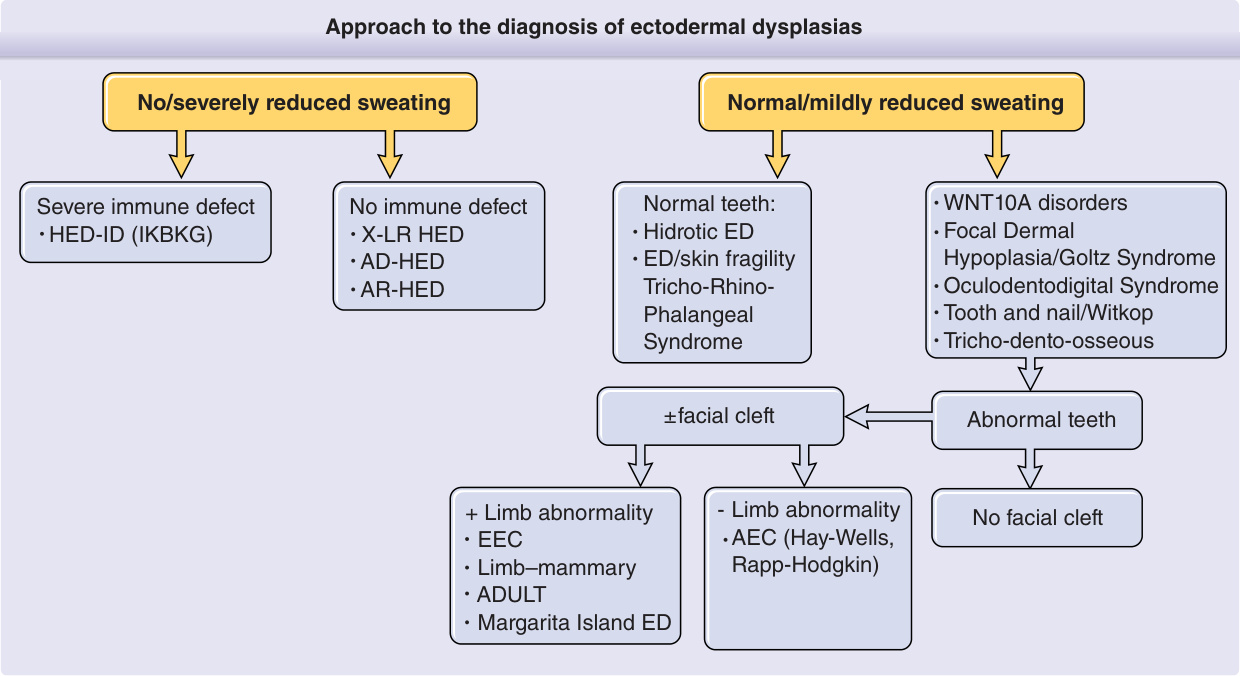
圖 131-1:外胚層發育不良的診斷演算法。
低汗性外胚層發育不良 (Hypohidrotic Ectodermal Dysplasia, HED):XLHED、ADHED、ARHED、HED-ID
- 分類與基因:
- XLHED (OMIM 305100):Xq12-q13.1 之 EDA1 的 XLR 突變(編碼 ectodysplasin-A);又稱 Christ–Siemens–Touraine 症候群,最常見的 ED,佔 NFED 登記家庭的 80%,約 70% 患病男性遺傳自帶因母親。
- ADHED (OMIM 129490)/ARHED (OMIM 224900):EDAR/EDARADD 突變,臨床似 XLHED 但較罕見,AD 型可能較輕。
- HED-ID (OMIM 300291):Xq28 之 IKBKG (NEMO) 的 XLR 突變,合併免疫缺乏;男性 HED 伴反覆/顯著感染時應考慮。
- 致病機轉:相關基因屬腫瘤壞死因子 α (TNF-α)/ectodysplasin 訊息傳遞路徑;突變中斷上皮與其下間質的交互作用。
- 臨床三主徵:毛髮稀疏 (hypotrichosis)、少汗 (hypohidrosis)、牙齒過少 (hypodontia)。
- 出生可有膠樣膜或明顯脫屑;頭皮毛髮稀疏、細軟、金黃。出汗顯著受損 → 耐熱不良、不明原因高燒(過往誤認的智能障礙實為高燒與癲癇之後遺)。
- 牙齒過少/寡牙/無牙為男性必有特徵,萌發者呈釘狀;齒槽嵴發育不全為早期線索。
- 顏面:額部隆突 (frontal bossing)、中臉部凹陷伴鞍鼻、豐厚外翻嘴唇;眼周皺紋與色素沉著常於出生即有。
