皮膚淋巴瘤 (Cutaneous Lymphoma)
PART 20
腫瘤學 (Neoplasia)
原發性皮膚淋巴瘤(primary cutaneous lymphomas)是一群異質性的結外非何杰金氏淋巴瘤(extranodal non-Hodgkin lymphomas),源自皮膚歸巢(skin homing)與/或皮膚常駐(skin resident)的 T 細胞或 B 淋巴球,以及血液皮膚性前驅腫瘤(hematodermic precursor neoplasias,即漿細胞樣樹突細胞腫瘤 plasmacytoid dendritic cell neoplasias)的惡性無性繁殖系(clonal)轉化。皮膚淋巴瘤(圖 119-1)被定義為一個在臨床表現、組織病理、免疫表型分析與預後上具有明顯變異的異質性族群。原發性皮膚淋巴瘤是具有明確定義的疾病實體,其臨床行為與預後均與結節性非何杰金氏淋巴瘤(nodal non-Hodgkin lymphomas)完全不同,且需要不同的治療策略。基於此理由,歐洲癌症研究與治療組織(European Organization for Research and Treatment of Cancer, EORTC)與世界衛生組織(World Health Organization, WHO)於 2005 年發表了皮膚淋巴瘤的共識分類。¹ 這個第一份共同分類(WHO-EORTC)依細胞譜系(lineage)先行分類,再依形態學、免疫表型、遺傳特徵與臨床症候群的組合分類,並構成了 2008 年 WHO 分類以及 2016 年修訂版淋巴樣腫瘤分類中皮膚淋巴瘤分類的基礎。²,³ 本章討論最常見的皮膚 T 細胞淋巴瘤(cutaneous T-cell lymphomas, CTCLs)——蕈樣肉芽腫(mycosis fungoides, MF)、Sézary 症候群(Sézary syndrome)、原發性皮膚退行性大細胞淋巴瘤(primary cutaneous anaplastic large-cell lymphoma)與淋巴瘤樣丘疹病(lymphomatoid papulosis)——以及最常見的皮膚 B 細胞淋巴瘤(cutaneous B-cell lymphomas, CBCLs)——原發性皮膚濾泡中心淋巴瘤(primary cutaneous follicle center lymphoma, PCFCL)、原發性皮膚邊緣區淋巴瘤(primary cutaneous marginal zone lymphoma, PCMZL)與原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型(primary cutaneous diffuse large B-cell lymphoma, PCLBCL, leg type)。這 7 種皮膚淋巴瘤約佔所有皮膚淋巴瘤的 90%。原發於皮膚的罕見疾病實體亦於本章描述。
流行病學 (EPIDEMIOLOGY)
CTCLs 是繼原發性胃腸道淋巴瘤之後第二常見的結外淋巴瘤族群。CTCLs 的發生率一直在上升,目前在美國,估計 1993 至 2002 年間為每百萬人 6.4 例,或 2001 至 2005 年間為每百萬人 7.7 例。CTCL 的發生率隨年齡顯著上升,診斷年齡中位數約在 50 多歲(mid-50s),且大於 70 歲的病人發生率上升約 4 倍。⁴⁻⁶
原發性皮膚 T 細胞淋巴瘤 (PRIMARY CUTANEOUS T-CELL LYMPHOMAS)
CTCLs 是非何杰金氏淋巴瘤,其特徵為表現 E-選擇素配體(E-selectin ligand)皮膚淋巴球抗原(cutaneous lymphocyte antigen)與趨化激素受體(chemokine receptors,例如 CCR4、CCR8、CCR10)之活化 T 細胞的無性繁殖系擴增;這些受體是其後續歸巢至皮膚所必需的。⁷⁻⁹ 無性繁殖系擴增之後,會分化為多種亞群的效應細胞(effector cells)與記憶細胞(memory cells)。人類皮膚受 4 個功能上有區別的 T 細胞族群保護,其中 2 群為常駐型、2 群為再循環型,具有不同的遷移領域與不同的功能活性。中央記憶細胞(central memory cells, TCM)保有進入周邊血液與淋巴結的能力。相對地,效應記憶細胞(effector memory cells, TEM)會遷移至結外部位,包括皮膚,其中一個亞群會以組織常駐記憶細胞(tissue-resident memory cells, TRM)的形式留存。皮膚中大多數 T 細胞為 TRM,表現高親和力抗原受體,並具有獨特的基因表現譜。MF 中的無性繁殖系 T 細胞通常源自 TRM,這解釋了其傾向侷限於皮膚的特性。相對地,在白血病型 CTCL 變異型(Sézary 症候群以及合併續發性白血病侵犯的 MF)病人中,腫瘤細胞表現 CCR7 與 L-選擇素(L-selectin),類似 TCM。Sézary 症候群(源自 TCM)與 MF(源自 TRM)兩者推定細胞起源上的此一根本差異,與其不同的臨床行為一致。在再循環細胞中,觀察到 2 個不同族群:CCR7⁺/L-選擇素⁺ 的 TCM 與 CCR7⁺/L-選擇素⁻ 的 T 細胞,後者被稱為遷移記憶 T 細胞(migratory memory T cells, TMM)。一個合併續發性白血病侵犯、邊界不清的斑塊/斑片、真皮侵犯與淋巴結病變的 MF 病人亞群,最可能帶有 TMM 無性繁殖系。¹⁰,¹¹
最常見的形式——約佔 CTCL 的 65%——是 MF 與 Sézary 症候群,年發生率為每百萬人 7.7 例。CTCL 涵蓋皮膚侷限型變異(如 MF)以及疾病的白血病型(包括 Sézary 症候群)。在 MF 與 Sézary 症候群之後,原發性皮膚 CD30⁺ 淋巴增生性疾病(包含淋巴瘤樣丘疹病與皮膚退行性大細胞淋巴瘤)是第二常見的 CTCLs 族群(約 27%)。¹² 表 119-1 概述了原發性皮膚淋巴瘤的 WHO 分類。
皮膚 T 細胞與自然殺手(NK)細胞淋巴瘤之分類
皮膚 T 細胞與自然殺手(natural killer, NK)細胞淋巴瘤包括:
- 蕈樣肉芽腫(mycosis fungoides)
- 蕈樣肉芽腫變異型與亞型(mycosis fungoides variants and subtypes)
- 親毛囊性蕈樣肉芽腫(folliculotropic mycosis fungoides)
- 派傑樣網狀細胞增生症(pagetoid reticulosis)
- 肉芽腫性鬆弛皮膚(granulomatous slack skin)
- Sézary 症候群(Sézary syndrome)
- 成人 T 細胞白血病/淋巴瘤(adult T-cell leukemia/lymphoma)
- 原發性皮膚 CD30⁺ T 細胞淋巴增生性疾病(primary cutaneous CD30⁺ T-cell lymphoproliferative disorders)
- 原發性皮膚退行性大細胞淋巴瘤(primary cutaneous anaplastic large-cell lymphoma)
- 淋巴瘤樣丘疹病(lymphomatoid papulosis)
- 皮下脂膜炎樣 T 細胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma)
- 結外 NK/T 細胞淋巴瘤,鼻型(extranodal NK/T-cell lymphoma, nasal type)
- 原發性皮膚周邊 T 細胞淋巴瘤,非特定型(primary cutaneous peripheral T-cell lymphoma, not otherwise specified)
- 原發性皮膚 γ/δ T 細胞淋巴瘤(primary cutaneous γ/δ T-cell lymphoma)
- 原發性皮膚 CD8⁺ 侵襲性親表皮細胞毒性 T 細胞淋巴瘤(primary cutaneous CD8⁺ aggressive epidermotropic cytotoxic T-cell lymphoma)
- 原發性皮膚肢端 CD8⁺ T 細胞淋巴瘤(primary cutaneous acral CD8⁺ T-cell lymphoma)
- 原發性皮膚 CD4⁺ 小至中型 T 細胞淋巴增生性疾病(primary cutaneous CD4⁺ small–medium T-cell lymphoproliferative disorder)
皮膚 B 細胞淋巴瘤之分類
皮膚 B 細胞淋巴瘤(cutaneous B-cell lymphomas)包括:
- 原發性皮膚邊緣區 B 細胞淋巴瘤(primary cutaneous marginal zone B-cell lymphoma)
- 原發性皮膚濾泡中心淋巴瘤(primary cutaneous follicle center lymphoma)
- 原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型(primary cutaneous diffuse large B-cell lymphoma, leg type)
- 原發性皮膚瀰漫性大 B 細胞淋巴瘤,其他(primary cutaneous diffuse large B-cell lymphoma, other)
- 血管內大 B 細胞淋巴瘤(intravascular large B-cell lymphoma)
- EB 病毒陽性黏膜皮膚潰瘍(Epstein-Barr virus–positive, mucocutaneous ulcer)
- 前驅血液腫瘤(precursor hematologic neoplasm)
- CD4⁺/CD56⁺ 血液皮膚性腫瘤(漿細胞樣樹突細胞腫瘤 plasmacytoid dendritic cell neoplasia)
病因 (ETIOLOGY)
人類成人的皮膚約含 200 億個記憶 T 細胞。儘管細胞與分子生物學的重大進展已揭示了關於淋巴球的許多細節,包括 T 細胞抗原受體驚人的多樣性、其作為 TCM、TEM 或 TRM 的特徵描述,以及環境與宿主遺傳因素在 CTCL 致病機轉中所扮演的角色,但這些仍不明確。一般而言,長期的抗原刺激被認為會誘發伴隨 T 細胞增生的發炎反應,進而導致具有持續擴增能力的惡性無性繁殖系 T 細胞。然而,近年來對分子致病機轉、訊息傳遞路徑與疾病相關免疫失調的理解進展,有助於我們了解此一複雜的致病機轉,以推進 CTCL 的治療。¹⁰⁻¹⁹
內生性因素 (ENDOGENOUS FACTORS)
基於上述抗原刺激的假說,數項研究分析了受影響個體的人類白血球抗原(human leukocyte antigen, HLA)背景。兩項獨立研究顯示特定 HLA class II 分子與 MF 或 Sézary 症候群之間的關聯;亦即等位基因 HLA-DRB1∗11 與 DQB1∗03 在這些病人中顯著過度表現(圖 119-2)。
促成皮膚 T 細胞淋巴瘤之內生性與外生性因素包括:
- 外生性因素(感染性):
- 細菌:金黃色葡萄球菌(Staphylococcus aureus)
- 病毒:HTLV;EBV
- 外生性因素(環境/職業性):
- 玻璃製造工(glass formers)
- 陶工(potters)
- 造紙工廠
- 木材工廠
- 內生性因素:
- HLA DRB1*11
- HLA DQB1*03
外生性因素 (EXOGENOUS FACTORS)
病毒已被確認為至少 2 種皮膚淋巴瘤的病因因素(人類 T 細胞嗜淋巴球病毒-1 [human T-cell lymphotropic virus-1, HTLV-1] 相關成人 T 細胞淋巴瘤/白血病,以及 EB 病毒 [Epstein-Barr virus, EBV] 相關自然殺手 [natural killer, NK]/T 細胞淋巴瘤),但 MF 或 Sézary 症候群尚未證實有此類關聯。所有這些資料顯示,在 HTLV-1 流行地區以外,HTLV 在 CTCLs 的病因中並不扮演重要角色,而為病人篩檢抗體的唯一理由是懷疑診斷為成人 T 細胞淋巴瘤/白血病而非 MF。EBV 與巨細胞病毒(cytomegalovirus)也曾被討論為致病病原。EBV 與 CD30 淋巴增生及免疫抑制有關。數項研究顯示 EBV 僅在少數百分比的 CTCL 病灶中可被偵測到。在這些研究中,EBV 的偵測與不良預後相關,且其存在更可能與疾病本身或治療所造成的免疫抑制有關,而非與 CTCL 的病因有關。然而,已觀察到 EBV 與一種具有種痘樣水疱病樣(hydroa vacciniforme–like)外觀的罕見皮膚淋巴增生性疾病有強烈關聯,此疾病大多發生於亞洲裔人群。細菌感染也被認為與 CTCLs 的病因有關。特別受到關注的是金黃色葡萄球菌(Staphylococcus aureus)的超級抗原(superantigens)可能負責慢性抗原刺激的假說。在數項研究中,於高腫瘤負荷的 CTCL 病人皮膚上偵測到高百分比的 S. aureus,而早期疾病病人則未與對照組顯示顯著差異。雖然這些研究確切證明了 S. aureus 參與疾病惡化,以及抗生素治療後的臨床改善,但 CTCL 早期與對照組之間 S. aureus 群落定植缺乏差異,使人質疑 S. aureus 或這些細菌所產生的超級抗原是否參與 CTCLs 的起始。然而,S. aureus 腸毒素 A(enterotoxin A)會刺激訊息傳導與轉錄活化因子(signal transducer and activator of transcription, STAT)3 的活化,以及皮膚 T 細胞淋巴瘤中介白素(interleukin, IL)-17 的表現,並可能在疾病的進展中扮演直接角色。除了感染性病原之外,也有人提出環境與職業風險因子在 CTCL 中扮演致病角色(見圖 119-2),因為診斷之前常先有一段惰性皮膚炎。工作環境中暴露於致癌物,可能提供了引發無性繁殖系擴增起始所推測的長期抗原刺激。在流行病學研究中,數種職業(如玻璃製造工、陶工、造紙與木材業工人)已與較高的 MF 發生風險相關。然而,不同研究的結果並不一致,且無法確認出如暴露於已知致癌物等共同因素。關於職業性接觸過敏原所造成的慢性抗原刺激,必須考慮到 MF 典型發生於工作時被衣物保護的身體區域(如側軀幹)。此外,其他環境風險因子(如飲酒、吸菸或暴露於紫外線 [ultraviolet, UV] 輻射)也未一致地被觀察到與 CTCLs 的風險增加相關。²⁰⁻³¹
發病機轉 (PATHOGENESIS)
皮膚 T 細胞淋巴瘤是一種皮膚歸巢 T 細胞的惡性腫瘤。病人典型表現為侷限性斑片與斑塊,分布於日照保護的皮膚。淋巴瘤細胞由這些病灶延伸至未受侵犯的皮膚,並累積於淺層真皮,導致斑片/斑塊與腫瘤。在進展期疾病中,惡性 T 細胞會播散至血液、淋巴結與內臟。在白血病型 CTCL(Sézary 症候群)中,惡性 T 細胞可佔循環中 T 淋巴球的 99% 以上。正常 T 細胞受體(T-cell receptor, TCR)的喪失與正常淋巴球的消失,導致免疫抑制與伺機性感染,這是與疾病相關最常見的死亡原因。「皮膚 T 細胞淋巴瘤」一詞所涵蓋的臨床疾病實體共享數個組成成分:表皮與/或真皮微環境、一個無性繁殖系 T 細胞族群,以及一個受調控的抗腫瘤反應。光譜核型分析(spectral karyotyping)與比較基因體雜交(comparative genomic hybridization)研究結合 TCRγ 聚合酶連鎖反應(polymerase chain reaction)已證明,即使在 MF 最早期,遺傳受損的惡性 T 細胞即已存在,確認 MF 即使在其最早期表現也是一種遺傳受損惡性 T 細胞的淋巴瘤。有逐漸增加的證據顯示,CTCLs 不同的臨床表現可能反映其源自不同亞群的皮膚歸巢 T 細胞。MF 中的惡性 T 細胞具有非再循環 TRM 的表面表型,而典型紅皮症型 Sézary 症候群的惡性 T 細胞則具有 TCM 的表面表型,分別與其傾向形成穩定的發炎性皮膚病灶相對於暫時性紅皮症的特性一致。然而,Sézary 細胞的表型比最初報導的更為異質,且 Sézary 細胞也可表現出表型可塑性(phenotypic plasticity)。¹⁰,³²⁻³⁷
