皮膚淋巴瘤 (Cutaneous Lymphoma) 精華筆記
總論與分類
- 原發性皮膚淋巴瘤 (primary cutaneous lymphomas) 是一群異質性結外非何杰金氏淋巴瘤 (extranodal non-Hodgkin lymphomas),源自皮膚歸巢/常駐 T 或 B 淋巴球及血液皮膚性前驅腫瘤的惡性無性繁殖系轉化。
- 與結節性非何杰金氏淋巴瘤臨床行為、預後完全不同,需不同治療策略。分類採 WHO-EORTC 共識(2005,後為 2008 WHO 與 2016 修訂版基礎):先依細胞譜系,再依形態、免疫表型、遺傳與臨床症候群分類。
- 最常見的 7 種(4 種 CTCL:蕈樣肉芽腫 MF、Sézary 症候群、原發性皮膚退行性大細胞淋巴瘤、淋巴瘤樣丘疹病;3 種 CBCL:PCFCL、PCMZL、PCLBCL 腿型)約佔所有皮膚淋巴瘤的百分之九十。
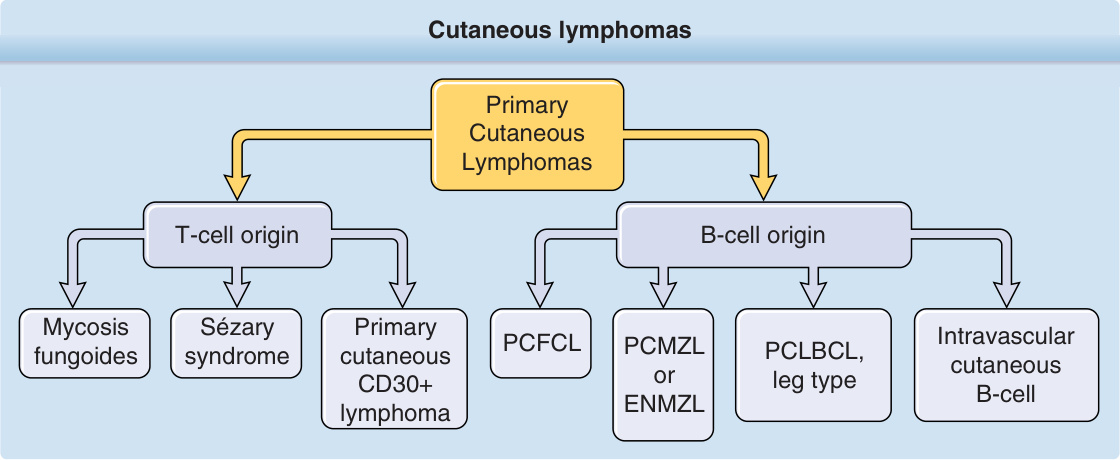
圖 119-1:皮膚淋巴瘤分類概觀(T 細胞與 B 細胞起源)。
流行病學
- CTCLs 是僅次於原發性胃腸道淋巴瘤的第二常見結外淋巴瘤群;發生率上升中,美國約每百萬人 6.4 例(1993–2002)至 7.7 例(2001–2005)。
- 發生率隨年齡顯著上升,診斷年齡中位數約 50 多歲,大於 70 歲者發生率上升約 4 倍。
- MF 與 Sézary 症候群約佔 CTCL 的百分之六十五;其次為原發性皮膚 CD30⁺ 淋巴增生性疾病(約百分之二十七)。
病因 (Etiology)
- 內生性:特定 HLA class II 等位基因(HLA-DRB111、DQB103)在 MF/Sézary 症候群顯著過度表現。
- 外生性(感染):HTLV-1(相關成人 T 細胞淋巴瘤/白血病)、EBV(相關 NK/T 細胞淋巴瘤、種痘樣水疱病樣淋巴增生,多見亞洲裔)已確認為部分皮膚淋巴瘤病因;MF/Sézary 症候群未證實病毒關聯。金黃色葡萄球菌 (Staphylococcus aureus) 超級抗原可能參與疾病惡化(其腸毒素 A 刺激 STAT3 活化與 IL-17 表現),但是否參與起始仍存疑。
- 外生性(環境/職業):玻璃製造工、陶工、造紙與木材業工人風險較高,但結果不一致。
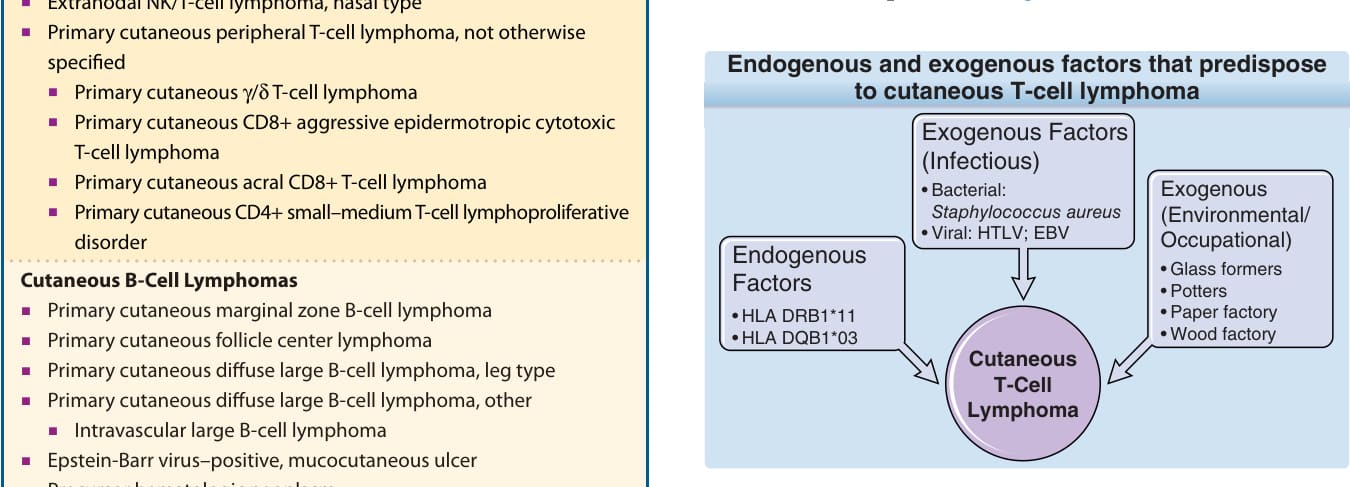
圖 119-2:促成皮膚 T 細胞淋巴瘤的內生性與外生性因素。
發病機轉 (Pathogenesis)
- CTCL 是皮膚歸巢 T 細胞的惡性腫瘤;淋巴瘤細胞累積於淺層真皮形成斑片/斑塊/腫瘤,進展期播散至血液、淋巴結與內臟。白血病型(Sézary 症候群)惡性 T 細胞可佔循環 T 淋巴球的百分之九十九以上。
