Cutaneous Pseudolymphoma
20
AT-A-GLANCE
■ Cutaneous pseudolymphomas are benign lymphoproliferations that clinically and/or histologically simulate cutaneous lymphomas.
■ They exhibit a wide range of clinical, histological, and immunophenotypic features and can be triggered by different infectious and non-infectious agents.
■ Clinicopathological correlation plays an important role in the differentiation from cutaneous lymphomas.
■ Therapy includes avoidance of exposure to the causative agent, immunomodulating agents or ablative approaches.
DEFINITION
The term cutaneous pseudolymphoma refers to a group of skin diseases that can be defined as benign lymphoproliferative processes that clinically and/or histologically simulate cutaneous lymphomas. These diseases differ in their clinical, histologic, and immunophenotypic presentation, and are of different etiologies (Table 120-1). A broad spectrum of causative factors known to induce cutaneous pseudolymphomas (PSLs) have been identified. Infectious agents, such as spirochetal bacteria (Borrelia burgdorferi sp., Treponema pallidum), viruses (eg, parapoxviruses), infestations (eg, scabies), insect bites, injection of vaccines or antigens for hyposensitization, foreign bodies such as tattoos and metals, and drugs have been identified as causative factors for PSL. All cases without identifiable cause are referred to as an idiopathic form of PSL.
HISTORICAL ASPECTS
Various synonyms have been introduced and several terms are still nowadays used to describe PSLs of the skin. For the first time the concept of PSL (pseudomalignancy) had been introduced by M. Kaposi in 1891 (Table 120-2).1 In 1923, Biberstein coined the term lymphocytoma cutis.2 Subsequently, in 1943, Bäfverstedt used the designation lymphadenosis benigna cutis.3 Lever introduced the term pseudolymphoma of Spiegler and Fendt in 1967, and in 1969, Caro and Helwig employed the term cutaneous lymphoid hyperplasia.4 Many other terms were proposed and referred mostly to B-cell PSLs. Among T-cell PSLs, actinic reticuloid was first described in 1969.5 In the 1980s the designation cutaneous PSL for B-cell–dominated and T-cell–dominated processes became more widely
used and accepted.6 Synonymously, the term atypical lymphoid proliferation is often employed by pathologists and dermatopathologists.
CLASSIFICATION
Various approaches have been proposed to categorize cutaneous PSL. For example, according to the etiology, the predominant lymphocytic component (T, B, or mixed cells) or distinct clinical features (eg, acral papular angiokeratoma of childhood).7-9
Categorization of PSL according to etiology or histology and phenotype into T-cell and B-cell PSL is widely used in textbooks. In daily work, clinicians or pathologists encountering infiltrates suspicious of being a PSL cannot recognize the etiology and the phenotype at first glance; further diagnostic workup is needed. Moreover, it has to be emphasized that the composition of the infiltrate is determined mostly by genetic and immunologic factors of the host rather than the causative agent per se, as the same agents can, in many instances, induce B-cell PSL and T-cell PSL as well. From a practical point of view, cutaneous PSL presenting as a solitary or multiple nodule(s), which clinically and histologically simulate lymphoma, can be distinguished from other forms of PSL, which mimic cutaneous T-cell lymphoma on histologic grounds alone, and are summarized under the term pseudo–mycosis fungoides (pseudo-MF). In addition, there are numerous infectious and noninfectious conditions that are characterized by a lymphocyte-rich infiltrate and are prone to be misinterpreted as cutaneous lymphoma primarily on histologic grounds. Thus, this chapter is divided into the following sections: (a) nodular PSL simulating clinically and histologically T-cell or B-cell lymphoma; (b) PSLs mimicking histologically mycosis fungoides (so called pseudo-MF); (c) other skin disorders with lymphocyte-rich infiltrates; and (d) benign intravascular proliferation of lymphocytes (simulating intravascular lymphoma) (see Table 120-1).
DIAGNOSTIC APPROACH
The clinical presentation of cutaneous PSL ranges from a solitary nodule to clustered or disseminated papules to erythroderma.8,10 The histologic analysis plays a crucial role in the diagnostic approach to cutaneous PSLs. Different infiltrate patterns (nodular infiltrate vs epidermotropic infiltrates), the size of the lymphocytes (mostly small cells; occasionally medium and large cells), immunophenotype (T-cell vs B-cell; CD4 vs CD8; CD30) can be distinguished. Molecular studies
- Nodular pseudolymphoma B-cell pseudolymphoma (PSL) T-cell PSL CD30+ T-cell PSL Mixed PSL Pseudolymphomatous folliculitis Causes: Infections: Borrelia burgdorferi
Treponema pallidum
α-Herpesviruses
Parapoxviruses
Molluscum contagiosum virus Drugs: Anticonvulsants, vaccines Arthropod bites, infestations, leeches (Hirudo medicinalis) Lymphomatoid reaction to tattoo and metals
-
PSLs—histologic simulators of mycosis fungoides (“pseudo-MF”) Lymphomatoid contact dermatitis Lymphomatoid drug reaction Actinic reticuloid Immunodeficiency-associated cutaneous CD8+ infiltrates B. burgdorferi–associated T-cell infiltrates Papuloerythroderma Ofuji Clonal dermatitis
-
Other disorders with lymphocyte-rich infiltrates Acral persistent angiokeratoma Lymphoplasmacytic plaque Cutaneous plasmacytosis Cutaneous inflammatory pseudotumor Angiolymphoid hyperplasia with eosinophilia T-cell–rich angiomatoid polypoid pseudolymphoma of the skin Castleman disease
Lymphocytic infiltration of the skin
Palpable arciforme migratory erythema
Lichen sclerosus, pityriasis lichenoides, vitiligo
Pigmented purpuric dermatoses
Morphea (inflammatory stage)
Lupus tumidus, lupus panniculitis
-
Benign intravascular proliferation of lymphocytes Benign intravascular proliferation of lymphoid blasts Benign atypical intravascular CD30+ T-cell proliferation
-
Benign intravascular proliferation of lymphocytes Benign intravascular proliferation of lymphoid blasts Benign atypical intravascular CD30+ T-cell proliferation
for clonality and infectious agents, especially Borrelia burgdorferi, are adjunctive diagnostic tools. It is important to emphasize that the detection of a clonal T-cell or B-cell population per se does not indicate the presence
1891 Sarcomatosis cutis (M. Kaposi)1
1910 Sarcoid Spiegler-Fendt (Darier)114
1923 Lymphocytoma cutis (Biberstein)2
1943 Lymphadenosis benigna cutis (Bäfverstedt)3
1969 Cutaneous lymphoid hyperplasia (Caro and Helwig)4
1982 Cutaneous pseudolymphoma (Burg et al)6
1982 Cutaneous pseudolymphoma (Burg et al)6
20
of malignant lymphoma. Moreover some PSL cases have been reported to harbor clonal T or B cells.11-14
Thus, both the histologic and the molecular findings always need to be interpreted in synopsis with the clinical context; that is, the clinicopathologic correlation is essential to achieve the final diagnosis. The diagnostic workup includes the medical history (particularly exposure to arthropods, allergens, and drugs) and physical examination including palpatory evaluation of lymph nodes, examination of peripheral blood (lymphocytosis, eosinophilia, serology for infectious agents, especially B. burgdorferi, syphilis, HIV). Because PSLs represent benign lymphocytic proliferations without the potential for extracutaneous spread, staging examinations are usually not indicated. In cases with unusual clinical manifestations and multiple nodular lesions, monotypic expression of immunoglobulin light chains, detection of T-cell or B-cell clonality, or other inconsistent or unexpected histologic or phenotypic findings, radiologic staging (computed tomography or positron emission tomography-computed tomography) should be considered to exclude primary or secondary cutaneous lymphoma mimicking PSL.
COURSE
The course of cutaneous PSLs is highly variable. Resolution may occur after biopsy, but some lesions may persist over several months or even years. Recurrences can be observed particularly after reexposure to the inducing agent in cases that are caused by drugs or allergens. Progression of PSL has been reported, but is a very rare event, if it exists at all.15 It remains unclear whether PSL in fact progressed to overt lymphoma (eg, by acquisition of chromosomal aberrations or persisting antigenic stimulus resulting in permanent proliferation of lymphocytes) or whether those cases represent cutaneous lymphomas from the very beginning, but could not be recognized as such by histologic, phenotypic, or molecular findings in their very early disease stages.
NODULAR PSEUDOLYMPHOMA
PSLs that present with a solitary nodule or with multiple nodules simulate cutaneous T-cell and B-cell lymphomas on clinical and histologic grounds. They represent one of the most common forms of PSL, but no detailed data on the overall prevalence of B-cell PSLs have been reported. Histologically they can be classified according to the predominant lymphocytic subset into B-cell, T-cell, and mixed (T-cell/B-cell) PSL.8,10 This classification is rather artificial as B-cell PSL always contains T cells and vice versa. T-cell PSL often harbors a variable number of B cells. Nevertheless, the distinction into the 3 histologic and phenotypic subtypes of nodular PSL remains useful for the differential diagnosis, which also has to be considered for the potential cause of the pseudolymphomatous reaction.
2109
20
-
Avoidance or elimination of the causative agent (eg, contact allergen)
-
Antibiotic treatment for infection-associated pseudolymphoma (eg, Borrelia-associated-cell pseudolymphoma)
-
Surgical excision, cryotherapy, laser treatment
-
Topical/intralesional steroids or interferon-α
-
Systemic steroids, hydroxychloroquine
-
Avoidance or elimination of the causative agent (eg, contact
allergen)
2. Antibiotic treatment for infection-associated pseudolymphoma
(eg, Borrelia-associated-cell pseudolymphoma)
3. Surgical excision, cryotherapy, laser treatment
4. Topical/intralesional steroids or interferon-α
5. Systemic steroids, hydroxychloroquine
Avoidance of reexposure to the inducing agent (eg, vaccines, allergen injection, other drugs, Hirudo medicinalis treatment, acupuncture, and tattoo) is the most important step to prevent persistence and recurrence of nodular PSL. Solitary lesions can be treated by complete surgical excision, topical or intralesional corticosteroids, cryotherapy, or laser therapy (Table 120-3). If those therapeutic approaches are not successful, radiation therapy may be considered. In patients with multiple PSL lesions, particularly those with idiopathic multifocal PSL, systemic corticosteroids, intralesional interferon-α, or hydroxychloroquine may be therapeutic options. Hydroxychloroquine inhibits the activity of plasmacytoid dendritic cells, which are found in the majority of B-cell PSLs as clusters in close vicinity to T cells and plasma cells, and may represent the driving force in induction and maintenance of the PSLs.16,17
CUTANEOUS B-CELL PSEUDOLYMPHOMA
CUTANEOUS B-CELL
PSEUDOLYMPHOMA
B-cell PSL is often also referred as lymphocytoma cutis or cutaneous lymphoid hyperplasia.
CLINICAL FEATURES
B-cell PSL most commonly presents with a nodule or plaque. The face, especially the nose and the cheeks, the upper trunk, and the arms are the most commonly involved sites. A male-to-female ratio of 2:1 has been described.18 Approximately 67% of the patients with B-cell PSLs are younger than age 40 years and less than 10% are children and adolescents.4
The localized form of B-cell PSL that presents with a solitary nodule measuring up to 4 cm, is the most common presentation. One-third of patients develop multiple nodules or a miliarial form in which the lesions are papules measuring only a few millimeters in diameter.19
HISTOLOGY
All nodular B-cell PSLs essentially share the same growth pattern and composition of the infiltrate. There is a dense nodular infiltrate, predominantly located in the reticular dermis and occasionally extending into
2110
the superficial parts of the subcutis. The infiltrate is mostly composed of small lymphocytes with chromatin dense nuclei and reactive germinal centers containing tingible body macrophages. The lymphocytes do not show nuclear atypia. There is an admixture of plasma cells, which usually do not form aggregates but are rather diffusely scattered throughout the infiltrate. Eosinophils and a granulomatous component can be observed in some cases. There is an admixture of a variable number of T cells, which usually account for less than 30% of the infiltrate. Immunophenotyping reveals that the majority of the infiltrate is represented by CD19+, CD20+, and CD79a+ B cells. The cells in the reactive follicles express bcl-6, but are negative for bcl-2, whereas the small B cells in the interfollicular area express bcl-2, but are negative for bcl-6. The networks of CD21+ follicular dendritic cells are sharply demarcated and regularly structured. Polytypic expression of immunoglobulin (Ig) light chains κ and λ by plasma cells is found by immunohistochemistry or in situ hybridization. Molecular studies demonstrate lack of monoclonal rearrangement of Ig heavy-chain genes by polymerase chain reaction (PCR) or Southern blot analysis.
