Cutaneous Lymphoma
20
Primary cutaneous lymphomas are a heterogeneous group of extranodal non-Hodgkin lymphomas arising from malignant clonal transformation of skin homing or/and skin resident T cells or B lymphocytes and hematodermic precursor neoplasias (plasmacytoid dendritic cell neoplasias). Cutaneous lymphomas (Fig. 119-1) are defined as a heterogeneous group with distinct variability in clinical presentation, histopathology, immunophenotyping, and prognosis. Primary cutaneous lymphomas are defined entities with a completely different clinical behavior and prognosis as nodal non-Hodgkin lymphomas and require different treatment approaches. For this reason the European Organization for Research and Treatment of Cancer (EORTC) and World Health Organization (WHO) published a consensus classification for cutaneous lymphomas in 2005.1 This first common classification (WHO-EORTC) categorizes the entities according to lineage and then according to a combination of morphology, immunophenotype, genetic features, and clinical syndromes, and constitutes the basis for the classification of cutaneous lymphomas in the WHO classification 2008 and the revised classification of lymphoid neoplasias in 2016.2,3 This chapter discusses the most frequent cutaneous T-cell lymphomas (CTCLs)—mycosis fungoides (MF), Sézary syndrome, primary cutaneous anaplastic large-cell lymphoma, and lymphomatoid papulosis— and the most frequent cutaneous B-cell lymphomas (CBCLs)—primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous marginal zone lymphoma (PCMZL), and primary cutaneous diffuse large B-cell lymphoma (PCLBCL), leg type. These 7 types of cutaneous lymphoma represent nearly 90% of all cutaneous lymphomas. Rare entities occurring primary in the skin are also described.
EPIDEMIOLOGY
CTCLs represent the second most common group of extranodal lymphomas after the primary GI lymphomas. The incidence of CTCLs has been increasing and is currently, in the United States, estimated to be 6.4 cases/million people between 1993 and 2002 or 7.7 cases/million people between 2001 and 2005. The incidence of CTCL increases significantly with age, with a median age at diagnosis in the mid-50s and a fourfold increase in incidence appreciated in patients older than age 70 years.4-6
PRIMARY CUTANEOUS T-CELL LYMPHOMAS
CTCLs are non-Hodgkin lymphomas characterized by clonal expansion of activated T-cells expressing the E-selectin ligand cutaneous lymphocyte antigen and chemokine receptors (eg, CCR4, CCR8, CCR10) that are required for their subsequent trafficking to the skin.7-9 Clonal expansion is followed by differentiation into multiple subsets of effector and memory cells. Human skin is protected by 4 functionally-distinct populations of T cells, 2 resident and 2 recirculating, with differing territories of migration and distinct functional activities. Central memory cells (TCM) retain the ability to access the peripheral blood and lymph nodes. Effector memory cells (TEM), in contrast, migrate into extranodal sites, including the skin, where a subset will remain as tissue-resident memory cells (TRM). The majority of T cells in the skin are TRM, express a high affinity antigen receptor, and have a distinct gene expression profile. Clonal T cells in MF are commonly TRM-derived, which explains their tendency to be confined to the skin. In contrast, in patients with leukemic CTCL variants (Sézary syndrome and MF with secondary leukemic involvement), tumor cells express CCR7 and L-selectin, resembling TCM. This fundamental difference in the putative cell origin between Sézary syndrome (TCM-derived) and MF (TRM-derived) is consistent with the distinct clinical behavior. Among the recirculating cells 2 distinct populations were observed, CCR7+/L-selectin+ TCM and CCR7+/L-selectin− T cells termed migratory memory T cells (TMM). A subset of MF patients with secondary leukemic involvement, poorly demarcated patches/ plaques, dermal involvement, and lymphadenopathy most probably harbor a TMM clone.10,11
The most common forms, representing approximately 65% of CTCL, are MF and Sézary syndrome, with an annual incidence of 7.7 cases/million people. CTCL encompasses skin-limited variants such as MF and leukemic forms of the disease, including Sézary syndrome. After MF and Sézary syndrome, the primary cutaneous CD30+ lymphoproliferative disorders, comprising lymphomatoid papulosis and cutaneous anaplastic large-cell lymphoma, represent the second most common group of CTCLs (approximately 27%).12 Table 119-1 outlines the WHO classification of primary cutaneous lymphomas.
20
Cutaneous lymphomas
Primary Cutaneous Lymphomas
T-cell origin
Sézary syndrome Primary cutaneous CD30+ lymphoma
Mycosis fungoides
PCFCL
B-cell origin
Intravascular cutaneous B-cell
PCMZL or ENMZL
PCLBCL, leg type
ETIOLOGY
ETIOLOGY
The skin of a human adult contains approximately 20 billion memory T cells. Despite major advances in cellular and molecular biology revealing many details about lymphocytes, including the incredible diversity
Cutaneous T-Cell and Natural Killer (NK)-Cell Lymphomas
■Mycosis fungoides
■Mycosis fungoides variants and subtypes
■Folliculotropic mycosis fungoides
■Pagetoid reticulosis
■Granulomatous slack skin
■Sézary syndrome
■Adult T-cell leukemia/lymphoma
■Primary cutaneous CD30+ T-cell lymphoproliferative disorders
■Primary cutaneous anaplastic large-cell lymphoma
■Lymphomatoid papulosis
■Subcutaneous panniculitis-like T-cell lymphoma
■Extranodal NK/T-cell lymphoma, nasal type
■Primary cutaneous peripheral T-cell lymphoma, not otherwise specified
■Primary cutaneous γ/δ T-cell lymphoma
■Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma
■Primary cutaneous acral CD8+ T-cell lymphoma
■Primary cutaneous CD4+ small–medium T-cell lymphoproliferative disorder
Cutaneous B-Cell Lymphomas
Cutaneous B-Cell Lymphomas
■Primary cutaneous marginal zone B-cell lymphoma
■Primary cutaneous marginal zone B-cell lymphoma
■Primary cutaneous follicle center lymphoma
■Primary cutaneous follicle center lymphoma
■Primary cutaneous diffuse large B-cell lymphoma, leg type
■Primary cutaneous diffuse large B-cell lymphoma, leg type
■Primary cutaneous diffuse large B-cell lymphoma, other
■Primary cutaneous diffuse large B-cell lymphoma, other
■Intravascular large B-cell lymphoma
■Intravascular large B-cell lymphoma
■Epstein-Barr virus–positive, mucocutaneous ulcer
■Epstein-Barr virus–positive, mucocutaneous ulcer
■Precursor hematologic neoplasm
■Precursor hematologic neoplasm
■CD4+/CD56+ hematodermic neoplasm (plasmacytoid dendritic cell neoplasia)
■CD4+/CD56+ hematodermic neoplasm (plasmacytoid dendritic cell
neoplasia)
of T-cell antigen receptors, their characterization as TCM or TEM or TRM, and the role of environmental and host genetic factors for the pathogenesis in CTCL remains unclear. In general, long-term antigen stimulation is thought to induce an inflammatory response with T-cell proliferation leading to clonal malignant T cells with continuous expansion. However, recent advances in the understanding of the molecular pathogenesis, signal transduction pathways, and disease-associated immune dysregulation helped to understand the complex pathogenesis to advance the treatment in CTCL.10-19
ENDOGENOUS FACTORS
ENDOGENOUS FACTORS
As a result of the above-mentioned hypothesis of antigen stimulation, several studies have analyzed the human leukocyte antigen (HLA) background of affected individuals. Two independent studies showed an association of distinct HLA class II molecules and MF or Sézary syndrome; that is, the alleles HLA- DRB1∗11 and DQB1∗03 are significantly overrepresented in these patients (Fig. 119-2).
Endogenous and exogenous factors that predispose
to cutaneous T-cell lymphoma
Exogenous Factors (Infectious)
Exogenous (Environmental/ Occupational)
• Bacterial: Staphylococcus aureus
• Viral: HTLV; EBV Endogenous Factors
• Glass formers
• Potters
• Paper factory
• Wood factory Cutaneous T-Cell Lymphoma
• HLA DRB111
• HLA DQB103
2073
20
EXOGENOUS FACTORS
EXOGENOUS FACTORS
Viruses have been identified as etiologic factors in at least 2 cutaneous lymphomas (human T-cell lymphotropic virus-1 [HTLV-1]-associated adult T-cell lymphoma/leukemia, and Epstein-Barr virus [EBV]- associated natural killer [NK]/T-cell lymphoma), whereas no such relation has been confirmed for MF or Sézary syndrome. All these data suggest that HTLV does not play an important role in the etiology of CTCLs, outside of HTLV-1 endemic regions, and that the only reason to screen patients for antibodies is the suspicion that the diagnosis is adult T-cell lymphoma/ leukemia rather than MF. EBV as well as cytomegalovirus have been discussed as causative pathogens. EBV is associated with CD30 lymphoproliferation and with immunosuppression. Several studies show that EBV is detectable only in a minor percentage of CTCL lesions. In these studies, EBV detection was related to a poor prognosis and its presence is more likely related to immunosuppression caused by either the disease or the therapy, rather than to the etiology of CTCL. However, a strong association of EBV in a rare cutaneous lymphoproliferative disease with a hydroa vacciniforme–like appearance, which occurs mostly in people of Asian origin, has been observed. Bacterial infections also have been implicated in the etiology of CTCLs. Of special interest has been the hypothesis that superantigens from Staphylococcus aureus may be responsible for chronic antigenic stimulation. In several studies, S. aureus has been detected in a high percentage on the skin of CTCL patients with a high tumor burden, while patients in early stage disease did not show significant differences to control groups. Although these studies conclusively demonstrate the involvement of S. aureus in disease exacerbation and clinical improvement following antibiotic treatment, the missing difference in S. aureus colonization in early stages of CTCL and control groups questions the involvement of S. aureus or superantigens produced by these bacteria in initiation of CTCLs. However, S. aureus enterotoxin A stimulates signal transducer and activator of transcription (STAT) 3 activation, and interleukin (IL)-17 expression in cutaneous T-cell lymphoma and may play a direct role in the progression of the disease. Besides infectious pathogens, it also has been suggested that environmental and occupational risk factors play a causative role in CTCL (see Fig. 119-2), because an indolent dermatitis often precedes the diagnosis. Exposure to carcinogens in the work environment could provide the suspected long-term antigenic stimulation for the initiation of the clonal expansion. In epidemiologic studies, several occupations, such as glass formers, potters, and paper and wood industry workers, have been associated with a higher risk for development of MF. However, the results of the different studies were not consistent, and a common denominator, like exposure to known carcinogens, could not be identified. With regard to chronic antigenic stimulation by occupational contact allergens, it has to be considered
2074
that MF arises typically on body areas like the lateral trunk that are protected by clothes during working time. Also, other environmental risk factors, like consumption of alcohol, smoking, or exposure to ultraviolet (UV) radiation, were not consistently observed in association with an increased risk for CTCLs.20-31
PATHOGENESIS
PATHOGENESIS
Cutaneous T-cell lymphoma is a malignancy of skin- homing T-cells. Patients typically present with localized patches and plaques in sun-protected skin. Lymphoma cells extend from these lesions to uninvolved skin and accumulate in the superficial dermis, leading to patches/plaques and tumors. In advanced disease, malignant T cells disseminate to blood, lymph nodes and viscera. In leukemic CTCL (Sézary syndrome), malignant T-cells can comprise greater than 99% of the circulating T lymphocytes. Loss of the normal T-cell receptor (TCR) and the disappearance of normal lymphocytes lead to immunosuppression and opportunistic infections, which are the most common disease-related causes of death. The clinical entities encompassed by the term cutaneous T-cell lymphoma share several components: the epidermal and/or dermal microenvironment, a clonal T-cell population, and a modulated antitumor response. A spectral karyotyping and comparative genomic hybridization studies combined with TCRγ polymerase chain reaction have demonstrated that genetically-damaged malignant T cells are present in even the earliest stages of MF, confirming that MF is a lymphoma of genetically-damaged malignant T cells even in its earliest manifestation. There is emerging evidence that the distinct clinical presentation of CTCLs may represent their derivation from different subsets of skin-homing T cells. Malignant T cells in MF have the surface phenotype of nonrecirculating TRM and classic erythrodermic Sézary syndrome has malignant T cells with a surface phenotype of TCM, consistent with their tendency to form stable inflammatory skin lesions versus transitory erythroderma, respectively. However, the phenotype of Sézary cells is more heterogeneous than initially reported, and Sézary cells can also present phenotypic plasticity.10,32-37
TMM are novel skin-homing T cells. These cells express CCR7 but lack L-selectin, are present in the blood and skin of healthy individuals, and recirculate more slowly out of skin than do TCM. In CTCL patients, malignant TMM give rise to discrete skin lesions with illdefined borders and peripheral blood disease, which, in the current classification, is referred to as MF with peripheral blood disease. The fundamental difference in the putative cell of origin between Sézary syndrome (TCM derived) and MF (TRM derived) is consistent with their distinct clinical behaviors, as TCM may be found in both, in the peripheral blood, lymph node, and skin, and are resistant to apoptosis, whereas resident TRM cells remain fixed within the skin. In addition, a population
of recirculating CCR7+ L-selectin− TMM has been described in the skin. The contention that MF subtypes and Sézary syndrome originate from different T-cell subsets is consistent with comparative genomic hybridization (CGH), a gene expression profiling data demonstrating that these CTCL subtypes are genetically distinct. Detection of these malignant T-cell clones is critical in making the diagnosis of CTCL.10,11
Emerging with new molecular technology using high-throughput TCR sequencing, it has been demonstrated that the malignant T-cell clones in MF and leukemic CTCL localized to different anatomic compartments in the skin could be discriminated from benign inflammatory skin diseases. Regulatory T cells expressing the transcription factor FOX-P3 are important in the maintenance of selftolerance and form a minor subset of skin-resident T cells. It is discussed that a subset of Sézary patients harbor a clone that is derived from resident regulatory T cells. However, regulatory T cells represent only a minority of skin-resident T cells; the majority of T cells in the skin produce cytokines characteristic of distinct effector T-cell subsets, including T-helper (Th) 1, Th2, and Th17 cells. MF and Sézary syndrome are associated with the expression of Th2-associated genes (eg, GATA-3) and the production of Th2-associated cytokines (eg, IL-4, IL-5, IL-13), raising the possibility that a significant subset of patients may harbor Th2-derived clones.38-44
Alternatively, recurrence mutations activating specific signaling pathways (nuclear factor of activated T cells [NFAT], nuclear factor κB [NFκB], Janus kinase [JAK]/STAT) may provoke the acquisition of a particular phenotype independent of the cell of origin.37
Recent molecular studies have advanced our understanding of the molecular pathogenesis of CTCL. Recurrent deletions of 10q and 17p and amplification of 8q and 17q have been identified with robust evidence implicating deletions of TP53 und CDKN2a and amplifications of 8q containing MYC. In a recent study of the genomic landscape of cutaneous lymphomas, somatic mutations in 17 genes in CTCL were described. Frequent deletion and damaging somatic copy number variants in chromatin-modifying genes (ARID1A [62.5%], CTCF [12.5%], and DNNT3A [42.5%]) were found. Many genes mutated in CTCL contribute to other T-cell neoplasms, including peripheral T-cell lymphoma (CD28, DNMT3A, and RHOA), underscoring the importance of these genes for the malignant transformation of T cells. Consistent with this notion, mutations were found in multiple components of the TCR signaling pathway, including CD28 and the genes for TCR-associated enzymes (PLCG1, PRKCQ, and TNFAIP3) and transcription factors (NFkB2, STAT5B, and ZEB1). These genes drive the Th2 differentiation (ZEB1) that facilitates escape from transforming growth factor-β–mediated growth suppression (ZEB1) and facilitates resistance to tumor necrosis factor receptor superfamily–mediated apoptosis (FAS and ARID1A).45,46
In CTCL, several cytokines play a role in disease manifestation as well as in the progression of this
20
disease. In early stages of CTCL, signaling of IL-2, IL-7, and IL-15, which all are γc-chain cytokines using JAK1/JAK3, drives the activation of STAT5 and STAT3. As a result of downstream processes of STAT3 and STAT5, a shift from Th1 toward a Th2 phenotype of the malignant T cells can be observed. This is achieved by the transcriptional activation of the micro-RNA (miRNA)-155 by STAT5. In turn, miRNA- 155 targets STAT4, leading to the downregulation of Th1 genes. In addition, STAT5 activates IL-4 expression, fostering the Th2 phenotype. This shift is associated with the progression of CTCL as Th2 responses (IL-4, IL-10) are well known mechanisms of tumorinduced immunosuppression. Furthermore, the activation of JAK3, STAT3, and STAT5 leads to a transcriptional repression of miRNA-22, a known tumor suppressor. For CTCL, malignant T cells—CTCL cell lines as well as peripheral blood Sézary CD4+ T cells—demonstrate a reduced expression of miRNA-22. The miRNA- 22 normally inhibits tumor growth and metastasis because it targets the transcription of a number of putative oncogenic genes (eg, MYCB, HDAC4, HDAC6, CDK6, and NcoA1). But in CTCL, a loss of these tumor-suppressive activities by miRNA-22 is observed, as the expression of miRNA-22 is directly downregulated by STAT5, leading to a faster progression of the malignant state of the T cells. These findings suggest that JAK/STAT signaling plays another key role in the pathogenesis and progression of CTCL and that JAK-inhibition could mediate a direct suppressive effect on tumor growth and metastasis (Fig. 119-3). In addition to this, the activation of STAT3 is a critical mediator of the transformation of malignant CTCL T cells, as well as an important mediator of plasticity.1,7
STAT3 activation leads to IL-17 production of CTCL T cells as demonstrated in vitro with CTCL cell lines, as well as by expression of IL-17 in neoplastic lymphocytes in CTCL skin lesions ex vivo. Receptors for IL-17 are expressed by various cells in the skin microenvironment of CTCL lesions, such as fibroblast, keratinocytes, and epithelial cells. Upon stimulation with IL-17 these cells produce other proinflammatory cytokines, chemokines, and angiogenic factors. It was shown that CTCL lesions exhibit increased angiogenesis, providing the suggestion that IL-17 of neoplastic T cells in CTCL influences tumorigenesis by modulating inflammation and angiogenesis in CTCL skin lesions. Therefore, targeting the initial STAT3 activation by JAK inhibition would also have beneficial effects on these indirect effects of aberrant T cells of the Th17 phenotype. STAT3 transcriptional activity also leads to the expression of IL-21 in the aberrant T cells, which drives an autocrine signaling loop, leading to constitutive signaling via JAK1/JAK3 and the activation of STAT3. This constitutive activation of STAT3 driven by IL-21 is essential for the progression of CTCL by several mechanisms: (a) it promotes the expression of antiapoptotic proteins like bcl-2, leading to survival of malignant T cells1; (b) STAT3 is involved in
2075
20
Signaling events in cutaneous T-cell lymphoma
JAK-STAT
CCR4 FAS TNFRSF10A
Type I/II-R
TRAIL-R
FAS
TNFRSF1B
CCR4
JAK
STAT
Skin homing Apoptosis
JAK1/3 STAT3/5B NFKB2
STAT IKBα
Proteasome
STAT
Bortezomib Tofacitinib Ruxolitinib
NF-jB
TCR
TCR/CD3
TNFR
IL-1R
CTLA-4
PRKCQ PDCD1 CD28
CD28
PTEN
PTEN
CARD11 PLCG1
IKKα
PI3K RAS
PLCG1
CARD11
RAF
NF-jB
KRAS BRAF MAPK1
AKT
MEK
mTOR
ERK
RHOA
Cytoskeletal Dynamics
Cell cycle ZEB1
Genome integrity
TH2 polarization Epigenetic regulators
TP53 ATM
ARID1A DNMT3A KMT2C
p16
Rb
CDKN2A RB1
the upregulation of the transcription of the angiogenic factor vascular endothelial growth factor; (c) it induces the expression of IL-5 and other cytokines involved in erythroderma and eosinophilia; (d) STAT3 transcriptional activities are involved in driving the plasticity of the T cells (Th2 and Th17 phenotypes) observed in advanced stages of CTCL; and (e) it induces the expression of an oncogenic miRNA, miRNA-21. The miRNA-21 is involved in the survival of malignant T cells due to antiapoptotic activities. All these reasons provide a strong rationale for targeting JAK/STAT pathways in CTCL. Other cytokines in the microenvironment of CTCL are also involved, like IL-13, a cytokine related to the γc-chain cytokines sharing the IL-4 receptor α subunit (IL-4Rα). IL-13 belongs, therefore, to the IL-4 family and is secreted by the transformed malignant Th2 cells in CTCL, as IL-13 is highly expressed in clinically involved skin of CTCL patients. IL-13 acts as an autocrine growth factor of CTCL cells, especially in the Sézary syndrome variant of CTCL. Blockade of JAK1/JAK3 would also block IL-13 cytokine signaling, leading to the inhibition of tumor cell proliferation.47-60
2076
In recent years, it has become evident that cytokine signaling plays a critical role in the pathogenesis of CTCL. In addition to multiple defects in apoptosis, aberrant cell-cycle regulation, including inactivation of the CDKN2A-CDKN2B locus, is frequently observed in CTCL. Cyclin upregulation, including cyclin D1, and loss of RB1 also have been described. As gene expression profiling and next-generation sequencing technologies are employed, additional pathogenic pathways, including those involved in transcription factors regulating T-cell differentiation and C-MYC, RAS, BRAF, and MEK signaling, are being identified in subsets of CTCL.13,34
In summary, recent research into CTCL has significantly advanced our knowledge of molecular pathogenesis, cellular origin, migratory behavior, and death signaling. As for other malignancies, the major challenge now will be to define meaningful molecular and/or phenotypic subgroups that relate to clinical behavior and/or treatment response to drugs that specifically interfere with disease-promoting signaling cascades or cellular interactions. As a result, promising treatment approaches will be based on our increasing knowledge about the molecular pathogenesis of CTCL.
MYCOSIS FUNGOIDES
MYCOSIS FUNGOIDES
DEFINITION
MF is the most common form of primary cutaneous lymphoma, accounting for approximately 40% of all cutaneous lymphomas, usually arising in mid to late adulthood (median age at diagnosis: 55-60 years) with a male predominance of 2:1. According to the WHO classification, MF is defined by its classic form, that is, by patches and plaques or variants.
