組織球增生症 (Histiocytosis)
20
組織球增生症的皮膚臨床特徵 (CUTANEOUS CLINICAL FEATURES OF HISTIOCYTOSIS)
重點一覽 (AT-A-GLANCE)
■ 蘭格罕細胞組織球增生症 (Langerhans cell histiocytosis):半透明、玫瑰黃色、結痂的丘疹或丘疹水疱、濕疹樣病灶、出血性丘疹與結節、瘀點 (petechiae)、結節潰瘍性黏膜病灶,以及甲侵犯。
■ Rosai-Dorfman 病 (Rosai-Dorfman disease):淋巴結病變 (lymphadenopathy)、皮膚結節與斑塊。
■ 噬血球性淋巴組織球增生症 (Hemophagocytic lymphohistiocytosis):各種皮膚表現,例如紅皮症 (erythroderma)、廣泛性紫癜性斑與丘疹,以及麻疹樣疹 (morbilliform eruptions)。
■ 幼年黃色肉芽腫 (Juvenile xanthogranuloma):單發以及多發(寡病灶 oligolesional)的丘疹或結節——早期病灶呈紅棕色;成熟病灶則呈紅黃色外觀。
■ 良性頭部組織球增生症 (Benign cephalic histiocytosis):類似幼年黃色肉芽腫;超微結構上可見蠕蟲樣小體 (wormlike bodies)。
■ 兒童期廣泛性發疹性組織球瘤 (Generalized eruptive histiocytoma of childhood):廣泛分布、紅斑性、基本上對稱的丘疹。
■ 成人黃色肉芽腫 (Adult xanthogranuloma):寡病灶、黃橙色丘疹,通常出現於顏面、頸部與下臂。
■ 丘疹性黃色瘤 (Papular xanthoma):單發的黃色丘疹。
■ 廣泛性發疹性組織球瘤 (Generalized eruptive histiocytoma):多發、無症狀且對稱分布。
蘭格罕細胞組織球增生症 (Langerhans cell histiocytoses, LCHs) 與非蘭格罕細胞組織球增生症 (non–Langerhans cell histiocytoses, N-LCHs) 構成一組罕見的腫瘤性與反應性疾病,其特徵為骨髓系細胞 (myeloid cells) 增生於
棕色紅斑性丘疹,特別侵犯軸向區域,例如軀幹、顏面與近端肢體,且經常發作。
■ 播散性黃色瘤 (Xanthoma disseminatum):小型、黃紅色至棕色的丘疹與結節,呈散在性並播散分布,好發於屈側 (flexural) 與間擦 (intertriginous) 區域,以及黏膜。
■ 多中心網狀組織球增生症 (Multicentric reticulohistiocytosis):堅實、黃棕色的丘疹或結節,可達數公分大小並緩慢增大;位於手指與手腕關節上方的病灶是典型表現;常侵犯黏膜與結膜。
■ Erdheim-Chester 病 (Erdheim-Chester disease):六分之一的病例出現黃斑瘤 (xanthelasma) 與黃色瘤 (xanthoma);長骨及其他皮膚外侵犯。
■ 壞死性黃色肉芽腫 (Necrobiotic xanthogranuloma):黃色斑塊與結節,可能潰瘍。
■ 遺傳性進行性黏液性組織球增生症 (Hereditary progressive mucinous histiocytosis):膚色至紅棕色丘疹,通常在十歲前發生於鼻部、手部、前臂與大腿,之後可能發展為持續且進行性的紅斑性丘疹。
■ 進行性結節性組織球增生症 (Progressive nodular histiocytosis):廣泛性、散在性的黃色至紅棕色丘疹與結節,大小數公分,顏面侵犯明顯。
各種器官部位,並以皮膚為好發處。如表 117-1 所示,它們被歸類為不同的亞群。
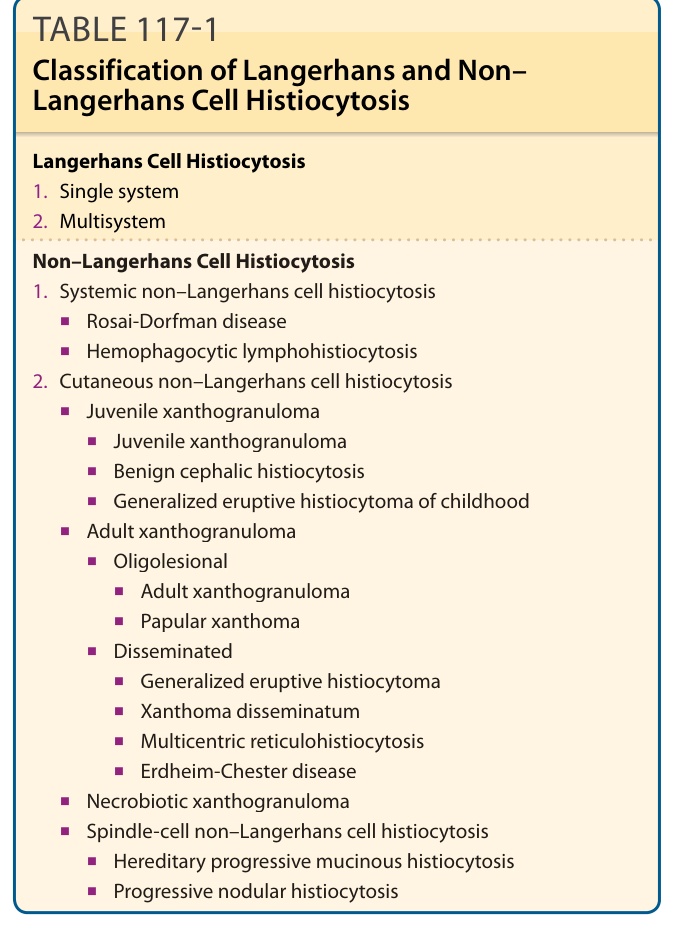
表 117-1:蘭格罕細胞與非蘭格罕細胞組織球增生症的分類 (Classification of Langerhans and Non–Langerhans Cell Histiocytosis)
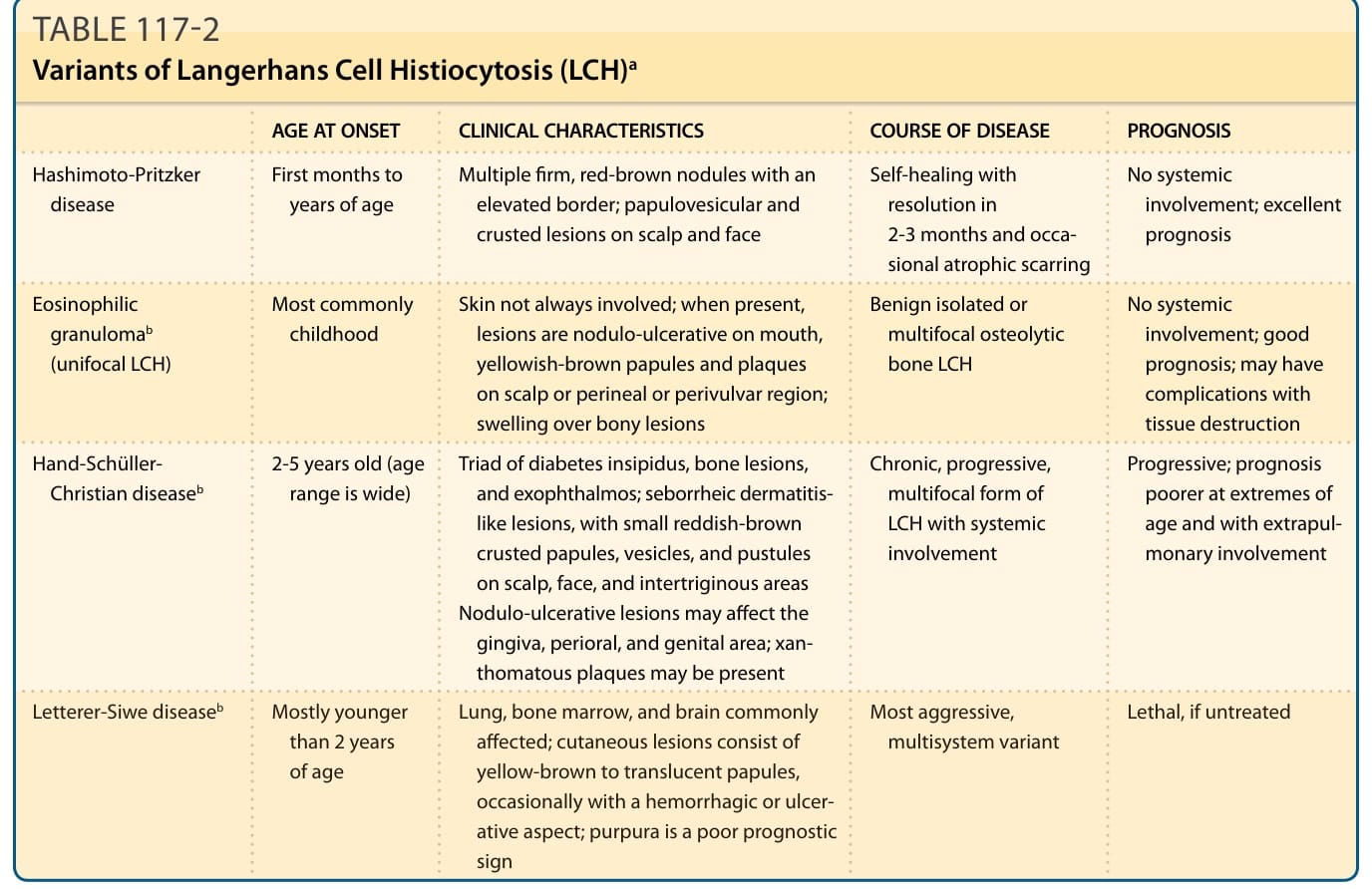
表 117-2:蘭格罕細胞組織球增生症 (LCH) 的變異型 (Variants of Langerhans Cell Histiocytosis)
蘭格罕細胞組織球增生症 (Langerhans Cell Histiocytosis)
- 單一系統 (Single system)
- 多系統 (Multisystem)
非蘭格罕細胞組織球增生症 (Non–Langerhans Cell Histiocytosis)
- 全身性非蘭格罕細胞組織球增生症 (Systemic non–Langerhans cell histiocytosis)
■ Rosai-Dorfman 病 (Rosai-Dorfman disease)
■ 噬血球性淋巴組織球增生症 (Hemophagocytic lymphohistiocytosis)
2. 皮膚性非蘭格罕細胞組織球增生症 (Cutaneous non–Langerhans cell histiocytosis)
■ 幼年黃色肉芽腫 (Juvenile xanthogranuloma)
■ 良性頭部組織球增生症 (Benign cephalic histiocytosis)
■ 兒童期廣泛性發疹性組織球瘤 (Generalized eruptive histiocytoma of childhood)
■ 成人黃色肉芽腫 (Adult xanthogranuloma)
■ 寡病灶型 (Oligolesional)
■ 播散型 (Disseminated)
■ 丘疹性黃色瘤 (Papular xanthoma)
■ 廣泛性發疹性組織球瘤 (Generalized eruptive histiocytoma)
■ 播散性黃色瘤 (Xanthoma disseminatum)
■ 多中心網狀組織球增生症 (Multicentric reticulohistiocytosis)
■ Erdheim-Chester 病 (Erdheim-Chester disease)
■ 壞死性黃色肉芽腫 (Necrobiotic xanthogranuloma)
■ 紡錘細胞非蘭格罕細胞組織球增生症 (Spindle-cell non–Langerhans cell histiocytosis)
■ 遺傳性進行性黏液性組織球增生症 (Hereditary progressive mucinous histiocytosis)
■ 進行性結節性組織球增生症 (Progressive nodular histiocytosis)
蘭格罕細胞組織球增生症 (LANGERHANS CELL HISTIOCYTOSIS)
重點一覽 (AT-A-GLANCE)
■ 蘭格罕細胞組織球增生症 (Langerhans cell histiocytosis, LCH) 的新定義:一種罕見的、骨髓樹突狀細胞 (myeloid-dendritic cells)(多為克隆性 clonal)的發炎性腫瘤 (inflammatory neoplasia),具有異質性的臨床表現。
■ 組織病理學 (Histopathology):
■ 器官受到不成熟、形態上呈圓形、具有腎形 (bean-shaped) 細胞核的骨髓樹突狀細胞浸潤;與成熟皮膚蘭格罕細胞共有的細胞表面抗原特徵(CD1a/S100B/CD207 陽性)。
■ 60% 的 LCH 切片標本帶有控制細胞生長之致癌基因 BRAF(v-Raf murine sarcoma viral oncogene homolog B)的 V600E 突變。
■ 高度多變的臨床表現,範圍從單一、溶骨性 (osteolytic) 骨病灶,到侵犯如脾臟、肝臟與造血系統等危險器官 (risk organs) 的侵襲性、危及生命之多系統疾病。
■ 39% 有皮膚侵犯(對早期診斷至關重要)。
20
■ 根據器官侵犯範圍的新(臨床)分類:
■ 單一器官系統 LCH (Single-organ system LCH)
■ 多系統 LCH (Multisystem LCH)
■ 治療 (Treatment):
■ 單一器官系統 LCH 採密切監測(「觀察與等待 watch and wait」)
■ 局部或全身性治療包括局部氮芥 (topical nitrogen mustard)、窄頻紫外線 B (narrowband ultraviolet light-B)、光化學治療 (photochemotherapy)、甲氨蝶呤 (methotrexate)、6-巰基嘌呤 (6-mercaptopurine) 與化學治療,視疾病範圍而定。
歷史觀點 (HISTORICAL PERSPECTIVE)
LCH 新近被定義為一種罕見、異質性的樹突狀細胞腫瘤 (neoplasm of dendritic cells)。特徵上,具有腎形細胞核的 CD1a/S100B/CD207 陽性單核細胞 (mononuclear cells) 浸潤單一器官系統(最常見為骨骼,但也包括皮膚)或多器官系統。關於本病腫瘤性本質長期存在爭議,因為有些論點支持其為免疫調節失衡所致的反應性病況,例如 LCH 中所見的調節性 T 細胞 (regulatory T-cell) 擴增,以及缺乏腫瘤抑制基因 (tumor-suppressor genes) 的突變。¹ 另一個論點是 LCH 廣泛的臨床表現範圍,從自癒性的單一骨病灶到具有致命結局的侵襲性多器官侵犯。因此,過去病人被分為四種主要臨床類型:Hashimoto-Pritzker 病、嗜酸性肉芽腫 (eosinophilic granuloma)、Hand-Schüller-Christian 病與 Letterer-Siwe 病(表 117-2 概述了 LCH 的傳統分類)。Hashimoto-Pritzker 病於 1973 年首次被描述,是一種良性的臨床變異型,典型表現為生命最初數月與數年內,多發的堅實紅棕色結節,邊緣隆起,或為丘疹水疱性與丘疹結痂性病灶,多位於頭皮與顏面,無全身侵犯徵象。這些病灶通常在 2 至 3 個月內癒合,偶爾留下白色萎縮性瘢痕 (whitish atrophic scars)。嗜酸性肉芽腫也是一種主要為良性的孤立性或多發性溶骨性骨 LCH,有時侵犯皮膚與黏膜。因此,其皮膚病灶常類似於 Hand-Schüller-Christian (HSC) 病所見者,在口腔有結節潰瘍性病灶,在頭皮或會陰或外陰周圍區域有黃棕色丘疹與斑塊。Thomas Smith 描述本病的首篇出版物見於 1865 年版的《American Journal of Pathology》。HSC 病是多系統 LCH 的慢性變異型,多見於 2 至 5 歲兒童,但年齡範圍很廣。它最早由 Alfred Hand 於 1893 年描述,隨後 Arthur Schüller 於 1915 年
2019
20
表 117-2 LCH 傳統分類(依發病年齡、臨床特徵、病程、預後)
發病年齡 (AGE AT ONSET) 臨床特徵 (CLINICAL CHARACTERISTICS) 病程 (COURSE OF DISEASE) 預後 (PROGNOSIS)
Hashimoto-Pritzker 病 生命最初數月至數年 多發、堅實、邊緣隆起的紅棕色結節;頭皮與顏面的丘疹水疱性與結痂性病灶
自癒,2-3 個月內消退,偶有萎縮性瘢痕
無全身侵犯;預後極佳
最常見於兒童期 皮膚不一定受侵;若有,病灶為口腔結節潰瘍性、頭皮或會陰或外陰周圍區域的黃棕色丘疹與斑塊;骨病灶上方腫脹
嗜酸性肉芽腫ᵇ(單灶性 LCH, unifocal LCH)
良性孤立性或多發性溶骨性骨 LCH
無全身侵犯;預後良好;可能有組織破壞之併發症
Hand-Schüller-Christian 病ᵇ 2-5 歲(年齡範圍廣) 尿崩症 (diabetes insipidus)、骨病灶與眼球突出 (exophthalmos) 三聯徵;脂漏性皮膚炎樣 (seborrheic dermatitis-like) 病灶,伴頭皮、顏面與間擦區域的小型紅棕色結痂丘疹、水疱與膿疱。結節潰瘍性病灶可侵犯牙齦、口周與生殖器區域;可能出現黃色瘤性斑塊 (xanthomatous plaques)
Letterer-Siwe 病ᵇ 多為小於 2 歲
慢性、進行性、多灶性的 LCH 形式,伴全身侵犯
進行性;在年齡極端與肺外侵犯時預後較差
常侵犯肺、骨髓與腦
最具侵襲性,
若不治療則致命
常侵犯肺、骨髓與腦;皮膚病灶為黃棕色至半透明丘疹,偶有出血性或潰瘍性外觀;紫癜 (purpura) 為不良預後徵象
最具侵襲性的多系統變異型
ᵃ此表為供讀者參考的 LCH 傳統分類;較新的分類(見圖 117-1)對治療與預後有更大的意義。
ᵇ這些變異型歸屬於單一疾病學實體,稱為組織球增生症 X (histiocytosis X)。
接續,Henry Christian 於 1920 年描述。它的特徵為尿崩症、骨病灶與眼球突出的三聯徵,並在約 30% 的病例中侵犯皮膚。病人常主訴慢性中耳炎 (chronic otitis media),此係乳突 (mastoid) 或顳骨岩部 (petrous temporal bones) 受侵的結果。皮膚病灶可類似脂漏性皮膚炎,伴小型紅棕色結痂丘疹,但也有頭皮、顏面與間擦區域的水疱與膿疱。此外,結節潰瘍性病灶可發生於黏膜,尤其是牙齦及口周與生殖器區域。也曾描述黃色瘤性的黃棕色或黃紅色丘疹與斑塊。Letterer-Siwe 病是最具侵襲性的急性 LCH 多系統變異型,具有明顯的全身症狀,例如發燒、肝脾腫大 (hepatosplenomegaly)、多發淋巴結病變 (polylymphadenopathy)、貧血、關節痛、倦怠與體重減輕。Letterer 於 1924 年首次描述本病,隨後 Siwe 於 1933 年描述。常受侵的器官為肺、骨髓與腦。皮膚病灶常見,為黃棕色、有時半透明的丘疹,可有出血性外觀並潰瘍,類似 HSC 病所見者。此外,紫癜常見,並為不良預後徵象。Letterer-Siwe 病若不治療則致命。雖然這些 LCH 類型呈現不同的臨床病程,但受侵器官中的細胞浸潤是相同的。因此,1953 年 Lichtenstein 提議將
2020
