脂肪源性腫瘤 (Lipogenic Neoplasms)
20
重點一覽 (AT-A-GLANCE)
■ 脂肪源性腫瘤 (lipogenic neoplasms) 是最常見的間葉性腫瘤 (mesenchymal neoplasms)。
■ 脂肪源性腫瘤包括良性腫瘤(如脂肪瘤 lipoma、血管脂肪瘤 angiolipoma)、中間型且局部侵襲性但不轉移的腫瘤(如非典型脂肪瘤樣腫瘤 atypical lipomatous tumor),以及明確惡性的病灶(如多形性脂肪肉瘤 pleomorphic liposarcoma)。
■ 解剖部位、深度與大小造成發生率、型態與預後上的顯著差異。
■ 脂肪源性腫瘤的臨床表現通常不具特異性。
■ 大多數脂肪源性腫瘤的特徵為獨特的細胞遺傳學異常 (cytogenetic abnormalities),可能具有診斷與治療價值。
■ 手術切除是首選治療;在無法完全切除的惡性腫瘤病例中,可使用放射治療 (radiation therapy)、化學治療 (chemotherapy) 或標靶類治療 (target-like therapy)。
雖然脂肪源性腫瘤是皮膚與軟組織中最大的單一類間葉性腫瘤,但淺表性與深部脂肪源性腫瘤在發生率、型態特徵與預後上存在顯著差異,了解這些差異對於避免誤診與治療錯誤相當重要。深部軟組織的脂肪肉瘤 (liposarcomas) 是成人最常見的肉瘤 (sarcomas),但在純真皮 (purely dermal) 位置則極為罕見。發生於皮下組織 (subcutis) 的梭形細胞/多形性脂肪瘤 (spindle-cell/pleomorphic lipoma) 病例具有包膜,且主要發生於老年男性病人的頸部與肩部區域。相對地,純真皮的梭形細胞/多形性脂肪瘤則為界線不清、浸潤性的病灶,男女發生率相等,並呈現廣泛的解剖分布。非典型脂肪瘤樣腫瘤(「高分化脂肪肉瘤 well-differentiated liposarcoma」)的預後與腫瘤的部位及大小密切相關。發生於後腹膜 (retroperitoneum)、腹腔內 (intraabdominal)、縱膈 (mediastinum) 與精索 (spermatic cord) 的非典型脂肪瘤樣腫瘤病例,臨床預後不佳。這些腫瘤常達到相當大的體積、反覆復發,並可能因無法控制的局部影響而在大量病例中導致死亡。形成鮮明對比的是,發生於四肢可手術處理之軟組織的非典型脂肪瘤樣腫瘤病例,在完全切除後通常不會復發,而一般而言,淺表性脂肪肉瘤的預後相對良好。
脂肪瘤 (LIPOMA)
重點一覽 (AT-A-GLANCE)
■ 脂肪瘤 (lipoma) 是成人最常見的良性間葉性腫瘤之一。
■ 脂肪瘤由成熟的白色脂肪細胞 (white adipocytes) 組成。
■ 脂肪瘤呈現不同的細胞遺傳學異常。
■ 雖然手術切除可治癒,但肌肉內脂肪瘤 (intramuscular lipoma) 病例顯示較高的復發率。
流行病學 (EPIDEMIOLOGY)
脂肪瘤是最常見的良性間葉性腫瘤,傾向發生於成人,無性別偏好。兒童期病例與多發性病灶罕見。
臨床特徵 (CLINICAL FEATURES)
脂肪瘤通常表現為侵犯軀幹、頸部或四肢皮下組織的無痛、緩慢生長腫塊;手部與足部受侵犯並不常見。淺表性脂肪瘤通常較小,測量小於 5 cm。相對地,深部肌肉內 (intramuscular) 與肌肉間 (intermuscular) 脂肪瘤可能達到相當大的體積。骨旁脂肪瘤 (Parosteal lipoma) 發生於骨表面,而所謂的樹枝狀脂肪瘤 (lipoma arborescens) 代表滑膜 (synovial membrane) 的絨毛狀脂肪瘤樣增生。¹ 脂肪瘤可作為加德納症候群 (Gardner syndrome) 的表現,而多發性脂肪瘤合併巨頭症 (macrocephaly)、淋巴管瘤 (lymphangiomas) 與血管瘤 (hemangiomas) 則見於班納揚症候群 (Bannayan syndrome)(表 122-1)。²
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
脂肪瘤在細胞遺傳學上具有異質性,最常見的異常包括涉及 12q13-15 的畸變、涉及 6p21-23 的重排,以及涉及 13q 的缺失。³ 位於 12q14.3 的 HMGA2 基因在某些脂肪瘤的致病機轉中扮演重要角色,且已有 HMGA2 與數個基因重組的報告。⁴
| 症候群 (Syndrome) | 相關腫瘤 (Associated Tumors) |
|---|---|
| 加德納症候群 (Gardner syndrome) | 家族性腺瘤性息肉病 (familial adenomatous polyposis)、硬纖維瘤病 (desmofibromatosis)、脂肪瘤 (lipomas)、骨瘤 (osteomas)、囊腫 (cysts) |
| 班納揚-萊利-魯瓦卡巴症候群 (Bannayan-Riley-Ruvalcaba syndrome) | 巨頭症 (macrocephaly)、脂肪瘤、淋巴管瘤 (lymphangiomas)、錯構瘤 (hamartomas)、腸道息肉病 (intestinal polyposis)、雀斑樣痣 (lentigines) |
| 考登症候群 (Cowden syndrome) | 錯構瘤 (hamartomas)、毛根鞘瘤 (trichilemmomas)、脂肪瘤、口腔乳頭狀瘤 (oral papillomas)、掌蹠角化 (palmoplantar keratoses) |
| 戈登哈-戈林症候群 (Goldenhar-Gorlin syndrome) | 顱內脂肪瘤 (intracranial lipomas)、水腦症 (hydrocephalus)、眼-耳-脊椎發育不良 (oculoauriculovertebral dysplasia) |
| 肌陣攣性癲癇與襤褸紅纖維症候群 (Myoclonus epilepsy and ragged-red fibers syndrome) | 對稱性脂肪瘤 (symmetric lipomas) |
| CLOVE 症候群 (CLOVE syndrome) | 先天性脂肪瘤樣過度生長 (congenital lipomatous overgrowth)、血管畸形 (vascular malformations)、表皮痣 (epidermal nevi) |
| 埃克博姆症候群 (Ekbom syndrome) | 頸部脂肪瘤 (cervical lipomas)、小腦性共濟失調 (cerebellar ataxia)、光肌陣攣 (photomyoclonus)、骨骼畸形 (skeletal deformities) |
組織學特徵 (HISTOLOGIC FEATURES)
典型位於皮下的脂肪瘤是界線清楚、有包膜且分葉狀的病灶,由成熟脂肪細胞組成,伴有含薄壁微血管的細薄、細胞稀少之纖維性間隔 (fibrous septa)(圖 122-1)。脂肪細胞的大小與形狀僅有輕微變異,且不存在細胞核非典型 (nuclear atypia)。免疫組織化學上,腫瘤細胞對 p16、MDM2 與 CDK4 呈陰性。脂肪瘤病例可能顯示伴有脂肪壞死 (fat necrosis) 與發炎的創傷後變化,且大量組織細胞 (histiocytes) 的存在——這些組織細胞含有增大的細胞核並對 p16、MDM2 與 CDK4 顯示細胞質染色——可能模擬較具侵襲性的非典型脂肪瘤樣腫瘤的特徵。脂肪瘤病例可能含有豐富的纖維組織(纖維脂肪瘤 fibrolipoma)、軟骨(軟骨脂肪瘤 chondrolipoma)或骨(骨脂肪瘤 osteolipoma)(圖 122-2)。純真皮脂肪瘤無包膜,且為界線相當不清的病灶(圖 122-3),若病灶內存在汗管與汗腺,則使用腺脂肪瘤 (adenolipoma) 一詞。肌肉內與肌肉間脂肪瘤是界線不清、浸潤性的腫瘤(圖 122-4),且原有的骨骼肌纖維常顯示萎縮 (atrophy) 的特徵。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