DIFFERENTIAL DIAGNOSES
The differential diagnosis of B-cell PSLs primarily includes other B-cell infiltrates with a follicular pattern; that is, primary cutaneous marginal zone lymphoma (PCMZL) and primary cutaneous follicle center lymphoma (PCFCL) or their nodal or other extranodal counterparts presenting with secondary cutaneous infiltrates (Table 120-4). The architecture and composition of the infiltrate in PCMZL is very similar to that of B-cell PSL as both present with nodular infiltrates in the reticular dermis and superficial subcutis.20 In comparison with B-cell PSL, the plasma cells in PCMZL are usually more prominent and found in sheets, particularly at the periphery of the infiltrates. The most important histopathologic diagnostic finding is the monotypic expression of Ig light chains in PCMZL with a ratio of at least 5:1 and ranging up to 10:1 for the expression of 1 of the 2 Ig light chains. The presence and number of eosinophils are not a useful finding for discrimination between B-cell PSL and PCMZL. Detection of a clonal B-cell population can be used as an additional diagnostic hint for PCMZL, but such deletion is only found in 50% to 70% of PCMZL cases, thereby limiting its diagnostic value for distinguishing between B-cell PSL and PCMZL. The other important differential diagnosis to be considered is PCFCL, which is characterized by the predominance of centrocyte-like differentiated tumor cells arranged in large neoplastic follicles. In addition, tingible body macrophages are only found in a small minority of PCFCLs compared to those found in B-cell PSLs with readily identifiable tingible body macrophages as part of the reactive follices.21 Furthermore, a low proliferative activity in the neoplastic follicles of PCFCL is a characteristic finding, which contrasts with the high proliferative activity in the reactive germinal
20
B-CELL PSEUDOLYMPHOMA PRIMARY CUTANEOUS MARGINAL ZONE LYMPHOMA PRIMARY CUTANEOUS FOLLICLE CENTER LYMPHOMA
Gender (male-to-female ratio) 3:1 1:2 1:1.5
Age <40 years 30-50 years >50 years
Localization Face > upper trunk > arms Upper trunk > arms > face/head Head/neck > upper trunk
Clinical features Solitary nodule Solitary or multiple nodules Solitary or multiple nodules and plaques
Histology Nodular infiltrate Nodular infiltrate Nodular infiltrate
Reactive germinal centers with tingible body macrophages Reactive germinal centers with tingible body macrophages Neoplastic germinal centers; lack of polarization
Small lymphocytes Tumor cells with lymphoplasmacytoid (or, rarely, with monocytoid) differentiation
Tumor cells with centrocyte-like differentiation
Plasma cells scattered Plasma cells in small sheets Few plasma cells
Phenotype Small lymphocytes: CD20+ B cells > CD3+ T cells Tumor cells: CD20+, bcl-2+, bcl-6− Tumor cells: CD20+, bcl-2−, bcl-6+
Germinal centers: bcl-2−, bcl-6+; high proliferative activity; sharply demarcated networks of follicular dendritic cells
Germinal centers: bcl-2−, bcl-6+; high proliferative activity; sharply demarcated networks of follicular dendritic cells
Germinal centers: bcl-2−, bcl-6+; low proliferative activity; irregular networks of follicular dendritic cells
Polytypic expression of immunoglobulin light chains Monotypic expression of immunoglobulin light chains Usually no significant expression of immunoglobulin light chains
Molecular findings Polyclonal Monoclonal (70% to 80%) Monoclonal (80% to 90%)
Molecular findings Polyclonal Monoclonal (70% to 80%) Monoclonal (80% to 90%)
centers of B-cell PSLs. The networks of CD21+ follicular dendritic cells in PCFCL are irregular and disrupted in contrast to the sharply demarcated and regularly structured networks in B-cell PSLs. A clonal B-cell population can be detected in the majority of PCFCL detected by PCR or Southern blot analysis. It should be mentioned that the vast majority of PCFCL do not express bcl-2 by the neoplastic centrocyte-like differentiated cells. Consequently, the expression of bcl-2 is not of diagnostic value for distinguishing PCFCL from B-cell PSL. In cases with expression of bcl-2 by the centrocyte-like tumor cells, secondary cutaneous infiltration by a nodal follicle center lymphoma has to be considered because nodal follicle center lymphoma exhibits expression of bcl-2 by the neoplastic cells as the result of an underlying t(14;18) translocation in the majority of the cases. Other differential diagnoses include cutaneous infiltrates of B-cell chronic lymphocytic leukemia or small cell lymphocytic lymphoma, although the cutaneous infiltrates of small cell lymphocytic lymphoma usually do not show reactive germinal centers. Clonality studies in B-cell PSLs are of limited value in distinguishing B-cell PSLs from cutaneous B-cell lymphomas as approximately 10% to 20% of PSLs harbor a clonal B-cell population.12,22 In some studies, an even higher percentage of cases with clonal B cells were detected in nodular PSL.23 In lesions with rather subtle infiltrates, pseudoclonality should always be ruled out as it represents a diagnostic pitfall.24
ETIOLOGY
A broad range of causes for B-cell PSLs has been identified. In addition to B-cell PSLs being caused by Borrelia burgdorferi sp. infection, insect bites, or injection of vaccines or antigens for hyposensitization, B-cell PSLs can be caused by acupuncture treatment, tattoos, metals in piercing rings and earrings, and drugs.
BORRELIA-ASSOCIATED B-CELL PSEUDOLYMPHOMA
BORRELIA
-ASSOCIATED
B-CELL PSEUDOLYMPHOMA
EPIDEMIOLOGY
The terms lymphocytoma cutis and lymphadenosis cutis benigna are used synonymously for cases caused by B. burgdorferi infection. Approximately 1% of clinically apparent Borrelia burgdorferi sp. infections manifest as B-cell PSLs. A female preponderance (male-to-female ratio: 2:1) is observed in some, but not all, studies. This form of B-cell PSLs has more often been reported in whites than in African Americans. Borrelia-associated B-cell PSLs affects typically children and occurs in early adulthood, but may be seen in all age groups, with most patients being younger than age 40 years.
2111
20
CLINICAL FINDINGS
A solitary red to violaceous dome-shaped nodule usually develops in Borrelia-induced B-cell PSLs, but in approximately 10% to 15% of patients, multifocal skin lesions can be observed. The face and scalp, and especially the earlobes, nipples, and scrotum, are the preferred sites for Borrelia-induced B-cell PSLs, but the trunk and extremities also may be involved (Fig. 120-1). Regional lymphadenopathy can be present.
LABORATORY TESTS
Histologically, the archetypic pattern of a dense dermal nodular infiltrate of small B cells and reactive germinal centers is found (Fig. 120-2).14 In Borrelia-associated B-cell PSL, the germinal centers tend to be larger and confluent with only a small mantle zone, and in some cases there is a lack of polarization.14,22 Because of the confluence of the large germinal centers, the lesions resemble the neoplastic follicles in PCFCL (follicular growth pattern).22 Plasma cells are almost always present and are found particularly at the periphery of the infiltrates. In rare cases of so-called large cell lymphocytoma associated with Borrelia infection, a predominance of blasts, resembling centroblasts and immunoblasts, are found simulating the findings in large B-cell lymphoma.25 Those cases are prone to be misdiagnosed as diffuse large B-cell lymphoma.
2112
A
B
In the vast majority of the cases of Borrelia-associated B-cell PSLs, molecular studies show a polyclonal rearrangement of IgH genes, but detection of monoclonal B cells has been observed and thus does not exclude the diagnosis of Borrelia-induced B-cell PSL.14
Serology shows antibodies against B. burgdorferi with variable pattern; that is, IgG and IgM or only 1 class of antibodies may be detectable and elevated. Nevertheless, cases with negative serologic findings can be seen14; consequently, negative serology does not exclude Borrelia-induced B-cell PSL. Molecular studies for the detection of B. burgdorferi DNA by PCR are a helpful adjunctive diagnostic tool with a sensitivity of approximately 70%.26
Diagnosis is based on histology, the clinical context (history of tick bite, localization at predilection site), serologic findings and/or detection of B. burgdorferi DNA in the tissue by PCR.
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
In Borrelia-associated B-cell PSLs, antibiotics (see Table 120-3) are the first-line therapy and prevent other complications from Borrelia infection, such as arthritis and carditis.
CUTANEOUS T-CELL AND MIXED PSEUDOLYMPHOMA
CUTANEOUS T-CELL AND
MIXED PSEUDOLYMPHOMA
Nodular T-cell PSL is characterized by a dense dermal T-cell–rich nodular infiltrate, which is accompanied by variable number of B cells, which can reach up to 30% of the entire infiltrate.27 Mixed forms of PSL contain an equal number of T-cells and B-cells.
EPIDEMIOLOGY
There are no detailed epidemiologic data on the prevalence of T-cell or mixed PSL. They affect patients of both genders and all age and ethnic groups.
ETIOLOGY AND PATHOGENESIS
All causes identified in B-cell PSLs also can be found as underlying stimuli in T-cell and mixed PSLs. In approximately 5% of cases, drugs are identified as the causative stimulus in T-cell PSL. Most cases, however, are without known cause and therefore referred to as idiopathic T-cell PSL or mixed PSL.
CLINICAL FEATURES
T-cell and mixed PSL usually presents with a solitary or multiple red to violaceous nodules similar to B-cell PSL (Fig. 120-3).
LABORATORY TESTS
Histologically, T-cell and mixed PSL present in most cases with a dense nodular infiltrate extending throughout the entire dermis and into the superficial parts of the subcutis (Fig. 120-4). The infiltrates
20
are predominantly composed of small lymphocytes with chromatin-dense nuclei, but a variable number of slightly enlarged (up to medium-sized) lymphocytes with chromatin-dense nuclei can be found. There is an admixture of a variable number of eosinophils, histiocytes, plasma cells (ranging from a few up to small clusters). The B cells can be arranged in small aggregates, but, rarely, germinal centers are found. Granuloma formation can be observed. There may be exocytosis of T lymphocytes into the epithelia of the hair follicles, but usually there is no significant exocytosis of lymphocytes into the overlying interfollicular epidermis (lack of epidermotropism). Immunohistochemistry shows that, in most cases, the majority of the small lymphocytes are CD4+ CD30− T cells. There may be an admixture of a few CD30+ blasts representing activated lymphocytes. Clonality studies reveal a polyclonal infiltrate in the majority of T-cell PSLs, but PSLs with clonal T cells have been reported and are referred to as clonal PSLs. Some of those cases may progress to overt lymphoma and may rather represent very early stages of lymphomagenesis than real PSLs.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis of nodular T-cell and mixed PSLs includes cutaneous CD4+ small-/medium-sized T-cell lymphoma/lymphoproliferative disorder (CD4+ SMT-LPD) according to the World Health Organization (WHO) 2008 classification and the revised WHO 2016 classification, which show overlapping histologic and immunophenotypic features.28,29 The CD4+ SMT- LPD also presents usually with a solitary lesion located mostly on the head and neck area and shows an indolent course (Fig. 120-5). Because nodular T-cell PSL and cutaneous CD4+ SMT-LPD cannot be distinguished with certainty based on clinical or histopathologic or phenotypic features, some authors consider them to represent the same process. Therefore, the encompassing term cutaneous CD4+ small/medium T-cell lymphoproliferative disorder has been introduced in the updated WHO 2016 classification to emphasize the indolent
2113
20
nature of this process. The expression of programmed cell death protein 1 (PD-1), originally thought to be a discriminative marker, is not of diagnostic value. Nodular T-cell PSLs should be differentiated from mycosis fungoides (MF) in tumor stage because MF tumor stage may rarely present with small to mediumsized T cells without a significant number of large cells. In MF, however, the small and medium-sized cells show a higher degree of nuclear atypia than in CD4+ SMT-LPD. The distinction between MF, CD4+ SMT-LPD, and T-cell PSL has to be based on the clinical presentation with patches and plaques preceding the tumors in MF in contrast to the mostly solitary nodule without preceding patch(es) and plaque(s) in T-cell PSL. The differential diagnosis further includes secondary cutaneous infiltrates of angioimmunoblastic T-cell lymphoma (AITL), in which the small CD4+ and PD-1+ T cells are accompanied by a significant number of B cells. The clinical context with B symptoms, serologic findings, the nodal involvement shown by radiologic staging examinations, a high proliferation rate in AITL infiltrates, and the association with Epstein-Barr virus in some of the cases of AITL, are useful findings for distinguishing AITL from nodular T-cell PSL. Among inflammatory skin disorders, lupus erythematosus (particularly the tumidus type) has to be considered, which also can present with dense dermal lymphocytic infiltrates. In those cases, however, vacuolization at the interface of the hair follicle epithelium and interstitial mucin deposits are present. In addition, the clinical presentation allows differentiation from nodular T-cell PSLs.
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
T-cell and mixed PSL can resolve spontaneously after withdrawal of the underlying cause, but may persist for longer periods (months or years). If no spontaneous regression is observed after biopsy, surgical excision, cryotherapy, laser treatment, topical/
2114
intralesional steroids, or interferon represent therapeutic options.30 T-cell PSL usually does not recur unless the underlying stimulus persists (eg, a drug). As in B-cell PSL, avoidance of the underlying stimulus is the effective preventive measure to avoid recurrence.
CD30+ T-CELL PSEUDOLYMPHOMA
CD30+ T-CELL
PSEUDOLYMPHOMA
CD30+ PSL represents a histologic subtype of T-cell PSL of the skin that is characterized by the presence of medium-sized to large atypical CD30+ T cells.31,32
T-cell–rich pseudolymphomatous infiltrates with admixture of CD30+ cells have been reported in the context of lymphomatoid drug eruptions, nodular scabies, and arthropod bite reactions, as well as viral infections, particularly parapoxvirus-associated disorders such as Orf disease33 and milker nodule, as well as molluscum contagiosum and herpes virus infections (for a review see reference 31). Among noninfectious disorders, CD30+ pseudolymphomatous infiltrates have been described in drug eruptions, hidradenitis, and injuries by corals.34
In CD30+ PSL, immunohistochemistry shows enlarged, that is, medium-sized to even large, CD30+ blast-like cells that are usually found as single units scattered throughout the infiltrate, which is otherwise dominated by small T cells (Fig. 120-6). In contrast to CD30+ lymphomas, CD30+ PSLs do not, in most of the cases, harbor a clonal T-cell population. Because of the presence of enlarged CD30+ cells, lymphomatoid papulosis (particularly histologic Type A) and cutaneous anaplastic large cell lymphoma (ALCL) have to be considered as differential diagnoses. In contrast to lymphomatoid papulosis and ALCL, the CD30+ cells in CD30+ PSL are usually not arranged in aggregates. Moreover, the significant number of B cells and plasma cells argues for a reactive process, that is, PSL, as they are usually absent or only present in a small number in lymphomatoid papulosis and ALCL. In addition, in contrast to lymphomatoid papulosis, expression of 5hmC in the CD30+ cells in CD30+ PSL is preserved and can serve as an additional diagnostic marker.35
Treatment of CD30+ PSL is directed against the underlying cause of the disease.