CLINICAL FINDINGS
Skin Signs: Clinically, MF is categorized as patch, plaque, or tumor stage, but patients may simultaneously have more than 1 type of lesion. In early patch stage MF (Figs. 119-4 and 119-5), there are single or multiple erythematous, scaly macules and patches that vary in size and are usually well defined. The color of the lesions may vary from orange to a dusky violet-red. The distribution classically favors non–sunexposed sites, with the “bathing trunk” and intertriginous areas predominant early in the course of the disease. The eruption may be intensely pruritic or asymptomatic, and occasionally may be transitory, disappearing spontaneously without scarring. Diagnosis at this stage may be difficult. Often a patient will recall a preceding “chronic dermatitis” for 10 to 20 years that may have been considered to be therapeutically-resistant contact dermatitis, atopic dermatitis, psoriasis, or eczema. In any patient with a dermatosis that is refractory to the usual modalities of treatment,
20
multiple biopsy specimens should be taken to pursue a diagnosis. Patches may last for months or years before progressing to plaque stage (Fig. 119-6), or plaques may arise de novo. Plaques appear as sharply demarcated, scaly, elevated lesions that are dusky red to violaceous and variably indurated (Figs. 119-6 and 119-7). Lesions in this stage may regress spontaneously or may coalesce to form large plaques with annular, arcuate, or serpiginous borders, and may clear centrally with disease activity remaining at the periphery of the lesion. There may be purpuric hyperpigmentation or hypopigmentation and poikiloderma. Tumors may occur anywhere on the body, but have a predilection for the face (Figs. 119-8 and 119-9) and body folds: axillae, groin, antecubital fossae, and, in
2077
20
women, the inframammary area. These usually occur in preexisting plaques or patches of MF; this coincides with an extension of these lesions in the vertical dimension (see Fig. 119-6). At this point, the neoplastic cells behave in a biologically-more aggressive manner, with pronounced tumor cell accumulation that leads to the clinical appearance of an expanding dermal nodule (see Fig. 119-9). De novo occurrence
2078
suggests metastatic spread by cells of a malignant T-cell clone. The nodules are reddish brown or purplish red and smooth surfaced, but they often ulcerate and may become secondarily infected. Growth rate is variable. Patients with tumors tend to have a more aggressive form of the disease than patients with patch and plaque disease. Erythroderma (Fig. 119-10A) may start de novo or develop in MF. The nomenclature for erythrodermic phases of CTCL varies. It has been proposed that erythroderma be defined as the involvement of 80% of body surface area with lesions of ill-defined borders and that patients with a history of preexisting MF be defined as having a separate syndrome of “erythrodermic MF.” The skin is diffusely bright red with readily apparent scaling, but there may be characteristic islands of uninvolved skin. There may be sparing of the areas of skin that are frequently folded, such as the abdomen and antecubital and axillary areas. This sparing produces a finding often called the deck chair or folded luggage sign. Some patients with the erythrodermic form of CTCL develop tumors.
Other Symptoms: Patients may complain of fever, chills, weight loss, malaise, insomnia secondary to the overwhelming pruritus, and poor body temperature homeostasis. There may be hyperkeratosis, scaling and fissuring of the palms and soles, alopecia, ectropion, nail dystrophy, and ankle edema, with the integument being shiny and hidebound. These changes result in pain on walking and extreme difficulty with tasks requiring manual dexterity. Such patients experience severe restrictions by the extent and localization of their skin manifestations. Pruritus is often intense, which results in excoriation, exudation, and secondary infection that may dominate the clinical picture.
A
20
B
C
Hypopigmentated Mycosis Fungoides: Patients with dark skin develop hypopigmented MF, a variant (Table 119-2) of patch MF. This form of MF must be differentiated from vitiligo. In darker-skinned individuals, this may be the most common presentation of the disease. Patients respond to therapy with repigmentation, and the reappearance of hypopigmented lesions often indicates a relapse.
HISTOPATHOLOGY
SUBTYPES
Erythrodermic mycosis fungoides (MF) Develops de novo or as a progression of MF; > 80% involvement of skin surface
Hypopigmented mycosis
Common in darkly pigmented
Hypopigmented mycosis fungoides Common in darkly pigmented individuals; response to therapy characterized by repigmentation. Vitiligo is a differential diagnosis
fungoides
individuals; response to therapy characterized by repigmentation. Vitiligo is a differential diagnosis
such as CD7 and CD3 may be a feature of MF, but is not pathognomonic of the disease. Analysis of TCR genes typically shows a clonal rearrangement as demonstrated by polymerase chain reaction or Southern blot techniques. However, a T-cell clone is only found in half the biopsies in early stages of disease. Thus, neither molecular tests for T-cell clonality nor phenotypic marker are of significant diagnostic value in early MF. Maybe in the near future modern molecular diagnostics, such as high-throughput technology, will be capable of detecting T-cell clones even in early MF.
DIFFERENTIAL DIAGNOSIS
TREATMENT AND PROGNOSIS
Treatment should be stage adapted. In early disease stages, treatment should be based on skin-directed
2079
20
Characteristic histology of mycosis fungoides
therapies alone or combined with systemic biologic response modifiers (eg, interferon α or γ, retinoids). Targeted therapies and small molecules are gaining favor as ways to debulk tumors and blood compartments as an alternative for chemotherapy approaches (see sections “Staging of Cutaneous T-Cell Lymphoma” and “Principles of Treatment of Cutaneous T-Cell Lymphoma”). Prognosis depends on the type and extent of skin involvement (plaque, tumor, or erythroderma), the presence of lymph node involvement, and the presence of visceral disease. Among early stage patients, 25% will progress to advanced stage. Overall, patients with MF limited to the skin have a 5-year survival rate of 80% to 100%. In contrast, patients with lymph node involvement show a 5-year survival rate of 40%.62
The Cutaneous Lymphoma International Consortium study published survival data and analysis based on 1275 patients with advanced MF and Sézary syndrome. The median overall survival was 63 months with 2-year
2080
Pautrier microabcesses
Pagetoid scatter
Atypical Iymphocytes
and 5-year survival rates of 77% and 52%, respectively. The median overall survival for patients with Stage IIB disease was 86 months, but patients diagnosed with Stage III disease have slightly improved survival compared with patients with Stage IIB disease. Patients
A
B
diagnosed with Stage IV disease had a significantly worse survival (48 months for Stage IVA and 33 months for Stage IVB). Of 10 variables tested, 4 (Stage IV, age >60 years, large-cell transformation, and increased lactate dehydrogenase) were independent prognostic markers for worse survival. Combining these 4 factors for a prognostic index model led to the identification of 3 risk groups across stages, with significantly different 5-year survival rates: low risk (0-1) 68%, intermediate risk (2) 44%, and high risk (3-4) 28%.63
CRITERIA SCORING SYSTEM
■Clinical
■Basic 2 points for basic criteria and 2 additional criteria
■Persistent and/or progressive patches/thin plaques 1 point for basic criteria and 1 additional criterion
■Additional
■Non–sun-exposed location
■Size/shape variation
■Poikiloderma
■Histopathologic
■Basic 2 points for basic criteria and 2 additional criteria
■Superficial lymphoid infiltrate 1 point for basic criteria and 1 additional criterion
■Additional
■Epidermotropism without spongiosis
■Lymphoid atypia
■Molecular biological
■Clonal TCR gene rearrangement 1 point for clonality
■Immunopathologic
■<50% CD2+, CD3+, and/or CD5+ T cells 1 point for 1 or more criteria
■<10% CD7+ T cells
■Epidermal/dermal discordance of CD2, CD3, CD5, or CD7
A total of 4 points is required for the diagnosis of MF based on any
A total of 4 points is required for the diagnosis of MF based on any combination of points from the clinical, histopathologic, molecular biologic, and immunopathologic criteria
combination of points from the clinical, histopathologic, molecular biologic, and immunopathologic criteria
From Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063, with permission. Copyright © American Academy of Dermatology.
20
■Evaluation of skin lesions; measure of the percentages of body surface area of patches, plaques, and tumors
■Evaluation of skin lesions; measure of the percentages of body
surface area of patches, plaques, and tumors
■Evaluation of localization and measures of lymph nodes
■Evaluation of localization and measures of lymph nodes
■Clinical identification of visceral disease
■Clinical identification of visceral disease
■Histology and immunohistology of skin lesions, and evaluation for T-cell clonality
■Histology and immunohistology of skin lesions, and evaluation for
T-cell clonality
■Histology and immunohistology of enlarged lymph nodes, and evaluation for T-cell clonality
■Histology and immunohistology of enlarged lymph nodes, and
evaluation for T-cell clonality
■Blood cell count, lactate dehydrogenase, liver function tests
■Blood cell count, lactate dehydrogenase, liver function tests
■Blood T-cell clonality
■Blood T-cell clonality
■Sézary cell count by cytomorphology and/or flow cytometric analysis of T-cell blood subpopulations (CD4+/CD7− and CD4+CD26−)
■Sézary cell count by cytomorphology and/or flow cytometric analysis
of T-cell blood subpopulations (CD4+/CD7− and CD4+CD26−)
■Early-stage patients: chest radiograph and ultrasound of abdomen
■Early-stage patients: chest radiograph and ultrasound of abdomen
■Late-stage patients: CT scan of chest, abdomen, and pelvis
■Late-stage patients: CT scan of chest, abdomen, and pelvis
MYCOSIS FUNGOIDES VARIANTS
MYCOSIS FUNGOIDES
VARIANTS
FOLLICULOTROPIC MYCOSIS FUNGOIDES
Compared to classic MF, follicular or folliculotropic MF has classically been considered to carry a worse prognosis with 5-year survival rates of approximately 60% (follicular MF) and 41% (folliculotropic MF) by 15 years.
Patch/Plaque Stage
■“Chronic dermatitis”
■Psoriasis
■Contact dermatitis
■Eczema
■Tinea corporis
■Vitiligo
Tumor Stage
Tumor Stage
■B-cell lymphoma
■B-cell lymphoma
■Carcinoma cutis
■Carcinoma cutis
■Sarcoidosis
■Sarcoidosis
■Deep fungal infection
■Deep fungal infection
■Atypical mycobacterial infection
■Atypical mycobacterial infection
■Leprosy
■Leprosy
■Leishmaniasis
■Leishmaniasis
■Erythroderma
■Erythroderma
■Pityriasis rubra pilaris
■Pityriasis rubra pilaris
■Psoriasis
■Psoriasis
■Atopic dermatitis
■Atopic dermatitis
■Drug eruption
■Drug eruption
2081
■Seborrheic dermatitis
■Seborrheic dermatitis
20
CLINICAL FINDINGS PROGNOSIS (5-YEAR SURVIVAL RATE)
Mycosis fungoides (skin limited) Mycosis fungoides (lymph node involvement)
Single-to-multiple erythematous scaly macules and patches on non– sun-exposed sites; plaques appear sharply demarcated, scaly, violaceous, and indurated; tumors have a predilection for the face and body folds
80%-100%
40%
Folliculotropic mycosis fungoides Characterized by patches, plaques, and hair loss within lesions; preferential involvement of head and neck More aggressive than early mycosis fungoides; 60%
Pagetoid reticulosis A solitary, hyperkeratotic plaque, usually on an extremity Generally indolent behavior, recurrences and relapses may occura,b
Granulomatous slack skin Localized areas of bulky folding of the skin, on axillae and groins Indolent course, slowly progressive with rare nodal involvementa
Sézary syndrome Diffuse erythroderma, generalized lymphadenopathy, presence of circulating Sézary cells 24%-43%
Sézary syndrome Diffuse erythroderma, generalized lymphadenopathy, presence of circulat-
ing Sézary cells
24%-43%
aMartinez-Escala ME, Gonzales BR, Guitart J. Mycosis fungoides variants. Surg Pathol Clin. 2014;7(2):169-89.
bHaghighi B, Smoller B, LeBoit P, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular and clinicopathologic study. Mod Pathol. 2000;13(5):502-10.
Clinically, folliculotropic MF presents with patches, plaques, and unusual hair loss within the lesions; occasionally, the disease may manifest with predominantly papular lesions. Folliculotropic MF preferentially involves the head and neck region and is characterized by folliculotropic T-cell infiltrates, with or without mucinous degeneration of the hair follicles. Previously, this variant was called follicular mucinosis or alopecia mucinosa. Folliculotropic MF affects mostly adults and is rarely observed in children and adolescents. Patients may have grouped follicular papules (Fig. 119-15A), acneiform lesions, indurated plaques, and, sometimes, tumors, which usually involve the head and neck region. The occurrence of hair loss within the lesions, most conspicuous on the eyebrows, an intense pruritus, and secondary bacterial infection are common. It is important to mention that 2 newer studies show that it is possible to distinguish 2 different types of folliculotropic MF, including a subtype with a favorable prognosis.64,65
Distinction of folliculotropic MF-associated follicular mucinosis from benign (idiopathic follicular
A
mucinosis) remains challenging. Although folliculotropic MF more probably displays a dense lymphocytic infiltrate with slight nuclear atypia, increased CD4-to-CD8 ratio, and a clonal rearrangement of TCR genes, the histologic or phenotypic features do not allow separating the 2 entities with certainty.
PAGETOID RETICULOSIS
Patients with pagetoid reticulosis present with a solitary psoriasiform or hyperkeratotic patch or plaque, which is usually localized on the extremities (Fig. 119-16) and is slowly progressive. Unlike in classic MF, extracutaneous dissemination has not been observed. Pagetoid reticulosis, listed as an MF subtype in the WHO classification, shows more prominent epidermotropism and nuclear pleomorphism compared with unilesional MF, and more commonly shows a CD8+ phenotype. Furthermore, pagetoid reticulosis manifests more often as a hyperkeratotic lesion.
B
2082
GRANULOMATOUS SLACK SKIN
Granulomatous slack skin is a rare subtype of MF characterized by localized areas of bulky folding of skin, with a predilection for the axillae and groin (Fig. 119-17). Light microscopy reveals a dense granulomatous infiltrate in the entire dermis. In addition to small, atypical cells with cerebriform nuclei, one finds macrophages and multinucleated giant cells
20
and loss of elastic fibres. The neoplastic cells express a CD3+CD4+CD8− phenotype.
SÉZARY SYNDROME
SÉZARY SYNDROME
DEFINITION
Sézary syndrome is characterized by the triad of diffuse erythroderma, generalized lymphadenopathy, and circulating malignant T cells with cerebriform nuclei, so-called Sézary cells. Sézary syndrome is a rare form of CTCL, accounting for 3% of all cutaneous lymphomas. In contrast to MF, Sézary syndrome carries an unfavorable prognosis, with a 5-year overall survival varying from 24% to 43%.1,63
CLINICAL FINDINGS
The erythroderma is often accompanied by severe scaling or fissuring of the palms and soles (see Fig. 119-10), alopecia, and onychodystrophy, and may be associated with marked exfoliation, edema, lichenification, and intense pruritus. In rare cases, hyperpigmentation occurs.
LABORATORY FINDINGS
Sézary syndrome demonstrates histologic features similar to those of MF, but repeated biopsies may be necessary as specimens often show nondiagnostic findings. The clonal T cells are generally CD3+, CD4+, and CD8− by multicolor flow cytometry. As in MF, the aberrant loss of T-cell antigens, including CD2, CD3, CD4, CD5, and CD7, is frequently observed. Of these, loss of CD7 expression is observed in approximately twothirds of cases. Loss of CD26 expression, observed in the majority of cases, is also useful in the identification of Sézary cells. The aberrant expression of the major histocompatibility complex class I-binding, immunoglobulin-like receptor (KIR) CD158k/KIR3DL2, normally expressed by NK cells, was described in the majority of patients with Sézary syndrome. In the current International Society for Cutaneous Lymphomas (ISCL)/EORTC TNMB staging classification, the diagnosis of Sézary syndrome requires an erythroderma with a positive T-cell clone in the peripheral blood associated with at least one B2 criterion, such as identification of more than 1000 Sézary cells/mm3 in the blood. Sézary cells were first described in 1938 by Sézary as large, atypical, mononuclear cells with lobulated, cerebriform nuclei. However, detection of Sézary cells by cytomorphology lacks specificity for the diagnosis of Sézary syndrome as they can be found in other inflammatory erythrodermas. Other diagnosis criteria include an expanded CD4+ T-cell population resulting in a CD4-to-CD8 ratio of more than 10; loss of any or all of the T-cell antigens CD2, CD3, CD4, or CD5; and lack of CD7 and CD26.66,67
2083
20
TREATMENT AND PROGNOSIS
Compared with patients with patch/plaque-stage MF, patients with Sézary syndrome have a markedly decreased 5-year survival rate. By the time the Sézary syndrome appears, there is very little normal immunity left. Indeed, Sézary syndrome patients often die because of infectious complications.
PRIMARY CUTANEOUS CD30+ LYMPHOPROLIFERATIVE DISORDERS
PRIMARY
CUTANEOUS CD30+
LYMPHOPROLIFERATIVE
DISORDERS
Primary cutaneous CD30+ lymphoproliferative disorders are the second most common form (20%-25%) of cutaneous lymphomas (CTCL). Primary cutaneous CD30+ lymphoproliferative disorders represent a spectrum of diseases, including lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma (ALCL).
LYMPHOMATOID PAPULOSIS Definition: Lymphomatoid papulosis was first described by Macaulay in 1968. It is an uncommon chronic disorder (prevalence of 1.2-1.9 cases per 1,000,000 persons) characterized by recurrent, selfhealing crops of papules and nodules.
A
Clinical Findings: Lymphomatoid papulosis is a chronic, recurrent, and self-healing papulonecrotic or papulonodular skin eruption (Fig. 119-18). The lesions typically involve the trunk and extremities, and lesions in various stages of evolution may be present concurrently.
Histopathology: Lymphomatoid papulosis is a clinically diverse disorder; in recent years a number of new pathologic and clinical variants have been described. The atypical cells express one or more T-cell antigens as well as the lymphoid activation antigen CD30 (Fig. 119-19). The WHO 2016 classification recognizes the original variants Types A, B, and C, as well as the more recently described Type D (mimics primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma) and the angioinvasive, angiocentric Type E. Lymphomatoid papulosis with chromosome 6p25 rearrangement (IRF4/DUSPP locus) was described by Karai et al, clinically characterized by localized papules and nodules, and histologically characterized by epidermotropic and nodular CD30+ cells. Appreciation of these variants is important, as histologically they can mimic very aggressive T-cell lymphomas, but they are clinically similar to other forms of lymphomatoid papulosis.68,69
Treatment and Prognosis: Because a curative therapy is not available and none of the available treatment modalities affects the natural course of the disease, the short-term benefits of active treatment should be balanced carefully against the potential side effects. Low-dose methotrexate (5-10 mg/week) is the most effective therapy to suppress development of new
B
C
2084
A
20
B
skin lesions. Treatment with PUVA has been reported to yield beneficial effects, but duration of response is often short-lived after discontinuation of treatment. Therefore, in patients with few, nonscarring lesions, long-term followup without active treatment should be considered.70
In general, lymphomatoid papulosis shows a benign clinical course and a favorable 10-year survival rate of nearly 100%. However, in a proportion of patients, estimated at 10% to 20% of cases, lymphomatoid papulosis can precede, coexist with, or follow malignant lymphoma, especially MF, Hodgkin lymphoma, and nodal ALCL. In many of these cases, the same clonal TCR rearrangements have been found in the lymphomatoid papulosis, as well as in the associated lymphoma. In the majority of lymphomatoid papulosis cases, despite the sometimes extremely long course of the disease, there is no evolution of a secondary lymphoma. Nevertheless, patients suffering from lymphomatoid papulosis should be monitored lifelong. In patients with lymphomatoid papulosis, monoclonal TCR rearrangement or histologic mixed-type may be prognostic for disease more likely to develop lymphomatoid papulosis–associated lymphomas.
CUTANEOUS ANAPLASTIC LARGE-CELL LYMPHOMA Definition: Cutaneous ALCL is characterized by large tumor cells, of which the majority express the CD30 antigen, with no evidence or history of MF or other type of primary CTCL. Regardless of the morphology of the tumor cells (eg. anaplastic, immunoblastic, or pleomorphic large cells), the clinical presentation and behavior are identical.
Clinical Findings: CD30+ cutaneous large-cell lymphomas occur in adults and rarely in children and adolescents, with a male-to-female ratio of 1.5:1. The clinical picture is characterized by the solitary or locoregional occurrence of reddish-to-brownish nodules and tumors, which frequently ulcerate (Fig. 119-20A). Cutaneous lesions may regress spontaneously.
Although secondary involvement of regional lymph nodes is observed in roughly 10% of patients, it is not necessarily associated with an unfavorable prognosis.
Histopathology: A nodular or diffuse nonepidermotropic infiltrate of large cells is seen in the dermis (see Fig. 119-20B). In the majority of cases, the neoplastic cells show an anaplastic morphology with oval or irregularly-shaped nuclei, prominent nucleoli, and an abundant cytoplasm. Less commonly, a pleomorphic or immunoblastic appearance is observed. Atypical mitotic figures are frequent. In the periphery of the lesions, inflammatory cells (eg. lymphocytes, eosinophils, and neutrophils) are present, sometimes mimicking the histologic picture seen in lymphomatoid papulosis. The most common phenotype in primary cutaneous ALCL is that of a CD4+ T-helper phenotype. In rare cases, tumor cells express CD8+, which does not seem to be associated with an impaired prognosis. By definition, more than 75% of the tumor cells express CD30 in cohesive sheets. In contrast to nodal ALCL, which expresses anaplastic lymphoma kinase (ALK) in approximately 60% of cases, primary cutaneous ALCLs are usually negative for this marker and lack the translocation t(2;5). Unusual cases of ALK+ primary cutaneous ALCL that are associated with a translocation variant and cytoplasmic staining for ALK have been reported. Improved criteria now exist for the recognition of ALK− ALCL in daily practice, and the actual WHO 2016 classification no longer considers this type provisional. Gene expression profiling studies show that ALK− ALCL has a signature quite close to that of ALK+ ALCL and distinct from NK/T-cell lymphomas. Newer studies illuminating the genetic landscape of ALK− ALCL have shown convergent mutations and kinase fusions that lead to constitutive activation of the JAK/STAT3 pathways. These studies provide a genetic rationale for the morphologic and phenotypic similarities between ALK+ and ALK− ALCL. However, not all cases of ALK− ALCL are created equal. A subset with rearrangements at the locus
2085
20
A
B
containing DUSP22-IRF4 in chromosome 6p25 tends to be relatively monomorphic, usually lacks cytotoxic granules, and is reported to have a better prognosis, whereas a small subset with TP63 rearrangement is very aggressive. Interestingly, the same locus in 6p25 is also implicated in lymphomatoid papulosis and primary cutaneous ALCL.3,69,71,72
Treatment and Prognosis: In cases of solitary or localized skin lesions, excision or radiotherapy is the treatment of choice. A successful treatment with PUVA in combination with interferon-α has been reported.
