The Porphyrias
21
INTRODUCTION
Porphyrias are metabolic diseases caused by abnormalities of the 8 enzymes in the heme biosynthetic pathway. All but one arise from mutation of a pathway enzyme; the exception is porphyria cutanea tarda (PCT), which develops as an acquired deficiency of the fifth enzyme in the pathway with or without a mutation. Porphyrias are classified as hepatic or erythropoietic based on whether heme precursors first accumulate in liver or bone marrow, the tissues that most actively synthesize heme. Porphyrias are categorized clinically based on their clinical features as either cutaneous or acute (Table 124-1). Cutaneous porphyrias are due to overproduction and accumulation of photosensitizing porphyrins. Most, as exemplified by PCT, cause chronic blistering and scarring on sun-exposed areas of skin, whereas protoporphyrias produce an acute, severe, and mostly nonblistering reaction to light, often leaving few if any chronic skin changes. Acute porphyrias are characterized by neurologic symptoms and elevated levels of the porphyrin precursors, δ-aminolevulinic acid (ALA) and porphobilinogen (PBG). Porphyrins also accumulate in acute porphyrias, and sometimes achieve levels in plasma sufficient to cause cutaneous blistering, as exemplified especially by variegate porphyria (VP). In some porphyrias, damage to other organs, such as liver and kidney, may also occur. Symptoms, signs, and histologic findings caused by porphyrias are nonspecific, whereas patterns of porphyrins and porphyrin precursors enable specific diagnoses and treatments (Table 124-2).
HISTORICAL PERSPECTIVE
HISTORICAL PERSPECTIVE
The first known description of porphyria was in 1874 by Schultz. He described a 33-year-old man with photosensitivity since 3 months of age with associated anemia, splenomegaly, and red urine.1,2
T. McCall Anderson in 1898 described 2 brothers with blistering of sun-exposed skin, extensive scarring of facial features, and red urine.3 These patients are thought to have had congenital erythropoietic porphyria (CEP), a very rare and severe cutaneous porphyria. Acute porphyria was first described by Stokvis in 1888 in an elderly woman with symptoms after taking sulphonal, a sedative related to barbiturates; she developed dark red urine and later died.4
In 1923, Archibald Garrod proposed the term inborn errors of metabolism to describe a number of inherited metabolic disorders, including the porphyrias.5
Porphyrias were first classified as hepatic or erythropoietic in 1954.6 Treatment of porphyria cutanea tarda by phlebotomy was introduced by Ippen in 1961.7 In 1970, an inherited enzyme deficiency was first described in acute intermittent porphyria,8 and hemin therapy was first introduced for this condition in 1971.9 In the decades that followed, the enzymes of the pathway and their genes were characterized and multiple mutations found in each of the porphyrias. Also, regulation of heme synthesis especially in liver and bone marrow has become better understood. Based on these advances, additional treatments have been introduced and are being developed.
HEME SYNTHESIS AND FUNCTIONS
HEME SYNTHESIS AND
FUNCTIONS
Heme, or iron protoporphyrin IX, is synthesized in eukaryotic cells in 8 steps, each catalyzed by a different enzyme (Fig. 124-1). The first and last 3 of these enzymes are in the mitochondria and the other 4 are cytosolic. The first enzyme, δ-aminolevulinic acid synthase (ALAS), combines glycine and succinyl-coenzyme A to produce the amino acid δ-aminolevulinic acid (ALA), also known as 5-aminolevulinic acid. Two molecules of ALA are then combined to form a pyrrole, porphobilinogen (PBG). Four molecules of PBG are joined to form a linear tetrapyrrole, hydroxymethylbilane (HMB). HMB is a substrate for the fourth enzyme, which inverts one of the pyrrole rings of HMB and cyclizes the molecule to create uroporphyrinogen III, the first porphyrin in the pathway. This asymmetric molecule undergoes a series of decarboxylations and an oxidation to form protoporphyrin IX. Iron is then inserted to form heme. With the exception of protoporphyrin IX, the porphyrin intermediates in this pathway are in their reduced forms (ie, porphyrinogens).10
The end product heme consists of an iron atom in the ferrous (reduced) state (Fe2+) bound to the 4 pyrrolic nitrogens of the porphyrin macrocycle (Fig. 124-1), leaving 2 unoccupied electron pairs. Heme is the prosthetic group for many essential hemoproteins. In hemoglobin, for example, one unoccupied electron pair is coordinated with a histidine residue of the globin chain while the other is available to bind molecular oxygen. Hemin is the chemical term for the oxidized form of heme, ferric protoporphyrin IX, which has only one residual positive charge, and can be isolated from blood and other tissues in the chloride form. Hemin also refers generically to biologics, namely,
21
PORPHYRIAa AFFECTED ENZYME KNOWN MUTATIONS INHERITANCE CLASSIFICATION PRINCIPAL CLINICAL FEATURES
X-linked protoporphyria (XLP) δ-Aminolevulinic acid (ALA) synthase– erythroid specific form (ALAS2)
4 (gain of function) Sex linked Erythropoietic Nonblistering photosensitivity
c-Aminolevulinic acid dehydratase porphyria (ADP) ALA dehydratase (ALAD) 10 Autosomal recessive Hepaticb Neurovisceral
Acute intermittent porphyria (AIP) Hydroxymethylbilane synthase (HMBS) >400 Autosomal dominant Hepatic Neurovisceral
Congenital erythropoietic porphyria (CEP) Uroporphyrinogen III synthase (UROS) 48 Autosomal recessive Erythropoietic Neurovisceral
Porphyria cutanea tarda (PCT) Uroporphyrinogen decarboxylase (UROD) 121 (includes HEP) Autosomal dominantc Hepatic Blistering photosensitivity
Hepatoerythropoietic porphyria (HEP) UROD — Autosomal recessive Hepaticb Blistering photosensitivity
Hereditary coproporphyria (HCP) Coproporphyrinogen oxidase (CPOX) 64 Autosomal dominant Hepatic Neurovisceral; blistering photosensitivity (uncommon)
Variegate porphyria (VP) Protoporphyrinogen oxidase (PPOX) 174 Autosomal dominant Hepatic Neurovisceral; blistering photosensitivity (common)
Erythropoietic protoporphyria
Ferrochelatase (FECH) 189 Autosomal
Erythropoietic Nonblistering
Erythropoietic protoporphyria (EPP) Ferrochelatase (FECH) 189 Autosomal recessive Erythropoietic Nonblistering photosensitivity
(EPP)
recessive
photosensitivity
aPorphyrias are listed in the order of the affected enzyme in the heme biosynthetic pathway.
bThese porphyrias also have erythropoietic features, including increases in erythrocyte zinc protoporphyrin.
cUROD inhibition in PCT is mostly acquired, but an inherited deficiency of the enzyme predisposes in familial (type 2) disease.
lyophilized hematin (heme hydroxide) and heme arginate, which are available for treatment of acute porphyrias. Approximately 85% of heme synthesis occurs in the bone marrow to support hemoglobin formation, with the remainder mostly in the liver, primarily for cytochrome P450 enzymes (CYPs) found in the endoplasmic reticulum.11 All other tissues synthesize heme in smaller amounts. Examples of the numerous other vital hemoproteins include myoglobin, mitochondrial respiratory cytochromes, nitric oxide synthase, and catalase. Heme synthesis in the liver is regulated primarily by the activity of δ-aminolevulinic acid synthase 1 (ALAS1, the housekeeping form of ALAS, the first enzyme in the pathway), with repression of synthesis of this enzyme and its import into mitochondria by heme, the end product of the pathway. In the marrow, heme and globin synthesis are closely coordinated during erythropoietin signaling. Expression of ALAS2, the erythroid-specific form of ALAS, and a number of other pathway enzymes, are stimulated by heme or iron and by erythroid-specific cis-acting elements including GATA-1 and NF-E2, culminating with phosphorylation of ferrochelatase (FECH), the final enzyme in heme synthesis.12
PORPHYRIA CUTANEA TARDA
AT-A-GLANCE
■ Characterized by skin friability and blistering lesions on sun-exposed areas of skin
■ Caused by inhibition of hepatic uroporphyrinogen decarboxylase (UROD) activity
■ This leads to excess amounts of highly carboxylated porphyrins in liver, plasma, urine, and feces in diagnostic patterns
■ Genetic factors that predispose may include mutations of UROD (heterozygous, only in ∼20% of cases) and hemochromatosis (HFE)
■ Acquired susceptibility factors (alcohol, smoking, secondary iron overload, chronic hepatitis C, HIV, and estrogen) are often multiple
■ Responds readily to repeated phlebotomy while following serum ferritin, or low-dose hydroxychloroquine
2235
21
Protoporphyrinogen IX
Protoporphyrin IX
Protoporphyrinogen oxidase
COOH HOOC
HOOC COOH
N
N N
N
N N
NH H H H
NH H
Coproporphyrinogen oxidase
Fe2+
Ferrochelatase
MITOCHONDRION
HOOC COOH
COOH
N
N N
Heme
Fe Fe
N
repression
Feedback
CYTOSOL
Glycine + succinyl CoA
δ-Aminolevulinic acid synthase
N H N H N H
Porphobilinogen deaminase
δ-Aminolevulinic acid dehydratase
COOH HOOC
COOH
N H
H2N
H2N
δ-Aminolevulinic acid
N H
Porphobilinogen
HO
Hydroxymethylbilane
COOH
N
N N
Uroporphyrinogen decarboxylase
COOH
COOH
COOH COOH
N N
NH H H H
HOOC
HOOC
HOOC
Uroporphyrinogen I
Uroporphyrinogen decarboxylase
Coproporphyrinogen I
21
PORPHYRIA ERYTHROCYTES PLASMA URINE STOOL
ADP∗
■Increased zinc protoporphyrin
■Increased ALA
■PBG and porphyrin levels normal or slightly increased
AIP
■PBGD usually decreased§
■Increased ALA and PBG
■Porphyrin levels normal or slightly increased
■Peak fluorescence ~620 nmb
CEP
■Uroporphyrin I; coproporphyrin I
■Uroporphyrin I; coproporphyrin I
■Peak fluorescence ~620 nmb
PCT
■Porphyrin levels normal or slightly increased
■Uroporphyrin, heptacarboxyl porphyrin
■Increased ALA
■Porphyrin levels normal or slightly increased
■Increased coproporphyrin III
■PBG and other porphyrin levels normal or slightly increased
■Increased ALA, PBG, and porphyrins (often mostly uroporphyrin)
■Porphyrin levels normal or slightly increased
■Uroporphyrin I; coproporphyrin I
■Coproporphyrin I
■Uroporphyrin, heptacarboxyl porphyrin
■Uroporphyrin, heptacarboxyl porphyrin, coproporphyrin III, isocoproporphyrins HEP
■Zinc protoporphyrin
■Peak fluorescence ~620 nmb
HCP
■Porphyrin levels normal or slightly increased
■Porphyrins normal or increasedc
■Peak fluorescence ~620 nmb
VP
■Porphyrin levels normal or slightly increased
■Protoporphyrins usually increased
■Peak fluorescence ~626 nmb
EPP and XLP
■Metal-free protoporphyrind
■Protoporphyrin
EPP and XLP ■Metal-free
■Protoporphyrin
■Peak fluorescence ~634 nmb
■Peak fluorescence ~634 nmb
protoporphyrind
aAbbreviations as in Table 124-1.
bFluorescence emission peak of diluted plasma at neutral Ph.
cIncreased in the presence of skin lesions.
■ALA, PBG, coproporphyrin III
■Coproporphyrin III
■ALA, PBG, coproporphyrin III
■Coproporphyrin III, protoporphyrin
■ALA, PBG, porphyrins normal
■Protoporphyrin normal or modestly increased
■ALA, PBG, porphyrins normal ■Protoporphyrin normal
or modestly increased
dMetal-free protoporphyrin approximately 85%-100% of the total in EPP, and 50%-85% in XLP.