PSEUDOLYMPHOMATOUS FOLLICULITIS
PSEUDOLYMPHOMATOUS
FOLLICULITIS
This PSL variant was first described in 1988 by Kibbi and coworkers.36 It presents with a solitary nodule located on the face that measures less than 1.5 cm and is similar or identical identical to the clinical manifestation found in cutaneous CD4+ SMT-LPD.37 A miliarial
A
B
form of B-cell pseudolymphomatous folliculitis has been described.19 Histologically, half of the cases carry the features of nodular B-cell PSLs and the remaining cases carry the features of nodular T-cell PSLs. The lymphocytic infiltrates are located throughout the entire dermis and may extend into the subcutis and are arranged around the hair follicles (Fig. 120-7). A Grenz zone may be present. There is exocytosis of lymphocytes into the hair follicles often showing broadened epithelia.38 In half of the cases, atypical lymphocytes are found. Half of the cases harbor aggregates of histiocytes. An admixture of numerous dendritic cells
20
with expression of CD1a and S-100 was identified in all cases.37 The course and treatment is similar to other forms of nodular PSL and CD4+ SMT-LPD.
OTHER FORMS OF NODULAR PSEUDOLYMPHOMA
OTHER FORMS OF NODULAR
PSEUDOLYMPHOMA
On clinical grounds alone T-cell, B-cell, or mixed PSL cannot be discerned. In addition, some agents may induce T-cell and B-cell PSL so that an unequivocal correlation between the causative factor and the composition of the PSL is not possible. Nevertheless, the clinical context may provide essential information regarding the etiology of PSL, especially for those cases resulting from tattoos or metals (piercing, earrings), or PSL caused by exposure to leaches and acupuncture.
PERSISTENT NODULAR ARTHROPOD BITE REACTIONS AND NODULAR SCABIES
In both conditions longstanding pruritic papules and nodules are found with a predilection for the elbows, trunk, genitalia, and axillary, as well as inguinal folds. The lesions tend to be red-brown and may be excoriated. Mites or scybala are rarely found in the epidermis overlying the dense dermal lymphocytic infiltrates. A delayed-type hypersensitivity reaction is considered to be the underlying trigger factor for the formation of the T-cell–rich infiltrates. Histologically the epidermis often shows features of pruriginous reaction with acanthosis and hyperparakeratosis. There is a wedge-shaped dermal infiltrate predominantly composed of small lymphocytes with admixture of eosinophils and occasional plasma cells (Fig. 120-8). Formation of flame figures can be seen particularly in cases with numerous eosinophils. In the center of the infiltrates, hypereosinophilic collagen bundles are found. The vessels show plump endothelia. In the majority of nodular arthropod bite
2115
20
reactions, the infiltrate is dominated by T cells. Rarely, B-cell–rich forms with germinal centers may be seen. There can be an admixture of medium to even large atypical-appearing CD30+ lymphocytes (see section “CD30+ pseudolymphomas”).31,32,39 In those cases, distinguishing CD30+ pseudolymphomas from lymphomatoid papulosis can be challenging. There is no clonal rearrangement of T cells in nodular arthropod bite reactions and nodular scabies. Persistent arthropod bite reactions have to be differentiated from exaggerated bite reactions, which can occur in patients with B-cell chronic lymphocytic leukemia and other leukemias.40 In those patients, hematologic examination may be considered. Moreover, lymphomatoid papulosis (Type A), Hodgkin lymphoma, and nodular secondary syphilis are histologically differential diagnoses. The lesions in nodular arthropod bite reactions and nodular scabies may be longstanding and persist for several months. Antiscabietic treatment is ineffective. Excision, intralesional corticosteroids, or topical immunomodulators may be considered if spontaneous regression does not occur or topical steroids are ineffective.41
PSEUDOLYMPHOMA CAUSED BY LEECHES (HIRUDO MEDICINALIS) THERAPY
The distribution of the lesions corresponds to the exposure to leeches (H. medicinalis) which are commonly found on the lower back or over areas that have been treated for hematoma after trauma or surgery (Fig. 120-9).42 A diffuse form has been described.43 Both T-cell and B-cell predominant forms were observed. Hypothetically bacteria residing in the leeches are transferred to the host during the blood meal and induce the pseudolymphomatous reaction.
NODULAR PSEUDOLYMPHOMA CAUSED BY DRUGS
PSL caused by drugs show a variety of clinical, histologic, and phenotypic manifestations. Some of the
2116
drugs, like anticonvulsants, may induce nodular PSL (Fig. 120-10), as well as other histologic manifestations (see below).27,44,45 Clinically, drug-related nodular PSL may present with a solitary or multiple nodules.44
Histologically, nodular drug-related PSL presents with the typical histologic features of nodular T-cell or B-cell PSL.46,47 The lymphocytes in nodular T-cell PSLs may show subtle nuclear atypia. Proliferation rate is low, approximately 10%. In B-cell PSLs, the typical immunophenotypic features as outlined above are found. In T-cell PSLs, immunohistochemistry reveals a predominance of CD4 lymphocytes and admixture of a variable number of CD30+ lymphocytes.48 Loss of pan–T-cell markers is not observed. In most cases, there is no monoclonal rearrangement of T-cell receptor γ genes or IgH genes. Diagnosis of lymphomatoid drug eruption is challenging, particularly as the latency between onset of medication and development of drug eruption can be very long (several months up to years). Differential diagnosis depends on the phenotype. In B-cell nodular PSL, differential diagnosis includes cutaneous marginal zone lymphoma and follicle center lymphoma. In CD4+ T-cell rich forms of drug-related nodular PSL, the differential diagnoses include cutaneous CD4+ SMT-LPD, MF, and Sézary syndrome. Withdrawal of the drug is the first step in the management of drug-related PSL. Surgical excision or intralesional injection of corticosteroids may be considered, if spontaneous regression is delayed. One has to be aware that drug-related nodular PSL may persist for several months even after withdrawal of the causative drug. Reexposure to the drug may elicit a relapse of the PSL.
PSEUDOLYMPHOMA AT DRUG AND VACCINE INJECTION SITES
Solitary or multiple PSL may also develop after injection of vaccines to prevent infectious diseases
A
B
or after injection of allergens for hyposensitization. They are typically found in the subcutaneous tissue at the injection site.49 The histologic features show B-cell–rich infiltrates with reactive germinal centers. In the center of the infiltrates, an accumulation of histiocytes can be seen (Fig. 120-11).50,51 The histiocytes show a cytoplasm with a granular gray-blue aspect, which seems to represent aluminium hydroxide– bound vaccines.52
PSEUDOLYMPHOMA IN ASSOCIATION WITH TATTOOS OR METALS
Pseudolymphomatous infiltrates arising in tattoos and caused by metals in earrings or piercings share similar features.53,54 In tattoos, the lymphocytic infiltrates mostly arise and remain limited to areas with red tattoo dye (Fig. 120-12). Cinnabar, a mercuric sulfide, is the most commonly used dye for red tattoos. In addition, PSLs also have been observed in blue or green tattoos, for which cobalt and chrome salts are employed. The PSL to the tattoo dye can be regarded
20
A
B
as a delayed-type hypersensitivity reaction. Clinically, mostly papules, nodules, or plaque-like infiltrates develop within 1 compartment of the tattoo. They are usually asymptomatic. The latency between application of the tattoo dye and occurrence of the PSL is highly variable, ranging from months to years.55 Histologically, a lichenoid component with a band-like superficial infiltrate, interface changes, and exocytosis of lymphocytes, as well as a deeper lymphocytic infiltrate in the mid and deep dermis with admixture of eosinophils, is present leading to a pseudolymphomatous appearance.56 The lymphocytes are small and do not display significant nuclear atypia. Patch test to mercury or other substances in the tattoo dyes may reveal delayed type hypersensitivity, but the test also may be negative.55 Therapy is challenging and includes intralesional injections of corticosteroids, laser therapy, or excision if small papulonodular lesions are present.
INFECTIONS WITH LYMPHOCYTE- RICH INFILTRATES SIMULATING LYMPHOMA
Various infections, particularly those caused by viruses and parasites, may show dense lymphocyte-rich
2117
20
infiltrates, thereby simulating a lymphoma. Among infections with parasites, cutaneous leishmaniasis histologically simulates lymphoma by its nodular infiltrate composed of numerous lymphocytes, including plasma cells and histiocytes. The lack of nuclear atypia and detection of the parasite either by special stains, immunohistochemistry or by molecular techniques enables cutaneous leishmaniasis to be differentiated from cutaneous lymphoma. Not only histologic can findings in cutaneous leishmaniasis simulate lymphoma, but the clinical aspects occasionally can resemble B-cell PSL.57,58
In infections with herpes simplex virus and varicella zoster virus, occasionally lymphocyte-rich infiltrates without the pathognomonic epithelial changes can be observed and have been referred to as herpes incognito. Both lymphocytes with slightly enlarged and atypical-appearing chromatin-dense nuclei and enlarged CD30+ lymphocytes may be found in herpes incognito, making those infiltrates subject to being mistaken for lymphoma infiltrates. Detection of viral antigens by immunohistochemistry and/or detection of viral DNA by PCR enable identification of those infiltrates as herpes virus–related T-cell reactions. Parapoxvirus infection may induce cytomorphologic changes and expression of CD30 by the infiltrating T cells, which makes distinguishing them from pleomorphic lymphocytes in the context of anaplastic largecell lymphoma challenging (see Fig. 120-6).31,32 The presence of epithelial changes with inclusion bodies typical for parapoxvirus infection, the absence of loss of T-cell markers, and the lack of monoclonal rearrangement of T-cell receptor–γ genes help to distinguish those infiltrates from cutaneous T-cell lymphoma. However, the diagnosis is based on the detection of the virus by immunohistochemistry, electron microscopy, or PCR.
PSEUDOLYMPHOMAS- HISTOLOGIC MYCOSIS FUNGOIDES SIMULATORS
The term pseudo-MF describes a group of disorders of different etiologies that histologically mimic MF. Because the histologic evaluation suggests MF or another form of epidermotropic cutaneous T-cell lymphoma, the clinicopathologic correlation is crucial to avoid misinterpreting the histologic findings as a lymphoma and for assigning the findings to a distinct reactive skin disease, such as PSL. Histologically, the diseases described in this section have in common a dermal, either band-like or perivascular, infiltrate of mostly small lymphocytes, which show exocytosis into the epidermis and may exhibit subtle nuclear atypia, thereby simulating epidermotropic cutaneous T-cell lymphoma (CTCL). Phenotyping reveals either a predominance of CD4+ or CD8+ cells. In addition, a variable expression of CD30 can be seen in some cases of pseudo-MF. As in other forms of PSL, the lymphocytes are polyclonal in the majority of
2118
cases. In some diseases, such as pityriasis lichenoides et varioliformis acuta, a significant percentage of clonal T cells is found, but does not indicate malignancy or a risk of progression to lymphoma. Differential diagnoses include primarily MF or Sézary syndrome. In cases with a predominantly CD8+ infiltrate, differential diagnosie include CD8+ MF, Sézary syndrome, cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma, and lymphomatoid papulosis (Types D and E). Profound nuclear atypia, predominance of medium-sized to large cells, loss of pan T-cell markers, and monoclonal rearrangement of T-cell receptor genes, are findings in favor of CTCL. As in other forms of PSL, the treatment focuses on avoiding exposure to the causative agents, such as allergens in lymphomatoid contact dermatitis and drugs in lymphomatoid drug eruption. In addition, topical corticosteroids or immunomodulators and ultraviolet (UV) light therapy are used. Systemic steroids and immunosuppressive drugs, such as cyclosporine, may be indicated in severe forms of PSL.
LYMPHOMATOID CONTACT DERMATITIS
LYMPHOMATOID CONTACT
DERMATITIS
Lymphomatoid contact dermatitis (LCD) is a chronic contact dermatitis that histologically simulates MF.59
Various antigens, including nickel sulfate, gold, zinc, paraphenylenediamine, textile dye, and several other antigens, have been identified as culprits.60 LCD occurs mostly in adults and affects both genders. Clinically, LCD manifests with eczematous erythematous and scaly papules, patches, plaques, and, in rare cases, erythroderma (Fig. 120-13). The lesions are pruritic. Histopathologically there is a superficial band-like infiltrate with variable exocytosis of lymphocytes into the epidermis, which may show spongiosis or spongiotic vesicles. Intraepidermal accumulations of Langerhans cells (so-called pseudo Pautrier collections) can be found. Some of the lymphocytes can show slightly convoluted nuclei, but nuclear atypia is not prominent.
There is an admixture of eosinophils. The ratio of CD4+ to CD8+ lymphocytes is inconspicuous, but there can be an admixture of slightly enlarged CD30+ cells representing activated lymphocytes. Differentiation of LCD from MF is based on the presence of variable degree of spongiosis, the lack of significant atypia of lymphocytes, an inconspicuous CD4- to-CD8 ratio, and the absence of loss of T-cell markers in LCD. Monoclonal T cells have been reported in LCD and do not indicate lymphoma. Sensitization against the allergen(s) is documented by patch test and is an essential diagnostic criterion for proving a diagnosis of LCD.61
The course of LCD is chronic, particularly if exposure to the allergen cannot be avoided. The treatment follows the general recommendation for the treatment of other forms of contact dermatitis with avoidance of exposure to the allergen and suppression of the immunologic reaction against the allergen mostly by topical corticosteroids or topical immunomodulators. In addition, UV light–based strategies or systemic immunosuppression may be effective.