TMM 是一種新發現的皮膚歸巢 T 細胞。這些細胞表現 CCR7 但缺乏 L-選擇素,存在於健康個體的血液與皮膚中,且比 TCM 更緩慢地由皮膚再循環出去。在 CTCL 病人中,惡性 TMM 引起邊界不清的散在性皮膚病灶以及周邊血液疾病,在目前的分類中,這被稱為合併周邊血液疾病的 MF(MF with peripheral blood disease)。Sézary 症候群(源自 TCM)與 MF(源自 TRM)兩者推定細胞起源上的根本差異,與其不同的臨床行為一致,因為 TCM 可在周邊血液、淋巴結與皮膚中發現,並對細胞凋亡有抗性,而常駐型 TRM 細胞則固定留在皮膚內。此外,皮膚中也已描述有一個再循環的 CCR7⁺ L-選擇素⁻ TMM 族群。MF 亞型與 Sézary 症候群源自不同 T 細胞亞群的論點,與比較基因體雜交(comparative genomic hybridization, CGH)以及基因表現譜資料一致,這些資料證明這些 CTCL 亞型在遺傳上是不同的。偵測這些惡性 T 細胞無性繁殖系對於 CTCL 的診斷至關重要。¹⁰,¹¹
隨著使用高通量 TCR 定序(high-throughput TCR sequencing)的新分子技術出現,已證明 MF 與白血病型 CTCL 中定位於皮膚不同解剖隔室的惡性 T 細胞無性繁殖系,可與良性發炎性皮膚疾病區分開來。表現轉錄因子 FOX-P3 的調節性 T 細胞(regulatory T cells)在維持自我耐受性方面很重要,且構成皮膚常駐 T 細胞的一個次要亞群。有人討論,一個 Sézary 病人亞群帶有源自常駐調節性 T 細胞的無性繁殖系。然而,調節性 T 細胞僅佔皮膚常駐 T 細胞的少數;皮膚中大多數 T 細胞產生不同效應 T 細胞亞群(包括 T 輔助細胞 [T-helper, Th] 1、Th2 與 Th17 細胞)特有的細胞激素。MF 與 Sézary 症候群與 Th2 相關基因(如 GATA-3)的表現以及 Th2 相關細胞激素(如 IL-4、IL-5、IL-13)的產生有關,提高了相當一部分病人可能帶有源自 Th2 之無性繁殖系的可能性。³⁸⁻⁴⁴
或者,活化特定訊息傳遞路徑的復發性突變(活化 T 細胞核因子 [nuclear factor of activated T cells, NFAT]、核因子 κB [nuclear factor κB, NFκB]、Janus 激酶 [Janus kinase, JAK]/STAT)可能引發特定表型的獲得,而與細胞起源無關。³⁷
近期的分子研究推進了我們對 CTCL 分子致病機轉的理解。已確認出 10q 與 17p 的復發性缺失以及 8q 與 17q 的擴增,並有確鑿證據指出 TP53 與 CDKN2a 的缺失以及含有 MYC 的 8q 擴增。在近期一項皮膚淋巴瘤基因體圖譜的研究中,描述了 CTCL 中 17 個基因的體細胞突變。發現染色質修飾基因(ARID1A [62.5%]、CTCF [12.5%] 與 DNNT3A [42.5%])的頻繁缺失與有害的體細胞拷貝數變異。許多在 CTCL 中突變的基因也促成其他 T 細胞腫瘤,包括周邊 T 細胞淋巴瘤(CD28、DNMT3A 與 RHOA),強調這些基因對 T 細胞惡性轉化的重要性。與此概念一致,在 TCR 訊息傳遞路徑的多個組成成分中發現突變,包括 CD28 以及 TCR 相關酵素的基因(PLCG1、PRKCQ 與 TNFAIP3)與轉錄因子(NFkB2、STAT5B 與 ZEB1)。這些基因驅動 Th2 分化(ZEB1),促進逃避轉化生長因子-β(transforming growth factor-β)介導的生長抑制(ZEB1),並促進對腫瘤壞死因子受體超家族(tumor necrosis factor receptor superfamily)介導之細胞凋亡的抗性(FAS 與 ARID1A)。⁴⁵,⁴⁶
在 CTCL 中,數種細胞激素在疾病表現以及此疾病的進展中扮演角色。在 CTCL 早期,IL-2、IL-7 與 IL-15 的訊息傳遞(這些都是使用 JAK1/JAK3 的 γc 鏈細胞激素)驅動 STAT5 與 STAT3 的活化。作為 STAT3 與 STAT5 下游過程的結果,可觀察到惡性 T 細胞由 Th1 朝向 Th2 表型的轉變。這是透過 STAT5 對微 RNA(micro-RNA, miRNA)-155 的轉錄活化來達成。接著,miRNA-155 標靶 STAT4,導致 Th1 基因的下調。此外,STAT5 活化 IL-4 的表現,促進 Th2 表型。此轉變與 CTCL 的進展相關,因為 Th2 反應(IL-4、IL-10)是腫瘤誘導免疫抑制的眾所周知機制。此外,JAK3、STAT3 與 STAT5 的活化導致已知腫瘤抑制因子 miRNA-22 的轉錄抑制。在 CTCL 中,惡性 T 細胞——CTCL 細胞株以及周邊血液 Sézary CD4⁺ T 細胞——顯示 miRNA-22 的表現降低。miRNA-22 正常會抑制腫瘤生長與轉移,因為它標靶數個推定致癌基因(如 MYCB、HDAC4、HDAC6、CDK6 與 NcoA1)的轉錄。但在 CTCL 中,觀察到 miRNA-22 此種腫瘤抑制活性的喪失,因為 miRNA-22 的表現被 STAT5 直接下調,導致 T 細胞惡性狀態的更快進展。這些發現顯示 JAK/STAT 訊息傳遞在 CTCL 的致病與進展中扮演另一個關鍵角色,且 JAK 抑制可能對腫瘤生長與轉移介導直接的抑制作用(圖 119-3)。除此之外,STAT3 的活化是惡性 CTCL T 細胞轉化的關鍵介導者,也是可塑性的重要介導者。¹,⁷
STAT3 的活化導致 CTCL T 細胞產生 IL-17,如以 CTCL 細胞株在體外(in vitro)所證明,以及由 CTCL 皮膚病灶中腫瘤性淋巴球在離體(ex vivo)的 IL-17 表現所證明。IL-17 的受體由 CTCL 病灶皮膚微環境中的各種細胞表現,例如纖維母細胞、角質形成細胞與上皮細胞。在受 IL-17 刺激時,這些細胞產生其他促發炎細胞激素、趨化激素與血管新生因子。已顯示 CTCL 病灶呈現增加的血管新生,提供了 CTCL 中腫瘤性 T 細胞的 IL-17 透過調節 CTCL 皮膚病灶中的發炎與血管新生而影響腫瘤發生的見解。因此,藉由 JAK 抑制來標靶最初的 STAT3 活化,也會對 Th17 表型異常 T 細胞的這些間接作用產生有益效果。STAT3 的轉錄活性也導致異常 T 細胞中 IL-21 的表現,這驅動了一個自分泌訊息傳遞迴路,導致透過 JAK1/JAK3 的持續訊息傳遞與 STAT3 的活化。此由 IL-21 驅動的 STAT3 持續活化,透過數種機制對 CTCL 的進展至關重要:(a) 它促進抗凋亡蛋白(如 bcl-2)的表現,導致惡性 T 細胞的存活¹;(b) STAT3 參與血管新生因子血管內皮生長因子(vascular endothelial growth factor)轉錄的上調;(c) 它誘導 IL-5 與其他參與紅皮症與嗜酸性球增多的細胞激素的表現;(d) STAT3 的轉錄活性參與驅動在 CTCL 進展期所觀察到的 T 細胞可塑性(Th2 與 Th17 表型);以及 (e) 它誘導一種致癌 miRNA(即 miRNA-21)的表現。miRNA-21 因抗凋亡活性而參與惡性 T 細胞的存活。所有這些理由都提供了在 CTCL 中標靶 JAK/STAT 路徑的強力依據。CTCL 微環境中的其他細胞激素也參與其中,如 IL-13,這是一種與 γc 鏈細胞激素相關、共享 IL-4 受體 α 次單元(IL-4Rα)的細胞激素。因此,IL-13 屬於 IL-4 家族,並由 CTCL 中轉化的惡性 Th2 細胞分泌,因為 IL-13 在 CTCL 病人臨床受侵犯的皮膚中高度表現。IL-13 作為 CTCL 細胞的自分泌生長因子,尤其在 CTCL 的 Sézary 症候群變異型中。阻斷 JAK1/JAK3 也會阻斷 IL-13 細胞激素訊息傳遞,導致腫瘤細胞增生的抑制。⁴⁷⁻⁶⁰
近年來,已逐漸明確細胞激素訊息傳遞在 CTCL 的致病中扮演關鍵角色。除了細胞凋亡的多重缺陷之外,異常的細胞週期調控(包括 CDKN2A-CDKN2B 基因座的失活)在 CTCL 中也頻繁觀察到。週期蛋白(cyclin,包括 cyclin D1)的上調以及 RB1 的喪失也已被描述。隨著基因表現譜分析與次世代定序技術的運用,更多致病路徑正在 CTCL 亞群中被確認,包括參與調控 T 細胞分化的轉錄因子以及 C-MYC、RAS、BRAF 與 MEK 訊息傳遞的路徑。¹³,³⁴
總而言之,近期對 CTCL 的研究顯著推進了我們對分子致病機轉、細胞起源、遷移行為與死亡訊息傳遞的知識。如同其他惡性腫瘤,現在的主要挑戰將是定義有意義的分子與/或表型亞群,使其與臨床行為、與/或對特異性干擾促疾病訊息傳遞級聯反應或細胞交互作用之藥物的治療反應相關。因此,有前景的治療策略將奠基於我們對 CTCL 分子致病機轉日益增加的知識。
CTCL 訊息傳遞事件(圖 119-3)所涉及的主要路徑與分子包括:JAK-STAT 路徑(JAK1/3、STAT3/5B、NFKB2,與皮膚歸巢、細胞凋亡相關;可被 Bortezomib、Tofacitinib、Ruxolitinib 標靶);CCR4、FAS、TNFRSF10A(TRAIL-R)、TNFRSF1B 等受體;NF-κB 路徑(TCR/CD3、TNFR、IL-1R、CTLA-4,涉及 PRKCQ、PDCD1、CD28、PTEN、CARD11、PLCG1、IKKα、IKBα、蛋白酶體 [proteasome]);PI3K/RAS/MAPK 路徑(KRAS、BRAF、MAPK1、AKT、MEK、mTOR、ERK、RHOA,涉及細胞骨架動力學);以及細胞週期與基因體完整性調控(TP53、ATM、CDKN2A [p16]、RB1 [Rb])、Th2 極化(ZEB1)與表觀遺傳調控因子(ARID1A、DNMT3A、KMT2C)。
蕈樣肉芽腫 (MYCOSIS FUNGOIDES)
定義 (DEFINITION)
MF 是原發性皮膚淋巴瘤最常見的形式,約佔所有皮膚淋巴瘤的 40%,通常發生於成年中至晚期(診斷年齡中位數:55-60 歲),男性佔優勢,男女比為 2:1。根據 WHO 分類,MF 由其典型形式定義,即以斑片與斑塊或變異型來定義。
臨床發現 (CLINICAL FINDINGS)
皮膚徵象:臨床上,MF 被分類為斑片期、斑塊期或腫瘤期,但病人可能同時有 1 種以上類型的病灶。在早期斑片期 MF(圖 119-4 與 119-5)中,有單個或多個紅斑性、脫屑性的斑(macules)與斑片(patches),大小不一且通常邊界清楚。病灶顏色可由橘色變化至暗紫紅色。其分布典型偏好非日照部位,疾病早期以「泳褲區(bathing trunk)」與間擦部位為主。皮疹可能極度搔癢或無症狀,偶爾可能是暫時性的,會自發消退而不留疤痕。此階段的診斷可能困難。病人常會回想起先前曾有 10 至 20 年的「慢性皮膚炎」病史,可能曾被視為對治療有抗性的接觸性皮膚炎、異位性皮膚炎、乾癬或濕疹。對於任何對常用治療方式有抗性的皮膚病病人,應採取多處切片標本以追查診斷。斑片可能持續數月或數年才進展至斑塊期(圖 119-6),或斑塊可能重新(de novo)出現。斑塊呈現為邊界銳利、脫屑、隆起的病灶,呈暗紅至紫紅色且硬化程度不一(圖 119-6 與 119-7)。此階段的病灶可能自發消退,或可能融合形成具有環形、弧形或匐行性邊緣的大斑塊,並可能中央清除而疾病活性留在病灶周邊。可能有紫斑性色素沉著或色素脫失以及異色症(poikiloderma)。腫瘤可發生於身體任何部位,但好發於臉部(圖 119-8 與 119-9)與身體皺褶處:腋窩、腹股溝、肘前窩,以及女性的乳房下區。這些通常發生於既存的 MF 斑塊或斑片中;這與這些病灶在垂直方向上的延伸相一致(見圖 119-6)。在此時,腫瘤細胞表現出生物學上更具侵襲性的行為,伴隨明顯的腫瘤細胞累積,導致擴大真皮結節的臨床外觀(見圖 119-9)。重新(de novo)發生則提示惡性 T 細胞無性繁殖系細胞的轉移性擴散。這些結節呈紅棕色或紫紅色且表面光滑,但常潰瘍化並可能續發感染。生長速率不一。有腫瘤的病人傾向於有比斑片與斑塊期病人更具侵襲性的疾病形式。紅皮症(圖 119-10A)可能重新發生或在 MF 中發展出來。CTCL 紅皮症期的命名各異。有人提議將紅皮症定義為 80% 體表面積受邊界不清病灶侵犯,並將具有先前 MF 病史的病人定義為患有一種獨立的「紅皮症型 MF(erythrodermic MF)」症候群。皮膚呈瀰漫性鮮紅色並有明顯可見的脫屑,但可能有特徵性的未受侵犯皮膚島嶼。經常摺疊的皮膚區域(如腹部、肘前與腋窩區)可能被倖免。這種倖免產生一個常被稱為躺椅徵或摺疊行李徵(deck chair or folded luggage sign)的發現。部分紅皮症型 CTCL 病人會發展出腫瘤。
其他症狀:病人可能抱怨發燒、寒顫、體重減輕、倦怠、續發於難以忍受搔癢的失眠,以及體溫恆定不良。可能有手掌與腳掌的角化過度、脫屑與龜裂、禿髮、眼瞼外翻(ectropion)、甲營養不良與踝部水腫,皮膚變得發亮且緊繃。這些變化導致行走疼痛以及執行需要手部靈巧度之工作的極度困難。此類病人因其皮膚表現的範圍與位置而經歷嚴重的活動限制。搔癢常很劇烈,導致抓傷、滲液與續發感染,可能主導整個臨床表現。
色素脫失型蕈樣肉芽腫 (Hypopigmented Mycosis Fungoides):深膚色病人會發展出色素脫失型 MF,這是斑片型 MF 的一種變異型(表 119-2)。此型 MF 必須與白斑症(vitiligo)區別。在較深膚色的個體中,這可能是此疾病最常見的表現。病人對治療有反應並伴隨復色(repigmentation),而色素脫失病灶的再次出現常表示復發。
組織病理 (HISTOPATHOLOGY)
圖 119-11 顯示 MF 的特徵性組織學。在斑片、斑塊以及紅皮症期,上真皮有帶狀浸潤,由反應性 T 細胞與腫瘤性 T 淋巴球組成,後者以高度迴旋的腦回狀(cerebriform)細胞核為特徵。腫瘤性 T 細胞顯示親表皮性(epidermotropism),形成表皮內 Pautrier 微膿瘍(Pautrier microabscesses)(圖 119-12 與 119-13)。在腫瘤期,真皮中可見結節性浸潤,表皮成分則明顯較不顯著(圖 119-14)。免疫組織學上,惡性細胞表現成熟周邊 T 細胞(CD4⁺)表型。全 T 細胞抗原(如 CD7 與 CD3)的部分喪失可能是 MF 的一個特徵,但並非此疾病的特異徵象(pathognomonic)。TCR 基因分析典型顯示無性繁殖系重排(clonal rearrangement),如聚合酶連鎖反應或南方墨點法(Southern blot)技術所證明。然而,在疾病早期僅在半數切片中發現 T 細胞無性繁殖系。因此,無論是 T 細胞無性繁殖性的分子檢測或表型標記,在早期 MF 中均無顯著的診斷價值。或許在不久的將來,現代分子診斷(如高通量技術)將能夠在早期 MF 中偵測到 T 細胞無性繁殖系。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
MF 的鑑別診斷(見表 119-5)依分期而異:
斑片/斑塊期:
- 「慢性皮膚炎」
- 乾癬(psoriasis)
- 接觸性皮膚炎(contact dermatitis)
- 濕疹(eczema)
- 體癬(tinea corporis)
- 白斑症(vitiligo)
腫瘤期:
- B 細胞淋巴瘤(B-cell lymphoma)
- 皮膚癌(carcinoma cutis)
- 類肉瘤病(sarcoidosis)
- 深部黴菌感染(deep fungal infection)
- 非典型分枝桿菌感染(atypical mycobacterial infection)
- 痲瘋(leprosy)
- 利什曼原蟲病(leishmaniasis)
紅皮症(erythroderma):
- 毛髮紅糠疹(pityriasis rubra pilaris)
- 乾癬(psoriasis)
- 異位性皮膚炎(atopic dermatitis)