嗜酸性肉芽腫、HSC 與 Letterer-Siwe 病合併為單一疾病學實體,稱為組織球增生症 X (histiocytosis X)。1973 年,Nezelof 根據電子顯微鏡協助下發現的伯貝克顆粒 (Birbeck granules),將病灶細胞稱為「類蘭格罕 (Langerhans-like)」細胞。1987 年,組織球協會 (Histiocyte Society) 發表了組織球性疾病的分類,最終鞏固了上述所有疾病皆屬於單一實體這一日益強化的立場。自此以後,關於 LCH 應歸類為腫瘤性或反應性疾病,一直存在熱烈的爭論。這在 2010 年隨著在約 60% 的 LCH 病人中發現 BRAF-V600E 體細胞點突變 (somatic point mutation) 而最終獲得解決²,
這導致了 LCH 是一種克隆性腫瘤性疾病 (clonal neoplastic disorder) 的結論。此爭議的解決意義重大,因為它將顯著加速本領域的研究與治療進展。
流行病學 (EPIDEMIOLOGY)
由於 LCH 是一種罕見的腫瘤性疾病,缺乏確切的流行病學資料。在小於 15 歲的兒童中,年發生率約為每 100 萬人口 0.7 至 4.1 例,診斷時的中位年齡為 30.2 個月至 5.9 歲。³
在成人中,發生率較低,據報告每年所有族裔的成人僅 1 至 2 例/100 萬成人。
在黑人病人中觀察到較低的發生率,而西班牙裔 (Hispanic) 族群則顯示略高的發生率。其他危險因子包括居住於擁擠環境、較低的教育程度,以及父母暴露於金屬、花崗岩、木屑或溶劑、甲狀腺疾病與癌症的家族史,以及圍產期感染 (perinatal infections),但這些危險因子應謹慎詮釋。³,⁴
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
從病因學觀點來看,LCH 是一種發炎性骨髓腫瘤 (inflammatory myeloid neoplasm),特徵為不同器官系統受到 CD1a/S100B/CD207 陽性單核細胞浸潤,這些細胞在形態學與免疫組織化學上類似皮膚中稱為蘭格罕細胞的特化樹突狀細胞 (specialized dendritic cells)。基因表現陣列 (gene expression arrays) 顯示,這些細胞與皮膚的常駐成熟蘭格罕細胞不同,因為它們表現不成熟的骨髓標記,且與骨髓衍生的前驅樹突狀細胞 (myeloid-derived precursor dendritic cell) 關係更為密切。⁵ 此外,約 60% 的 LCH 細胞帶有 BRAF(v-Raf murine sarcoma viral oncogene homolog B)致癌基因的 V600E 突變,而 33% 的 BRAF 野生型 (wild-type) 病灶帶有 MAP2K1(mitogen-activated protein kinase kinase 1)基因突變,導致普遍的 MEK(mitogen activated protein/extracellular signal-related kinase kinase)與 ERK(extracellular signal-regulated kinase)活化(圖 117-1A)。基於這些結果,有人提出「誤導的骨髓樹突狀細胞前驅物模型 (Misguided Myeloid Dendritic Cell Precursor Model)」,其中骨髓衍生的前驅骨髓樹突狀細胞 (precursor bone marrow–derived myeloid dendritic cells) 獲得病理性 ERK 活化,並表現典型見於成熟蘭格罕細胞的抗原,例如 CD207 與 CD1a(圖 117-1B)。這些細胞分泌如骨橋蛋白 (osteopontin)、vanin 與不同促發炎因子 (proinflammatory factors),以及轉化生長因子-β (transforming growth factor-β),並吸引(除巨噬細胞與嗜酸性球等其他發炎細胞外)活化的 T 細胞,尤其是活化的調節性 FOXP3/CTLA4 與 SPP 陽性 T 細胞。⁵
器官侵犯的嚴重度與範圍據認為取決於 ERK 被活化時所處的分化階段:在造血幹細胞 (hematopoietic stem cell) 或未分化的骨髓樹突狀細胞中活化導致多系統疾病,而在較分化的骨髓前驅物中 ERK 活化則僅導致多灶性或單灶性疾病。¹
需要更多研究以進一步洞察 LCH 的致病機轉,因為仍有許多懸而未決的問題,包括:為何在成人肺部 LCH 中,與其他器官不同,浸潤的單核細胞似乎大多為非克隆性的成熟樹突狀細胞?另一個有趣的問題是,LCH 中 PD-L1(programmed death ligand 1)免疫檢查點蛋白的高表現是否可作為新的治療標靶⁶;在撰寫本文時,現在下定論仍言之過早。
20
臨床特徵 (CLINICAL FEATURES)
分類 (CLASSIFICATION)
LCH 分為 Hashimoto-Pritzker 病、嗜酸性肉芽腫、HSC 與 Letterer-Siwe 病的傳統分類已被棄用,因為疾病譜比傳統分類所涵蓋者更廣,且許多病人不符合這些臨床亞型。此外,臨床病程可能隨時間改變。新分類考量器官侵犯的範圍,因為這對治療與預後有重大意義(見圖 117-1)。此外,LCH 或許最好被描述為一種連續譜 (continuum),如圖 117-2 所示。病人被分為:
■ 單一系統 LCH (Single-system LCH):約 55% 至 65% 的 LCH 病人表現為單一系統 LCH,其中僅 1 個器官系統受侵。最常受侵的器官系統為骨骼、皮膚、淋巴結、肺與中樞神經系統 (CNS)。⁷
■ 多系統 LCH (Multisystem LCH):在多系統 LCH 中,疾病侵犯 1 個以上的器官系統。重要的是要區分是否有高危險器官 (high-risk organs) 受侵。
危險器官 (at-risk organs) 包括造血系統 (hematopoietic system)、肝臟與脾臟。肺已不再被視為高危險器官,因為近期研究無法證實肺侵犯為負面預後因子。表 117-1 提供目前公認分類的概覽。
皮膚表現 (CUTANEOUS FINDINGS)
LCH 的皮膚表現可見於超過三分之一的 LCH。它是繼骨病灶後第二常見的受侵器官,並可為疾病的最早徵象。只有 12% 的單一系統疾病兒童與 53% 的多系統 LCH 病人顯示皮膚侵犯。⁷ 因此,年齡是重要的危險因子,因為小於 1 歲的兒童更常顯示真正的僅皮膚 LCH (skin-only LCH),而大於 18 個月者有較高的多系統 LCH 風險。因此,皮膚科醫師在做出早期診斷上的角色至關重要。病人可表現出多種多樣的皮膚表現。最典型者為軀幹、間擦區域與頭皮上小型、半透明的玫瑰黃色結痂丘疹或丘疹水疱,伴隨類似「念珠菌間擦疹 (candida intertrigo)」或「脂漏性皮膚炎 (seborrheic dermatitis)」的濕疹樣脫屑(圖 117-3、117-4 與 117-5)。病灶也可表現為出血性丘疹與結節,伴隨瘀點,使人聯想到血管性病灶或「水痘樣疹 (varicella-like eruptions)」(圖 117-6)。也曾描述水疱、膿疱與甲侵犯。甲侵犯可表現為甲溝炎 (paronychia)、甲褶破壞 (nailfold destruction)、甲剝離 (onycholysis)、甲下角化過度 (subungual hyperkeratosis)、縱向溝紋 (longitudinal grooving),以及
2021
20
報告於蘭格罕細胞組織球增生症中的突變 (Reported mutations in Langerhans cell histiocytosis)
BRAF 50%-65%
BRAF-V600E、BRAF-V600D、BRAF-600DLAT、BRAF-T599A
MAP2K1 0%-20%
未知 (Unknown) 15%-40%
MAP3K 10%
A
RTK(受體酪胺酸激酶 receptor tyrosine kinase)
Ras
Raf
MEK
ERK1/ERK2
臨床表現嚴重度的風險 (Risk of severity of clinical manifestation)
高風險 (High risk)
造血幹細胞 (Hematopoietic stem cells)(CD34+)
樹突狀前驅細胞 (Dendritic precursor cells)
組織樹突狀前驅細胞 (Tissue dendritic precursor cells)
低風險 (Low risk)
CD1a+/CD207+ LCH 病灶樹突狀細胞 (LCH lesion dendritic cells)
ERK ERK ERK
ERK
+ 巨噬細胞、T 細胞與嗜酸性球 (Macrophages, T-cells, and Eosinophils)
多系統 多灶性 多系統 多灶性 多灶性 單灶性
B
2022
20
LCH 的臨床譜 (Clinical spectrum of LCH)
多骨性骨侵犯 (Polyostotic bone involvement) 多灶性骨疾病(2 或更多不同骨骼) 多發淋巴結侵犯
播散性疾病,侵犯低危險器官(皮膚、骨骼、淋巴結、腦下垂體 pituitary)
多部位 低危險組 (Multiple site, Low-risk group)
單骨性骨侵犯 (Monostotic bone involvement) 孤立性皮膚侵犯 單一淋巴結受侵
播散性疾病,侵犯 1 或更多高危險器官(造血系統、肺、肝與脾)
單一部位 高危險組 (Single site, High-risk group)
單一系統疾病 症狀:無或輕微 多系統疾病 症狀:存在且嚴重
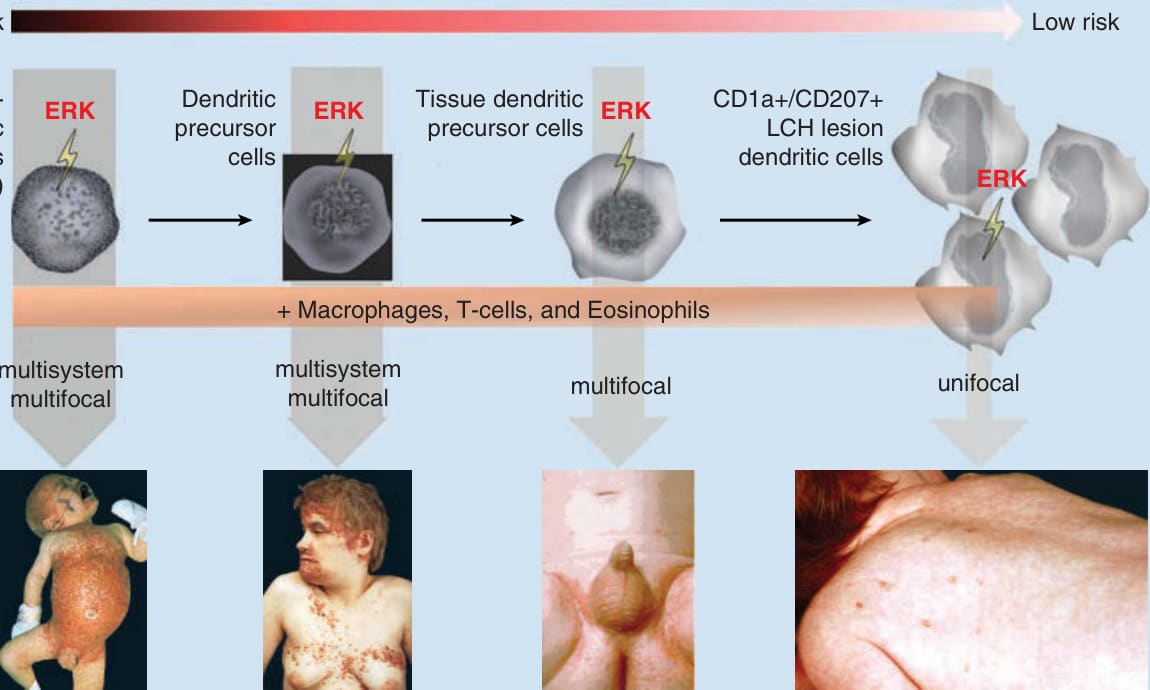
圖 117-1 A,報告於蘭格罕細胞組織球增生症 (LCH) 中的突變。此圖說明 LCH 中已辨識突變的頻率,這些突變導致轉錄因子 ERK(extracellular signal-regulated kinase)的下游活化。在 50% 至 65% 的 LCH 病例中發現 BRAF(v-Raf murine sarcoma viral oncogene homolog B)基因突變。BRAF-V600E 突變是迄今為止最常見的突變,但也曾描述 BRAF-V600D、BRAF-600DLAT 與 BRAF-T599A 突變。MAP2K1,mitogen-activated protein kinase kinase 1;MAP3K,mitogen-activated protein kinase kinase kinase;MEK,mitogen-activated protein/extracellular signal-related kinase kinase;RTK,receptor tyrosine kinase。B,「誤導的骨髓樹突狀細胞模型 (Misguided Myeloid Dendritic Cell Model)」。根據「誤導的骨髓樹突狀細胞模型」,臨床表現取決於 ERK 活化所發生的造血細胞分化階段。在造血幹細胞與樹突狀前驅細胞中的 ERK 活化導致廣泛性 LCH,而在組織樹突狀前驅細胞與 CD1a+/CD207+ LCH 病灶樹突狀細胞中的 ERK 活化則導致單一系統 LCH。
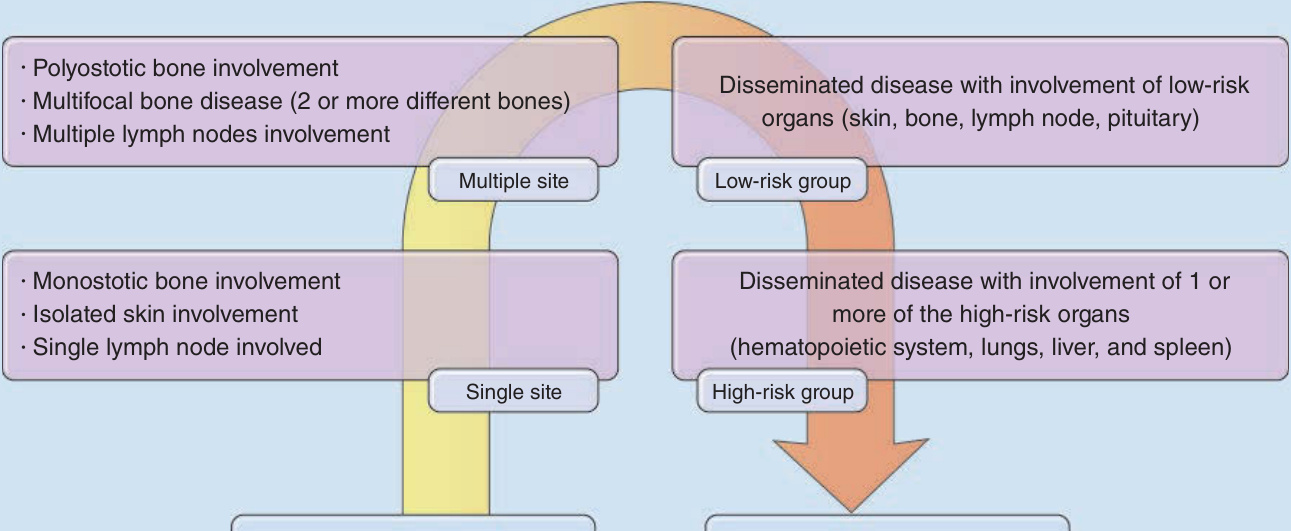
圖 117-2:蘭格罕細胞組織球增生症 (LCH) 的臨床譜。任何單一 LCH 病人都可被定位在箭頭上的某一特定點。
甲床的色素性與紫癜性條紋 (pigmented and purpuric striae)。黏膜病灶最常為結節潰瘍性,並侵犯口周、生殖器周圍與肛周區域以及牙齦。黏膜病灶與外耳道中耳炎 (external otitis media) 似乎與較高的多系統 LCH 風險相關。⁷ 伴甲侵犯的紫癜可能是不良預後徵象,但這應在更大型的臨床試驗中評估。這種多樣的皮膚與黏膜表現經常導致診斷延誤,因為皮膚病灶被誤判為濕疹、痱子 (miliaria)、疥瘡 (scabies)、水痘 (varicella)、脂漏性皮膚炎、毛囊炎 (folliculitis) 或念珠菌病 (candidiasis)。當看到上述病灶時,尤其當它們對治療有抗藥性並正在擴散時,應將 LCH 謹記為一個罕見但重要的鑑別診斷。
非皮膚表現 (NONCUTANEOUS FINDINGS)
LCH 中最常受侵的非皮膚器官為骨骼(77% 的病例),其次為淋巴結(19%)、肝臟(16%)、脾臟(13%)、肺(10%)與 CNS(6%)。在疾病的侵襲性表現中
2023
20
2024
中,病人表現出多種臨床症狀,例如倦怠、發燒、噁心、體重減輕、肌痛與關節痛,以及記憶問題。骨病灶 (Bone Lesions):雖然 LCH 可侵犯體內任何骨骼,但它最常侵犯顱骨、頜骨、股骨、肋骨、椎骨與肱骨。病人主訴觸痛性腫塊,影像學檢查顯示受侵骨骼有「穿鑿狀 (punched out)」外觀的溶解區域。尤其在兒童中,頸椎常受侵,可導致扁平椎 (vertebra plana)。在成人中,較常見椎體的不對稱塌陷,可引發神經缺損。頜骨侵犯可導致牙齒鬆動。椎體塌陷可為 LCH 最顯著的診斷線索之一。顱底侵犯可能導致聽力喪失、復發性外耳炎 (otitis externa)、尿崩症或顱神經麻痺。顏面骨與前顱窩或中顱窩 (anterior or middle cranial fossae) 的病灶被歸類為「CNS 風險 (CNS-risk)」病灶,因為它們與發展 CNS 疾病(如尿崩症)的風險增加 3 倍相關,而尿崩症在診斷時多為不可逆的病況。淋巴結 (Lymph Nodes):頸部淋巴結最常受 LCH 侵犯,但 LCH 也可侵犯縱膈 (mediastinum) 淋巴結,這可能被誤診為淋巴瘤,並因氣道系統受壓而引起類似氣喘的症狀。為正確診斷,切片檢查是必須的。骨髓 (Bone Marrow):當其他危險器官(如肝臟與脾臟)受侵時,骨髓侵犯常侵犯幼童。過去只有在出現顯著貧血、血小板減少 (thrombocytopenia) 或嗜中性球減少 (neutropenia) 時才懷疑骨髓侵犯。近期一項研究發現,即使無法偵測到血液學異常,在單一系統 LCH 的骨髓中也可發現 CD1a+ 細胞。當出現血小板減少與貧血,尤其合併低白蛋白血症 (hypoalbuminemia) 時,LCH 與不良結局相關。⁸
