脂肪源性腫瘤 (Lipogenic Neoplasms) 精華筆記
總論
- 脂肪源性腫瘤 (lipogenic neoplasms) 是皮膚與軟組織中最常見、最大宗的間葉性腫瘤 (mesenchymal neoplasms),分為良性(脂肪瘤 lipoma、血管脂肪瘤 angiolipoma)、中間型/局部侵襲性但不轉移(非典型脂肪瘤樣腫瘤 atypical lipomatous tumor)與明確惡性(多形性脂肪肉瘤 pleomorphic liposarcoma)。
- 部位、深度、大小是決定發生率、型態與預後的關鍵。深部軟組織脂肪肉瘤 (liposarcoma) 是成人最常見的肉瘤 (sarcoma),但純真皮位置極罕見。
- 臨床表現多不具特異性;多數腫瘤具獨特細胞遺傳學異常 (cytogenetic abnormalities),具診斷與治療價值。
- 治療首選手術切除;無法完整切除的惡性腫瘤可用放射治療、化學治療或標靶類治療。
脂肪瘤 (Lipoma)
- 成人最常見的良性間葉性腫瘤,由成熟白色脂肪細胞 (white adipocytes) 組成,無性別偏好,兒童與多發病灶罕見。
- 臨床:軀幹/頸/四肢無痛、緩慢生長腫塊;淺表者通常小於 5 cm,深部肌肉內 (intramuscular)/肌肉間脂肪瘤可很大。可為加德納症候群 (Gardner syndrome)、班納揚症候群 (Bannayan syndrome) 的表現(見表 122-1)。
- 致病機轉:最常見異常為 12q13-15 畸變、6p21-23 重排、13q 缺失;位於 12q14.3 的 HMGA2 基因扮演重要角色。
- 病理:界線清楚、有包膜、分葉狀,成熟脂肪細胞+薄纖維間隔,無細胞核非典型;p16/MDM2/CDK4 陰性。受創脂肪瘤可有富含 p16/MDM2/CDK4 細胞質染色之組織細胞,須與非典型脂肪瘤樣腫瘤鑑別。亞型:纖維脂肪瘤、軟骨脂肪瘤、骨脂肪瘤、腺脂肪瘤 (adenolipoma)。
