Ectodermal Dysplasias
21
AT-A-GLANCE
■ A group of inherited disorders characterized by developmental abnormalities in 2 or more ectodermal structures.
■ These include hair, teeth, nails, and sebaceous and sweat glands.
■ There may be abnormalities in nonectodermal structures and functions.
■ Distinction is based on clinical features, mode of inheritance, and molecular/genetic findings.
■ Clinically distinct disorders may be due to different mutations in the same gene (allelic heterogeneity), and clinically similar conditions may be due to mutations in different genes (locus heterogeneity).
INTRODUCTION
The ectodermal dysplasias (EDs) are a complex and heterogenous group of inherited disorders that share developmental defects involving at least 2 of the major structures classically known to derive from the embryonic ectoderm: hair, teeth, nails, and sweat and other eccrine and sebaceous glands. Developmental disorders involving only one type of ectodermal structure, even if associated with other congenital malformations, are not classified as EDs. There are almost 200 conditions classified as EDs, and given the varied clinical presentations and clinical overlap, it can be difficult to diagnose them precisely.1
Various classification schemes have been proposed over the past several decades to help in ordering and thinking about these disorders. Freire-Maia and Pinheiro published an exhaustive review and classification system, with later updates, for these disorders using a numeric system.2 Although this system developed a rational approach to a previously chaotic field, it has little use in clinical practice and does not account for the pathogenesis or genetics of specific conditions. Over the past few decades, many of the genes responsible for these disorders have been identified, leading to several attempts to reclassify the EDs based on molecular and developmental biopsy. In 2008, an international ED classification conference consisting
of health care providers, researchers, patient advocate representatives, and administrators convened in an attempt to move toward a more useful and unified system. In 2009, a conference report was released outlining the goal of creating an integrated ED classification system based on the most recent clinical and molecular information available.3 Given the rate of discovery of new molecular information, this has been challenging. Thus, in 2012, a second international conference was held resulting in the development of a multi-axis model approach to EDs.4 This model will be based on a clinical/phenotype axis, a gene-based axis, and a functional/pathways axis and is still in the process of creation. This chapter will only cover the EDs that are more common and most likely to present to a dermatologist for diagnosis and medical attention. As much as possible, it will be organized in concordance with the ED international classification conference, limited by the complexities discussed above. Given the evolving landscape of EDs, the reader is directed to the following resources for the most current information on EDs: https://omim.org (Online Catalog of Mendelian Inheritance in Man) and http://www.ncbi.nlm.nih. gov/gtr/ (an up-to-date listing of laboratories offering molecular testing).
21
Approach to the diagnosis of ectodermal dysplasias
No/severely reduced sweating Normal/mildly reduced sweating
Severe immune defect HED-ID (IKBKG) No immune defect X-LR HED AD-HED AR-HED
- Limb abnormality EEC Limb–mammary ADULT Margarita Island ED
WNT10A disorders Focal Dermal Hypoplasia/Goltz Syndrome Oculodentodigital Syndrome Tooth and nail/Witkop Tricho-dento-osseous
Normal teeth: Hidrotic ED ED/skin fragility Tricho-Rhino- Phalangeal Syndrome
Abnormal teeth ±facial cleft
- Limb abnormality AEC (Hay-Wells, Rapp-Hodgkin)
No facial cleft
HYPOHIDROTIC ECTODERMAL DYSPLASIA: XLHED, ADHED, ARHED, HED-ID
AT-A-GLANCE
■ Variants:
■ XLHED (OMIM 305100): XLR mutation of EDA1 on Xq12-q13.1 encoding the triggering ligand molecule ectodysplasin-A
■ ADHED (OMIM 129490): AD mutation of EDARADD/EDAR on 1q42.2-q43/2q11-q13; the intracellular molecule adaptor of EDAR death domain/transmembrane receptor of EDA
■ ARHED (OMIM 224900): AR mutation of EDARADD/EDAR on 1q42.2-q43/2q11-q13; the intracellular molecule adaptor of EDAR death domain/transmembrane receptor of EDA
■ HED with immune deficiency (HED-ID; OMIM 300291): XLR mutation of IKBKG on Xq28 encoding NF-κB cytoplasmic inhibitor (also referred to as NF-κB essential modulator/NEMO)
■ Ectodermal features: characterized by hypotrichosis, hypohidrosis, and hypodontia.
■ Systemic features: characteristic facial features with frontal bossing and depressed midface. Often associated with atopic dermatitis, asthma, upper respiratory tract infections, pneumonias
INTRODUCTION
INTRODUCTION
One of the earliest descriptions of hypohidrotic ectodermal dysplasia (HED) was in 1848, and involved male first cousins and their grandmother with sparse hair, missing teeth, and dry skin.8 HED is the most common ED,9 specifically the X-linked recessive (XLHED) form of HED, and accounts for 80% of the families registered with NFED. There are several forms of HED, including XLHED (also known as Christ–Siemens–Touraine syndrome), autosomal dominant HED (ADHED), autosomal recessive HED (ARHED), and HED with immunodeficiency (HED-ID). The autosomal dominant and autosomal recessive forms of HED are similar to XLHED, although the autosomal dominant form may be milder.10 HED-ID should be considered in a male with HED and recurrent or significant infections.11,12
EPIDEMIOLOGY
EPIDEMIOLOGY
XLHED occurs in all racial groups and is thought to have an incidence at birth of anywhere from 1 in 5,000 to 100,000 births.10,13,14 Accurate prevalence and incidence data are not available for the other forms of HED.
CLINICAL FEATURES
CLINICAL FEATURES
Clinical features across the variants are similar. HED is characterized by hypotrichosis, hypohidrosis,
2373
21
and hypodontia. Affected males with XLHED may present at birth with a collodion membrane or with marked scaling of the skin,15 similar to congenital ichthyosis. Scalp hair is usually sparse, fine, and blonde. It may thicken and darken at puberty, and secondary sexual hair is typically normal. Other body hair is usually sparse or absent. The ability to sweat is significantly compromised, and most affected males with XLHED have marked heat intolerance.16 The inability to sweat adequately in response to environmental heat results in an elevation of core temperature and bouts of unexplained high fevers, usually leading to an extensive workup for infectious disease, malignancy, or autoimmune disease before the correct diagnosis is recognized. In an older series of patients, intellectual disability was reported as a feature of XLHED. Currently, this is believed to
A B
C
have been due to damage from prolonged high fevers and seizures and not to be an intrinsic feature of the disorder.17 Individuals with ADHED appear to have a milder defect in the ability to sweat. The nails are usually normal, although there are reports of thin, fragile nails.18 Fingerprint ridges are effaced. Periorbital wrinkling and hyperpigmentation are typical and often present at birth (Fig. 131-2). Eczema affects more than two-thirds of affected males and almost half of affected females.19 Hyperplasia of sebaceous glands, particularly on the face, can develop over time and appear as small, pearly, skin-colored to white papules that may resemble milia. Hypodontia, oligodontia, or anodontia are invariable features of XLHED in affected males. Hypoplastic alveolar ridges in an affected infant can be an early clue to the diagnosis of the disorder. Teeth that do erupt are usually peg-shaped and small (Fig. 131-2B).
2374
A B
21
The facial features of the disorder are characterized by frontal bossing and a depressed midface with a saddle nose and full, everted lips. Otolaryngologic and pulmonary manifestations include thick nasal secretions and impaction, ozena (atrophic rhinitis), sinusitis, recurrent upper respiratory tract infections and pneumonias, decreased saliva production, hoarse voice, and an increased frequency of asthma. The increased frequency of respiratory tract infections has been attributed to hypoplastic or absent mucus-secreting glands in the bronchial tree.20 Gastroesophageal reflux and feeding difficulties may be a problem in infancy. The basis for this is unknown. Preliminary studies suggest that there may be failure to thrive in infancy and early childhood in as many as 20% to 40% of affected boys, with catch-up growth seen later.21
Female carriers for XLHED may be affected as severely as males or may show few, if any, signs of the disorder (Fig. 131-3). Between 60% and 80% of carrier females express some clinical signs of the disorder; the most frequent are patchy hypotrichosis and hypodontia or peg-shaped teeth. Heat intolerance, if present, is usually mild. Adult carrier women comment that they do not sweat much or that they do not like very warm weather, but it is unusual for a female to experience fever because of inability to sweat.22
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
HED is genetically heterogenous, with several forms related to different genes involved in the tumor necrosis factor α (TNF-α) signaling pathway.9 Mutations in this pathway lead to interruption of the interaction between epithelial cells and underlying mesenchyme, leading to the clinically observed features that are similar between the different forms of
HED. Figure 131-4 demonstrates the ectodysplasin signal transduction pathway and how it relates to each form of HED. XLHED results from alterations in the gene EDA1 encoding ectodysplasin-A located at Xq12-13.1.10
Approximately 70% of affected males inherited the mutation from a carrier mother. EDA1 codes for a
Ectodysplasin signal transduction pathway
EDA XL-HED
EDAR AR-HED, AD-HED
EDARADD
AR-HED, AD-HED
TAB2 TRAF6
cytoplasm
TAK1
IKK1 IKK2
IKBKG (NEMO)
HED-ID
I-κBα NF-κB
HED-ID
transcription of target genes
NF-κB
nucleus
2375
21
transmembrane protein, ectodysplasin, which plays a role in regulation of the formation of ectodermal structures. It forms trimers and is expressed in keratinocytes, the outer root sheath of hair follicles, and sweat glands. It localizes to the lateral and apical surfaces of cells. A multitude of mutations in this gene causing XLHED have been identified. Interfamilial and intrafamilial variation occurs, but there are reports of genotype-phenotype correlation of skin and hair findings.23
Mutations in the autosomal genes, EDAR, located at 2q11-q13 and EDARADD, located at 1q42.2-q43, have been implicated in both an autosomal dominant form of hypohidrotic ectodermal dysplasia (ADHED; OMIM
129490) and in an autosomal recessive form (ARHED; OMIM 224900). As discussed above, these entities are clinically similar to XLHED, but are rarer. EDAR acts as a receptor for ectodysplasin and EDARADD acts as an intracellular adaptor protein that assists in transmitting the signal from the activated EDA receptor to the nucleus of the cell.10
Certain mutations in the X-linked IKBKG gene, which causes incontinentia pigmenti (IP) in females have been shown to result in HED-ID in males. There are 2 similar OMIM defined syndromes, HED-ID (OMIM#300291) and ectodermal dysplasia, anhidrotic with immunodeficiency, osteopetrosis, and lymphedema (OLEDAID; OMIM#300301) that describe males with ED and immune deficiency with mutations in IKBKG.11,12 These patients demonstrate ED along with dysgammaglobulinemia and significant early morbidity and mortality. The complexity of the NFKB signaling pathway accounts for the diverse genotypes of HED-ID and immunologic phenotypes.