肝臟與脾臟 (Liver and Spleen):由 LCH 肝臟侵犯所誘發的膽汁鬱積 (cholestasis) 與硬化性膽管炎 (sclerosing cholangitis) 是 LCH 最嚴重的併發症之一。在大多數情況下,硬化性膽管炎對化學治療無反應,肝臟移植仍是唯一可能的治療選擇。臨床上,病人表現為肝脾腫大與肝臟酵素升高,例如肝轉胺酶 (liver transaminases)、γ-麩胺醯轉移酶 (γ-glutamyltransferase) 與/或鹼性磷酸酶 (alkaline phosphatase) 升高。此外,可能出現伴腹水 (ascites) 與凝血缺陷的低白蛋白血症。脾臟侵犯伴大量脾腫大可因脾功能亢進 (hypersplenism) 而導致血球減少 (cytopenias) 與呼吸功能受損,顯著惡化臨床結局。肺 (Lungs):肺侵犯在成人比在兒童中更常見,吸菸已被確認為主要危險因子。LCH 誘發上肺野與中肺野肺組織的囊性與/或結節性破壞,可導致自發性氣胸 (spontaneous pneumothorax)、呼吸急促 (tachypnea) 與/或呼吸困難⁹,最終導致嚴重的肺功能不全。
中樞神經系統 (Central Nervous System):LCH 可直接侵犯腦的任何部分。CNS 侵犯的危險因子(25% 風險)為顏面骨或前顱窩或中顱窩骨骼的 LCH 病灶(「CNS 風險」病灶)。CNS 侵犯最常見的表現包括由大型腦下垂體腫瘤所致的內分泌異常,最常見為尿崩症,以及神經退化性症狀,例如運動失調 (ataxia)、構音障礙 (dysarthria)、認知功能障礙與行為改變。影像學變化可早於這些症狀許多年。有趣的是,在神經退化性病灶中組織學上並未發現 CD1a+ 細胞;取而代之的是發現淋巴細胞與活化的微膠細胞 (microglia cells),這引出此可能為一種副腫瘤性發炎反應 (paraneoplastic inflammatory response) 的推測。
內分泌病變 (Endocrinopathies):尿崩症是 LCH 中最常見的內分泌病變。病人表現出多尿、煩渴與夜尿。這是對分泌抗利尿激素 (antidiuretic hormone) 之腦下垂體後葉細胞受損的徵象。在約 4% 的病人中,特發性尿崩症可早於 LCH 的診斷。¹⁰ 在已診斷 LCH 的病人中,發展尿崩症的風險約為 24%。在大多數情況下,尿崩症儘管接受治療仍持續存在。其他與腦下垂體前葉相關的內分泌異常,例如性腺功能低下 (hypogonadism)、生長遲滯、甲狀腺激素功能障礙與葡萄糖代謝異常,皆可能表現出來。因此,建議進行完整的內分泌檢查。
胃腸系統 (Gastrointestinal System):有胃腸侵犯的病人臨床上可表現為腹瀉、血便 (hematochezia)、肛周瘻管 (perianal fistulas) 與/或吸收不良 (malabsorption),但此器官系統罕見受侵。若病人表現出胃腸症狀,需要多處切片,因為在大多數情況下胃腸侵犯為斑塊狀分布 (patchy)。
診斷 (DIAGNOSIS)
LCH 的診斷基於受侵器官的臨床病理學與影像學特徵。
完整病史 (COMPLETE HISTORY)
應評估症狀的性質與持續時間。此外,根據組織球協會 (Histiocyte Society) 的建議,完整病史應包括關於疼痛、腫脹、皮疹、耳漏 (otorrhea)、易怒 (irritability)、發燒、食慾不振、腹瀉、體重減輕、生長遲滯、煩渴、多尿、活動程度改變、呼吸困難、菸害暴露,以及行為與神經學改變的問題。
20
完整身體檢查 (COMPLETE PHYSICAL EXAMINATION)
應測量體溫、身高與體重。此外,組織球協會建議評估青春期狀態 (pubertal status);徹底的皮膚與黏膜評估,檢查是否有黃疸 (jaundice)、蒼白 (pallor)、水腫、淋巴結病變、耳分泌物 (ear discharge)、眼眶異常、異常黏膜病灶、異常牙列 (dentation) 與軟組織腫脹;並評估任何呼吸急促、肋間凹陷 (intercostal retractions) 與腹水;以及肝臟與脾臟大小評估。此外,完整的神經學評估是必須的。
實驗室檢查與影像學檢查 (LABORATORY TESTING AND IMAGING STUDIES)
組織病理學 (HISTOPATHOLOGY)
組織病理學診斷是迄今為止最可靠、最準確的 LCH 確定診斷工具,且只要不會使病人面臨更高風險,就應始終進行。組織病理學上,皮膚切片的典型表現顯示乳突真皮 (papillary dermis) 有 LCH 細胞的緻密帶狀浸潤(圖 117-7)。這些細胞呈卵圓形,
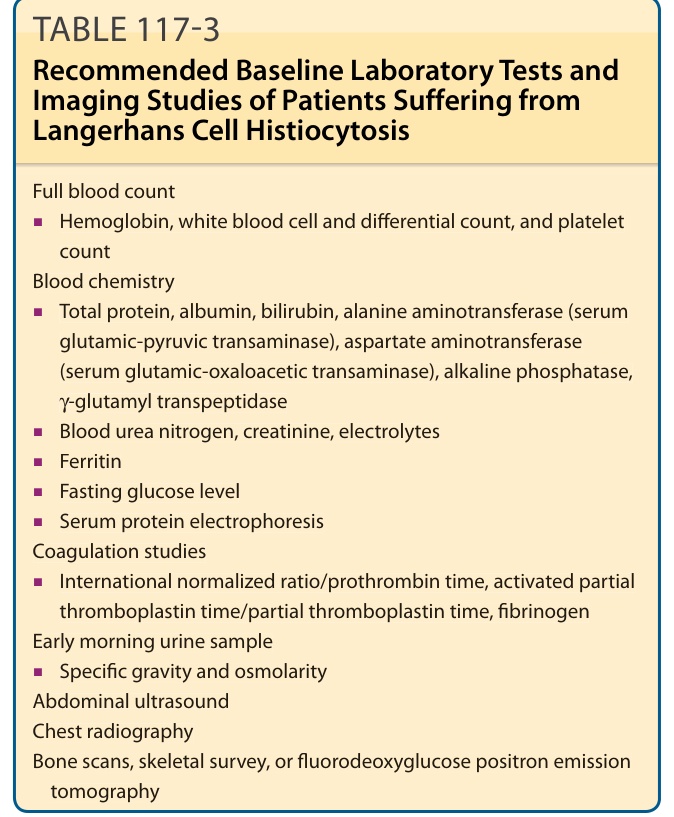
表 117-3 列出針對 LCH 病人建議的實驗室檢查與影像學檢查。表 117-4 列出針對特定臨床情境的建議。
全血球計數 (Full blood count)
■ 血紅素 (Hemoglobin)、白血球與分類計數,以及血小板計數
血液生化 (Blood chemistry)
■ 總蛋白 (Total protein)、白蛋白、膽紅素 (bilirubin)、丙胺酸轉胺酶(alanine aminotransferase,血清麩胺酸-丙酮酸轉胺酶 serum glutamic-pyruvic transaminase)、天門冬胺酸轉胺酶(aspartate aminotransferase,血清麩胺酸-草醋酸轉胺酶 serum glutamic-oxaloacetic transaminase)、鹼性磷酸酶、γ-麩胺醯轉肽酶 (γ-glutamyl transpeptidase)
■ 血尿素氮 (Blood urea nitrogen)、肌酸酐 (creatinine)、電解質
■ 鐵蛋白 (Ferritin)
■ 空腹血糖值
■ 血清蛋白電泳 (Serum protein electrophoresis)
凝血檢查 (Coagulation studies)
■ 國際標準化比值/凝血酶原時間(INR/prothrombin time)、活化部分凝血活酶時間/部分凝血活酶時間(activated partial thromboplastin time/partial thromboplastin time)、纖維蛋白原 (fibrinogen)
清晨尿液檢體 (Early morning urine sample)
■ 比重 (Specific gravity) 與滲透壓 (osmolarity)
腹部超音波 (Abdominal ultrasound)
胸部 X 光 (Chest radiography)
骨掃描、骨骼系列攝影,或氟去氧葡萄糖正子斷層掃描(bone scans, skeletal survey, or fluorodeoxyglucose positron emission tomography)
2025
改編自組織球協會 (Histiocyte Society) 的建議 (https://histiocytesociety.org/)。
20
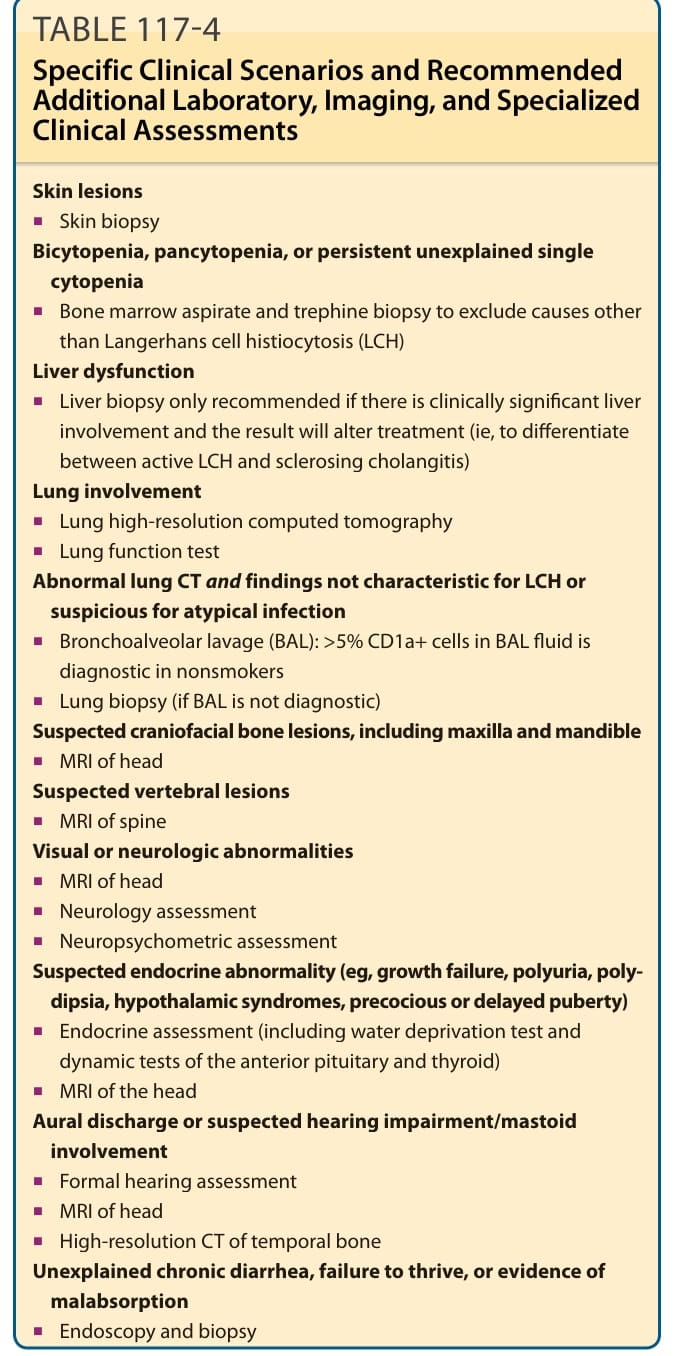
表 117-4:特定臨床情境與建議的額外實驗室、影像學及專科臨床評估 (Specific Clinical Scenarios and Recommended Additional Laboratory, Imaging, and Specialized Clinical Assessments)
皮膚病灶 (Skin lesions)
■ 皮膚切片 (Skin biopsy)
雙血球減少、全血球減少,或持續無法解釋的單一血球減少 (Bicytopenia, pancytopenia, or persistent unexplained single cytopenia)
■ 骨髓抽吸與環鑽切片 (bone marrow aspirate and trephine biopsy),以排除蘭格罕細胞組織球增生症 (LCH) 以外的原因
肝功能障礙 (Liver dysfunction)
■ 只有在有臨床上顯著的肝臟侵犯且結果將改變治療時,才建議進行肝臟切片(即用以區分活動性 LCH 與硬化性膽管炎)
肺侵犯 (Lung involvement)
■ 肺高解析度電腦斷層 (Lung high-resolution computed tomography)
■ 肺功能檢查 (Lung function test)
肺 CT 異常且發現不具 LCH 特徵或疑似非典型感染 (Abnormal lung CT and findings not characteristic for LCH or suspicious for atypical infection)
■ 支氣管肺泡灌洗(bronchoalveolar lavage, BAL):BAL 液中 >5% 的 CD1a+ 細胞在非吸菸者中具診斷性
■ 肺切片(若 BAL 不具診斷性)
疑似顱顏骨病灶,包括上頜骨與下頜骨 (Suspected craniofacial bone lesions, including maxilla and mandible)
■ 頭部 MRI
疑似椎體病灶 (Suspected vertebral lesions)
■ 脊椎 MRI
視覺或神經學異常 (Visual or neurologic abnormalities)
■ 頭部 MRI
■ 神經科評估 (Neurology assessment)
■ 神經心理測量評估 (Neuropsychometric assessment)
疑似內分泌異常(如生長遲滯、多尿、煩渴、下視丘症候群、性早熟或青春期延遲)(Suspected endocrine abnormality)
■ 內分泌評估(包括禁水試驗 water deprivation test,以及腦下垂體前葉與甲狀腺的動態檢查)
■ 頭部 MRI
耳分泌物或疑似聽力損害/乳突侵犯 (Aural discharge or suspected hearing impairment/mastoid involvement)
■ 正式聽力評估 (Formal hearing assessment)
■ 頭部 MRI
■ 顳骨高解析度 CT (High-resolution CT of temporal bone)
無法解釋的慢性腹瀉、生長遲滯,或吸收不良的證據 (Unexplained chronic diarrhea, failure to thrive, or evidence of malabsorption)
■ 內視鏡檢查與切片 (Endoscopy and biopsy)
改編自組織球協會 (Histiocyte Society) 的建議 (https://histiocytesociety.org/)。
具有嗜酸性細胞質 (eosinophilic cytoplasm),並典型地呈現不規則、囊泡狀、內折(腎形 kidney-shaped)的細胞核(圖 117-8)。在某些情況下,可見縱向核溝 (longitudinal nuclear grooves),使這些細胞具有咖啡豆樣 (coffee bean-like) 外觀。引人注目的是,LCH 細胞比淋巴細胞大 4 至 5 倍,並混雜著數量不等的嗜中性球、嗜酸性球、淋巴細胞、漿細胞 (plasma cells) 與組織球,尤其在早期病灶中。在較晚期的病灶中,泡沫狀組織球 (foamy histiocytes) 與真皮顯著的纖維化 (fibrosis) 佔主導。LCH 細胞常顯示明顯的表皮趨向性 (epidermotropism),形成表皮內微膿瘍 (intraepidermal microabscesses) 與附屬器周圍分布 (periappendageal distribution)(圖 117-7 與 117-9)。表皮趨向性可能極為明顯,以致真皮表皮交界 (dermoepidermal junction) 變得模糊,表皮變薄甚至遭破壞。此外,偶可發現
2026
乳突真皮的水腫與多核巨細胞 (multinucleated giant cells)。LCH 細胞的典型免疫組織化學標記為 CD1a(圖 117-9)、S100B、CD207(Langerin)與 fascin。Stabilin-1 與 CD34 不表現(表 117-5)。LCH 細胞的典型發現為細胞質中存在伯貝克顆粒 (Birbeck granules),可經由電子顯微鏡辨識。它們是類似網球拍 (tennis racquet) 的胞器,沿「球拍柄」有拉鍊樣 (zipper-like) 外觀(圖 117-10)。過去,以電子顯微鏡偵測伯貝克顆粒是 LCH 的診斷黃金標準。然而現今,以 CD207(一種辨識與伯貝克顆粒相關之 C 型凝集素 C-type lectin 的抗體)進行免疫組織化學染色,是最敏感的標記。電子顯微鏡已不再為特異性診斷所需。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
LCH 是一種異質性疾病。病人可表現為侷限於單一器官系統的病灶,呈惰性自癒病程,或表現為不同器官(尤其是危險器官)中的多發病灶,伴致命結局。因此,風險分層 (risk stratification)
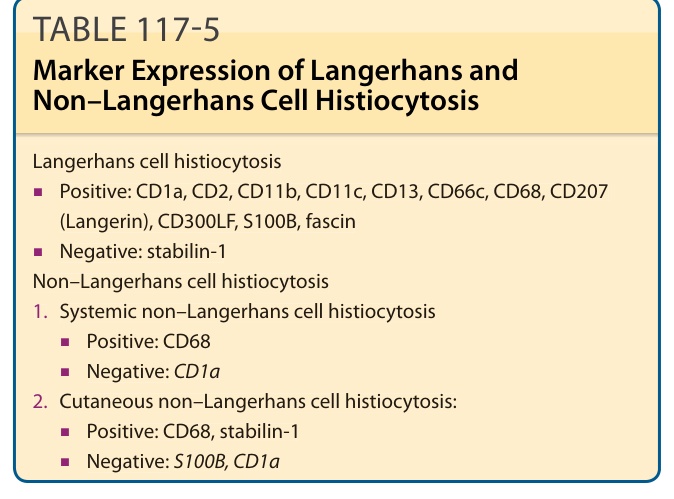
表 117-5:蘭格罕細胞與非蘭格罕細胞組織球增生症的標記表現 (Marker Expression of Langerhans and Non–Langerhans Cell Histiocytosis)
蘭格罕細胞組織球增生症 (Langerhans cell histiocytosis)
■ 陽性:CD1a、CD2、CD11b、CD11c、CD13、CD66c、CD68、CD207(Langerin)、CD300LF、S100B、fascin
■ 陰性:stabilin-1
非蘭格罕細胞組織球增生症 (Non–Langerhans cell histiocytosis)
- 全身性非蘭格罕細胞組織球增生症 (Systemic non–Langerhans cell histiocytosis)
■ 陽性:CD68
■ 陰性:CD1a
2. 皮膚性非蘭格罕細胞組織球增生症 (Cutaneous non–Langerhans cell histiocytosis):
■ 陽性:CD68、stabilin-1
■ 陰性:S100B、CD1a
20
對於為每位病人提供最佳治療方案至關重要。預後取決於多種因素。近期一項多中心 LCH 第 III 期試驗研究了為期 12 個月的長春花鹼 (vinblastine) 與培尼皮質醇 (prednisolone) 治療,顯示在 6 週誘導期 (induction phase) 後達到疾病完全緩解、對治療呈中間反應,以及疾病進展的病人中,5 年存活率分別為 95%、83% 與 57%。¹¹ 因此,多器官侵犯病人結局最重要的預測因子之一,是疾病在最初 6 週內對全身治療的反應。¹¹ 其他先前與不良結局相關的預測因子,例如小於 2 歲或肺侵犯,現在已不再被視為不良結局的危險因子。危險器官(如肝臟、脾臟與骨髓)的侵犯也與不良結局相關。肝臟在鎖骨中線 (midclavicular line) 肋緣下增大超過 3 cm;高膽紅素血症 (hyperbilirubinemia) 大於正常值的 3 倍;低白蛋白血症小於 30 g/dL;γ-麩胺醯轉肽酶增加超過正常值的 2 倍;丙胺酸轉胺酶與天門冬胺酸轉胺酶大於正常值的 3 倍;腹水;水腫;或肝內結節性腫塊,都是肝臟侵犯的徵象,需經超音波,以及若可能經切片確認。若脾臟在鎖骨中線肋緣下可觸及超過 3 cm,則應懷疑脾臟侵犯。造血系統侵犯可分為輕度型,血紅素介於 10 與 7 g/dL 之間(非其他原因所致),與/或血小板減少