- 病程/治療:完全良性、極少復發;肌肉內/肌肉間型局部復發率較高(高達 20%)。手術切除可治癒。
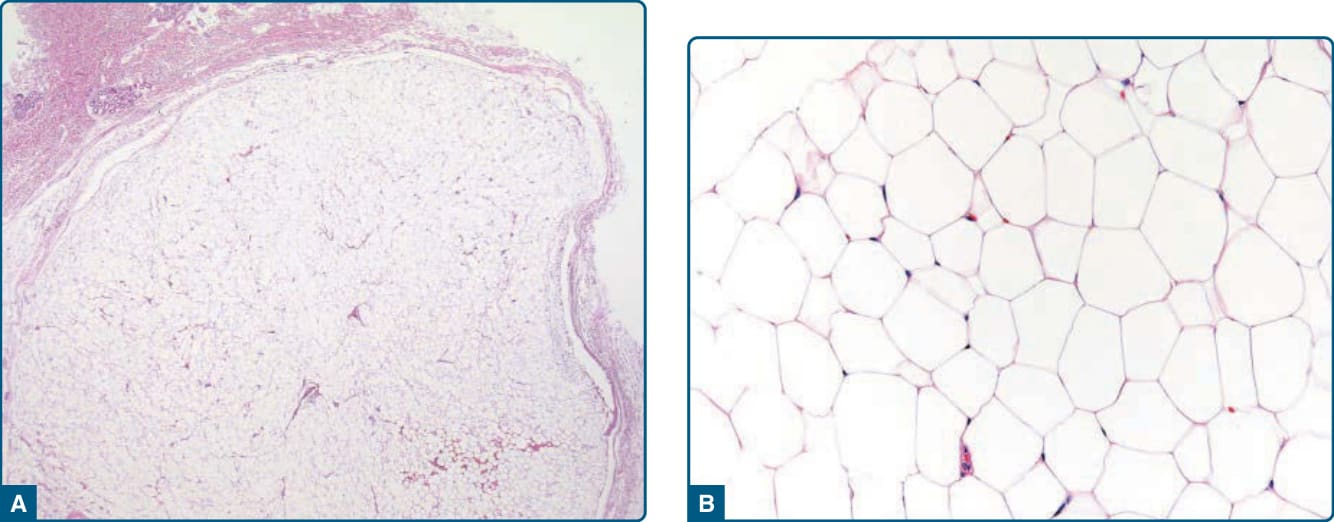
圖 122-1:脂肪瘤——界線清楚、有包膜的脂肪源性病灶,由成熟單空泡化脂肪細胞組成。
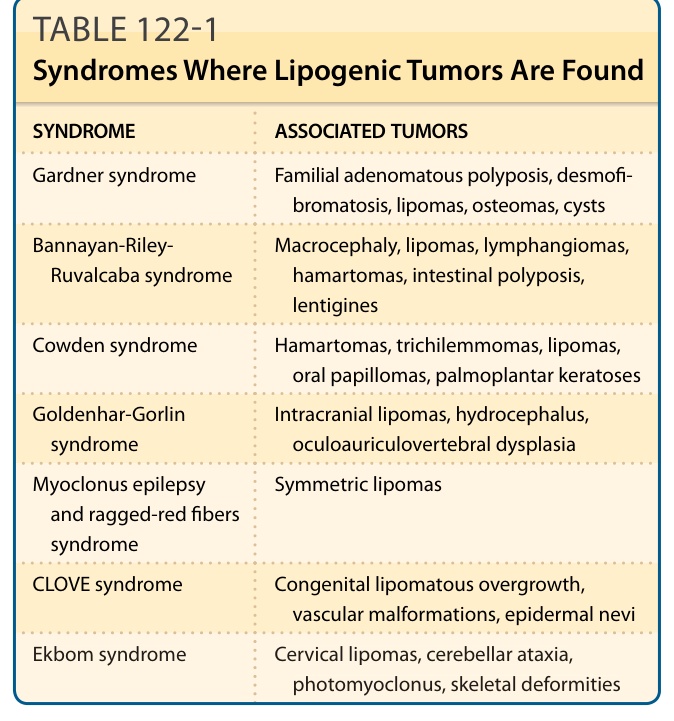
表 122-1:發現脂肪源性腫瘤的症候群(加德納、班納揚-萊利-魯瓦卡巴、考登、戈登哈-戈林、CLOVE、埃克博姆等)。
脂肪瘤病 (Lipomatosis)
- 成熟脂肪組織瀰漫性過度生長、浸潤原有結構。瀰漫型主要見於兒童;骨盆脂肪瘤病多見於黑人男性;對稱性脂肪瘤病(馬德隆病 Madelung disease)見於地中海血統中年男性(好發頸、肩、上肢),常合併周邊神經病變。HIV 接受蛋白酶抑制劑者可有脂肪失養症;類固醇脂肪瘤病見於荷爾蒙治療者。
- 機轉未明,與粒線體功能障礙相關(mtDNA 多重缺失、點突變)。
- 各亞型病理相同:界線不清、浸潤性、柔軟黃色脂肪。
- 姑息性手術切除為首選,但各型皆易復發。
神經脂肪瘤病 (Lipomatosis of Nerve)
- 又稱神經的纖維脂肪瘤性錯構瘤;多於出生時或兒童早期,好發手部,可伴巨指(趾)症 (macrodactyly)。最常侵犯正中神經 (median nerve) 及其分支,其次尺神經。
- 臨床:緩慢生長腫塊,伴疼痛、感覺異常、感覺/運動缺損。
- 病理:神經梭形腫大,神經外膜 (epineurium)/神經束膜 (perineurium) 被成熟脂肪與膠原浸潤,同心圓狀神經束膜纖維化。
- 完全良性;手術切除可能嚴重損傷神經。