2086
If skin lesions are generalized, systemic therapy with methotrexate (20 mg/week) is preferred; vinblastine is an alternative option. In the case of extracutaneous dissemination, treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) is the most frequently chosen option. Brentuximab vedotin can also be regarded as a therapy of choice. This immunoconjugate is an anti-CD30 monoclonal antibody linked to monomethyl auristatin, a spindlecell toxin that induces cell-cycle arrest. A 100% response of primary cutaneous ALCL was observed with this treatment. In contrast to nodal ALCL, cutaneous CD30+ large- cell lymphomas have a favorable prognosis, with a disease-related 5-year survival rate of 90%.
SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA Definition: Subcutaneous panniculitis-like T-cell lymphoma is defined as a cytotoxic T-cell lymphoma characterized by the presence of primarily subcutaneous infiltrates of small, medium, or large pleomorphic αβ T cells and many macrophages that predominantly affect the legs and are occasionally complicated by a hemophagocytic syndrome. Subcutaneous lymphomas with a γδ phenotype of the TCR show a more aggressive course and are classified within the cutaneous γδ T-cell lymphomas.
Clinical Findings: This lymphoma accounts for 1% of all cutaneous lymphoma and for 75% of all subcutaneous forms of T-cell lymphoma. In the revised WHO 2016 classification, this term is, by definition, restricted to cases expressing a TCR α/β phenotype. Subcutaneous panniculitis-like T-cell lymphoma is characterized by subcutaneous nodules and plaques, which usually involve the extremities, the trunk, and, less commonly, the face. Patients may present with “B” symptoms, that is, weight loss, fever, and fatigue.
Histopathology: Histologic examination shows subcutaneous infiltrates simulating a lobular panniculitis. Infiltrates contain a mixture of neoplastic pleomorphic cells of various sizes and macrophages. Rimming of individual fat cells by neoplastic T cells is a helpful diagnostic feature. Immunophenotyping shows that the neoplastic cells express CD3+, CD4−, CD8+, CD56−, TIA-1+, granzyme-β+, and βF1+. The expression of βF1 (TCR α/β) by immunohistochemistry is a pivotal diagnostic marker for this entity.
Treatment and Prognosis: The α/β type of the subcutaneous panniculitis-like T-cell lymphoma responds well to systemic corticosteroids with an excellent prognosis (a 5-year survival rate of 85%), which justifies an initial treatment approach with corticosteroids alone.73-75
EXTRANODAL NATURAL KILLER/ T-CELL LYMPHOMA, NASAL TYPE Definition: Extranodal NK/T-cell lymphoma, nasal type, is a rare, aggressive form of primary cutaneous lymphoma that shares immunophenotypical characteristics with normal NK cells and characteristically displays a strong expression of CD56 and cytotoxic proteins, such as perforin, granzyme B, and TIA-1. This lymphoma is nearly always EBV+. The tumor cells are small, medium, or large, and usually have an NK cell or, more rarely, a cytotoxic T-cell phenotype. The skin is the second most common site of presentation after the nasal cavity.
Clinical Findings: Extranodal NK/T-cell lymphoma either affects the nasopharynx, which leads to destruction of the nasal region (formerly described as lethal midline granuloma), or manifests in skin, subcutis, lungs, viscera, and testes. Skin lesions comprise subcutaneous tumors, erythematous plaques, ulcers, or an exanthematous eruption with macules and papules (Fig. 119-21). The clinical course is often worsened by a hemophagocytic syndrome with pancytopenia.
Histopathology: This type of lymphoma shows dense infiltrates involving the dermis and often the subcutis. Epidermotropism may be present. Prominent angiocentricity and angiodestruction are often accompanied by extensive necrosis (Fig. 119-22). Immunophenotypically, the neoplastic cells express CD56 and cytotoxic proteins (TIA-1, granzyme B, perforin), and are characteristically positive for EBV. The TCR–CD3 complex is not expressed on the surface. Clonal episome presence of EBV is typically found. TCR genes are usually in germline configuration.3,76
20
Treatment and Prognosis: Even with aggressive polychemotherapy, the disease is often lethal within months. A study of the EORTC cutaneous lymphoma group suggested that bone marrow transplantation may be the treatment of choice.
PROVISIONAL ENTITIES OF CUTANEOUS T-CELL LYMPHOMA
PROVISIONAL ENTITIES
OF CUTANEOUS T-CELL
LYMPHOMA
DEFINITION
In addition to the diseases discussed in preceding sections, a number of provisional entities are included in the revised WHO classification system. These primary CTCLs display characteristic clinical and histologic features, but the series reported remain limited and do not allow definition of a precise outcome.
PRIMARY CUTANEOUS CD4+ SMALL AND MEDIUM T-CELL LYMPHOPROLIFERATIVE DISORDER Definition: This entity is defined clinically by papules and nodules, and histologically by a skin infiltrate composed of pleomorphic, small- and medium-sized T cells. The outcome is usually favorable but limited series have been reported.
Clinical Findings: Patients have one or several red-purplish papules or nodules with a predilection for the head and neck area. Because histologic differentiation from MF and MF-associated follicular mucinosis can raise problems, the absence of patches and plaques in pleomorphic small- and medium-sized CTCL is an important criterion.
Histopathology: Histologically, a dense, diffuse or nodular infiltrate containing small- to mediumsized pleomorphic cells is observed within the dermis and, sometimes, the subcutis. Epidermotropism may
2087
20
be present. The neoplastic cells express a T-helper cell phenotype with frequent loss of pan–T-cell markers. Demonstration of an aberrant phenotype and of T-cell clonality, as well as predominance of pleomorphic T cells in the infiltrate, serve as useful criteria for the exclusion of pseudolymphomas, which often show an identical histologic pattern. MF is excluded by the absence of a dominant cerebriform tumor cell population.
Treatment and Prognosis: Solitary lesions are often excised for diagnostic purposes. If excision is not possible or lesions are localized, radiotherapy is the preferred mode of treatment. PUVA therapy, possibly in combination with interferon (IFN)-α, is useful in cases with disseminated lesions. A 5-year survival rate between 60% and 90% is reported for this type of lymphoma.77-79
PRIMARY CUTANEOUS AGGRESSIVE EPIDERMOTROPIC CD8+ CYTOTOXIC T-CELL LYMPHOMA Definition: Primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma is a CTCL characterized by a proliferation of CD8+ cytotoxic T cells that exhibit a strong epidermotropism and aggressive clinical behavior. Differentiation from other types of CTCL expressing a CD8+ cytotoxic T-cell phenotype, as observed in pagetoid reticulosis and rare cases of MF, lymphomatoid papulosis, and cutaneous ALCL, is based on clinical presentation, histopathology, and clinical behavior.
Clinical Findings: Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma presents with hyperkeratotic patches and plaques, well-demarcated papules and tumors, or ulcerations. A metastatic spread to unusual sites, such as the lung, testis, CNS, and oral cavity, but not to the lymph nodes, is often observed.
Histopathology: Histologically, band-like infiltrates consisting of pleomorphic lymphocytes or immunoblasts are observed, displaying diffuse infiltration of the epidermis with variable degrees of spongiosis, intraepidermal blistering, and necrosis. The neoplastic cells express the Ki67 antigen at high frequency and are positive for CD3, CD8, CD45RA, and TIA, whereas CD2 and CD5 are frequently lost. Expression of TIA identifies these lymphomas derived from a cytotoxic T-cell subset.
Treatment and Prognosis: Even with multiagent chemotherapy, the disease shows an aggressive course and median survival is 32 months.76,79,80
CUTANEOUS γ/δ T-CELL LYMPHOMA Definition: Cutaneous γ/δ T-cell lymphoma comprises the peripheral T-cell lymphomas with a clonal proliferation of mature, activated γ/δ T cells with a
2088
cytotoxic phenotype. This group includes cases previously termed subcutaneous panniculitis-like T-cell lymphoma with a γ/δ phenotype.3
Clinical Findings: Patients have disseminated ulceronecrotic nodules or tumors, particularly on the extremities, but other sites also may be affected. Involvement of mucosal and other extranodal sites is frequent, but involvement of lymph nodes, spleen, or bone marrow is uncommon. A hemophagocytic syndrome may occur.
Histopathology: Histologically, 3 major patterns of involvement can be present in the skin: epidermotropic, dermal, and subcutaneous. The neoplastic cells are generally medium to large with coarsely clumped chromatin. Large blastic cells with vesicular nuclei and prominent nucleoli are infrequent. Apoptosis and necrosis are common, often with vessel invasion. Immunohistologically, the tumor cells have a βF1−, CD3+, CD2+, CD5−, CD7+/−, CD56+ phenotype with strong expression of cytotoxic proteins. Most cases are CD4 or lack both CD4 and CD8, although CD8 may be expressed in some cases. In frozen sections, the cells are strongly positive for TCRδ (antibody testing is not available for paraffin sections). If only paraffin sections are available, the absence of βF1 may be used to conclude a γ/δ origin.74,75
Treatment and Prognosis: Most patients have aggressive disease resistant to multiagent chemotherapy and/or radiation therapy. Median survival is 15 months.
PRIMARY CUTANEOUS ACRAL CD8+ T-CELL LYMPHOPROLIFERATION Definition: This entity is characterized by indolent cutaneous, CD8+ lymphoid proliferation originating predominately in the ear, with a favorable prognosis.
Clinical Features: Indolent cutaneous, CD8+ lymphoid proliferation, typically presents with solitary skin lesions on the face or at acral sites. Solitary papules or, in some cases, bilateral plaques, have been described on the feet.
Histopathology: Histologically, indolent CD8+ lymphoid proliferations are characterized by a dense dermal infiltrate of nonepidermotropic, medium-sized pleomorphic clonal CD8+ T cells, mostly of the nonactivated cytotoxic phenotype, showing, in the majority of cases, a clear cut grenz zone and a low proliferation index. Differentiation from otherwise aggressive T-cell lymphomas bearing a cytotoxic CD8+ phenotype is fundamental to avoid unnecessary anxiety for the patient and unwarranted aggressive treatment that would be considered as part of the therapeutic algorithm for the CD8+ phenotype. It has recently been suggested that CD68 could be a new discriminative marker, helpful in distinguishing
indolent CD8+ lymphoid proliferation from other CD8+ cutaneous lymphomas in ambiguous cases.
Treatment and Prognosis: Treatment is based on local excision or radiation therapy. Complete remission lasts for years and the prognosis is excellent.81
STAGING OF CUTANEOUS T-CELL LYMPHOMA
STAGING OF CUTANEOUS
T-CELL LYMPHOMA
After establishing the diagnosis of a CTCL, appropriate staging investigations are mandatory to exclude secondary involvement of the skin by an extracutaneous lymphoma and to determine the extent of disease. The first classification and staging system of CTCLs was published in 1979 by the MF Cooperative Group. It is recognized that this staging system does not apply to all CTCL types listed in the current WHO classification. Furthermore, the Ann Arbor system, commonly used for staging of nodal non-Hodgkin lymphomas, is not suitable for all CTCL types. Because of these facts and new data on prognostic factors, both revisions of the staging and classification for MF and Sézary syndrome and a TNM (tumor, node, metastasis) classification system for cutaneous lymphomas other than MF and Sézary syndrome have been proposed by the EORTC and the ISCL. It has to be mentioned that staging according to the TNM system has been proven to be useful for choosing an appropriate therapy for patients with MF and Sézary syndrome, but data correlating results of the TNM staging and prognosis are missing for some types of CTCLs. Staging examination for all types of CTCLs includes examination of the entire skin, chest radiography, and ultrasonography of abdominal organs and peripheral lymph nodes (cervical, axillary, and inguinal). Blood investigations should include complete blood cell count, clinical chemistry with liver enzymes, kidney function tests, and lactate dehydrogenase level, as well as T-cell clonality. Staging may be completed by CT scan and/or histologic and molecular (TCR rearrangement) investigations of suspicious lymph nodes and/or visceral organs. Staging examination should be repeated at relapse or progression of disease. A bone marrow examination is only recommended at a B2 blood rating (Table 119-7) or where there are unexplained hematologic abnormalities. However, this procedure is not of direct clinical relevance, as detection of atypical cells in the bone marrow is not an independent prognostic factor. The aforementioned investigations allow for classification according to the TNM system (see Tables 119-4 and 119-7). Although the prognostic value and applicability of TNM staging for different CTCLs is controversial, the TNM scheme directs the decision-making process toward an appropriate therapeutic regimen for most CTCLs.
20
STAGE T (TUMOR) N (LYMPH NODE) M (METASTASES) B (BLOOD)
IA T1 N0 M0 B0 or B1
IB T2 N0 M0 B0 or B1
IIA T1 or T2 N1 or N2 M0 B0 or B1
IIB T3 N0-N2 M0 B0 or B1
III T4 N0-N2 M0 B0 or B1
IIIA T4 N0-N2 M0 B0
IIIB T4 N0-N2 M0 B1
IVA1 T1-T4 N0-N2 M0 B2
IVA2 T1-T4 N3 M0 B0-B2
IVB T1-T4 N0-N3 M1 B0-B2
IVB T1-T4 N0-N3 M1 B0-B2
T1, patch/plaque on ≤10% of body surface; T2, patch/plaque on ≥10% of body surface; T3, skin tumor(s); T4, erythroderma; N0, normal nodes; N1, palpable nodes without clear histologic evidence of lymphoma (for N1 and N2, “a” or “b” may be added for either no [a] or detection [b] of a T-cell clone by Southern blot or polymerase chain reaction [PCR] analysis); N2, palpable nodes, histologic evidence of lymphoma, node architecture preserved; N3, palpable nodes with histologic evidence of lymphoma, effacement of node architecture; M0, no visceral involvement; M1, histologically confirmed visceral involvement. B0, ≤5% Sézary cells (for B0 and B1, “a” or “b” may be added for either no [a] or detection [b] of a T-cell clone by Southern blot or PCR analysis); B1, >5% Sézary cells but either less than 1.0 K/µL absolute Sézary cells or absence of a clonal rearrangement of the TCR or both; clonal rearrangement of the TCR in the blood and either 1.0 K/µL or more Sézary cells or one of the following 2: (a) increased CD4+ or CD3+ cells with CD4/CD8 of 10 or more or (b) increase in CD4+ cells with an abnormal phenotype (>40% CD4+/CD7− or >30% CD4+/CD26−).
PRINCIPLES OF TREATMENT OF CUTANEOUS T-CELL LYMPHOMA
PRINCIPLES OF TREATMENT
OF CUTANEOUS T-CELL
LYMPHOMA
Every successful strategy in managing CTCL begins with the correct diagnosis and staging. The prognosis and survival of patients does not only vary based on the type of cutaneous lymphoma but also on the stage; each lymphoma has its own best treatment to date, which is primarily stage based. Current staging is based on the proposal of the ISCL and the Cutaneous Lymphoma Task Force of the EORTC as published in the journal Blood in 2007.66
2089
20
T (Tumor)
■T1: solitary skin involvement
■T1a: a solitary lesion with a diameter <5 cm
■T1b: a solitary lesion with a diameter >5 cm
■T2: regional skin involvement: multiple lesions limited to 1 body region or 2 contiguous body regionsa
■T2a: all-disease-encompassing in a circular area with a diameter <15 cm
■T2b: all-disease-encompassing in a circular area with a diameter >15 but <30 cm
■T2c: all-disease-encompassing in a circular area with a diameter >30 cm
■T3: generalized skin involvement
■T3a: multiple lesions involving 2 noncontiguous body regions
■T3b: multiple lesions involving ≥3 body regions
N (Lymph Node)
■N0: No clinical or pathologic lymph node involvement
■N1: Involvement of 1 peripheral lymph node regionb that drains an area of current or prior skin involvement
■N2: Involvement of 2 or more peripheral lymph node regionsb or involvement of any lymph node region that does not drain an area of current or prior skin involvement
■N3: Involvement of central lymph nodes
M (Metastases)
M (Metastases)
■M0: No evidence of extracutaneous non–lymph node disease
■M0: No evidence of extracutaneous non–lymph node disease
■M1: Extracutaneous non–lymph node disease present
■M1: Extracutaneous non–lymph node disease present
aDefinition of body regions.61
bDefinition of lymph node regions is consistent with the Ann Arbor system: Peripheral sites: antecubital, cervical, supraclavicular, axillary, inguinal-femoral, and popliteal. Central sites: mediastinal, pulmonary hilar, paraortic, iliac.
the Cutaneous Lymphoma Task Force of the EORTC, and published in 2011.82 A global response score for MF and Sézary syndrome was established that addresses the entire TNMB spectrum, not only the response in the skin. However, as of this writing, there are still some problems regarding staging and prediction of survival. Indeed, survival does not always correlate with conventional staging: there is an overlap between Stages IIB versus III, and IB with folliculotropic MF. Many prognostic factors are still outside the staging system and have to be evaluated in prognostic trials. As of this writing, the most valid prognostic factors are age older than 60 years, tumor burden, and largecell transformation. A prognostic index model was published by Scarisbrick et al.63
The chronic disease course of the most frequently occurring CTCL subtypes, MF and Sézary syndrome, makes surrogate markers necessary and tumor burden is still the best surrogate marker for survival. Additional measures of symptoms, like pruritus or qualityof-life assessments, are commonly used; skin scores provide a measure of objective responses to therapy; and questionnaires guide the assessment of subjective responses to therapy. It has not been shown that
2090
reducing disease in a patient from T3 to T1 is accompanied by any survival benefit, yet it is also recognized that a cure is unattainable unless the patient is first in remission with a skin score of 0 by whatever skin scoring system is used. Thus, remission is the first step toward cure. Treatment of CTCL ideally requires a multidisciplinary team that includes dermatologists, radiation oncologists, and hematological-medical oncologists. The National Comprehensive Cancer Network, the EORTC, and the European Medical Society of Oncology have developed treatment guidelines that might be helpful in guiding therapeutic strategies. With a chronic disease like CTCL, supportive care, addressing quality of life, and reducing symptoms like pruritus and skin infections are mandatory for improving the quality of life of patients with CTCL.83,84
TREATMENT OF EARLY STAGE MYCOSIS FUNGOIDES
Treatment of early stage MF usually involves skindirected therapy, with or without systemic therapy. Established skin-directed therapy approaches consist of UVB phototherapy, PUVA phototherapy, topical corticosteroids, topical chlormethine, topical retinoids (eg, bexarotene), and radiation (eg, external beam radiotherapy and total skin electron beam therapy).