DIAGNOSIS
DIAGNOSIS
The diagnosis of HED is recognized readily when expected, such as when a child is born into a family with a known history of HED. However, without advance knowledge, the diagnosis can initially be clinically challenging. As the patient ages, the characteristic features of hypotrichosis, hypohidrosis, and hypodontia become more evident.22 Hypoplastic alveolar ridges, indicating lack of teeth, can be an early diagnostic clue. A Panorex view of the jaw can assist in making the correct diagnosis. Although rarely necessary, evaluation of sweating by examination for sweat pores with iodine solution, quantification of pilocarpine-induced sweating,23 or skin biopsy to assess for the absence of eccrine structures in the scalp and/or palmar region may help confirm a diagnosis.24 In an isolated, fully expressing female, the autosomal dominant and recessive forms of HED need to be considered. Family history review is necessary, and mothers always should be examined fully to detect mild manifestations of the X-linked form. With the recent expansion and development of molecular genetic testing, definitive genetic diagnosis is becoming more readily available. Referral
2376
■Collodion membrane
■Collodion membrane
■Self-healing collodion baby
■Self-healing collodion baby
■Neutral lipid storage disease
■Neutral lipid storage disease
■Autosomal recessive congenital ichthyosis
■Autosomal recessive congenital ichthyosis
■Nonbullous congenital ichthyosiform erythroderma
■Nonbullous congenital ichthyosiform erythroderma
■Lamellar ichthyosis
■Lamellar ichthyosis
■Trichothiodystrophy
■Trichothiodystrophy
■Storage diseases (eg, Gaucher)
■Storage diseases (eg, Gaucher)
■Chondrodysplasia punctata
■Chondrodysplasia punctata
■Ankyloblepharon filiforme adnatum (AFA)
■Ankyloblepharon filiforme adnatum (AFA)
■Lethal popliteal pterygium syndrome
■Lethal popliteal pterygium syndrome
■Popliteal pterygium syndrome
■Popliteal pterygium syndrome
■Isolated AFA
■Isolated AFA
■AFA and cleft palate
■AFA and cleft palate
■Hypodontia
■Hypodontia
■Isolated hypodontia
■Isolated hypodontia
■Incontinentia pigmenti
■Incontinentia pigmenti
■Skin fragility/erosions
■Skin fragility/erosions
■Epidermolysis bullosa (all subtypes)
■Epidermolysis bullosa (all subtypes)
■Incontinentia pigmenti
■Incontinentia pigmenti
■Acrodermatitis enteropathica
■Acrodermatitis enteropathica
■Congenital erythropoietic porphyria
■Congenital erythropoietic porphyria
■Atrophic streaks
■Atrophic streaks
■Incontinentia pigmenti—stage 4
■Incontinentia pigmenti—stage 4
■MIDAS (microphthalmia, dermal aplasia, and sclerocornea)
■MIDAS (microphthalmia, dermal aplasia, and sclerocornea)
■Focal dermal hypoplasia
■Focal dermal hypoplasia
to a pediatric dermatologist or geneticist can assist in diagnosis. On histopathology, the epidermis is thinned and flattened. There is a reduction in the number of sebaceous glands and hair follicles. Eccrine glands are absent or incompletely developed. Histologic evaluation of the skin is usually not necessary.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Maintenance of cool ambient temperatures is vital to prevent hyperpyrexia. Most children do well with simple measures, such as wet T-shirts and headbands, air conditioning in home and school, etc. Occasionally, cooling vests allow a broader range of
participation in sports and vigorous physical activity in warm climates. Dental restoration is of primary importance, and early implementation of dentures and ultimate use of dental implants are mainstays of treatment. Management of otolaryngologic complications, asthma, and recurrent infections needs to be individualized. The eczema may be quite refractory to care and difficult to manage. Infants with HED are at increased risk of death due to hyperthermia and potentially other features of the disorder, such as infection.25,26 In a survey of the Ectodermal Dysplasia International Registry (EDIR), 21% of XLHED reported a family history of infant or childhood death.19
Although infancy and childhood are complicated by many problems, most individuals with HED lead adult lives that allow them to function successfully in society. Heat intolerance seems to decrease because of the development of some ability to sweat in adolescence or to the development adaptation of lifestyle, or both. Recombinant ectodysplasin protein injections of pregnant affected mice led to permanent rescue of the phenotypic features in the offspring.27 Further, early postnatal injections were also shown to ameliorate the developmental defects. Similar results have been noted in affected canine models who were infused with recombinant ectodysplasin protein in the postnatal period, with resultant normalization of teeth, lacrimation, and sweating ability, as well as decreased eye and respiratory infections.28,29 These developments in animal models have been encouraging. There are currently human trials under way.
A
21
HIDROTIC ECTODERMAL DYSPLASIA (CLOUSTON)
AT-A-GLANCE
■ OMIM 129500, AD mutation of GJB6 on 13q12 which encodes connexin 30, a connexin protein of the intercellular junction
■ Sparse, wiry pale hair with progressive alopecia; variable onychodystrophy; progressive palmoplantar hyperkeratosis
■ Normal sweating and teeth
EPIDEMIOLOGY
EPIDEMIOLOGY
Hidrotic ED was first described in a French–Canadian kindred.30 It has been reported in other ethnic groups, but the majority of affected individuals can trace their ancestry back to an original French–Canadian settler.
CLINICAL FEATURES
CLINICAL FEATURES
The scalp hair is wiry, brittle, and pale, and there is often patchy alopecia (Fig. 131-5). This progresses in adult life and may lead to total alopecia. Body and facial hair are affected. The nails may be milky white
B
C
2377
21
in infancy and early childhood, gradually thickening and becoming dystrophic. The nail plates in adults are thick, short, and slow growing. They separate distally from the nail bed (Fig. 131-5C), and may cause pain. Anonychia has been reported. Not all the nails are necessarily affected to the same degree. Progressive palmar/plantar hyperkeratosis is common (Fig. 131-5B). In contrast to HED, sweating is normal, as are the teeth. Oral leukoplakia has been reported. Conjunctivitis and blepharitis, possibly due to poor function of sparse eyelashes, are common.31
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Hidrotic ED is autosomal dominant with variable expression, and the degree of severity can vary within and between families. Males and females are affected in equal numbers and to equal degree. The disorder is caused by mutations in a connexin gene, GJB6, which encodes the intercellular junction protein connexin 30.32 Different mutations in the same gene are responsible for a form of nonsyndromic autosomal dominant deafness. Connexin mutations are involved in several other genodermatoses, such as keratitisichthyosis-deafness (KID) syndrome (Chaps. 47 and 48).
DIAGNOSIS
DIAGNOSIS
The diagnosis is straightforward with time. The involvement of nails and hair and palmar/plantar thickening, in the absence of other signs of ED, are reasonably specific. Again, genetic testing can be confirmatory.31
On histopathology, the thickened palms and soles show orthohyperkeratosis with a normal granular layer. On electron microscopy, an increase in the number of desmosomes in the cells of the stratum corneum is found. The hair shows nonspecific changes.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
Other palmar/plantar hyperkeratoses do not have similar hair changes. Orofacial clefting differentiates other forms of autosomal dominant hidrotic EDs, such as ankyloblepharon–ED–cleft palate (AEC) syndrome. Although the nail changes are similar to those of pachyonychia congenita, the hair changes are distinctive.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Occasionally, ablation of the nail matrix is necessary for relief of pain. Wigs may provide cosmetic benefit.
2378
Treatment of the palmoplantar keratoderma is not specific and minimally successful.
P63-RELATED ECTODERMAL DYSPLASIA SYNDROMES: AEC (HAY–WELLS/RAPP– HODGKIN); EEC; LIMB– MAMMARY SYNDROME; ACRO-DERMATO-UNGUAL– LACRIMAL–TOOTH (ADULT) SYNDROME; ISOLATED SPLIT HAND–SPLIT FOOT MALFORMATION
AT-A-GLANCE
■ Variants: all of the conditions below are AD with a mutation of TP63 on 3q27, which encodes the p63 transcription protein
■ Ankyloblepharon-ectodermal defects-clefting (AEC)/Hay–Wells/Rapp–Hodgkin syndrome (AEC/Hay–Wells OMIM 106260; Rapp– Hodgkin syndrome OMIM 129400)
■ Limb–mammary syndrome (OMIM 603543)
■ Acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome (OMIM 103285)
■ Ectrodactyly-ED-cleft lip/palate syndrome 3 (EEC3; OMIM 604292)
■ Ectodermal features: collodion membrane at birth, dry thin skin, erosive scalp dermatitis, patchy alopecia, coarse light hair, dyspigmentation, variable nail dystrophy.
■ Systemic features: Ankyloblepharon filiforme adnatum, lacrimal duct atresia, cleft lip/palate, hypodontia, syndactyly, ectopic breast tissue and hypospadias.
INTRODUCTION
INTRODUCTION
Mutations in p63, a tumor-suppressor gene mapped to 3q27, have been found in most, but not all, individuals with ankyloblepharon-ectodermal defects-clefting (AEC; Hay–Wells; Rapp–Hodgkin) syndrome.33 The gene is expressed widely, including in the basal cells of proliferating epithelial tissues. Mutations in the same gene cause some cases of isolated split hand–split foot malformation, limb–mammary syndrome, and acro-dermato-unguallacrimal-tooth (ADULT) syndrome. There appear to be some genotype–phenotype correlations.34
AEC (HAY–WELLS/RAPP– HODGKIN) SYNDROME
AEC (HAY–WELLS/RAPP–
HODGKIN) SYNDROME
EPIDEMIOLOGY
Hay–Wells syndrome was first described in 1976,35
and has been found in ethnically and geographically disparate families. Rapp–Hodgkin syndrome was first described in 1968,36 and although it had many clinical similarities to Hay–Wells syndrome, it was once thought to be a distinct clinical entity. More recently, it has become evident that Rapp–Hodgkin syndrome and Hay–Wells syndrome are allelic and should be considered variants of the same disorder, AEC syndrome.37,38
CLINICAL FEATURES
Eighty to 90% of affected infants present at birth with shiny red, cracking, peeling skin, and superficial erosions, similar to the appearance of a collodion
A B
C
21
membrane (Fig. 131-6A).18 This sheds within a few weeks, and the skin underneath is dry and thin. The scalp is almost invariably affected. Many individuals have chronic erosive dermatitis with abnormal granulation tissue on the scalp and recurrent bacterial infections (Fig. 131-6B).39 There is alopecia of the scalp of variable extent along with hair that is often wiry, coarse, and light in color with an uncombable appearance. Sparseness to absence of body hair is typical. Cutaneous dyspigmentation, both hypo- and hyperpigmentation, can be quite striking and is seen universally in affected patients.40,41 The nails may be normal, hyperconvex and thickened, absent, or partially dystrophic, and all changes can be found in a single individual and can worsen with age. Effaced dermatoglyphics and palmoplantar erosive changes are also quite common. Sweating may be normal to slightly decreased. Although most affected individuals describe subjective heat intolerance, frank hyperpyrexia is uncommon. Ankyloblepharon filiforme adnatum (AFA), the term for strands of skin between the eyelids, is seen in approximately 70% of affected infants (Fig. 131-6C).
2379
21
These may tear spontaneously prior to birth, and minimally involve the lateral eyelids or require surgical lysis. Lacrimal duct atresia or obstruction is common. Supernumerary nipples and ectopic breast tissue are seen occasionally, as is mild cutaneous syndactyly of the second and third toes. More prominent limb defects, including ectrodactyly (abnormal development of the median rays of the hands and feet), have been observed.42
Historically, the main clinical differences between Hay–Wells and Rapp–Hodgkin syndrome included characteristic facial features with a short nasal columella and maxillary hypoplasia, thin upper lip, and full lower lip associated with Rapp–Hodgkin syndrome (Fig. 131-7A,B). Also, with Rapp–Hodgkin syndrome, the teeth may be conical and prone to caries (Fig. 131-7D), lacrimal puncta are aplastic in almost one-third of affected individuals, but AFA is rare, and scalp involvement with breakdown and granulation tissue formation is far less common. Cleft palate, with or without cleft lip, occurs in 80% to 100% of reported cases, with some cases displaying submucosal cleft palate alone.43 There is typically hypodontia with missing or misshapen teeth, and
A
maxillary hypoplasia is also common.44 Malformed auricles have been described in some. Recurrent otitis media and secondary conductive hearing loss are common and may be consequences of the cleft palate. Hypospadias has been described in affected males, and labial hypoplasia with absence of the opening of the vagina has been reported in a single female. These features are also reminiscent of the EEC syndrome.
DIAGNOSIS
See discussion of etiology and pathogenesis for information on genetic testing. On histopathology, consistent changes include mild epidermal atrophy, focal orthokeratosis, prominent superficial vascular plexus, and pigment incontinence with melanophages.45 Electron microscopy of hairs shows a defective cuticular structure, atrophy and loss of melanin, along with structural abnormalities including pili torti and pili trianguli et canaliculi. There is a decrease in the keratins of the basal and suprabasal layers of the epidermis and disorganized keratin filaments in the stratum corneum.