血管脂肪瘤 (Angiolipoma) 的特徵為含有纖維蛋白血栓 (fibrin thrombi) 之血管數目不等地增加,且常表現為疼痛性多發病灶。非典型脂肪瘤樣腫瘤病例的特徵為脂肪源性細胞在大小與形狀上的顯著變異,以及增大且深染 (hyperchromatic) 之細胞核的存在。非典型脂肪瘤樣腫瘤中的纖維性間隔常含有非典型細胞,且可能存在脂肪母細胞 (lipoblasts)。免疫組織化學上,非典型脂肪瘤樣腫瘤可見 p16 的局部表現,且在若干病例中可見 MDM2 與 CDK4 的局部核染色。螢光原位雜交 (fluorescence in situ hybridization, FISH) 分析在非典型脂肪瘤樣腫瘤病例中可揭示 MDM2 與 CDK4 的擴增。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
典型脂肪瘤完全為良性,且極少復發;相對地,肌肉內與肌肉間脂肪瘤顯示較高的局部復發率(高達 20%)。⁵ 原有脂肪瘤轉變為非典型脂肪瘤樣腫瘤是極為罕見的現象。⁶ 手術切除可治癒。
脂肪瘤病 (LIPOMATOSIS)
重點一覽 (AT-A-GLANCE)
■ 脂肪瘤病 (lipomatosis) 的特徵為成熟脂肪組織瀰漫性過度生長,浸潤穿過原有結構。
■ 已知有不同的臨床表現。
流行病學與臨床發現 (EPIDEMIOLOGY AND CLINICAL FINDINGS)
瀰漫性脂肪瘤病 (Diffuse lipomatosis) 主要發生於兒童,在成人罕見。其特徵為侵犯軀幹與四肢之皮下組織與骨骼肌的成熟脂肪源性組織瀰漫性過度生長;此外,可能見到骨性 (osseous) 侵犯。骨盆脂肪瘤病 (Pelvic lipomatosis) 較常見於黑人男性,而對稱性脂肪瘤病(馬德隆病 Madelung disease)則發生於地中海血統的中年男性,並好發於頸部、肩部與上肢近端。此外,周邊神經病變 (peripheral neuropathy) 是這些病人的常見發現。接受蛋白酶抑制劑 (protease inhibitors) 治療的 HIV 陽性病人可能發展出脂肪失養症 (lipodystrophy),於頸部、乳房與內臟器官有脂肪組織增加,而類固醇脂肪瘤病 (steroid lipomatosis) 則見於接受荷爾蒙治療或內源性腎上腺皮質類固醇 (adrenocortical steroids) 生成增加的病人。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
一般而言,脂肪瘤病的基本機轉尚未充分了解;然而,已有與粒線體功能障礙 (mitochondrial dysfunction) 相關的報告,並已發現粒線體 DNA 的多重缺失以及粒線體基因的點突變。⁷
組織學特徵 (HISTOLOGIC FEATURES)
所有不同亞型脂肪瘤病的大體外觀與組織學特徵都相同。可見界線不清、浸潤性、柔軟、黃色、與正常脂肪相同的脂肪組織。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
脂肪組織的姑息性手術切除是首選治療;然而,所有形式的脂肪瘤病都傾向復發,且脂肪組織的大量堆積可能造成相當大的臨床問題。
神經脂肪瘤病 (LIPOMATOSIS OF NERVE)
重點一覽 (AT-A-GLANCE)
■ 神經脂肪瘤病 (lipomatosis of nerve) 是見於嬰兒與兒童的罕見病灶。
■ 神經脂肪瘤病是一種生長中的腫塊,好發於手部。
■ 神經脂肪瘤病顯示脂肪與纖維組織在神經外膜 (epineurium) 與神經束膜 (perineurium) 內的增生,主要發生於正中神經 (median nerve)。
流行病學 (EPIDEMIOLOGY)
神經脂肪瘤病,亦稱為神經的纖維脂肪瘤性錯構瘤 (fibrolipomatous hamartoma of nerve),最常發生於出生時或兒童早期,並可能與受影響神經所支配之指(趾)的巨指(趾)症 (macrodactyly) 相關。⁸
臨床特徵 (CLINICAL FEATURES)
臨床上,病人表現為緩慢生長的腫塊,常伴隨疼痛、感覺異常 (paresthesia),以及感覺或運動缺損。正中神經及其分支,其次是尺神經 (ulnar nerve),最常受影響,而顱神經 (cranial nerves) 與臂神經叢 (brachial plexus) 的受侵犯則僅罕見。⁹,¹⁰
組織學特徵 (HISTOLOGIC FEATURES)
大體上,可見受影響神經呈梭形腫大,組織學上,受影響神經的神經外膜與神經束膜間隔被成熟脂肪組織與膠原性纖維組織浸潤(圖 122-5)。可見同心圓狀的神經束膜纖維化 (perineurial fibrosis),且罕見地有化生性骨形成 (metaplastic bone formation) 的報告。¹¹
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
神經脂肪瘤病是完全良性的病灶;手術切除可能造成受影響神經的嚴重損傷。
淺表性脂肪瘤樣痣 (NEVUS LIPOMATOSIS SUPERFICIALIS)
重點一覽 (AT-A-GLANCE)
■ 淺表性脂肪瘤樣痣(霍夫曼-楚爾黑勒 Hoffmann-Zurhelle)代表一種罕見類型的結締組織痣 (connective tissue nevus),侵犯兒童與年輕成人。
■ 淺表性脂肪瘤樣痣通常表現為斑塊狀、單發的病灶,主要發生於臀部、腰背部與大腿後側。
■ 病灶由成熟脂肪組織與結締組織成分組成。
流行病學 (EPIDEMIOLOGY)
淺表性脂肪瘤樣痣 (Nevus lipomatosus superficialis) 是一種罕見形式的結締組織痣,通常侵犯生命前幾十年的兒童與年輕成人,性別分布相等。
臨床特徵 (CLINICAL FEATURES)
淺表性脂肪瘤樣痣病例表現為斑塊、丘疹或單發病灶,好發於臀部、大腿後上側與腰背部。全身性形式極為罕見。病灶為單側性,有時可見線狀 (linear) 或帶狀皰疹樣 (zosteriform) 排列。
組織學特徵 (HISTOLOGIC FEATURES)
組織學上,這些界線不清的病灶由取代真皮的成熟脂肪組織組成。此外,可見膠原蛋白束 (collagen bundles) 的增厚,以及真皮較深部彈性纖維 (elastic fibers) 的增加與纖維母細胞 (fibroblasts) 的增加。覆蓋其上的表皮可能顯示輕微的棘層增厚 (acanthosis) 與過度色素沉著 (hyperpigmentation)(圖 122-6)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
罕見的真皮梭形細胞脂肪瘤 (dermal spindle-cell lipoma) 由成熟脂肪組織合併溫和的梭形腫瘤細胞組成,這些細胞對 CD34 呈陽性染色、顯示 Rb-1 表現的喪失,且常為黏液樣 (myxoid) 的基質含有玻璃樣變 (hyalinized) 膠原纖維與肥大細胞 (mast cells)。
脂肪母細胞瘤/脂肪母細胞瘤病 (LIPOBLASTOMA/LIPOBLASTOMATOSIS)
重點一覽 (AT-A-GLANCE)
■ 脂肪母細胞瘤/脂肪母細胞瘤病 (lipoblastomas/lipoblastomatosis) 是主要發生於嬰兒期與兒童早期的良性脂肪源性腫瘤。
■ 脂肪母細胞瘤/脂肪母細胞瘤病是界線清楚、分葉狀的病灶,由脂肪源性細胞組成,顯示從原始間葉細胞 (primitive mesenchymal cells) 與脂肪母細胞 (lipoblasts) 到成熟脂肪細胞的成熟譜系。
■ 脂肪母細胞瘤/脂肪母細胞瘤病顯示特徵性的遺傳學變化。
流行病學 (EPIDEMIOLOGY)
脂肪母細胞瘤/脂肪母細胞瘤病是罕見的、良性的胚胎性白色脂肪 (embryonal white fat) 腫瘤,主要發生於嬰兒與幼童,以男性為主。¹²⁻¹⁴ 腫瘤偶見為先天性或發生於年輕成人。¹⁵
臨床特徵 (CLINICAL FEATURES)
脂肪母細胞瘤/脂肪母細胞瘤病病例傾向發生於軀幹、四肢以及頭頸部區域,而內臟侵犯以及後腹膜、骨盆、縱膈與腹腔的侵犯則罕見。大多數病例表現為無痛、緩慢生長的病灶。脂肪母細胞瘤是界線相當清楚的腫瘤,而脂肪母細胞瘤病的特徵為浸潤性生長,常延伸至較深的結構。相當數目的病人患有中樞神經系統 (CNS) 疾患,例如癲癇發作 (seizures)、自閉症 (autism)、發展遲緩 (developmental delay)、先天性異常,以及/或史特格-韋伯症候群 (Sturge-Weber syndrome)。¹⁴
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在細胞遺傳學上,大多數經分析的脂肪母細胞瘤/脂肪母細胞瘤病病例顯示涉及第 8 號染色體的畸變,以及一個涉及染色體帶 8q11-13、伴有 PLAG1 基因重排的反覆性易位 (translocation)。¹⁶