淺表性脂肪瘤樣痣 (Nevus Lipomatosus Superficialis)
- 霍夫曼-楚爾黑勒 (Hoffmann-Zurhelle);罕見結締組織痣,侵犯兒童與年輕成人,性別相等。
- 斑塊/丘疹/單發病灶,好發臀部、大腿後上側、腰背部;單側,可呈線狀或帶狀皰疹樣排列。
- 病理:成熟脂肪取代真皮,膠原束增厚、彈性纖維與纖維母細胞增加,表皮可有棘層增厚與過度色素沉著。
- 鑑別:真皮梭形細胞脂肪瘤(CD34⁺、Rb-1 喪失、黏液樣基質含肥大細胞)。
脂肪母細胞瘤/脂肪母細胞瘤病 (Lipoblastoma/Lipoblastomatosis)
- 嬰幼兒良性胚胎性白色脂肪腫瘤,男性為主;好發軀幹、四肢、頭頸。脂肪母細胞瘤界線清楚,脂肪母細胞瘤病為浸潤性。相當比例病人有 CNS 疾患(癲癇、自閉、發展遲緩、史特格-韋伯症候群)。
- 致病機轉:第 8 號染色體畸變,8q11-13 易位伴 PLAG1 基因重排。
- 病理:界線清楚、分葉狀,成熟與未成熟脂肪源性細胞+纖維血管間隔,黏液樣基質含原始梭形細胞與叢狀血管(似黏液樣脂肪肉瘤)。S-100、PLAG1、CD34 陽性;可 p16 陽性(與非典型脂肪瘤樣腫瘤鑑別之陷阱)。
- 鑑別:黏液樣脂肪肉瘤(<10 歲極罕見);顯著分葉+無深染細胞核支持脂肪母細胞瘤。
- 預後極佳,局部復發率高達 46%(因不完全切除或瀰漫型),無進展、轉移或惡性轉化風險。
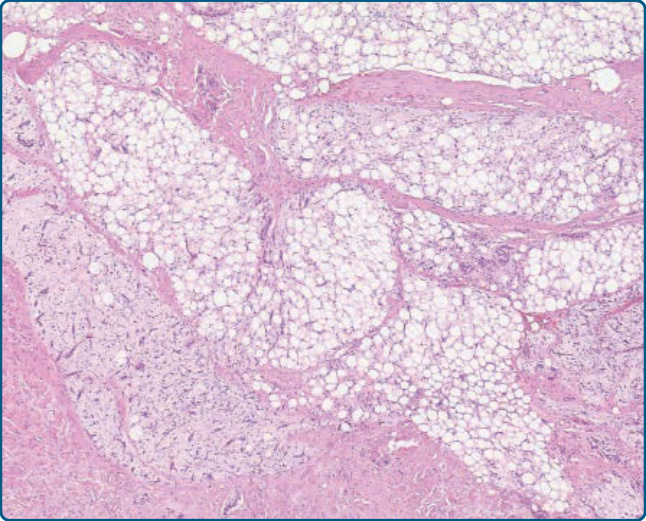
圖 122-7:脂肪母細胞瘤——界線清楚、多分葉、具膠原性纖維間隔的脂肪源性腫瘤。
血管脂肪瘤 (Angiolipoma)
- 常見、常疼痛、完全良性;多見於年輕男性,常多發,可家族性。好發前臂,其次軀幹、上臂、腿。
- 與脂肪瘤不同,核型正常;細胞性血管脂肪瘤可幾乎全由血管組成(推測為血管瘤)。