- 正常 TCR 喪失與正常淋巴球消失導致免疫抑制與伺機性感染——最常見的疾病相關死因。
- 細胞起源差異:MF 惡性 T 細胞源自非再循環的組織常駐記憶細胞 (TRM),傾向侷限皮膚;Sézary 症候群源自中央記憶細胞 (TCM),可在血液/淋巴結/皮膚再循環並抗凋亡。
- 分子致病:復發性缺失 10q、17p(含 TP53、CDKN2a)與擴增 8q(含 MYC)、17q;染色質修飾基因頻繁缺失(ARID1A、CTCF、DNMT3A);TCR 訊息傳遞路徑多處突變(CD28、PLCG1、PRKCQ、NFkB2、STAT5B、ZEB1 等)。
- JAK/STAT 路徑為核心:早期 γc 鏈細胞激素(IL-2、IL-7、IL-15 經 JAK1/JAK3)驅動 STAT3/STAT5 活化 → 經 miRNA-155 致 Th1 朝 Th2 轉變、抑制腫瘤抑制因子 miRNA-22。STAT3 活化促 IL-17、IL-21(自分泌迴路)、抗凋亡 bcl-2 與致癌 miRNA-21,為標靶 JAK/STAT 提供依據。IL-13 為 CTCL(尤其 Sézary 變異型)的自分泌生長因子。
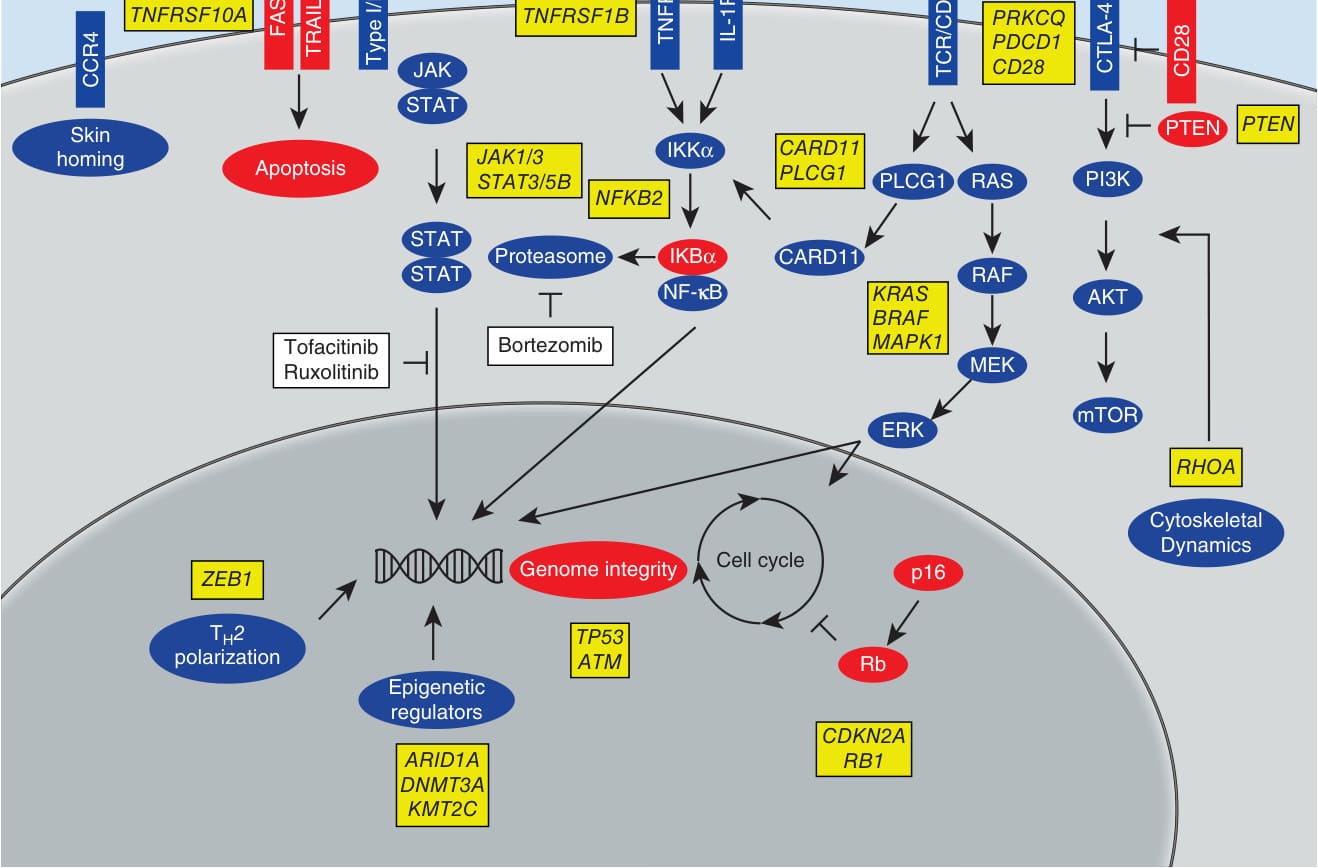
圖 119-3:皮膚 T 細胞淋巴瘤的訊息傳遞事件(JAK-STAT、NF-κB、PI3K/RAS/MAPK 等路徑)。
蕈樣肉芽腫 (Mycosis Fungoides, MF)
- 定義/流病:最常見原發性皮膚淋巴瘤,約佔所有皮膚淋巴瘤的百分之四十;中至晚成年發病(診斷中位數 55–60 歲),男女比 2:1。
- 臨床(分期):斑片期、斑塊期、腫瘤期可並存。
- 斑片期:紅斑脫屑斑/斑片,邊界清楚,色橘至暗紫紅;好發非日照部位(「泳褲區」、間擦部位)。常先有 10–20 年「慢性皮膚炎」病史,易誤為接觸性/異位性皮膚炎、乾癬、濕疹。對常規治療有抗性者應多處切片。
- 斑塊期:邊界銳利、脫屑、隆起、暗紅至紫紅、不等硬化;可融合呈環形/弧形/匐行性邊緣或中央清除。
- 腫瘤期:好發臉部與身體皺褶(腋窩、腹股溝、肘前窩、女性乳房下);常潰瘍、續發感染;行為較具侵襲性。獅面 (leonine facies) 可見。
- 紅皮症:定義為 ≥80% 體表面積受邊界不清病灶侵犯;可見躺椅徵/摺疊行李徵(皺褶處倖免)。
- 其他症狀:發燒、寒顫、體重減輕、劇癢致失眠、掌蹠角化過度龜裂、禿髮、眼瞼外翻、甲營養不良、踝水腫。
- 色素脫失型 MF:深膚色者常見,須與白斑症 (vitiligo) 鑑別;復色表治療反應,色素脫失復發表復發。