LYMPHOMATOID DRUG REACTION
LYMPHOMATOID
DRUG REACTION
Apart from its nodular form, drug-related PSL commonly presents with maculopapular or papular eruptions. A large number of drug classes that induce various forms of lymphomatoid drug eruptions have been described.8,62
In this form of lymphomatoid drug reaction, a band-like infiltrate in the upper dermis, with variable degrees of exocytosis of lymphocytes, is found.63
Vacuolar alteration at the dermoepidermal junction and apoptotic keratinocytes may be present. The lymphocytes may show an irregular nuclear contour (ie, nuclear atypia) (Fig. 120-14 A, B). Eosinophils are commonly found, but also may be absent. Immunohistochemistry reveals either a predominance of CD4+ or CD8+ lymphocytes and an admixture of a variable number of CD30+ lymphocytes (Fig. 120-14C).48 Loss of pan T-cell markers is not observed. Differential diagnosis includes epidermotropic CTCL, particularly MF and Sézary syndrome. Lining up of lymphocytes along the junctional zone, nuclear atypia of the lymphocytes, loss of pan T-cell markers, and monoclonal rearrangement of T-cell receptor-γ genes favor MF or Sézary syndrome. Moreover, expression of PD-1 and TOX (thymocyte selection-associated high-mobility group box factor) by the majority of the lymphocytes was found in Sézary syndrome, but not in inflammatory erythrodermas.64 Cases with CD8+ infiltrates showing exocytosis of lymphocytes have to be distinguished from CD8+ MF (patches and plaques), CD8+ lymphomatoid papulosis (especially Type D or Type E), and pityriasis lichenoides et varioliformis acuta (PLEVA). Drug-related CD30+ PSL simulates lymphomatoid papulosis (Type A or Type B).65 Occasionally,
20
A
B
the lymphocytes in lymphomatoid drug eruptions exhibit remarkable nuclear atypia so that distinction from lymphoma is very challenging or even impossible by histology alone. Clinicopathologic correlation and history are mandatory to reach the final diagnosis in drug-related PSL. As in the nodular form of drug-related PSL, withdrawal of the drug is the essential step in the management. Lymphomatoid drug eruption can persist for several months and reexposure to the drug may elicit a relapse of the PSL. Systemic, intralesional, or topical steroids are used for treatment.
ACTINIC RETICULOID
ACTINIC RETICULOID
Actinic reticuloid represents a chronic multifactorial dermatitis with severe photosensitivity to a broad spectrum of wavelengths, which histologically mimics epidermotropic CTCL.5
CLINICAL FINDINGS
It affects mostly middle-aged and older men.66 It presents with persistent erythematous lichenoid papules and plaques on light-exposed skin areas, particularly on the face and neck. In some patients infiltration on the face leads to a facies leonina-like aspect (Fig. 120-15). Progression into erythroderma can be observed. Lichenification and erosions usually occur
2119
20
A
B
over time. The skin lesions are accompanied by intense pruritus.
LABORATORY TESTS
Histologically, there is psoriasiform hyperplasia of the epidermis, slight spongiosis, and compact orthokeratosis with focal parakeratosis. In the upper dermis there is an infiltrate arranged in a perivascular, but also interstitial, distribution and composed of small lymphocytes, eosinophils, and plasma cells. Coarse
2120
bundles of collagen arranged in vertical streaks are found in the papillary dermis. Multinucleated fibroblasts may be present. The lymphocytes may show slightly atypical nuclei and exocytosis into the overlying epidermis. Immunohistochemistry reveals a predominance of CD8+ T cells.67
In the peripheral blood, an increased number of CD8+ T cells (reversed CD4-to-CD8 ratio) is characteristic for actinic reticuloid, particularly in erythrodermic patients.66 The atypical circulating lymphocytes show indented nuclei. Phototesting, including positive photo patch tests, reveals sensitization to 1 or multiple allergens.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The diagnosis of actinic reticuloid is based on the presence of persistent infiltrated plaques and papules on light-exposed skin areas, photo sensitivity to a broad spectrum of wavelengths, and histologic findings with a predominance of CD8+ slightly atypical lymphocytes and variable degree of exocytosis, as well as the presence of circulating CD8+ lymphocytes in the peripheral blood.67
Based on clinical and/or histologic grounds, MF and/or Sézary syndrome have to be considered. CD8+ MF shows a more pronounced epidermotropism and nuclear atypia of the lymphocytes. Moreover, CD8+ MF is usually located on UV-protected body regions in contrast to actinic reticuloid displaying a distribution in light-exposed areas. Particularly in patients with actinic reticuloid presenting with erythroderma, Sézary syndrome represents an important differential diagnosis. In contrast to actinic reticuloid with a predominance of CD8+ lymphocytes, Sézary syndrome is characterized by clonal CD4+ atypical lymphocytes in the skin and the peripheral blood, lymphadenopathy, alopecia, and palmoplantar hyperkeratosis.
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
Actinic reticuloid shows a longstanding highly chronic course. In addition to UV light protection with sunscreens, topical steroids and topical immunomodulators can be used, but show variable efficacy. Among systemic drugs, cyclosporine may be effective. Photo hardening with UV-B irradiation to induce photo tolerance was reported to produce a good response in one series.67
CD8+ T-CELL PSEUDOLYMPHOMA IN IMMUNODEFICIENCY
CD8+ T-CELL
PSEUDOLYMPHOMA IN
IMMUNODEFICIENCY
In patients with immunodeficiency, in particular HIV infection, infiltrates of CD8+ lymphocytes mimicking MF may develop. Fewer than 50 cases of CD8+
infiltrates in HIV-infected patients have been reported as of this writing. The etiology and pathogenesis of these infiltrates is largely unknown, but may be related to severe immunodeficiency as the cutaneous CD8+ infiltrates occur in advanced HIV infection with low numbers of CD4+ cells. A variety of clinical presentations have been described, including disseminated, often pruritic papules, or a diffuse, mildly pruritic papular eruption.68,69 In addition, palmoplantar hyperkeratosis and erythroderma were observed. The dermal infiltrates of CD8+ lymphocytes are accompanied by exocytosis of CD8+ lymphocytes into the epidermis. The lymphocytes may show subtle nuclear atypia. Molecular studies revealed a polyclonal nature. The course of the cutaneous CD8+ infiltrates per se is indolent in most patients, but is considered as a sign for bad overall prognosis, which is mostly determined by the severe lymphopenia of CD4+ T cells. In most patients remission was achieved when receiving highly active antiretroviral therapy, and in moderately immunosuppressed patients methotrexate was shown to be effective.70,71
This condition seems not to be exclusively limited to patients with HIV infection, as similar features were recently described in a renal transplant recipient.72
BORRELIA-ASSOCIATED T-CELL PSEUDOLYMPHOMA
BORRELIA
-ASSOCIATED
T-CELL PSEUDOLYMPHOMA
With the exception of erythema migrans, Borrelia infection of the skin most commonly manifests with B-cell– predominant infiltrates, including plasma cells, such as lymphocytoma cutis. Recently B. burgdorferi infections with T-cell–rich infiltrates have been reported.73,74
The dermal T-cell infiltrate was either band-like or diffuse and displayed focal epidermotropism with lining-up of lymphocytes along the junctional zone. The lymphocytes show slight nuclear atypia. In addition, a minor interstitial histiocytic component of the infiltrate can be observed. Both the histologic features that mimic MF and the clinical aspect with patches resembling MF represent a diagnostic pitfall in some of the affected individuals. Other clinical presentations included the typical findings of erythema migrans or acrodermatitis chronica atrophicans. Treatment with antibiotics results in remission.
PAPULOERYTHRODERMA OFUJI
PAPULOERYTHRODERMA
OFUJI
Papuloerythroderma Ofuji is a rare pruritic erythrodermic dermatosis that clinically can simulate CTCL.75 Association with drugs, Hodgkin lymphoma, visceral malignancies, and immunodeficiency syndromes have been reported. In some patients,76,77 papuloerythroderma Ofuji was described as a manifestation of MF, in others as a disease accompanying MF.78
20
Papuloerythroderma Ofuji was originally described in Japanese patients, but similar cases have been reported from other geographic regions. The median age is 70 years. It occurs more frequently in males than in females. Clinically, papuloerythroderma Ofuji manifests with disseminated solid papules. The coalescing brownish papules are flat-topped and observed mainly on the flexor surfaces of the extremities. The axillae, inguinal regions, antecubital and popliteal fossae, and big furrows on the abdomen are spared (“deckchair” sign; Fig. 120-16).79
Histology shows eczematous features with acanthosis, spongiosis and hyperparakeratosis, and a dermal lymphocytic infiltrate with admixture of plasma cells, eosinophils, and neutrophils is found. Exocytosis of neutrophils and eosinophils may be observed.80 Immunohistochemistry shows numerous dendritic cells and mature T cells in the dermis. The most common abnormal laboratory findings were eosinophilia and an elevated serum IgE level. There should be a search for immunodeficiency, especially HIV infection. The clinical differential diagnoses include Sézary syndrome. Papuloerythroderma Ofuji can be distinguished from Sézary syndrome because, unlike Sézary syndrome, papuloerythroderma Ofuji lacks nuclear atypia of the lymphocytes and circulating atypical lymphocytes. The prognosis of papuloerythroderma Ofuji is good, if there is no underlying malignancy. UV light treatment including psoralen and ultraviolet A (PUVA), bath PUVA, UV-B in combination with topical steroids, etretinate, Re-PUVA, oral steroids, and cyclosporine have been described as effective therapeutic approaches.11-14
INFLAMMATORY DISORDERS AS CTCL AND CLONAL DERMATITIS
INFLAMMATORY
DISORDERS AS CTCL AND
CLONAL DERMATITIS
Various disorders, which are discussed in other chapters, are characterized by a T-cell–rich infiltrate and exocytosis of lymphocytes into the epidermis, making
2121
20
the disorders prone to be misinterpreted as epidermotropic CTCL. These disorders include lichen planus, lichen sclerosus et atrophicans, pigmented purpuric dermatitis, and pityriasis lichenoides.65,81-83 Furthermore, inflammatory diseases with lymphocyte-rich dermal and/or subcutaneous infiltrates, such as lupus erythematosus (particularly the tumid type) and lupus panniculitis, have to be differentiated from other forms of CTCL including subcutaneous panniculitis-like T-cell lymphoma.
DETECTION OF T-CELL CLONES IN INFLAMMATORY SKIN DISEASES
DETECTION OF T-CELL
CLONES IN INFLAMMATORY
SKIN DISEASES
Highly sensitive methods to assess clonality, such as PCR combined with temperature gradient gel electrophoresis or denaturing gradient gel electrophoresis and automated high-resolution fragment analysis, allow detection of clonal T cells, which may comprise as little as 1% of all infiltrating cells. Clonal T-cell populations have been found in some cases in the above mentioned inflammatory skin conditions, for example, in lichen planus and lichen sclerosus et atrophicans, in which clonal T cells were found in 6% (lichen planus) and 13% (lichen sclerosus et atrophicans) of the cases. Remarkably, a monoclonal rearrangement of TCR genes is commonly found in pityriasis lichenoides harboring clonal T cells in up to 60% of cases.82,84 The significance of these T-cell clones is unclear. As a consequence for the diagnostic workup of lymphocyte-rich infiltrates, detection of a clonal T-cell population cannot be used as a sufficient finding to diagnose CTCL.13
The presence of clonal T cells within an infiltrate may merely represent a predominance of 1 or more T-cell clones in the context of an immunologic reaction that may be induced and maintained by certain antigens. In contrast to cutaneous lymphomas with persistence of a single T-cell clone during disease evolution, the T-cell clones in inflammatory skin disorders are transient and change over time. As a consequence the detection of clonal T-cells in inflammatory disorders underlines the necessity to correlate the results of clonality assays with the clinical, histologic, and immunophenotypic findings to achieve the final diagnosis.85
CLONAL DERMATITIS
CLONAL DERMATITIS
Apart from the above-mentioned well-characterized and distinct inflammatory skin diseases that may harbor clonal T cells in some cases, clonality studies employing PCR techniques led to the identification of cases with chronic, nonspecific dermatitis harboring T-cell clones. Those cases have been referred to as “clonal dermatitis.”86 Approximately 25% of clonal dermatitis cases progress to overt CTCL within 5 years.86 These observations indicate that at least some
2122
cases of “clonal dermatitis” may represent CTCL precursor lesions. Clonal dermatitis would thus be one of the earliest manifestations of CTCL, harboring a dominant T-cell clone but lacking histologic features diagnostic for CTCL.
LYMPHOCYTIC INFILTRATION OF THE SKIN AND PALPABLE ARCIFORME MIGRATORY ERYTHEMA
LYMPHOCYTIC
INFILTRATION OF THE SKIN
AND PALPABLE ARCIFORME
MIGRATORY ERYTHEMA
Lymphocytic infiltration of the skin Jessner-Kanof (LIS) and palpable arciform migratory erythema of Clark (PAME) have been regarded by some authors as T-cell PSLs, and by others as lupus-like eruptions. The clinical presentation in PAME led to its designation with infiltrated annular erythema developing into large migrating lesions.87 The trunk is the predilection site. Histology shows dense perivascular and periadnexal predominantly lymphocytic infiltrate. There is no admixture of plasma cells and interstitial mucin deposits such as in lupus erythematosus are absent. Immunohistochemistry reveals an infiltrate dominated by T cells with admixture of B cells and histiocytes. The lymphocytes are polyclonal. The histology is very similar to the findings in LIS.88 Phenotypically, the infiltrate in LIS is mostly composed of CD8+ lymphocytes.89 We consider PAME and LIS to represent the same process because of the similarities in clinical presentation, histology, and course. Both processes may respond to topical steroids, oral antibiotics, and UV-A1.88 There is a tendency for relapses.