B
C D
2380
DIFFERENTIAL DIAGNOSIS
Among the autosomal dominant EDs associated with clefting, EEC syndrome is characterized by bony hand and foot abnormalities not typically seen in AEC and also lacks the ankyloblepharon. The peeling, eroded skin of the newborn can lead to misdiagnosis of epidermolysis bullosa or congenital ichthyosis. AFA can occur in the absence of syndromic associations, and strands of tissue between the eyelids have been seen in several forms of arthrogryposis and in CHANDS (curly hair, ankyloblepharon, and nail dysplasia syndrome), an autosomal recessive form of ED.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
Light emollients should be used until the collodion membrane sheds. Ankyloblepharon may require surgical lysis. Ongoing ocular hygiene is important. The skin erosions, especially of the scalp, are difficult to manage and prone to excessive granulation tissue and secondary infection. They should be managed with gentle wound care, dilute bleach or other antimicrobial soaks. Occlusive dressings should be avoided.40
Grafting of skin to the scalp has not proven successful in most instances. Clefting requires a team approach for repair and followup for secondary issues, such as feeding difficulties, speech defects, orthodontia, and ear infections.
EEC SYNDROME (OMIM 129900,604292)
EEC syndrome is an ED classified as a multiple congenital anomaly syndrome because it has major involvement of structures other than those derived from the ectoderm. There are 3 variants of EEC syndrome, EEC1 (OMIM 129900), EEC2 (no OMIM number) and EEC3 (OMIM 604292). EEC1 and EEC2 have unknown gene product mutations, but EEC1 has been linked to a mutation on chromosome 7. EEC3 is caused by an autosomal dominant mutation in the tumor-suppressor gene TP63, designating the EECs as p63-related EDs. EEC syndrome has occurred in all racial groups worldwide, and all 3 forms share clinical manifestations.
CLINICAL FEATURES
CLINICAL FEATURES
The ectodermal dysplasia manifestations may be quite mild. The hair is usually blonde, coarse, and dry. It may be sparse and slow growing. Axillary and pubic hair also may be affected. The nails are dystrophic in approximately four-fifths of individuals with transverse ridging, pitting, and slow growth. Dry skin and thickening of the palms and soles can occur. Sweating is usually normal.46
21
The major distinguishing feature of EEC syndrome is ectrodactyly (Figs. 131-8A-C). The feet are involved more frequently than the hands, and there may be asymmetry of involvement. Cleft palate, with or without cleft lip, occurs in 70% to 100% of affected individuals.46,47 Hypodontia and premature loss of secondary teeth and the dental abnormalities associated with clefting are found in most affected individuals. Lacrimal gland abnormalities and secondary conductive hearing loss are common. Genitourinary abnormalities that include hydronephrosis and structural renal or genital malformations affect one-third or more of persons with EEC syndrome. Although intellectual disability has been reported, it is not believed to be an inherent feature of the disorder.
DIFFERENTIAL DIAGNOSIS
Disorders with limb defects that need to be considered in the differential diagnosis of EEC syndrome are odontotrichomelic syndrome (OMIM 273400), in which there are severe absence deformities of the limbs, and aplasia cutis congenita with limb defects (Adams–Oliver syndrome, OMIM 100300), which does not have clefting or ectodermal defects other than absence of skin. Cutis marmorata telangiectatica congenita (Chap. 103) has been described in some individuals with Adams–Oliver syndrome. Other EDs with clefting include AEC and limb–mammary syndrome, all of which are allelic to EEC syndrome. Ectrodactyly with cleft palate without ED (OMIM 129830) may be a distinct entity. There appear to be some families with EEC in whom linkage studies have suggested other causal genes located at different chromosomal loci. Prenatal diagnosis by ultrasound for detection of limb abnormalities is unreliable; genetic testing may prove useful in some families.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
As for other EDs with orofacial clefting and ophthalmologic involvement, management requires a team approach. Similarly, treatment for the limb defects must be individualized. Renal ultrasonography and a high index of suspicion for urinary tract problems are appropriate and warranted.
P63-RELATED ECTODERMAL DYSPLASIA SYNDROMES
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
AEC, EEC, split hand–split foot malformation, limb– mammary syndrome, and ADULT syndrome are
2381
21
A
C
D
B
caused by mutations in the tumor-suppressor gene TP63 mapped to 3q27. Different mutations within this gene have been implicated in the pathogenesis of these syndromes.34,48 TP63 is expressed widely, including in the basal cells of proliferating epithelial tissues. AEC syndrome is an autosomal dominant disorder with complete penetrance and variable expression. The majority of alterations identified thus far in AEC syndrome have been missense mutations in the sterile α motif (SAM) domain of p63. EEC syndrome is an autosomal dominant disorder with variable expression and reduced penetrance. Mutations in TP63 have been found in most, but not all, individuals with EEC syndrome. The majority of mutations result in single–amino acid substitutions in the DNA-binding domain of p63.33
Similar to EEC, mutations in the DNA-binding domain of TP63 are also found in split hand–split foot malformation.49 Frameshift mutations in TP63 cause limb–mammary syndrome, in which ectodermal structures other than the mammary gland often, but not always, appear normal. Among 3 unrelated families with ADULT syndrome, all shared the same point mutation in p63.50
2382
WNT10A DISORDERS: ODONTO-ONYCHO- DERMAL DYSPLASIA (OODD), SCHOPF– SCHULZ–PASSARGE SYNDROME (SSPS)
AT-A-GLANCE
■ Variants: all of the below are AR with a mutation of WNT10A (wingless-type MMTV integration site family, member 10A) on 2q35, which mediates β catenin–mediated specific intracellular signaling
■ Odonto-onycho-dermal dysplasia (OODD; OMIM 257980)
■ Schopf–Schulz–Passarge Syndrome (SSPS; OMIM 224750)
(Continued)
AT-A-GLANCE (Continued)
Continued
AT A GLANCE (
)
■ Both OODD and SSPS demonstrate variable degrees of hypodontia, dystrophic nails, hypotrichosis, palmar plantar hyperkeratosis with hyperhidrosis, hypohidrosis on rest of body.
■ SSPS is also characterized by eyelid hidrocystomas
INTRODUCTION
INTRODUCTION
Odonto-onycho-dermal dysplasia (OODD) and Schopf–Schulz–Passarge Syndrome (SSPS) are both forms of ED caused by mutations in the WNT10A gene.
EPIDEMIOLOGY
EPIDEMIOLOGY
Mutations in WNT10A have been reported in about 9% of EDs and in 25% of hypohidrotic ED (HED) patients who do not have mutations in EDA.51,52 With recent advances in molecular genetic testing, WNT10A mutations have been found in about 16% of suspected cases of HED.9
21
CLINICAL FEATURES
CLINICAL FEATURES
OODD has a broad spectrum of clinical features. The most consistent finding is severe hypodontia to anodontia of the permanent teeth. Primary teeth are almost always normal in number but may be small and widely spaced (Fig. 131-9A).53-55 The tongue may be smooth, with decreased fungiform and filiform papillae. The nails are dystrophic, and there can be congenital anonychia. Hair is absent at birth, progressing to dry, thin hair in older individuals. Eyebrows may be sparse. Skin changes include palmar erythema, palmar plantar hyperkeratosis (Fig. 131-9B),56 and keratosis pilaris. The palms and soles commonly have hyperhidrosis, although there is hypohidrosis elsewhere on the body. SSPS is characterized by eyelid hidrocystomas; in addition, the findings described above in OODD.57
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
OODD and SSPS are caused by mutations in WNT10A (wingless-type MMTV integrations site family, member 10A) gene.57 Most cases are autosomal recessive, but up to 50% of heterozygotes may display clinical features.
B
A
2383
21
DIAGNOSIS
DIAGNOSIS
On histopathology, the skin shows orthokeratosis, hyperkeratosis, hypergranulosis, and mild acanthosis. When examined by EM, the hairs of OODD have longitudinal depressions. Again, genetic testing can be helpful.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Restorative dentistry is important. Hyperkeratosis management is nonspecific and the nails usually require no treatment. There is potential increased risk of nonmelanoma skin cancer in SSP, and patients should be monitored appropriately.
FOCAL DERMAL HYPOPLASIA (GOLTZ SYNDROME; GOLTZ- GORLIN SYNDROME)
AT-A-GLANCE
■ OMIM 305600
■ XLD, PORCN, Xp11.23, involved in membrane targeting and secretion of Wnt proteins necessary for embryonic tissue development
■ Cutaneous features: Blaschkoid dermal atrophy with fat herniations; progressive papillomas; patchy alopecia
■ Extracutaneous features: microphthalmia and colobomas; oligodontia and enamel defects; numerous skeletal anomalies including syndactyly and osteopathia striata; developmental delays in 15%
INTRODUCTION
INTRODUCTION
Focal dermal hypoplasia (FDH) is a rare ectodermal dysplasia with multisystem involvement, including skin, teeth, bones, and eyes with variable clinical manifestations.58
EPIDEMIOLOGY
EPIDEMIOLOGY
FDH was first described in 1934.59 Subsequently, there have been more than 200 case reports published.
2384
CLINICAL FEATURES
CLINICAL FEATURES
The skin changes of FDH are the primary diagnostic features with considerable variability because of both postzygotic somatic mosaicism in both males and females and random X-chromosome inactivation in females. There is linear, punctate, streaky cribriform atrophy (Fig. 131-10), with telangiectasia distributed along the lines of Blaschko. The cribriform atrophy is marked by tiny ice pick–like depressions in the skin. Areas of thinned to absent dermis are irregularly distributed and the resultant herniations of fat appear as yellow–pink excrescences that are easily depressed on the skin surface (Fig. 131-10). Papillomas that may be raspberry-like or vascular develop throughout life and favor the perigenital, perioral, intertriginous, and mucosal surfaces. Other dermatologic features include pigmentary changes, patchy alopecia, brittle or sparse hair, and palmar and plantar hyperkeratoses. Some individuals have had hyperhidrosis and some have had aplasia cutis congenita. The other organ systems most frequently involved in FDH are the skeleton, CNS, teeth, and eyes. Microphthalmia and colobomas are common, and the diagnosis of FDH should prompt a full ophthalmologic evaluation. Oligodontia, tooth dysplasia, and enamel defects are common. The skeletal abnormalities are too numerous to list; the most common are vertical banding of the bones (osteopathia striata), syndactyly (both cutaneous and bony), ectrodactyly, asymmetry, oligodactyly, and short stature. Intellectual disability has been reported in approximately 15% of cases. Defects in other organ systems have been described in a minority of cases, including cardiac defects, abdominal wall defects, and renal malformations.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Focal dermal hypoplasia is an X-linked dominant disorder, usually lethal in males. Case reports of males with the condition are believed to be due to mosaicism for postzygotic somatic mutations (as is true for incontinentia pigmenti); the presence of some normal cells allows survival in the male. The mutated gene is PORCN, the human homolog of the porcupine gene in Drosophila. PORCN is thought to be important for palmitoylation and secretion of Wnt protein, a key regulator of the development of skin and bone.60,61
It is important to inquire about the family history of lost pregnancies and a skewed male–female ratio in offspring, as these are clues to the mother being a carrier. Both mothers and fathers should be examined carefully; fathers may have subtle features and, presumably, represent individuals with postzygotic mutations for the FDH gene.
21
B
A
C
D
DIAGNOSIS
DIAGNOSIS
The diagnosis of FDH is challenging given the rarity of the syndrome. Biopsy can be helpful in demonstrating the dermal hypoplasia and increased capillaries in the papillary dermis.58 Additionally, genetic testing is commercially available, but can be challenging in patients with postzygotic mutations.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
Cribriform atrophy has been described in X-linked dominant Conradi–Hünermann syndrome (chondrodysplasia punctata), but ichthyosis is not a feature of FDH, and fat herniation is not part of Conradi–Hünermann. The
Blaschkoid distribution of the atrophic lesions of IP is similar, but the blistering and hyperkeratosis of IP are not found in FDH. In microphthalmia and linear skin defects/ microphthalmia, dermal aplasia, sclerocornea (MIDAS; OMIM 309801), the skin defects are limited to the head and neck; there is atrophy and scarring of the skin more similar to aplasia cutis congenita and not dermal atrophy alone. The disorders do share similar ocular abnormalities.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
There is no specific treatment for the dermatologic and systemic features of FDH. Areas of atrophy may
2385
21
be prone to infection or become erosive. Papillomas can be excised if they interfere with function. Referral to otolaryngology may be necessary for management of papillomas of the larynx or trachea and preoperative evaluation for these lesions should be considered.62 Use of vascular lasers to decrease the erythema of telangiectatic areas may have cosmetic benefit. As with most X-linked dominant disorders, clinical involvement varies considerably, and the range in severity is marked. This makes prognostic counseling difficult early in infancy, and usually it is wise to counsel patience and reassurance until the extent to which there is systemic involvement becomes clear.
