紫質症 (The Porphyrias)
PART 21
代謝、遺傳與系統性疾病 (METABOLIC, GENETIC, AND SYSTEMIC DISEASES)
前言 (INTRODUCTION)
紫質症 (porphyrias) 是由血基質 (heme) 生合成途徑中 8 種酵素異常所引起的代謝性疾病。除一種以外,所有紫質症皆源自途徑酵素的突變;唯一的例外是遲發性皮膚紫質症 (porphyria cutanea tarda, PCT),它是該途徑第五個酵素的後天性缺乏所致,可伴有或不伴有突變。紫質症依血基質前驅物最初於肝臟或骨髓(最活躍合成血基質的組織)累積,而被分類為肝性 (hepatic) 或紅血球生成性 (erythropoietic)。在臨床上,紫質症依其臨床特徵被歸類為皮膚型 (cutaneous) 或急性型 (acute)(表 124-1)。皮膚型紫質症係因光敏性紫質 (photosensitizing porphyrins) 過量產生與累積所致。多數皮膚型紫質症(以 PCT 為代表)在曝曬陽光的皮膚區域造成慢性水疱與疤痕,而原紫質症 (protoporphyrias) 則對光線產生急性、嚴重、且多為非水疱性的反應,往往幾乎不留下慢性皮膚變化。急性紫質症 (acute porphyrias) 的特徵為神經學症狀,以及紫質前驅物 δ-胺基乙醯丙酸 (δ-aminolevulinic acid, ALA) 與膽色素原 (porphobilinogen, PBG) 的濃度升高。急性紫質症中紫質也會累積,有時血漿中濃度高到足以引起皮膚水疱,尤其以斑斕型紫質症 (variegate porphyria, VP) 為典型代表。在某些紫質症中,肝臟與腎臟等其他器官也可能受損。紫質症所引起的症狀、徵象與組織學表現皆無特異性,而紫質與紫質前驅物的模式則可達成特異性診斷與治療(表 124-2)。
歷史觀點 (HISTORICAL PERSPECTIVE)
已知對紫質症的首次描述是在 1874 年由 Schultz 提出。他描述了一名 33 歲男性,自 3 個月大起即有光敏感,並合併貧血、脾腫大與紅色尿液。1,2
T. McCall Anderson 於 1898 年描述了 2 名兄弟,他們在曝曬陽光的皮膚出現水疱、臉部廣泛疤痕與紅色尿液。3 這些病人被認為患有先天性紅血球生成性紫質症 (congenital erythropoietic porphyria, CEP),這是一種非常罕見且嚴重的皮膚型紫質症。急性紫質症最早由 Stokvis 於 1888 年描述,患者為一名年長女性,於服用磺胺 (sulphonal,一種與巴比妥類相關的鎮靜劑) 後出現症狀;她出現深紅色尿液,後來死亡。4
1923 年,Archibald Garrod 提出「先天性代謝異常 (inborn errors of metabolism)」一詞,用以描述包括紫質症在內的多種遺傳性代謝疾病。5
紫質症於 1954 年首次被分類為肝性或紅血球生成性。6 以放血 (phlebotomy) 治療遲發性皮膚紫質症由 Ippen 於 1961 年引入。7 1970 年,急性間歇性紫質症 (acute intermittent porphyria) 中首次描述了一種遺傳性酵素缺乏,8 而血基質 (hemin) 療法則於 1971 年首次用於此疾病。9 在隨後的數十年中,途徑中的各酵素及其基因被加以鑑定,並在每一種紫質症中發現多個突變。此外,血基質合成(尤其在肝臟與骨髓中)的調控也獲得更深入的理解。基於這些進展,更多治療方法被引入並持續開發中。
血基質的合成與功能 (HEME SYNTHESIS AND FUNCTIONS)
血基質 (heme),即鐵原紫質 IX (iron protoporphyrin IX),在真核細胞中經 8 個步驟合成,每一步皆由不同的酵素催化(圖 124-1)。這些酵素中的第一個與最後三個位於粒線體中,其餘 4 個則位於細胞質中。第一個酵素,δ-胺基乙醯丙酸合成酶 (δ-aminolevulinic acid synthase, ALAS),將甘胺酸 (glycine) 與琥珀醯輔酶 A (succinyl-coenzyme A) 結合,產生胺基酸 δ-胺基乙醯丙酸 (δ-aminolevulinic acid, ALA),又稱 5-胺基乙醯丙酸。接著兩分子 ALA 結合形成吡咯 (pyrrole),即膽色素原 (porphobilinogen, PBG)。四分子 PBG 連接形成線狀四吡咯 (linear tetrapyrrole),即羥甲基膽色烷 (hydroxymethylbilane, HMB)。HMB 是第四個酵素的受質,該酵素將 HMB 的其中一個吡咯環反轉並使分子環化,形成尿紫質原 III (uroporphyrinogen III),這是該途徑中的第一個紫質。此不對稱分子經歷一系列去羧基化與一次氧化,形成原紫質 IX (protoporphyrin IX)。接著鐵被嵌入,形成血基質。除了原紫質 IX 之外,此途徑中的紫質中間產物皆為其還原形式(即紫質原 porphyrinogens)。10
最終產物血基質由一個處於亞鐵(還原)狀態 (Fe2+) 的鐵原子與紫質大環的 4 個吡咯氮 (pyrrolic nitrogens) 結合而成(圖 124-1),留下 2 對未占據的電子對。血基質是許多必需血基質蛋白 (hemoproteins) 的輔基 (prosthetic group)。例如在血紅素 (hemoglobin) 中,一對未占據的電子對與球蛋白鏈的一個組胺酸 (histidine) 殘基配位,另一對則可與分子氧結合。Hemin 是血基質氧化形式的化學名稱,即鐵原紫質 IX (ferric protoporphyrin IX),它只有一個殘餘正電荷,可自血液及其他組織中以氯化物形式分離。Hemin 也泛指生物製劑,即凍乾血基質 (lyophilized hematin,血基質氫氧化物 heme hydroxide) 與血基質精胺酸鹽 (heme arginate),這些可供治療急性紫質症之用。約 85% 的血基質合成發生在骨髓中以支持血紅素形成,其餘大多在肝臟中,主要供應位於內質網的細胞色素 P450 酵素 (cytochrome P450 enzymes, CYPs)。11 所有其他組織則以較少量合成血基質。眾多其他重要血基質蛋白的例子包括肌紅素 (myoglobin)、粒線體呼吸鏈細胞色素、一氧化氮合成酶 (nitric oxide synthase) 與過氧化氫酶 (catalase)。肝臟中的血基質合成主要受 δ-胺基乙醯丙酸合成酶 1 (ALAS1,即 ALAS 的管家型,途徑中的第一個酵素) 的活性所調控,並由途徑最終產物血基質抑制此酵素的合成及其進入粒線體。在骨髓中,血基質與球蛋白的合成於紅血球生成素 (erythropoietin) 訊號傳遞期間緊密協調。ALAS2(ALAS 的紅血球特異型)及其他多個途徑酵素的表現,受血基質或鐵以及包括 GATA-1 與 NF-E2 在內的紅血球特異性順式作用元件 (cis-acting elements) 所刺激,最終以血基質合成最後一個酵素——亞鐵螯合酶 (ferrochelatase, FECH) 的磷酸化作為終點。12
表 124-1:人類紫質症的酵素分類總表
| 紫質症ª | 受影響的酵素 | 已知突變數 | 遺傳方式 | 分類 | 主要臨床特徵 |
|---|---|---|---|---|---|
| X 染色體連鎖原紫質症 (X-linked protoporphyria, XLP) | δ-胺基乙醯丙酸合成酶—紅血球特異型 (ALAS2) | 4(功能增益型) | 性聯遺傳 | 紅血球生成性 | 非水疱性光敏感 |
| δ-胺基乙醯丙酸脫水酶紫質症 (ALA dehydratase porphyria, ADP) | ALA 脫水酶 (ALAD) | 10 | 體染色體隱性 | 肝性ᵇ | 神經內臟型 |
| 急性間歇性紫質症 (acute intermittent porphyria, AIP) | 羥甲基膽色烷合成酶 (HMBS) | >400 | 體染色體顯性 | 肝性 | 神經內臟型 |
| 先天性紅血球生成性紫質症 (congenital erythropoietic porphyria, CEP) | 尿紫質原 III 合成酶 (UROS) | 48 | 體染色體隱性 | 紅血球生成性 | 神經內臟型 |
| 遲發性皮膚紫質症 (porphyria cutanea tarda, PCT) | 尿紫質原脫羧酶 (UROD) | 121(含 HEP) | 體染色體顯性ᶜ | 肝性 | 水疱性光敏感 |
| 肝紅血球生成性紫質症 (hepatoerythropoietic porphyria, HEP) | UROD | — | 體染色體隱性 | 肝性ᵇ | 水疱性光敏感 |
| 遺傳性糞紫質症 (hereditary coproporphyria, HCP) | 糞紫質原氧化酶 (CPOX) | 64 | 體染色體顯性 | 神經內臟型;水疱性光敏感(少見) | |
| 斑斕型紫質症 (variegate porphyria, VP) | 原紫質原氧化酶 (PPOX) | 174 | 體染色體顯性 | 肝性 | 神經內臟型;水疱性光敏感(常見) |
| 紅血球生成性原紫質症 (erythropoietic protoporphyria, EPP) | 亞鐵螯合酶 (FECH) | 189 | 體染色體隱性 | 紅血球生成性 | 非水疱性光敏感 |
ª紫質症按其受影響酵素於血基質生合成途徑中的順序排列。