- 組織病理:上真皮帶狀浸潤,腫瘤性 T 細胞具腦回狀 (cerebriform) 核、親表皮性 (epidermotropism)、形成表皮內 Pautrier 微膿瘍。免疫表型成熟周邊 T 細胞 (CD4⁺);可部分喪失全 T 細胞抗原(CD7、CD3),但非特異。早期僅半數切片可測得 T 細胞無性繁殖系,分子與表型在早期 MF 診斷價值有限。
- 鑑別診斷:斑片/斑塊期—慢性皮膚炎、乾癬、接觸性皮膚炎、濕疹、體癬、白斑症;腫瘤期—B 細胞淋巴瘤、皮膚癌、類肉瘤病、深部黴菌/非典型分枝桿菌感染、痲瘋、利什曼原蟲病;紅皮症—毛髮紅糠疹、乾癬、異位性皮膚炎、藥物疹、脂漏性皮膚炎。
- 治療與預後:依分期調整,早期以皮膚導向治療為主,可合併全身性生物反應調節劑(IFN-α/γ、retinoids)。預後依皮膚侵犯類型/範圍、淋巴結與內臟侵犯而定。早期 25% 進展至晚期;侷限皮膚者 5 年存活率 80%–100%,有淋巴結侵犯者 40%。
- Cutaneous Lymphoma International Consortium(1275 名晚期病人):整體存活中位數 63 個月,2 年/5 年存活率 77%/52%;4 項獨立不良預後標記(Stage IV、年齡 >60 歲、大細胞轉化、乳酸去氫酶升高)組合分 3 風險群(低 0–1 為 68%、中 2 為 44%、高 3–4 為 28%)。
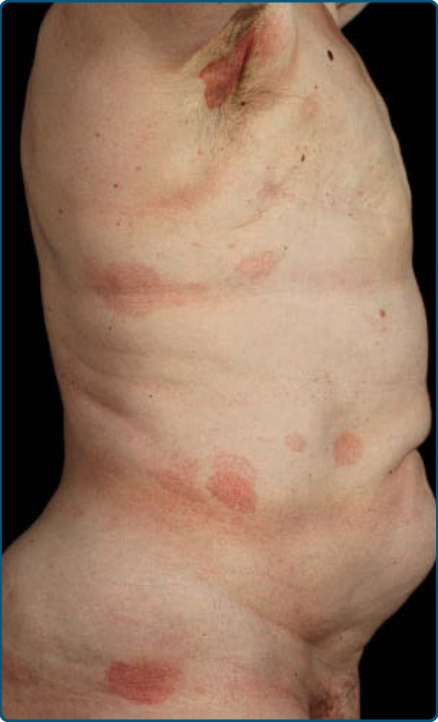
圖 119-4:蕈樣肉芽腫的斑片病灶(側軀幹典型位置)。
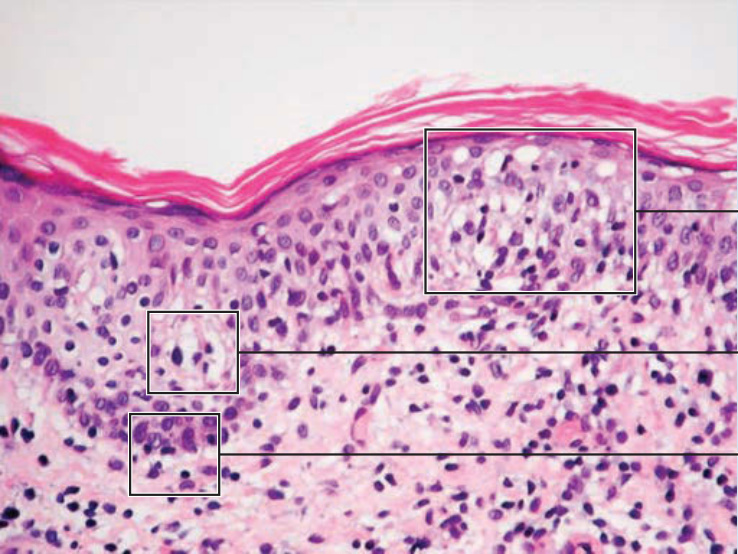
圖 119-11:蕈樣肉芽腫的特徵性組織學(帶狀浸潤、Pautrier 微膿瘍)。
蕈樣肉芽腫變異型
- 親毛囊性 MF (folliculotropic):斑片、斑塊及病灶內毛髮脫落,優先侵犯頭頸部,伴或不伴毛囊黏液性變性(舊稱毛囊性黏液病/黏液性禿髮)。傳統認預後較差,15 年時 5 年存活率約 60%(濾泡型)與 41%(親毛囊型);近年可分出預後良好亞型。
- 派傑樣網狀細胞增生症 (pagetoid reticulosis):單發乾癬樣/角化過度斑塊,多在四肢;無皮膚外播散;親表皮性與核多形性更顯著,常 CD8⁺。惰性。
- 肉芽腫性鬆弛皮膚 (granulomatous slack skin):罕見 MF 亞型,腋窩腹股溝皮膚大塊摺疊;真皮緻密肉芽腫浸潤、多核巨細胞、彈性纖維喪失;CD3⁺CD4⁺CD8⁻。惰性。
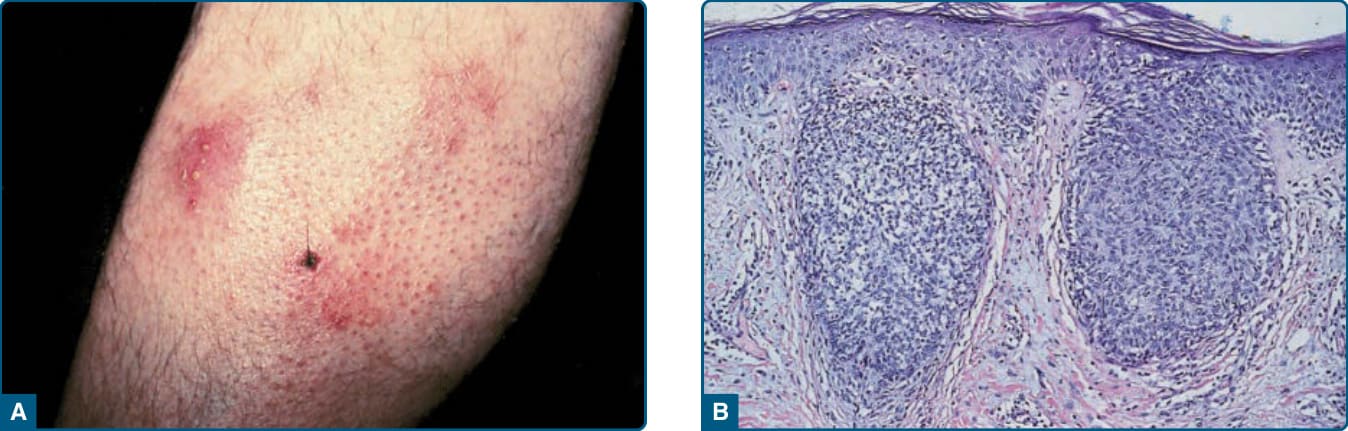
圖 119-15:親毛囊性蕈樣肉芽腫,毛囊定位與毛髮脫落。
Sézary 症候群
- 定義:三聯徵—瀰漫性紅皮症、全身性淋巴結病變、循環中具腦回狀核的惡性 T 細胞(Sézary 細胞)。罕見(佔皮膚淋巴瘤 3%),預後不佳,5 年整體存活率 24%–43%。