PCT is the most common porphyria, and is characterized by the development of skin friability and chronic, blistering lesions on the dorsal aspects of the hands and other sun-exposed areas of skin usually in mid- or late life.13 The underlying cause is deficient uroporphyrinogen deaminase (UROD) activity in hepatocytes, where uroporphyrinogen and other highly carboxylated porphyrinogens accumulate and are oxidized to the corresponding porphyrins. In active cases, hepatic UROD activity is reduced to less than ∼20% of normal. PCT is an iron-related disorder and develops only in the presence of normal or increased amounts of hepatic iron. Multiple susceptibility factors contribute to iron accumulation, oxidative stress, and generation of a UROD inhibitor in hepatocytes, and are important to identify in individual patients. This is the only porphyria that can develop in absence of the mutation of the affected enzyme.14 Heterozygous UROD mutations are found in ∼20% of patients, but these do not cause PCT in the absence of other susceptibility factors. PCT is the most readily treated porphyria, responding well to either phlebotomy or low-dose hydroxychloroquine. But the disease must first be differentiated from other less common porphyrias that cause identical skin lesions but are unresponsive to these treatments.
CLINICAL FEATURES
CLINICAL FEATURES
PCT usually develops in the fourth or fifth decade of life, most commonly in males. Fluid-filled blisters and bullae are found especially on the dorsal hands (Fig. 124-2), the most sun-exposed areas of the body, and often arise after minor or inapparent trauma as a result of increased skin friability. Blisters may also occur on the forearms, face, ears, neck, legs, and feet. These rupture easily, leaving erosions that may become dry and crusted and heal slowly (Fig. 124-2). Eroded areas of skin are prone to bacterial infection. Residual scarring and hyper- and hypopigmentation are prominent with prolonged disease. Milia may precede or follow vesicle formation. Facial hypertrichosis and hyperpigmentation (Fig. 124-3) are common, may even occur in the absence of blistering, and are cosmetically problematic, especially in women.16 Severe thickening of affected areas of skin, sometimes with associated calcification, can resemble systemic sclerosis and is termed pseudoscleroderma. Neurologic symptoms characteristic of the acute porphyrias are not seen in PCT. Rare childhood cases have often been associated with UROD mutations and cancer chemotherapy15 (Fig. 124-3). The disease may develop during
2237
21
A
B
pregnancy, possibly related to effects of increased estrogen. Reported associations with cutaneous and systemic lupus erythematosus are unexplained. PCT associated with end-stage renal disease is usually more severe, because urinary excretion of porphyrins is impaired, and porphyrins are poorly dialyzed. The resulting plasma porphyrin levels can equal those seen in congenital erythropoietic porphyria (CEP) and be associated with severe bacterial infections and mutilation.17
2238
Mild abnormalities in liver function tests are found in almost all cases. Fresh hepatic tissue exhibits strong red fluorescence on exposure to long-wave ultraviolet light (Fig. 124-4), reflecting massive accumulation of porphyrins. Liver histopathology is nonspecific and usually includes increased iron, increased fat, hepatocyte necrosis and inflammation, which in many cases reflects the effects of alcohol or hepatitis C infection. Cirrhosis is unusual at the onset of PCT. The risk of HCC is increased, especially with more prolonged disease, perhaps due partly to concomitant susceptibility factors that themselves can cause chronic liver damage and fibrosis.18-20
SUSCEPTIBILITY FACTORS
SUSCEPTIBILITY FACTORS
PCT is a multifactorial disorder, with many common susceptibility factors contributing to the disease, none of which is present in every patient. These include genetic factors, viral infections, and chemical exposures. Patients almost always possess multiple such factors, which have other health implications.21 For example, in a large series of 143 patients with PCT in the United States, the most common susceptibility factors were ethanol use (87%), smoking (81%), chronic hepatitis C (69%), and HFE (hemochromatosis) mutations (53%).22 These factors are common in the general population but do not by themselves cause PCT to develop. Additional susceptibility factors are likely to be identified in the future.
UROD MUTATIONS
Most PCT patients do not have a UROD mutation and are said to have sporadic (type 1) disease. Approximately 20% of patients have a heterozygous predisposing UROD mutation and are labeled as familial (type 2) PCT. Such mutations are inherited as autosomal
A
Analysis of liver porphyrins
URO-I
Total porphyrins: 176 nmol/g tissue (ref<1)
URO-III
HEPTA-III
PROTO (?)
B
dominant traits, but with low penetrance, so most type 2 patients present sporadically, having no known relatives with PCT. Having such a mutation is a susceptibility factor that does not cause PCT in the absence of other acquired or inherited susceptibility factors. HEP is the homozygous form of type 2 PCT, and resembles CEP clinically. At least 100 different mutations of the UROD gene, mostly missense mutations occurring in 1
21
or a few families, have been identified in type 2 PCT and HEP (Table 124-1). Type 3 refers to rare families with more than 1 affected individuals but no UROD mutation. All 3 PCT types are clinically identical, although disease onset is sometimes earlier in type 2.23
IRON AND HEMOCHROMATOSIS GENE (HFE) MUTATIONS
Iron stores are always normal or increased in PCT, whereas iron deficiency is protective. Iron provides an oxidative environment in hepatocytes and facilitates generation of a UROD inhibitor, but does not itself directly inhibit UROD. The C282Y mutation of the HFE gene, the major mutation causing hemochromatosis in whites, is more prevalent in both sporadic and familial PCT than in unaffected individuals. Up to 10% to 20% of patients may be C282Y homozygotes, and these may experience earlier onset of disease.23,24 In southern Europe, where the C282Y is less prevalent, the H63D mutation is more commonly associated with PCT.25 HFE mutations impair sensing of serum iron, thereby reducing hepatic hepcidin production. Because circulating levels of hepcidin are inappropriately low, downregulation of enterocyte ferroportin by hepcidin is impaired, leading to inappropriately high intestinal iron absorption. Hepatic hepcidin expression is also reduced in PCT patients without HFE mutations, because some other susceptibility factors reduce expression of this hormone, as noted below.26
ALCOHOL
PCT has long been associated with alcohol abuse. Alcohol and its metabolites may predispose to onset of PCT by inducing hepatic ALAS1 and CYPs, generating reactive oxygen species, and causing mitochondrial injury, lipid deposition, depletion of reduced glutathione and other antioxidant defenses, increasing production of endotoxin and activating Küpffer cells. Alcohol intake can also reduce hepcidin expression.26
SMOKING AND CYTOCHROME P450 ENZYMES
Smoking is frequently associated with alcohol use but is regarded as an independent risk factor in PCT.21 Smoking may increase oxidative stress in hepatocytes and induces hepatic CYPs, including CYP1A2 which is important for development of uroporphyria in rodent models of PCT.27,28 Hepatic CYPs are often increased in human PCT,29 but it is unclear which CYPs contribute to the disease. However, a more inducible CYP1A2 variant was found to be more common in PCT than in normal subjects.30,31
ESTROGENS
Estrogen use is common in women with PCT.21,32,33 The disease has also occurred in some men treated with
2239
21
estrogen for prostate cancer.32 Female rats or males receiving estrogens are more susceptible to development of chemically-induced uroporphyria than untreated males.34 The mechanism for this predisposition is not certain, but may be secondary to generation of reactive oxygen species.14,35
HEPATITIS C
Hepatitis C promotes hepatocyte steatosis, iron accumulation, mitochondrial dysfunction, oxidative stress and dysregulation of hepcidin expression.36,37 The prevalence of chronic hepatitis C in PCT ranges from 21% to 92% in various countries, and always exceeds the prevalence of this viral infection in the general population. Only an estimated 0.05% of individuals with chronic hepatitis C develop PCT.38 Large differences in the prevalence of this viral infection in PCT in different countries reflects the considerable geographic variation in prevalence of this infection in atrisk populations.
HIV
PCT is less commonly associated with HIV infection, which may be present with or without HCV coinfection.39 PCT can be the initial manifestation of HIV infection. The mechanism for the association with HIV infection and possible relationships to specific antiretroviral therapies are not established.
ANTIOXIDANTS
Reduced plasma levels of ascorbate and carotenoids have been noted in some patients with PCT.40-42
Rodents with ascorbate deficiency are more susceptible to development of uroporphyria.43
CHEMICAL EXPOSURE AND DRUGS
A large outbreak of PCT occurred in eastern Turkey in the 1950s during a period of food shortage when the population consumed seed wheat treated with the fungicide hexachlorobenzene.44 Smaller outbreaks and individual cases of the disease have occurred after exposures to other chemicals such as 2,3,7,8- tetrachlorodibenzo-p-dioxin (TCDD, dioxin).45 These and related chemicals cause biochemical features of PCT in laboratory animals and cultured hepatocytes.10,14
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
PCT develops when hepatic UROD activity is reduced to ∼20% of normal.14 With enzyme inhibition, the amount of UROD protein remains at its genetically determined level in the liver.46,47 As previously discussed, about 20% of patients are heterozygous for
2240
UROD mutations and are more susceptible to developing PCT, because their UROD activity in the liver (as well as other tissues) is half-normal from birth. PCT does not manifest in these individuals unless further reduction of UROD activity occurs in the liver. A UROD inhibitor, characterized as a uroporphomethene, was identified in a mouse model that spontaneously develops biochemical features of PCT. This inhibitor is a product of partial oxidation of uroporphyrinogen, possibly generated by 1 or more CYPs (Fig. 124-5).49
UROD sequentially decarboxylates uroporphyrinogen I and III (each with 8 carboxyl side groups) to coproporphyrinogen I and III (each with 4 carboxyl
Generation of a hepatic UROD inhibitor in PCT
Glycine + succinyl CoA
ALAS1
δ-Aminolevulinic acid
CYP1A2
- Fe2+
Uroporphyrinogen
Uroporphyrin
Specific inhibitor
UROD
Coproporphyrinogen
Fe2+
Heme
A
Uroporphomethene inhibitor
COOH COOH
COOH
N
N
HOOC
H H H
HOOC
N N
COOH
HOOC COOH
B
groups), respectively. Both isomers are substrates for UROD, but uroporphyrinogen III is preferred. (The next enzyme in the pathway is specific for coproporphyrinogen III as a substrate, so uroporphyrinogen I and coproporphyrin I are not heme precursors.) When hepatic UROD is inhibited, uroporphyrinogen I and III and the intermediates in the reaction (ie, I and III isomers of hepta-, hexa-, and pentacarboxyl porphyrinogen) accumulate as the corresponding oxidized porphyrins, and are eventually transported in plasma to the skin, causing photosensitivity, and to the kidneys for excretion. Biliary and fecal porphyrins are also increased. Porphyrins in their oxidized state are reddish, fluorescent, and photosensitizing, whereas porphyrinogens do not have these properties. The polycyclic aromatic structure of oxidized porphyrins contains delocalized electrons, which increase in energy level on exposure to violet light (at a wavelength of ∼410 nm). This energy may be released at a higher wavelength as red fluorescent light or transferred to molecular oxygen, forming reactive singlet state oxygen and other reactive oxygen species. These may damage cutaneous cellular constituents or cause mast cell degranulation and complement activation.50,51 Damage to the subepidermal layers of skin makes the skin friable and prone to blistering.50,51
DIAGNOSIS
DIAGNOSIS
PCT develops most commonly in adult males in association with factors such as excess alcohol use, smoking and chronic hepatitis C, and in females, particularly with estrogen use. The presentation is usually characteristic, but it must be remembered that adults with variegate porphyria (VP), hereditary coproporphyria (HCP) or pseudoporphyria, and children or adults with congenital erythropoietic porphyria (CEP) or hepatoerythropoietic porphyria (HEP) can present with identical skin lesions. Therefore, it is essential to establish a laboratory diagnosis of PCT before initiating treatment. First-line testing (ie, screening) for PCT is measurement of total plasma or urine porphyrins. Normal results may indicate a diagnosis of pseudoporphyria. Because elevated porphyrins, especially in urine, is common in liver disease and other medical conditions, a finding of increased porphyrins and does not itself support a diagnosis of porphyria. Therefore, if firstline testing is positive, the following are suggested to establish a diagnosis and exclude other less common porphyrias that can cause similar skin manifestations and often be misdiagnosed as PCT:
■ Fluorescence scanning of diluted plasma at neutral pH.52 The porphyria most commonly misdiagnosed as PCT is VP,53 and is rapidly and reliably recognized by plasma scanning and finding an emission peak at ∼626 nm, in contrast to ∼620 nm for PCT and other blistering cutaneous porphyrias.