INCONTINENTIA PIGMENTI
AT-A-GLANCE
■ OMIM 308300
■ XLD, IKBKG (also called NEMO), Xp28, encodes the I-kappa-B kinase gamma subunit protein involved in cell protection from apoptosis
■ Cutaneous features: 4 characteristic stages involving (1) vesicobullous lesions; (2) verrucous lesions; (3) hyperpigmentation; and (4) hypopigmentation and atrophy. May also see hypotrichosis, cicatricial alopecia, and tooth defects.
■ Extracutaneous features: ophthalmologic complications, CNS manifestations (seizures, developmental delay)
INTRODUCTION
INTRODUCTION
Incontinentia pigmenti (IP), also known as male-lethal type Bloch–Sulzberger syndrome, was first described in 1906 by Garrod.63 IP is an ectodermal dysplasia with highly variable involvement of skin, teeth, hair, nails, eyes, and the CNS.64 The skin findings are the first clinical manifestation.65
EPIDEMIOLOGY
EPIDEMIOLOGY
IP occurs in 1 in 50,000 to 150,000 newborns.65 It is X-linked dominant and usually lethal in male fetuses; however, similar to FDH, there are reports of male cases in the setting of late postzygotic mutations, segmental involvement, or XXY genotype.
CLINICAL FEATURES
CLINICAL FEATURES
There are 4 characteristic stages of skin lesions, all of which occur along the lines of Blaschko.
2386
Not every stage may occur, and some stages may overlap.
■ Stage I: Perinatal inflammatory stage with erythema, vesicles, and pustules with individual lesions lasting for several days to weeks (Fig. 131-11A). Present at birth in 50% and by 2 weeks of age in 90%. Typically resolves by 4 to 6 months, but may have recurrences with illnesses.66
■ Stage II: Verrucous and hyperkeratotic papules persistent for several weeks to months, with reports of it lasting up to years (Fig. 131-11B).
■ Stage III: Hyperpigmentation that usually presents around 6 months of age and persists for several years, with reports of persistence into adulthood. Highly variable extent, often unrelated to the distribution in the previous stages. Most frequently involves the groin and axillae.64
■ Stage IV: Hypopigmentation and atrophy in previously affected areas. Starts with the resolution of the previous stages and lasting into adulthood.
Many of the cutaneous features resolve or become very subtle by the 20 years of age. Cicatricial and patchy alopecia are common and tend to be persistent. In adult females, areas of hypotrichosis on the legs and whorled alopecia of the scalp can be helpful clinically.67
Nail dystrophy is common during the second stage, but often mild.64 There is a wide range of teeth findings, including hypodontia, microdontia, adontia, and conical teeth. Ophthalmological involvement is present in 20% to 77%64,65 of affected individuals and includes retinal neovascularization, strabismus, cataracts, optic atrophy, and retinal pigmentary abnormalities. It is important to screen for eye findings at the time of diagnosis because retinal neovascularization can result in retinal detachment, with the highest risk in the neonatal period and up to the first 6 years of life.64 Given the risk of decreased visual acuity, repeat eye examinations are necessary. CNS involvement is estimated at 10% to 30%, although it may be lower.64,65 CNS abnormalities range from a single seizure to encephalopathy, ischemic stroke, and significant developmental delay. Symptoms most often present in the first year of life, which is consistent with the hypothesis that neurologic manifestations are a result of inflammation and vasculopathy/ischemia.68 CNS manifestations highly impact morbidity and mortality, and predictive features, such as retinal neovascularization and male gender, are helpful for assessing risk.68
Unilateral breast aplasia is uncommon, but should be alert one to the possible diagnosis of IP.69
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
IP is an X-linked dominant disorder, typically lethal in males (see discussion regarding mosaicism for
A
21
B
postzygotic mutations above in FDH). About 80% of IP are due to mutations in the IKBKG gene, which encodes the I-kappa-B kinase gamma subunit protein (IKK-gamma). More than 50 distinct postzygotic somatic mutation variants in the IKBKG gene have been reported. The IKK-gamma protein is widely expressed and binds to the IKK-alpha and IKK-beta proteins to activate the KF-kappa-B complex. This complex protects cells from TNF-induced apoptosis. The different stages of IP represent cell death of affected cells that are unable to activate this protective complex. It is proposed that after cells with the IKBKG mutation undergo apoptosis, they are replaced by unaffected cells. Late recurrences may be due to persistence of affected keratinocytes in the sites of previous lesions.65
DIAGNOSIS
DIAGNOSIS
In 2014, Minic et al updated the diagnostic criteria. Major criteria include any of the 4 stages of skin lesions. Minor criteria include dental, ocular, CNS, hair, nail, palate, breast, and nipple anomalies; family history; multiple male miscarriages; and histopathologic skin findings.64,69
Skin biopsy greatly aids in diagnosis, especially in suspected males, and histopathology is stage
specific. Biopsy of vesiculobullous lesions reveals a spongiotic epidermis with eosinophilic infiltrates and microabscesses, and dyskeratosis. Verrucous lesions demonstrate hyperkeratosis, papillomatosis, and dyskeratosis. Genetic testing is commercially available for IKBKG common deletion/duplication and gene sequencing, which identifies IP and HED-ID. About 75% of affected patients have the common 11.7kb deletion of exons 4 to 10.65 Testing can confirm a clinical diagnosis, detect female carriers, and evaluate at-risk pregnancies in families carrying the IKBKG mutation. Without family history, prenatal diagnosis is difficult as there are no characteristic ultrasound features other than intrauterine growth retardation.64
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
The differential for IP depends on what point in the time course the patient is presenting. In the neonatal period, vesiculobullous disorders, including HSV, VZV, bullous impetigo, bullous mastocytosis, and epidermolysis bullosa, and other mosaic disorders, such as focal dermal hypoplasia and epidermal nevus syndrome, may be considered. Later on, linear epidermal nevus, ILVEN, and pigmentary mosaicism, can appear similar.
2387
21
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Most affected individuals without significant complications in infancy do well with a normal life expectancy. Treatment is symptom directed. The skin changes do not require specific treatment other than wound care of blisters to prevent secondary infection.64 There are reports of topical corticosteroids minimizing inflammation and accelerating healing in the first stage.66 As with other EDs, dental evaluation and restoration is important. A baseline eye examination at diagnosis and following the recommended guidelines for repeat examinations during the first 3 years of life is critical. Peripheral retinal photocoagulation can reduce the risk of retinal detachment. Referral to neurology at time of diagnosis for a comprehensive examination and anticipatory guidance should be considered and is imperative for the management of seizures, spasticity, or other deficits if needed.64 If neurologic symptoms are present, MRI and EEG should be performed. MRI can be considered in affected individuals with retinal neovascularization.64
TOOTH AND NAIL SYNDROME (WITKOP SYNDROME)
AT-A-GLANCE
■ OMIM 189500, AD mutation of MSX1on 4p16.1, which encodes the transcription factor Msx1
■ Characterized by small, friable nails at birth that may improve over time; may have fine, sparse hair. Primary teeth are usually unaffected, but secondary teeth fail to erupt or have partial absence
EPIDEMIOLOGY
EPIDEMIOLOGY
The earliest case was reported by Witkop in 1965, when he described a pedigree demonstrating autosomal dominant inheritance. Along with Hudson, he later went on to describe 23 cases in 6 families in 1975.70 All demonstrated hypoplastic nails and hypodontia; the latter manifested as failure of permanent teeth to erupt.
CLINICAL FEATURES
CLINICAL FEATURES
In tooth and nail syndrome, the nails are thin, small, and friable and may show koilonychia at birth. Toenails
2388
A
B
are usually more severely involved than fingernails (Fig. 131-12). Nail changes improve with age and may be unappreciated in affected adults. A few individuals have reported thin, fine hair.71
The primary teeth usually are unaffected, although they may be small. The secondary teeth may fail to erupt, and there can be partial or total absence (Fig. 131-12A). The mandibular incisors, second molars, and maxillary canines are missing most often.70
No other ectodermal structures are affected.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
A nonsense mutation in MSX1, a gene expressed in the developing teeth and nail beds, was first identified in 2001 in a 3-generation family.72 In 2013, an unrelated patient was found to have an MSX1 mutation as well.73
Other mutations in MSX1 have been associated with isolated tooth agenesis or tooth agenesis with cleft palate, and mouse models have demonstrated the role of MSX1 in tooth and nail development.72 Tooth and nail syndrome is autosomal dominant, with variable expression and intrafamilial variability.
DIAGNOSIS
DIAGNOSIS
This is an easy condition to miss. The nail changes may be subtle. The tooth abnormalities may be mild enough to escape detection by a physician. The lack of associated features, either dermatologic or systemic, readily distinguish Witkop syndrome from other EDs. Genetic testing is available.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
There is a presumed autosomal recessive disorder termed taurodontia, absent teeth, and sparse hair (OMIM 272980) that appears similar.74 Taurodontia refers to teeth with an elongated body and pulp chamber and short roots.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
The nails usually require no treatment. Restorative dentistry is important.
TRICHO-DENTO-OSSEOUS (TDO) SYNDROME
AT-A-GLANCE
■ OMIM 190320, AD mutation of DLX3 on 17q21.3-q22, which encodes the transcription factor homeobox protein DLX-3
■ Ectodermal features: teeth, nail, and hair changes in the setting of normal sweating and skin
■ Systemic features: increased bone density
INTRODUCTION
INTRODUCTION
Tricho-dento-osseous (TDO) syndrome is a rare autosomal dominant ED that involves the teeth, hair, nails, and bone.75 It has been described in several families, although exact epidemiologic information is lacking.
CLINICAL FEATURES
CLINICAL FEATURES
TDO involves the hair, teeth, and nails. Hair is kinky or extremely curly, especially during the neonatal period. Dental findings include enamel hypoplasia, hypocalcification, and taurodontism (enlargement of the body
21
of the tooth and pulp chamber) in both primary and secondary teeth. Nails are often thickened with splitting in the superficial layers. Sweat gland function and skin are unaffected. Skeletal findings included increased bone density of the long bones and skull. There can be craniosynostosis resulting in dolichocephaly.76
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
TDO is an autosomal dominant disorder caused by mutations in DLX3, which encodes the human distal-less homeobox. Depending on the mutation, there are different severities in phenotype, but penetrance is usually complete.77 The DLX3 protein is a transcriptional activator in the development and differentiation of epithelial tissue and has been shown to regulate hair follicles.78,79
DIAGNOSIS
DIAGNOSIS
Clinical molecular genetic testing is available through several labs.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
TDO has the same dental findings as amelogenesis imperfecta (hypomaturation-hypoplasia type) with taurodontism (AIHHT), but AIHHT lacks hair and nail findings.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Management includes dental care for the enamel hypoplasia. Given its autosomal dominant nature, genetic counseling should be provided.
OCULO-DENTO-DIGITAL DYSPLASIA (ODDD)
AT A GLANCE
■ OMIM 164200
■ AD mutation of GJA1 on 6p21-23.2, which encodes connexin 43, a connexin protein of the intercellular junction
■ Characterized by sparse hair, enamel hypoplasia, camptodactyly, and small eyes
2389
21
INTRODUCTION
INTRODUCTION
Oculo-dento-digital dysplasia (ODDD) is an autosomal dominant ED with variable expression.
