Histiocytosis
20
CUTANEOUS CLINICAL FEATURES OF HISTIOCYTOSIS
AT-A-GLANCE
■ Langerhans cell histiocytosis: Translucent, roseyellowish, crusted papules or papulovesicles, eczematous lesions, hemorrhagic papules and nodules, petechiae, noduloulcerative mucosal lesions, and nail involvement
■ Rosai-Dorfman disease: Lymphadenopathy, cutaneous nodules, and plaques
■ Hemophagocytic lymphohistiocytosis: Various cutaneous manifestations such as erythroderma, generalized purpuric macules and papules, and morbilliform eruptions
■ Juvenile xanthogranuloma: Solitary as well as multiple (oligolesional) papules or nodules—early lesions show a reddish-brown color; mature lesions have a reddish-yellow appearance
■ Benign cephalic histiocytosis: Like juvenile xanthogranuloma; ultrastructural presence of wormlike bodies
■ Generalized eruptive histiocytoma of childhood: Widespread, erythematous, essentially symmetrical papules
■ Adult xanthogranuloma: Oligolesional, yellow-orange papules that usually appear on the face, neck, and lower arms
■ Papular xanthoma: Solitary yellowish papule
■ Generalized eruptive histiocytoma: Multiple asymptomatic and symmetrically distributed
Langerhans cell histiocytoses (LCHs) and non– Langerhans cell histiocytoses (N-LCHs) present a rare group of neoplastic and reactive diseases characterized by the proliferation of myeloid cells in
brownish erythematous papules, particularly involving the axial regions such as the trunk, face, and proximal extremities, frequently flares
■ Xanthoma disseminatum: Small, yellow-red to brown papules and nodules that are discrete and are disseminated with a predilection for the flexural and intertriginous areas, as well as for mucous membranes
■ Multicentric reticulohistiocytosis: Firm, yellowbrownish papules or nodules that reach a size of several centimeters and progress slowly in size; lesions over the joints of fingers and wrists are typical; often involvement of mucosae and conjunctivae
■ Erdheim-Chester disease: Xanthelasma and xanthoma are present in one-sixth of cases; long bone and other extracutaneous involvement
■ Necrobiotic xanthogranuloma: Yellowish plaques and nodules that can ulcerate
■ Hereditary progressive mucinous histiocytosis: Skincolored to red-brown papules that usually develop in the first decade on the nose, hands, forearms, and thighs that can later develop into persistent and progressive erythematous papules
■ Progressive nodular histiocytosis: Generalized, discrete yellow to red-brown papules and nodules measuring a few centimeters in size with prominent facial involvement
various organ sites with a predilection to the skin. As shown in Table 117-1, they are categorized in different subgroups.
Langerhans Cell Histiocytosis
- Single system
- Multisystem
Non–Langerhans Cell Histiocytosis
- Systemic non–Langerhans cell histiocytosis
Non–Langerhans Cell Histiocytosis
- Systemic non–Langerhans cell histiocytosis
■Rosai-Dorfman disease
■Rosai-Dorfman disease
■Hemophagocytic lymphohistiocytosis
2. Cutaneous non–Langerhans cell histiocytosis
■Hemophagocytic lymphohistiocytosis
2. Cutaneous non–Langerhans cell histiocytosis
■Juvenile xanthogranuloma
■Juvenile xanthogranuloma
■Juvenile xanthogranuloma
■Juvenile xanthogranuloma
■Benign cephalic histiocytosis
■Benign cephalic histiocytosis
■Generalized eruptive histiocytoma of childhood
■Generalized eruptive histiocytoma of childhood
■Adult xanthogranuloma
■Adult xanthogranuloma
■Oligolesional
■Oligolesional
■Adult xanthogranuloma
■Adult xanthogranuloma
■Papular xanthoma
■Papular xanthoma
■Disseminated
■Disseminated
■Generalized eruptive histiocytoma
■Generalized eruptive histiocytoma
■Xanthoma disseminatum
■Xanthoma disseminatum
■Multicentric reticulohistiocytosis
■Multicentric reticulohistiocytosis
■Erdheim-Chester disease
■Erdheim-Chester disease
■Necrobiotic xanthogranuloma
■Necrobiotic xanthogranuloma
■Spindle-cell non–Langerhans cell histiocytosis
■Spindle-cell non–Langerhans cell histiocytosis
■Hereditary progressive mucinous histiocytosis
■Hereditary progressive mucinous histiocytosis
■Progressive nodular histiocytosis
■Progressive nodular histiocytosis
LANGERHANS CELL HISTIOCYTOSIS
AT-A-GLANCE
■ New definition of Langerhans cell histiocytosis (LCH) as rare, inflammatory neoplasia of myeloiddendritic (mostly clonal) cells with heterogeneous clinical manifestation
■ Histopathology:
■ Infiltration of the organs with immature, morphologically rounded myeloid dendritic cells with bean-shaped nuclei; shared characteristics of cell surface antigens (CD1a/ S100B/CD207-positive) with mature skin Langerhans cells
■ 60% of LCH biopsy specimens bear the V600E mutation in cell-growth-directing oncogene BRAF (v-Raf murine sarcoma viral oncogene homolog B)
