皮膚假性淋巴瘤 (Cutaneous Pseudolymphoma) 精華筆記
總論
- 定義:皮膚假性淋巴瘤 (cutaneous pseudolymphoma, PSL) 是一群在臨床上與/或組織學上模擬皮膚淋巴瘤的良性淋巴增生過程 (benign lymphoproliferative processes),臨床、組織學與免疫表型表現各異。
- 病因:感染(螺旋體:伯氏疏螺旋體 Borrelia burgdorferi、梅毒螺旋體 Treponema pallidum;病毒:副痘病毒 parapoxviruses;寄生蟲:疥瘡 scabies)、昆蟲叮咬、疫苗/去敏化抗原注射、異物(刺青 tattoos、金屬)、藥物;無法辨識病因者稱特發性 (idiopathic)。
- 分類(表 120-1):(a) 結節性 PSL(模擬 T/B 細胞淋巴瘤);(b) 模擬蕈狀肉芽腫的 pseudo-MF;(c) 其他富淋巴細胞浸潤疾病;(d) 淋巴細胞的良性血管內增生(模擬血管內淋巴瘤)。
診斷與病程
- 診斷核心:臨床病理相關性 (clinicopathologic correlation) 至關重要。組織學分析(浸潤模式、淋巴細胞大小、免疫表型 T/B、CD4/CD8、CD30)為關鍵;克隆性 (clonality) 與感染原(尤其 B. burgdorferi)的分子研究為輔助。
- 重要觀念:偵測到克隆性 T 或 B 細胞族群本身不代表惡性淋巴瘤;部分 PSL 病例帶有克隆性 T/B 細胞。
- 檢查:病史(節肢動物、過敏原、藥物暴露)、淋巴結觸診、周邊血液(淋巴球增多、嗜酸性球增多、B. burgdorferi/梅毒/HIV 血清學)。PSL 為良性、無皮外擴散潛能,通常不需分期;僅在不尋常表現、輕鏈單型表現、克隆性或非預期發現時,考慮放射學分期 (CT 或 PET-CT) 以排除淋巴瘤。
- 病程:變化極大;切片後可能消退,部分持續數月至數年;再次暴露誘發病原可復發。進展為明顯淋巴瘤極為罕見。
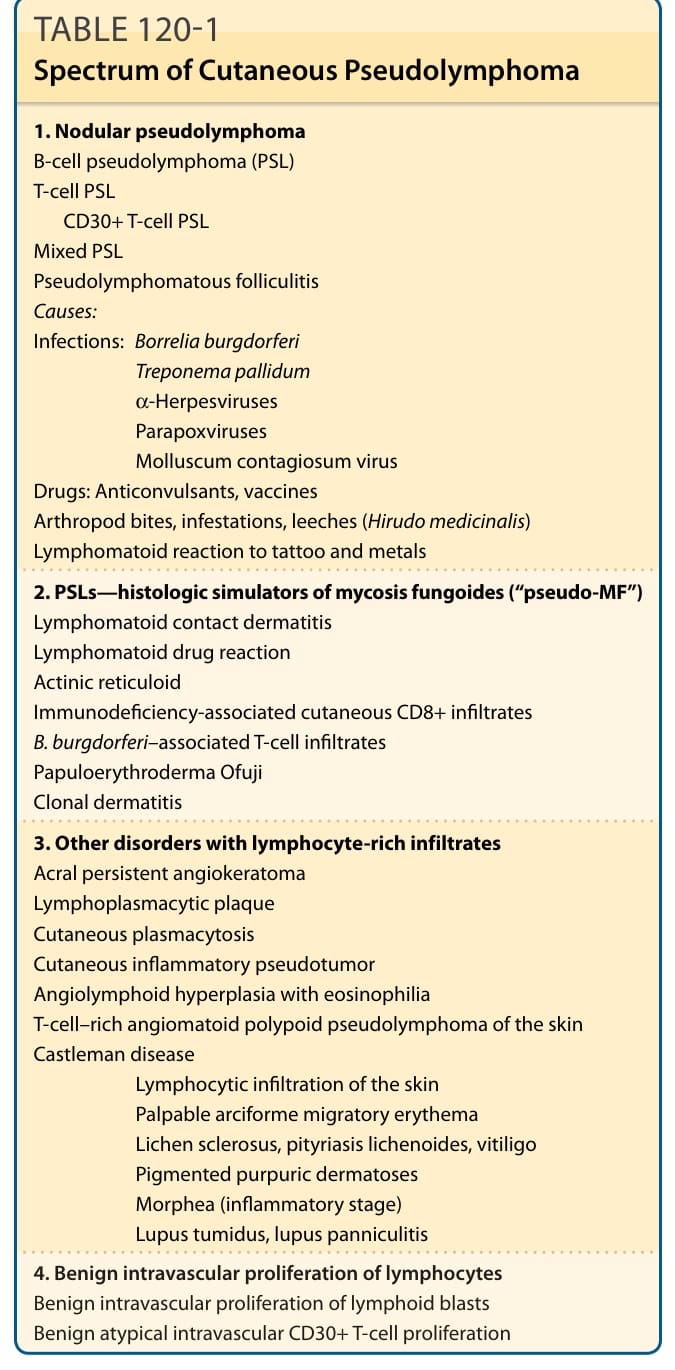