- 常合併濕疹(>2/3 男性)、反覆上呼吸道感染與肺炎、氣喘、胃食道逆流;20%–40% 嬰幼兒生長遲滯後追趕。
- 女性帶因者:60%–80% 有臨床徵象,最常為斑塊狀毛髮稀疏與牙齒過少/釘狀牙。
- 診斷:年齡增長後三主徵更明顯;齒槽嵴發育不全、顎部全景 X 光 (Panorex) 有助診斷;分子基因檢測漸普及。組織病理見表皮變薄平坦、皮脂腺與毛囊減少、外分泌腺缺如,但通常無需切片。
- 病程/預後/處置:
- 維持涼爽環境最關鍵(濕 T 恤、頭帶、空調、降溫背心)以防高熱。
- 牙齒修復為首要:早期假牙、最終牙科植體。
- 嬰兒因高熱與感染死亡風險增加(EDIR 調查 21% XLHED 有嬰兒/兒童死亡家族史);成年多能正常生活,耐熱不良隨青春期出汗能力與生活適應而減輕。
- 重組 ectodysplasin 蛋白於動物模型可挽救表現型,目前有人體試驗進行中。
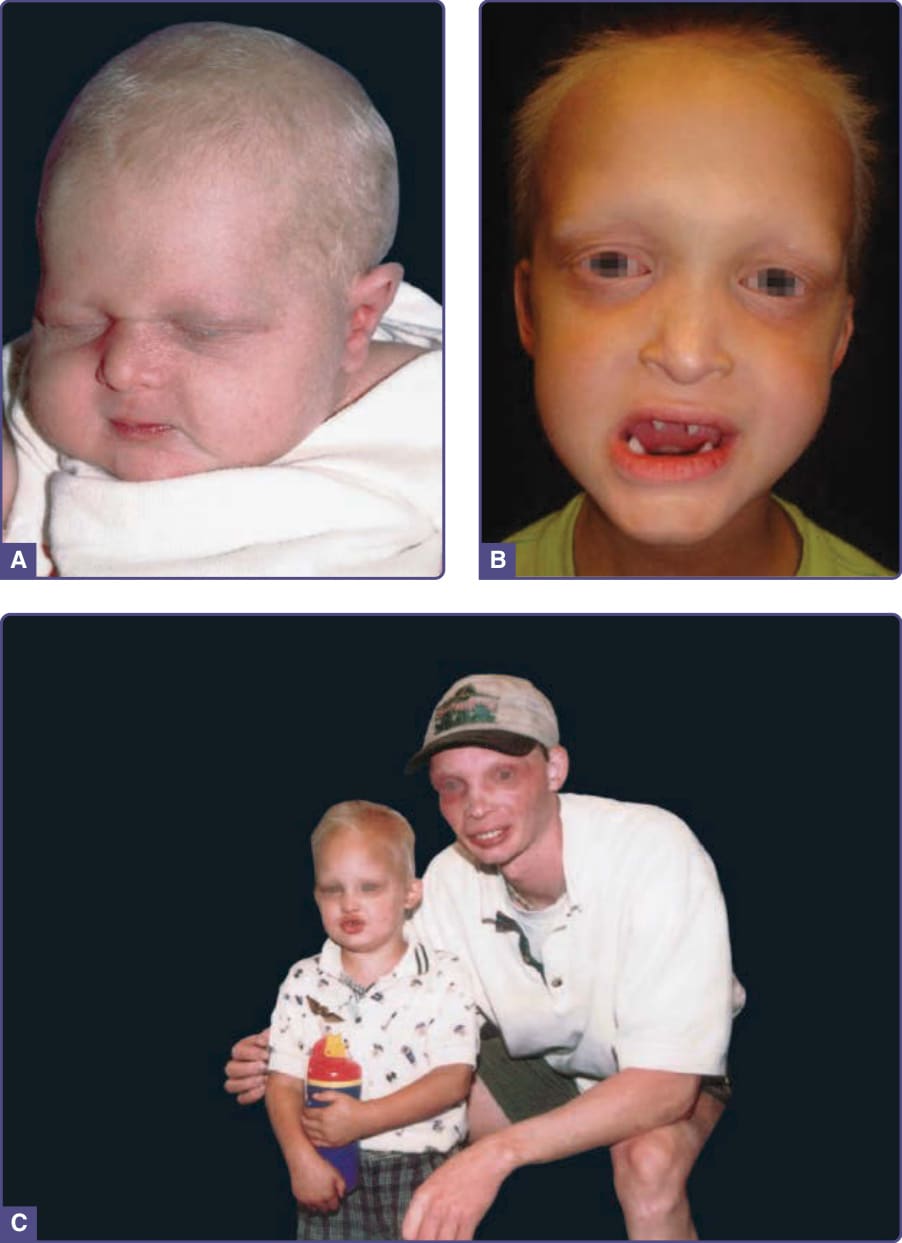
圖 131-2:低汗性外胚層發育不良(眼周皺紋、釘狀牙、成人配戴假牙)。
汗性外胚層發育不良(Clouston 症候群)(Hidrotic Ectodermal Dysplasia)
- 基因:OMIM 129500,13q12 之 GJB6 的 AD 突變,編碼細胞間連接蛋白 connexin 30;具可變表現度,男女受影響相等。最早見於法裔加拿大家系。
- 臨床三特徵(與 HED 相對:出汗與牙齒正常):
- 毛髮硬、脆、蒼白,斑塊狀禿髮可進展至完全禿髮;體毛與顏面毛亦受影響。
- 指甲嬰兒期乳白,漸增厚、營養不良、遠端與甲床分離、可疼痛,曾報告無甲症。
- 進行性掌蹠角化過度。常見結膜炎與眼瞼炎。