■ Highly variable clinical manifestations ranging from single, osteolytic bone lesions to an aggressive, life-threatening multisystem disease affecting risk organs such as the spleen, the liver, and the hematopoietic system
■ Skin involvement in 39% (crucial for early diagnosis)
20
■ New (clinical) classification according to the extent of organ involvement:
■ Single-organ system LCH
■ Multisystem LCH
■ Treatment:
■ Close monitoring for single-organ system LCH (“watch and wait”)
■ Topical or systemic treatments include topical nitrogen mustard, narrowband ultraviolet light-B, photochemotherapy, methotrexate, 6-mercaptopurine, and chemotherapy, depending on the extent of the disease
HISTORICAL PERSPECTIVE
HISTORICAL PERSPECTIVE
LCH is newly defined as a rare, heterogeneous neoplasm of dendritic cells. Characteristically, CD1a/ S100B/CD207-positive mononuclear cells with beanshaped nuclei infiltrate single-organ systems, most commonly the bone, but also the skin, or multipleorgan systems. A long-term controversy regarding the neoplastic nature of this disease exists as there are some arguments in support of a reactive condition resulting from an immunologic dysregulation such as a regulatory T-cell expansion found in LCH and the lack of mutations in tumor-suppressor genes.1 Another argument is the broad spectrum of clinical manifestations of LCH, ranging from a self-healing single bone lesion to aggressive multiorgan involvement with a lethal outcome. Thus, in the past, patients were divided into 4 main clinical types: Hashimoto-Pritzker disease, eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease (Table 117-2 outlines the traditional classification of LCH). Hashimoto-Pritzker disease, first described in 1973, is the benign clinical variant typically presenting with multiple firm red-brown nodules with an elevated border or papulovesicular and papulocrusted lesions mostly on the scalp and face in the first few months and years of life, with no signs of systemic involvement. These lesions usually heal within 2 to 3 months and occasionally leave whitish atrophic scars. Eosinophilic granuloma is also a mainly benign isolated or multifocal osteolytic bone LCH, which sometimes affects the skin and mucous membranes. Consequently, skin lesions often resemble those seen in Hand-Schüller-Christian (HSC) disease with nodulo-ulcerative lesions in the mouth and yellowishbrown papules and plaques on the scalp or in the perineal or perivulvar region. The first publication by Thomas Smith describing this disease can be found in the 1865 edition of the American Journal of Pathology. HSC disease is the chronic variant of a multisystem LCH mostly found in children 2 to 5 years old, but the age-range is wide. It was first described by Alfred Hand in 1893, followed by Arthur Schüller in 1915
2019
20
AGE AT ONSET CLINICAL CHARACTERISTICS COURSE OF DISEASE PROGNOSIS
Hashimoto-Pritzker disease First months to years of age Multiple firm, red-brown nodules with an elevated border; papulovesicular and crusted lesions on scalp and face
Self-healing with resolution in 2-3 months and occasional atrophic scarring
No systemic involvement; excellent prognosis
Most commonly childhood Skin not always involved; when present, lesions are nodulo-ulcerative on mouth, yellowish-brown papules and plaques on scalp or perineal or perivulvar region; swelling over bony lesions
Eosinophilic granulomab (unifocal LCH)
Benign isolated or multifocal osteolytic bone LCH
No systemic involvement; good prognosis; may have complications with tissue destruction
Hand-Schüller- Christian diseaseb 2-5 years old (age range is wide) Triad of diabetes insipidus, bone lesions, and exophthalmos; seborrheic dermatitislike lesions, with small reddish-brown crusted papules, vesicles, and pustules on scalp, face, and intertriginous areas Nodulo-ulcerative lesions may affect the gingiva, perioral, and genital area; xanthomatous plaques may be present
Letterer-Siwe diseaseb Mostly younger than 2 years of age
Letterer-Siwe diseaseb Mostly younger
Chronic, progressive, multifocal form of LCH with systemic involvement
Progressive; prognosis poorer at extremes of age and with extrapulmonary involvement
Lung, bone marrow, and brain commonly
Most aggressive,
Lethal, if untreated
Lung, bone marrow, and brain commonly affected; cutaneous lesions consist of yellow-brown to translucent papules, occasionally with a hemorrhagic or ulcerative aspect; purpura is a poor prognostic sign
than 2 years of age
Most aggressive, multisystem variant Lethal, if untreated
affected; cutaneous lesions consist of yellow-brown to translucent papules, occasionally with a hemorrhagic or ulcerative aspect; purpura is a poor prognostic sign
multisystem variant
aThis table represents the traditional classification of LCH for reader reference; newer classifications (see Fig. 117-1) have greater implications for treatment and prognosis.