CLINICAL FEATURES
CLINICAL FEATURES
ODDD patients have sparse, dry, slow-growing scalp hair with absent or sparse eyelashes. Both the primary and secondary teeth have enamel hypoplasia, resulting in small, yellow, and friable teeth prone to decay. Sweat glands and skin are normal. Although vision is normal, the eyes have short palpebral fissures with microcornea and/or microphthalmia. Digital changes include camptodactyly of the fourth and fifth fingers in more than 80% of cases, with possible syndactyly of the fourth and fifth digits. Other skeletal anomalies include hyperostosis of the skull, broad ribs and clavicles, and abnormal trabeculation of the long bones.80
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
ODDD is autosomal dominant and caused by mutations in the gene for connexin-43 (GJA1).81 Clinical molecular genetic testing is available.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Management includes dental care of the enamel hypoplasia. Although vision is normal, patients should be followed by ophthalmology as 10% to 15% develop glaucoma.82
TRICHO-RHINO- PHALANGEAL SYNDROME
AT-A-GLANCE
■ Variants:
■ TRPS I (OMIM 190350): AD mutation of TRPS1 on 8q23.2; mutation in the same gene causes TRPS III (OMIM 190351)
■ TRPS II (Langer–Giedion Syndrome; OMIM 150230): AD, contiguous gene syndrome involving TRPS1 and EXT1, 8q24.11-q24.13
■ Characterized by sparse hair, prominent nose, receding chin, and cone-shaped epiphyses leading to deviation of the middle phalanges
2390
INTRODUCTION
INTRODUCTION
Tricho-rhino-phalangeal (TRP) syndrome is an AD malformation syndrome characterized by distinctive craniofacial and skeletal anomalies. Although it is not considered a typical ED given the involvement of hair alone without involvement of the teeth, nails, or sweat/sebaceous glands, this chapter best encompasses its other features.
CLINICAL FEATURES
CLINICAL FEATURES
TRPS I patients have sparse, brittle, slow-growing scalp hair. On light microscopy, pili torti can be appreciated. They have a prominent, pear-shaped nose with a broad high nasal bridge and bulbous tip. The philtrum is long and flat with a thin upper lip and receding chin (Fig. 131-13).83
Digital changes may not appear until several years of age and include cone-shaped epiphyses and deviation of the middle phalanges giving the appearance of crooked fingers. Other skeletal abnormalities include hip malformations and short stature.84
TRPS III has the features above in addition to the presence of severe brachydactyly and more pronounced short stature.85 TRPSII combines the features of TRPS1 and multiple exostoses type 1. Additionally, these patients often have intellectual disabilities.86
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
TRPS I and TRPS III are autosomal dominant and caused by a mutation in TRPS1, a transcription factor zinc finger protein.83 TRPS II is caused by a deletion that includes both TRPS1 and EXT1. Genetic testing is available to assist with diagnosis.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
For short stature, consideration should be given to growth hormone treatment on an individual basis.87
The skeletal changes should be monitored and managed appropriately for pain if needed. Otoplasty and rhinoplasty can provide aesthetic correction.88
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Alanna F. Bree, Nnenna Agim, and Virginia P. Sybert, authors of prior versions of this chapter.
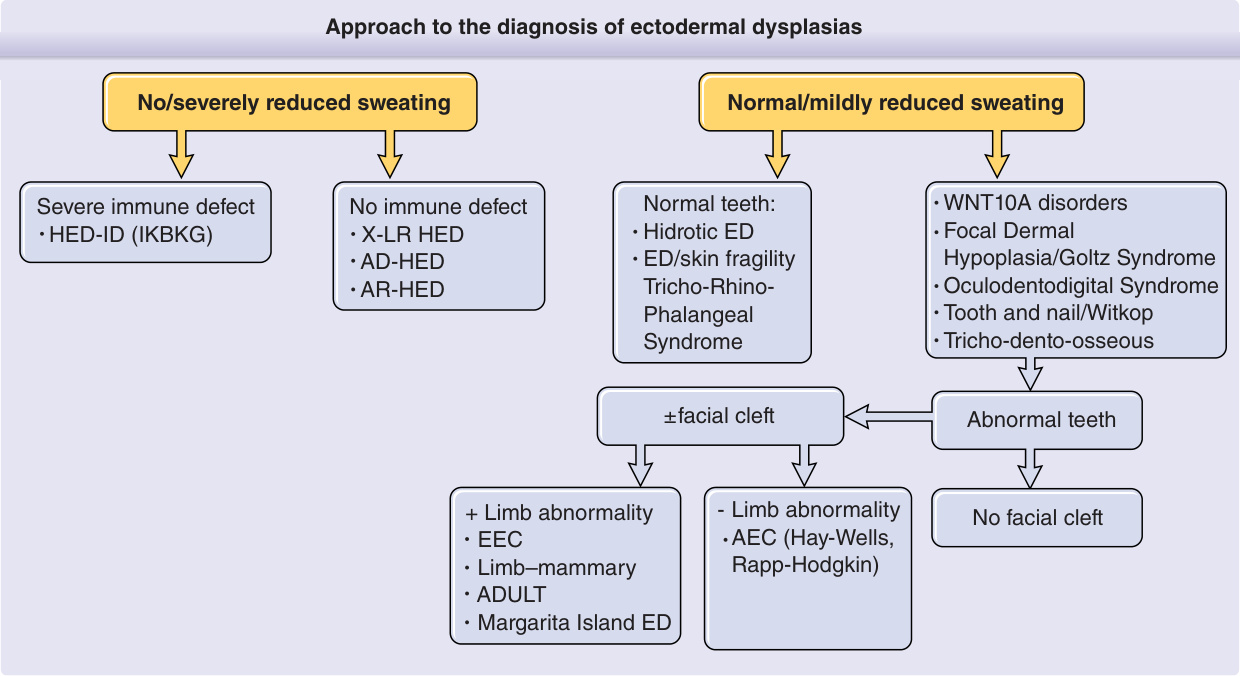
Figure 131-1 is an algorithm showing the clinical approach to the diagnosis of EDs. The first step in the algorithm for making a specific diagnosis of an ED is to determine the presence of sweating (hidrotic) or absence of sweating (hypohidrotic/anhidrotic). The involvement of other ectodermal structures and of non–ectodermal derived tissues provides further branching points in a diagnostic hierarchy. Mode of inheritance may differ within a seemingly uniform diagnostic group, and care must be taken in evaluating family members before providing genetic counseling. The National Foundation for Ectodermal Dysplasias (NFED; http://www.nfed.org) is a lay support group that has numerous informative pamphlets for families and physicians, as well as a strong advocacy program for dental care, insurance coverage, and research. With the knowledge gained from studies on the molecular pathways leading to EDs, there is hope that molecularbased therapies will be able to treat or replace defective or missing teeth, hair follicles, or eccrine sweat glands.5-7
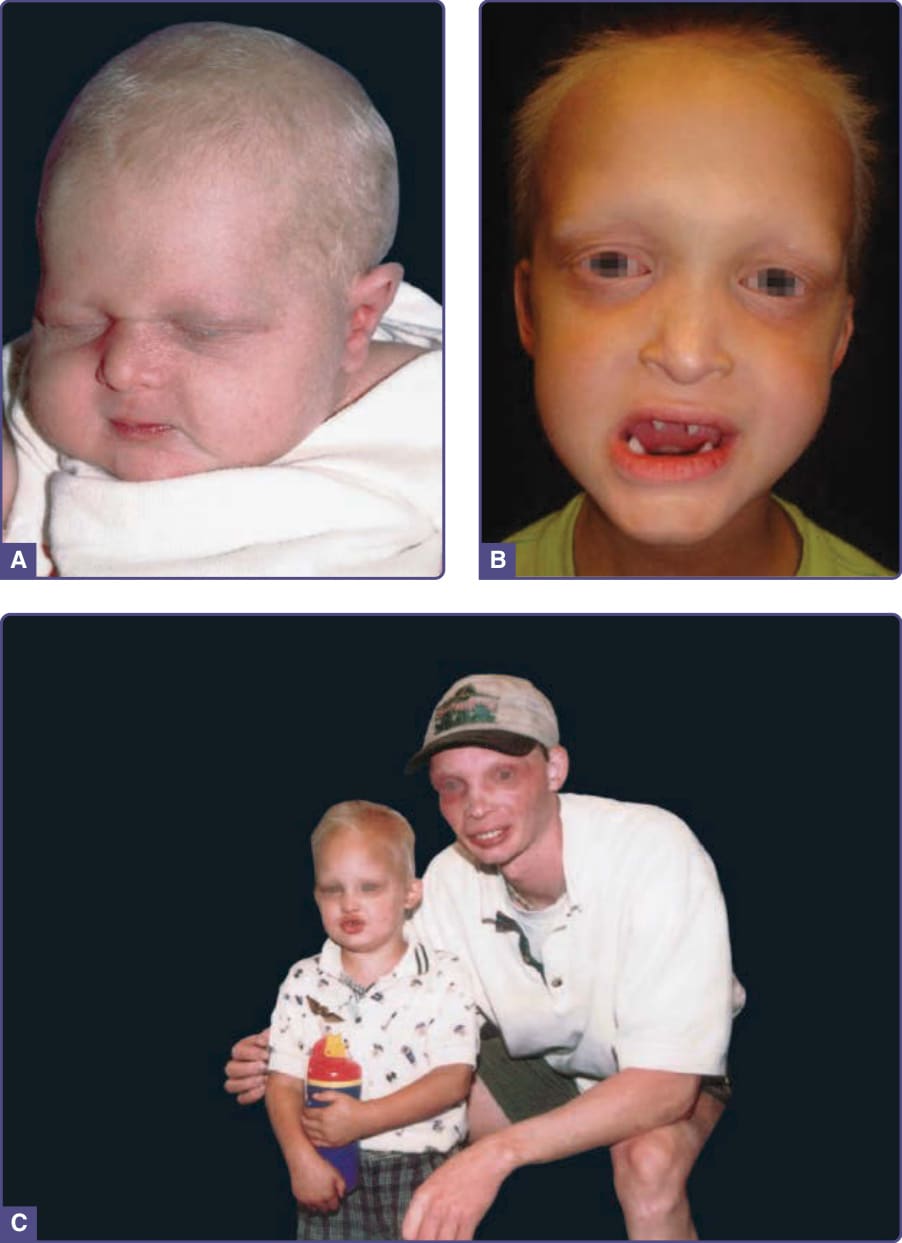
Figure 131-2 Hypohidrotic ectodermal dysplasia. A, Newborn with periorbital wrinkling, beaked nose. Diagnosis would not be suspected unless there was a positive family history. B, Peg-shaped teeth; fine periorbital wrinkling can be appreciated. C, Two unrelated males with X-linked hypohidrotic ectodermal dysplasia; adult is wearing dentures; periorbital wrinkling and hyperpigmentation are evident. (Used with permission from the National Foundation for Ectodermal Dysplasias.)
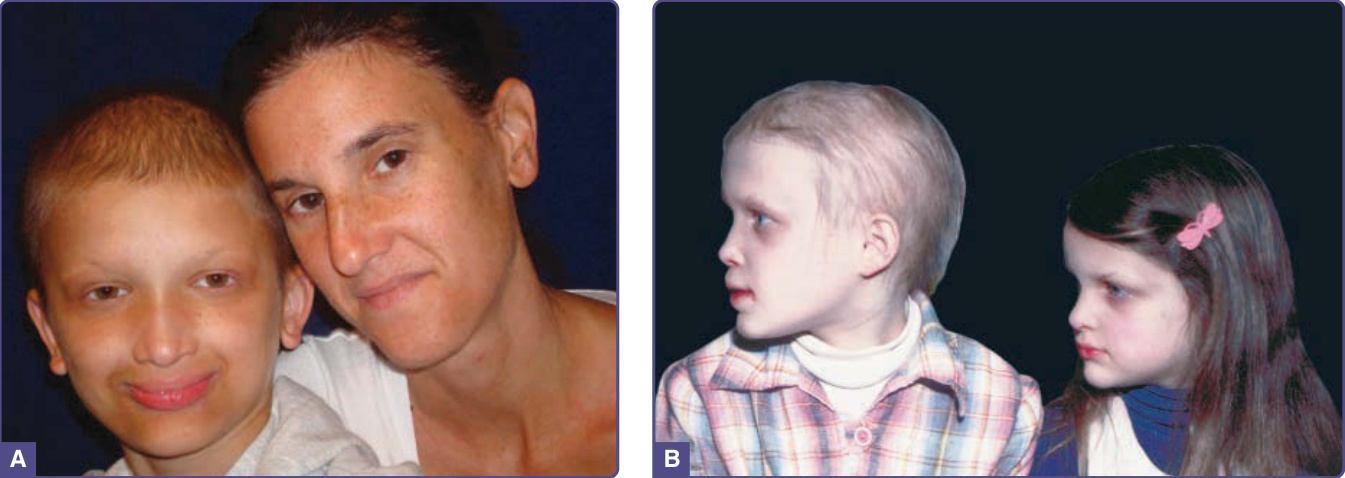
Figure 131-3 Hypohidrotic ectodermal dysplasia. A, Female carrier of X-linked hypohidrotic ectodermal dysplasia with her affected son. B, Two sisters with X-linked hypohidrotic ectodermal dysplasia manifesting to different degrees. Note periorbital hyperpigmentation, full everted lips, and sculpted noses. (Reproduced from Sybert V. Hypohidrotic ectodermal dysplasia: Argument against an autosomal recessive form clinically indistinguishable from X-linked hypohidrotic ectodermal dysplasia (Christ-Siemens-Touraine syndrome). Pediatr Dermatol. 1989;6:76-81; with permission. Copyright © 1989 John Wiley & Sons.)
