組織球增生症 (Histiocytosis)
總論與分類
- 組織球增生症分為蘭格罕細胞組織球增生症 (Langerhans cell histiocytosis, LCH) 與非蘭格罕細胞組織球增生症 (non–Langerhans cell histiocytosis, N-LCH),為一組罕見的腫瘤性與反應性疾病,特徵為骨髓系細胞 (myeloid cells) 在多種器官增生,以皮膚為好發處。
- N-LCH 再分為全身性(Rosai-Dorfman 病、噬血球性淋巴組織球增生症)與皮膚性(幼年黃色肉芽腫、良性頭部組織球增生症、成人黃色肉芽腫、丘疹性黃色瘤、廣泛性發疹性組織球瘤、播散性黃色瘤、多中心網狀組織球增生症、Erdheim-Chester 病、壞死性黃色肉芽腫等)。
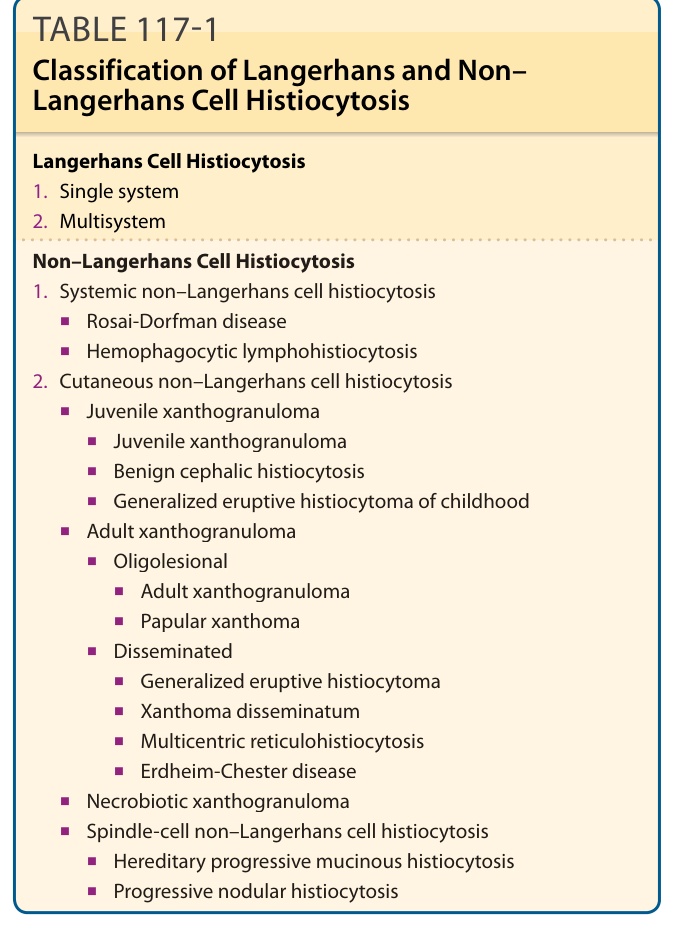
表 117-1:蘭格罕細胞與非蘭格罕細胞組織球增生症的分類
蘭格罕細胞組織球增生症 (LCH)
定義與致病機轉
- 新定義為罕見、異質性的骨髓樹突狀細胞 (myeloid-dendritic cells)(多為克隆性 clonal)發炎性腫瘤 (inflammatory neoplasia)。
- 浸潤細胞為 CD1a/S100B/CD207 陽性、具腎形 (bean-shaped) 細胞核的單核細胞,類似皮膚蘭格罕細胞,但屬不成熟骨髓標記,與骨髓衍生前驅樹突狀細胞關係較近。
- 約 60% 的 LCH 帶有 BRAF(v-Raf murine sarcoma viral oncogene homolog B)的 V600E 突變;33% 的 BRAF 野生型帶有 MAP2K1 突變,導致 MEK 與 ERK 活化。2010 年發現 BRAF-V600E 體細胞突變後確立其為克隆性腫瘤性疾病。
- 「誤導的骨髓樹突狀細胞前驅物模型」:ERK 活化發生的分化階段決定疾病範圍——在造血幹細胞/未分化細胞活化導致多系統疾病,在較分化前驅物活化僅造成多灶或單灶疾病。
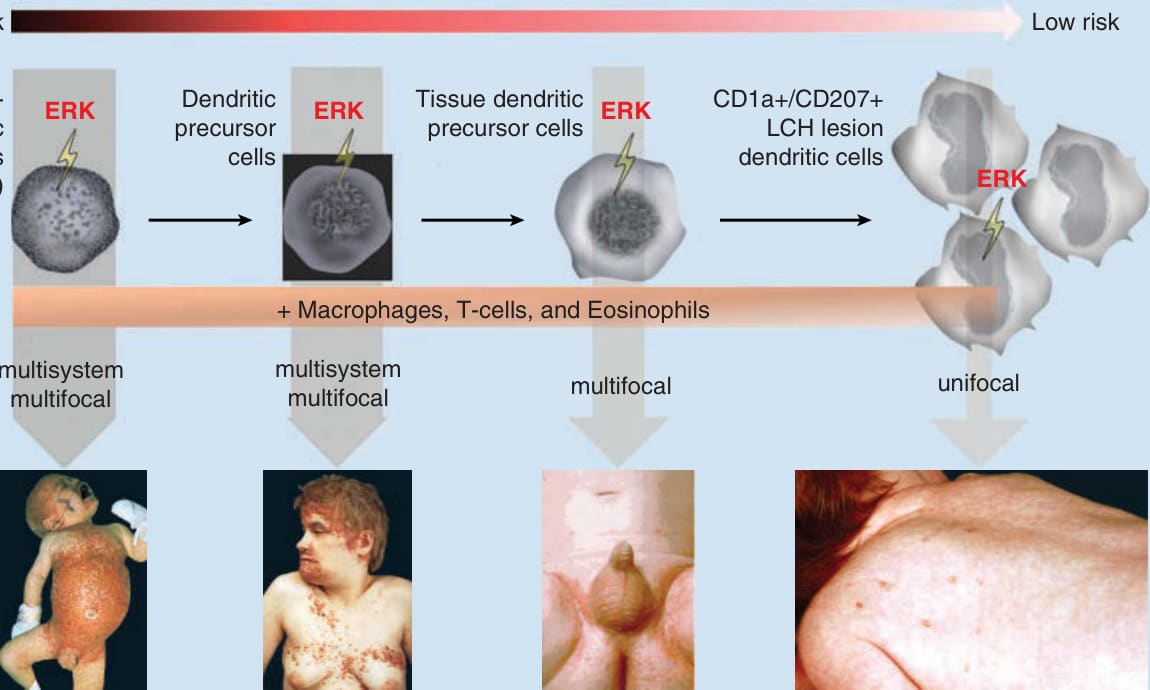