bThese variants fall under a single nosologic entity called histiocytosis X.
and Henry Christian in 1920. It is characterized by the triad of diabetes insipidus, bone lesions, and exophthalmos and affects the skin in approximately 30% of cases. Patients frequently complain about a chronic otitis media that is the result of an involvement of the mastoid or the petrous temporal bones. Cutaneous lesions can resemble seborrheic dermatitis with small reddish-brown crusted papules, but also vesicles and pustules on the scalp and face and in intertriginous areas. Also, nodulo-ulcerative lesions can develop in mucous membranes, especially in the gingiva and the perioral and genital areas. Xanthomatous yellowishbrown or yellowish-red papules and plaques also have been described. Letterer-Siwe disease is the most aggressive acute LCH multisystem variant with apparent systemic symptoms such as fever, hepatosplenomegaly, polylymphadenopathy, anemia, arthralgia, malaise, and weight loss. Letterer first described the disease in 1924, followed by Siwe in 1933. Frequently affected organs are the lung, the bone marrow, and the brain. Skin lesions are common and consist of yellow-brown, sometimes translucent papules, which can have a hemorrhagic aspect and ulcerate similar to the ones seen in HSC disease. Also, purpura is frequently found and is a poor prognostic sign. Untreated, Letterer-Siwe disease is lethal. Although these LCH types show a distinct clinical course, the cellular infiltrate in the affected organs is the same. Thus, in 1953, Lichtenstein proposed to
2020
combine eosinophilic granuloma, HSC, and Letterer- Siwe disease into a single nosologic entity called histiocytosis X. In 1973, Nezelof termed the lesional cell as a “Langerhans-like” cell based on the presence of Birbeck granules found with the help of the electron microscope. In 1987, the Histiocyte Society published the classification of the histiocytic disorders, which finally consolidated the strenghtened position that all the above-mentioned diseases are part of a single entity. Since then there has been a vivid dispute about whether LCH should be classified as a neoplastic or a reactive disease. This was finally resolved in 2010 with the discovery of the BRAF-V600E somatic point mutation in approximately 60% of patients with LCH,2
which led to the conclusion that LCH is a clonal neoplastic disorder. The resolution of this controversy is important as it will significantly accelerate research in this field and progress in treatment.
EPIDEMIOLOGY
EPIDEMIOLOGY
Because LCH is a rare neoplastic disease, exact epidemiologic data is missing. In children younger than 15 years of age, the annual incidence is approximately 0.7 to 4.1 cases per 1 million population with a median age at diagnosis of 30.2 months to 5.9 years.3
In adults, the incidence is lower with only 1 to 2 cases per 1 million adults of all ethnicities per year reported.