OTHER DISORDERS WITH LYMPHOCYTE-RICH INFILTRATES
ACRAL PSEUDOLYMPHOMATOUS ANGIOKERATOMA
ACRAL
PSEUDOLYMPHOMATOUS
ANGIOKERATOMA
Acral pseudolymphomatous angiokeratoma (APA) was originally described as occurring in children (hence the original name acral pseudolymphomatous angiokeratoma of childhood [APACHE]), but it has been shown that it also affects adults.90-92 Some authors consider the lesions to represent persistent arthropod bite reactions, whereas others categorize APA as a benign vascular process with a prominent lymphocytic infiltrate, that is, a form of cutaneous PSL.90,92 Consequently, the term papular pseudolymphoma has been proposed.92
Clinically, APA manifests as a unilateral eruption of clustered red to violaceous angiomatous papules (diameter: 1 to 5 mm) on acral sites, that is, hands and
feet (Fig. 120-17).90 Coalescence of the lesions can be seen. Longitudinal splitting of the nails, onycholysis, and nail deformities may occur. The histology of APA shows a dense infiltrate of small lymphocytes, eosinophils, plasma cells, and histiocytes. Within the infiltrate there are thick-walled vessels lined by plump endothelia. Diagnosis is based on clinicopathologic correlation. The accompanying infiltrate is composed of small, well-differentiated lymphocytes admixed with a few plasma cells and histiocytes, including histiocytic giant cells. In some instances, a few eosinophils may be present. Immunohistochemistry demonstrates that the lymphocytes are polyclonal T cells and B cells, with B cells occasionally forming small aggregates. The histologic differential diagnoses of APACHE include lymphomatoid drug eruptions and arthropod bite reaction. Similar histologic findings, but clinically manifesting as a solitary, polypoid, erythematous papule, was described under the term T-cell–rich angiomatoid polypoid PSL of the skin.93 Its nosologic relationship to APACHE remains to be determined. APACHE is a benign process. No reoccurrences have been reported following treatment. When left untreated, the lesions can regress, show a waning-andwaxing course, or remain unchanged for months or years. Destruction of the lesions by curettage, intralesional corticosteroids, or high-potency topical corticosteroids
20
under occlusion has been used for treatment and resulted in complete remission.91
LYMPHOPLASMACYTIC PLAQUE
LYMPHOPLASMACYTIC
PLAQUE
Lymphoplasmacytic plaque (LPP) is a recently described rare skin disease that is considered to be a form of PSL of unknown etiology. Originally it was reported in children with the pretibial region as predilection site, and referred to as isolated primary cutaneous plasmacytosis in children and pretibial LPP.94,95 A recent study indicates that LPP can also affect adults and involve the trunk and arms. Consequently, we prefer the term lymphoplasmacytic plaque.96 There is a female preponderance. Clinically, LPP shows a distinct presentation characterized by a longstanding plaque or circumscribed, often linear arranged, reddish and brownish papules and plaques (Fig. 120-18).96,97 Histology reveals a dense dermal lymphohistiocytic infiltrate with numerous polyclonal plasma cells accounting for up to 25% of the entire infiltrate. The infiltrate is superficial, bandlike, or deep nodular and interstitial, often accentuated around adnexal structures or blood vessels (Fig. 120-19). Interstitial histiocytic granulomas around sclerotic collagen bundles (so-called pseudorosettes) with histiocytic giant cells and an increased number of vessels can be seen.96
LPP have to be differentiated from other conditions containing plasma cells and histiocytes, such
2123
20
as primary cutaneous plasmocytosis, lymphocytoma cutis, cutaneous marginal zone lymphoma, primary and secondary cutaneous plasmocytoma, and infections (eg, fungal, mycobacterial, treponemal). An infectious process should always be ruled out by serology, special stains, and molecular pathologic techniques. The diagnosis of LPP is based on clinicopathologic correlation. LPP and APA show overlapping clinical and histologic features, such as female predominance and a predilection for the extremities, especially the legs, as well as a lymphohistiocytic infiltrate and polyclonal plasma cells. It has been postulated that both entities (APACHE and LPP) belong to the same spectrum of diseases and represent a plasma cell–rich PSL with a prominent vascular component. The course of LPP is longstanding up to several years. Surgical excision is the first-line treatment, especially as other therapies are not effective.96
CUTANEOUS PLASMOCYTOSIS
CUTANEOUS
PLASMOCYTOSIS
Cutaneous plasmocytosis is a rare disease that has been reported in Asian countries, especially Japan. It mostly affects adults.98,99 It is characterized by multiple, brownish, small plaques and nodules occurring all over the body. Histology shows dermal infiltrates composed predominantly of mature polyclonal plasma cells.98,99 In some patients, signs of a systemic involvement (eg, lymphadenopathy, hepatosplenomegaly, hypergammaglobulinemia, increased levels of interleukin-6 in the serum, and elevated erythrocyte sedimentation rate) can be found. Treatment includes PUVA, steroids, and chemotherapeutic approaches.
CUTANEOUS INFLAMMATORY PSEUDOTUMOR
CUTANEOUS
INFLAMMATORY
PSEUDOTUMOR
The term inflammatory pseudotumor has been used to describe 2 conditions that today are considered to be distinct entities, namely, plasma cell granuloma and inflammatory myofibroblastic tumor.100 Inflammatory pseudotumor is a benign process of unknown etiology. Clinically, it manifests as longstanding dermal or subcutaneous nodules of firm consistency. Histologically, a circumscribed nodular infiltrate of small lymphocytes, numerous plasma cells arranged in sheets, and histiocytes is found in plasma cell granuloma. Reactive germinal centers may be found. In inflammatory myofibroblastic tumor, a prominent spindle-cell component of myofibroblasts with expression of ALK (anaplastic lymphoma kinase) and a lymphocytic infiltrate with plasma cells are the characteristic findings. Surgical excision results in complete remission.
2124
ANGIOLYMPHOID HYPERPLASIA WITH EOSINOPHILIA
ANGIOLYMPHOID
HYPERPLASIA WITH
EOSINOPHILIA
Angiolymphoid hyperplasia with eosinophilia (ALHE) represents a proliferation of blood vessels with prominent endothelia accompanied by a dense infiltrate of T-cells and B-cells in conjunction with eosinophils. It was first described in 1969 by Wells and Whimster.101 Nowadays ALHE is commonly regarded to be an angioproliferative process resulting from the presence of prominent, bizarrely shaped blood vessels and epithelioid endothelia leading to its alternative synonymous designation as epithelioid hemangioma.102,103 Most authors consider ALHE as a hyperplastic process in response to tissue damage and formation of vascular shunts. The relationship of ALHE to Kimura disease is a matter of debate, but most experts consider them as distinct clinicopathologic entities. The lesions in Kimura disease tend to be larger and deeper and are accompanied by peripheral blood eosinophilia as well as enlarged lymph nodes.
EPIDEMIOLOGY AND PATHOGENESIS
ALHE and Kimura disease affect both genders without sex predominance.103 Most patients are in the third or fourth decade of life. Arteriovenous shunts may underlie the formation of blood vessels seen in ALHE and Kimura disease.
CLINICAL FEATURES
ALHE presents as angiomatous pink to red-brown papules or nodule(s) most commonly found on the head and neck, especially on the face and ears, but also occurring at the extremities and genital area (Fig. 120-20). The lesions may be asymptomatic or be associated with pruritus, pain, or bleeding.
HISTOPATHOLOGY, IMMUNOPHENOTYPING, AND MOLECULAR TESTS
The dermal and/or subcutaneous nodules are composed of a proliferation of postcapillary vessels and a dense lymphocytic infiltrate (Fig. 120-21). The vessels are lined by prominent endothelial cells with a round or oval nucleus, an abundant eosinophilic cytoplasm, and an epithelioid or hobnail appearance. The infiltrate contains small lymphocytes, reactive germinal centers, and eosinophils.104 Immunohistochemistry shows expression of CD31 and ERG by the endothelia, but no reactivity for podoplanin/D2-40. Immunohistochemical studies demonstrate that the majority of lymphocytes are of T-cell lineage. Admixed B cells may form lymphoid follicles. In Kimura disease the lymphoid component with germinal centers predominates over the proliferation of vessels. Clonal T cells have been detected in both ALHE and in Kimura disease.105,106 In ALHE, the majority of those cases showed a protracted and therapy-resistant course with recurrences.
20
DIFFERENTIAL DIAGNOSIS
Differential diagnosis includes pyogenic granuloma, which shows lobularly proliferation of vessels accompanied by a mixed-cellular infiltrate. Among lymphomas, specific infiltrates of adult T-cell lymphoma/ leukemia (AITL) should be considered as they also show an increased number of vessels as well as an infiltrate composed of T cells and B cells with slight nuclear atypia. The T cells in AITL, however, are atypical, proliferatively active, and express markers of follicular helper T cells such as PD-1, ICOS, bcl-6, CD10, and CXCL-13.
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
There is no spontaneous regression of ALHE lesions. Recurrences are common, particularly after surgical resection.103,107 Surgery, cryotherapy, and laser ablation, as well as methotrexate and interferon-α, have been used to treat ALHE.108 If an arteriovenous shunt can be identified as an underlying triggering factor, the shunt should be removed by, for example, embolization.
CASTLEMAN DISEASE
CASTLEMAN DISEASE
Castleman disease is a benign lymphoproliferative disorder with 2 variants: the hyaline vascular type and the plasma cell variant. The plasma cell variant can be associated with POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, m protein, and skin changes).109 Castleman disease most commonly is located in the lymph nodes (mediastinal or generalized) and rarely affects the skin.110 Histologically, the hyaline vascular type is more common with small lymphocytes that surround the germinal centers in a concentrically whorled pattern. This type is more common in younger patients. The plasma cell type shows large follicles and an interfollicular zone rich in blood vessels and plasma cells. The prognosis of Castleman disease with localized extranodal lesions is favorable. Therapy depends on the extent of the disease and includes surgical excision, radiation, and chemotherapy.
BENIGN INTRAVASCULAR PROLIFERATION OF LYMPHOCYTES
Recently, benign intravascular proliferation of lymphoid blasts with or without expression of CD30 has been reported. This condition arises in areas with inflammatory skin diseases or trauma of the skin.111-113
Pathogenetically, obstruction of lymphatics by lichen sclerosus with disrupted immune cell trafficking may result in the accumulation of activated CD30+ lymphocytes. The lymphocytes are large and have
2125
20
A
B
a blast-like morphology (Fig. 120-22). They express T-cell markers (CD3, CD4) and, in some cases, CD30. There is no association with Epstein-Barr virus infection. Clonality studies reveal the polyclonal nature of the process. Intravascular lymphoma is the most important differential diagnosis as it is an aggressive lymphoma with various phenotypic forms (B cell, T cell, or natural killer/T cell). In addition, benign intravascular proliferation of lymphoid cells needs to be distinguished from intralymphatic histiocytosis representing a reactive proliferation of histiocytes in the lumina of lymphatics in patients with rheumatoid arthritis or orthopedic metal implants.
SUMMARY
Cutaneous PSL refers to a group of lymphocyte-rich infiltrates that clinically and/or histologically simulate cutaneous lymphomas.115 Clinicopathologic correlation is essential to achieve the final diagnosis in cutaneous PSL and to differentiate it from cutaneous lymphomas. Elimination of infectious agents (eg, by antibiotics or antiviral drugs) or avoidance of exposure to the causative agent (eg, LCD and drug reactions) are the first step in the management in addition to immunomodulating agents (eg, corticosteroids, UV light treatment) or ablative approaches (eg, surgical excision, cryotherapy).
2126
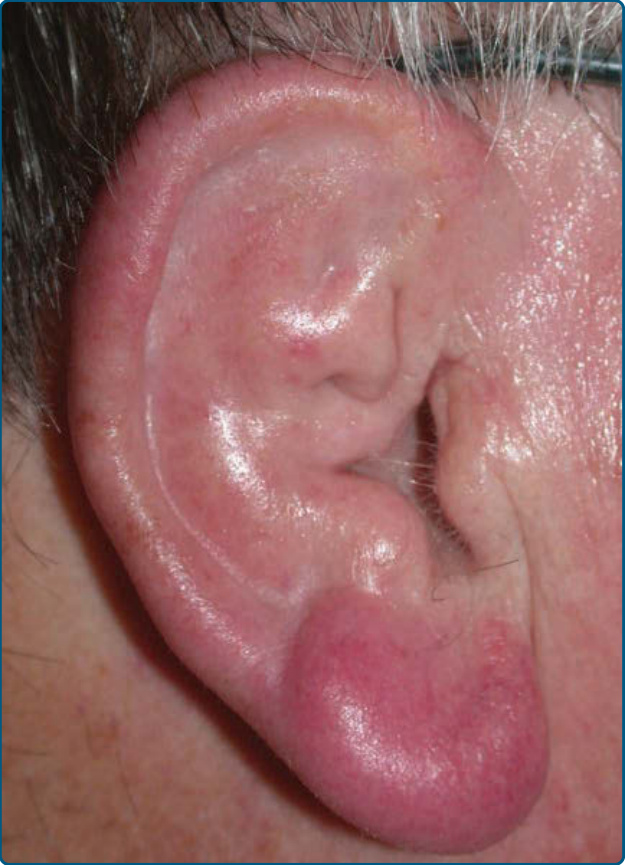
Figure 120-1 Borrelia-associated nodular B-cell lymphoma (lymphocytoma cutis, lymphadenosis cutis benigna): violaceous nodule on the earlobe.