■ Fractionation of urine or plasma porphyrins, which will show a predominance of uroporphyrin, and
21
hepta-, hexa-, and pentacarboxyl, porphyrins in PCT. This pattern of predominance of highly carboxylated porphyrins is not fully diagnostic, as it can occur in other much less common porphyrias that can be misdiagnosed as PCT especially when they present in adults.
■ Total erythrocyte porphyrins, which are normal or modestly elevated in PCT, but markedly elevated in rare cases of CEP, HEP, or homozygous HCP or VP. These usually present in infancy, but can first become manifest in adults, sometimes in associated with a clonal myeloproliferative or myelodysplastic disorder.
■ Fecal porphyrins may be normal or modestly increased in PCT, with a complex pattern that includes isocoproporphyrins. These atypical tetracarboxylporphyrins are formed when pentacarboxylporphyrinogen accumulates in liver as a result of UROD inhibition and undergoes premature decarboxylation by CPOX, the next enzyme in the pathway, forming dehydroisocoproporphyrinogen. The latter is excreted in bile and undergoes oxidation by intestinal bacteria to isocoproporphyrins.54 In contrast to PCT, fecal porphyrins are markedly elevated in CEP, HCP, and VP, with a predominance of coproporphyrin I in CEP, coproporphyrin III in HCP, and both coproporphyrin III and protoporphyrin in VP.
■ Urine ALA is normal or modestly increased in PCT, and PBG is always normal. Levels of these porphyrin precursors may be elevated in the acute hepatic porphyrias, AIP, HCP and VP.
An increase of plasma porphyrins and demonstration of a predominance of highly carboxylated porphyrins is essential for diagnosis of PCT in patients with advanced renal disease, although the reference range is higher in this population.55 Advanced renal disease is commonly associated with altered erythropoiesis and resulting increases in erythrocyte zinc protoporphyrin, which should not be attributed to PCT or other porphyria. Patients with AIP and end-stage renal disease sometimes present with blistering skin lesions that resemble PCT.56
Although not required for diagnosis of PCT, skin biopsy reveals characteristic but nonspecific findings such as subepidermal blistering and deposition of periodic acid–Schiff-positive material around blood vessels and fine fibrillar material in the upper dermis and at the dermoepithelial junction. Deposits of immunoglobulins and complement are also found.57
These histologic changes are also seen in other cutaneous porphyrias as well as pseudoporphyria, a poorly understood condition that presents with lesions that closely resemble PCT, but with plasma porphyrins that are not significantly increased.58
IDENTIFICATION OF SUSCEPTIBILITY FACTORS
IDENTIFICATION OF
SUSCEPTIBILITY FACTORS
All PCT patients should be questioned or examined for the following susceptibility factors, some of which
2241
21
are modifiable: alcohol and estrogen use, smoking, hepatitis C and HIV infection, and HFE and UROD mutations. Use of drugs that exacerbate acute porphyrias are seldom implicated in PCT. Although familial (type 2) cases of PCT can be identified by half-normal erythrocyte UROD activity, UROD mutation analysis is more dependable. This full analysis of susceptibility factors helps explain the disease in individual cases and may affect management. Moreover, some of these factors also have medical implications of their own. Serum ferritin should be measured and may influence choice of treatment.
THERAPY
THERAPY
Treatment with phlebotomy or low-dose hydroxychloroquine is highly effective in both sporadic and familial forms of PCT. These should be initiated only after the diagnosis is certain, because they are not effective in other porphyrias. It may be reasonable to start treatment after plasma porphyrin results, including fluorescence scanning,52 are consistent with PCT and have excluded VP and pseudoporphyria. Patients should be advised to stop smoking and alcohol consumption. Use of estrogen and drugs known to induce hepatic heme synthesis should be discontinued if possible. Estrogen replacement therapy, preferably transdermal, can be considered, if needed, after PCT is in remission.59 Adequate intake of ascorbic acid and other nutrients should be established. Removal of 1 or more susceptibility factors can lead to improvement, but the response is slow or unpredictable.60
Repeated phlebotomy to reduce hepatic iron is the preferred treatment at most institutions. Removal of 450 mL blood at 2-week intervals is guided by the serum ferritin level, with a target of 15 to 20 ng/mL (ie, near the lower limit of normal). Measurement of hematocrit and ferritin at each session allows monitoring to prevent symptomatic anemia and assess progress toward the target ferritin level. Phlebotomies are stopped when the ferritin from the previous visit is 25 to 30 ng/L, and ferritin is measured to confirm that the target level was reached. At that point, porphyrin levels may not have become completely normal and skin lesions are not completely resolved. Additional iron depletion is not beneficial and leads to anemia. Most patients require 6 to 8 phlebotomies for biochemical and clinical remission, but additional sessions may be necessary, especially if the baseline serum ferritin level is markedly elevated. Decreases in plasma (or serum) porphyrin levels tend to lag behind serum ferritin, but will normalize within weeks after phlebotomies are completed.61,62 Friability of skin may persist for some time after porphyrin levels are normal, until healing and repair of the subepidermal layer of the skin is complete. Subsequent phlebotomies are generally not needed, with the exception of patients homozygous or compound heterozygous for the C282Y HFE mutation, as current guidelines for hemochromatosis recommend maintaining a ferritin level between 50 and
2242
100 ng/mL.63 Relapse of PCT may occur, often related to resumption of alcohol use, but usually responds to retreatment. Following porphyrin levels after remission can detect recurrences early so that retreatment can be reinstituted promptly. A low-dose regimen of hydroxychloroquine (or chloroquine) is an effective alternative to phlebotomies, and is the preferred approach at some institutions.14,64-70 These 4-aminoquinoline antimalarials do not appear to deplete hepatic iron, but mobilize porphyrins that have accumulated in lysosomes and other intracellular organelles in hepatocytes. Full therapeutic doses of these drugs rapidly mobilize porphyrins and induce an acute hepatitis, characterized by fever, malaise, nausea, and marked increases in urinary and plasma porphyrins and photosensitivity, but followed by remission.71 Unmasking of previously unrecognized PCT with use of chloroquine for malaria prophylaxis has been described.72 A low-dose regimen (hydroxychloroquine 100 mg or chloroquine 125 mg [one half of a standard tablet] twice weekly) is recommended to achieve remission and avoid the side effects of full doses of these drugs.64,67,68 Treatment is continued until plasma or urine porphyrins are normalized for at least a month. Patients who respond poorly may require alternate therapy with phlebotomy.71 These medications are associated with a small risk of retinopathy,73 which may be lower with hydroxychloroquine. Therefore, patients should be screened by an ophthalmologist before treatment.74 Comparison of treatment with phlebotomy or hydroxychloroquine (100 mg twice weekly), showed that time to remission (normal plasma porphyrin levels) was comparable with these treatments.70 Treatment with an iron chelator such as desferrioxamine is much less efficient for removal of iron than phlebotomy, but can be considered if both phlebotomy and low-dose hydroxychloroquine are contraindicated.75
Treatment of hepatitis C with interferon-based regimens is lengthy and often not successful. Therefore we have first treated PCT, which is usually more symptomatic, and after achieving remission, then treated coexisting hepatitis C. Interferon-based treatment regimens commonly cause anemia, which usually precludes phlebotomy for PCT, and are sometimes associated with relapse of previously treated PCT.76
Experience with low-dose hydroxychloroquine during concurrent treatment of hepatitis C is limited. The new direct-acting antivirals can treat this infection rapidly and dependably, and studies are under way to collect evidence whether they should be used instead of phlebotomy or low-dose hydroxychloroquine as initial treatment of PCT associated with hepatitis C.76
PCT associated with end-stage renal disease is often difficult to treat. Phlebotomy is effective if supported, as needed, by starting or increasing the dose of erythropoietin.17,77,78 High-flux hemodialysis may provide some benefit.79 Renal transplantation can lead to remission presumably because of resumption of endogenous erythropoietin production and reduced hepcidin levels.80
Periodic screening for hepatocellular carcinoma should include liver imaging by abdominal ultrasonography or computed tomography. As in other liver
diseases, this should be guided by evidence of cirrhosis or hepatic fibrosis, which may be assessed by liver biopsy or indirectly by elastography, and the presence of other causes of liver disease.
HEPATOERYTHROPOIETIC PORPHYRIA
AT-A-GLANCE
■ HEP is the homozygous form of familial (type 2) PCT, with mutation of both UROD alleles resulting in severely deficient UROD activity
■ HEP resembles CEP, with blistering lesions on sunexposed areas of skin, usually starting in childhood
■ Biochemical findings resemble PCT, but with substantial elevation of erythrocyte zinc protoporphyrin
■ Patients should avoid sunlight. Treatment with phlebotomy or low-dose hydroxychloroquine is not effective.
This very rare form of porphyria is the homozygous (or compound heterozygous) form of familial (type 2) PCT and is characterized by blistering skin lesions from chronic, severe photosensitivity.
CLINICAL FEATURES
CLINICAL FEATURES
HEP presents with onset of blistering skin lesions, hypertrichosis, scarring, hemolytic anemia, and red urine typically in early childhood. Sclerodermoid skin changes are sometimes prominent. The cutaneous manifestations resemble those found in PCT, but are usually much more severe. Unusually mild cases and onset during adult life have been reported.81
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Patients with HEP have inherited a UROD mutation from each parent. Numerous UROD mutations associated with PCT or UROD have been identified. Erythrocyte UROD activity is 5% to 30% of normal in HEP. As homozygosity for a UROD null allele is lethal, at least one of the mutant UROD alleles must produce some enzyme protein with catalytic activity. Expression studies in eukaryotic cells suggest that some mutations may destabilize the enzyme protein in a tissue-specific manner.82 Excess porphyrins are primarily generated in the liver. Zinc protoporphyrin accumulates in the marrow and is markedly elevated in erythrocytes.
21
DIAGNOSIS
DIAGNOSIS
Biochemical findings in HEP resemble PCT, with predominant accumulation and excretion of uroporphyrin, heptacarboxyl porphyrin, and isocoproporphyrins. In contrast to PCT, erythrocyte zinc protoporphyrin is substantially increased. Erythrocyte UROD activity is markedly diminished, and molecular studies should demonstrate mutations affecting both UROD alleles.
THERAPY
THERAPY
Patients with HEP should be advised to avoid sunlight. Phlebotomy and hydroxychloroquine have shown little or no benefit.83 Patients should avoid susceptibility factors known to be important in PCT. Oral charcoal was helpful in a severe case associated with dyserythropoiesis, likely due to trapping of porphyrins in the intestinal lumen and preventing their enterohepatic circulation.84 Retrovirus-mediated gene transfer studies suggest that correction of the HEP defect through gene replacement therapy may be applicable in the future.85
CONGENITAL ERYTHROPOIETIC PORPHYRIA
AT-A-GLANCE
■ As a result of mutation of both uroporphyrinogen III synthase (UROS) alleles resulting in severe loss of activity of UROS and elevations of uroporphyrin I and coproporphyrin I
■ Characterized by subepidermal scarring and bullous lesions affecting light-exposed areas
■ Diagnosis is established by markedly elevated uroporphyrin I and coproporphyrin I in erythrocytes, urine, plasma, and feces and identification of causative mutations
■ Hematopoietic stem cell transplantation at a young age is potentially curative
■ Strict light avoidance can prevent chronic, disfiguring skin disease
Cases of CEP, also known as Günther disease, were first described in 1874 and 1898.3 CEP is due to substantial deficiency of uroporphyrinogen III synthase (UROS), which results in accumulation of isomer I porphyrinogens, especially uroporphyrinogen I and coproporphyrinogen I, in the marrow during hemoglobin synthesis. Porphyrins are usually markedly elevated in erythrocytes, plasma and urine and cause chronic, severe photosensitivity
2243
21
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
UROS catalyzes the formation of uroporphyrinogen III from hydroxymethylbilane (HMB) in the cytosol (Fig. 124-1). This reaction involves inverting one of the 4 pyrroles (ring D) of the linear tetrapyrrole and formation of a porphyrin macrocycle with an asymmetric arrangement of side chains. In the absence of this enzyme, HMB spontaneously cyclizes to form uroporphyrinogen I, which is symmetrical. UROD, the next enzyme in the pathway, will metabolize both uroporphyrinogen I and III to coproporphyrinogen I and III, respectively. However, coproporphyrinogen I is a dead-end product because the next enzyme in the heme biosynthetic pathway accepts only coproporphyrinogen III as a substrate. More than 48 mutations of the UROS gene have been identified, and more severe mutations are associated with more severe clinical manifestations.86 A GATA-1 mutation described in one case associated with betathalassemia indicates that a genetic defect outside the heme biosynthetic pathway can cause CEP.87
Marrow normoblasts in CEP display marked microscopic fluorescence due to porphyrin accumulation.88 Increased porphyrin concentrations in circulating erythrocytes cause cell damage due to exposure to light in the dermal capillaries. Hemolysis and increased uptake of damaged cells by the spleen leads to splenomegaly as a common finding in CEP. Ineffective erythropoiesis in the marrow contributes to anemia. Excess porphyrins are transported in erythrocytes and plasma to the skin and lead to chronic and usually severe photosensitivity.