表 120-1:皮膚假性淋巴瘤的譜系 (Spectrum of Cutaneous Pseudolymphoma)
結節性假性淋巴瘤 (Nodular Pseudolymphoma)
- 以單一或多發結節表現,模擬 T/B 細胞淋巴瘤;依主要淋巴細胞亞群分為 B 細胞、T 細胞、混合型(此分類偏人為,B 細胞 PSL 必含 T 細胞,反之亦然)。
- 治療原則(表 120-3):
- 避免/消除致病病原(如接觸性過敏原、疫苗、過敏原注射、水蛭、針灸、刺青)——是預防持續與復發最重要的步驟。
- 感染相關者用抗生素(如 Borrelia 相關 PSL)。
- 單一病灶:手術切除、局部/病灶內皮質類固醇、冷凍療法 (cryotherapy)、雷射;無效時考慮放射治療 (radiation therapy)。
- 多發/特發性多灶性者:全身性皮質類固醇、病灶內干擾素-α (interferon-α)、羥氯奎寧 (hydroxychloroquine)(抑制漿細胞樣樹突狀細胞)。
皮膚 B 細胞假性淋巴瘤
- 又稱皮膚淋巴細胞瘤 (lymphocytoma cutis)、皮膚淋巴樣增生 (cutaneous lymphoid hyperplasia)。
- 臨床:最常為結節或斑塊,好發臉部(鼻、頰)、上軀幹、手臂;男女比 2:1;約 67% 病人 < 40 歲。局限型單一結節(最大 4 cm)最常見;三分之一發展多發結節或粟粒狀 (miliarial) 型。
- 組織學:網狀真皮緻密結節性浸潤,主要為小淋巴細胞與含可染體巨噬細胞 (tingible body macrophages) 的反應性生發中心;無核異型;漿細胞瀰漫散布。免疫表型:CD19+/CD20+/CD79a+ B 細胞為主;反應性濾泡 bcl-6+/bcl-2−,濾泡間區小 B 細胞 bcl-2+/bcl-6−;CD21+ 濾泡樹突狀細胞網絡界限分明、規則;輕鏈 κ/λ 多型 (polytypic) 表現;無 IgH 單株重排。