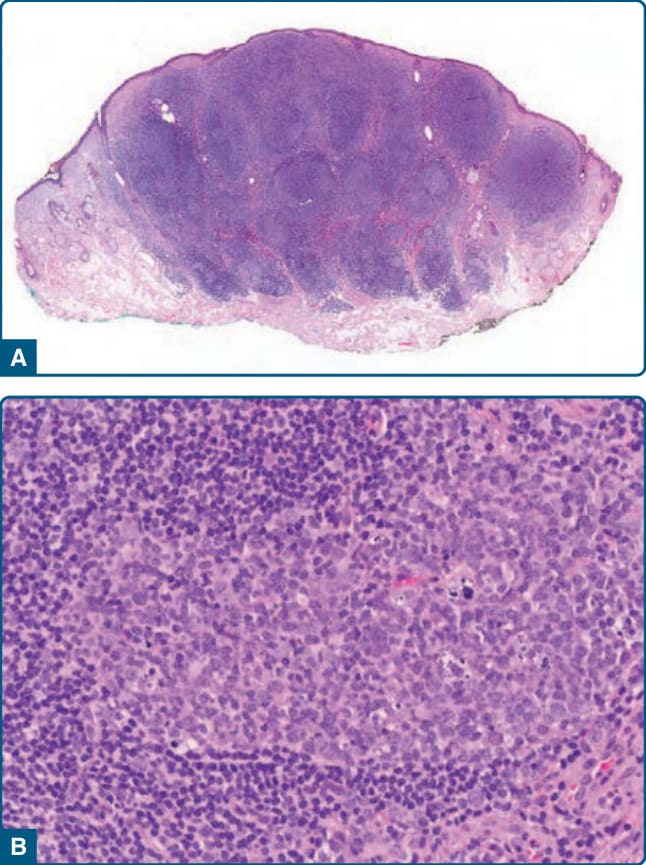
Figure 120-2 Borrelia-associated B-cell pseudolymphoma, histology: nodular lymphocytic infiltrate (A) with reactive germinal centers containing tingible body macrophages (B).
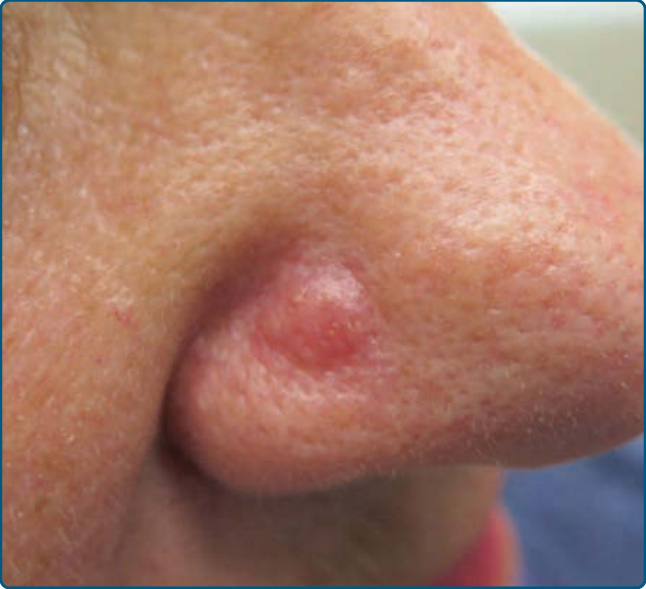
Figure 120-3 Nodular T-cell pseudolymphoma: erythematous nodule on the nose.
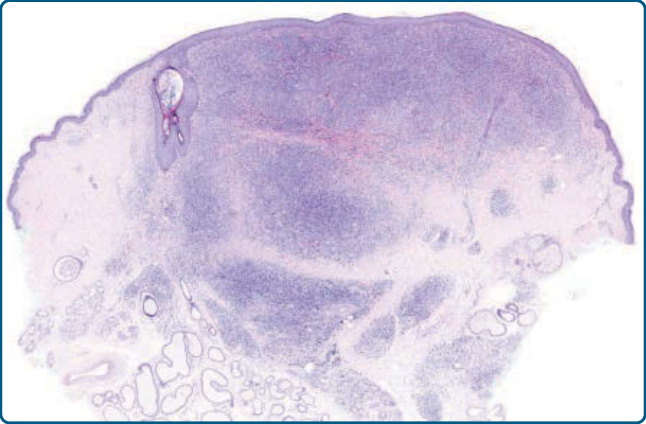
Figure 120-4 Nodular T-cell pseudolymphoma, histology: nodular infiltrate composed mostly of small lymphocytes. No follicles are present.
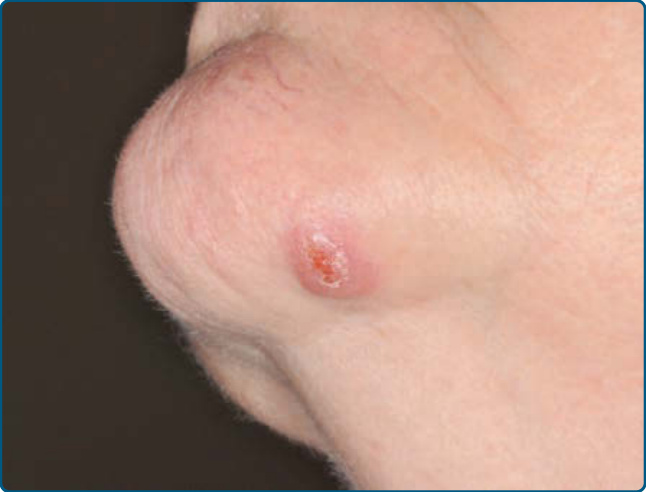
Figure 120-5 Cutaneous CD4+ small-/medium-sized T-cell lymphoproliferative disorder: nodular lesion on the chin.
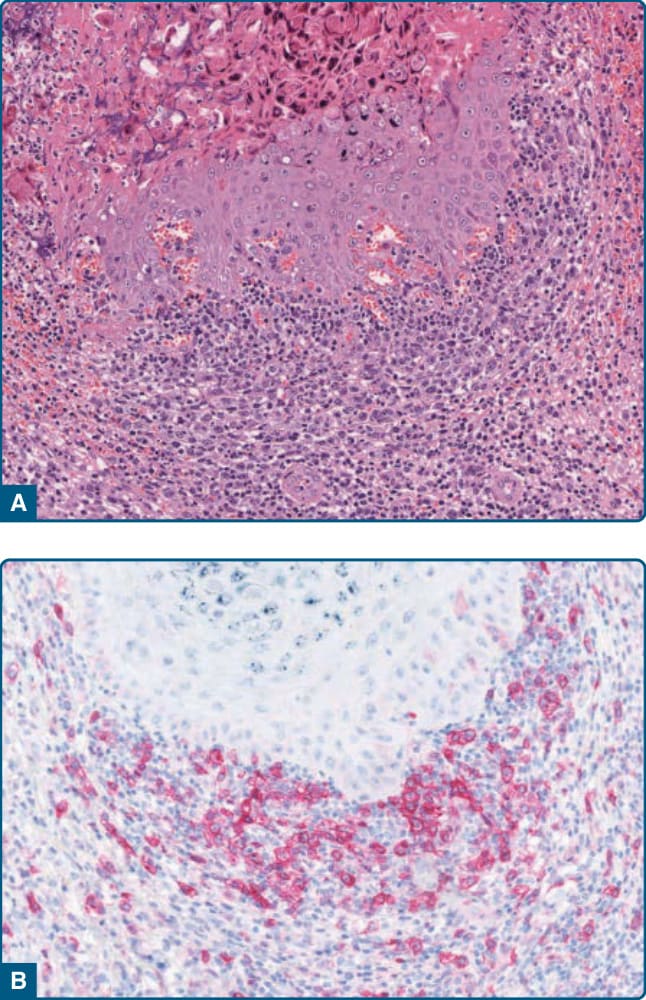
Figure 120-6 CD30+ pseudolymphoma. A, Infiltrates of atypical lymphocytes in molluscum contagiosum. B, Expression of CD30 by the medium-sized to large atypical lymphocytes.
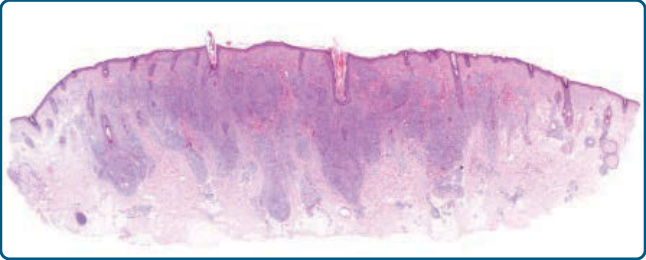
Figure 120-7 Pseudolymphomatous folliculitis, histology: dense dermal lymphocytic infiltrates with perifollicular accentuation.
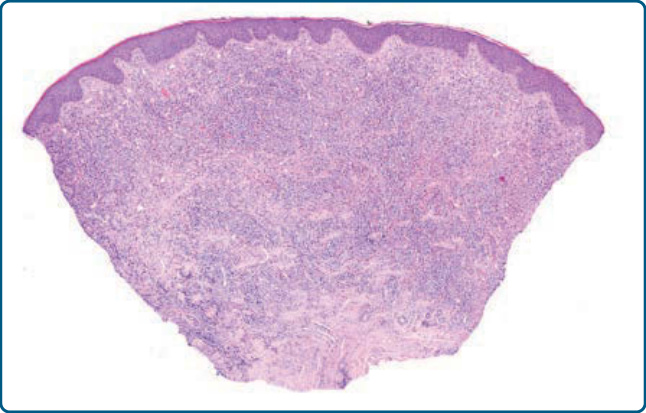
Figure 120-8 Nodular arthropod bite reaction, histology: wedge-shaped dermal infiltrate composed of lymphocytes and numerous eosinophils.
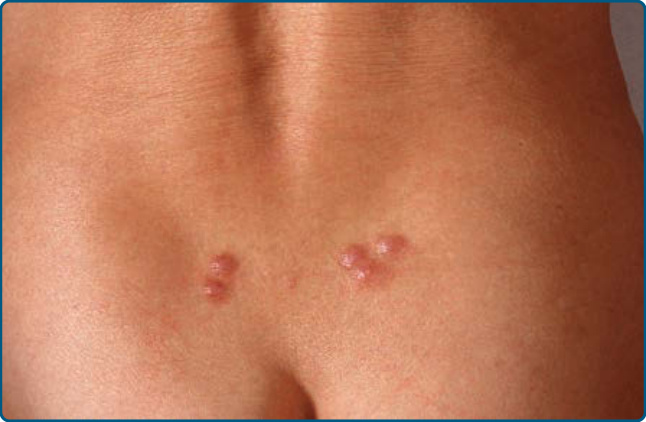
Figure 120-9 Pseudolymphoma caused by leeches (Hirudo medicinalis): characteristic localization of the pseudolymphoma.
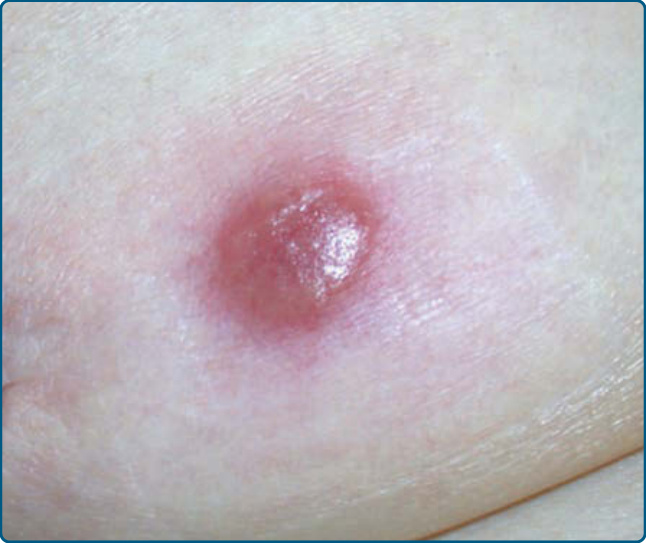
Figure 120-10 Nodular T-cell pseudolymphoma caused by anticonvulsant (phenytoin): erythematous nodule on the right breast.
