外胚層發育不良 (Ectodermal Dysplasias)
PART 21
重點一覽 (AT-A-GLANCE)
■ 一群遺傳性疾病,特徵為 2 個或更多外胚層結構出現發育異常。
■ 這些結構包括毛髮、牙齒、指甲,以及皮脂腺與汗腺。
■ 也可能出現非外胚層結構與功能的異常。
■ 各疾病的區別依據臨床特徵、遺傳模式,以及分子/基因學發現。
■ 臨床上不同的疾病可能源自同一基因的不同突變(對偶基因異質性, allelic heterogeneity),而臨床上相似的狀況也可能源自不同基因的突變(基因座異質性, locus heterogeneity)。
前言 (Introduction)
外胚層發育不良 (ectodermal dysplasias, EDs) 是一群複雜且異質性高的遺傳性疾病,共同特徵是侵犯至少 2 種傳統上認為源自胚胎外胚層的主要結構:毛髮、牙齒、指甲,以及汗腺與其他外分泌腺和皮脂腺的發育缺陷。僅侵犯單一類型外胚層結構的發育疾病,即使合併其他先天畸形,亦不歸類為 EDs。被歸類為 EDs 的疾病將近有 200 種,由於其臨床表現多樣且彼此重疊,要精確診斷可能相當困難。1
過去數十年間,已有人提出多種分類架構,以協助整理與思考這些疾病。Freire-Maia 與 Pinheiro 曾發表一套詳盡的綜述與分類系統,並以數字系統加以後續更新。2 雖然此系統為過去混亂的領域建立了一套合理的方法,但在臨床實務上用途不大,且未涵蓋特定狀況的致病機轉或遺傳學。過去數十年間,許多造成這些疾病的基因已被鑑定出來,因而促成數次嘗試,以分子與發育生物學為基礎重新分類 EDs。2008 年,一場國際 ED 分類會議召開,與會者包括醫療提供者、研究人員、病友倡議代表與行政人員,試圖朝向一套更實用且統一的系統邁進。2009 年發布了一份會議報告,概述了以最新臨床與分子資訊為基礎,建立整合性 ED 分類系統的目標。3 鑑於新分子資訊的發現速度,這項工作具有挑戰性。因此,2012 年舉行了第二次國際會議,發展出針對 EDs 的多軸模型 (multi-axis model) 方法。4 此模型將以臨床/表現型軸、基因軸,以及功能/路徑軸為基礎,目前仍在建立過程中。本章僅涵蓋較常見且最可能因診斷與就醫而求診於皮膚科醫師的 EDs。在上述複雜性所限的範圍內,本章將盡可能依照 ED 國際分類會議的架構來編排。鑑於 EDs 領域不斷演變,建議讀者參考以下資源以取得 EDs 的最新資訊:https://omim.org (線上人類孟德爾遺傳目錄, Online Catalog of Mendelian Inheritance in Man)與 http://www.ncbi.nlm.nih.gov/gtr/ (提供分子檢測之實驗室的最新清單)。
外胚層發育不良的診斷方法 (Approach to the diagnosis of ectodermal dysplasias)
- 無汗/嚴重減少出汗(No/severely reduced sweating):
- 有嚴重免疫缺損 → HED-ID (IKBKG)
- 無免疫缺損 → X-LR HED、AD-HED、AR-HED
- 合併肢體異常(+ Limb abnormality)→ EEC、肢體–乳腺症候群 (Limb–mammary)、ADULT、Margarita Island ED
- 正常/輕微減少出汗(Normal/mildly reduced sweating):
- WNT10A 疾病、局灶性真皮發育不良/Goltz 症候群 (Focal Dermal Hypoplasia/Goltz Syndrome)、眼齒指症候群 (Oculodentodigital Syndrome)、牙甲/Witkop 症候群 (Tooth and nail/Witkop)、毛齒骨症候群 (Tricho-dento-osseous)
- 牙齒正常(Normal teeth):汗性 ED (Hidrotic ED)、ED/皮膚脆弱 (ED/skin fragility)、毛鼻指(趾)骨症候群 (Tricho-Rhino-Phalangeal Syndrome)
- 牙齒異常 ± 顏面裂(Abnormal teeth ±facial cleft):
- 合併肢體異常(Limb abnormality)→ AEC (Hay-Wells, Rapp-Hodgkin)
- 無顏面裂(No facial cleft)
低汗性外胚層發育不良:XLHED、ADHED、ARHED、HED-ID (Hypohidrotic Ectodermal Dysplasia)
重點一覽 (AT-A-GLANCE)
■ 變異型:
■ XLHED (OMIM 305100):位於 Xq12-q13.1 之 EDA1 的 XLR 突變,該基因編碼觸發性配體分子 ectodysplasin-A。
■ ADHED (OMIM 129490):位於 1q42.2-q43/2q11-q13 之 EDARADD/EDAR 的 AD 突變;分別為 EDAR 死亡結構域的細胞內分子轉接蛋白/EDA 的跨膜受體。
■ ARHED (OMIM 224900):位於 1q42.2-q43/2q11-q13 之 EDARADD/EDAR 的 AR 突變;分別為 EDAR 死亡結構域的細胞內分子轉接蛋白/EDA 的跨膜受體。
■ 合併免疫缺乏的 HED (HED-ID; OMIM 300291):位於 Xq28 之 IKBKG 的 XLR 突變,該基因編碼 NF-κB 細胞質抑制蛋白(又稱 NF-κB 必需調節因子/NEMO)。
■ 外胚層特徵:以毛髮稀疏 (hypotrichosis)、少汗 (hypohidrosis) 與牙齒過少 (hypodontia) 為特徵。
■ 全身性特徵:具特徵性的顏面外觀,包括額部隆突 (frontal bossing) 與中臉部凹陷 (depressed midface)。常合併異位性皮膚炎 (atopic dermatitis)、氣喘、上呼吸道感染與肺炎。
前言 (Introduction)
低汗性外胚層發育不良 (hypohidrotic ectodermal dysplasia, HED) 最早的描述之一出現在 1848 年,內容涉及具有毛髮稀疏、缺牙與皮膚乾燥的男性表兄弟及其祖母。8 HED 是最常見的 ED,9 尤其是其 X 染色體隱性遺傳 (XLHED) 型,佔向 NFED 登記之家庭的 80%。HED 有數種型態,包括 XLHED(又稱 Christ–Siemens–Touraine 症候群)、體染色體顯性 HED (ADHED)、體染色體隱性 HED (ARHED),以及合併免疫缺乏的 HED (HED-ID)。HED 的體染色體顯性與隱性型態與 XLHED 相似,但顯性型可能較輕微。10 對於患有 HED 並伴有反覆或顯著感染的男性,應考慮 HED-ID。11,12
流行病學 (Epidemiology)
XLHED 發生於所有種族族群,其出生時發生率推估介於每 5,000 至 100,000 名新生兒中有 1 例。10,13,14 其他型態 HED 的盛行率與發生率則無確切資料。
臨床特徵 (Clinical Features)
各變異型的臨床特徵相似。HED 以毛髮稀疏 (hypotrichosis)、少汗 (hypohidrosis) 與牙齒過少 (hypodontia) 為特徵。患有 XLHED 的男性可能在出生時即出現膠樣膜 (collodion membrane) 或皮膚明顯脫屑,15 類似先天性魚鱗癬。頭皮毛髮通常稀疏、細軟且呈金黃色。毛髮在青春期可能變粗、變深,而第二性徵毛髮通常正常。其他體毛通常稀疏或缺如。出汗能力顯著受損,多數患有 XLHED 的男性有明顯的耐熱不良 (heat intolerance)。16 對環境熱無法充分出汗,會導致核心體溫升高與不明原因的高燒反覆發作,通常會在診斷正確之前,引發針對感染性疾病、惡性腫瘤或自體免疫疾病的廣泛檢查。在較早的病例系列中,智能障礙曾被報告為 XLHED 的特徵之一。目前認為這是長期高燒與癲癇所造成的損害,而非此疾病本身固有的特徵。17 ADHED 患者的出汗能力缺陷似乎較輕微。指甲通常正常,但也有薄、脆指甲的報告。18 指紋脊紋消失。眼周皺紋與色素沉著為典型表現,且常於出生時即存在(圖 131-2)。濕疹影響超過三分之二的患病男性,以及將近半數的患病女性。19 皮脂腺增生(尤其在臉部)可能隨時間發展,呈現為小型、珍珠樣、膚色至白色的丘疹,可能類似粟丘疹 (milia)。牙齒過少 (hypodontia)、寡牙 (oligodontia) 或無牙 (anodontia) 是患病男性 XLHED 不變的特徵。患病嬰兒的齒槽嵴發育不全可能是診斷此疾病的早期線索。確實萌發的牙齒通常呈釘狀且細小(圖 131-2B)。
此疾病的顏面特徵為額部隆突 (frontal bossing) 與中臉部凹陷,伴鞍鼻 (saddle nose) 及豐厚外翻的嘴唇。耳鼻喉與肺部表現包括濃稠的鼻分泌物與阻塞、臭鼻症 (ozena, 萎縮性鼻炎)、鼻竇炎、反覆上呼吸道感染與肺炎、唾液分泌減少、聲音沙啞,以及氣喘的發生率增加。呼吸道感染發生率增加,被歸因於支氣管樹中分泌黏液的腺體發育不全或缺如。20 胃食道逆流與餵食困難在嬰兒期可能是個問題,其原因不明。初步研究顯示,多達 20% 至 40% 的患病男孩在嬰兒期與兒童早期可能出現生長遲滯 (failure to thrive),之後可見追趕性生長 (catch-up growth)。21
XLHED 的女性帶因者受影響的嚴重程度可能與男性相當,也可能僅顯現少數(甚至沒有)此疾病的徵象(圖 131-3)。介於 60% 至 80% 的女性帶因者會表現出此疾病的某些臨床徵象;最常見的是斑塊狀毛髮稀疏與牙齒過少或釘狀牙。耐熱不良若存在,通常輕微。成年女性帶因者表示她們不太出汗,或不喜歡非常炎熱的天氣,但女性因無法出汗而發燒的情況並不常見。22
病因與致病機轉 (Etiology and Pathogenesis)
HED 具基因異質性,有數種型態與腫瘤壞死因子 α (tumor necrosis factor α, TNF-α) 訊息傳遞路徑中所涉及的不同基因有關。9 此路徑中的突變會導致上皮細胞與其下方間質 (mesenchyme) 間交互作用的中斷,進而造成不同型態 HED 之間相似的臨床觀察特徵。圖 131-4 展示 ectodysplasin 訊息傳導路徑及其與各型 HED 的關係。XLHED 源自位於 Xq12-13.1 之 EDA1 基因的改變,該基因編碼 ectodysplasin-A。10
大約 70% 的患病男性是從帶因母親處遺傳到此突變。EDA1 編碼一種跨膜蛋白 ectodysplasin,其在外胚層結構形成的調控中扮演角色。它會形成三聚體 (trimers),並表現於角質細胞 (keratinocytes)、毛囊外根鞘 (outer root sheath) 與汗腺中。它定位於細胞的側面與頂端表面。已鑑定出此基因中造成 XLHED 的眾多突變。家族間與家族內的變異均會發生,但有皮膚與毛髮表現之基因型–表現型相關性的報告。23
位於 2q11-q13 的 EDAR 基因與位於 1q42.2-q43 的 EDARADD 基因之突變,已被認為與低汗性外胚層發育不良的體染色體顯性型 (ADHED; OMIM 129490) 及體染色體隱性型 (ARHED; OMIM 224900) 皆有關。如上所述,這些疾病在臨床上與 XLHED 相似,但較為罕見。EDAR 作為 ectodysplasin 的受體,而 EDARADD 作為細胞內轉接蛋白,協助將訊號從活化的 EDA 受體傳遞至細胞核。10
造成女性色素失禁症 (incontinentia pigmenti, IP) 的 X 染色體連鎖 IKBKG 基因的某些突變,已被證實會在男性中造成 HED-ID。有 2 個相似且由 OMIM 定義的症候群:HED-ID (OMIM#300291) 與「合併免疫缺乏、骨硬化症與淋巴水腫之無汗性外胚層發育不良」(OLEDAID; OMIM#300301),二者皆描述具有 IKBKG 突變、患有 ED 與免疫缺乏的男性。11,12 這些患者表現出 ED 並伴有異常丙種球蛋白血症 (dysgammaglobulinemia) 及顯著的早期病態與死亡。NFKB 訊息傳遞路徑的複雜性,可解釋 HED-ID 的多樣基因型與免疫學表現型。