組織學特徵 (HISTOLOGIC FEATURES)
脂肪母細胞瘤代表一個界線清楚、分葉狀的腫瘤,由成熟與未成熟脂肪源性細胞的混合組成,並由纖維血管性間隔 (fibrovascular septa) 分隔(圖 122-7)。在顯示浸潤性生長模式的脂肪母細胞瘤病病例中,分葉較不明顯。脂肪母細胞處於不同的發育階段,且可能存在數量不等的黏液樣基質,含有原始梭形細胞與叢狀血管模式 (plexiform vascular pattern)(讓人聯想到黏液樣脂肪肉瘤 myxoid liposarcoma 的血管模式)(圖 122-8 與 122-9)。即使某些腫瘤主要由成熟脂肪細胞組成(作為成熟的表現),其特徵性的分葉狀生長與纖維性間隔的存在仍是診斷的有用線索。偶爾可見纖維母細胞性增生、軟骨樣化生 (chondroid metaplasia) 或髓外造血 (extramedullary hematopoiesis)。脂肪源性細胞顯示 S-100 蛋白、PLAG1 與 CD34 的表現,¹⁴ 梭形細胞常對結蛋白 (desmin) 呈陽性染色。¹⁷ 有趣的是,脂肪母細胞瘤病例可能對 p16 呈陽性染色,這在與非典型脂肪瘤樣腫瘤的區分上代表一個診斷陷阱。¹⁸
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
黏液樣脂肪肉瘤 (Myxoid liposarcoma) 是脂肪母細胞瘤/脂肪母細胞瘤病最重要的鑑別診斷之一;然而,此診斷在小於 10 歲的病人中極為罕見。在較大的兒童、青少年與年輕成人中,鑑別可能相當棘手。顯著的分葉與缺乏深染細胞核支持脂肪母細胞瘤/脂肪母細胞瘤病的診斷;此外,不同的分子變化有助於正確診斷。進一步的鑑別診斷包括脂肪瘤、脂肪纖維瘤病 (lipofibromatosis)、冬眠瘤 (hibernoma) 與非典型脂肪瘤樣腫瘤。罕見的外陰脂肪母細胞瘤樣腫瘤 (lipoblastoma-like tumor of the vulva) 發生於成人;該腫瘤缺乏 PLAG1 表現且顯示 Rb-1 的喪失。¹⁹
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
脂肪母細胞瘤/脂肪母細胞瘤病儘管具有局部侵犯的潛力以及在若干病例中所見的相當大體積,預後仍極佳。已報告的局部復發率高達 46%,¹⁴ 這是由不完全切除所致,或局限於瀰漫型病灶(脂肪母細胞瘤病)。沒有腫瘤進展、轉移或惡性轉化的風險。
血管脂肪瘤 (ANGIOLIPOMA)
重點一覽 (AT-A-GLANCE)
■ 血管脂肪瘤 (angiolipoma) 是一種常見、常為疼痛性、完全良性的脂肪源性病灶。
■ 血管脂肪瘤侵犯年輕成人;常見多發病灶。
■ 血管脂肪瘤含有數量不等的薄壁血管,這些血管可能含有纖維蛋白血栓 (fibrin thrombi)。
流行病學 (EPIDEMIOLOGY)
皮下血管脂肪瘤是常見的間葉性腫瘤,主要發生於年輕男性;已有家族性發生的描述。²⁰
臨床特徵 (CLINICAL FEATURES)
血管脂肪瘤傾向為多病灶,常表現為疼痛且觸痛的皮下病灶,主要發生於前臂,其次為軀幹、上臂與腿部。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
與脂肪瘤相反,所有被研究的血管脂肪瘤都具有正常核型 (normal karyotype),而由於細胞性血管脂肪瘤 (cellular angiolipoma) 病例可能幾乎完全由血管組成,因此有人推測這些病灶代表血管瘤 (hemangiomas) 而非脂肪瘤。²¹
組織學特徵 (HISTOLOGIC FEATURES)
組織學上,血管脂肪瘤是界線清楚、有包膜的結節狀病灶,由成熟脂肪細胞、數量不等的薄壁微血管(可能含有纖維蛋白血栓)以及基質梭形細胞 (stromal spindled cells) 組成(圖 122-10 與 122-11)。在細胞性血管脂肪瘤病例中,血管占主導地位,而脂肪源性細胞可能缺如(圖 122-12)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
纖維蛋白血栓的存在與臨床特徵有助於將血管脂肪瘤與血管數目增加的脂肪瘤病例區分。細胞性血管脂肪瘤必須與血管性腫瘤區分,例如卡波西肉瘤 (Kaposi sarcoma)、卡波西樣血管內皮瘤 (kaposiform hemangioendothelioma)、梭形細胞血管瘤 (spindle-cell hemangioma) 與血管肉瘤 (angiosarcoma)。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
血管脂肪瘤的臨床行為完全為良性,單純切除即可治癒。
梭形細胞脂肪瘤與多形性脂肪瘤 (SPINDLE-CELL LIPOMA AND PLEOMORPHIC LIPOMA)
重點一覽 (AT-A-GLANCE)
■ 梭形細胞脂肪瘤 (spindle-cell lipoma) 與多形性脂肪瘤 (pleomorphic lipoma) 代表單一臨床病理實體的型態學連續譜。
■ 梭形細胞脂肪瘤與多形性脂肪瘤與乳腺型肌纖維母細胞瘤 (mammary myofibroblastoma) 及細胞性血管纖維瘤 (cellular angiofibroma) 相關。
■ 典型的梭形細胞脂肪瘤與多形性脂肪瘤病例以有包膜的皮下病灶形式出現,主要發生於老年男性的頸後部、肩部與上背部。
■ 純真皮的梭形細胞脂肪瘤與多形性脂肪瘤代表界線不清、浸潤性的病灶,呈現廣泛的解剖分布,且無性別偏好。
■ 梭形細胞脂肪瘤由成熟脂肪細胞與 CD34⁺、溫和的梭形細胞組成;多形性脂肪瘤中存在多核 (multinucleated)、CD34⁺ 的巨細胞。
■ 非典型梭形細胞脂肪瘤樣腫瘤 (atypical spindle cell lipomatous tumor) 與非典型多形性脂肪瘤樣腫瘤 (atypical pleomorphic lipomatous tumor) 代表中間型、局部侵襲性、不轉移的腫瘤。
流行病學 (EPIDEMIOLOGY)
梭形細胞脂肪瘤與多形性脂肪瘤構成單一臨床病理實體的型態學譜系,在臨床、型態與細胞遺傳學上有顯著重疊。大多數這類罕見腫瘤見於老年男性;極罕見地有報告指出受影響病人有多發病灶的家族性梭形細胞脂肪瘤。²² 相對地,純真皮脂肪瘤無性別偏好。²³
臨床特徵 (CLINICAL FEATURES)
梭形細胞脂肪瘤與多形性脂肪瘤通常表現為皮下組織中無症狀、常為長期存在的可移動腫瘤。大多數此類病灶見於頸後部、肩部與上背部。臉部、口腔與四肢的受影響並不常見。²⁴,²⁵ 純真皮的梭形細胞脂肪瘤與多形性脂肪瘤呈現廣泛的解剖分布。²³
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
梭形細胞脂肪瘤與多形性脂肪瘤的核型複雜,常為亞二倍體 (hypodiploid),伴有頻繁的部分缺失與少數平衡性重排。許多病例顯示涉及第 13 與 16 號染色體的單體 (monosomy) 與部分缺失。³ 鑑於梭形細胞脂肪瘤、細胞性血管纖維瘤與乳腺型肌纖維母細胞瘤之間在臨床、組織學與分子特徵上的重疊,這些腫瘤可能代表單一臨床病理實體譜系上的不同點。²⁶,²⁷
組織學特徵 (HISTOLOGIC FEATURES)
皮下梭形細胞脂肪瘤代表一個有包膜的病灶,由成熟脂肪細胞與數量不等、細胞學上溫和的梭形腫瘤細胞組成,這些細胞位於含有血管、玻璃樣變繩索狀 (rope-like) 膠原纖維與常見肥大細胞的膠原性與黏液樣基質中(圖 122-13)。有絲分裂活性 (mitotic activity) 幾乎缺如。梭形細胞可能占主導地位,甚至有報告指出沒有脂肪源性細胞的病例,可能造成顯著的診斷問題(圖 122-14)。²⁸ 偶爾可見明顯的黏液樣基質變化,且在所謂的假血管瘤樣變異型 (pseudoangiomatous variant) 中存在許多類似血管腔的裂隙狀空間 (slit-like spaces)(圖 122-15)。²⁹