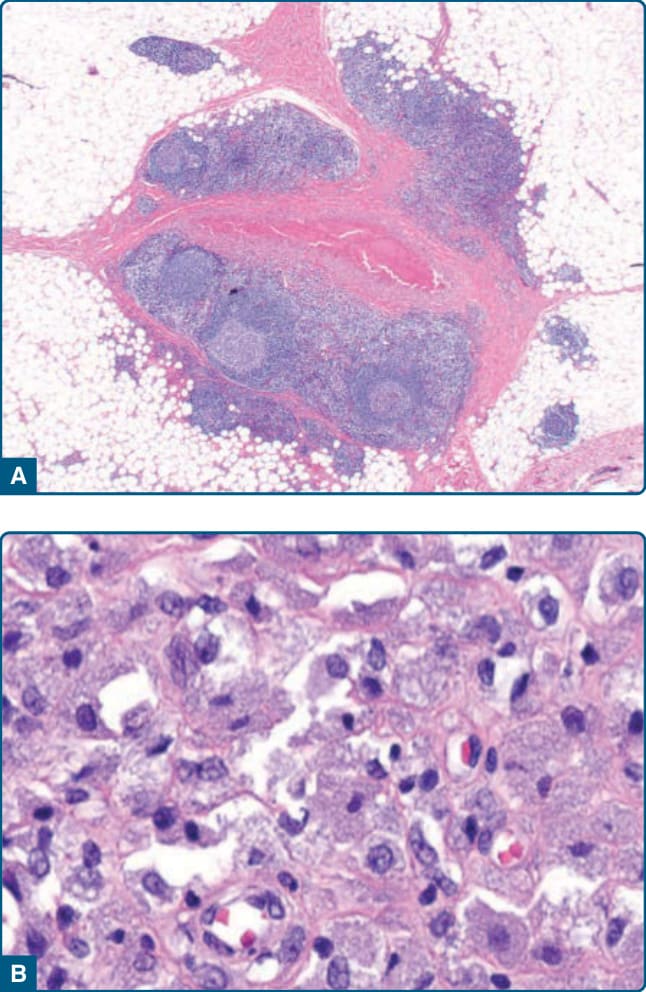
Figure 120-11 Pseudolymphoma caused by allergen injection: A, Subcutaneous B-cell–rich infiltrate with formation of follicles. Note the central area with the histiocyte-rich infiltrate. B, The histiocytes show a grayish granular cytoplasm containing aluminum hydroxide–bound vaccine.
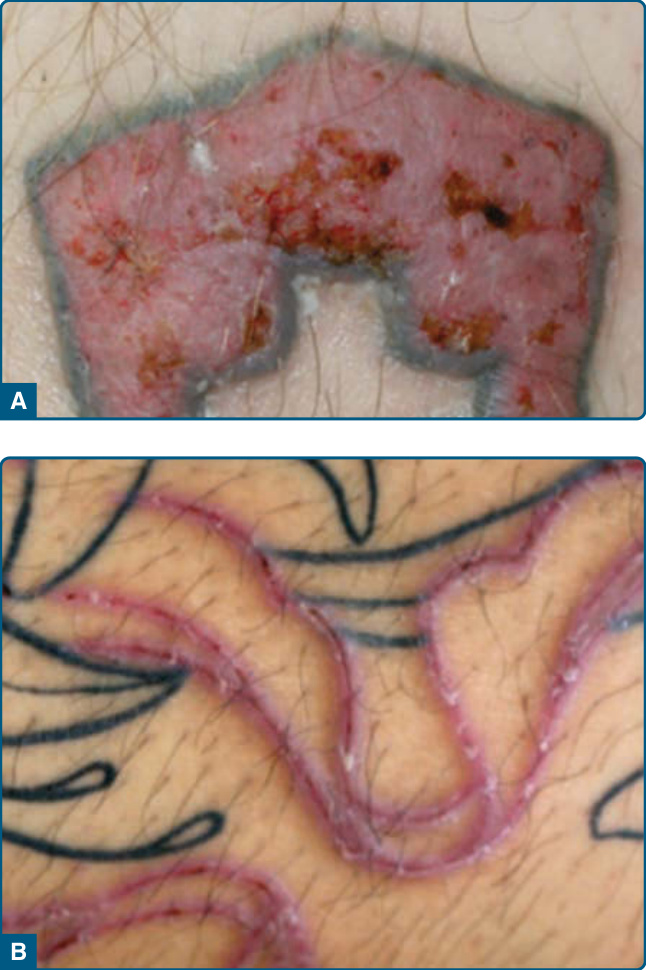
Figure 120-12 A and B, Pseudolymphoma in association with tattoo: Infiltration within the area of red tattoo component.
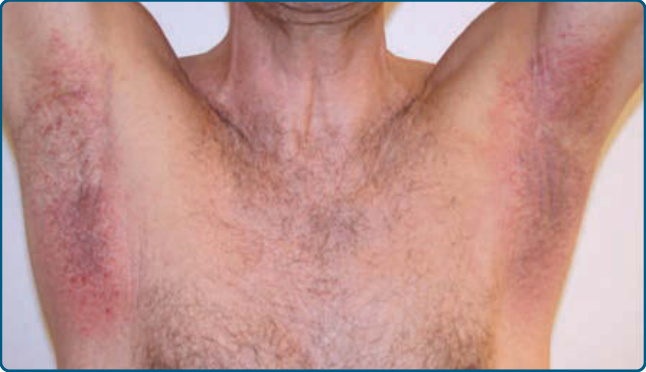
Figure 120-13 Lymphomatoid contact dermatitis: eczematous lesions in both axillae caused by contact allergy to fragrance components.
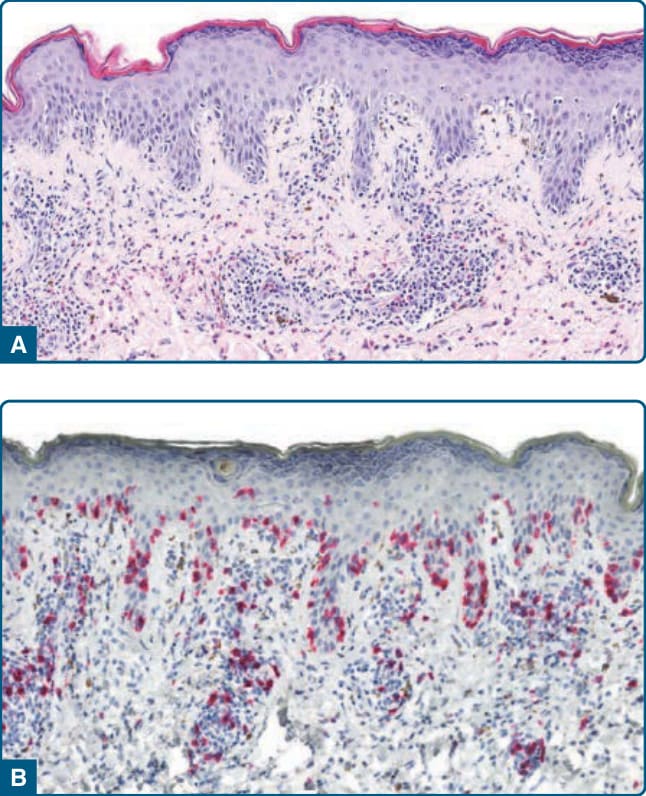
Figure 120-14 Lymphomatoid drug reaction, histology: lymphocytic infiltrate with exocytosis of lymphocytes into the epidermis and nuclear atypia of the lymphocytes (A) with expression of CD8 (B).
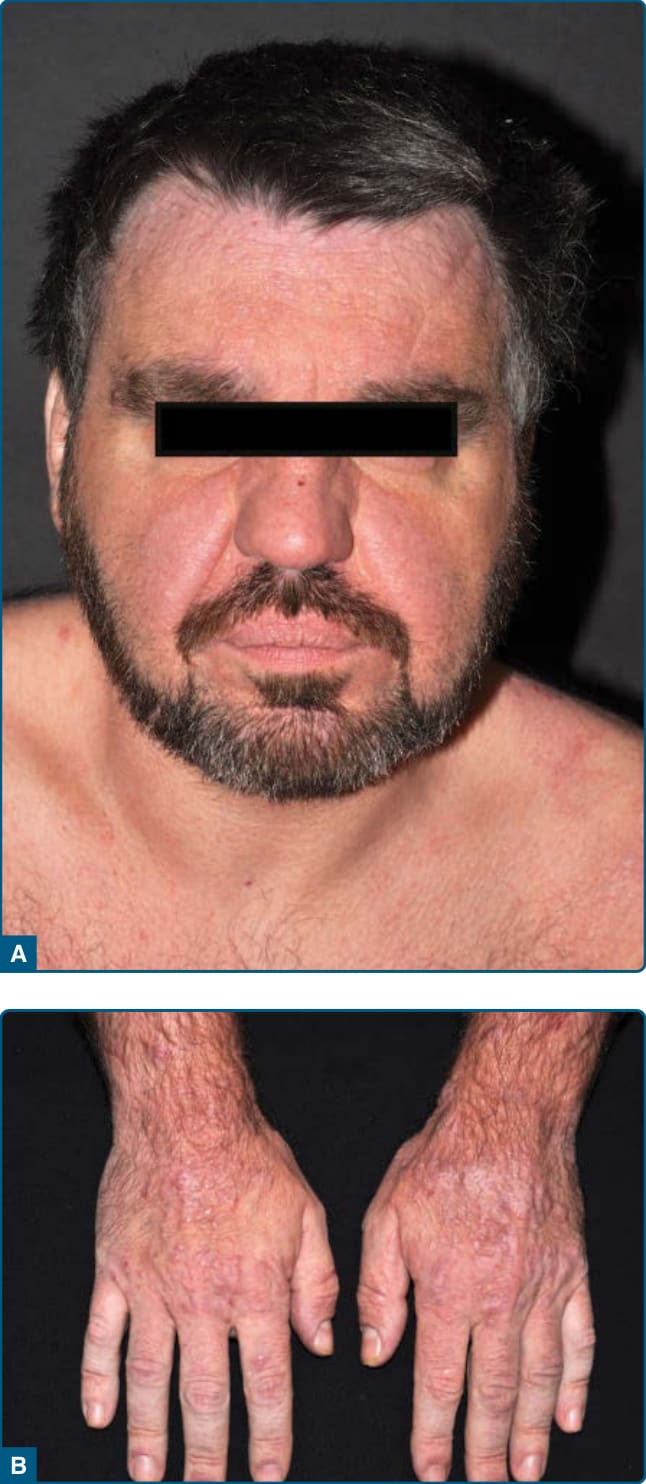
Figure 120-15 Actinic reticuloid: diffuse infiltration and erythema and papules on the face (A) and papules on the back of the hands (B).
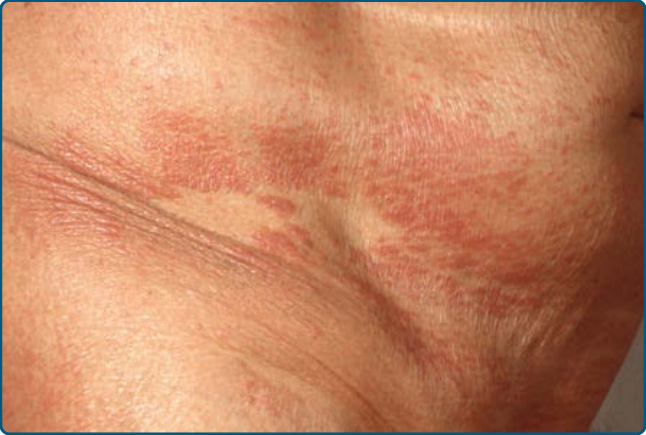
Figure 120-16 Papuloerythroderma Ofuji: papules with confluence to plaques and spared areas.
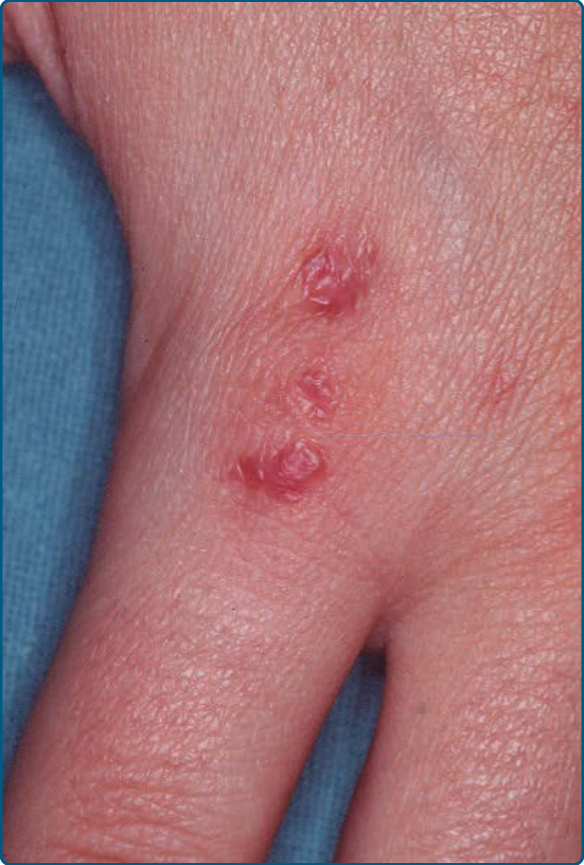
Figure 120-17 Acral pseudolymphomatous angiokeratoma of childhood (APACHE): grouped papules on the hand of an 8-year old girl. (Used with permission from Prof. Dr. S. Lautenschlager, Zurich, Switzerland.)
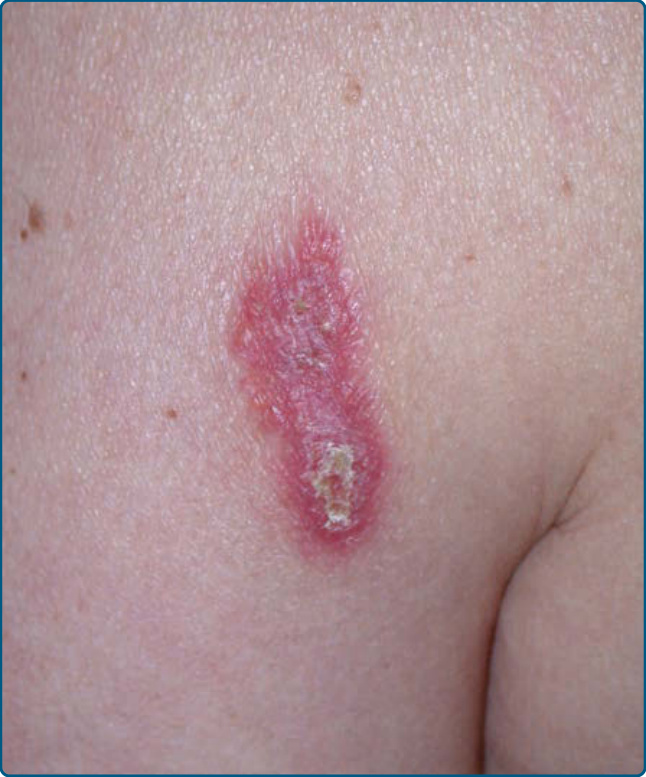
Figure 120-18 Lymphoplasmacytoid plaque: longstanding erythematous plaque on the lateral aspect of the left upper arm.