- 鑑別診斷(表 120-4):主要與原發性皮膚邊緣區淋巴瘤 (PCMZL)、原發性皮膚濾泡中心淋巴瘤 (PCFCL) 區分。關鍵點:PCMZL 漿細胞呈片狀、輕鏈單型 (monotypic)(比例 ≥ 5:1,可達 10:1);PCFCL 為腫瘤性濾泡、低增生活性、濾泡樹突狀細胞網絡不規則斷裂。約 10%–20% PSL 帶克隆性 B 細胞,故克隆性研究價值有限;輕微浸潤須排除假性克隆性 (pseudoclonality)。
Borrelia 相關 B 細胞假性淋巴瘤
- 約 1% 臨床明顯 B. burgdorferi 感染表現為 B 細胞 PSL;白人較非裔美國人多;多 < 40 歲。
- 臨床:單一紅至紫紅色 (violaceous) 圓頂狀結節,10%–15% 多灶;好發臉部、頭皮,尤其耳垂、乳頭、陰囊;可有區域性淋巴結病變。
- 組織學/檢查:緻密真皮結節性浸潤伴反應性生發中心,生發中心傾向大而融合、套區小,類似 PCFCL。多株 IgH 重排為主(偵測單株不排除診斷)。血清學抗 B. burgdorferi 抗體型態多變(陰性也不排除);PCR 偵測 DNA 敏感度約 70%。診斷依組織學+臨床(蜱叮咬史 history of tick bite、好發部位)+血清學/PCR。
- 治療:抗生素為第一線,並預防關節炎、心肌炎等併發症。
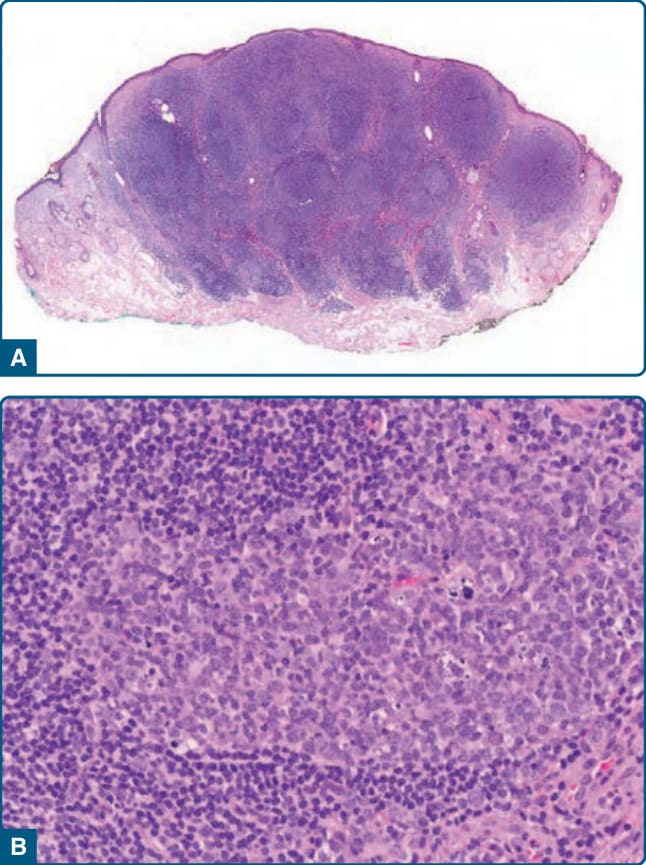
圖 120-2:Borrelia 相關 B 細胞假性淋巴瘤組織學——結節性淋巴細胞浸潤伴含可染體巨噬細胞的反應性生發中心。
皮膚 T 細胞與混合型假性淋巴瘤
- T 細胞 PSL 為緻密真皮富 T 細胞結節浸潤(B 細胞可達 30%);混合型含相等 T/B 細胞。約 5% 由藥物引起,多數為特發性。
- 臨床:單一或多發紅至紫紅色結節,類似 B 細胞 PSL。
- 組織學:緻密結節性浸潤遍及真皮並入淺層皮下;小淋巴細胞為主(偶見略增大至中型);混雜嗜酸性球、組織球、漿細胞;可有毛囊外滲但缺乏表皮趨向性 (lack of epidermotropism)。多為 CD4+ CD30− T 細胞,偶見少數 CD30+ 母細胞。多株為主;部分為克隆性 PSL(可能為極早期淋巴瘤生成)。