- 致病機轉:同一 GJB6 不同突變可致非症候群型 AD 耳聾;connexin 突變亦見於 KID 症候群等。
- 鑑別:其他掌蹠角化過度無類似毛髮變化;指甲似先天性厚甲症 (pachyonychia congenita) 但毛髮變化具區別性。
- 處置:偶需去除甲基質緩解疼痛;假髮有美容益處;掌蹠角皮症治療無特異性、成效甚微。
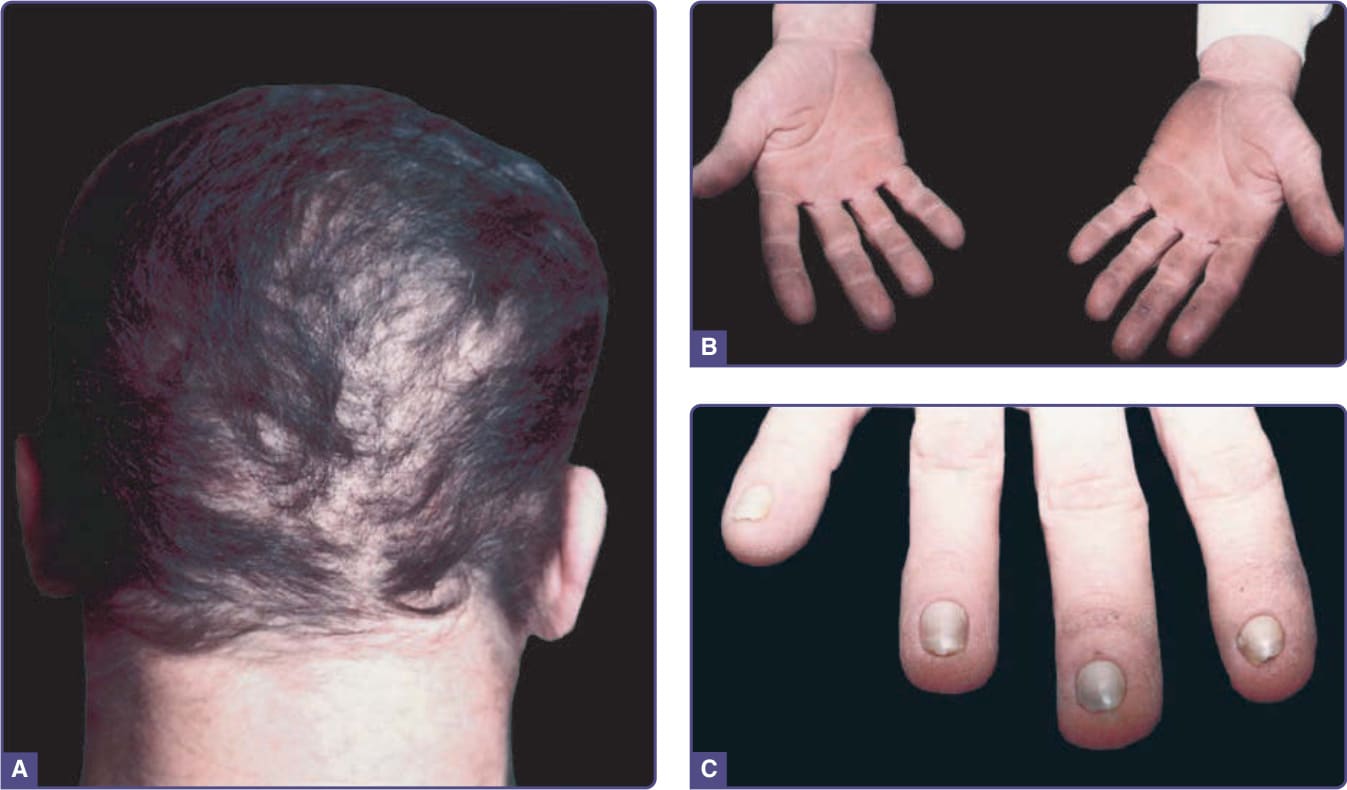
圖 131-5:汗性外胚層發育不良(斑塊狀禿髮、掌部角化過度、甲營養不良)。
p63 相關外胚層發育不良症候群 (P63-Related EDs)
- 共同基因:全部為 AD,3q27 之 TP63(腫瘤抑制基因)突變;廣泛表現於增生性上皮基底細胞,有基因型–表現型相關性。涵蓋 AEC、EEC、肢體–乳腺症候群、ADULT 症候群、裂手–裂足畸形。
AEC(Hay–Wells/Rapp–Hodgkin)症候群
- AEC/Hay–Wells (OMIM 106260)、Rapp–Hodgkin (OMIM 129400);後二者為對偶基因,視為同一疾病變異型。
- 臨床:80%–90% 嬰兒出生時呈發亮、發紅、龜裂脫皮皮膚與表淺糜爛(似膠樣膜),數週脫落;頭皮幾乎一律受影響,常慢性糜爛性皮膚炎伴異常肉芽組織與反覆細菌感染;禿髮、硬粗淡色「難梳理」毛髮;色素異常普遍。指甲多變(正常/增厚/缺如/部分營養不良)。先天性絲狀眼瞼粘連 (AFA) 見於約 70%。淚管閉鎖/阻塞常見。
- Rapp–Hodgkin 特點:鼻小柱短、上頜發育不全、上唇薄下唇厚、圓錐易蛀牙;顎裂(伴或不伴唇裂)見於 80%–100%。
- 病理:輕度表皮萎縮、局灶正角化、伴噬黑素細胞之色素失禁;毛髮可見捻轉發 (pili torti)、三角溝狀毛。