IMIQUIMOD AND RESIQUIMOD
Imiquimod and resiquimod are novel topical immune response modifiers belonging to the imidazoquinoline family of drugs. They are Toll-like receptor agonists that, when used topically or injected into lesions or tumors, may cause systemic effects. Both imiquimod and resiquimod induce synthesis and release of the cytokines IFN-α, tumor necrosis factor-α, IL-6, and IL-12 that all activate the adaptive immune response toward Th1 or the cell-mediated pathway, while inhibiting the Th2 pathway. Promising results have been reported with resiquimod.85,86
LOCAL AND TOTAL SKIN ELECTRON BEAM THERAPY
Total skin electron beam is perhaps the most effective of all skin-directed therapies. It is often used locally in patients with skin-limited disease, especially resistant plaques and tumors. Combined initial data of 11,065 patients with total skin electron beam therapy showed complete response rates close to 70%. Complete response rates are higher in patients with T1-limited disease where use of early, low-dose radiation toward solitary lesions may lead to a cure. However, total skin electron beam is usually reserved for patients with greater skin involvement than IA, especially
20
LEVEL OF EVIDENCE
Recommendations for First-Line Treatment of Mycosis Fungoides (MF) Stages IA, IB, and IIA
Expectant policy (mainly T1a) Level 4
SDTa Topical corticosteroids (mainly T1a and T2a) Level 3
UVB (ultraviolet B) (mainly T1a and T2a) Level 2
PUVA (psoralen and ultraviolet A) Level 2
Localized radiotherapy (RT) (for localized MF including pagetoid reticulosis) Level 4
Recommendations for Second-Line Treatment of MF Stages IA, IB, and IIA
Systemic therapiesb
Retinoidsc Level 2
Interferon (IFN)-α Level 2
Total skin electron beam (TSEB) (mainly T2b)
Level 2
Low-dose methotrexate (MTX)
Level 4
Recommendations for First-Line Treatment of MF Stage IIB
Systemic therapiesb
Retinoidsc Level 2
IFN-α Level 2
TSEB
Level 2
Monochemotherapy (gemcitabine, pegylated liposomal doxorubicin)
Level 4
Low-dose MTX
Level 4
Localized RTd
Level 4
Recommendations for Second-Line Treatment of MF Stage IIB
Polychemotherapye
Level 3
Allogeneic stem cell transplantationf
Level 3
Recommendations for First-Line Treatment of MF Stages IIIA and IIIB
Systemic therapiesb
Retinoidsc Level 2
IFN-α Level 2
Extracorporeal photochemotherapy (ECP)g
Level 3
Low-dose MTX
Level 4
TSEB
Level 2
Recommendations for Second-Line Treatment of MF Stages IIIA and IIIB
Monochemotherapy (gemcitabine, pegylated liposomal doxorubicin)
Level 3
Allogeneic stem cell transplantationh
Level 3
Recommendations for Treatment of MF Stages IVA and IVBi
Chemotherapy (gemcitabine, pegylated liposomal doxorubicin, CHOP, and CHOP-like polychemotherapy)j
Level 3
Radiotherapy (TSEB and localized)k
Level 4
Alemtuzumab (mainly in B2)
Level 4
Allogeneic stem cell transplantation
Level 3
Recommendations for First-Line Treatment of Sézary Syndrome
ECPp
Level 3
Chlorambucil + prednisone
Level 3
Systemic therapies in combination with ECP or PUVA
Retinoidsc Level 3
IFN-α Level 3
Low-dose MTX
Level 4
2091
(Continued)
20
(Continued)
LEVEL OF EVIDENCE
Recommendations for Second-Line Treatment of Sézary Syndrome
Chemotherapy (gemcitabine, pegylated liposomal doxorubicin, CHOP, and CHOP-like polychemotherapy)
Level 3
Alemtuzumab
Level 4
Allogeneic stem cell transplantationh
Level 3
Agents That Can Be Used for Maintenance After Remission Has Been Achieved in MF and Sézary Syndromel
ECP
IFN-α
Low-dose MTX
Mechlorethamine
PUVA
Retinoids
Topical corticosteroids
UVB
UVB
aSkin-directed therapy.
bThe following agents are most commonly combined with PUVA; combinations with other modalities and with each other are also widely used.
cIncluding retinoic acid receptor (RAR) and retinoid X receptor (RXR) agonists.
dUsed as add-on treatment in combination with systemic and other SDTs.
eCHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) is the most widely used regimen with a number of variants and other combinations available.
fShould be restricted to exceptional patients, see text for details.
gECP can be used alone or in combination with skin-directed and other systemic therapies.
hShould be restricted to exceptional patients.
iFor treatment of MF Stage IVA1, recommendations for Sézary syndrome might apply.
jMonochemotherapy should be preferentially used.
kUsed alone or in combination with systemic therapies.
lOptions are listed alphabetically and should be chosen to be effective, tolerable, easy to use, and efficient. Oxford Center for Evidence-Based Medicine levels are generally 5. Adapted from Trautinger F, Eder J, Assaf C, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update
2017. Eur J Cancer. 2017;77:57-74.
for extensive plaques or for palliation of Sézary syndrome or prior to nonablative allogeneic stem cell transplantation. In solitary nodules in Stage IIB disease, control may be achieved using a lower dose of 4 to 8 Gy administered for single refractory lesions or using low-dose 12 Gy total-body administration. Lower doses are effective and provide an opportunity for multiple treatments to be given without undue toxicity.87-89
MAINTENANCE THERAPY AND TOPICAL STEROID THERAPY
The concept of treating normal skin evolved from the clinical experiences with skin-directed therapies. There are 2 components to this approach: the treatment of normal skin during remission-induction skin-directed therapy and the treatment of normal skin during the remission-maintenance phase. Topical
2092
chemotherapy, PUVA, and total skin electron beam radiation all involve the exposure of normal skin as an integral component of their success in achieving remission. This success reflects the ability of the therapy to interrupt the critical skin-based phase of the lifecycle of a recirculating CTCL cell. Once remission has been achieved, normal skin can be maintained with lower doses and frequencies of the therapies used to clear it. Maintenance therapies have been described with PUVA, total skin application of nitrogen mustard, extracorporeal photochemotherapy (ECP), and IFN. The most commonly used maintenance therapy is PUVA or UVB irradiation. As a maintenance therapy, PUVA is initially administered at once-weekly intervals for 1 year. Beginning with the second year of treatment, the schedule is changed to every other week for another year, to every third week for the following year, and, finally, to every fourth week for 2 years. At this point, the patient should have been in remission for 5 years. Consideration should be given to stopping therapy at this point. A cure is defined as freedom from
disease for 8 years off all therapy. This definition arose from the experience with nitrogen mustard treatment and total skin electron beam radiotherapy showing that after a patient achieves a remission off therapy for 5 to 8 years, late relapse is extremely rare. This would imply that malignant cutaneous T cells recirculate without causing lesions for up to 5 years. With 5 years of intermittent PUVA, it is less likely that one of these cells will survive, but it is still possible. After therapy has been discontinued, patients should not be considered cured unless they remain clear of disease for 8 years.88,89
The management of suspected relapse often includes the use of topical glucocorticoids and reflects the critical role this modality can play in the treatment of suspicious lesions. Early in the course of CTCL and in a relapse of the disease in a patient in remission, the T-cell activation process can be blunted by the aggressive use of topical glucocorticoids. Indeed, most patients have a history of using these agents before a firm diagnosis is made. A regimen for treating early lesions of MF is twice-daily applications of a class I topical glucocorticoid for 8 weeks. This regimen is one of the first-line modalities for suspected relapse, and it can help to identify those patients who need to undergo a 4-week “washout” before repeated biopsies are performed.
ERYTHRODERMA
In erythrodermic CTCLs, immune dysfunction and inflammatory processes initiated by the malignant cells result in total skin redness, scaling, and discomfort. It is not surprising that immune-based therapies take the forefront in the management of these disorders. The 3 major biologic response modifiers (BRMs) used in the treatment of erythrodermic CTCLs are (a)
20
oral retinoids, (b) ECP via an intravenous route, and (c) subcutaneous injections of IFN-α (Table 119-10). In the clinical trials of these agents, patients have undergone monotherapy for what has usually been heavily pretreated refractory disease. In practice, these treatments are often used as first-line monotherapy in erythroderma, and other agents are incorporated for combination therapies if the response is incomplete. With these agents, partial responses are more common than complete responses. Thus, if the goal is remission, combination therapy is used more commonly than monotherapy. If the goal is palliation, monotherapy with a BRM is often sufficient. The BRMs differ in terms of their administration, side effects, interactions with other therapy modalities, and availability.83
RETINOID THERAPY
First-generation retinoids, such as isotretinoin, have limited effect on CTCL. The synthetic retinoid bexarotene binds the retinoid X receptor with high selectivity, whereas the other available retinoids have less-specific binding patterns. In monotherapy trials, bexarotene was used at 300 mg/m2. Responses were seen in patients in all stages of the disease: plaques, erythroderma, and tumors. Responses paralleled the secondary end points: decreases in overall body surface area involved and in overall tumor aggregate area, and improvement in pruritus. Erythrodermic patients may experience increased desquamation during the first few weeks of oral bexarotene therapy. Improvement typically starts by week 12 of therapy. Although there appeared to be a dose–response relationship with respect to efficacy, the higher dosages were also associated with a higher rate of adverse events and dose-limiting toxicities. Of these, hypertriglyceridemia, hypercholesterolemia, neutropenia, and
DOSE OPTIONS TIME TO EXPECTED CLINICAL RESPONSE SIDE EFFECTS AND ADVERSE REACTIONS
Retinoid therapy Bexarotene, 300 mg/m2 12 weeks Hypertriglyceridemia, hypercholesterolemia, neutropenia, central hypothyroidism, pancreatitis (reversible)
Extracorporeal photochemotherapy Photopheresis, with 8-methoxypsoralen and ultraviolet A, on 2 consecutive days every 4 weeks
Interferon-α therapy 3 million units, 3×/week, increasing to a maximally tolerated dose (∼9 million units/day)
Targeted monoclonal antibodies
4-6 months See Chap. 198 (“Phototherapy”)
3-6 months Flu-like symptoms, chronic fatigue; risk for long-term neurologic toxicity (depression, neuropathy, dementia, and myelopathy); thyroiditis, liver, bone marrow toxicity
Alemtuzumab Varies, according to protocol (see text) Cytomegalovirus reactivation, opportunistic infections at higher doses
Brentuximab
1.2-1.8 mg/kg every 3 weeks 12 weeks Peripheral neuropathy (reversible in most)
Brentuximab vedotin 1.2-1.8 mg/kg every 3 weeks 12 weeks Peripheral neuropathy (reversible in most)
vedotin
2093
20
central hypothyroidism were most common. Elevations in lipid levels occurred rapidly, within 2 to 4 weeks, and were associated with serious, but reversible, pancreatitis. Monitoring of lipids and the use of lipid-lowering drugs were helpful in controlling the lipid levels. Dosage reduction of bexarotene capsules was also required in some patients. Drug interactions of oral bexarotene with gemfibrozil and warfarin have been observed. Patients started on bexarotene therapy develop central hypothyroidism with low levels of thyroidstimulating hormone and free thyroxine within weeks of starting the medication. Symptoms of hypothyroidism may be subtle and include feeling fatigued and feeling cold, which may wrongly be attributed to the disease itself. Supplementation with levothyroxine while taking bexarotene alleviates the symptoms and improves tolerance of treatment. The condition is reversible within weeks of stopping therapy. There is no immunosuppression with bexarotene therapy. Patients taking bexarotene typically have monthly monitoring visits to follow lipid, liver, and thyroid parameters.90-94
EXTRACORPOREAL PHOTOCHEMOTHERAPY
Refer to Chap. 199, “Photochemotherapy and Photodynamic Therapy” for an in-depth discussion of ECP. ECP involves the treatment of a portion of a patient’s lymphocyte compartment with 8-methoxypsoralen in the presence of UVA light, followed by reinfusion of these cells. The treatment is performed via an intravenous line that feeds into an UVA-radiation device, and the procedure typically requires the patient to remain recumbent for 3 hours. Treatments are conducted on 2 consecutive days every 4 weeks. Erythrodermic CTCLs can be managed with ECP monotherapy, but treatment of other disease stages with monotherapy has not been rigorously studied. In a multicenter study involving erythrodermic CTCL patients, approximately onefourth had a complete response, one-fourth had no response, and the remainder had partial responses. However, it is clear that even a partial response can improve the quality of life of these patients. Improvement sometimes began as early as 6 weeks into therapy, but some patients did not show complete lesion clearance until 12 months after starting therapy. There were occasional temporary responses immediately after a 2-day cycle of therapy. On average, after 4 to 6 months there was typically a gradual and permanent decrease in erythema, scaling, and pruritus. Patients often notice more subtle changes, such as the return of body hair, loss of rigors, and a return of the ability to sweat. Partial responses may also decrease the morbidity these patients experience in terms of infectious complications. More heavily involved and inflamed skin is more readily colonized, providing both a reservoir and access point for microbes to invade the host. Thus, cutaneous improvement can also minimize complications of CTCLs.
2094
The clinical experiences with ECP for erythrodermic MF and Sézary syndrome suggest that the therapeutic response may well be based in the immune system. One feature is that when less than 5% of the malignant lymphocyte pool is photoinactivated with 8-methoxypsoralen and UVA light, clinical responses can be seen, with more than 95% of the malignant lymphocytes disappearing over time. It also appears that most immunocompetent patients respond. Patients, who were heavily pretreated, with longer disease durations, were less responsive. Also, patients with normal or only slightly decreased CD8+ lymphocyte levels were responsive to ECP. In one study, total skin electron beam radiotherapy was combined with ECP in patients with Stage T3 and Stage T4 disease. Comparison of patients receiving skin-directed radiotherapy plus BRM with historic controls who underwent electron beam radiotherapy at the same institution demonstrated the impact of ECP, because patients in the skin-directed therapy plus BRM group showed significantly longer survival.95,96
INTERFERON-α THERAPY
In the treatment of CTCLs, the most-studied IFN is IFN-α; however, clinical studies regarding the use of IFN-β and IFN-γ in the treatment of CTCLs have also been conducted. The initial studies using IFN-α as monotherapy showed rates of complete responses that varied from 10% to 27% with treatment durations of less than 6 months. Again, the heterogeneity of the disease and the pretreatments patients underwent before may affect outcomes and make comparisons with other modalities impossible. IFN-α is typically started at 3 million units, 3 times a week, and can be increased to a maximally tolerated dose, typically in the range of 9 million units/day. As with the other BRMs, the response to IFN is gradual, and 3 to 6 months are needed to determine the maximal response. After patients achieve a maximal response, IFN dosage can be lowered to a maintenance level. All IFNs have similar toxicities. Initially IFN therapy is complicated by a flu-like illness that is characterized by fever, headache, myalgia, and fatigue. As this wears off, patients are often left with a slight feeling of chronic fatigue. The long-term toxicity that causes most concern is neurologic: depression, neuropathy, dementia, and myelopathy. Autoimmune phenomena, such as thyroiditis, may occur. Furthermore, toxic effects of the liver and bone marrow may occur. Monitoring of IFN therapy includes blood counts along with questionnaires assessing the impact on the patient’s quality of life.97
TARGETED MONOCLONAL ANTIBODIES Alemtuzumab: Alemtuzumab is a humanized monoclonal antibody that targets CD52, expressed on most T and B lymphocytes and NK cells. Lundin et al97A reported a response rate of 50% to 70% in patients with CTCL. Because of prolonged depression of T, B, and NK cells in the original dosage schedule and
immunosuppression, this treatment led to reactivation of cytomegalovirus and other opportunistic infections. Alternative dosage schedules with lower doses and subcutaneous administration have been investigated. Querfeld et al97B reported a favorable response when intravenous alemtuzumab was followed with lowerdose subcutaneous antibody. A different schedule was proposed by Bernengo et al99; 4 patients received 3 mg alemtuzumab on day 1, 10 mg on day 3, and then 15 mg on alternate days. Bernengo et al reported 86% response rates in 12 of 14 refractory Sézary syndrome patients, including 3 complete remissions. However, Rei Watanabe98 showed that low-dose alemtuzumab is a highly effective and generally well tolerated therapy for refractory CTCL (ie, CTCL patients with peripheral blood disease). Low-dose alemtuzumab is effective in patients with blood involvement (leukemic disease) but ineffective in MF, reflecting the fact that MF derives from distinct T-cell subsets. Patients with malignant T cells with blood disease have the phenotype of CCR7+ L-selectin+ TCM that are migratory and recirculate between skin, blood, and lymph nodes, whereas MF T cells are derived from nonmigratory skin-resident TRM. Low-dose alemtuzumab depletes all circulating T cells and depletes skin in recirculating malignant T cells, leading to complete, and often durable, remissions in 50% of patients while sparing benign T cells in skin. Based on the clinical experience of Rachael Clark,98A alemtuzumab should be used in patients with or without adjuvant skin-directed therapy and in patients with diffuse cutaneous erythema, but some investigators caution against its use in patients with preexisting plaques and/or tumors.98,99
Brentuximab Vedotin: The antibody drug conjugate brentuximab vedotin is an anti-CD30 monoclonal antibody conjugated via a highly stable protease cleavable linker with monomethyl auristatin E, an antitubulin agent. It is U.S. Food and Drug Administration approved for the treatment of relapsed-refractory Hodgkin lymphoma and ALCL. In the past, CD30 has been identified as an excellent therapeutic target in lymphomas that express CD30. Histologically it is uniquely expressed in Hodgkin lymphoma, ALCL, and normal activated T, B, and NK cells, but is not expressed in normal tissues. Monoclonal antibodies that interact with CD30 are thought to induce apoptosis by initiating CD30 signaling. CD30 is highly expressed on the surface of primary cutaneous ALCL and subtypes of lymphomatoid papulosis and variably expressed in MF morphologic subtypes, including large-cell transformed MF. The first results of Phase II clinical trials in CTCL were published recently. In the Phase II trial of brentuximab vedotin in MF or Sézary syndrome by Kim et al,101 the overall global response was 70% among the 30 evaluable patients. CD30 expression assessed by immunohistochemistry was highly variable with a median CD30 maximum expression of 13% (range: 0%-100%); those with less than 5% CD30 expression had a lower likelihood of global response than those with greater
20
than or equal to 5% CD30 expression. The detection of abundant macrophages CD163+ M2 type in the tumor milieu suggested that brentuximab may target these macrophages in addition to the malignant T cells and disrupt their tumor-promoting function. The detection also suggested that these CD30-bearing macrophages may offer an additional source of monomethyl auristatin E to the nearby malignant T cells. The other Phase II study was published by Duvic et al100 in patients with CD30 lymphoproliferative disorder or CD30 MF/Sézary syndrome. The evaluable 48 patients showed an overall response rate of 73% and a complete response rate of 35%. The median time to response was 12 weeks, and duration of response was 32 weeks (range: 3-93 weeks). All patients with lymphomatoid papulosis or primary cutaneous ALCL responded. The most common side effect is the induction of peripheral neuropathy. Although reversible in most of the cases, some experienced irreversible neuropathy. The recommended dose of brentuximab vedotin is 1.8 mg/kg every 3 weeks; to overcome its side effects, a lower dosage of 1.2 mg/kg is recommended or prolongation of the interval between doses. Currently, brentuximab vedotin in the treatment of CTCL is being studied in an open-label Phase III ALCANZA Study Group trial,100A which is comparing brentuximab vedotin with physicians’ choices, either bexarotene or methotrexate, in patients with relapsed CD30+ MF or primary cutaneous ALCL.100,101
Mogamulizumab: Mogamulizumab is a defucosylated anti-CCR4 monoclonal antibody. It targets CCR4, a chemokine receptor that is preferentially expressed by Th2 and regulatory T cells in MF and Sézary syndrome patients. In response to its ligands, CCL17 (TARC) and CCL22 (MDC), CCR4 promotes T-cell migration to the skin. These CCR4 ligands are produced by keratinocytes, dendritic cells, and macrophages, and are abundant within MF/Sézary syndrome–involved skin. A recently described subset of peripheral T-cell lymphoma expresses CCR4 as part of the transcriptional repertoire of GATA-3, the master transcriptional regulator driving Th2 differentiation. GATA-3 is not only expressed by regulatory T cells, residing within various sites such as the skin, but also by MF/Sézary syndrome cells, and may drive CCR4 expression in these cells. In addition to its role in regulating cell homing and trafficking, CCR4 engagement may also promote cell growth and survival. However, cells can become desensitized to CCR4 through receptor internalization, a homeostatic regulatory mechanism. Clearly, CCR4 has a pathogenic role in MF/Sézary syndrome and other T-cell lymphoproliferative disorders and is an attractive therapeutic target. Mogamulizumab depletes CCR4-expressing cells by antibody-dependent cell-mediated cytotoxicity. Targeting the chemokine receptor CCR4, Duvic et al demonstrated that mogamulizumab is well tolerated and has significant clinical activity, with an overall response rate of 36% and a median duration of time to response of 1.4 months in heavily pretreated patients with MF
2095
20
and Sézary syndrome. The same group showed that in addition to antibody-dependent cell-mediated cytotoxicity, mogamulizumab depletes regulatory T cells, an important therapeutic target in many human cancers because of the role of regulatory T cells in suppressing host antitumor immunity. Mogamulizumab is currently being evaluated worldwide in a Phase III trial comparing mogamulizumab with the histone deacetylase inhibitor vorinostat.102,102A
SINGLE-AGENT CHEMOTHERAPY
More than 20 years ago, Bunn et al103 at the National Institutes of Health concluded that multiagent chemotherapy was not superior to sequential conservative therapies with respect to overall survival in advanced CTCL. Therefore, the therapeutic approach was more concentrated to reduce systemic side effects to a singleagent chemotherapy shown to be as effective. Choice of therapy is based on stage, concomitant medical condition, and prior treatment, as each agent has unique side effects and efficacy profile. Methotrexate, pegylated liposomal doxorubicin, gemcitabine, and pentostatin have all been studied in Phase II trials of CTCL patients.84
Gemcitabine: Gemcitabine hydrochloride, a nucleoside analog of deoxycytidine that inhibits DNA synthesis has shown activity against solid tumors as well as hematologic malignancies. In a number of small studies, gemcitabine, given as a dosage of 1200 mg/m2 administered for 3 or 4 weeks for 3 courses, has shown overall response rates of 70.5% with a median duration of 15 months. Duvic et al demonstrated that a lower dose of gemcitabine (1000 mg/m2 once per week for 3 weekly cycles) produced an overall response rate of 68% in 25 patients with advanced stage and refractory MF. It was especially active in MF patients with cutaneous tumors. Gemcitabine can be used in combination with bexarotene maintenance therapy to manage the plaques and patches of MF. It can also be alternated with liposomal doxorubicin infusion to prolong the duration of chemotherapy. The adverse effects of gemcitabine have most frequently involved bone marrow suppression (leukopenia), anemia, and, especially, thrombocytopenia.104,105
Pegylated Liposomal Doxorubicin: Pegylated liposomal doxorubicin is a new formulation of doxorubicin in which the drug is encapsulated in liposomes and stabilized by the attachment of polyethylene glycol (ie, pegylation) to the liposomal surface, resulting in increased half-life and improved accumulation in tumor tissues. Toxicity profile is characterized by dose-limiting mucosal and cutaneous adverse effects, in particular palmar-plantar erythrodysesthesia syndrome reporting up to 20% of treated patients. In a prospective multicenter controlled trial by the EORTC, patients with Stage IIB, IIIB, or IVA MF, refractory or recurrent after at least 2 previous systemic therapies, were treated with 6 cycles of pegylated liposomal doxorubicin 20 mg/m2 on days 1 and 15 over a single 28-day cycle. The primary end point was response rate.
2096
Among the 49 patients, the overall response rate was 48% with 6.1% complete remission. The median progression-free survival was 6.2 months. Toxicity (grades III to IV) was observed in 20% of the treated patients.106
The trial has produced benchmark data in a defined population of patients with MF in need of cytotoxic therapy. The efficacy is reasonable, and patients are allowed to use liposomal encapsulated doxorubicin as a debulking agent.