- 臨床:紅皮症伴掌蹠嚴重脫屑/龜裂、禁髮、甲營養不良、水腫、苔癬化、劇癢。
- 實驗室:無性繁殖系 T 細胞多為 CD3⁺CD4⁺CD8⁻;常見 T 細胞抗原異常喪失(約三分之二喪失 CD7,多數喪失 CD26);KIR CD158k/KIR3DL2 異常表現。診斷需紅皮症 + 周邊血液陽性 T 細胞無性繁殖系 + 至少一項 B2 標準(如血液 Sézary 細胞 >1000/mm³)。其他標準:CD4/CD8 比值 >10。
- 治療與預後:5 年存活率明顯低於斑片/斑塊期 MF;常因感染性併發症死亡。
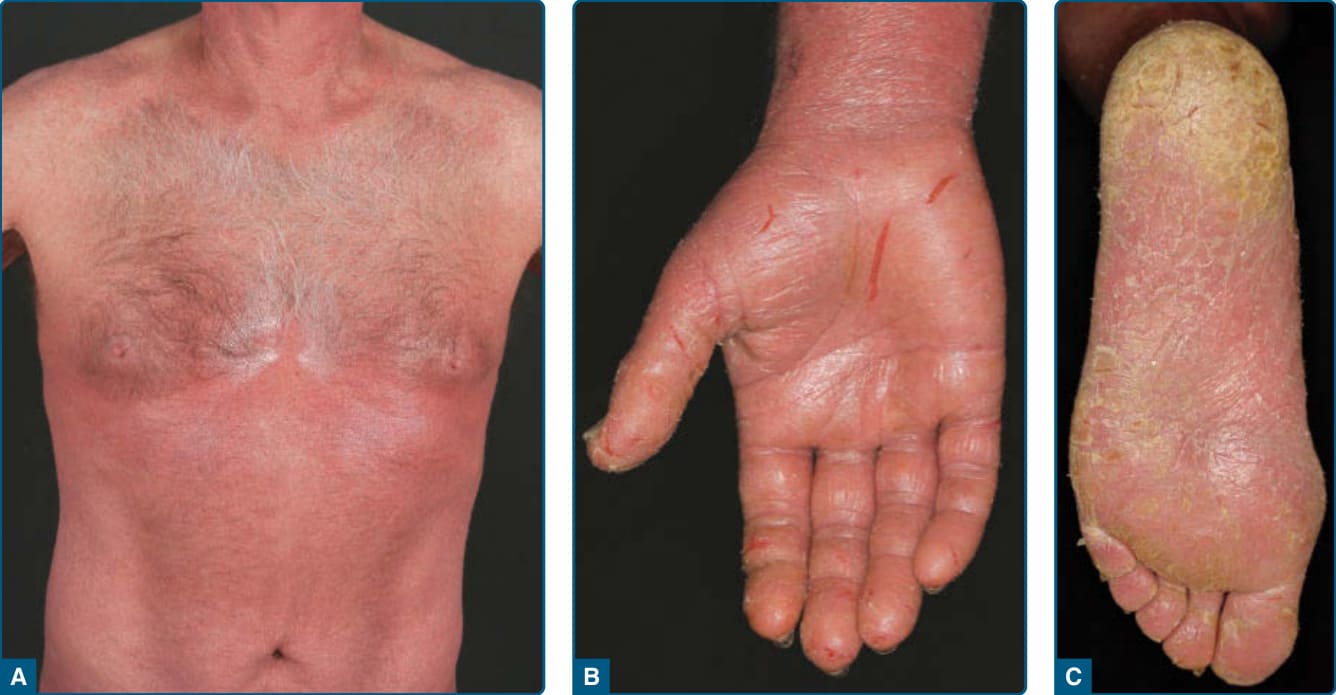
圖 119-10:Sézary 症候群—紅皮症、手掌龜裂與腳掌角化過度。
原發性皮膚 CD30⁺ 淋巴增生性疾病
- 第二常見 CTCL(20%–25%),涵蓋淋巴瘤樣丘疹病與原發性皮膚 ALCL。
淋巴瘤樣丘疹病 (Lymphomatoid Papulosis)
- 慢性、反覆、自癒性的丘疹壞死性/丘疹結節性皮疹,侵犯軀幹四肢,不同演化階段病灶並存。
- 組織學:非典型細胞表現 T 細胞抗原與 CD30;WHO 2016 承認 Type A、B、C,及 Type D、Type E、6p25 重排型。
- 治療:無治癒性療法。低劑量 methotrexate (5–10 mg/week) 是抑制新病灶最有效療法;PUVA 反應短暫;病灶少者可長期追蹤不積極治療。
- 預後:良性病程,10 年存活率近百分之百;惟 10%–20% 可先於/共存/續發惡性淋巴瘤(MF、何杰金氏淋巴瘤、結節性 ALCL),應終身監測。
皮膚退行性大細胞淋巴瘤 (Cutaneous ALCL)
- 大型腫瘤細胞、多數表現 CD30,無 MF 病史。成人,男女比 1.5:1;單發或局部紅至棕色結節腫瘤、常潰瘍,可自發消退;約 10% 區域淋巴結侵犯(不一定預後差)。
- 組織學:真皮結節性/瀰漫性非親表皮浸潤,退行性型態;>75% 腫瘤細胞以黏聚成片表現 CD30;多為 CD4⁺;通常 ALK 陰性、缺 t(2;5)(與結節性 ALCL 相反)。6p25 含 DUSP22-IRF4 重排亞群預後較佳,TP63 重排亞群極具侵襲性。
- 治療:單發/局部—切除或放射治療(可 PUVA + IFN-α);廣泛性—全身性 methotrexate (20 mg/week),vinblastine 為替代;皮膚外播散—CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone);Brentuximab vedotin(抗 CD30 結合 monomethyl auristatin)反應達百分之百。預後良好,疾病相關 5 年存活率 90%。