- 鑑別:EEC 有手足骨性異常且無眼瞼粘連;新生兒脫皮糜爛易誤為表皮鬆解水疱症或先天性魚鱗癬。
- 處置:膠樣膜脫落前用輕質潤膚劑;眼瞼粘連可需手術鬆解;頭皮糜爛以溫和傷口照護、稀釋漂白水浸泡,避免封閉性敷料;裂需團隊修復。
EEC 症候群 (OMIM 129900, 604292)
- 分 EEC1、EEC2、EEC3;EEC3 由 TP63 突變所致。屬多發性先天異常症候群。
- 主要區別特徵:缺指(趾)(ectrodactyly),足部多於手部、可不對稱。
- 顎裂(伴或不伴唇裂)70%–100%;牙齒過少、恆齒過早脫落;淚腺異常、續發性傳導性聽力喪失常見;1/3 以上有泌尿生殖異常(腎水腫、腎或生殖器畸形)。ED 表現可輕微(約 4/5 指甲營養不良)。
- 處置:團隊處理口面裂與眼科問題;肢體缺陷個別化;應做腎臟超音波並警覺泌尿道問題。
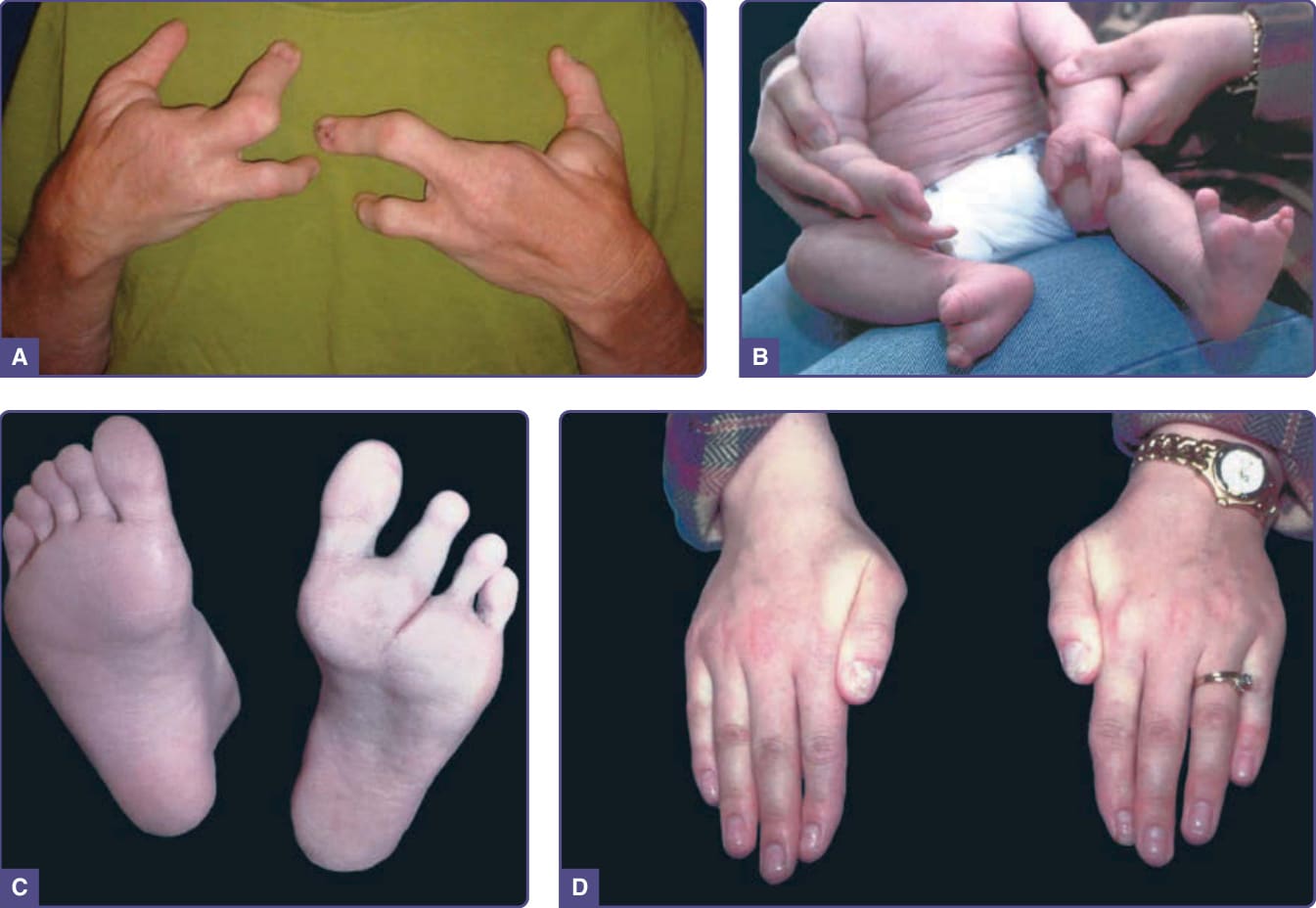
圖 131-8:缺指(趾)–外胚層發育不良–裂 (EEC) 症候群。
- p63 路徑機轉重點:AEC 多為 SAM 結構域之錯義突變(完全外顯率、可變表現度);EEC 多為 DNA 結合結構域之單一胺基酸取代(可變表現度、降低外顯率);裂手–裂足亦在 DNA 結合域;肢體–乳腺症候群為移碼突變。
WNT10A 疾病:OODD、SSPS (WNT10A Disorders)
- 基因:全部為 AR,2q35 之 WNT10A 突變(介導 β-catenin 訊息)。WNT10A 突變見於約 9% EDs、約 16% 疑似 HED 病例。
- OODD (OMIM 257980):最一致為恆齒嚴重牙齒過少至無牙(乳齒數目多正常但細小、間隙寬);指甲營養不良/先天性無甲;毛髮出生缺如;掌蹠角化過度與毛孔角化;掌蹠多汗、身體其餘部位少汗。