- 藥物疹(drug eruption)
- 脂漏性皮膚炎(seborrheic dermatitis)
治療與預後 (TREATMENT AND PROGNOSIS)
治療應依分期調整。在疾病早期,治療應以皮膚導向治療(skin-directed therapies)為基礎,可單獨使用或合併全身性生物反應調節劑(biologic response modifiers,如干擾素 α 或 γ、類視黃酸 retinoids)。標靶治療與小分子藥物作為清除腫瘤與血液隔室、替代化療策略的方式,正逐漸受到青睞(見「皮膚 T 細胞淋巴瘤分期」與「皮膚 T 細胞淋巴瘤治療原則」兩節)。預後取決於皮膚侵犯的類型與範圍(斑塊、腫瘤或紅皮症)、淋巴結侵犯的存在,以及內臟疾病的存在。在早期病人中,25% 將進展至進展期。整體而言,MF 侷限於皮膚的病人有 80% 至 100% 的 5 年存活率。相對地,有淋巴結侵犯的病人顯示 40% 的 5 年存活率。⁶²
皮膚淋巴瘤國際聯盟(Cutaneous Lymphoma International Consortium)研究發表了基於 1275 名進展期 MF 與 Sézary 症候群病人的存活資料與分析。整體存活中位數為 63 個月,2 年與 5 年存活率分別為 77% 與 52%。Stage IIB 疾病病人的整體存活中位數為 86 個月,但診斷為 Stage III 疾病的病人,與 Stage IIB 疾病病人相比,存活略有改善。診斷為 Stage IV 疾病的病人存活顯著較差(Stage IVA 為 48 個月,Stage IVB 為 33 個月)。在所測試的 10 個變項中,有 4 個(Stage IV、年齡 >60 歲、大細胞轉化 large-cell transformation,以及乳酸去氫酶 lactate dehydrogenase 升高)是較差存活的獨立預後標記。將這 4 個因子組合成一個預後指數模型,可在各分期間辨識出 3 個風險族群,其 5 年存活率有顯著差異:低風險(0-1)68%、中風險(2)44%,以及高風險(3-4)28%。⁶³
早期蕈樣肉芽腫診斷的評分系統標準(表 119-3)
(根據 Pimpinelli 等人的早期 MF 定義;診斷 MF 須臨床、組織病理、分子生物學與免疫病理標準任意組合共計達 4 分)
- 臨床(Clinical)
- 基本(Basic):持續性與/或進展性斑片/薄斑塊
- 附加(Additional):非日照部位;大小/形狀變異;異色症(poikiloderma)
- (基本標準 2 分搭配 2 項附加標準;或基本標準 1 分搭配 1 項附加標準)
- 組織病理(Histopathologic)
- 基本:淺層淋巴樣浸潤
- 附加:無海綿狀水腫(spongiosis)的親表皮性;淋巴樣非典型(lymphoid atypia)
- (基本標準 2 分搭配 2 項附加標準;或基本標準 1 分搭配 1 項附加標準)
- 分子生物學(Molecular biological)
- 無性繁殖系 TCR 基因重排:無性繁殖性 1 分
- 免疫病理(Immunopathologic)(符合 1 項以上標準計 1 分)
- <50% 的 CD2⁺、CD3⁺ 與/或 CD5⁺ T 細胞
- <10% 的 CD7⁺ T 細胞
- CD2、CD3、CD5 或 CD7 的表皮/真皮不一致(discordance)
(資料來源:Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063,經授權。Copyright © American Academy of Dermatology。)
MF/Sézary 症候群病人的建議評估/初步分期(表 119-4)
- 皮膚病灶的評估;測量斑片、斑塊與腫瘤的體表面積百分比
- 淋巴結位置的評估與測量
- 內臟疾病的臨床鑑別
- 皮膚病灶的組織學與免疫組織學,以及 T 細胞無性繁殖性的評估
- 腫大淋巴結的組織學與免疫組織學,以及 T 細胞無性繁殖性的評估
- 血球計數、乳酸去氫酶、肝功能檢查
- 血液 T 細胞無性繁殖性
- 以細胞型態學與/或 T 細胞血液亞群(CD4⁺/CD7⁻ 與 CD4⁺CD26⁻)的流式細胞術分析計算 Sézary 細胞數
- 早期病人:胸部 X 光與腹部超音波
- 晚期病人:胸部、腹部與骨盆腔的電腦斷層(CT)掃描
蕈樣肉芽腫變異型 (MYCOSIS FUNGOIDES VARIANTS)
MF 及其變異型的臨床發現與預後(5 年存活率)(表 119-6)
| 疾病 | 臨床發現 | 預後(5 年存活率) |
|---|---|---|
| 蕈樣肉芽腫(皮膚侷限) | 單個至多個紅斑性脫屑斑與斑片,位於非日照部位;斑塊呈邊界銳利、脫屑、紫紅色且硬化;腫瘤好發於臉部與身體皺褶處 | 80%-100% |
| 蕈樣肉芽腫(淋巴結侵犯) | 同上 | 40% |
| 親毛囊性蕈樣肉芽腫 | 以斑片、斑塊及病灶內毛髮脫落為特徵;優先侵犯頭頸部 | 比早期蕈樣肉芽腫更具侵襲性;60% |
| 派傑樣網狀細胞增生症 | 單發、角化過度的斑塊,通常位於肢端 | 一般為惰性行為,可能復發與再發ᵃ,ᵇ |
| 肉芽腫性鬆弛皮膚 | 皮膚局部大塊摺疊,位於腋窩與腹股溝 | 惰性病程,緩慢進展,罕見淋巴結侵犯ᵃ |
| Sézary 症候群 | 瀰漫性紅皮症、全身性淋巴結病變、循環中 Sézary 細胞的存在 | 24%-43% |
ᵃMartinez-Escala ME, Gonzales BR, Guitart J. Mycosis fungoides variants. Surg Pathol Clin. 2014;7(2):169-89.
ᵇHaghighi B, Smoller B, LeBoit P, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular and clinicopathologic study. Mod Pathol. 2000;13(5):502-10.
親毛囊性蕈樣肉芽腫 (FOLLICULOTROPIC MYCOSIS FUNGOIDES)
與典型 MF 相比,濾泡型或親毛囊性 MF 傳統上被認為預後較差,至 15 年時 5 年存活率約為 60%(濾泡型 MF)與 41%(親毛囊性 MF)。
臨床上,親毛囊性 MF 表現為斑片、斑塊以及病灶內不尋常的毛髮脫落;偶爾,疾病可能以主要為丘疹性病灶表現。親毛囊性 MF 優先侵犯頭頸部區域,並以親毛囊性 T 細胞浸潤為特徵,伴或不伴有毛囊的黏液性變性(mucinous degeneration)。先前,此變異型被稱為毛囊性黏液病(follicular mucinosis)或黏液性禿髮(alopecia mucinosa)。親毛囊性 MF 主要影響成人,罕見於兒童與青少年。病人可能有成群的毛囊性丘疹(圖 119-15A)、痤瘡樣病灶、硬化斑塊,有時還有腫瘤,通常侵犯頭頸部區域。病灶內毛髮脫落(以眉毛最為明顯)、劇烈搔癢與續發性細菌感染很常見。值得一提的是,有 2 項較新的研究顯示,可區分出 2 種不同類型的親毛囊性 MF,包括一個預後良好的亞型。⁶⁴,⁶⁵
要將親毛囊性 MF 相關的毛囊性黏液病與良性(特發性毛囊性黏液病 idiopathic follicular mucinosis)區別仍具挑戰性。雖然親毛囊性 MF 更可能呈現緻密的淋巴球浸潤,伴有輕微的核非典型、增加的 CD4 對 CD8 比值,以及 TCR 基因的無性繁殖系重排,但組織學或表型特徵並不足以確定地將兩個疾病實體分開。
派傑樣網狀細胞增生症 (PAGETOID RETICULOSIS)
派傑樣網狀細胞增生症病人表現為單發的乾癬樣(psoriasiform)或角化過度的斑片或斑塊,通常位於四肢(圖 119-16)且緩慢進展。與典型 MF 不同,未觀察到皮膚外播散。派傑樣網狀細胞增生症在 WHO 分類中列為 MF 的一個亞型,與單一病灶 MF 相比顯示更顯著的親表皮性與核多形性,且更常顯示 CD8⁺ 表型。此外,派傑樣網狀細胞增生症更常以角化過度病灶表現。
肉芽腫性鬆弛皮膚 (GRANULOMATOUS SLACK SKIN)
肉芽腫性鬆弛皮膚是一種罕見的 MF 亞型,特徵為皮膚局部大塊摺疊,好發於腋窩與腹股溝(圖 119-17)。光學顯微鏡顯示整個真皮有緻密的肉芽腫性浸潤。除了具有腦回狀細胞核的小型非典型細胞外,還可見巨噬細胞與多核巨細胞,以及彈性纖維的喪失。腫瘤細胞表現 CD3⁺CD4⁺CD8⁻ 表型。
Sézary 症候群 (SÉZARY SYNDROME)
定義 (DEFINITION)
Sézary 症候群以三聯徵為特徵:瀰漫性紅皮症、全身性淋巴結病變,以及循環中具有腦回狀細胞核的惡性 T 細胞(即所謂 Sézary 細胞)。Sézary 症候群是一種罕見的 CTCL 形式,佔所有皮膚淋巴瘤的 3%。與 MF 相反,Sézary 症候群預後不佳,5 年整體存活率介於 24% 至 43% 之間。¹,⁶³
臨床發現 (CLINICAL FINDINGS)
紅皮症常伴隨手掌與腳掌的嚴重脫屑或龜裂(見圖 119-10)、禿髮與甲營養不良,並可能伴有明顯的脫屑、水腫、苔癬化與劇烈搔癢。在罕見情況下,會發生色素沉著。
實驗室發現 (LABORATORY FINDINGS)
Sézary 症候群顯示與 MF 相似的組織學特徵,但可能需要重複切片,因為標本常顯示非診斷性的發現。無性繁殖系 T 細胞透過多色流式細胞術一般為 CD3⁺、CD4⁺ 與 CD8⁻。如同 MF,常觀察到 T 細胞抗原(包括 CD2、CD3、CD4、CD5 與 CD7)的異常喪失。其中,約三分之二的病例觀察到 CD7 表現的喪失。CD26 表現的喪失(在大多數病例中觀察到)也有助於 Sézary 細胞的鑑別。正常由 NK 細胞表現的主要組織相容性複合體 class I 結合、免疫球蛋白樣受體(immunoglobulin-like receptor, KIR)CD158k/KIR3DL2 的異常表現,在大多數 Sézary 症候群病人中被描述。在目前國際皮膚淋巴瘤學會(International Society for Cutaneous Lymphomas, ISCL)/EORTC TNMB 分期分類中,Sézary 症候群的診斷需要紅皮症,且周邊血液有陽性 T 細胞無性繁殖系,並至少合併一項 B2 標準,例如血液中鑑別出超過 1000 個 Sézary 細胞/mm³。Sézary 細胞於 1938 年首次由 Sézary 描述為大型、非典型、單核細胞,具有分葉、腦回狀的細胞核。然而,以細胞型態學偵測 Sézary 細胞對 Sézary 症候群的診斷缺乏特異性,因為它們也可在其他發炎性紅皮症中發現。其他診斷標準包括:擴增的 CD4⁺ T 細胞族群導致 CD4 對 CD8 比值超過 10;任一或全部 T 細胞抗原 CD2、CD3、CD4 或 CD5 的喪失;以及 CD7 與 CD26 的缺乏。⁶⁶,⁶⁷
治療與預後 (TREATMENT AND PROGNOSIS)
與斑片/斑塊期 MF 病人相比,Sézary 症候群病人的 5 年存活率明顯降低。當 Sézary 症候群出現時,正常免疫力所剩無幾。事實上,Sézary 症候群病人常因感染性併發症而死亡。
原發性皮膚 CD30⁺ 淋巴增生性疾病 (PRIMARY CUTANEOUS CD30⁺ LYMPHOPROLIFERATIVE DISORDERS)
原發性皮膚 CD30⁺ 淋巴增生性疾病是第二常見的皮膚淋巴瘤(CTCL)形式(20%-25%)。原發性皮膚 CD30⁺ 淋巴增生性疾病代表一個疾病譜,包括淋巴瘤樣丘疹病與原發性皮膚退行性大細胞淋巴瘤(anaplastic large-cell lymphoma, ALCL)。
淋巴瘤樣丘疹病 (LYMPHOMATOID PAPULOSIS)
定義:淋巴瘤樣丘疹病於 1968 年由 Macaulay 首次描述。它是一種不常見的慢性疾病(盛行率為每 1,000,000 人 1.2-1.9 例),特徵為反覆、自癒性成批出現的丘疹與結節。
臨床發現:淋巴瘤樣丘疹病是一種慢性、反覆性、自癒性的丘疹壞死性或丘疹結節性皮膚疹(圖 119-18)。病灶典型侵犯軀幹與四肢,且不同演化階段的病灶可能同時存在。
組織病理:淋巴瘤樣丘疹病是一種臨床上多樣化的疾病;近年來描述了許多新的病理與臨床變異型。非典型細胞表現一種或多種 T 細胞抗原以及淋巴樣活化抗原 CD30(圖 119-19)。WHO 2016 分類承認原始的 Type A、B 與 C 變異型,以及較近期描述的 Type D(模擬原發性皮膚侵襲性親表皮 CD8⁺ 細胞毒性 T 細胞淋巴瘤)與血管侵襲性、血管中心性的 Type E。具有第 6p25 號染色體重排(IRF4/DUSPP 基因座)的淋巴瘤樣丘疹病由 Karai 等人描述,臨床上以侷限性丘疹與結節為特徵,組織學上以親表皮性與結節性 CD30⁺ 細胞為特徵。對這些變異型的認識很重要,因為它們在組織學上可模擬非常具侵襲性的 T 細胞淋巴瘤,但在臨床上與其他形式的淋巴瘤樣丘疹病相似。⁶⁸,⁶⁹
治療與預後:由於目前沒有治癒性療法,且現有的治療方式皆無法影響疾病的自然病程,積極治療的短期效益應與潛在的副作用謹慎權衡。低劑量 methotrexate(5-10 mg/week)是抑制新皮膚病灶發展最有效的療法。以 PUVA 治療曾被報導產生有益效果,但停止治療後反應持續時間常很短暫。因此,對於病灶少且不留疤痕的病人,應考慮長期追蹤而不積極治療。⁷⁰
一般而言,淋巴瘤樣丘疹病顯示良性的臨床病程與接近 100% 的有利 10 年存活率。然而,在一部分病人(估計為 10% 至 20% 的病例)中,淋巴瘤樣丘疹病可能先於、共存於或繼發於惡性淋巴瘤,尤其是 MF、何杰金氏淋巴瘤與結節性 ALCL。在許多此類病例中,於淋巴瘤樣丘疹病以及相關淋巴瘤中發現了相同的無性繁殖系 TCR 重排。在大多數淋巴瘤樣丘疹病病例中,儘管疾病有時病程極長,但並無續發性淋巴瘤的演化。然而,淋巴瘤樣丘疹病病人應終身監測。在淋巴瘤樣丘疹病病人中,單株 TCR 重排或組織學混合型可能可預測較可能發展出淋巴瘤樣丘疹病相關淋巴瘤的疾病。
皮膚退行性大細胞淋巴瘤 (CUTANEOUS ANAPLASTIC LARGE-CELL LYMPHOMA)
定義:皮膚 ALCL 以大型腫瘤細胞為特徵,其中大多數表現 CD30 抗原,且無 MF 或其他類型原發性 CTCL 的證據或病史。無論腫瘤細胞的型態為何(例如退行性、免疫母細胞性或多形性大細胞),其臨床表現與行為皆相同。
臨床發現:CD30⁺ 皮膚大細胞淋巴瘤發生於成人,罕見於兒童與青少年,男女比為 1.5:1。臨床表現以單發或局部區域性出現的紅至棕色結節與腫瘤為特徵,常潰瘍化(圖 119-20A)。皮膚病灶可能自發消退。雖然約 10% 的病人觀察到區域淋巴結的續發侵犯,但這不一定與不良預後相關。
組織病理:真皮中可見大細胞的結節性或瀰漫性非親表皮浸潤(圖 119-20B)。在大多數病例中,腫瘤細胞顯示退行性型態,具有卵圓形或不規則形狀的細胞核、顯著的核仁與豐富的細胞質。較少見的情況下,可觀察到多形性或免疫母細胞性外觀。非典型有絲分裂像常見。在病灶周邊,存在發炎細胞(如淋巴球、嗜酸性球與嗜中性球),有時模擬淋巴瘤樣丘疹病的組織學表現。原發性皮膚 ALCL 最常見的表型是 CD4⁺ T 輔助細胞表型。在罕見病例中,腫瘤細胞表現 CD8⁺,這似乎與預後受損無關。根據定義,超過 75% 的腫瘤細胞以黏聚成片(cohesive sheets)的方式表現 CD30。與結節性 ALCL(約 60% 病例表現退行性淋巴瘤激酶 anaplastic lymphoma kinase, ALK)相反,原發性皮膚 ALCL 通常此標記為陰性,且缺乏 t(2;5) 易位。已有報導罕見的 ALK⁺ 原發性皮膚 ALCL 病例,與一種易位變異型及 ALK 的細胞質染色相關。目前已存在改良的標準,可在日常實務中辨識 ALK⁻ ALCL,且現行 WHO 2016 分類不再將此型視為暫定的(provisional)。基因表現譜分析研究顯示 ALK⁻ ALCL 具有與 ALK⁺ ALCL 相當接近、而與 NK/T 細胞淋巴瘤不同的特徵譜。闡明 ALK⁻ ALCL 遺傳圖譜的較新研究顯示有趨同的突變與激酶融合,導致 JAK/STAT3 路徑的持續活化。這些研究為 ALK⁺ 與 ALK⁻ ALCL 之間型態與表型的相似性提供了遺傳學依據。然而,並非所有 ALK⁻ ALCL 病例都相同。在第 6p25 號染色體含有 DUSP22-IRF4 的基因座有重排的一個亞群,傾向於相對單形性,通常缺乏細胞毒性顆粒,且據報導預後較佳,而一個帶有 TP63 重排的小亞群則非常具侵襲性。有趣的是,6p25 的同一基因座也與淋巴瘤樣丘疹病及原發性皮膚 ALCL 有關。³,⁶⁹,⁷¹,⁷²