診斷 (Diagnosis)
當預期到 HED 時(例如孩子出生於已知有 HED 病史的家庭),診斷可輕易辨識。然而,若無事先的認知,初期診斷在臨床上可能具挑戰性。隨著患者年齡增長,毛髮稀疏、少汗與牙齒過少的特徵性表現會變得更加明顯。22 齒槽嵴發育不全(顯示缺牙)可作為早期診斷線索。顎部的全景 X 光片 (Panorex view) 有助於做出正確診斷。雖然極少有必要,但以碘溶液檢查汗孔來評估出汗、量化毛果芸香鹼 (pilocarpine) 誘發的出汗,23 或皮膚切片以評估頭皮和/或手掌區域是否缺乏外分泌腺結構,可能有助於確認診斷。24 在一名孤立、完全表現的女性中,需考慮 HED 的體染色體顯性與隱性型態。家族史的回顧是必要的,且應始終對母親進行完整檢查,以偵測 X 染色體連鎖型的輕微表現。隨著近來分子基因檢測的擴展與發展,確定性的基因診斷正變得更易取得。轉介至小兒皮膚科醫師或遺傳學家有助於診斷。組織病理學上,表皮變薄且平坦。皮脂腺與毛囊的數量減少。外分泌腺缺如或發育不完全。皮膚的組織學評估通常並非必要。
鑑別診斷 (Differential Diagnosis)
(依原文圖表化的鑑別診斷列表,按臨床表現分組整理如下)
- 膠樣膜 (Collodion membrane):
- 自癒型膠樣兒 (Self-healing collodion baby)
- 中性脂質儲積病 (Neutral lipid storage disease)
- 體染色體隱性先天性魚鱗癬 (Autosomal recessive congenital ichthyosis)
- 非水疱性先天性魚鱗癬樣紅皮症 (Nonbullous congenital ichthyosiform erythroderma)
- 層狀魚鱗癬 (Lamellar ichthyosis)
- 毛髮硫營養不良症 (Trichothiodystrophy)
- 儲積病(如高雪氏病, Gaucher)(Storage diseases, eg, Gaucher)
- 點狀軟骨發育不良 (Chondrodysplasia punctata)
- 先天性絲狀眼瞼粘連 (Ankyloblepharon filiforme adnatum, AFA):
- 致死性膕翼狀胬肉症候群 (Lethal popliteal pterygium syndrome)
- 膕翼狀胬肉症候群 (Popliteal pterygium syndrome)
- 孤立性 AFA (Isolated AFA)
- AFA 合併顎裂 (AFA and cleft palate)
- 牙齒過少 (Hypodontia):
- 孤立性牙齒過少 (Isolated hypodontia)
- 色素失禁症 (Incontinentia pigmenti)
- 皮膚脆弱/糜爛 (Skin fragility/erosions):
- 表皮鬆解水疱症(所有亞型)(Epidermolysis bullosa, all subtypes)
- 色素失禁症 (Incontinentia pigmenti)
- 腸病性肢端皮膚炎 (Acrodermatitis enteropathica)
- 先天性紅血球生成性紫質症 (Congenital erythropoietic porphyria)
- 萎縮性條紋 (Atrophic streaks):
- 色素失禁症—第 4 期 (Incontinentia pigmenti—stage 4)
- MIDAS(小眼症、真皮發育不全與鞏膜角膜化)(microphthalmia, dermal aplasia, and sclerocornea)
- 局灶性真皮發育不良 (Focal dermal hypoplasia)
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
維持涼爽的環境溫度對於預防高熱 (hyperpyrexia) 至關重要。多數兒童透過簡單措施即可良好控制,例如濕 T 恤與頭帶、家中與學校的空調等。偶爾,降溫背心 (cooling vests) 能讓患者在溫暖氣候中更廣泛地參與運動與劇烈體能活動。牙齒修復至為重要,早期裝置假牙與最終使用牙科植體 (dental implants) 是治療的主軸。耳鼻喉併發症、氣喘與反覆感染的處置須個別化。濕疹可能對治療相當頑固而難以控制。患有 HED 的嬰兒因高熱及此疾病潛在的其他特徵(如感染)而死亡的風險增加。25,26 在一項對外胚層發育不良國際登記處 (Ectodermal Dysplasia International Registry, EDIR) 的調查中,21% 的 XLHED 患者報告有嬰兒或兒童期死亡的家族史。19
雖然嬰兒期與兒童期因諸多問題而複雜,但多數患有 HED 的個體成年後能過著讓他們在社會中成功運作的生活。耐熱不良似乎會減輕,原因是青春期發展出某些出汗能力、或生活型態的適應發展,或二者兼具。對患病的懷孕母鼠注射重組 ectodysplasin 蛋白,使其後代的表現型特徵獲得永久性的挽救。27 此外,出生後早期注射也被證實能改善發育缺陷。在以重組 ectodysplasin 蛋白於出生後期注入的患病犬類模型中,也觀察到相似結果,使牙齒、淚液分泌與出汗能力恢復正常,並減少眼部與呼吸道感染。28,29 動物模型的這些進展令人振奮。目前已有人體試驗正在進行中。
汗性外胚層發育不良(Clouston 症候群)(Hidrotic Ectodermal Dysplasia)
重點一覽 (AT-A-GLANCE)
■ OMIM 129500,位於 13q12 之 GJB6 的 AD 突變,該基因編碼 connexin 30,一種細胞間連接 (intercellular junction) 的 connexin 蛋白。
■ 稀疏、硬而蒼白的毛髮,伴進行性禿髮;不一的甲營養不良 (onychodystrophy);進行性掌蹠角化過度。
■ 出汗與牙齒正常。
流行病學 (Epidemiology)
汗性 ED 最早描述於一個法裔加拿大家系。30 它也曾在其他族裔群體中被報告,但多數患病個體的祖先可追溯回一名最初的法裔加拿大移民。
臨床特徵 (Clinical Features)
頭皮毛髮硬、脆且蒼白,並常有斑塊狀禿髮(圖 131-5)。此情況在成年生活中進展,可能導致完全禿髮。體毛與顏面毛髮亦受影響。指甲在嬰兒期與兒童早期可能呈乳白色,逐漸增厚並變得營養不良。成人的甲板厚、短且生長緩慢。它們從遠端與甲床分離(圖 131-5C),並可能造成疼痛。曾有無甲症 (anonychia) 的報告。並非所有指甲都必然受到相同程度的影響。進行性掌/蹠角化過度很常見(圖 131-5B)。與 HED 相比,出汗正常,牙齒亦正常。曾有口腔白斑 (oral leukoplakia) 的報告。結膜炎與眼瞼炎(可能因稀疏睫毛功能不良所致)很常見。31
病因與致病機轉 (Etiology and Pathogenesis)
汗性 ED 為體染色體顯性,具可變的表現度 (variable expression),嚴重程度在家族內與家族間皆可不同。男性與女性受影響的人數與程度相等。此疾病由一個 connexin 基因 GJB6 的突變所致,該基因編碼細胞間連接蛋白 connexin 30。32 同一基因的不同突變造成一種非症候群型體染色體顯性耳聾。Connexin 突變也涉及數種其他遺傳性皮膚病,例如角膜炎–魚鱗癬–耳聾 (keratitis-ichthyosis-deafness, KID) 症候群(第 47 與 48 章)。
診斷 (Diagnosis)
隨時間推移,診斷相當直接。在無其他 ED 徵象的情況下,指甲與毛髮的侵犯及掌/蹠增厚相當具特異性。同樣地,基因檢測可作為確認之用。31
組織病理學上,增厚的手掌與足底呈現正角化過度 (orthohyperkeratosis) 並具正常的顆粒層。電子顯微鏡下可見角質層 (stratum corneum) 細胞中橋粒 (desmosomes) 數量增加。毛髮顯示非特異性變化。
鑑別診斷 (Differential Diagnosis)
其他掌/蹠角化過度並無相似的毛髮變化。口面裂 (orofacial clefting) 可區分其他型態的體染色體顯性汗性 ED,例如眼瞼粘連–ED–顎裂 (ankyloblepharon–ED–cleft palate, AEC) 症候群。雖然指甲變化與先天性厚甲症 (pachyonychia congenita) 相似,但毛髮變化具有區別性。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
偶爾需要去除甲基質 (nail matrix) 以緩解疼痛。假髮可提供美容上的益處。掌蹠角皮症 (palmoplantar keratoderma) 的治療並無特異性,且成效甚微。
p63 相關之外胚層發育不良症候群:AEC(Hay–Wells/Rapp–Hodgkin)、EEC、肢體–乳腺症候群、肢端–皮膚–甲–淚腺–牙齒 (ADULT) 症候群、孤立性裂手–裂足畸形 (P63-Related Ectodermal Dysplasia Syndromes)
重點一覽 (AT-A-GLANCE)
■ 變異型:以下所有狀況皆為 AD,伴位於 3q27 之 TP63 的突變,該基因編碼 p63 轉錄蛋白。
■ 眼瞼粘連–外胚層缺損–裂 (Ankyloblepharon-ectodermal defects-clefting, AEC)/Hay–Wells/Rapp–Hodgkin 症候群(AEC/Hay–Wells OMIM 106260;Rapp–Hodgkin 症候群 OMIM 129400)。
■ 肢體–乳腺症候群 (Limb–mammary syndrome; OMIM 603543)。
■ 肢端–皮膚–甲–淚腺–牙齒 (Acro-dermato-ungual-lacrimal-tooth, ADULT) 症候群 (OMIM 103285)。
■ 缺指(趾)–ED–唇/顎裂症候群 3 (Ectrodactyly-ED-cleft lip/palate syndrome 3, EEC3; OMIM 604292)。
■ 外胚層特徵:出生時膠樣膜、乾燥而薄的皮膚、糜爛性頭皮皮膚炎、斑塊狀禿髮、粗糙的淡色毛髮、色素異常、不一的甲營養不良。
■ 全身性特徵:先天性絲狀眼瞼粘連 (Ankyloblepharon filiforme adnatum)、淚管閉鎖 (lacrimal duct atresia)、唇/顎裂、牙齒過少、併指(趾)(syndactyly)、異位乳房組織與尿道下裂 (hypospadias)。
前言 (Introduction)
p63(一個對應至 3q27 的腫瘤抑制基因)的突變,已在大多數(但非全部)患有眼瞼粘連–外胚層缺損–裂(AEC;Hay–Wells;Rapp–Hodgkin)症候群的個體中發現。33 此基因廣泛表現,包括在增生性上皮組織的基底細胞中。同一基因的突變也造成某些孤立性裂手–裂足畸形、肢體–乳腺症候群,以及肢端–皮膚–甲–淚腺–牙齒 (ADULT) 症候群的病例。似乎存在某些基因型–表現型相關性。34
AEC(Hay–Wells/Rapp–Hodgkin)症候群
流行病學 (Epidemiology)
Hay–Wells 症候群最早描述於 1976 年,35 並已在族裔與地理上分散的家族中被發現。Rapp–Hodgkin 症候群最早描述於 1968 年,36 雖然它與 Hay–Wells 症候群有許多臨床相似之處,但曾一度被認為是獨立的臨床實體。近來已逐漸明確,Rapp–Hodgkin 症候群與 Hay–Wells 症候群為對偶基因 (allelic) 關係,應被視為同一疾病(AEC 症候群)的變異型。37,38
臨床特徵 (Clinical Features)