- SSPS (OMIM 224750):上述 + 眼瞼汗腺囊瘤 (hidrocystomas)。
- 機轉:多為 AR,但高達 50% 異型合子可顯現臨床特徵。
- 處置:修復性牙科重要;角化過度處置無特異性;SSPS 有非黑色素瘤皮膚癌風險增加,須監測。
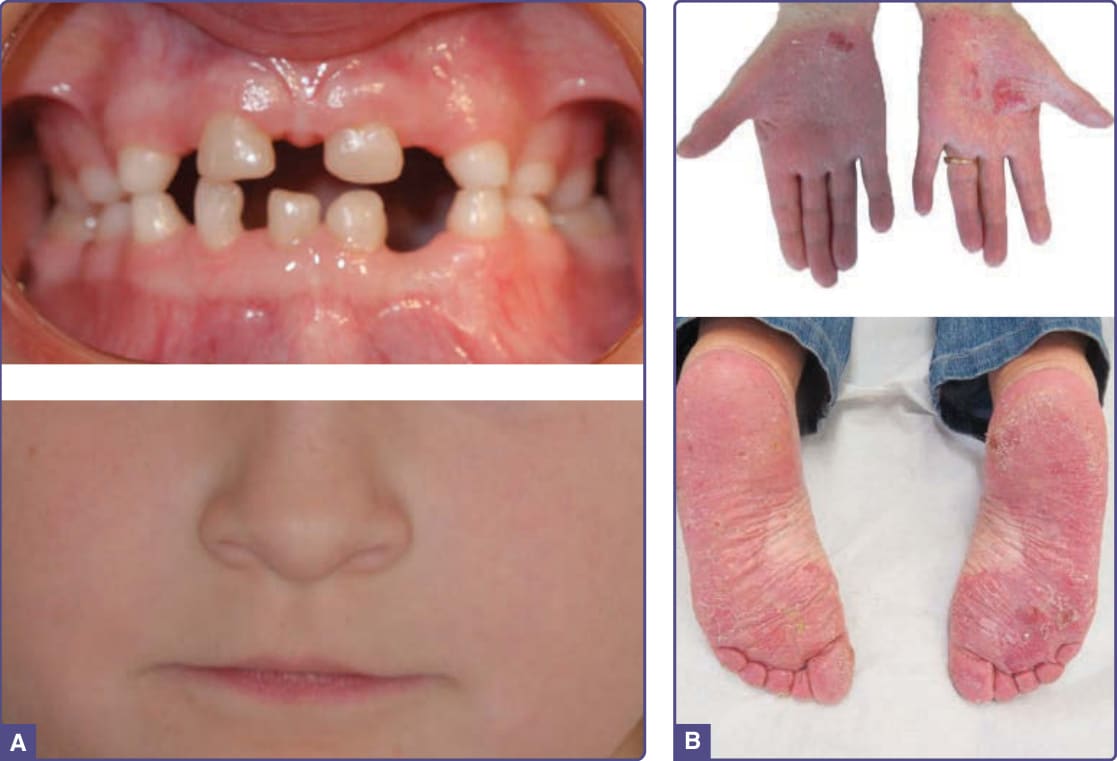
圖 131-9:WNT10A 疾病(散在缺失乳齒、掌蹠紅斑性角化過度)。
局灶性真皮發育不良(Goltz/Goltz-Gorlin 症候群)(Focal Dermal Hypoplasia, FDH)
- 基因:OMIM 305600,XLD,PORCN (Xp11.23),參與 Wnt 蛋白的棕櫚醯化與分泌;男性通常致死,存活男性多因合子後體細胞鑲嵌。
- 皮膚(主要診斷特徵):沿 Blaschko 線之線狀/點狀/條紋狀篩狀萎縮 (cribriform atrophy) 伴微血管擴張;真皮缺如處之脂肪疝出呈黃–粉紅色易壓陷贅生物;終生發展之乳頭狀瘤(好發生殖器周圍、口周、間擦、黏膜);斑塊狀禿髮、掌蹠角化過度。
- 皮膚外:小眼症與虹膜缺損(應做完整眼科評估);寡牙與琺瑯質缺陷;眾多骨骼異常(骨紋狀症 osteopathia striata、併指、缺指、身材矮小);約 15% 智能障礙。
- 機轉提示:應問流產家族史與後代男女比例偏斜(母親帶因線索)。
- 鑑別:Conradi–Hünermann(有魚鱗癬、無脂肪疝出)、IP(有水疱與角化過度)、MIDAS(皮膚缺損限於頭頸、似先天性皮膚發育不全)。
