紫質症 (The Porphyrias) 精華筆記
總論
- 紫質症 (porphyrias) 是血基質 (heme) 生合成途徑中 8 種酵素異常所致的代謝病;除遲發性皮膚紫質症 (porphyria cutanea tarda, PCT) 為後天性酵素缺乏外,其餘皆源自酵素突變。
- 依血基質前驅物最初累積部位分為肝性 (hepatic) 或紅血球生成性 (erythropoietic);依臨床表現分為皮膚型 (cutaneous) 與急性型 (acute)。
- 皮膚型源自光敏性紫質過量累積:多數(以 PCT 為代表)造成曝光區慢性水疱與疤痕;原紫質症 (protoporphyrias) 則為急性、非水疱性反應。
- 急性紫質症以神經學症狀為特徵,伴前驅物 δ-胺基乙醯丙酸 (δ-aminolevulinic acid, ALA) 與膽色素原 (porphobilinogen, PBG) 升高。
- 症狀、徵象與組織學皆無特異性;診斷依賴紫質與前驅物的模式。
- 血基質合成約 85% 在骨髓(供血紅素),其餘多在肝臟(供細胞色素 P450, CYPs)。肝臟以 ALAS1(管家型)受最終產物血基質回饋抑制為主要調控;骨髓則用 ALAS2(紅血球特異型)。

圖 124-1:血基質生合成途徑、8 個酵素的定位,及肝臟 ALAS1 受血基質回饋控制。
遲發性皮膚紫質症 (Porphyria Cutanea Tarda, PCT)
- 最常見的紫質症;因肝臟尿紫質原脫羧酶 (uroporphyrinogen decarboxylase, UROD) 活性降至正常約 20% 以下所致,高羧基化紫質累積。
- 是與鐵相關的疾病,僅在肝鐵正常或增加時發生;鐵缺乏具保護作用。
- 唯一可在酵素無突變下發生的紫質症;約 20% 病人帶異型合子 UROD 突變(家族性/第 2 型),但單獨此突變不致病。