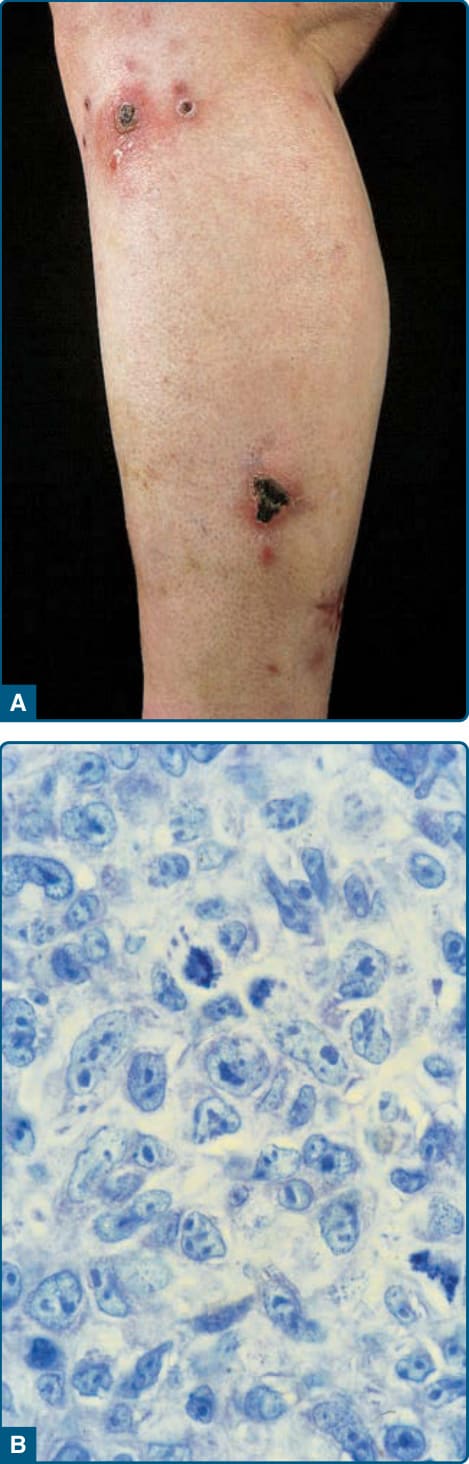
圖 119-20:原發性皮膚退行性大細胞淋巴瘤—侷限性結節(部分潰瘍)與多形性大細胞浸潤。
其他 CTCL 與罕見疾病實體
- 皮下脂膜炎樣 T 細胞淋巴瘤:αβ 細胞毒性 T 細胞,佔皮膚淋巴瘤 1%;皮下結節斑塊(四肢、軀幹),可伴噬血症候群與「B」症狀;腫瘤 T 細胞環繞脂肪細胞 (rimming)、βF1⁺、CD8⁺、TIA-1⁺、granzyme-β⁺。對全身性皮質類固醇反應良好,預後極佳(5 年存活率 85%)。
- 結外 NK/T 細胞淋巴瘤,鼻型:罕見侵襲性,幾乎總是 EBV⁺;CD56⁺ 與細胞毒性蛋白(perforin、granzyme B、TIA-1)強陽;血管中心性、血管破壞、廣泛壞死;常伴噬血症候群。即使積極多藥化療常數月內致死,骨髓移植可能為首選。
- 原發性皮膚 CD4⁺ 小至中型 T 細胞淋巴增生性疾病:丘疹結節(頭頸部),無斑片/斑塊;T 輔助表型;5 年存活率 60%–90%。
- 原發性皮膚侵襲性親表皮 CD8⁺ 細胞毒性 T 細胞淋巴瘤:角化過度斑片斑塊、丘疹、腫瘤或潰瘍;轉移至肺、睪丸、CNS、口腔(不至淋巴結);CD3⁺CD8⁺TIA⁺。侵襲性,存活中位數 32 個月。
- 皮膚 γ/δ T 細胞淋巴瘤:播散性潰瘍壞死性結節/腫瘤(四肢);βF1⁻、CD56⁺、強表現細胞毒性蛋白。侵襲性,存活中位數 15 個月。
- 原發性皮膚肢端 CD8⁺ T 細胞淋巴增生:惰性,主要起源耳部,預後佳;局部切除或放射治療。
CTCL 分期與治療原則
- 採 ISCL/EORTC 之 MF/Sézary 症候群 TNMB 分期;其他皮膚淋巴瘤採 TNM 系統。分期檢查:全身皮膚、胸部 X 光、腹部/淋巴結超音波、血液(CBC、肝酵素、腎功能、乳酸去氫酶、T 細胞無性繁殖性);可疑處加 CT 與組織學/分子。骨髓檢查僅在 B2 血液評級或不明血液異常時建議。
- 腫瘤負荷為存活最佳替代標記;緩解是邁向治癒的第一步;治療理想需多專科團隊。最有效預後因子:年齡 >60 歲、腫瘤負荷、大細胞轉化。
早期 MF 治療
- 皮膚導向治療:UVB、PUVA、外用皮質類固醇、外用 chlormethine、外用類視黃酸 (bexarotene)、放射(含全皮膚電子束)。
- Imiquimod/resiquimod(類鐸受體致效劑,誘導 IFN-α、TNF-α、IL-6、IL-12,朝 Th1 偏移)。
- 全皮膚電子束或為最有效皮膚導向治療:11,065 名病人合併資料完全反應率近 70%,T1 侷限性更高。Stage IIB 單發結節可低劑量 4–8 Gy 局部或低劑量 12 Gy 全身。