- 處置:無特異性治療;乳頭狀瘤妨礙功能可切除(喉/氣管者轉耳鼻喉並術前評估);血管雷射減紅斑;嚴重度差異大,宜延後預後諮詢。
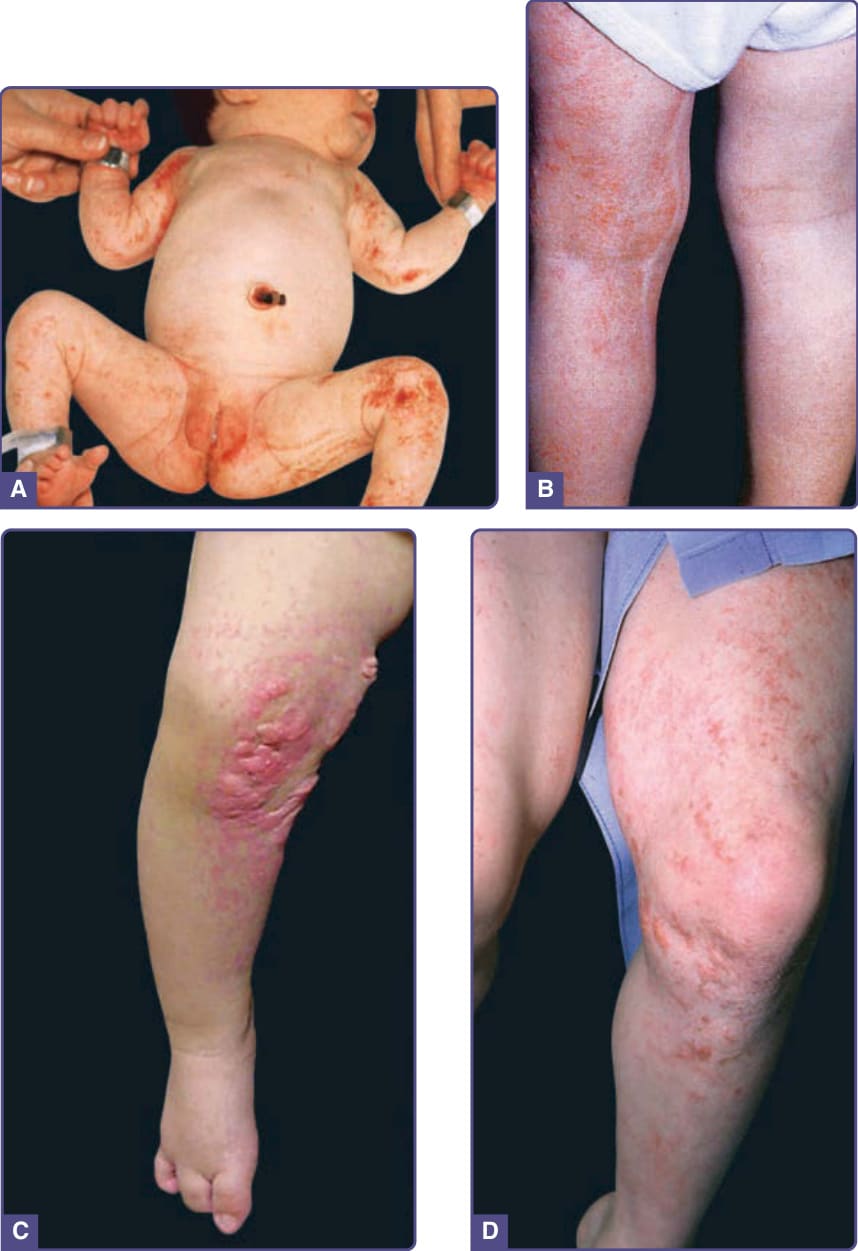
圖 131-10:局灶性真皮發育不良(條紋狀萎縮與脂肪疝出)。
色素失禁症 (Incontinentia Pigmenti, IP)
- 基因:OMIM 308300,XLD,IKBKG (NEMO),Xp28,保護細胞免於凋亡;男性胎兒通常致死(男性病例見於合子後晚期突變、節段性、XXY)。發生率 1/50,000–150,000。約 80% 源自 IKBKG 突變,約 75% 為外顯子 4–10 的 11.7 kb 常見缺失。
- 皮膚四期(皆沿 Blaschko 線,未必全出現、可重疊):
- 第 I 期:圍產期發炎,紅斑、水疱、膿疱;50% 出生即有、90% 於 2 週齡;多 4–6 個月消退。
- 第 II 期:疣狀/角化過度丘疹,持續數週至數月(可達數年)。
- 第 III 期:色素沉著,約 6 個月齡起,最常侵犯腹股溝與腋窩。
- 第 IV 期:色素減退與萎縮,持續至成年。
- 多在 20 歲變細微;瘢痕性與斑塊狀禿髮常見且持續。
- 皮膚外:甲營養不良(第二期,多輕微);牙齒多變(過少、小牙、無牙、圓錐狀);眼科侵犯 20%–77%(視網膜新生血管可致剝離,新生兒期至 6 歲風險最高,須篩檢與重複檢查);CNS 侵犯 10%–30%(癲癇至腦病變、缺血性中風、發育遲緩,多於第一年出現)。
- 診斷:2014 Minic 準則——主要準則為 4 期皮膚病灶任一;次要含牙齒/眼/CNS/毛髮/指甲等異常、家族史、多次男性流產、病理。切片具分期特異性(水疱期見海綿狀水腫、嗜酸性球、微膿瘍、角化不良);基因檢測可查 IKBKG 缺失/重複。
