皮膚假性淋巴瘤 (Cutaneous Pseudolymphoma)
重點一覽 (AT-A-GLANCE)
■ 皮膚假性淋巴瘤 (cutaneous pseudolymphomas) 是在臨床上與/或組織學上模擬皮膚淋巴瘤的良性淋巴增生 (benign lymphoproliferations)。
■ 它們表現出廣泛的臨床、組織學與免疫表型 (immunophenotypic) 特徵,並可由不同的感染性與非感染性病原所觸發。
■ 臨床病理相關性 (Clinicopathological correlation) 在與皮膚淋巴瘤的鑑別中扮演重要角色。
■ 治療包括避免暴露於致病病原、免疫調節劑 (immunomodulating agents) 或破壞性 (ablative) 方法。
定義 (DEFINITION)
皮膚假性淋巴瘤 (cutaneous pseudolymphoma) 一詞指的是一群皮膚疾病,可被定義為在臨床上與/或組織學上模擬皮膚淋巴瘤的良性淋巴增生過程 (benign lymphoproliferative processes)。這些疾病在臨床、組織學與免疫表型表現上各不相同,且具有不同的病因(表 120-1)。已辨識出一系列廣泛的致病因子,已知能誘發皮膚假性淋巴瘤 (cutaneous pseudolymphomas, PSLs)。感染性病原(如螺旋體細菌 spirochetal bacteria〔伯氏疏螺旋體 Borrelia burgdorferi sp.、梅毒螺旋體 Treponema pallidum〕)、病毒(如副痘病毒 parapoxviruses)、寄生蟲感染(如疥瘡 scabies)、昆蟲叮咬、注射疫苗或用於去敏化 (hyposensitization) 的抗原、異物(如刺青 tattoos 與金屬),以及藥物,都已被辨識為 PSL 的致病因子。所有無法辨識病因的病例都被稱為 PSL 的特發性 (idiopathic) 型。
歷史觀點 (HISTORICAL ASPECTS)
已有各種同義詞被引入,且至今仍有數個名詞被用來描述皮膚的 PSLs。PSL(假性惡性 pseudomalignancy)的概念最早於 1891 年由 M. Kaposi 提出(表 120-2)。¹ 1923 年,Biberstein 創造了 lymphocytoma cutis(皮膚淋巴細胞瘤)一詞。² 隨後於 1943 年,Bäfverstedt 使用 lymphadenosis benigna cutis(皮膚良性淋巴腺病)這一名稱。³ Lever 於 1967 年引入 pseudolymphoma of Spiegler and Fendt(Spiegler-Fendt 假性淋巴瘤)一詞,而 1969 年 Caro 與 Helwig 採用 cutaneous lymphoid hyperplasia(皮膚淋巴樣增生)一詞。⁴ 還有許多其他名詞被提出,且大多指 B 細胞 PSLs。在 T 細胞 PSLs 中,光化性網狀細胞增多症 (actinic reticuloid) 最早於 1969 年被描述。⁵ 在 1980 年代,cutaneous PSL(皮膚假性淋巴瘤)用於指 B 細胞主導與 T 細胞主導過程的名稱變得更廣泛使用與被接受。⁶ 病理學家與皮膚病理學家常以同義方式使用 atypical lymphoid proliferation(非典型淋巴樣增生)一詞。
分類 (CLASSIFICATION)
已提出各種方法來分類皮膚 PSL。例如,根據病因、主要的淋巴細胞成分(T、B 或混合細胞)或獨特的臨床特徵(如兒童肢端丘疹性血管角化瘤 acral papular angiokeratoma of childhood)。⁷⁻⁹
根據病因或組織學與表型將 PSL 分類為 T 細胞與 B 細胞 PSL,在教科書中被廣泛使用。在日常工作中,臨床醫師或病理學家遇到疑似為 PSL 的浸潤時,無法在第一眼就辨認出病因與表型;需要進一步的診斷檢查。此外,必須強調,浸潤的組成主要由宿主的遺傳與免疫因子決定,而非致病病原本身,因為相同的病原在許多情況下既能誘發 B 細胞 PSL,也能誘發 T 細胞 PSL。從實務觀點來看,表現為單一或多發結節、在臨床與組織學上模擬淋巴瘤的皮膚 PSL,可以與其他僅憑組織學基礎模擬皮膚 T 細胞淋巴瘤的 PSL 形式相區分,後者統稱為類蕈狀肉芽腫 (pseudo–mycosis fungoides, pseudo-MF)。此外,有許多感染性與非感染性病況,其特徵為富含淋巴細胞的浸潤,且容易主要因組織學基礎而被誤判為皮膚淋巴瘤。因此,本章分為以下幾個部分:(a) 在臨床與組織學上模擬 T 細胞或 B 細胞淋巴瘤的結節性 PSL;(b) 在組織學上模擬蕈狀肉芽腫的 PSLs(即所謂的 pseudo-MF);(c) 其他具有富含淋巴細胞浸潤的皮膚疾病;以及 (d) 淋巴細胞的良性血管內增生(模擬血管內淋巴瘤 intravascular lymphoma)(見表 120-1)。
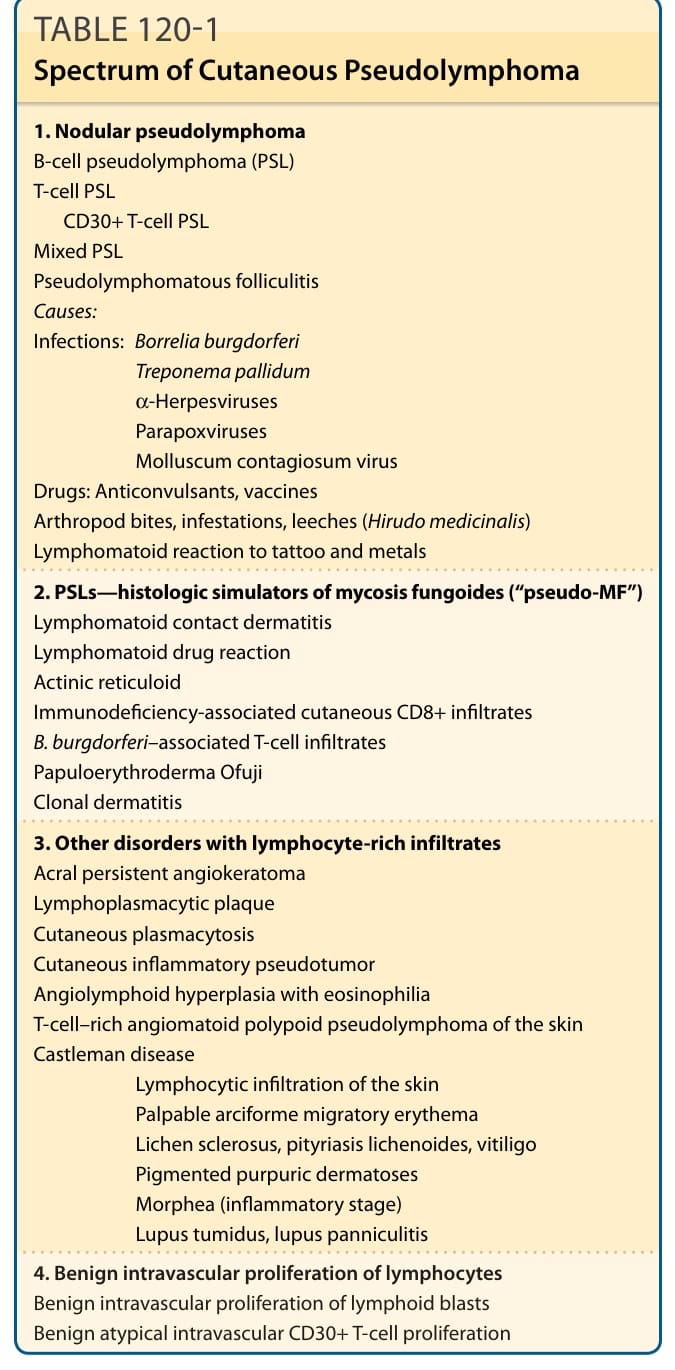
表 120-1:皮膚假性淋巴瘤的譜系 (Spectrum of Cutaneous Pseudolymphoma)
表 120-1 的內容摘錄如下:
-
結節性假性淋巴瘤 (Nodular pseudolymphoma):B 細胞假性淋巴瘤 (PSL)、T 細胞 PSL、CD30+ T 細胞 PSL、混合型 PSL、假性淋巴瘤性毛囊炎 (Pseudolymphomatous folliculitis)。病因——感染:伯氏疏螺旋體 (Borrelia burgdorferi)、梅毒螺旋體 (Treponema pallidum)、α 疱疹病毒 (α-Herpesviruses)、副痘病毒 (Parapoxviruses)、傳染性軟疣病毒 (Molluscum contagiosum virus);藥物:抗痙攣藥 (Anticonvulsants)、疫苗 (vaccines);節肢動物叮咬、寄生蟲感染、水蛭 (leeches, Hirudo medicinalis);對刺青與金屬的淋巴瘤樣反應。
-
PSLs——蕈狀肉芽腫的組織學模擬者(“pseudo-MF”):淋巴瘤樣接觸性皮膚炎 (Lymphomatoid contact dermatitis)、淋巴瘤樣藥物反應 (Lymphomatoid drug reaction)、光化性網狀細胞增多症 (Actinic reticuloid)、免疫缺陷相關之皮膚 CD8+ 浸潤 (Immunodeficiency-associated cutaneous CD8+ infiltrates)、B. burgdorferi 相關之 T 細胞浸潤、Ofuji 丘疹性紅皮症 (Papuloerythroderma Ofuji)、克隆性皮膚炎 (Clonal dermatitis)。
-
其他具富含淋巴細胞浸潤的疾病:肢端持續性血管角化瘤 (Acral persistent angiokeratoma)、淋巴漿細胞性斑塊 (Lymphoplasmacytic plaque)、皮膚漿細胞增多症 (Cutaneous plasmacytosis)、皮膚發炎性假瘤 (Cutaneous inflammatory pseudotumor)、伴嗜酸性球增多之血管淋巴樣增生 (Angiolymphoid hyperplasia with eosinophilia)、皮膚富 T 細胞血管瘤樣息肉狀假性淋巴瘤 (T-cell–rich angiomatoid polypoid pseudolymphoma of the skin)、Castleman 病、皮膚淋巴細胞浸潤 (Lymphocytic infiltration of the skin)、可觸及之弓形遊走性紅斑 (Palpable arciforme migratory erythema)、硬化性苔癬 (Lichen sclerosus)、苔癬樣糠疹 (pityriasis lichenoides)、白斑 (vitiligo)、色素性紫癜性皮膚病 (Pigmented purpuric dermatoses)、硬斑病(發炎期 Morphea, inflammatory stage)、腫脹型紅斑性狼瘡 (Lupus tumidus)、狼瘡性脂膜炎 (lupus panniculitis)。
-
淋巴細胞的良性血管內增生 (Benign intravascular proliferation of lymphocytes):淋巴母細胞的良性血管內增生 (Benign intravascular proliferation of lymphoid blasts)、良性非典型血管內 CD30+ T 細胞增生 (Benign atypical intravascular CD30+ T-cell proliferation)。
診斷方法 (DIAGNOSTIC APPROACH)
皮膚 PSL 的臨床表現範圍從單一結節到成簇或播散性丘疹,乃至紅皮症 (erythroderma)。⁸,¹⁰ 組織學分析在皮膚 PSLs 的診斷方法中扮演關鍵角色。可區分不同的浸潤模式(結節性浸潤 vs 表皮趨向性浸潤 epidermotropic infiltrates)、淋巴細胞的大小(大多為小細胞;偶爾為中型與大型細胞)、免疫表型(T 細胞 vs B 細胞;CD4 vs CD8;CD30)。針對克隆性 (clonality) 與感染性病原(尤其是伯氏疏螺旋體 Borrelia burgdorferi)的分子研究是輔助性的診斷工具。重要的是要強調,偵測到克隆性 T 細胞或 B 細胞族群本身並不代表存在惡性淋巴瘤。此外,已有報告指出某些 PSL 病例帶有克隆性 T 或 B 細胞。¹¹⁻¹⁴
表 120-2 的歷史名詞列出如下:
- 1891 年 皮膚肉瘤病 (Sarcomatosis cutis)(M. Kaposi)¹
- 1910 年 Spiegler-Fendt 類肉瘤 (Sarcoid Spiegler-Fendt)(Darier)¹¹⁴
- 1923 年 皮膚淋巴細胞瘤 (Lymphocytoma cutis)(Biberstein)²
- 1943 年 皮膚良性淋巴腺病 (Lymphadenosis benigna cutis)(Bäfverstedt)³