紅皮症型 CTCL — 三大生物反應調節劑 (BRM)
- 類視黃酸(Bexarotene 300 mg/m²):選擇性結合 RXR;反應約治療第 12 週起;副作用—高三酸甘油酯血症、高膽固醇血症、嗜中性球減少、中樞性甲狀腺低能症、可逆胰臟炎;需監測脂質/肝/甲狀腺,補 levothyroxine。
- 體外光化學治療 (ECP):8-methoxypsoralen + UVA,每 4 週連續 2 天;反應 4–6 個月;可單一療法管理紅皮症型 CTCL。
- IFN-α:3 百萬單位每週 3 次,增至最大耐受劑量(典型範圍 9 百萬單位/day);反應 3–6 個月;副作用—類流感症狀、慢性疲勞、長期神經毒性(憂鬱、神經病變、失智、脊髓病變)、甲狀腺炎、肝與骨髓毒性。
標靶單株抗體
- Alemtuzumab(抗 CD52):反應率 50%–70%;致 CMV 與伺機性感染再活化;低劑量對白血病型/血液侵犯病人有效,對 MF 無效。
- Brentuximab vedotin(抗 CD30 + monomethyl auristatin E):建議劑量 1.8 mg/kg 每 3 週(為減副作用可降至 1.2 mg/kg);第 II 期整體反應率 70%–73%、完全反應 35%;主要副作用周邊神經病變(多可逆)。
- Mogamulizumab(去岩藻糖基化抗 CCR4):經抗體依賴性細胞媒介細胞毒性清除 CCR4⁺ 細胞與調節性 T 細胞;整體反應率 36%,反應中位數 1.4 個月。
單藥化療
- Gemcitabine:1200 mg/m²(3 個療程)整體反應率 70.5%(中位 15 個月);較低劑量 1000 mg/m² 每週 1 次共 3 週期反應率 68%;對皮膚腫瘤特別有活性;主要副作用骨髓抑制、貧血、血小板減少。
- Pegylated liposomal doxorubicin:20 mg/m²(28 天週期第 1、15 天,共 6 週期)整體反應率 48%、完全緩解 6.1%、無惡化存活中位數 6.2 個月;劑量限制性掌蹠紅斑感覺異常症候群(達 20%)。
- Pralatrexate:抗葉酸劑(高親和 RFC-1);CTCL 最佳劑量 15 mg/m²(每 4 週中 3 週每週)反應率 43%;常規補葉酸與維生素 B12 減少黏膜炎。
異體幹細胞移植
- 適用第一線無反應的較年輕晚期 CTCL(Stage IIB 以上),基於移植物抗 T 細胞淋巴瘤效應;5 年整體存活率 46%、7 年 44%、5 年無惡化存活率 32%。需於完全/接近完全緩解後進行。
原發性皮膚 B 細胞淋巴瘤 (CBCLs)