在其他方面典型的梭形細胞脂肪瘤病例中可能存在散在的脂肪母細胞。多形性脂肪瘤的特徵性特徵是額外存在帶有放射狀排列細胞核的多核巨細胞(花環狀巨細胞 floret-like giant cells;圖 122-16);亦已有低脂肪型與無脂肪型多形性脂肪瘤的報告。³⁰ 相對地,純真皮的梭形細胞脂肪瘤與多形性脂肪瘤病例無包膜且為浸潤性,模擬較具侵襲性的腫瘤(圖 122-17)。少數情況下,梭形細胞脂肪瘤與多形性脂肪瘤病例發生於不尋常的解剖部位,並顯示浸潤性模式與脂肪源性細胞、梭形細胞及多形性巨細胞的顯著非典型(圖 122-18)。這些病例被指定為非典型梭形細胞脂肪瘤樣腫瘤與非典型多形性脂肪瘤樣腫瘤,並顯示與非典型脂肪瘤樣腫瘤相當的臨床行為。³¹,³²,³²ᴬ,³²ᴮ 免疫組織化學上,梭形細胞與多形性腫瘤巨細胞對 CD34 呈陽性染色(圖 122-19),並顯示 Rb-1 表現的喪失(圖 122-20)。³³ 極罕見地,有梭形細胞表現結蛋白 (desmin) 與 S-100 蛋白的報告。³⁴,³⁵
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
非典型脂肪瘤樣腫瘤顯示脂肪源性細胞在大小與形狀上的顯著變異,以及伴有增大且深染細胞核存在的顯著非典型。此外,可能見到 MDM2 與 CDK4 的局部核表現;FISH 分析揭示 MDM2 與 CDK4 的擴增。孤立性纖維性腫瘤 (solitary fibrous tumor) 病例可能含有脂肪細胞,且腫瘤細胞同樣對 CD34 呈陽性染色。然而,孤立性纖維性腫瘤的特徵為細胞密度不等、存在血管外皮細胞瘤樣 (hemangiopericytoma-like) 血管,以及顯示 STAT6 核表現的腫瘤細胞。隆突性皮膚纖維肉瘤 (Dermatofibrosarcoma protuberans) 由 CD34⁺ 梭形細胞組成,然而,這些腫瘤顯示皮下組織瀰漫性蜂窩狀 (honey-comb) 浸潤與不同的遺傳學變化。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
梭形細胞脂肪瘤與多形性脂肪瘤完全為良性,雖然局部復發罕見地發生。非典型梭形細胞脂肪瘤樣腫瘤與多形性脂肪瘤樣腫瘤病例是局部侵襲性腫瘤,並顯示增加的局部復發率。³¹,³²
軟骨樣脂肪瘤 (CHONDROID LIPOMA)
重點一覽 (AT-A-GLANCE)
■ 軟骨樣脂肪瘤 (chondroid lipoma) 代表一種罕見的良性脂肪源性腫瘤。
■ 軟骨樣脂肪瘤較常見於女性病人。
■ 軟骨樣脂肪瘤主要發生於四肢的深部軟組織。
■ 軟骨樣脂肪瘤由脂肪細胞、脂肪母細胞與小型空泡化細胞 (vacuolated cells) 組成。
■ 軟骨樣脂肪瘤中的腫瘤細胞位於黏液樣–軟骨樣基質 (myxoid–chondroid matrix) 中。
■ 軟骨樣脂肪瘤病例中可見反覆性的 t(11;16)(q13;p13)。
流行病學 (EPIDEMIOLOGY)
軟骨樣脂肪瘤於 1993 年首次描述,³⁶ 是一種非常罕見的脂肪源性腫瘤,主要發生於成人,好發於女性。
臨床特徵 (CLINICAL FEATURES)
大多數軟骨樣脂肪瘤病例表現為無痛、深部的腫塊,可能顯示近期生長的病史。四肢近端與肢帶 (limb girdles) 是最常受影響的解剖部位,³⁶,³⁷ 但軀幹與頭頸部區域(包括口腔)³⁸ 也可能受影響。這些腫瘤通常發生於深部皮下組織、筋膜 (fascia) 與骨骼肌。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
超微結構研究確認了軟骨樣脂肪瘤病例中的脂肪源性分化,並已報告一個從原始間葉細胞(具有前脂肪母細胞 prelipoblasts 的特徵)到脂肪母細胞、前脂肪細胞 (preadipocytes) 與成熟脂肪細胞的分化譜系。³⁶,³⁹ 在細胞遺傳學上,已發現涉及第 11 與 16 號染色體的平衡性易位 t(11;16)(q13;p12-13),伴有 C11orf95-MKL2 融合基因產物,⁴⁰ 並已報告週期素 D1 (cyclin D1, CCND1) 的表現而無 CCND1 基因座的異常。⁴¹
組織學特徵 (HISTOLOGIC FEATURES)
軟骨樣脂肪瘤通常是深部、界線清楚、有包膜、結節狀且分葉狀的腫瘤。腫瘤由成熟脂肪細胞、具有增大且深染細胞核的多空泡化脂肪母細胞 (multivacuolated lipoblasts),以及含有嗜酸性 (eosinophilic) 且空泡化細胞質、顯示一系列脂肪母細胞分化並含有小型脂滴與過碘酸-希夫陽性 (periodic acid–Schiff–positive) 肝醣的小型圓形細胞,以數量不等的混合組成(圖 122-21、122-22 與 122-23)。腫瘤細胞位於黏液樣–軟骨樣基質中,低 pH 下的陽性愛爾遜藍染色 (Alcian blue stainings) 顯示硫酸軟骨素 (sulfated chondroitin) 的存在。病灶含有許多血管,且可能存在出血、纖維化與鈣化區域。成熟脂肪源性細胞對 S-100 蛋白呈陽性染色,且偶爾可見脂肪源性細胞對全細胞角蛋白 (pancytokeratin) 的局部表現。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑於病灶的深度與大小,再加上細胞核非典型與脂肪母細胞的存在,良性軟骨樣脂肪瘤必須與較具侵襲性的腫瘤區分,包括非典型脂肪瘤樣腫瘤、黏液樣脂肪肉瘤、骨骼外黏液樣軟骨肉瘤 (extraskeletal myxoid chondrosarcoma) 與軟組織肌上皮瘤 (myoepithelioma of soft tissues)。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
軟骨樣脂肪瘤代表一種完全良性的脂肪源性腫瘤,單純手術切除即可治癒。局部復發罕見,且截至本文撰寫時,尚未有惡性轉化或轉移的報告。
肌脂肪瘤 (MYOLIPOMA)
重點一覽 (AT-A-GLANCE)
■ 肌脂肪瘤 (myolipoma) 是一種非常罕見的深部軟組織良性間葉性腫瘤。
■ 肌脂肪瘤主要發生於女性。
■ 肌脂肪瘤由成熟脂肪細胞與平滑肌細胞 (smooth muscle cells) 的不規則混合組成。
流行病學 (EPIDEMIOLOGY)
肌脂肪瘤代表一種非常罕見的軟組織腫瘤,發生於成人病人,以女性為主。⁴²
臨床特徵 (CLINICAL FEATURES)
肌脂肪瘤病例傾向發生於後腹膜、腹腔與腹股溝 (inguinal) 區域的深部軟組織,而四肢與軀幹皮下組織的受侵犯則僅罕見。⁴² 雖然這些病灶可能達到相當大的體積,但常為偶然發現。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
肌脂肪瘤病例中已有 HMGA2 改變的報告,最近並偵測到 HMGA2 與 C9orf92 基因的融合,伴有 t(9;12)(p22;q14)。⁴³
組織學特徵 (HISTOLOGIC FEATURES)
軟組織肌脂肪瘤是界線清楚或有包膜的病灶,由細胞學上溫和的平滑肌細胞束 (fascicles) 與數量不等的成熟脂肪相關聯組成(圖 122-24)。不存在細胞學非典型、有絲分裂或厚壁血管。免疫組織化學上,平滑肌細胞對 α-平滑肌肌動蛋白 (alpha-smooth muscle actin)、結蛋白 (desmin) 與 h-鈣調素結合蛋白 (h-caldesmon) 呈陽性染色(圖 122-25),並已有雌激素 (estrogen) 與黃體素 (progesterone) 受體表現的報告。⁴⁴
平滑肌細胞不表現 HMB-45,脂肪源性細胞不表現 MDM2 與 CDK4。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
軟組織肌脂肪瘤的鑑別診斷包括非典型脂肪瘤樣腫瘤、去分化脂肪肉瘤 (dedifferentiated liposarcoma)、高分化平滑肌肉瘤 (well-differentiated leiomyosarcoma) 與血管肌脂肪瘤 (angiomyolipoma)。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
儘管體積龐大,軟組織肌脂肪瘤代表一種完全良性的間葉性腫瘤,完全切除即可治癒。
冬眠瘤 (HIBERNOMA)
重點一覽 (AT-A-GLANCE)
■ 冬眠瘤 (hibernoma) 代表一種棕色脂肪 (brown fat) 的良性腫瘤。
■ 冬眠瘤通常發生於成人,最常見的解剖部位是大腿。
■ 非典型脂肪瘤樣腫瘤與黏液樣脂肪肉瘤病例可能含有冬眠瘤樣區域。
流行病學 (EPIDEMIOLOGY)