ᵇ這些紫質症亦具有紅血球生成性特徵,包括紅血球鋅原紫質 (zinc protoporphyrin) 增加。
ᶜPCT 中的 UROD 抑制大多為後天性,但酵素的遺傳性缺乏會使罹患家族性(第 2 型)疾病的風險增加。
遲發性皮膚紫質症 (PORPHYRIA CUTANEA TARDA)
重點一覽 (AT-A-GLANCE)
■ 特徵為曝曬陽光皮膚區域的皮膚脆弱與水疱病灶
■ 由肝臟尿紫質原脫羧酶 (uroporphyrinogen decarboxylase, UROD) 活性受抑制所致
■ 此導致肝臟、血漿、尿液與糞便中出現過量、高羧基化紫質,呈現具診斷性的模式
■ 易感的遺傳因素可能包括 UROD 突變(異型合子,僅見於約 20% 的病例)與血色素沉著症 (hemochromatosis, HFE)
■ 後天易感因素(酒精、抽菸、續發性鐵質過載、慢性 C 型肝炎、HIV 與雌激素)常為多重併存
■ 在追蹤血清鐵蛋白 (serum ferritin) 的情況下,對反覆放血反應良好;或對低劑量羥氯奎寧 (hydroxychloroquine) 反應良好
表 124-2:人類紫質症的主要生化異常(粗體)與其他鑑別性檢測值ª
| 紫質症 | 紅血球 | 血漿 | 尿液 | 糞便 |
|---|---|---|---|---|
| ADP* | 鋅原紫質增加 | — | ALA 增加;PBG 與紫質濃度正常或略增 | 糞紫質 III 增加 |
| AIP | PBGD 通常降低§ | 紫質濃度正常或略增;螢光峰約 620 nmᵇ | ALA 與 PBG 增加 | ALA、PBG、糞紫質 III |
| CEP | 尿紫質 I;糞紫質 I | 尿紫質 I;糞紫質 I;螢光峰約 620 nmᵇ | 尿紫質 I;糞紫質 I | 糞紫質 I |
| PCT | 紫質濃度正常或略增 | 尿紫質、七羧基紫質;螢光峰約 620 nmᵇ | 尿紫質、七羧基紫質;ALA 增加 | 尿紫質、七羧基紫質、糞紫質 III、異糞紫質 (isocoproporphyrins) |
| HEP | 鋅原紫質;螢光峰約 620 nmᵇ | 尿紫質、七羧基紫質 | 尿紫質、七羧基紫質 | 同 PCT 模式 |
| HCP | 紫質濃度正常或略增 | 紫質正常或增加ᶜ;螢光峰約 620 nmᵇ | ALA、PBG、糞紫質 III | 糞紫質 III |
| VP | 紫質濃度正常或略增 | 原紫質通常增加;螢光峰約 626 nmᵇ | ALA、PBG、糞紫質 III | 糞紫質 III、原紫質 |
| EPP 與 XLP | **不含金屬原紫質 (metal-free protoporphyrin)**ᵈ | 原紫質;螢光峰約 634 nmᵇ | ALA、PBG、紫質正常 | 原紫質正常或中度增加 |
ªP 縮寫同表 124-1。
ᵇ稀釋血漿於中性 pH 的螢光發射峰。
ᶜ於有皮膚病灶時增加。
ᵈ不含金屬原紫質在 EPP 中約佔總量的 85%–100%,在 XLP 中約佔 50%–85%。
PCT 是最常見的紫質症,其特徵為皮膚脆弱以及手背及其他曝曬陽光皮膚區域出現慢性、水疱性病灶,通常發生於中年或晚年。13 其根本原因為肝細胞中尿紫質原脫羧酶 (UROD) 活性不足,使得尿紫質原與其他高羧基化紫質原於肝細胞中累積,並被氧化為相對應的紫質。在活動性病例中,肝臟 UROD 活性降至正常的約 20% 以下。PCT 是一種與鐵相關的疾病,僅在肝臟鐵量正常或增加的情況下發生。多重易感因素促成肝細胞中鐵質累積、氧化壓力,以及一種 UROD 抑制物的生成,在個別病人身上辨識出這些因素相當重要。這是唯一一種可在受影響酵素無突變的情況下發生的紫質症。14 約 20% 的病人帶有異型合子的 UROD 突變,但在缺乏其他易感因素時,這些突變並不會導致 PCT。PCT 是最易於治療的紫質症,對放血或低劑量羥氯奎寧反應良好。但此疾病必須先與其他較不常見、會造成相同皮膚病灶但對這些治療無反應的紫質症加以區別。
臨床特徵 (CLINICAL FEATURES)
PCT 通常發生於人生的第四或第五個十年,以男性最為常見。充滿液體的水疱與大疱尤其見於手背(圖 124-2,身體最常曝曬陽光的區域),且常因皮膚脆弱性增加而於輕微或不明顯的創傷後發生。水疱也可能出現於前臂、臉部、耳朵、頸部、腿部與足部。這些水疱容易破裂,留下糜爛 (erosions),可能變得乾燥結痂並緩慢癒合(圖 124-2)。皮膚糜爛區域容易發生細菌感染。隨病程延長,殘留的疤痕與色素過度沉著及色素減退會相當顯著。粟粒疹 (milia) 可能在水疱形成之前或之後出現。臉部多毛 (hypertrichosis) 與色素過度沉著(圖 124-3)很常見,甚至可能在無水疱的情況下發生,並造成美觀上的困擾,尤其是女性。16 受影響皮膚區域的嚴重增厚,有時伴隨鈣化,可類似於全身性硬化症 (systemic sclerosis),稱為假性硬皮病 (pseudoscleroderma)。急性紫質症特徵性的神經學症狀在 PCT 中並不出現。罕見的兒童病例常與 UROD 突變及癌症化學治療相關15(圖 124-3)。此疾病可能於懷孕期間發生,可能與雌激素增加的影響有關。與皮膚型及全身性紅斑性狼瘡 (lupus erythematosus) 的相關性報告原因不明。與末期腎臟疾病相關的 PCT 通常更為嚴重,因為紫質的尿液排泄受損,且紫質難以透析清除。由此產生的血漿紫質濃度可等同於先天性紅血球生成性紫質症 (CEP) 所見的濃度,並可能伴隨嚴重的細菌感染與毀損 (mutilation)。17
幾乎所有病例都可發現肝功能檢查的輕度異常。新鮮肝臟組織在暴露於長波紫外光下呈現強烈紅色螢光(圖 124-4),反映紫質的大量累積。肝臟組織病理學無特異性,通常包括鐵質增加、脂肪增加、肝細胞壞死與發炎,在許多病例中反映出酒精或 C 型肝炎感染的影響。在 PCT 發病初期,肝硬化並不常見。肝細胞癌 (HCC) 的風險增加,尤其在病程較長的情況下,部分原因可能在於併存的易感因素本身即可造成慢性肝損傷與纖維化。18-20
易感因素 (SUSCEPTIBILITY FACTORS)
PCT 是一種多因子疾病,有許多常見的易感因素促成此疾病,但沒有任何一個因素存在於每位病人身上。這些因素包括遺傳因素、病毒感染與化學物質暴露。病人幾乎總是同時具有多個此類因素,且這些因素還具有其他健康意涵。21 例如,在美國一個包含 143 名 PCT 病人的大型系列研究中,最常見的易感因素為飲酒 (87%)、抽菸 (81%)、慢性 C 型肝炎 (69%) 與 HFE(血色素沉著症)突變 (53%)。22 這些因素在一般族群中很常見,但其本身並不會導致 PCT 發生。未來可能還會辨識出更多的易感因素。
UROD 突變 (UROD MUTATIONS)
多數 PCT 病人並無 UROD 突變,被稱為散發型(第 1 型)疾病。約 20% 的病人帶有異型合子的易感性 UROD 突變,被歸類為家族性(第 2 型)PCT。此類突變以體染色體顯性方式遺傳,但外顯率低,因此多數第 2 型病人以散發方式表現,並無已知的 PCT 親屬。帶有此類突變是一種易感因素,在缺乏其他後天或遺傳易感因素時並不會導致 PCT。HEP 是第 2 型 PCT 的同型合子形式,臨床上類似於 CEP。在第 2 型 PCT 與 HEP 中已辨識出至少 100 種不同的 UROD 基因突變,大多為發生於 1 或少數幾個家族中的錯義突變 (missense mutations)(表 124-1)。第 3 型指的是罕見的家族,其中有超過 1 名受影響的個體但無 UROD 突變。所有 3 種 PCT 型別在臨床上完全相同,不過第 2 型的發病有時較早。23
鐵與血色素沉著症基因 (HFE) 突變 (IRON AND HEMOCHROMATOSIS GENE [HFE] MUTATIONS)
PCT 中鐵儲存量總是正常或增加,而鐵缺乏則具有保護作用。鐵在肝細胞中提供氧化環境並促進 UROD 抑制物的生成,但其本身並不直接抑制 UROD。HFE 基因的 C282Y 突變是白人血色素沉著症的主要致病突變,在散發型與家族型 PCT 中的盛行率皆高於未受影響的個體。多達 10% 至 20% 的病人可能是 C282Y 同型合子,而這些病人可能會經歷較早的發病。23,24 在南歐,C282Y 較不普遍,H63D 突變則與 PCT 較常相關。25 HFE 突變損害對血清鐵的感應,從而減少肝臟鐵調素 (hepcidin) 的生成。由於循環中鐵調素濃度不適當地偏低,鐵調素對腸細胞鐵轉運蛋白 (ferroportin) 的下調作用受損,導致腸道鐵吸收不適當地偏高。在無 HFE 突變的 PCT 病人中,肝臟鐵調素表現也降低,因為某些其他易感因素會降低此荷爾蒙的表現,如下所述。26
酒精 (ALCOHOL)
PCT 長期以來即與酒精濫用相關。酒精及其代謝產物可能透過誘導肝臟 ALAS1 與 CYPs、產生活性氧物種、造成粒線體損傷、脂質沉積、耗竭還原型麩胱甘肽 (glutathione) 與其他抗氧化防禦、增加內毒素生成及活化庫佛細胞 (Küpffer cells),而使 PCT 易於發病。攝取酒精也可降低鐵調素表現。26
抽菸與細胞色素 P450 酵素 (SMOKING AND CYTOCHROME P450 ENZYMES)
抽菸經常與飲酒併存,但被視為 PCT 的一個獨立危險因子。21 抽菸可能增加肝細胞中的氧化壓力並誘導肝臟 CYPs,包括在 PCT 嚙齒類動物模型中對尿紫質症 (uroporphyria) 發展很重要的 CYP1A2。27,28 人類 PCT 中肝臟 CYPs 常增加,29 但尚不清楚哪些 CYPs 促成此疾病。然而,一種較易被誘導的 CYP1A2 變異型,被發現在 PCT 中比在正常受試者中更為常見。30,31