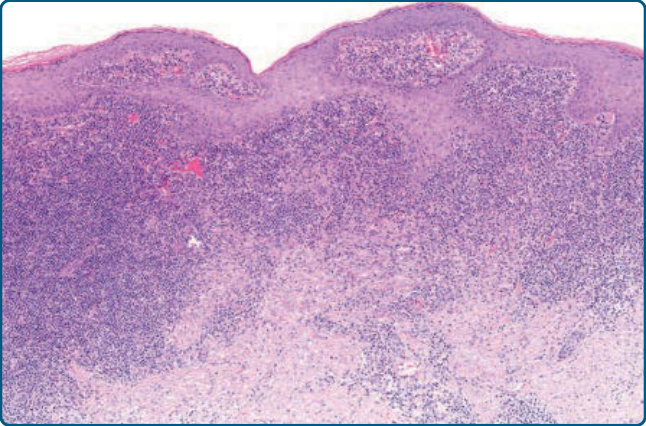
Figure 120-19 Lymphoplasmacytoid plaque, histology: epidermal hyperplasia and dense dermal infiltrate of lymphocytes and histiocytes.
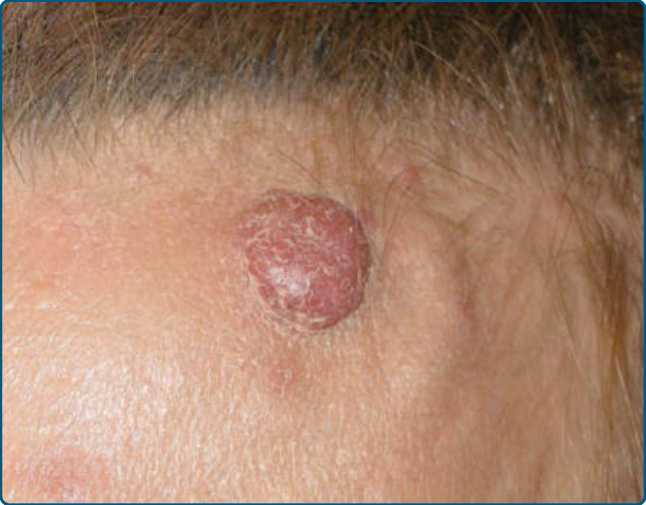
Figure 120-20 Angiolymphoid hyperplasia with eosinophilia: nodular infiltrate on the left forehead.
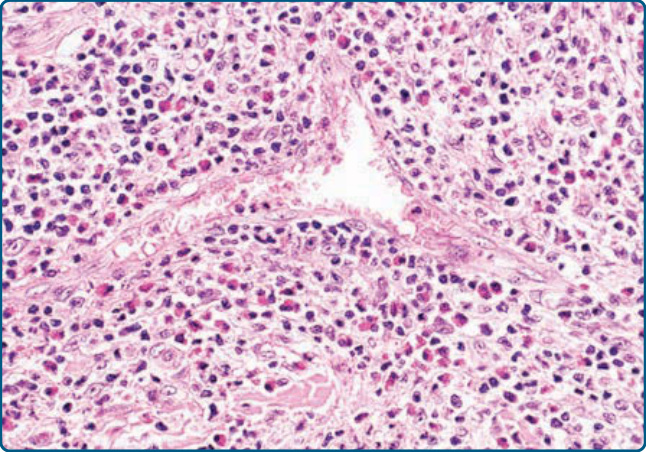
Figure 120-21 Angiolymphoid hyperplasia with eosinophilia, histology: vessels with prominent endothelia embedded in a dense infiltrate of lymphocytes and eosinophils.
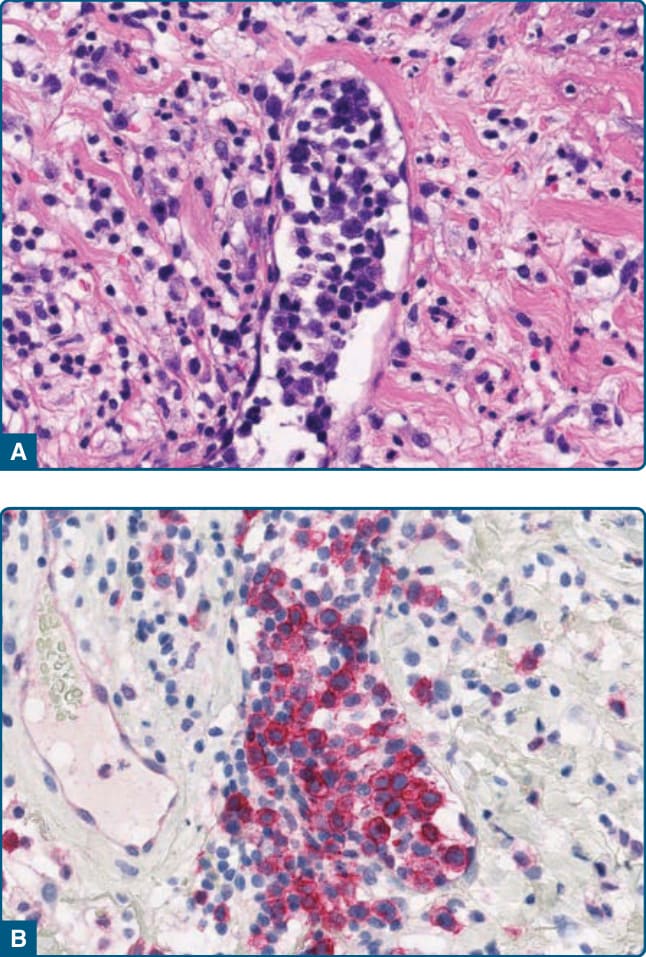
Figure 120-22 Intravascular lymphoid proliferation, histology: Intralymphatic accumulation of blast-like lymphoid cells (A) with expression of CD30 (B).
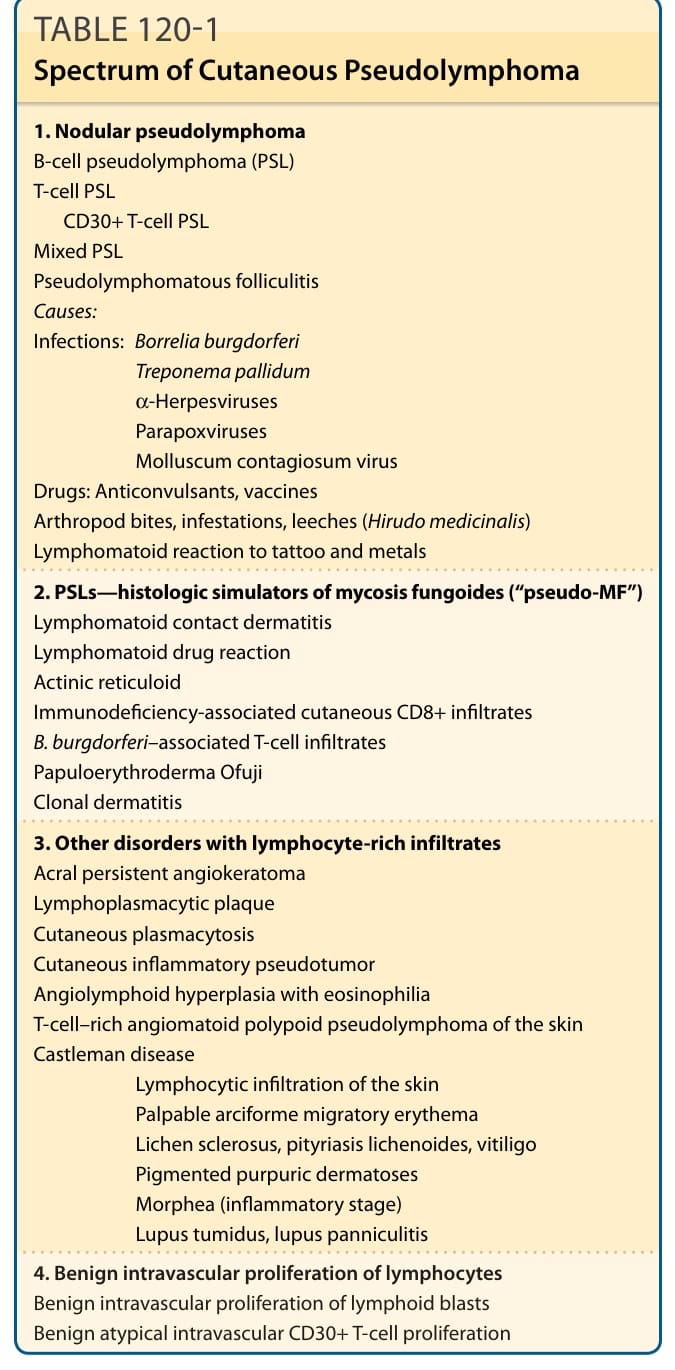
TABLE 120-1 Spectrum of Cutaneous Pseudolymphoma

TABLE 120-2 Terminology of Cutaneous Pseudolymphoma— Historical Aspect

TABLE 120-3 Treatment of Nodular Pseudolymphoma
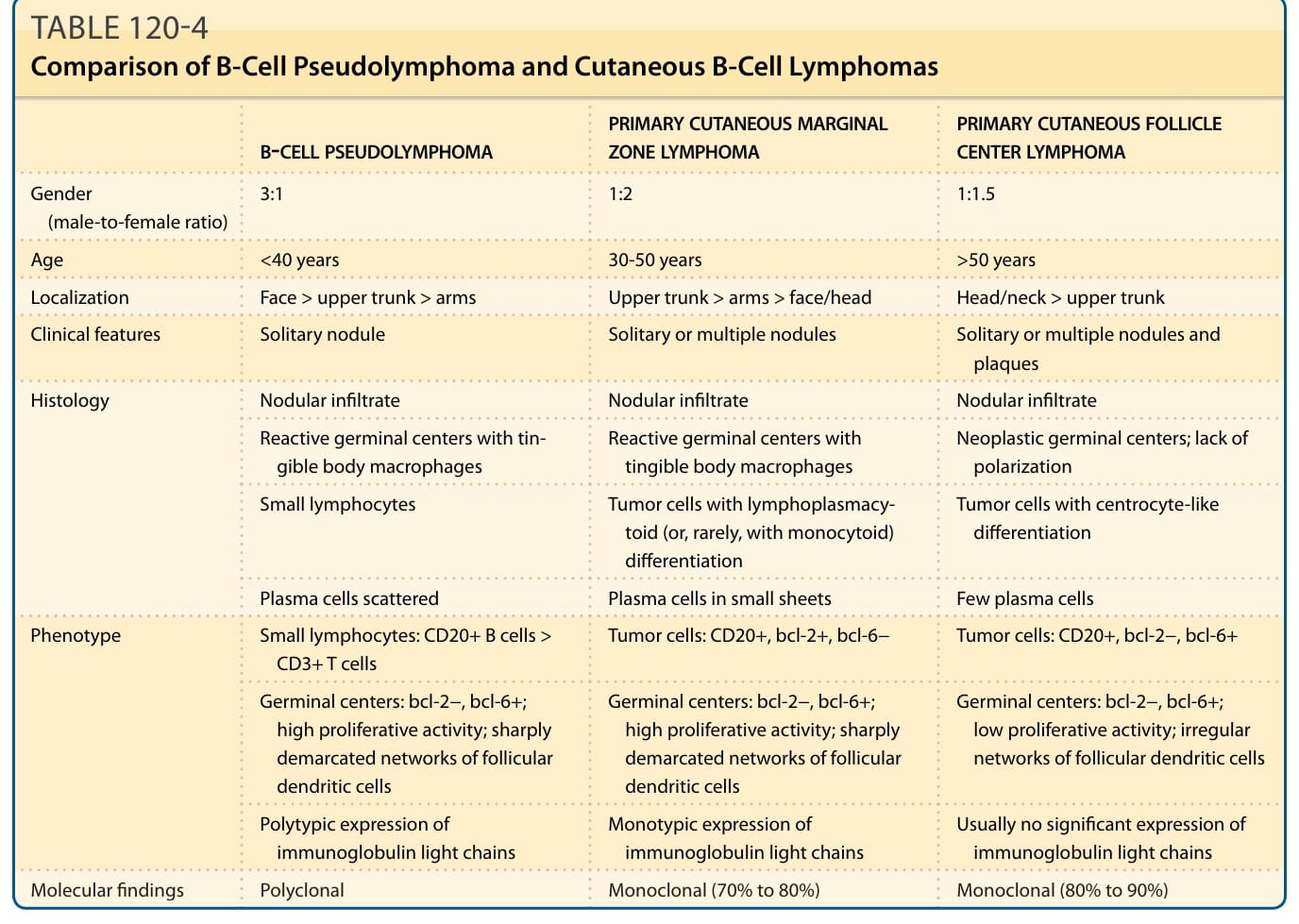
TABLE 120-4 Comparison of B-Cell Pseudolymphoma and Cutaneous B-Cell Lymphomas