- 病理:界線清楚、有包膜結節,成熟脂肪細胞+薄壁微血管(可含纖維蛋白血栓 fibrin thrombi)+基質梭形細胞。
- 鑑別:纖維蛋白血栓助與脂肪瘤區分;細胞性者須與卡波西肉瘤、血管肉瘤等鑑別。單純切除可治癒。
梭形細胞脂肪瘤與多形性脂肪瘤 (Spindle-Cell / Pleomorphic Lipoma)
- 單一臨床病理實體的型態學連續譜,與乳腺型肌纖維母細胞瘤、細胞性血管纖維瘤相關。
- 典型為有包膜皮下病灶,好發老年男性的頸後、肩、上背;純真皮型界線不清、浸潤性、無性別偏好、分布廣。
- 致病機轉:核型複雜、常亞二倍體,第 13、16 號染色體單體與部分缺失。
- 病理:梭形細胞脂肪瘤=成熟脂肪細胞+CD34⁺溫和梭形細胞,含玻璃樣變繩索狀膠原與肥大細胞,幾無有絲分裂;多形性脂肪瘤另有花環狀巨細胞 (floret-like giant cells)。CD34 陽性、Rb-1 喪失。出現顯著非典型者為非典型梭形細胞/多形性脂肪瘤樣腫瘤(中間型、局部侵襲性)。
- 病程:典型者完全良性、罕復發;非典型者局部復發率升高。
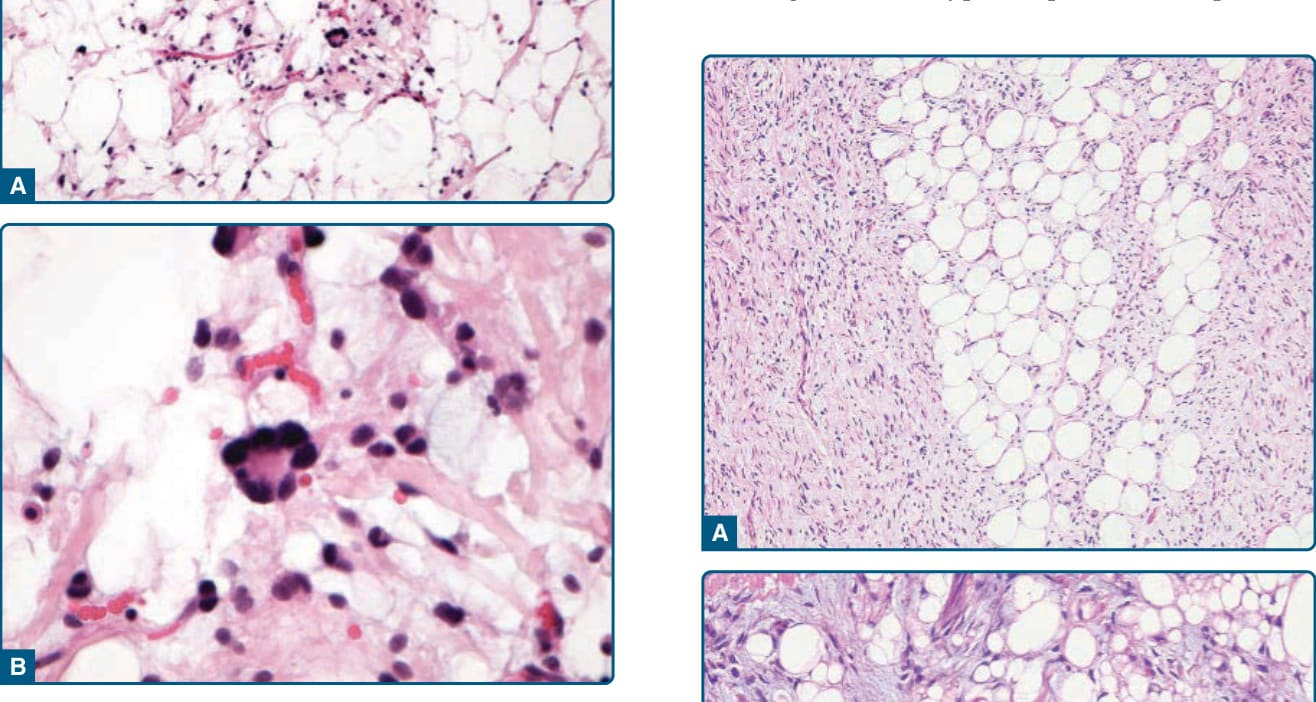
圖 122-16:多形性脂肪瘤——特徵性花環狀多核巨細胞,細胞核呈放射狀排列。
軟骨樣脂肪瘤 (Chondroid Lipoma)
- 罕見良性,好發女性、四肢深部軟組織(近端與肢帶)。
- 由成熟脂肪細胞、多空泡化脂肪母細胞、含嗜酸性空泡化細胞質之小圓細胞組成,位於黏液樣–軟骨樣基質中(低 pH 愛爾遜藍陽性,示硫酸軟骨素)。
- 致病機轉:t(11;16)(q13;p12-13),C11orf95-MKL2 融合基因;CCND1 表現。
- 完全良性,單純切除可治癒,無惡性轉化或轉移報告。
肌脂肪瘤 (Myolipoma)
- 罕見深部軟組織良性腫瘤,好發女性、後腹膜/腹腔/腹股溝,常偶然發現。