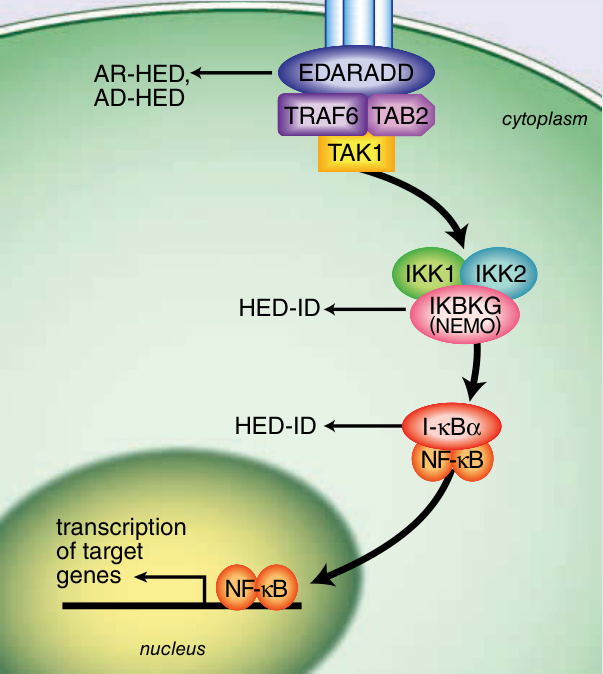
Figure 131-4 Ectodysplasin signal transduction pathway. (Image B, Reproduced from Rimoin D, Pyeritz R, Korf B, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics, 6th ed. Oxford, UK: Academic Press; 2013; Fig. 148-5; with permission. Copyright © Elsevier.)
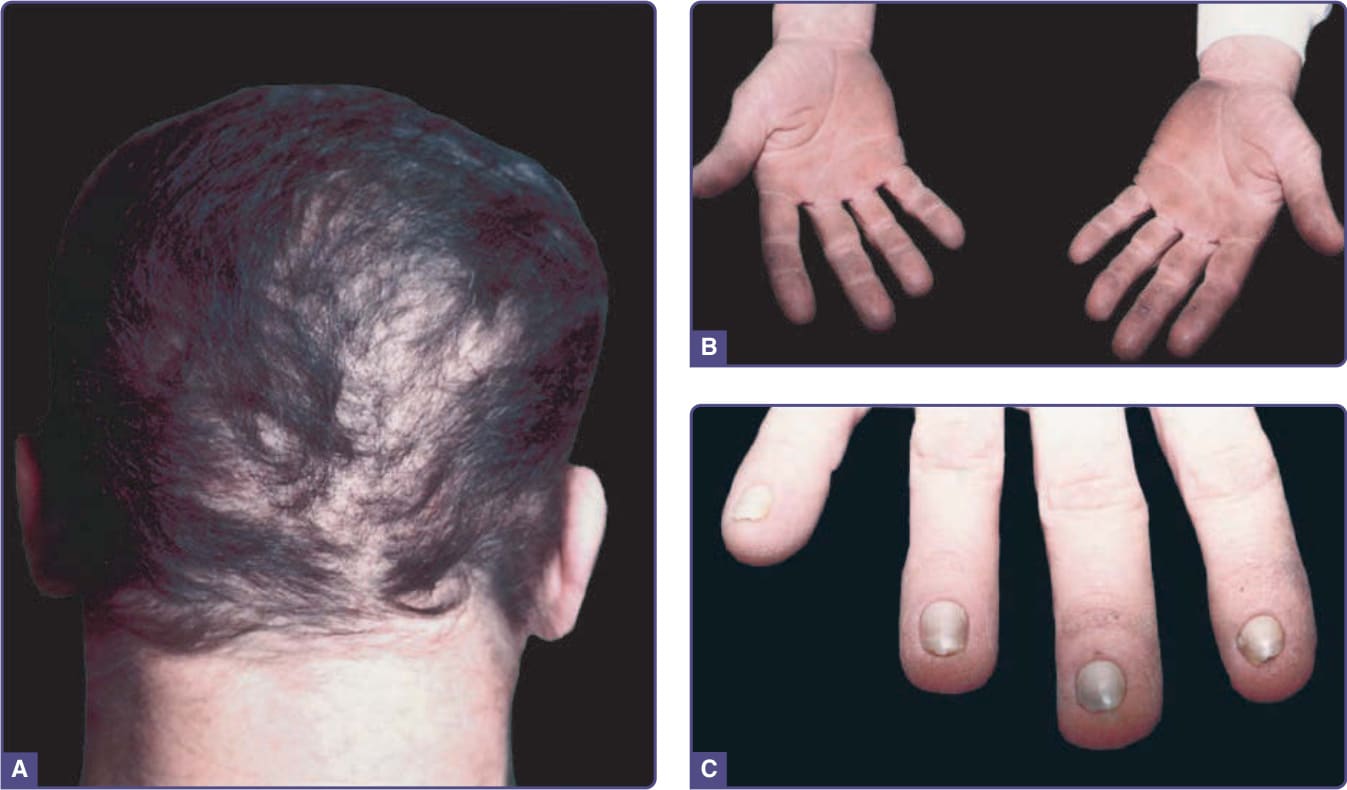
Figure 131-5 Hidrotic ectodermal dysplasia (Clouston syndrome). A, Patchy alopecia in adult. Coarseness of hair can be appreciated. B, Palmar hyperkeratosis. C, Nail dystrophy.
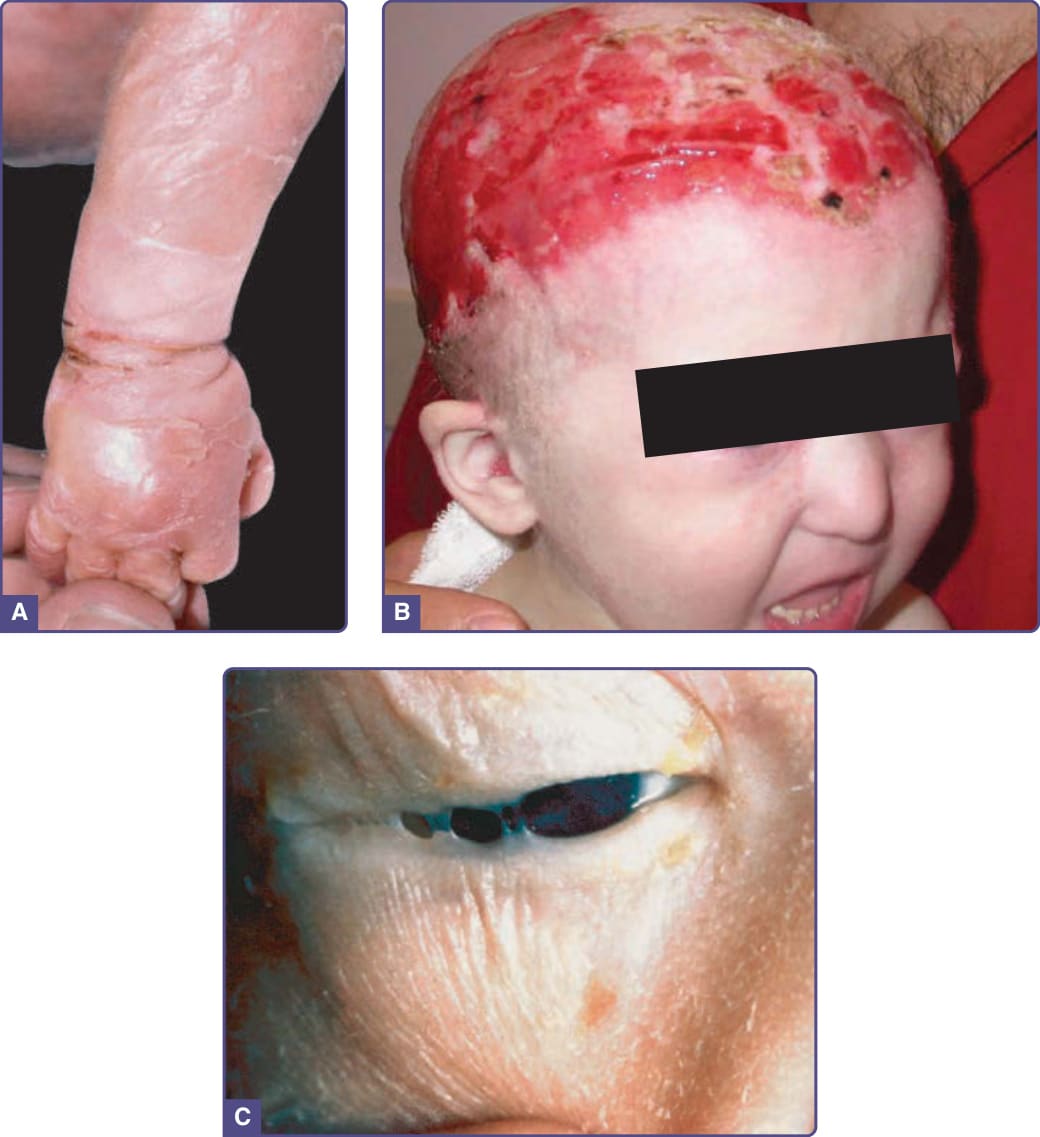
Figure 131-6 Ankyloblepharon filiforme adnatum–ectodermal dysplasia–cleft palate (Hay–Wells) syndrome. A, Newborn with peeling collodion membrane. B, Scalp erosions. C, Fine strands of tissue (ankyloblepharon filiforme adnatum) between eyelids.