2027
20
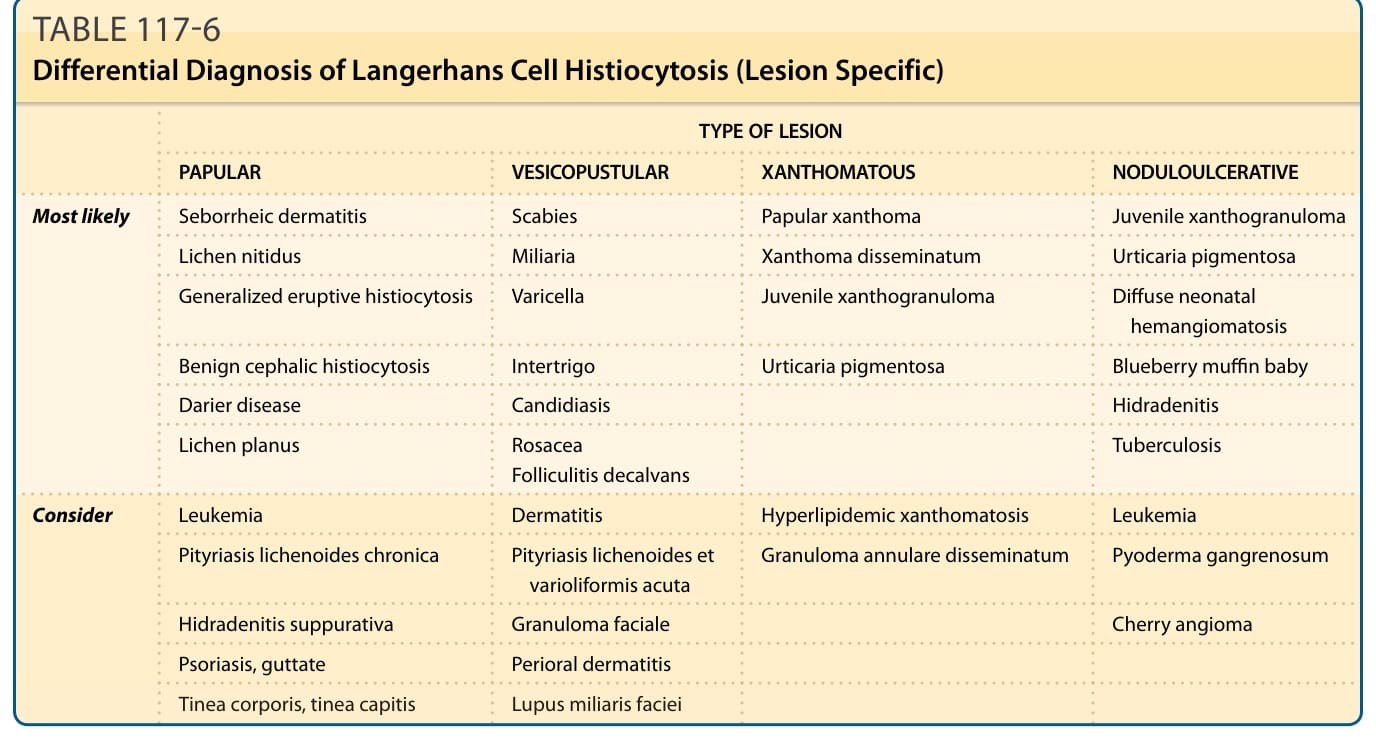
表 117-6 列出 LCH 在皮膚中不同臨床表現的可能鑑別診斷。
病灶類型 (TYPE OF LESION)
丘疹性 (PAPULAR) 水疱膿疱性 (VESICOPUSTULAR) 黃色瘤性 (XANTHOMATOUS) 結節潰瘍性 (NODULOULCERATIVE)
最可能 (Most likely) 脂漏性皮膚炎 (Seborrheic dermatitis) 疥瘡 (Scabies) 丘疹性黃色瘤 (Papular xanthoma) 幼年黃色肉芽腫 (Juvenile xanthogranuloma)
光澤苔癬 (Lichen nitidus) 痱子 (Miliaria) 播散性黃色瘤 (Xanthoma disseminatum) 色素性蕁麻疹 (Urticaria pigmentosa)
廣泛性發疹性組織球增生症 (Generalized eruptive histiocytosis) 水痘 (Varicella) 幼年黃色肉芽腫 (Juvenile xanthogranuloma) 瀰漫性新生兒血管瘤病 (Diffuse neonatal hemangiomatosis)
良性頭部組織球增生症 (Benign cephalic histiocytosis) 間擦疹 (Intertrigo) 色素性蕁麻疹 (Urticaria pigmentosa) 藍莓鬆餅嬰兒 (Blueberry muffin baby)
毛囊角化病 (Darier disease) 念珠菌病 (Candidiasis)
化膿性汗腺炎 (Hidradenitis)
扁平苔癬 (Lichen planus)
酒糟 (Rosacea) 禿髮性毛囊炎 (Folliculitis decalvans)
結核 (Tuberculosis)
考慮 (Consider) 白血病 (Leukemia) 皮膚炎 (Dermatitis) 高血脂性黃色瘤病 (Hyperlipidemic xanthomatosis) 白血病 (Leukemia)
慢性苔癬樣糠疹 (Pityriasis lichenoides chronica) 急性痘瘡樣苔癬樣糠疹 (Pityriasis lichenoides et varioliformis acuta) 播散性環狀肉芽腫 (Granuloma annulare disseminatum) 壞疽性膿皮症 (Pyoderma gangrenosum)
化膿性汗腺炎 (Hidradenitis suppurativa) 顏面肉芽腫 (Granuloma faciale)
櫻桃血管瘤 (Cherry angioma)
點滴狀乾癬 (Psoriasis, guttate) 口周皮膚炎 (Perioral dermatitis)
體癬、頭癬 (Tinea corporis, tinea capitis) 顏面粟粒性狼瘡 (Lupus miliaris faciei)
且血小板介於 20,000 與 100,000/mm³ 之間;以及重度型,血紅素低於 7 g/dL 且血小板少於 20,000/mm³。¹² 迄今,尚無資料證明尿崩症為不良結局的預測因子。病灶數目少、病灶迅速消退與單一器官侵犯通常與良好結局相關。然而,預後不僅取決於疾病本身,也取決於相關發現。LCH 病人傾向罹患間發性感染 (intercurrent infections),多為輕度者,例如念珠菌病與皮癬菌病 (dermatophytosis),但他們也易發生更嚴重的全身性感染,可導致敗血症與死亡。此外,LCH 與其他惡性腫瘤相關,這些惡性腫瘤可能早於、與之同時,或在 LCH 診斷之後發生。已描述的相關惡性腫瘤為實體腫瘤(肺腫瘤、腹腔腸繫膜神經母細胞瘤 celiomesenteric neuroblastoma)、惡性淋巴瘤與白血病,尤其是急性淋巴母細胞白血病 (acute lymphoblastic leukemia) 與骨髓性白血病 (myeloid leukemias)。其中一些惡性腫瘤可能與用於 LCH 的烷化劑化療方案 (alkylating chemotherapeutic regimens) 及放射治療有關。在每次臨床就診時都應考慮這些可能的併發症與/或關聯。
治療 (MANAGEMENT)
治療策略取決於疾病的範圍與位置以及病人的年齡。侷限於骨骼或皮膚之較侷限疾病的病人通常不接受全身治療,但「特殊部位 (special site)」病灶除外,例如齒突 (odontoid peg)、伴脊髓內軟組織延伸的椎體病灶,以及解剖功能上關鍵的部位。所有治療建議皆基於
2028
組織球協會 (Histiocyte Society) 的建議。對於多灶性骨病灶與「CNS 風險」病灶,也可考慮全身治療。在 LCH 侷限於皮膚的兒童中,觀察與等待 (watch-and-wait) 策略是最佳方法。可嘗試以皮質類固醇藥膏 (corticosteroid ointments) 進行局部治療,但局部類固醇顯示療效甚微。對局部類固醇無反應的皮疹被視為 LCH 的線索。其他局部治療選擇包括 imiquimod 與 tacrolimus,以及病灶內皮質類固醇注射 (intralesional corticosteroid injections)、CO2 雷射治療,或切除單一 LCH 結節。氮芥藥膏 (Nitrogen mustard ointment) 可用於治療成人的皮膚病灶。有數篇病例報告顯示,成人與兒童在窄頻紫外線 B (narrowband ultraviolet B) 照射後有顯著改善。此治療對丘疹與濕疹樣病灶可能效果良好,但在有結節存在時則否。光化學治療 (Photochemotherapy) 對某些成人病人有效。在局部治療無效的情況下,可嘗試全身性糖皮質素 (systemic glucocorticoids)、沙利竇邁 (thalidomide),或抗有絲分裂藥物 (antimitotic drugs),例如低劑量甲氨蝶呤 (low-dose methotrexate)。沙利竇邁可改善皮膚病灶,但此治療與神經毒性 (neurologic toxicity) 與疲勞相關。若皮膚是唯一受侵的器官,則不應在有生育能力的女性中使用沙利竇邁。有 1 篇病例報告中,病人在口服異維 A 酸 (oral isotretinoin) 治療(1.5 mg/day 持續 8 個月)後進入完全緩解。表 117-7 概述了一些治療選擇。對於單一骨病灶,也可採用觀察與等待策略。其他選擇包括單純刮除 (simple curettage)、完全切除小病灶(病灶 <2 cm 直徑)與病灶內類固醇注射(見表 117-7)。多虧成立於 1985 年的組織球協會的努力,
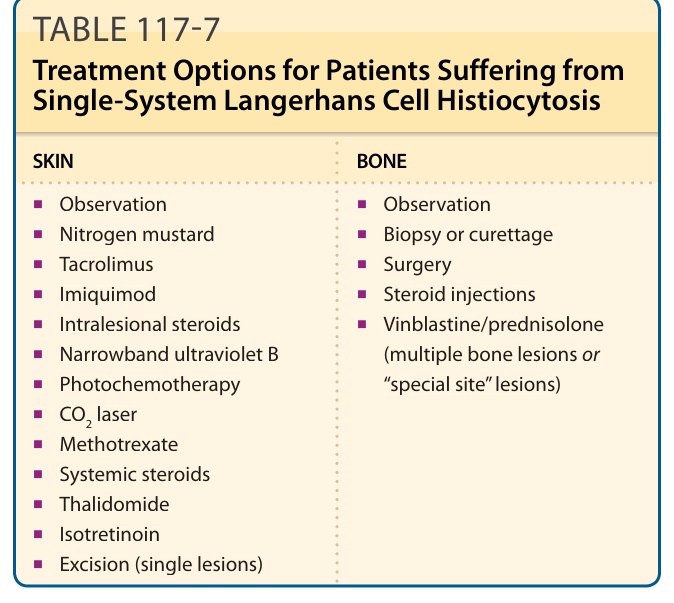
表 117-7:單一系統蘭格罕細胞組織球增生症病人的治療選擇 (Treatment Options for Patients Suffering from Single-System Langerhans Cell Histiocytosis)
皮膚 (SKIN)
■ 觀察 (Observation)
■ 氮芥 (Nitrogen mustard)
■ Tacrolimus
■ Imiquimod
■ 病灶內類固醇 (Intralesional steroids)
■ 窄頻紫外線 B (Narrowband ultraviolet B)
■ 光化學治療 (Photochemotherapy)
■ CO2 雷射
■ 甲氨蝶呤 (Methotrexate)
■ 全身性類固醇 (Systemic steroids)
■ 沙利竇邁 (Thalidomide)
■ 異維 A 酸 (Isotretinoin)
■ 切除(單一病灶)(Excision, single lesions)
骨骼 (BONE)
■ 觀察 (Observation)
■ 切片或刮除 (Biopsy or curettage)
■ 手術 (Surgery)
■ 類固醇注射 (Steroid injections)
■ 長春花鹼/培尼皮質醇(多發骨病灶或「特殊部位」病灶)(Vinblastine/prednisolone)
LCH 多系統疾病治療的科學證據,根據多中心試驗(LCH-I、LCH-II、LCH-III)所報告的結果,遠優於單一系統 LCH。根據最新 LCH-III 試驗的結果,病人應接受 12 個月的全身治療,因為僅治療 6 個月的病人有較高的早期復發頻率。治療方案包括 6 週的誘導化療期,使用長春花鹼(vinblastine,6 mg/m²
每週靜脈推注 weekly intravenous bolus)合併培尼皮質醇(prednisolone,40 mg/m²/day 口服 4 週後於 2 週內逐漸減量),隨後依 6 週後的治療反應與危險器官的侵犯情況,進行含或不含巰基嘌呤 (mercaptopurine) 的長春花鹼/培尼皮質醇治療(圖 117-11)。在此治療方案下,86% 無危險器官侵犯的病人與 66% 有危險器官侵犯的病人在 6 週後對治療顯示出反應——這是結局的重要預後因子。儘管如此,本研究中仍有 50% 的病人對治療無反應¹¹,說明了對額外治療策略的需求。其他用作搶救策略 (salvage strategies)、對多系統 LCH 已證實具有療效(儘管有嚴重副作用)的化療藥物為阿糖胞苷 (cytarabine)、克拉屈濱 (cladribine) 與氯法拉濱 (clofarabine)。同種異體骨髓移植 (Allogeneic bone marrow transplantation) 已在 87 名高風險 LCH 病人中嘗試。在接受移植的病人中,77% 在採用清髓性 (myeloablative) 與減低強度調理方案 (reduced-intensity conditioning regimens) 後存活 3 年。¹³ 基於近期研究結果,這些結果提供了證據顯示 LCH 中絲裂原活化蛋白激酶 (mitogen-activated protein kinase, MAPK) 訊號途徑的體細胞突變產生了強烈的 ERK 活化,新型治療選擇包括 BRAF 與 MAPK 抑制劑。雖然這些治療方案的資料有限,但首批報告令人鼓舞。¹⁴,¹⁵ 此外,已描述 LCH 中有高度的 programmed death ligand 1 表現⁶,這顯示抗 programmed death 1 與抗 programmed death ligand 1 治療可能成功。
20
非蘭格罕細胞組織球增生症 (NON–LANGERHANS CELL HISTIOCYTOSIS)
重點一覽 (AT-A-GLANCE)
■ 非蘭格罕細胞組織球增生症 (Non–Langerhans cell histiocytosis, N-LCH) 代表一組以組織球增生為特徵的不同疾病,這些組織球不符合被診斷為蘭格罕細胞的標準。
■ N-LCH 在免疫組織化學上對 CD68 呈陽性,對 S100B 與 CD1a 呈陰性。
■ Stabilin-1(原稱 MS-1 抗原或 MS-1-HMWP)可區分 N-LCH 與 LCH 及其他肉芽腫性疾病 (granulomatous diseases)。
■ N-LCH 可發生全身性表現,例如尿崩症或眼部侵犯,以及與惡性腫瘤的關聯。
■ 疾病的局部皮膚表現可藉由切除、雷射治療或病灶內類固醇注射治療。放射治療也用於治療皮膚病灶與腦部病灶。對於有內臟病灶的病人,全身性糖皮質素與化學治療,以及在某些情況下標靶治療,例如甲磺酸伊馬替尼 (imatinib mesylate) 或 BRAF 與 MEK 抑制劑,皆有用。
N-LCH 是一組以蘭格罕細胞以外之組織球增生為特徵的不同疾病。N-LCH 可分類為全身性或皮膚性 N-LCH(見表 117-1)。皮膚性 N-LCH 可進一步分類為幼年型、成人型與壞死性黃色肉芽腫,以及紡錘細胞 N-LCH。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
N-LCH 的全身型被視為典型活化巨噬細胞 (classically activated macrophages, Mφ1) 的累積。相反地,皮膚性 N-LCH 的病灶以替代活化巨噬細胞 (alternatively activated macrophages, Mφ2) 的存在為特徵。在單一病灶的形式中,局部創傷可能扮演致病角色。在瀰漫型中,與惡性腫瘤及自體免疫疾病的關聯顯示其有免疫學病因。已知 Mφ1 會在對促發炎刺激(例如 T 輔助細胞 (T-helper, Th) 1 型細胞激素〔干擾素-γ 或細菌產物(脂多醣 lipopolysaccharides)〕)的反應中發展出來。它們的特徵為分泌促發炎細胞激素,例如白血球介素 (interleukin, IL)-1、
2029
20
多系統蘭格罕細胞組織球增生症的治療演算法 (Treatment algorithm for multisystem Langerhans cell histiocytosis)
多系統疾病(切片證實)(Multisystem disease, biopsy proven)
誘導:6 週治療 長春花鹼加培尼皮質醇 (Induction 6-week treatment vinblastine plus prednisolone)
治療反應 (Treatment response)
進展 (Progression) 不完全消退 (Incomplete resolution) 完全消退 (Complete resolution)
第二次 6 週治療 長春花鹼加培尼皮質醇 (Second 6-week treatment vinblastine plus prednisolone)
危險器官受侵?(肝、脾、造血系統)(Risk organ involved? liver, spleen, hematopoietic system)
是 (yes)
否 (no)
改善?(Improved?)