- 成熟脂肪+溫和平滑肌細胞 (smooth muscle cells) 不規則混合;無非典型/有絲分裂。平滑肌對 α-平滑肌肌動蛋白、結蛋白 (desmin)、h-鈣調素結合蛋白陽性,HMB-45 陰性;脂肪源性細胞 MDM2/CDK4 陰性。
- 致病機轉:HMGA2 改變,HMGA2-C9orf92 融合伴 t(9;12)(p22;q14)。
- 完全良性,完全切除可治癒。
冬眠瘤 (Hibernoma)
- 棕色脂肪 (brown fat) 良性腫瘤;好發年輕成人,最常見部位為大腿,男性略多。
- 病理:界線清楚、分葉狀,棕色脂肪細胞(多角形、多空泡化、顆粒狀細胞質、中央核)+白色脂肪細胞。變異型:嗜酸性、蒼白細胞、混合、黏液樣、梭形細胞、脂肪瘤樣。S-100 與解偶聯蛋白 (uncoupling protein) 陽性。
- 完全良性,完全切除可治癒。
非典型脂肪瘤樣腫瘤/高分化脂肪肉瘤 (Atypical Lipomatous Tumor / Well-Differentiated Liposarcoma)
- 兩者為同義詞,型態與細胞遺傳學相同;屬局部侵襲性、不轉移腫瘤。部位、大小、深度為重要預後因子。
- 流行病學:臨床侵襲性脂肪源性腫瘤中最大宗,脂肪肉瘤為成人最常見肉瘤;發生率高峰在第六個十年(60 多歲)。
- 臨床:多在深部軟組織,好發四肢肌肉(尤大腿),其次後腹膜、腹腔、腹股溝、睪丸旁、縱膈;表現為不斷增大的無痛腫塊。
- 致病機轉:源自 12q13-15 的環狀/巨大標記染色體,導致 MDM2 與 CDK4 一致性擴增。
- 病理:大型、界線清楚、分葉狀。三大亞型——脂肪細胞型(脂肪瘤樣,最常見)、硬化型(好發後腹膜/睪丸旁)、發炎性;另有高分化梭形細胞脂肪肉瘤。p16 局部表現、MDM2/CDK4 核染色;高分化者免疫染色可陰性,此時 FISH 偵測 MDM2/CDK4 擴增很有幫助。