A lower incidence has been observed in black patients, whereas the Hispanic population shows a slightly higher incidence. Other risk factors include living in crowded conditions and lower education level as well as exposure to metal, granite, wood dust, or solvents in parents, a family history of thyroid disease and cancer, and perinatal infections, but these risk factors should be interpreted with caution.3,4
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
From an etiologic point of view, LCH is an inflammatory myeloid neoplasm characterized by infiltrations of different organ systems with CD1a/S100B/ CD207-positive mononuclear cells, which morphologically and immunohistochemically resemble specialized dendritic cells of the skin known as Langerhans cells. Gene expression arrays revealed that these cells are different from resident mature Langerhans cells of the skin as they express immature myeloid markers and are more closely related to a myeloid-derived precursor dendritic cell.5 Also, approximately 60% of LCH-cells bear a V600E mutation in the BRAF (v-Raf murine sarcoma viral oncogene homolog B) oncogene, and 33% of BRAF wild-type lesions harbor mutations in the MAP2K1 (mitogen-activated protein kinase kinase 1) gene leading to universal MEK (mitogen activated protein/extracellular signal-related kinase kinase) and ERK (extracellular signal-regulated kinase) activation (Fig. 117-1A). Based on these results the “Misguided Myeloid Dendritic Cell Precursor Model,” in which precursor bone marrow–derived myeloid dendritic cells acquire a pathologic ERK activation and express antigens typically found in mature Langerhans cells such as CD207 and CD1a (Fig. 117-1B), has been proposed. These cells secrete factors such as osteopontin, vanin, and different proinflammatory factors, as well as transforming growth factor-β and attract, among other inflammatory cells such as macrophages and eosinophils, activated T cells, especially activated regulatory FOXP3/CTLA4, and SPP-positive T cells.5
The severity and the extent of the organ involvement is supposed to depend on the stage of differentiation in which ERK gets activated: whereas an activation in a hematopoietic stem cell or an undifferentiated myeloid dendritic cell leads to multisystem disease, ERK activation in more differentiated myeloid precursors only results in multifocal or unifocal disease.1
More studies are needed to gain further insights into the pathogenesis of LCH as there are still many open questions, including: Why is it that in adult pulmonary LCH, unlike in the other organs, the infiltrating mononuclear cells seem to be mostly nonclonal mature dendritic cells? Another interesting question is whether a high expression of PD-L1 (programmed death ligand 1) immune checkpoint protein in LCH could serve as a new therapeutic target6; as of this writing, it is still too early to tell.
20
CLINICAL FEATURES
CLINICAL FEATURES
CLASSIFICATION
The traditional classification of LCH in Hashimoto- Pritzker disease, eosinophilic granuloma, HSC, and Letterer-Siwe disease has been abandoned because the disease spectrum is broader than that of the traditional classification and many patients do not fit into these clinical subtypes. Also, the clinical course can change with time. The new classification considers the extent of organ involvement as this has significant implications for treatment and prognosis (see Fig. 117-1). In addition, LCH might best be described as a continuum as illustrated in Figure 117-2. Patients are divided into:
■ Single-system LCH: Approximately 55% to 65% of the LCH patients present with single-system LCH, in which only 1 organ system is involved. Organ systems most commonly affected are bone, skin, lymph nodes, lungs, and the CNS.7
■ Multisystem LCH: In multisystem LCH, more than 1 organ system is affected by the disease. It is important to distinguish whether high-risk organs are involved.
At-risk organs include the hematopoietic system, the liver, and the spleen. The lung is no longer regarded as a high-risk organ, as recent studies could not confirm lung involvement as a negative prognostic factor. Table 117-1 provides an overview of the currently accepted classification.