是 (yes)
延續治療(完成共 12 個月的治療)長春花鹼加培尼皮質醇 (Continuation therapy to complete 12-month total treatment, vinblastine plus prednisolone)
二線治療 (Second-line treatment)
加巰基嘌呤 (plus mercaptopurine)
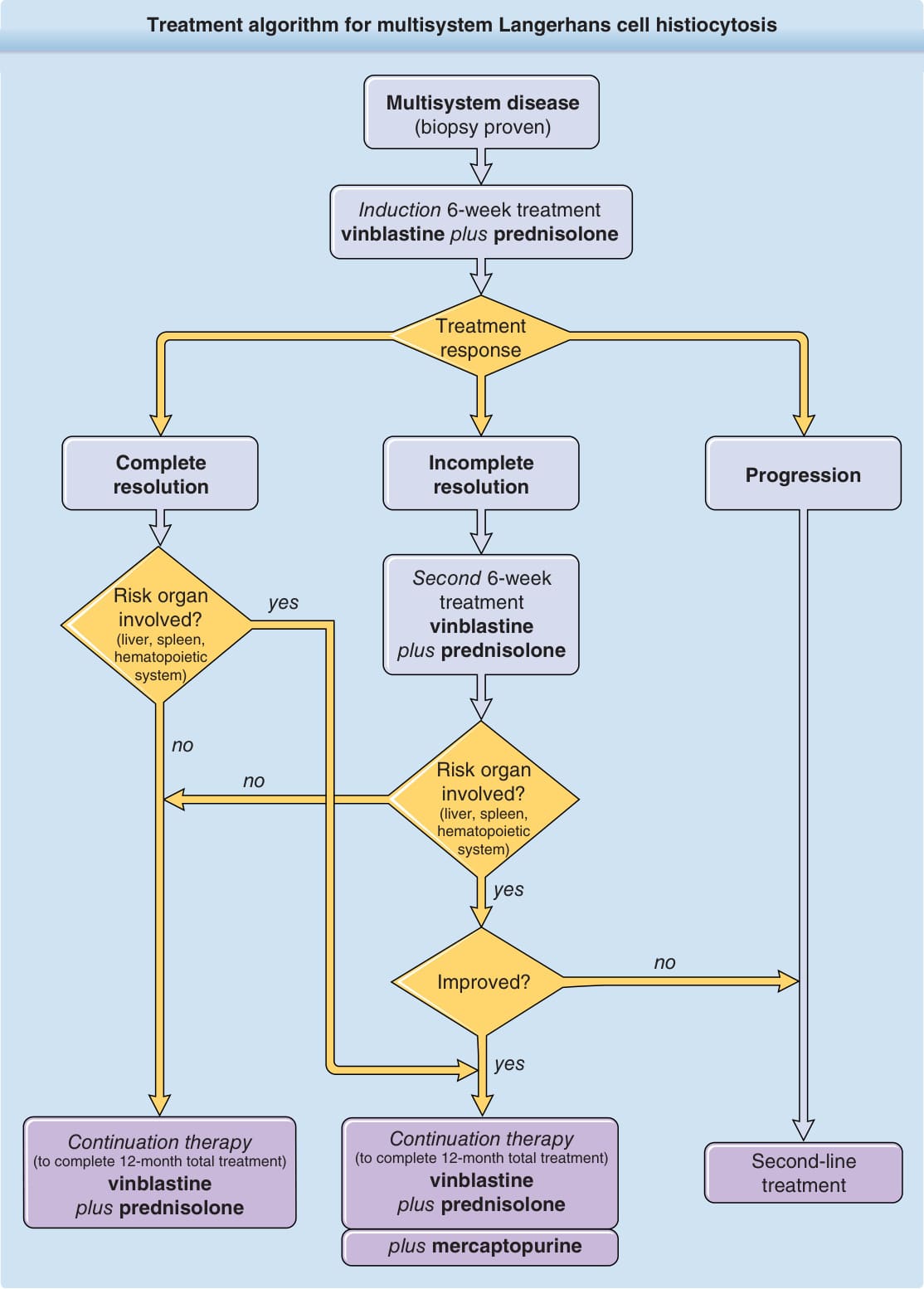
圖 117-11:多系統蘭格罕細胞組織球增生症的治療演算法 (Treatment algorithm for multisystem Langerhans cell histiocytosis)。
IL-6、IL-1 與腫瘤壞死因子-α (tumor necrosis factor-α),並具有強烈的氧化爆發 (oxidative burst) 與顯著的抗菌活性。與全身性 N-LCH 相反,皮膚性 N-LCH 的病灶顯示替代活化的效應巨噬細胞 (alternatively activated effector macrophages, Mφ2)。Mφ2 由 Th2 型細胞激素誘導,包括 IL-4、IL-10、IL-13 與轉化生長因子-β,或由抗發炎介質(例如糖皮質素)誘導。它們表現抗發炎細胞激素,例如 IL-1R 拮抗劑與 IL-10、
2030
趨化激素受體拮抗劑(如 AMAC-1)、先天免疫的廣譜受體(例如巨噬細胞甘露糖受體 macrophage mannose receptor 與血基質結合蛋白受體 haptoglobin receptor CD163)。N-LCH 對 CD68 呈陽性,但對 CD1a 呈陰性。Stabilin-1(原稱 MS-1 抗原或 MS-1-HMWP)表現於某些 N-LCH 上。由於它在 N-LCH 中的特異性表現,它可作為區分 N-LCH 與 LCH 或肉芽腫性疾病的標記之一(見表 117-5)。
Rosai-Dorfman 病(伴大量淋巴結病變之竇組織球增生症)(ROSAI-DORFMAN DISEASE [SINUS HISTIOCYTOSIS WITH MASSIVE LYMPHADENOPATHY])
病理生理面向與臨床特徵 (PATHOPHYSIOLOGIC ASPECTS AND CLINICAL FEATURES)
1969 年,伴大量淋巴結病變之竇組織球增生症 (sinus histiocytosis with massive lymphadenopathy) 由 Juan Rosai 與 Ronald Dorfman 描述。Rosai-Dorfman 病 (Rosai-Dorfman disease, RDD) 是一種特發性疾病,但其發生常在傳染病之後被觀察到。數位作者曾提出可能的病毒病因,例如 Epstein-Barr 病毒、人類疱疹病毒 6 (human herpesvirus 6)、小病毒 B19 (parvovirus B19) 與多瘤病毒 (polyomavirus)。¹⁶,¹⁷ 淋巴結病變是主要的臨床表現。頸部淋巴結是組織球累積最常見的部位,儘管淋巴結外的累積也可能發生。在此,皮膚(圖 117-12)與其他器官,例如乳房、腎臟、甲狀腺、睪丸與 CNS,可能受侵。¹⁷ 與其他局部腫瘤類似,本病的症狀隨累積部位而異。儘管 RDD 罕見,但已有與 LCH、淋巴瘤或自體免疫疾病共同發生的報告。¹⁷ 由於組織球表現 S100B,S100B 可用作血清標記 (serum marker),以監測疾病進展或治療反應。
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
RDD 細胞特徵上為 S100B+(圖 117-13A)與 CD1a−。RDD 細胞也對 CD68、CD163、α1-抗胰蛋白酶 (α1-antitrypsin)、α1-抗胰凝乳蛋白酶 (α1-antichymotrypsin)、fascin、HAM-56(human alveolar macrophage 56)與 stabilin-1(圖 117-13B)呈陽性。RDD 的標誌是伸入現象 (emperipolesis),其中不同類型的骨髓細胞(例如淋巴細胞或嗜中性球)被發現於組織球的細胞質內,背景為成熟的淋巴細胞與漿細胞。¹⁷
20
圖 117-12:Rosai-Dorfman 病 (Rosai-Dorfman disease)。
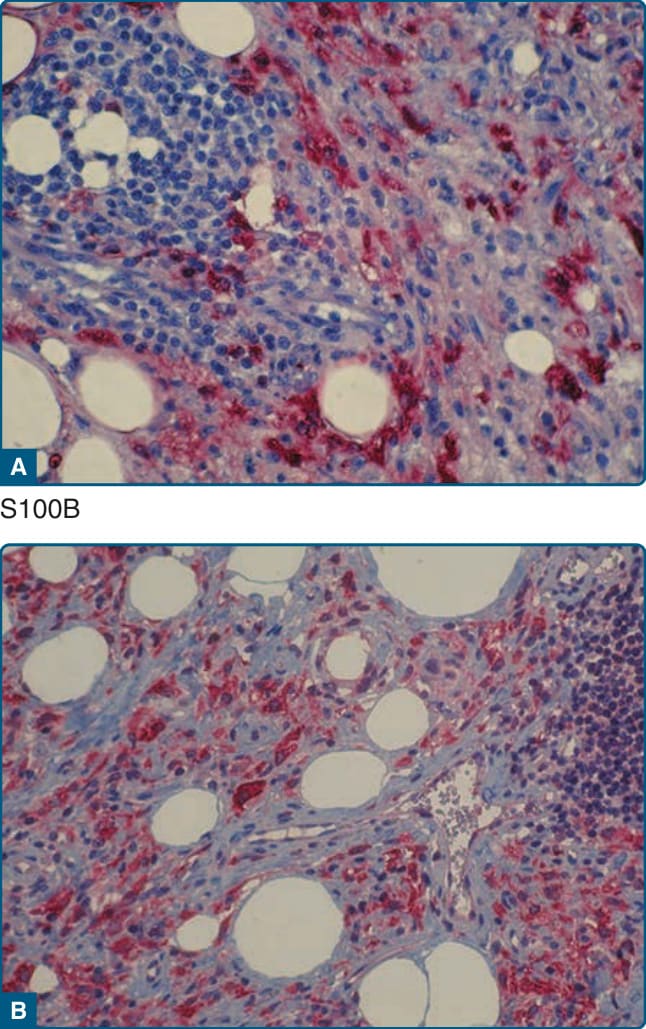
圖 117-13:Rosai-Dorfman 病病灶的顯微照片。組織球對 S100B (A) 與 stabilin-1 (B) 呈陽性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
由於腫大且大量的單側或雙側淋巴結是在孤立或多個區域的常見表現,臨床上可能懷疑為淋巴瘤或膿瘍 (abscesses)。淋巴結外 RDD (Extranodal RDD) 也可模擬其他疾病,例如腦膜瘤 (meningioma)。¹⁷
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
曾有 RDD 病人自發消退的報告。有報告指出,受侵淋巴結或皮膚病灶的手術切除以及放射治療已使 RDD 完全消失。口服培尼皮質醇 (Oral prednisone) 可有顯著有利的反應,主要在有淋巴結侵犯的病人中。然而,病灶內皮質類固醇或口服培尼皮質醇並非總能使皮膚病灶消退。以化療藥物治療一般而言令人失望。然而,有報告指出某些病人可從甲氨蝶呤/6-巰基嘌呤/長春花鹼/6-硫鳥嘌呤(methotrexate/6-mercaptopurine/vinblastine/6-thioguanine)的組合中獲益。¹⁷
2031
20
酪胺酸激酶抑制劑 (Tyrosine kinase inhibitors),例如甲磺酸伊馬替尼 (imatinib mesylate),對某些 RDD 病人有效。¹⁸
噬血球性淋巴組織球增生症 (HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS)
臨床特徵 (CLINICAL FEATURES)
噬血球性淋巴組織球增生症 (Hemophagocytic lymphohistiocytosis, HLH) 於 1952 年由 Farquhar 與 Claireaux 首次描述,是一種罕見疾病。70% 的 HLH 病例為小於 1 歲的兒童。HLH 的特徵為持續發燒、伴血球減少的脾腫大、高三酸甘油脂血症 (hypertriglyceridemia) 與低纖維蛋白原血症 (hypofibrinogenemia)。組織球的浸潤通常見於網狀內皮系統 (reticuloendothelial systems),包括骨髓與 CNS。已描述各種皮膚表現,例如紅皮症、廣泛性紫癜性斑與丘疹,以及麻疹樣疹,可見於高達 65% 的 HLH 病人。HLH 被歸類為細胞激素風暴症候群 (cytokine storm syndromes) 之一,因為會分泌大量的發炎細胞激素。有 2 種主要形式:原發性 (primary) 與繼發性 (secondary)。原發性 HLH 包括家族性 HLH (familial HLH) 與數種原發性免疫缺陷 (primary immunodeficiencies),這些呈現遺傳性遺傳,通常發生於嬰兒期。已辨識出原發性 HLH 中的數個基因缺陷——尤其是家族性 HLH 基因,例如 HPLH1、perforin、UNC13D、STX11 與 STXBP2。繼發性 HLH 與感染(如 Epstein-Barr 病毒)、自體免疫疾病(如幼年特發性關節炎 juvenile idiopathic arthritis)與惡性腫瘤(主要為非何杰金氏淋巴瘤 non-Hodgkin lymphoma)相關。各種細胞激素與可溶性 IL-2 受體 (soluble IL-2 receptor) 濃度升高是 HLH 的生物學標記。¹⁹
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
HLH 的鑑別診斷包括自體免疫淋巴增生症候群 (autoimmune lymphoproliferative syndrome)、Griscelli 症候群 (Griscelli syndrome)、巨噬細胞活化症候群 (macrophage activation syndrome),以及其他以 HLH 表現的原發性免疫缺陷,例如 X 連鎖淋巴增生疾病 (X-linked lymphoproliferative disease)。¹⁹
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
預後不甚樂觀,整體死亡率為 50%。若 HLH 與惡性腫瘤相關,則為不良預後因子。²⁰ 在某些個體中,繼發性 HLH 可能為自限性。治療選擇包括高劑量皮質類固醇、環孢素 (cyclosporine)、依託泊苷 (etoposide)、甲氨蝶呤與長春新鹼 (vincristine)。也曾描述使用靜脈注射免疫球蛋白 (intravenous immunoglobulin)。在大多數情況下,採取積極的治療方法是合理的,包括免疫化療 (immunochemotherapy) 與造血幹細胞移植 (hematopoietic stem cell transplantation)。¹⁹
2032
幼年黃色肉芽腫 (JUVENILE XANTHOGRANULOMA)
臨床特徵 (CLINICAL FEATURES)