- 鑑別診斷:皮膚 CD4+ 小型/中型 T 細胞淋巴增生性疾病 (CD4+ SMT-LPD)(WHO 2016 涵蓋性名詞,強調惰性本質,PD-1 無診斷價值);腫瘤期蕈狀肉芽腫 (MF,靠先前斑片/斑塊區別);血管免疫母細胞性 T 細胞淋巴瘤 (AITL) 續發性皮膚浸潤;腫脹型紅斑性狼瘡 (lupus tumidus,有交界空泡化與間質黏蛋白)。
- 治療/預後:去除病因後可自發消退或持續數月至數年;手術切除、冷凍、雷射、局部/病灶內類固醇或干擾素;避免刺激因子預防復發。
CD30+ T 細胞假性淋巴瘤
- T 細胞 PSL 的組織學亞型,特徵為中型至大型非典型 CD30+ T 細胞。
- 見於淋巴瘤樣藥物疹、結節性疥瘡、節肢動物叮咬、病毒感染(副痘病毒——羊痘 Orf、擠奶者結節、傳染性軟疣、疱疹病毒),及化膿性汗腺炎、珊瑚傷害。
- 組織學:增大的母細胞樣 CD30+ 細胞單個散布於小 T 細胞為主的浸潤中;多數無克隆性 T 細胞。鑑別淋巴瘤樣丘疹病 (A 型)、皮膚退行性大細胞淋巴瘤 (ALCL):CD30+ PSL 之 CD30+ 細胞不成聚集體、含顯著 B 細胞與漿細胞、5hmC 表現保留。治療針對潛在病因。
假性淋巴瘤性毛囊炎
- 1988 年 Kibbi 描述;臉部單一結節 < 1.5 cm,似 CD4+ SMT-LPD。半數具 B 細胞、半數具 T 細胞 PSL 特徵;毛囊周圍排列、可有無浸潤帶 (Grenz zone)、毛囊淋巴細胞外滲;混雜大量 CD1a+ S-100+ 樹突狀細胞。
其他結節性 PSL
- 持續性結節性節肢動物叮咬反應與結節性疥瘡:長期搔癢丘疹/結節,好發肘、軀幹、生殖器、腋窩/鼠蹊皺褶。組織學楔形真皮浸潤、嗜酸性球、火焰圖形 (flame figures)、高度嗜酸性膠原束、飽滿內皮;T 細胞主導、無克隆性。須與誇大叮咬反應(B-CLL 等白血病)、淋巴瘤樣丘疹病、何杰金氏淋巴瘤、結節性續發性梅毒區分。抗疥瘡治療無效;可切除、病灶內類固醇或局部免疫調節劑。