治療與預後:在單發或局部皮膚病灶的病例中,切除或放射治療是首選治療。曾有報導以 PUVA 合併干擾素-α 成功治療。若皮膚病灶為廣泛性,則首選以 methotrexate(20 mg/week)的全身性治療;vinblastine 是另一種選擇。在皮膚外播散的情況下,以 cyclophosphamide、doxorubicin、vincristine 與 prednisone(CHOP)治療是最常選擇的選項。Brentuximab vedotin 也可被視為首選療法。此免疫結合物是一種抗 CD30 單株抗體,連接 monomethyl auristatin(一種誘導細胞週期停滯的紡錘體細胞毒素)。以此治療觀察到原發性皮膚 ALCL 達 100% 的反應。與結節性 ALCL 相反,皮膚 CD30⁺ 大細胞淋巴瘤預後良好,與疾病相關的 5 年存活率為 90%。
皮下脂膜炎樣 T 細胞淋巴瘤 (SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA)
定義:皮下脂膜炎樣 T 細胞淋巴瘤被定義為一種細胞毒性 T 細胞淋巴瘤,特徵為主要位於皮下的小、中或大型多形性 αβ T 細胞浸潤以及許多巨噬細胞,主要影響腿部,偶爾併發噬血症候群(hemophagocytic syndrome)。具有 γδ TCR 表型的皮下淋巴瘤顯示更具侵襲性的病程,被歸類於皮膚 γδ T 細胞淋巴瘤之內。
臨床發現:此淋巴瘤佔所有皮膚淋巴瘤的 1%,佔所有皮下型 T 細胞淋巴瘤的 75%。在修訂的 WHO 2016 分類中,此名詞根據定義限定於表現 TCR α/β 表型的病例。皮下脂膜炎樣 T 細胞淋巴瘤以皮下結節與斑塊為特徵,通常侵犯四肢、軀幹,較少侵犯臉部。病人可能出現「B」症狀,即體重減輕、發燒與疲勞。
組織病理:組織學檢查顯示模擬小葉性脂膜炎(lobular panniculitis)的皮下浸潤。浸潤含有不同大小的腫瘤性多形性細胞與巨噬細胞的混合物。腫瘤性 T 細胞環繞個別脂肪細胞(rimming)是一個有幫助的診斷特徵。免疫表型分析顯示腫瘤細胞表現 CD3⁺、CD4⁻、CD8⁺、CD56⁻、TIA-1⁺、granzyme-β⁺ 與 βF1⁺。以免疫組織化學表現 βF1(TCR α/β)是此疾病實體的關鍵診斷標記。
治療與預後:α/β 型的皮下脂膜炎樣 T 細胞淋巴瘤對全身性皮質類固醇反應良好,預後極佳(5 年存活率為 85%),這證明了單獨以皮質類固醇作為初始治療策略是合理的。⁷³⁻⁷⁵
結外自然殺手/T 細胞淋巴瘤,鼻型 (EXTRANODAL NATURAL KILLER/T-CELL LYMPHOMA, NASAL TYPE)
定義:結外 NK/T 細胞淋巴瘤,鼻型,是一種罕見、侵襲性的原發性皮膚淋巴瘤形式,與正常 NK 細胞共享免疫表型特徵,並特徵性地顯示 CD56 與細胞毒性蛋白(如 perforin、granzyme B 與 TIA-1)的強烈表現。此淋巴瘤幾乎總是 EBV⁺。腫瘤細胞為小、中或大型,且通常具有 NK 細胞表型,或較罕見地具有細胞毒性 T 細胞表型。皮膚是繼鼻腔之後第二常見的表現部位。
臨床發現:結外 NK/T 細胞淋巴瘤要不影響鼻咽部,導致鼻部區域的破壞(以前被描述為致死性中線肉芽腫 lethal midline granuloma),要不則表現於皮膚、皮下、肺、內臟與睪丸。皮膚病灶包含皮下腫瘤、紅斑性斑塊、潰瘍,或伴有斑與丘疹的疹樣皮疹(圖 119-21)。臨床病程常因伴隨全血球減少的噬血症候群而惡化。
組織病理:此型淋巴瘤顯示侵犯真皮且常侵犯皮下的緻密浸潤。可能存在親表皮性。顯著的血管中心性(angiocentricity)與血管破壞常伴隨廣泛的壞死(圖 119-22)。免疫表型上,腫瘤細胞表現 CD56 與細胞毒性蛋白(TIA-1、granzyme B、perforin),並特徵性地對 EBV 呈陽性。TCR–CD3 複合體不在表面表現。典型可發現 EBV 的無性繁殖系游離基因體(clonal episome)存在。TCR 基因通常呈生殖系(germline)構型。³,⁷⁶
治療與預後:即使採用積極的多藥化療,此疾病常於數月內致死。EORTC 皮膚淋巴瘤組的一項研究建議骨髓移植可能是首選治療。
皮膚 T 細胞淋巴瘤的暫定疾病實體 (PROVISIONAL ENTITIES OF CUTANEOUS T-CELL LYMPHOMA)
定義 (DEFINITION)
除了前述各節討論的疾病之外,修訂的 WHO 分類系統中還納入了若干暫定疾病實體。這些原發性 CTCL 顯示特徵性的臨床與組織學特徵,但報導的病例系列仍有限,無法定義精確的預後。
原發性皮膚 CD4⁺ 小型與中型 T 細胞淋巴增生性疾病 (PRIMARY CUTANEOUS CD4⁺ SMALL AND MEDIUM T-CELL LYMPHOPROLIFERATIVE DISORDER)
定義:此疾病實體臨床上由丘疹與結節定義,組織學上由小型與中型多形性 T 細胞組成的皮膚浸潤定義。結果通常良好,但報導的病例系列有限。
臨床發現:病人有一個或數個紅紫色丘疹或結節,好發於頭頸部區域。由於與 MF 及 MF 相關毛囊性黏液病的組織學區別可能有問題,多形性小型與中型 CTCL 中斑片與斑塊的缺如是一個重要標準。
組織病理:組織學上,真皮內(有時皮下)可觀察到含有小型至中型多形性細胞的緻密、瀰漫性或結節性浸潤。可能存在親表皮性。腫瘤細胞表現 T 輔助細胞表型,常伴有全 T 細胞標記的喪失。證明異常表型與 T 細胞無性繁殖性,以及浸潤中以多形性 T 細胞為主,可作為排除假性淋巴瘤(pseudolymphomas,常顯示相同的組織學模式)的有用標準。MF 因缺乏主導性的腦回狀腫瘤細胞族群而被排除。
治療與預後:單發病灶常為診斷目的而切除。若無法切除或病灶為侷限性,放射治療是首選治療方式。PUVA 治療(可能合併干擾素 [interferon, IFN]-α)對播散性病灶的病例有用。此型淋巴瘤的 5 年存活率據報導介於 60% 與 90% 之間。⁷⁷⁻⁷⁹
原發性皮膚侵襲性親表皮 CD8⁺ 細胞毒性 T 細胞淋巴瘤 (PRIMARY CUTANEOUS AGGRESSIVE EPIDERMOTROPIC CD8⁺ CYTOTOXIC T-CELL LYMPHOMA)
定義:原發性皮膚侵襲性親表皮 CD8⁺ 細胞毒性 T 細胞淋巴瘤是一種 CTCL,特徵為 CD8⁺ 細胞毒性 T 細胞的增生,呈現強烈的親表皮性與侵襲性的臨床行為。與其他表現 CD8⁺ 細胞毒性 T 細胞表型的 CTCL(如派傑樣網狀細胞增生症以及罕見的 MF、淋巴瘤樣丘疹病與皮膚 ALCL 病例)的區別,是基於臨床表現、組織病理與臨床行為。
臨床發現:原發性皮膚侵襲性親表皮 CD8⁺ T 細胞淋巴瘤表現為角化過度的斑片與斑塊、邊界清楚的丘疹與腫瘤,或潰瘍。常觀察到轉移性擴散至不尋常部位,如肺、睪丸、中樞神經系統與口腔,但不轉移至淋巴結。
組織病理:組織學上,可觀察到由多形性淋巴球或免疫母細胞組成的帶狀浸潤,呈現對表皮的瀰漫性浸潤,伴有不同程度的海綿狀水腫、表皮內水疱形成與壞死。腫瘤細胞高頻率地表現 Ki67 抗原,並對 CD3、CD8、CD45RA 與 TIA 呈陽性,而 CD2 與 CD5 常喪失。TIA 的表現可鑑別這些源自細胞毒性 T 細胞亞群的淋巴瘤。
治療與預後:即使採用多藥化療,此疾病仍顯示侵襲性病程,存活中位數為 32 個月。⁷⁶,⁷⁹,⁸⁰
皮膚 γ/δ T 細胞淋巴瘤 (CUTANEOUS γ/δ T-CELL LYMPHOMA)
定義:皮膚 γ/δ T 細胞淋巴瘤涵蓋具有成熟、活化 γ/δ T 細胞無性繁殖系增生並具有細胞毒性表型的周邊 T 細胞淋巴瘤。此族群包括以前被稱為具有 γ/δ 表型的皮下脂膜炎樣 T 細胞淋巴瘤的病例。³
臨床發現:病人有播散性的潰瘍壞死性結節或腫瘤,特別在四肢,但其他部位也可能受影響。黏膜與其他結外部位的侵犯常見,但淋巴結、脾臟或骨髓的侵犯不常見。可能發生噬血症候群。
組織病理:組織學上,皮膚中可呈現 3 種主要的侵犯模式:親表皮性、真皮性與皮下性。腫瘤細胞一般為中至大型,染色質呈粗塊狀。具有泡狀核與顯著核仁的大型母細胞性細胞不常見。細胞凋亡與壞死常見,常伴有血管侵犯。免疫組織學上,腫瘤細胞具有 βF1⁻、CD3⁺、CD2⁺、CD5⁻、CD7⁺/⁻、CD56⁺ 表型,並強烈表現細胞毒性蛋白。大多數病例為 CD4 或缺乏 CD4 與 CD8,雖然部分病例可能表現 CD8。在冷凍切片中,細胞對 TCRδ 強烈陽性(石蠟切片無抗體檢測可用)。若只有石蠟切片可用,可利用 βF1 的缺如來推斷 γ/δ 起源。⁷⁴,⁷⁵
治療與預後:大多數病人有對多藥化療與/或放射治療具抗性的侵襲性疾病。存活中位數為 15 個月。
原發性皮膚肢端 CD8⁺ T 細胞淋巴增生 (PRIMARY CUTANEOUS ACRAL CD8⁺ T-CELL LYMPHOPROLIFERATION)
定義:此疾病實體以惰性皮膚 CD8⁺ 淋巴樣增生為特徵,主要起源於耳部,預後良好。
臨床特徵:惰性皮膚 CD8⁺ 淋巴樣增生,典型表現為臉部或肢端部位的單發皮膚病灶。已描述足部出現單發丘疹,或在某些病例為雙側斑塊。
組織病理:組織學上,惰性 CD8⁺ 淋巴樣增生以非親表皮性、中型多形性無性繁殖系 CD8⁺ T 細胞(大多為非活化細胞毒性表型)的緻密真皮浸潤為特徵,在大多數病例中顯示清楚的 grenz zone 與低增生指數。要與其他帶有細胞毒性 CD8⁺ 表型的侵襲性 T 細胞淋巴瘤區別,是根本之務,以避免病人不必要的焦慮以及不應有的積極治療(這些治療會被視為 CD8⁺ 表型治療演算法的一部分)。近期有人建議 CD68 可作為一個新的鑑別標記,有助於在模稜兩可的病例中區分惰性 CD8⁺ 淋巴樣增生與其他 CD8⁺ 皮膚淋巴瘤。
治療與預後:治療以局部切除或放射治療為基礎。完全緩解可持續數年,預後極佳。⁸¹
皮膚 T 細胞淋巴瘤的分期 (STAGING OF CUTANEOUS T-CELL LYMPHOMA)
確立 CTCL 的診斷後,必須進行適當的分期檢查,以排除結外淋巴瘤對皮膚的續發侵犯,並確定疾病的範圍。CTCLs 的第一個分類與分期系統於 1979 年由 MF 合作組(MF Cooperative Group)發表。人們認識到此分期系統並不適用於目前 WHO 分類中所列的所有 CTCL 類型。此外,常用於結節性非何杰金氏淋巴瘤分期的 Ann Arbor 系統,也不適合所有 CTCL 類型。基於這些事實以及關於預後因子的新資料,EORTC 與 ISCL 已提出 MF 與 Sézary 症候群的分期與分類修訂版,以及 MF 與 Sézary 症候群以外皮膚淋巴瘤的 TNM(tumor, node, metastasis)分類系統。必須提及的是,依 TNM 系統分期已被證明對選擇 MF 與 Sézary 症候群病人的適當治療有用,但某些類型 CTCLs 缺乏關聯 TNM 分期結果與預後的資料。所有類型 CTCLs 的分期檢查包括檢查全身皮膚、胸部 X 光攝影,以及腹部器官與周邊淋巴結(頸部、腋窩與腹股溝)的超音波檢查。血液檢查應包括全血球計數、含肝酵素的臨床化學、腎功能檢查與乳酸去氫酶值,以及 T 細胞無性繁殖性。分期可由可疑淋巴結與/或內臟器官的 CT 掃描與/或組織學與分子(TCR 重排)檢查來完成。分期檢查應在疾病復發或進展時重複進行。骨髓檢查僅在 B2 血液評級(表 119-7)或有無法解釋的血液學異常時建議進行。然而,此程序並無直接的臨床相關性,因為在骨髓中偵測到非典型細胞並非獨立的預後因子。前述檢查可依 TNM 系統進行分類(見表 119-4 與 119-7)。雖然 TNM 分期對不同 CTCLs 的預後價值與適用性仍有爭議,但 TNM 方案引導大多數 CTCLs 朝向適當治療方案的決策過程。
皮膚淋巴瘤的國際分期系統(MF 與 Sézary 症候群)(表 119-7/119-8)
| 分期 | T(腫瘤) | N(淋巴結) | M(轉移) | B(血液) |
|---|---|---|---|---|
| IA | T1 | N0 | M0 | B0 或 B1 |
| IB | T2 | N0 | M0 | B0 或 B1 |
| IIA | T1 或 T2 | N1 或 N2 | M0 | B0 或 B1 |
| IIB | T3 | N0-N2 | M0 | B0 或 B1 |
| III | T4 | N0-N2 | M0 | B0 或 B1 |
| IIIA | T4 | N0-N2 | M0 | B0 |
| IIIB | T4 | N0-N2 | M0 | B1 |
| IVA1 | T1-T4 | N0-N2 | M0 | B2 |
| IVA2 | T1-T4 | N3 | M0 | B0-B2 |
| IVB | T1-T4 | N0-N3 | M1 | B0-B2 |
T1,斑片/斑塊佔 ≤10% 體表面積;T2,斑片/斑塊佔 ≥10% 體表面積;T3,皮膚腫瘤;T4,紅皮症。N0,正常淋巴結;N1,可觸及淋巴結但無明確淋巴瘤組織學證據(N1 與 N2 可加註「a」或「b」分別表示以南方墨點法或聚合酶連鎖反應 [PCR] 分析未偵測 [a] 或偵測 [b] 到 T 細胞無性繁殖系);N2,可觸及淋巴結,有淋巴瘤組織學證據,淋巴結結構保留;N3,可觸及淋巴結,有淋巴瘤組織學證據,淋巴結結構消失。M0,無內臟侵犯;M1,組織學確認的內臟侵犯。B0,≤5% Sézary 細胞(B0 與 B1 可加註「a」或「b」分別表示以南方墨點法或 PCR 分析未偵測 [a] 或偵測 [b] 到 T 細胞無性繁殖系);B1,>5% Sézary 細胞但絕對 Sézary 細胞數少於 1.0 K/µL,或缺乏 TCR 無性繁殖系重排,或兩者皆是;B2,血液中有 TCR 無性繁殖系重排,且 Sézary 細胞 ≥1.0 K/µL,或符合下列 2 者之一:(a) CD4⁺ 或 CD3⁺ 細胞增加且 CD4/CD8 ≥10,或 (b) CD4⁺ 細胞增加並具異常表型(>40% CD4⁺/CD7⁻ 或 >30% CD4⁺/CD26⁻)。
皮膚 T 細胞淋巴瘤的治療原則 (PRINCIPLES OF TREATMENT OF CUTANEOUS T-CELL LYMPHOMA)
每個成功的 CTCL 管理策略都始於正確的診斷與分期。病人的預後與存活不僅依皮膚淋巴瘤的類型而異,也依分期而異;每種淋巴瘤迄今都有其各自的最佳治療,主要以分期為基礎。目前的分期是基於 ISCL 與 EORTC 皮膚淋巴瘤工作小組(Cutaneous Lymphoma Task Force)於 2007 年發表於《Blood》期刊的提案。⁶⁶
MF 與 Sézary 症候群以外皮膚淋巴瘤的 TNM 分類(表 119-4)
T(腫瘤)
- T1:單發皮膚侵犯
- T1a:直徑 <5 cm 的單發病灶
- T1b:直徑 >5 cm 的單發病灶
- T2:區域性皮膚侵犯:限於 1 個身體區域或 2 個相鄰身體區域的多個病灶ᵃ
- T2a:所有疾病涵蓋於直徑 <15 cm 的圓形區域內
- T2b:所有疾病涵蓋於直徑 >15 但 <30 cm 的圓形區域內
- T2c:所有疾病涵蓋於直徑 >30 cm 的圓形區域內
- T3:全身性皮膚侵犯
- T3a:侵犯 2 個不相鄰身體區域的多個病灶
- T3b:侵犯 ≥3 個身體區域的多個病灶
N(淋巴結)
- N0:無臨床或病理性淋巴結侵犯
- N1:侵犯引流目前或先前皮膚侵犯區域的 1 個周邊淋巴結區域ᵇ
- N2:侵犯 2 個以上周邊淋巴結區域ᵇ,或侵犯任何不引流目前或先前皮膚侵犯區域的淋巴結區域
- N3:侵犯中央淋巴結
M(轉移)
- M0:無皮膚外非淋巴結疾病的證據
- M1:存在皮膚外非淋巴結疾病
ᵃ身體區域的定義。⁶¹
ᵇ淋巴結區域的定義與 Ann Arbor 系統一致:周邊部位:肘前、頸部、鎖骨上、腋窩、腹股溝-股部與膕窩。中央部位:縱膈、肺門、主動脈旁、髂部。