雌激素 (ESTROGENS)
雌激素使用在 PCT 女性中很常見。21,32,33 此疾病也曾發生於一些因前列腺癌而接受雌激素治療的男性。32 雌性大鼠或接受雌激素的雄性大鼠,比未經處理的雄性大鼠更容易發生化學誘導的尿紫質症。34 此易感性的機轉並不確定,但可能續發於活性氧物種的生成。14,35
C 型肝炎 (HEPATITIS C)
C 型肝炎促進肝細胞脂肪變性、鐵質累積、粒線體功能障礙、氧化壓力與鐵調素表現的失調。36,37 PCT 中慢性 C 型肝炎的盛行率在不同國家介於 21% 至 92%,且總是超過一般族群中此病毒感染的盛行率。估計只有 0.05% 的慢性 C 型肝炎個體會發展為 PCT。38 不同國家 PCT 中此病毒感染盛行率的巨大差異,反映出此感染在高風險族群中盛行率的顯著地理變異。
HIV (HIV)
PCT 與 HIV 感染的相關性較不常見,HIV 感染可伴有或不伴有 HCV 共同感染。39 PCT 可為 HIV 感染的初始表現。其與 HIV 感染相關的機轉以及與特定抗反轉錄病毒療法可能的關係尚未確立。
抗氧化劑 (ANTIOXIDANTS)
在某些 PCT 病人中已注意到血漿抗壞血酸 (ascorbate) 與類胡蘿蔔素 (carotenoids) 濃度降低。40-42 抗壞血酸缺乏的嚙齒類動物更容易發生尿紫質症。43
化學物質暴露與藥物 (CHEMICAL EXPOSURE AND DRUGS)
1950 年代土耳其東部曾發生一次大規模 PCT 爆發,當時正值糧食短缺,民眾食用了以殺真菌劑六氯苯 (hexachlorobenzene) 處理過的種用小麥。44 較小規模的爆發與個別病例則發生於暴露於其他化學物質後,例如 2,3,7,8-四氯雙苯并戴奧辛 (TCDD, dioxin)。45 這些及相關化學物質會在實驗動物與培養的肝細胞中造成 PCT 的生化特徵。10,14
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
當肝臟 UROD 活性降至正常的約 20% 時,PCT 即會發生。14 在酵素受抑制的情況下,肝臟中 UROD 蛋白的量仍維持在其基因決定的水平。46,47 如前所述,約 20% 的病人為 UROD 突變的異型合子,因其肝臟(及其他組織)中的 UROD 活性自出生起即為正常的一半,故較易發展為 PCT。除非肝臟中的 UROD 活性進一步降低,否則這些個體不會表現出 PCT。一種被鑑定為尿紫質甲烯 (uroporphomethene) 的 UROD 抑制物,是在一個會自發產生 PCT 生化特徵的小鼠模型中被辨識出來。此抑制物是尿紫質原部分氧化的產物,可能由 1 種或多種 CYPs 所生成(圖 124-5)。49
UROD 依序將尿紫質原 I 與 III(各帶有 8 個羧基側基)脫羧為糞紫質原 I 與 III(各帶有 4 個羧基側基)。兩種異構物皆為 UROD 的受質,但較偏好尿紫質原 III。(途徑中的下一個酵素僅以糞紫質原 III 為受質,因此尿紫質原 I 與糞紫質 I 並非血基質前驅物。)當肝臟 UROD 受抑制時,尿紫質原 I 與 III 及反應中的中間產物(即七羧基、六羧基與五羧基紫質原的 I 與 III 異構物)會以相對應的氧化型紫質形式累積,最終經血漿運送至皮膚而引起光敏感,並運送至腎臟排泄。膽汁與糞便中的紫質也增加。處於氧化狀態的紫質呈紅色、具螢光且具光敏性,而紫質原則不具這些特性。氧化型紫質的多環芳香族結構含有非定域電子 (delocalized electrons),在暴露於紫光(波長約 410 nm)時其能階升高。此能量可以較高波長以紅色螢光的形式釋放,或轉移至分子氧,形成具反應性的單態氧 (singlet state oxygen) 與其他活性氧物種。這些物質可能損害皮膚的細胞組成,或造成肥大細胞去顆粒化與補體活化。50,51 對皮膚表皮下層的損傷使皮膚變得脆弱且易於形成水疱。50,51
診斷 (DIAGNOSIS)
PCT 最常發生於成年男性,並與過量飲酒、抽菸與慢性 C 型肝炎等因素相關;在女性則特別與雌激素使用相關。其表現通常具特徵性,但必須記住,患有斑斕型紫質症 (VP)、遺傳性糞紫質症 (HCP) 或假性紫質症 (pseudoporphyria) 的成人,以及患有先天性紅血球生成性紫質症 (CEP) 或肝紅血球生成性紫質症 (HEP) 的兒童或成人,都可能以相同的皮膚病灶表現。因此,在開始治療前建立 PCT 的實驗室診斷至關重要。PCT 的第一線檢測(即篩檢)為測量血漿或尿液中的總紫質。結果正常可能提示假性紫質症的診斷。由於紫質升高(尤其是尿液中)在肝臟疾病與其他醫學狀況中很常見,因此發現紫質增加本身並不足以支持紫質症的診斷。因此,若第一線檢測為陽性,建議進行以下檢查以建立診斷並排除其他較不常見、可造成類似皮膚表現且常被誤診為 PCT 的紫質症:
■ 稀釋血漿於中性 pH 下的螢光掃描。52 最常被誤診為 PCT 的紫質症是 VP,53 可藉由血漿掃描迅速且可靠地辨識出來,其發射峰約在 626 nm,與 PCT 及其他水疱性皮膚紫質症的約 620 nm 形成對比。
■ 尿液或血漿紫質的分餾 (fractionation),在 PCT 中將顯示以尿紫質以及七羧基、六羧基與五羧基紫質為主。此種以高羧基化紫質為主的模式並非完全具診斷性,因為它也可能發生於其他遠較少見、且尤其在成人發病時可能被誤診為 PCT 的紫質症中。
■ 紅血球總紫質,在 PCT 中正常或中度升高,但在罕見的 CEP、HEP 或同型合子 HCP 或 VP 病例中則顯著升高。這些疾病通常於嬰兒期表現,但也可能首次於成人期顯現,有時與克隆性骨髓增生 (myeloproliferative) 或骨髓化生不良 (myelodysplastic) 疾病相關。
■ 糞便紫質在 PCT 中可能正常或中度增加,呈現包含異糞紫質 (isocoproporphyrins) 的複雜模式。這些非典型的四羧基紫質,是在 UROD 受抑制導致五羧基紫質原於肝臟累積、並被途徑中的下一個酵素 CPOX 過早脫羧形成去氫異糞紫質原 (dehydroisocoproporphyrinogen) 時所形成。後者經膽汁排泄,並被腸道細菌氧化為異糞紫質。54 與 PCT 相反,糞便紫質在 CEP、HCP 與 VP 中顯著升高,其中 CEP 以糞紫質 I 為主、HCP 以糞紫質 III 為主、VP 則以糞紫質 III 與原紫質兩者為主。
■ 尿液 ALA 在 PCT 中正常或中度增加,而 PBG 則總是正常。這些紫質前驅物的濃度在急性肝性紫質症(AIP、HCP 與 VP)中可能升高。
對於患有進行性腎臟疾病的病人,血漿紫質增加並證明以高羧基化紫質為主,對於診斷 PCT 至關重要,不過此族群的參考範圍較高。55 進行性腎臟疾病常與紅血球生成改變相關,從而導致紅血球鋅原紫質增加,但這不應歸因於 PCT 或其他紫質症。患有 AIP 與末期腎臟疾病的病人有時會以類似 PCT 的水疱性皮膚病灶表現。56
雖然診斷 PCT 並不需要皮膚切片,但切片會顯示特徵性但無特異性的表現,例如表皮下水疱形成、血管周圍過碘酸-希夫染色 (periodic acid–Schiff) 陽性物質的沉積,以及真皮上層與真皮表皮交界處的細纖維狀物質沉積。也可發現免疫球蛋白與補體的沉積。57 這些組織學變化也見於其他皮膚型紫質症以及假性紫質症——後者是一種了解尚不充分的狀況,表現出與 PCT 極為相似的病灶,但血漿紫質並無顯著增加。58
易感因素的辨識 (IDENTIFICATION OF SUSCEPTIBILITY FACTORS)
所有 PCT 病人都應就以下易感因素進行詢問或檢查,其中部分因素是可改變的:飲酒與雌激素使用、抽菸、C 型肝炎與 HIV 感染,以及 HFE 與 UROD 突變。會使急性紫質症惡化的藥物使用很少與 PCT 有關。雖然家族性(第 2 型)PCT 病例可藉由紅血球 UROD 活性為正常的一半來辨識,但 UROD 突變分析更為可靠。這種對易感因素的完整分析有助於解釋個別病例的疾病,並可能影響治療處置。此外,其中部分因素本身也具有醫學意涵。應測量血清鐵蛋白,其結果可能影響治療的選擇。
治療 (THERAPY)
以放血或低劑量羥氯奎寧治療,對散發型與家族型 PCT 皆高度有效。這些治療應僅在診斷確定後才開始,因為它們對其他紫質症無效。在血漿紫質檢測結果(包括螢光掃描52)與 PCT 一致並已排除 VP 與假性紫質症之後,開始治療可能是合理的。應建議病人戒菸及戒酒。若可能,應停用雌激素以及已知會誘導肝臟血基質合成的藥物。若有需要,可在 PCT 緩解後考慮使用雌激素補充療法,最好採經皮 (transdermal) 給藥。59 應確保攝取足量的抗壞血酸與其他營養素。去除 1 個或多個易感因素可帶來改善,但反應緩慢或難以預測。60
反覆放血以降低肝臟鐵質是多數機構偏好的治療。每 2 週移除 450 mL 血液,並以血清鐵蛋白濃度為指引,目標為 15 至 20 ng/mL(即接近正常下限)。每次放血時測量血比容 (hematocrit) 與鐵蛋白,可進行監測以預防症狀性貧血並評估朝目標鐵蛋白濃度的進展。當前次回診的鐵蛋白為 25 至 30 ng/L 時即停止放血,並測量鐵蛋白以確認達到目標濃度。此時,紫質濃度可能尚未完全恢復正常,皮膚病灶也未完全消退。額外的鐵質耗竭並無益處且會導致貧血。多數病人需要 6 至 8 次放血才能達到生化與臨床緩解,但可能還需要額外的療程,尤其當基線血清鐵蛋白濃度顯著升高時。血漿(或血清)紫質濃度的下降往往落後於血清鐵蛋白,但在放血完成後數週內將恢復正常。61,62 皮膚的脆弱性在紫質濃度正常後可能仍持續一段時間,直到皮膚表皮下層的癒合與修復完成為止。之後通常不需再放血,但 C282Y HFE 突變的同型合子或複合異型合子病人例外,因為目前的血色素沉著症指引建議將鐵蛋白濃度維持在 50 至 100 ng/mL 之間。63 PCT 可能復發,通常與恢復飲酒有關,但對重新治療通常有反應。緩解後追蹤紫質濃度可早期偵測復發,以便迅速重啟治療。