圖 117-1:LCH 中報告的突變 (A) 與「誤導的骨髓樹突狀細胞模型」(B)
流行病學
- 罕見。小於 15 歲兒童年發生率約每百萬人口 0.7–4.1 例,診斷中位年齡 30.2 個月至 5.9 歲;成人每年僅 1–2 例/百萬。
- 黑人發生率較低,西班牙裔 (Hispanic) 略高。
臨床分類(新)
- 傳統四型分類(Hashimoto-Pritzker 病、嗜酸性肉芽腫、Hand-Schüller-Christian 病、Letterer-Siwe 病,合稱組織球增生症 X)已棄用。
- 新分類依器官侵犯範圍:
- 單一系統 LCH (single-system):約 55%–65%,僅 1 個器官系統受侵(骨、皮膚、淋巴結、肺、CNS)。
- 多系統 LCH (multisystem):超過 1 個器官系統受侵;須區分是否有高危險器官(造血系統、肝、脾)侵犯。肺已不再視為高危險器官。
皮膚表現
- 超過三分之一 LCH 有皮膚侵犯,為骨之後第二常見部位,常是最早徵象。小於 1 歲多為僅皮膚 LCH,大於 18 個月者多系統風險較高。
- 最典型:軀幹、間擦區與頭皮上小型半透明玫瑰黃色結痂丘疹或丘疹水疱,伴濕疹樣脫屑,類似念珠菌間擦疹或脂漏性皮膚炎。也可見出血性丘疹結節伴瘀點、水疱膿疱、甲侵犯(甲溝炎、甲剝離、甲下角化過度等)。
- 黏膜病灶多為結節潰瘍性(口周、生殖器周、肛周、牙齦),與外耳道中耳炎一樣與較高多系統風險相關。常被誤診為濕疹、疥瘡、水痘、脂漏性皮膚炎、念珠菌病等。
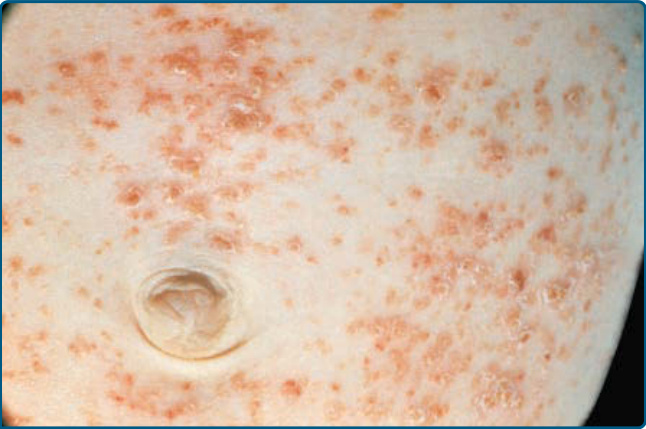
圖 117-3:廣泛性 LCH 嬰兒腹部的玫瑰黃色丘疹
非皮膚表現
- 最常侵犯骨(77%)、淋巴結(19%)、肝(16%)、脾(13%)、肺(10%)、CNS(6%)。
- 骨:好發顱骨、頜骨、股骨、肋骨、椎骨、肱骨,呈「穿鑿狀」溶骨;兒童頸椎受侵可致扁平椎;顏面骨與前/中顱窩病灶屬「CNS 風險」病灶(CNS 疾病風險增 3 倍)。
- 肝脾:膽汁鬱積與硬化性膽管炎為最嚴重併發症之一,多對化療無反應,肝移植為唯一選擇。
- 肺:成人較常見,吸菸為主要危險因子,可致自發性氣胸。