臨床特徵
- 多於第四、五個十年發病,男性常見;手背等曝光區出現水疱、大疱,因皮膚脆弱於輕微創傷後形成,破裂留糜爛、結痂緩慢癒合。
- 可見粟粒疹 (milia)、臉部多毛 (hypertrichosis) 與色素過度沉著;嚴重增厚可類似假性硬皮病 (pseudoscleroderma)。
- 無急性紫質症的神經學症狀。新鮮肝組織於長波紫外光下呈紅色螢光。肝細胞癌 (HCC) 風險增加。
易感因素(常多重併存)
- 飲酒、抽菸、慢性 C 型肝炎、血色素沉著症基因 (HFE) 突變(C282Y 為白人主要突變)、雌激素、HIV、化學物質(如六氯苯 hexachlorobenzene、戴奧辛 TCDD)。
- 機轉多為促進肝細胞鐵累積、氧化壓力與 UROD 抑制物生成;HFE 突變降低肝臟鐵調素 (hepcidin) 致鐵吸收增加。
致病機轉
- UROD 受抑制時,尿紫質原 I/III 及中間產物累積、氧化為紫質,經血漿運至皮膚致光敏感。
- 一種 UROD 抑制物為尿紫質甲烯 (uroporphomethene),是尿紫質原部分氧化產物,可能由 CYPs 生成。
診斷
- 第一線:測血漿或尿液總紫質(正常提示假性紫質症 pseudoporphyria)。
- 確診與鑑別:稀釋血漿中性 pH 螢光掃描(PCT 約 620 nm,VP 約 626 nm,為最常被誤診的鑑別點);尿/血漿紫質分餾以尿紫質與七、六、五羧基紫質為主;糞便含異糞紫質 (isocoproporphyrins)。
- 紅血球總紫質正常或中度升高(CEP、HEP、同型合子 HCP/VP 則顯著升高)。尿 ALA 正常或中度增、PBG 總是正常。
治療
- 戒菸、戒酒、停雌激素,補充抗壞血酸。診斷確定後才可治療(對其他紫質症無效)。