低劑量羥氯奎寧(或氯奎寧)療程是放血的有效替代方案,也是某些機構偏好的方法。14,64-70 這些 4-胺基喹啉類抗瘧藥似乎不會耗竭肝臟鐵質,而是動員累積於肝細胞溶酶體 (lysosomes) 及其他細胞內胞器中的紫質。這些藥物的足量治療劑量會迅速動員紫質並誘發急性肝炎,特徵為發燒、倦怠、噁心,以及尿液與血漿紫質與光敏感的顯著增加,但隨後即進入緩解。71 曾有報告指出,使用氯奎寧作為瘧疾預防時會揭露先前未被辨識的 PCT。72 建議採低劑量療程(羥氯奎寧 100 mg 或氯奎寧 125 mg〔半顆標準錠劑〕每週兩次)以達成緩解並避免這些藥物足量劑量的副作用。64,67,68 治療持續至血漿或尿液紫質恢復正常至少一個月為止。反應不佳的病人可能需要以放血進行替代治療。71 這些藥物與一個小的視網膜病變 (retinopathy) 風險相關,73 此風險在使用羥氯奎寧時可能較低。因此,病人應在治療前接受眼科醫師的篩檢。74 比較放血與羥氯奎寧(100 mg 每週兩次)治療的研究顯示,達到緩解(血漿紫質濃度正常)的時間在這兩種治療間相當。70 以鐵螯合劑(如去鐵胺 desferrioxamine)治療在移除鐵質方面的效率遠不及放血,但若放血與低劑量羥氯奎寧皆為禁忌時,可加以考慮。75
以干擾素 (interferon) 為基礎的療程治療 C 型肝炎曠日費時且常不成功。因此我們會先治療 PCT(通常症狀較明顯),在達成緩解後,再治療併存的 C 型肝炎。以干擾素為基礎的治療療程常造成貧血,這通常排除了以放血治療 PCT 的可能,且有時與先前已治療之 PCT 的復發相關。76
在 C 型肝炎同時治療期間使用低劑量羥氯奎寧的經驗有限。新型直接作用抗病毒藥物 (direct-acting antivirals) 可迅速且可靠地治療此感染,目前正進行研究以蒐集證據,判斷在與 C 型肝炎相關的 PCT 中,是否應以其取代放血或低劑量羥氯奎寧作為初始治療。76
與末期腎臟疾病相關的 PCT 通常難以治療。若在需要時藉由開始使用或增加紅血球生成素 (erythropoietin) 劑量加以支持,放血是有效的。17,77,78 高通量血液透析 (high-flux hemodialysis) 可能提供部分益處。79 腎臟移植可帶來緩解,推測是因為恢復了內源性紅血球生成素的產生並降低了鐵調素濃度。80
肝細胞癌的定期篩檢應包括以腹部超音波或電腦斷層進行的肝臟影像檢查。如同其他肝臟疾病,這應以肝硬化或肝纖維化的證據為指引,後者可藉由肝臟切片或間接以彈性造影 (elastography) 評估,並考量其他肝臟疾病病因的存在。
肝紅血球生成性紫質症 (HEPATOERYTHROPOIETIC PORPHYRIA)
重點一覽 (AT-A-GLANCE)
■ HEP 是家族性(第 2 型)PCT 的同型合子形式,UROD 兩個對偶基因皆突變,導致 UROD 活性嚴重不足
■ HEP 類似於 CEP,在曝曬陽光皮膚區域出現水疱病灶,通常始於兒童期
■ 生化發現類似 PCT,但紅血球鋅原紫質有顯著升高
■ 病人應避免日曬。以放血或低劑量羥氯奎寧治療無效。
這種非常罕見的紫質症是家族性(第 2 型)PCT 的同型合子(或複合異型合子)形式,其特徵為因慢性、嚴重光敏感所致的水疱性皮膚病灶。
臨床特徵 (CLINICAL FEATURES)
HEP 通常於幼兒早期表現出水疱性皮膚病灶、多毛、疤痕、溶血性貧血與紅色尿液。硬皮病樣 (sclerodermoid) 皮膚變化有時相當顯著。其皮膚表現類似 PCT 所見,但通常嚴重得多。曾有報告罕見的輕症病例以及成年期發病的案例。81
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
HEP 病人從父母雙方各遺傳到一個 UROD 突變。已辨識出眾多與 PCT 或 UROD 相關的 UROD 突變。HEP 中的紅血球 UROD 活性為正常的 5% 至 30%。由於 UROD 無效對偶基因 (null allele) 的同型合子具致死性,因此至少其中一個突變的 UROD 對偶基因必須能產生一些具催化活性的酵素蛋白。真核細胞中的表現研究顯示,某些突變可能以組織特異性的方式使酵素蛋白不穩定。82 過量的紫質主要在肝臟中生成。鋅原紫質在骨髓中累積,並在紅血球中顯著升高。
診斷 (DIAGNOSIS)
HEP 的生化發現類似 PCT,以尿紫質、七羧基紫質與異糞紫質的累積與排泄為主。與 PCT 相反,HEP 中紅血球鋅原紫質顯著增加。紅血球 UROD 活性顯著降低,且分子研究應能證明影響 UROD 兩個對偶基因的突變。
治療 (THERAPY)
應建議 HEP 病人避免日曬。放血與羥氯奎寧顯示出極少或毫無益處。83 病人應避免已知在 PCT 中很重要的易感因素。在一個與紅血球生成異常 (dyserythropoiesis) 相關的嚴重病例中,口服活性碳 (oral charcoal) 有所助益,可能是由於將紫質困於腸腔內並防止其進行腸肝循環。84 反轉錄病毒介導的基因轉移研究顯示,未來透過基因置換療法矯正 HEP 缺陷可能可行。85
先天性紅血球生成性紫質症 (CONGENITAL ERYTHROPOIETIC PORPHYRIA)
重點一覽 (AT-A-GLANCE)
■ 由於尿紫質原 III 合成酶 (uroporphyrinogen III synthase, UROS) 兩個對偶基因皆突變,導致 UROS 活性嚴重喪失以及尿紫質 I 與糞紫質 I 升高
■ 特徵為影響曝光區域的表皮下疤痕與大疱性病灶
■ 診斷藉由紅血球、尿液、血漿與糞便中尿紫質 I 與糞紫質 I 的顯著升高以及致病突變的辨識來建立
■ 在年幼時進行造血幹細胞移植具潛在治癒性
■ 嚴格避光可預防慢性、毀容性的皮膚疾病
CEP(又稱 Günther 病)的病例最早於 1874 年與 1898 年被描述。3 CEP 係由尿紫質原 III 合成酶 (UROS) 的顯著缺乏所致,導致異構物 I 型紫質原(尤其是尿紫質原 I 與糞紫質原 I)於血紅素合成期間在骨髓中累積。紫質通常在紅血球、血漿與尿液中顯著升高,並造成慢性、嚴重的光敏感。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
UROS 催化在細胞質中由羥甲基膽色烷 (HMB) 形成尿紫質原 III(圖 124-1)。此反應涉及將線狀四吡咯的 4 個吡咯之一(D 環)反轉,並形成側鏈呈不對稱排列的紫質大環。在缺乏此酵素時,HMB 會自發環化形成對稱的尿紫質原 I。途徑中的下一個酵素 UROD 會將尿紫質原 I 與 III 分別代謝為糞紫質原 I 與 III。然而,糞紫質原 I 是一個死路產物 (dead-end product),因為血基質生合成途徑中的下一個酵素僅接受糞紫質原 III 為受質。已辨識出超過 48 種 UROS 基因突變,較嚴重的突變與較嚴重的臨床表現相關。86 在一個與 β-地中海型貧血 (beta-thalassemia) 相關的病例中描述的 GATA-1 突變顯示,血基質生合成途徑以外的基因缺陷也可導致 CEP。87
CEP 中骨髓的有核紅血球 (normoblasts) 因紫質累積而呈現顯著的顯微螢光。88 循環紅血球中紫質濃度增加,在真皮微血管中暴露於光線而造成細胞損傷。溶血以及脾臟對受損細胞攝取增加,導致脾腫大成為 CEP 的常見發現。骨髓中無效的紅血球生成促成貧血。過量紫質經紅血球與血漿運送至皮膚,導致慢性且通常嚴重的光敏感。
臨床特徵 (CLINICAL FEATURES)
CEP 的特徵為影響曝光區域(如手背、手指與臉部)的表皮下大疱性病灶(圖 124-6),常導致疤痕以及色素過度沉著與色素減退區域。其皮膚病灶類似 PCT 所見,但因紫質濃度高得多,故造成更顯著的色素過度沉著、疤痕、攣縮,以及手指與臉部特徵的喪失。較輕的疾病常被誤診為 PCT,而罕見的成人發病可能與骨髓化生不良或骨髓增生疾病相關,其中帶有 UROS 突變的紅血球前驅細胞克隆擴增。90
此疾病可能在子宮內即被辨識為胎兒水腫 (fetal hydrops) 的病因。91 多數病例中,光敏感在出生後不久即被注意到。紅牙症 (erythrodontia,牙齒的棕色染色) 是由於紫質於子宮內沉積於發育中的牙齒所致。紫質也沉積於骨骼中。92 因骨髓擴張以及因避光所致的維生素 D 缺乏,可能導致病理性骨折與其他骨骼異常。因血管內溶血與無效紅血球生成所致的貧血可能很嚴重,需要反覆輸注紅血球。未矯正的貧血可進一步刺激紅血球生成,並促成紫質產生增加。周邊血液抹片顯示多染色性 (polychromasia)、異形紅血球症 (poikilocytosis)、紅血球大小不均 (anisocytosis) 與紅血球嗜鹼性點彩 (basophilic stippling),以及網狀紅血球與有核紅血球的增加。CEP 中肝臟與神經系統不受影響。一名患有 CEP 的母親成功生下一名健康、未受影響但帶有紅牙症的嬰兒,係因紫質穿過胎盤所致。93
診斷 (DIAGNOSIS)