- 病程/預後:高度取決於部位。四肢等可切除部位手術可治癒,進展為去分化脂肪肉瘤比率小於 2%、死亡率近零。後腹膜、縱膈、腹腔、睪丸旁等深部者易局部復發、去分化風險超過 20%,後腹膜整體死亡率超過 80%。治療以手術為主,晚期加輔助化療與放療。
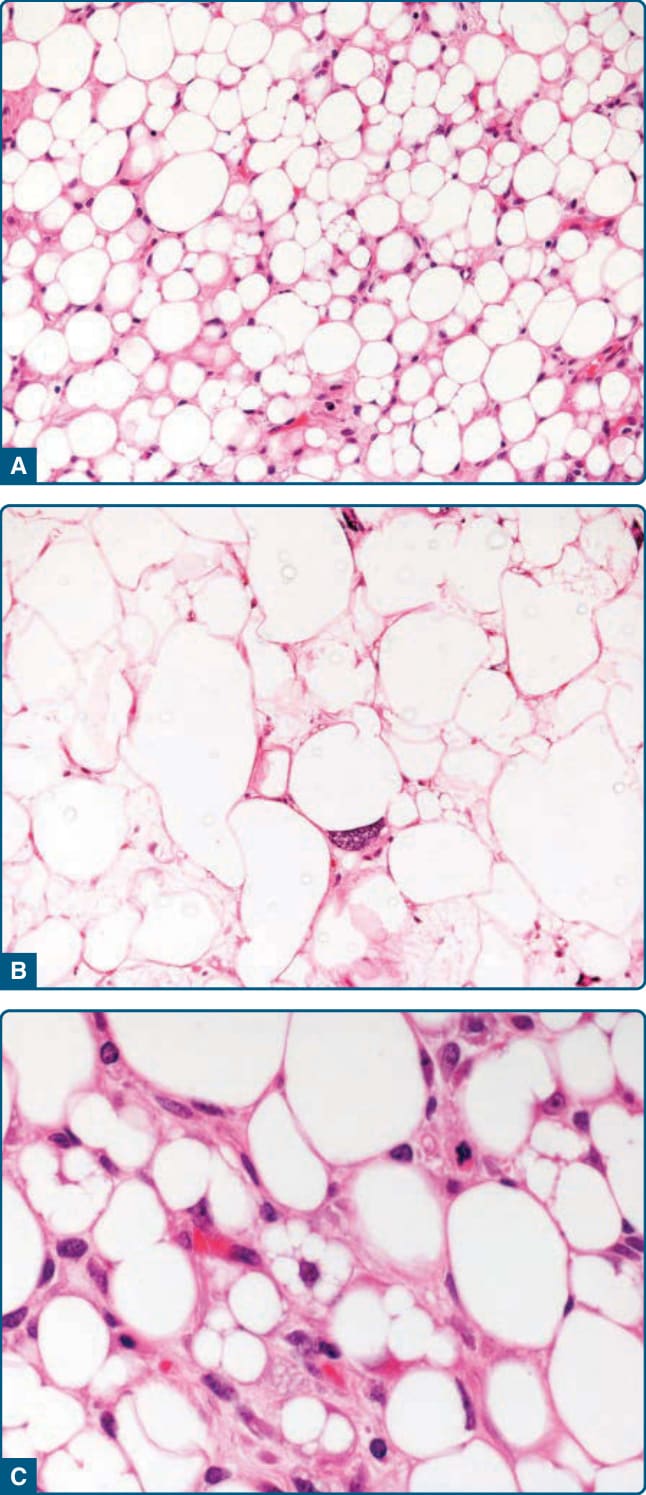
圖 122-28:非典型脂肪瘤樣腫瘤——脂肪瘤樣變異型,脂肪源性細胞大小形狀顯著變異,散在增大深染細胞核與多空泡化脂肪母細胞。
去分化脂肪肉瘤 (Dedifferentiated Liposarcoma)
- 非典型脂肪瘤樣腫瘤的型態學進展形式;最常見於後腹膜與腹腔,呈大型腫塊。約 90% 為新發 (de novo),10% 在局部復發中發展。最佳視為中度惡性(grade 2)肉瘤。
- 病理:從非典型脂肪瘤樣腫瘤區域到非脂肪源性肉瘤區的突然或漸進過渡;非脂肪源性成分多為高度惡性未分化多形性肉瘤,後腹膜常見黏液纖維肉瘤;約 10% 有異源性分化(肌源性、骨肉瘤樣/軟骨肉瘤樣最常見)。p16/MDM2/CDK4 強表現(尤非脂肪源性成分)。
- 致病機轉:核型同非典型脂肪瘤樣腫瘤,另有 1p32 與 6q23 共擴增伴 JUN、ASK2 活化。
- 病程/預後:病程漫長、局部復發率高;遠端轉移風險約 15-20%(較其他高度惡性肉瘤低,推測與缺乏複雜核型畸變及 p53 突變罕見有關)。部位為最重要預後因子,後腹膜/腹腔內者預後差;肌源性分化與非脂肪源性成分惡性度為重要預後參數。完全切除為首選但常不可能;不可切除者討論用第 12 號染色體基因產物標靶或 Akt-mTOR、MAPK 途徑抑制劑。
黏液樣脂肪肉瘤 (Myxoid Liposarcoma)