CUTANEOUS FINDINGS
Cutaneous findings in LCH can be identified in more than one-third of LCH. It is the second most common organ involved after bone lesions and can be the earliest sign of disease. Only 12% of children with single-system disease and 53% of patients affected by multisystem LCH show a skin involvement.7 Consequently, age is an important risk factor as children younger than the age of 1 year more frequently show a true skin-only LCH, while those older than the age of 18 months have a higher risk for multisystem LCH. The role of the dermatologist in making an early diagnosis is, therefore, crucial. Patients can present with a broad variety of skin manifestations. The most typical ones are small, translucent rose-yellowish crusted papules or papulovesicles on the trunk, in the intertriginous areas and the scalp, associated with eczematous scaling which resembles “candida intertrigo” or “seborrheic dermatitis” (Figs. 117-3, 117-4, and 117-5). Lesions can also present as hemorrhagic papules and nodules associated with petechiae reminiscent of vascular lesions or “varicella-like eruptions” (Fig. 117-6). Vesicles, pustules, and nail involvement have been described. Nail involvement can present as paronychia, nailfold destruction, onycholysis, subungual hyperkeratosis, longitudinal grooving, and
2021
20
Reported mutations in Langerhans cell histiocytosis
BRAF 50%-65%
BRAF-V600E BRAF-V600D BRAF-600DLAT BRAF-T599A
MAP2K1 0%-20%
Unknown 15%-40%
MAP3K 10%
A
RTK
Ras
Raf
MEK
ERK1/ERK2
Risk of severity of clinical manifestation
High risk
Hematopoietic stem cells (CD34+)
Dendritic precursor cells
Tissue dendritic precursor cells
Low risk
CD1a+/CD207+ LCH lesion dendritic cells
ERK ERK ERK
ERK
- Macrophages, T-cells, and Eosinophils
multisystem multifocal multisystem multifocal multifocal unifocal
B
2022
20
Clinical spectrum of LCH
Polyostotic bone involvement Multifocal bone disease (2 or more different bones) Multiple lymph nodes involvement
Disseminated disease with involvement of low-risk organs (skin, bone, lymph node, pituitary)
Multiple site Low-risk group
Monostotic bone involvement Isolated skin involvement Single lymph node involved
Disseminated disease with involvement of 1 or more of the high-risk organs (hematopoietic system, lungs, liver, and spleen)
Single site High-risk group
Single-system disease Symptoms: absent or mild Multisystem disease Symptoms: present and severe
pigmented and purpuric striae of the nail bed. Mucosal lesions most commonly are nodulo-ulcerative and involve the perioral, the perigenital, and the perianal areas as well as the gingiva. Mucosal lesions and external otitis media seem to be associated with a higher risk for multisystem LCH.7 Purpura with nail involvement might be a poor prognostic sign, but this should be evaluated in larger clinical trials. This broad variety of skin and mucosal manifestations frequently leads to a delayed diagnosis as skin lesions are misinterpreted as eczema, miliaria, scabies, varicella, seborrheic dermatitis, folliculitis, or candidiasis. LCH should be kept in mind as a rare, but important, differential diagnosis when the above-mentioned lesions are seen, especially if they are resistant to therapy and are spreading.
NONCUTANEOUS FINDINGS
The most common noncutaneous organ involved in LCH is bone (77% of cases), followed by lymph nodes (19%), liver (16%), spleen (13%), lung (10%), and the CNS (6%). In aggressive manifestations
2023
20
2024
of the disease, patients present with a variety of clinical symptoms such as malaise, fever, nausea, weight loss, myalgia and arthralgia, and memory problems. Bone Lesions: Although LCH can affect any bone in the body, it most frequently affects the skull, jaw, femur, rib, vertebra, and humerus. Patients complain about tender masses, and the radiologic workup shows lytic areas in affected bones with a “punched out” appearance. Particularly in children, the cervical vertebrae are commonly involved, which can lead to vertebra plana. In adults, an asymmetric collapse of the vertebrae is more common, which can provoke neurologic defects. Involvement of the jaw can cause loose teeth. Vertebral collapse can be one of the most prominent diagnostic hints for LCH. Involvement of the base of the skull might result in hearing loss, recurrent otitis externa, diabetes insipidus, or cranial nerve palsies. Lesions of the facial bones and anterior or middle cranial fossae classify as “CNS-risk” lesions as they are associated with a 3-fold increased risk for developing CNS diseases such as diabetes insipidus, which at diagnosis is mostly an irreversible condition. Lymph Nodes: Cervical lymph nodes are the most frequently affected by LCH, but LCH can also involve lymph nodes of the mediastinum, which could be misdiagnosed as lymphoma, and can cause asthma-like symptoms because of airway system compression. A biopsy for proper diagnosis is mandatory. Bone Marrow: Bone marrow involvement frequently affects young children when other risk organs, such as liver and spleen, are involved. In the past, bone marrow involvement was suspected only if significant anemia, thrombocytopenia, or neutropenia was present. A recent study found CD1a+ cells in the bone marrow in single-system LCH even if no hematologic abnormalities could be detected. When thrombocytopenia and anemia, especially in combination with hypoalbuminemia, are present, LCH was associated with a poor outcome.8
Liver and Spleen: Cholestasis and sclerosing cholangitis induced by hepatic involvement of LCH are among the most serious complications of LCH. In most cases, sclerosing cholangitis will not respond to chemotherapy and liver transplantation remains the only possible treatment option. Clinically, patients present with hepatosplenomegaly and elevated liver enzymes such as elevated liver transaminases, γ-glutamyltransferase, and/or alkaline phosphatase. Also, hypoalbuminemia with ascites and clotting deficiencies can appear. Involvement of the spleen with massive splenomegaly can significantly worsen the clinical outcome because of hypersplenism with resulting cytopenias and respiratory compromise. Lungs: Involvement of the lungs is more frequently seen in adults than in children, and smoking has been identified as a major risk factor. LCH induces a cystic and/or nodular destruction of lung tissue of the upper
and middle lung fields and can result in a spontaneous pneumothorax, tachypnea, and/or dyspnea,9 ultimately leading to severe pulmonary insufficiency.