- 水蛭 (Hirudo medicinalis) 引起:分布對應水蛭暴露處(下背、血腫治療區);推測水蛭內細菌轉移誘發反應。
- 藥物引起的結節性 PSL:抗痙攣藥等;單一或多發結節;組織學為典型 T/B 細胞 PSL(T 細胞增生率約 10%);潛伏期可數月至數年;停藥為第一步,再暴露可復發。
- 疫苗/藥物注射部位:注射部位皮下富 B 細胞浸潤伴反應性生發中心;中央組織球聚集,胞質灰藍顆粒狀(氫氧化鋁 aluminium hydroxide 結合疫苗)。
- 刺青/金屬相關:浸潤多侷限於紅色刺青染料區(硃砂 cinnabar,硫化汞),藍/綠刺青用鈷與鉻鹽;屬遲發型過敏反應,潛伏期數月至數年;苔癬樣+深層混嗜酸性球浸潤;貼布試驗 (patch test) 可陽性或陰性。
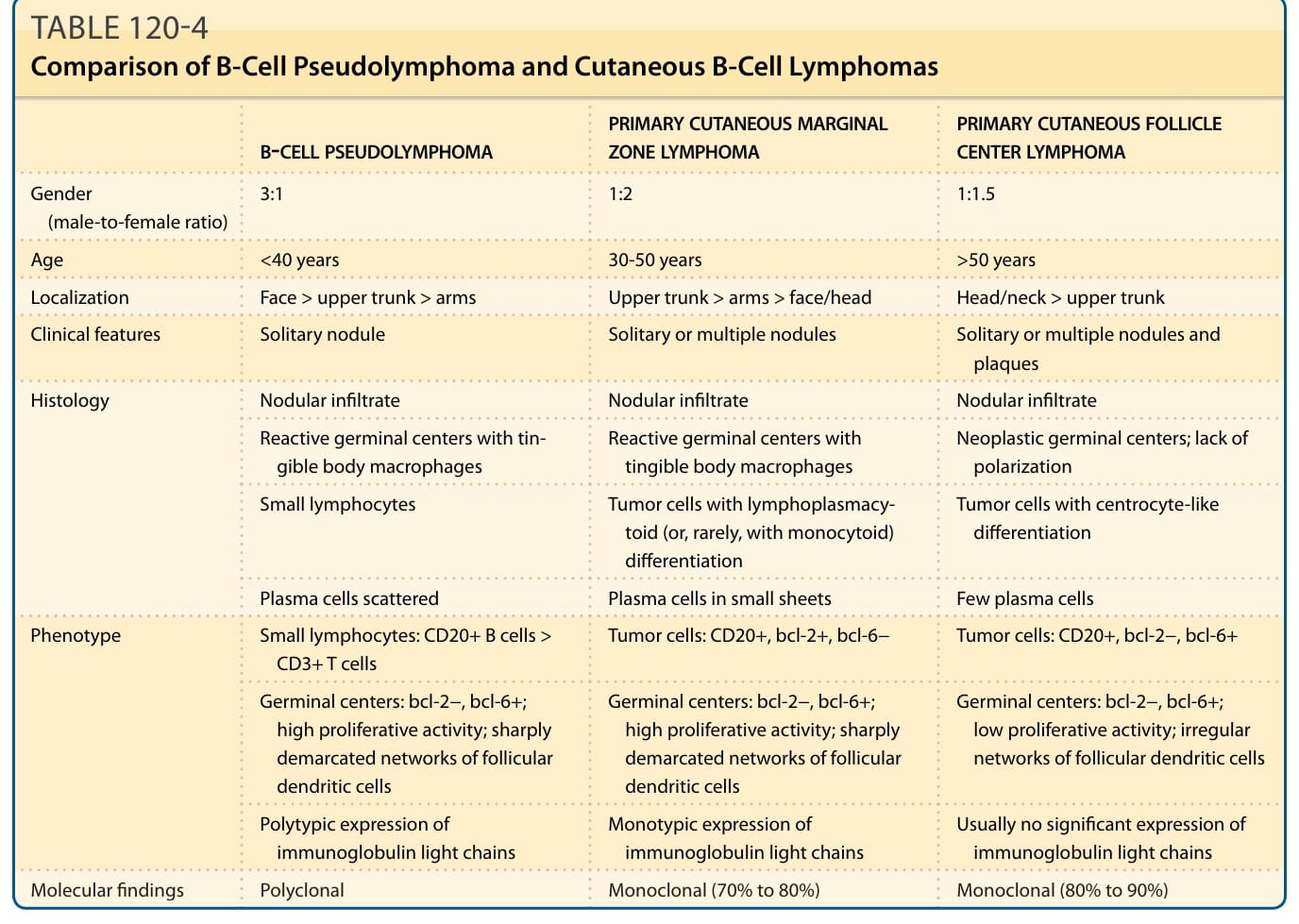
表 120-4:B 細胞假性淋巴瘤與皮膚 B 細胞淋巴瘤的比較。
模擬淋巴瘤的感染
- 皮膚利什曼病 (cutaneous leishmaniasis):結節性浸潤含漿細胞、組織球;靠無核異型+特殊染色/免疫組化/分子偵測寄生蟲區分。
- 隱匿性疱疹 (herpes incognito):HSV/VZV 富淋巴細胞浸潤無典型上皮變化,可見增大 CD30+ 細胞;靠免疫組化/PCR 偵測病毒辨識。
- 副痘病毒:誘發 CD30 表現;靠包涵體 (inclusion bodies)、無 T 細胞標記喪失、無 TCR-γ 單株重排與病毒偵測區分。
假性淋巴瘤——蕈狀肉芽腫的組織學模擬者 (Pseudo-MF)
- 共通:真皮帶狀或血管周圍小淋巴細胞浸潤,外滲入表皮、可有輕微核異型,模擬表皮趨向性 CTCL;CD4+ 或 CD8+ 為主,多株為主。
- 支持 CTCL(淋巴瘤)的發現:深層核異型、中至大型細胞為主、全 T 細胞標記喪失、TCR 基因單株重排。
- 治療:避免致病病原;局部類固醇/免疫調節劑+UV 光療;嚴重者全身性類固醇與免疫抑制劑(如環孢素 cyclosporine)。