CEP 可藉由測量羊水或胎兒血液中大量的紫質而在子宮內被辨識出來。CEP 常於出生後不久,當注意到尿布呈粉紅至棕色染色、且在長波紫外光下呈現紅色螢光時被診斷出來。尿液、紅血球與血漿紫質顯著增加,以尿紫質 I 與糞紫質 I 為主。糞便紫質顯著增加,且主要為糞紫質 I。原紫質 IX 有時為紅血球中主要的紫質,尤其在較輕的病例中。用以辨識致病突變的 DNA 研究,對於確認診斷、遺傳諮詢以及後續懷孕的產前診斷很重要。
治療 (THERAPY)
需要採取個別化、多專科的方法。99 應建議病人避免日曬、皮膚創傷與感染,以避免嚴重疤痕以及臉部特徵與手指的喪失。吸收紫外線 A 與 B 的局部防曬劑助益甚微,因為有害的光線大多為可見光;口服 β-胡蘿蔔素亦無效。94
病人可能因嚴重貧血而需要紅血球輸注,95 並需鐵螯合劑以避免鐵質過載的後遺症。96 亦可考慮使用羥基脲 (hydroxyurea) 以減少紅血球生成與紫質產生。97 若脾功能亢進 (hypersplenism) 促成顯著貧血、白血球減少或血小板減少,脾臟切除可能有益。在一名病人身上,抗壞血酸與 α-生育酚 (α-tocopherol) 改善了貧血。98 在另一名病人身上,口服活性碳據報有益。84 在一個大型病例系列中,使用口服活性碳、脾臟切除與慢性高量輸血與多種併發症相關,並無明顯臨床益處,且對健康相關生活品質有負面影響。99
造血幹細胞移植具治癒性,是患有嚴重疾病的年輕病人的首選治療。100 成功的移植可帶來顯著的臨床改善與紫質濃度的降低。目前正在探索使用反轉錄病毒與慢病毒 (lentiviral) 載體及造血幹細胞於 CEP 病人的基因療法。101,102
原紫質症 (PROTOPORPHYRIAS)
重點一覽 (AT-A-GLANCE)
■ EPP 是第三常見的紫質症,也是兒童中最常見的紫質症
■ 它源自亞鐵螯合酶 (FECH) 兩個對偶基因的突變;一個帶有嚴重突變,而在多數家族中另一個帶有族群中常見的易感性改變
■ XLP 較不常見,源自 ALAS2 的功能增益型突變
■ 兩者皆以急性、多為非水疱性的皮膚光敏感以及肝損傷風險為特徵。
■ 診斷藉由紅血球原紫質的顯著升高來建立,其中以不含金屬原紫質(而非鋅原紫質)為主
■ 治療處置強調避光與光防護
紅血球生成性原紫質症 (erythropoietic protoporphyria, EPP) 是兒童中最常見的紫質症類型,在成人中為第三常見,報告的盛行率介於每 100,000 人 0.5 至 1.5 例之間。103-105
它源自亞鐵螯合酶 (FECH)(血基質生合成途徑的最後一個酵素)的功能喪失型突變。但原紫質症的表型也可由其他基因的突變所致。X 染色體連鎖原紫質症 (X-linked protoporphyria, XLP) 源自 ALAS2(途徑中第一個酵素的紅血球特異型)的功能增益型突變,約佔原紫質症病例的 5%。106 伴有 ALAS2 活性增加的原紫質症也可源自粒線體 CLPX 的突變。107 除罕見例外,所有原紫質症的症狀皆於幼兒早期開始,表現為急性、非水疱性的皮膚光敏感。原紫質症性肝病 (protoporphyric hepatopathy) 是一種潛在致命的併發症,發生於不到 5% 的病人。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
EPP 由亞鐵螯合酶 (FECH) 的突變所致,FECH 催化將鐵嵌入原紫質 IX,即血基質生合成途徑的最後一步(圖 124-1)。FECH 缺乏導致受質原紫質的累積,尤其在骨髓網狀紅血球 (reticulocytes) 中。EPP 的遺傳曾被描述為帶有可變外顯率的體染色體顯性。但自從發現需要 FECH 兩個對偶基因皆突變後,現在最好將其描述為體染色體隱性。在多數家族中,一個嚴重的 FECH 突變(其中已描述至少 189 種)與一個低表現(次型 hypomorphic)變異對偶基因 (IVS3-48C/T) 互為反式 (trans)。108-110 低表現對偶基因中 DNA 序列的改變導致一個異常剪接位點的使用增加,產生一種更易降解的 mRNA,並使野生型 FECH mRNA 的穩態濃度降低。110 此 FECH 對偶基因出現於 10% 的白人,且其本身並無表型。它在其他族群中的頻率差異甚大;在東亞人中盛行率較高,但在非洲非常低,這解釋了 EPP 盛行率的地理差異。103-110
曾描述帶有 2 個嚴重 FECH 突變(從父母雙方各遺傳 1 個)但無低表現對偶基因的病例。至少 1 個對偶基因必須能合成足夠的 FECH 酵素以充分合成血基質。這些病例有時與季節性掌部角皮症 (seasonal palmar keratoderma)、神經學症狀、紅血球原紫質低於預期的增加,以及無肝功能障礙相關。111
EPP 的成年期發病病例曾於患有骨髓增生或骨髓化生不良症候群的病人中被描述,其中帶有 FECH 突變的造血細胞克隆擴增。112,113 例如,一名患有骨髓增生疾病的病人,因帶有 FECH 缺失且與一個次型 FECH 對偶基因互為反式的紅血球前驅細胞克隆擴增,而發展出嚴重的 EPP,並死於 EPP 誘發的肝臟疾病。114
在某些缺乏 FECH 突變的原紫質症病人中,X 染色體連鎖的遺傳模式導致發現了 ALAS2 的功能增益型突變(ALAS2 是血基質生合成途徑中唯一位於 X 染色體上的酵素編碼基因)。106 這些突變導致 ALAS 蛋白的酵素活性增加約 3 倍以及原紫質 IX 的累積,這顯示 FECH 活性是紅血球血基質合成中的下一個速率限制步驟。106 法國一個家族中最近發現有 2 名成員患有原紫質症,但無 FECH 或 ALAS2 突變,而是帶有以體染色體顯性方式遺傳的 CLPX 功能喪失型突變,導致 ALAS2 的降解減少以及 ALA 與原紫質的過度產生。107
在 EPP 中,循環紅血球中發現的過量原紫質主要為不含金屬,而非與鋅複合,這與許多其他會增加紅血球原紫質(大多為鋅原紫質)的狀況(如鉛中毒、鐵缺乏、慢性病貧血、溶血狀態)形成對比。血漿中過量的不含金屬原紫質源自血紅素合成活躍的骨髓以及循環紅血球,被肝細胞攝取,排泄至膽汁與糞便中,並可能進行腸肝再循環。原紫質具疏水性,因為它只擁有 2 個羥基,且不經尿液排泄。XLP 以及因 CLPX 突變所致原紫質症的紅血球,含有增加量的不含金屬原紫質與鋅複合原紫質。紫質受紫外光激發會產生自由基與單態氧,115 在 EPP 中可導致補體活化、脂質過氧化、116 膜蛋白交聯,117 與多形核細胞趨化,造成皮膚病理變化。118 皮膚組織病理學的非特異性發現包括乳頭狀真皮中增厚的微血管壁,其周圍有無定形玻璃樣與過碘酸-希夫 (PAS) 陽性黏多醣、補體與免疫球蛋白的沉積。119 基底膜異常較其他形式的皮膚型紫質症不明顯。120
原紫質症性肝病發生於不到 5% 的病人。它可能造成肝功能檢查的慢性異常並進展為肝硬化,或急性表現並快速進展而需要緊急肝臟移植。其他肝損傷病因(如酒精性或非酒精性脂肪肝炎或病毒性肝炎)可藉由損害原紫質排泄、從而將循環原紫質增加至加速肝臟損害的濃度,而使病人易於發生此狀況。過量原紫質具膽汁鬱積性,在肝細胞中形成結晶結構,並損害粒線體功能。121,122 累積的原紫質可能以棕色色素出現於肝細胞、庫佛細胞與膽小管中,並在偏光顯微鏡下以呈「馬爾他十字 (Maltese cross)」外觀的雙折射內含物出現。123
表 124-3:32 例紅血球生成性原紫質症的臨床表現
| 症狀與發現 | 發生率(佔總數百分比) |
|---|---|
| 灼燒感 (Burning) | 97 |
| 水腫 (Edema) | 94 |
| 搔癢 (Itching) | 88 |
| 紅斑 (Erythema) | 69 |
| 疤痕 (Scarring) | 19 |
| 水疱 (Vesicles) | 3 |
| 貧血 (Anemia) | 27 |
| 膽石症 (Cholelithiasis) | 12 |
| 肝功能檢查異常 | 4 |
資料來自 Bloomer J, Wang Y, Singhal A, et al. Molecular studies of liver disease in erythropoietic protoporphyria. J Clin Gastroenterol. 2005;39(4 suppl 2):S167-S175.124
臨床特徵 (CLINICAL FEATURES)
一個包含 32 名 EPP 病人系列的疾病表現摘要於表 124-3。急性皮膚光敏感(醫師很少見到)是最顯著的症狀,被描述為刺痛、灼燒或刺麻感,可能在曝曬陽光後數分鐘內發生,隨後出現紅斑與水腫(被描述為日光性蕁麻疹 solar urticaria)(圖 124-7),以及可能持續數天的全身性表現。長時間曝曬可能出現瘀點 (petechiae) 或紫斑 (purpura)。兒童無法描述這些症狀,但父母可能觀察到曝曬陽光後的哭鬧、皮膚腫脹與紅斑。手部與臉部最常受影響,且症狀在春夏季較為嚴重。病人對陽光(直接或穿過窗玻璃的)以及人工光源(包括手術室燈光)皆敏感。125 在北美 226 名病人中,症狀發病的平均年齡為 4.4 歲,且較高的原紫質濃度與較早的發病年齡、較差的陽光耐受度以及較常出現肝功能異常的報告相關。平均而言,患有 XLP 的男性病人比 EPP 病人有較高的紅血球原紫質濃度(3574 vs 1669 µg/dL;P <.001)。由於 X 染色體的隨機去活化 (random X-chromosomal inactivation),女性 XLP 病人之間的症狀嚴重度與原紫質濃度差異顯著。126