- CNS:「CNS 風險」病灶有 25% CNS 侵犯風險;尿崩症最常見。
- 內分泌:尿崩症為最常見內分泌病變;已診斷 LCH 者發展尿崩症風險約 24%,多持續不可逆。
診斷
- 基於受侵器官的臨床病理與影像特徵;組織病理為最可靠的確定診斷工具。
- 組織學:乳突真皮緻密帶狀浸潤的 LCH 細胞,卵圓形、嗜酸性細胞質、腎形(咖啡豆樣)細胞核,比淋巴細胞大 4 至 5 倍;常見明顯表皮趨向性與表皮內微膿瘍。標記 CD1a、S100B、CD207(Langerin)、fascin 陽性,stabilin-1 與 CD34 陰性。
- 伯貝克顆粒 (Birbeck granules)(網球拍狀、拉鍊樣胞器)曾為診斷黃金標準;現以 CD207 免疫染色為最敏感標記,電子顯微鏡已非必須。
預後
- 異質性疾病;多器官侵犯者最重要預後因子為最初 6 週對全身治療的反應。多中心第 III 期試驗(長春花鹼 vinblastine + 培尼皮質醇 prednisolone 治療 12 個月)顯示完全緩解、中間反應、疾病進展者 5 年存活率分別為 95%、83%、57%。
- 危險器官(肝、脾、骨髓)侵犯與不良結局相關。造血系統侵犯分輕度型(血紅素 10–7 g/dL,血小板 20,000–100,000/mm³)與重度型(血紅素低於 7 g/dL,血小板少於 20,000/mm³)。
- 小於 2 歲或肺侵犯已不再視為不良結局危險因子;尿崩症尚無證據為不良預後因子。LCH 與其他惡性腫瘤(實體腫瘤、淋巴瘤、白血病)相關。
治療
- 取決於疾病範圍、位置與年齡。侷限於骨或皮膚者通常不全身治療(「特殊部位」病灶除外,如齒突、伴脊髓內延伸的椎體病灶)。
- 皮膚侷限(兒童):以觀察與等待 (watch-and-wait) 為最佳策略;局部類固醇療效甚微(對局部類固醇無反應為 LCH 線索)。其他局部選擇:imiquimod、tacrolimus、病灶內類固醇注射、CO2 雷射、切除。成人皮膚病灶可用氮芥藥膏 (nitrogen mustard ointment)。窄頻紫外線 B (narrowband ultraviolet B) 對丘疹與濕疹樣病灶可能有效(有結節時否);光化學治療 (photochemotherapy) 對某些成人有效。