百分之八十至 90% 的患病嬰兒出生時呈現發亮、發紅、龜裂、脫皮的皮膚與表淺糜爛,類似膠樣膜的外觀(圖 131-6A)。18 此情況在數週內脫落,其下的皮膚乾燥而薄。頭皮幾乎一律受影響。許多個體有慢性糜爛性皮膚炎,伴頭皮上異常的肉芽組織與反覆細菌感染(圖 131-6B)。39 頭皮有不一程度的禿髮,毛髮常硬、粗且色淺,外觀難以梳理 (uncombable)。體毛稀疏至缺如為典型表現。皮膚色素異常(低色素與高色素皆有)可相當顯著,且在患病者中普遍可見。40,41 指甲可能正常、過度凸起並增厚、缺如,或部分營養不良,且所有變化可同時見於單一個體,並隨年齡而惡化。皮紋消失 (effaced dermatoglyphics) 與掌蹠糜爛性變化也相當常見。出汗可能正常至輕微減少。雖然多數患病個體描述主觀的耐熱不良,但明顯的高熱並不常見。先天性絲狀眼瞼粘連 (ankyloblepharon filiforme adnatum, AFA),即眼瞼間皮膚束帶的稱呼,見於約 70% 的患病嬰兒(圖 131-6C)。
這些束帶可能在出生前即自發撕裂,僅輕微侵犯外側眼瞼,或需手術鬆解。淚管閉鎖或阻塞很常見。偶爾可見多乳頭 (supernumerary nipples) 與異位乳房組織,第二、三趾的輕度皮膚併趾亦然。也曾觀察到更明顯的肢體缺陷,包括缺指(趾)(ectrodactyly,手足正中列的異常發育)。42
歷史上,Hay–Wells 與 Rapp–Hodgkin 症候群之間的主要臨床差異,包括 Rapp–Hodgkin 症候群所伴隨的特徵性顏面外觀,即鼻小柱短 (short nasal columella) 與上頜發育不全 (maxillary hypoplasia)、上唇薄、下唇豐厚(圖 131-7A,B)。此外,在 Rapp–Hodgkin 症候群中,牙齒可能呈圓錐狀且易蛀(圖 131-7D),近三分之一的患病個體淚點 (lacrimal puncta) 發育不全,但 AFA 罕見,且伴隨皮膚崩解與肉芽組織形成的頭皮侵犯則少見得多。顎裂(伴或不伴唇裂)發生於 80% 至 100% 的報告病例,部分病例僅顯現黏膜下顎裂 (submucosal cleft palate)。43 典型表現為牙齒過少,伴缺牙或牙齒形狀異常,且上頜發育不全也很常見。44 部分病例曾描述耳廓畸形。反覆中耳炎與續發性傳導性聽力喪失很常見,可能是顎裂的後果。患病男性曾被描述有尿道下裂,並有單一女性病例報告陰唇發育不全伴陰道開口缺如。這些特徵也讓人聯想到 EEC 症候群。
診斷 (Diagnosis)
關於基因檢測的資訊,見病因與致病機轉之討論。組織病理學上,一致的變化包括輕度表皮萎縮、局灶性正角化 (focal orthokeratosis)、明顯的表淺血管叢,以及伴噬黑素細胞 (melanophages) 的色素失禁 (pigment incontinence)。45 毛髮的電子顯微鏡顯示有缺陷的角質層結構、萎縮與黑色素流失,以及包括捻轉發 (pili torti) 與三角溝狀毛 (pili trianguli et canaliculi) 在內的結構異常。表皮基底層與基底上層的角蛋白 (keratins) 減少,且角質層中角蛋白絲排列紊亂。
鑑別診斷 (Differential Diagnosis)
在伴隨裂 (clefting) 的體染色體顯性 EDs 中,EEC 症候群的特徵是手足骨性異常,這在 AEC 中通常不會見到,且 EEC 也缺少眼瞼粘連。新生兒脫皮、糜爛的皮膚可能導致誤診為表皮鬆解水疱症或先天性魚鱗癬。AFA 可在無症候群關聯的情況下發生,眼瞼間的組織束帶也曾見於數種型態的關節攣縮 (arthrogryposis) 與 CHANDS(捲髮、眼瞼粘連與甲發育不良症候群, curly hair, ankyloblepharon, and nail dysplasia syndrome)這種體染色體隱性型 ED。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
在膠樣膜脫落前應使用輕質潤膚劑 (light emollients)。眼瞼粘連可能需要手術鬆解。持續的眼部衛生很重要。皮膚糜爛(尤其是頭皮)難以處置,易產生過度肉芽組織與續發性感染。應以溫和的傷口照護、稀釋漂白水或其他抗菌浸泡來處置。應避免封閉性敷料。40
將皮膚移植至頭皮在多數情況下並未被證實成功。裂的修復需要團隊合作,並對續發問題(如餵食困難、語言缺陷、齒列矯正與耳部感染)進行追蹤。
EEC 症候群 (EEC Syndrome) (OMIM 129900, 604292)
EEC 症候群是一種 ED,被歸類為多發性先天異常症候群 (multiple congenital anomaly syndrome),因為它對外胚層衍生結構以外的結構也有主要侵犯。EEC 症候群有 3 種變異型:EEC1 (OMIM 129900)、EEC2(無 OMIM 編號)與 EEC3 (OMIM 604292)。EEC1 與 EEC2 的基因產物突變不明,但 EEC1 已被連鎖至第 7 號染色體上的一個突變。EEC3 由腫瘤抑制基因 TP63 的體染色體顯性突變所致,因而將 EECs 歸為 p63 相關的 EDs。EEC 症候群已發生於全球所有種族族群,且 3 種型態皆共有相同的臨床表現。
臨床特徵 (Clinical Features)
外胚層發育不良的表現可能相當輕微。毛髮通常呈金黃色、粗且乾燥。它可能稀疏且生長緩慢。腋毛與陰毛也可能受影響。約五分之四的個體指甲營養不良,伴橫向脊紋 (transverse ridging)、點狀凹陷 (pitting) 與生長緩慢。可能出現皮膚乾燥與掌蹠增厚。出汗通常正常。46
EEC 症候群的主要區別特徵是缺指(趾)(ectrodactyly)(圖 131-8A-C)。足部的侵犯比手部更為常見,且侵犯可能不對稱。顎裂(伴或不伴唇裂)發生於 70% 至 100% 的患病個體。46,47 牙齒過少、續發齒(恆齒)過早脫落,以及與裂相關的牙齒異常,見於大多數患病個體。淚腺異常與續發性傳導性聽力喪失很常見。包括腎水腫 (hydronephrosis) 與結構性腎或生殖器畸形在內的泌尿生殖系統異常,影響三分之一或更多的 EEC 症候群患者。雖然曾有智能障礙的報告,但一般認為它並非此疾病的固有特徵。
鑑別診斷 (Differential Diagnosis)
在 EEC 症候群的鑑別診斷中,需考慮具有肢體缺陷的疾病包括:齒–毛–肢症候群 (odontotrichomelic syndrome; OMIM 273400),其有嚴重的肢體缺失畸形;以及合併肢體缺陷的先天性皮膚發育不全(Adams–Oliver 症候群, OMIM 100300),其除了皮膚缺失外並無裂或外胚層缺陷。先天性大理石樣毛細血管擴張症 (Cutis marmorata telangiectatica congenita)(第 103 章)曾被描述於某些 Adams–Oliver 症候群的個體。其他伴隨裂的 EDs 包括 AEC 與肢體–乳腺症候群,二者皆與 EEC 症候群為對偶基因關係。不伴 ED 的缺指(趾)合併顎裂 (Ectrodactyly with cleft palate without ED; OMIM 129830) 可能是一個獨立的實體。似乎有某些 EEC 家族,其連鎖研究提示有位於不同染色體基因座的其他致病基因。透過超音波進行產前診斷以偵測肢體異常並不可靠;基因檢測在某些家族中可能有用。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
如同其他伴有口面裂與眼科侵犯的 EDs,處置需要團隊合作的方法。同樣地,肢體缺陷的治療必須個別化。腎臟超音波檢查與對泌尿道問題的高度警覺是適當且有其必要的。
p63 相關之外胚層發育不良症候群 (P63-Related Ectodermal Dysplasia Syndromes)
病因與致病機轉 (Etiology and Pathogenesis)
AEC、EEC、裂手–裂足畸形、肢體–乳腺症候群與 ADULT 症候群,皆由對應至 3q27 之腫瘤抑制基因 TP63 的突變所致。此基因內不同的突變已被認為涉及這些症候群的致病機轉。34,48 TP63 廣泛表現,包括在增生性上皮組織的基底細胞中。AEC 症候群是一種體染色體顯性疾病,具完全外顯率 (complete penetrance) 與可變的表現度。迄今在 AEC 症候群中所鑑定出的改變多為 p63 之無菌 α 模體 (sterile α motif, SAM) 結構域中的錯義突變 (missense mutations)。EEC 症候群是一種體染色體顯性疾病,具可變的表現度與降低的外顯率。TP63 的突變已在大多數(但非全部)EEC 症候群個體中被發現。多數突變導致 p63 之 DNA 結合結構域 (DNA-binding domain) 中單一胺基酸的取代。33
與 EEC 相似,TP63 之 DNA 結合結構域中的突變也見於裂手–裂足畸形。49 TP63 的移碼突變 (frameshift mutations) 造成肢體–乳腺症候群,在此症候群中,乳腺以外的外胚層結構通常(但非總是)顯得正常。在 3 個不相關的 ADULT 症候群家族中,全部共有 p63 上相同的點突變 (point mutation)。50
WNT10A 疾病:齒–甲–皮膚發育不良 (OODD)、Schopf–Schulz–Passarge 症候群 (SSPS) (WNT10A Disorders)
重點一覽 (AT-A-GLANCE)
■ 變異型:以下所有狀況皆為 AR,伴位於 2q35 之 WNT10A(無翅型 MMTV 嵌入位點家族成員 10A, wingless-type MMTV integration site family, member 10A)的突變,該基因調控 β-catenin 介導的特異性細胞內訊息傳遞。
■ 齒–甲–皮膚發育不良 (Odonto-onycho-dermal dysplasia, OODD; OMIM 257980)。
■ Schopf–Schulz–Passarge 症候群 (SSPS; OMIM 224750)。
■ OODD 與 SSPS 皆顯現不一程度的牙齒過少、甲營養不良、毛髮稀疏、掌蹠角化過度伴多汗 (hyperhidrosis),而身體其餘部位則少汗 (hypohidrosis)。
■ SSPS 另以眼瞼汗腺囊瘤 (eyelid hidrocystomas) 為特徵。
前言 (Introduction)
齒–甲–皮膚發育不良 (OODD) 與 Schopf–Schulz–Passarge 症候群 (SSPS) 皆為由 WNT10A 基因突變所致的 ED 型態。
流行病學 (Epidemiology)
WNT10A 的突變已被報告於約 9% 的 EDs,以及 25% 不具 EDA 突變的低汗性 ED (HED) 患者中。51,52 隨著分子基因檢測的近期進展,WNT10A 突變已在約 16% 的疑似 HED 病例中被發現。9
臨床特徵 (Clinical Features)
OODD 具有廣泛的臨床特徵譜。最一致的發現是恆齒嚴重牙齒過少至無牙。乳齒在數量上幾乎總是正常,但可能細小且間隙寬大(圖 131-9A)。53-55 舌頭可能光滑,伴蕈狀乳頭 (fungiform papillae) 與絲狀乳頭 (filiform papillae) 減少。指甲營養不良,且可能有先天性無甲症。毛髮在出生時缺如,於較年長個體進展為乾燥、稀薄的毛髮。眉毛可能稀疏。皮膚變化包括手掌紅斑、掌蹠角化過度(圖 131-9B)56 與毛孔角化 (keratosis pilaris)。手掌與足底常有多汗 (hyperhidrosis),但身體其他部位則為少汗。SSPS 以眼瞼汗腺囊瘤為特徵;此外,還有上述 OODD 中所描述的發現。57
病因與致病機轉 (Etiology and Pathogenesis)
OODD 與 SSPS 由 WNT10A(無翅型 MMTV 嵌入位點家族成員 10A)基因的突變所致。57 多數病例為體染色體隱性,但高達 50% 的異型合子 (heterozygotes) 可能顯現臨床特徵。
診斷 (Diagnosis)