由於病人因疼痛而被促使並學會避免日曬,慢性皮膚變化並不常見,且通常輕微不明顯。這些變化可能包括手背與手指關節上革樣 (leathery) 的角化過度皮膚、輕度疤痕、唇部溝紋 (labial grooving),以及隨頻繁曝曬陽光而發生的甲板分離(甲剝離 onycholysis)(圖 124-8)。如其他皮膚型紫質症所見的大疱、皮膚脆弱、多毛、色素過度沉著、嚴重疤痕與毀損則屬罕見。據報告,懷孕會稍微降低紅血球原紫質濃度並增加對陽光的耐受度。128
在 20% 至 50% 的 EPP 病例中可見輕度缺鐵性貧血伴小球性 (microcytosis)、運鐵蛋白飽和度 (transferrin saturation) 降低,以及血清鐵蛋白位於正常範圍的低端。129,130 幾乎沒有證據顯示紅血球生成受損或鐵吸收或代謝異常,131-133 且溶血缺如或輕微。在無併發症的 EPP 中,神經內臟表現缺如,但原紫質症性肝病可能併發類似急性紫質症所見的嚴重運動神經病變。134 與掌部角皮症相關的體染色體隱性 EPP 也與原因不明的神經學症狀相關。111
含大量原紫質的膽結石很常見,係由排泄至膽汁中的大量水不溶性原紫質所形成。此類結石可能有症狀,並需在年幼時進行膽囊切除術。135 EPP 中的肝功能與肝臟原紫質含量通常正常。原紫質症性肝病係因過量呈現於肝臟的原紫質的膽汁鬱積效應所致。罕見情況下,這是 EPP 的主要表現特徵。136 隨著膽汁排泄受損的進展,循環原紫質濃度顯著增加。肝臟移植或其他長時間手術期間的手術室燈光,尤其在患有肝病的病人中,可造成顯著的光敏感,伴隨皮膚與腹膜的廣泛灼傷,並可藉由使用特殊濾光片來避免。137,138
診斷 (DIAGNOSIS)
任何年齡發生、伴有極少慢性皮膚變化的疼痛性、非水疱性光敏感,應提示原紫質症的診斷。測量紅血球總原紫質,若升高,再測量不含金屬與鋅原紫質的比例,即可建立或排除此診斷。紅血球總原紫質升高對 EPP 並無特異性,因為它在許多狀況中皆升高且以紅血球鋅原紫質為主,例如同型合子紫質症(多數 CEP 病例除外)、鐵缺乏、鉛中毒、慢性病貧血、139 溶血狀況140 與許多其他紅血球疾病。但紅血球原紫質增加且主要由不含金屬原紫質構成,則是原紫質症獨有的發現。FECH 催化鋅原紫質與血基質(鐵原紫質)兩者的形成,當此酵素缺乏時,兩者的形成皆受損,且累積的原紫質大多維持不含金屬。在 XLP 中,FECH 活性正常但其容量被過量的受質所超出,因此累積的原紫質通常大多不含金屬。希望篩檢 EPP 與 XLP 的臨床醫師應注意為此目的測量紅血球原紫質的重大陷阱,並應使用能正確報告「紅血球總原紫質」與「不含金屬原紫質」結果的實驗室。現已過時的「游離紅血球原紫質 (free erythrocyte protoporphyrin)」一詞(縮寫為「FEP」),在 1970 年代以前廣泛用於描述不含鐵的原紫質。這是在發現此增加在鉛中毒、鐵缺乏及許多其他狀況中係由鋅原紫質所致、而在 EPP 中則由不含金屬原紫質所致之前的事。某些實驗室(Quest 與 LabCorp)以血液螢光測定法 (hematofluorometry) 測量紅血球原紫質,該方法僅測量鋅原紫質,且是為篩檢鉛中毒而非紫質症所開發;它們使用誤導性且過時的「游離原紫質」與「FEP」等詞,錯誤地暗示測量的是不含金屬原紫質。ARUP 正確測量總量,但若該量升高,則不會分餾不含金屬與鋅原紫質,而後者對於證明此增加係由原紫質症所致至關重要。在美國,原紫質症的檢測由德州大學醫學分部 (University of Texas Medical Branch) 的紫質症實驗室以及 Mayo Medical Laboratories 正確執行。141
血漿紫質在多數但非所有 EPP 病例中增加,且在樣本處理期間特別容易光降解 (photodegradation)。142 稀釋血漿於中性 pH 下接近 634 nm 的血漿螢光峰,可協助確認 EPP 的診斷。53 糞便紫質濃度正常或稍增,且主要由原紫質構成。尿液紫質正常,但患有原紫質症性肝病的病人例外,後者會造成尿液糞紫質增加,如同任何病因的肝功能障礙之典型表現。
治療 (THERAPY)
光防護,尤其是避免曝曬陽光,是 EPP 的主要治療介入。這需要改變生活方式與工作環境以及其他避光行為,而這些大幅損害生活品質。多數病人在戶外時,防護衣物與帽子是必需的。含氧化鋅或二氧化鈦的局部防曬乳可提供部分益處。吸收 UV 波長的防曬劑則無益。口服 β-胡蘿蔔素被認為可淬熄活化的氧自由基,143 可增加對陽光的耐受度。144 建議每日劑量 120 至 180 mg 或更高,以達到血清 β-胡蘿蔔素濃度 600 至 800 µg/dL,此可在劑量改變後 3 至 4 週評估。125 然而,因類胡蘿蔔素血症 (carotenemia) 所致的皮膚變色在美觀上可能造成困擾,尤其對兒童而言。口服半胱胺酸 (cysteine) 500 mg 每日兩次,也可能淬熄受激發的氧物種並增加 EPP 對陽光的耐受度。145 阿法諾肽 (Afamelanotide) 是 α-黑色素細胞刺激素 (α-melanocyte–stimulating hormone) 的合成類似物,模擬天然存在的荷爾蒙並增加黑色素細胞中的黑色素產生,使 EPP 或 XLP 病人的皮膚色素增加並改善對陽光的耐受度。146,147 阿法諾肽目前在歐盟與瑞士獲准限制性使用,並正由美國食品藥物管理局審查用於 EPP 與 XLP 病人。希美替定 (Cimetidine) 可抑制肝臟 CYPs,據報告可降低 3 名 EPP 兒童的光敏感。148 但不建議使用,因為其所提出的抑制 ALAS2 與降低原紫質濃度的作用尚未獲得證實。149
原紫質症中的輕度鐵缺乏原因不明,部分人認為其有益(藉由限制鐵對 ALAS2 的上調),但也具有潛在害處(藉由進一步限制鐵嵌入原紫質)。一些病人注意到補充鐵質會增加光敏感,但據我們所知,紅血球或血漿原紫質濃度的增加尚未獲得證實。另一方面,在某些 EPP 與 XLP 的個別病例中,曾注意到補充鐵質後的臨床與生化改善。150,151
對於原紫質症且血清鐵蛋白濃度偏低的病人補充鐵質的效果,目前正由美國的紫質症聯盟 (Porphyrias Consortium) 進行研究。應預防性避免暴露於酒精、肝毒性藥物,或會增加肝臟紫質與血基質合成或損害肝臟排泄功能的藥物與荷爾蒙。152,153 應每年監測紅血球與血漿紫質濃度、肝功能檢查、血清鐵蛋白與血清維生素 D 濃度。由於 EPP 病人必須避免曝曬陽光,建議每日攝取 800 國際單位 (international units) 的維生素 D 與 1000 mg 的鈣。原紫質症性肝病可能自發地或經治療而緩解,尤其在它由可逆的肝功能障礙病因(如病毒感染或酒精)所引發時。120,154 此危及生命的併發症通常以合併多種介入來治療,包括膽固醇胺 (cholestyramine)、122,155,156 熊去氧膽酸 (ursodeoxycholic acid)、157 維生素 E、紅血球輸注、158 血漿置換 (plasma exchange) 與靜脈注射 hemin,目的在於降低血漿與紅血球原紫質濃度以及輸送至肝臟的原紫質量。這可作為病人接受肝臟移植甚至自發性改善的橋接。159 肝臟移植與其他肝臟疾病一樣成功,不過據報告在新肝臟中 5 年的復發率為 69%。160,161 一些患有原紫質症性肝病的病人在輸血162 或肝臟移植後發生急性運動神經病變,134,163 有時可在移植前給予 hemin 與血漿置換 (plasmapheresis) 而逆轉。164 骨髓移植在 EPP 中有效,165 且在肝臟移植後接續進行骨髓移植可預防肝臟疾病的復發。166
急性肝性紫質症 (ACUTE HEPATIC PORPHYRIAS)
重點一覽 (AT-A-GLANCE)
■ 急性紫質症的特徵為間歇性急性神經學症狀,但其中 3 種狀況(VP、HCP 與 AIP〔於進行性腎臟疾病時〕)可發展出慢性水疱病灶。
■ 神經內臟表現包括腹痛、嘔吐、四肢疼痛、癲癇發作,以及因運動神經病變所致的肌肉無力。
■ 尿液膽色素原 (porphobilinogen) 升高,尤其在急性發作期間。血漿紫質的螢光掃描以及糞便紫質的模式,對於區別特定的急性紫質症最為有用。
■ 皮膚表現可藉由避免日曬來預防。
■ 急性神經內臟發作可藉由避免惡化因素來預防,並以輸注 hemin 治療最佳。
4 種急性紫質症——δ-胺基乙醯丙酸脫水酶缺乏紫質症 (ADP)、急性間歇性紫質症 (AIP)、遺傳性糞紫質症 (HCP) 與斑斕型紫質症 (VP)——分別源自血基質生合成途徑中第二、第三、第六與第七個酵素的功能喪失型突變,其特徵為主要以間歇性急性惡化形式發生的神經學症狀。然而,其中 3 種狀況可發生類似 PCT 的慢性水疱性皮膚病灶。這些病灶在 VP 中常見、在 HCP 中不常見、在 AIP 中僅於進行性腎臟疾病時發生,而在 ADP 中則未見描述。167
ADP 極為罕見,僅有 6 例報告;與其他急性紫質症不同,其遺傳為體染色體隱性。168(本章不再進一步討論。)其他急性紫質症的酵素缺乏以帶有低外顯率的體染色體顯性方式遺傳(見表 124-1)。AIP 是全球最常見的急性肝性紫質症。AIP 的盛行率在歐洲估計為每 100,000 人 1 至 2 例,169 HCP 在丹麥估計為每 100,000 人 0.2 例,170 而 VP 在芬蘭報告為每 100,000 人 1.3 例。171 VP 在南非荷蘭裔白人中特別常見,係因奠基者效應 (founder effect),估計盛行率為每 100,000 人 300 例,172 且幾乎所有病例皆共有相同的 PPOX 突變 (R59W)。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在 AIP 中已辨識出至少 400 種 PBGD/HMBS 突變、HCP 中 64 種 CPOX 突變、173 VP 中 174 種 PPOX 突變86(表 124-1)。突變類型包括錯義、無義、剪接、缺失與插入突變。PBGD 催化由 4 分子 PBG 組裝出線狀四吡咯 HMB。CPOX 在單一活性位點催化糞紫質原 III 的兩步驟氧化脫羧,生成原紫質原 IX,並中間形成三羧基紫質原——硬羥紫質原 (harderoporphyrinogen)。PPOX 催化原紫質原 IX 脫氫形成原紫質 IX。急性肝性紫質症的神經內臟症狀與紫質前驅物 δ-胺基乙醯丙酸 (ALA) 與膽色素原 (PBG) 的累積相關。症狀可能源自 ALA 的神經毒性效應,及/或源自神經元或血管組織中的血基質缺乏。174 紫質在所有這些疾病中皆會累積,且最常以足夠量累積而在 VP 中造成慢性水疱性皮膚表現。