Pralatrexate: Pralatrexate, a novel antifolate with a high affinity for the reduced folate carrier (RFC-1), a novel mechanism of resistance when compared with methotrexate, was associated with an overall response rate of 29% in patients with relapsed or refractory peripheral T-cell lymphoma. A total of 12 patients with transformed MF were included in the study. Many of these patients had received more than 5 prior systemic therapies, including CHOP or CHOP-like regimens. However, results of a dose-finding study were reported in a larger cohort of CTCL patients. In this study, published by Horwitz et al,106A the optimal dose was identified as 15 mg/m2, given weekly 3 weeks out of 4, and was associated with an overall response rate of 43%. In an effort to reduce the incidence of mucositis, folic acid and vitamin B12 supplementation was routinely provided to these patients. However, pralatrexate has not been studied in a randomized trial against established compounds in CTCL.107
ALLOGENEIC STEM CELL TRANSPLANTATION
Younger patients with advanced stage CTCL (Stage IIB or greater) who fail to respond to first-line therapy are now being considered for nonablative allogeneic hematopoietic stem cell transplantation. Patients need to have a related or unrelated matched donor and be physically and emotionally able to undergo the procedure. The therapeutic concept is based on the existence of a graft-versus-T-cell-lymphoma effect particularly using nonmyeloablative conditions. Selected patients have achieved long-term remission and curative responses. Duarte et al reported on the long-term outcome of allogeneic hematopoietic stem cell transplantation for patients with MF and Sézary syndrome. These data show that patients with advanced stage MF and Sézary syndrome continue to benefit from allogenic hematopoietic cell transplantation over time, with an overall survival of 46% at 5 years and 44% at 7 years, and progression-free survival of 32% at 5 years and 30% at 7 years. Disease-relapse progression is a main cause of posttransplantation failure; a total of 45% of patients experience relapse progression at a medium of 3.8 months after hematopoietic stem cell transplantation. In a multivariable analysis, a number of disease and transplantation factors showed an independent impact on patient outcomes. The analysis focused on nonrelapse mortality, which was borderline associated with transplants from unrelated donors. Relapse
was highly significantly associated with myeloablative conditioning versus nonmyeloablative and with a poor performance score at hematopoietic cell transplantation (Karnofsky score <70). Patients must be treated with allogeneic transplantation after having achieved complete or nearly complete remission.108,109
PRIMARY CUTANEOUS B-CELL LYMPHOMAS
Primary CBCLs (Table 119-11) are B-lymphocyte– derived malignancies that develop in the skin without extracutaneous involvement at the time of diagnosis and account for 20% to 25% of primary cutaneous lymphomas. The incidence of CBCL is estimated at 3 cases per 1,000,000 persons per year based on surveillance epidemiology and end results registry data.110
The PCMZL/extranodal marginal zone lymphoma and the PCFCL are indolent subtypes, whereas the PCLBCL, leg type has an intermediate to aggressive clinical behavior. These 3 entities (ie, PCMZL, PCFCL, and PCLBCL) together comprise 97% of the CBCL.111-113
ETIOLOGY
ETIOLOGY
As for CTCL, an infectious trigger has been hypothesized to be involved in the etiology of CBCL. A strong
20
association of gastric extranodal marginal zone lymphomas and Helicobacter pylori infection is well known. In a minority of PCMZLs from European patients, but not in American or Asian patients, Borrelia burgdorferi has been detected. A newer study suggests that, based on immunoglobulin expression, 2 types of PCMZLs can be distinguished. In the most frequent PCMZL subtype, B cells expressing class-switched immunoglobulin (Ig) heavy chains, including IgG and to a lesser extend IgA or IgE, are observed. In a majority of cases, diffuse proliferation of IgM+ MCXCR3-expressing B cells is observed, which is associated with fewer infiltrating T cells and a skewed CD4-to-CD8 ratio toward an increased number of CD4+ T cells. In IgM+ cases, extracutaneous localization of disease is frequently observed, suggesting that cutaneous localizations of extranodal mucosal-associated lymphoid tissue from class-switched and non–classswitched immunoglobulin heavy chains result from distinct pathologic processes. Detailed studies on immunoglobulin heavychain expression showed that PCMZL showed IgG4 expression in 39% of cases. There was no evidence of IgG4-related disease in any of these patients pointing to a localized immunologic IgG4-driven process. In clinical management, these observations are helpful, as the marginal zone lymphomas presenting skin that expresses IgG4 will invariably be PCMZL. A viral agent, in particular hepatitis C virus, also is suspected to be relevant in the etiology of CBCL, but the results of the studies are contradictory.110-116
AGE AT DIAGNOSIS NATURE OF DISEASE CLINICAL FINDINGS CELL OF ORIGIN IMMUNOHISTOCHEMICAL CHARACTERISTICS TREATMENT
Primary cutaneous follicle center lymphoma (PCFCL)
Median age at diagnosis is 58 years
Indolent Solitary or grouped firm, painless erythematous plaques and tumors, head and trunk commonly affected
Extranodal marginal zone lymphoma (EMZL)/primary cutaneous marginal zone lymphoma (PCMZL)
Median age at diagnosis is 55 years; female predominant
Indolent Violaceous papules, plaques, or nodules; occur on multiple locations
Primary cutane-
Median age
Intermedi-
Solitary or clus-
Primary cutaneous diffuse large B-cell lymphoma, leg type
Median age at diagnosis is 65 years; female predominant
Intermediate to aggressive
Solitary or clustered bluish erythematous plaques and tumors located on 1 or both legs
ous diffuse large B-cell lymphoma, leg type
at diagnosis is 65 years; female predominant
ate to aggressive
tered bluish erythematous plaques and tumors located on 1 or both legs
aMay be Bcl2+ with nodal disease.
CD19+, CD20+, CD79a, CD10, Bcl6+a Radiotherapy; immune therapy for disseminated lesions
Clonal centrocytes and centroblasts
Small B lymphocytes, marginal zone cells, lymphoplasmacytoid cells, plasma cells
CD20, CD79a, Bcl2+; plasma cells may express CD138, CD79a
Radiotherapy; systemic antibiotics if Borrelia burgdorferi associated; chlorambucil, rituximab, intralesional interferon
Large B cells CD20+, CD79a, IRF4-
Rituximab, cyclo-
Large B cells CD20+, CD79a, IRF4- MUM-1+, FOXP1+, cytoplasmic immunoglobulin M ± D, Bcl2+, Bcl6+
Rituximab, cyclophosphamide, hydroxydaunorubicin, Oncovin, prednisolone, bendamustine + rituximab regimen
MUM-1+, FOXP1+, cytoplasmic immunoglobulin M ± D, Bcl2+, Bcl6+
phosphamide, hydroxydaunorubicin, Oncovin, prednisolone, bendamustine + rituximab regimen
2097
20
PATHOGENESIS
PATHOGENESIS
Because of the rarity of the CBCL subtypes, studies investigating pathogenetic events in CBCL have been mainly conducted on a small number of cases. However, in recent years, considerable progress in understanding the pathogenesis of CBCL has been made. A phenomenon called aberrant somatic hypermutation, which has been reported in nodal B-cell lymphomas, has been detected in the 3 main types of CBCL. This term describes the activity of the enzyme activation– induced deaminase, which contributes to the process of affinity maturation of immunoglobulins by somatic hypermutation, in regions of the genome that do not encode immunoglobulin genes. If this process occurs in oncogene-containing gene loci, in association with the loss of the physiologically high-fidelity DNA repair mechanisms, tumorigenic mutations may occur and contribute to lymphomagenesis.79
Furthermore, genetic investigations demonstrated distinct differences of CBCL subtypes. A genetic basis for differentiation of histologic skin PCFCL with a diffuse growth pattern of large cells and PCLBCL, leg type, was shown. The gene expression profiles of these entities were consistent with either germinal center B cells for PCFCL or activated B cells for PCLBCL, leg type. Similar results were found in a study focusing on proapoptotic and antiapoptotic genes. While PCLBCL, leg type with a poor prognosis had a genetic profile called activate apoptosis cascade, PCFCL and cases of PCLBCL, leg type with a favorable prognosis had a high expression level for genes that are associated with an antitumoral cytotoxic immune response. These findings also explain why PCFCL has a more favorable prognosis than PCLBCL, leg type. Furthermore, PCMZLs were found to arise in a different pathogenetic background than extranodal marginal zone lymphomas. The main differences were the high percentage of CXCR3− PCMZLs, which also, in
contrast to other extranodal marginal zone lymphoma, exhibited an immunoglobulin class switch and a Th2 cytokine milieu. It remains to be investigated how this relates to the presence of plasmacytoid dendritic cells in PCMZL, which were also found in cutaneous pseudolymphomas, but not in diffuse large B-cell lymphomas and only rarely in PCFCL.113,117-124
PRIMARY CUTANEOUS FOLLICULAR CENTER LYMPHOMA
PRIMARY CUTANEOUS
FOLLICULAR CENTER
LYMPHOMA
DEFINITION
PCFCL can be defined as a neoplasm of clonal centrocytes (small and large cleaved follicle center cells) and centroblasts (large follicle center cells with prominent nucleoli) with or without formation of follicles.
CLINICAL FINDINGS
PCFCL usually presents with solitary or grouped firm, painless, erythematous plaques and tumors that are preferentially located on the head and trunk, and rarely present on the leg. Patients with PCFCL have a median age of 58 years at diagnosis, and adult males are affected nearly twice as often as adult females. Occasionally, an annular erythema can be observed in the surrounding area (Fig. 119-23A). A typical finding is the occurrence of lesions in a circumscribed area of the head and neck region or the trunk, but rarely on the legs.
HISTOPATHOLOGY
These tumors are composed of medium to large follicle center cells containing a mixture of centrocytes and centroblasts arranged in a follicular or diffuse
A B
2098
growth pattern (see Fig. 119-23B). The neoplastic cells express the pan–B-cell markers CD19, CD20, and CD79a, and express the germinal center marker Bcl6. Expression of CD10 is particularly observed in cases with a follicular growth pattern. Unlike primary cutaneous B-cell lymphoma, leg type, expression of IRF4-MUM-1, FOXP1, and cytoplasmic immunoglobulin is generally not observed. In contrast to nodal follicular center lymphomas, in most studies PCFCLs uncommonly express Bcl2 and generally do not contain the translocation t(14;18). Gene expression studies demonstrate that PCFCLs have a gene expression profile resembling germinal center B-cell–like diffuse large B-cell lymphomas. Genomic studies found c-REL amplification in 63% of cases and deletions of 14q32.33 in 68% of cases.
TREATMENT AND PROGNOSIS
PCFCLs are indolent lymphomas that rarely metastasize to extracutaneous localizations and their prognosis is excellent with a 5-year survival of greater than 95%. Approximately 5% of PCFCLs develop on the legs and these cases have a more aggressive behavior. Radiotherapy (30 Gy) is the preferred treatment for PCFCL (Table 119-12). Relapses are observed in 30% of cases, but these do not herald an adverse prognosis and can be retreated with radiotherapy. For relapses a palliative dose of 4 Gy can be used, which will result in effective local control in 90% of cases. Immune therapy, such as administration of IFN-α or monoclonal antibodies against CD20, may be beneficial in cases with disseminated lesions.124,125
EXTRANODAL MARGINAL ZONE B-CELL LYMPHOMA
EXTRANODAL MARGINAL
ZONE B-CELL LYMPHOMA
DEFINITION
PCMZL or extranodal marginal zone lymphoma is an indolent B-cell lymphoma composed of small B lymphocytes, marginal zone cells, lymphoplasmacytoid cells, and plasma cells, which are initially localized in the marginal zone of a follicular center. In the current WHO classification, it is classified within the extranodal marginal zone lymphoma of mucosa-associated lymphatic tissue. In a subgroup of PCMZL, B. burgdorferi is thought to have an etiologic role.
CLINICAL FINDINGS
PCMZLs are seen most commonly on the upper extremities or the trunk and occur at a median age of 55 years; they predominate in females. Patients have small red-to-violaceous papules, plaques, or nodules that, in contrast to the lesions of PCFCL, frequently occur in multiple locations. PCMZL accounts for 25% of primary CBCLs.
20
DISEASE TYPE AND EXTENT FIRST-LINE THERAPY ALTERNATIVE THERAPIES
Primary Cutaneous Marginal Zone Lymphoma
Solitary/localized Local radiotherapy Interferon (IFN)-α intralesional
Excision Rituximab intralesional
Antibioticsa Intralesional steroids
Multifocal Wait-and-see IFN-α intralesional
Local radiotherapy Rituximab intralesional
Chlorambucilb Topical or intralesional steroids
Rituximab IV
Antibioticsa
Primary Cutaneous Follicle Center Lymphoma
Solitary/localized Local radiotherapy IFN-α intralesional
Excision Rituximab intralesional
Multifocal Wait-and-see R-CVP/CHOPc
Local radiotherapy
Rituximab IV
Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
Solitary/localized R-CHOP ± IFRT Local radiotherapy
Rituximab IV
Multifocal R-CHOP Rituximab IV
Multifocal R-CHOP Rituximab IV
Abbreviations: CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; IFRT, involved-field radiotherapy; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-CVP, rituximab, cyclophosphamide, vincristine, and prednisone.
aIn case of evidence for Borrelia burgdorferi infection.
bOr other single or combination regimens appropriate for low-grade B-cell lymphomas.
cIn exceptional cases or for patients developing extracutaneous disease. Adapted from Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112(5):1600-09, with permission.
HISTOPATHOLOGY
The histologic infiltrate includes marginal zone (centrocyte-like) cells, plasmacytoid cells, and plasma cells. Typically, the marginal zone cells express CD20, CD79a, and Bcl2, but are negative for CD5, CD10, and Bcl6. Plasma cells express CD138 and CD79a, and show monotypic cytoplasmic immunoglobulin lightchain expression on paraffin sections.118,119,126,127
TREATMENT AND PROGNOSIS
PCMZLs have been described in relation to tick bites and antigen injection, which suggests that chronic antigenic stimulation may play a role in the
2099
20
pathogenesis of these lymphomas. PCMZLs are indolent lymphomas and scattered lesions can be treated with radiotherapy (see Table 119-12). In case of detection of B. burgdorferi, a systemic antibiotic treatment should be given first. For widespread lesions a waitand-see approach can be adopted and symptomatic lesions can be treated with surgery, topical or intralesional steroids, or low-dose radiotherapy. In addition, systemic therapy with chlorambucil or rituximab and intralesional treatment with IFN-α or rituximab are reported to lead to complete responses in a majority of patients.117
PRIMARY CUTANEOUS DIFFUSE LARGE B-CELL LYMPHOMA, LEG TYPE
PRIMARY CUTANEOUS
DIFFUSE LARGE B-CELL
LYMPHOMA, LEG TYPE
DEFINITION
PCLBCL, leg type, has been identified as a distinct clinical entity because of its perceived poor outcome compared with the indolent subtypes described above. This entity shows an intermediate, and in some patients aggressive, clinical course. It is defined by tumors composed of large B cells that present in the overwhelming majority of cases on the legs, but can also arise at other locations. The majority of patients with PCLBCL, leg type, have aberrations on chromosome 9p21, and loss of this region, which contains the CDKN2A gene, is associated with a worse prognosis.128,129
A B
CLINICAL FINDINGS
PCLBCL, leg type affects elderly patients (older than age 65 years), with a predominance in females. Typically, patients have solitary or clustered bluish erythematous plaques and tumors located on one or, sometimes, both legs (Fig. 119-24A); approximately 10% of patients present with lesions at other sites than the legs. Ulceration is common and sometimes leads to the misdiagnosis of an ulcer from chronic venous insufficiency.
HISTOPATHOLOGY
The diffuse infiltrate shows sheets of immunoblasts and centroblasts with few mixed reactive cells (see Fig. 119-24B). The neoplastic cells express CD20 and CD79a, and, unlike PCFCL, are strongly positive for Bcl2, IRF4-MUM1, and FOXP1, and have cytoplasmic expression of IgM ± IgD. Bcl6 is expressed in most cases, whereas CD10 is generally absent. In line with the immunophenotypic profile, PCLBCL, leg type has a gene expression profile resembling activated B cells diffuse large B-cell lymphoma. In addition, PCLBCL, leg type expresses activation-induced cytidine desaminase, solves thermotic hypermutation of the immunoglobulin genes, and harbors mutations in the Bcl6, Myc, Rho-TTF, and PAX5 genes, which are indicative of ectopic somatic hypermutation. Translocations have been observed for Myc in up to 43% of PCLBCL, leg type cases, and Bcl6 in up to 46% of PCLBCL, leg type cases. Studies on copy number alterations describe high-level amplifications for the Bcl2 gene in 67% of cases and loss of CDKN2a in 23% to 42% of patients.
2100
Loss of CDKN2a expression either by gene deletion or promoter methylation correlates with an adverse prognosis.
TREATMENT AND PROGNOSIS
PCLBCL, leg type, belongs to the intermediateaggressive group of CBCLs and should preferentially be treated with rituximab, cyclophosphamide, hydroxydaunorubicin, Oncovin (vincristine), and prednisolone (R-CHOP) (see Table 119-12). In general, age-adapted polychemotherapy combined with rituximab showed a better outcome than polychemotherapy regimens alone.125 Because PCLBCL, leg type, occurs in elderly women (older than age 80 years), less-toxic regimens like bendamustine combined with rituximab could also be recommended. Despite this treatment, recurrences are observed in the majority of patients, and disease-related 5-year survival is approximately 50%.
PRIMARY CUTANEOUS DIFFUSE LARGE B-CELL LYMPHOMA, OTHER
PRIMARY CUTANEOUS
DIFFUSE LARGE B-CELL
LYMPHOMA, OTHER
PCLBCL, other, covers diffuse large B-cell lymphomas that do not belong to either PCLBCL, leg type, or PCFCL. These cases may represent a skin manifestation of systemic lymphomas. T-cell/histocyte-rich B-cell lymphomas with skin lesions only are also included; these cases show, in contrast to their nodal counterparts, an excellent prognosis.
INTRAVASCULAR CUTANEOUS B-CELL LYMPHOMA
INTRAVASCULAR
CUTANEOUS B-CELL
LYMPHOMA
Intravascular CBCL is characterized by clusters of large neoplastic B cells within dermal and subcutaneous blood vessels. Occasionally, slight extravascular infiltrates of atypical cells are observed. Clinically, red to bluish, indurated plaques occur on the legs or trunk. Sometimes, a panniculitis-like pattern can be seen. Multiagent chemotherapy is the preferred mode of treatment.130,131
STAGING OF CUTANEOUS B-CELL LYMPHOMA
STAGING OF CUTANEOUS
B-CELL LYMPHOMA
The International Society for Cutaneous Lymphoma and the Cutaneous Lymphoma Task Force of the EORTC developed a proposal for a tumor-node-metastases (TNM) classification for cutaneous lymphomas,
20
other than MF and Sézary syndrome (see Table 119-4). Recommended staging procedures for primary cutaneous B-cell lymphomas (PCBL) included thorough physical examination, laboratory studies, including a complete blood cell count, blood chemistry, including lactate dehydrogenase level, and, if indicated, a serum electrophoresis to exclude monoclonal gammopathy and/or flow cytometry on peripheral blood. In endemic regions, Borrelia serologic testing and polymerase chain reaction of skin biopsy specimens should be performed. Imaging studies include a contrast-enhanced CT scan with or without positron emission tomography, for chest, abdomen, and pelvis, and if lesions arose on the head and neck area, of the neck. Bone marrow biopsy and aspirate are required in PCFCL and PCLBCL, leg type, but are not required in patients presenting with a B-cell lymphoma in the skin with histologic features suggesting a follicular center lymphoma or marginal zone lymphoma, unless indicated by other staging assessments. However, a study by Senff et al131A
demonstrated bone marrow involvement at diagnosis in 22 (11%) of 193 PCFCLs in 9 patients. Bone marrow involvement was the only extracutaneous localization. Because these patients with skin and bone marrow localization had significantly worse prognoses than patients with only skin lesions, it was suggested that bone marrow examination should be considered in these patients. The proposed staging system for CBCL has been investigated in retrospective studies, demonstrating that the TNM system is a useful tool when documenting the extent of disease in CBCLs and for providing prognostic information in PCLBCL, leg type.
PRINCIPLES OF TREATMENT OF CUTANEOUS B-CELL LYMPHOMA
PRINCIPLES OF TREATMENT
OF CUTANEOUS B-CELL
LYMPHOMA
Treatment of primary CBCLs (see Table 119-12) should be adapted to the favorable prognosis of these lymphomas, in particular of PCFCL and PCMZL. Because no curative regimen has been defined as of this writing, therapy depends on the lymphoma and the dissemination of cutaneous lesions. In the case of solitary lesions, complete excision of the tumor may be proposed. Alternatively, or in the case of few localized lesions, local irradiation (a single dose of 3-4 Gy; total dose of 30-40 Gy) by X-ray or electron beam is effective. When this regimen is used, long-lasting remissions can be achieved.125,132
Systemic treatment is recommended for disseminated PCMZL or PCFCL with anti-CD20 antibodies. For relapse of indolent CBCLs with disseminated lesions a wait-and-see strategy combined with treatment of symptomatic lesions may be followed. Systemic treatment regimens are recommended in cases of PCLBCL, leg type, and in PCFCL with localization on the legs, as these PCFCLs have a worse
2101
20
prognosis, as well as when secondary extracutaneous manifestations are present. Polychemotherapy (6 cycles of CHOP or COP [cyclophosphamide, vincristine, and prednisone]) in combination with an anti-CD20 antibody is recommended. In patients who would not tolerate such an aggressive treatment, local radiotherapy or rituximab monotherapy may be considered.
PRECURSOR NEOPLASMS
BLASTIC PLASMACYTOID DENDRITIC CELL NEOPLASM
BLASTIC PLASMACYTOID
DENDRITIC CELL NEOPLASM
DEFINITION
According to the current WHO classification, blastic plasmacytoid dendritic cell neoplasm (BPDCN) is classified as an acute myeloid leukemia–related precursor neoplasm that derives from precursors of plasmacytoid dendritic cells. BPDCN is an orphan disease with a very aggressive clinical course that results in median survival times of 12 to 14 months.
CLINICAL FINDINGS
BPDCN usually occurs in elderly patients with a median age between 60 and 70 years. However, BPDCN can present at any age, even in children. More often BPDCN occurs in men (male-to-female ratio: 3:1) but has no known racial or ethnic predilection. Patients typically present with asymptomatic solitary or multiple skin lesions, such as nodules, plaques, or bruise-like lesions that can range in size from a few millimeters to 10 cm (Fig. 119-25). The skin lesions can be associated with erythema, hyperpigmentation, purpura, or ulceration. Extracutaneous disease is present in most patients at diagnosis, often involving the regional lymph nodes. As the disease continues to progress, the peripheral blood and bone marrow become involved.
2102
In 10% to 20% of patients with BPDCN, coincident myelodysplasia that can subsequently lead to the development of acute myelomonocytic leukemia is identified.