淋巴瘤樣接觸性皮膚炎 (LCD)
- 慢性接觸性皮膚炎,組織學模擬 MF;元兇含硫酸鎳 (nickel sulfate)、金、鋅、對苯二胺 (paraphenylenediamine)、紡織染料。濕疹性紅斑脫屑丘疹/斑塊、搔癢。組織學淺層帶狀浸潤伴海綿狀水腫、表皮內蘭格罕細胞聚集(假性 Pautrier 聚集體);核異型不顯著、CD4/CD8 比例不明顯、無 T 細胞標記喪失。貼布試驗證明致敏化為必要診斷標準。治療同接觸性皮膚炎。
淋巴瘤樣藥物反應
- 結節型外常為斑丘疹/丘疹疹;上層真皮帶狀浸潤伴外滲,可有交界空泡變化與凋亡角質細胞、核異型;CD4+ 或 CD8+ 為主+CD30+;無全 T 細胞標記喪失。鑑別 MF/塞扎里症候群(沿交界排列、核異型、標記喪失、TCR-γ 單株重排支持淋巴瘤;塞扎里 PD-1/TOX 表現)。停藥為必要步驟;可全身/病灶內/局部類固醇。
光化性網狀細胞增多症 (Actinic Reticuloid)
- 慢性多因子皮膚炎、對廣泛波長嚴重光敏感,組織學模擬表皮趨向性 CTCL;多見中老年男性;光照部位(臉、頸)持續性紅斑苔癬樣丘疹/斑塊,可獅面樣 (facies leonina-like),劇癢。
- 組織學:表皮乾癬樣增生、輕微海綿狀水腫、緻密正角化;上真皮血管周圍+間質浸潤含小淋巴細胞、嗜酸性球、漿細胞;乳頭真皮粗膠原束垂直條紋、可有多核纖維母細胞;以 CD8+ T 細胞為主。
- 周邊血 CD8+ 增加(CD4/CD8 比例倒置),循環淋巴細胞鋸齒狀核;光照測試/光貼布試驗陽性。
- 鑑別 CD8+ MF(更強表皮趨向性與核異型、位於 UV 保護區)、塞扎里症候群(克隆性 CD4+、淋巴結病變、禿髮、掌蹠角化過度)。
- 治療:防曬、局部類固醇/免疫調節劑(療效不一)、環孢素;UV-B 光硬化可良好反應。
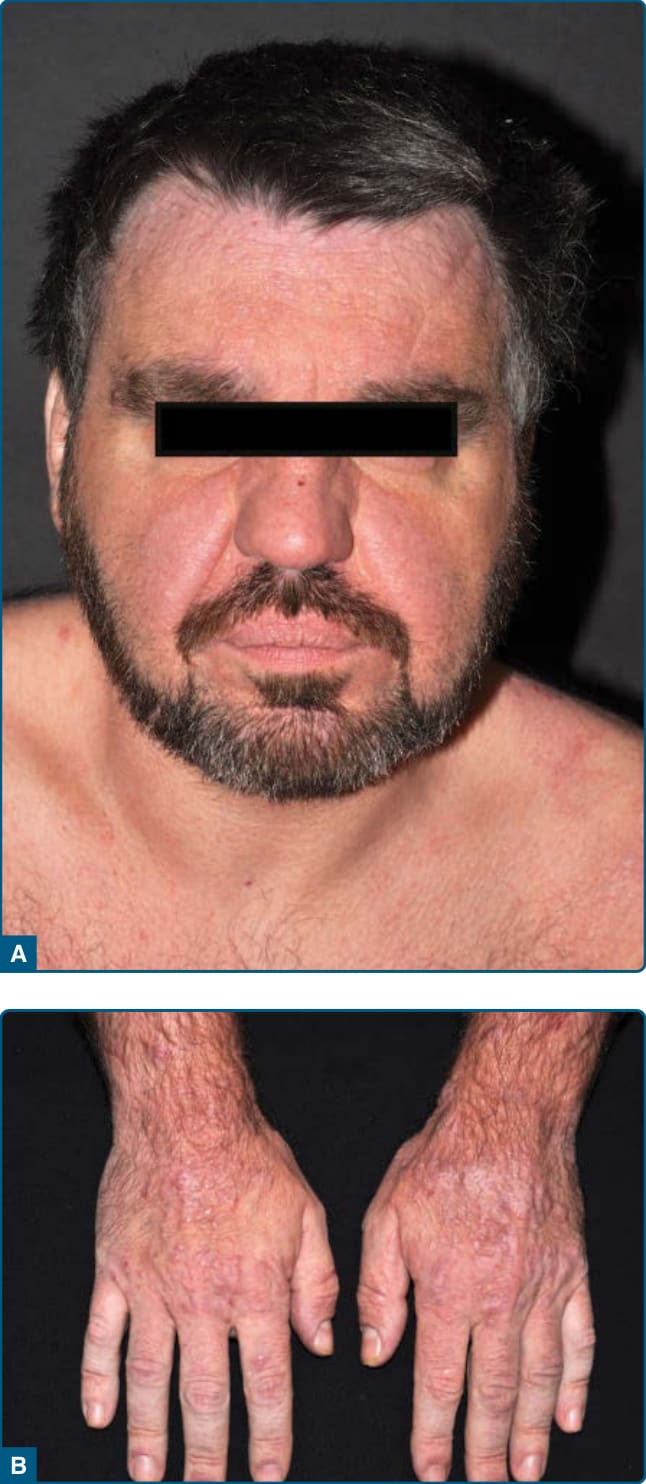
圖 120-15:光化性網狀細胞增多症——臉部瀰漫性浸潤、紅斑與丘疹。
其他 pseudo-MF