- 反覆放血:每 2 週移除 450 mL,以血清鐵蛋白為指引,目標 15–20 ng/mL;多數需 6–8 次達緩解。C282Y 同型合子例外,依血色素沉著症指引維持鐵蛋白 50–100 ng/mL。
- 低劑量羥氯奎寧 (hydroxychloroquine) 100 mg 或氯奎寧 125 mg(半顆),每週兩次,至紫質正常滿一個月;治療前需眼科篩檢視網膜病變。兩法達緩解時間相當。
- 鐵螯合劑(去鐵胺 desferrioxamine)效率較差,僅於前述禁忌時考慮。末期腎病相關 PCT 難治療,放血合併紅血球生成素 (erythropoietin) 支持有效,腎移植可緩解。
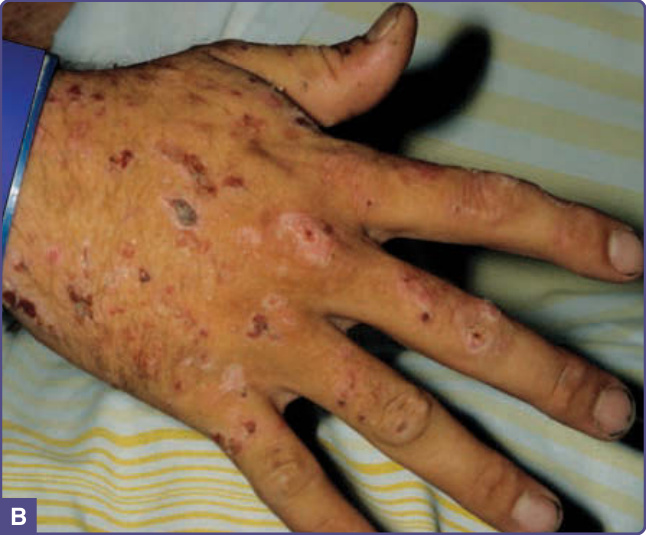
圖 124-2:PCT 特徵性手背水疱、糜爛、結痂與疤痕。
肝紅血球生成性紫質症 (Hepatoerythropoietic Porphyria, HEP)
- 為家族性(第 2 型)PCT 的同型合子(或複合異型合子)形式,UROD 兩對偶基因皆突變,活性嚴重不足。
- 臨床類似 CEP,常於幼兒早期出現水疱病灶、多毛、疤痕、溶血性貧血與紅色尿液,較 PCT 嚴重。
- 生化類似 PCT,但紅血球鋅原紫質顯著升高。
- 治療:避光;放血與羥氯奎寧無效。口服活性碳於部分病例有益。
先天性紅血球生成性紫質症 (Congenital Erythropoietic Porphyria, CEP)
- 又稱 Günther 病;因尿紫質原 III 合成酶 (uroporphyrinogen III synthase, UROS) 兩對偶基因突變、活性嚴重喪失,致異構物 I 型紫質原(尿紫質原 I、糞紫質原 I)於骨髓累積。