HISTOPATHOLOGY
In cutaneous lesions, BPDCN typically infiltrates the dermis but spares the epidermis. As the disease progresses, it frequently extends into the subcutaneous fat. The neoplastic cells tend to aggregate in the superficial to mid dermis in a perivascular and/or periadnexal distribution, although, less frequently, they may be seen as a lichenoid infiltrate in the superficial dermis. At high magnification, BPDCN is characterized by a monotonous population of small to medium cells with irregular nuclear contours, fine to evenly dispersed chromatin, 1 to 3 small nucleoli, and scant to moderate amounts of cytoplasm. By immunohistochemistry, the BPDCN cells typically express CD56, CD4, CD123, and T-cell leukemia/ lymphoma 1 (TCL1). They can also express other plasmacytoid dendritic cell–associated antigens, such as blood dendritic cell antigen 2 (BDCA-2)/CD303 and the IFN-α–dependent molecule MxA.
PATHOGENESIS
BPDCN cells demonstrate a recurrent recombination of deletions in several tumor-suppressor genes, including RB1, CDKN1B, CDKN2A, and tumor protein P53 (TP53). The TET2 gene (ten-eleven translocation-2) located on band 4q24 is mutated in BPDCN, myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myelomonocytic leukemia, which provides addition evidence that BPDCN is a myeloidrelated neoplasm. Targeted ultradeep sequencing revealed recurrent and mutually exclusive mutation of cancer genes in BPDCN. In 33 BPDCN samples, point mutations in NRAS (27.3%), ATM (21.2%), MET, KRAS, IDH2, and KIT (9.1% each) occurred. Moreover, NRAS, KRAS, and ATM mutations were found to be mutually exclusive. CDKN2A deletions were detected in 27.3% of the cases, followed by deletion of RB1 (9.1%). The mutual exclusive distribution of some mutations may point to different subgroups of BPDCN, the biologic significance of which remains to be explored.131,133,134
TREATMENT AND PROGNOSIS
BPDCN is associated with a highly aggressive clinical course and thus a poor prognosis (median survival of 14 months). Although systemic chemotherapy is the first choice for treatment of this disease, gemcitabine may be useful to control the initial disease so that the patient can be referred as fast as possible for bone marrow transplantation.135,136
Another possibly promising approach is to address the IL-3 receptor CD123 by an immunoconjugate.137
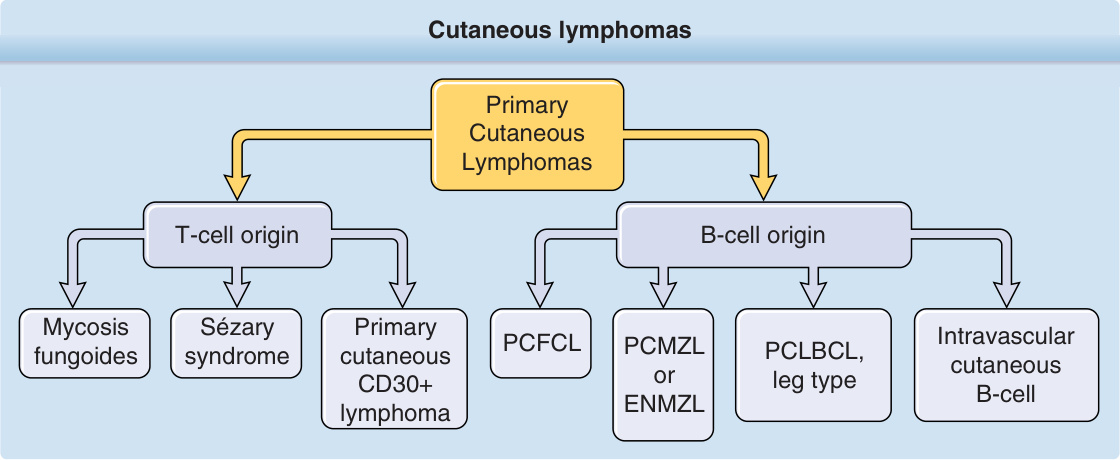
Figure 119-1 Cutaneous lymphomas. ENMZL, extranodal marginal zone lymphoma; PCFCL, primary cutaneous follicle center lymphoma; PCLBCL, primary cutaneous diffuse large B-cell lymphoma; PCMZL, primary cutaneous marginal zone lymphoma.
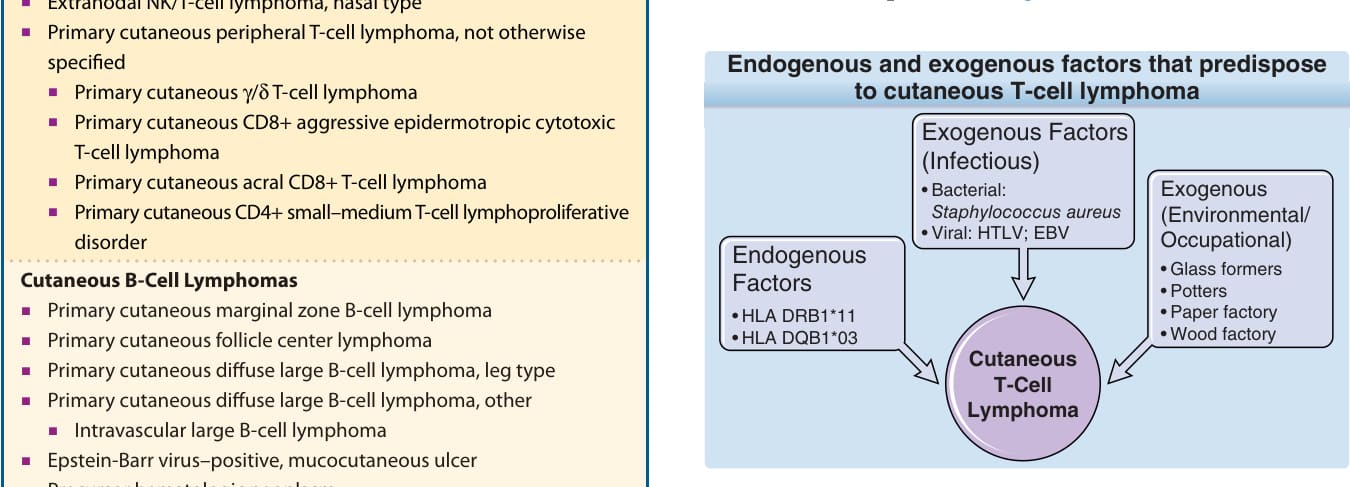
Figure 119-2 Endogenous and exogenous factors that predispose to cutaneous T-cell lymphoma. EBV, Epstein- Barr virus; HTLV, human T-cell lymphotropic virus.
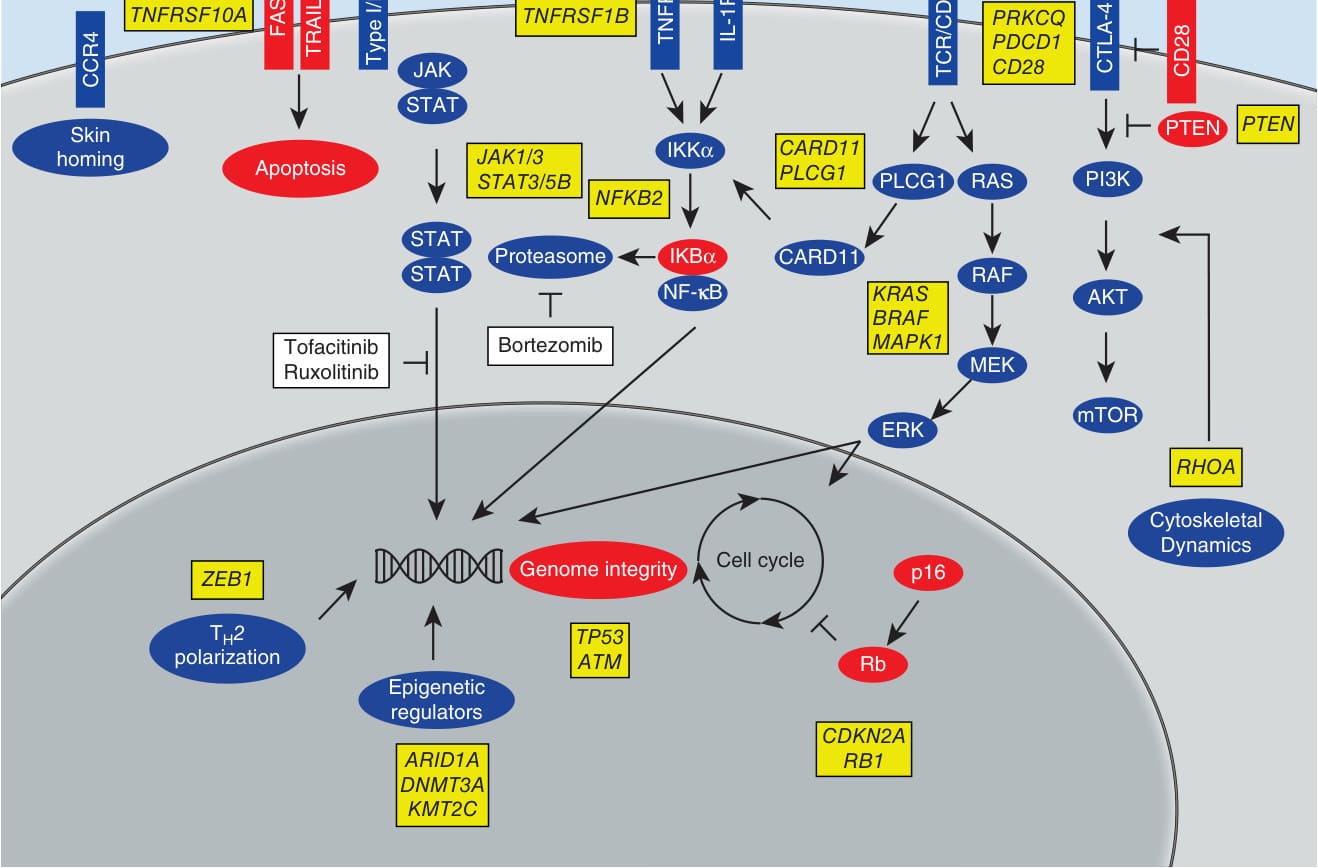
Figure 119-3 Signaling events in cutaneous T-cell lymphoma. (From Damsky WE, Choi J. Genetics of cutaneous T cell lymphoma: from bench to bedside. Curr Treat Options Oncol. 2016;17(7):33, with permission from Springer. Copyright © 2016.)
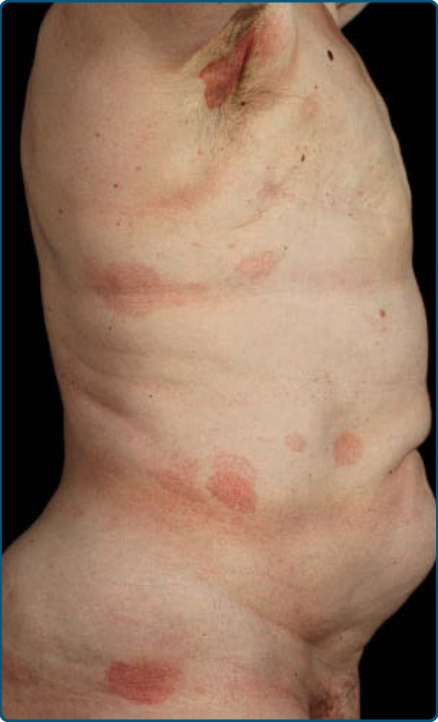
Figure 119-4 Patch lesions of mycosis fungoides in typical locations on the lateral trunk.
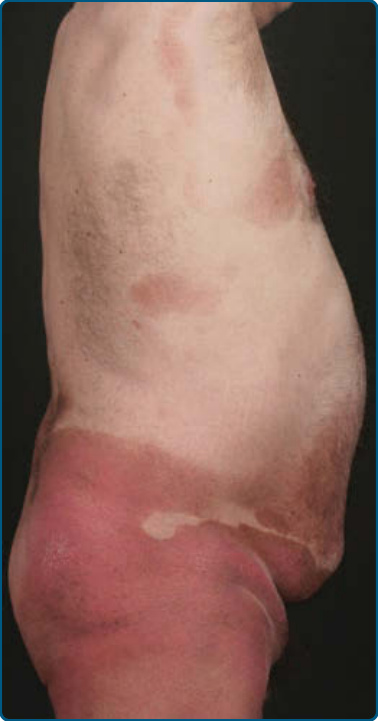
Figure 119-5 Large patch lesions of mycosis fungoides surrounding areas of uninvolved skin.
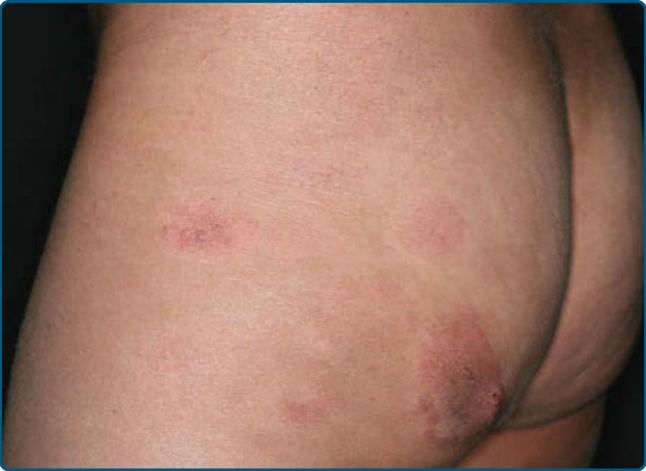
Figure 119-6 Polymorphic nature of mycosis fungoides. Patches and a plaque with a developing nodule on the left buttock.
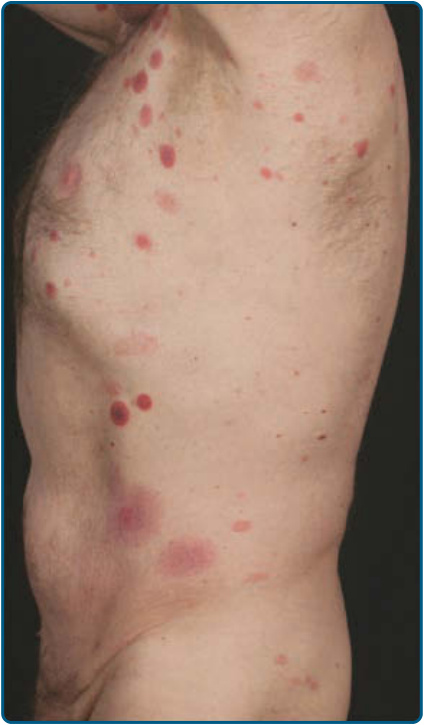
Figure 119-7 Multiple patches and plaques of mycosis fungoides on the lateral trunk. In this patient, the plaques developed rapidly and show partial central necrosis.
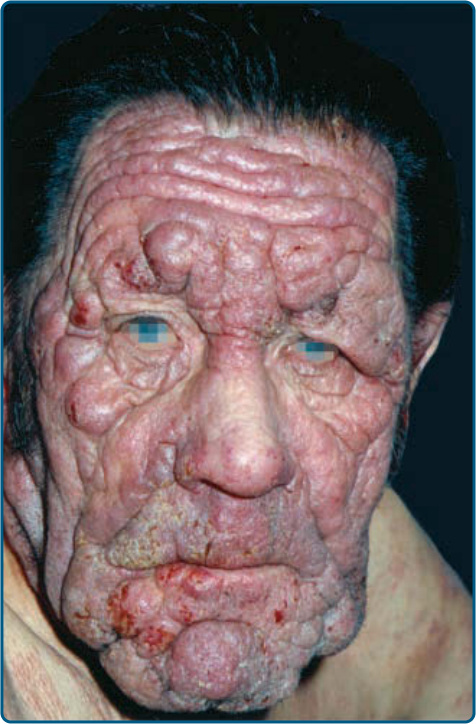
Figure 119-8 Leonine facies characterized by infiltrated plaques and tumors of cutaneous T-cell lymphoma.
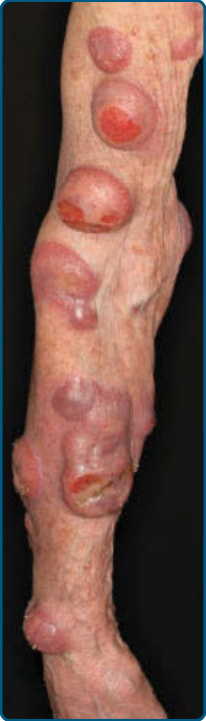
Figure 119-9 Tumor lesions of mycosis fungoides on the right arm, which are partially eroded and ulcerated.
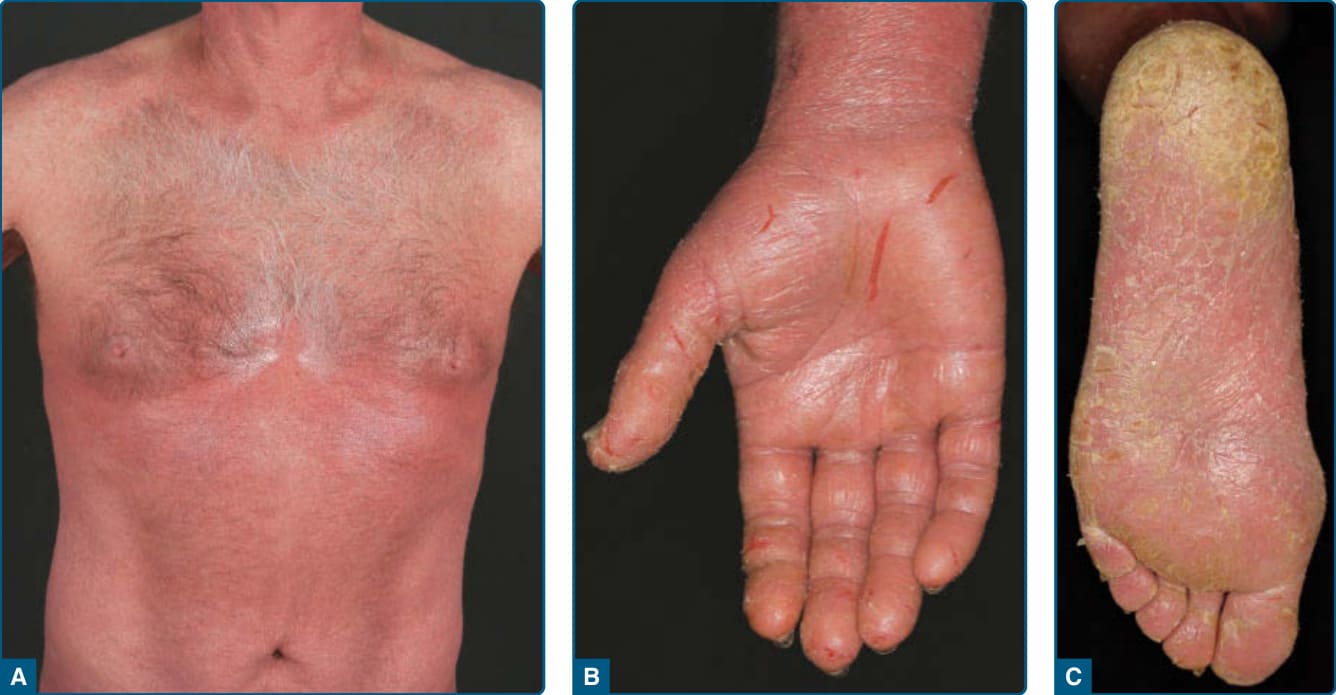
Figure 119-10 Sézary syndrome patient with erythroderma (A), palmar fissuring (B), and plantar hyperkeratosis (C).
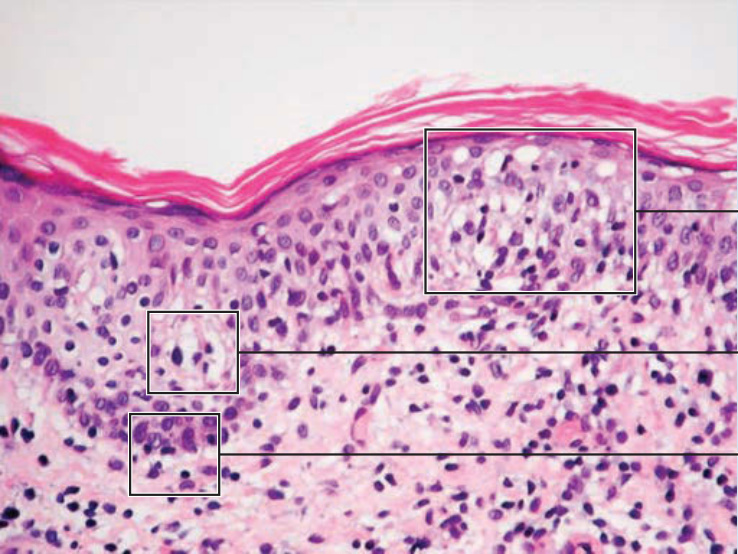
Figure 119-11 shows the characteristic histology of MF. In the patch, plaque, and also in the erythrodermic stage, there is a band-like infiltrate in the upper dermis composed of reactive T cells and neoplastic T lymphocytes, which are characterized by hyperconvoluted cerebriform nuclei. The neoplastic T cells show an epidermotropism with formation of intraepidermal Pautrier microabscesses (Figs. 119-12 and 119-13). In the tumor stage, a nodular infiltrate in the dermis is found, and the epidermal component is much less pronounced (Fig. 119-14). Immunohistologically, the malignant cells express a mature peripheral T-cell (CD4+) phenotype. Partial loss of pan–T-cell antigens
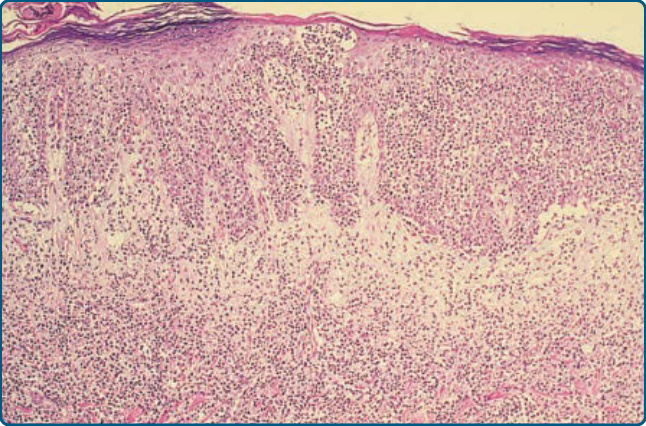
Figure 119-12 Dense mononuclear cell infiltrate extending from the papillary dermis into the epidermis. The epidermis is completely permeated by these cells, which form a Pautrier abscess.