於 2011 年由 EORTC 皮膚淋巴瘤工作小組發表了 MF 與 Sézary 症候群的整體反應評分。⁸² 此評分涵蓋整個 TNMB 譜,而不僅是皮膚的反應。然而,於本文撰寫時,分期與存活預測方面仍有若干問題。事實上,存活並不總是與傳統分期相關:Stage IIB 與 III、IB 與親毛囊性 MF 之間存在重疊。許多預後因子仍在分期系統之外,必須在預後試驗中加以評估。於本文撰寫時,最有效的預後因子為年齡大於 60 歲、腫瘤負荷與大細胞轉化。Scarisbrick 等人發表了一個預後指數模型。⁶³
最常見的 CTCL 亞型(MF 與 Sézary 症候群)的慢性疾病病程,使替代標記(surrogate markers)成為必要,而腫瘤負荷仍是存活的最佳替代標記。其他症狀的測量(如搔癢或生活品質評估)也常被使用;皮膚評分提供對治療客觀反應的測量;問卷則引導對治療主觀反應的評估。尚未證明將病人疾病由 T3 降至 T1 會伴隨任何存活效益,但也被認識到,除非病人首先達到緩解、且以任何皮膚評分系統評為皮膚評分 0,否則治癒是無法達成的。因此,緩解是邁向治癒的第一步。CTCL 的治療理想上需要一個多專科團隊,包括皮膚科醫師、放射腫瘤科醫師與血液-內科腫瘤科醫師。美國國家綜合癌症網絡(National Comprehensive Cancer Network)、EORTC 與歐洲腫瘤內科醫學會(European Medical Society of Oncology)已制定治療指引,可能有助於引導治療策略。對於像 CTCL 這樣的慢性疾病,支持性照護、處理生活品質,以及減輕搔癢與皮膚感染等症狀,對於改善 CTCL 病人的生活品質是必要的。⁸³,⁸⁴
早期蕈樣肉芽腫的治療 (TREATMENT OF EARLY STAGE MYCOSIS FUNGOIDES)
早期 MF 的治療通常涉及皮膚導向治療,伴或不伴全身性治療。已確立的皮膚導向治療策略包括 UVB 光照治療、PUVA 光照治療、外用皮質類固醇、外用 chlormethine、外用類視黃酸(如 bexarotene)與放射(如體外放射線治療與全皮膚電子束治療)。
Imiquimod 與 Resiquimod
Imiquimod 與 resiquimod 是新型外用免疫反應調節劑,屬於 imidazoquinoline 類藥物。它們是類鐸受體致效劑(Toll-like receptor agonists),當外用或注射至病灶或腫瘤時,可能造成全身性效果。Imiquimod 與 resiquimod 皆誘導細胞激素 IFN-α、腫瘤壞死因子-α、IL-6 與 IL-12 的合成與釋放,這些都將適應性免疫反應活化朝向 Th1 或細胞媒介路徑,同時抑制 Th2 路徑。Resiquimod 已被報導有令人鼓舞的結果。⁸⁵,⁸⁶
局部與全皮膚電子束治療
全皮膚電子束或許是所有皮膚導向治療中最有效者。它常用於皮膚侷限型疾病的病人局部使用,尤其是抗性斑塊與腫瘤。11,065 名接受全皮膚電子束治療病人的合併初始資料顯示完全反應率接近 70%。在 T1 侷限性疾病的病人中完全反應率更高,對單發病灶使用早期、低劑量放射可能導致治癒。然而,全皮膚電子束通常保留給比 IA 期皮膚侵犯更廣泛的病人,尤其是廣泛斑塊,或用於 Sézary 症候群的緩和治療,或非清髓性異體幹細胞移植之前。在 Stage IIB 疾病的單發結節中,可使用較低劑量 4 至 8 Gy 對單一抗性病灶投予,或使用低劑量 12 Gy 全身投予來達到控制。較低劑量有效,並提供在無過度毒性下進行多次治療的機會。⁸⁷⁻⁸⁹
蕈樣肉芽腫各分期的治療建議(依證據等級)(表 119-9)
MF Stage IA、IB 與 IIA 的第一線治療建議
- 觀察策略(主要 T1a):Level 4
- 皮膚導向治療(SDTᵃ)外用皮質類固醇(主要 T1a 與 T2a):Level 3
- UVB(紫外線 B)(主要 T1a 與 T2a):Level 2
- PUVA(psoralen 與紫外線 A):Level 2
- 侷限性放射治療(RT)(用於侷限性 MF 包括派傑樣網狀細胞增生症):Level 4
MF Stage IA、IB 與 IIA 的第二線治療建議(全身性治療ᵇ)
- 類視黃酸ᶜ:Level 2
- 干擾素(interferon, IFN)-α:Level 2
- 全皮膚電子束(TSEB)(主要 T2b):Level 2
- 低劑量 methotrexate(MTX):Level 4
MF Stage IIB 的第一線治療建議(全身性治療ᵇ)
- 類視黃酸ᶜ:Level 2
- IFN-α:Level 2
- TSEB:Level 2
- 單藥化療(gemcitabine、pegylated liposomal doxorubicin):Level 4
- 低劑量 MTX:Level 4
- 侷限性 RTᵈ:Level 4
MF Stage IIB 的第二線治療建議
- 多藥化療ᵉ:Level 3
- 異體幹細胞移植ᶠ:Level 3
MF Stage IIIA 與 IIIB 的第一線治療建議(全身性治療ᵇ)
- 類視黃酸ᶜ:Level 2
- IFN-α:Level 2
- 體外光化學治療(extracorporeal photochemotherapy, ECP)ᵍ:Level 3
- 低劑量 MTX:Level 4
- TSEB:Level 2
MF Stage IIIA 與 IIIB 的第二線治療建議
- 單藥化療(gemcitabine、pegylated liposomal doxorubicin):Level 3
- 異體幹細胞移植ʰ:Level 3
MF Stage IVA 與 IVB 的治療建議ⁱ
- 化療(gemcitabine、pegylated liposomal doxorubicin、CHOP 與 CHOP 樣多藥化療)ʲ:Level 3
- 放射治療(TSEB 與侷限性)ᵏ:Level 4
- Alemtuzumab(主要在 B2):Level 4
- 異體幹細胞移植:Level 3
Sézary 症候群的第一線治療建議
- ECPᵖ:Level 3
- Chlorambucil + prednisone:Level 3
- 與 ECP 或 PUVA 合併的全身性治療:
- 類視黃酸ᶜ:Level 3
- IFN-α:Level 3
- 低劑量 MTX:Level 4
Sézary 症候群的第二線治療建議
- 化療(gemcitabine、pegylated liposomal doxorubicin、CHOP 與 CHOP 樣多藥化療):Level 3
- Alemtuzumab:Level 4
- 異體幹細胞移植ʰ:Level 3
MF 與 Sézary 症候群達到緩解後可用於維持治療的藥劑ˡ
- ECP
- IFN-α
- 低劑量 MTX
- Mechlorethamine
- PUVA
- 類視黃酸
- 外用皮質類固醇
- UVB
ᵃ皮膚導向治療。ᵇ下列藥劑最常與 PUVA 合併;與其他治療方式以及彼此合併也被廣泛使用。ᶜ包括類視黃酸受體(RAR)與類視黃酸 X 受體(RXR)致效劑。ᵈ作為與全身性及其他 SDT 合併的附加治療。ᵉCHOP(cyclophosphamide、doxorubicin、vincristine 與 prednisone)是最廣泛使用的方案,有許多變異與其他組合可用。ᶠ應限於特殊病人,詳見內文。ᵍECP 可單獨使用或與皮膚導向及其他全身性治療合併。ʰ應限於特殊病人。ⁱ對於 MF Stage IVA1 的治療,可能適用 Sézary 症候群的建議。ʲ應優先使用單藥化療。ᵏ單獨使用或與全身性治療合併。ˡ各選項按字母順序列出,應選擇有效、可耐受、易於使用且有效率者。牛津實證醫學中心等級一般為 5。(改編自 Trautinger F, Eder J, Assaf C, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74。)
維持治療與外用類固醇治療 (MAINTENANCE THERAPY AND TOPICAL STEROID THERAPY)
治療正常皮膚的概念是由皮膚導向治療的臨床經驗演化而來。此策略有 2 個組成部分:在緩解誘導皮膚導向治療期間對正常皮膚的治療,以及在緩解維持期對正常皮膚的治療。外用化療、PUVA 與全皮膚電子束放射,皆涉及將正常皮膚暴露作為其達成緩解成功的整體組成部分。此成功反映了治療能夠中斷再循環 CTCL 細胞生命週期中關鍵之以皮膚為基礎的階段。一旦達成緩解,正常皮膚可用較低劑量與頻率的清除療法維持。已描述以 PUVA、全皮膚施用氮芥(nitrogen mustard)、體外光化學治療(ECP)與 IFN 進行的維持治療。最常使用的維持治療是 PUVA 或 UVB 照射。作為維持治療,PUVA 初始以每週 1 次的間隔施行 1 年。從治療的第二年開始,排程改為每隔 1 週 1 次再進行 1 年,接下來那一年每隔 2 週(every third week)1 次,最後在 2 年內每隔 3 週(every fourth week)1 次。在此時,病人應已緩解 5 年。應考慮在此時停止治療。治癒被定義為停止所有治療後 8 年無疾病。此定義源自氮芥治療與全皮膚電子束放射治療的經驗,顯示病人在停止治療後達到緩解 5 至 8 年後,晚期復發極為罕見。這意味著惡性皮膚 T 細胞可能再循環長達 5 年而不引起病灶。以 5 年間歇性 PUVA,這些細胞之一存活的可能性較低,但仍有可能。停止治療後,病人除非保持無疾病 8 年,否則不應被視為治癒。⁸⁸,⁸⁹
疑似復發的管理常包括外用糖皮質類固醇的使用,反映此治療方式在可疑病灶治療中可扮演的關鍵角色。在 CTCL 病程早期,以及在緩解病人疾病復發時,T 細胞活化過程可藉由積極使用外用糖皮質類固醇而鈍化。事實上,大多數病人在確診之前都有使用這些藥劑的病史。治療 MF 早期病灶的一個療法是每日兩次施用 class I 外用糖皮質類固醇,持續 8 週。此療法是疑似復發的第一線方式之一,並有助於辨識出那些在重複切片前需要進行 4 週「洗清期(washout)」的病人。
紅皮症 (ERYTHRODERMA)
在紅皮症型 CTCLs 中,由惡性細胞所啟動的免疫功能障礙與發炎過程導致全皮膚發紅、脫屑與不適。免疫為基礎的治療在這些疾病的管理中居於首要地位,並不令人意外。用於治療紅皮症型 CTCLs 的 3 種主要生物反應調節劑(biologic response modifiers, BRMs)為:(a) 口服類視黃酸、(b) 經靜脈途徑的 ECP,以及 (c) 皮下注射 IFN-α(表 119-10)。在這些藥劑的臨床試驗中,病人對通常為大量預先治療過的抗性疾病接受了單一療法。在實務中,這些治療常作為紅皮症的第一線單一療法使用,若反應不完全,則納入其他藥劑作為合併治療。使用這些藥劑時,部分反應比完全反應更常見。因此,若目標是緩解,合併治療比單一療法更常使用。若目標是緩和治療,單獨使用一種 BRM 的單一療法常已足夠。BRMs 在投藥、副作用、與其他治療方式的交互作用以及可得性方面各有不同。⁸³
紅皮症型皮膚 T 細胞淋巴瘤治療之生物反應調節劑(表 119-10)
| 治療 | 劑量選項 | 預期臨床反應的時間 | 副作用與不良反應 |
|---|---|---|---|
| 類視黃酸治療 | Bexarotene,300 mg/m² | 12 週 | 高三酸甘油酯血症、高膽固醇血症、嗜中性球減少、中樞性甲狀腺低能症、胰臟炎(可逆) |
| 體外光化學治療 | 光分離術(photopheresis),使用 8-methoxypsoralen 與紫外線 A,每 4 週連續 2 天 | 4-6 個月 | 見第 198 章(「光照治療」) |
| 干擾素-α 治療 | 3 百萬單位,每週 3 次,增加至最大耐受劑量(約 9 百萬單位/day) | 3-6 個月 | 類流感症狀、慢性疲勞;長期神經毒性風險(憂鬱、神經病變、失智與脊髓病變);甲狀腺炎、肝臟、骨髓毒性 |
| 標靶單株抗體 — Alemtuzumab | 依方案而異(見內文) | — | 巨細胞病毒再活化、較高劑量時的伺機性感染 |
| Brentuximab vedotin | 1.2-1.8 mg/kg 每 3 週 | 12 週 | 周邊神經病變(大多可逆) |
類視黃酸治療 (RETINOID THERAPY)
第一代類視黃酸(如 isotretinoin)對 CTCL 的效果有限。合成類視黃酸 bexarotene 以高選擇性結合類視黃酸 X 受體,而其他可用的類視黃酸則具有較不專一的結合模式。在單一療法試驗中,bexarotene 以 300 mg/m² 使用。在疾病所有分期(斑塊、紅皮症與腫瘤)的病人中都見到反應。反應與次級終點平行:整體受侵犯體表面積與整體腫瘤聚集面積的減少,以及搔癢的改善。紅皮症病人在口服 bexarotene 治療的最初數週可能經歷脫屑增加。改善通常在治療第 12 週開始。雖然在療效方面似乎存在劑量-反應關係,但較高劑量也與較高的不良事件率與劑量限制性毒性相關。其中,高三酸甘油酯血症、高膽固醇血症、嗜中性球減少與中樞性甲狀腺低能症最為常見。脂質值的升高發生迅速,在 2 至 4 週內,並與嚴重但可逆的胰臟炎相關。脂質的監測與降血脂藥物的使用有助於控制脂質值。部分病人也需要減少 bexarotene 膠囊的劑量。已觀察到口服 bexarotene 與 gemfibrozil 及 warfarin 的藥物交互作用。開始 bexarotene 治療的病人在開始用藥後數週內會發展出中樞性甲狀腺低能症,伴有低甲狀腺刺激素與游離甲狀腺素值。甲狀腺低能症的症狀可能不明顯,包括感到疲倦與怕冷,這可能被錯誤歸因於疾病本身。服用 bexarotene 期間補充 levothyroxine 可緩解症狀並改善治療的耐受性。此狀況在停止治療後數週內可逆。Bexarotene 治療無免疫抑制。服用 bexarotene 的病人通常每月有監測就診,以追蹤脂質、肝臟與甲狀腺參數。⁹⁰⁻⁹⁴
體外光化學治療 (EXTRACORPOREAL PHOTOCHEMOTHERAPY)
關於 ECP 的深入討論請參閱第 199 章「光化學治療與光動力治療」。ECP 涉及在 UVA 光存在下以 8-methoxypsoralen 治療病人淋巴球隔室的一部分,接著將這些細胞回輸。治療透過一條餵入 UVA 放射裝置的靜脈導管進行,此程序通常需要病人保持平躺 3 小時。治療每 4 週連續 2 天進行。紅皮症型 CTCLs 可用 ECP 單一療法管理,但以單一療法治療其他疾病分期尚未經嚴格研究。在一項涉及紅皮症型 CTCL 病人的多中心研究中,約四分之一有完全反應,四分之一無反應,其餘有部分反應。然而,很明顯即使是部分反應也可改善這些病人的生活品質。改善有時早在治療第 6 週開始,但部分病人直到開始治療後 12 個月才顯示完全的病灶清除。偶有在 2 天治療週期後立即出現的暫時性反應。平均而言,在 4 至 6 個月後,紅斑、脫屑與搔癢通常逐漸且永久地減少。病人常注意到更細微的變化,如體毛的恢復、寒顫的消失與出汗能力的恢復。部分反應也可能減少這些病人在感染性併發症方面的罹病率。受侵犯與發炎更嚴重的皮膚更容易被定植,提供微生物入侵宿主的儲存庫與進入點。因此,皮膚的改善也可使 CTCLs 的併發症減至最少。
紅皮症型 MF 與 Sézary 症候群的 ECP 臨床經驗顯示,治療反應很可能奠基於免疫系統。一個特徵是,當以 8-methoxypsoralen 與 UVA 光使少於 5% 的惡性淋巴球池失活時,即可見臨床反應,超過 95% 的惡性淋巴球隨時間消失。也似乎大多數免疫功能正常的病人有反應。經大量預先治療、疾病病程較長的病人,反應較差。此外,CD8⁺ 淋巴球值正常或僅輕微降低的病人對 ECP 有反應。在一項研究中,於 Stage T3 與 Stage T4 疾病病人中將全皮膚電子束放射治療與 ECP 合併。將接受皮膚導向放射治療加 BRM 的病人,與在同一機構接受電子束放射治療的歷史對照組相比,證明了 ECP 的影響,因為皮膚導向治療加 BRM 組的病人顯示明顯較長的存活。⁹⁵,⁹⁶
干擾素-α 治療 (INTERFERON-α THERAPY)