幼年黃色肉芽腫 (Juvenile xanthogranuloma, JXG) 於 1905 年由 Adamson 首次描述。它是一種良性、自癒性的皮膚疾病,主要侵犯小於 1 歲的嬰兒,但也可見於較大的兒童。出生時,5% 至 17% 的兒童已有皮膚病灶。在受 JXG 侵犯的兒童中,40% 至 70% 在生命第一年內表現出本病。JXG 通常表現為單發以及多發(寡病灶)的丘疹或結節,通常位於顏面、頸部與上軀幹,以及其他身體部位,包括肺、骨、心臟與胃腸道。早期病灶呈紅棕色(圖 117-14A)。成熟病灶呈紅黃色外觀(圖 117-14B)。可能存在毛細血管擴張 (telangiectasia)。眼部病灶 (Ocular lesions) 發生於高達 10% 的 JXG 兒童,並可能影響其視力。JXG 常伴隨其他疾病,例如第 1 型神經纖維瘤病 (neurofibromatosis Type 1) 與幼年慢性骨髓性白血病 (juvenile chronic myelogenous leukemia)。²¹
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
在早期病灶中,顯微鏡檢查顯示真皮中單形性、不含脂質的組織球浸潤 (non–lipid-containing histiocytic infiltrates)(圖 117-15A)。相反地,成熟病灶以淺層真皮與浸潤邊緣的泡沫細胞 (foam cells)、Touton 巨細胞 (Touton giant cells) 與異物巨細胞 (foreign-body giant cells) 為特徵(圖 117-15B)。此外,可見纖維化。病灶組織球對 stabilin-1 呈陽性。細胞對巨噬細胞標記呈染色,例如 CD163、CD11b、CD11c、CD36、CD68、factor XIIIa 與 vimentin。S100B 與 CD1a 不表現。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
JXG 可藉由 stabilin-1 的表現以及 CD1a 與 CD207 的缺乏而與 LCH 區分。其他鑑別診斷包括,例如傳染性軟疣 (molluscum contagiosum)、血管瘤 (hemangioma) 與神經纖維瘤 (neurofibroma)(表 117-8)。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
JXG 通常在 1 至 5 年內自發消退。然而,眼部病灶罕能自發改善,需要治療。治療包括手術切除、CO2 雷射治療、病灶內類固醇注射、冷凍治療 (cryotherapy) 與低劑量放射治療。在病灶具抗藥性或復發的情況下,應考慮化學治療。²¹
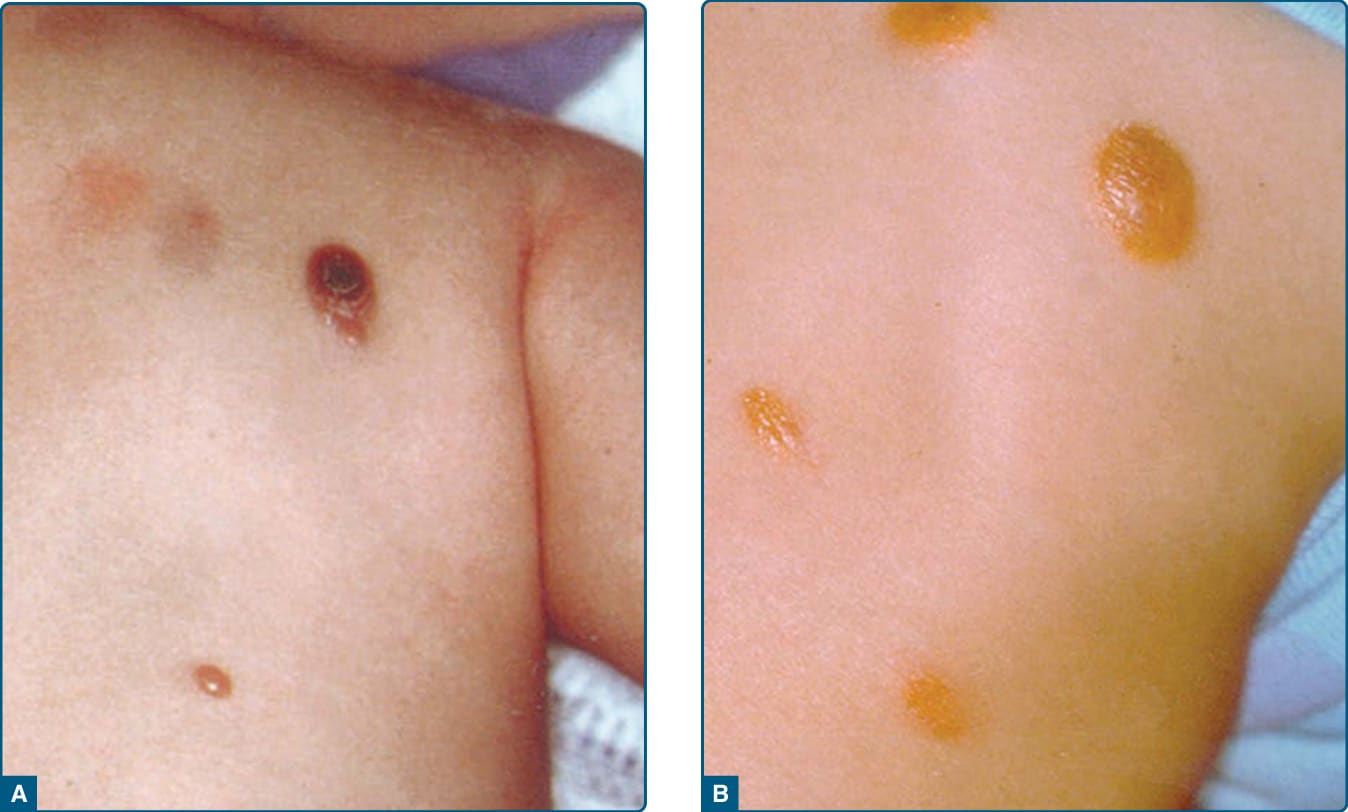
圖 117-14:幼年黃色肉芽腫 (Juvenile xanthogranuloma)。A,早期表現為紅色丘疹。B,成熟期顯示紅黃色調。
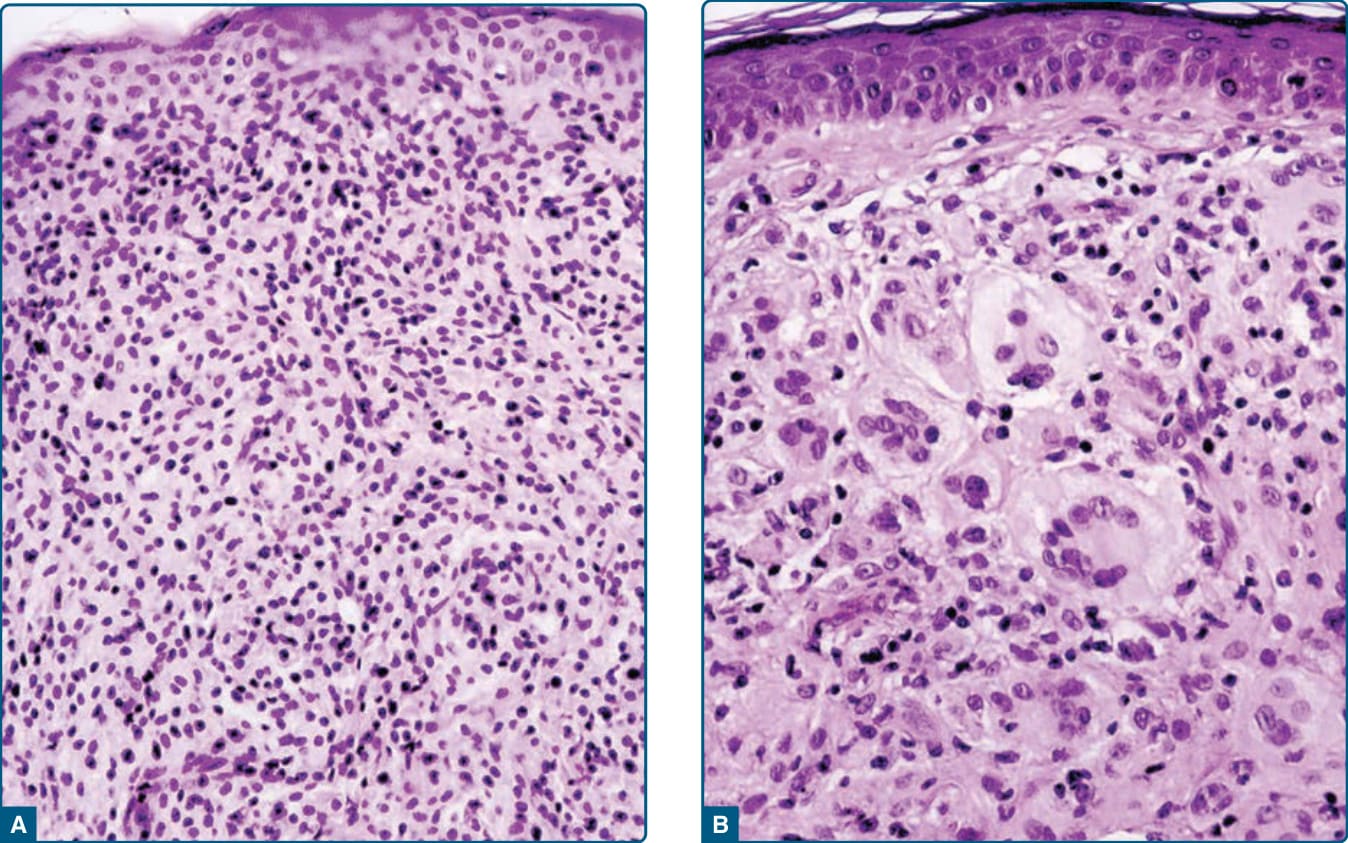
圖 117-15:幼年黃色肉芽腫病灶的顯微照片。A,早期以小型、多為單核、具有小細胞質與較少脂質、到達表皮且無任何 Grenz 帶 (Grenz zone) 的組織球為特徵。B,成熟期顯示大型、富含細胞質、脂質化的組織球、大型泡沫細胞與典型的 Touton 巨細胞。
20

表 117-8:幼年黃色肉芽腫的鑑別診斷 (Differential Diagnosis of Juvenile Xanthogranuloma)
■ 黃色瘤 (Xanthoma)
■ 丘疹性肥大細胞增生症 (Papular mastocytosis)
■ 神經纖維瘤 (Neurofibroma)
■ 類肉瘤病 (Sarcoidosis)
■ 血管瘤 (Hemangioma)
■ 皮膚纖維瘤 (Dermatofibroma)
■ 傳染性軟疣 (Molluscum contagiosum)
■ 皮膚白血病 (Cutaneous leukemia)
■ Spitz 痣 (Spitz nevus)
■ 其他形式的非蘭格罕細胞組織球增生症與蘭格罕細胞組織球增生症 (Other forms of non–Langerhans cell histiocytosis and Langerhans cell histiocytosis)
良性頭部組織球增生症 (BENIGN CEPHALIC HISTIOCYTOSIS)
臨床特徵 (CLINICAL FEATURES)
良性頭部組織球增生症 (Benign cephalic histiocytosis) 最初於 1971 年由 Gianotti 報告,並從 JXG 中區分出來。由於超微結構上存在蠕蟲樣小體 (worm-like bodies),它也被稱為「具有胞質內蠕蟲樣小體的組織球增生症 (histiocytosis with intracytoplasmic worm-like bodies)」。然而,這些小體也可見於其他形式的 N-LCH。已有與其他形式 N-LCH(尤其是 JXG)重疊的報告。良性頭部組織球增生症通常表現為小型、黃紅色或黃棕色、無症狀的丘疹,位於幼童的頭頸部,並有自發緩解的傾向。²²
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
皮膚樣本的組織學檢查顯示組織球浸潤,緊鄰表皮,並伴隨散在的淋巴細胞與嗜酸性球。組織球表現典型的巨噬細胞標記 CD68,而蘭格罕細胞標記(例如 CD1a 與 CD207)的免疫染色為陰性。²² 超微結構上存在蠕蟲樣小體是典型的。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
很難將良性頭部組織球增生症與其他形式的 N-LCH(尤其是 JXG)區分。因此,它可被討論為 JXG 的一種變異型。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
良性頭部組織球增生症通常自發消退。治療選擇包括 CO2 雷射治療。
2034
兒童期廣泛性發疹性組織球瘤 (GENERALIZED ERUPTIVE HISTIOCYTOMA OF CHILDHOOD)
臨床特徵 (CLINICAL FEATURES)
廣泛性發疹性組織球瘤 (Generalized eruptive histiocytoma, GEH)(也稱為「發疹性組織球瘤 eruptive histiocytoma」或「廣泛性發疹性組織球增生症 generalized eruptive histiocytosis」)最初於 1963 年由 Winkelmann 與 Muller 報告。這種罕見疾病的特徵為廣泛分布、紅斑性、基本上對稱的丘疹,特別侵犯近端肢體以及軀幹。與 JXG 不同,棕色丘疹與結節不會發展出來;然而,黏膜可能受侵。皮膚病灶自發消退,留下色素沉著斑 (hyperpigmented maculae)。也有一篇報告將 GEH 與風濕熱 (rheumatic fever) 相關聯。GEH 可被視為 JXG 的一種變異型。
成人黃色肉芽腫 (ADULT XANTHOGRANULOMA)
臨床特徵 (CLINICAL FEATURES)
成人黃色肉芽腫 (Adult xanthogranuloma) 最初於 1963 年由 Gartmann 與 Tritsch 描述。單發以及寡病灶的黃橙色丘疹通常出現於顏面、頸部與下臂。與 JXG 相反,成人黃色肉芽腫不顯示自發緩解。尚未有與全身性疾病(例如神經纖維瘤病或白血病)相關的報告。本病成人型與幼年型的組織學發現幾乎相同。通常在成人黃色肉芽腫中有較多的巨細胞。治療選擇包括病灶切除或 CO2 雷射治療。
丘疹性黃色瘤 (PAPULAR XANTHOMA)
臨床特徵 (CLINICAL FEATURES)
丘疹性黃色瘤 (Papular xanthoma) 於 1981 年由 Winkelmann 首次報告。它是發生於血脂正常 (normolipemic) 病人的罕見疾病。它主要表現為單發的黃色丘疹,且似乎較常出現於男性。它在年輕青少年與中年人中顯示出雙相發生 (biphasic occurrence)。曾有先天型 (congenital form) 的報告。某些病例中黏膜受侵,但無全身侵犯。丘疹性黃色瘤的斑塊樣形式 (plaquelike form) 可被視為一種變異型。²³,²⁴
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
組織學顯示正常表皮,以及由眾多 Touton 型巨細胞 (Touton-type giant cells) 散布其間的黃色瘤化巨噬細胞 (xanthomatized macrophages) 的緻密浸潤。免疫組織化學上,單核與多核巨噬細胞對 KiM1p 呈陽性。只有巨細胞對 CD68 呈陽性。高達 50% 的黃色瘤化細胞對凝集素花生凝集素 (peanut agglutinin) 呈陽性。factor XIIIa 與 CD1a 的染色為陰性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括黃色瘤、粉瘤 (atheroma)、蟹足腫 (keloid)、組織球瘤 (histiocytoma)、Spitz 痣、透明細胞棘皮瘤 (clear cell acanthoma),以及其他良性與惡性皮膚腫瘤。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
丘疹性黃色瘤通常自發消退。治療選擇包括手術切除與 CO2 雷射治療。²²,²³
廣泛性發疹性組織球瘤 (GENERALIZED ERUPTIVE HISTIOCYTOMA)
臨床特徵 (CLINICAL FEATURES)
GEH 於 1963 年由 Winkelmann 與 Muller 首次描述。全世界有少於 50 篇病例報告,且主要侵犯成人。²⁵ GEH 的特徵為多發、無症狀且對稱分布的棕色紅斑性丘疹,侵犯軸向區域——軀幹、顏面與近端肢體——並經常演變為發作 (flares)。大屈側 (big flexures) 不受侵犯(圖 117-16)。曾報告某些病例中黏膜受侵。²⁶ 雖然本病通常呈良性臨床病程,但有 2 篇報告指出一種與急性單核球性白血病 (acute monocytic leukemia) 相關的非典型形式。