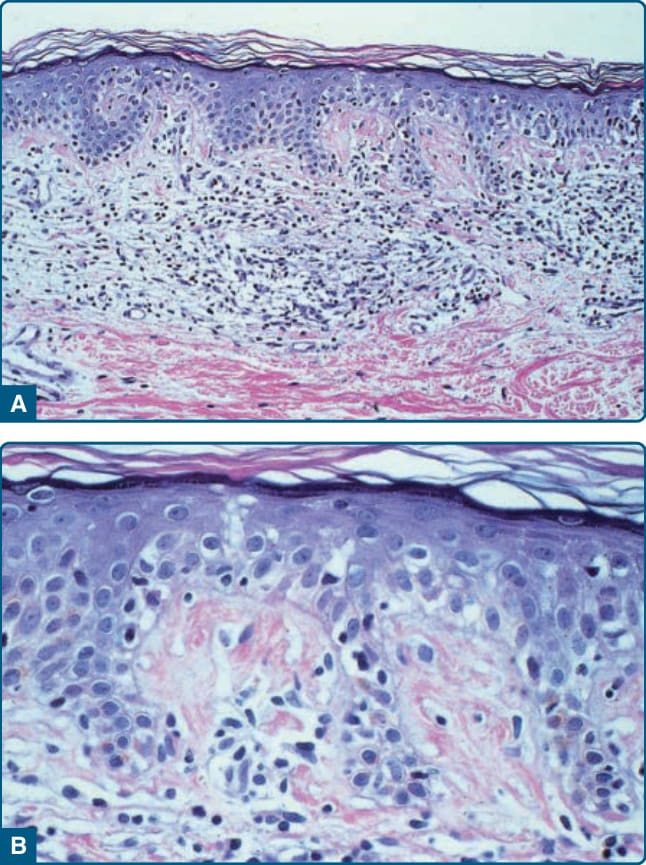
Figure 119-13 Patch stage of mycosis fungoides. A, Single atypical mononuclear cells in epidermis with sparse superficial perivascular infiltrate in the papillary dermis. (Hematoxylin and eosin-stained section.) B, High-power view of atypical cells in epidermis of same section.
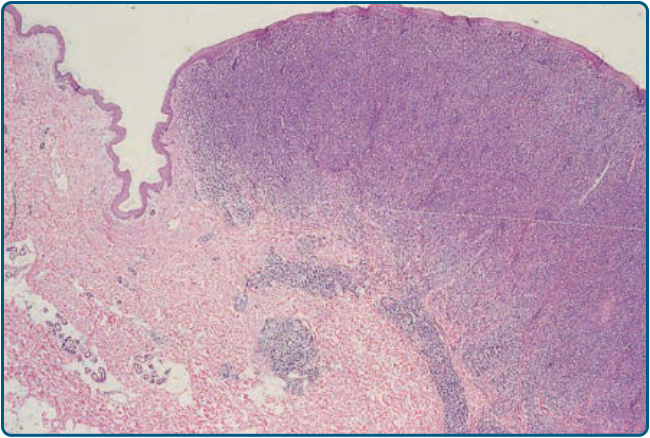
Figure 119-14 Low-power view of a mycosis fungoides tumor. The dense infiltrate extends deep into the dermis.
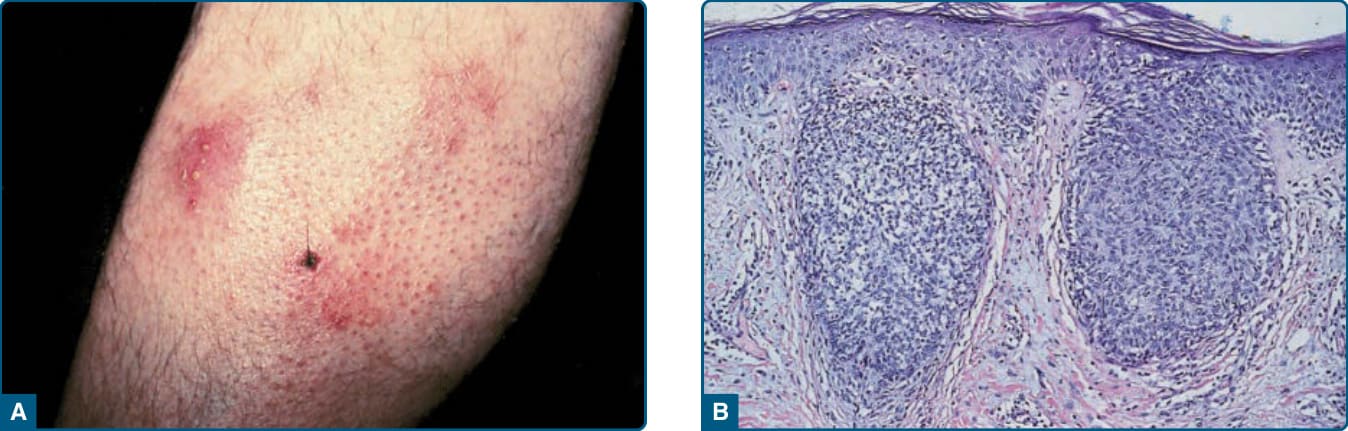
Figure 119-15 A, Folliculotropic mycosis fungoides. Note follicular localization and resulting hair loss. B, Outer root sheath of hair follicle is disrupted by T cells and mucin deposition, which leads to small cystic spaces.
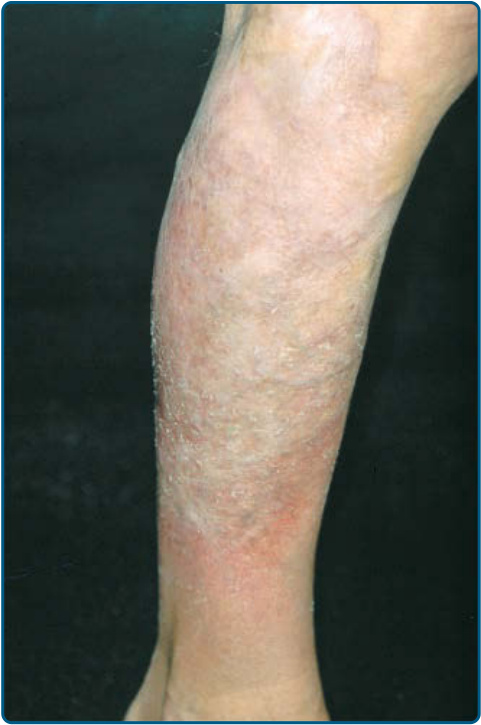
Figure 119-16 Pagetoid reticulosis. Hyperkeratotic plaque localized on the leg of a male patient.
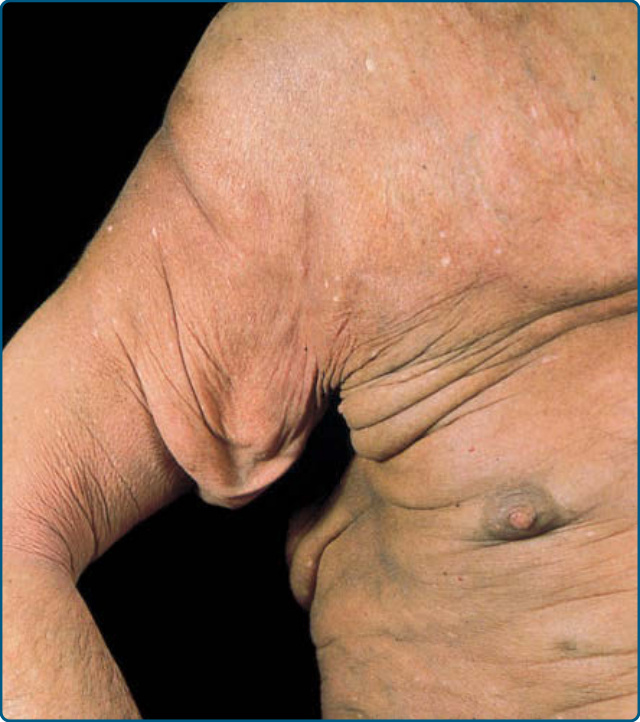
Figure 119-17 Granulomatous slack skin. Note skinfolds caused by secondary elastolysis.
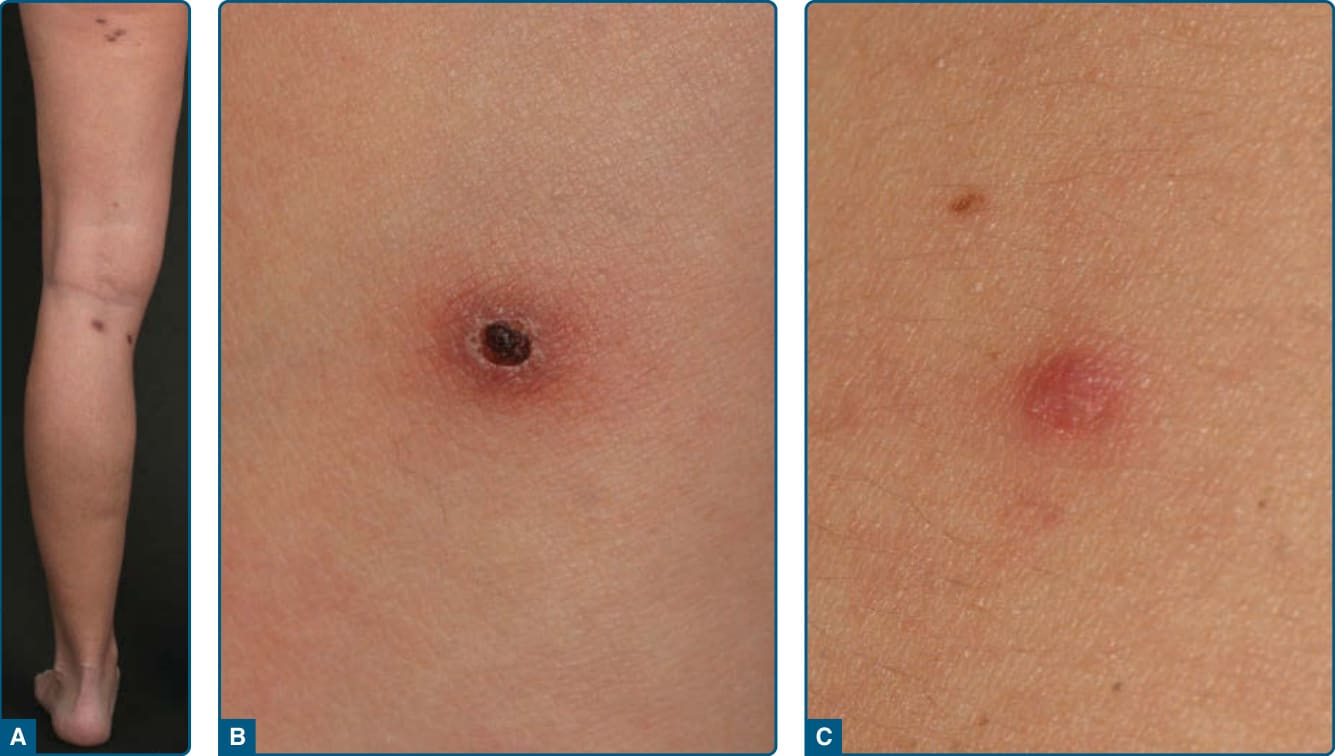
Figure 119-18 Lymphomatoid papulosis. A, Papular skin lesions on the right leg. The lesions may appear disseminated and grouped. B, Lymphomatoid papulosis papulonecrotic lesion. C, Lymphomatoid papulosis erythematous papulonodule.
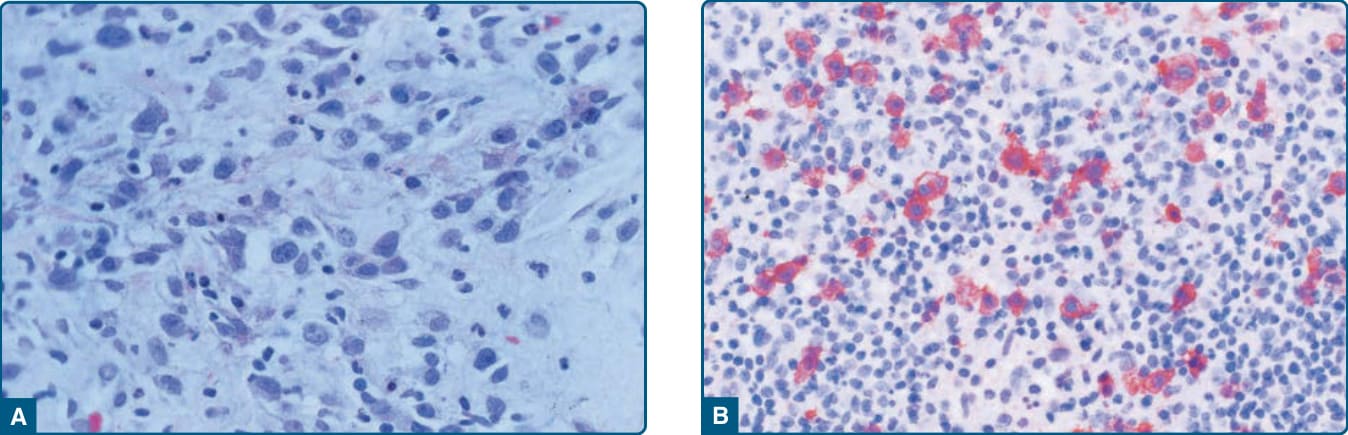
Figure 119-19 A, Lymphomatoid papulosis. Dermal infiltrate contains several large lymphoid cells with nuclei showing evenly dispersed chromatin and variably prominent nucleoli (so-called Type A cells). B, Type A with large CD30+ T cells (red) among admixed inflammatory cells.
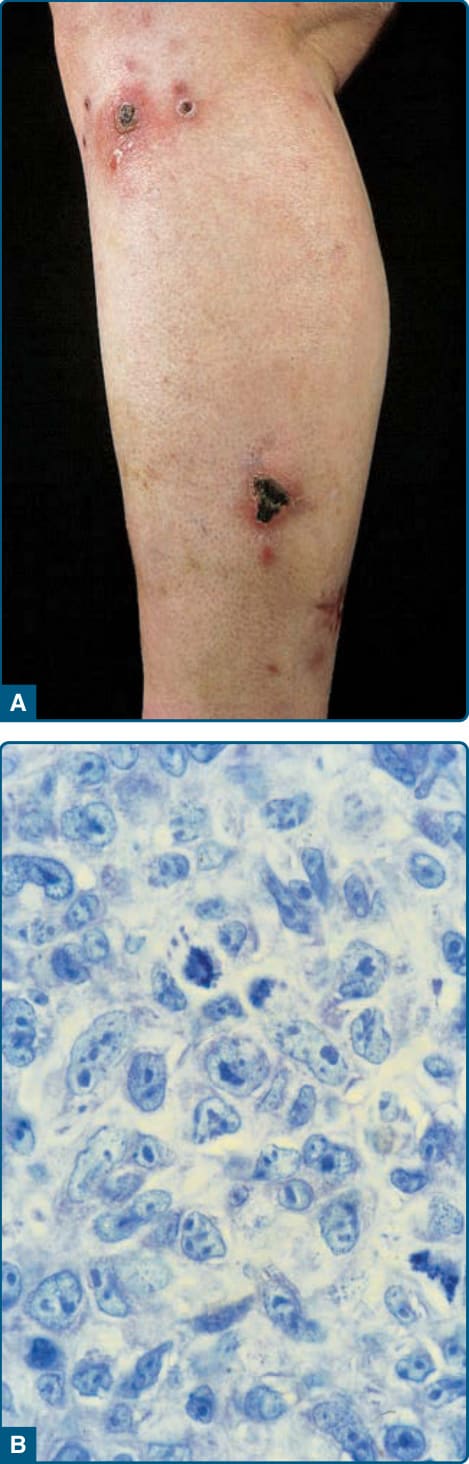
Figure 119-20 Primary cutaneous anaplastic large-cell lymphoma. A, Localized nodules, some with ulcerations. B, Pleomorphic large-cell lymphoma T-cell infiltrate.
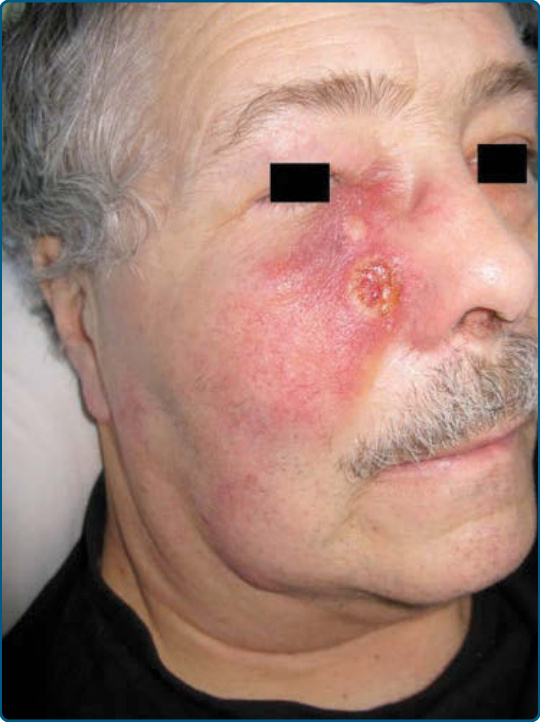
Figure 119-21 Extranodal natural killer/T-cell lymphoma, nasal type. Infiltrated and ulcerated plaque.
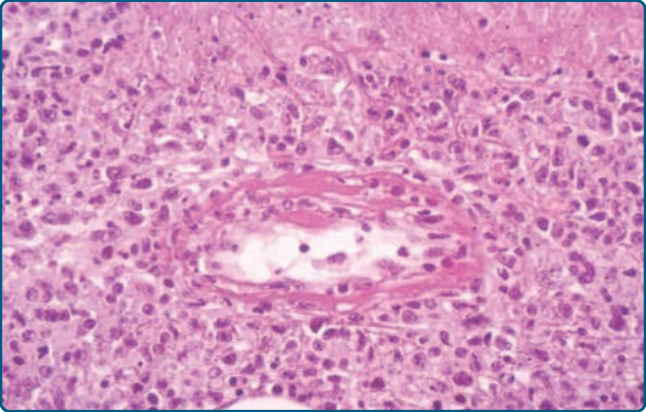
Figure 119-22 Extranodal natural killer/T-cell lymphoma, nasal type. Dense angiocentric infiltrate.
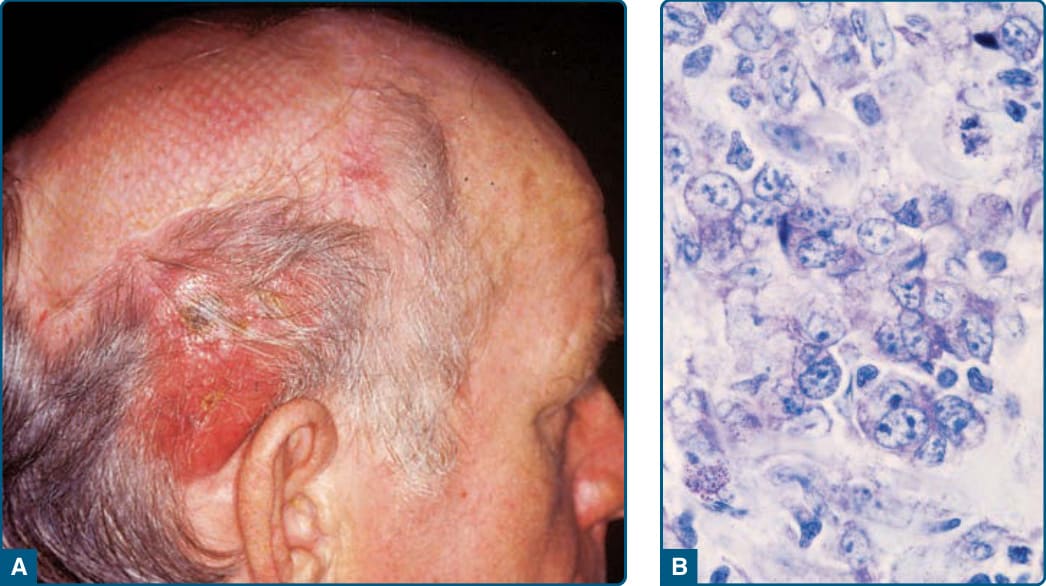
Figure 119-23 Primary cutaneous follicle center lymphoma. A, Tumor in the head area surrounded by erythema. B, The cellular infiltrate consists of a mixture of centrocytes, a number of centroblasts, and reactive T cells.