- 源自 B 淋巴球、診斷時無皮膚外侵犯;佔原發性皮膚淋巴瘤的 20%–25%(發生率約每年每百萬人 3 例)。PCMZL、PCFCL(惰性)與 PCLBCL 腿型(中等-侵襲性)合計佔 CBCL 的 97%。
- 病因:胃結外邊緣區淋巴瘤與幽門螺旋桿菌強關聯;歐洲少數 PCMZL 偵測到伯氏疏螺旋體 (Borrelia burgdorferi);皮膚表現 IgG4 的邊緣區淋巴瘤必為 PCMZL。
原發性皮膚濾泡中心淋巴瘤 (PCFCL)
- 無性繁殖系中心細胞 (centrocytes) 與中心母細胞 (centroblasts) 構成的腫瘤;單發或成群、堅實、無痛紅斑性斑塊與腫瘤,優先頭部與軀幹,罕見於腿部;診斷中位數 58 歲,男約為女兩倍。
- 免疫表型:CD19⁺、CD20⁺、CD79a⁺、Bcl6⁺,濾泡型可 CD10⁺;多數不表現 Bcl2、無 t(14;18)(與結節性相反)。
- 治療與預後:惰性,5 年存活率 >95%;首選放射治療 (30 Gy),復發可 4 Gy 緩和;播散性病灶用免疫治療(IFN-α 或抗 CD20)。腿部 PCFCL 行為較侵襲。
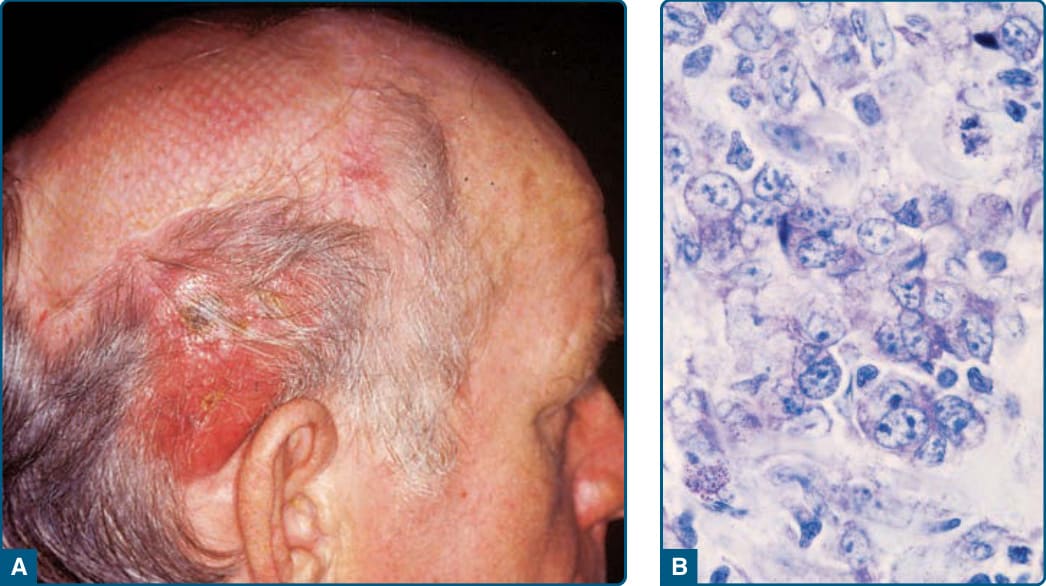
圖 119-23:原發性皮膚濾泡中心淋巴瘤—頭部被紅斑環繞的腫瘤與細胞浸潤。
原發性皮膚邊緣區淋巴瘤 (PCMZL/結外邊緣區淋巴瘤)
- 惰性 B 細胞淋巴瘤,由小型 B 淋巴球、邊緣區細胞、淋巴漿細胞樣細胞與漿細胞組成;上肢或軀幹常見、多部位,紫紅色丘疹/斑塊/結節;診斷中位數 55 歲,女性佔優勢;佔 CBCL 的 25%。
- 免疫表型:邊緣區細胞 CD20⁺、CD79a⁺、Bcl2⁺,CD5/CD10/Bcl6 陰性;漿細胞 CD138⁺、CD79a⁺,單型性輕鏈。
- 治療:偵測到 B. burgdorferi 應先全身性抗生素;散在病灶放射治療;廣泛者觀察等待 + 症狀治療;全身性 chlorambucil 或 rituximab,病灶內 IFN-α 或 rituximab。
原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型 (PCLBCL, leg type)
- 大型 B 細胞腫瘤,老年(>65 歲)女性佔優勢;單發/成簇藍色紅斑性斑塊與腫瘤,多在單側/雙側腿部,常潰瘍(易誤為靜脈潰瘍)。
- 免疫表型:CD20⁺、CD79a⁺、Bcl2⁺、IRF4-MUM1⁺、FOXP1⁺、細胞質 IgM ± IgD;類活化 B 細胞表型。Myc 易位達 43%、Bcl6 易位達 46%;CDKN2a 喪失(23%–42%)與不良預後相關。