- 臨床:曝光區表皮下大疱,常致疤痕、攣縮、手指與臉部特徵喪失(較 PCT 嚴重)。可於子宮內表現為胎兒水腫 (fetal hydrops);出生後不久即光敏感。
- 紅牙症 (erythrodontia,牙齒棕染) 與骨骼紫質沉積;溶血與無效紅血球生成致脾腫大與貧血。肝臟與神經不受影響。
- 診斷:尿/紅血球/血漿/糞便紫質顯著升高,以尿紫質 I 與糞紫質 I 為主;尿布粉至棕色、紫外光下紅螢光。
- 治療:嚴格避光(有害光多為可見光,防曬劑與 β-胡蘿蔔素無效);輸血加鐵螯合、羥基脲 (hydroxyurea)、脾切除。造血幹細胞移植具治癒性,為嚴重病童首選。
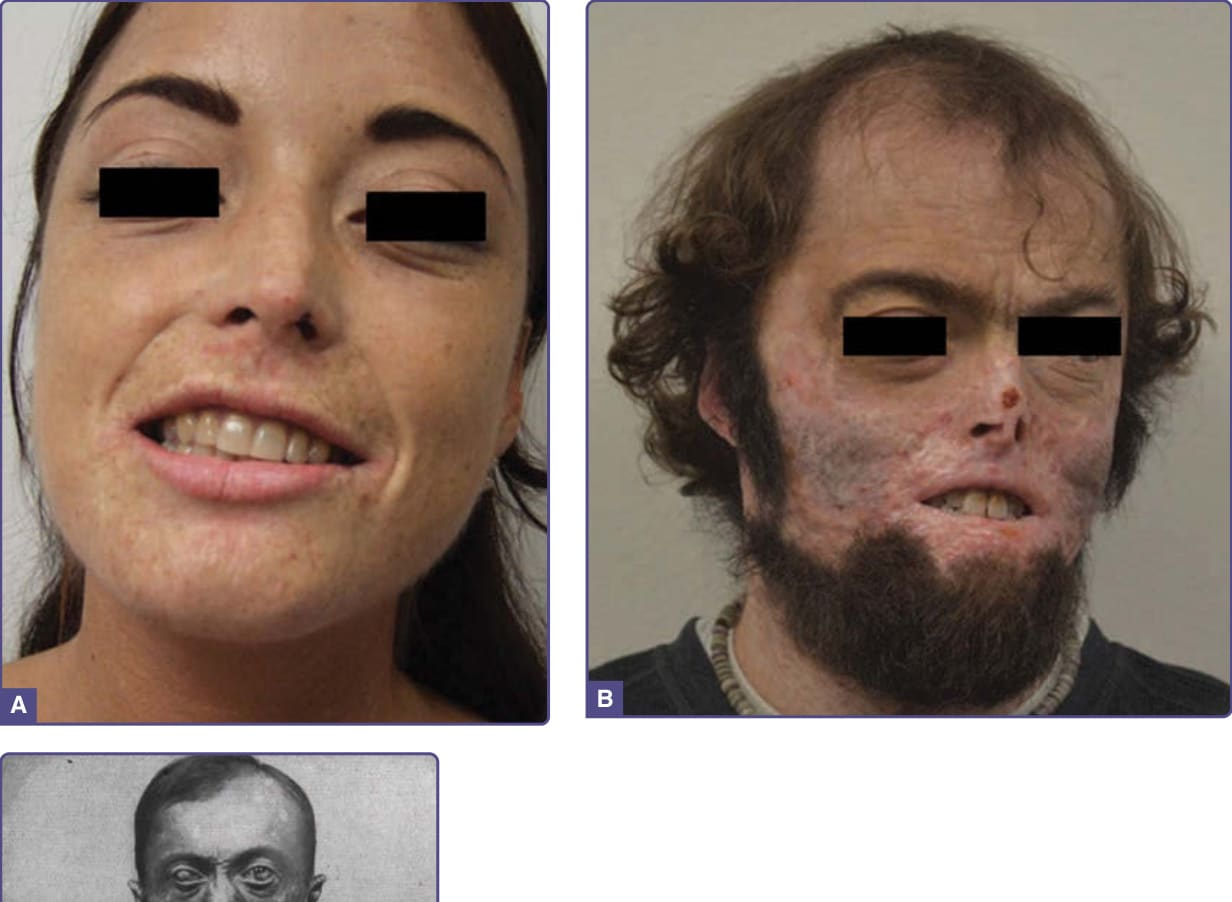
圖 124-6:三例嚴重度不同的先天性紅血球生成性紫質症。
原紫質症 (Protoporphyrias, EPP / XLP)
- 紅血球生成性原紫質症 (erythropoietic protoporphyria, EPP):兒童中最常見的紫質症、成人第三常見;盛行率每 100,000 人 0.5–1.5 例。
- 源自亞鐵螯合酶 (ferrochelatase, FECH) 功能喪失型突變;多數家族為一嚴重突變與一低表現對偶基因 (IVS3-48C/T) 互為反式(出現於 10% 白人,本身無表型)。
- X 染色體連鎖原紫質症 (X-linked protoporphyria, XLP):源自 ALAS2 功能增益型突變,約佔原紫質症 5%;亦可由 CLPX 突變所致。
臨床特徵
- 急性、非水疱性光敏感:曝曬後數分鐘內刺痛、灼燒、刺麻,續以紅斑與水腫(似日光性蕁麻疹 solar urticaria),症狀多於幼兒早期始(北美平均發病年齡 4.4 歲)。
- 慢性皮膚變化少且輕微(革樣角化、輕度疤痕、唇部溝紋、甲剝離 onycholysis)。