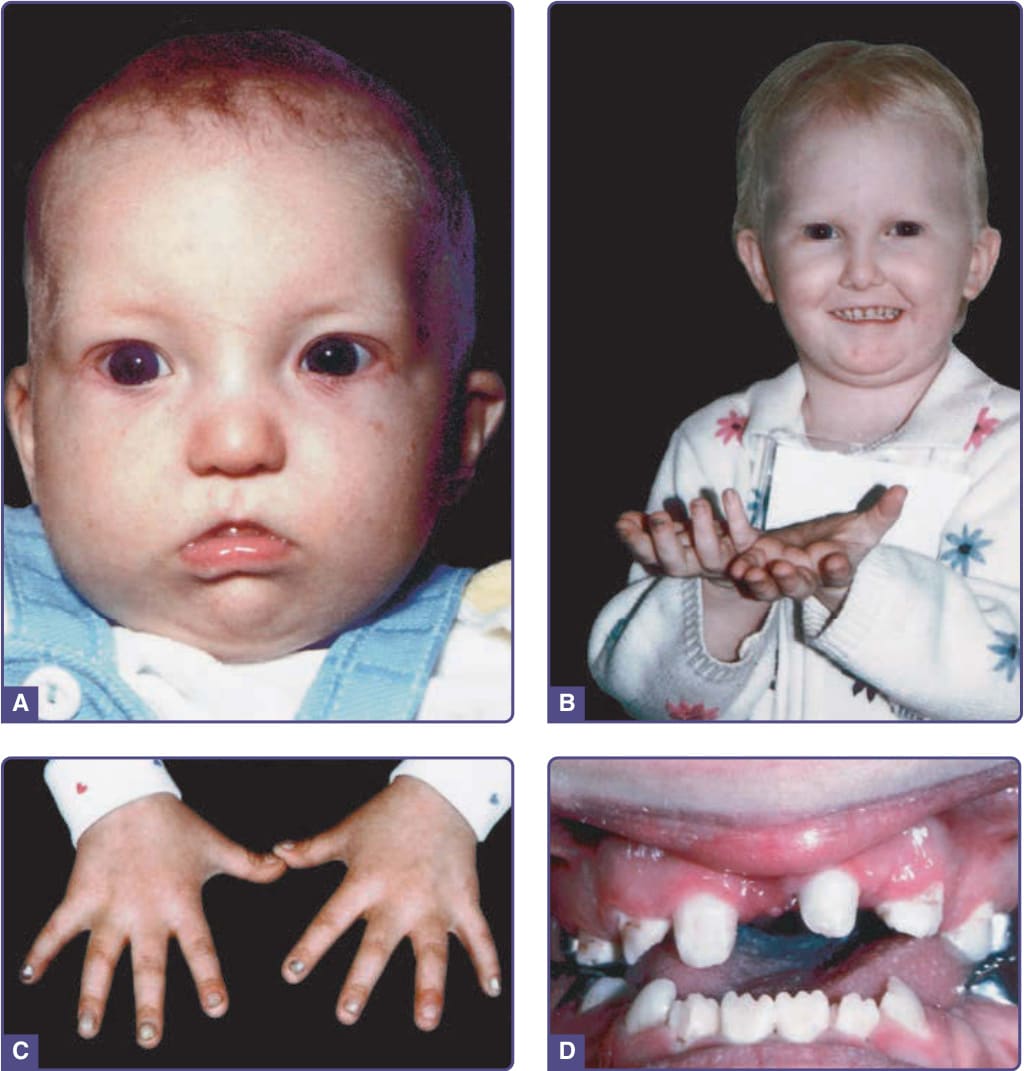
Figure 131-7 Rapp–Hodgkin syndrome. A, Affected infant at 5 months. B, Fine, blond, sparse hair; beginnings of nail changes on middle finger of right hand. C, Abnormal nails in same patient as (A), at age 4½ years with thickened and friable nail plates. D, Abnormal dentition, missing teeth, and peg teeth. (Images A and C, From Schroeder HW Jr, Sybert VP. Rapp-Hodgkin ectodermal dysplasia. J Pediatr. 1987;110(1):72-75; with permission. Copyright © Elsevier. Image B, Used with permission from the National Foundation for Ectodermal Dysplasias.)
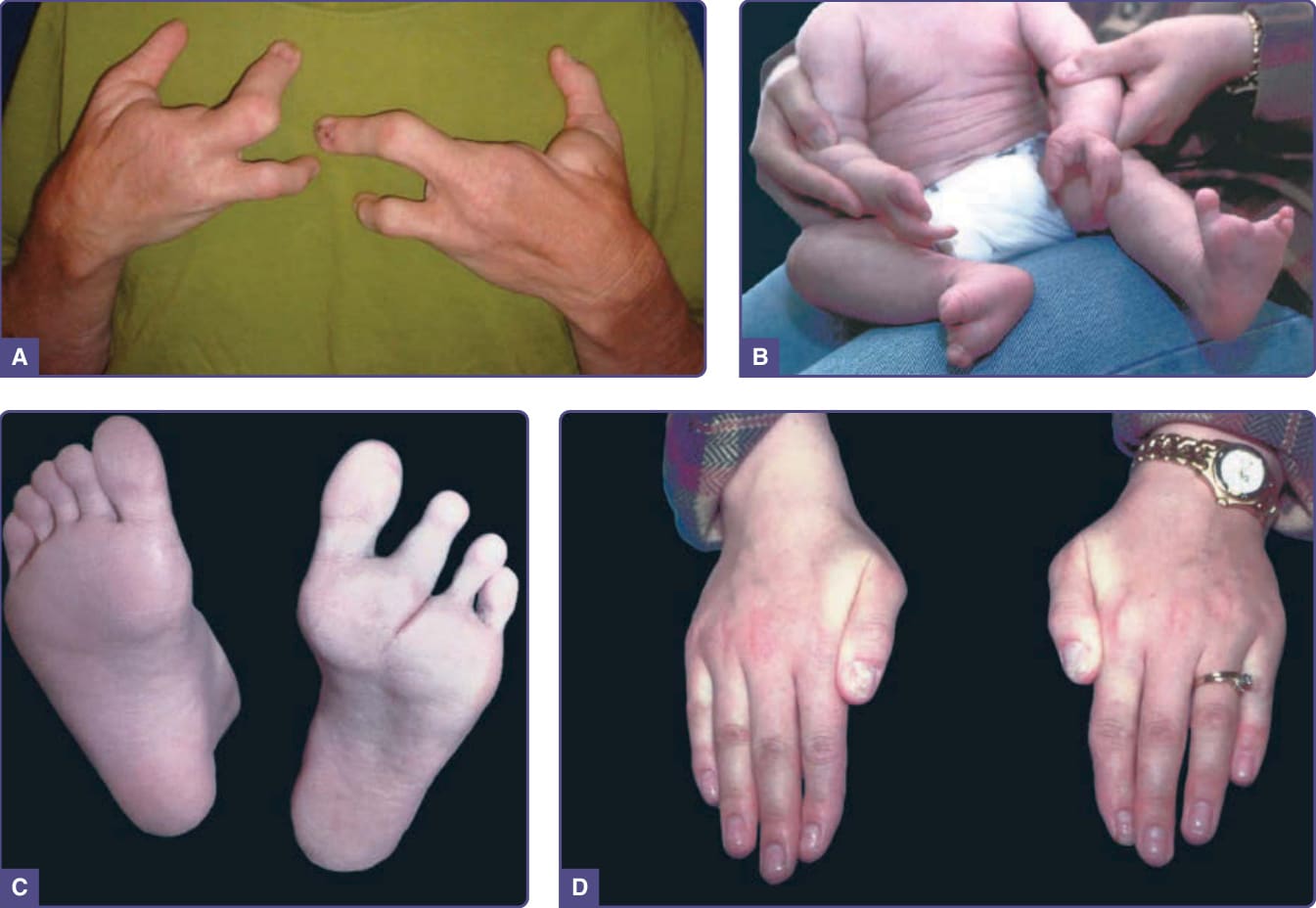
Figure 131-8 Ectrodactyly–ectodermal dysplasia–clefting syndrome. A, Hands of an affected adult. B, Hands and feet of affected infant. C and D, Feet and hands of parent of infant in (B) demonstrating variability of expression both among limbs and between family members. Note the nail dystrophy in the mildly affected mother, especially evident on the thumbs.
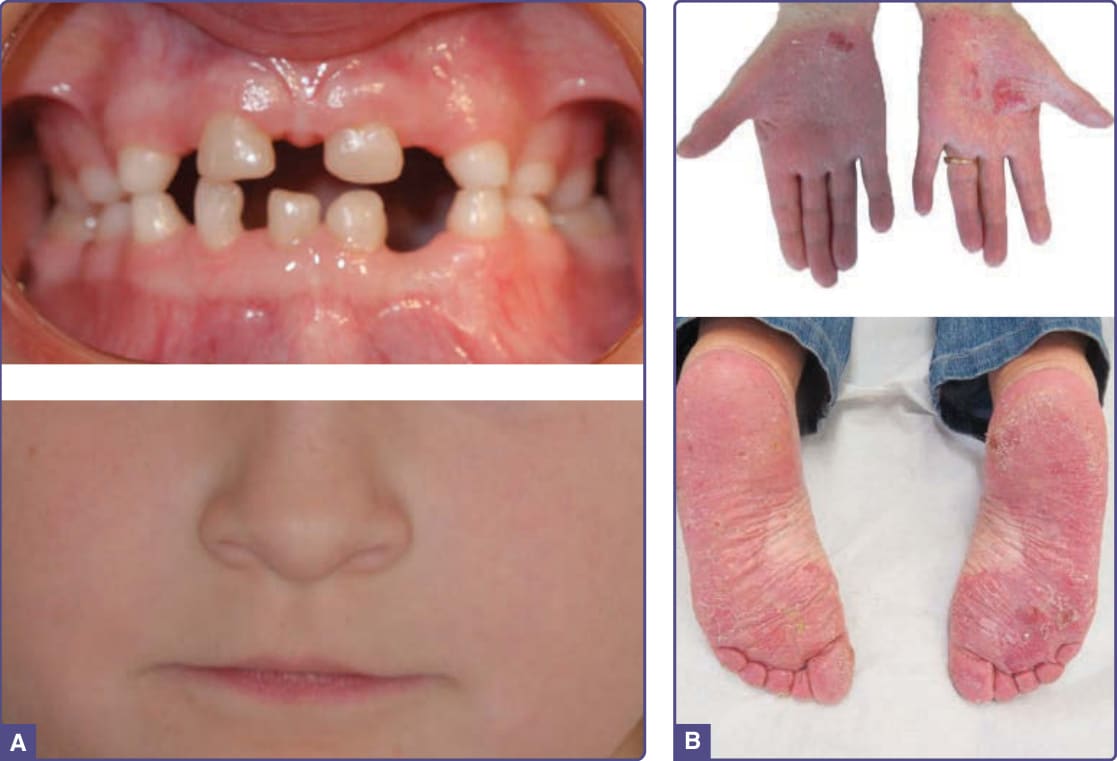
Figure 131-9 WNT10A disorders. A, Note the widely spaced and scattered missing deciduous teeth with the characteristic thin upper lip. (Image reused from Bergendal B, Norderyd J, Zhou X, et al. Abnormal primary and permanent dentitions with ectodermal symptoms predict WNT10A deficiency. BMC Med Genet. 2016;17(1):88 with permission from BioMed Central.) B, Erythematous hyperkeratosis with erosions and fissures of the palmar hands and the plantar feet. (Images reused from Krøigård AB, Clemmensen O, Gjørup H, et al. Odonto-onycho-dermal dysplasia in a patient homozygous for a WNT10A nonsense mutation and mild manifestations of ectodermal dysplasia in carriers of the mutation. BMC Dermatol. 2016;16:3, with permission from BioMed Central.)
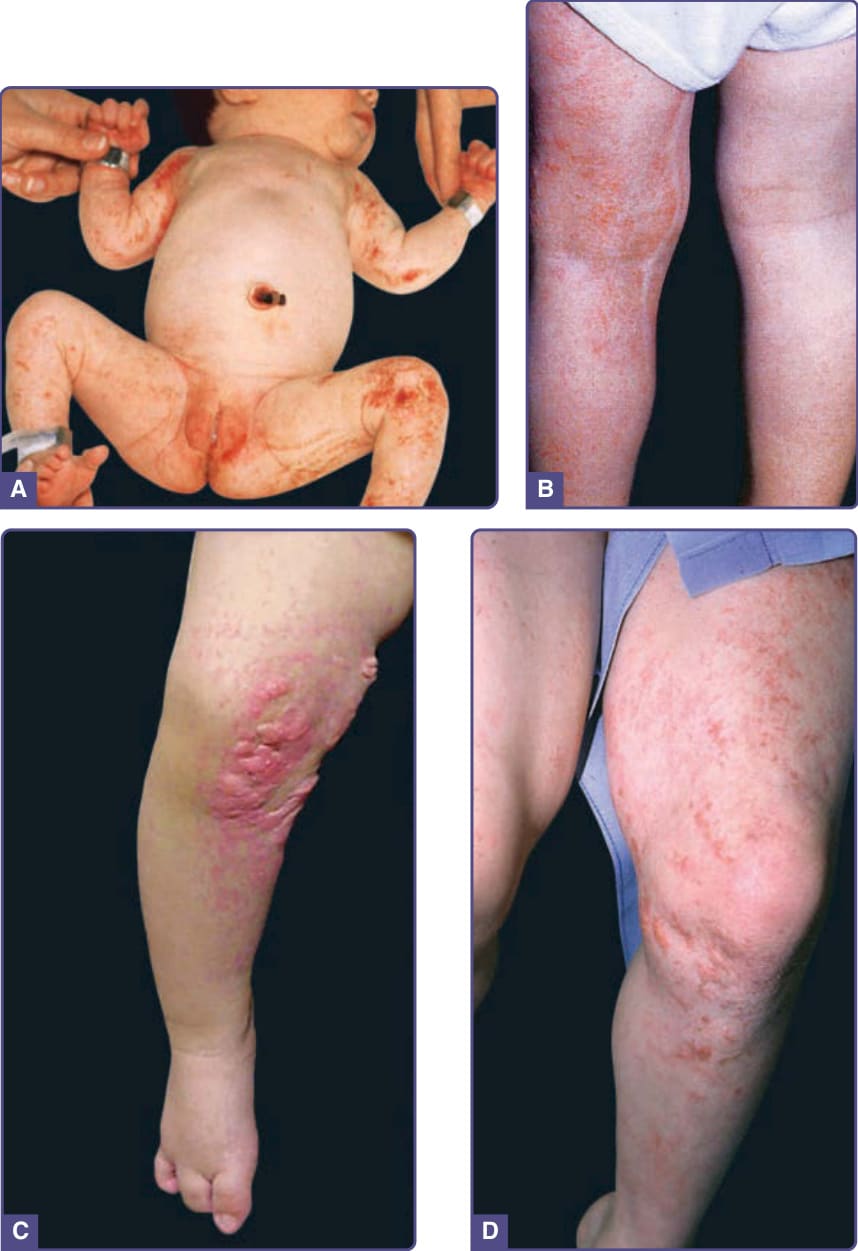
Figure 131-10 Focal dermal hypoplasia. A, Streaky and patchy involvement in the newborn. Note similarity to the distribution of lesions in incontinentia pigmenti. B and C, Same girl at age 2 showing atrophy and fat herniation. D, Now a young adult, erythema and atrophy, with dryness and scale, predominate. (Image A, From Sybert VP. Genetic Skin Disorders, 3rd ed. New York, NY: Oxford University Press; 2017; with permission.)