- 第二常見脂肪肉瘤變異型,約占成人所有軟組織肉瘤 5%;好發年輕至中年成人四肢深部軟組織(尤大腿),亦為兒童/青少年最常見脂肪肉瘤變異型。
- 病理:小型原始間葉細胞+不同分化階段脂肪母細胞,位於明顯黏液樣基質中,伴特徵性分枝狀薄壁血管。圓細胞成分 (round cell component) 出現具不良預後意義。S-100 在高惡性圓細胞成分不等程度陽性。
- 致病機轉:t(12;16)(q13;p11) 伴 FUS-DDIT3 融合(多數);較罕見 t(12;22) 伴 EWSR1-DDIT3。
- 病程/預後:易復發,高達 40% 病人發生遠端轉移(常在軟組織或骨等不尋常部位)。表現時即多病灶者預後不佳;不良組織學特徵包括圓細胞分化(>5%)、腫瘤壞死、p53 過度表現、CDKN2A 畸變。晚期可用曲貝替定 (trabectedin) 成功治療。
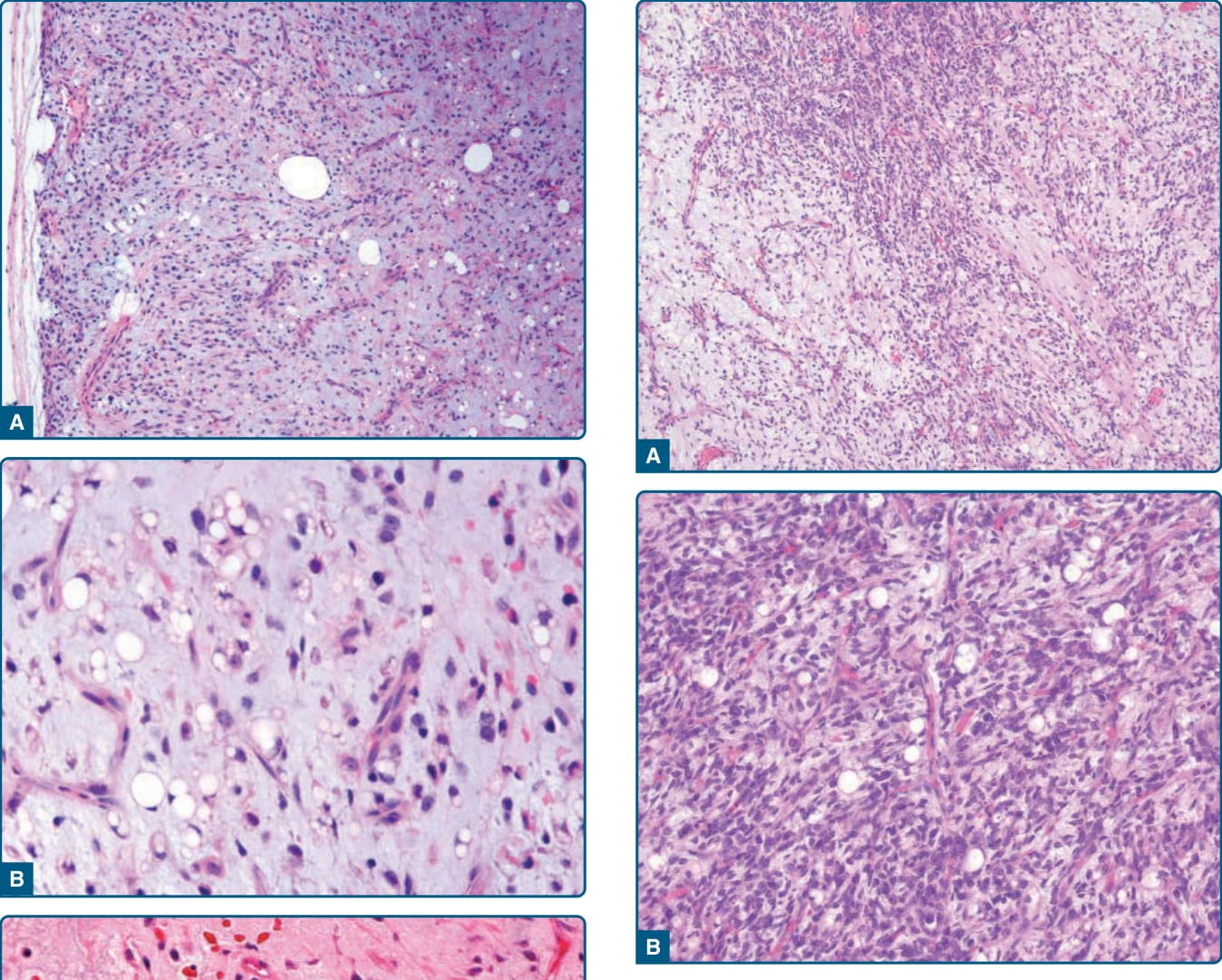
圖 122-36:黏液樣脂肪肉瘤——分葉狀黏液樣腫瘤,含脂肪母細胞與纖細分枝狀微血管。
多形性脂肪肉瘤 (Pleomorphic Liposarcoma)
- 脂肪肉瘤最罕見亞型(5%),高度惡性,好發老年人四肢(下肢 > 上肢),略以男性為主。可新發或為非典型脂肪瘤樣腫瘤/去分化脂肪肉瘤之進展。
- 病理:高度惡性多形性肉瘤(高度惡性纖維肉瘤或「惡性纖維組織細胞瘤」)含多形性梭形細胞與多核巨細胞,混有多形性脂肪母細胞(細胞核增大怪異深染、被脂滴扇形圍繞)。上皮樣變異型似腎透明細胞癌/腎上腺皮質癌。腫瘤細胞可表現肌動蛋白、細胞角蛋白、CD34、罕見結蛋白。
- 致病機轉:多與其他多形性肉瘤相似(複雜結構重排);少數有 MDM2/CDK4 擴增。
- 病程/預後:高度惡性,局部復發與轉移率 30-50%,整體 5 年存活率約 50-60%,肺為優先轉移部位。治療為手術切除,晚期加放療與化療。