組織病理學上,皮膚顯示正角化 (orthokeratosis)、角化過度 (hyperkeratosis)、顆粒層增厚 (hypergranulosis) 與輕度棘層肥厚 (acanthosis)。以電子顯微鏡檢查時,OODD 的毛髮有縱向凹陷。同樣地,基因檢測可能有所助益。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
修復性牙科治療很重要。角化過度的處置並無特異性,指甲通常無需治療。SSP 有非黑色素瘤皮膚癌 (nonmelanoma skin cancer) 風險增加的可能,患者應接受適當監測。
局灶性真皮發育不良(Goltz 症候群;Goltz-Gorlin 症候群)(Focal Dermal Hypoplasia)
重點一覽 (AT-A-GLANCE)
■ OMIM 305600。
■ XLD,PORCN,Xp11.23,參與胚胎組織發育所需之 Wnt 蛋白的膜定位與分泌。
■ 皮膚特徵:沿 Blaschko 線分布的真皮萎縮伴脂肪疝出 (fat herniations);進行性乳頭狀瘤 (papillomas);斑塊狀禿髮。
■ 皮膚外特徵:小眼症 (microphthalmia) 與虹膜缺損 (colobomas);寡牙與琺瑯質缺陷;眾多骨骼異常,包括併指(趾)與骨紋狀症 (osteopathia striata);15% 有發育遲緩。
前言 (Introduction)
局灶性真皮發育不良 (focal dermal hypoplasia, FDH) 是一種罕見的外胚層發育不良,具多系統侵犯,包括皮膚、牙齒、骨骼與眼睛,臨床表現多變。58
流行病學 (Epidemiology)
FDH 最早描述於 1934 年。59 此後已發表超過 200 篇病例報告。
臨床特徵 (Clinical Features)
FDH 的皮膚變化是主要的診斷特徵,因男性與女性皆有合子後體細胞鑲嵌 (postzygotic somatic mosaicism),以及女性的隨機 X 染色體去活化 (random X-chromosome inactivation),故變異性相當大。可見線狀、點狀、條紋狀的篩狀萎縮 (cribriform atrophy)(圖 131-10),伴微血管擴張 (telangiectasia),沿 Blaschko 線分布。篩狀萎縮的特徵是皮膚上有微小的冰鑿樣 (ice pick–like) 凹陷。真皮變薄至缺如的區域呈不規則分布,所造成的脂肪疝出呈現為黃–粉紅色的贅生物,在皮膚表面易被壓陷(圖 131-10)。乳頭狀瘤(可能呈覆盆子樣或血管性)終生發展,好發於生殖器周圍、口周、間擦部位 (intertriginous) 與黏膜表面。其他皮膚特徵包括色素變化、斑塊狀禿髮、脆弱或稀疏的毛髮,以及掌蹠角化過度。某些個體曾有多汗,某些則有先天性皮膚發育不全 (aplasia cutis congenita)。FDH 中最常受侵犯的其他器官系統為骨骼、中樞神經系統 (CNS)、牙齒與眼睛。小眼症與虹膜缺損很常見,FDH 的診斷應促使進行完整的眼科評估。寡牙、牙齒發育不良與琺瑯質缺陷很常見。骨骼異常多到無法一一列舉;最常見的是骨骼的垂直條帶 (vertical banding, 骨紋狀症 osteopathia striata)、併指(趾)(皮膚性與骨性皆有)、缺指(趾)、不對稱、寡指(趾)(oligodactyly) 與身材矮小。約 15% 的病例曾報告智能障礙。少數病例曾描述其他器官系統的缺陷,包括心臟缺損、腹壁缺損與腎臟畸形。
病因與致病機轉 (Etiology and Pathogenesis)
局灶性真皮發育不良是一種 X 染色體顯性疾病,在男性通常為致死性。患有此疾病的男性病例報告,一般認為是源自合子後體細胞突變的鑲嵌現象(如同色素失禁症的情況);某些正常細胞的存在使男性得以存活。突變的基因是 PORCN,即果蠅 (Drosophila) porcupine 基因的人類同源基因。PORCN 一般認為對 Wnt 蛋白(皮膚與骨骼發育的關鍵調節因子)的棕櫚醯化 (palmitoylation) 與分泌很重要。60,61
詢問流產家族史與後代男女比例偏斜很重要,因為這些是母親為帶因者的線索。母親與父親都應仔細檢查;父親可能有細微特徵,且推測代表具有 FDH 基因合子後突變的個體。
診斷 (Diagnosis)
鑑於此症候群的罕見,FDH 的診斷具挑戰性。切片有助於顯示真皮發育不全與乳頭層真皮 (papillary dermis) 微血管增加。58 此外,基因檢測已可商業取得,但在具有合子後突變的患者中可能具挑戰性。
鑑別診斷 (Differential Diagnosis)
篩狀萎縮曾被描述於 X 染色體顯性的 Conradi–Hünermann 症候群(點狀軟骨發育不良),但魚鱗癬並非 FDH 的特徵,且脂肪疝出並非 Conradi–Hünermann 的一部分。IP 之萎縮病灶的 Blaschko 樣分布與其相似,但 IP 的水疱與角化過度在 FDH 中不會見到。在小眼症與線狀皮膚缺損/小眼症、真皮發育不全、鞏膜角膜化 (MIDAS; OMIM 309801) 中,皮膚缺損侷限於頭頸部;其有皮膚的萎縮與瘢痕,較類似先天性皮膚發育不全,而非單純的真皮萎縮。這些疾病確實共有相似的眼部異常。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
FDH 的皮膚與全身性特徵並無特異性治療。萎縮區域可能易感染或變得糜爛。乳頭狀瘤若妨礙功能可予以切除。喉部或氣管乳頭狀瘤的處置可能需轉介耳鼻喉科,並應考慮針對這些病灶進行術前評估。62 使用血管雷射以減少微血管擴張區域的紅斑可能有美容上的益處。如同多數 X 染色體顯性疾病,臨床侵犯程度差異甚大,嚴重度範圍很廣。這使得嬰兒期早期的預後諮詢困難,通常明智的做法是在全身侵犯的程度明朗化之前,建議耐心等待與安心保證。
色素失禁症 (Incontinentia Pigmenti)
重點一覽 (AT-A-GLANCE)
■ OMIM 308300。
■ XLD,IKBKG(又稱 NEMO),Xp28,編碼 I-kappa-B 激酶 gamma 次單元蛋白,參與細胞免於凋亡 (apoptosis) 的保護。
■ 皮膚特徵:4 個特徵性分期,分別涉及(1)水疱大疱性病灶;(2)疣狀病灶;(3)色素沉著;以及(4)色素減退與萎縮。也可能見到毛髮稀疏、瘢痕性禿髮 (cicatricial alopecia) 與牙齒缺陷。
■ 皮膚外特徵:眼科併發症、CNS 表現(癲癇、發育遲緩)。
前言 (Introduction)
色素失禁症 (incontinentia pigmenti, IP),又稱男性致死型 Bloch–Sulzberger 症候群,最早於 1906 年由 Garrod 描述。63 IP 是一種外胚層發育不良,對皮膚、牙齒、毛髮、指甲、眼睛與 CNS 的侵犯高度多變。64 皮膚發現是最早的臨床表現。65
流行病學 (Epidemiology)
IP 發生於每 50,000 至 150,000 名新生兒中有 1 例。65 它為 X 染色體顯性,且在男性胎兒通常為致死性;然而,如同 FDH,亦有男性病例的報告,發生於合子後晚期突變、節段性侵犯,或 XXY 基因型的情況下。
臨床特徵 (Clinical Features)
皮膚病灶有 4 個特徵性分期,全部沿 Blaschko 線發生。
並非每一期都會出現,且某些期別可能重疊。
■ 第 I 期:圍產期發炎期 (perinatal inflammatory stage),伴紅斑、水疱與膿疱,個別病灶持續數日至數週(圖 131-11A)。50% 於出生時即存在,90% 於 2 週齡時出現。通常於 4 至 6 個月時消退,但可能因疾病而復發。66
■ 第 II 期:疣狀與角化過度的丘疹,持續數週至數月,亦有報告持續長達數年(圖 131-11B)。
■ 第 III 期:色素沉著,通常於約 6 個月齡時出現,並持續數年,亦有報告持續至成年。範圍高度多變,常與先前各期的分布無關。最常侵犯腹股溝與腋窩。64
■ 第 IV 期:先前受影響區域的色素減退與萎縮。隨先前各期的消退而開始,並持續至成年。
許多皮膚特徵在 20 歲時消退或變得非常細微。瘢痕性與斑塊狀禿髮很常見,且傾向持續存在。在成年女性中,腿部的毛髮稀疏區域與頭皮的渦旋狀禿髮 (whorled alopecia) 在臨床上可能有所助益。67
甲營養不良在第二期很常見,但通常輕微。64 牙齒發現範圍廣泛,包括牙齒過少、小牙症 (microdontia)、無牙與圓錐狀牙。眼科侵犯見於 20% 至 77% 的患病個體,64,65 包括視網膜新生血管 (retinal neovascularization)、斜視、白內障、視神經萎縮,以及視網膜色素異常。在診斷時篩檢眼部發現很重要,因為視網膜新生血管可導致視網膜剝離,風險在新生兒期至出生後最初 6 年內最高。64 鑑於視力降低的風險,重複的眼科檢查是必要的。CNS 侵犯估計為 10% 至 30%,但可能更低。64,65 CNS 異常範圍從單次癲癇發作到腦病變、缺血性中風與顯著的發育遲緩。症狀最常於出生後第一年出現,這與「神經學表現是發炎及血管病變/缺血之結果」的假說一致。68 CNS 表現對病態與死亡影響甚大,而預測性特徵(如視網膜新生血管與男性性別)有助於評估風險。68
單側乳房發育不全 (Unilateral breast aplasia) 不常見,但應警覺到 IP 可能的診斷。69
病因與致病機轉 (Etiology and Pathogenesis)
IP 是一種 X 染色體顯性疾病,在男性通常為致死性(關於合子後突變鑲嵌的討論見上文 FDH)。約 80% 的 IP 源自 IKBKG 基因的突變,該基因編碼 I-kappa-B 激酶 gamma 次單元蛋白 (IKK-gamma)。已報告超過 50 種不同的 IKBKG 基因合子後體細胞突變變異型。IKK-gamma 蛋白廣泛表現,並與 IKK-alpha 及 IKK-beta 蛋白結合,以活化 NF-kappa-B 複合體。此複合體保護細胞免於 TNF 誘發的凋亡。IP 的不同分期代表了無法活化此保護性複合體之受影響細胞的死亡。有人提出,帶有 IKBKG 突變的細胞經歷凋亡後,會被未受影響的細胞所取代。晚期復發可能源自受影響角質細胞在先前病灶部位的持續存在。65
診斷 (Diagnosis)
2014 年,Minic 等人更新了診斷準則。主要準則包括 4 個皮膚病灶分期中的任一個。次要準則包括牙齒、眼部、CNS、毛髮、指甲、顎部、乳房與乳頭異常;家族史;多次男性流產;以及組織病理學皮膚發現。64,69
皮膚切片對診斷大有助益,尤其在疑似男性病例中,且組織病理學具分期特異性。水疱大疱性病灶的切片顯示海綿狀水腫 (spongiotic) 的表皮,伴嗜酸性球浸潤與微膿瘍 (microabscesses),以及角化不良 (dyskeratosis)。疣狀病灶顯示角化過度、乳頭瘤樣增生 (papillomatosis) 與角化不良。針對 IKBKG 常見缺失/重複與基因定序的基因檢測已可商業取得,可鑑定出 IP 與 HED-ID。約 75% 的患病者有外顯子 (exons) 4 至 10 的常見 11.7 kb 缺失。65 檢測可確認臨床診斷、偵測女性帶因者,並評估帶有 IKBKG 突變家族中的高風險妊娠。在無家族史的情況下,產前診斷困難,因為除子宮內生長遲滯 (intrauterine growth retardation) 外並無特徵性的超音波表現。64
鑑別診斷 (Differential Diagnosis)
IP 的鑑別診斷取決於患者在病程的哪個時間點呈現。在新生兒期,可能需考慮水疱大疱性疾病,包括 HSV、VZV、大疱性膿痂疹、大疱性肥大細胞增生症 (bullous mastocytosis) 與表皮鬆解水疱症,以及其他鑲嵌型疾病,如局灶性真皮發育不良與表皮母斑症候群 (epidermal nevus syndrome)。之後,線狀表皮母斑、ILVEN 與色素鑲嵌 (pigmentary mosaicism) 可能外觀相似。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