冬眠瘤是一種罕見的腫瘤,主要發生於年輕成人,而在兒童與老年人中僅罕見。男性受影響的頻率略高於女性。
臨床特徵 (CLINICAL FEATURES)
冬眠瘤代表一種無痛、緩慢生長、可移動的腫瘤,發生於皮下或深部軟組織。冬眠瘤最常見的解剖部位是大腿,其次是軀幹、胸壁、肩部區域、上肢與頭頸部區域。⁴⁵ 罕見地,冬眠瘤發生於腹腔內、後腹膜、縱膈或骨內 (intraosseous)。⁴⁶,⁴⁷
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
冬眠瘤已報告的核型似乎比其他良性脂肪源性腫瘤的核型更為複雜,並已偵測到涉及染色體帶 11q13-21 的結構性重排。⁴⁸
組織學特徵 (HISTOLOGIC FEATURES)
冬眠瘤是界線清楚、分葉狀的腫瘤,含有微血管網,並由數量不等的棕色脂肪細胞與白色脂肪細胞及基質細胞混合組成。典型冬眠瘤由大型、多角形、多空泡化、具有顆粒狀細胞質與小型、位於中央之細胞核的棕色脂肪細胞,與成熟白色脂肪細胞混合組成(圖 122-26)。嗜酸性變異型 (eosinophilic variant) 主要由具有深嗜酸性與顆粒狀細胞質的棕色脂肪細胞組成,蒼白細胞變異型 (pale cell variant) 主要含有具有淺染空泡化細胞質的大型棕色脂肪細胞,而混合亞型則介於兩者之間。此外,已知有黏液樣、梭形細胞與脂肪瘤樣變異型。⁴⁵
不存在明顯的細胞學非典型與增加的增生活性。免疫組織化學上,腫瘤細胞對 S-100 蛋白與解偶聯蛋白 (uncoupling protein)(一種棕色脂肪細胞粒線體特有的蛋白)呈陽性染色。⁴⁹ 梭形細胞可能表現 CD34。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
散在的棕色脂肪細胞以及冬眠瘤樣區域可能存在於黏液樣脂肪肉瘤與非典型脂肪瘤樣腫瘤病例中,必須與冬眠瘤區分。進一步的鑑別診斷包括顆粒細胞瘤 (granular cell tumor) 與梭形細胞脂肪瘤。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
冬眠瘤是一種完全良性的脂肪源性腫瘤,完全切除即可治癒。
非典型脂肪瘤樣腫瘤與高分化脂肪肉瘤 (ATYPICAL LIPOMATOUS TUMOR AND WELL-DIFFERENTIATED LIPOSARCOMA)
重點一覽 (AT-A-GLANCE)
■ 非典型脂肪瘤樣腫瘤 (atypical lipomatous tumor) 與高分化脂肪肉瘤 (well-differentiated liposarcoma) 是同義詞,描述具有相同型態與細胞遺傳學特徵的脂肪源性腫瘤。
■ 非典型脂肪瘤樣腫瘤代表一種局部侵襲性、不轉移的腫瘤。
■ 病灶的解剖位置、大小與深度是重要的預後預測因子。
■ 發生於可手術切除部位(即四肢)的腫瘤顯示低局部復發率、低去分化 (dedifferentiation) 風險與低死亡率。
■ 形成鮮明對比的是,發生於後腹膜、腹腔內或睪丸旁 (paratesticular) 位置——這些部位難以或無法完全切除——的腫瘤,其特徵為高局部復發率、高去分化風險與不良的臨床結果。
■ 已知的型態學變異型沒有預後價值。
流行病學 (EPIDEMIOLOGY)
非典型脂肪瘤樣腫瘤代表臨床上侵襲性脂肪源性腫瘤中最大的單一類別,而脂肪肉瘤整體而言是成人最常見的肉瘤。大多數腫瘤發生於中年成人,發生率高峰在第六個十年(60 多歲)。⁵⁰⁻⁵⁴ 兒童病例極為罕見,但已有描述。⁵⁵,⁵⁶
臨床特徵 (CLINICAL FEATURES)
非典型脂肪瘤樣腫瘤病例通常發生於深部軟組織。皮下組織偶爾受侵犯,但可能表現為皮膚贅生物 (skin tag) 的純真皮病灶則極為罕見(圖 122-27)。⁵⁷
主要受影響的部位是四肢的肌肉組織,尤其是大腿,其次是後腹膜、腹腔、腹股溝、睪丸旁區域與縱膈。然而,其他解剖位置也罕見地受侵犯,如口腔、眼眶 (orbit)、喉部 (larynx) 與外陰。⁵⁸⁻⁶¹ 不斷增大的無痛腫塊是典型的表現徵象,腫瘤在出現症狀之前可能生長至非常大的體積,尤其在後腹膜與腹腔內位置。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
非典型脂肪瘤樣腫瘤病例在細胞遺傳學上的特徵為衍生自 12q13-15 區域的額外環狀與巨大標記染色體 (supernumerary ring and giant marker chromosomes),並導致 MDM2 與 CDK4 基因的一致性擴增。⁶²,⁶³
組織學特徵 (HISTOLOGIC FEATURES)
非典型脂肪瘤樣腫瘤通常是大型、界線清楚且分葉狀的病灶,在後腹膜與腹腔內位置,可能存在多個不連續的腫瘤。型態上,非典型脂肪瘤樣腫瘤可分為 3 種主要亞型;然而,尤其在後腹膜的大型腫瘤中,可能存在超過 1 種變異型。最常見的脂肪細胞型(脂肪瘤樣 lipoma-like)變異型由相對成熟、在大小與形狀上有顯著變異、顯示散在增大且深染細胞核的脂肪細胞性細胞組成。此外,可能存在位於纖維性間隔中具有增大細胞核的非典型基質細胞,以及多空泡化脂肪母細胞;然而,脂肪母細胞可能完全缺如(圖 122-28)。第二常見的硬化型 (sclerosing variant) 常見於後腹膜與睪丸旁位置,其特徵為存在具有大型且深染細胞核的增大基質細胞,位於細胞稀少、纖維狀、膠原性的基質中,以及散在的非典型脂肪源性細胞與脂肪母細胞(圖 122-29)。罕見的發炎性非典型脂肪瘤樣腫瘤 (inflammatory atypical lipomatous tumors) 主要見於後腹膜與腹腔內位置,並顯示明顯的發炎性浸潤,模擬淋巴瘤 (lymphoma) 或發炎性假瘤 (inflammatory pseudotumor) 的特徵。⁶⁴ 所謂的高分化梭形細胞脂肪肉瘤 (well-differentiated spindle-cell liposarcoma)⁶⁵ 好發於肩帶 (shoulder girdle) 與四肢的皮下組織,由略具非典型、具有增大、深染細胞核並排列成束與漩渦狀、位於纖維性與/或黏液樣基質中、與非典型脂肪細胞性細胞相關聯的梭形細胞組成。遺傳學研究強調這些腫瘤最可能代表非典型梭形細胞脂肪瘤樣腫瘤,其臨床行為幾乎與非典型脂肪瘤樣腫瘤相同。³¹,³²,⁶⁶ 極罕見地,可見伴有骨性或肌源性成分的異源性分化 (heterologous differentiation)。脂肪平滑肌肉瘤 (lipoleiomyosarcoma) 一詞曾被用於顯示非典型脂肪瘤樣腫瘤與高分化平滑肌肉瘤型態特徵的病灶。⁶⁷ 免疫組織化學上,p16 的局部表現以及 MDM2 和/或 CDK4 的核染色,有助於與其他脂肪源性與非脂肪源性病灶的鑑別診斷。然而,尤其是非常高分化的非典型脂肪瘤樣腫瘤範例,免疫組織化學上可能不顯示 MDM2 和/或 CDK4 表現。在這些病例中,FISH 分析可能非常有幫助。⁶⁸,⁶⁹
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
與非典型脂肪瘤樣腫瘤相反,典型的脂肪瘤病例由成熟脂肪細胞組成,沒有細胞核非典型與脂肪母細胞,且對 p16、MDM2 與 CDK4 不呈陽性染色。然而,受創的脂肪瘤可能含有大量具增大細胞核的組織細胞,這些細胞顯示對 p16、MDM2 與 CDK4 的細胞質染色,並可能模擬較具侵襲性的腫瘤。脂肪母細胞瘤由數量不等的脂肪母細胞組成,並可能對 p16 呈陽性染色,¹⁸ 但具有特徵性間隔的分葉狀病灶的存在以及缺乏 MDM2/CDK4 表現有助於鑑別診斷。梭形細胞脂肪瘤與多形性脂肪瘤可能含有散在的脂肪母細胞,但缺乏非典型,且腫瘤細胞對 MDM2 和/或 CDK4 呈陰性。去分化脂肪肉瘤含有強烈表現 p16、MDM2 與 CDK4 的非脂肪源性成分。血管肌脂肪瘤的特徵為存在厚壁血管與一個血管周圍肌源性成分,後者對肌源性標記與 HMB-45 呈陽性染色。罕見的巨大局限性淋巴水腫 (massive localized lymphedema) 病例在臨床與組織學上模擬非典型脂肪瘤樣腫瘤,⁷⁰ 但這些病灶中沒有明顯的非典型,也沒有 p16、MDM2 與 CDK4 的表現。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