- 處置:多數預後良好、壽命正常;治療以症狀為導向(水疱傷口照護防感染,第一期可局部類固醇);牙科修復;診斷時基線眼科檢查並依指引於前 3 年重複,周邊視網膜光凝固術可降剝離風險;轉神經科,有神經症狀做 MRI 與 EEG。
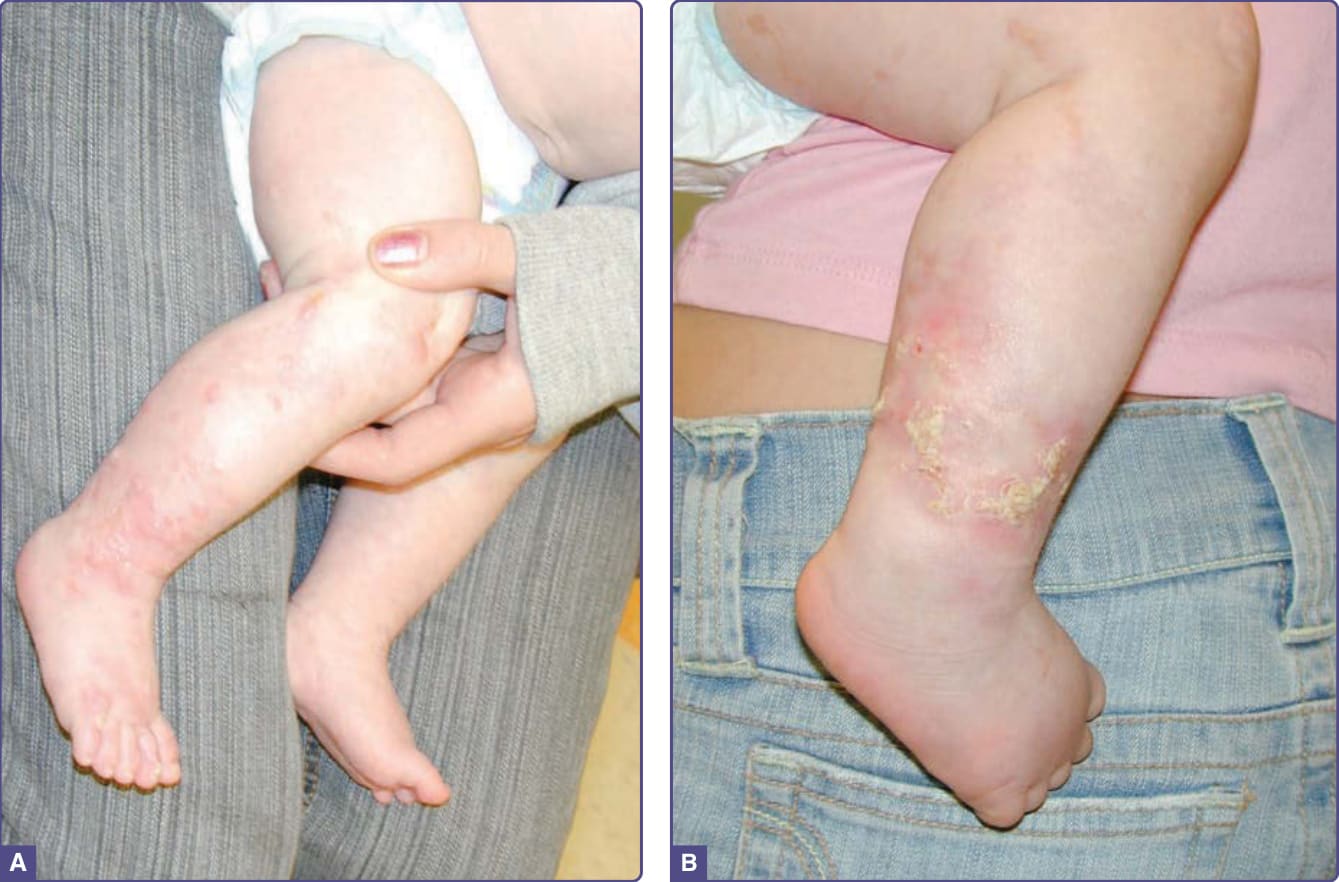
圖 131-11:色素失禁症(第 I 期水疱、第 II 期疣狀病灶)。
牙甲症候群(Witkop 症候群)(Tooth and Nail Syndrome)
- 基因:OMIM 189500,4p16.1 之 MSX1 的 AD 突變(編碼轉錄因子 Msx1),具可變表現度。
- 臨床:指甲薄、小、易碎,出生可有匙狀甲,趾甲較指甲嚴重,隨年齡改善;少數有細稀毛髮。乳齒多正常(可細小),恆齒可無法萌發或部分/完全缺如(最常缺下頜門齒、第二臼齒、上頜犬齒)。無其他外胚層結構受影響。
- 診斷:易被遺漏;缺乏其他皮膚/全身特徵可與其他 ED 區分;基因檢測可用。
- 鑑別:巨牙症、缺牙與毛髮稀疏 (OMIM 272980)。
- 處置:指甲多無需治療;修復性牙科重要。
毛–齒–骨 (TDO) 症候群 (Tricho-Dento-Osseous Syndrome)
- 基因:OMIM 190320,17q21.3-q22 之 DLX3 的 AD 突變,外顯率通常完全。
- 臨床:毛髮捲曲/極度蜷曲(尤其新生兒期);牙齒琺瑯質發育不全、鈣化不足、巨牙症(乳恆齒皆有);指甲增厚伴表層分裂;出汗與皮膚正常;骨密度增加(長骨與顱骨),可有顱縫早閉致長頭畸形。
- 鑑別:AIHHT 有相同牙齒發現但無毛髮與指甲變化。
- 處置:琺瑯質牙科照護;提供遺傳諮詢。