- 過量原紫質主要為不含金屬 (metal-free) 而非鋅複合(與鉛中毒、鐵缺乏等以鋅原紫質為主者區別)。
- 多有輕度缺鐵性貧血伴小球性。原紫質症性肝病 (protoporphyric hepatopathy) 發生於不到 5% 病人,因原紫質的膽汁鬱積效應,可致肝硬化或急速惡化需緊急肝移植;含原紫質的膽結石常見。
- XLP 男性紅血球原紫質高於 EPP(3574 vs 1669 µg/dL;P <.001)。
診斷
- 測紅血球總原紫質,升高再測不含金屬與鋅原紫質比例;紅血球原紫質增加且以不含金屬為主為原紫質症獨有發現。
- 血漿螢光峰約 634 nm(須注意血漿樣本易光降解)。應使用能正確分餾的實驗室(陷阱:hematofluorometry 僅測鋅原紫質、過時的「游離原紫質 FEP」用詞會誤導)。
治療
- 光防護與避光為主要介入(防護衣物、帽子、含氧化鋅或二氧化鈦防曬乳;吸收 UV 的防曬劑無益)。
- 口服 β-胡蘿蔔素每日 120–180 mg 或更高(目標血清 β-胡蘿蔔素 600–800 µg/dL);口服半胱胺酸 (cysteine) 500 mg 每日兩次。
- 阿法諾肽 (Afamelanotide)(α-黑色素細胞刺激素類似物)增加皮膚色素、改善陽光耐受度(歐盟、瑞士已核准)。
- 避免酒精與肝毒性藥物;每年監測紫質、肝功能、鐵蛋白與維生素 D;建議每日維生素 D 800 國際單位 (international units) 與鈣 1000 mg。
- 原紫質症性肝病:膽固醇胺 (cholestyramine)、熊去氧膽酸 (ursodeoxycholic acid)、維生素 E、輸血、血漿置換、靜脈 hemin;嚴重者肝移植(新肝 5 年復發率 69%),肝移植後接續骨髓移植可防復發。