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
皮膚切片顯示正常表皮。乳突真皮被大多為小型、非脂質化的細胞與少數淋巴細胞浸潤,其下方有一 Grenz 帶 (Grenz zone),這是 GEH 的特徵(圖 117-17)。缺乏巨細胞。巨噬細胞對 CD68 與 stabilin-1 呈陽性,對 CD1a、CD34 與 S100B 呈陰性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括 LCH、其他形式的 N-LCH 與色素性蕁麻疹。其他 N-LCH 容易藉由組織學區分。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
GEH 通常自發消退。治療選擇包括手術切除與 CO2 雷射
20
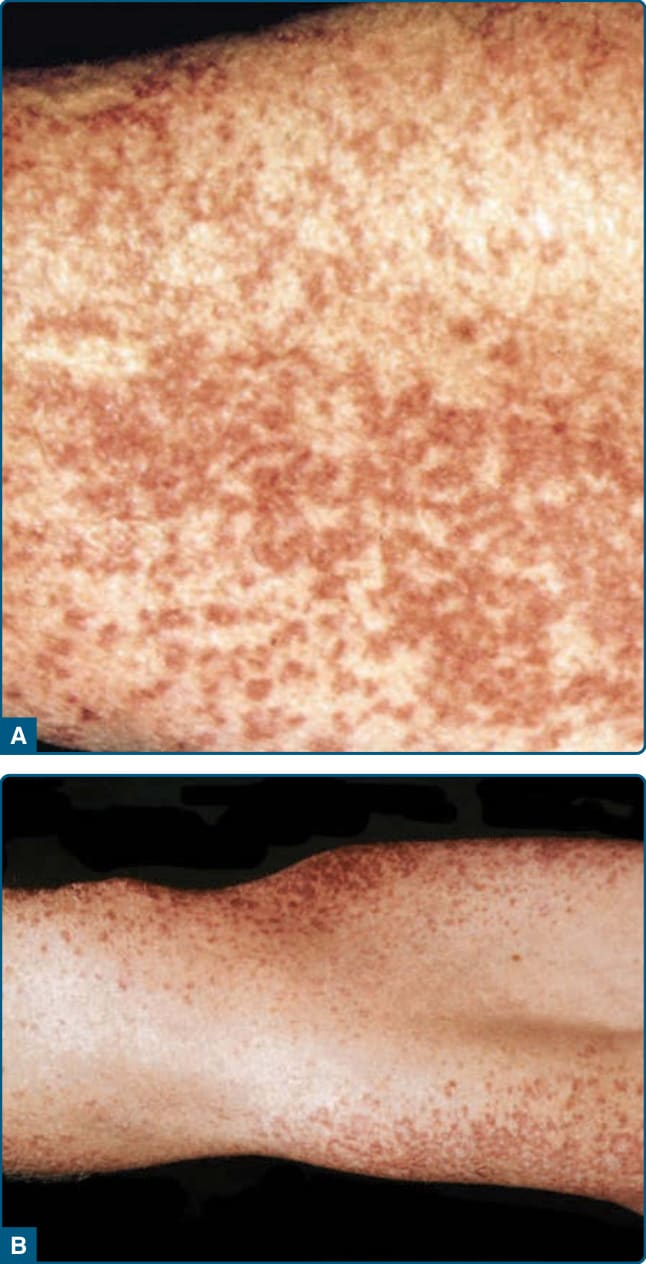
圖 117-16:廣泛性發疹性組織球瘤 (Generalized eruptive histiocytoma)。注意大屈側不受侵犯,如圖中 B 部分所示。
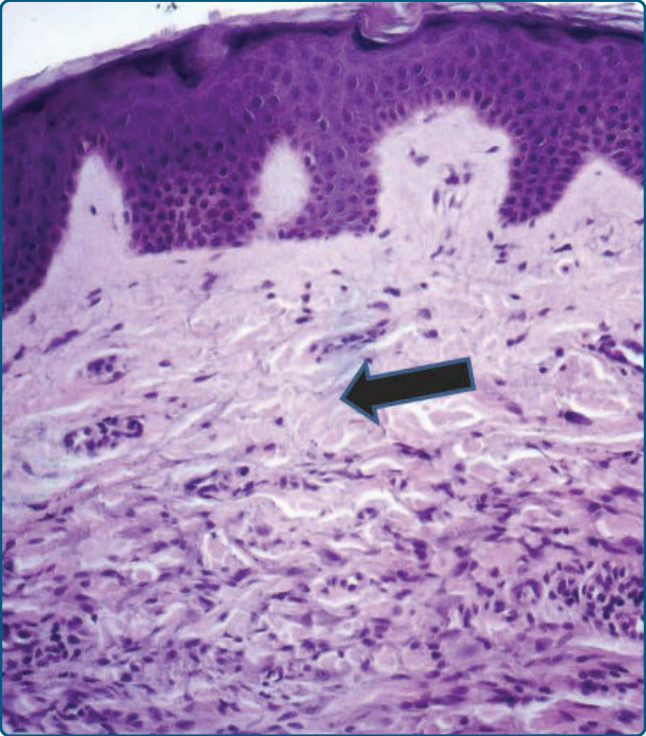
圖 117-17:廣泛性發疹性組織球瘤病灶的顯微照片。在乳突真皮中,可見大多為小型、非脂質化細胞與少數淋巴細胞的浸潤,其下方有一 Grenz 帶(箭頭)。這是廣泛性發疹性組織球瘤的典型徵象。缺乏巨細胞。
治療。據報告,全身性補骨脂素與紫外線 A 治療 (systemic psoralen and ultraviolet A therapy) 可使皮膚病灶迅速消退。鑑於可能發展急性單核球性白血病,建議對 GEH 病人進行密切追蹤。²⁵,²⁶
播散性黃色瘤 (XANTHOMA DISSEMINATUM)
臨床特徵 (CLINICAL FEATURES)
播散性黃色瘤 (Xanthoma disseminatum) 於 1938 年由 Montgomery 描述。它好發於男童。本病的特徵為突然出現小型、黃紅色至棕色的丘疹與結節,呈散在性並播散分布,好發於屈側與間擦區域以及黏膜
2035
20
(圖 117-18)。黏膜與多種內臟器官的侵犯可導致顯著的病態與死亡。全身性關聯包括因腦下垂體柄 (pituitary stalk) 侵犯所致的中樞性尿崩症 (central diabetes insipidus),以及副蛋白血症 (paraproteinemias),例如多發性骨髓瘤 (multiple myeloma)。²⁷
播散性黃色瘤的變異型包括播散性黃色含鐵血黃素組織球增生症 (disseminated xanthosiderohistiocytosis),於 1960 年由 Halprin 與 Lorincz 描述,並與多發性骨髓瘤相關。1998 年,Ferrando 描述了一例
2036
全身性黃色組織球瘤 (systemic xanthohistiocytoma),也可被視為播散性黃色瘤的一種變異型。
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
播散性黃色瘤早期病灶的組織病理學檢查顯示以扇貝狀組織球 (scalloped histiocytes) 為主,而較成熟的病灶主要由泡沫狀組織球與少數扇貝狀細胞組成。在最成熟或成熟期的病灶中,可見扇貝狀細胞、泡沫細胞、淋巴細胞與 Touton 巨細胞的混合。這些細胞對 stabilin-1、HAM-56、HHF35、KP1、KiM1P、factor XIIIa 與 vimentin 呈陽性,對 S100B 與 CD1a 呈陰性(圖 117-19)。²⁷
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
主要的鑑別診斷包括發疹性黃色瘤 (eruptive xanthomas)、其他 N-LCH 與 LCH。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
播散性黃色瘤是一種緩慢進行的疾病。皮膚病灶可藉由例如 CO2 雷射治療來治療。病灶對放射線不太敏感。已嘗試數種藥物,包括 statins、fibrates、glitazones、培尼皮質醇、azathioprine、cyclophosphamide 與 thalidomide,結果不一。一種似乎成功的治療方法是投予 2-氯去氧腺苷 (2-chlorodeoxyadenosine)。²⁷,²⁸
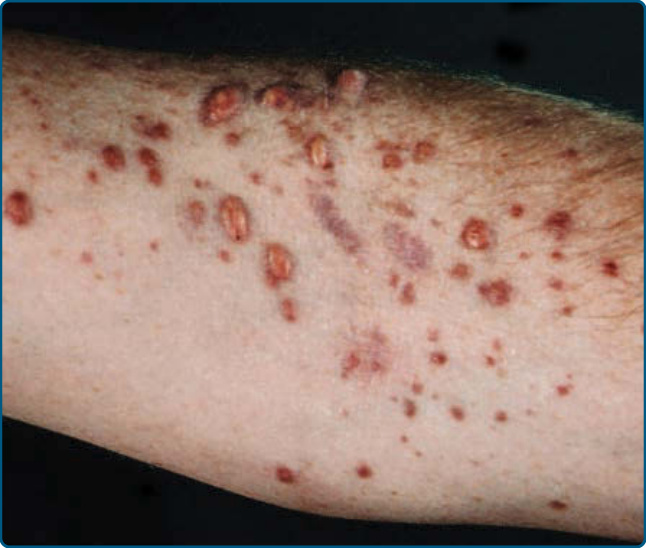
圖 117-18:播散性黃色瘤 (Xanthoma disseminatum)。
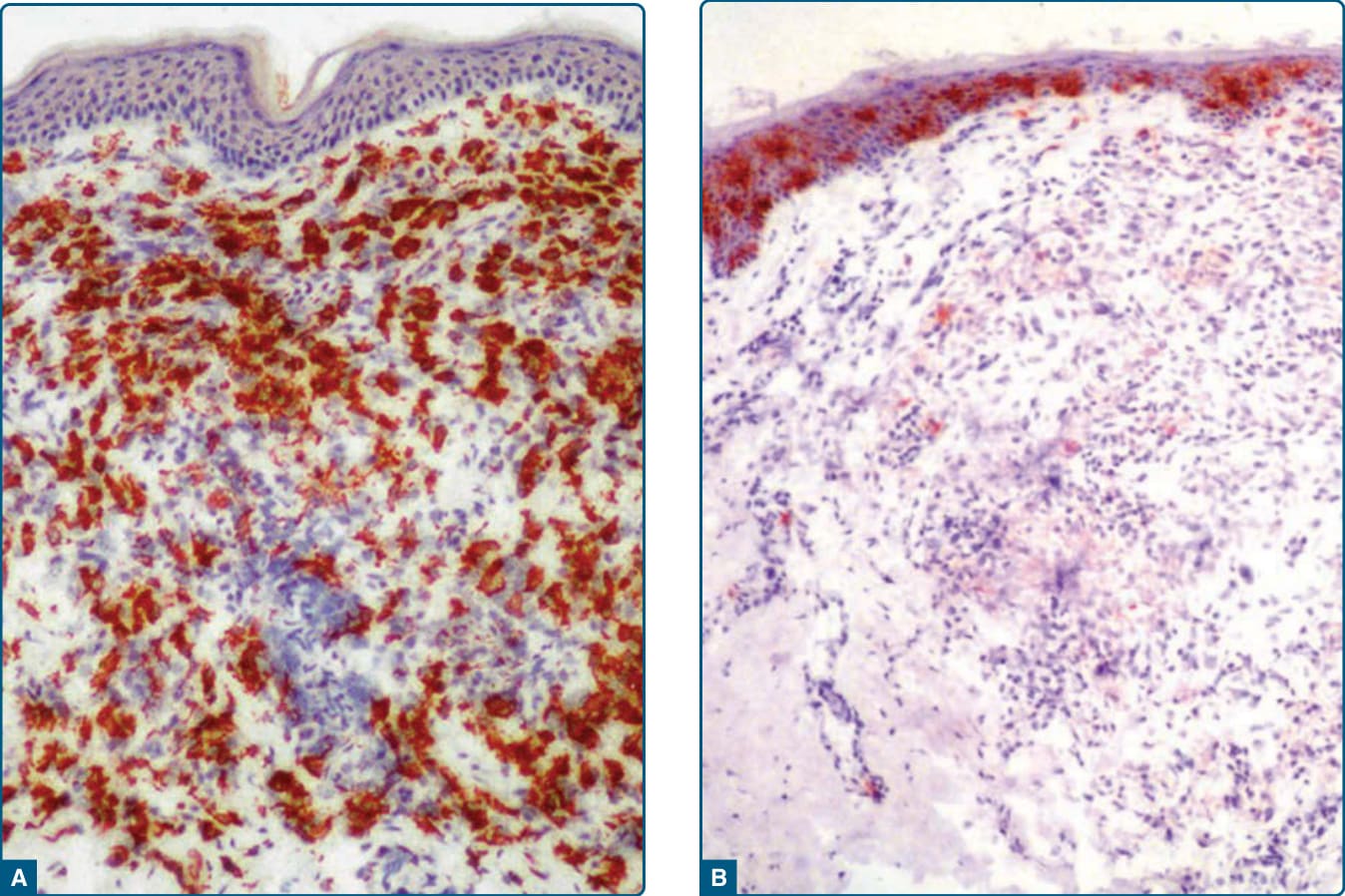
圖 117-19:播散性黃色瘤早期病灶的顯微照片。A,Stabilin-1 在病灶組織球中顯示強烈表現。B,CD1a 染色主要侷限於表皮蘭格罕細胞。
多中心網狀組織球增生症 (MULTICENTRIC RETICULOHISTIOCYTOSIS)
臨床特徵 (CLINICAL FEATURES)
多中心網狀組織球增生症 (Multicentric reticulohistiocytosis, MRH) 於 1937 年由 Weber 與 Freudenthal 首次報告。此名詞由 Goltz 與 Laymon 於 1954 年所創。它是一種罕見的多系統發炎性疾病,通常出現於 50 歲左右,但有少數於兒童期發生的報告。它在女性中的發生率為兩倍。皮膚病灶為堅實、黃棕色的丘疹或結節,可達數公分大小並緩慢增大。位於手指與手腕關節上方的病灶是典型的(圖 117-20 與 117-21)。可因顏面病灶融合而出現獅面 (leonine facies)。每兩名病人中即有一名可見黏膜與結膜的侵犯。MRH 可侵犯任何器官。然而,最常見的臨床表現為皮膚疹與對稱性發炎性多關節炎 (symmetric inflammatory polyarthritis)。已描述內臟器官受 MRH 侵犯,例如肺(導致胸膜積液 pleural effusion)與心臟(心包積液 pericardial effusion 與充血性心衰竭 congestive heart failure)。此外,已報告腸繫膜淋巴結病變 (mesenteric lymphadenopathy) 與
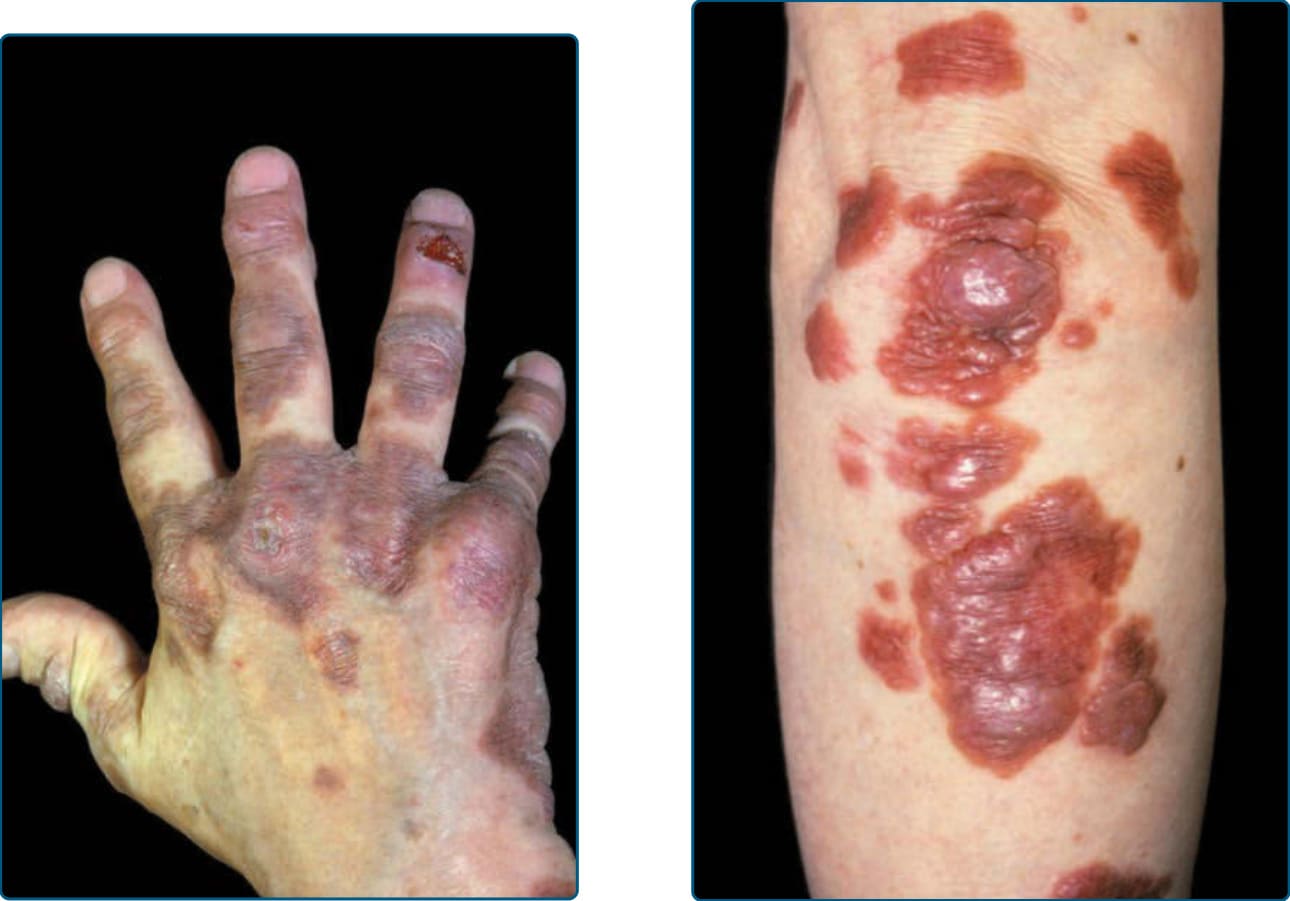
圖 117-20:多中心網狀組織球增生症 (Multicentric reticulohistiocytosis)。
20
泌尿生殖系統病灶 (urogenital lesions)。²⁹ 可觀察到全身性症狀,例如發燒、體重減輕與倦怠。³⁰ 文獻中已報告 133 例 MRH 中有 33 例(25%)的惡性腫瘤率。這些惡性腫瘤最常為血液學、乳房或胃癌。³¹
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
組織學顯示真皮中層浸潤著單核組織球與多核組織球,具有毛玻璃樣外觀 (ground-glass appearance),以及數量不等的空泡狀、紡錘狀與黃色瘤化的單核組織球。在 MRH 的情況下,免疫組織化學分析通常對 CD45、CD68 與 HLA-DR(human leukocyte antigen-D related)呈陽性,但對 S100B、CD1a 或 HHF-35 actin 呈陰性。²⁹,³⁰
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括 François 症候群(皮膚軟骨角膜失養症 dermochondrocorneal dystrophy)、Farber 病(播散性脂肪肉芽腫病 lipogranulomatosis disseminata),以及不同的代謝疾病,例如痛風 (gout)。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
本病可為自限性。然而,通常會導致嚴重的關節破壞。手術切除、脈衝染料雷射 (pulsed-dye laser)、口服皮質類固醇、甲氨蝶呤、cyclophosphamide 與托珠單抗 (tocilizumab) 在治療廣泛性 MRH 病灶上已顯示成功。一篇對 17 例以腫瘤壞死因子拮抗劑 (tumor necrosis factor antagonists) 治療之 MRH 的系統性回顧顯示,抗腫瘤壞死因子治療在 MRH 中具有療效。³⁰
Erdheim-Chester 病 (ERDHEIM-CHESTER DISEASE)
臨床特徵 (CLINICAL FEATURES)
本病的首次報告由 Chester 於 1930 年與 Erdheim 合作完成。1972 年,Jaffe 報告了一個類似的病例,並將本病命名為 Erdheim-Chester 病。Erdheim-Chester 病是一種罕見疾病,發病於中年。長骨侵犯在 Erdheim-Chester 病病人中幾乎是普遍性的,且本質上為雙側性與對稱性。它涵蓋一個疾病譜,範圍從無症狀的骨病灶到多系統、危及生命的變異型。超過 50% 的病例有某種形式的骨骼外侵犯。這可包括腎臟、皮膚、腦與肺侵犯;較少見地,可觀察到眼眶後組織 (retro-orbital tissue)、腦下垂體與心臟侵犯。³²,³³