- 免疫缺陷 (HIV) 中的 CD8+ T 細胞 PSL:晚期 HI(低 CD4) 出現模擬 MF 之 CD8+ 浸潤;播散性搔癢丘疹、掌蹠角化過度、紅皮症;多株性;病程惰性但預後不良(取決於 CD4 淋巴球減少);HAART 多可緩解,中度免疫抑制者甲胺喋呤 (methotrexate) 有效。
- Borrelia 相關 T 細胞 PSL:帶狀/瀰漫 T 細胞浸潤、局部表皮趨向性、輕微核異型,模擬 MF;抗生素可緩解。
- Ofuji 丘疹性紅皮症:罕見搔癢性紅皮症,可與藥物、何杰金氏淋巴瘤、內臟惡性腫瘤、免疫缺陷相關;中位年齡 70 歲、男性多;播散性平頂棕色丘疹,皺褶不受侵犯(「躺椅 deckchair」徵);嗜酸性球增多、IgE 升高;缺核異型可與塞扎里區分;應查 HIV;PUVA、UV-B+局部類固醇、環孢素等有效。
- 作為 CTCL 模擬者的發炎性疾病:扁平苔癬、硬化萎縮性苔癬、色素性紫癜性皮膚炎、苔癬樣糠疹、腫脹型狼瘡、狼瘡性脂膜炎。
- 發炎性皮膚病的 T 細胞克隆:扁平苔癬 6%、硬化萎縮性苔癬 13%、苔癬樣糠疹高達 60% 帶克隆性 T 細胞;克隆性偵測不足以診斷 CTCL;發炎性疾病之克隆短暫且隨時間改變。
- 克隆性皮膚炎 (clonal dermatitis):帶 T 細胞克隆的慢性非特異性皮膚炎,約 25% 在 5 年內進展為 CTCL,可能為 CTCL 前驅病灶。
- 皮膚淋巴細胞浸潤 (Jessner-Kanof, LIS) 與可觸及之弓形遊走性紅斑 (PAME):被視為 T 細胞 PSL 或狼瘡樣疹;軀幹好發;血管周圍/附屬器周圍 T 細胞浸潤,無漿細胞與黏蛋白;LIS 以 CD8+ 為主、多株;對局部類固醇、口服抗生素、UV-A1 有反應,易復發。
其他具富淋巴細胞浸潤的疾病
- 肢端假性淋巴瘤性血管角化瘤 (APA/APACHE):兒童(亦見成人)肢端單側群聚紅至紫紅色血管瘤樣丘疹(1–5 mm);可甲變化;緻密淋巴細胞浸潤+厚壁血管伴飽滿內皮;多株 T/B 細胞;良性,刮除術、病灶內或封閉高效價局部類固醇可緩解。