- 1969 年 皮膚淋巴樣增生 (Cutaneous lymphoid hyperplasia)(Caro 與 Helwig)⁴
- 1982 年 皮膚假性淋巴瘤 (Cutaneous pseudolymphoma)(Burg 等人)⁶

表 120-2:皮膚假性淋巴瘤的術語——歷史觀點 (Terminology of Cutaneous Pseudolymphoma— Historical Aspect)
因此,組織學與分子發現都必須結合臨床背景一併解讀;亦即,臨床病理相關性 (clinicopathologic correlation) 對於達成最終診斷至關重要。診斷檢查包括病史(尤其是暴露於節肢動物、過敏原與藥物)以及體格檢查,包括淋巴結的觸診評估、周邊血液檢查(淋巴球增多 lymphocytosis、嗜酸性球增多 eosinophilia、感染性病原的血清學檢查,尤其是 B. burgdorferi、梅毒 syphilis、HIV)。由於 PSLs 代表良性的淋巴細胞增生,無皮膚外擴散的潛能,通常不需進行分期 (staging) 檢查。在具有不尋常臨床表現與多發結節性病灶、免疫球蛋白輕鏈的單型 (monotypic) 表現、偵測到 T 細胞或 B 細胞克隆性,或其他不一致或非預期的組織學或表型發現的病例中,應考慮放射學分期(電腦斷層攝影 computed tomography 或正子發射斷層攝影-電腦斷層攝影 positron emission tomography-computed tomography),以排除模擬 PSL 的原發性或續發性皮膚淋巴瘤。
病程 (COURSE)
皮膚 PSLs 的病程變化極大。切片後可能消退,但某些病灶可能持續數個月甚至數年。再次暴露於誘發病原後可觀察到復發,特別是在由藥物或過敏原所引起的病例中。已有 PSL 進展的報告,但這是極其罕見的事件,如果它確實存在的話。¹⁵ 目前仍不清楚 PSL 是否真的進展為明顯的淋巴瘤(例如透過獲得染色體異常 chromosomal aberrations,或持續的抗原刺激導致淋巴細胞永久性增生),或者這些病例從一開始就是皮膚淋巴瘤,只是在其極早期疾病階段無法藉由組織學、表型或分子發現被辨識出來。
結節性假性淋巴瘤 (NODULAR PSEUDOLYMPHOMA)
以單一結節或多發結節表現的 PSLs,在臨床與組織學基礎上模擬皮膚 T 細胞與 B 細胞淋巴瘤。它們代表最常見的 PSL 形式之一,但尚無關於 B 細胞 PSLs 整體盛行率的詳細數據被報告。組織學上,它們可根據主要的淋巴細胞亞群分類為 B 細胞、T 細胞與混合型(T 細胞/B 細胞)PSL。⁸,¹⁰ 此分類相當人為,因為 B 細胞 PSL 總是含有 T 細胞,反之亦然。T 細胞 PSL 常帶有數量不一的 B 細胞。儘管如此,將結節性 PSL 區分為 3 種組織學與表型亞型,對於鑑別診斷仍然有用,這也必須考量假性淋巴瘤反應的潛在病因。
結節性假性淋巴瘤的治療(表 120-3):
- 避免或消除致病病原(如接觸性過敏原)
- 對感染相關之假性淋巴瘤進行抗生素治療(如 Borrelia 相關細胞性假性淋巴瘤)
- 手術切除、冷凍療法 (cryotherapy)、雷射治療
- 局部/病灶內類固醇或干擾素-α (interferon-α)
- 全身性類固醇、羥氯奎寧 (hydroxychloroquine)
避免再次暴露於誘發病原(如疫苗、過敏原注射、其他藥物、水蛭 Hirudo medicinalis 治療、針灸與刺青)是預防結節性 PSL 持續存在與復發最重要的步驟。單一病灶可藉由完整的手術切除、局部或病灶內皮質類固醇、冷凍療法或雷射治療來處理(表 120-3)。若這些治療方法不成功,可考慮放射治療 (radiation therapy)。在具有多發 PSL 病灶的病人中,尤其是那些特發性多灶性 PSL (idiopathic multifocal PSL) 的病人,全身性皮質類固醇、病灶內干擾素-α 或羥氯奎寧可能是治療選項。羥氯奎寧抑制漿細胞樣樹突狀細胞 (plasmacytoid dendritic cells) 的活性,這些細胞以群聚形式存在於大多數 B 細胞 PSLs 中,緊鄰 T 細胞與漿細胞,可能代表誘發與維持 PSLs 的驅動力。¹⁶,¹⁷

表 120-3:結節性假性淋巴瘤的治療 (Treatment of Nodular Pseudolymphoma)
皮膚 B 細胞假性淋巴瘤 (CUTANEOUS B-CELL PSEUDOLYMPHOMA)
B 細胞 PSL 常也被稱為皮膚淋巴細胞瘤 (lymphocytoma cutis) 或皮膚淋巴樣增生 (cutaneous lymphoid hyperplasia)。
臨床特徵 (CLINICAL FEATURES)
B 細胞 PSL 最常以結節或斑塊表現。臉部(尤其是鼻部與臉頰)、上軀幹與手臂是最常受侵犯的部位。已描述男女比為 2:1。¹⁸ 約 67% 的 B 細胞 PSLs 病人年齡小於 40 歲,而兒童與青少年佔比少於 10%。⁴
以單一結節(最大可達 4 cm)表現的局限型 B 細胞 PSL 是最常見的表現。三分之一的病人會發展出多發結節,或一種粟粒狀 (miliarial) 型,其病灶為直徑僅數毫米的丘疹。¹⁹
組織學 (HISTOLOGY)
所有結節性 B 細胞 PSLs 基本上具有相同的生長模式與浸潤組成。存在一個緻密的結節性浸潤,主要位於網狀真皮 (reticular dermis),偶爾延伸至皮下組織 (subcutis) 的淺層部分。浸潤主要由具有染色質緻密細胞核的小淋巴細胞與含有可染體巨噬細胞 (tingible body macrophages) 的反應性生發中心 (reactive germinal centers) 組成。淋巴細胞不顯示核異型 (nuclear atypia)。有漿細胞 (plasma cells) 混雜其中,這些漿細胞通常不形成聚集體,而是相當瀰漫地散布於整個浸潤中。在某些病例中可觀察到嗜酸性球與肉芽腫成分 (granulomatous component)。混雜有數量不一的 T 細胞,通常佔浸潤的不到 30%。免疫表型分析顯示,大部分浸潤由 CD19+、CD20+ 與 CD79a+ B 細胞代表。反應性濾泡 (reactive follicles) 中的細胞表現 bcl-6,但 bcl-2 為陰性,而濾泡間區 (interfollicular area) 的小 B 細胞表現 bcl-2,但 bcl-6 為陰性。CD21+ 濾泡樹突狀細胞 (follicular dendritic cells) 的網絡界限分明且結構規則。藉由免疫組織化學或原位雜交 (in situ hybridization),可發現漿細胞對免疫球蛋白 (Ig) 輕鏈 κ 與 λ 的多型 (polytypic) 表現。分子研究藉由聚合酶連鎖反應 (polymerase chain reaction, PCR) 或南方墨點分析 (Southern blot analysis) 證明缺乏 Ig 重鏈基因的單株重排 (monoclonal rearrangement)。
鑑別診斷 (DIFFERENTIAL DIAGNOSES)
B 細胞 PSLs 的鑑別診斷主要包括其他具有濾泡模式的 B 細胞浸潤;亦即原發性皮膚邊緣區淋巴瘤 (primary cutaneous marginal zone lymphoma, PCMZL) 與原發性皮膚濾泡中心淋巴瘤 (primary cutaneous follicle center lymphoma, PCFCL),或其表現為續發性皮膚浸潤的淋巴結性或其他結外 (extranodal) 對應疾病(表 120-4)。PCMZL 浸潤的結構與組成與 B 細胞 PSL 非常相似,因為兩者都表現為網狀真皮與淺層皮下組織中的結節性浸潤。²⁰ 與 B 細胞 PSL 相比,PCMZL 中的漿細胞通常更為顯著且以片狀 (sheets) 出現,特別是在浸潤的周邊。最重要的組織病理診斷發現是 PCMZL 中 Ig 輕鏈的單型 (monotypic) 表現,兩種 Ig 輕鏈之一的表現比例至少為 5:1,最高可達 10:1。嗜酸性球的存在與數量並非鑑別 B 細胞 PSL 與 PCMZL 的有用發現。偵測到克隆性 B 細胞族群可作為 PCMZL 的額外診斷線索,但這種偵測僅在 50% 至 70% 的 PCMZL 病例中發現,因此限制了它在鑑別 B 細胞 PSL 與 PCMZL 上的診斷價值。另一個需要考慮的重要鑑別診斷是 PCFCL,其特徵為以排列於大型腫瘤性濾泡中、具有中心細胞樣 (centrocyte-like) 分化的腫瘤細胞為主。此外,與在 B 細胞 PSLs 中容易辨識到作為反應性濾泡一部分的可染體巨噬細胞相比,可染體巨噬細胞僅在少數 PCFCLs 中發現。²¹ 此外,PCFCL 腫瘤性濾泡中的低增生活性 (low proliferative activity) 是一個特徵性發現,這與 B 細胞 PSLs 反應性生發中心中的高增生活性形成對比。PCFCL 中 CD21+ 濾泡樹突狀細胞的網絡不規則且斷裂,與 B 細胞 PSLs 中界限分明且結構規則的網絡形成對比。藉由 PCR 或南方墨點分析,可在大多數 PCFCL 中偵測到克隆性 B 細胞族群。應提及的是,絕大多數 PCFCL 的腫瘤性中心細胞樣分化細胞不表現 bcl-2。因此,bcl-2 的表現對於鑑別 PCFCL 與 B 細胞 PSL 沒有診斷價值。在中心細胞樣腫瘤細胞表現 bcl-2 的病例中,必須考慮淋巴結性濾泡中心淋巴瘤的續發性皮膚浸潤,因為淋巴結性濾泡中心淋巴瘤在大多數病例中由於潛在的 t(14;18) 易位 (translocation) 而表現 bcl-2。其他鑑別診斷包括 B 細胞慢性淋巴球性白血病 (B-cell chronic lymphocytic leukemia) 或小細胞淋巴球性淋巴瘤 (small cell lymphocytic lymphoma) 的皮膚浸潤,雖然小細胞淋巴球性淋巴瘤的皮膚浸潤通常不顯示反應性生發中心。B 細胞 PSLs 中的克隆性研究在鑑別 B 細胞 PSLs 與皮膚 B 細胞淋巴瘤上價值有限,因為約 10% 至 20% 的 PSLs 帶有克隆性 B 細胞族群。¹²,²² 在某些研究中,於結節性 PSL 中偵測到克隆性 B 細胞的病例比例甚至更高。²³ 在浸潤相當輕微的病灶中,應始終排除假性克隆性 (pseudoclonality),因為它代表一個診斷陷阱 (pitfall)。²⁴