- 局部無效時可試全身性糖皮質素、沙利竇邁 (thalidomide,有神經毒性與疲勞;皮膚為唯一器官時不用於育齡女性)、低劑量甲氨蝶呤 (low-dose methotrexate)。一篇病例報告口服異維 A 酸 (oral isotretinoin) 1.5 mg/day 持續 8 個月達完全緩解。
- 單一骨病灶:觀察等待、單純刮除、小病灶(直徑 <2 cm)完全切除、病灶內類固醇注射。
- 多系統疾病:應全身治療 12 個月(僅 6 個月者早期復發較高)。方案為 6 週誘導化療(長春花鹼 6 mg/m² 每週靜脈推注 + 培尼皮質醇 40 mg/m²/day 口服 4 週後 2 週內減量),後續依反應與危險器官侵犯給予含或不含巰基嘌呤 (mercaptopurine) 的長春花鹼/培尼皮質醇。6 週後無危險器官侵犯者 86%、有侵犯者 66% 有反應。搶救藥物:阿糖胞苷 (cytarabine)、克拉屈濱 (cladribine)、氯法拉濱 (clofarabine);高風險者可考慮同種異體骨髓移植(87 例中 77% 存活 3 年)。新型選擇:BRAF 與 MAPK 抑制劑、抗 PD-1/PD-L1 治療。
非蘭格罕細胞組織球增生症 (N-LCH)
- 一組以蘭格罕細胞以外組織球增生為特徵的疾病;免疫組化 CD68 陽性,S100B 與 CD1a 陰性。Stabilin-1 可區分 N-LCH 與 LCH 及肉芽腫性疾病。
- 全身型為典型活化巨噬細胞 (Mφ1) 累積;皮膚型以替代活化巨噬細胞 (Mφ2) 為特徵。
- 局部皮膚病灶可用切除、雷射、病灶內類固醇注射、放射治療;內臟病灶用全身性糖皮質素、化療,部分用標靶治療(甲磺酸伊馬替尼 imatinib mesylate、BRAF 與 MEK 抑制劑)。
Rosai-Dorfman 病(伴大量淋巴結病變之竇組織球增生症, RDD)
- 特發性,常於感染後發生(可能病毒病因:EB 病毒、HHV-6、parvovirus B19、polyomavirus)。淋巴結病變為主要表現,頸部淋巴結最常見;皮膚與其他器官(乳房、腎、甲狀腺、睪丸、CNS)可受侵。