CLINICAL FEATURES
CLINICAL FEATURES
CEP is characterized by subepidermal bullous lesions affecting light-exposed areas such as the dorsal hands, fingers and face (Fig. 124-6), which often leads to scarring and areas of hyper- and hypopigmentation. Cutaneous lesions resemble those found in PCT, but with much higher porphyrin levels causing more prominent hyperpigmentation, scarring, contraction, and loss of fingers and facial features. Milder disease is often misdiagnosed as PCT, and rare onset in adults may be associated with myelodysplastic or myeloproliferative disorders with expansion of a clone of erythrocyte precursors carrying a UROS mutation.90
The disease may be recognized in utero as a cause of fetal hydrops.91 In most cases, photosensitivity is noted soon after birth. Erythrodontia (brown staining of the teeth) is caused by porphyrin deposition in developing teeth in utero. Porphyrins are also deposited in bone.92 Pathologic fractures and other bony abnormalities may result from bone marrow expansion and vitamin D deficiency resulting from avoidance of sunlight. Anemia resulting from intravascular hemolysis and ineffective erythropoiesis can be severe and require
2244
repeated red blood cell transfusions. Uncorrected anemia can further stimulate erythropoiesis and contribute to increased porphyrin production. Peripheral blood smears reveal polychromasia, poikilocytosis, anisocytosis, and basophilic stippling of erythrocytes and increases in reticulocytes and nucleated red blood cells. The liver and nervous system are not affected in CEP. A mother with CEP successfully delivered a healthy, unaffected infant with erythrodontia as a result of porphyrins crossing placenta.93
DIAGNOSIS
DIAGNOSIS
CEP can be recognized in utero by large amounts of porphyrins measured in amniotic fluid or fetal blood. CEP is often diagnosed shortly after birth when pink to brownish staining of the diapers is noted, with red fluorescence under long-wave ultraviolet light. Urinary, erythrocyte, and plasma porphyrins are markedly increased, with a predominance of uroporphyrin I and coproporphyrin I. Fecal porphyrins are markedly increased and are predominantly coproporphyrin I. Protoporphyrin IX is sometimes the predominant porphyrin in erythrocytes, especially in milder cases. DNA studies to identify causative mutations are important for confirming the diagnosis, genetic counseling, and prenatal diagnosis in subsequent pregnancies.
THERAPY
THERAPY
An individualized, multidisciplinary approach is required.99 Patients should be advised to avoid sunlight, skin trauma, and infections to avoid severe scarring and loss of facial features and digits. Topical sunscreens that absorb ultraviolet A and B light are marginally beneficial because the harmful light is mostly visible; oral beta-carotene is also not effective.94
Patients may require erythrocyte transfusions for severe anemia95 and an iron chelator to avoid the sequelae of iron overload.96 Hydroxyurea to reduce erythropoiesis and porphyrin production also may be considered.97 Splenectomy may be beneficial if hypersplenism is contributing to significant anemia, leucopenia or thrombocytopenia. Ascorbic acid and α-tocopherol improved anemia in one patient.98 Oral charcoal was reportedly beneficial in another.84 Use of oral charcoal, splenectomy, and chronic hypertransfusion were associated with complications, without appreciable clinical benefit and negative impact on health-related quality of life in a large case series.99
Hematopoietic stem cell transplantation is curative and is the treatment of choice for young patients with severe disease.100 Successful transplantation results in marked clinical improvement and reduction in porphyrin levels. Gene therapy is being explored using retroviral and lentiviral vectors and hematopoietic stem cells in patients with CEP.101,102
B
A
21
C
PROTOPORPHYRIAS
AT-A-GLANCE
■ EPP is the third most common porphyria and the most common in children
■ It results from mutation of both ferrochelatase (FECH) alleles; one with a severe mutation and in most families the other with a predisposing alteration that is common in the population
■ XLP is less common and results from gain-offunction mutations of ALAS2
■ Both are characterized by an acute, mostly nonblistering type of cutaneous photosensitivity and risk of liver damage.
■ Diagnosis is established by marked elevation of erythrocyte protoporphyrin, which is predominantly metal-free protoporphyrin (not zinc protoporphyrin)
■ Management emphasizes light avoidance and photoprotection
2245
21
Erythropoietic protoporphyria (EPP) is the most common type of porphyria in children and the third most common in adults, with reported prevalence ranging between 0.5 and 1.5 cases per 100,000 individuals.103-105
It results from loss-of-function mutation of ferrochelatase (FECH), the last enzyme of the heme biosynthetic pathway. But the protoporphyria phenotype can result from mutation of other genes. X-linked protoporphyria (XLP) results from gain-of-function mutation of ALAS2, the erythroid-specific form of the first enzyme in the pathway, and comprises ∼5% of protoporphyria cases.106 Protoporphyria with increased ALAS2 activity can also result from mutation of mitochondrial CLPX.107 With rare exceptions, symptoms in all protoporphyrias begin in early childhood with an acute and nonblistering type of cutaneous photosensitivity. Protoporphyric hepatopathy is a potentially fatal complication occurring in less than 5% of patients.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
EPP is caused by mutation of ferrochelatase (FECH, which catalyzes insertion of iron into protoporphyrin IX, the last step of the heme biosynthetic pathway; Fig. 124-1). FECH deficiency results in the accumulation of the substrate protoporphyrin particularly in marrow reticulocytes. Inheritance of EPP has been described as autosomal dominant with variable penetrance. But since the discovery that mutation of both FECH alleles is required, it is now best described as autosomal recessive. In most families, a severe FECH mutation, of which at least 189 have been described, is trans to a low-expression (hypomorphic) variant allele (IVS3-48C/T).108-110 The change in DNA sequence in the low-expression allele results in increased use of an aberrant splice site that produces an mRNA more prone to degradation and a decreased steady-state level of wildtype FECH mRNA.110 This FECH allele occurs in 10% of whites, and by itself has no phenotype. Its frequency varies widely in other populations; its prevalence is higher in east Asians but very low in Africa, accounting for geographic differences in the prevalence of EPP.103-110
Cases with 2 severe FECH mutations, 1 inherited from each parent, but without the low-expression allele, have been described. At least 1 allele must allow enough FECH enzyme to be synthesized for adequate synthesis of heme. These cases are sometimes associated with seasonal palmar keratoderma, neurologic symptoms, less-than-expected increases in erythrocyte protoporphyrin, and absence of liver dysfunction.111
Adult-onset cases of EPP have been described in patients with a myeloproliferative or myelodysplastic syndrome, in which there is expansion of a clone of hematopoietic cells carrying a FECH mutation.112,113
For example, a patient with a myeloproliferative disorder developed severe EPP due to clonal expansion of
2246
erythroid precursor cells with a FECH deletion trans to a hypomorphic FECH allele, and died of EPP-induced liver disease.114
An X-linked pattern of inheritance in some patients with protoporphyria who lacked FECH mutations led to discovery of gain-of-function mutations of ALAS2 (the only gene encoding an enzyme in the heme biosynthetic pathway found on the X chromosome).106 These mutations lead to an approximately 3-fold increase in enzymatic activity of the ALAS protein and accumulation of protoporphyrin IX, which indicates that FECH activity is the next rate-limiting step in erythroid heme synthesis.106 Two members of a family in France were recently found to have protoporphyria without mutation of FECH or ALAS2, but with a loss-of-function mutation of CLPX inherited as an autosomal dominant trait, resulting in decreased degradation of ALAS2 and overproduction of ALA and protoporphyrin.107
In EPP, the excess protoporphyrin found in circulating erythrocytes is primarily metal-free rather complexed with zinc, in contrast to many other conditions that increase erythrocyte protoporphyrin that is mostly zinc protoporphyrin (eg, lead intoxication, iron deficiency, anemia of chronic disease, hemolytic states). Excess metal-free protoporphyrin in plasma, which originates from the marrow, where hemoglobin synthesis is active, as well as from circulating erythrocytes, is taken up by hepatocytes, excreted in bile and feces, and may undergo enterohepatic recirculation. Protoporphyrin is hydrophobic as it only possesses 2 hydroxyl groups and is not excreted in urine. Erythrocytes in XLP and protoporphyria due to a CLPX mutation contain increased amounts of both metal-free and zinc-complexed protoporphyrin. Excitation of porphyrins by ultraviolet light generates free radicals and singlet oxygen,115 which in EPP can lead to complement activation, peroxidation of lipids,116 crosslinking of membrane proteins,117 and polymorphonuclear chemotaxis, resulting in skin pathology.118 Nonspecific findings on skin histopathology include thickened capillary walls in the papillary dermis surrounded by deposition of amorphous hyalinelike and periodic acid–Schiff (PAS)-positive mucopolysaccharides, complement, and immunoglobulins.119 Basement membrane abnormalities are less marked than in other forms of cutaneous porphyria.120
Protoporphyric hepatopathy occurs in less than 5% of patients. It may cause chronic abnormalities in liver function tests with progression to cirrhosis, or present acutely with rapid progression and require urgent liver transplantation. Other causes of liver damage, such as alcoholic or nonalcoholic steatohepatitis or viral hepatitis, can predispose patients to this condition by impairing protoporphyrin excretion and thereby increasing circulating protoporphyrin to levels that accelerate damage to the liver. Excess protoporphyrin is cholestatic, forms crystalline structures in hepatocytes, and impairs mitochondrial function.121,122 Accumulated protoporphyrin may appear as brown pigment in hepatocytes, Küpffer cells, and biliary canaliculi and as doubly refractive inclusions with a “Maltese cross” appearance under polarizing microscopy.123
SYMPTOMS AND FINDINGS INCIDENCE (% OF TOTAL)
Burning 97
Edema 94
Itching 88
Erythema 69
Scarring 19
Vesicles 3
Anemia 27
Cholelithiasis 12
Abnormal liver function results 4
Abnormal liver function results 4
Data from Bloomer J, Wang Y, Singhal A, et al. Molecular studies of liver disease in erythropoietic protoporphyria. J Clin Gastroenterol. 2005;39(4 suppl 2):S167-S175.124
CLINICAL FEATURES
CLINICAL FEATURES
Disease manifestations in a series of 32 patients with EPP are summarized in Table 124-3. Acute cutaneous photosensitivity, which is seldom seen by physicians, is the most prominent symptom described as stinging, burning, or tingling pain that may develop within minutes of sunlight exposure, followed by erythema
A
21
and edema (described as solar urticaria) (Fig. 124-7), and systemic manifestations that may last for several days. Petechiae or purpura may occur with prolonged exposure. Children are unable to describe these symptoms, but parents may observe crying, skin swelling, and erythema after exposure to sunlight. Hands and face are most often affected, and symptoms are worse during spring and summer. Patients are sensitive to sunlight, either direct or passing through window glass, and also to artificial light, including operating room lights.125 Among 226 patients in North America, the mean age at symptom onset was 4.4 years, and higher protoporphyrin levels were associated with earlier age of onset, less sun tolerance, and more frequent reports of liver function abnormalities. On average, male patients with XLP had higher erythrocyte protoporphyrin levels than patients with EPP (3574 vs 1669 µg/dL; P <.001). Symptom severity and protoporphyrin levels varied markedly among female XLP patients owing to random X-chromosomal inactivation.126
Because patients are prompted by pain and learn to avoid sunlight, chronic skin changes are uncommon and usually subtle. These may include leathery hyperkeratotic skin on the dorsae of the hands and finger joints, mild scarring, labial grooving, and separation of the nail plate (onycholysis), developing with frequent sun exposure (Fig. 124-8). Bullae, skin fragility, hypertrichosis, hyperpigmentation, severe scarring, and mutilation, as seen in other cutaneous porphyrias, are rare. Pregnancy is reported to lower erythrocyte
B
2247
21
A
B
protoporphyrin levels somewhat and increase tolerance to sunlight.128