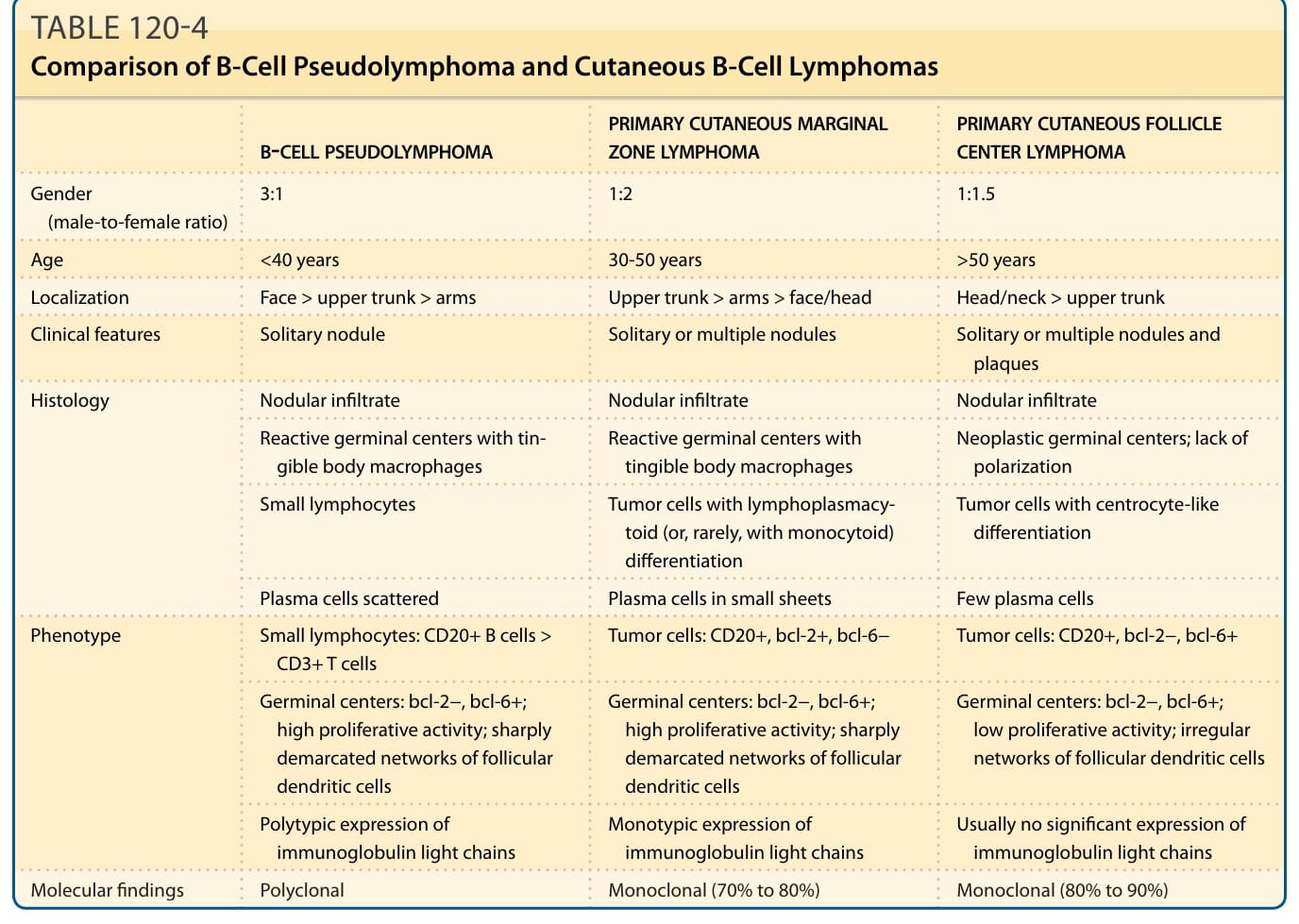
表 120-4:B 細胞假性淋巴瘤與皮膚 B 細胞淋巴瘤的比較 (Comparison of B-Cell Pseudolymphoma and Cutaneous B-Cell Lymphomas)
表 120-4 的比較內容摘錄如下(B 細胞假性淋巴瘤 vs 原發性皮膚邊緣區淋巴瘤 vs 原發性皮膚濾泡中心淋巴瘤):
- 性別(男女比):3:1 | 1:2 | 1:1.5
- 年齡:<40 歲 | 30-50 歲 | >50 歲
- 位置:臉部 > 上軀幹 > 手臂 | 上軀幹 > 手臂 > 臉部/頭部 | 頭/頸 > 上軀幹
- 臨床特徵:單一結節 | 單一或多發結節 | 單一或多發結節與斑塊
- 組織學:結節性浸潤 | 結節性浸潤 | 結節性浸潤
- 反應性生發中心伴可染體巨噬細胞 | 反應性生發中心伴可染體巨噬細胞 | 腫瘤性生發中心;缺乏極化 (polarization)
- 小淋巴細胞 | 具淋巴漿細胞樣(或罕見單核樣 monocytoid)分化的腫瘤細胞 | 具中心細胞樣分化的腫瘤細胞
- 漿細胞散布 | 漿細胞呈小片狀 | 少量漿細胞
- 表型:小淋巴細胞:CD20+ B 細胞 > CD3+ T 細胞 | 腫瘤細胞:CD20+、bcl-2+、bcl-6− | 腫瘤細胞:CD20+、bcl-2−、bcl-6+
- 生發中心:bcl-2−、bcl-6+;高增生活性;界限分明的濾泡樹突狀細胞網絡 | 生發中心:bcl-2−、bcl-6+;高增生活性;界限分明的濾泡樹突狀細胞網絡 | 生發中心:bcl-2−、bcl-6+;低增生活性;不規則的濾泡樹突狀細胞網絡
- 免疫球蛋白輕鏈的多型表現 | 免疫球蛋白輕鏈的單型表現 | 通常無顯著的免疫球蛋白輕鏈表現
- 分子發現:多株性 (Polyclonal) | 單株性(70% 至 80%)| 單株性(80% 至 90%)
病因 (ETIOLOGY)
已辨識出 B 細胞 PSLs 的廣泛病因。除了由伯氏疏螺旋體 (Borrelia burgdorferi sp.) 感染、昆蟲叮咬,或注射疫苗或用於去敏化的抗原所引起的 B 細胞 PSLs 之外,B 細胞 PSLs 還可由針灸治療、刺青、穿洞環與耳環中的金屬,以及藥物所引起。
Borrelia 相關 B 細胞假性淋巴瘤 (BORRELIA-ASSOCIATED B-CELL PSEUDOLYMPHOMA)
流行病學 (EPIDEMIOLOGY)
皮膚淋巴細胞瘤 (lymphocytoma cutis) 與皮膚良性淋巴腺病 (lymphadenosis cutis benigna) 這些名詞被同義地用於由 B. burgdorferi 感染所引起的病例。約 1% 臨床上明顯的伯氏疏螺旋體 (Borrelia burgdorferi sp.) 感染表現為 B 細胞 PSLs。在某些(但非全部)研究中觀察到女性偏多(男女比:2:1)。此型 B 細胞 PSLs 在白人中比在非裔美國人 (African Americans) 中更常被報告。Borrelia 相關 B 細胞 PSLs 典型侵犯兒童並發生於成年早期,但可見於所有年齡層,大多數病人年齡小於 40 歲。
臨床發現 (CLINICAL FINDINGS)
在 Borrelia 誘發的 B 細胞 PSLs 中,通常發展出單一的紅色至紫紅色 (violaceous) 圓頂狀結節,但在約 10% 至 15% 的病人中,可觀察到多灶性皮膚病灶。臉部與頭皮,尤其是耳垂、乳頭與陰囊,是 Borrelia 誘發的 B 細胞 PSLs 的好發部位,但軀幹與四肢也可能受侵犯(圖 120-1)。可能出現區域性淋巴結病變 (Regional lymphadenopathy)。
實驗室檢查 (LABORATORY TESTS)
組織學上,可發現緻密真皮結節性浸潤的原型模式 (archetypic pattern),由小 B 細胞與反應性生發中心組成(圖 120-2)。¹⁴ 在 Borrelia 相關 B 細胞 PSL 中,生發中心傾向較大且融合,僅有小的套區 (mantle zone),在某些病例中缺乏極化。¹⁴,²² 由於大型生發中心的融合,病灶類似 PCFCL 中的腫瘤性濾泡(濾泡性生長模式 follicular growth pattern)。²² 漿細胞幾乎總是存在,且特別見於浸潤的周邊。在罕見的所謂與 Borrelia 感染相關的大細胞淋巴細胞瘤 (large cell lymphocytoma) 病例中,可發現以類似中心母細胞 (centroblasts) 與免疫母細胞 (immunoblasts) 的母細胞 (blasts) 為主,模擬大 B 細胞淋巴瘤的發現。²⁵ 這些病例容易被誤診為瀰漫性大 B 細胞淋巴瘤 (diffuse large B-cell lymphoma)。
在絕大多數 Borrelia 相關 B 細胞 PSLs 的病例中,分子研究顯示 IgH 基因的多株重排 (polyclonal rearrangement),但已觀察到偵測出單株 B 細胞的情況,因此並不排除 Borrelia 誘發 B 細胞 PSL 的診斷。¹⁴
血清學顯示針對 B. burgdorferi 的抗體,型態多變;亦即 IgG 與 IgM 或僅一類抗體可能被偵測到並升高。儘管如此,仍可見到血清學發現陰性的病例¹⁴;因此,陰性血清學並不排除 Borrelia 誘發的 B 細胞 PSL。藉由 PCR 偵測 B. burgdorferi DNA 的分子研究是一個有幫助的輔助診斷工具,敏感度約為 70%。²⁶
診斷基於組織學、臨床背景(蜱叮咬病史 history of tick bite、好發部位的定位)、血清學發現與/或藉由 PCR 在組織中偵測到 B. burgdorferi DNA。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
在 Borrelia 相關 B 細胞 PSLs 中,抗生素(見表 120-3)是第一線治療,並可預防 Borrelia 感染的其他併發症,例如關節炎 (arthritis) 與心肌炎 (carditis)。
皮膚 T 細胞與混合型假性淋巴瘤 (CUTANEOUS T-CELL AND MIXED PSEUDOLYMPHOMA)
結節性 T 細胞 PSL 的特徵為緻密的真皮富 T 細胞結節性浸潤,伴隨數量不一的 B 細胞,這些 B 細胞可達整個浸潤的 30%。²⁷ 混合型 PSL 含有相等數量的 T 細胞與 B 細胞。
流行病學 (EPIDEMIOLOGY)
關於 T 細胞或混合型 PSL 盛行率,並無詳細的流行病學數據。它們侵犯兩種性別、所有年齡層與所有族裔群體的病人。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在 B 細胞 PSLs 中辨識出的所有病因,也可作為 T 細胞與混合型 PSLs 的潛在刺激因子。在約 5% 的病例中,藥物被辨識為 T 細胞 PSL 的致病刺激因子。然而,大多數病例沒有已知病因,因此被稱為特發性 T 細胞 PSL (idiopathic T-cell PSL) 或混合型 PSL。
臨床特徵 (CLINICAL FEATURES)
T 細胞與混合型 PSL 通常以單一或多發的紅色至紫紅色結節表現,類似 B 細胞 PSL(圖 120-3)。
實驗室檢查 (LABORATORY TESTS)
組織學上,T 細胞與混合型 PSL 在大多數病例中表現為緻密的結節性浸潤,延伸遍及整個真皮並進入皮下組織的淺層部分(圖 120-4)。浸潤主要由具有染色質緻密細胞核的小淋巴細胞組成,但可發現數量不一、略微增大(最大可達中型)且具有染色質緻密細胞核的淋巴細胞。混雜有數量不一的嗜酸性球、組織球 (histiocytes)、漿細胞(從少數到小群聚不等)。B 細胞可排列成小聚集體,但罕見發現生發中心。可觀察到肉芽腫形成 (Granuloma formation)。可能有 T 淋巴細胞外滲 (exocytosis) 進入毛囊的上皮,但通常沒有淋巴細胞顯著外滲進入上方的濾泡間表皮(缺乏表皮趨向性 lack of epidermotropism)。免疫組織化學顯示,在大多數病例中,大部分小淋巴細胞為 CD4+ CD30− T 細胞。可能混雜有少數代表活化淋巴細胞的 CD30+ 母細胞。克隆性研究顯示大多數 T 細胞 PSLs 為多株浸潤,但已有具克隆性 T 細胞的 PSLs 被報告,並稱為克隆性 PSLs (clonal PSLs)。其中某些病例可能進展為明顯的淋巴瘤,可能更代表淋巴瘤生成 (lymphomagenesis) 的極早期階段,而非真正的 PSLs。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
結節性 T 細胞與混合型 PSLs 的鑑別診斷包括依據世界衛生組織 (World Health Organization, WHO) 2008 年分類及修訂後的 WHO 2016 年分類的皮膚 CD4+ 小型/中型 T 細胞淋巴瘤/淋巴增生性疾病 (cutaneous CD4+ small-/medium-sized T-cell lymphoma/lymphoproliferative disorder, CD4+ SMT-LPD),後者顯示重疊的組織學與免疫表型特徵。²⁸,²⁹ CD4+ SMT-LPD 通常也以單一病灶表現,大多位於頭頸部區域,並顯示惰性 (indolent) 病程(圖 120-5)。由於結節性 T 細胞 PSL 與皮膚 CD4+ SMT-LPD 無法基於臨床、組織病理或表型特徵確切區分,某些作者認為它們代表同一過程。因此,在更新的 WHO 2016 年分類中引入了涵蓋性名詞「皮膚 CD4+ 小型/中型 T 細胞淋巴增生性疾病」,以強調此過程的惰性本質。原本被認為是區別性標記的程序性細胞死亡蛋白 1(programmed cell death protein 1, PD-1)的表現,並無診斷價值。結節性 T 細胞 PSLs 應與腫瘤期 (tumor stage) 的蕈狀肉芽腫 (mycosis fungoides, MF) 相區別,因為腫瘤期 MF 罕見可能以小型至中型 T 細胞表現,而無顯著數量的大型細胞。然而,在 MF 中,小型與中型細胞顯示出比 CD4+ SMT-LPD 更高程度的核異型。MF、CD4+ SMT-LPD 與 T 細胞 PSL 之間的區別必須基於臨床表現:MF 在腫瘤之前有斑塊 (patches) 與斑片 (plaques),相對地,T 細胞 PSL 大多為單一結節而無先前的斑塊與斑片。鑑別診斷還包括血管免疫母細胞性 T 細胞淋巴瘤 (angioimmunoblastic T-cell lymphoma, AITL) 的續發性皮膚浸潤,其中小型 CD4+ 與 PD-1+ T 細胞伴隨顯著數量的 B 細胞。具有 B 症狀 (B symptoms) 的臨床背景、血清學發現、放射學分期檢查所示的淋巴結侵犯、AITL 浸潤中的高增生率,以及在某些 AITL 病例中與 Epstein-Barr 病毒的關聯,都是鑑別 AITL 與結節性 T 細胞 PSL 的有用發現。在發炎性皮膚疾病中,必須考慮紅斑性狼瘡 (lupus erythematosus)(尤其是腫脹型 tumidus type),它也可表現為緻密的真皮淋巴細胞浸潤。然而,在這些病例中,存在毛囊上皮交界處的空泡化 (vacuolization) 與間質性黏蛋白沉積 (interstitial mucin deposits)。此外,臨床表現也可與結節性 T 細胞 PSLs 相區別。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