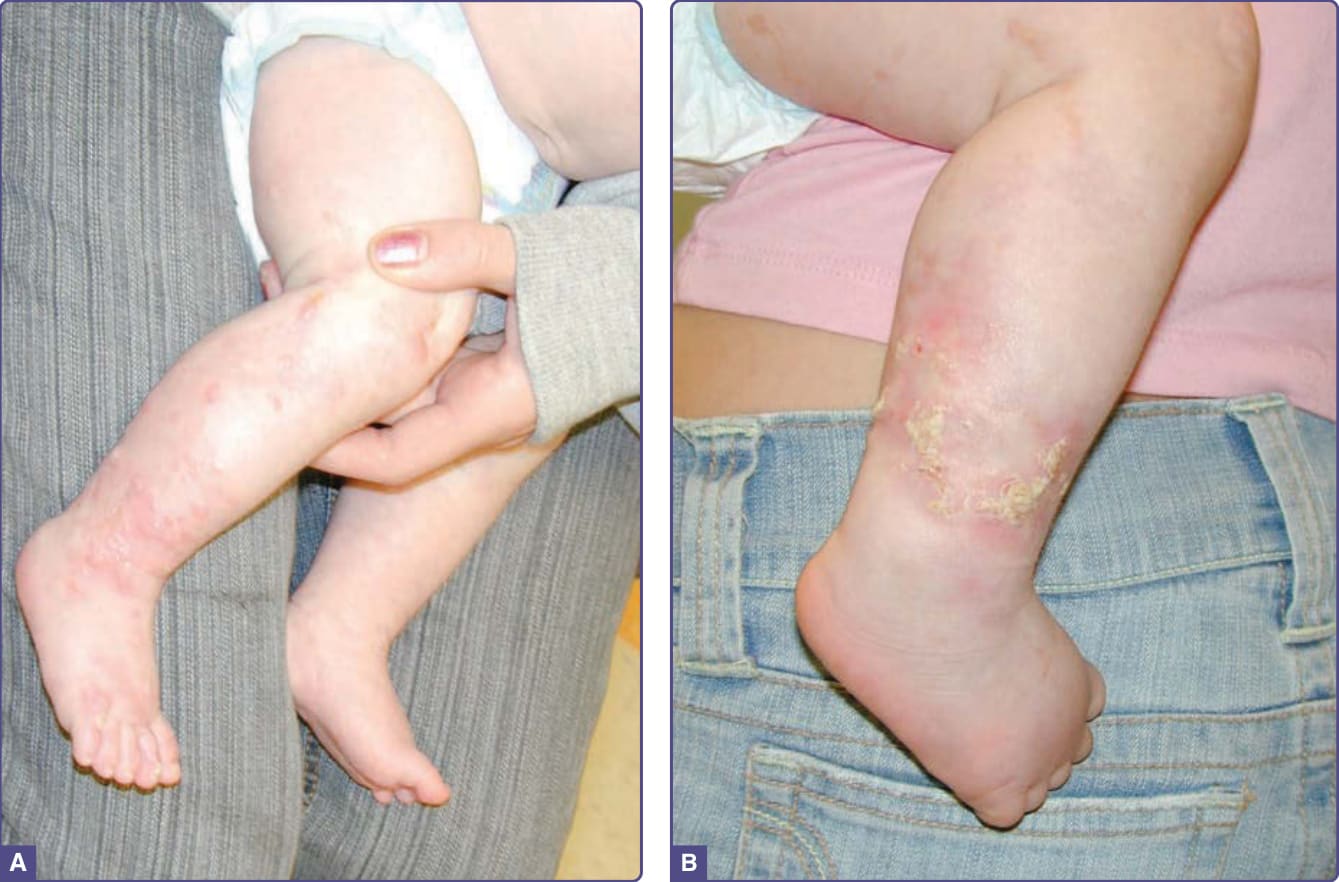
Figure 131-11 Incontinentia pigmenti. A, Stage I. Edematous erythematous papules and vesicles following the lines of Blaschko on the lower leg. Note the early stage II verrucous papules on the toes. B, Stage II. Hyperkeratotic papules and papules and plaques with surrounding erythema. (Reproduced from Rimoin D, Pyeritz R, Korf B, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics, 6th ed. Oxford, UK: Academic Press; 2013; Figure 148-15; with permission. Copyright © Elsevier.)
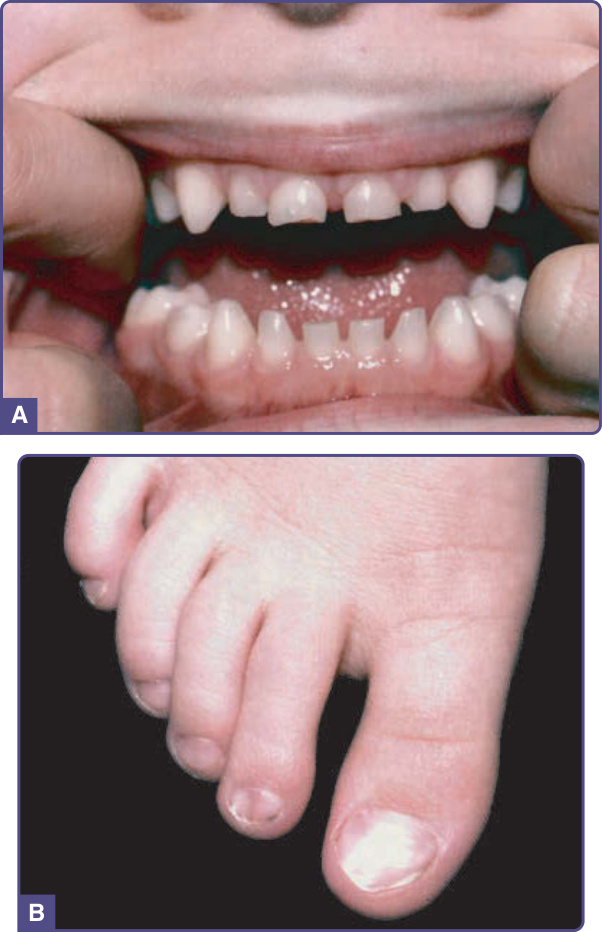
Figure 131-12 Tooth–nail syndrome. A, Primary teeth still in place; failure of adult teeth to erupt. B, Dystrophic toenails with flattening of nail plates.
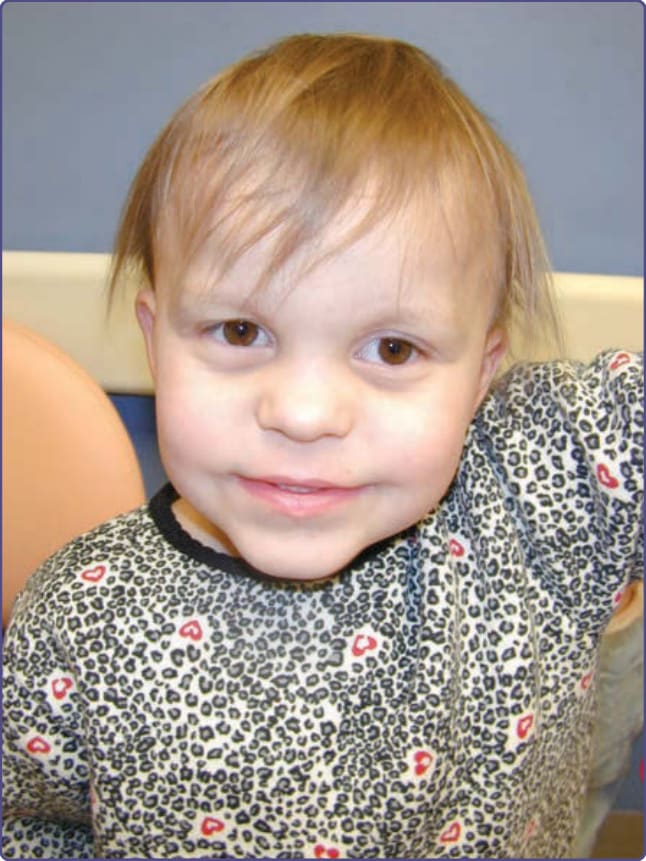
Figure 131-13 Tricho-rhino-phalangeal syndrome. Note the sparse hair and eyebrows, bulbous nose and thin upper lip.
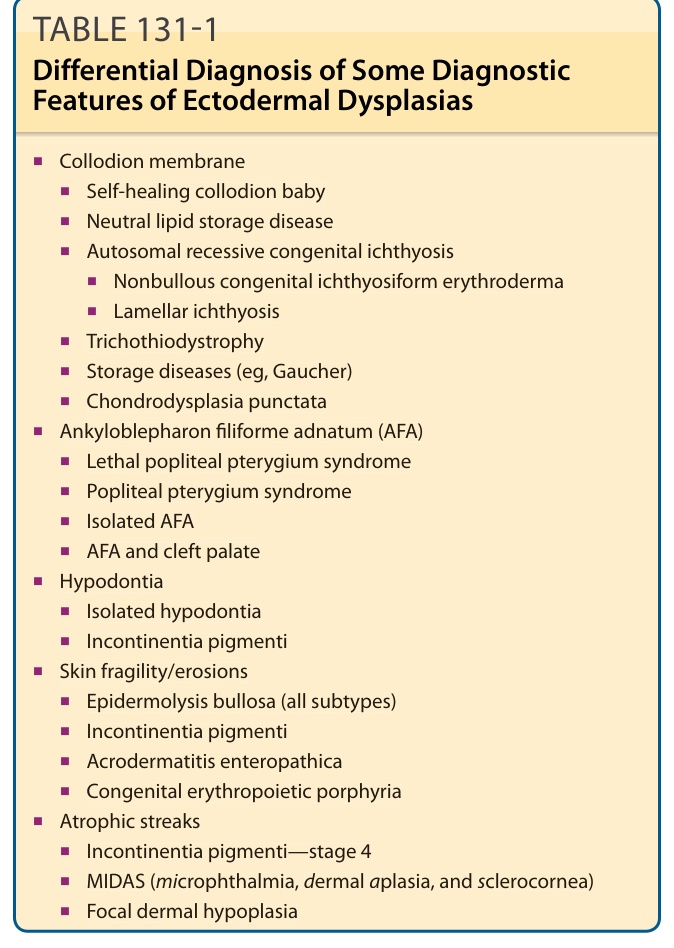
TABLE 131-1 Differential Diagnosis of Some Diagnostic Features of Ectodermal Dysplasias