- 治療與預後:中等-侵襲性,首選 R-CHOP(rituximab + cyclophosphamide, hydroxydaunorubicin, Oncovin/vincristine, prednisolone);老年(>80 歲)可用 bendamustine + rituximab;多復發,疾病相關 5 年存活率約 50%。
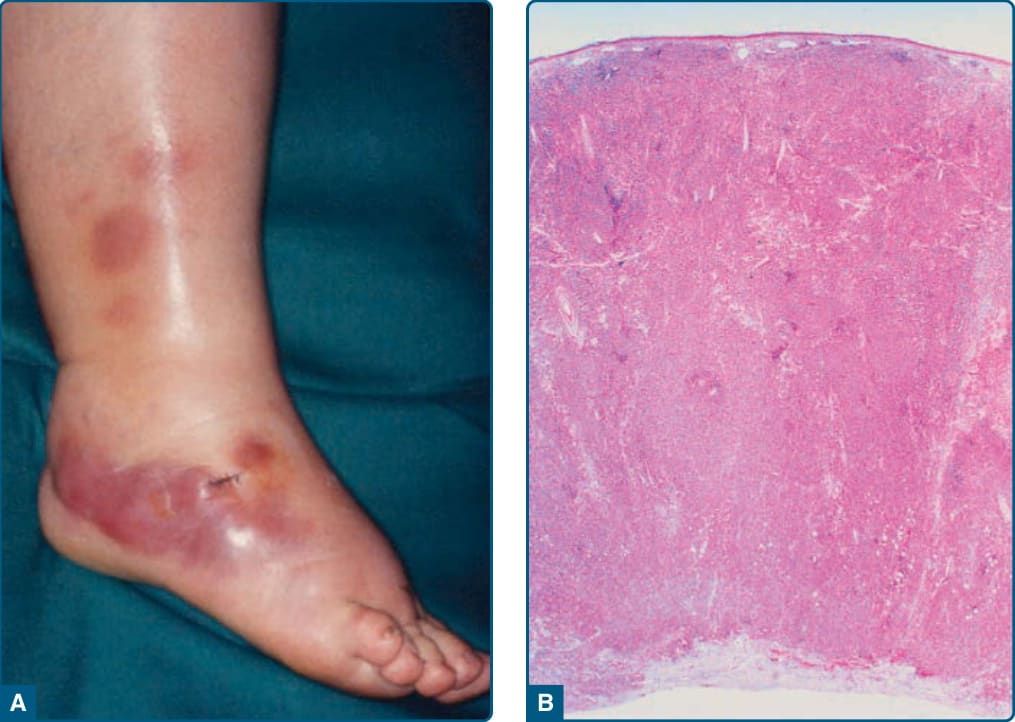
圖 119-24:原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型—右腿結節腫瘤與大型 B 細胞瀰漫浸潤。
其他 CBCL 與分期/治療原則
- PCLBCL, other:不屬腿型或 PCFCL 者;T 細胞/組織球豐富型 B 細胞淋巴瘤僅皮膚病灶者預後極佳。
- 血管內 CBCL:真皮/皮下血管內成簇大型腫瘤性 B 細胞;腿部或軀幹紅至藍色硬化斑塊;多藥化療為首選。
- 分期/治療:採 TNM 分類;PCFCL 與 PCLBCL 腿型需骨髓切片/抽吸。單發病灶可完全切除或局部放射(單次 3–4 Gy;總劑量 30–40 Gy);播散性 PCMZL/PCFCL 用抗 CD20 抗體;PCLBCL 腿型與腿部 PCFCL 用多藥化療(6 週期 CHOP 或 COP)+ 抗 CD20 抗體。
前驅腫瘤 — 母細胞性漿細胞樣樹突細胞腫瘤 (BPDCN)
- WHO 分類列為急性骨髓性白血病相關前驅腫瘤,源自漿細胞樣樹突細胞前驅;罕見、高度侵襲性,存活中位數 12–14 個月。
- 老年(中位 60–70 歲,可任何年齡),男女比 3:1;無症狀單/多發皮膚病灶(結節、斑塊、瘀傷樣,數毫米至 10 cm),多於診斷時即有皮膚外疾病;10%–20% 合併骨髓化生不良,可進展為急性骨髓單核球性白血病。
- 組織病理:浸潤真皮倖免表皮;單調小至中型細胞;免疫組化 CD56⁺、CD4⁺、CD123⁺、TCL1⁺。
- 致病:腫瘤抑制基因(RB1、CDKN1B、CDKN2A、TP53)復發性缺失,TET2 突變支持骨髓相關起源;NRAS(27.3%)、ATM(21.2%)等點突變。
- 治療:全身性化療為首選;gemcitabine 可控制初始疾病以利盡早骨髓移植;標靶 IL-3 受體 CD123 之免疫結合物為可能前景策略。
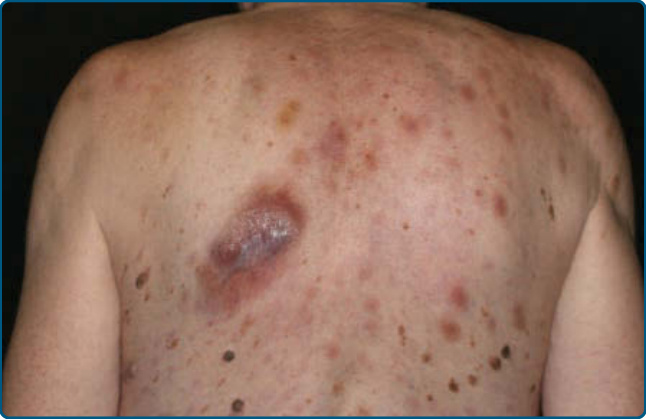
圖 119-25:母細胞性漿細胞樣樹突細胞腫瘤—背部多發病灶。
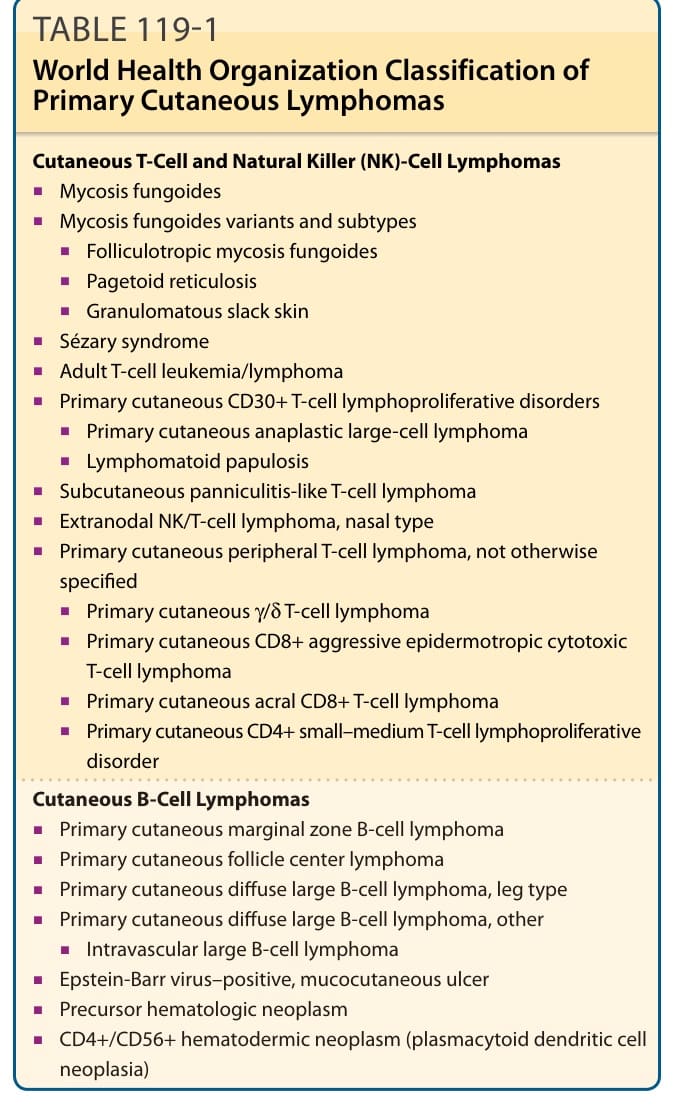
表 119-1:世界衛生組織原發性皮膚淋巴瘤分類。
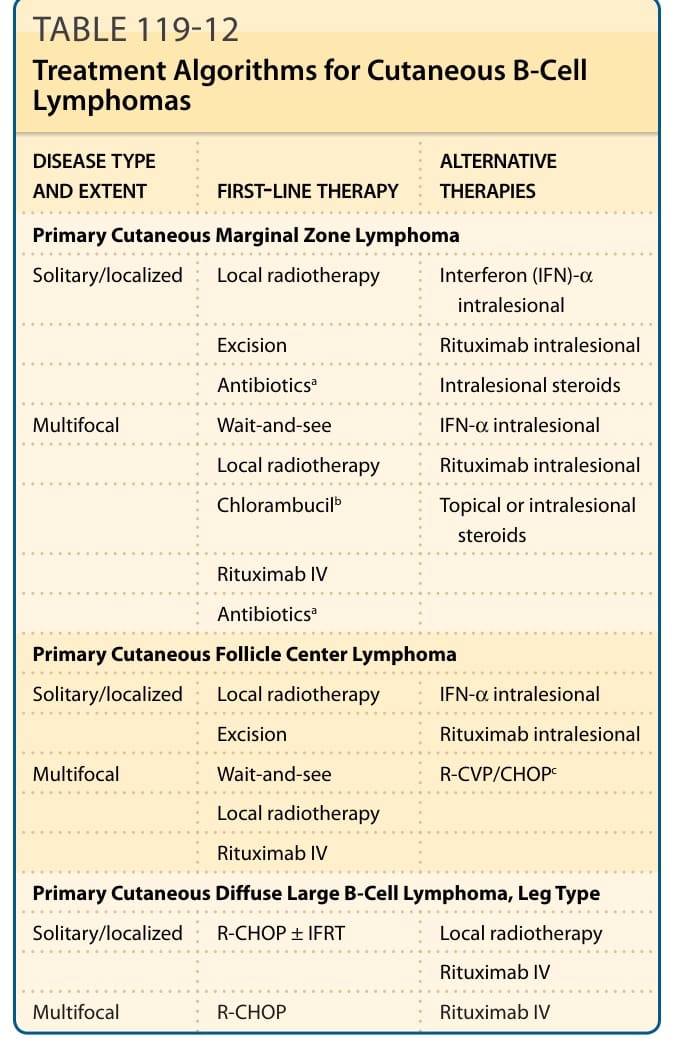
表 119-12:皮膚 B 細胞淋巴瘤的治療演算法。