T 細胞與混合型 PSL 在去除潛在病因後可能自發消退,但可能持續較長時間(數個月或數年)。若切片後未觀察到自發消退,則手術切除、冷凍療法、雷射治療、局部/病灶內類固醇或干擾素代表治療選項。³⁰ T 細胞 PSL 通常不會復發,除非潛在刺激因子持續存在(如藥物)。如同 B 細胞 PSL,避免潛在刺激因子是避免復發的有效預防措施。
CD30+ T 細胞假性淋巴瘤 (CD30+ T-CELL PSEUDOLYMPHOMA)
CD30+ PSL 代表皮膚 T 細胞 PSL 的一種組織學亞型,其特徵為存在中型至大型非典型 CD30+ T 細胞。³¹,³²
富 T 細胞的假性淋巴瘤性浸潤伴隨混雜 CD30+ 細胞,已在淋巴瘤樣藥物疹 (lymphomatoid drug eruptions)、結節性疥瘡 (nodular scabies) 與節肢動物叮咬反應,以及病毒感染(尤其是副痘病毒相關疾病,如羊痘 Orf disease³³ 與擠奶者結節 milker nodule),以及傳染性軟疣 (molluscum contagiosum) 與疱疹病毒 (herpes virus) 感染的背景下被報告(綜述見參考文獻 31)。在非感染性疾病中,CD30+ 假性淋巴瘤性浸潤已被描述於藥物疹、化膿性汗腺炎 (hidradenitis) 與珊瑚 (corals) 造成的傷害中。³⁴
在 CD30+ PSL 中,免疫組織化學顯示增大(即中型至甚至大型)、母細胞樣 (blast-like) 的 CD30+ 細胞,通常作為單個單位散布於整個浸潤中,該浸潤在其他方面則由小型 T 細胞主導(圖 120-6)。與 CD30+ 淋巴瘤相比,CD30+ PSLs 在大多數病例中不帶有克隆性 T 細胞族群。由於存在增大的 CD30+ 細胞,必須將淋巴瘤樣丘疹病 (lymphomatoid papulosis)(尤其是組織學 A 型)與皮膚退行性大細胞淋巴瘤 (cutaneous anaplastic large cell lymphoma, ALCL) 列為鑑別診斷。與淋巴瘤樣丘疹病與 ALCL 相比,CD30+ PSL 中的 CD30+ 細胞通常不排列成聚集體。此外,顯著數量的 B 細胞與漿細胞支持反應性過程,即 PSL,因為在淋巴瘤樣丘疹病與 ALCL 中它們通常缺如或僅以少量存在。此外,與淋巴瘤樣丘疹病相比,CD30+ PSL 中 CD30+ 細胞的 5hmC 表現得以保留,並可作為額外的診斷標記。³⁵
CD30+ PSL 的治療針對疾病的潛在病因。
假性淋巴瘤性毛囊炎 (PSEUDOLYMPHOMATOUS FOLLICULITIS)
此 PSL 變異型於 1988 年由 Kibbi 及其同事首次描述。³⁶ 它表現為位於臉部的單一結節,直徑小於 1.5 cm,與皮膚 CD4+ SMT-LPD 中所見的臨床表現相似或完全相同。³⁷ 已描述一種粟粒狀型的 B 細胞假性淋巴瘤性毛囊炎。¹⁹ 組織學上,半數病例帶有結節性 B 細胞 PSLs 的特徵,其餘病例帶有結節性 T 細胞 PSLs 的特徵。淋巴細胞浸潤位於整個真皮,可能延伸至皮下組織,並排列於毛囊周圍(圖 120-7)。可能存在無浸潤帶 (Grenz zone)。有淋巴細胞外滲進入毛囊,常顯示增寬的上皮。³⁸ 在半數病例中,可發現非典型淋巴細胞。半數病例帶有組織球的聚集體。在所有病例中都辨識出混雜大量表現 CD1a 與 S-100 的樹突狀細胞。³⁷ 其病程與治療類似其他形式的結節性 PSL 與 CD4+ SMT-LPD。
其他形式的結節性假性淋巴瘤 (OTHER FORMS OF NODULAR PSEUDOLYMPHOMA)
僅憑臨床基礎無法辨別 T 細胞、B 細胞或混合型 PSL。此外,某些病原可能誘發 T 細胞與 B 細胞 PSL,因此致病因子與 PSL 組成之間無法建立明確的對應關係。儘管如此,臨床背景可能提供關於 PSL 病因的重要資訊,尤其是對於那些由刺青或金屬(穿洞、耳環)所致的病例,或由暴露於水蛭與針灸所引起的 PSL。
持續性結節性節肢動物叮咬反應與結節性疥瘡 (PERSISTENT NODULAR ARTHROPOD BITE REACTIONS AND NODULAR SCABIES)
在這兩種病況中,都可發現長期存在、搔癢的丘疹與結節,好發於肘部、軀幹、生殖器,以及腋窩與鼠蹊皺褶 (inguinal folds)。病灶傾向呈紅棕色,可能有抓痕 (excoriated)。在緻密真皮淋巴細胞浸潤上方的表皮中,罕見發現蟎蟲或糞粒 (scybala)。遲發型過敏反應 (delayed-type hypersensitivity reaction) 被認為是富 T 細胞浸潤形成的潛在觸發因子。組織學上,表皮常顯示伴有棘層肥厚 (acanthosis) 與角化不全過度 (hyperparakeratosis) 的癢疹性反應 (pruriginous reaction) 特徵。有一個楔形 (wedge-shaped) 真皮浸潤,主要由小淋巴細胞組成,混雜有嗜酸性球與偶見的漿細胞(圖 120-8)。火焰圖形 (flame figures) 的形成可見於特別是含有大量嗜酸性球的病例。在浸潤的中心,可發現高度嗜酸性的膠原束 (hypereosinophilic collagen bundles)。血管顯示飽滿的內皮 (plump endothelia)。在大多數結節性節肢動物叮咬反應中,浸潤由 T 細胞主導。罕見可見富 B 細胞型伴生發中心。可能混雜有中型甚至大型、外觀非典型的 CD30+ 淋巴細胞(見「CD30+ 假性淋巴瘤」一節)。³¹,³²,³⁹ 在這些病例中,區分 CD30+ 假性淋巴瘤與淋巴瘤樣丘疹病可能具挑戰性。結節性節肢動物叮咬反應與結節性疥瘡中沒有 T 細胞的克隆性重排。持續性節肢動物叮咬反應必須與誇大叮咬反應 (exaggerated bite reactions) 相區別,後者可發生於 B 細胞慢性淋巴球性白血病及其他白血病的病人。⁴⁰ 在這些病人中,可考慮進行血液學檢查。此外,淋巴瘤樣丘疹病(A 型)、何杰金氏淋巴瘤 (Hodgkin lymphoma) 與結節性續發性梅毒 (nodular secondary syphilis) 是組織學上的鑑別診斷。結節性節肢動物叮咬反應與結節性疥瘡的病灶可能長期存在並持續數個月。抗疥瘡治療無效。若未發生自發消退或局部類固醇無效,可考慮切除、病灶內皮質類固醇或局部免疫調節劑。⁴¹
水蛭 (Hirudo medicinalis) 治療引起的假性淋巴瘤 (PSEUDOLYMPHOMA CAUSED BY LEECHES)
病灶的分布對應於暴露於水蛭 (H. medicinalis) 的位置,水蛭常用於下背部或外傷或手術後曾治療血腫 (hematoma) 的區域(圖 120-9)。⁴² 已描述一種瀰漫型。⁴³ 觀察到 T 細胞與 B 細胞為主的型態。假設上,棲息於水蛭中的細菌在吸血餐 (blood meal) 期間轉移至宿主,並誘發假性淋巴瘤反應。
藥物引起的結節性假性淋巴瘤 (NODULAR PSEUDOLYMPHOMA CAUSED BY DRUGS)
由藥物引起的 PSL 顯示出多樣的臨床、組織學與表型表現。某些藥物(如抗痙攣藥)可能誘發結節性 PSL(圖 120-10),以及其他組織學表現(見下文)。²⁷,⁴⁴,⁴⁵ 臨床上,藥物相關的結節性 PSL 可能以單一或多發結節表現。⁴⁴
組織學上,藥物相關的結節性 PSL 表現為結節性 T 細胞或 B 細胞 PSL 的典型組織學特徵。⁴⁶,⁴⁷ 結節性 T 細胞 PSLs 中的淋巴細胞可能顯示輕微的核異型。增生率低,約為 10%。在 B 細胞 PSLs 中,可發現上述典型的免疫表型特徵。在 T 細胞 PSLs 中,免疫組織化學顯示以 CD4 淋巴細胞為主,並混雜數量不一的 CD30+ 淋巴細胞。⁴⁸ 未觀察到全 T 細胞標記 (pan–T-cell markers) 的喪失。在大多數病例中,沒有 T 細胞受體 γ 基因或 IgH 基因的單株重排。淋巴瘤樣藥物疹的診斷具挑戰性,尤其因為用藥開始至藥物疹發生之間的潛伏期可能非常長(數個月至數年)。鑑別診斷取決於表型。在 B 細胞結節性 PSL 中,鑑別診斷包括皮膚邊緣區淋巴瘤與濾泡中心淋巴瘤。在富 CD4+ T 細胞型的藥物相關結節性 PSL 中,鑑別診斷包括皮膚 CD4+ SMT-LPD、MF 與塞扎里症候群 (Sézary syndrome)。停用藥物是藥物相關 PSL 處置的第一步。若自發消退延遲,可考慮手術切除或病灶內注射皮質類固醇。必須注意的是,藥物相關的結節性 PSL 即使在停用致病藥物後仍可能持續數個月。再次暴露於該藥物可能引發 PSL 的復發。
藥物與疫苗注射部位的假性淋巴瘤 (PSEUDOLYMPHOMA AT DRUG AND VACCINE INJECTION SITES)