Mild iron-deficiency anemia with microcytosis, decreased transferrin saturation, in the low part of the normal range of serum ferritin is seen in 20% to 50% of cases of EPP.129,130 There is little evidence for impaired erythropoiesis or abnormal iron absorption or metabolism,131-133 and hemolysis is absent or mild. Neurovisceral manifestations are absent in uncomplicated EPP, but protoporphyric hepatopathy may be complicated by severe motor neuropathy similar to that seen in the acute porphyrias.134 Autosomal recessive EPP associated with palmar keratoderma also has been associated with unexplained neurologic symptoms.111
Gallstones containing large amounts of protoporphyrin are common, and form from excretion of large amounts of water-insoluble protoporphyrin in bile. Such stones may be symptomatic and require
2248
cholecystectomy at an early age.135 Liver function and liver protoporphyrin content are usually normal in EPP. Protoporphyric hepatopathy results from the cholestatic effects of protoporphyrin presented in excess amounts to the liver. Rarely, this is the major presenting feature of EPP.136 Circulating protoporphyrin levels increase markedly as impairment of biliary excretion progresses. Operating room lights during liver transplantation or other prolonged surgery, especially in patients with hepatopathy, can cause marked photosensitivity with extensive burns of the skin and peritoneum, and can be avoided by use of special filters.137,138
DIAGNOSIS
DIAGNOSIS
Painful, nonblistering photosensitivity occurring at any age with few chronic skin changes should suggest a diagnosis of protoporphyria. Measurement of total erythrocyte protoporphyrin and, if elevated, measurement of the proportions of metal-free and zinc protoporphyrin will establish or exclude this diagnosis. Elevation of total erythrocyte protoporphyrin is not specific for EPP, because it is increased, with a predominance of erythrocyte zinc protoporphyrin, in many conditions such as homozygous porphyrias (other than most cases of CEP), iron deficiency, lead poisoning, anemia of chronic disease,139 hemolytic conditions,140 and many other erythrocyte disorders. But an increase in erythrocyte protoporphyrin comprised predominantly of metal-free protoporphyrin is a finding unique to protoporphyrias. FECH catalyzes the formation of both zinc protoporphyrin and heme (iron protoporphyrin), and when this enzyme is deficient, formation of both is impaired and the accumulating protoporphyrin remains mostly metal free. In XLP, FECH activity is normal but its capacity is exceeded by an excess amount of substrate, so the protoporphyrin that accumulates is usually mostly metal free. Clinicians who wish to screen for EPP and XLP should be aware of major pitfalls in measuring erythrocyte protoporphyrin for this purpose and should use a laboratory that report results correctly as “total erythrocyte protoporphyrin” and “metal-free protoporphyrin.” The now obsolete term “free erythrocyte protoporphyrin,” abbreviated “FEP,” was widely used to describe ironfree protoporphyrin before the 1970s. This was before it was discovered that this increase is due to zinc protoporphyrin in lead poisoning, iron deficiency, and the many other conditions, but to metal-free protoporphyrin in EPP. Some laboratories (Quest and LabCorp) measure erythrocyte protoporphyrin by hematofluorometry, which only measures zinc protoporphyrin and was developed for screening for lead poisoning and not protoporphyrias; their use of the misleading and obsolete terms “free protoporphyrin” and “FEP” falsely indicate that metal-free protoporphyrin was measured. ARUP measures the total amount correctly but if that is elevated does not fractionate metal-free and zinc protoporphyrin, which is essential to show that the increase is due to protoporphyrias. Testing for protoporphyrias
is carried out correctly in the United States by the Porphyria Laboratory at the University of Texas Medical Branch and Mayo Medical Laboratories.141
Plasma porphyrins are increased in most but not all cases of EPP and are particularly susceptible to photodegradation during sample processing.142 A plasma fluorescence peak of diluted plasma at neutral pH near 634 nm can help confirm a diagnosis of EPP.53 Fecal porphyrin levels are normal or somewhat increased and consist mostly of protoporphyrin. Urine porphyrins are normal, except in patients with protoporphyric hepatopathy, which causes increases in urinary coproporphyrin, as is typical for hepatic dysfunction of any cause.
THERAPY
THERAPY
Photoprotection, especially avoidance of sunlight exposure, is the primary therapeutic intervention in EPP. This necessitates changes in lifestyle and the working environment and other avoidance behaviors that greatly impair quality of life. Protective clothing and hats are essential for most patients when outdoors. Topical sunblocks containing zinc oxide or titanium dioxide can provide some benefit. Sunscreens that absorb UV wavelengths are not beneficial. Orally administered beta-carotene, thought to quench activated oxygen radicals,143 can increase sunlight tolerance.144 A daily dose of 120 to 180 mg or higher is recommended to achieve serum beta-carotene levels of 600 to 800 µg/dL, which can be assessed 3 to 4 weeks after a dose change.125 However, skin discoloration from carotenemia may be difficult cosmetically especially for children. Oral cysteine at doses of 500 mg twice daily may also quench excited oxygen species and increase tolerance to sunlight in EPP.145 Afamelanotide, a synthetic analog of α-melanocyte–stimulating hormone mimics the naturally occurring hormone and increases melanin production in melanocytes, resulting in increased skin pigmentation and better sunlight tolerance in patients with EPP or XLP.146,147 Afamelanotide is currently approved for restricted use in the European Union and Switzerland and is under review by the US Food and Drug Administration for use in patients with EPP and XLP. Cimetidine, which inhibits hepatic CYPs, was reported to decrease photosensitivity in 3 children with EPP.148 This is not recommended because the proposed inhibition of ALAS2 and lowering of protoporphyrin levels have not been documented.149
Mild iron deficiency in protoporphyrias is unexplained and is thought by some to be beneficial (by limiting up-regulation of ALAS2 by iron), but is also potentially detrimental (by further limiting iron incorporation into protoporphyrin). Some patients have noted that iron supplementation increases photosensitivity, but increases in erythrocyte or plasma protoporphyrin levels have not been documented, to our knowledge. On the other hand, clinical and biochemical improvement have been noted in some individual cases of EPP and XLP with iron supplementation.150,151
The effects of iron supplementation in patients with protoporphyrias and low serum ferritin levels is
21
currently under study by the Porphyrias Consortium in the United States. Exposure to alcohol, hepatotoxic drugs, or drugs and hormones that increase hepatic porphyrin and heme synthesis or impair hepatic excretory function should be avoided as a precaution.152,153 Erythrocyte and plasma porphyrin levels, liver function tests, serum ferritin, and serum vitamin D levels should be monitored yearly. Daily intake of 800 international units of vitamin D and 1000 mg of calcium is recommended because EPP patients must avoid sunlight exposure. Protoporphyric hepatopathy may resolve spontaneously or with treatment, especially if it was triggered by a reversible cause of liver dysfunction, such as viral infection or alcohol.120,154 This life-threatening complication is usually treated with a combination of interventions including cholestyramine,122,155,156
ursodeoxycholic acid,157 vitamin E, red blood cell transfusions,158 plasma exchange, and intravenous hemin with the aim to reduce plasma and erythrocyte protoporphyrin levels and the amount of protoporphyrin delivered to the liver. This may bridge patients to liver transplantation or even spontaneous improvement.159 Liver transplantation is as successful as in other liver diseases, although a recurrence rate in the new liver of 69% at 5 years has been reported.160,161 Acute motor neuropathy has developed in some patients with protoporphyric liver disease after transfusion162
or liver transplantation,134,163 and is sometimes reversible with administration of hemin and plasmapheresis prior to transplantation.164 Bone marrow transplantation is effective in EPP,165 and sequential bone marrow transplantation after liver transplantation can prevent recurrence of liver disease.166
ACUTE HEPATIC PORPHYRIAS
AT-A-GLANCE
■ The acute porphyrias are characterized by intermittent acute neurologic symptoms, but can develop chronic blistering lesions in 3 of these conditions, VP, HCP, and AIP (in advanced renal disease).
■ Neurovisceral manifestations include abdominal pain, vomiting, extremity pain, seizures, and muscle weakness due to motor neuropathy.
■ Urine porphobilinogen is elevated especially during acute attacks. Fluorometric scanning of plasma porphyrin and patterns of fecal porphyrins are most useful for distinguishing the specific acute porphyria.
■ Cutaneous manifestations are prevented by avoidance of sunlight.
■ Acute neurovisceral attacks are prevented by avoiding exacerbating factors and best treated with hemin infusion
2249
21
The 4 acute porphyrias, δ-aminolevulinate dehydratase deficiency porphyria (ADP), acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP) result from loss-of-function mutation of the second, third, sixth, and seventh enzymes in the heme biosynthetic pathway, respectively, and are characterized by neurologic symptoms occurring mostly as intermittent acute exacerbations. However, chronic blistering skin lesions resembling PCT can occur in 3 of these conditions. They are common in VP, uncommon in HCP, in AIP occur only with advanced renal disease, and are not described in ADP.167
ADP is extremely rare, with only 6 cases reported; unlike the other acute porphyrias, its inheritance is autosomal recessive.168 (It will not be discussed further in this chapter.) The enzyme deficiencies in the other acute porphyrias are inherited as autosomal dominant traits with low penetrance (see Table 124-1). AIP is the most common acute hepatic porphyria worldwide. The prevalence of AIP has been estimated to be 1 to 2 per 100,000 in Europe,169 that of HCP was estimated to be 0.2 per 100,000 in Denmark,170 and that of VP in Finland reported at 1.3 per 100,000.171 VP is especially common in South Africa among whites of Dutch descent due a founder effect, with an estimated prevalence of 300 per 100,000172, and almost all cases share the same PPOX mutation (R59W).
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
At least 400 PBGD/HMBS mutations have been identified in AIP, 64 CPOX mutations in HCP173 and 174 PPOX mutations in VP86 (Table 124-1). Mutation types include missense, nonsense, splicing, deletion, and insertion mutations. PBGD catalyzes the assembly of HMBS, a linear tetrapyrrole, from 4 molecules of PBG. CPOX catalyzes the 2-step oxidative decarboxylation at a single active site of coproporphyrinogen III to yield protoporphyrinogen IX, with intermediate formation of harderoporphyrinogen, a tricarboxyl porphyrinogen. PPOX catalyzes the dehydrogenation of protoporphyrinogen IX to form protoporphyrin IX. Neurovisceral symptoms in the acute hepatic porphyrias are associated with accumulation of the porphyrin precursors delta-aminolevulinic acid (ALA) and porphobilinogen (PBG). Symptoms may result from neurotoxic effects of ALA and/or from heme deficiency in neuronal or vascular tissue.174 Porphyrins accumulate in all of these disorders and most commonly in sufficient amounts to cause chronic blistering skin manifestations in VP.
CLINICAL FEATURES
CLINICAL FEATURES
Typical neurovisceral manifestations of the acute porphyrias include abdominal pain, vomiting, extremity
2250
pain, seizures, and muscle weakness due to motor neuropathy. Several factors are recognized to contribute to attacks in AIP, HCP, and VP, including drugs, hormones, and dietary factors. Risk of chronic hypertension, renal disease, and hepatocellular carcinoma are increased. Very rare cases of homozygous AIP, HCP, and VP have presented with severe photosensitivity and neurologic impairment early in life but without acute attacks.174 A variant form of homozygous HCP termed harderoporphyria is due to CPOX mutations that prematurely release harderoporphyrinogen from the enzyme before the second decarboxylation occurs.175
PRECIPITATING FACTORS
PRECIPITATING FACTORS
Known endogenous and exogenous factors can precipitate acute attacks in heterozygotes and are additive in individual patients. Many known factors cause induction of hepatic ALAS1, and this leads to accumulation of pathway intermediates. Some individuals remain susceptible to repeated attacks despite avoidance of known precipitants. Additional unknown factors, including undiscovered modifying genes, are likely to contribute.