非典型脂肪瘤樣腫瘤的臨床行為與預後高度取決於腫瘤的解剖部位、深度與大小。對於發生於淺表處以及可手術處理之解剖部位(如四肢)的病灶,手術治療可治癒。對於發生於這些解剖位置的病例,進展為去分化脂肪肉瘤的估計比率小於 2%,死亡率幾乎為零。形成鮮明對比的是,發生於後腹膜、縱膈、腹腔與睪丸旁位置等深部軟組織的非典型脂肪瘤樣腫瘤病例——這些部位的完全手術切除困難且不可能——傾向局部復發,可能因無法控制的局部影響而導致死亡,且去分化風險超過 20%。後腹膜非典型脂肪瘤樣腫瘤的整體死亡率超過 80%。⁵³,⁷¹ 手術切除是非典型脂肪瘤樣腫瘤病例的主要治療方法;在晚期病例中可使用輔助性化學治療與放射治療。
去分化脂肪肉瘤 (DEDIFFERENTIATED LIPOSARCOMA)
重點一覽 (AT-A-GLANCE)
■ 去分化脂肪肉瘤 (dedifferentiated liposarcoma) 代表非典型脂肪瘤樣腫瘤的型態學進展形式。
■ 去分化脂肪肉瘤最常以大型腫塊的形式發生於後腹膜與腹腔。
■ 去分化脂肪肉瘤中可見從非典型脂肪瘤樣腫瘤到非脂肪源性肉瘤成分的突然或漸進性過渡。
■ 非脂肪源性成分顯示多變的型態,且在相當數目的病例中可見異源性分化。
■ 去分化脂肪肉瘤顯示與非典型脂肪瘤樣腫瘤相同的核型變化。
■ 去分化脂肪肉瘤最好被視為中度惡性肉瘤(惡性度第 2 級 grade 2)。
流行病學 (EPIDEMIOLOGY)
去分化脂肪肉瘤代表後腹膜最常見的多形性肉瘤 (pleomorphic sarcoma),典型地影響第六個十年(60 多歲)的成人病人。⁷² 去分化作為腫瘤進展的一種型態學形式,代表一種時間依賴性現象,約 90% 的病例以新發 (de novo) 表現出現,而 10% 在局部復發中發展出來。
臨床特徵 (CLINICAL FEATURES)
大多數去分化脂肪肉瘤發生於後腹膜、腹腔內與睪丸旁位置,而四肢深部軟組織的受侵犯則較罕見。其他罕見位置包括軀幹與頭頸部區域,在皮下組織的表現則非常罕見。⁷³ 典型地,去分化脂肪肉瘤病例表現為大型無痛腫塊,並在後腹膜與腹腔內位置偶然被發現。四肢中長期存在之深部腫塊近期體積增大,可能指示非典型脂肪瘤樣腫瘤中的去分化。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
去分化脂肪肉瘤的核型發現與非典型脂肪瘤樣腫瘤所見的相似;然而,已有涉及 1p32 與 6q23、伴有 JUN 與 ASK2 基因活化的共擴增 (coamplification) 報告。⁷⁴
組織學特徵 (HISTOLOGIC FEATURES)
組織學上,從顯示非典型脂肪瘤樣腫瘤特徵的區域到非脂肪源性肉瘤區域的突然或漸進性過渡是去分化脂肪肉瘤的特徵(圖 122-30)。非脂肪源性成分的範圍與型態是多變的,但在大多數病例中,去分化成分顯示高度惡性、未分化、多形性肉瘤的特徵(圖 122-31)。尤其在後腹膜與腹腔內位置,存在中度惡性或高度惡性黏液纖維肉瘤 (myxofibrosarcoma) 區域。偶爾可見低度惡性去分化,模擬低度惡性纖維肉瘤 (low-grade fibrosarcoma) 或硬纖維瘤病 (desmoid fibromatosis) 的特徵(圖 122-32)。異源性分化見於約 10% 的病例,且最常偵測到肌源性或骨肉瘤樣/軟骨肉瘤樣 (osteosarcomatous/chondrosarcomatous) 分化(圖 122-33)。較罕見地,已有血管肉瘤樣 (angiosarcomatous) 與腦膜上皮樣 (meningothelial-like) 漩渦的報告。⁷⁵
有趣的是,去分化成分可能顯示類似多形性脂肪肉瘤的脂肪母細胞性分化,但分子變化與典型去分化脂肪肉瘤相同。⁷⁶
免疫組織化學上,可見 p16、MDM2 與 CDK4 的強烈表現,尤其在非脂肪源性成分中(圖 122-34)。異源性肌源性分化反映在肌源性(肌動蛋白 actin、結蛋白 desmin、h-鈣調素結合蛋白 h-caldesmon、肌生成蛋白 myogenin)、骨肉瘤樣(SATB2)、軟骨肉瘤樣(S-100 蛋白)或血管肉瘤樣(CD31、ERG)免疫組織化學標記的陽性染色上(圖 122-35)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑於非脂肪源性成分的多變型態,去分化脂肪肉瘤的鑑別診斷範圍廣泛,包括多形性脂肪肉瘤、黏液纖維肉瘤、胃腸道間質瘤 (GI stromal tumor)、平滑肌肉瘤 (leiomyosarcoma)、橫紋肌肉瘤 (rhabdomyosarcoma)、惡性孤立性纖維性腫瘤、罕見的纖維肉瘤與硬纖維瘤病。此外,必須考慮具有細胞學非典型的大型脂肪瘤以及非典型梭形細胞脂肪瘤樣腫瘤與非典型多形性脂肪瘤樣腫瘤,尤其在處理小型切片時。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
去分化脂肪肉瘤的特徵為病程漫長,伴有高局部復發風險與局部侵襲性生長。幾乎所有發生於後腹膜與腹腔內位置的病例,若追蹤足夠久都會復發。然而,與許多其他高度惡性肉瘤相反,遠端轉移擴散的整體風險估計約為 15% 至 20%。⁷²,⁷⁷ 可推測,複雜核型畸變的缺乏以及去分化脂肪肉瘤病例中 p53 突變的罕見⁷⁸ 可能解釋了較佳的結果。如同非典型脂肪瘤樣腫瘤病例,解剖位置是去分化脂肪肉瘤中最重要的預後因子,而發生於後腹膜與腹腔內位置的病例與不良預後相關。已顯示肌源性分化與非脂肪源性腫瘤成分的組織學惡性度是重要的預後參數。⁷⁹ 去分化的範圍在預後上不具預測性。完全手術切除代表首選治療;然而,尤其在後腹膜與腹腔內位置,這常是不可能的。針對第 12 號染色體基因產物的新型治療選擇,或 Akt-mTOR 與 MAPK(促分裂原活化蛋白激酶 mitogen-activated protein kinase)途徑的抑制劑,正被討論用於無法切除的病例。⁸⁰,⁸¹
黏液樣脂肪肉瘤 (MYXOID LIPOSARCOMA)
重點一覽 (AT-A-GLANCE)
■ 黏液樣脂肪肉瘤 (myxoid liposarcoma) 通常發生於中年成人四肢的深部軟組織,尤其是大腿。
■ 黏液樣脂肪肉瘤由小型、原始的間葉細胞與處於不同分化階段的脂肪母細胞組成,具有特徵性的血管模式。
■ 圓細胞成分 (round cell component) 的存在具有顯著的不良預後重要性。
■ 黏液樣脂肪肉瘤的特徵為 FUS-DDIT3 或 EWSR1-DDIT3 重排。
流行病學 (EPIDEMIOLOGY)
黏液樣脂肪肉瘤代表脂肪肉瘤的第二常見變異型,約占成人所有軟組織肉瘤的 5%。大多數黏液樣脂肪肉瘤病例發生於年輕至中年成人;然而,相當數目的病例見於兒童與青少年,代表此年齡組中最常見的脂肪肉瘤變異型,且這些病例可能顯示不尋常的型態特徵。⁸² 此外,可見癌睪丸抗原 (cancer testis antigens)(即 PRAME)的表現。⁸²ᴬ
臨床特徵 (CLINICAL FEATURES)
黏液樣脂肪肉瘤病例表現為四肢深部軟組織中的大型無痛腫塊,且大多數腫瘤發生於大腿。黏液樣脂肪肉瘤僅罕見地見於皮下組織以及後腹膜或腹腔內位置。多病灶黏液樣脂肪肉瘤病例的單株起源 (monoclonal origin) 證實了這些病例中不尋常的轉移模式。⁸³
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在細胞遺傳學上,黏液樣脂肪肉瘤在大多數病例中的特徵為反覆性易位 t(12;16)(q13;p11),伴有 FUS-DDIT3 融合;較罕見地,存在 t(12;22)(q13:q12) 伴有 EWSR1-DDIT3 融合。⁸⁴,⁸⁵
組織學特徵 (HISTOLOGIC FEATURES)
這些結節狀腫瘤由一致的、細胞學上溫和的、小型、卵圓形的間葉腫瘤細胞,以及小型、單空泡化或雙空泡化的脂肪母細胞混合組成,位於明顯的黏液樣基質中,伴有微囊狀空間 (microcystic spaces) 與特徵性的分枝狀薄壁血管(圖 122-36)。這些高分化腫瘤缺乏明顯的細胞學非典型、有絲分裂與腫瘤壞死。黏液樣/圓細胞脂肪肉瘤 (Myxoid/round-cell liposarcoma) 以及主要為圓細胞脂肪肉瘤 (predominantly round-cell liposarcoma) 的特徵為進展為更具細胞密度的區域,含有具增大且重疊之圓形細胞核的增大圓形腫瘤細胞(圖 122-37)。罕見地,黏液樣脂肪肉瘤可能顯示明顯的脂肪細胞性分化,模擬非典型脂肪瘤樣腫瘤的型態特徵(圖 122-38),⁸⁶ 這在接受新輔助治療 (neoadjuvant therapy) 的病例中也曾被報告。⁸⁷ 此外,黏液樣脂肪肉瘤病例可能顯示明顯的梭形細胞成分(梭形細胞黏液樣脂肪肉瘤 spindle-cell myxoid liposarcoma)或多形性成分(多形性黏液樣脂肪肉瘤 pleomorphic myxoid liposarcoma),這些特徵主要見於年輕病人。⁸² 免疫組織化學上,S-100 蛋白在高度惡性的圓細胞成分中呈不等程度的陽性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括良性病灶如脂肪母細胞瘤與軟骨樣脂肪瘤,以及不同分化系列的惡性腫瘤(黏液纖維肉瘤、低度惡性纖維黏液樣肉瘤 low-grade fibromyxoid sarcoma、骨骼外黏液樣軟骨肉瘤、黏液樣惡性黑色素瘤 myxoid malignant melanoma)。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