眼–齒–指(趾)發育不良 (Oculo-Dento-Digital Dysplasia, ODDD)
- 基因:OMIM 164200,6p21-23.2 之 GJA1 的 AD 突變(編碼 connexin 43)。
- 臨床:毛髮稀疏、乾燥、生長緩慢,睫毛缺如/稀疏;乳恆齒琺瑯質發育不全(牙細小、發黃、易碎、易蛀);汗腺與皮膚正常;短瞼裂伴小角膜/小眼症(視力多正常);>80% 有第四、五指屈指畸形 (camptodactyly),可併指;其他骨骼異常(顱骨增生、寬肋骨鎖骨)。
- 處置:琺瑯質牙科照護;眼科追蹤——10%–15% 發展青光眼。
毛–鼻–指(趾)骨症候群 (Tricho-Rhino-Phalangeal Syndrome, TRPS)
- 分類:TRPS I (OMIM 190350) 與 TRPS III (OMIM 190351)——8q23.2 之 TRPS1 AD 突變;TRPS II(Langer–Giedion,OMIM 150230)——含 TRPS1 與 EXT1 的連續基因缺失。
- 非典型 ED(僅侵犯毛髮,不及牙齒/指甲/汗腺)。
- 臨床:TRPS I——毛髮稀疏、脆、生長慢,可見捻轉發;突出梨形鼻、寬高鼻樑、球狀鼻尖、人中長而平、上唇薄、下巴後縮;錐形骨骺 (cone-shaped epiphyses) 致中節指骨偏斜(手指彎曲外觀)、髖部畸形、身材矮小。TRPS III 另有嚴重短指與更矮;TRPS II 合併多發性外生骨疣與常見智能障礙。
- 處置:身材矮小可個別考慮生長激素;骨骼變化監測與止痛;耳廓整形與鼻整形可美觀矯正。
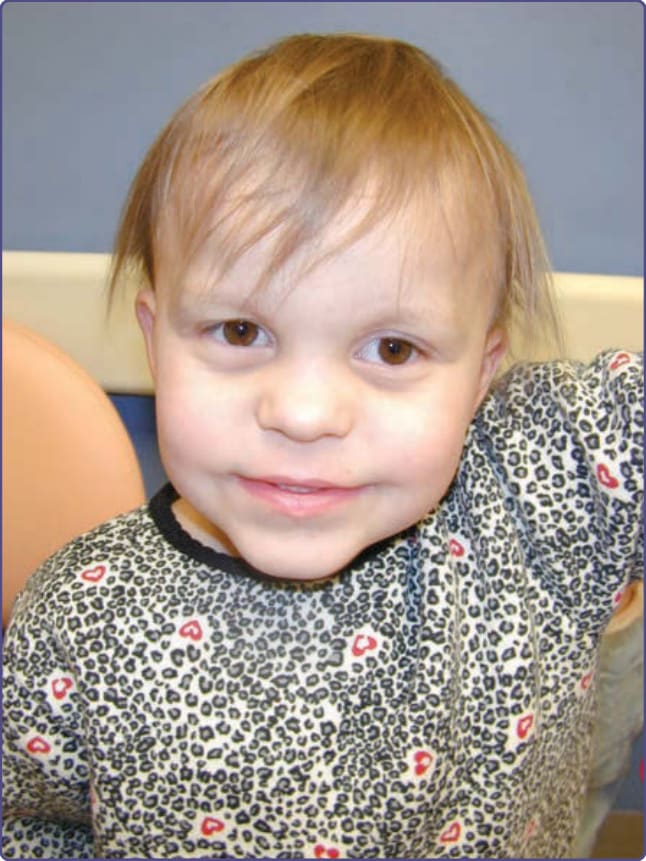
圖 131-13:毛–鼻–指(趾)骨症候群(稀疏毛髮與眉毛、球狀鼻、薄上唇)。