DRUGS AND OTHER EXOGENOUS CHEMICALS
Most drugs that are harmful in acute porphyrias are known to induce hepatic CYPs and ALAS1. Induction of CYPs (the most abundant hemoproteins in liver) increases demand for newly synthesized heme.176 Examples of drugs known to be harmful and safe are shown in Table 124-4. Updated drug safety databases are found at the websites of the American Porphyria Foundation (www.porphyriafoundation.com) and the European Porphyria Network (www.porphyria-europe .com). Smoking is known to increase hepatic CYPs in humans, probably from effects of inhaled polycyclic aromatic hydrocarbons, and is associated with more frequent symptoms.177 Ethanol and other alcohols also induce ALAS1 and some CYPs, and have been implicated in causing attacks.178,179
ENDOCRINE FACTORS
Symptoms often develop after puberty and occur more commonly in women, suggesting that endogenous female hormones contribute to the onset of symptoms. Progesterone, synthetic progestins, and certain metabolites of testosterone are potent inducers of ALAS1 and CYPs and have been associated with acute attacks of porphyria. Increases in progesterone in the luteal phase of the menstrual cycle are most likely responsible for cyclic attacks in AIP, HCP, and VP. Estrogens have been considered harmful, but do not have the inducing effects of progestins on ALAS1 and CYPs, and reports of attacks when they are administered without a progestin are few. But as discussed
UNSAFE SAFE
Meprobamatea (also mebutamate,a tybutamatea) Methyprylon Metoclopramidea
Alcohol Barbituratesa
Acetaminophen Aspirin Atropine Bromides Cimetidine Erythropoietina,b
Alcohol Barbituratesa
Meprobamatea (also
Acetaminophen Aspirin Atropine Bromides Cimetidine Erythropoietina,b
mebutamate,a
Carbamazepinea
Carbamazepinea
tybutamatea) Methyprylon Metoclopramidea
Carisoprodola
Carisoprodola
Clonazepam (high doses) Danazola
Clonazepam
Phenytoina
(high doses) Danazola
Phenytoina
Primidonea
Gabapentin Glucocorticoids Insulin Levetiracetam Narcotic analgesics Penicillin and derivatives Phenothiazines Ranitidinea,b
Primidonea
Gabapentin Glucocorticoids Insulin Levetiracetam Narcotic
Diclofenaca and
Diclofenaca and possibly other NSAIDs Ergots Estrogensa,c
Progesterone and synthetic progestinsa
Progesterone and synthetic
possibly other NSAIDs Ergots Estrogensa,c
progestinsa
Pyrazinamidea
Pyrazinamidea
Pyrazolones (aminopyrine, antipyrine) Rifampina
Pyrazolones (aminopyrine,
antipyrine) Rifampina
analgesics Penicillin and
Ethchlorvynola
Ethchlorvynola
Glutethimidea
Succinimides (ethosuximide, methsuximide) Sulfonamide antibioticsa
Glutethimidea
Succinimides (ethosuximide,
derivatives Phenothiazines Ranitidinea,b
Griseofulvina
Griseofulvina
methsuximide) Sulfonamide antibioticsa
Mephenytoin
Mephenytoin
Valproic acida
Streptomycin Vigabatrin
Valproic acida
Streptomycin Vigabatrin
aPorphyria is listed as a contraindication, warning, precaution, or adverse effect in US labeling for these drugs.
bAlthough porphyria is listed as a precaution in US labeling; these drugs are regarded as safe by other sources.
cEstrogens–unsafe for porphyria cutanea tarda but can be used with caution in the acute porphyrias. Note: More complete and frequently updates sources, such as the websites of the American Porphyria Foundation (www.porphyriafoundation.com/) and the European Porphyria Initiative (www.porphyria-europe.com/), should be consulted before using drugs not listed here. Modified from Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439-450; with permission.167
earlier, estrogen use is a common susceptibility factor in women with PCT. Diabetes mellitus may decrease the frequency of attacks and lower porphyrin precursor levels, likely because of high glucose levels.180
PREGNANCY
Most women with AIP report that pregnancy is well tolerated.181 However, attacks sometimes become more frequent during pregnancy, which may be due to harmful drugs (eg, metoclopramide)182,183 or reduced caloric intake with hyperemesis gravidarum. Attacks during pregnancy should be treated with hemin, unless they are mild. Termination of pregnancy is rarely indicated for an acute attack of porphyria.
NUTRITION
Reduced intake of calories and carbohydrate to lose weight or during an illness or surgery can precipitate attacks. In these circumstances, peroxisomal proliferator-activated cofactor 1α (PGC-1α) is up-regulated in the liver, which induces ALAS1 and increases ALA and PBG. These effects can be prevented or reversed
21
by administration of carbohydrate.184,185 Starvation may also induce hepatic heme oxygenase,186 which may deplete hepatic heme and contribute to ALAS1 induction.
DIAGNOSIS
DIAGNOSIS
Urinary PBG is elevated during acute attacks of AIP, HCP and VP, but often less so, and more transiently, in HCP and VP than in AIP; further testing readily differentiates these 3 conditions. Skin lesions resembling PCT are rare in AIP and occur only in the presence of advanced renal disease with increases in plasma levels of both PBG and porphyrins. In HCP and VP, urine porphyrins (especially uroporphyrin and coproporphyrin) are also elevated, but this finding by itself is nonspecific and does not readily differentiate these porphyrias from each other. Fecal porphyrin levels are markedly increased in both HCP and VP and are predominantly coproporphyrin III in HCP, and both coproporphyrin III and protoporphyrin in VP. The fecal coproporphyrin III/I ratio is sensitive for diagnosis of HCP, even in asymptomatic stages of the disease.187
Plasma porphyrin concentration is commonly elevated in VP and seldom increased in AIP and HCP, unless there are cutaneous manifestations. A specific feature of VP is a plasma porphyrin fluorescence maximum at neutral pH of ∼626 nm, which represents protoporphyrin bound covalently to plasma proteins.188 This fluorometric scanning method is more effective than examination of fecal porphyrins for detecting asymptomatic VP,189 and is especially useful for rapidly differentiating VP from PCT, which displays a fluorescence peak at ∼620 nm. Erythrocyte PBGD activity is decreased in most AIP patients and normal in HCP and VP. Assays for CPOX and PPOX require cells containing mitochondria and are not widely available. DNA studies are most reliable for identifying asymptomatic carriers, once the familial mutation is identified. Homozygous cases of AIP, HCP, and VP demonstrate more severe increases in porphyrin precursors and porphyrins, including erythrocyte zinc protoporphyrin. Harderoporphyria is identified by elevations in both coproporphyrin III and harderoporphyrin III in feces and erythrocytes.190
THERAPY
THERAPY
Cutaneous manifestations of VP and HCP, though identical to the blistering skin lesions seen in PCT, do not respond to phlebotomies or low-dose hydroxychloroquine. Therefore, avoidance of sunlight and use of protective clothing is most important. Long-term avoidance of precipitating factors may lower chronically elevated porphyrin levels, but to our knowledge this has not been documented. Patients with AIP, renal failure, and associated cutaneous manifestations
2251
21
should be considered for renal or combined hepatic and renal transplantation.191
For acute attacks, hospitalization is usually advisable for treatment of severe symptoms, intravenous therapies and monitoring of respiration, electrolytes, and nutritional status. Milder recurring attacks that respond rapidly to treatment are sometimes managed as outpatients. Precipitating factors, such as harmful drugs are removed. Pain, nausea, and vomiting usually require narcotic analgesics, chlorpromazine or another phenothiazine, or ondansetron. Short-acting benzodiazepines are given for anxiety and insomnia.167
β-Adrenergic-blocking agents can be used to control tachycardia and hypertension, unless contraindicated by hypovolemia or heart failure.192 Treatment for seizures should focus on correcting hyponatremia, if present. Most anticonvulsant drugs are potentially harmful in acute porphyrias. Clonazepam may be less harmful than phenytoin, barbiturates, or valproic acid.193,194
The identification and long-term avoidance of factors precipitating acute neurovisceral attacks is essential. Yearly screening for hepatocellular carcinoma is recommended after 50 years of age, especially in those with persistent increases in porphyrin precursors or porphyrins. Hemin is the most effective treatment for acute attacks, and is available in the United States as lyophilized hematin (Panhematin, Recordati Rare Diseases), and as heme arginate (Normosang, Orphan Europe) in Europe and South Africa.195,196 Intravenously infused hemin is taken up primarily by hepatocytes where it reconstitutes the regulatory heme pool and represses the synthesis of ALAS1, leading to dramatic reduction in ALA, PBG, and porphyrins, and more rapid resolution of symptoms.197
Hemin therapy should be initiated for moderate to severe attacks, and for mild attacks unresponsive to carbohydrate loading.167,195,198 Infusion of 3 to 4 mg/kg daily for 4 days is standard therapy and may be extended if a response is incomplete within this time. Hemin has been administered safely during pregnancy.167,195,196 Reconstitution of hematin with 25% human albumin is recommended, as degradation products that form when using sterile water may cause infusion site phlebitis and limit future venous access.167,199 A transient coagulopathy with thrombocytopenia and prolongation of prothrombin and partial thromboplastin times is also prevented with use of albumin.200,201 Side effects of hemin may include fever, aching, malaise, and rarely hemolysis, anaphylaxis, and circulatory collapse.202,203 Hemin administered once or twice weekly is an option for preventing frequent, noncyclic attacks.204,205
CARBOHYDRATE LOADING
Administration of carbohydrates downregulates PGC-1α and the induction of hepatic ALAS1, thereby decreasing production of porphyrin precursors. However, this is less effective than hemin and should be used only for mild attacks (without severe pain requiring opioids, paresis, seizures or hyponatremia),167 or
2252
until hemin becomes available. Intravenous treatment with 300 to 500 g of IV glucose is usually administered as a 10% solution, with careful monitoring to avoid hyponatremia due to administration of large volumes of free water.167 Oral glucose polymer solutions may be given if tolerated.

Figure 124-1 The sequence and chemical structures of intermediates in the heme biosynthetic pathway and localization of the 8 pathway enzymes in mitochondria or the cytosol. δ-Aminolevulinic acid synthase exists as a house-keeping form (ALAS1), found in all tissues, and an erythroid-specific form (ALAS2) found only in marrow erythroid cells. As shown here, the synthesis of ALAS1 in liver is under sensitive feedback control by the end product heme.
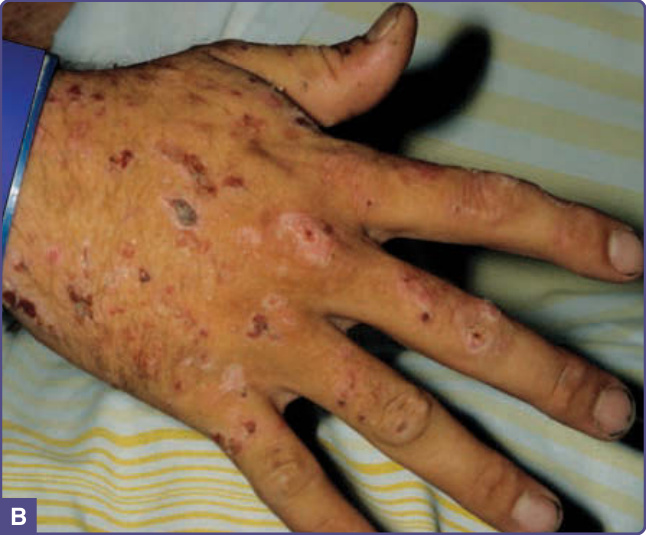
Figure 124-2 Characteristic skin lesions in PCT occur most commonly on the dorsal hands and are indistinguishable from those seen in other blistering cutaneous porphyrias (especially variegate porphyria) and pseudoporphyria. A, Blisters (short black arrow) often form after minor trauma, rupture to leave superficial ulcerations that may crust over (short white arrows) and leave atropic areas (long arrows) that heal slowly. B, Blisters are fragile, so often the only lesions present are chronic, such as erosions, crusts and scarring.