多數在嬰兒期無顯著併發症的患病個體預後良好,具有正常的預期壽命。治療以症狀為導向。皮膚變化除了水疱的傷口照護(以預防續發性感染)外,無需特異性治療。64 有報告指出,局部皮質類固醇可在第一期減輕發炎並加速癒合。66 如同其他 EDs,牙科評估與修復很重要。在診斷時進行基線眼科檢查,並遵循出生後前 3 年重複檢查的建議指引至關重要。周邊視網膜光凝固術 (retinal photocoagulation) 可降低視網膜剝離的風險。應考慮在診斷時轉介神經科進行全面檢查與預先指引,且這對於癲癇、痙攣 (spasticity) 或其他必要缺陷之處置而言是必要的。64 若出現神經學症狀,應進行 MRI 與 EEG。對於有視網膜新生血管的患病個體,可考慮 MRI。64
牙甲症候群(Witkop 症候群)(Tooth and Nail Syndrome)
重點一覽 (AT-A-GLANCE)
■ OMIM 189500,位於 4p16.1 之 MSX1 的 AD 突變,該基因編碼轉錄因子 Msx1。
■ 特徵為出生時細小、易碎的指甲,可能隨時間改善;可能有細而稀疏的毛髮。乳齒通常不受影響,但恆齒無法萌發或部分缺如。
流行病學 (Epidemiology)
最早的病例由 Witkop 於 1965 年報告,當時他描述了一個顯示體染色體顯性遺傳的家系。他後來於 1975 年與 Hudson 一同描述了 6 個家族中的 23 例。70 全部皆顯現甲發育不全與牙齒過少;後者表現為恆齒無法萌發。
臨床特徵 (Clinical Features)
在牙甲症候群中,指甲薄、小且易碎,出生時可能顯現匙狀甲 (koilonychia)。趾甲通常比指甲受影響更嚴重(圖 131-12)。甲變化隨年齡改善,在患病成人中可能不被察覺。少數個體曾報告有細而稀薄的毛髮。71
乳齒通常不受影響,但可能細小。恆齒可能無法萌發,且可有部分或完全缺如(圖 131-12A)。最常缺如的是下頜門齒、第二臼齒與上頜犬齒。70
沒有其他外胚層結構受到影響。
病因與致病機轉 (Etiology and Pathogenesis)
MSX1(一個在發育中牙齒與甲床表現的基因)的無義突變 (nonsense mutation),於 2001 年首次在一個三代家族中被鑑定出來。72 2013 年,一名不相關的患者也被發現有 MSX1 突變。73
MSX1 的其他突變已與孤立性牙齒缺失或合併顎裂的牙齒缺失有關,且小鼠模型已證實 MSX1 在牙齒與甲發育中的角色。72 牙甲症候群為體染色體顯性,具可變的表現度與家族內變異性。
診斷 (Diagnosis)
這是一個容易被遺漏的狀況。甲變化可能很細微。牙齒異常可能輕微到逃過醫師的偵測。缺乏相關特徵(無論是皮膚或全身性),可輕易將 Witkop 症候群與其他 EDs 區分開來。基因檢測可供使用。
鑑別診斷 (Differential Diagnosis)
有一種推測為體染色體隱性的疾病,稱為「巨牙症、缺牙與毛髮稀疏」(taurodontia, absent teeth, and sparse hair; OMIM 272980),外觀相似。74 巨牙症 (Taurodontia) 指牙齒具有伸長的牙體與牙髓腔,以及短的牙根。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
指甲通常無需治療。修復性牙科治療很重要。
毛–齒–骨 (TDO) 症候群 (Tricho-Dento-Osseous Syndrome)
重點一覽 (AT-A-GLANCE)
■ OMIM 190320,位於 17q21.3-q22 之 DLX3 的 AD 突變,該基因編碼轉錄因子同源框蛋白 DLX-3。
■ 外胚層特徵:在出汗與皮膚正常的情況下,出現牙齒、指甲與毛髮變化。
■ 全身性特徵:骨密度增加。
前言 (Introduction)
毛–齒–骨 (tricho-dento-osseous, TDO) 症候群是一種罕見的體染色體顯性 ED,侵犯牙齒、毛髮、指甲與骨骼。75 它已在數個家族中被描述,但確切的流行病學資訊付之闕如。
臨床特徵 (Clinical Features)
TDO 侵犯毛髮、牙齒與指甲。毛髮捲曲或極度蜷曲 (kinky),尤其在新生兒期。牙齒發現包括琺瑯質發育不全、鈣化不足 (hypocalcification),以及巨牙症 (taurodontism, 牙體與牙髓腔的擴大),乳齒與恆齒皆有。指甲常增厚,並在表層出現分裂。汗腺功能與皮膚不受影響。骨骼發現包括長骨與顱骨的骨密度增加。可能有顱縫早閉 (craniosynostosis),導致長頭畸形 (dolichocephaly)。76
病因與致病機轉 (Etiology and Pathogenesis)
TDO 是一種體染色體顯性疾病,由 DLX3 的突變所致,該基因編碼人類 distal-less 同源框。視突變而定,表現型有不同的嚴重程度,但外顯率通常完全。77 DLX3 蛋白是上皮組織發育與分化中的轉錄活化因子,並已被證實調控毛囊。78,79
診斷 (Diagnosis)
臨床分子基因檢測可透過數個實驗室取得。
鑑別診斷 (Differential Diagnosis)
TDO 與「合併巨牙症之琺瑯質發生不全(成熟不全–發育不全型)」(amelogenesis imperfecta, hypomaturation-hypoplasia type, with taurodontism, AIHHT) 有相同的牙齒發現,但 AIHHT 缺乏毛髮與指甲的發現。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
處置包括針對琺瑯質發育不全的牙科照護。鑑於其體染色體顯性性質,應提供遺傳諮詢。
眼–齒–指(趾)發育不良 (ODDD) (Oculo-Dento-Digital Dysplasia)
重點一覽 (AT-A-GLANCE)
■ OMIM 164200。
■ 位於 6p21-23.2 之 GJA1 的 AD 突變,該基因編碼 connexin 43,一種細胞間連接的 connexin 蛋白。
■ 特徵為毛髮稀疏、琺瑯質發育不全、屈指畸形 (camptodactyly) 與小眼。
前言 (Introduction)
眼–齒–指(趾)發育不良 (oculo-dento-digital dysplasia, ODDD) 是一種體染色體顯性 ED,具可變的表現度。
臨床特徵 (Clinical Features)
ODDD 患者有稀疏、乾燥、生長緩慢的頭皮毛髮,伴睫毛缺如或稀疏。乳齒與恆齒皆有琺瑯質發育不全,導致牙齒細小、發黃且易碎,易蛀。汗腺與皮膚正常。雖然視力正常,但眼睛有短的瞼裂 (palpebral fissures),伴小角膜 (microcornea) 和/或小眼症。指(趾)變化包括超過 80% 的病例有第四與第五指的屈指畸形,並可能有第四與第五指的併指。其他骨骼異常包括顱骨增生 (hyperostosis)、寬肋骨與鎖骨,以及長骨的異常骨小樑形成 (trabeculation)。80
病因與致病機轉 (Etiology and Pathogenesis)
ODDD 為體染色體顯性,由 connexin-43 基因 (GJA1) 的突變所致。81 臨床分子基因檢測可供使用。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
處置包括針對琺瑯質發育不全的牙科照護。雖然視力正常,但患者應由眼科追蹤,因為 10% 至 15% 會發展出青光眼。82
毛–鼻–指(趾)骨症候群 (Tricho-Rhino-Phalangeal Syndrome)
重點一覽 (AT-A-GLANCE)
■ 變異型:
■ TRPS I (OMIM 190350):位於 8q23.2 之 TRPS1 的 AD 突變;同一基因的突變造成 TRPS III (OMIM 190351)。
■ TRPS II(Langer–Giedion 症候群; OMIM 150230):AD,為涉及 TRPS1 與 EXT1(8q24.11-q24.13)的連續基因症候群 (contiguous gene syndrome)。
■ 特徵為毛髮稀疏、鼻部突出、下巴後縮,以及錐形骨骺 (cone-shaped epiphyses) 導致中節指骨偏斜。
前言 (Introduction)
毛–鼻–指(趾)骨 (tricho-rhino-phalangeal, TRP) 症候群是一種 AD 畸形症候群,特徵為獨特的顱顏與骨骼異常。雖然它因僅侵犯毛髮而不侵犯牙齒、指甲或汗腺/皮脂腺,而不被視為典型 ED,但本章最能涵蓋其其他特徵。
臨床特徵 (Clinical Features)
TRPS I 患者有稀疏、脆弱、生長緩慢的頭皮毛髮。在光學顯微鏡下,可見捻轉發 (pili torti)。他們有突出的梨形鼻 (pear-shaped nose),伴寬而高的鼻樑與球狀鼻尖。人中 (philtrum) 長而平坦,伴薄的上唇與後縮的下巴(圖 131-13)。83
指(趾)變化可能要到數歲時才出現,包括錐形骨骺與中節指骨偏斜,使手指呈現彎曲的外觀。其他骨骼異常包括髖部畸形與身材矮小。84
TRPS III 除上述特徵外,另有嚴重的短指(趾)(brachydactyly) 與更顯著的身材矮小。85 TRPS II 結合了 TRPS I 與第 1 型多發性外生骨疣 (multiple exostoses type 1) 的特徵。此外,這些患者常有智能障礙。86
病因與致病機轉 (Etiology and Pathogenesis)
TRPS I 與 TRPS III 為體染色體顯性,由 TRPS1(一種轉錄因子鋅指蛋白)的突變所致。83 TRPS II 由一個同時包含 TRPS1 與 EXT1 的缺失所致。基因檢測可供使用以協助診斷。
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
對於身材矮小,應就個別情況考慮生長激素治療。87
骨骼變化應加以監測,並在必要時針對疼痛適當處置。耳廓整形術 (Otoplasty) 與鼻整形術 (rhinoplasty) 可提供美觀上的矯正。88
誌謝 (Acknowledgments)
作者感謝 Alanna F. Bree、Nnenna Agim 與 Virginia P. Sybert 對本章先前版本的貢獻。
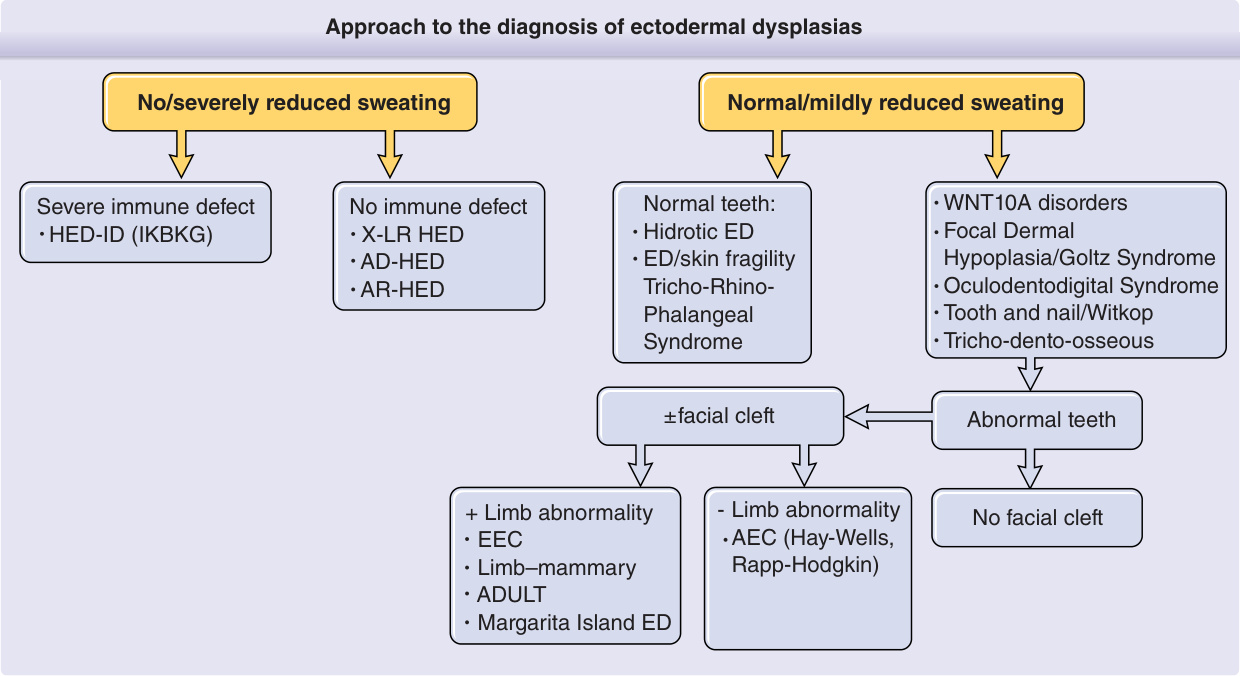
圖 131-1 為一演算法,呈現診斷 EDs 的臨床方法。在此演算法中,做出特定 ED 診斷的第一步是判定有出汗(汗性, hidrotic)或無出汗(低汗/無汗, hypohidrotic/anhidrotic)。其他外胚層結構與非外胚層衍生組織的侵犯,在診斷階層中提供進一步的分支點。遺傳模式在看似一致的診斷群組內可能有所不同,在提供遺傳諮詢前評估家族成員時必須謹慎。美國外胚層發育不良基金會 (National Foundation for Ectodermal Dysplasias, NFED; http://www.nfed.org) 是一個外行支持團體,為家庭與醫師提供眾多資訊性手冊,並有強而有力的倡議計畫,致力於牙科照護、保險給付與研究。隨著對導致 EDs 之分子路徑研究所獲得的知識,人們期望以分子為基礎的療法將能治療或替換有缺陷或缺失的牙齒、毛囊或外分泌汗腺。5-7
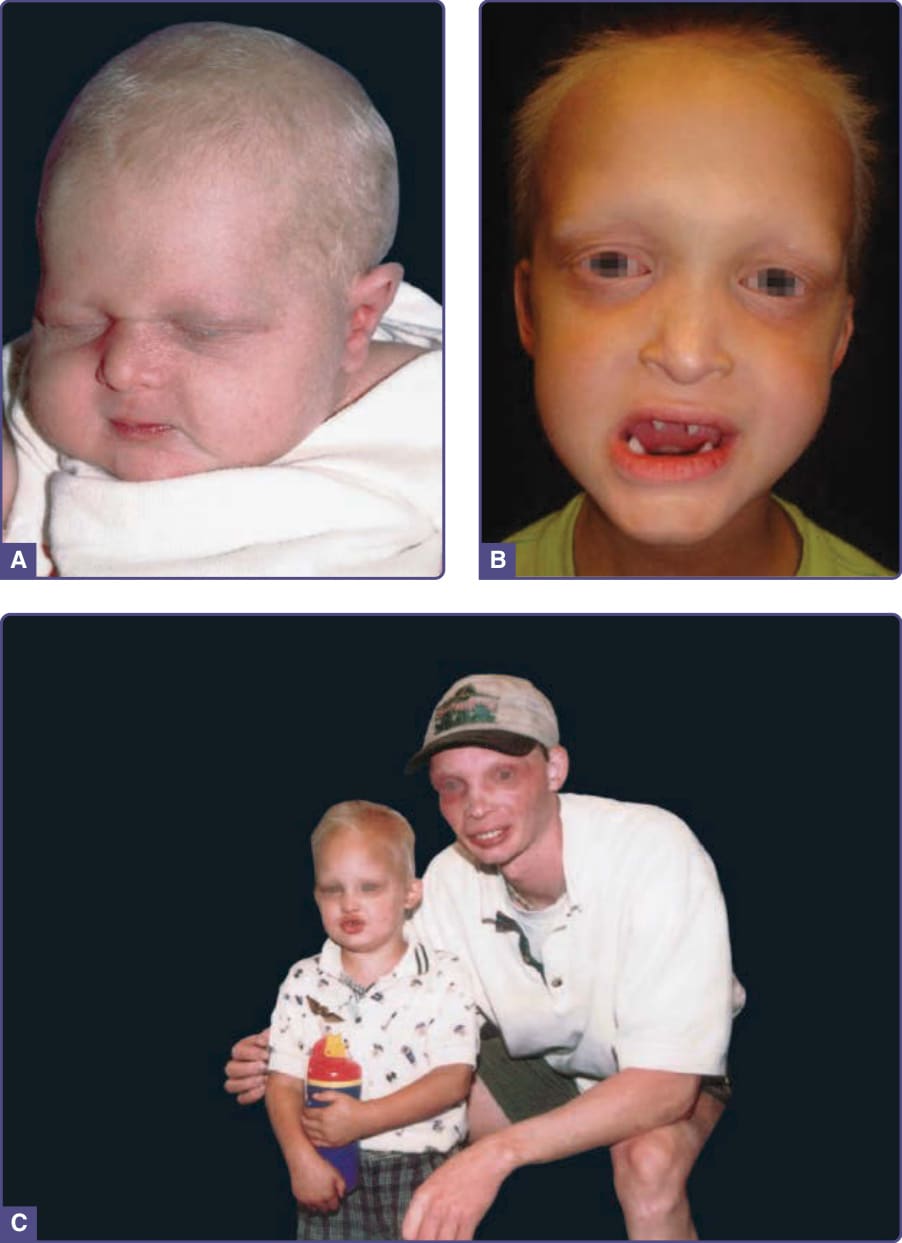
圖 131-2 低汗性外胚層發育不良 (Hypohidrotic ectodermal dysplasia)。A,新生兒,伴眼周皺紋與喙狀鼻 (beaked nose)。若無陽性家族史,不會懷疑此診斷。B,釘狀牙;可見細緻的眼周皺紋。C,兩名不相關、患有 X 染色體連鎖低汗性外胚層發育不良的男性;成人配戴假牙;眼周皺紋與色素沉著明顯可見。(Used with permission from the National Foundation for Ectodermal Dysplasias.)