在 CTCLs 的治療中,研究最多的 IFN 是 IFN-α;然而,也已進行關於使用 IFN-β 與 IFN-γ 治療 CTCLs 的臨床研究。使用 IFN-α 作為單一療法的初始研究顯示完全反應率介於 10% 至 27%,治療持續時間少於 6 個月。同樣地,疾病的異質性以及病人先前接受的預先治療可能影響結果,並使與其他治療方式的比較變得不可能。IFN-α 通常以 3 百萬單位、每週 3 次開始,可增加至最大耐受劑量,典型範圍為 9 百萬單位/day。如同其他 BRMs,對 IFN 的反應是漸進的,需要 3 至 6 個月才能確定最大反應。病人達到最大反應後,IFN 劑量可降至維持水準。所有 IFNs 有相似的毒性。最初 IFN 治療會併發以發燒、頭痛、肌痛與疲勞為特徵的類流感疾病。當此症狀消退後,病人常留有輕微的慢性疲勞感。最令人擔憂的長期毒性是神經學方面:憂鬱、神經病變、失智與脊髓病變。可能發生自體免疫現象,如甲狀腺炎。此外,可能發生肝臟與骨髓的毒性效果。IFN 治療的監測包括血球計數以及評估對病人生活品質影響的問卷。⁹⁷
標靶單株抗體 (TARGETED MONOCLONAL ANTIBODIES)
Alemtuzumab:Alemtuzumab 是一種人源化單株抗體,標靶 CD52,CD52 表現於大多數 T 與 B 淋巴球及 NK 細胞。Lundin 等人⁹⁷ᴬ 報導 CTCL 病人的反應率為 50% 至 70%。由於原始劑量排程對 T、B 與 NK 細胞造成長時間抑制以及免疫抑制,此治療導致巨細胞病毒與其他伺機性感染的再活化。已研究較低劑量與皮下投予的替代劑量排程。Querfeld 等人⁹⁷ᴮ 報導,靜脈注射 alemtuzumab 後接續較低劑量皮下抗體時有良好反應。Bernengo 等人⁹⁹ 提出不同的排程;4 名病人在第 1 天接受 3 mg alemtuzumab、第 3 天 10 mg,然後隔日 15 mg。Bernengo 等人報導 14 名抗性 Sézary 症候群病人中 12 名(86%)有反應,包括 3 例完全緩解。然而,Rei Watanabe⁹⁸ 顯示低劑量 alemtuzumab 是對抗性 CTCL(即合併周邊血液疾病的 CTCL 病人)高度有效且一般耐受良好的療法。低劑量 alemtuzumab 對有血液侵犯(白血病型疾病)的病人有效,但對 MF 無效,反映了 MF 源自不同 T 細胞亞群的事實。具有血液疾病惡性 T 細胞的病人具有 CCR7⁺ L-選擇素⁺ TCM 的表型,這些細胞會遷移並在皮膚、血液與淋巴結之間再循環,而 MF T 細胞則源自非遷移的皮膚常駐 TRM。低劑量 alemtuzumab 清除所有循環中的 T 細胞,並清除皮膚中再循環的惡性 T 細胞,在 50% 的病人中導致完全、且常持久的緩解,同時倖免皮膚中的良性 T 細胞。基於 Rachael Clark 的臨床經驗⁹⁸ᴬ,alemtuzumab 應用於有或無輔助皮膚導向治療的病人,以及有瀰漫性皮膚紅斑的病人,但某些研究者告誡不要用於既存有斑塊與/或腫瘤的病人。⁹⁸,⁹⁹
Brentuximab Vedotin:抗體藥物結合物 brentuximab vedotin 是一種抗 CD30 單株抗體,透過高度穩定的蛋白酶可切割連接子與 monomethyl auristatin E(一種抗微管蛋白藥劑)結合。它經美國食品藥物管理局核准用於治療復發-抗性的何杰金氏淋巴瘤與 ALCL。過去,CD30 已被鑑別為表現 CD30 之淋巴瘤的極佳治療標靶。組織學上,它獨特地表現於何杰金氏淋巴瘤、ALCL 與正常活化的 T、B 與 NK 細胞,但不表現於正常組織。與 CD30 交互作用的單株抗體被認為藉由啟動 CD30 訊息傳遞而誘導細胞凋亡。CD30 高度表現於原發性皮膚 ALCL 與淋巴瘤樣丘疹病亞型的表面,並在 MF 型態亞型(包括大細胞轉化 MF)中表現程度不一。CTCL 中 brentuximab vedotin 第 II 期臨床試驗的首批結果近期發表。在 Kim 等人¹⁰¹ 對 MF 或 Sézary 症候群進行的 brentuximab vedotin 第 II 期試驗中,30 名可評估病人的整體整體反應為 70%。以免疫組織化學評估的 CD30 表現高度變異,CD30 最大表現中位數為 13%(範圍:0%-100%);CD30 表現低於 5% 者,整體反應的可能性低於 CD30 表現大於或等於 5% 者。在腫瘤微環境中偵測到豐富的 CD163⁺ M2 型巨噬細胞,提示 brentuximab 除了惡性 T 細胞外,也可能標靶這些巨噬細胞並破壞其促腫瘤功能。此偵測也提示這些帶有 CD30 的巨噬細胞可能為鄰近的惡性 T 細胞提供額外的 monomethyl auristatin E 來源。另一項第 II 期研究由 Duvic 等人¹⁰⁰ 在 CD30 淋巴增生性疾病或 CD30 MF/Sézary 症候群病人中發表。可評估的 48 名病人顯示整體反應率為 73%,完全反應率為 35%。反應時間中位數為 12 週,反應持續時間為 32 週(範圍:3-93 週)。所有淋巴瘤樣丘疹病或原發性皮膚 ALCL 病人皆有反應。最常見的副作用是誘發周邊神經病變。雖然大多數病例可逆,但部分病人經歷不可逆的神經病變。Brentuximab vedotin 的建議劑量為 1.8 mg/kg 每 3 週;為克服其副作用,建議較低劑量 1.2 mg/kg 或延長劑量間隔。目前,brentuximab vedotin 在 CTCL 治療中正於一項開放標籤第 III 期 ALCANZA 研究組試驗¹⁰⁰ᴬ 中研究,該試驗比較 brentuximab vedotin 與醫師選擇(bexarotene 或 methotrexate),用於復發 CD30⁺ MF 或原發性皮膚 ALCL 的病人。¹⁰⁰,¹⁰¹
Mogamulizumab:Mogamulizumab 是一種去岩藻糖基化(defucosylated)的抗 CCR4 單株抗體。它標靶 CCR4,這是一種優先由 MF 與 Sézary 症候群病人的 Th2 及調節性 T 細胞表現的趨化激素受體。在其配體 CCL17(TARC)與 CCL22(MDC)的反應下,CCR4 促進 T 細胞遷移至皮膚。這些 CCR4 配體由角質形成細胞、樹突細胞與巨噬細胞產生,並在 MF/Sézary 症候群侵犯的皮膚中大量存在。近期描述的一個周邊 T 細胞淋巴瘤亞群將 CCR4 表現為 GATA-3(驅動 Th2 分化的主要轉錄調控因子)轉錄譜的一部分。GATA-3 不僅由位於皮膚等各種部位的調節性 T 細胞表現,也由 MF/Sézary 症候群細胞表現,並可能驅動這些細胞的 CCR4 表現。除了在調控細胞歸巢與運輸的角色外,CCR4 的接合也可能促進細胞生長與存活。然而,細胞可透過受體內化(receptor internalization)這種恆定調控機制而對 CCR4 去敏感化。顯然,CCR4 在 MF/Sézary 症候群與其他 T 細胞淋巴增生性疾病中具有致病角色,且是有吸引力的治療標靶。Mogamulizumab 藉由抗體依賴性細胞媒介細胞毒性(antibody-dependent cell-mediated cytotoxicity)清除表現 CCR4 的細胞。藉由標靶趨化激素受體 CCR4,Duvic 等人證明 mogamulizumab 耐受良好且有顯著的臨床活性,在大量預先治療過的 MF 與 Sézary 症候群病人中整體反應率為 36%,反應時間中位數為 1.4 個月。同一團隊顯示,除了抗體依賴性細胞媒介細胞毒性外,mogamulizumab 也清除調節性 T 細胞——由於調節性 T 細胞在抑制宿主抗腫瘤免疫中的角色,這是許多人類癌症的重要治療標靶。Mogamulizumab 目前正於全球第 III 期試驗中評估,比較 mogamulizumab 與組織蛋白去乙醯酶抑制劑(histone deacetylase inhibitor)vorinostat。¹⁰²,¹⁰²ᴬ
單藥化療 (SINGLE-AGENT CHEMOTHERAPY)
20 多年前,美國國立衛生研究院的 Bunn 等人¹⁰³ 結論認為,在進展期 CTCL 中,多藥化療在整體存活方面並不優於依序進行的保守治療。因此,治療策略更集中於減少全身性副作用,採用已證明同樣有效的單藥化療。治療的選擇基於分期、共病的醫療狀況與先前治療,因為每種藥劑都有獨特的副作用與療效特徵。Methotrexate、pegylated liposomal doxorubicin、gemcitabine 與 pentostatin 都已在 CTCL 病人的第 II 期試驗中研究。⁸⁴
Gemcitabine:Gemcitabine hydrochloride 是 deoxycytidine 的核苷類似物,可抑制 DNA 合成,已顯示對實體腫瘤以及血液惡性腫瘤具有活性。在若干小型研究中,gemcitabine 以 1200 mg/m² 的劑量投予、進行 3 或 4 週共 3 個療程,顯示整體反應率為 70.5%,持續時間中位數為 15 個月。Duvic 等人證明較低劑量 gemcitabine(1000 mg/m² 每週 1 次共 3 個每週週期)在 25 名進展期與抗性 MF 病人中產生 68% 的整體反應率。它在有皮膚腫瘤的 MF 病人中特別有活性。Gemcitabine 可與 bexarotene 維持治療合併使用,以管理 MF 的斑塊與斑片。它也可與 liposomal doxorubicin 輸注交替使用,以延長化療的持續時間。Gemcitabine 的不良反應最常涉及骨髓抑制(白血球減少)、貧血,尤其是血小板減少。¹⁰⁴,¹⁰⁵
Pegylated Liposomal Doxorubicin:Pegylated liposomal doxorubicin 是 doxorubicin 的新劑型,其中藥物被包封於微脂體(liposomes)中,並藉由在微脂體表面附著聚乙二醇(即 pegylation)而穩定化,導致半衰期增加並改善在腫瘤組織中的累積。其毒性特徵以劑量限制性的黏膜與皮膚不良反應為特徵,尤其是手掌-腳掌紅斑感覺異常症候群(palmar-plantar erythrodysesthesia syndrome),據報導發生於高達 20% 的接受治療病人。在 EORTC 的一項前瞻性多中心對照試驗中,Stage IIB、IIIB 或 IVA MF、在至少 2 次先前全身性治療後抗性或復發的病人,接受 6 個週期的 pegylated liposomal doxorubicin 20 mg/m²,於單一 28 天週期的第 1 天與第 15 天投予。主要終點為反應率。在 49 名病人中,整體反應率為 48%,完全緩解為 6.1%。無惡化存活中位數為 6.2 個月。在 20% 的接受治療病人中觀察到毒性(grades III 至 IV)。¹⁰⁶
此試驗在需要細胞毒性治療的明確 MF 病人族群中產生了基準資料。其療效合理,且病人可使用微脂體包封 doxorubicin 作為清除腫瘤的藥劑。
Pralatrexate:Pralatrexate 是一種新型抗葉酸劑,對還原葉酸載體(reduced folate carrier, RFC-1)具有高親和力,這是與 methotrexate 相比的一種新型抗藥機制,在復發或抗性周邊 T 細胞淋巴瘤病人中與 29% 的整體反應率相關。共有 12 名轉化型 MF 病人納入研究。其中許多病人曾接受超過 5 次先前的全身性治療,包括 CHOP 或 CHOP 樣方案。然而,一項較大世代 CTCL 病人的劑量探索研究結果已被報導。在此項由 Horwitz 等人¹⁰⁶ᴬ 發表的研究中,最佳劑量被確認為 15 mg/m²,每 4 週中有 3 週每週投予,並與 43% 的整體反應率相關。為了減少黏膜炎的發生率,這些病人常規補充葉酸與維生素 B12。然而,pralatrexate 尚未在針對 CTCL 既有藥物的隨機試驗中研究。¹⁰⁷
異體幹細胞移植 (ALLOGENEIC STEM CELL TRANSPLANTATION)
對第一線治療無反應的較年輕進展期 CTCL(Stage IIB 或更高)病人,現在正被考慮進行非清髓性異體造血幹細胞移植。病人需要有相關或不相關的相符捐贈者,且在生理與情緒上能夠承受此程序。其治療概念基於移植物抗 T 細胞淋巴瘤效應(graft-versus-T-cell-lymphoma effect)的存在,特別是使用非清髓性條件。經選擇的病人已達到長期緩解與治癒性反應。Duarte 等人報導了 MF 與 Sézary 症候群病人異體造血幹細胞移植的長期結果。這些資料顯示進展期 MF 與 Sézary 症候群病人隨時間持續從異體造血細胞移植中獲益,5 年整體存活率為 46%,7 年為 44%,5 年無惡化存活率為 32%,7 年為 30%。疾病復發進展是移植後失敗的主要原因;共有 45% 的病人在造血幹細胞移植後中位 3.8 個月經歷復發進展。在多變項分析中,若干疾病與移植因子顯示對病人結果有獨立影響。分析聚焦於非復發死亡率,其與來自不相關捐贈者的移植呈臨界相關。復發與清髓性條件(相對於非清髓性)以及造血細胞移植時較差的體能評分(Karnofsky 評分 <70)高度顯著相關。病人必須在達到完全或接近完全緩解後才接受異體移植治療。¹⁰⁸,¹⁰⁹
原發性皮膚 B 細胞淋巴瘤 (PRIMARY CUTANEOUS B-CELL LYMPHOMAS)
原發性 CBCLs(表 119-11)是源自 B 淋巴球的惡性腫瘤,於診斷時在無皮膚外侵犯的情況下發生於皮膚,佔原發性皮膚淋巴瘤的 20% 至 25%。根據監測流行病學與最終結果登錄(surveillance epidemiology and end results registry)資料,CBCL 的發生率估計為每年每 1,000,000 人 3 例。¹¹⁰
PCMZL/結外邊緣區淋巴瘤與 PCFCL 是惰性亞型,而 PCLBCL, leg type 則具有中等至侵襲性的臨床行為。這 3 個疾病實體(即 PCMZL、PCFCL 與 PCLBCL)合計佔 CBCL 的 97%。¹¹¹⁻¹¹³
病因 (ETIOLOGY)
如同 CTCL,感染性觸發因子被假設參與 CBCL 的病因。胃結外邊緣區淋巴瘤與幽門螺旋桿菌(Helicobacter pylori)感染的強烈關聯廣為人知。在歐洲病人少數的 PCMZLs 中(但在美國或亞洲病人中則否),曾偵測到伯氏疏螺旋體(Borrelia burgdorferi)。一項較新的研究顯示,根據免疫球蛋白表現,可區分出 2 種類型的 PCMZLs。在最常見的 PCMZL 亞型中,可觀察到表現類別轉換(class-switched)免疫球蛋白(Ig)重鏈(包括 IgG,以及較少的 IgA 或 IgE)的 B 細胞。在大多數病例中,可觀察到 IgM⁺ MCXCR3 表現 B 細胞的瀰漫性增生,這與較少的浸潤性 T 細胞以及朝向 CD4⁺ T 細胞數目增加偏斜的 CD4 對 CD8 比值相關。在 IgM⁺ 病例中,常觀察到疾病的皮膚外定位,提示來自類別轉換與非類別轉換免疫球蛋白重鏈的結外黏膜相關淋巴組織之皮膚定位,是由不同的病理過程所造成。關於免疫球蛋白重鏈表現的詳細研究顯示,PCMZL 在 39% 的病例中顯示 IgG4 表現。這些病人中沒有 IgG4 相關疾病的證據,指向一個侷限性的免疫學 IgG4 驅動過程。在臨床管理中,這些觀察很有幫助,因為皮膚表現 IgG4 的邊緣區淋巴瘤將必然是 PCMZL。病毒因子(特別是 C 型肝炎病毒)也被懷疑與 CBCL 的病因有關,但研究結果相互矛盾。¹¹⁰⁻¹¹⁶
皮膚 B 細胞淋巴瘤摘要(表 119-11)
| 類型 | 診斷年齡 | 疾病性質 | 臨床發現 | 細胞起源 | 免疫組織化學特徵 | 治療 |
|---|---|---|---|---|---|---|
| 原發性皮膚濾泡中心淋巴瘤(PCFCL) | 診斷年齡中位數 58 歲 | 惰性 | 單發或成群、堅實、無痛的紅斑性斑塊與腫瘤,常侵犯頭部與軀幹 | 無性繁殖系中心細胞(centrocytes)與中心母細胞(centroblasts) | CD19⁺、CD20⁺、CD79a、CD10、Bcl6⁺ᵃ | 放射治療;播散性病灶用免疫治療 |