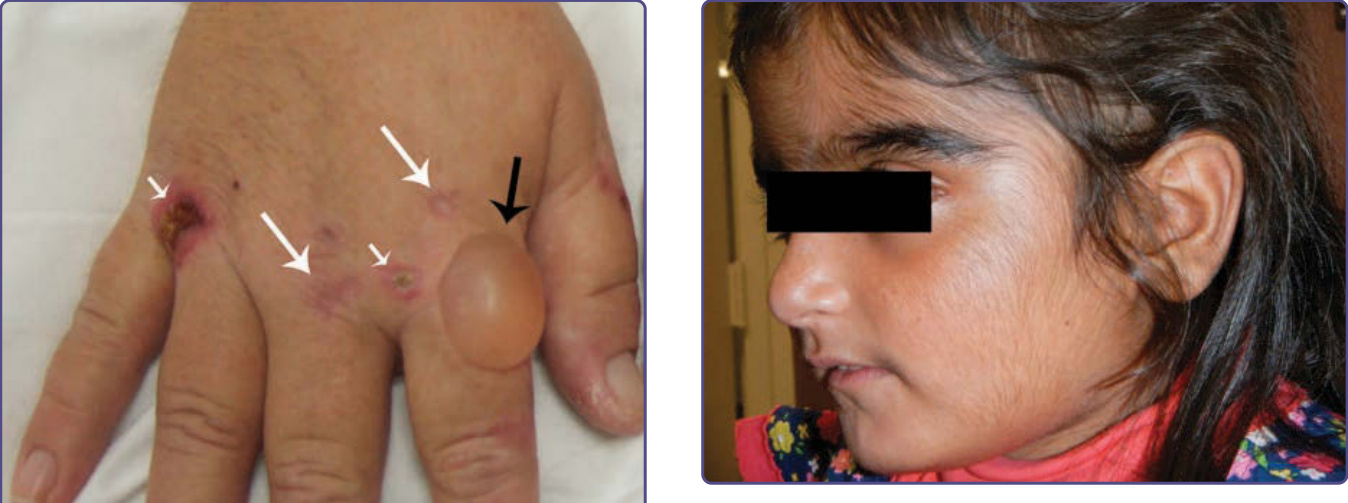
Figure 124-3 Marked hypertrichosis and hyperpigmentation due to porphyria cutanea tarda that developed in a child with an inherited UROD mutation after chemotherapy for leukemia, as previously reported.15
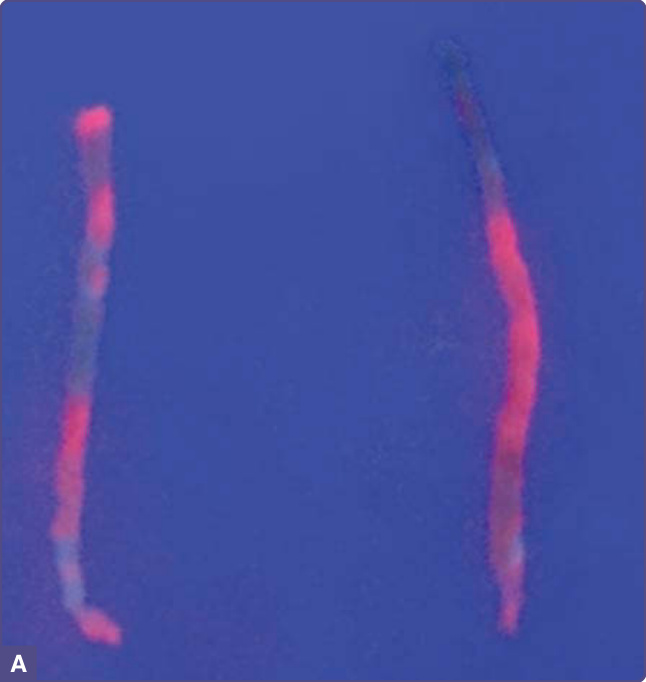
Figure 124-4 A, Fresh liver biopsy tissue showing bright red fluorescence under longwave ultraviolet light (Wood lamp) reflecting marked accumulation of highly carboxylated porphyrins in hepatocytes. B, Analysis of liver porphyrins from the same biopsy (total porphyrins 176 nmol/g tissue, ref <1) with a predominance of uroporphyrins I and III and heptacarboxyl porphyrin III on separation of porphyrins by high-performance liquid chromatography (HPLC); this pattern resembles that found in plasma and urine. (Image used with permission from Heather Stevenson-Lerner, MD, PhD; HPLC used with permission from V.M.S. Ramanujam, PhD).
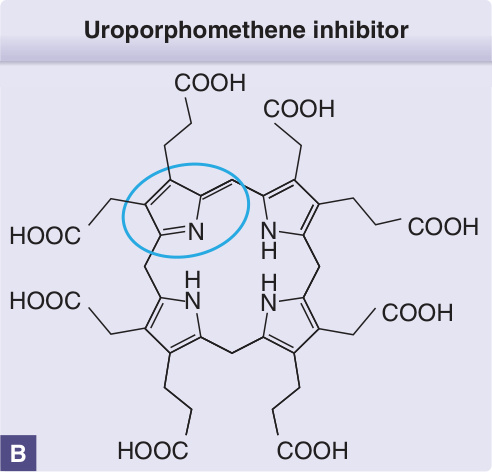
Figure 124-5 A, Schema for generation of a specific inhibitor of uroporphyrinogen decarboxylase in the liver in the presence of iron, cytochrome P450 enzymes (especially CYP1A2) and oxidative stress in PCT. (Modified from Anderson KE. Porphyria cutanea tarda: A possible role for ascorbic acid. Hepatology. 2007;45(1):6-8; with permission. Copyright © 2007, John Wiley & Sons.48) B, A UROD inhibitor has been isolated as a uroporphomethene, a uroporphyrinogen molecule that is partially oxidized at one ring (circled in blue) and its adjacent methyl bridge.
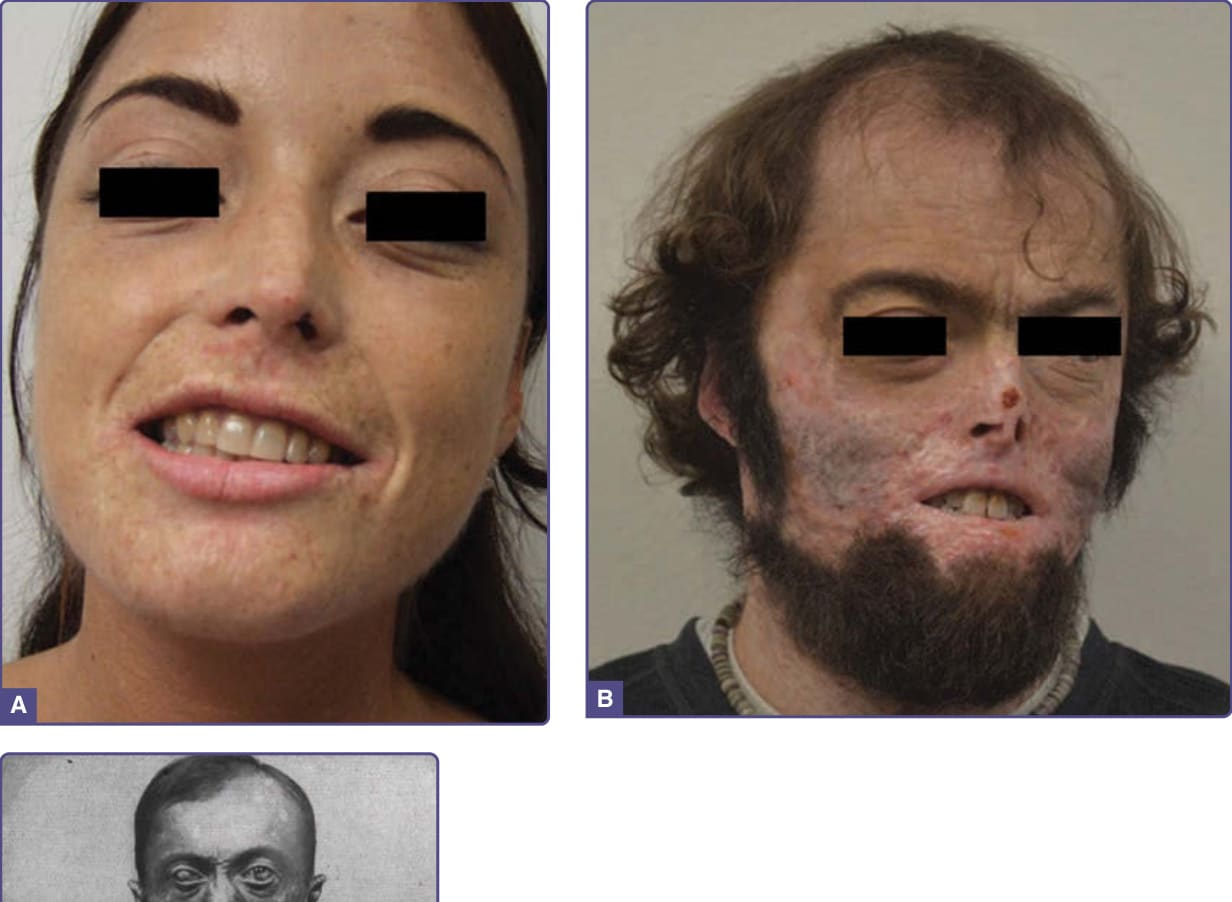
Figure 124-6 Three cases of congenital erythropoietic porphyria of different severity. Patient A had mild disease, strictly avoided sunlight, and manifested only mild skin contraction of the skin on the face, perioral scarring, and mild scarring of the dorsal hands and fingers with developmentally small, shortened fingers (not shown). Patient B also had mild disease with progressive scarring of the face, hands, and fingers from not avoiding sunlight. (Reprinted from Gou E, Phillips JD, Anderson KE. The porphyrias. In: Gilbert-Barness E, Barness L, Farrell PM, eds. Metabolic Diseases. Amsterdam: IOS Press BV; 2017:543-575,10 Copyright 2017, with permission from IOS Press. The publication is available at IOS Press through https://www.iospress.nl/book/metabolic-diseases/. Patient C is Mr. Petry, who was historically important having worked in the laboratory of the porphyrin chemist Hans Fischer, where he provided samples for early studies of porphyrin chemistry; he had severe disease and survived to age 34. From Günther H. In: Schittenhelm A, ed. Handbuch der Krankheiten der Blutes und der Blutbildenden Organe. Vol 2. Berlin: Springer; 1925; with permission. Copyright © 1925.89)
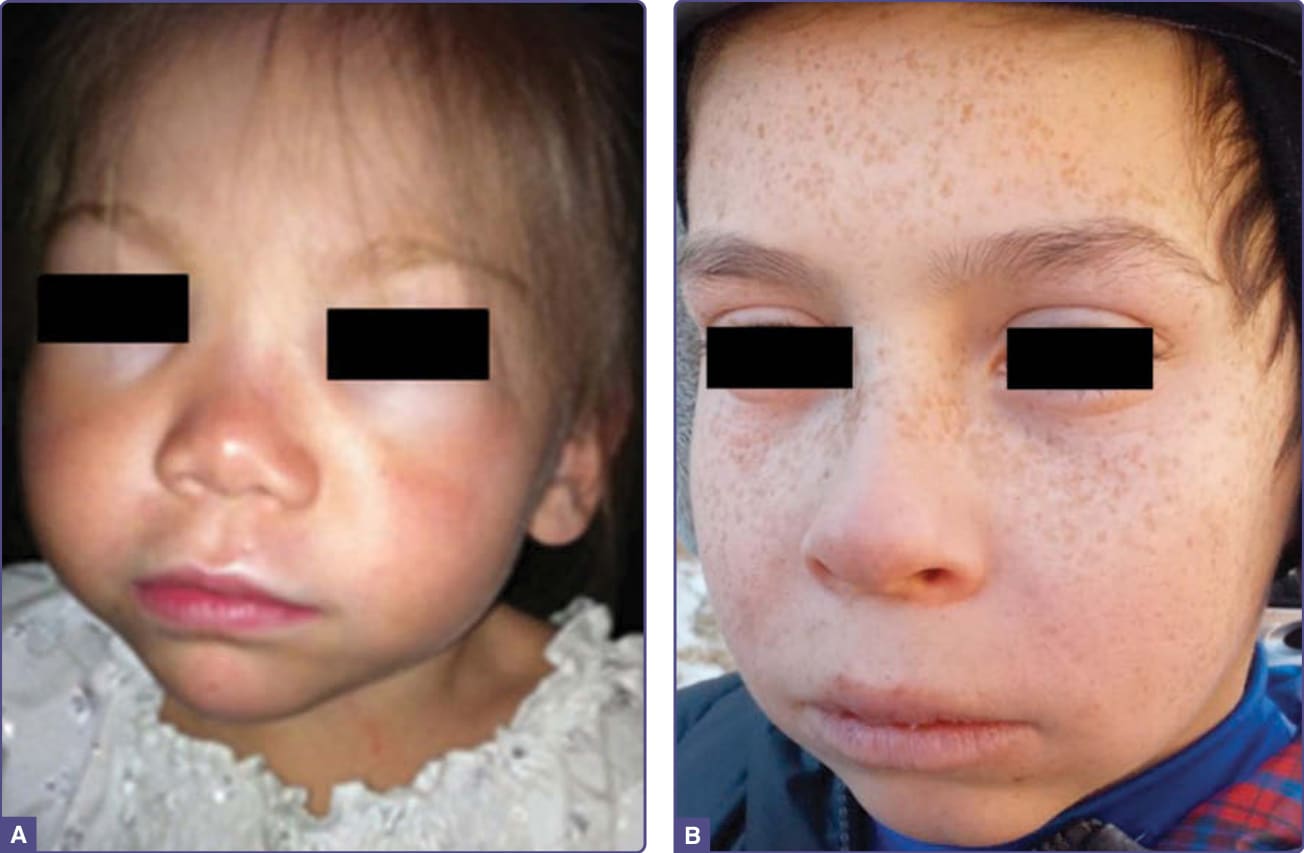
Figure 124-7 Acute swelling and erythema of the face in 2 children with EPP after exposure to sunlight. In the second child, only the lower face, which was not shielded by a ski mask, was affected.