黏液樣脂肪肉瘤傾向復發,並可能在高達 40% 的病人中發展出遠端轉移,且常觀察到軟組織或骨骼中不尋常位置的轉移。在表現時即有「多病灶 (multifocal)」疾病的病人預後不佳,而不良的組織學特徵包括圓細胞分化(>5%)的存在、腫瘤壞死、p53 過度表現與 CDKN2A 畸變。⁸⁸,⁸⁹ 已顯示晚期黏液樣脂肪肉瘤病例可用曲貝替定 (trabectedin) 成功治療。⁹⁰⁻⁹²
多形性脂肪肉瘤 (PLEOMORPHIC LIPOSARCOMA)
重點一覽 (AT-A-GLANCE)
■ 多形性脂肪肉瘤 (pleomorphic liposarcoma) 代表一種含有數量不等之多形性脂肪母細胞的高度惡性肉瘤。
■ 多形性脂肪肉瘤好發於成人的四肢。
■ 多形性脂肪肉瘤可為新發,或代表非典型脂肪瘤樣腫瘤/去分化脂肪肉瘤的型態學進展形式。
流行病學 (EPIDEMIOLOGY)
多形性脂肪肉瘤代表脂肪肉瘤最罕見的亞型(5%),主要發生於老年人,略以男性為主。⁹³,⁹⁴
臨床特徵 (CLINICAL FEATURES)
臨床上,多形性脂肪肉瘤通常表現為四肢深部軟組織中快速生長的腫瘤(下肢 > 上肢),而軀幹、後腹膜、腹腔、頭頸部區域與骨盆則僅罕見地受侵犯。少數但相當數目的多形性脂肪肉瘤病例發生於皮下組織,而純真皮腫瘤則極為罕見(圖 122-39)。⁹⁴⁻⁹⁶
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在細胞遺傳學上,許多多形性脂肪肉瘤病例與其他多形性肉瘤極為相似,並顯示複雜的結構性重排。然而,已顯示少數但相當數目的病例揭示 MDM2 與 CDK4 擴增,這顯示這些腫瘤代表來自非典型脂肪瘤樣腫瘤/去分化脂肪肉瘤的腫瘤進展。⁷⁶,⁹⁷
組織學特徵 (HISTOLOGIC FEATURES)
這些界線清楚或浸潤性的腫瘤顯示高度惡性多形性肉瘤的特徵(高度惡性纖維肉瘤或「惡性纖維組織細胞瘤 malignant fibrous histiocytoma」),含有多形性梭形細胞與多核腫瘤巨細胞,與數量不等的多形性脂肪母細胞混合。這些多形性脂肪母細胞含有增大且怪異、深染、被細胞質脂滴扇形圍繞 (scalloped) 的細胞核,並散布於整個多形性肉瘤中或排列成較大的片狀(圖 122-40)。常可見細胞內與細胞外的嗜酸性小滴。許多病例顯示與中度惡性或高度惡性黏液纖維肉瘤相似的型態特徵,伴有散在的多形性脂肪母細胞(圖 122-41)。多形性脂肪肉瘤的上皮樣變異型 (epithelioid variant) 的特徵為大型、密集排列、具有豐富嗜酸性細胞質與泡狀細胞核 (vesicular nuclei) 的上皮樣腫瘤細胞,類似腎透明細胞癌 (renal clear cell carcinoma) 或腎上腺皮質癌 (adrenal cortical carcinoma)。⁹⁸ 免疫組織化學上,多形性脂肪肉瘤中的腫瘤細胞可能對肌動蛋白 (actins)、細胞角蛋白 (cytokeratins)、CD34 與罕見地對結蛋白 (desmin) 呈陽性染色。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
去分化脂肪肉瘤含有非典型脂肪瘤樣腫瘤成分且沒有多形性脂肪母細胞。多形性脂肪肉瘤中至少散在的多形性脂肪母細胞的存在,在與其他多形性、高度惡性肉瘤(黏液纖維肉瘤、平滑肌肉瘤、橫紋肌肉瘤)以及低分化、肉瘤樣黑色素瘤與癌的鑑別診斷中相當重要。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
多形性脂肪肉瘤代表一種高度惡性肉瘤,局部復發與轉移率為 30-50%,估計整體 5 年存活率為 50% 至 60%。肺臟是優先的轉移部位。治療為手術切除,晚期病例加上放射治療與化學治療。
圖表 (FIGURES AND TABLES)
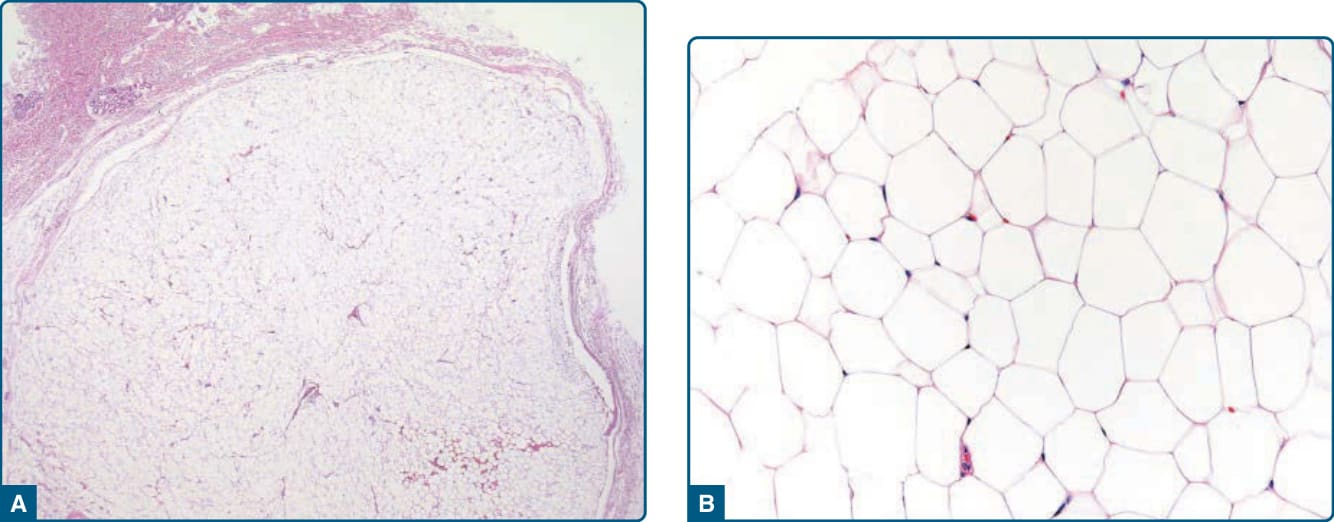
圖 122-1:脂肪瘤 (Lipoma)。位於皮下的脂肪瘤是界線清楚、常有包膜的脂肪源性病灶 (A),由僅顯示輕微大小與形狀變異的成熟單空泡化脂肪細胞 (univacuolated adipocytes) 組成 (B)。
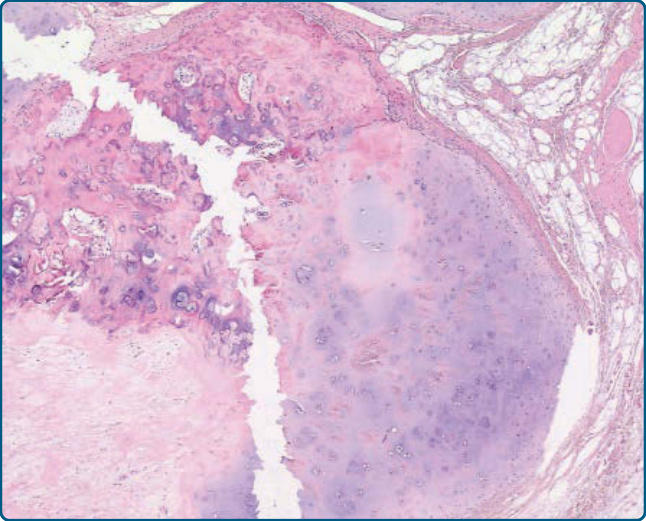
圖 122-2:軟骨脂肪瘤 (Chondrolipoma)。一個伴有明顯軟骨化生 (chondrous metaplasia) 的脂肪瘤範例。
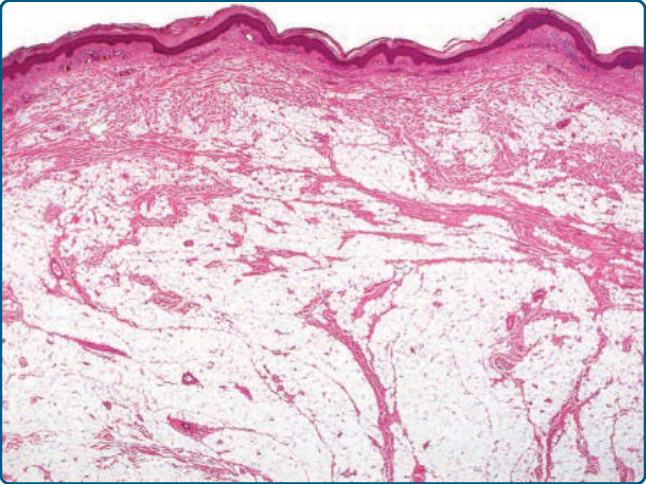
圖 122-3:真皮內脂肪瘤 (Intradermal lipoma)。成熟脂肪源性細胞瀰漫性浸潤原有的真皮膠原蛋白束。
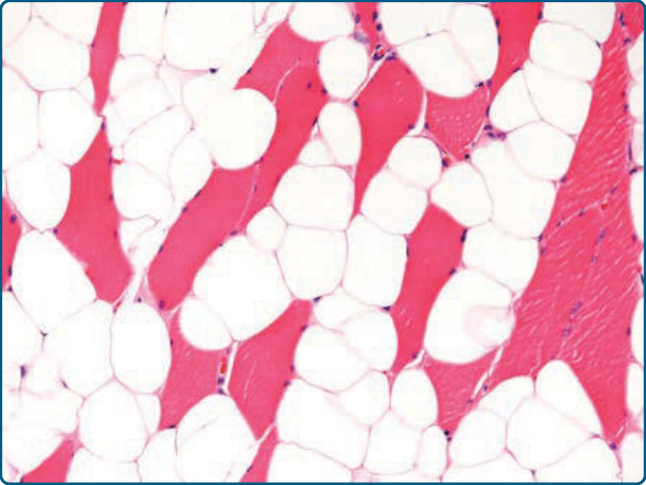
圖 122-4:肌肉內脂肪瘤 (Intramuscular lipoma)。成熟脂肪源性細胞在骨骼肌纖維內顯示浸潤性生長模式。
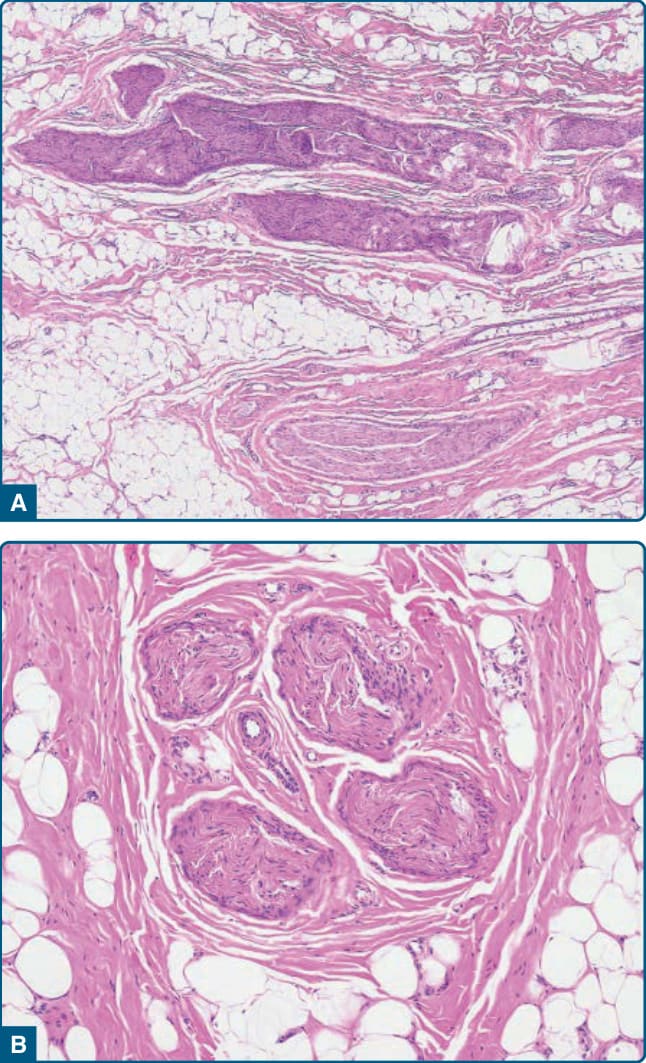
圖 122-5:神經脂肪瘤病 (Lipomatosis of nerve)。A,脂肪源性細胞與膠原性纖維組織的神經外膜浸潤,分隔神經束。B,注意神經束膜纖維化 (perineural fibrosis)。
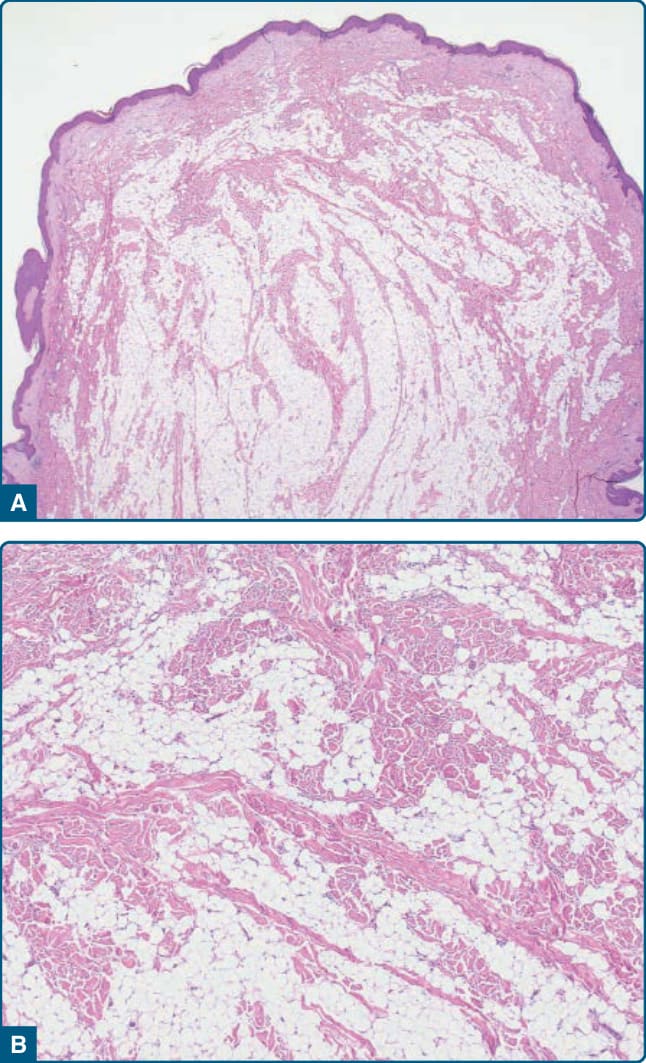
圖 122-6:淺表性脂肪瘤樣痣 (Nevus lipomatosus superficialis)。息肉狀病灶,伴有成熟脂肪源性細胞對真皮的瀰漫性浸潤 (A),並具有典型的玻璃樣變膠原蛋白束 (B)。
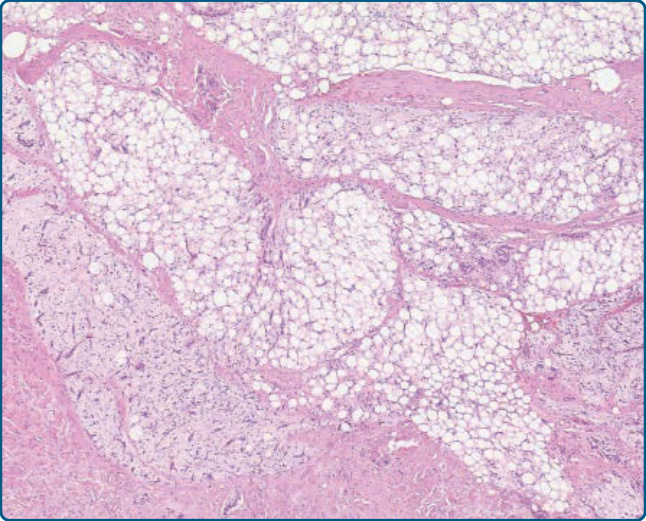
圖 122-7:脂肪母細胞瘤 (Lipoblastoma):脂肪母細胞瘤代表一個界線清楚的脂肪源性腫瘤,顯示多分葉 (multilobulation) 與膠原性纖維間隔。
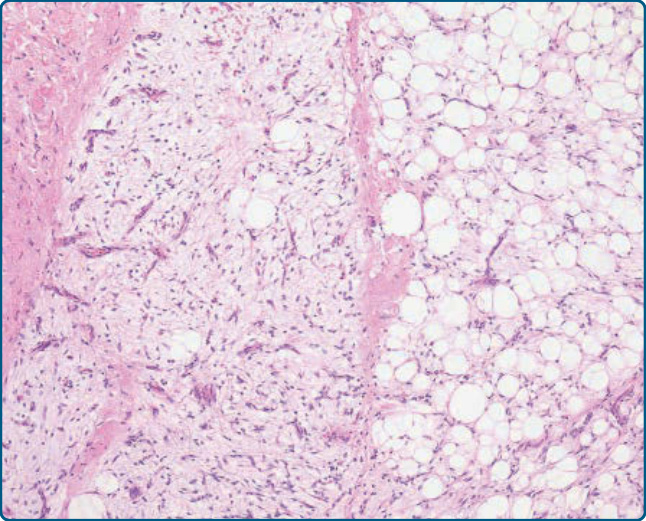
圖 122-8:脂肪母細胞瘤 (Lipoblastoma)。脂肪源性細胞區域與顯示叢狀血管模式的黏液樣區域混合。
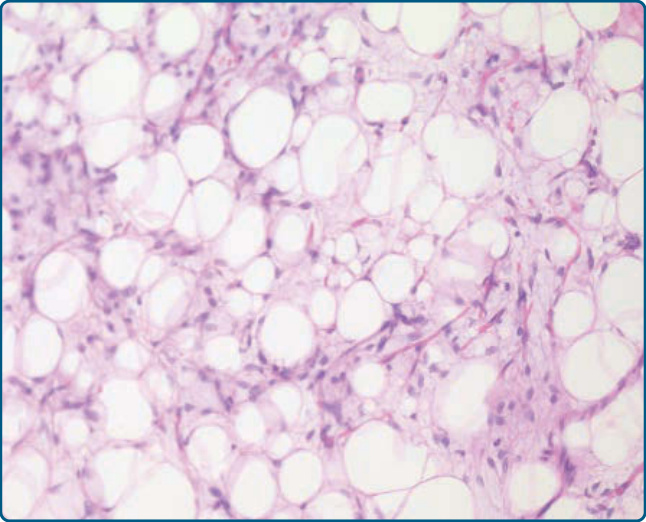
圖 122-9:脂肪母細胞瘤 (Lipoblastoma)。存在處於不同發育階段的脂肪源性細胞。
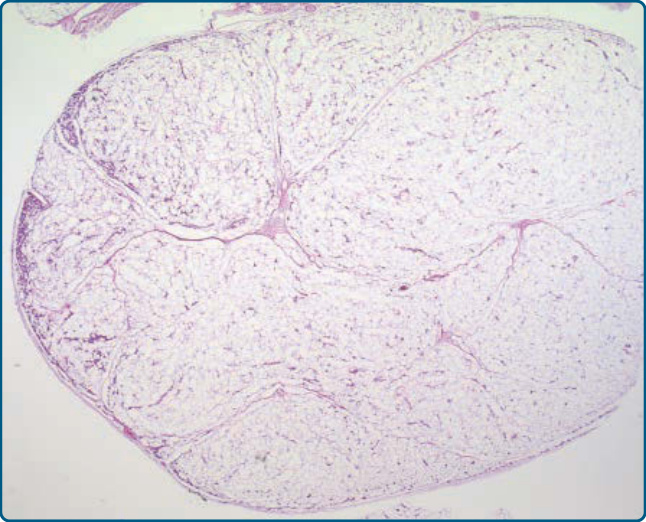
圖 122-10:血管脂肪瘤 (Angiolipoma)。低倍視野顯示一個有包膜、位於皮下的脂肪源性腫瘤,周邊有數目增加的血管。
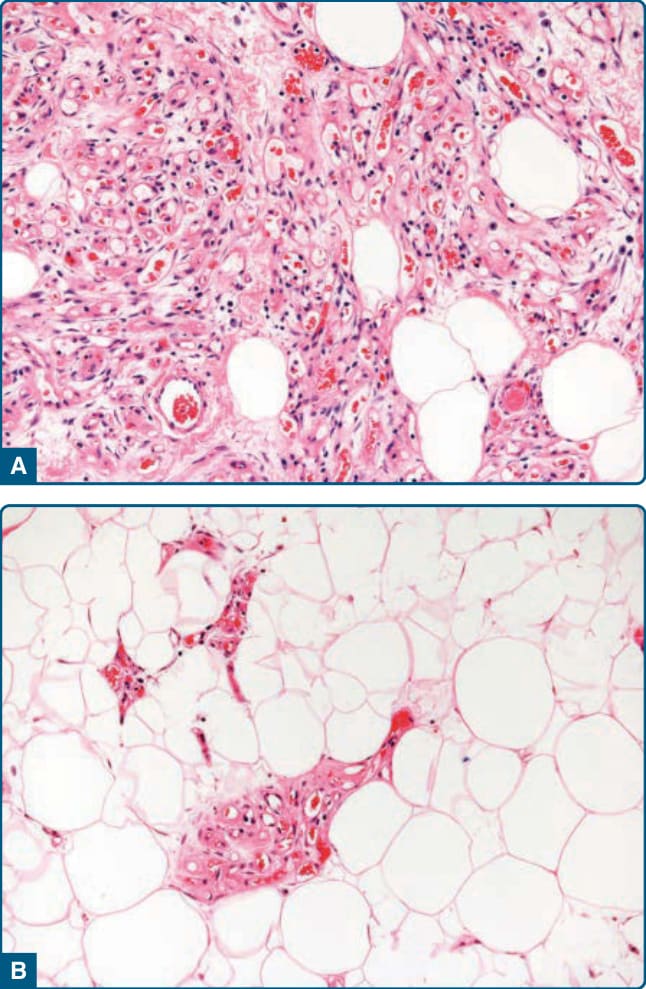
圖 122-11:血管脂肪瘤 (Angiolipoma)。A,狹窄與擴張的薄壁微血管與成熟脂肪細胞組織混合。B,脂肪細胞成分可占主導地位。
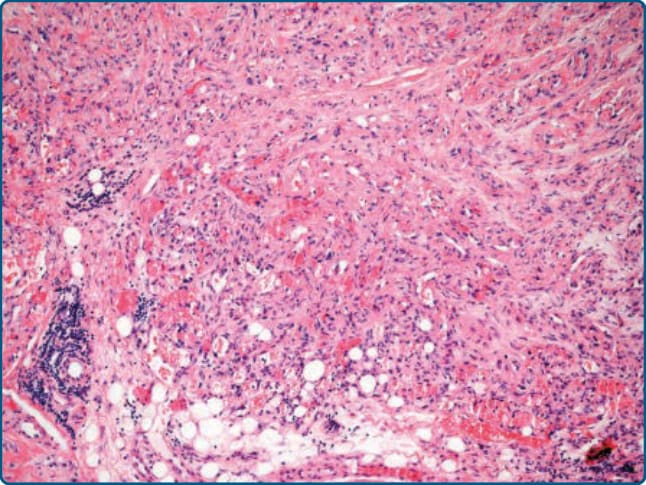
圖 122-12:細胞性血管脂肪瘤 (Cellular angiolipoma)。占主導地位的血管成分模擬血管性腫瘤。
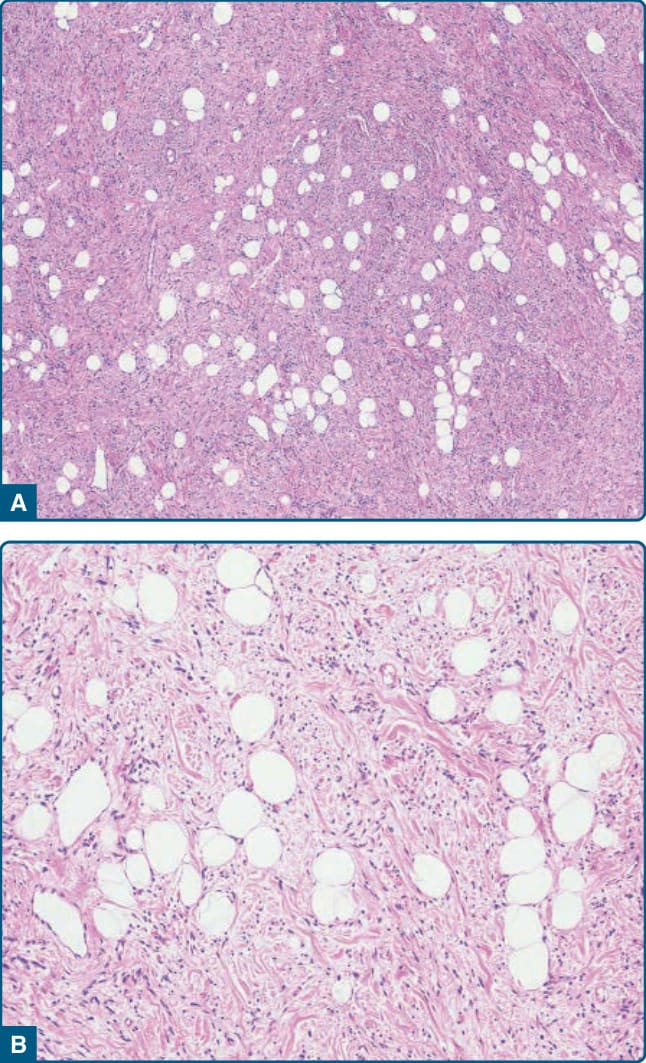
圖 122-13:梭形細胞脂肪瘤 (Spindle-cell lipoma)。A,成熟脂肪細胞與短的、鬆散排列的梭形腫瘤細胞混合。B,存在玻璃樣變的繩索狀膠原纖維。
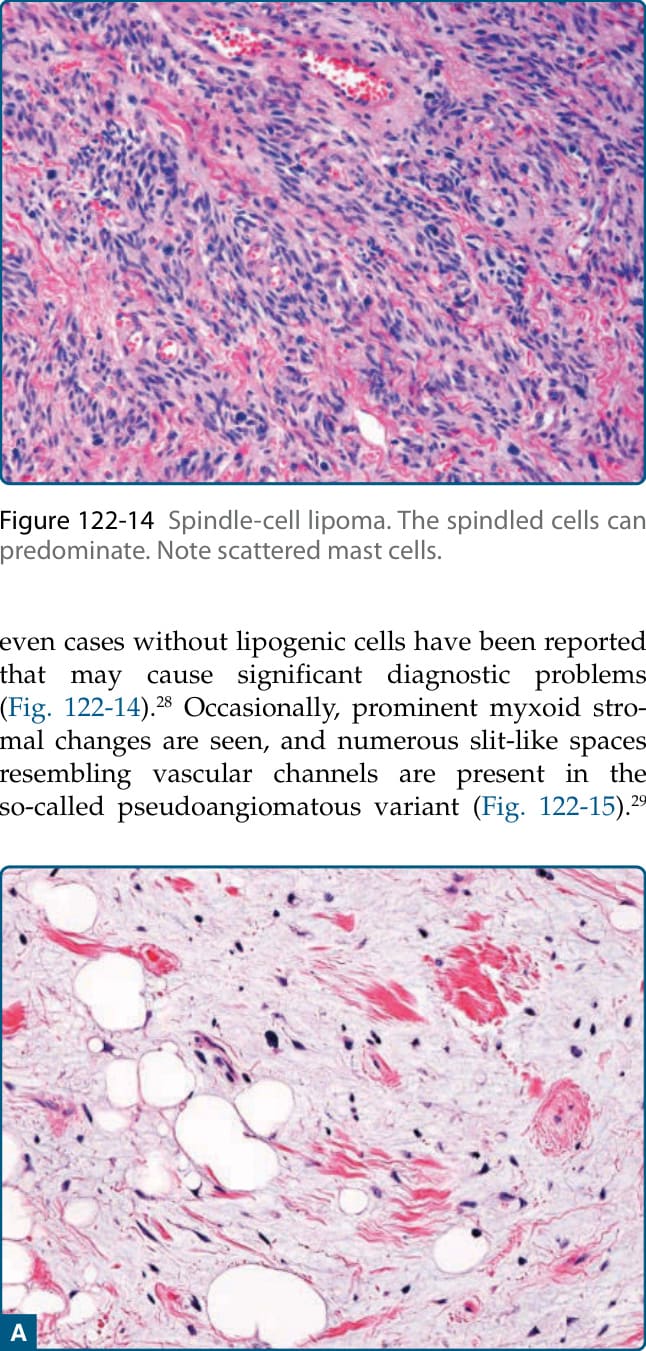
圖 122-14:梭形細胞脂肪瘤 (Spindle-cell lipoma)。梭形細胞可占主導地位。注意散在的肥大細胞。
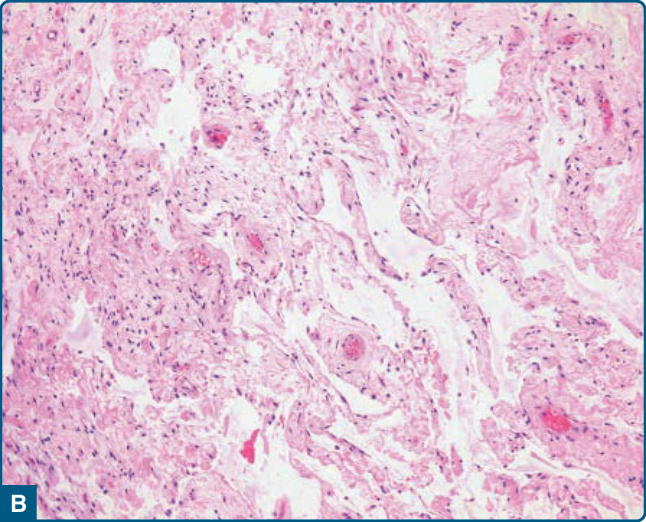