臨床特徵 (CLINICAL FEATURES)
急性紫質症典型的神經內臟表現包括腹痛、嘔吐、四肢疼痛、癲癇發作,以及因運動神經病變所致的肌肉無力。已知有數個因素促成 AIP、HCP 與 VP 的發作,包括藥物、荷爾蒙與飲食因素。慢性高血壓、腎臟疾病與肝細胞癌的風險增加。極為罕見的同型合子 AIP、HCP 與 VP 病例曾於生命早期表現出嚴重光敏感與神經損害,但無急性發作。174 一種同型合子 HCP 的變異形式,稱為硬羥紫質症 (harderoporphyria),係由 CPOX 突變所致,這些突變在第二次脫羧發生前即過早地從酵素釋放出硬羥紫質原。175
誘發因素 (PRECIPITATING FACTORS)
已知的內源性與外源性因素可在異型合子中誘發急性發作,且在個別病人身上具加成性。許多已知因素會造成肝臟 ALAS1 的誘導,進而導致途徑中間產物的累積。某些個體儘管避免已知的誘發因素,仍易於反覆發作。其他未知因素,包括尚未發現的修飾基因,可能也有所促成。
藥物與其他外源性化學物質 (DRUGS AND OTHER EXOGENOUS CHEMICALS)
多數在急性紫質症中有害的藥物,已知會誘導肝臟 CYPs 與 ALAS1。CYPs(肝臟中含量最豐富的血基質蛋白)的誘導增加了對新合成血基質的需求。176 已知有害與安全藥物的例子列於表 124-4。最新的藥物安全資料庫可於美國紫質症基金會 (American Porphyria Foundation, www.porphyriafoundation.com) 與歐洲紫質症網絡 (European Porphyria Network, www.porphyria-europe.com) 的網站找到。抽菸已知會增加人類的肝臟 CYPs,可能是吸入的多環芳香烴 (polycyclic aromatic hydrocarbons) 的效應,並與較頻繁的症狀相關。177 乙醇與其他醇類也會誘導 ALAS1 與某些 CYPs,並被認為與發作有關。178,179
內分泌因素 (ENDOCRINE FACTORS)
症狀常於青春期後發展,且較常發生於女性,顯示內源性女性荷爾蒙促成症狀的發生。黃體素 (progesterone)、合成黃體製劑 (synthetic progestins) 與睪固酮的某些代謝產物是 ALAS1 與 CYPs 的強力誘導物,並與紫質症的急性發作相關。月經週期黃體期 (luteal phase) 黃體素的增加,最可能是 AIP、HCP 與 VP 中週期性發作的原因。雌激素曾被認為有害,但其對 ALAS1 與 CYPs 並無黃體製劑般的誘導效應,且在未併用黃體製劑而給予雌激素時發生發作的報告甚少。但如前所述,雌激素使用是 PCT 女性中常見的易感因素。糖尿病可能降低發作頻率並降低紫質前驅物濃度,可能是因為高血糖濃度所致。180
表 124-4:急性紫質症中已知不安全或安全藥物的部分清單
| 不安全 (UNSAFE) | 安全 (SAFE) |
|---|---|
| 酒精 (Alcohol) | 對乙醯胺酚 (Acetaminophen) |
| 巴比妥類 (Barbiturates)ª | 阿斯匹靈 (Aspirin) |
| 卡馬西平 (Carbamazepine)ª | 阿托品 (Atropine) |
| 卡立普多 (Carisoprodol)ª | 溴化物 (Bromides) |
| 可氯氮平 (Clonazepam,高劑量) | 希美替定 (Cimetidine) |
| 達那唑 (Danazol)ª | 紅血球生成素 (Erythropoietin)ª,ᵇ |
| 雙氯芬酸 (Diclofenac)ª 及可能其他 NSAIDs | 加巴噴丁 (Gabapentin) |
| 麥角類 (Ergots) | 糖皮質素 (Glucocorticoids) |
| 雌激素 (Estrogens)ª,ᶜ | 胰島素 (Insulin) |
| 乙氯維諾 (Ethchlorvynol)ª | 左乙拉西坦 (Levetiracetam) |
| 戊四烯酮/格魯米特 (Glutethimide)ª | 麻醉性鎮痛劑 (Narcotic analgesics) |
| 灰黃黴素 (Griseofulvin)ª | 青黴素及其衍生物 (Penicillin and derivatives) |
| 美芬妥英 (Mephenytoin) | 吩噻嗪類 (Phenothiazines) |
| 甲丙胺酯 (Meprobamate)ª(亦含 mebutamateª、tybutamateª) | 雷尼替定 (Ranitidine)ª,ᵇ |
| 甲乙哌酮 (Methyprylon) | 鏈黴素 (Streptomycin) |
| 滅吐靈 (Metoclopramide)ª | 氨己烯酸 (Vigabatrin) |
| 苯妥英 (Phenytoin)ª | |
| 普力米酮 (Primidone)ª | |
| 吡嗪醯胺 (Pyrazinamide)ª | |
| 吡唑酮類 (Pyrazolones,aminopyrine、antipyrine) | |
| 利福平 (Rifampin)ª | |
| 琥珀醯亞胺類 (Succinimides,ethosuximide、methsuximide) | |
| 磺胺類抗生素 (Sulfonamide antibiotics)ª | |
| 丙戊酸 (Valproic acid)ª |
ªP 在美國藥品標示中,紫質症被列為這些藥物的禁忌、警告、注意事項或不良反應。
ᵇ雖然紫質症在美國藥品標示中被列為注意事項;但其他來源認為這些藥物是安全的。
ᶜ雌激素——對遲發性皮膚紫質症不安全,但在急性紫質症中可謹慎使用。註:在使用此處未列出的藥物前,應參考更完整且更頻繁更新的來源,例如美國紫質症基金會 (www.porphyriafoundation.com/) 與歐洲紫質症倡議 (European Porphyria Initiative, www.porphyria-europe.com/) 的網站。修改自 Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439-450;經授權使用。167
懷孕 (PREGNANCY)
多數患有 AIP 的女性報告懷孕的耐受度良好。181 然而,發作有時會在懷孕期間變得較頻繁,這可能是因有害藥物(如滅吐靈 metoclopramide)182,183 或因妊娠劇吐 (hyperemesis gravidarum) 而減少熱量攝取所致。懷孕期間的發作除非輕微,否則應以 hemin 治療。因紫質症的急性發作而終止懷孕的指徵甚少。
營養 (NUTRITION)
為減重或於疾病或手術期間減少熱量與碳水化合物的攝取,可誘發發作。在這些情況下,肝臟中過氧化體增殖物活化共同活化因子 1α (peroxisomal proliferator-activated cofactor 1α, PGC-1α) 被上調,誘導 ALAS1 並增加 ALA 與 PBG。這些效應可藉由給予碳水化合物來預防或逆轉。184,185 飢餓也可能誘導肝臟血基質氧化酶 (heme oxygenase),186 此可能耗竭肝臟血基質並促成 ALAS1 的誘導。
診斷 (DIAGNOSIS)
尿液 PBG 在 AIP、HCP 與 VP 的急性發作期間升高,但在 HCP 與 VP 中升高程度往往較 AIP 為低且較為短暫;進一步檢測可輕易區別這 3 種狀況。類似 PCT 的皮膚病灶在 AIP 中罕見,且僅在伴有進行性腎臟疾病、血漿中 PBG 與紫質濃度皆增加時才發生。在 HCP 與 VP 中,尿液紫質(尤其是尿紫質與糞紫質)也升高,但此發現本身無特異性,無法輕易將這些紫質症彼此區別。糞便紫質濃度在 HCP 與 VP 中皆顯著增加,HCP 以糞紫質 III 為主,VP 則以糞紫質 III 與原紫質兩者為主。糞便糞紫質 III/I 比值對於診斷 HCP 具敏感性,即使在疾病無症狀的階段亦然。187
血漿紫質濃度在 VP 中常升高,而在 AIP 與 HCP 中除非有皮膚表現,否則甚少增加。VP 的一個特異性特徵是血漿紫質於中性 pH 下螢光最大值約在 626 nm,代表與血漿蛋白共價結合的原紫質。188 此螢光掃描方法在偵測無症狀 VP 方面比檢查糞便紫質更有效,189 並特別有助於迅速將 VP 與 PCT 區別,後者顯示約 620 nm 的螢光峰。紅血球 PBGD 活性在多數 AIP 病人中降低,而在 HCP 與 VP 中正常。CPOX 與 PPOX 的檢測需要含粒線體的細胞,且並未廣泛可得。一旦家族突變被辨識,DNA 研究對於辨識無症狀帶因者最為可靠。同型合子的 AIP、HCP 與 VP 病例顯示紫質前驅物與紫質(包括紅血球鋅原紫質)更嚴重的增加。硬羥紫質症藉由糞便與紅血球中糞紫質 III 與硬羥紫質 III (harderoporphyrin III) 兩者的升高來辨識。190