在皮膚上,六分之一的病例存在黃斑瘤與黃色瘤。某些病人已描述黃棕色丘疹性與廣泛浸潤性的病灶。較新的研究證明,約 50% 的 Erdheim-Chester 病病人在早期多潛能骨髓單核細胞前驅物 (early multipotent myelomonocytic precursors) 或組織常駐組織球 (tissue-resident histiocytes) 中有 BRAF 突變。罕見地,MAPK 訊號途徑中涉及的其他基因也會突變。³²
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
組織樣本顯示由富含脂質或泡沫狀組織球進行的黃色瘤性或黃色肉芽腫性浸潤,這些組織球通常被纖維化所包圍,尤其在較長期的病灶中。骨切片顯示富含脂質的巨噬細胞、多核巨細胞、淋巴細胞的發炎性浸潤與組織球的浸潤,以及長骨的廣泛性硬化 (generalized sclerosis)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷主要包括 LCH。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
干擾素-α (Interferon-α) 是 Erdheim-Chester 病的一線治療,因為已明確證明它能提高整體存活率。Anakinra 與 infliximab 也帶來令人鼓舞的結果,當干擾素-α 治療失敗時應予考慮。最近,BRAF 抑制劑已被證明在所有接受治療的病例中皆有效。³³
壞死性黃色肉芽腫 (NECROBIOTIC XANTHOGRANULOMA)
臨床特徵 (CLINICAL FEATURES)
壞死性黃色肉芽腫 (Necrobiotic xanthogranuloma) 於 1980 年由 Kossard 與 Winkelmann 首次報告。然而,伴副蛋白血症的壞死合併黃色肉芽腫早在 1960 年代就已被觀察到。它是一種侵犯較年長成人的多系統疾病,表現為可能潰瘍的黃色斑塊與結節。壞死性黃色肉芽腫主要位於軀幹、四肢與顏面(眼眶周圍病灶 periorbital lesions)。也曾描述皮膚外侵犯,例如眼、心臟、骨骼肌、喉、脾與卵巢。在 80% 的壞死性黃色肉芽腫病人中可見一種全身性關聯,形式為通常為免疫球蛋白 G κ 與 λ 型的血清單株丙種球蛋白病 (serum monoclonal gammopathy)。並發血液學與淋巴增生性惡性腫瘤的風險增加。³⁴
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
壞死性黃色肉芽腫的特徵為典型的壞死區域 (necrobiotic areas),周圍環繞著肉芽腫
由淋巴細胞、Touton 巨細胞、泡沫狀組織球與異物型多核巨細胞組成,分布於真皮以及皮下組織。與其他 N-LCH 一樣,無 CD1a 抗原或 CD207 的表現。無伯貝克顆粒。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
類脂質壞死症 (Necrobiosis lipoidica)、皮下環狀肉芽腫 (subcutaneous granuloma annulare) 與類風濕結節 (noduli rheumatica) 可被視為鑑別診斷。在只有輕度壞死的病例中,應考慮與副蛋白血症及淋巴瘤相關的黃色瘤。在有眼眶周圍皮膚病灶的病例中,黃斑瘤 (xanthelasma) 可為鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
預後不良,數種治療顯示出不一的結果。以烷化劑治療或不治療單株丙種球蛋白病不一定會影響皮膚疾病的活性。沒有隨機對照試驗,也不存在長期結局的研究。治療選擇主要在病例報告中描述,包括局部與全身性皮質類固醇、thalidomide、高劑量靜脈注射免疫球蛋白、chlorambucil、cyclophosphamide、fludarabine、rituximab、melphalan、infliximab、干擾素-α、cladribine、hydroxychloroquine、azathioprine 與 methotrexate。此外,也描述了雷射治療、放射治療、手術、補骨脂素與紫外線 A、血漿置換術 (plasmapheresis) 與體外光分離術 (extracorporeal photopheresis)(在 Miguel 等人的系統性回顧中³⁵)。
遺傳性進行性黏液性組織球增生症 (HEREDITARY PROGRESSIVE MUCINOUS HISTIOCYTOSIS)
臨床特徵 (CLINICAL FEATURES)
遺傳性進行性黏液性組織球增生症 (Hereditary progressive mucinous histiocytosis) 於 1988 年由 Bork 與 Hoede 描述。它是一種罕見、可能為體染色體顯性遺傳 (autosomal dominant inherited) 的疾病。自我消退的膚色至紅棕色丘疹的進行性發疹,通常在十歲前發生於鼻部、手部、前臂與大腿(圖 117-22)。這些病灶之後可發展為持續且進行性的紅斑性丘疹。尚未有內臟侵犯的報告。
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
在早期病灶的上真皮中可見上皮樣 (epithelioid) S100B/CD1a− 與 CD68 以及弱 stabilin-1+ 的組織球聚集,以及毛細血管擴張性血管 (telangiectatic vessels)。在早期與發展良好病灶的真皮中層,存在緊密排列之紡錘狀細胞的結節性聚集。在上皮樣組織球與紡錘狀細胞中證明有中度至大量的黏液產生 (mucin production)(圖 117-23)。³⁶
20
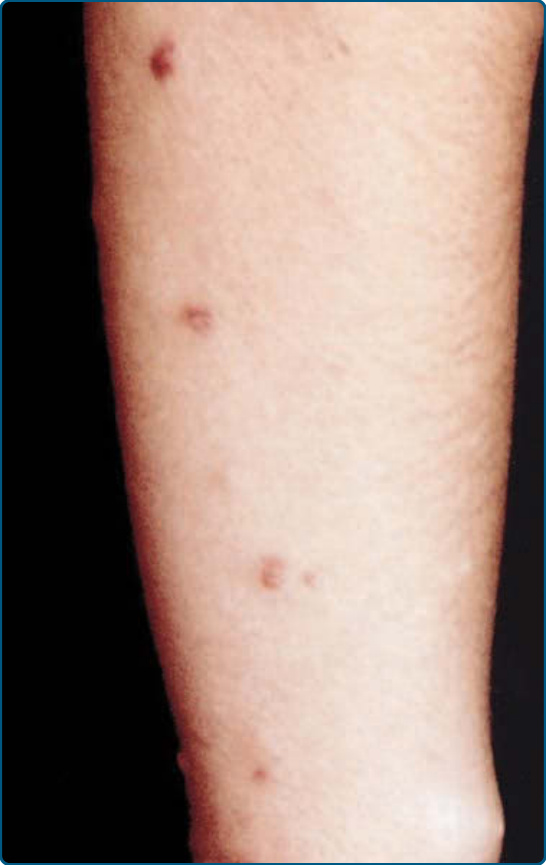
圖 117-22:遺傳性進行性黏液性組織球增生症 (Hereditary progressive mucinous histiocytosis)。
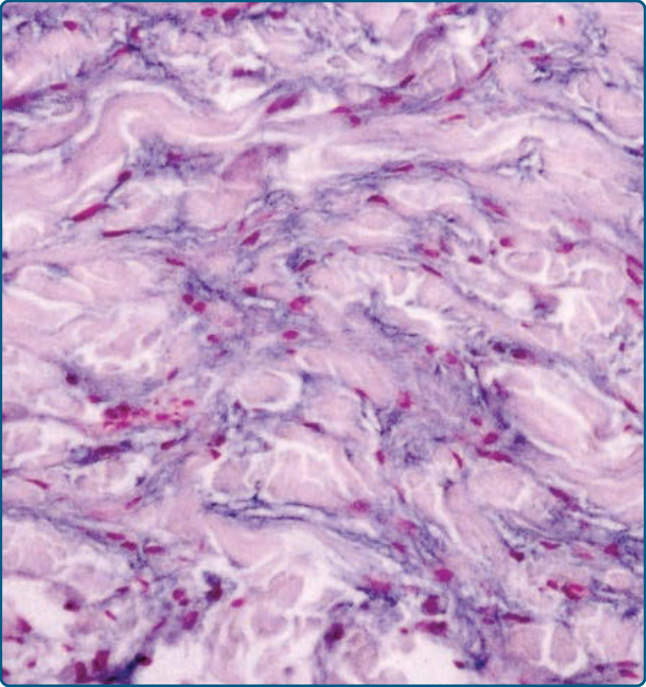
圖 117-23:遺傳性進行性黏液性組織球增生症病灶的顯微照片。真皮中組織球產生黏液是本病的典型特徵。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
肢端持續性丘疹性黏蛋白沉積症 (Acral persistent papular mucinosis)、硬化性黏液水腫 (scleromyxedema)、環狀肉芽腫、皮膚纖維瘤與 GEH 可藉由組織學區分。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
對單一病灶進行消融性雷射治療 (Ablative laser therapy) 是一種選擇。
進行性結節性組織球增生症 (PROGRESSIVE NODULAR HISTIOCYTOSIS)
臨床特徵 (CLINICAL FEATURES)
進行性結節性組織球增生症 (Progressive nodular histiocytosis) 於 1978 年由 Taunton 及其同事描述。本病的特徵為廣泛性、散在性的黃色至紅棕色丘疹與結節,大小數公分。已描述顯著的顏面侵犯。其他器官不受侵犯。它與慢性骨髓性白血病 (chronic myeloid leukemia) 以及下視丘腫瘤 (tumors of the hypothalamus) 相關。³⁷
組織病理學發現 (HISTOPATHOLOGIC FINDINGS)
真皮含有大量紡錘狀組織球,有些具有泡沫狀細胞質。細胞對 CD68、CD163、vimentin 與 fascin 呈陽性,而對 CD1a 與 S100B 呈陰性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
MRH 是鑑別診斷之一,它通常顯示出其他器官部位的侵犯。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
以例如 CO2 雷射對所有可見病灶進行手術消融 (Surgical ablation) 是一種治療選擇。
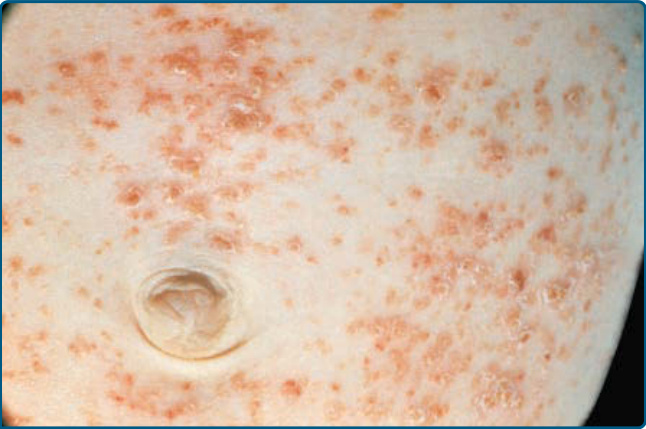
圖 117-3:一名患有廣泛性蘭格罕細胞組織球增生症之嬰兒腹部區域的玫瑰黃色丘疹。
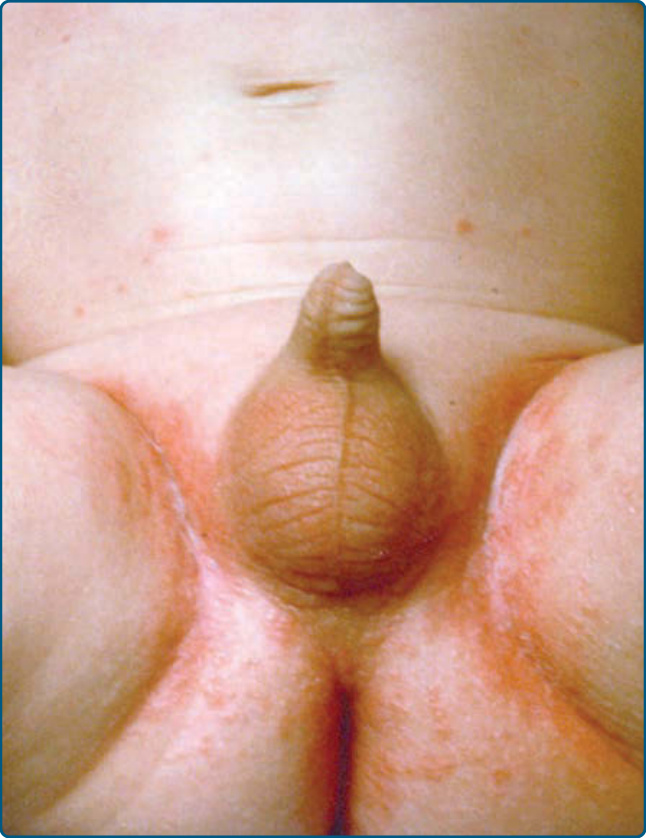
圖 117-4:一名蘭格罕細胞組織球增生症嬰兒生殖器區域的「念珠菌間擦疹 (candida intertrigo)」樣疹。
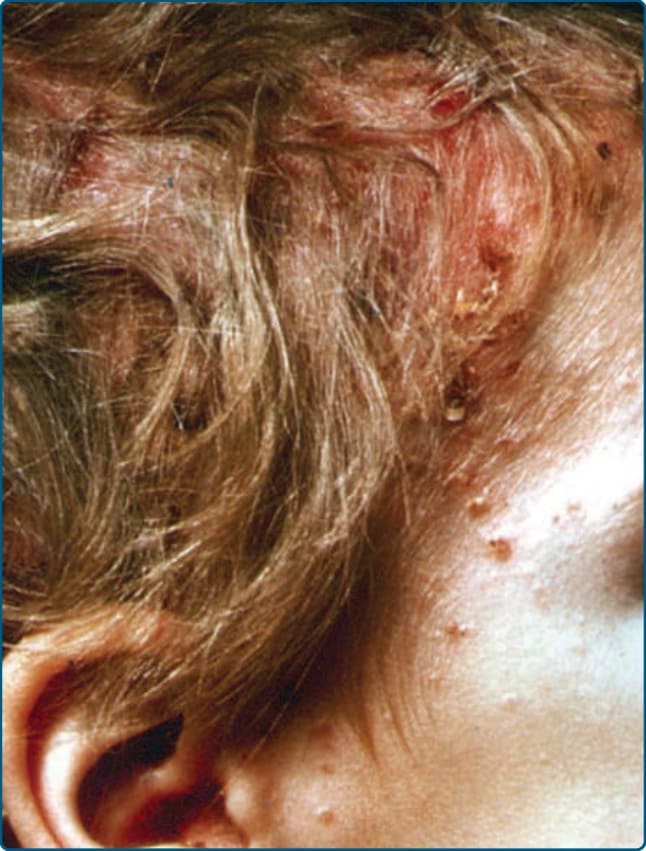
圖 117-5:一名蘭格罕細胞組織球增生症嬰兒頭皮上的「脂漏性皮膚炎 (seborrheic dermatitis)」樣疹。
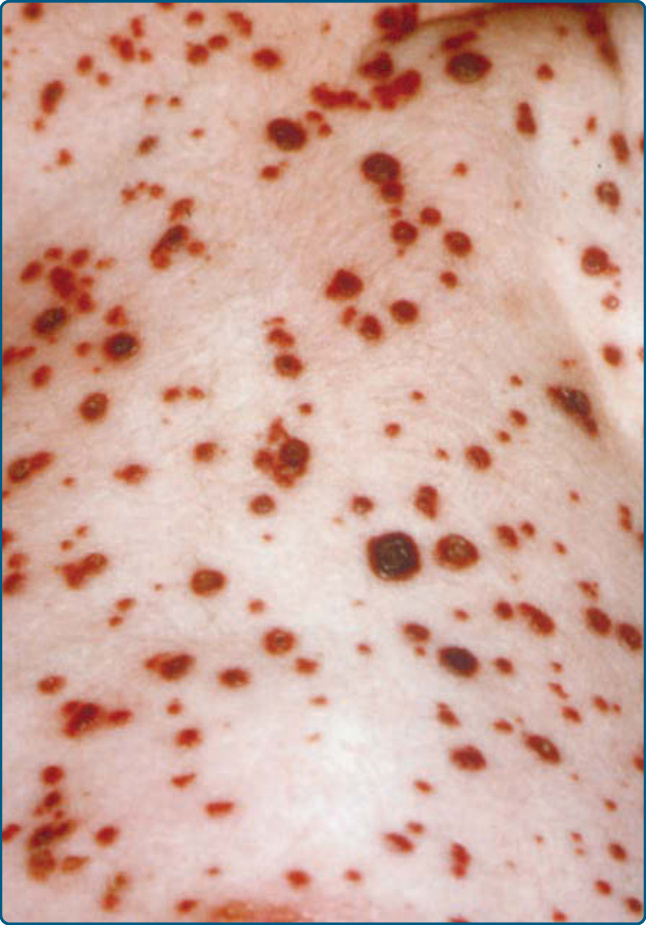
圖 117-6:一名患有蘭格罕細胞組織球增生症之病人腹部區域的出血性丘疹與結節。
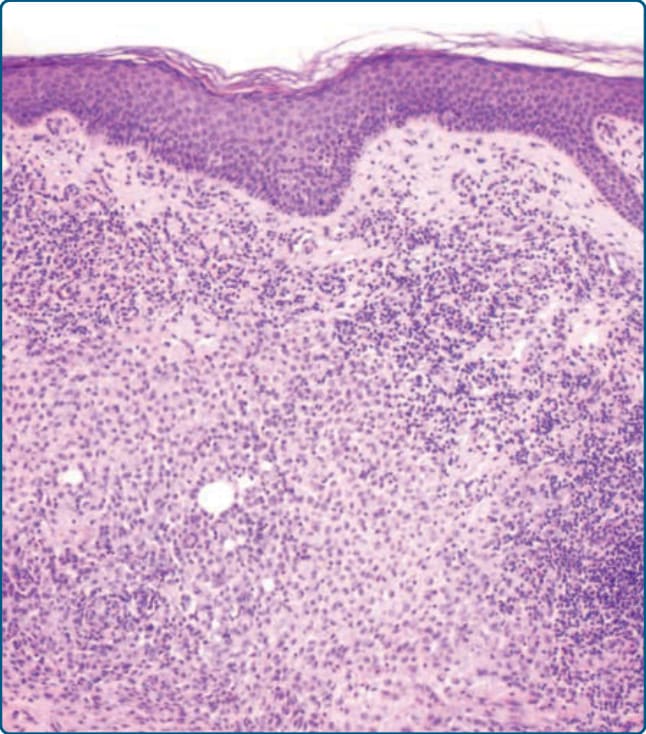
圖 117-7:蘭格罕細胞組織球增生症 (LCH) 的組織學,顯示真皮的緻密單核細胞浸潤,含卵圓形至圓形的 LCH 細胞與淋巴細胞,以及一些嗜酸性球,並有表皮侵犯。
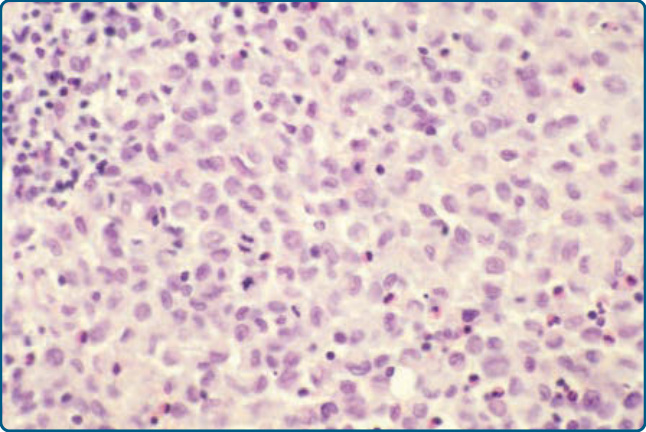
圖 117-8:蘭格罕細胞組織球增生症 (LCH) 的組織學。LCH 細胞具有腎形細胞核。
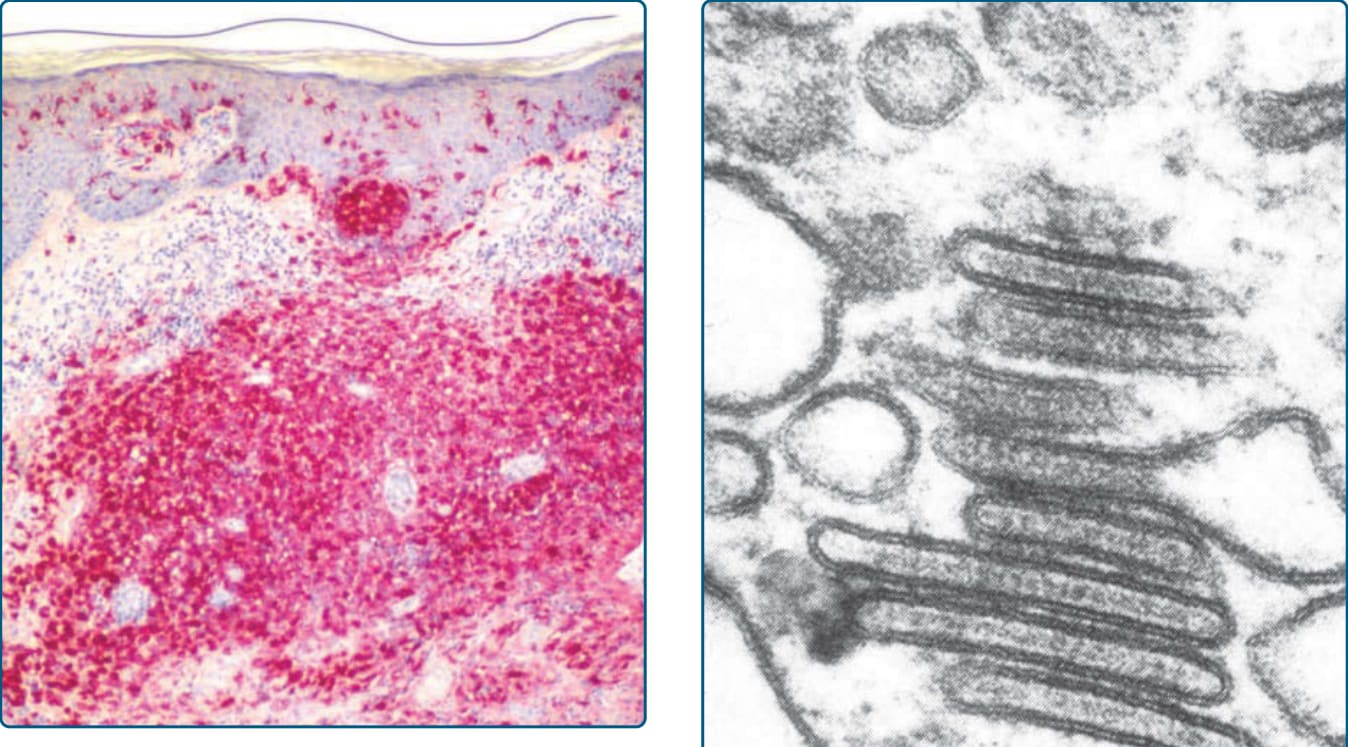
圖 117-9:蘭格罕細胞組織球增生症 (LCH) 的組織學。LCH 細胞對 CD1a 染色(紅色)並顯示明顯的表皮趨向性。
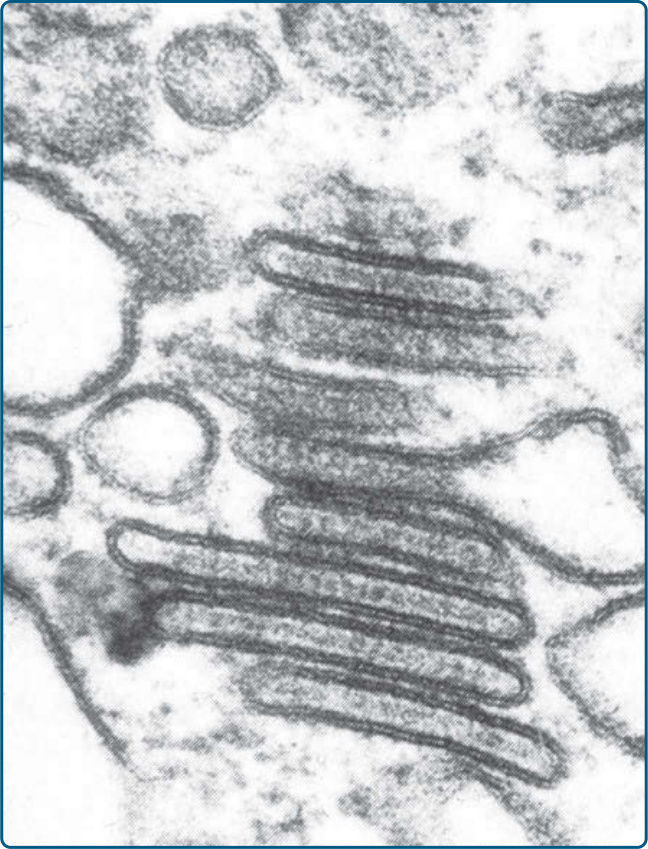
圖 117-10:蘭格罕細胞組織球增生症細胞的電子顯微鏡圖片顯示典型的伯貝克顆粒 (Birbeck granules)。