| 結外邊緣區淋巴瘤(EMZL)/原發性皮膚邊緣區淋巴瘤(PCMZL) | 診斷年齡中位數 55 歲;女性佔優勢 | 惰性 | 紫紅色丘疹、斑塊或結節;發生於多個部位 | 小型 B 淋巴球、邊緣區細胞、淋巴漿細胞樣細胞、漿細胞 | CD20、CD79a、Bcl2⁺;漿細胞可能表現 CD138、CD79a | 放射治療;若與伯氏疏螺旋體相關則用全身性抗生素;chlorambucil、rituximab、病灶內干擾素 |
| 原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型 | 診斷年齡中位數 65 歲;女性佔優勢 | 中等至侵襲性 | 單發或成簇的藍色紅斑性斑塊與腫瘤,位於單側或雙側腿部 | 大型 B 細胞 | CD20⁺、CD79a、IRF4-MUM-1⁺、FOXP1⁺、細胞質免疫球蛋白 M ± D、Bcl2⁺、Bcl6⁺ | Rituximab、cyclophosphamide、hydroxydaunorubicin、Oncovin、prednisolone;bendamustine + rituximab 方案 |
ᵃ若有結節性疾病可能為 Bcl2⁺。
發病機轉 (PATHOGENESIS)
由於 CBCL 亞型的罕見性,調查 CBCL 致病事件的研究主要在少數病例中進行。然而,近年來在理解 CBCL 的致病機轉方面已取得相當進展。一種稱為異常體細胞超突變(aberrant somatic hypermutation)的現象(曾在結節性 B 細胞淋巴瘤中報導),已在 3 種主要類型的 CBCL 中被偵測到。此名詞描述活化誘導去胺酶(activation-induced deaminase)的活性,此酵素藉由體細胞超突變促成免疫球蛋白的親和力成熟過程,但發生於不編碼免疫球蛋白基因的基因體區域。若此過程發生於含致癌基因的基因座,並伴隨生理性高保真 DNA 修復機制的喪失,則可能發生致腫瘤突變並促成淋巴瘤生成。⁷⁹
此外,遺傳調查證明了 CBCL 亞型的明顯差異。已顯示在組織學上區分具有大細胞瀰漫性生長模式的皮膚 PCFCL 與 PCLBCL, leg type 的遺傳基礎。這些疾病實體的基因表現譜與 PCFCL 的生發中心 B 細胞(germinal center B cells)或 PCLBCL, leg type 的活化 B 細胞(activated B cells)一致。在一項聚焦於促凋亡與抗凋亡基因的研究中發現了相似的結果。預後不良的 PCLBCL, leg type 具有稱為「活化凋亡級聯(activate apoptosis cascade)」的遺傳譜,而 PCFCL 與預後良好的 PCLBCL, leg type 病例則對與抗腫瘤細胞毒性免疫反應相關的基因有高表現水準。這些發現也解釋了為何 PCFCL 比 PCLBCL, leg type 預後更佳。此外,發現 PCMZLs 在與結外邊緣區淋巴瘤不同的致病背景下產生。主要差異是高百分比的 CXCR3⁻ PCMZLs,這與其他結外邊緣區淋巴瘤相反,也呈現免疫球蛋白類別轉換與 Th2 細胞激素環境。這如何與 PCMZL 中漿細胞樣樹突細胞的存在相關仍待調查;這些細胞也在皮膚假性淋巴瘤中發現,但不在瀰漫性大 B 細胞淋巴瘤中,且僅罕見於 PCFCL。¹¹³,¹¹⁷⁻¹²⁴
原發性皮膚濾泡中心淋巴瘤 (PRIMARY CUTANEOUS FOLLICULAR CENTER LYMPHOMA)
定義 (DEFINITION)
PCFCL 可被定義為由無性繁殖系中心細胞(小型與大型裂解濾泡中心細胞)與中心母細胞(具有顯著核仁的大型濾泡中心細胞)組成的腫瘤,伴或不伴濾泡形成。
臨床發現 (CLINICAL FINDINGS)
PCFCL 通常表現為單發或成群、堅實、無痛的紅斑性斑塊與腫瘤,優先位於頭部與軀幹,罕見於腿部。PCFCL 病人的診斷年齡中位數為 58 歲,成年男性受影響的頻率接近成年女性的兩倍。偶爾,可在周圍區域觀察到環狀紅斑(圖 119-23A)。典型發現是病灶出現於頭頸部區域或軀幹的局限區域,但罕見於腿部。
組織病理 (HISTOPATHOLOGY)
這些腫瘤由中至大型濾泡中心細胞組成,含有中心細胞與中心母細胞的混合物,以濾泡性或瀰漫性生長模式排列(圖 119-23B)。腫瘤細胞表現全 B 細胞標記 CD19、CD20 與 CD79a,並表現生發中心標記 Bcl6。CD10 的表現特別在具有濾泡性生長模式的病例中觀察到。與原發性皮膚 B 細胞淋巴瘤,腿型不同,一般不觀察到 IRF4-MUM-1、FOXP1 與細胞質免疫球蛋白的表現。與結節性濾泡中心淋巴瘤相反,在大多數研究中 PCFCLs 不常表現 Bcl2,且一般不含 t(14;18) 易位。基因表現研究證明 PCFCLs 具有類似生發中心 B 細胞樣瀰漫性大 B 細胞淋巴瘤的基因表現譜。基因體研究在 63% 的病例中發現 c-REL 擴增,在 68% 的病例中發現 14q32.33 的缺失。
治療與預後 (TREATMENT AND PROGNOSIS)
PCFCLs 是惰性淋巴瘤,罕見轉移至皮膚外定位,其預後極佳,5 年存活率大於 95%。約 5% 的 PCFCLs 發生於腿部,這些病例具有更具侵襲性的行為。放射治療(30 Gy)是 PCFCL 的首選治療(表 119-12)。在 30% 的病例中觀察到復發,但這些並不預示不良預後,且可用放射治療再次治療。對於復發,可使用 4 Gy 的緩和劑量,這將在 90% 的病例中達到有效的局部控制。免疫治療(如投予 IFN-α 或抗 CD20 單株抗體)對播散性病灶的病例可能有益。¹²⁴,¹²⁵
結外邊緣區 B 細胞淋巴瘤 (EXTRANODAL MARGINAL ZONE B-CELL LYMPHOMA)
定義 (DEFINITION)
PCMZL 或結外邊緣區淋巴瘤是一種惰性 B 細胞淋巴瘤,由小型 B 淋巴球、邊緣區細胞、淋巴漿細胞樣細胞與漿細胞組成,這些細胞最初定位於濾泡中心的邊緣區。在目前的 WHO 分類中,它被歸類於黏膜相關淋巴組織(mucosa-associated lymphatic tissue)的結外邊緣區淋巴瘤之內。在 PCMZL 的一個亞群中,B. burgdorferi 被認為具有病因角色。
臨床發現 (CLINICAL FINDINGS)
PCMZLs 最常見於上肢或軀幹,發生於診斷年齡中位數 55 歲;它們在女性中佔優勢。病人有小型紅至紫紅色的丘疹、斑塊或結節,與 PCFCL 的病灶相反,常發生於多個部位。PCMZL 佔原發性 CBCLs 的 25%。
皮膚 B 細胞淋巴瘤的治療演算法(表 119-12)
原發性皮膚邊緣區淋巴瘤(Primary Cutaneous Marginal Zone Lymphoma)
| 疾病類型與範圍 | 第一線治療 | 替代治療 |
|---|---|---|
| 單發/侷限 | 局部放射治療;切除;抗生素ᵃ | 病灶內干擾素(IFN)-α;病灶內 rituximab;病灶內類固醇 |
| 多病灶 | 觀察等待(wait-and-see);局部放射治療;chlorambucilᵇ;靜脈 rituximab;抗生素ᵃ | 病灶內 IFN-α;病灶內 rituximab;外用或病灶內類固醇 |
原發性皮膚濾泡中心淋巴瘤(Primary Cutaneous Follicle Center Lymphoma)
| 疾病類型與範圍 | 第一線治療 | 替代治療 |
|---|---|---|
| 單發/侷限 | 局部放射治療;切除 | 病灶內 IFN-α;病灶內 rituximab |
| 多病灶 | 觀察等待;局部放射治療;靜脈 rituximab | R-CVP/CHOPᶜ |
原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型(Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type)
| 疾病類型與範圍 | 第一線治療 | 替代治療 |
|---|---|---|
| 單發/侷限 | R-CHOP ± IFRT;靜脈 rituximab | 局部放射治療 |
| 多病灶 | R-CHOP;靜脈 rituximab | — |
縮寫:CHOP,cyclophosphamide、doxorubicin、vincristine 與 prednisone;IFRT,受累區域放射治療(involved-field radiotherapy);R-CHOP,rituximab、cyclophosphamide、doxorubicin、vincristine 與 prednisone;R-CVP,rituximab、cyclophosphamide、vincristine 與 prednisone。
ᵃ若有伯氏疏螺旋體感染的證據。ᵇ或其他適合低惡性度 B 細胞淋巴瘤的單一或合併方案。ᶜ用於特殊病例或發展出皮膚外疾病的病人。(改編自 Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112(5):1600-09,經授權。)
組織病理 (HISTOPATHOLOGY)
組織學浸潤包括邊緣區(中心細胞樣 centrocyte-like)細胞、漿細胞樣細胞與漿細胞。典型地,邊緣區細胞表現 CD20、CD79a 與 Bcl2,但對 CD5、CD10 與 Bcl6 為陰性。漿細胞表現 CD138 與 CD79a,並在石蠟切片上顯示單型性細胞質免疫球蛋白輕鏈表現。¹¹⁸,¹¹⁹,¹²⁶,¹²⁷
治療與預後 (TREATMENT AND PROGNOSIS)
PCMZLs 曾被描述與蜱叮咬及抗原注射有關,這提示慢性抗原刺激可能在這些淋巴瘤的致病中扮演角色。PCMZLs 是惰性淋巴瘤,散在病灶可用放射治療(見表 119-12)。在偵測到 B. burgdorferi 的情況下,應首先給予全身性抗生素治療。對於廣泛性病灶,可採用觀察等待策略,有症狀的病灶可用手術、外用或病灶內類固醇,或低劑量放射治療。此外,以 chlorambucil 或 rituximab 的全身性治療,以及以 IFN-α 或 rituximab 的病灶內治療,據報導可在大多數病人中導致完全反應。¹¹⁷
原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型 (PRIMARY CUTANEOUS DIFFUSE LARGE B-CELL LYMPHOMA, LEG TYPE)
定義 (DEFINITION)
PCLBCL, leg type 因其相對於上述惰性亞型被認為預後不佳,而被鑑別為一個獨立的臨床疾病實體。此疾病實體顯示中等、且在某些病人為侵襲性的臨床病程。它由大型 B 細胞組成的腫瘤定義,在絕大多數病例中表現於腿部,但也可發生於其他部位。大多數 PCLBCL, leg type 病人在第 9p21 號染色體上有異常,而此區域(含有 CDKN2A 基因)的喪失與較差的預後相關。¹²⁸,¹²⁹
臨床發現 (CLINICAL FINDINGS)
PCLBCL, leg type 影響老年病人(大於 65 歲),女性佔優勢。典型地,病人有單發或成簇的藍色紅斑性斑塊與腫瘤,位於單側或有時雙側腿部(圖 119-24A);約 10% 的病人在腿部以外的部位出現病灶。潰瘍化常見,有時導致誤診為慢性靜脈功能不全所致的潰瘍。
組織病理 (HISTOPATHOLOGY)
瀰漫性浸潤顯示成片的免疫母細胞與中心母細胞,混有少數反應性細胞(圖 119-24B)。腫瘤細胞表現 CD20 與 CD79a,且與 PCFCL 不同,對 Bcl2、IRF4-MUM1 與 FOXP1 強烈陽性,並有 IgM ± IgD 的細胞質表現。Bcl6 在大多數病例中表現,而 CD10 一般缺如。與免疫表型特徵一致,PCLBCL, leg type 具有類似活化 B 細胞瀰漫性大 B 細胞淋巴瘤的基因表現譜。此外,PCLBCL, leg type 表現活化誘導胞苷去胺酶(activation-induced cytidine deaminase),進行免疫球蛋白基因的體細胞超突變,並帶有 Bcl6、Myc、Rho-TTF 與 PAX5 基因的突變,這些指示異位性體細胞超突變。在高達 43% 的 PCLBCL, leg type 病例中觀察到 Myc 易位,在高達 46% 的病例中觀察到 Bcl6 易位。關於拷貝數變異的研究描述 67% 的病例有 Bcl2 基因的高度擴增,23% 至 42% 的病人有 CDKN2a 的喪失。CDKN2a 表現的喪失(無論是藉由基因缺失或啟動子甲基化)與不良預後相關。
治療與預後 (TREATMENT AND PROGNOSIS)
PCLBCL, leg type 屬於 CBCLs 的中等-侵襲性族群,應優先以 rituximab、cyclophosphamide、hydroxydaunorubicin、Oncovin(vincristine)與 prednisolone(R-CHOP)治療(見表 119-12)。一般而言,依年齡調整的多藥化療合併 rituximab 顯示比單獨多藥化療方案更好的結果。¹²⁵ 由於 PCLBCL, leg type 發生於老年女性(大於 80 歲),毒性較低的方案(如 bendamustine 合併 rituximab)也可被建議。儘管有此治療,大多數病人仍觀察到復發,與疾病相關的 5 年存活率約為 50%。
原發性皮膚瀰漫性大 B 細胞淋巴瘤,其他 (PRIMARY CUTANEOUS DIFFUSE LARGE B-CELL LYMPHOMA, OTHER)
PCLBCL, other 涵蓋不屬於 PCLBCL, leg type 或 PCFCL 的瀰漫性大 B 細胞淋巴瘤。這些病例可能代表全身性淋巴瘤的皮膚表現。僅有皮膚病灶的 T 細胞/組織球豐富型 B 細胞淋巴瘤(T-cell/histiocyte-rich B-cell lymphomas)也包括在內;這些病例與其結節性對應物相反,具有極佳的預後。
血管內皮膚 B 細胞淋巴瘤 (INTRAVASCULAR CUTANEOUS B-CELL LYMPHOMA)
血管內 CBCL 以真皮與皮下血管內成簇的大型腫瘤性 B 細胞為特徵。偶爾可觀察到非典型細胞的輕微血管外浸潤。臨床上,紅至藍色、硬化的斑塊發生於腿部或軀幹。有時可見脂膜炎樣模式。多藥化療是首選的治療方式。¹³⁰,¹³¹
皮膚 B 細胞淋巴瘤的分期 (STAGING OF CUTANEOUS B-CELL LYMPHOMA)
國際皮膚淋巴瘤學會與 EORTC 皮膚淋巴瘤工作小組為 MF 與 Sézary 症候群以外的皮膚淋巴瘤制定了腫瘤-淋巴結-轉移(TNM)分類提案(見表 119-4)。原發性皮膚 B 細胞淋巴瘤(PCBL)的建議分期程序包括徹底的身體檢查、實驗室檢查(包括全血球計數、含乳酸去氫酶值的血液化學),以及在有指徵時,進行血清電泳以排除單株免疫球蛋白病(monoclonal gammopathy),與/或周邊血液的流式細胞術。在流行地區,應進行伯氏疏螺旋體血清學檢測與皮膚切片標本的聚合酶連鎖反應。影像學檢查包括胸部、腹部與骨盆腔的對比增強 CT 掃描(伴或不伴正子放射斷層攝影 positron emission tomography),且若病灶起於頭頸部區域則加做頸部。PCFCL 與 PCLBCL, leg type 需要骨髓切片與抽吸,但對於皮膚 B 細胞淋巴瘤、組織學特徵提示濾泡中心淋巴瘤或邊緣區淋巴瘤的病人則不需要,除非其他分期評估有指徵。然而,Senff 等人¹³¹ᴬ 的一項研究在 193 例 PCFCLs 中證明 9 名病人於診斷時有骨髓侵犯(22 例,11%)。骨髓侵犯是唯一的皮膚外定位。由於這些有皮膚與骨髓定位的病人預後明顯比僅有皮膚病灶的病人差,因此建議這些病人應考慮骨髓檢查。提議的 CBCL 分期系統已在回溯性研究中調查,證明 TNM 系統在記錄 CBCLs 疾病範圍以及為 PCLBCL, leg type 提供預後資訊時,是一個有用的工具。
皮膚 B 細胞淋巴瘤的治療原則 (PRINCIPLES OF TREATMENT OF CUTANEOUS B-CELL LYMPHOMA)
原發性 CBCLs 的治療(見表 119-12)應依這些淋巴瘤的良好預後而調整,尤其是 PCFCL 與 PCMZL。由於於本文撰寫時尚未定義治癒性方案,治療取決於淋巴瘤以及皮膚病灶的播散。在單發病灶的情況下,可提議腫瘤的完全切除。或者,或在少數侷限性病灶的情況下,以 X 光或電子束進行局部照射(單次劑量 3-4 Gy;總劑量 30-40 Gy)是有效的。使用此方案時,可達到持久的緩解。¹²⁵,¹³²