圖 122-15:梭形細胞脂肪瘤 (Spindle-cell lipoma)。A,明顯的黏液樣基質變化;注意腫瘤細胞核略為增大且深染。B,在假血管瘤樣變異型中可見裂隙狀空間。
圖 122-16:多形性脂肪瘤 (Pleomorphic lipoma)。A,特徵性的花環狀多核巨細胞含有以圓形或半圓形排列的重疊深染細胞核。B,位於黏液樣基質中、伴有散在肥大細胞的花環狀腫瘤巨細胞之高倍視野。
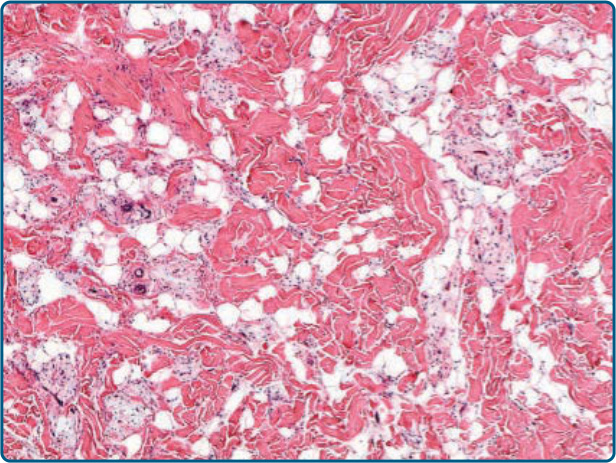
圖 122-17:真皮梭形細胞脂肪瘤 (Dermal spindle-cell lipoma)。真皮內病灶界線較不清楚,並顯示對原有真皮膠原蛋白束的瀰漫性浸潤。
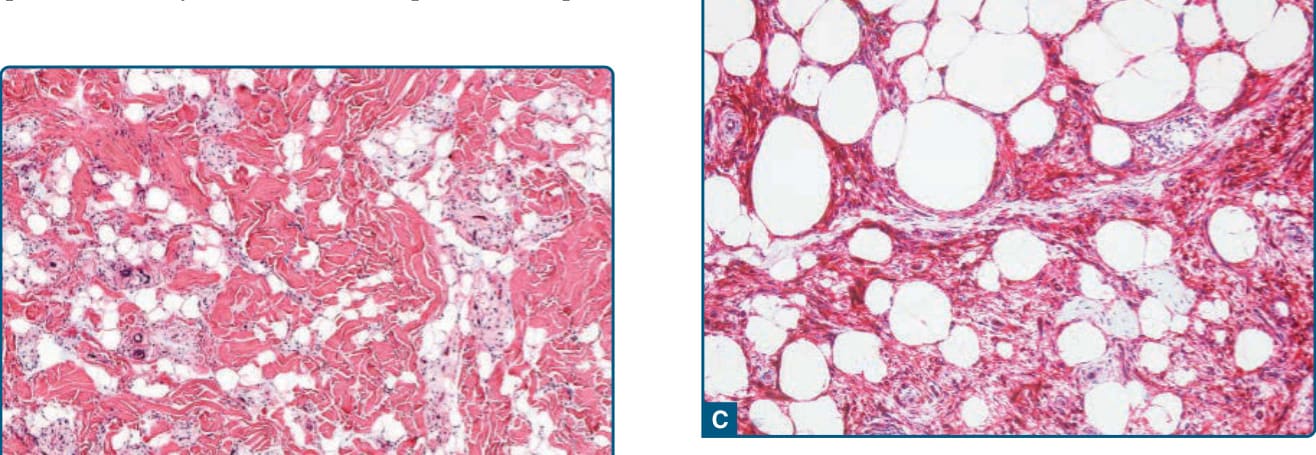
圖 122-18:非典型梭形細胞脂肪瘤 (Atypical spindle cell lipoma)。A,脂肪源性細胞與梭形腫瘤細胞的不規則混合。B,兩種成分都顯示至少輕微的細胞核非典型,伴有增大且深染的細胞核;此外可見脂肪母細胞性細胞。C,免疫組織化學上,梭形細胞對 CD34 呈陽性染色。
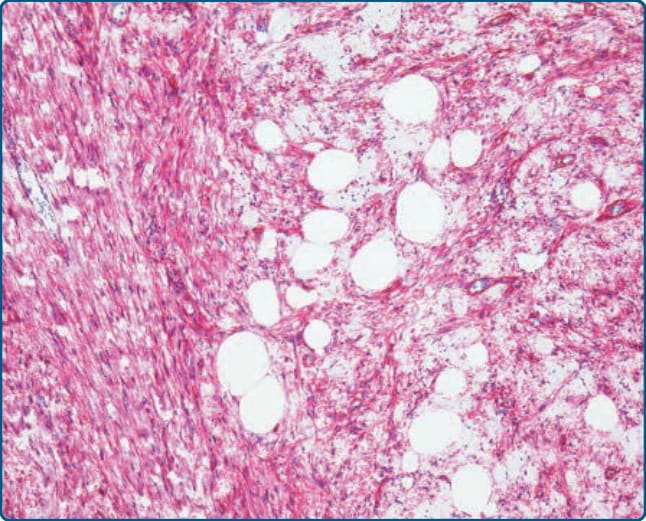
圖 122-19:梭形細胞脂肪瘤 (Spindle-cell lipoma)。腫瘤細胞對 CD34 呈陽性染色。
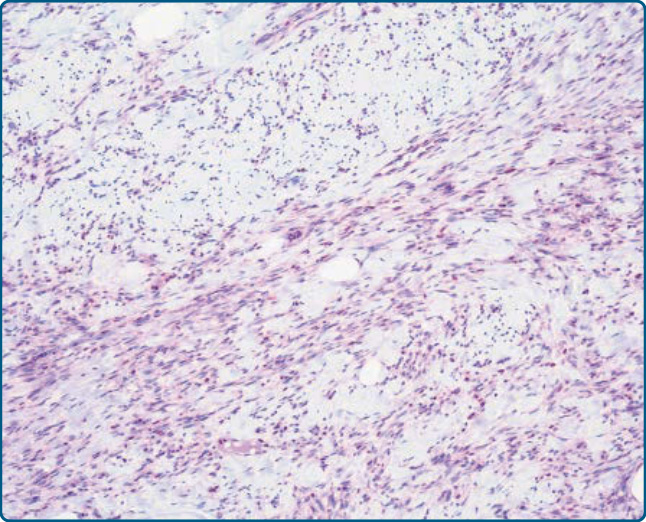
圖 122-20:梭形細胞脂肪瘤 (Spindle-cell lipoma)。腫瘤細胞顯示 Rb1 表現的喪失。
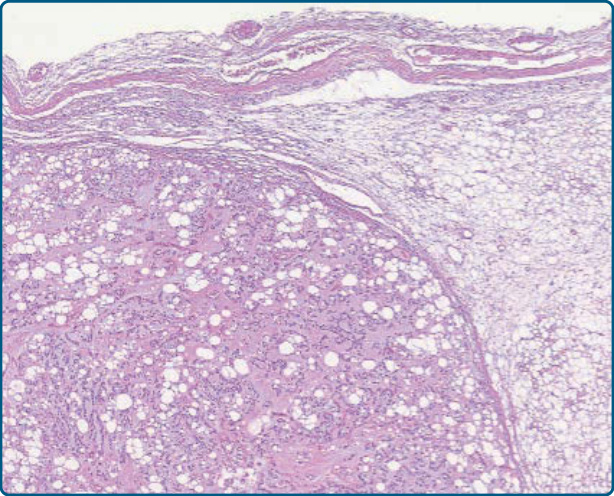
圖 122-21:軟骨樣脂肪瘤 (Chondroid lipoma)。有包膜、具有分葉狀生長的脂肪源性腫瘤。
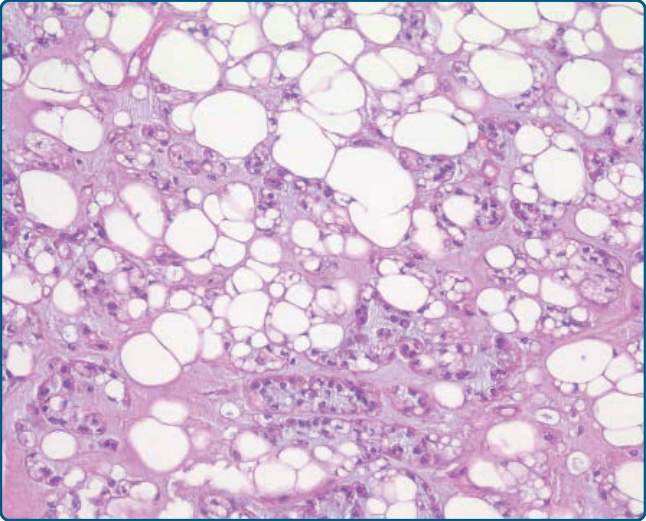
圖 122-22:軟骨樣脂肪瘤 (Chondroid lipoma)。單空泡化與多空泡化脂肪母細胞與成熟脂肪細胞混合。
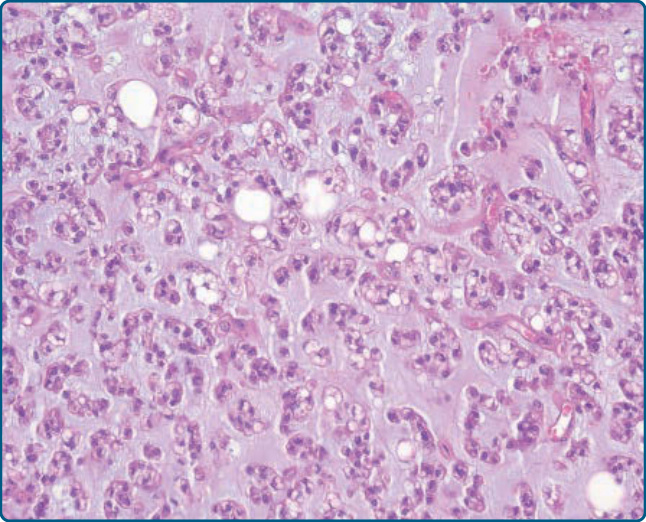
圖 122-23:軟骨樣脂肪瘤 (Chondroid lipoma)。此外,可見含有嗜酸性與空泡化細胞質的小型圓形腫瘤細胞。
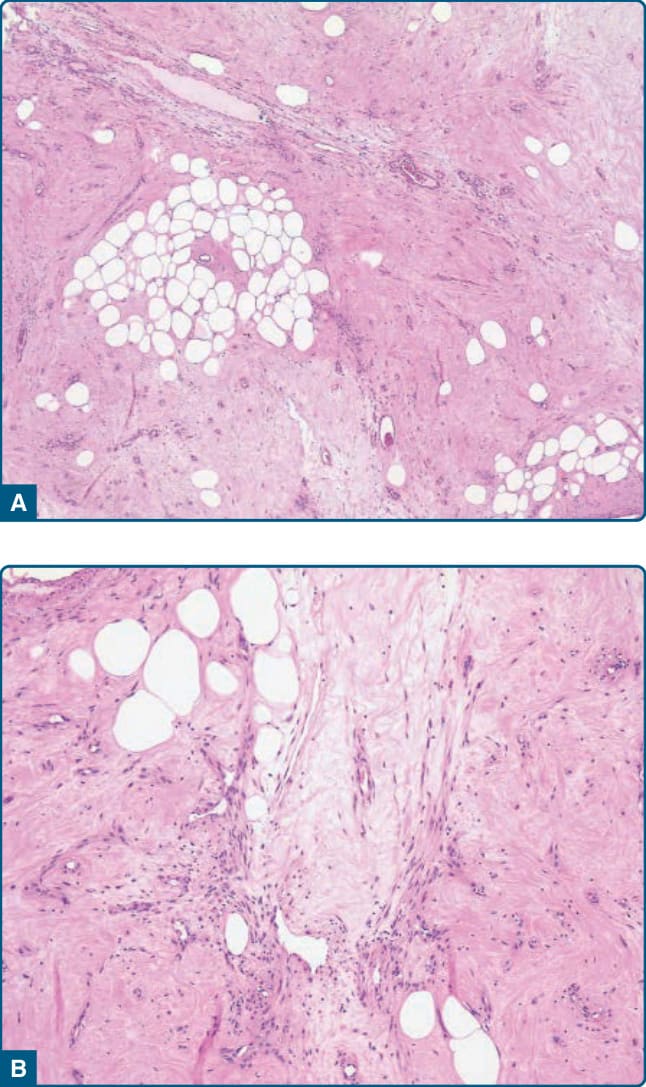
圖 122-24:肌脂肪瘤 (Myolipoma)。A,成熟脂肪細胞性細胞與溫和的梭形細胞混合。B,肌源性梭形細胞含有嗜酸性細胞質與溫和的梭形細胞核。
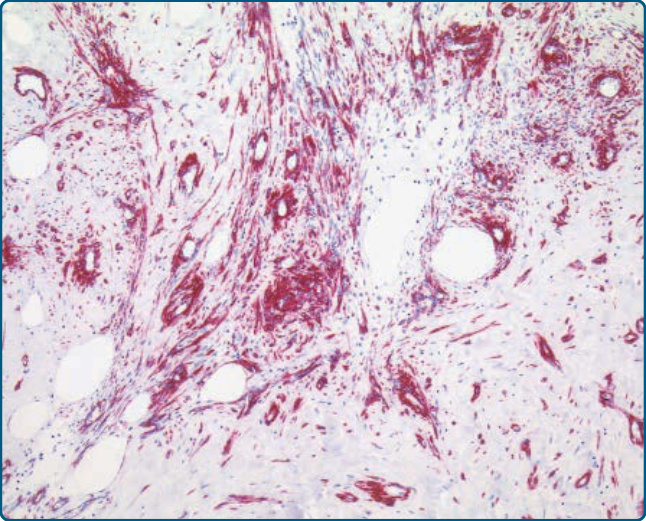
圖 122-25:肌脂肪瘤 (Myolipoma)。嗜酸性梭形腫瘤細胞對 h-鈣調素結合蛋白 (h-caldesmon) 呈陽性染色,確認其平滑肌分化。
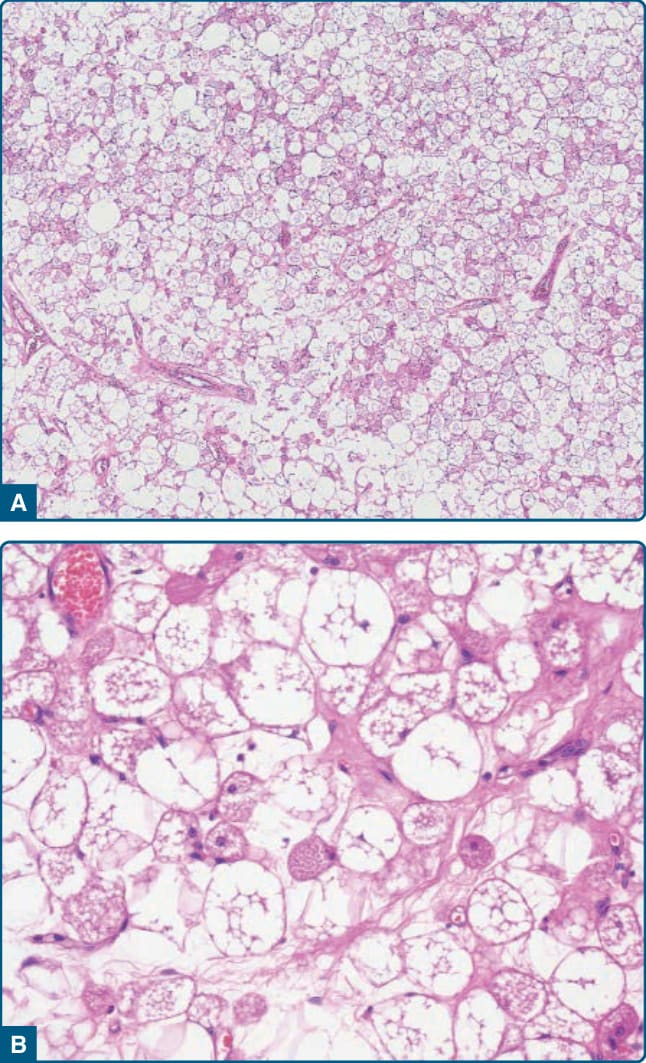
圖 122-26:冬眠瘤 (Hibernoma)。A,成熟脂肪細胞與大型多空泡化細胞及較小的嗜酸性細胞混合。B,具有位於中央之細胞核與透明或嗜酸性顆粒狀細胞質的空泡化腫瘤細胞。
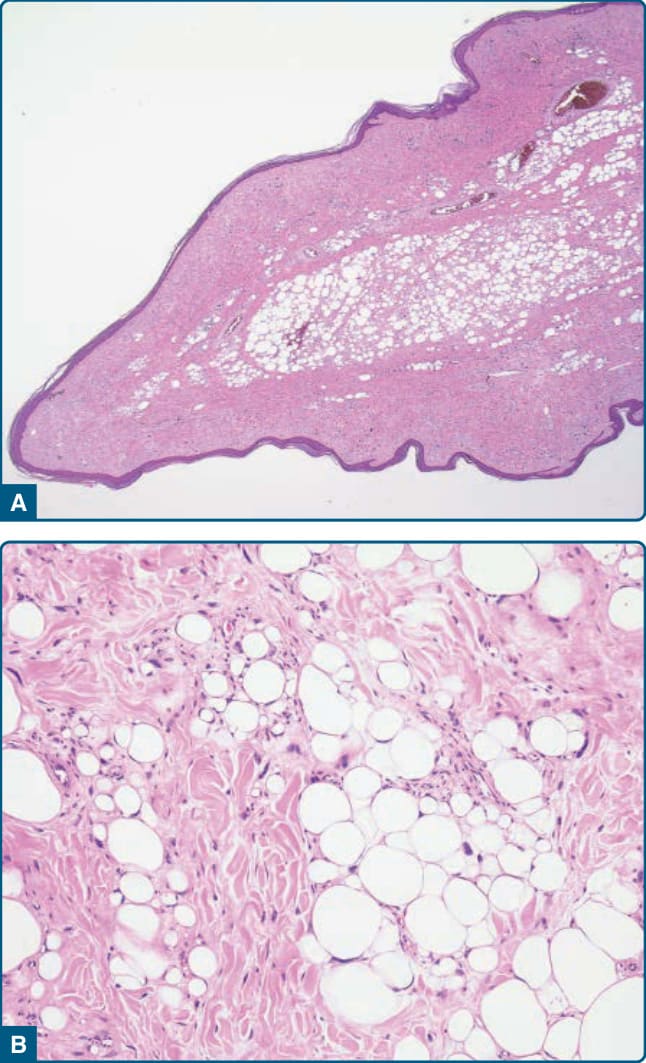
圖 122-27:真皮非典型脂肪瘤樣腫瘤 (Dermal atypical lipomatous tumor)。A,一個息肉狀病灶,臨床診斷為皮膚贅生物 (skin tag),已被切除。B,組織學上,可見一個由非典型基質細胞與具有增大深染細胞核及散在脂肪母細胞之非典型脂肪源性細胞組成的非典型脂肪瘤樣腫瘤。
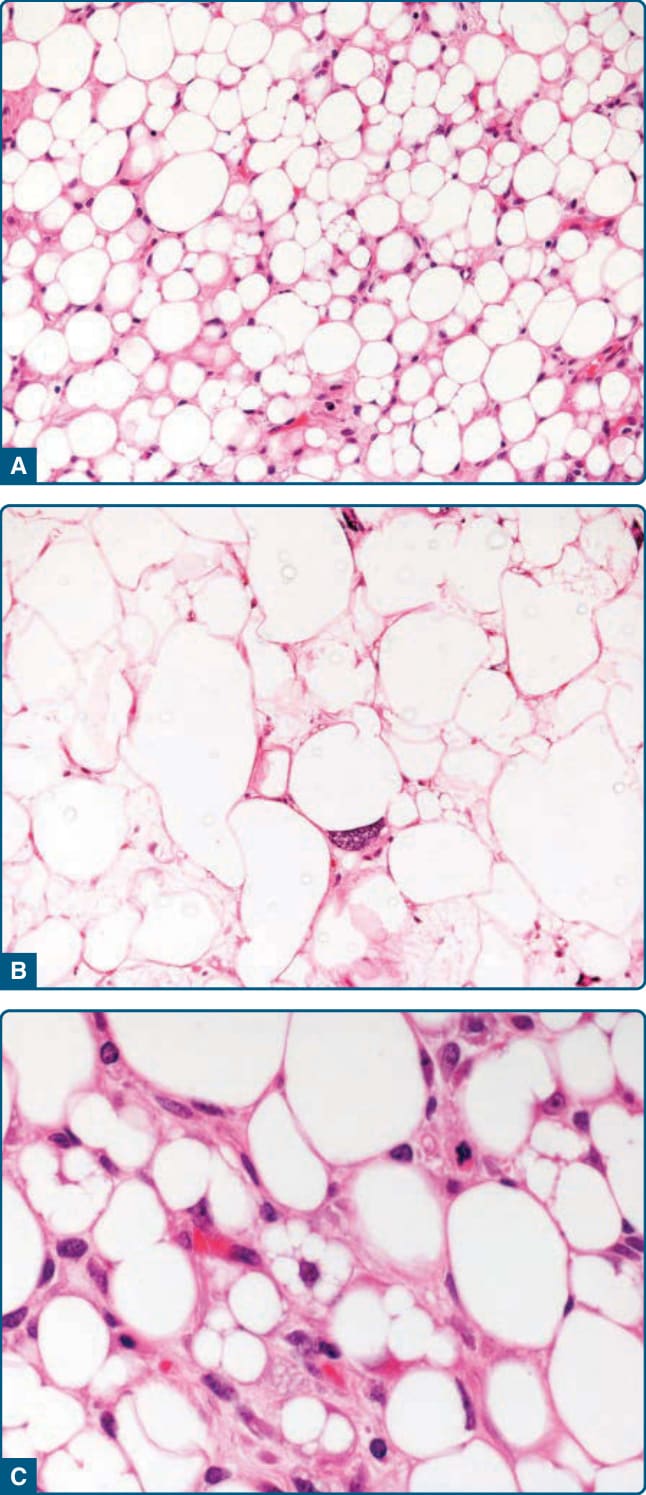
圖 122-28:非典型脂肪瘤樣腫瘤 (Atypical lipomatous tumor)。A,脂肪瘤樣變異型由顯示大小與形狀顯著變異的脂肪源性細胞組成。可見散在的、增大的、深染的細胞核與空泡化的脂肪源性細胞。高倍視野揭示增大且深染的細胞核 (B),以及多空泡化脂肪母細胞 (C)。
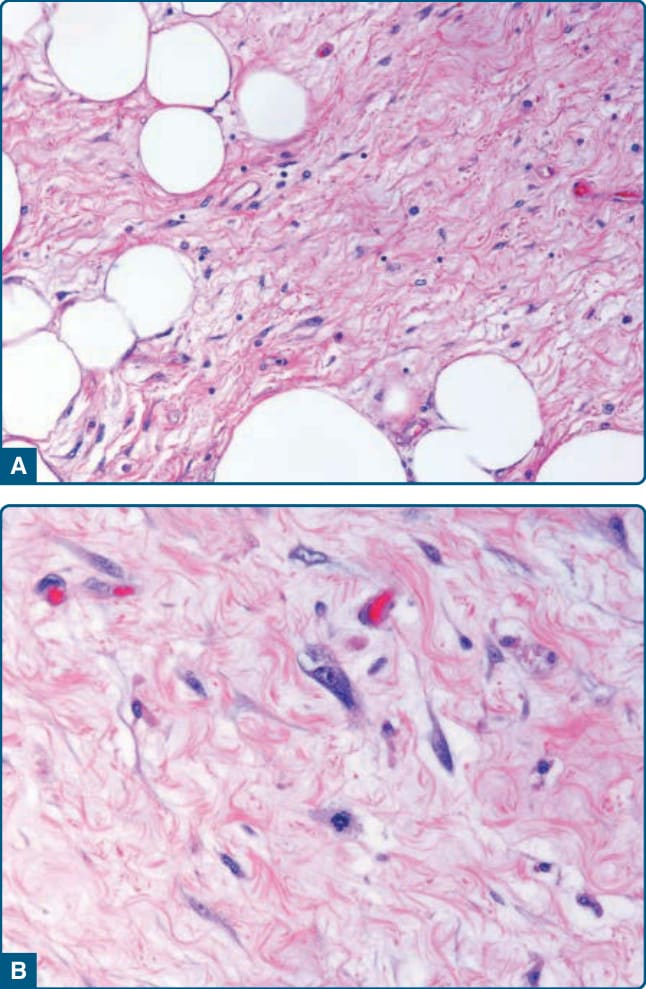
圖 122-29:非典型脂肪瘤樣腫瘤 (Atypical lipomatous tumor)。A,伴有混合脂肪細胞之細胞稀少基質的存在是硬化型的特徵。B,注意位於纖維狀基質中的怪異外觀基質細胞的存在。
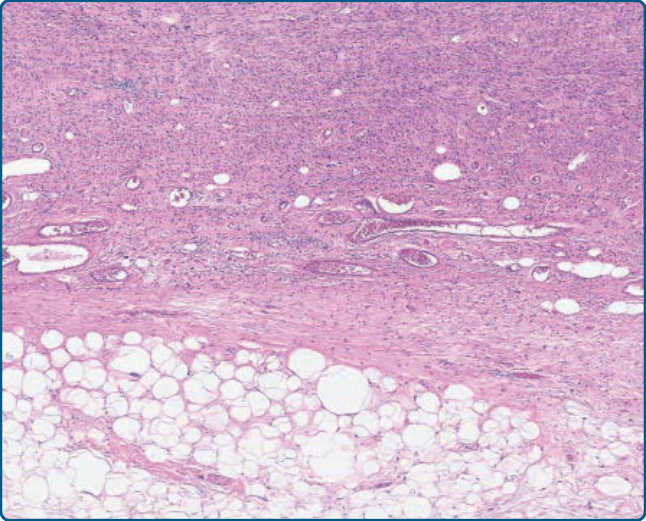
圖 122-30:去分化脂肪肉瘤 (Dedifferentiated liposarcoma)。可見從顯示非典型脂肪瘤樣腫瘤特徵的區域(按鈕狀 button)到非脂肪源性肉瘤的相當突然的過渡。
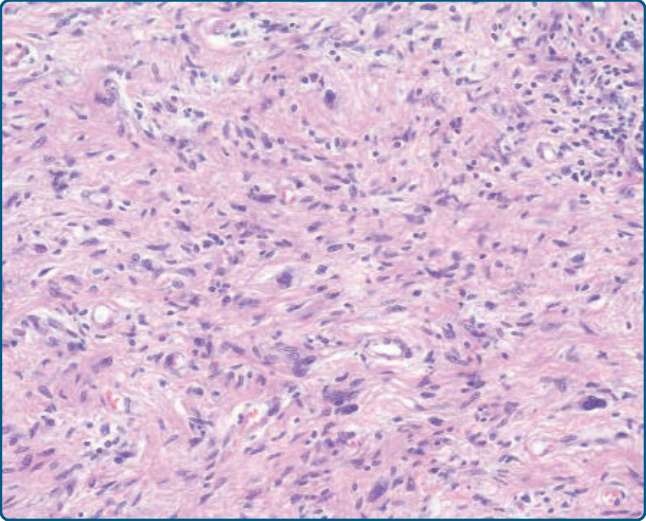
圖 122-31:去分化脂肪肉瘤 (Dedifferentiated liposarcoma)。非脂肪源性成分常顯示相當未分化、多形性肉瘤的特徵。
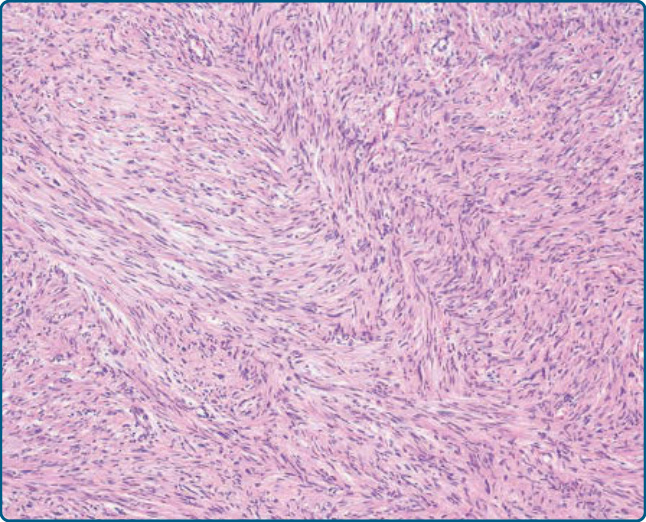
圖 122-32:去分化脂肪肉瘤 (Dedifferentiated liposarcoma)。少數情況下,可見伴有低度惡性纖維母細胞性肉瘤特徵的低度惡性去分化。
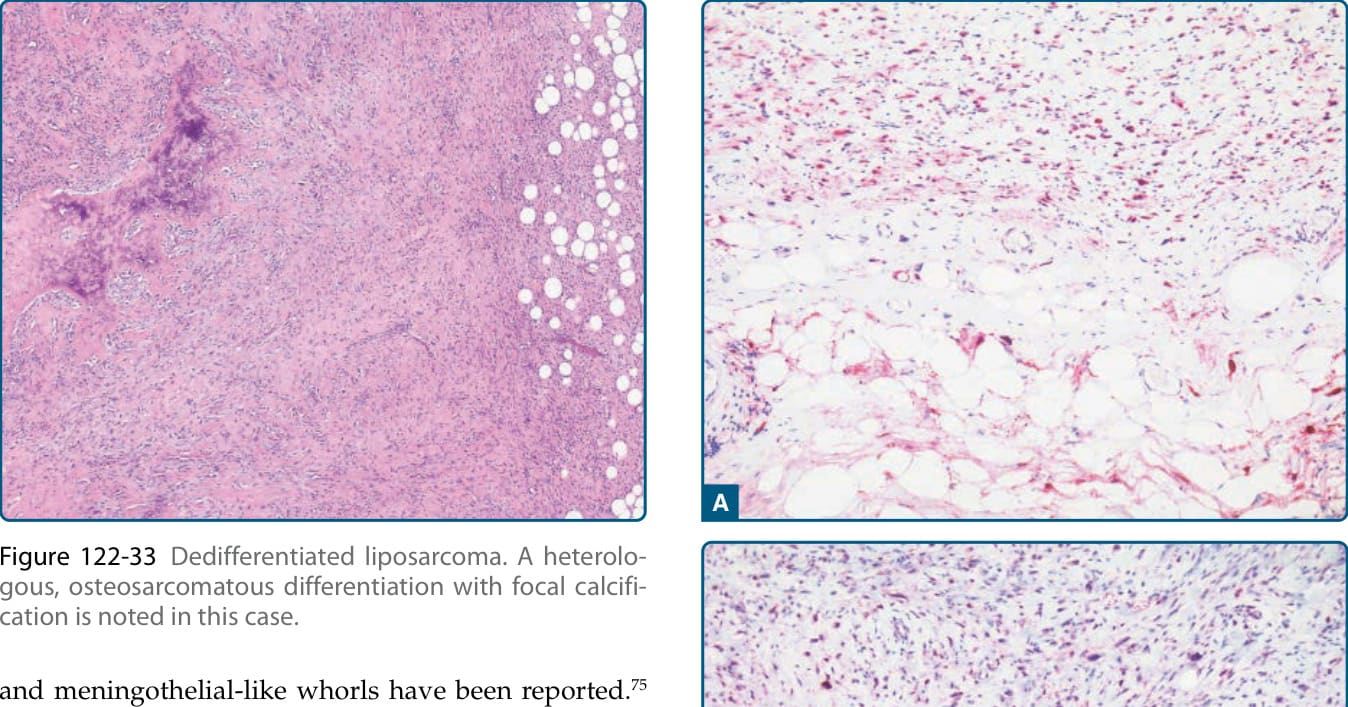
圖 122-33:去分化脂肪肉瘤 (Dedifferentiated liposarcoma)。本病例可見伴有局部鈣化的異源性骨肉瘤樣分化。
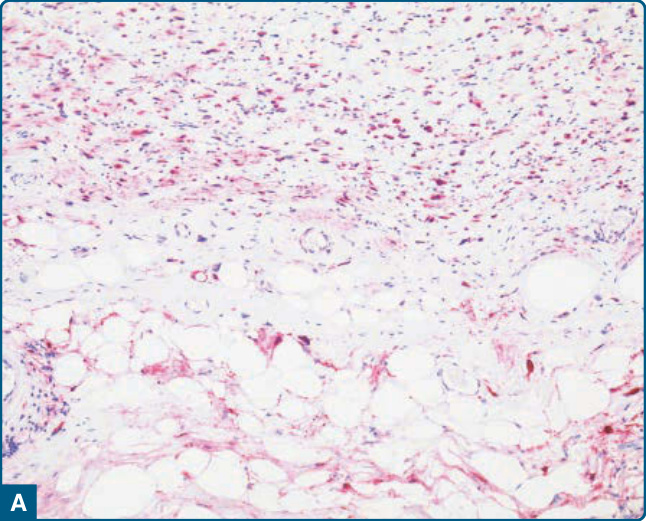
圖 122-34:去分化脂肪肉瘤 (Dedifferentiated liposarcoma)。免疫組織化學上,在脂肪源性與非脂肪源性成分中均可見 p16 (A) 與 MDM2 (B) 的表現。
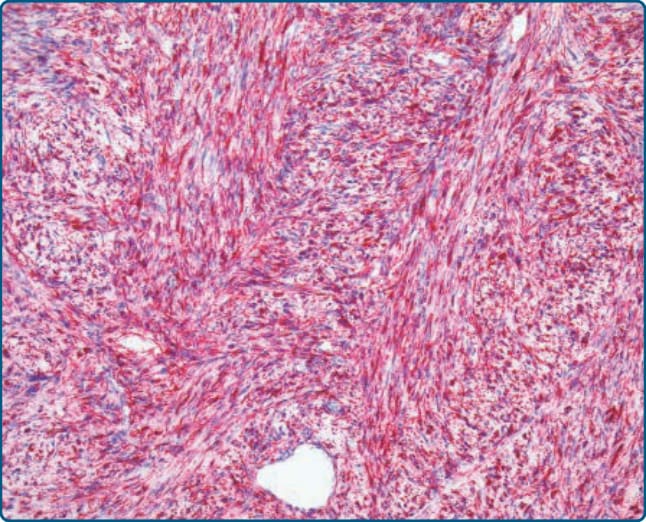
圖 122-35:去分化脂肪肉瘤 (Dedifferentiated liposarcoma)。異源性肌源性分化,伴有非脂肪源性梭形腫瘤細胞對結蛋白 (desmin) 的強烈表現。
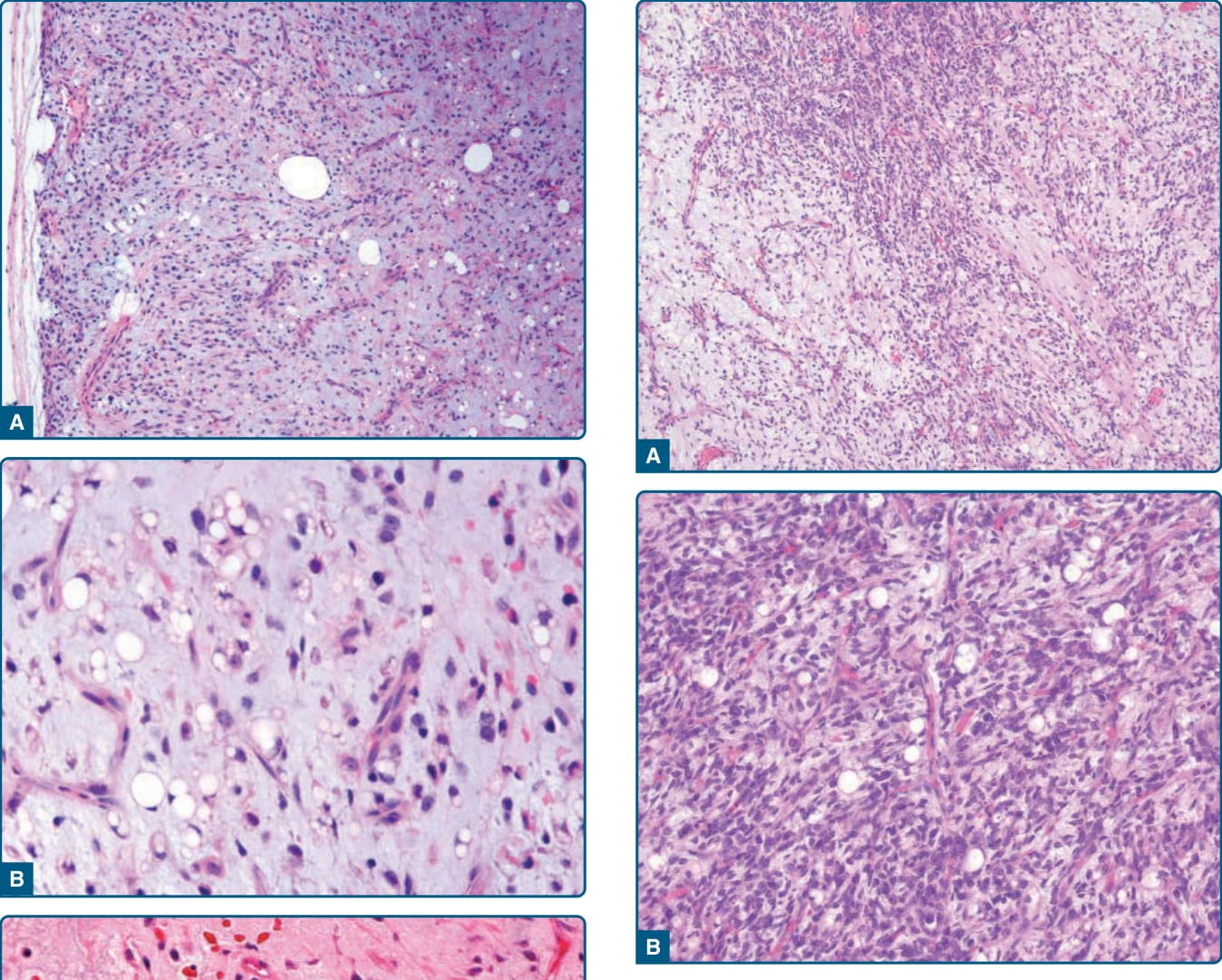
圖 122-36:黏液樣脂肪肉瘤 (Myxoid liposarcoma)。A,黏液樣腫瘤呈分葉狀,由未成熟間葉細胞與非典型脂肪源性細胞組成。注意周邊細胞密度增加。B,位於黏液樣基質中的單空泡化、雙空泡化與多空泡化脂肪母細胞以及細胞學上溫和的卵圓形細胞的存在是一個特徵性特徵。C,注意纖細的分枝狀、微血管大小的血管。
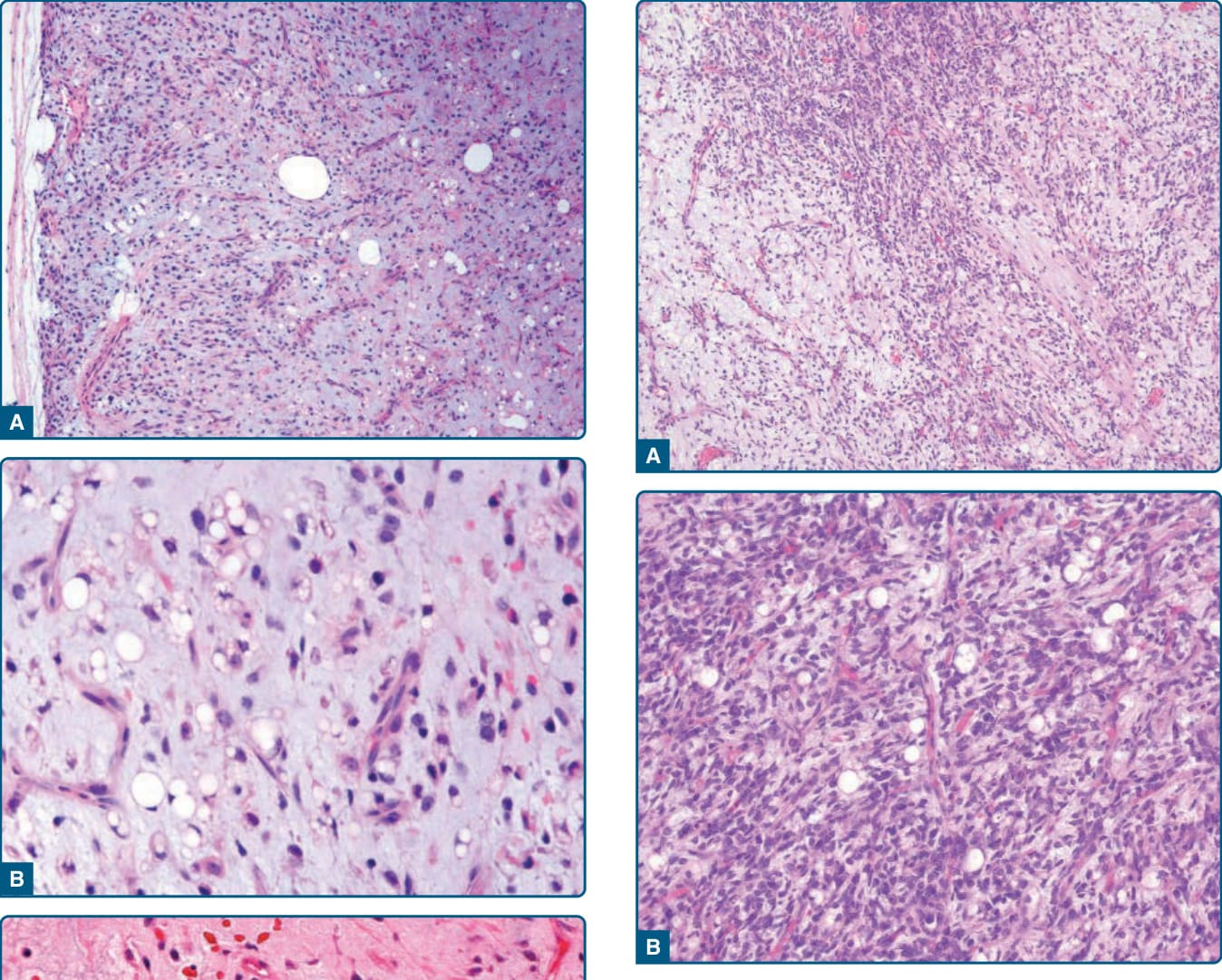
圖 122-37:黏液樣/圓細胞脂肪肉瘤 (Myxoid/round cell liposarcoma)。A,可見細胞密度增加、伴有含圓形細胞核之略增大圓形腫瘤細胞的區域。B,高度惡性圓細胞脂肪肉瘤主要由增大的圓形腫瘤細胞組成,無明顯多形性。注意特徵性的血管模式與脂肪母細胞的存在。
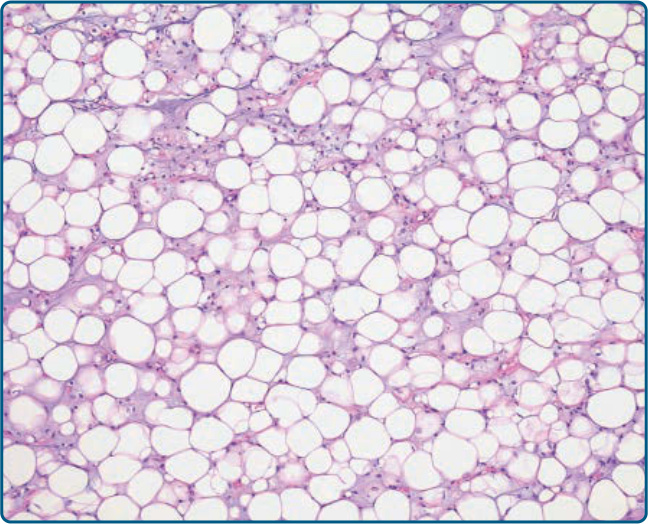
圖 122-38:脂肪源性黏液樣脂肪肉瘤 (Lipogenic myxoid liposarcoma)。在某些黏液樣脂肪肉瘤病例中,可見主要的脂肪源性分化,模擬非典型脂肪瘤樣腫瘤。
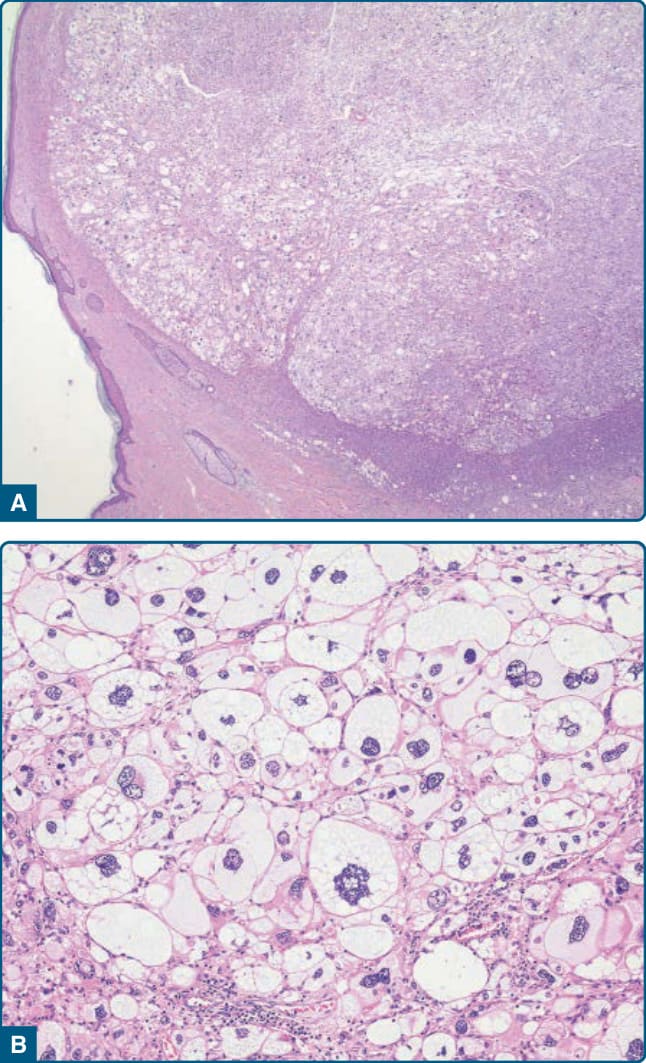
圖 122-39:淺表性多形性脂肪肉瘤 (Superficial pleomorphic liposarcoma)。A,一個外生性生長、純真皮的多形性脂肪肉瘤。B,注意具有怪異、深染細胞核的多形性脂肪母細胞的存在。
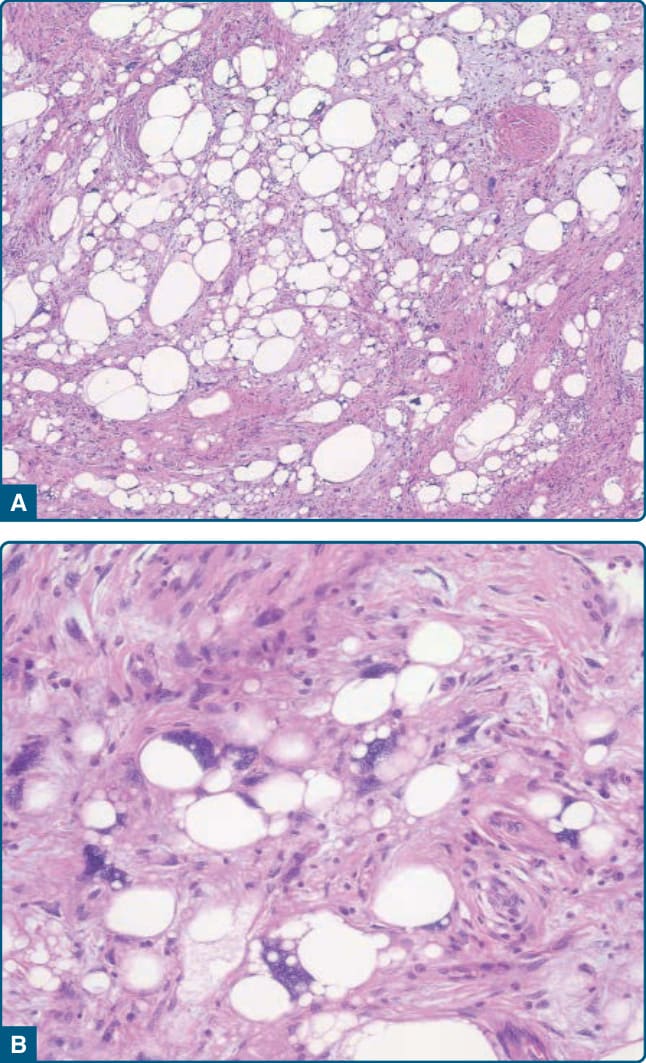
圖 122-40:多形性脂肪肉瘤 (Pleomorphic liposarcoma)。A 與 B,多形性脂肪母細胞與非典型脂肪源性細胞與肉瘤細胞不規則地混合。
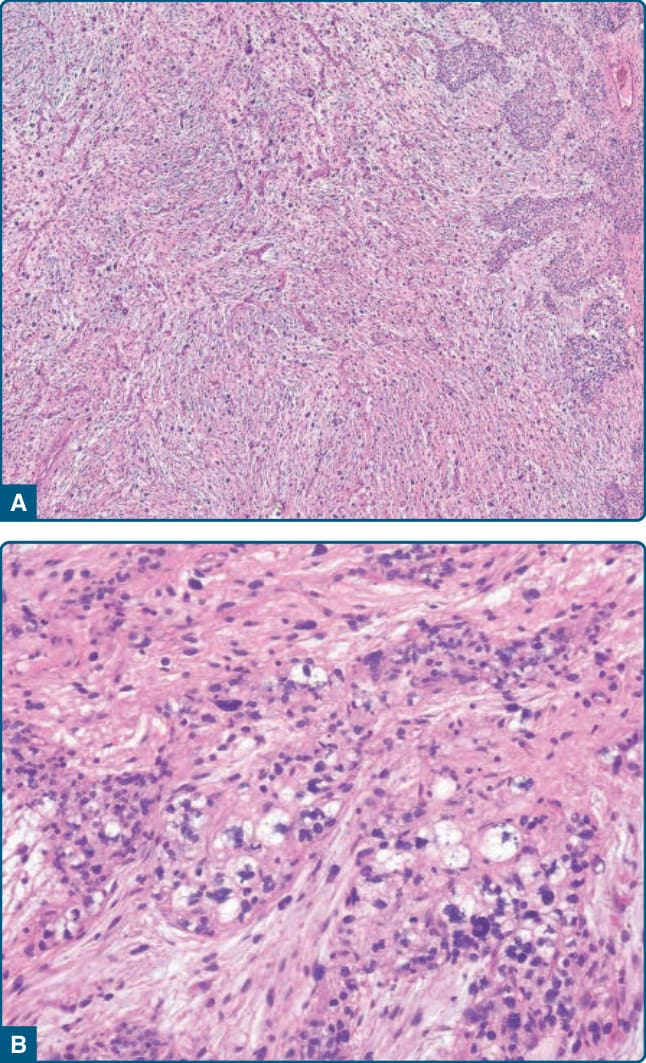
圖 122-41:多形性脂肪肉瘤 (Pleomorphic liposarcoma)。A,腫瘤顯示較高度惡性黏液纖維肉瘤的特徵。B,右側存在多形性脂肪母細胞。
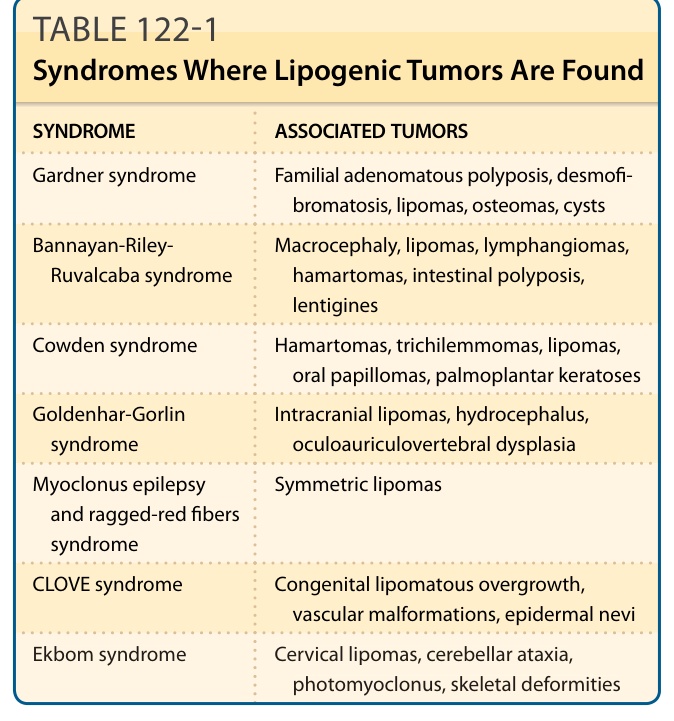
表 122-1:發現脂肪源性腫瘤的症候群 (Syndromes Where Lipogenic Tumors Are Found)