單一或多發 PSL 也可能在注射用以預防傳染病的疫苗後,或在注射用於去敏化的過敏原後發展出來。它們通常見於注射部位的皮下組織。⁴⁹ 組織學特徵顯示富 B 細胞浸潤伴反應性生發中心。在浸潤的中心,可見組織球的聚集。⁵⁰,⁵¹ 組織球顯示具有顆粒狀灰藍色外觀的細胞質,這似乎代表氫氧化鋁 (aluminium hydroxide) 結合的疫苗(圖 120-11)。⁵²
與刺青或金屬相關的假性淋巴瘤 (PSEUDOLYMPHOMA IN ASSOCIATION WITH TATTOOS OR METALS)
在刺青中產生並由耳環或穿洞中的金屬所引起的假性淋巴瘤性浸潤,具有相似的特徵。⁵³,⁵⁴ 在刺青中,淋巴細胞浸潤大多產生並侷限於紅色刺青染料 (red tattoo dye) 的區域(圖 120-12)。硃砂 (Cinnabar),一種硫化汞 (mercuric sulfide),是紅色刺青最常用的染料。此外,也已在藍色或綠色刺青中觀察到 PSLs,這些刺青使用鈷與鉻鹽 (cobalt and chrome salts)。對刺青染料的 PSL 可被視為一種遲發型過敏反應。臨床上,大多在刺青的一個區塊內發展出丘疹、結節或斑塊樣浸潤。它們通常無症狀。刺青染料的施用與 PSL 發生之間的潛伏期變化極大,範圍從數個月到數年。⁵⁵ 組織學上,存在一個苔癬樣 (lichenoid) 成分,伴有帶狀的淺層浸潤、交界處變化與淋巴細胞外滲,以及在中層與深層真皮中混雜嗜酸性球的較深淋巴細胞浸潤,導致假性淋巴瘤性外觀。⁵⁶ 淋巴細胞為小型,不顯示顯著的核異型。對汞或刺青染料中其他物質的貼布試驗 (Patch test) 可能顯示遲發型過敏反應,但試驗也可能為陰性。⁵⁵ 治療具挑戰性,包括病灶內注射皮質類固醇、雷射治療,或在存在小型丘疹結節性病灶時進行切除。
具富淋巴細胞浸潤而模擬淋巴瘤的感染 (INFECTIONS WITH LYMPHOCYTE-RICH INFILTRATES SIMULATING LYMPHOMA)
各種感染,尤其是由病毒與寄生蟲引起者,可能顯示緻密的富淋巴細胞浸潤,從而模擬淋巴瘤。在寄生蟲感染中,皮膚利什曼病 (cutaneous leishmaniasis) 藉由其由大量淋巴細胞(包括漿細胞與組織球)組成的結節性浸潤,在組織學上模擬淋巴瘤。缺乏核異型,以及藉由特殊染色、免疫組織化學或分子技術偵測到寄生蟲,使皮膚利什曼病能與皮膚淋巴瘤相區別。皮膚利什曼病不僅組織學發現可模擬淋巴瘤,臨床方面偶爾也可類似 B 細胞 PSL。⁵⁷,⁵⁸
在單純疱疹病毒 (herpes simplex virus) 與水痘帶狀疱疹病毒 (varicella zoster virus) 感染中,偶爾可觀察到富淋巴細胞浸潤而無特徵性 (pathognomonic) 的上皮變化,並被稱為隱匿性疱疹 (herpes incognito)。在隱匿性疱疹中可發現具有略微增大且外觀非典型染色質緻密細胞核的淋巴細胞,以及增大的 CD30+ 淋巴細胞,使這些浸潤容易被誤認為淋巴瘤浸潤。藉由免疫組織化學偵測病毒抗原與/或藉由 PCR 偵測病毒 DNA,可將這些浸潤辨識為疱疹病毒相關的 T 細胞反應。副痘病毒 (Parapoxvirus) 感染可能誘發細胞形態變化以及浸潤 T 細胞表現 CD30,這使得將它們與退行性大細胞淋巴瘤背景下的多形性淋巴細胞 (pleomorphic lymphocytes) 相區別具挑戰性(見圖 120-6)。³¹,³² 副痘病毒感染典型的上皮變化伴包涵體 (inclusion bodies)、T 細胞標記未喪失,以及缺乏 T 細胞受體 γ 基因的單株重排,有助於將這些浸潤與皮膚 T 細胞淋巴瘤相區別。然而,診斷基於藉由免疫組織化學、電子顯微鏡 (electron microscopy) 或 PCR 偵測到病毒。
假性淋巴瘤——蕈狀肉芽腫的組織學模擬者 (PSEUDOLYMPHOMAS- HISTOLOGIC MYCOSIS FUNGOIDES SIMULATORS)
pseudo-MF 一詞描述一群病因不同、在組織學上模擬 MF 的疾病。由於組織學評估提示 MF 或另一種形式的表皮趨向性皮膚 T 細胞淋巴瘤,臨床病理相關性對於避免將組織學發現誤判為淋巴瘤,並將發現歸屬於一種獨特的反應性皮膚疾病(如 PSL)至關重要。組織學上,本節描述的疾病共同具有一個真皮的、帶狀或血管周圍的浸潤,主要由小淋巴細胞組成,這些細胞顯示外滲進入表皮並可能表現出輕微的核異型,從而模擬表皮趨向性皮膚 T 細胞淋巴瘤 (cutaneous T-cell lymphoma, CTCL)。表型分析顯示以 CD4+ 或 CD8+ 細胞為主。此外,在某些 pseudo-MF 病例中可見 CD30 的不同表現。如同其他形式的 PSL,淋巴細胞在大多數病例中為多株性。在某些疾病(如急性痘瘡樣苔癬樣糠疹 pityriasis lichenoides et varioliformis acuta)中,可發現相當比例的克隆性 T 細胞,但這並不代表惡性或進展為淋巴瘤的風險。鑑別診斷主要包括 MF 或塞扎里症候群。在以 CD8+ 浸潤為主的病例中,鑑別診斷包括 CD8+ MF、塞扎里症候群、皮膚 CD8+ 侵襲性表皮趨向性細胞毒性 T 細胞淋巴瘤 (cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma),以及淋巴瘤樣丘疹病(D 型與 E 型)。深層核異型、以中型至大型細胞為主、全 T 細胞標記喪失,以及 T 細胞受體基因的單株重排,是支持 CTCL 的發現。如同其他形式的 PSL,治療著重於避免暴露於致病病原,例如淋巴瘤樣接觸性皮膚炎中的過敏原與淋巴瘤樣藥物疹中的藥物。此外,使用局部皮質類固醇或免疫調節劑與紫外線 (ultraviolet, UV) 光療法。全身性類固醇與免疫抑制藥物(如環孢素 cyclosporine)可能適用於嚴重形式的 PSL。
淋巴瘤樣接觸性皮膚炎 (LYMPHOMATOID CONTACT DERMATITIS)
淋巴瘤樣接觸性皮膚炎 (Lymphomatoid contact dermatitis, LCD) 是一種在組織學上模擬 MF 的慢性接觸性皮膚炎。⁵⁹
各種抗原,包括硫酸鎳 (nickel sulfate)、金 (gold)、鋅 (zinc)、對苯二胺 (paraphenylenediamine)、紡織染料 (textile dye) 與數種其他抗原,已被辨識為元兇。⁶⁰ LCD 大多發生於成人,並侵犯兩種性別。臨床上,LCD 表現為濕疹性紅斑與脫屑性丘疹、斑塊、斑片,以及罕見情況下的紅皮症(圖 120-13)。病灶會搔癢。組織病理學上,存在一個淺層帶狀浸潤,伴有不同程度的淋巴細胞外滲進入表皮,表皮可能顯示海綿狀水腫 (spongiosis) 或海綿狀水疱 (spongiotic vesicles)。可發現表皮內蘭格罕細胞 (Langerhans cells) 的聚集(即所謂的假性 Pautrier 聚集體 pseudo Pautrier collections)。某些淋巴細胞可顯示略微迴旋的細胞核 (slightly convoluted nuclei),但核異型不顯著。
混雜有嗜酸性球。CD4+ 與 CD8+ 淋巴細胞的比例不明顯,但可混雜略微增大的 CD30+ 細胞,代表活化淋巴細胞。LCD 與 MF 的鑑別基於:存在不同程度的海綿狀水腫、缺乏淋巴細胞的顯著異型、CD4 與 CD8 比例不明顯,以及 LCD 中無 T 細胞標記喪失。已有 LCD 中單株 T 細胞的報告,但並不代表淋巴瘤。藉由貼布試驗記錄對過敏原的致敏化 (Sensitization),是證明 LCD 診斷的必要診斷標準。⁶¹
LCD 的病程為慢性,尤其在無法避免暴露於過敏原時。治療遵循其他形式接觸性皮膚炎治療的一般建議,避免暴露於過敏原,並大多藉由局部皮質類固醇或局部免疫調節劑抑制對過敏原的免疫反應。此外,以 UV 光為基礎的策略或全身性免疫抑制可能有效。
淋巴瘤樣藥物反應 (LYMPHOMATOID DRUG REACTION)
除了其結節型外,藥物相關的 PSL 常以斑丘疹 (maculopapular) 或丘疹性疹表現。已描述大量誘發各種形式淋巴瘤樣藥物疹的藥物類別。⁸,⁶²
在此形式的淋巴瘤樣藥物反應中,可發現上層真皮的帶狀浸潤,伴有不同程度的淋巴細胞外滲。⁶³
真皮表皮交界處 (dermoepidermal junction) 可能存在空泡變化 (Vacuolar alteration) 與凋亡的角質細胞 (apoptotic keratinocytes)。淋巴細胞可能顯示不規則的核輪廓(即核異型)(圖 120-14 A, B)。嗜酸性球常見,但也可能缺如。免疫組織化學顯示以 CD4+ 或 CD8+ 淋巴細胞為主,並混雜數量不一的 CD30+ 淋巴細胞(圖 120-14C)。⁴⁸ 未觀察到全 T 細胞標記的喪失。鑑別診斷包括表皮趨向性 CTCL,尤其是 MF 與塞扎里症候群。淋巴細胞沿交界帶 (junctional zone) 排列、淋巴細胞的核異型、全 T 細胞標記喪失,以及 T 細胞受體 γ 基因的單株重排,支持 MF 或塞扎里症候群。此外,在塞扎里症候群中發現大部分淋巴細胞表現 PD-1 與 TOX(胸腺細胞選擇相關高遷移率族盒因子 thymocyte selection-associated high-mobility group box factor),但在發炎性紅皮症 (inflammatory erythrodermas) 中則沒有。⁶⁴ 顯示淋巴細胞外滲的 CD8+ 浸潤病例,必須與 CD8+ MF(斑片與斑塊)、CD8+ 淋巴瘤樣丘疹病(尤其是 D 型或 E 型)與急性痘瘡樣苔癬樣糠疹 (pityriasis lichenoides et varioliformis acuta, PLEVA) 相區別。藥物相關的 CD30+ PSL 模擬淋巴瘤樣丘疹病(A 型或 B 型)。⁶⁵ 偶爾,淋巴瘤樣藥物疹中的淋巴細胞表現出顯著的核異型,以致僅憑組織學要與淋巴瘤區別非常具挑戰性甚至不可能。臨床病理相關性與病史對於在藥物相關 PSL 中達成最終診斷是必要的。如同藥物相關 PSL 的結節型,停用藥物是處置的必要步驟。淋巴瘤樣藥物疹可能持續數個月,再次暴露於該藥物可能引發 PSL 的復發。全身性、病灶內或局部類固醇用於治療。
光化性網狀細胞增多症 (ACTINIC RETICULOID)
光化性網狀細胞增多症 (Actinic reticuloid) 代表一種慢性多因子皮膚炎,對廣泛波長譜系具有嚴重的光敏感性 (photosensitivity),在組織學上模擬表皮趨向性 CTCL。⁵