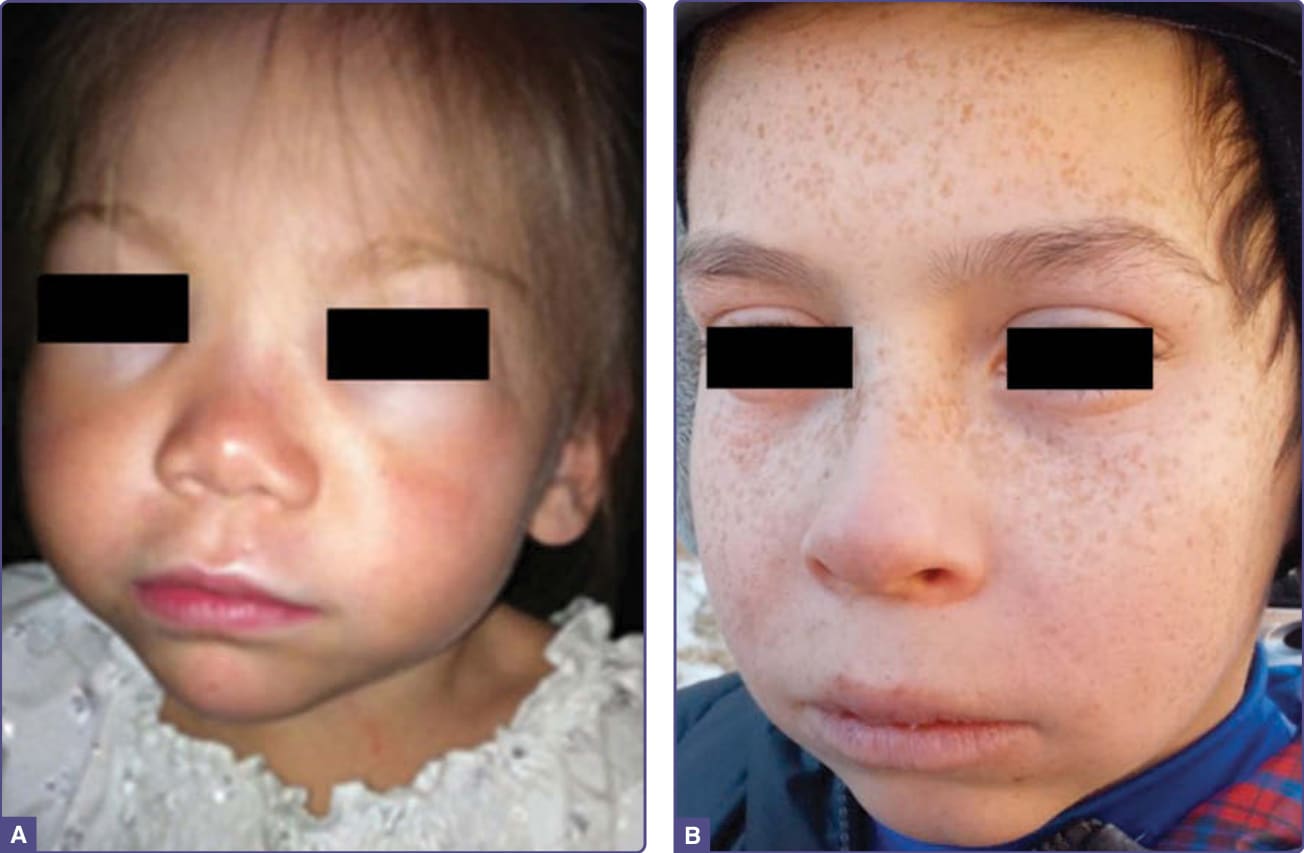
圖 124-7:2 名 EPP 兒童曝曬陽光後臉部急性腫脹與紅斑。
急性肝性紫質症 (Acute Hepatic Porphyrias, ADP / AIP / HCP / VP)
- 4 種(ALA 脫水酶缺乏紫質症 ADP、急性間歇性紫質症 AIP、遺傳性糞紫質症 HCP、斑斕型紫質症 VP)分別源自途徑第二、三、六、七酵素的功能喪失型突變,以間歇性急性神經學症狀為特徵。
- 其中 3 種可發生似 PCT 的慢性水疱病灶:VP 常見、HCP 不常見、AIP 僅於進行性腎病時。ADP 極罕見(僅 6 例、體染色體隱性,本章不深論)。
- AIP 為全球最常見的急性肝性紫質症;VP 在南非荷蘭裔白人因奠基者效應 (founder effect) 特別常見(盛行率每 100,000 人 300 例,多共有 PPOX R59W 突變)。
臨床與機轉
- 神經內臟表現:腹痛、嘔吐、四肢疼痛、癲癇發作、運動神經病變致肌肉無力;慢性高血壓、腎病與 HCC 風險增加。
- 症狀與前驅物 ALA、PBG 累積相關,源自 ALA 神經毒性或神經/血管組織血基質缺乏。
誘發因素
- 多數有害藥物會誘導肝臟 CYPs 與 ALAS1。內分泌:黃體素 (progesterone)、合成黃體製劑與睪固酮代謝物為強誘導物(月經黃體期 luteal phase 黃體素增加為週期性發作主因);雌激素對急性紫質症害處小。
- 抽菸、酒精;減重或疾病/手術期間減少熱量與碳水化合物(上調 PGC-1α 誘導 ALAS1,可由碳水化合物逆轉)。
- 不安全藥物:巴比妥類、卡馬西平、達那唑、雌激素、灰黃黴素、苯妥英、利福平、磺胺類、丙戊酸等。安全藥物:對乙醯胺酚、阿斯匹靈、加巴噴丁、糖皮質素、胰島素、左乙拉西坦、麻醉性鎮痛劑、青黴素類等。