- 組織學:細胞 S100B+、CD1a−,也對 CD68、CD163、stabilin-1 等陽性;標誌為伸入現象 (emperipolesis)。S100B 可作血清標記監測病情。
- 病程:可自發消退。口服培尼皮質醇對淋巴結侵犯反應佳;化療一般令人失望。酪胺酸激酶抑制劑(甲磺酸伊馬替尼)對某些病人有效。
噬血球性淋巴組織球增生症 (HLH)
- 70% 為小於 1 歲兒童。特徵為持續發燒、伴血球減少的脾腫大、高三酸甘油脂血症、低纖維蛋白原血症;高達 65% 有皮膚表現(紅皮症、廣泛性紫癜性斑與丘疹、麻疹樣疹)。屬細胞激素風暴症候群。
- 分原發性(家族性 HLH,基因 HPLH1、perforin、UNC13D、STX11、STXBP2)與繼發性(感染如 EB 病毒、自體免疫、惡性腫瘤如非何杰金氏淋巴瘤)。
- 預後不佳,整體死亡率 50%;合併惡性腫瘤為不良因子。治療:高劑量皮質類固醇、環孢素、依託泊苷、甲氨蝶呤、長春新鹼、靜脈注射免疫球蛋白,必要時免疫化療與造血幹細胞移植。
幼年黃色肉芽腫 (JXG)
- 良性自癒性,主要侵犯小於 1 歲嬰兒;單發或多發(寡病灶)丘疹結節,好發顏面、頸部、上軀幹。早期病灶紅棕色,成熟病灶紅黃色。眼部病灶見於高達 10% 兒童。常伴第 1 型神經纖維瘤病與幼年慢性骨髓性白血病。
- 組織學:早期為單形性、不含脂質組織球浸潤;成熟期見泡沫細胞、Touton 巨細胞、異物巨細胞。stabilin-1 陽性,S100B 與 CD1a 陰性(可與 LCH 區別)。
- 通常 1 至 5 年內自發消退;眼部病灶需治療(手術切除、CO2 雷射、病灶內類固醇、冷凍、低劑量放射)。
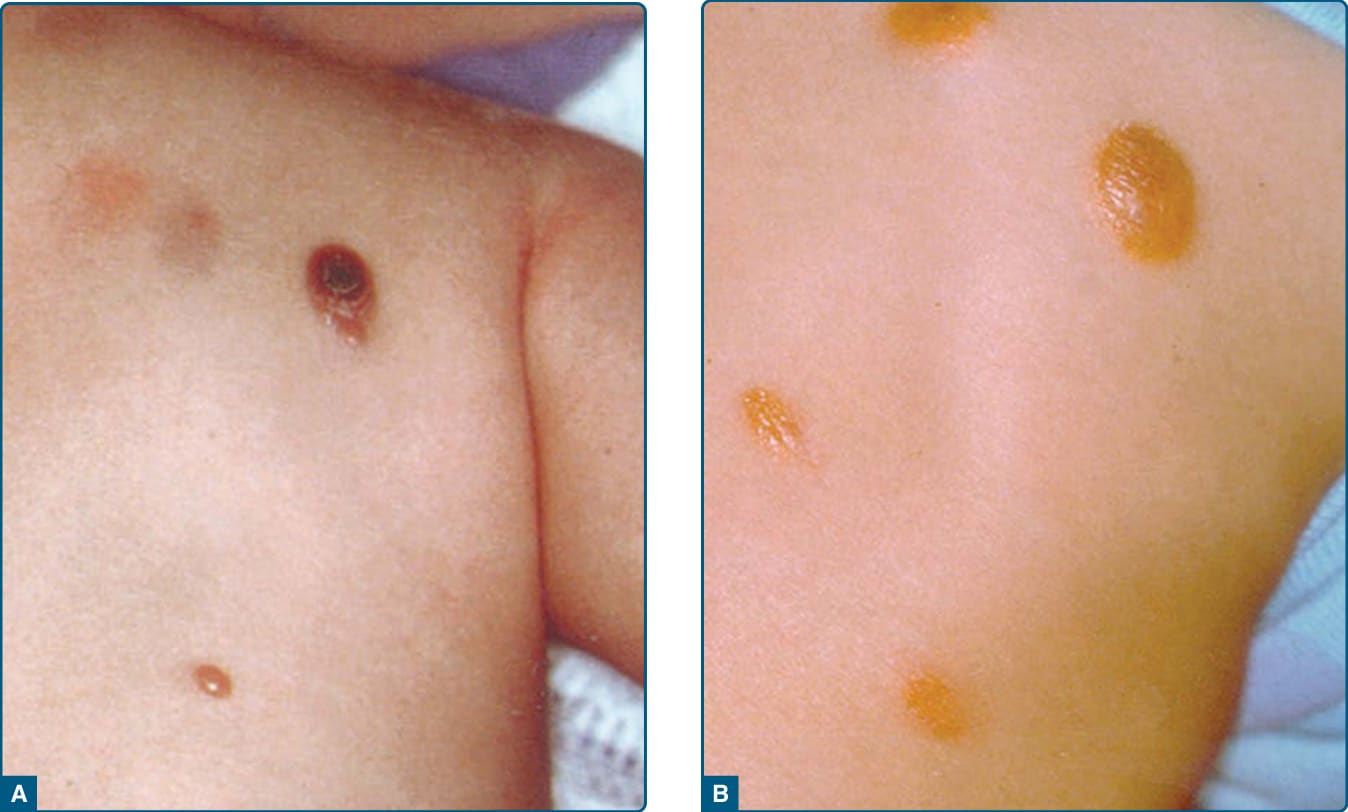
圖 117-14:幼年黃色肉芽腫——A 早期紅色丘疹,B 成熟期紅黃色調
其他皮膚性 N-LCH(要點)
- 良性頭部組織球增生症:幼童頭頸部小型黃紅/黃棕色丘疹,超微結構有蠕蟲樣小體 (worm-like bodies);CD68 陽性、CD1a/CD207 陰性;多自發消退(CO2 雷射)。