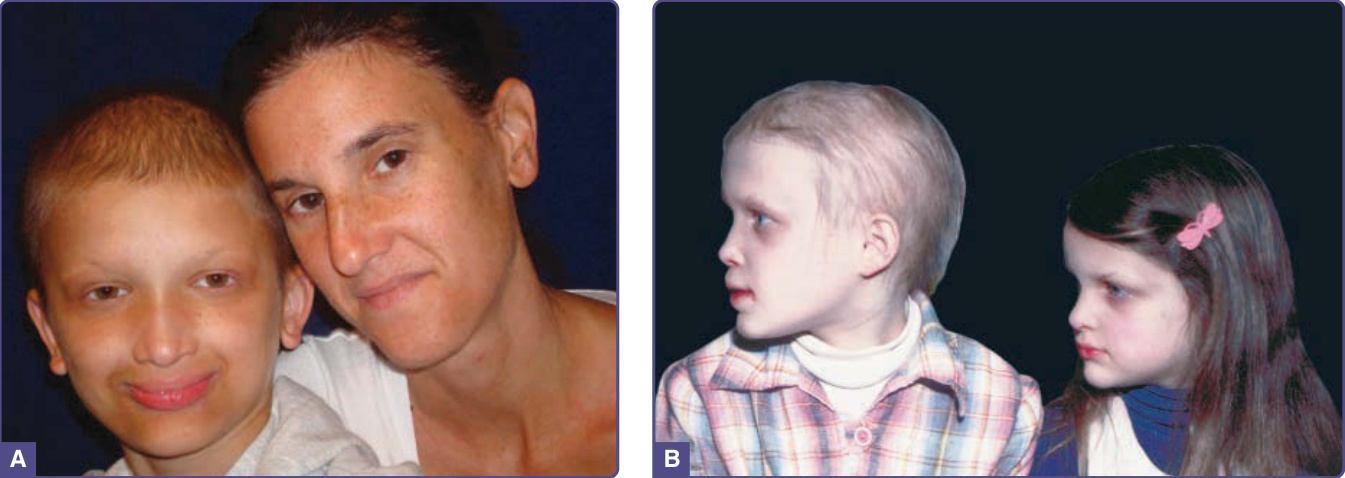
圖 131-3 低汗性外胚層發育不良 (Hypohidrotic ectodermal dysplasia)。A,X 染色體連鎖低汗性外胚層發育不良的女性帶因者及其患病的兒子。B,兩姊妹患有 X 染色體連鎖低汗性外胚層發育不良,表現程度不同。注意眼周色素沉著、豐厚外翻的嘴唇與雕塑般的鼻子。(Reproduced from Sybert V. Hypohidrotic ectodermal dysplasia: Argument against an autosomal recessive form clinically indistinguishable from X-linked hypohidrotic ectodermal dysplasia (Christ-Siemens-Touraine syndrome). Pediatr Dermatol. 1989;6:76-81; with permission. Copyright © 1989 John Wiley & Sons.)
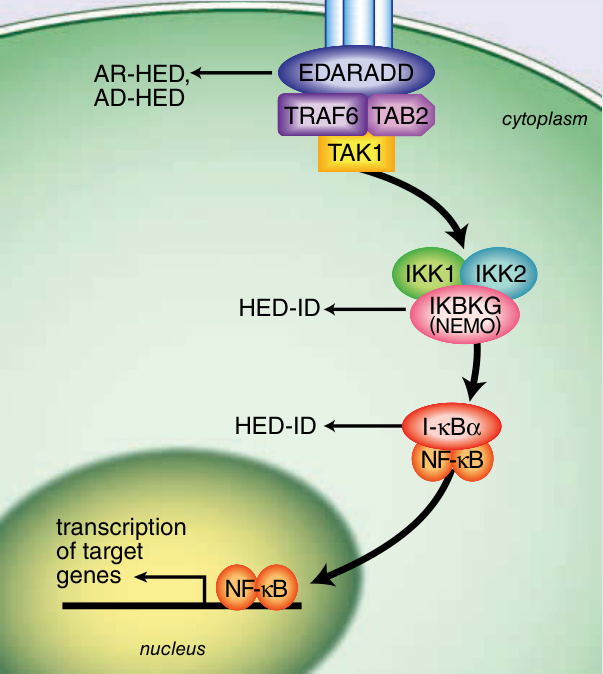
圖 131-4 Ectodysplasin 訊息傳導路徑。(Image B, Reproduced from Rimoin D, Pyeritz R, Korf B, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics, 6th ed. Oxford, UK: Academic Press; 2013; Fig. 148-5; with permission. Copyright © Elsevier.)
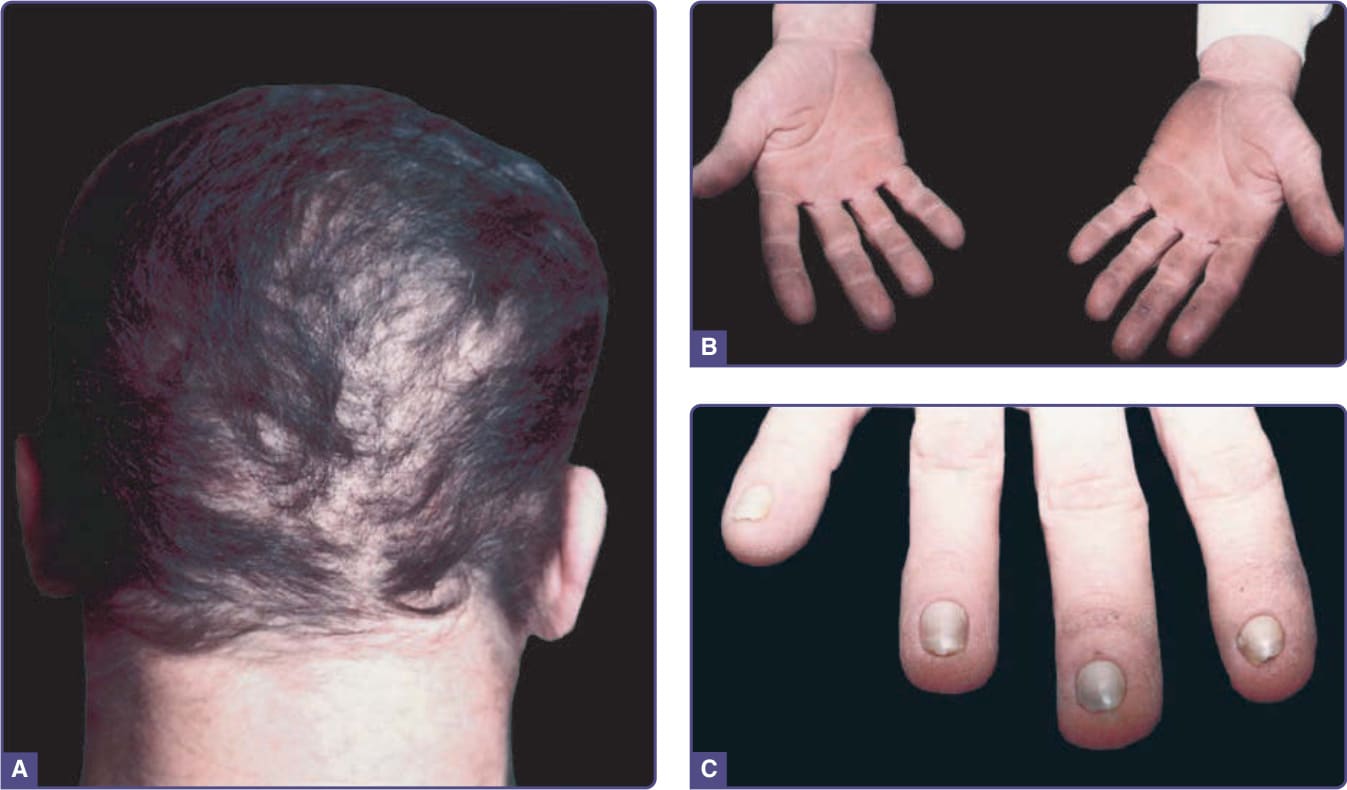
圖 131-5 汗性外胚層發育不良(Clouston 症候群)(Hidrotic ectodermal dysplasia, Clouston syndrome)。A,成人的斑塊狀禿髮。可見毛髮的粗糙度。B,手掌角化過度。C,甲營養不良。
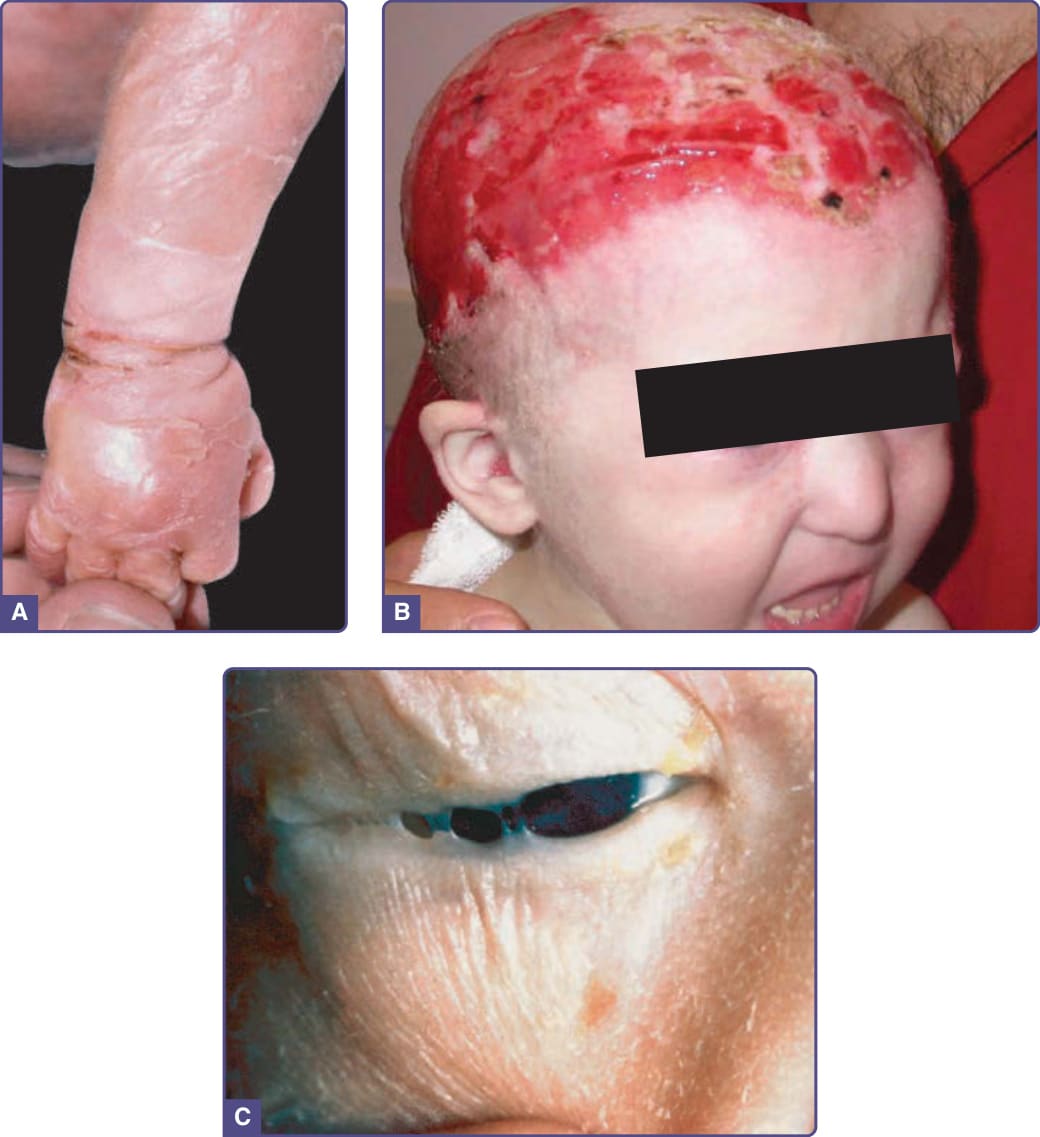
圖 131-6 先天性絲狀眼瞼粘連–外胚層發育不良–顎裂(Hay–Wells)症候群 (Ankyloblepharon filiforme adnatum–ectodermal dysplasia–cleft palate, Hay–Wells syndrome)。A,新生兒,伴脫皮的膠樣膜。B,頭皮糜爛。C,眼瞼間的細小組織束帶(先天性絲狀眼瞼粘連)。
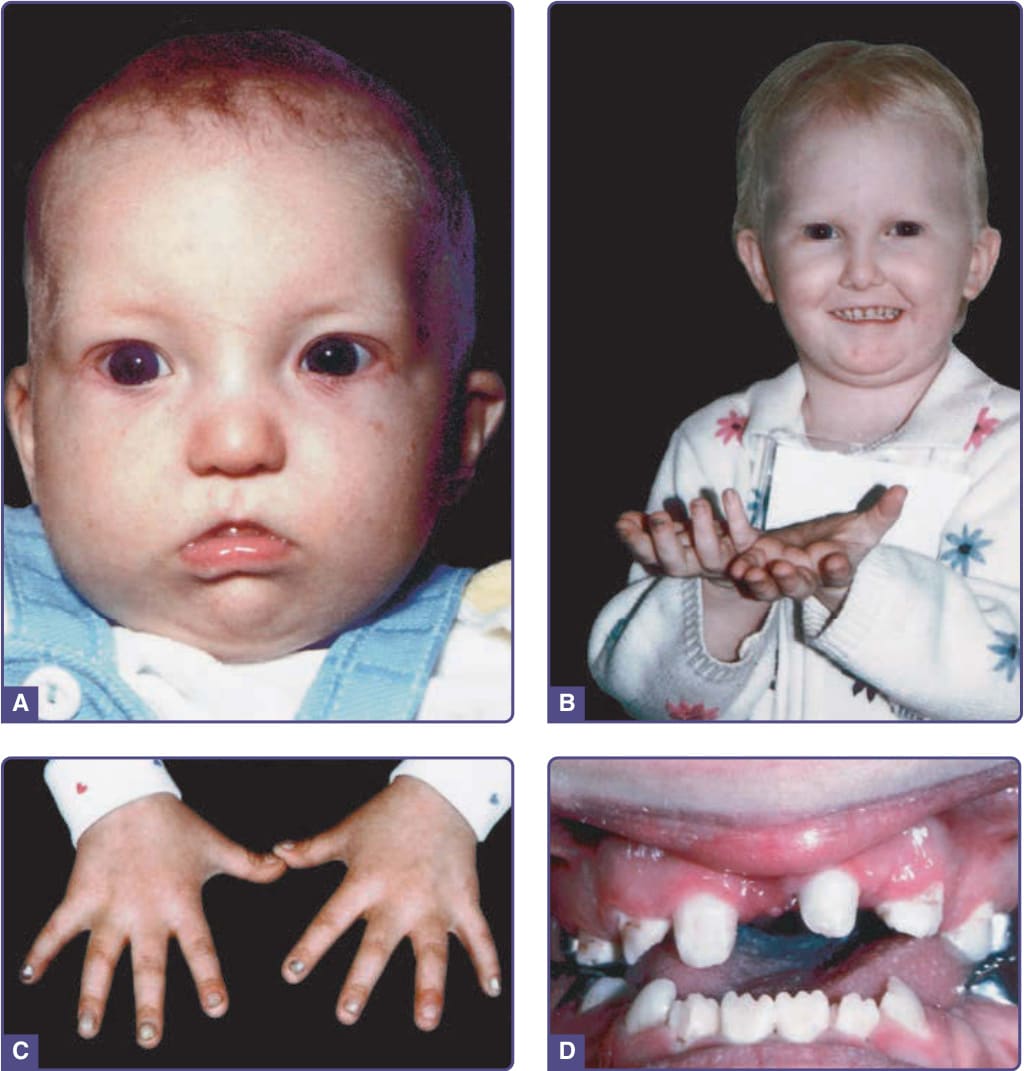