- 淋巴漿細胞性斑塊 (LPP):罕見,多見兒童脛前區(亦見成人軀幹/手臂)、女性偏多;長期斑塊或線狀紅棕丘疹/斑塊;真皮淋巴組織球浸潤伴大量多株漿細胞(達 25%)、假性玫瑰花結 (pseudorosettes);須排除感染;手術切除為第一線。
- 皮膚漿細胞增多症:亞洲(日本)多見、成人;多發棕色斑塊/結節;成熟多株漿細胞浸潤;可有全身侵犯(淋巴結病變、肝脾腫大、高丙種球蛋白血症、IL-6 升高);PUVA、類固醇、化療。
- 皮膚發炎性假瘤:含漿細胞肉芽腫與發炎性肌纖維母細胞瘤(後者紡錘細胞+ALK 表現);手術切除可緩解。
- 伴嗜酸性球增多之血管淋巴樣增生 (ALHE):血管增生伴上皮樣內皮(亦稱上皮樣血管瘤),緻密 T/B 細胞+嗜酸性球浸潤;好發頭頸(臉、耳);CD31/ERG+、D2-40−;與木村病 (Kimura disease) 多視為不同實體(木村病病灶較大較深、周邊嗜酸性球增多、淋巴結腫大);遷延抗治療、復發常見;手術、冷凍、雷射、甲胺喋呤、干擾素-α;有動靜脈分流者可栓塞。
- Castleman 病:良性淋巴增生,透明血管型(同心螺旋狀、年輕病人)與漿細胞變異型(可伴 POEMS 症候群);多在淋巴結,罕見皮膚;局限結外者預後良好。
淋巴細胞的良性血管內增生
- 淋巴母細胞良性血管內增生(伴或不伴 CD30),產生於發炎性皮膚病或皮膚外傷區;硬化性苔癬阻塞淋巴管擾亂免疫細胞運輸,致活化 CD30+ 淋巴細胞累積。
- 大型母細胞樣淋巴細胞,表現 CD3/CD4、部分 CD30;與 EB 病毒無關;多株性。
- 最重要鑑別:血管內淋巴瘤 (intravascular lymphoma)(侵襲性);亦須與淋巴管內組織球增多症(類風濕性關節炎、骨科金屬植入物)區分。
總結
- 皮膚 PSL 為一群在臨床/組織學上模擬皮膚淋巴瘤的良性富淋巴細胞浸潤;臨床病理相關性是達成最終診斷並與淋巴瘤區別的關鍵。處置首步為消除感染原(抗生素/抗病毒)或避免致病病原,輔以免疫調節劑(皮質類固醇、UV 光療)或破壞性方法(手術切除、冷凍療法)。