治療 (THERAPY)
VP 與 HCP 的皮膚表現雖與 PCT 所見的水疱性皮膚病灶相同,但對放血或低劑量羥氯奎寧無反應。因此,避免日曬與使用防護衣物最為重要。長期避免誘發因素可能降低慢性升高的紫質濃度,但據我們所知,這尚未獲得證實。患有 AIP、腎衰竭及相關皮膚表現的病人,應考慮進行腎臟移植或合併肝腎移植。191
對於急性發作,通常建議住院以治療嚴重症狀、進行靜脈療法並監測呼吸、電解質與營養狀態。較輕、反覆發作且對治療反應迅速者,有時可在門診處理。應去除誘發因素,例如有害藥物。疼痛、噁心與嘔吐通常需要麻醉性鎮痛劑、氯丙嗪 (chlorpromazine) 或另一種吩噻嗪類,或恩丹西酮 (ondansetron)。短效苯二氮平類 (benzodiazepines) 用於焦慮與失眠。167
β-腎上腺素阻斷劑 (β-Adrenergic-blocking agents) 可用於控制心搏過速與高血壓,除非因血容量不足或心臟衰竭而為禁忌。192 癲癇發作的治療應著重於矯正低血鈉症 (hyponatremia)(若存在)。多數抗癲癇藥物在急性紫質症中具潛在害處。可氯氮平 (Clonazepam) 可能比苯妥英 (phenytoin)、巴比妥類或丙戊酸 (valproic acid) 害處較小。193,194
辨識並長期避免誘發急性神經內臟發作的因素至關重要。建議在 50 歲以後進行肝細胞癌的每年篩檢,尤其是那些紫質前驅物或紫質持續增加的病人。Hemin 是急性發作最有效的治療,在美國以凍乾 hematin (Panhematin, Recordati Rare Diseases) 形式提供,並在歐洲與南非以血基質精胺酸鹽 (heme arginate, Normosang, Orphan Europe) 形式提供。195,196 靜脈注射的 hemin 主要被肝細胞攝取,在此重建調節性血基質池並抑制 ALAS1 的合成,導致 ALA、PBG 與紫質的顯著減少,以及症狀更迅速的緩解。197
Hemin 療法應用於中度至重度發作,以及對碳水化合物負荷無反應的輕度發作。167,195,198 每日輸注 3 至 4 mg/kg、連續 4 天是標準療法,若在此時間內反應不完全,可加以延長。Hemin 已被安全地用於懷孕期間。167,195,196 建議以 25% 人類白蛋白 (human albumin) 重新調配 hematin,因為使用無菌水時形成的降解產物可能造成輸注部位靜脈炎並限制未來的靜脈通路。167,199 使用白蛋白也可預防伴有血小板減少以及凝血酶原時間 (prothrombin) 與部分凝血活酶時間 (partial thromboplastin times) 延長的短暫凝血功能障礙。200,201 Hemin 的副作用可能包括發燒、痠痛、倦怠,以及罕見的溶血、過敏性反應與循環衰竭。202,203 每週給予一或兩次 hemin 是預防頻繁、非週期性發作的一個選項。204,205
碳水化合物負荷 (CARBOHYDRATE LOADING)
給予碳水化合物可下調 PGC-1α 與肝臟 ALAS1 的誘導,從而減少紫質前驅物的產生。然而,此法不如 hemin 有效,且應僅用於輕度發作(無需要鴉片類藥物的嚴重疼痛、無輕癱、癲癇發作或低血鈉症),167 或用於 hemin 可取得之前的過渡期。靜脈治療通常以 10% 溶液給予 300 至 500 g 的靜脈葡萄糖,並謹慎監測以避免因給予大量自由水所致的低血鈉症。167 若可耐受,可給予口服葡萄糖聚合物溶液。
圖表 (FIGURES AND TABLES)

圖 124-1:血基質生合成途徑中間產物的順序與化學結構,以及 8 個途徑酵素於粒線體或細胞質中的定位。δ-胺基乙醯丙酸合成酶存在管家型 (ALAS1,見於所有組織) 與紅血球特異型 (ALAS2,僅見於骨髓紅血球細胞) 兩種形式。如此處所示,肝臟中 ALAS1 的合成受最終產物血基質的敏感回饋控制。
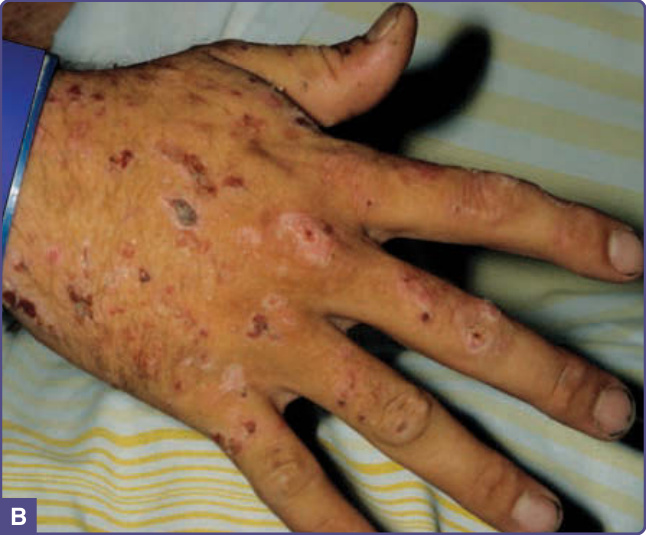
圖 124-2:PCT 的特徵性皮膚病灶最常發生於手背,與其他水疱性皮膚紫質症(尤其是斑斕型紫質症)及假性紫質症所見者無法區別。A,水疱(短黑箭頭)常於輕微創傷後形成,破裂後留下可結痂的表淺潰瘍(短白箭頭),並留下緩慢癒合的萎縮性區域(長箭頭)。B,水疱脆弱,因此常見的唯一病灶為慢性者,如糜爛、結痂與疤痕。
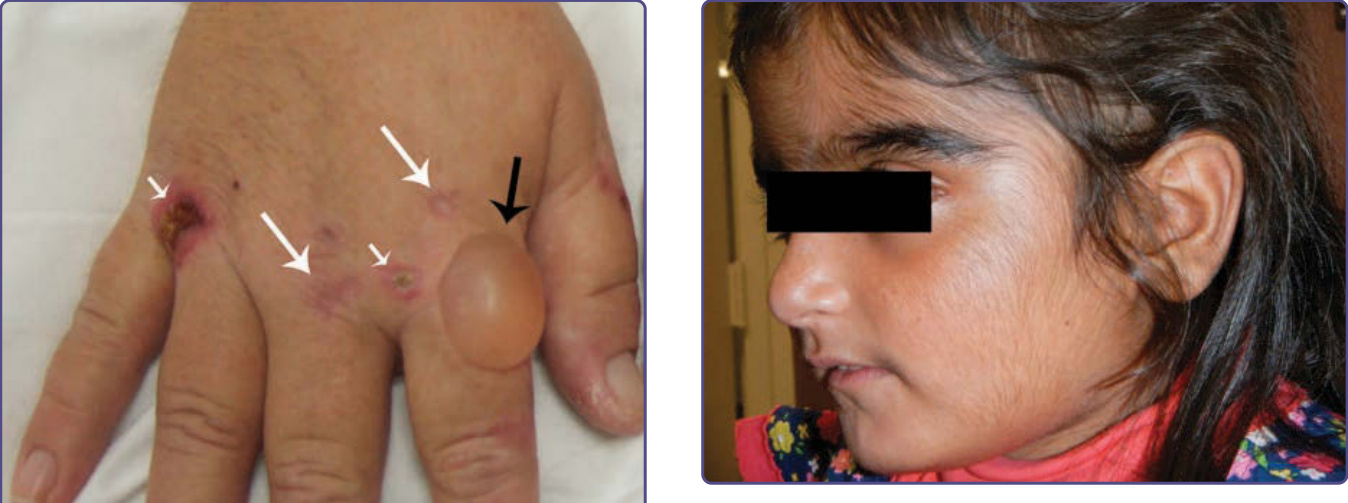
圖 124-3:一名帶有遺傳性 UROD 突變的兒童,在因白血病接受化學治療後發展出遲發性皮膚紫質症所致的顯著多毛與色素過度沉著,如先前所報告。15
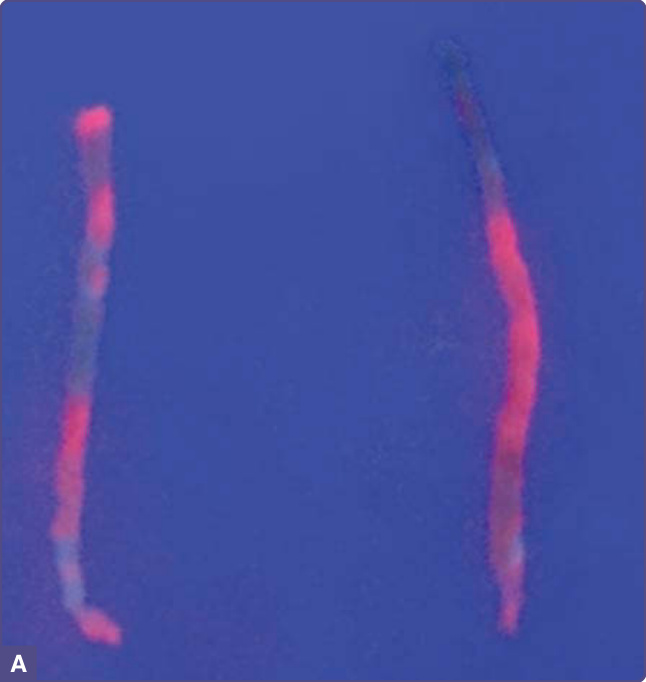
圖 124-4:A,新鮮肝臟切片組織在長波紫外光(伍氏燈 Wood lamp)下顯示明亮的紅色螢光,反映高羧基化紫質在肝細胞中的顯著累積。B,同一切片肝臟紫質的分析(總紫質 176 nmol/g 組織,參考值 <1),以高效液相層析 (HPLC) 分離紫質後,以尿紫質 I 與 III 及七羧基紫質 III 為主;此模式類似於血漿與尿液中所見者。(影像經 Heather Stevenson-Lerner, MD, PhD 授權使用;HPLC 經 V.M.S. Ramanujam, PhD 授權使用。)
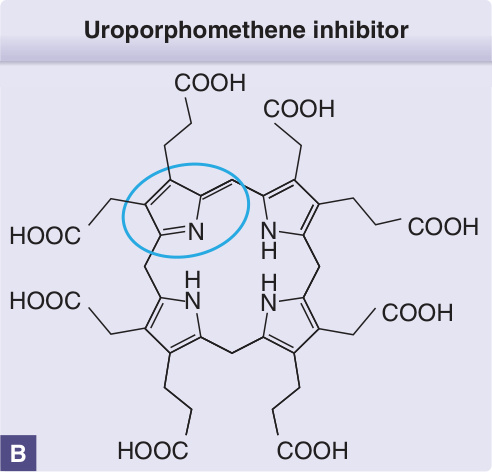
圖 124-5:A,PCT 中於肝臟在鐵、細胞色素 P450 酵素(尤其 CYP1A2)與氧化壓力存在下生成尿紫質原脫羧酶特異性抑制物的示意圖。(修改自 Anderson KE. Porphyria cutanea tarda: A possible role for ascorbic acid. Hepatology. 2007;45(1):6-8;經授權使用。Copyright © 2007, John Wiley & Sons.48)B,一種 UROD 抑制物已被分離為尿紫質甲烯 (uroporphomethene),這是一個於一個環(藍色圈出)及其相鄰甲基橋處被部分氧化的尿紫質原分子。
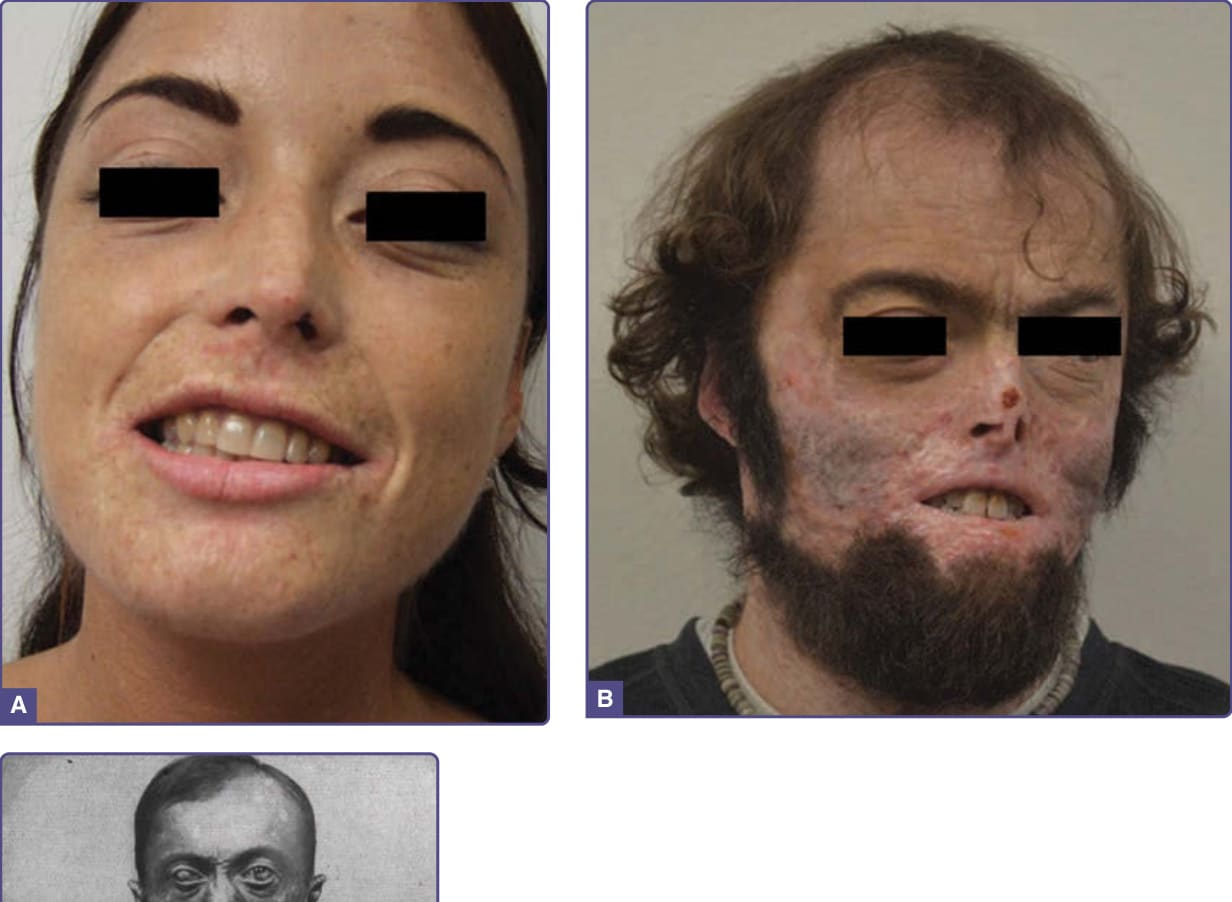
圖 124-6:三例嚴重度不同的先天性紅血球生成性紫質症。病人 A 為輕症,嚴格避免日曬,僅表現出臉部皮膚的輕度攣縮、口周疤痕,以及手背與手指的輕度疤痕,並伴發育上偏小、縮短的手指(未顯示)。病人 B 亦為輕症,因未避免日曬而臉部、手部與手指有進行性疤痕。(重印自 Gou E, Phillips JD, Anderson KE. The porphyrias. In: Gilbert-Barness E, Barness L, Farrell PM, eds. Metabolic Diseases. Amsterdam: IOS Press BV; 2017:543-575,10 Copyright 2017,經 IOS Press 授權。本出版品可於 IOS Press 透過 https://www.iospress.nl/book/metabolic-diseases/ 取得。病人 C 為 Petry 先生,他在歷史上具重要意義,曾在紫質化學家 Hans Fischer 的實驗室工作,提供樣本供早期紫質化學研究使用;他患有嚴重疾病並存活至 34 歲。出自 Günther H. In: Schittenhelm A, ed. Handbuch der Krankheiten der Blutes und der Blutbildenden Organe. Vol 2. Berlin: Springer; 1925;經授權使用。Copyright © 1925.89)
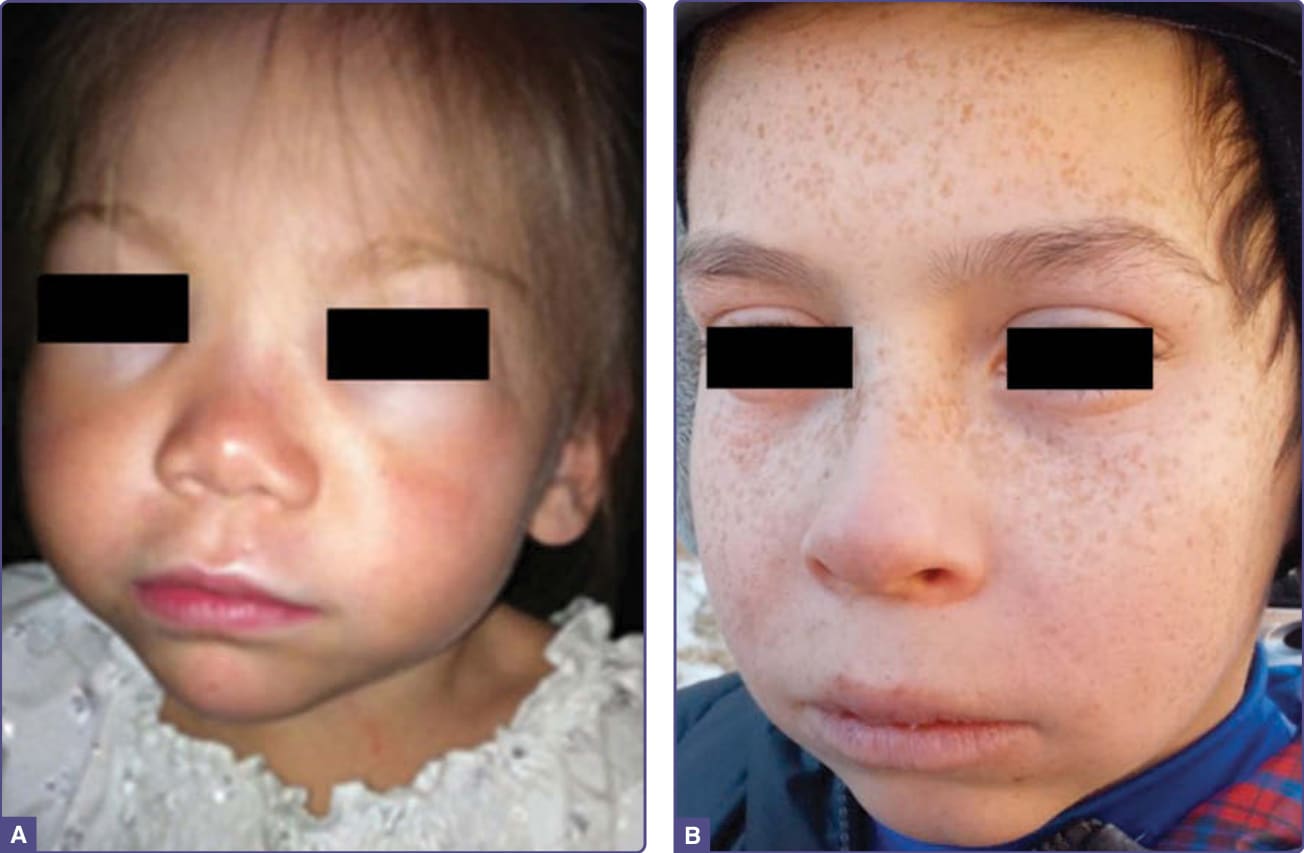
圖 124-7:2 名 EPP 兒童在曝曬陽光後臉部的急性腫脹與紅斑。在第二名兒童中,僅未被滑雪面罩遮蔽的下臉部受影響。
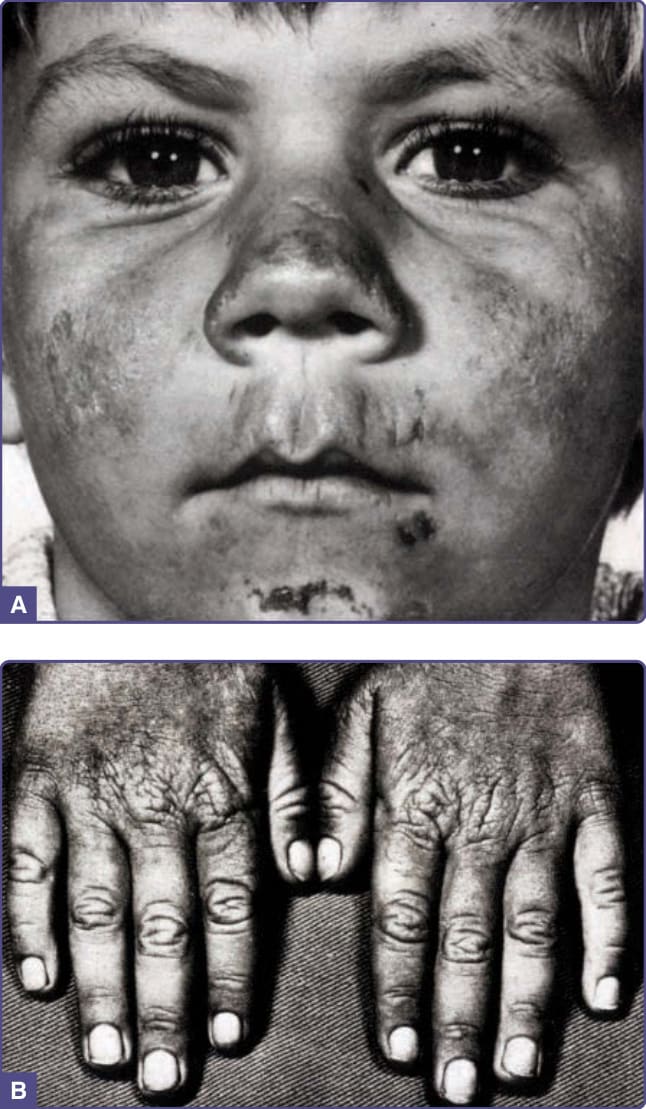
圖 124-8:一名 6 歲 EPP 兒童因反覆光線反應所致的慢性皮膚表現,包括臉部潰瘍與疤痕、唇部溝紋,以及手背皮膚增厚與指甲甲弧 (lunulae) 的喪失。(重製自 Schmidt H, Snitker G, Thomsen K, Lintrup J. Erythropoietic protoporphyria. A clinical study based on 29 cases in 14 families. Arch Dermatol. 1974;110(1):58-64;經授權使用。Copyright © 1974 American Medical Association. All rights reserved.127)
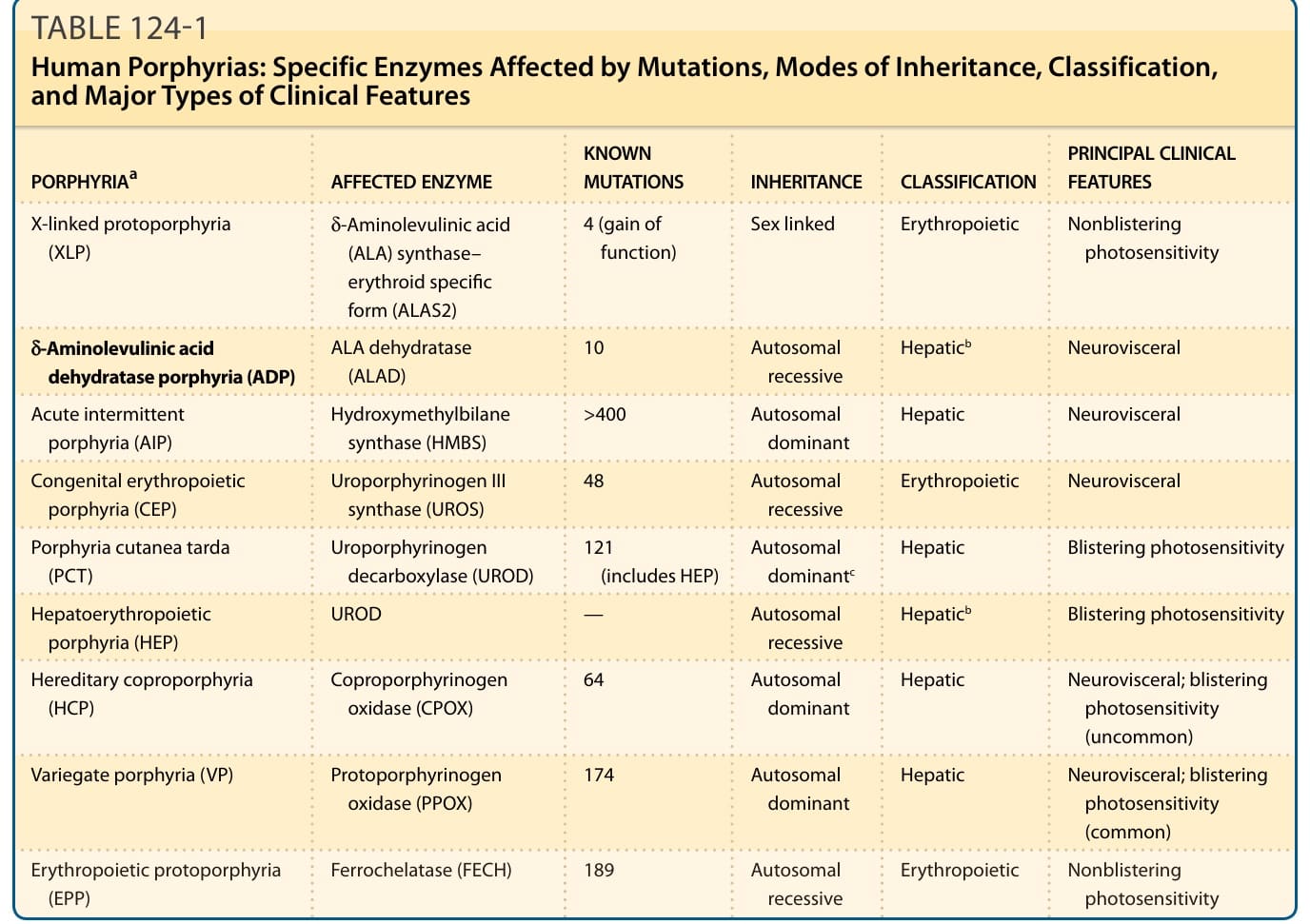
表 124-1:人類紫質症:突變所影響的特定酵素、遺傳方式、分類與主要臨床特徵類型。
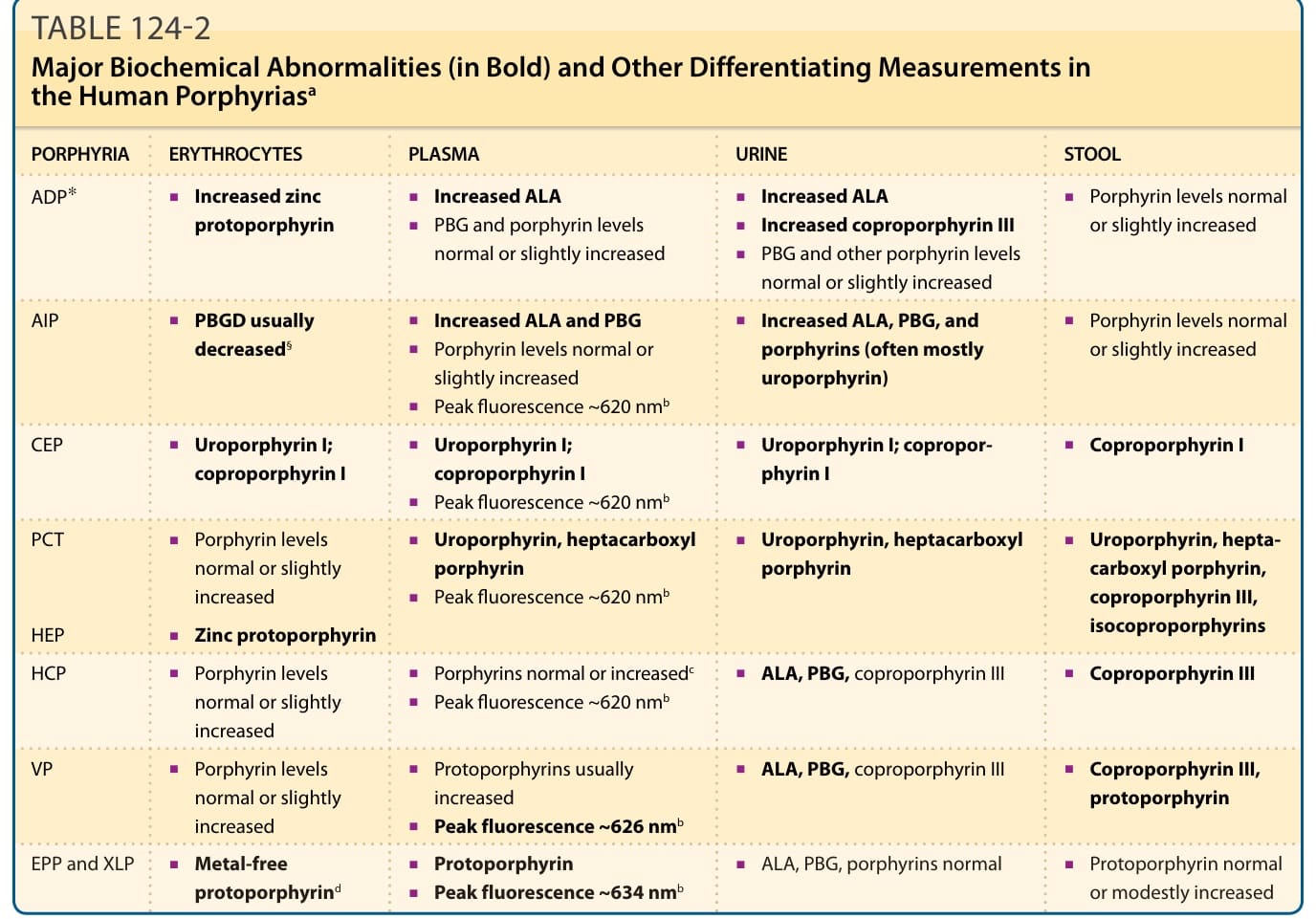
表 124-2:人類紫質症的主要生化異常(以粗體表示)與其他鑑別性檢測值。ª
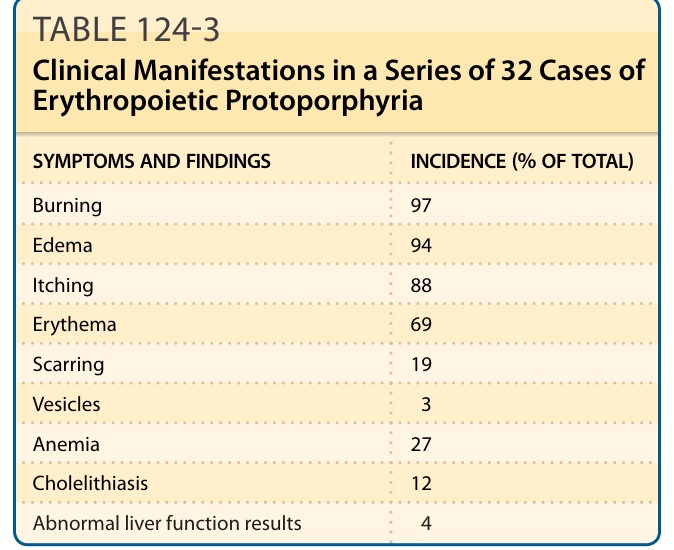
表 124-3:32 例紅血球生成性原紫質症的臨床表現。
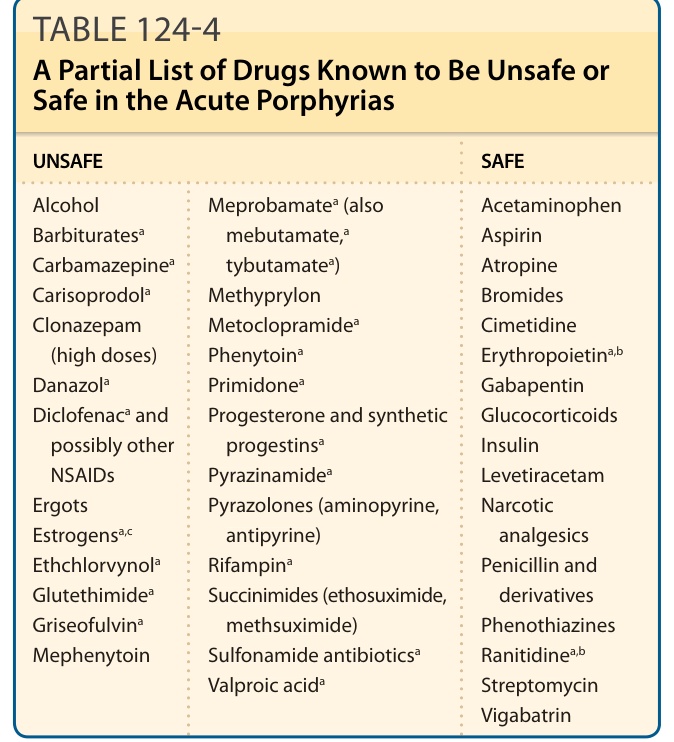
表 124-4:急性紫質症中已知不安全或安全藥物的部分清單。