圖 131-7 Rapp–Hodgkin 症候群 (Rapp–Hodgkin syndrome)。A,5 個月大的患病嬰兒。B,細、金黃、稀疏的毛髮;右手中指可見甲變化的開端。C,與 (A) 同一患者,4 歲半時的異常指甲,甲板增厚且易碎。D,異常齒列、缺牙與釘狀牙。(Images A and C, From Schroeder HW Jr, Sybert VP. Rapp-Hodgkin ectodermal dysplasia. J Pediatr. 1987;110(1):72-75; with permission. Copyright © Elsevier. Image B, Used with permission from the National Foundation for Ectodermal Dysplasias.)
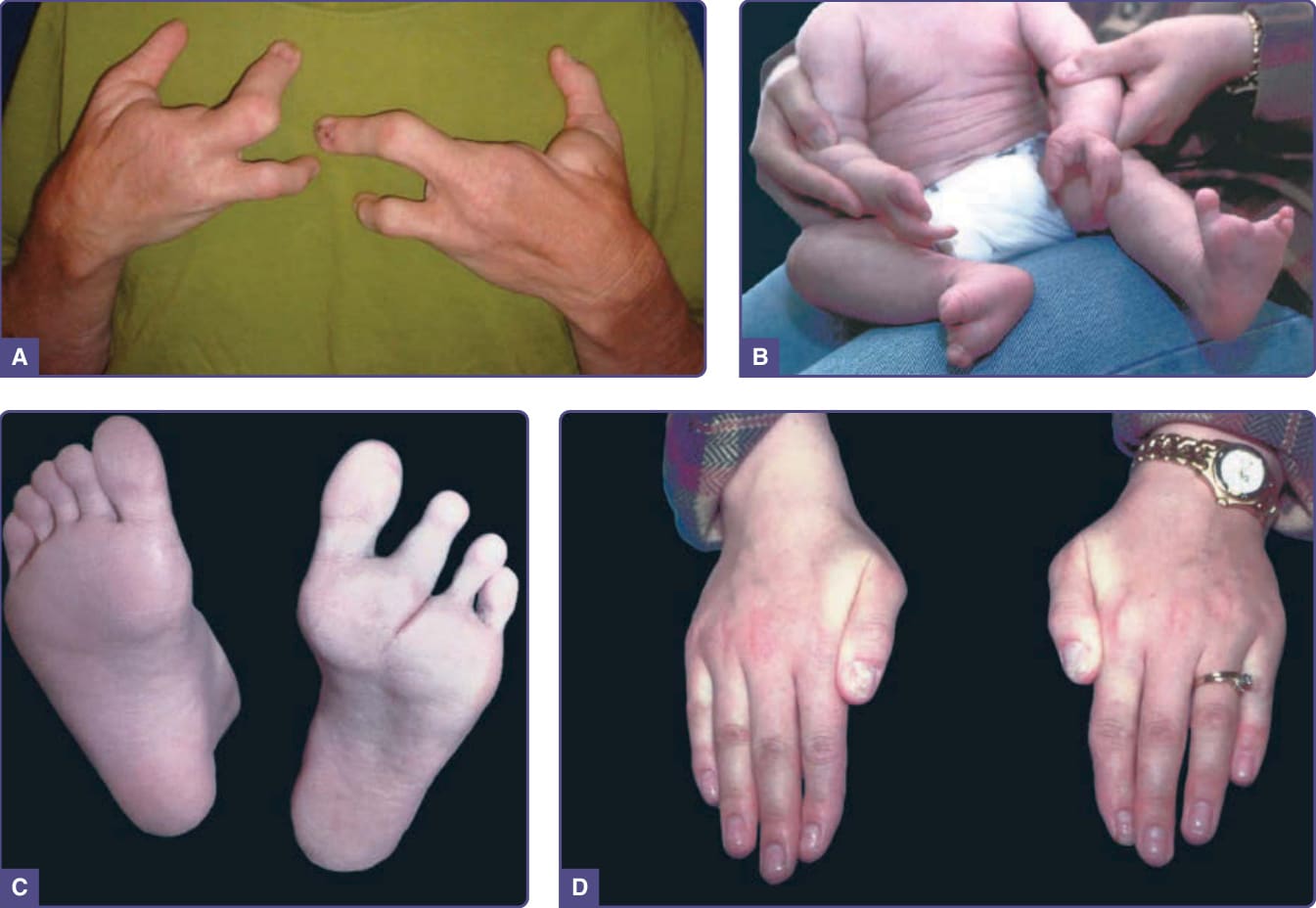
圖 131-8 缺指(趾)–外胚層發育不良–裂症候群 (Ectrodactyly–ectodermal dysplasia–clefting syndrome)。A,一名患病成人的手。B,一名患病嬰兒的手與足。C 與 D,(B) 中嬰兒之雙親的足與手,展現肢體間與家族成員間表現的變異性。注意輕度患病母親的甲營養不良,尤其在拇指上明顯可見。
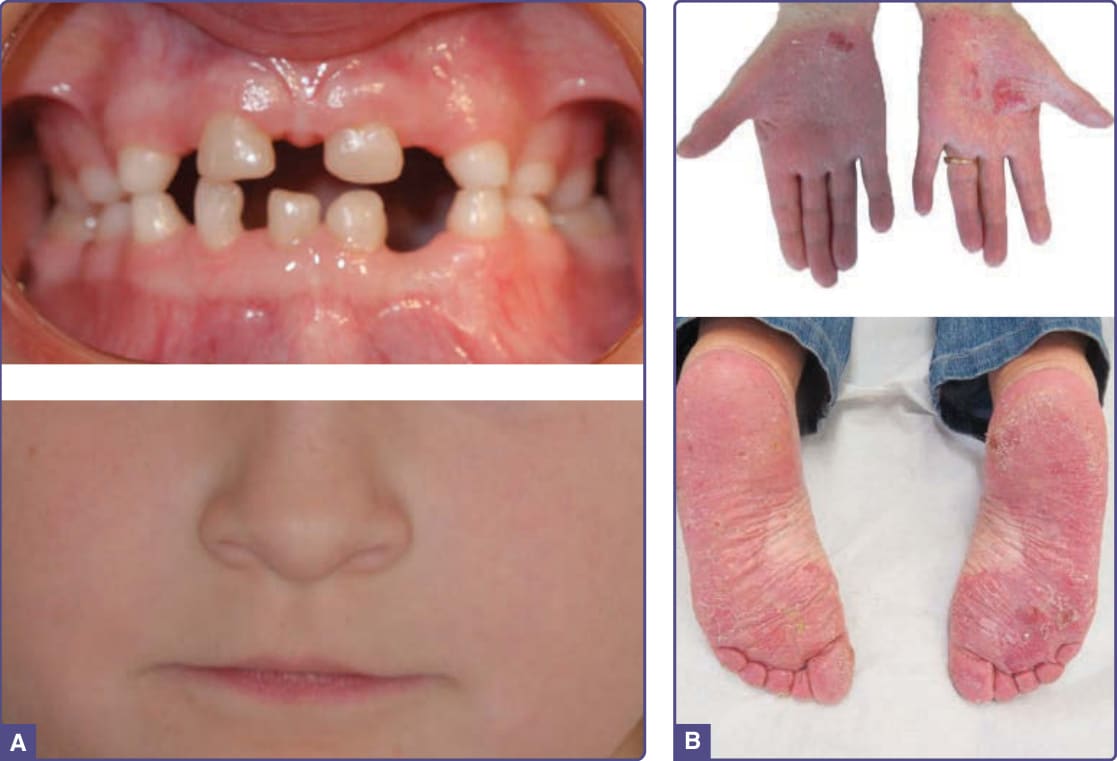
圖 131-9 WNT10A 疾病 (WNT10A disorders)。A,注意間隙寬大且散在缺失的乳齒,以及特徵性的薄上唇。(Image reused from Bergendal B, Norderyd J, Zhou X, et al. Abnormal primary and permanent dentitions with ectodermal symptoms predict WNT10A deficiency. BMC Med Genet. 2016;17(1):88 with permission from BioMed Central.) B,手掌與足底的紅斑性角化過度,伴糜爛與龜裂。(Images reused from Krøigård AB, Clemmensen O, Gjørup H, et al. Odonto-onycho-dermal dysplasia in a patient homozygous for a WNT10A nonsense mutation and mild manifestations of ectodermal dysplasia in carriers of the mutation. BMC Dermatol. 2016;16:3, with permission from BioMed Central.)