臨床發現 (CLINICAL FINDINGS)
它大多侵犯中年與老年男性。⁶⁶ 它表現為持續性的紅斑性苔癬樣丘疹與斑塊,位於光照部位的皮膚,特別是臉部與頸部。在某些病人中,臉部的浸潤導致獅面樣 (facies leonina-like) 外觀(圖 120-15)。可觀察到進展為紅皮症。隨著時間,通常會發生苔癬化 (Lichenification) 與糜爛 (erosions)。皮膚病灶伴隨劇烈搔癢。
實驗室檢查 (LABORATORY TESTS)
組織學上,存在表皮的乾癬樣增生 (psoriasiform hyperplasia)、輕微海綿狀水腫,以及伴有局部角化不全 (focal parakeratosis) 的緻密正角化 (compact orthokeratosis)。在上層真皮中,有一個以血管周圍但也呈間質性分布排列的浸潤,由小淋巴細胞、嗜酸性球與漿細胞組成。在乳頭狀真皮 (papillary dermis) 中可發現排列成垂直條紋的粗膠原束。可能存在多核纖維母細胞 (Multinucleated fibroblasts)。淋巴細胞可能顯示略微非典型的細胞核並外滲進入上方表皮。免疫組織化學顯示以 CD8+ T 細胞為主。⁶⁷
在周邊血液中,CD8+ T 細胞數量增加(CD4 與 CD8 比例倒置 reversed CD4-to-CD8 ratio)是光化性網狀細胞增多症的特徵,尤其在紅皮症病人中。⁶⁶ 循環的非典型淋巴細胞顯示鋸齒狀細胞核 (indented nuclei)。光照測試 (Phototesting),包括陽性的光貼布試驗 (photo patch tests),顯示對一種或多種過敏原的致敏化。
診斷與鑑別診斷 (DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS)
光化性網狀細胞增多症的診斷基於:在光照部位皮膚存在持續性浸潤的斑塊與丘疹、對廣泛波長譜系的光敏感性,以及組織學發現以 CD8+ 略微非典型淋巴細胞為主與不同程度的外滲,以及周邊血液中存在循環的 CD8+ 淋巴細胞。⁶⁷
基於臨床與/或組織學基礎,必須考慮 MF 與/或塞扎里症候群。CD8+ MF 顯示更顯著的表皮趨向性與淋巴細胞的核異型。此外,CD8+ MF 通常位於 UV 保護的身體部位,與光化性網狀細胞增多症分布於光照部位形成對比。特別是在以紅皮症表現的光化性網狀細胞增多症病人中,塞扎里症候群代表一個重要的鑑別診斷。與以 CD8+ 淋巴細胞為主的光化性網狀細胞增多症相比,塞扎里症候群的特徵為皮膚與周邊血液中的克隆性 CD4+ 非典型淋巴細胞、淋巴結病變、禿髮 (alopecia) 與掌蹠角化過度 (palmoplantar hyperkeratosis)。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
光化性網狀細胞增多症顯示長期高度慢性的病程。除了以防曬乳進行 UV 光防護外,可使用局部類固醇與局部免疫調節劑,但顯示出不一的療效。在全身性藥物中,環孢素可能有效。已有報告以 UV-B 照射進行光硬化 (Photo hardening) 以誘發光耐受性,在一個系列中產生良好反應。⁶⁷
免疫缺陷中的 CD8+ T 細胞假性淋巴瘤 (CD8+ T-CELL PSEUDOLYMPHOMA IN IMMUNODEFICIENCY)
在免疫缺陷病人,特別是 HIV 感染病人中,可能發展出模擬 MF 的 CD8+ 淋巴細胞浸潤。截至本文撰寫時,已報告少於 50 例 HIV 感染病人的 CD8+ 浸潤。這些浸潤的病因與致病機轉大多未知,但可能與嚴重的免疫缺陷有關,因為皮膚 CD8+ 浸潤發生於 CD4+ 細胞數量低的晚期 HIV 感染。已描述各種臨床表現,包括播散性、常為搔癢的丘疹,或瀰漫性、輕度搔癢的丘疹性疹。⁶⁸,⁶⁹ 此外,觀察到掌蹠角化過度與紅皮症。CD8+ 淋巴細胞的真皮浸潤伴隨 CD8+ 淋巴細胞外滲進入表皮。淋巴細胞可能顯示輕微的核異型。分子研究顯示多株性本質。皮膚 CD8+ 浸潤本身的病程在大多數病人中為惰性,但被視為整體預後不良的徵象,這主要由 CD4+ T 細胞的嚴重淋巴球減少 (lymphopenia) 所決定。在大多數病人中,接受高效能抗反轉錄病毒療法 (highly active antiretroviral therapy) 時可達到緩解,而在中度免疫抑制的病人中,已顯示甲胺喋呤 (methotrexate) 有效。⁷⁰,⁷¹
此病況似乎不僅限於 HIV 感染病人,因為近期在一名腎臟移植受贈者中也描述了類似的特徵。⁷²
Borrelia 相關 T 細胞假性淋巴瘤 (BORRELIA-ASSOCIATED T-CELL PSEUDOLYMPHOMA)
除了遊走性紅斑 (erythema migrans) 之外,皮膚的 Borrelia 感染最常以 B 細胞為主的浸潤(包括漿細胞)表現,例如皮膚淋巴細胞瘤。近期已報告具有富 T 細胞浸潤的 B. burgdorferi 感染。⁷³,⁷⁴
真皮 T 細胞浸潤呈帶狀或瀰漫狀,並顯示局部表皮趨向性,淋巴細胞沿交界帶排列。淋巴細胞顯示輕微的核異型。此外,可觀察到浸潤的次要間質性組織球成分。模擬 MF 的組織學特徵與類似 MF 的斑片臨床表現,在某些受影響的個體中都代表一個診斷陷阱。其他臨床表現包括遊走性紅斑或慢性萎縮性肢端皮膚炎 (acrodermatitis chronica atrophicans) 的典型發現。以抗生素治療導致緩解。
Ofuji 丘疹性紅皮症 (PAPULOERYTHRODERMA OFUJI)
Ofuji 丘疹性紅皮症 (Papuloerythroderma Ofuji) 是一種罕見的搔癢性紅皮症性皮膚病,臨床上可模擬 CTCL。⁷⁵ 已報告與藥物、何杰金氏淋巴瘤、內臟惡性腫瘤 (visceral malignancies) 與免疫缺陷症候群的關聯。在某些病人中,Ofuji 丘疹性紅皮症被描述為 MF 的一種表現⁷⁶,⁷⁷,在其他病人中則作為伴隨 MF 的疾病。⁷⁸
Ofuji 丘疹性紅皮症最初在日本病人中被描述,但類似的病例已從其他地理區域被報告。中位年齡為 70 歲。它在男性中比女性中更常發生。臨床上,Ofuji 丘疹性紅皮症表現為播散性實心丘疹。融合的棕色丘疹呈平頂狀 (flat-topped),主要見於四肢的屈側面。腋窩、鼠蹊區、肘前窩與膕窩 (antecubital and popliteal fossae) 以及腹部的大皺褶不受侵犯(「躺椅 deckchair」徵;圖 120-16)。⁷⁹
組織學顯示濕疹性特徵,伴有棘層肥厚、海綿狀水腫與角化不全過度,並可發現混雜漿細胞、嗜酸性球與嗜中性球的真皮淋巴細胞浸潤。可觀察到嗜中性球與嗜酸性球的外滲。⁸⁰ 免疫組織化學顯示真皮中有大量樹突狀細胞與成熟 T 細胞。最常見的異常實驗室發現為嗜酸性球增多與血清 IgE 濃度升高。應搜尋免疫缺陷,特別是 HIV 感染。臨床鑑別診斷包括塞扎里症候群。Ofuji 丘疹性紅皮症可與塞扎里症候群區別,因為與塞扎里症候群不同,Ofuji 丘疹性紅皮症缺乏淋巴細胞的核異型與循環的非典型淋巴細胞。若無潛在惡性腫瘤,Ofuji 丘疹性紅皮症的預後良好。已描述包括補骨脂素加紫外線 A(psoralen and ultraviolet A, PUVA)、浴 PUVA (bath PUVA)、UV-B 合併局部類固醇、依曲替酯 (etretinate)、Re-PUVA、口服類固醇與環孢素在內的有效治療方法。¹¹⁻¹⁴
作為 CTCL 模擬者的發炎性疾病與克隆性皮膚炎 (INFLAMMATORY DISORDERS AS CTCL AND CLONAL DERMATITIS)
在其他章節討論的各種疾病,其特徵為富 T 細胞浸潤與淋巴細胞外滲進入表皮,使這些疾病容易被誤判為表皮趨向性 CTCL。這些疾病包括扁平苔癬 (lichen planus)、硬化萎縮性苔癬 (lichen sclerosus et atrophicans)、色素性紫癜性皮膚炎 (pigmented purpuric dermatitis) 與苔癬樣糠疹。⁶⁵,⁸¹⁻⁸³ 此外,具有富淋巴細胞真皮與/或皮下浸潤的發炎性疾病,例如紅斑性狼瘡(尤其是腫脹型)與狼瘡性脂膜炎 (lupus panniculitis),必須與其他形式的 CTCL(包括皮下脂膜炎樣 T 細胞淋巴瘤 subcutaneous panniculitis-like T-cell lymphoma)相區別。
發炎性皮膚疾病中 T 細胞克隆的偵測 (DETECTION OF T-CELL CLONES IN INFLAMMATORY SKIN DISEASES)
評估克隆性的高度敏感方法,例如 PCR 結合溫度梯度凝膠電泳 (temperature gradient gel electrophoresis) 或變性梯度凝膠電泳 (denaturing gradient gel electrophoresis) 與自動化高解析度片段分析 (automated high-resolution fragment analysis),能夠偵測克隆性 T 細胞,這些細胞可能僅佔所有浸潤細胞的 1% 之少。在上述發炎性皮膚病況中,已在某些病例中發現克隆性 T 細胞族群,例如在扁平苔癬與硬化萎縮性苔癬中,分別在 6%(扁平苔癬)與 13%(硬化萎縮性苔癬)的病例中發現克隆性 T 細胞。值得注意的是,在帶有克隆性 T 細胞的苔癬樣糠疹中,常發現 TCR 基因的單株重排,高達 60% 的病例。⁸²,⁸⁴ 這些 T 細胞克隆的意義尚不清楚。作為富淋巴細胞浸潤診斷檢查的結果,偵測到克隆性 T 細胞族群不能作為診斷 CTCL 的充分發現。¹³
浸潤中存在克隆性 T 細胞可能僅代表在某種免疫反應背景下一個或多個 T 細胞克隆的優勢,此免疫反應可能由某些抗原誘發與維持。與皮膚淋巴瘤在疾病演進過程中持續存在單一 T 細胞克隆相比,發炎性皮膚疾病中的 T 細胞克隆是短暫的,並隨時間改變。因此,在發炎性疾病中偵測到克隆性 T 細胞強調了將克隆性檢測結果與臨床、組織學與免疫表型發現相關聯以達成最終診斷的必要性。⁸⁵
克隆性皮膚炎 (CLONAL DERMATITIS)