對於播散性 PCMZL 或 PCFCL,建議以抗 CD20 抗體進行全身性治療。對於有播散性病灶的惰性 CBCLs 復發,可採用觀察等待策略合併有症狀病灶的治療。對於 PCLBCL, leg type 病例,以及病灶位於腿部的 PCFCL(因為這些 PCFCLs 預後較差),以及當有續發性皮膚外表現時,建議全身性治療方案。建議以多藥化療(6 個週期的 CHOP 或 COP [cyclophosphamide、vincristine 與 prednisone])合併抗 CD20 抗體。在無法耐受此類積極治療的病人中,可考慮局部放射治療或 rituximab 單一療法。
前驅腫瘤 (PRECURSOR NEOPLASMS)
母細胞性漿細胞樣樹突細胞腫瘤 (BLASTIC PLASMACYTOID DENDRITIC CELL NEOPLASM)
定義 (DEFINITION)
根據目前的 WHO 分類,母細胞性漿細胞樣樹突細胞腫瘤(blastic plasmacytoid dendritic cell neoplasm, BPDCN)被歸類為一種急性骨髓性白血病相關的前驅腫瘤,源自漿細胞樣樹突細胞的前驅細胞。BPDCN 是一種罕見疾病(orphan disease),具有非常侵襲性的臨床病程,導致存活時間中位數為 12 至 14 個月。
臨床發現 (CLINICAL FINDINGS)
BPDCN 通常發生於老年病人,年齡中位數介於 60 至 70 歲之間。然而,BPDCN 可在任何年齡出現,甚至兒童。BPDCN 較常發生於男性(男女比:3:1),但無已知的種族或族群好發性。病人典型表現為無症狀的單發或多發皮膚病灶,如結節、斑塊或瘀傷樣病灶,大小可由數毫米至 10 cm(圖 119-25)。皮膚病灶可能伴隨紅斑、色素沉著、紫斑或潰瘍。大多數病人於診斷時即有皮膚外疾病,常侵犯區域淋巴結。隨著疾病持續進展,周邊血液與骨髓會受侵犯。在 10% 至 20% 的 BPDCN 病人中,可鑑別出同時發生的骨髓化生不良(myelodysplasia),其後可能導致急性骨髓單核球性白血病(acute myelomonocytic leukemia)的發展。
組織病理 (HISTOPATHOLOGY)
在皮膚病灶中,BPDCN 典型浸潤真皮但倖免表皮。隨著疾病進展,它常延伸至皮下脂肪。腫瘤細胞傾向於在淺層至中層真皮以血管周圍與/或附屬器周圍分布聚集,雖然較少見地,它們可能以淺層真皮的苔癬樣(lichenoid)浸潤呈現。在高倍率下,BPDCN 以單調的小至中型細胞族群為特徵,具有不規則的核輪廓、細緻至均勻分散的染色質、1 至 3 個小核仁,以及少量至中量的細胞質。以免疫組織化學,BPDCN 細胞典型表現 CD56、CD4、CD123 與 T 細胞白血病/淋巴瘤 1(T-cell leukemia/lymphoma 1, TCL1)。它們也可表現其他漿細胞樣樹突細胞相關抗原,如血液樹突細胞抗原 2(blood dendritic cell antigen 2, BDCA-2)/CD303 與 IFN-α 依賴性分子 MxA。
發病機轉 (PATHOGENESIS)
BPDCN 細胞顯示數個腫瘤抑制基因(包括 RB1、CDKN1B、CDKN2A 與腫瘤蛋白 P53 [TP53])缺失的復發性重組。位於 4q24 帶的 TET2 基因(ten-eleven translocation-2)在 BPDCN、骨髓化生不良症候群、慢性骨髓單核球性白血病與急性骨髓單核球性白血病中發生突變,這提供了 BPDCN 是骨髓相關腫瘤的額外證據。標靶超深度定序(targeted ultradeep sequencing)揭示了 BPDCN 中癌症基因的復發性且相互排斥的突變。在 33 個 BPDCN 樣本中,NRAS(27.3%)、ATM(21.2%)、MET、KRAS、IDH2 與 KIT(各 9.1%)發生點突變。此外,發現 NRAS、KRAS 與 ATM 突變相互排斥。在 27.3% 的病例中偵測到 CDKN2A 缺失,其次是 RB1 缺失(9.1%)。某些突變的相互排斥分布可能指向 BPDCN 的不同亞群,其生物學意義仍有待探討。¹³¹,¹³³,¹³⁴
治療與預後 (TREATMENT AND PROGNOSIS)
BPDCN 與高度侵襲性的臨床病程相關,因此預後不佳(存活中位數 14 個月)。雖然全身性化療是此疾病的首選治療,但 gemcitabine 可能有助於控制初始疾病,以便病人能盡快被轉介進行骨髓移植。¹³⁵,¹³⁶
另一個可能有前景的策略是藉由免疫結合物來標靶 IL-3 受體 CD123。¹³⁷
圖表 (Figures and Tables)
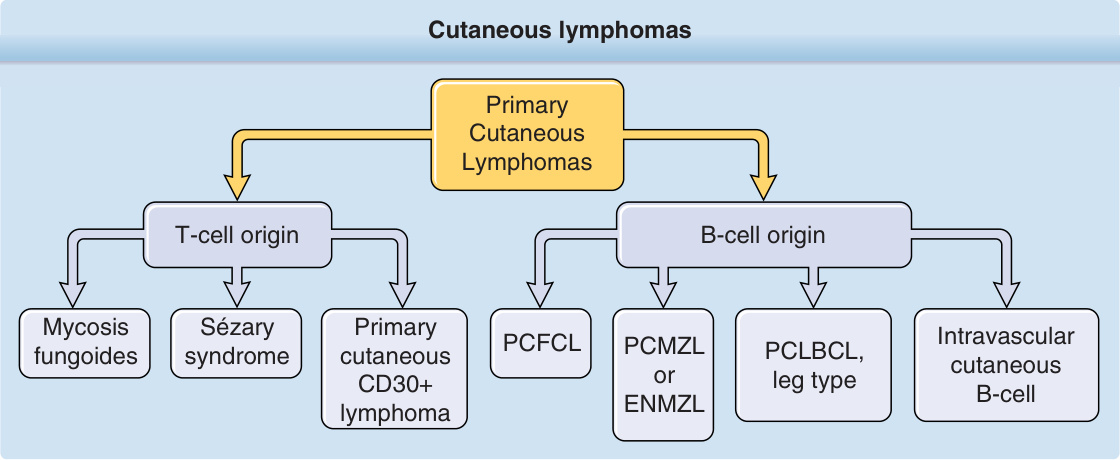
圖 119-1:皮膚淋巴瘤。ENMZL,結外邊緣區淋巴瘤 (extranodal marginal zone lymphoma);PCFCL,原發性皮膚濾泡中心淋巴瘤 (primary cutaneous follicle center lymphoma);PCLBCL,原發性皮膚瀰漫性大 B 細胞淋巴瘤 (primary cutaneous diffuse large B-cell lymphoma);PCMZL,原發性皮膚邊緣區淋巴瘤 (primary cutaneous marginal zone lymphoma)。
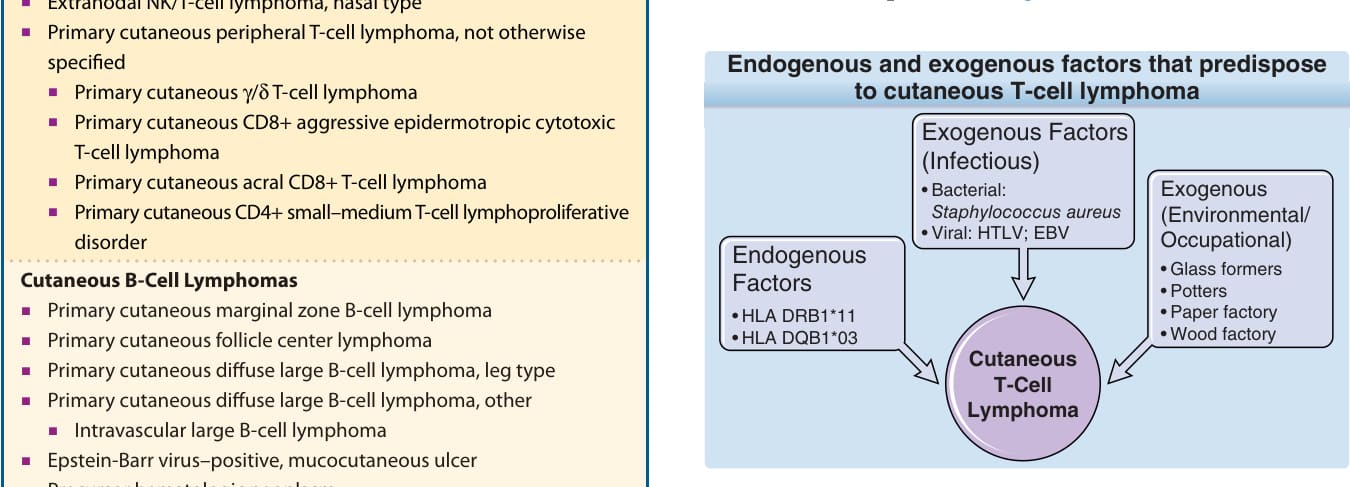
圖 119-2:促成皮膚 T 細胞淋巴瘤的內生性與外生性因素。EBV,EB 病毒 (Epstein-Barr virus);HTLV,人類 T 細胞嗜淋巴球病毒 (human T-cell lymphotropic virus)。
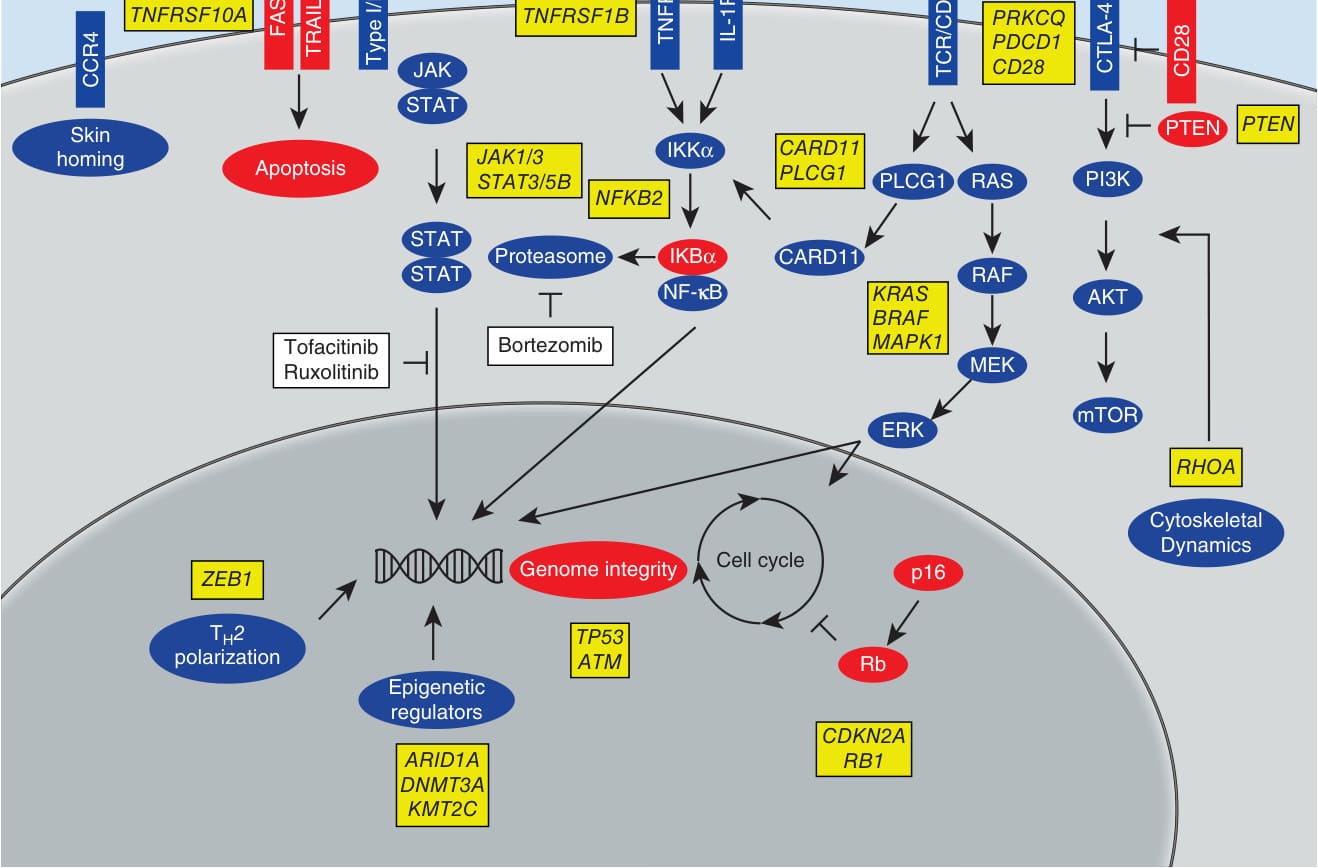
圖 119-3:皮膚 T 細胞淋巴瘤的訊息傳遞事件。(取自 Damsky WE, Choi J. Genetics of cutaneous T cell lymphoma: from bench to bedside. Curr Treat Options Oncol. 2016;17(7):33,經 Springer 授權。Copyright © 2016。)
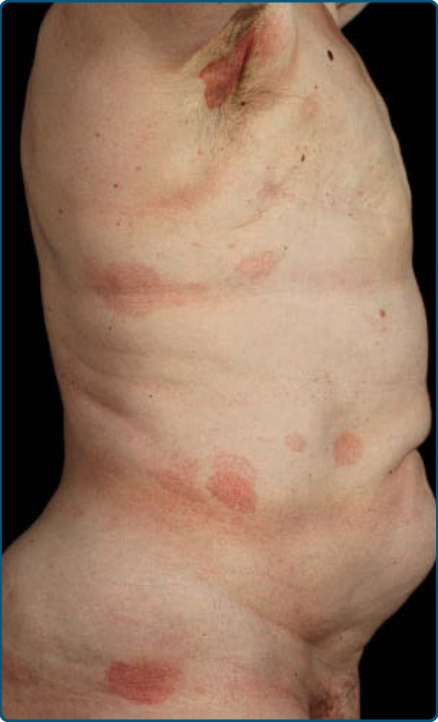
圖 119-4:蕈樣肉芽腫的斑片病灶,位於側軀幹的典型位置。
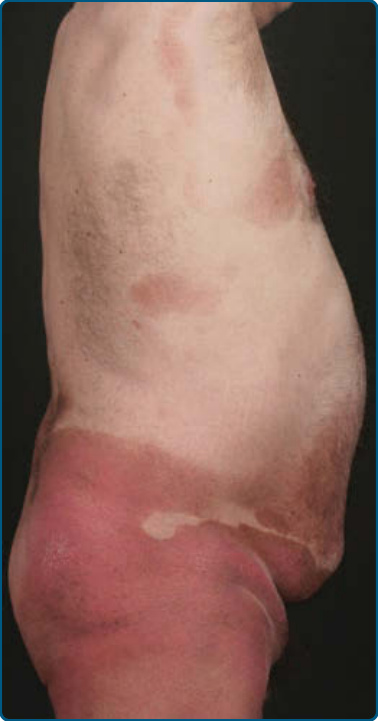
圖 119-5:蕈樣肉芽腫的大型斑片病灶,環繞著未受侵犯的皮膚區域。
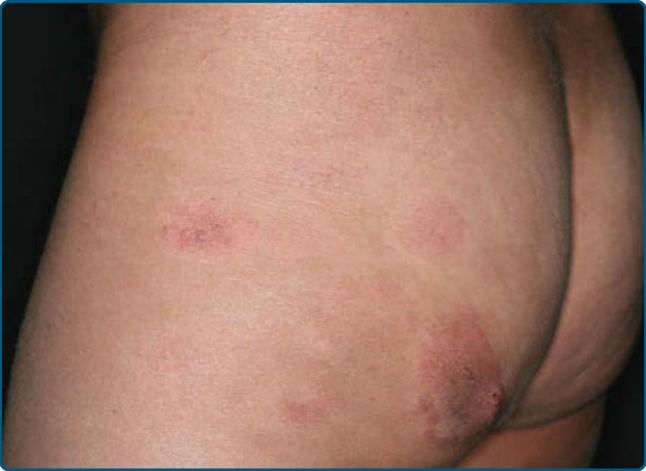
圖 119-6:蕈樣肉芽腫的多形性特徵。左臀有斑片與一個正在發展出結節的斑塊。
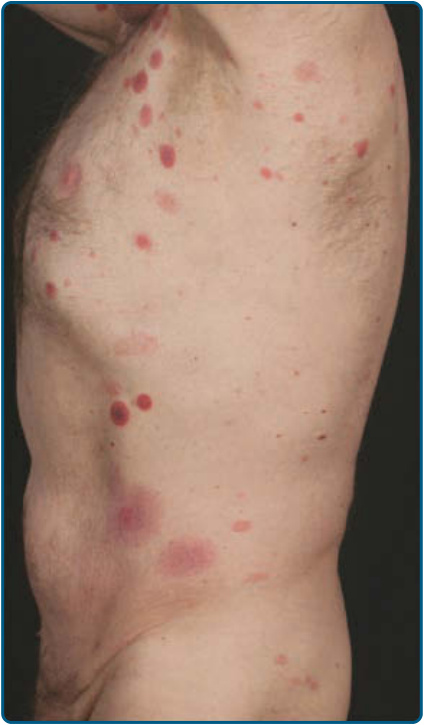
圖 119-7:側軀幹上多個蕈樣肉芽腫的斑片與斑塊。在此病人中,斑塊快速發展並顯示部分中央壞死。
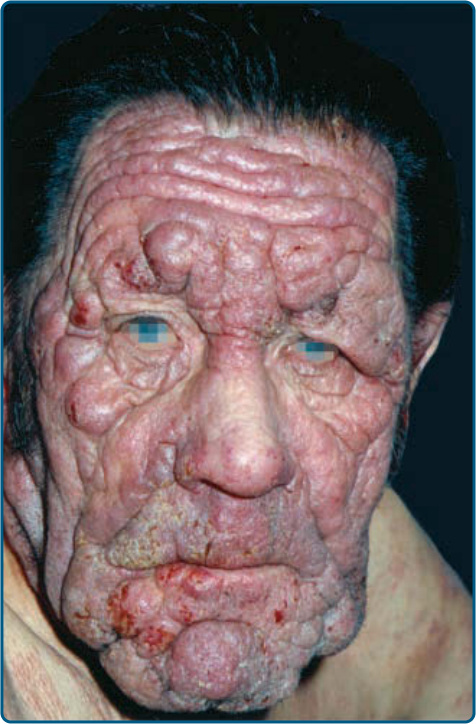
圖 119-8:以皮膚 T 細胞淋巴瘤的浸潤性斑塊與腫瘤為特徵的獅面 (leonine facies)。
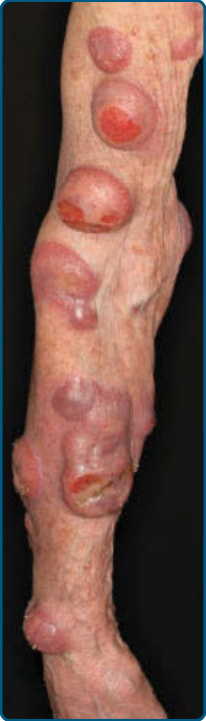
圖 119-9:右臂上的蕈樣肉芽腫腫瘤病灶,部分糜爛與潰瘍化。
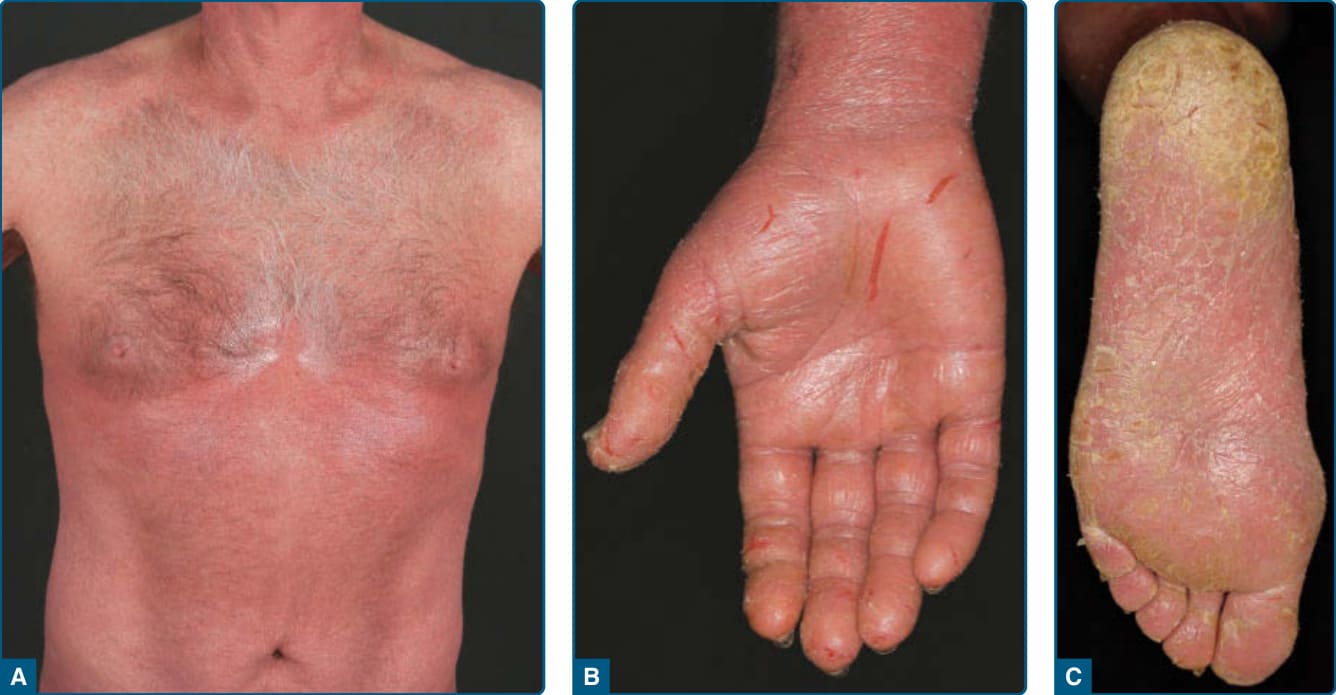
圖 119-10:Sézary 症候群病人,具有紅皮症 (A)、手掌龜裂 (B) 與腳掌角化過度 (C)。
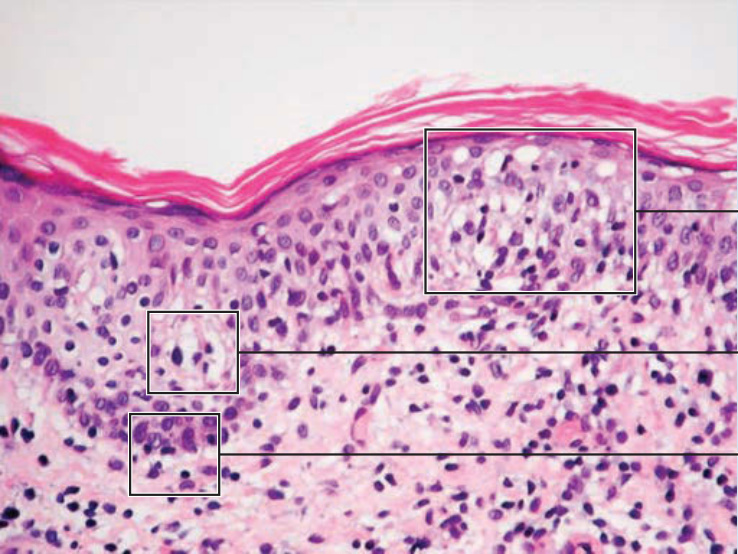
圖 119-11:顯示 MF 的特徵性組織學。在斑片、斑塊以及紅皮症期,上真皮有由反應性 T 細胞與腫瘤性 T 淋巴球組成的帶狀浸潤,後者以高度迴旋的腦回狀 (cerebriform) 細胞核為特徵。腫瘤性 T 細胞顯示親表皮性 (epidermotropism),形成表皮內 Pautrier 微膿瘍 (Pautrier microabscesses)(圖 119-12 與 119-13)。在腫瘤期,真皮中可見結節性浸潤,表皮成分則明顯較不顯著(圖 119-14)。免疫組織學上,惡性細胞表現成熟周邊 T 細胞 (CD4⁺) 表型。全 T 細胞抗原的部分喪失……
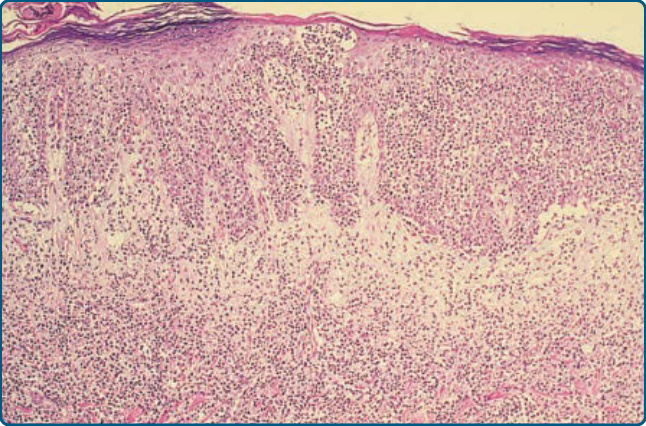
圖 119-12:緻密的單核細胞浸潤由乳頭真皮延伸至表皮。表皮完全被這些細胞滲透,形成一個 Pautrier 膿瘍 (Pautrier abscess)。
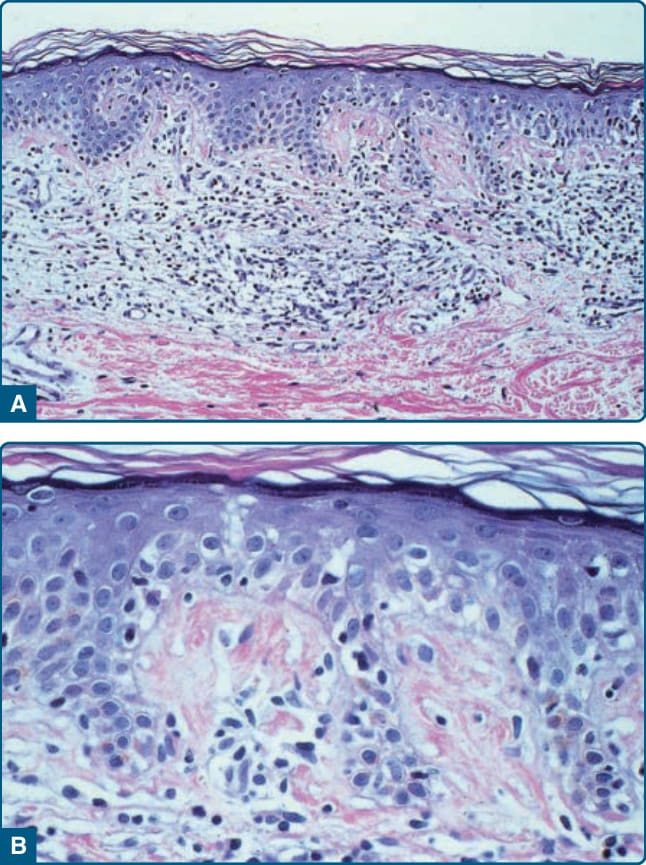
圖 119-13:蕈樣肉芽腫的斑片期。A,表皮中的單個非典型單核細胞,伴有乳頭真皮中稀疏的淺層血管周圍浸潤。(蘇木精與伊紅染色切片 Hematoxylin and eosin-stained section。) B,同一切片表皮中非典型細胞的高倍率視野。
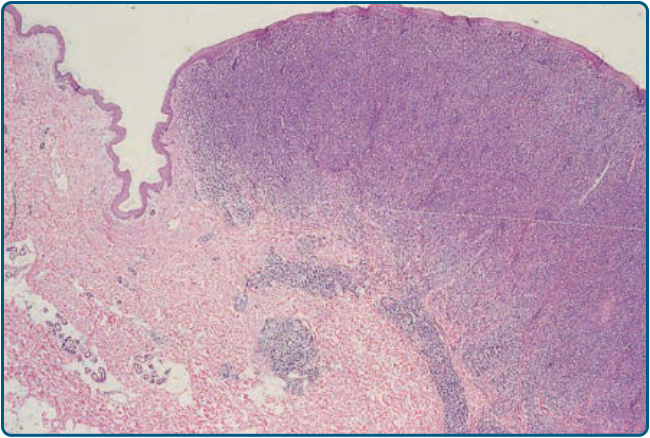
圖 119-14:蕈樣肉芽腫腫瘤的低倍率視野。緻密的浸潤深入真皮。
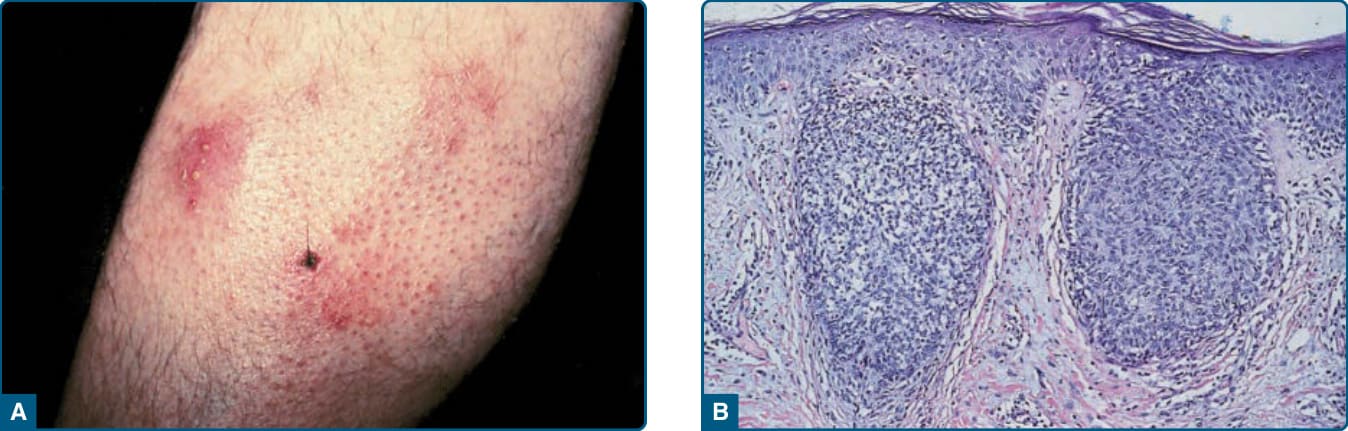
圖 119-15:A,親毛囊性蕈樣肉芽腫 (Folliculotropic mycosis fungoides)。注意毛囊性定位與所致的毛髮脫落。B,毛囊的外根鞘 (outer root sheath) 被 T 細胞與黏液 (mucin) 沉積所破壞,導致小囊狀空間。
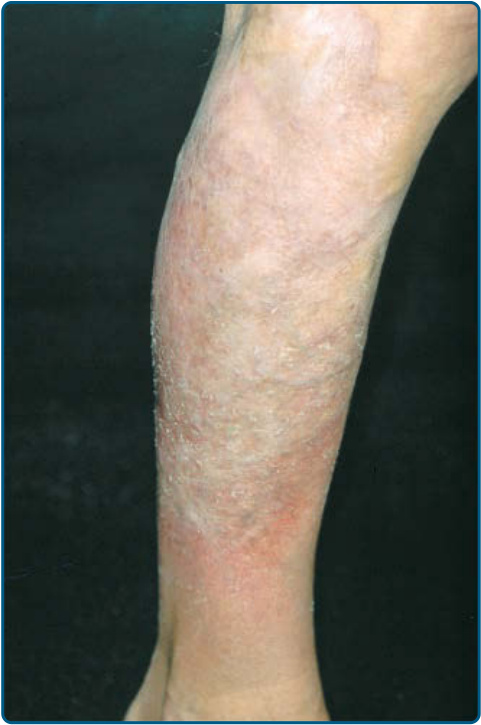
圖 119-16:派傑樣網狀細胞增生症 (Pagetoid reticulosis)。位於一名男性病人腿部的角化過度斑塊。
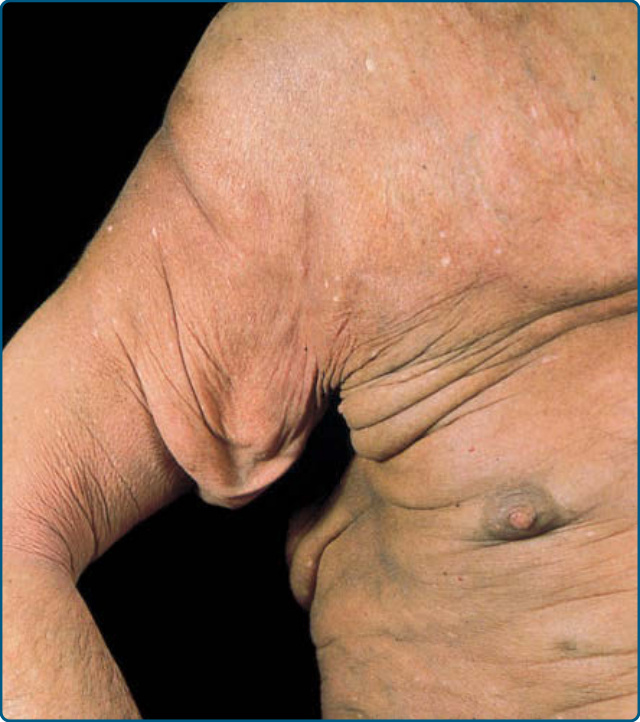
圖 119-17:肉芽腫性鬆弛皮膚 (Granulomatous slack skin)。注意續發性彈性纖維溶解 (elastolysis) 所致的皮膚皺褶。
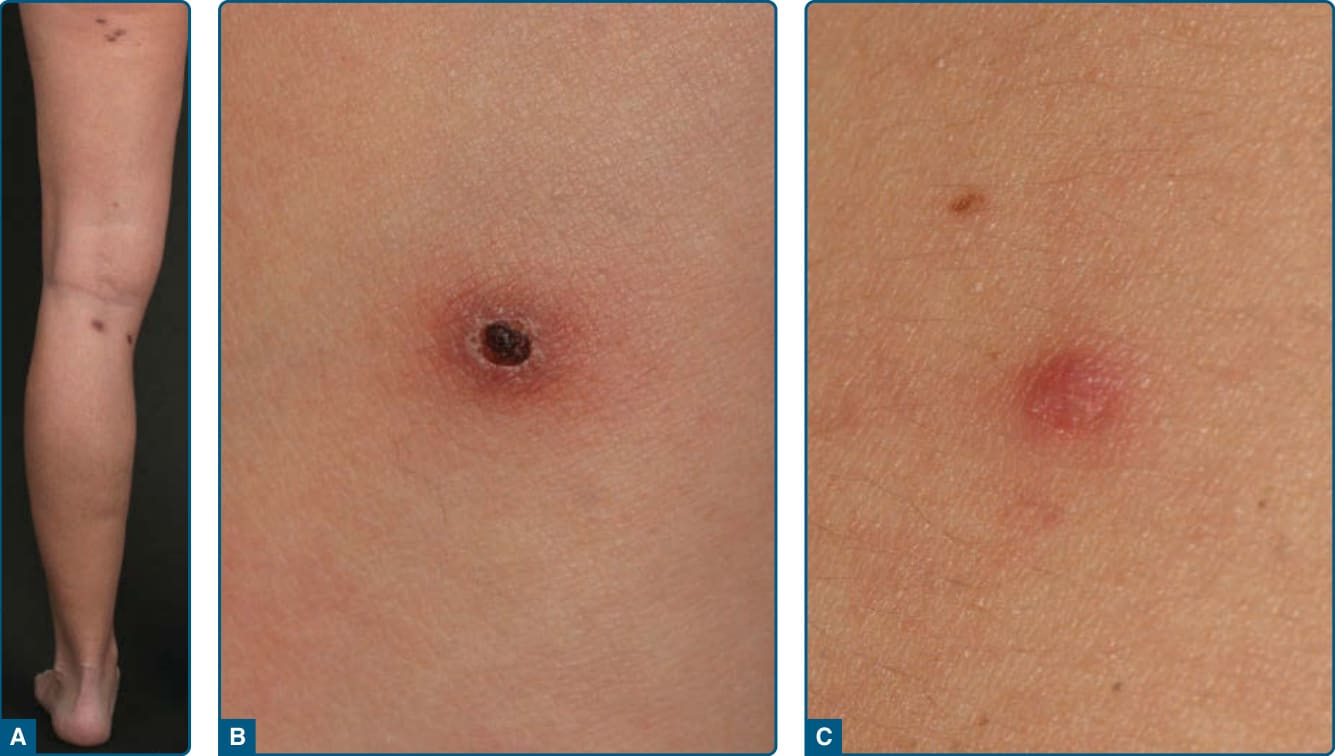
圖 119-18:淋巴瘤樣丘疹病 (Lymphomatoid papulosis)。A,右腿上的丘疹性皮膚病灶。病灶可能呈播散與成群。B,淋巴瘤樣丘疹病的丘疹壞死性病灶。C,淋巴瘤樣丘疹病的紅斑性丘疹結節。
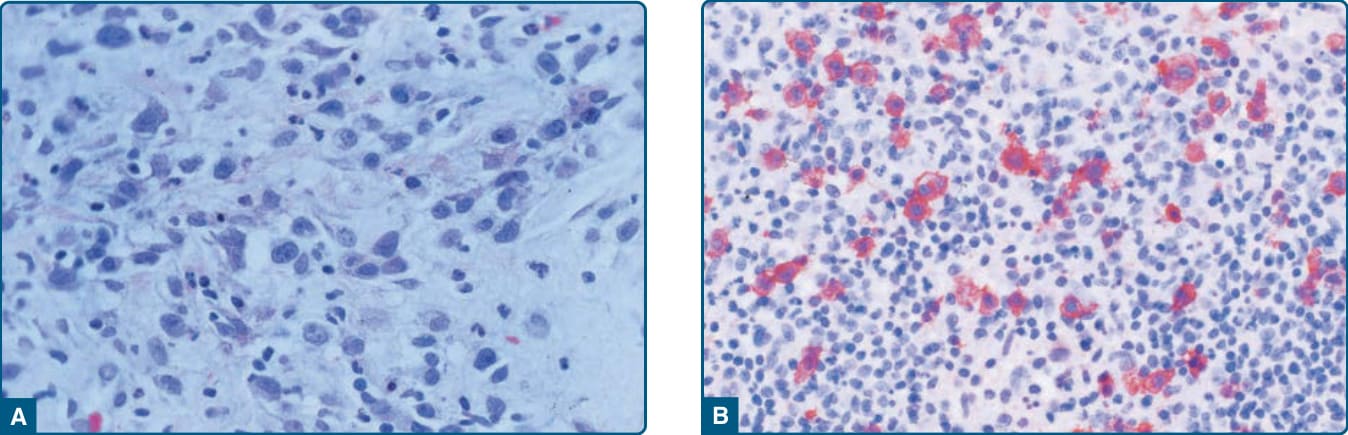
圖 119-19:A,淋巴瘤樣丘疹病。真皮浸潤含有數個大型淋巴樣細胞,其細胞核顯示均勻分散的染色質與程度不一的顯著核仁(所謂 Type A 細胞)。B,Type A,在混雜的發炎細胞中有大型 CD30⁺ T 細胞(紅色)。
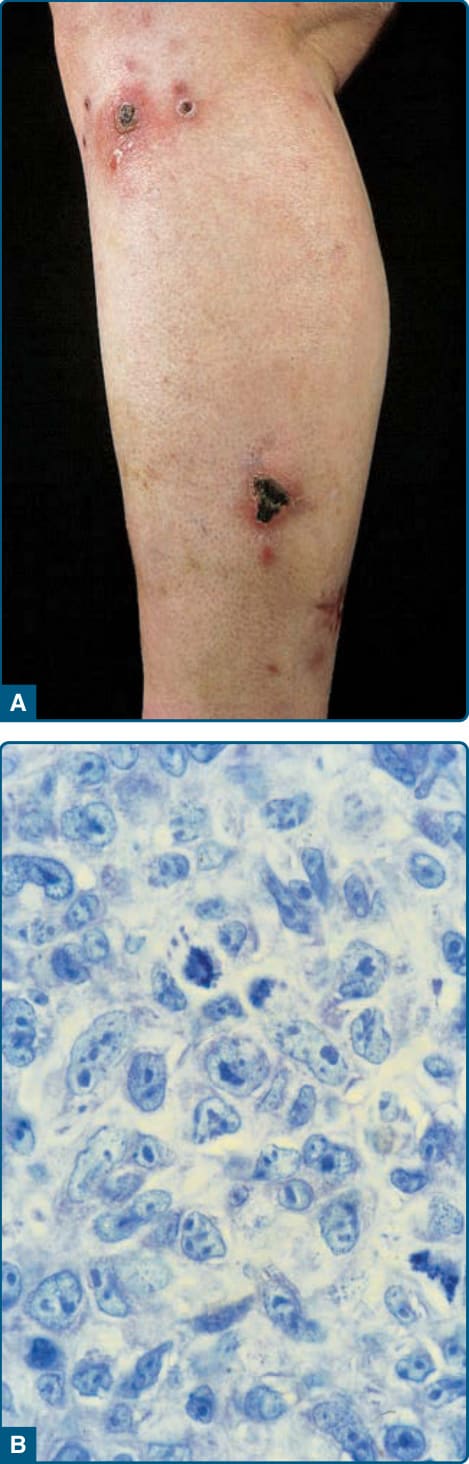
圖 119-20:原發性皮膚退行性大細胞淋巴瘤 (Primary cutaneous anaplastic large-cell lymphoma)。A,侷限性結節,部分有潰瘍。B,多形性大細胞淋巴瘤 T 細胞浸潤。
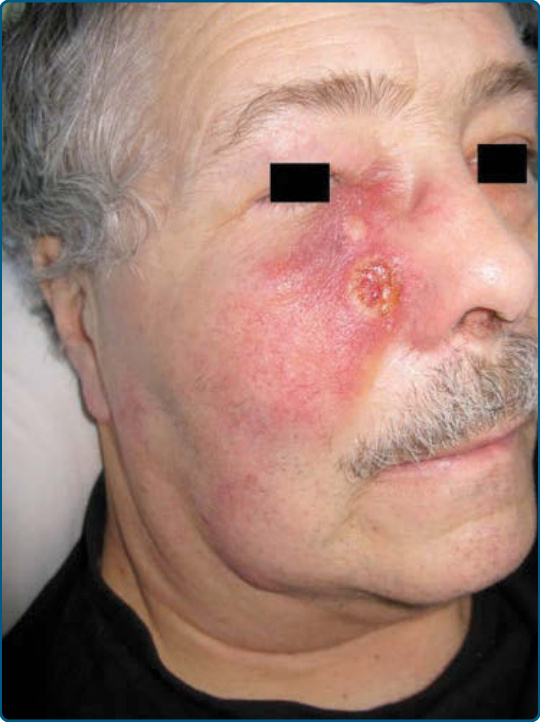
圖 119-21:結外自然殺手/T 細胞淋巴瘤,鼻型 (Extranodal natural killer/T-cell lymphoma, nasal type)。浸潤與潰瘍化的斑塊。
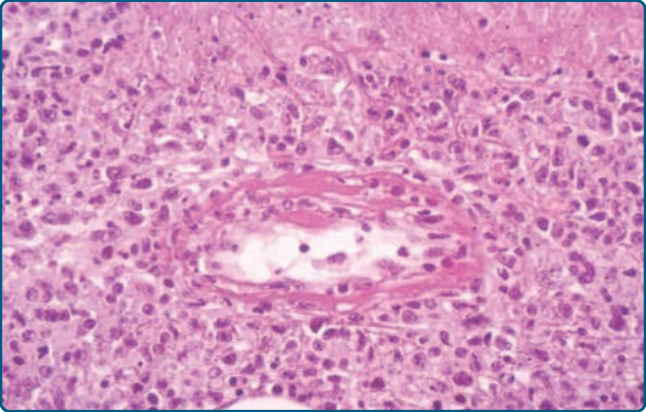
圖 119-22:結外自然殺手/T 細胞淋巴瘤,鼻型。緻密的血管中心性 (angiocentric) 浸潤。
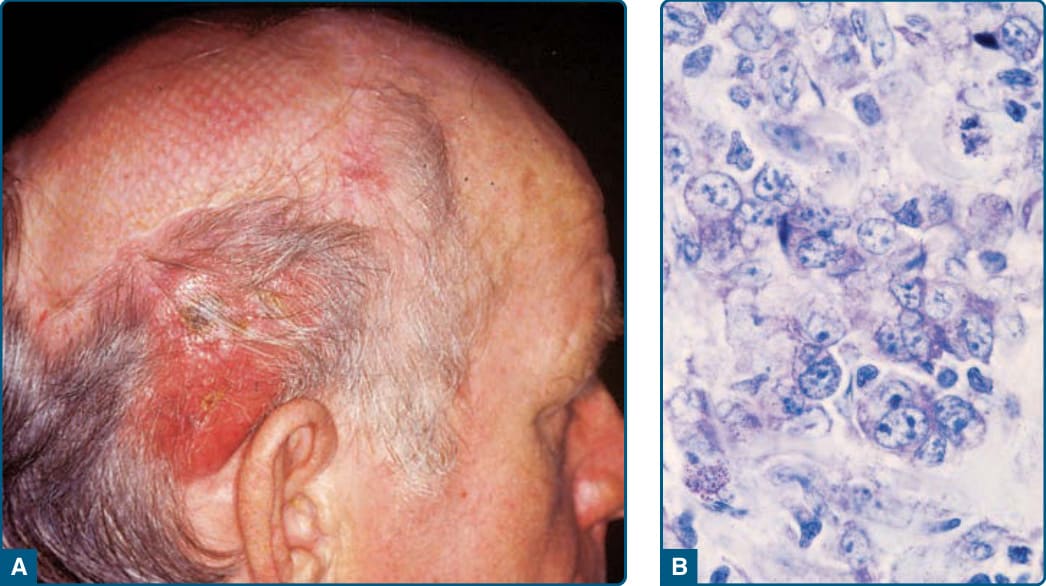
圖 119-23:原發性皮膚濾泡中心淋巴瘤 (Primary cutaneous follicle center lymphoma)。A,頭部區域被紅斑環繞的腫瘤。B,細胞浸潤由中心細胞 (centrocytes)、若干中心母細胞 (centroblasts) 與反應性 T 細胞的混合物組成。
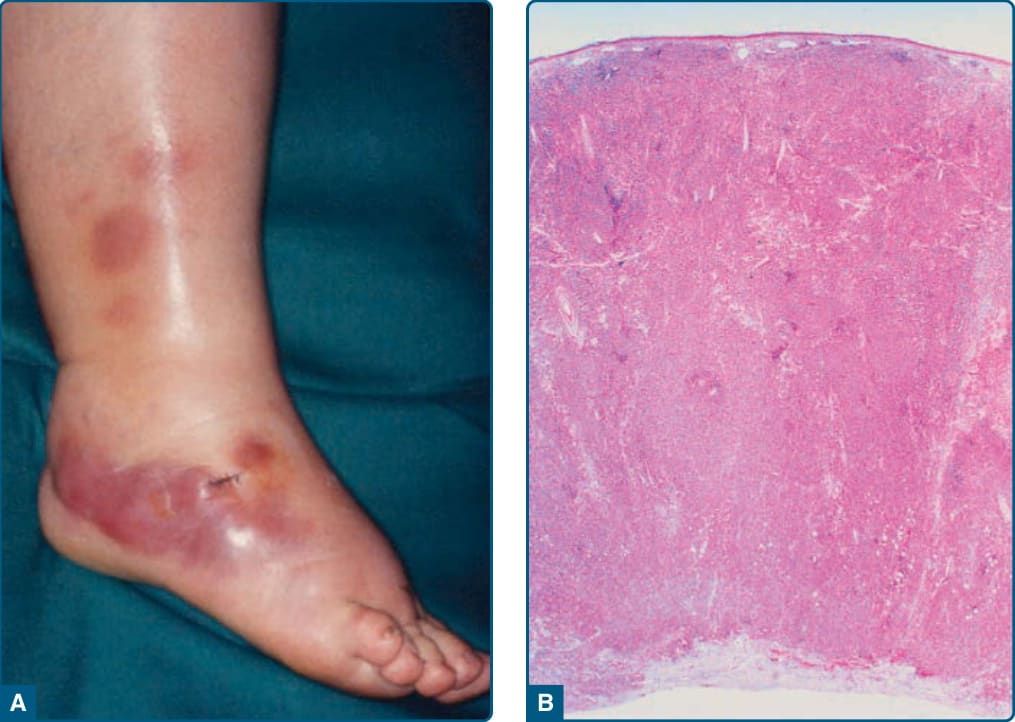
圖 119-24:原發性皮膚瀰漫性大 B 細胞淋巴瘤,腿型 (Primary cutaneous diffuse large B-cell lymphoma, leg type)。A,右腿上的結節與腫瘤。B,組織學顯示大型 B 細胞的瀰漫性浸潤,含有中心母細胞、大型中心細胞與眾多免疫母細胞 (immunoblasts)。
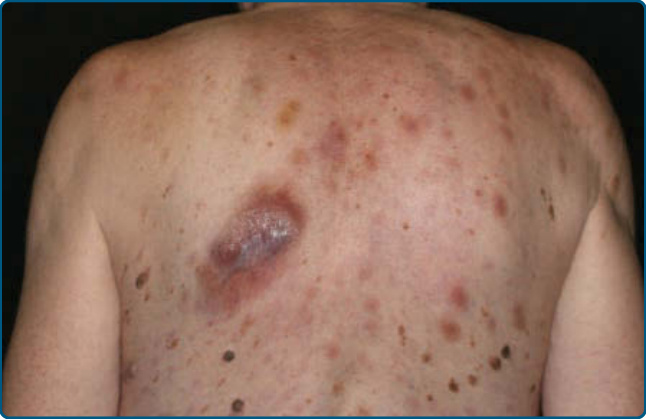
圖 119-25:母細胞性漿細胞樣樹突細胞腫瘤 (Blastic plasmacytoid dendritic cell neoplasm)。背部多發病灶。大型腫瘤為最初的表現部位(中央位置為切片的疤痕)。多個小病灶在短時間內發展出來。
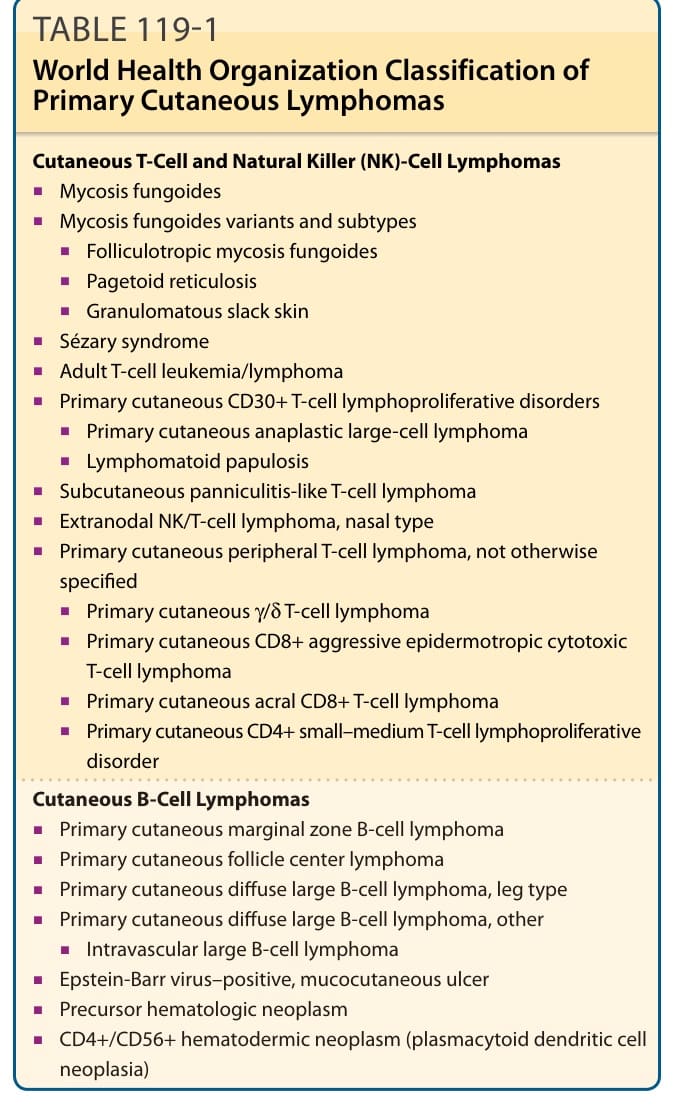
表 119-1:世界衛生組織原發性皮膚淋巴瘤分類 (World Health Organization Classification of Primary Cutaneous Lymphomas)。

表 119-2:蕈樣肉芽腫的亞型與變異型 (Subtypes and Variants of Mycosis Fungoides)。

表 119-3:呈現早期 MF 診斷的演算法。TOX(胸腺細胞選擇相關高遷移率族群盒因子 thymocyte selection-associated high-mobility group box factor)與胸腺中 CD4⁺、CD8⁻ T 細胞的發展相關,但在成熟 CD4⁺ T 細胞中被抑制。它在 MF 與 Sézary 症候群中異常表現。曾被認為有助於區別早期 MF 病灶與發炎性皮膚疾病的切片。根據近期資料,它可能有助於 CTCL 的診斷;然而,TOX 表現並不限於 CD4⁺、CD8⁻ 腫瘤性 T 細胞。⁶¹

表 119-4:摘要 MF/Sézary 症候群病人的建議評估/初步分期。
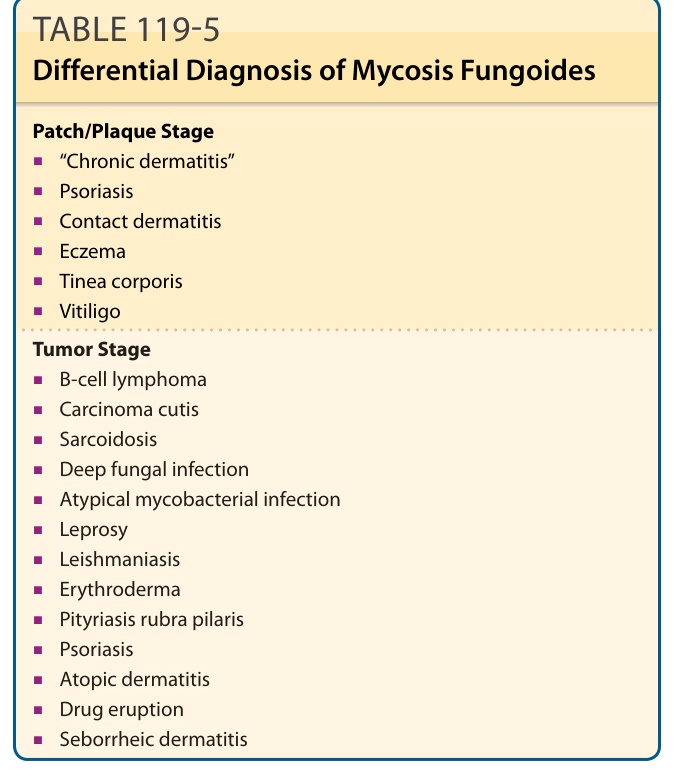
表 119-5:摘要 MF 的鑑別診斷。

表 119-6:概述 MF 及其變異型的臨床發現與預後。
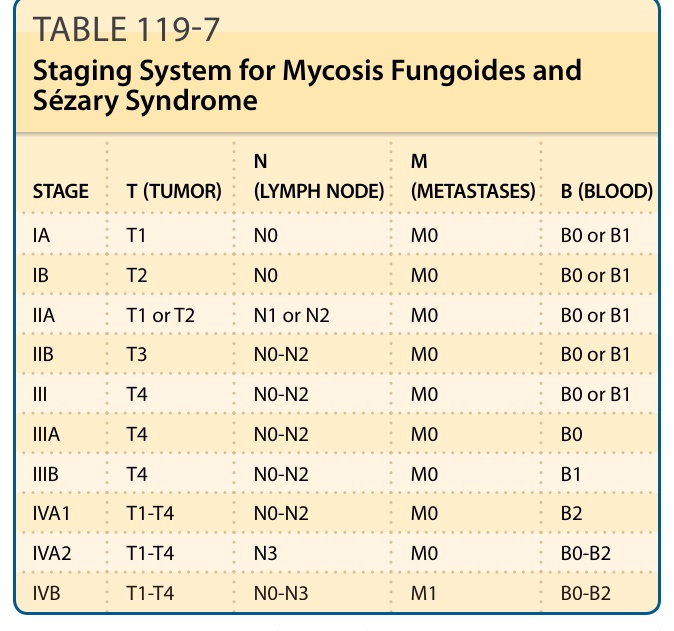
表 119-7:蕈樣肉芽腫與 Sézary 症候群的分期系統 (Staging System for Mycosis Fungoides and Sézary Syndrome)。
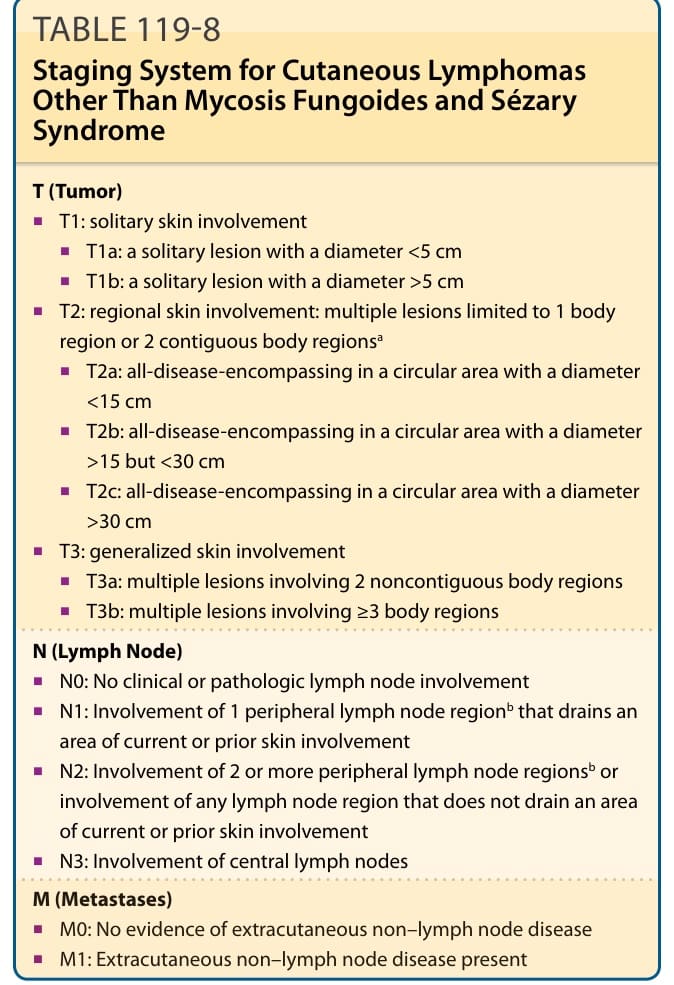
表 119-8:概述 MF 與 Sézary 症候群以外皮膚淋巴瘤的國際分期系統。一旦個別的 TNMB(腫瘤、淋巴結、轉移、血液 tumor, node, metastasis, blood)分類已確定,這些病例便可納入 MF 與 Sézary 症候群的分期系統。為評估對特定治療的反應,由國際皮膚 T 細胞淋巴瘤學會、美國皮膚淋巴瘤聯盟,以及……建立了一份標準共識聲明。
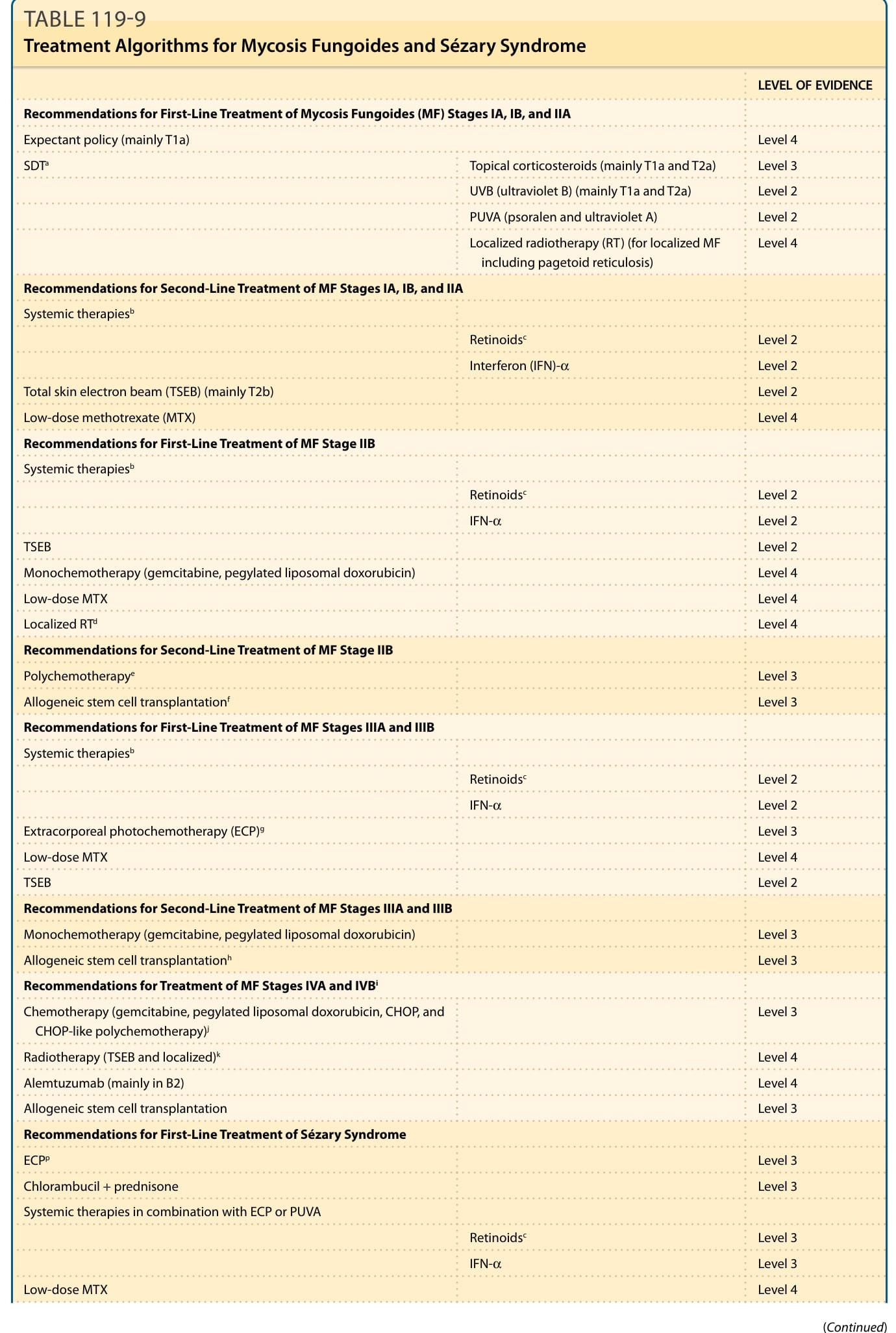
表 119-9:概述 MF 與 Sézary 症候群的治療演算法。

表 119-10:用於治療紅皮症型皮膚 T 細胞淋巴瘤的生物反應調節劑 (Biologic Response Modifiers for the Treatment of Erythrodermic Cutaneous T-cell Lymphomas)。
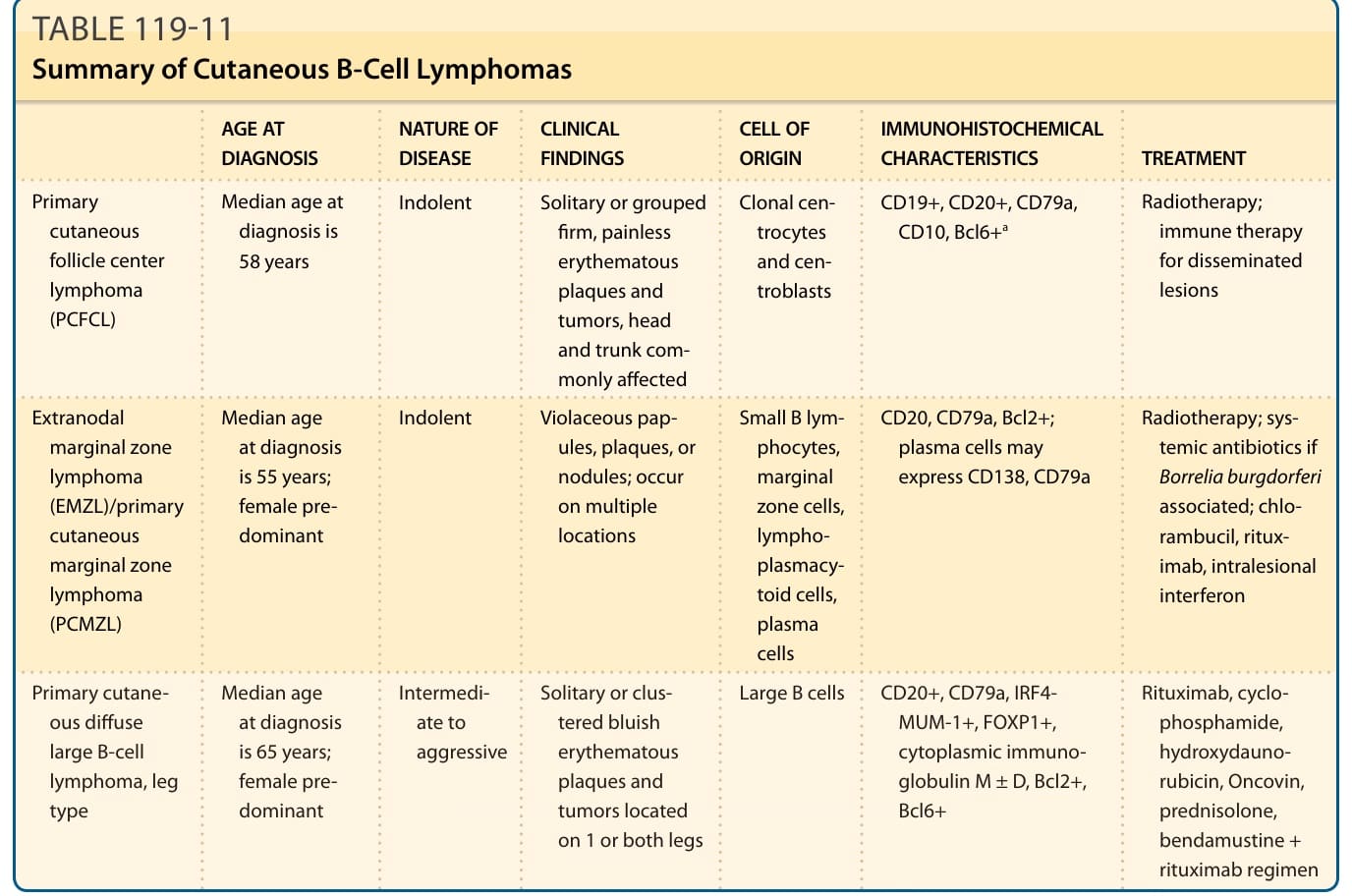
表 119-11:皮膚 B 細胞淋巴瘤摘要 (Summary of Cutaneous B-Cell Lymphomas)。
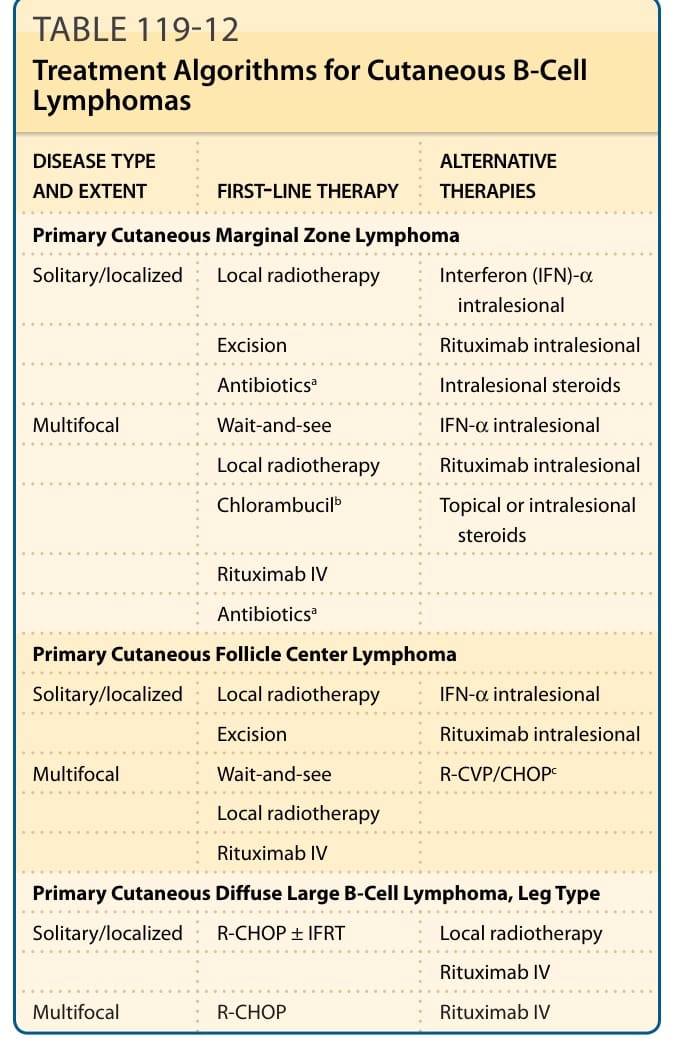
表 119-12:皮膚 B 細胞淋巴瘤的治療演算法 (Treatment Algorithms for Cutaneous B-Cell Lymphomas)。