診斷
- 急性發作時尿 PBG 升高(AIP 最顯著,HCP/VP 較低且短暫)。
- HCP/VP 尿紫質與糞便紫質升高(HCP 以糞紫質 III 為主,VP 以糞紫質 III 與原紫質兩者為主);糞便糞紫質 III/I 比值對診斷 HCP 敏感。
- VP 特異性:血漿螢光峰約 626 nm(代表與血漿蛋白共價結合的原紫質),優於糞便檢查偵測無症狀 VP。紅血球 PBGD 在多數 AIP 降低、HCP/VP 正常。
治療
- VP/HCP 皮膚表現對放血、羥氯奎寧無反應;以避光與防護衣物為主。
- 急性發作:多需住院;去除誘發因素;麻醉性鎮痛劑、氯丙嗪 (chlorpromazine) 或恩丹西酮 (ondansetron) 止痛止吐;短效苯二氮平類用於焦慮失眠;β-腎上腺素阻斷劑控制心搏過速與高血壓;癲癇先矯正低血鈉症,可氯氮平 (clonazepam) 害處較小。
- Hemin 為最有效治療(美國 Panhematin、歐洲與南非 heme arginate):用於中重度發作及對碳水化合物無反應的輕度發作;每日 3–4 mg/kg 連續 4 天,必要時延長;建議以 25% 人類白蛋白重新調配以防靜脈炎與凝血障礙;可安全用於懷孕。每週一或兩次給予可預防頻繁發作。
- 碳水化合物負荷:僅用於輕度發作或 hemin 取得前過渡;靜脈葡萄糖以 10% 溶液給 300–500 g,謹慎監測避免低血鈉症。
- 50 歲後建議每年篩檢 HCC。
關鍵表
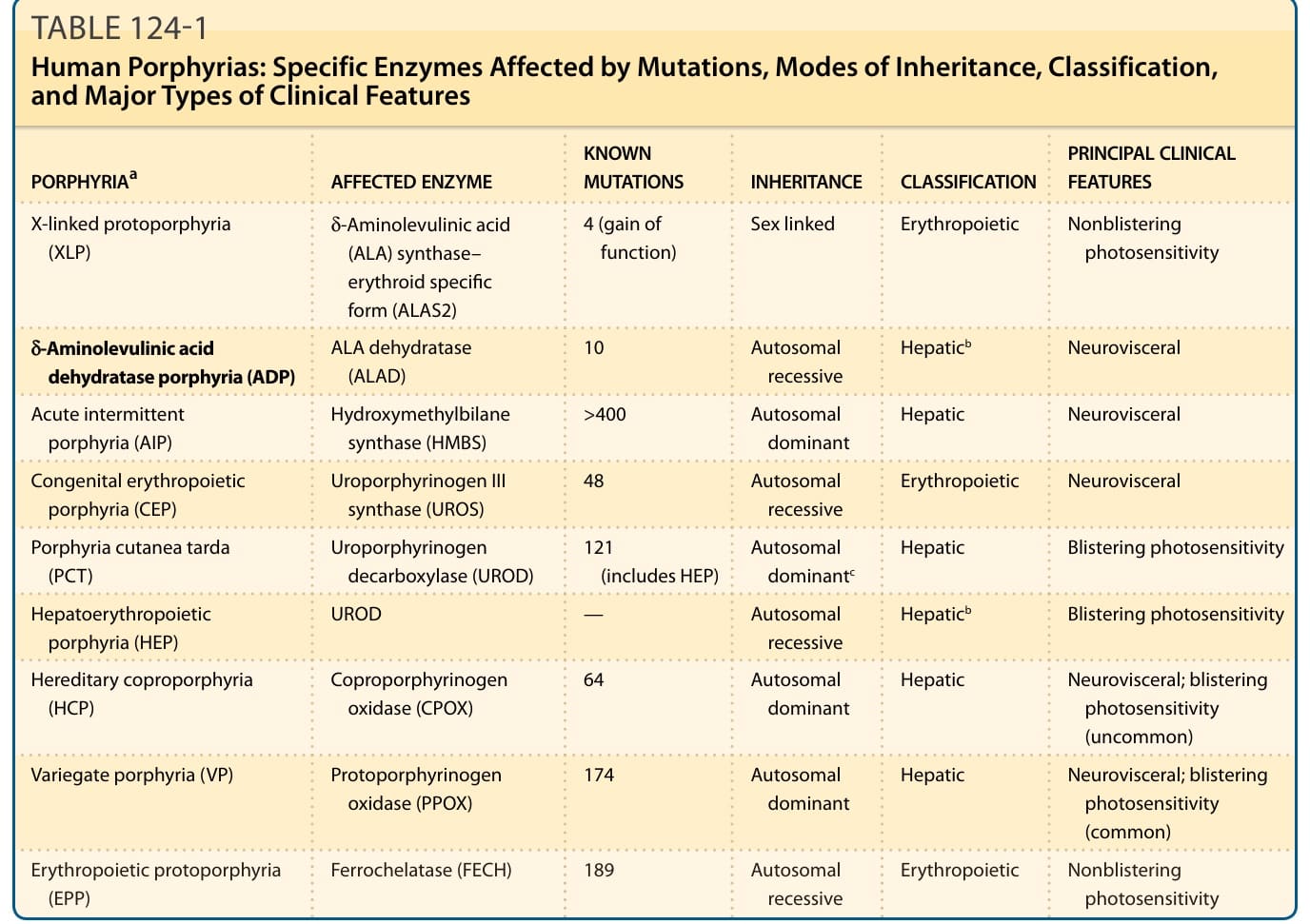
表 124-1:人類紫質症的酵素分類、遺傳方式與主要臨床特徵。
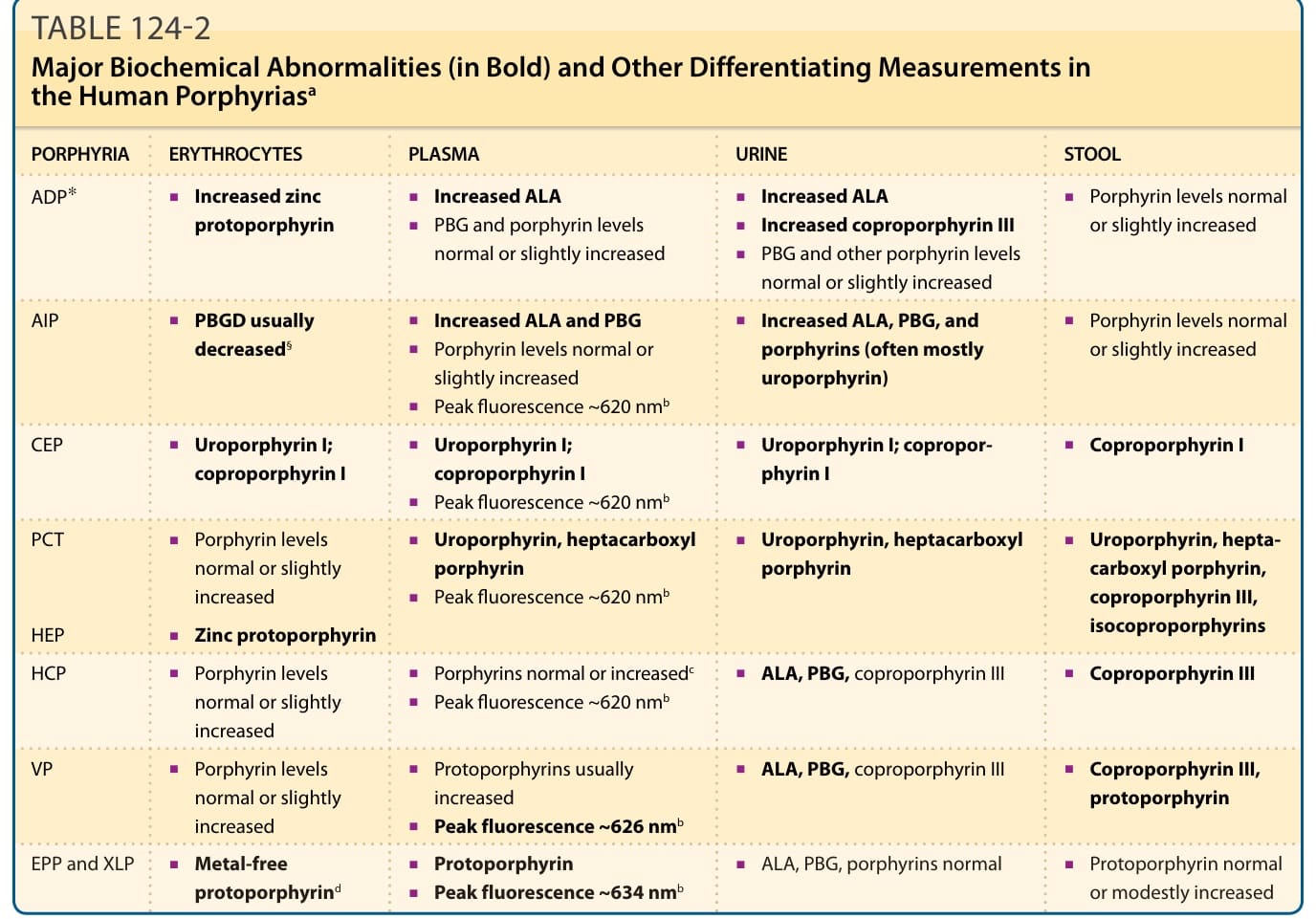
表 124-2:人類紫質症的主要生化異常與鑑別性檢測值。