除了上述特徵明確且獨特、在某些病例中可能帶有克隆性 T 細胞的發炎性皮膚疾病之外,採用 PCR 技術的克隆性研究導致辨識出帶有 T 細胞克隆的慢性非特異性皮膚炎病例。這些病例被稱為「克隆性皮膚炎 (clonal dermatitis)」。⁸⁶ 約 25% 的克隆性皮膚炎病例在 5 年內進展為明顯的 CTCL。⁸⁶ 這些觀察顯示,至少某些「克隆性皮膚炎」病例可能代表 CTCL 的前驅病灶。因此,克隆性皮膚炎將是 CTCL 最早的表現之一,帶有優勢 T 細胞克隆,但缺乏 CTCL 的診斷性組織學特徵。
皮膚淋巴細胞浸潤與可觸及之弓形遊走性紅斑 (LYMPHOCYTIC INFILTRATION OF THE SKIN AND PALPABLE ARCIFORME MIGRATORY ERYTHEMA)
皮膚淋巴細胞浸潤 Jessner-Kanof (lymphocytic infiltration of the skin Jessner-Kanof, LIS) 與 Clark 可觸及之弓形遊走性紅斑 (palpable arciform migratory erythema of Clark, PAME) 被某些作者視為 T 細胞 PSLs,被其他作者視為狼瘡樣疹 (lupus-like eruptions)。PAME 的臨床表現導致其命名為「浸潤性環狀紅斑發展為大型遷移性病灶」。⁸⁷ 軀幹是好發部位。組織學顯示緻密的血管周圍與附屬器周圍 (periadnexal) 主要為淋巴細胞的浸潤。無漿細胞混雜,且缺乏如紅斑性狼瘡中所見的間質性黏蛋白沉積。免疫組織化學顯示由 T 細胞主導的浸潤,混雜有 B 細胞與組織球。淋巴細胞為多株性。組織學與 LIS 的發現非常相似。⁸⁸ 表型上,LIS 中的浸潤大多由 CD8+ 淋巴細胞組成。⁸⁹ 由於 PAME 與 LIS 在臨床表現、組織學與病程上的相似性,我們認為它們代表同一過程。兩種過程都可能對局部類固醇、口服抗生素與 UV-A1 有反應。⁸⁸ 有復發的傾向。
其他具富淋巴細胞浸潤的疾病 (OTHER DISORDERS WITH LYMPHOCYTE-RICH INFILTRATES)
肢端假性淋巴瘤性血管角化瘤 (ACRAL PSEUDOLYMPHOMATOUS ANGIOKERATOMA)
肢端假性淋巴瘤性血管角化瘤 (Acral pseudolymphomatous angiokeratoma, APA) 最初被描述為發生於兒童(因此原名為兒童肢端假性淋巴瘤性血管角化瘤 acral pseudolymphomatous angiokeratoma of childhood [APACHE]),但已顯示它也侵犯成人。⁹⁰⁻⁹² 某些作者認為這些病灶代表持續性節肢動物叮咬反應,而其他作者將 APA 歸類為具有顯著淋巴細胞浸潤的良性血管過程,即一種形式的皮膚 PSL。⁹⁰,⁹² 因此,已提出丘疹性假性淋巴瘤 (papular pseudolymphoma) 一詞。⁹²
臨床上,APA 表現為單側性爆發、成簇的紅色至紫紅色血管瘤樣丘疹(直徑:1 至 5 mm),位於肢端部位,即手與腳(圖 120-17)。⁹⁰ 可見病灶的融合。可能發生指甲縱裂 (longitudinal splitting of the nails)、甲剝離 (onycholysis) 與指甲變形。APA 的組織學顯示小淋巴細胞、嗜酸性球、漿細胞與組織球的緻密浸潤。在浸潤內有由飽滿內皮所襯覆的厚壁血管 (thick-walled vessels)。診斷基於臨床病理相關性。伴隨的浸潤由小型、分化良好的淋巴細胞組成,混雜少數漿細胞與組織球,包括組織球性巨細胞 (histiocytic giant cells)。在某些情況下,可能存在少數嗜酸性球。免疫組織化學證明淋巴細胞為多株性 T 細胞與 B 細胞,B 細胞偶爾形成小聚集體。APACHE 的組織學鑑別診斷包括淋巴瘤樣藥物疹與節肢動物叮咬反應。具有相似組織學發現但臨床上表現為單一、息肉狀、紅斑性丘疹者,已在皮膚富 T 細胞血管瘤樣息肉狀 PSL (T-cell–rich angiomatoid polypoid PSL of the skin) 一詞下被描述。⁹³ 它與 APACHE 的疾病分類學關係仍有待確定。APACHE 是一個良性過程。治療後未報告復發。若未治療,病灶可能消退、顯示消長 (waning-and-waxing) 病程,或維持數個月或數年不變。已使用刮除術 (curettage)、病灶內皮質類固醇,或封閉下的高效價局部皮質類固醇來破壞病灶以進行治療,並導致完全緩解。⁹¹
淋巴漿細胞性斑塊 (LYMPHOPLASMACYTIC PLAQUE)
淋巴漿細胞性斑塊 (Lymphoplasmacytic plaque, LPP) 是一種近期描述的罕見皮膚疾病,被認為是一種病因不明的 PSL 形式。它最初在兒童中被報告,以脛前區 (pretibial region) 為好發部位,並被稱為兒童孤立性原發性皮膚漿細胞增多症 (isolated primary cutaneous plasmacytosis in children) 與脛前 LPP (pretibial LPP)。⁹⁴,⁹⁵ 近期一項研究指出,LPP 也可侵犯成人並侵犯軀幹與手臂。因此,我們偏好使用淋巴漿細胞性斑塊一詞。⁹⁶ 有女性偏多現象。臨床上,LPP 顯示獨特的表現,特徵為長期存在的斑塊,或界限分明、常呈線狀排列的紅色與棕色丘疹與斑塊(圖 120-18)。⁹⁶,⁹⁷ 組織學顯示緻密的真皮淋巴組織球性浸潤 (lymphohistiocytic infiltrate),伴有大量多株漿細胞,佔整個浸潤的高達 25%。浸潤呈淺層、帶狀或深層結節狀與間質性,常在附屬器結構或血管周圍增強(圖 120-19)。可見硬化膠原束 (sclerotic collagen bundles) 周圍的間質性組織球性肉芽腫(即所謂的假性玫瑰花結 pseudorosettes),伴有組織球性巨細胞與血管數量增加。⁹⁶
LPP 必須與其他含有漿細胞與組織球的病況相區別,例如原發性皮膚漿細胞增多症 (primary cutaneous plasmocytosis)、皮膚淋巴細胞瘤、皮膚邊緣區淋巴瘤、原發性與續發性皮膚漿細胞瘤 (cutaneous plasmocytoma),以及感染(如黴菌、分枝桿菌 mycobacterial、密螺旋體 treponemal)。應始終藉由血清學、特殊染色與分子病理技術排除感染過程。LPP 的診斷基於臨床病理相關性。LPP 與 APA 顯示重疊的臨床與組織學特徵,例如女性偏多與好發於四肢(尤其是腿部),以及淋巴組織球性浸潤與多株漿細胞。已有人推測這兩種疾病(APACHE 與 LPP)屬於同一疾病譜系,並代表一種具有顯著血管成分的富漿細胞 PSL。LPP 的病程長期存在,可達數年。手術切除是第一線治療,尤其因為其他治療無效。⁹⁶
皮膚漿細胞增多症 (CUTANEOUS PLASMOCYTOSIS)
皮膚漿細胞增多症 (Cutaneous plasmocytosis) 是一種罕見疾病,已在亞洲國家(特別是日本)被報告。它大多侵犯成人。⁹⁸,⁹⁹ 其特徵為遍布全身的多發、棕色小斑塊與結節。組織學顯示主要由成熟多株漿細胞組成的真皮浸潤。⁹⁸,⁹⁹ 在某些病人中,可發現全身性侵犯的徵象(如淋巴結病變、肝脾腫大 hepatosplenomegaly、高丙種球蛋白血症 hypergammaglobulinemia、血清中第 6 型介白素 interleukin-6 濃度升高,以及紅血球沉降率 erythrocyte sedimentation rate 升高)。治療包括 PUVA、類固醇與化療方法。
皮膚發炎性假瘤 (CUTANEOUS INFLAMMATORY PSEUDOTUMOR)
發炎性假瘤 (inflammatory pseudotumor) 一詞已被用來描述 2 種如今被認為是不同疾病實體的病況,即漿細胞肉芽腫 (plasma cell granuloma) 與發炎性肌纖維母細胞瘤 (inflammatory myofibroblastic tumor)。¹⁰⁰ 發炎性假瘤是一種病因不明的良性過程。臨床上,它表現為質地堅實的長期存在真皮或皮下結節。組織學上,在漿細胞肉芽腫中可發現小淋巴細胞、排列成片狀的大量漿細胞與組織球的界限分明結節性浸潤。可能發現反應性生發中心。在發炎性肌纖維母細胞瘤中,特徵性發現為顯著的肌纖維母細胞紡錘細胞 (spindle-cell) 成分,伴有 ALK(退行性淋巴瘤激酶 anaplastic lymphoma kinase)的表現,以及伴漿細胞的淋巴細胞浸潤。手術切除導致完全緩解。
伴嗜酸性球增多之血管淋巴樣增生 (ANGIOLYMPHOID HYPERPLASIA WITH EOSINOPHILIA)
伴嗜酸性球增多之血管淋巴樣增生 (Angiolymphoid hyperplasia with eosinophilia, ALHE) 代表伴有顯著內皮的血管增生,伴隨緻密的 T 細胞與 B 細胞浸潤連同嗜酸性球。它於 1969 年由 Wells 與 Whimster 首次描述。¹⁰¹ 如今 ALHE 普遍被視為一種血管增生過程,源於存在顯著、形狀怪異的血管與上皮樣內皮 (epithelioid endothelia),導致其別名同義名稱為上皮樣血管瘤 (epithelioid hemangioma)。¹⁰²,¹⁰³ 大多數作者認為 ALHE 是一種對組織損傷與血管分流 (vascular shunts) 形成的反應性增生過程。ALHE 與木村病 (Kimura disease) 的關係仍有爭議,但大多數專家認為它們是不同的臨床病理實體。木村病的病灶傾向較大且較深,並伴隨周邊血液嗜酸性球增多以及淋巴結腫大。
流行病學與致病機轉 (EPIDEMIOLOGY AND PATHOGENESIS)
ALHE 與木村病侵犯兩種性別,無性別優勢。¹⁰³ 大多數病人處於生命的第三或第四個十年。動靜脈分流 (Arteriovenous shunts) 可能是 ALHE 與木村病中所見血管形成的潛在原因。
臨床特徵 (CLINICAL FEATURES)
ALHE 表現為血管瘤樣的粉紅色至紅棕色丘疹或結節,最常見於頭頸部,尤其是臉部與耳朵,但也發生於四肢與生殖器區域(圖 120-20)。病灶可能無症狀,或伴隨搔癢、疼痛或出血。
組織病理學、免疫表型分析與分子檢查 (HISTOPATHOLOGY, IMMUNOPHENOTYPING, AND MOLECULAR TESTS)