- 兒童期廣泛性發疹性組織球瘤:廣泛、對稱紅斑性丘疹,自發消退留色素沉著;可視為 JXG 變異型。
- 成人黃色肉芽腫:顏面、頸部、下臂的黃橙色丘疹,不自發緩解;無全身關聯;組織學近似 JXG(巨細胞較多)。
- 丘疹性黃色瘤:血脂正常 (normolipemic) 病人單發黃色丘疹;組織學見 Touton 型巨細胞與黃色瘤化巨噬細胞;通常自發消退。
- 廣泛性發疹性組織球瘤 (GEH):成人多發、無症狀、對稱棕色紅斑性丘疹,侵犯軸向區,大屈側不受侵;組織學有特徵性 Grenz 帶、缺乏巨細胞;CD68/stabilin-1 陽性。因可能發展急性單核球性白血病,建議密切追蹤。
- 播散性黃色瘤:好發男童,小型黃紅至棕色丘疹結節散布於屈側與間擦區及黏膜;可伴中樞性尿崩症與副蛋白血症(多發性骨髓瘤)。2-氯去氧腺苷 (2-chlorodeoxyadenosine) 似有效。
- 多中心網狀組織球增生症 (MRH):約 50 歲、女性兩倍;手指與手腕關節上方堅實黃棕色丘疹結節,可致獅面與對稱性發炎性多關節炎,常侵黏膜結膜;25% 伴惡性腫瘤(血液、乳房、胃)。CD68/HLA-DR 陽性。治療:手術切除、脈衝染料雷射、口服類固醇、甲氨蝶呤、cyclophosphamide、托珠單抗、抗腫瘤壞死因子。
- Erdheim-Chester 病:中年發病,長骨侵犯幾乎普遍(雙側對稱);六分之一有黃斑瘤與黃色瘤;約 50% 帶 BRAF 突變。一線治療為干擾素-α(提高存活率);BRAF 抑制劑在所有治療病例皆有效。
- 壞死性黃色肉芽腫:較年長成人,可潰瘍的黃色斑塊結節,好發軀幹、四肢、顏面(眼眶周圍);80% 有血清單株丙種球蛋白病(多為 IgG κ 與 λ)。組織學見壞死區域與 Touton 巨細胞。預後不良,治療結果不一。
- 遺傳性進行性黏液性組織球增生症:罕見、可能體染色體顯性;十歲前鼻、手、前臂、大腿出現膚色至紅棕色丘疹;組織學有黏液產生 (mucin production) 特徵;消融性雷射為一選擇。
- 進行性結節性組織球增生症:廣泛散在黃色至紅棕色丘疹結節,顯著顏面侵犯;與慢性骨髓性白血病及下視丘腫瘤相關;CO2 雷射手術消融為選擇。