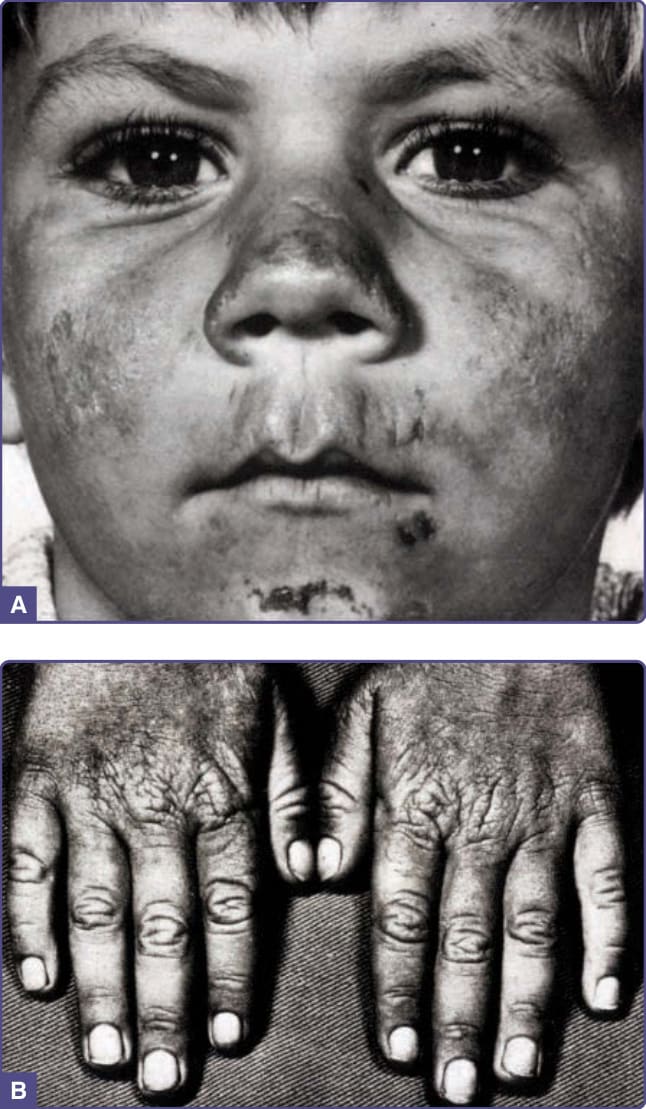
Figure 124-8 Chronic skin manifestations in a 6-year-old child with EPP resulting from repeated light reactions, including facial ulcerations and scarring, labial grooving, and thickened skin on the dorsal hands and loss of lanulae of the fingernails. (Reproduced from Schmidt H, Snitker G, Thomsen K, Lintrup J. Erythropoietic protoporphyria. A clinical study based on 29 cases in 14 families. Arch Dermatol. 1974;110(1):58-64; with permission. Copyright © 1974 American Medical Association. All rights reserved.127)
TABLE 124-1 Human Porphyrias: Specific Enzymes Affected by Mutations, Modes of Inheritance, Classification, and Major Types of Clinical Features
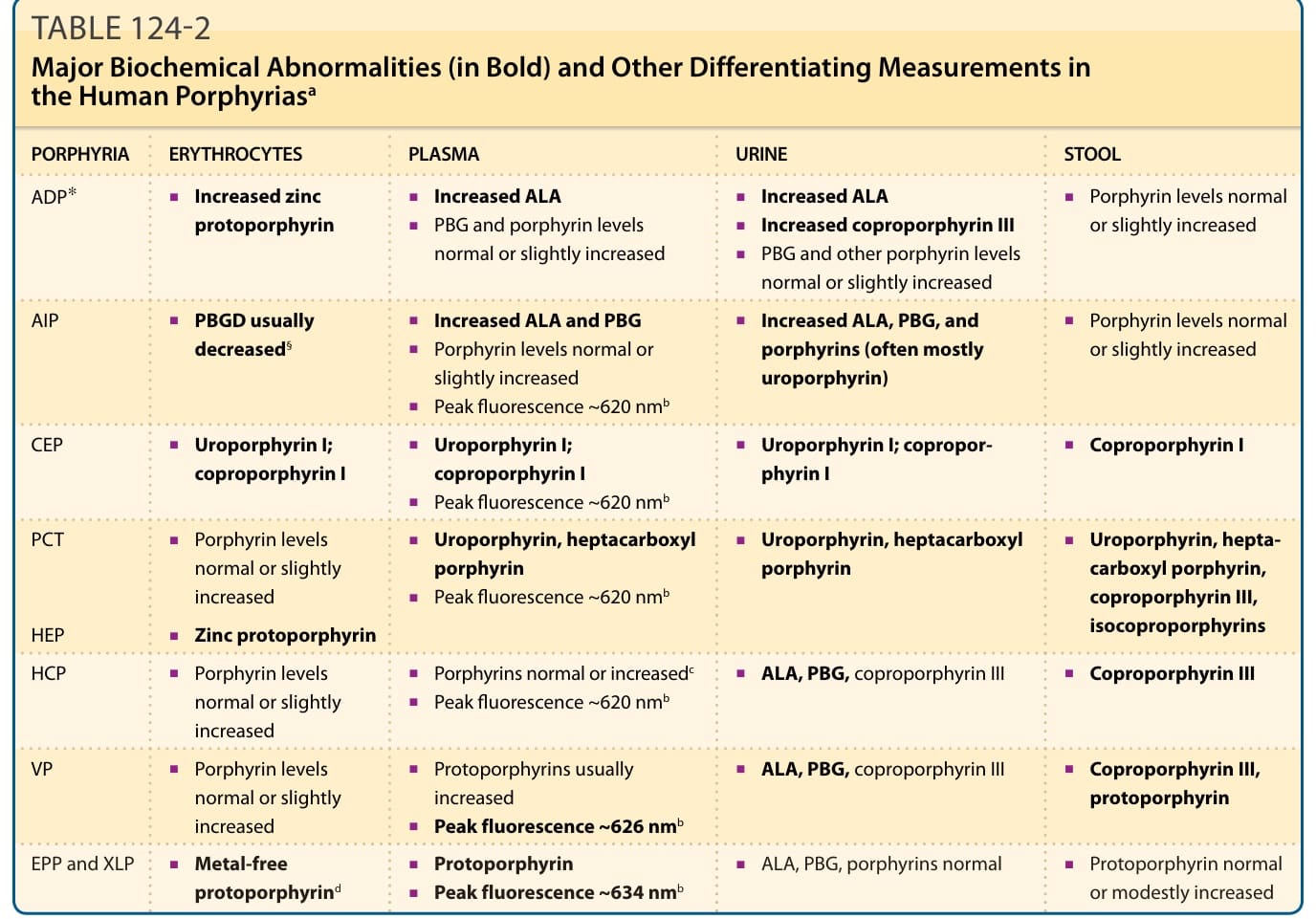
TABLE 124-2 Major Biochemical Abnormalities (in Bold) and Other Differentiating Measurements in the Human Porphyriasa
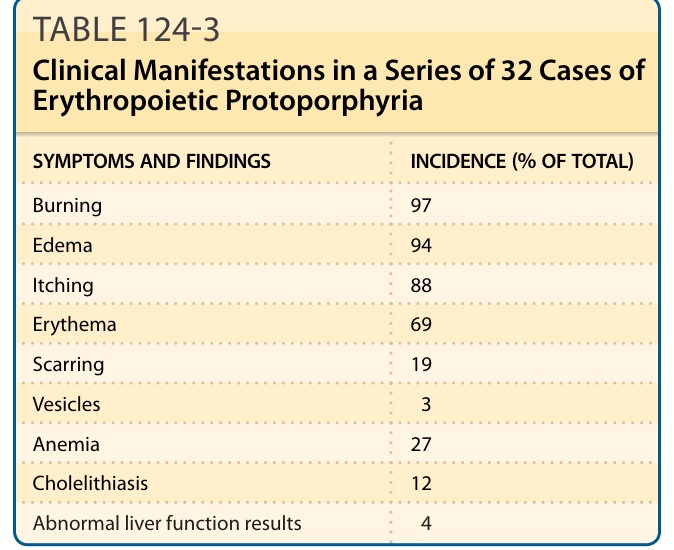
TABLE 124-3 Clinical Manifestations in a Series of 32 Cases of Erythropoietic Protoporphyria
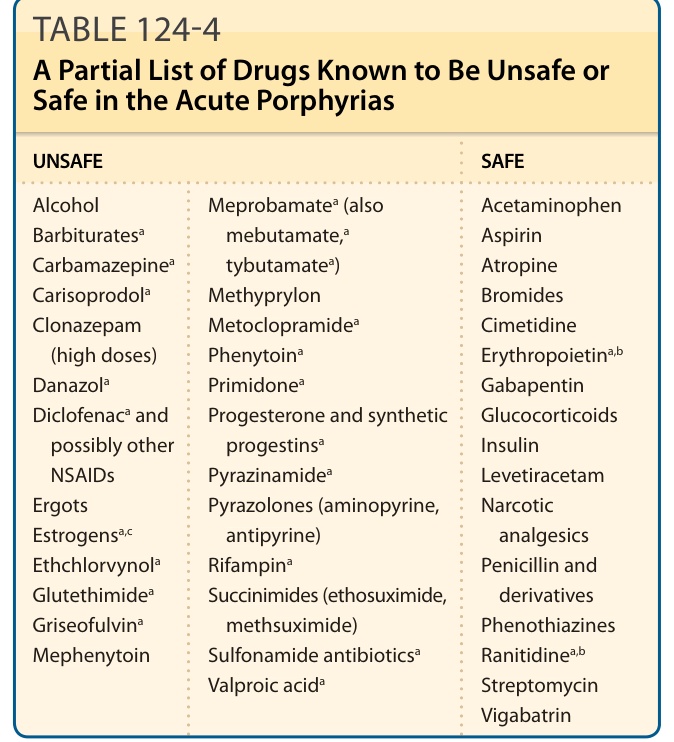
TABLE 124-4 A Partial List of Drugs Known to Be Unsafe or Safe in the Acute Porphyrias