真皮與/或皮下結節由微血管後血管 (postcapillary vessels) 的增生與緻密的淋巴細胞浸潤組成(圖 120-21)。血管由顯著的內皮細胞所襯覆,這些細胞具有圓形或卵圓形細胞核、豐富的嗜酸性細胞質,以及上皮樣或鞋釘樣 (hobnail) 外觀。浸潤含有小淋巴細胞、反應性生發中心與嗜酸性球。¹⁰⁴ 免疫組織化學顯示內皮表現 CD31 與 ERG,但對足糖萼蛋白 (podoplanin/D2-40) 無反應性。免疫組織化學研究證明大部分淋巴細胞為 T 細胞譜系。混雜的 B 細胞可能形成淋巴濾泡。在木村病中,伴有生發中心的淋巴樣成分超過血管的增生。在 ALHE 與木村病中都已偵測到克隆性 T 細胞。¹⁰⁵,¹⁰⁶ 在 ALHE 中,大多數這些病例顯示出遷延 (protracted) 且抗治療的病程,伴有復發。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括化膿性肉芽腫 (pyogenic granuloma),其顯示血管的小葉狀增生,伴隨混合細胞浸潤。在淋巴瘤中,應考慮成人 T 細胞淋巴瘤/白血病 (adult T-cell lymphoma/leukemia, AITL) 的特異性浸潤,因為它們也顯示血管數量增加,以及由具有輕微核異型的 T 細胞與 B 細胞組成的浸潤。然而,AITL 中的 T 細胞為非典型、增生活躍,並表現濾泡輔助 T 細胞 (follicular helper T cells) 的標記,例如 PD-1、ICOS、bcl-6、CD10 與 CXCL-13。
臨床病程、預後與治療 (CLINICAL COURSE, PROGNOSIS, AND TREATMENT)
ALHE 病灶無自發消退。復發常見,特別是在手術切除後。¹⁰³,¹⁰⁷ 已使用手術、冷凍療法與雷射燒蝕,以及甲胺喋呤與干擾素-α 來治療 ALHE。¹⁰⁸ 若可辨識出動靜脈分流為潛在觸發因子,則應藉由例如栓塞術 (embolization) 移除分流。
Castleman 病 (CASTLEMAN DISEASE)
Castleman 病是一種良性淋巴增生性疾病,有 2 種變異型:透明血管型 (hyaline vascular type) 與漿細胞變異型 (plasma cell variant)。漿細胞變異型可與 POEMS 症候群(多發性神經病變 polyneuropathy、器官腫大 organomegaly、內分泌病變 endocrinopathy、M 蛋白 m protein 與皮膚變化 skin changes)相關。¹⁰⁹ Castleman 病最常位於淋巴結(縱隔 mediastinal 或全身性),罕見侵犯皮膚。¹¹⁰ 組織學上,透明血管型較常見,以同心螺旋狀 (concentrically whorled) 模式圍繞生發中心的小淋巴細胞為特徵。此型在較年輕病人中較常見。漿細胞型顯示大濾泡與富含血管與漿細胞的濾泡間區。具有局限性結外病灶的 Castleman 病預後良好。治療取決於疾病的範圍,包括手術切除、放射與化療。
淋巴細胞的良性血管內增生 (BENIGN INTRAVASCULAR PROLIFERATION OF LYMPHOCYTES)
近期已報告淋巴母細胞的良性血管內增生(伴或不伴 CD30 表現)。此病況產生於具有發炎性皮膚疾病或皮膚外傷的區域。¹¹¹⁻¹¹³
致病機轉上,硬化性苔癬 (lichen sclerosus) 阻塞淋巴管並擾亂免疫細胞運輸 (immune cell trafficking),可能導致活化 CD30+ 淋巴細胞的累積。淋巴細胞為大型並具有母細胞樣形態(圖 120-22)。它們表現 T 細胞標記(CD3、CD4),在某些病例中表現 CD30。與 Epstein-Barr 病毒感染無關聯。克隆性研究顯示此過程的多株性本質。血管內淋巴瘤 (Intravascular lymphoma) 是最重要的鑑別診斷,因為它是一種具有各種表型形式(B 細胞、T 細胞或自然殺手/T 細胞)的侵襲性淋巴瘤。此外,淋巴樣細胞的良性血管內增生需與淋巴管內組織球增多症 (intralymphatic histiocytosis) 相區別,後者代表類風濕性關節炎 (rheumatoid arthritis) 或骨科金屬植入物 (orthopedic metal implants) 病人淋巴管腔內組織球的反應性增生。
總結 (SUMMARY)
皮膚 PSL 指的是一群在臨床上與/或組織學上模擬皮膚淋巴瘤的富淋巴細胞浸潤。¹¹⁵ 臨床病理相關性對於在皮膚 PSL 中達成最終診斷並將其與皮膚淋巴瘤相區別至關重要。消除感染性病原(如藉由抗生素或抗病毒藥物)或避免暴露於致病病原(如 LCD 與藥物反應),是處置的第一步,此外還有免疫調節劑(如皮質類固醇、UV 光治療)或破壞性方法(如手術切除、冷凍療法)。
圖譜 (FIGURES)
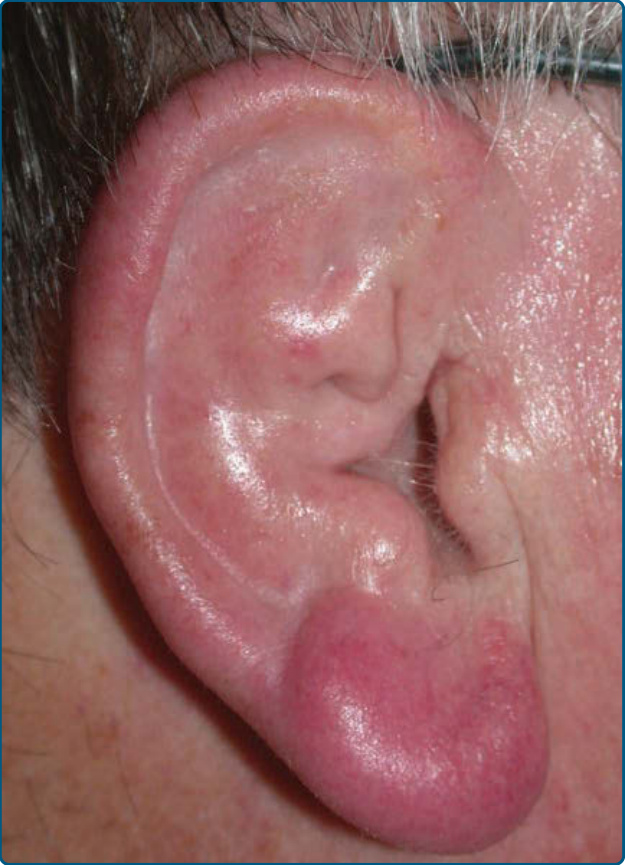
圖 120-1:Borrelia 相關之結節性 B 細胞淋巴瘤(皮膚淋巴細胞瘤 lymphocytoma cutis、皮膚良性淋巴腺病 lymphadenosis cutis benigna):耳垂上的紫紅色 (violaceous) 結節。
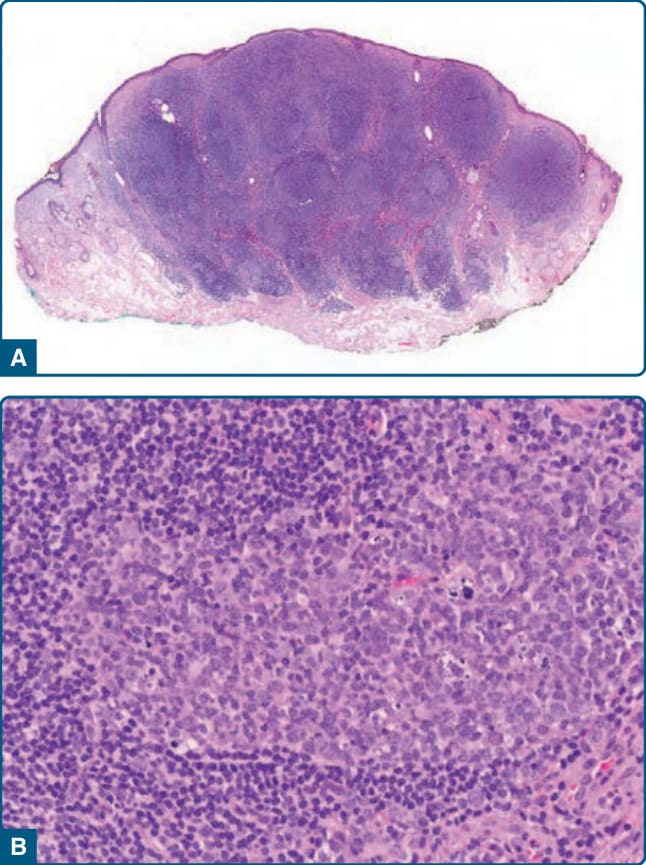
圖 120-2:Borrelia 相關 B 細胞假性淋巴瘤,組織學:結節性淋巴細胞浸潤 (A),伴有含可染體巨噬細胞 (tingible body macrophages) 的反應性生發中心 (B)。
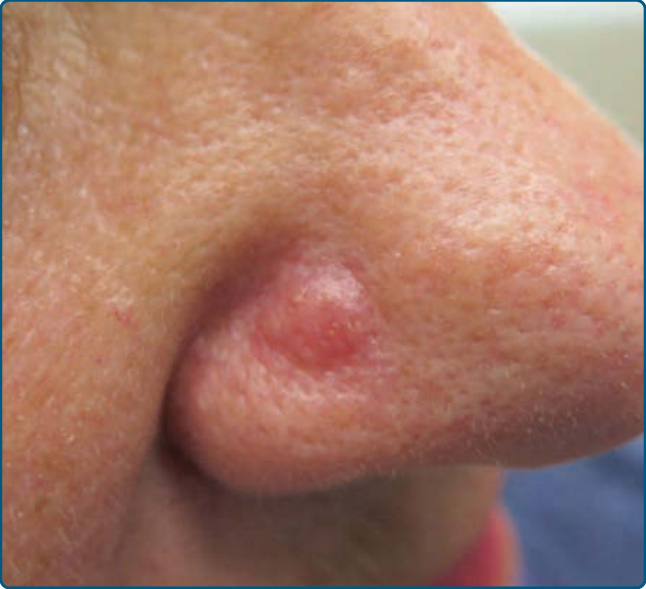
圖 120-3:結節性 T 細胞假性淋巴瘤:鼻部的紅斑性結節。
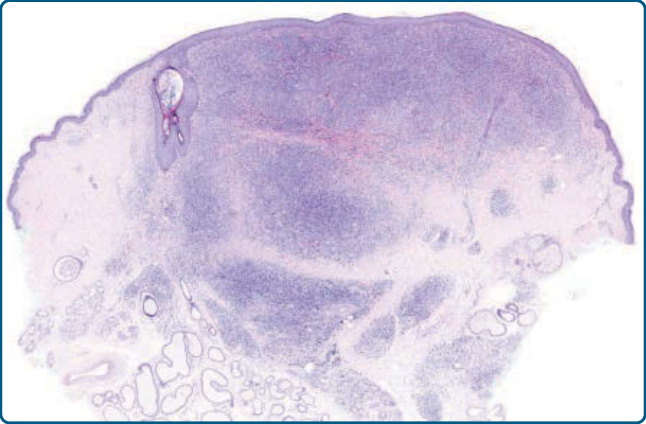
圖 120-4:結節性 T 細胞假性淋巴瘤,組織學:主要由小淋巴細胞組成的結節性浸潤。無濾泡存在。
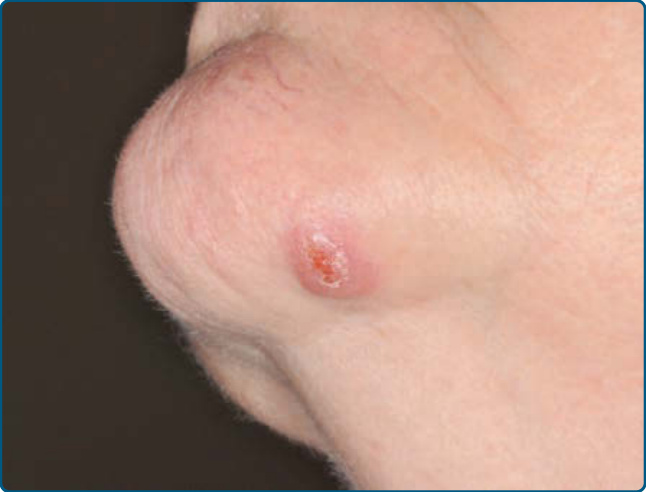
圖 120-5:皮膚 CD4+ 小型/中型 T 細胞淋巴增生性疾病 (cutaneous CD4+ small-/medium-sized T-cell lymphoproliferative disorder):下巴上的結節性病灶。
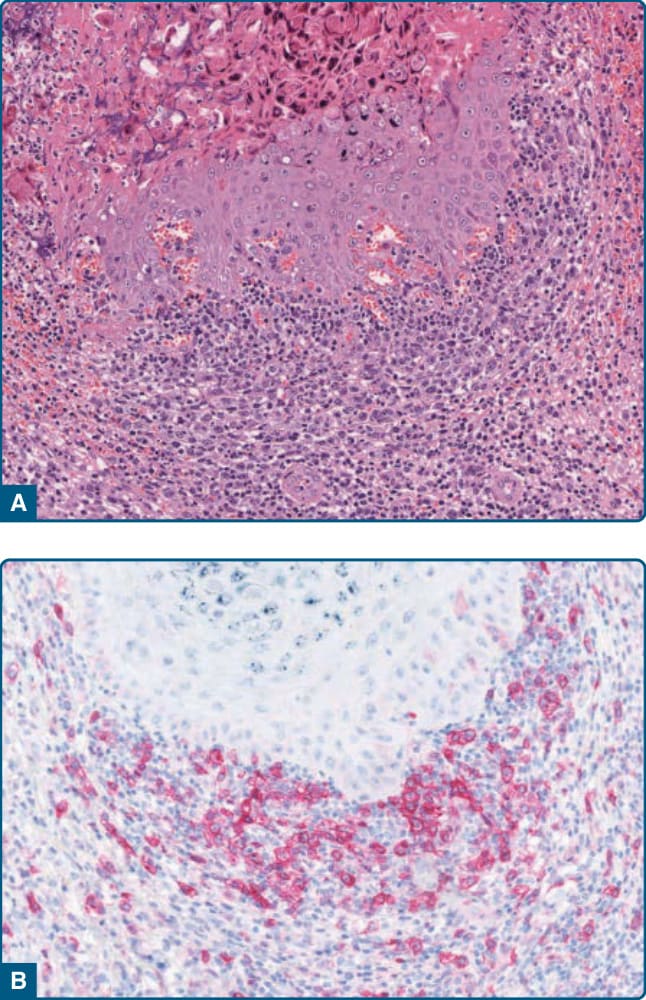
圖 120-6:CD30+ 假性淋巴瘤。A,傳染性軟疣 (molluscum contagiosum) 中非典型淋巴細胞的浸潤。B,中型至大型非典型淋巴細胞表現 CD30。
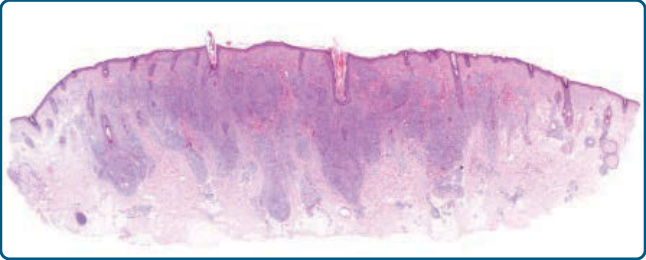
圖 120-7:假性淋巴瘤性毛囊炎,組織學:緻密的真皮淋巴細胞浸潤伴毛囊周圍增強 (perifollicular accentuation)。
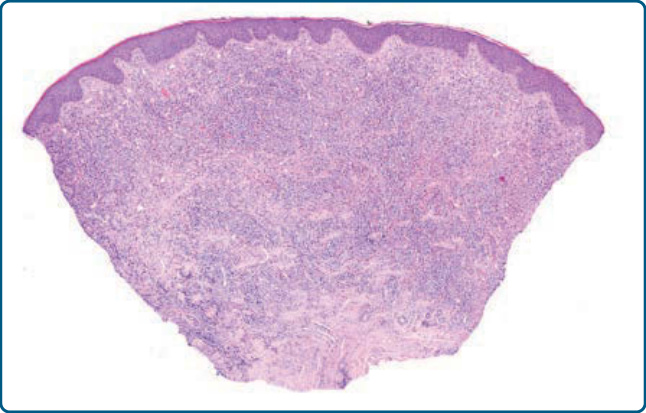
圖 120-8:結節性節肢動物叮咬反應,組織學:由淋巴細胞與大量嗜酸性球組成的楔形 (wedge-shaped) 真皮浸潤。
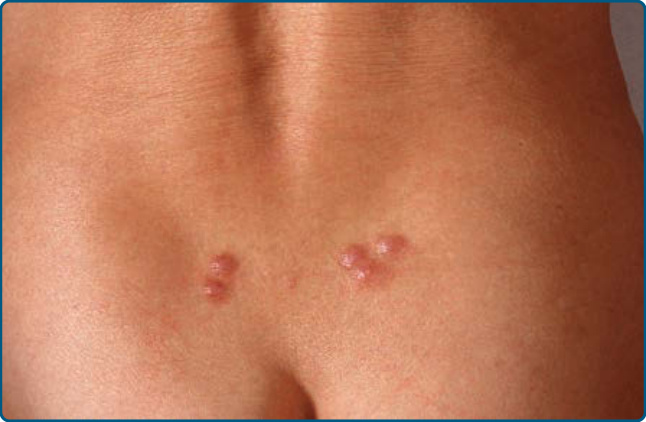
圖 120-9:水蛭 (Hirudo medicinalis) 引起的假性淋巴瘤:假性淋巴瘤的特徵性定位。
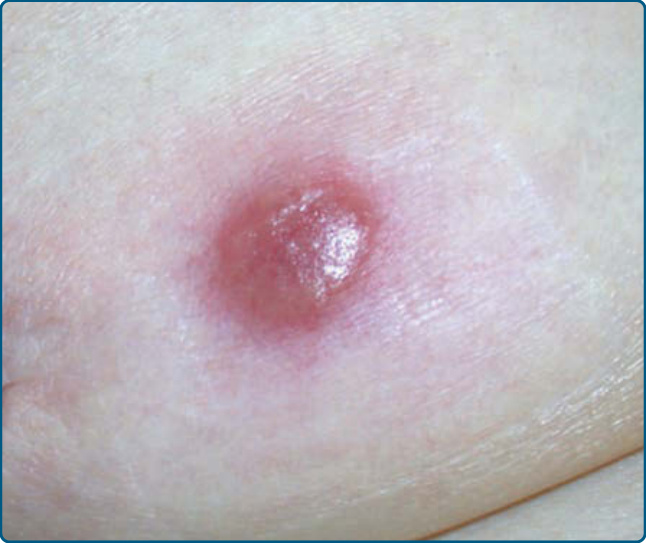
圖 120-10:抗痙攣藥(phenytoin)引起的結節性 T 細胞假性淋巴瘤:右乳房上的紅斑性結節。
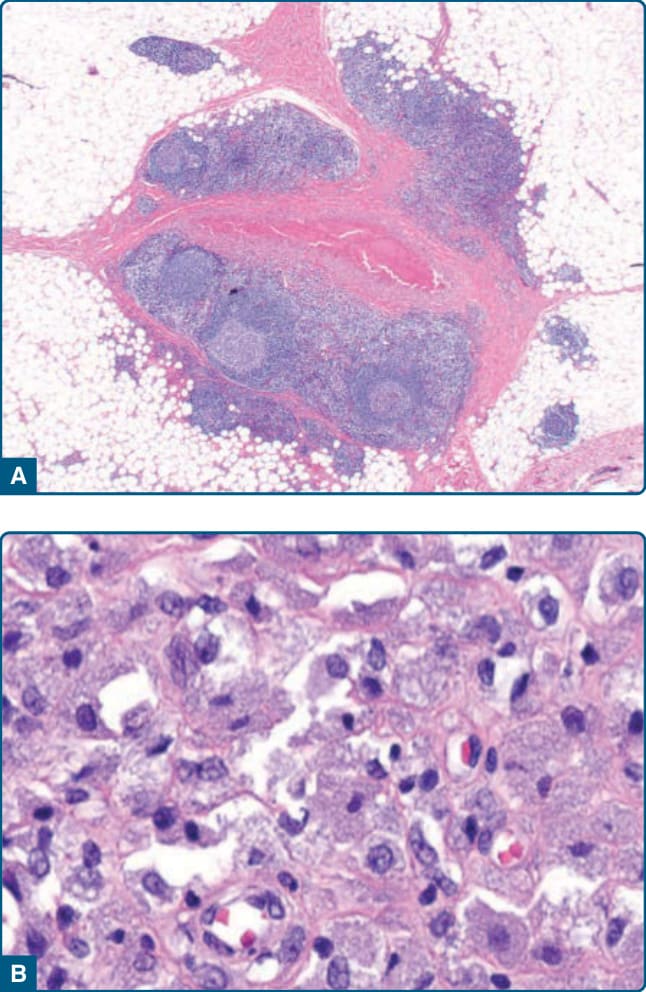
圖 120-11:過敏原注射引起的假性淋巴瘤:A,皮下富 B 細胞浸潤伴濾泡形成。注意中央富組織球浸潤的區域。B,組織球顯示含有氫氧化鋁 (aluminum hydroxide) 結合疫苗的灰色顆粒狀細胞質。
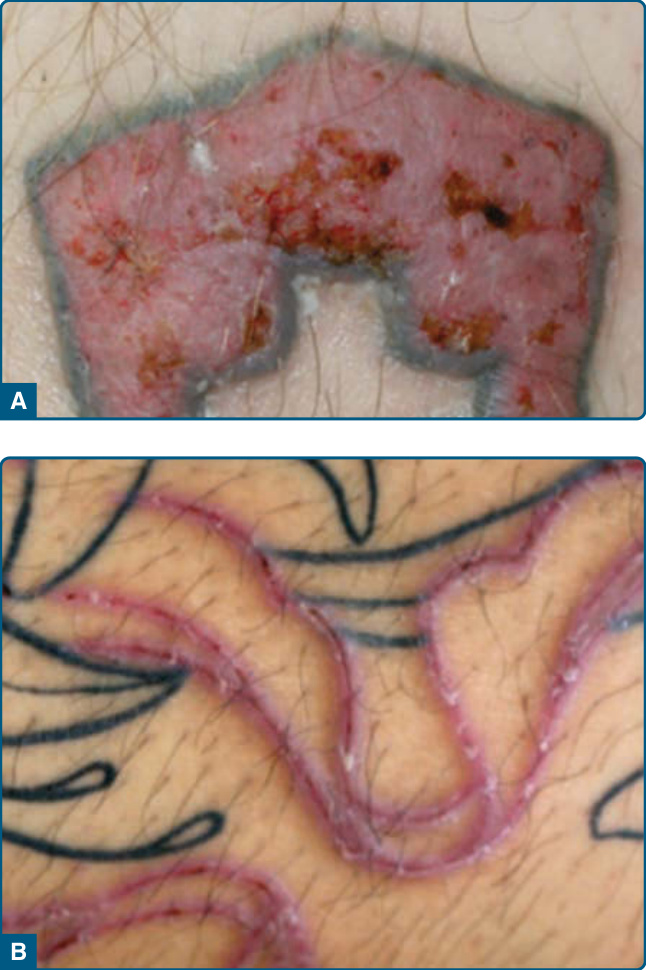
圖 120-12:A 與 B,與刺青相關的假性淋巴瘤:紅色刺青成分區域內的浸潤。
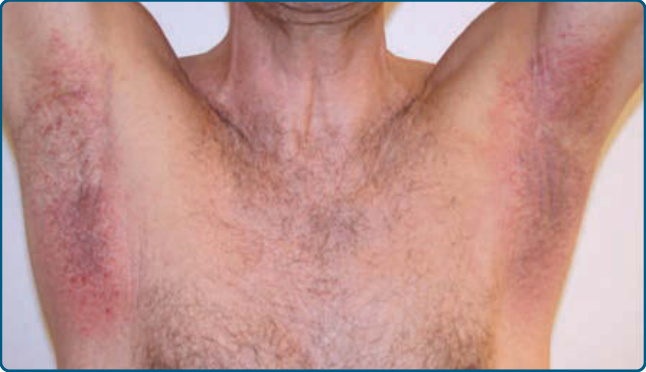
圖 120-13:淋巴瘤樣接觸性皮膚炎:由對香料成分的接觸性過敏所引起的雙側腋窩濕疹性病灶。
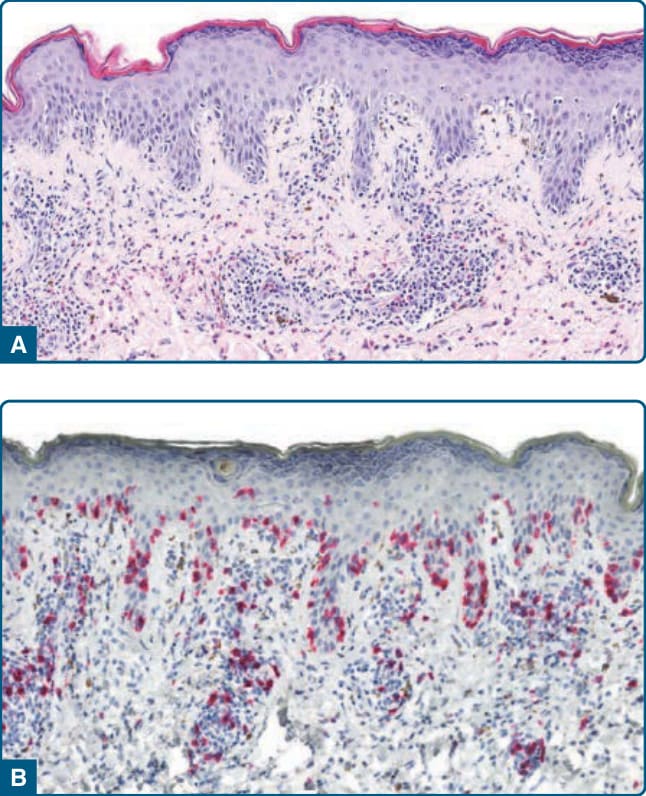
圖 120-14:淋巴瘤樣藥物反應,組織學:淋巴細胞浸潤伴淋巴細胞外滲進入表皮與淋巴細胞核異型 (A),伴 CD8 的表現 (B)。
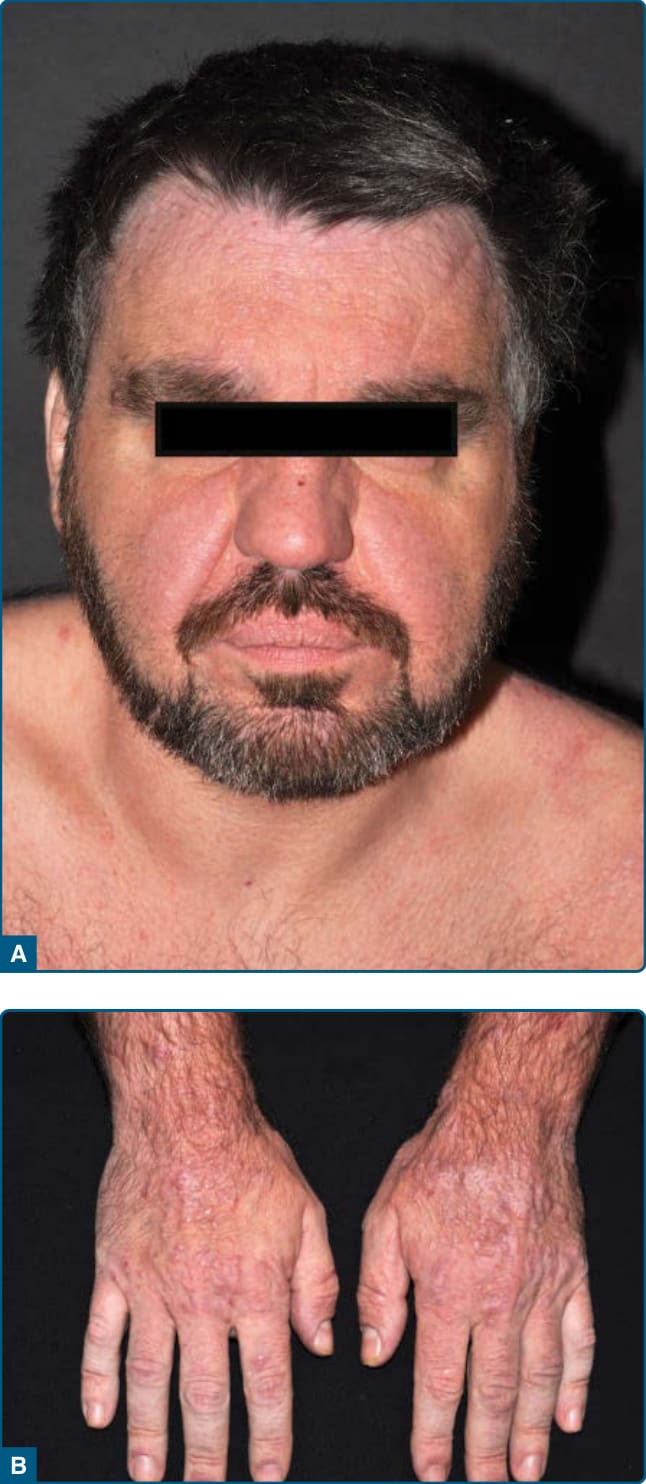
圖 120-15:光化性網狀細胞增多症:臉部的瀰漫性浸潤、紅斑與丘疹 (A) 以及手背上的丘疹 (B)。
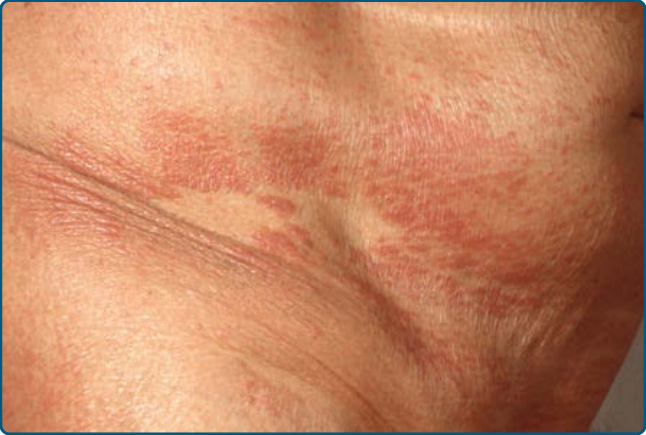
圖 120-16:Ofuji 丘疹性紅皮症:丘疹融合成斑塊與不受侵犯的區域。
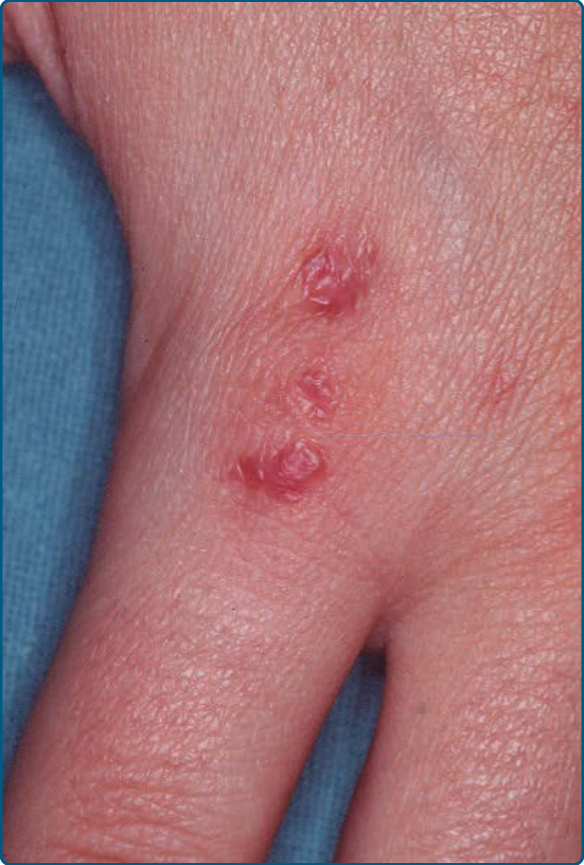
圖 120-17:兒童肢端假性淋巴瘤性血管角化瘤 (acral pseudolymphomatous angiokeratoma of childhood, APACHE):一名 8 歲女孩手部的群聚丘疹。(經 Prof. Dr. S. Lautenschlager, Zurich, Switzerland 許可使用。)
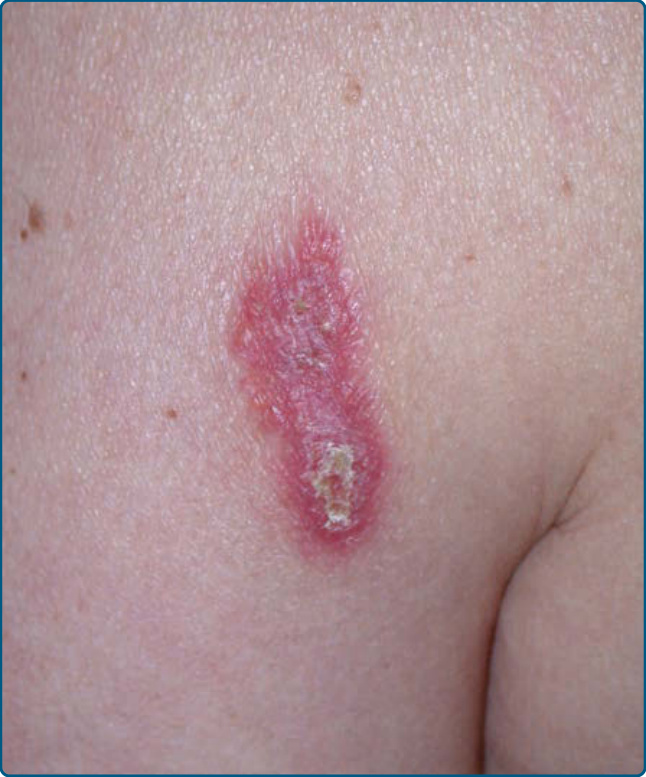
圖 120-18:淋巴漿細胞樣斑塊 (Lymphoplasmacytoid plaque):左上臂外側面的長期存在紅斑性斑塊。
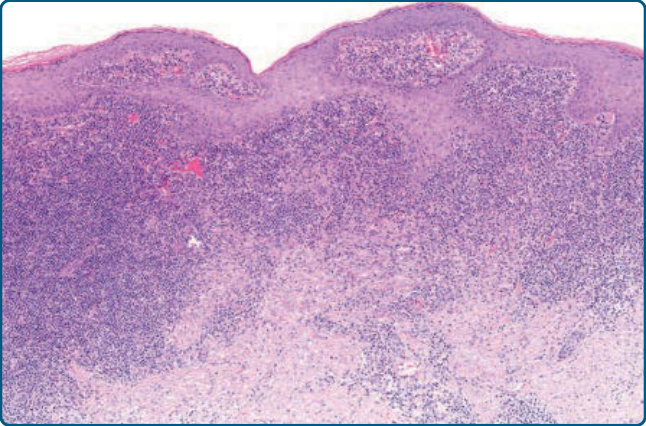
圖 120-19:淋巴漿細胞樣斑塊,組織學:表皮增生與淋巴細胞及組織球的緻密真皮浸潤。
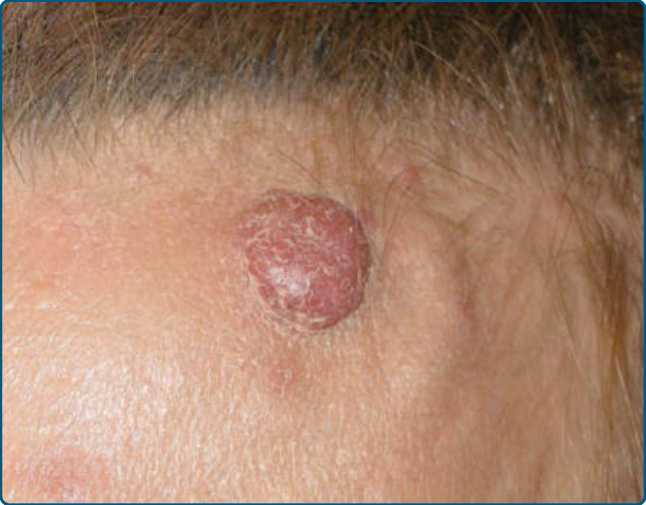
圖 120-20:伴嗜酸性球增多之血管淋巴樣增生:左前額的結節性浸潤。
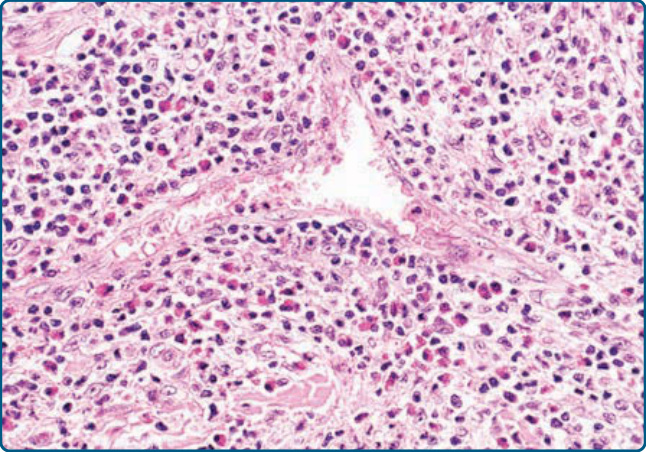
圖 120-21:伴嗜酸性球增多之血管淋巴樣增生,組織學:嵌於淋巴細胞與嗜酸性球緻密浸潤中、具有顯著內皮的血管。
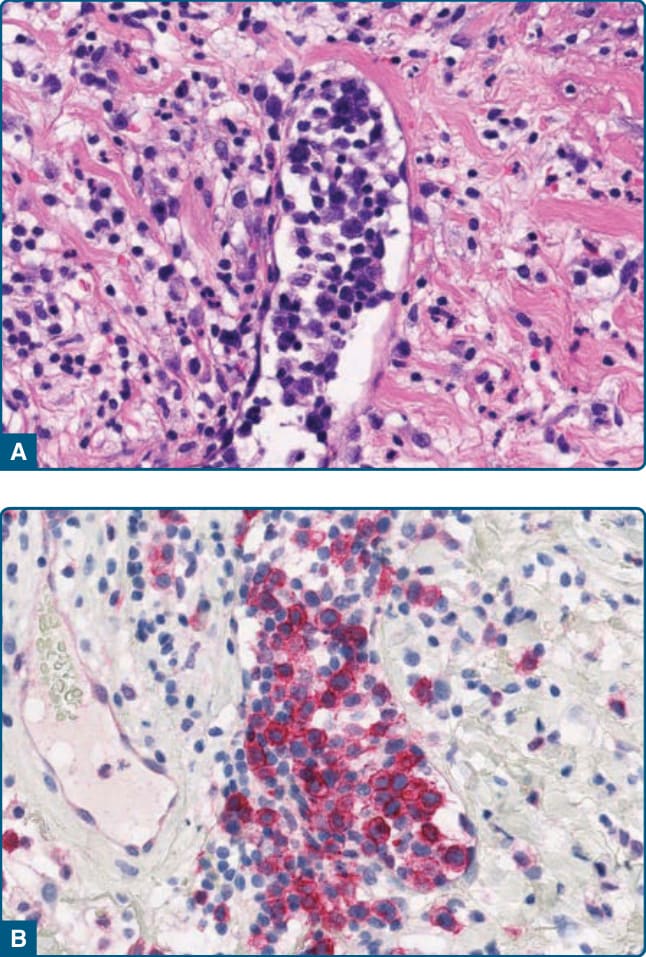
圖 120-22:血管內淋巴樣增生,組織學:淋巴管內母細胞樣淋巴樣細胞的累積 (A),伴 CD30 的表現 (B)。