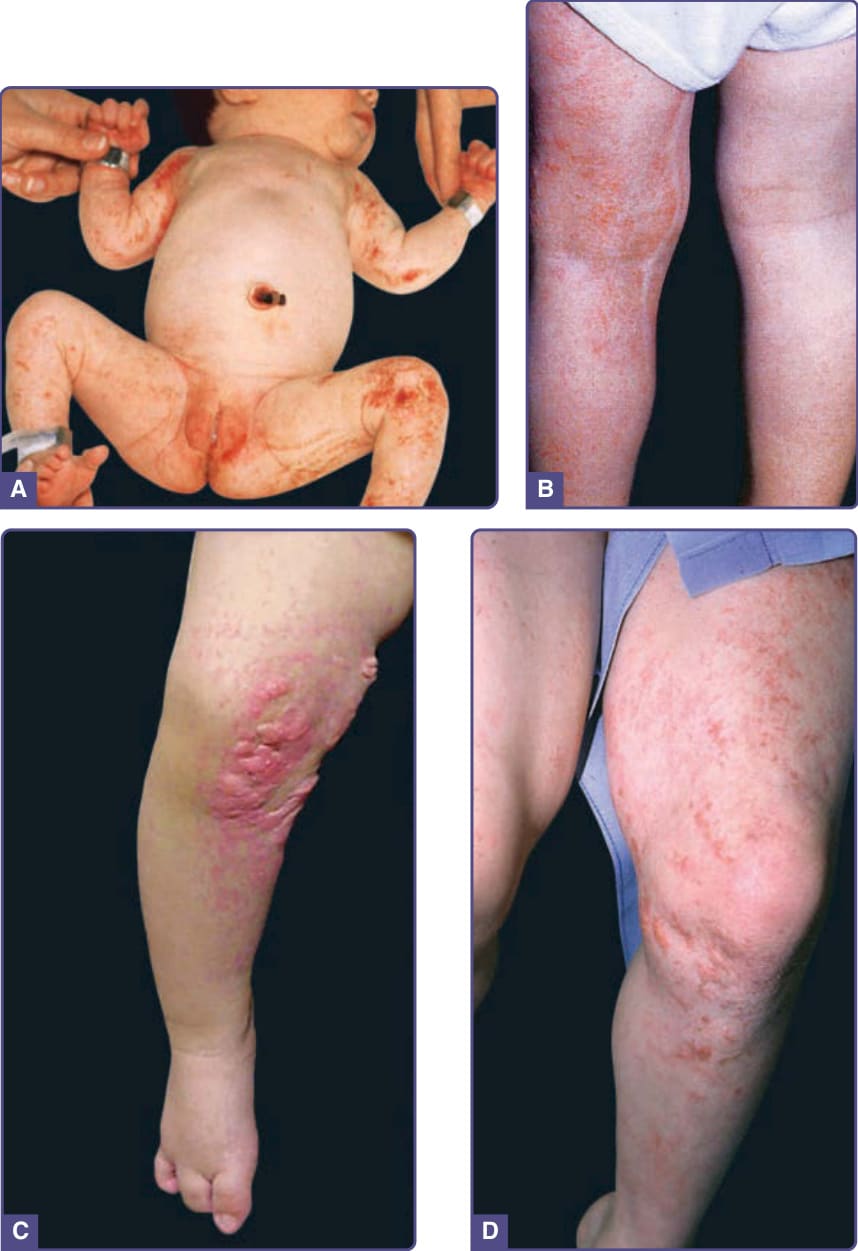
圖 131-10 局灶性真皮發育不良 (Focal dermal hypoplasia)。A,新生兒的條紋狀與斑塊狀侵犯。注意與色素失禁症病灶分布的相似性。B 與 C,同一女孩 2 歲時,顯示萎縮與脂肪疝出。D,現為年輕成人,以紅斑與萎縮為主,伴乾燥與鱗屑。(Image A, From Sybert VP. Genetic Skin Disorders, 3rd ed. New York, NY: Oxford University Press; 2017; with permission.)
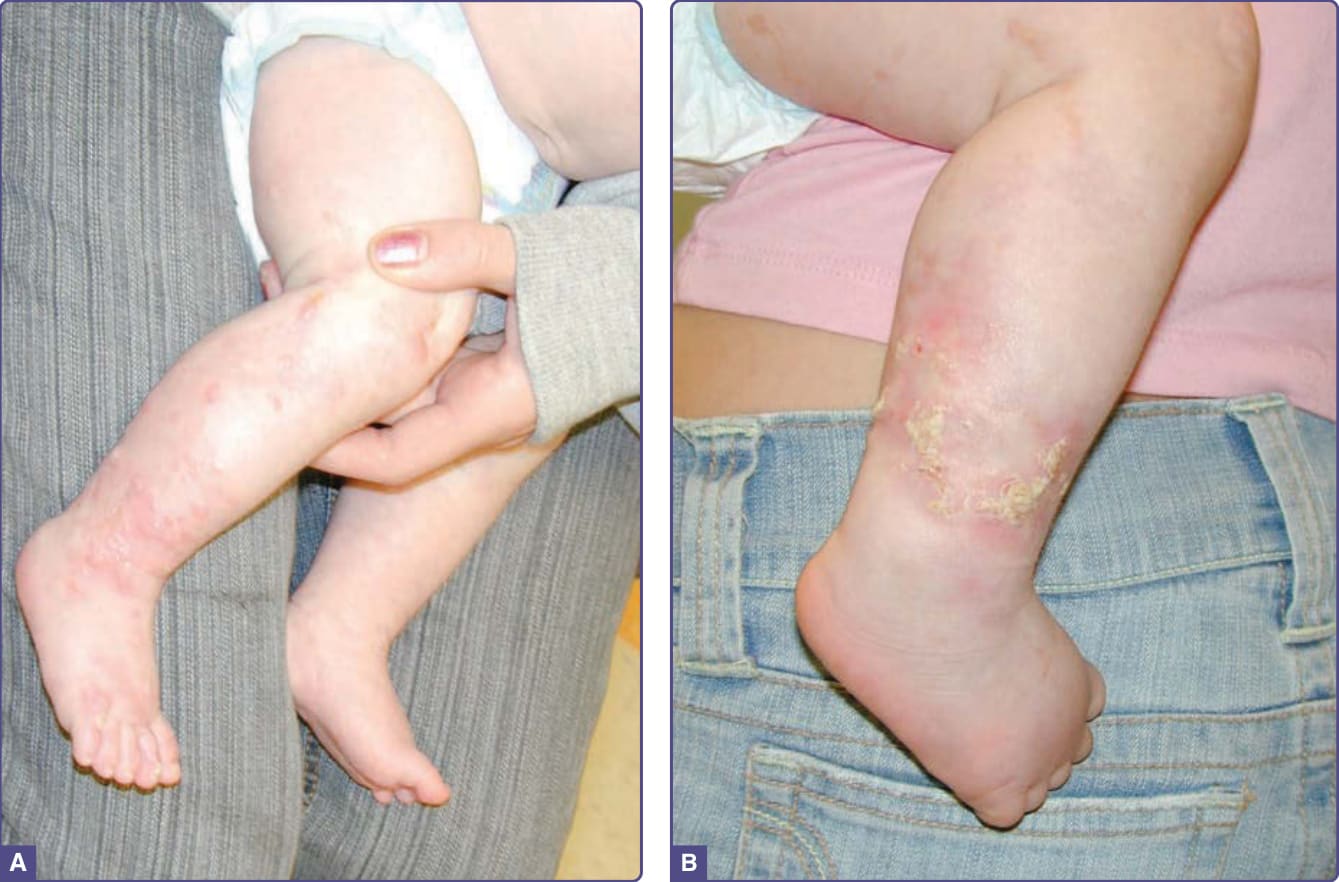
圖 131-11 色素失禁症 (Incontinentia pigmenti)。A,第 I 期。沿 Blaschko 線分布於小腿的水腫性紅斑丘疹與水疱。注意腳趾上早期第 II 期的疣狀丘疹。B,第 II 期。角化過度的丘疹,以及伴周圍紅斑的丘疹與斑塊。(Reproduced from Rimoin D, Pyeritz R, Korf B, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics, 6th ed. Oxford, UK: Academic Press; 2013; Figure 148-15; with permission. Copyright © Elsevier.)
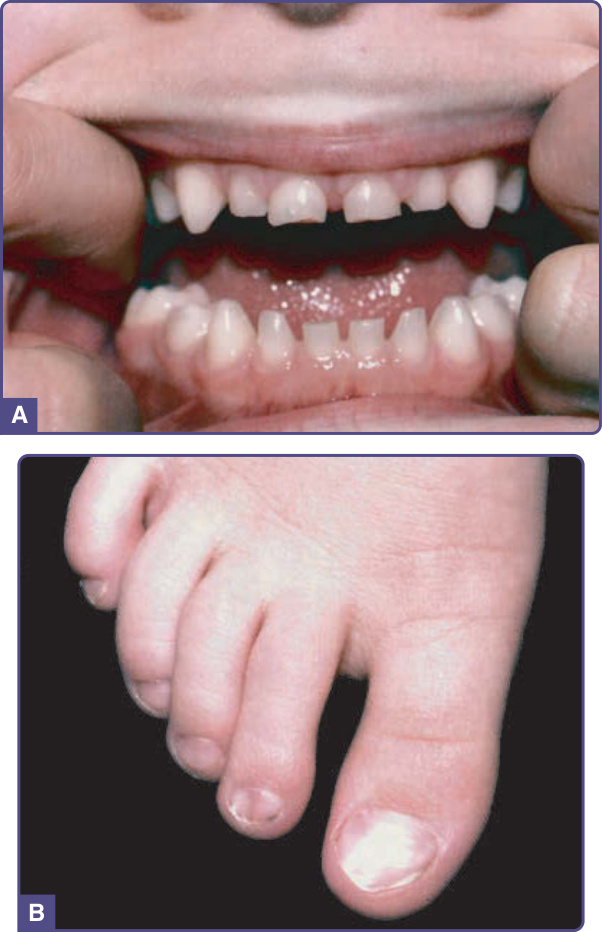
圖 131-12 牙–甲症候群 (Tooth–nail syndrome)。A,乳齒仍在位;恆齒無法萌發。B,營養不良的趾甲,伴甲板扁平化。
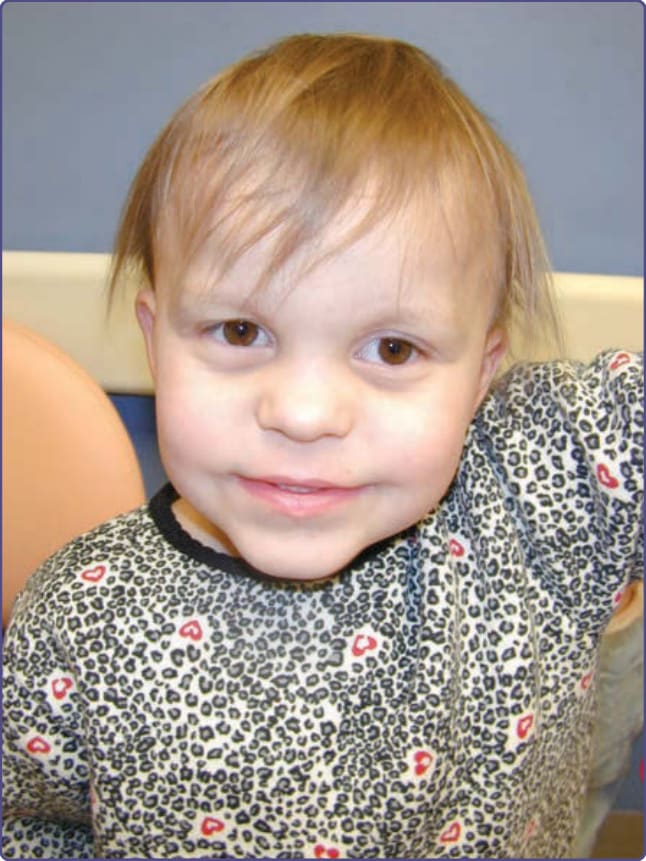
圖 131-13 毛–鼻–指(趾)骨症候群 (Tricho-rhino-phalangeal syndrome)。注意稀疏的毛髮與眉毛、球狀鼻與薄的上唇。
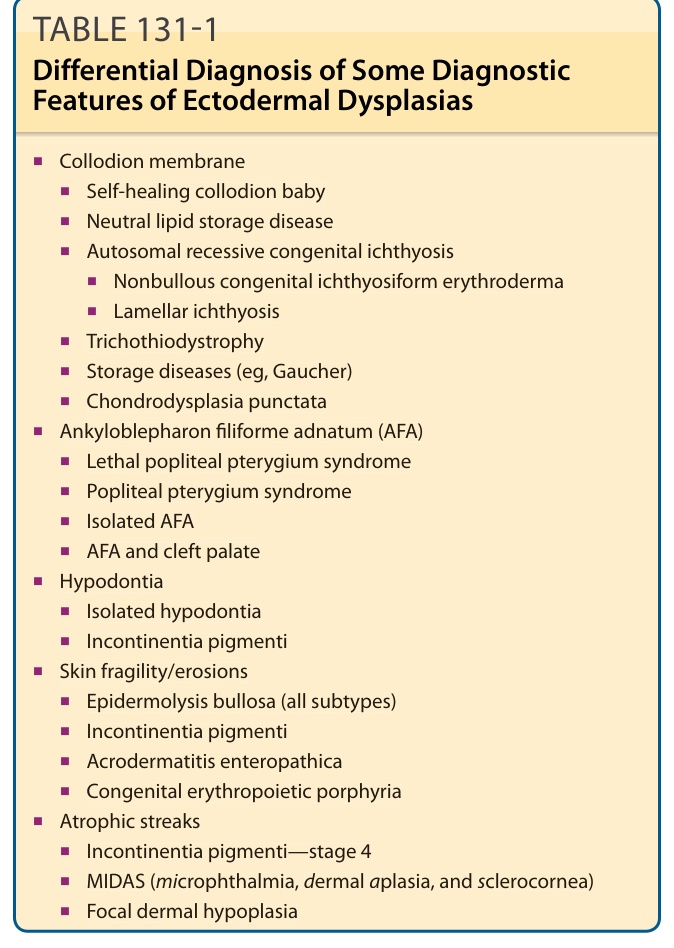
表 131-1 部分外胚層發育不良診斷特徵之鑑別診斷 (Differential Diagnosis of Some Diagnostic Features of Ectodermal Dysplasias)