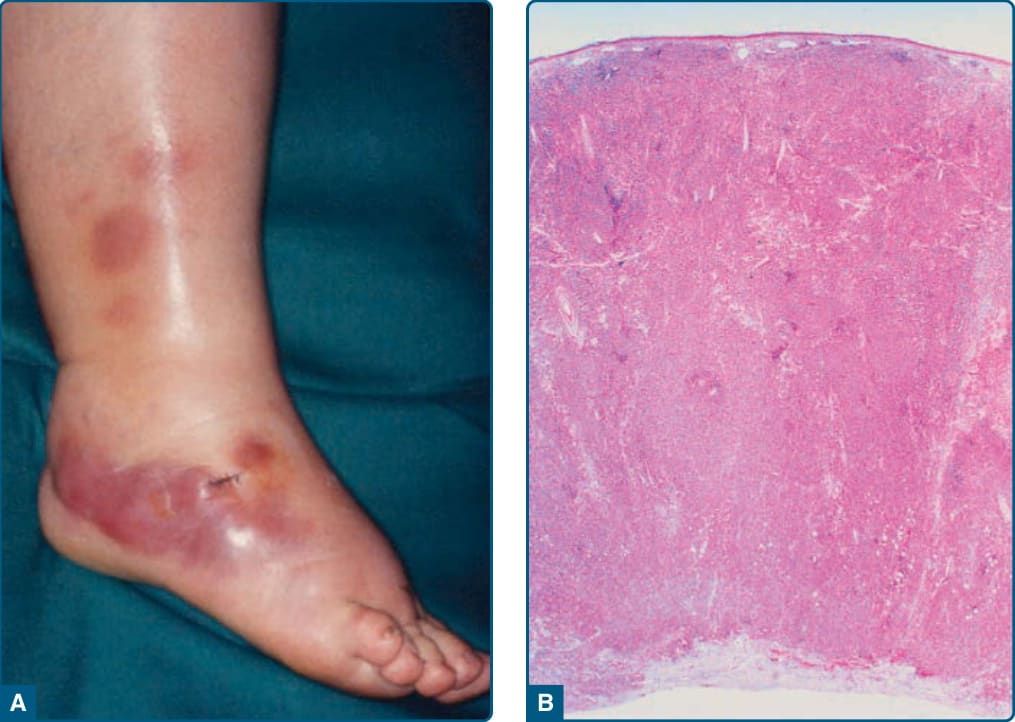
Figure 119-24 Primary cutaneous diffuse large B-cell lymphoma, leg type. A, Nodules and tumors on the right leg. B, Histology shows a diffuse infiltrate of large B cells with centroblasts, large centrocytes, and numerous immunoblasts.
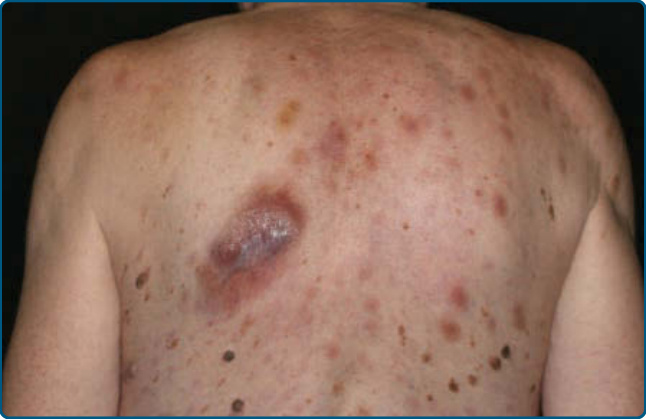
Figure 119-25 Blastic plasmacytoid dendritic cell neoplasm. Multiple lesions on the back. The large tumor was the initial site of manifestation (centrally located is the scar of a biopsy). The multiple small lesions developed in a short period of time.
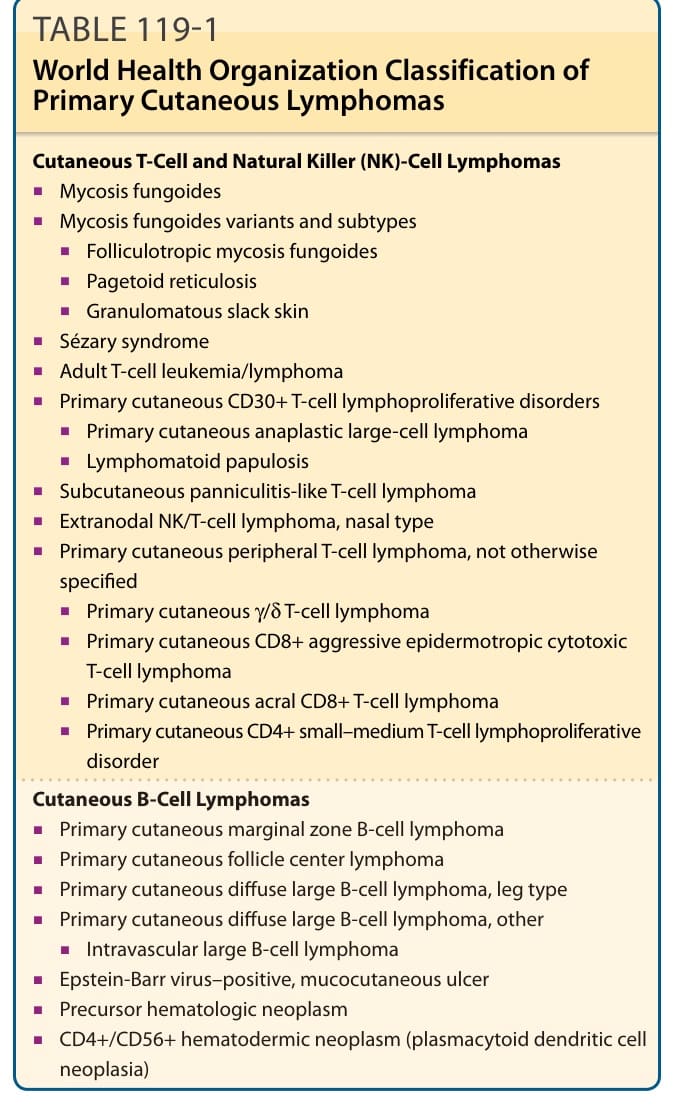
TABLE 119-1 World Health Organization Classification of Primary Cutaneous Lymphomas

TABLE 119-2 Subtypes and Variants of Mycosis Fungoides

Table 119-3 presents an algorithm for diagnosis of early MF. TOX (thymocyte selection-associated high-mobility group box factor) is associated with development of CD4+, CD8− T cells in the thymus, but is suppressed in mature CD4+ T cells. It is aberrantly expressed in MF and Sézary syndrome. It was thought to help in discriminating between early MF lesions and biopsy from inflammatory skin disorders. According to recent data it could contribute the diagnosis of CTCL; however, TOX expression is not restricted to CD4+, CD8− neoplastic T cells.61

Table 119-4 summarizes the recommended evaluation/initial staging of the patient with MF/Sézary syndrome.
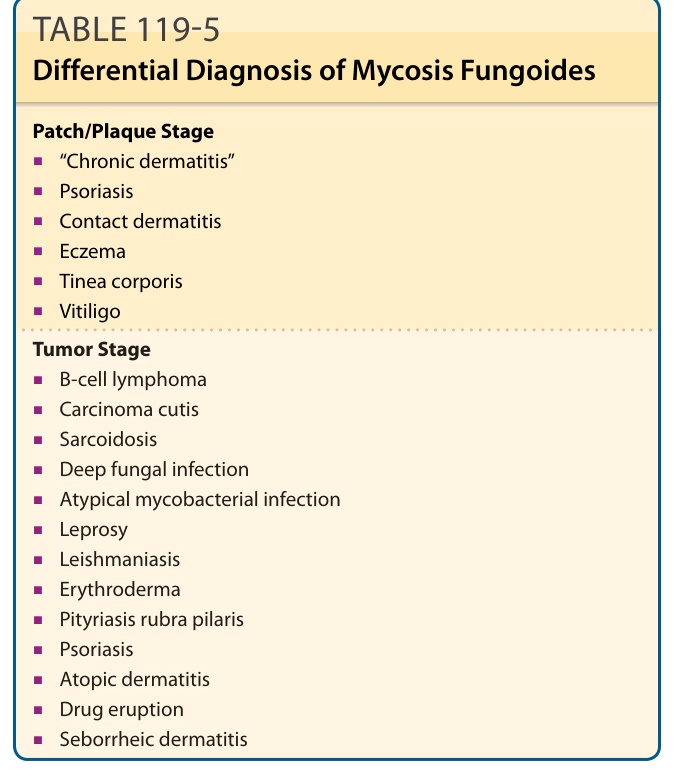
Table 119-5 summarizes the differential diagnosis of MF.

Table 119-6 outlines the clinical findings and prognosis for MF and variants.
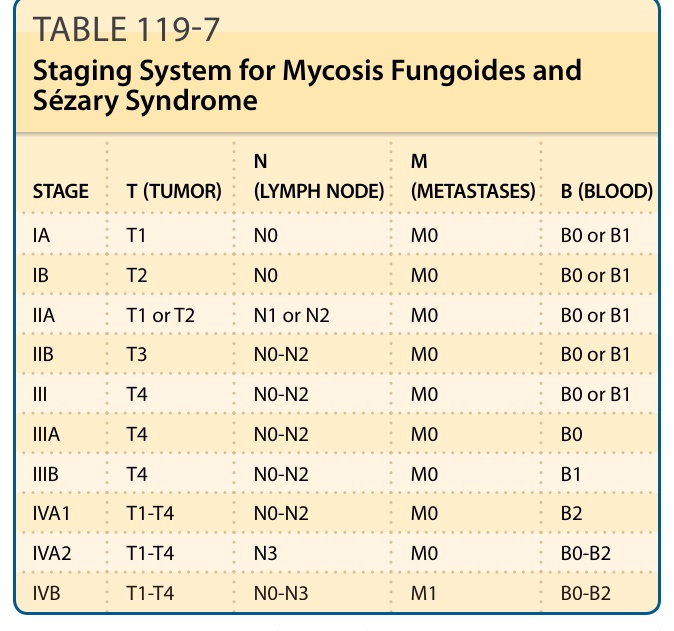
TABLE 119-7 Staging System for Mycosis Fungoides and Sézary Syndrome
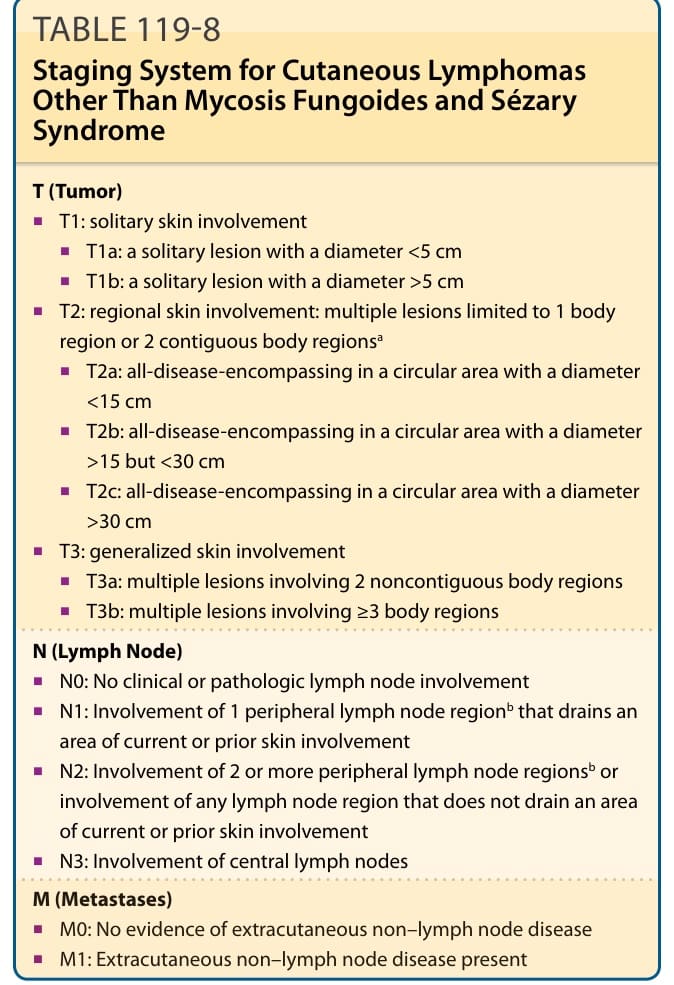
Table 119-8 outlines the international staging system for cutaneous lymphomas other than MF and Sézary syndrome. Once the individual classification of TNMB (tumor, node, metastasis, blood) has been determined, then these cases can be rolled into the staging system for MF and Sézary syndrome. To assess the response to a given treatment, a consensus statement of criteria was established by the International Society of Cutaneous T-cell Lymphomas, the United States Cutaneous Lymphoma Consortium, and
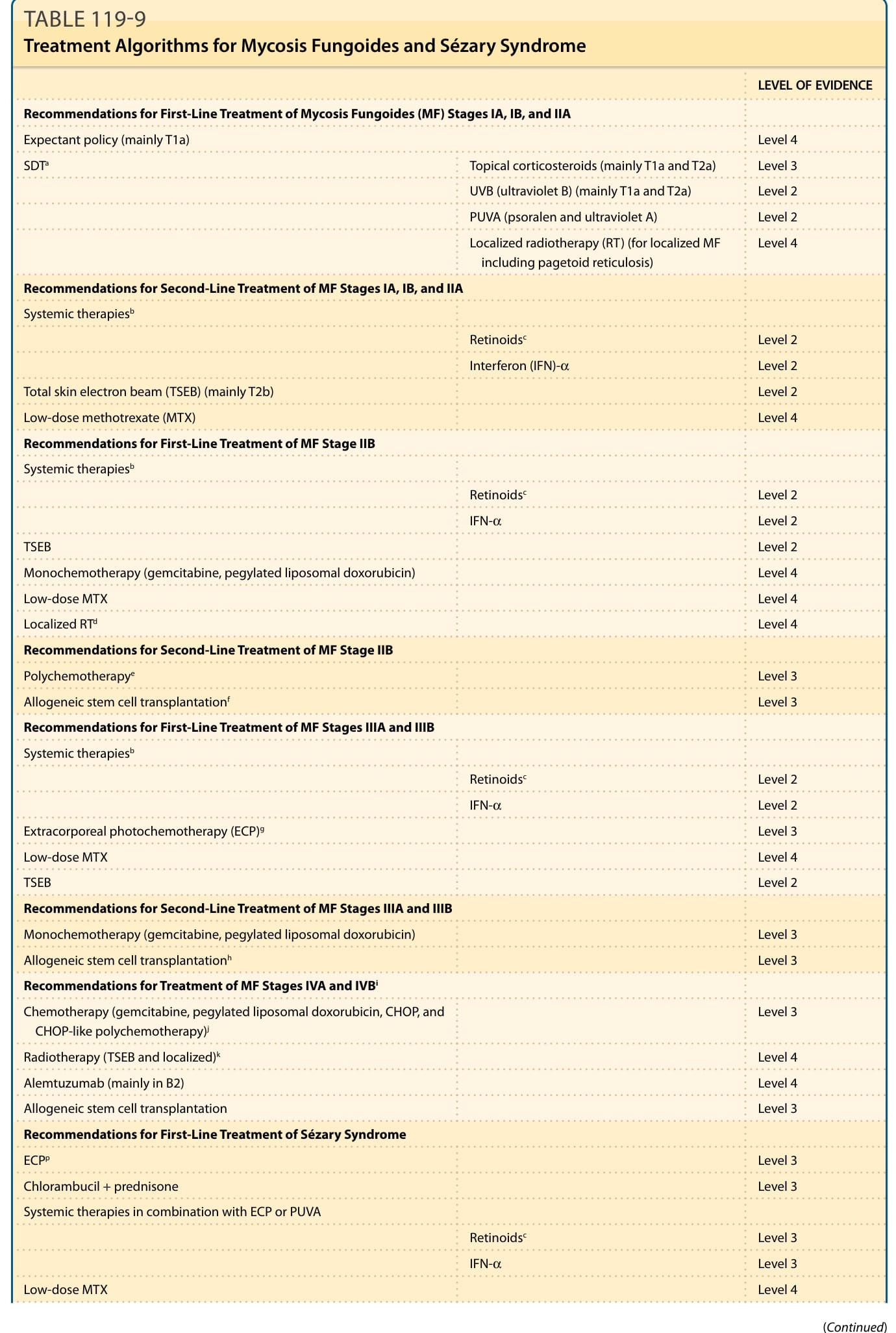
Table 119-9 outlines treatment algorithms for MF and Sézary syndrome.

TABLE 119-10 Biologic Response Modifiers for the Treatment of Erythrodermic Cutaneous T-cell Lymphomas
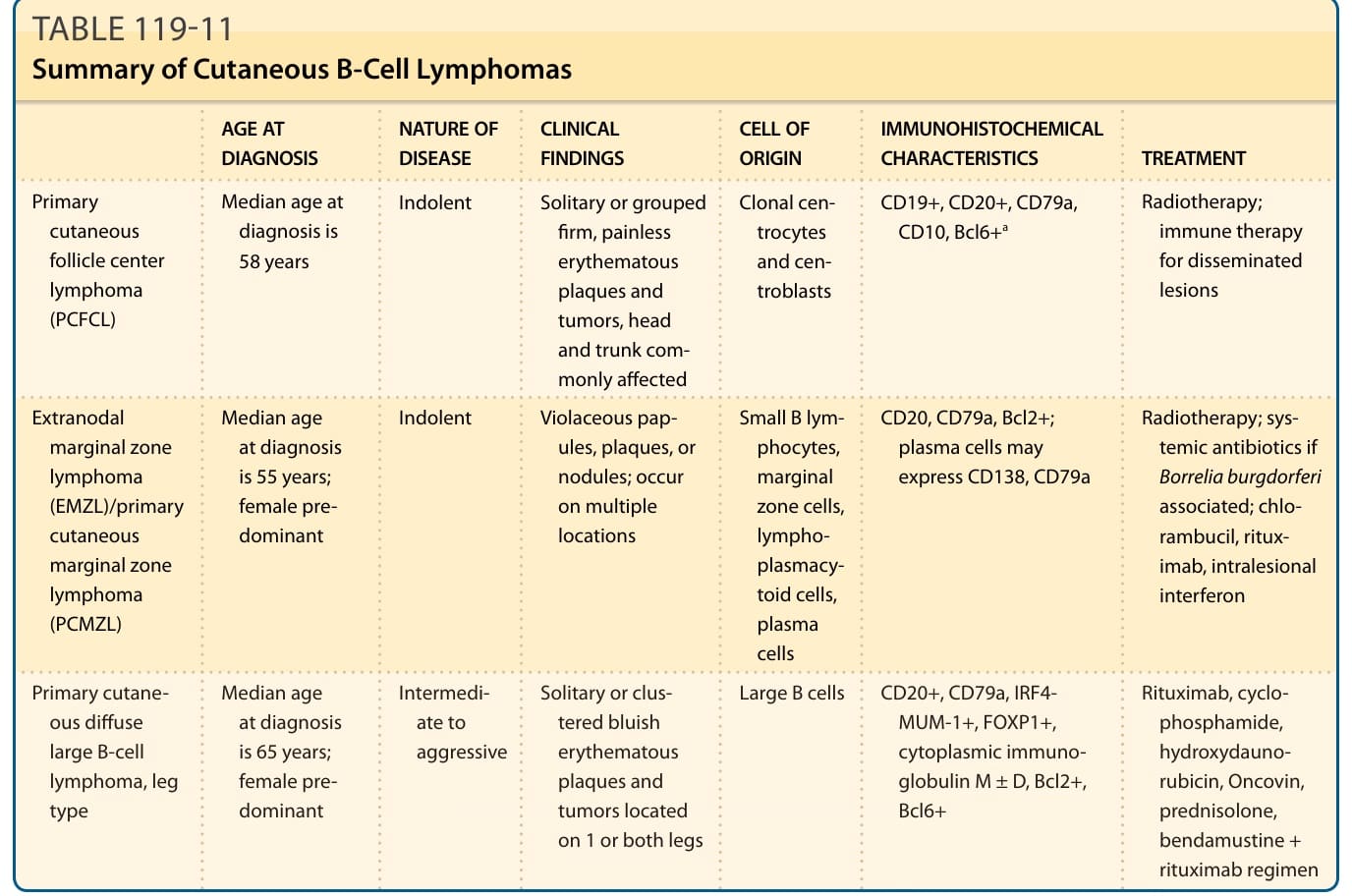
TABLE 119-11 Summary of Cutaneous B-Cell Lymphomas
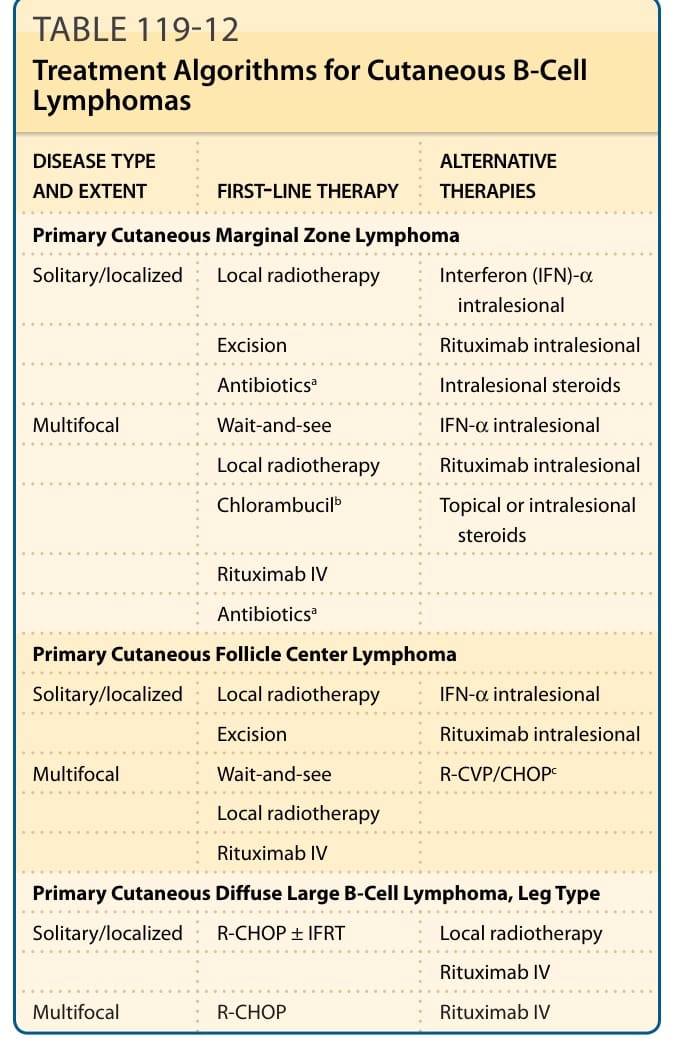
TABLE 119-12 Treatment Algorithms for Cutaneous B-Cell Lymphomas