Central Nervous System: LCH can directly affect any part of the brain. A risk factor for CNS involvement (25% risk) is LCH lesions of the facial bones or bones of the anterior or middle cranial fossae (“CNS-risk” lesions). The most common manifestations of CNS involvement include endocrine abnormalities resulting from large pituitary tumors, most frequently diabetes insipidus, and neurodegenerative symptoms such as ataxia, dysarthria, cognitive dysfunction, and behavior changes. Radiologic changes can precede these symptoms by many years. Interestingly, histologically in neurodegenerative lesions, no CD1a+ cells are found; instead, lymphocytes and activated microglia cells are found, which leads to the speculation that it could be a paraneoplastic inflammatory response.
Endocrinopathies: Diabetes insipidus is the most common endocrinopathy encountered in LCH. Patients present with polyuria, polydipsia, and nocturia. This is a sign of damage to the antidiuretic hormone-secreting cells of the posterior pituitary. In approximately 4% of patients, idiopathic diabetes insipidus can precede the diagnosis of LCH.10 In patients with diagnosed LCH, the risk of developing diabetes insipidus is approximately 24%. In most cases, diabetes insipidus persists despite treatment. Other endocrine abnormalities associated with the anterior pituitary, such as hypogonadism, growth failure, thyroid hormone dysfunctions, and abnormalities in glucose metabolism, can manifest. Therefore, a complete endocrine work-up is recommended.
Gastrointestinal System: Patients with GI involvement can present clinically with diarrhea, hematochezia, perianal fistulas, and/or malabsorption, but this organ system is rarely affected. If patients present with GI symptoms, multiple biopsies are required, as in most cases GI involvement is patchy.
DIAGNOSIS
DIAGNOSIS
The diagnosis of LCH is based on clinicopathologic and radiologic features of affected organs.
COMPLETE HISTORY
Nature and duration of symptoms should be assessed. In addition based on the recommendations of the Histiocyte Society, a complete history should include questions regarding pain, swelling, skin rash, otorrhea, irritability, fever, loss of appetite, diarrhea, weight loss, growth failure, polydipsia, polyuria, changes in activity level, dyspnea, smoke exposure, and behavioral and neurologic changes.
20
COMPLETE PHYSICAL EXAMINATION
Temperature, height, and weight should be measured. In addition, the Histiocyte Society recommends assessment of pubertal status; for thorough skin and mucous membrane evaluation, for presence of jaundice, pallor, edema, lymphadenopathy, ear discharge, orbital abnormalities, abnormal mucosal lesions, abnornal dentation, and soft-tissue swelling, as well as evaluation of any tachypnea, intercostal retractions, and ascites; and evaluation of liver and spleen size evaluation. Also, a complete neurologic evaluation is mandatory.
LABORATORY TESTING AND IMAGING STUDIES
HISTOPATHOLOGY
A histopathologic diagnosis is by far the most reliable and accurate diagnostic tool for a definitive diagnosis of LCH and should always be performed if it doesn’t put the patient at an increased risk. Histopathologically, typical findings in a skin biopsy show a dense and band-like infiltration of the papillary dermis with LCH cells (Fig. 117-7). These cells are oval shaped
Full blood count
Full blood count
■Hemoglobin, white blood cell and differential count, and platelet count Blood chemistry
■Hemoglobin, white blood cell and differential count, and platelet
count Blood chemistry
■Total protein, albumin, bilirubin, alanine aminotransferase (serum glutamic-pyruvic transaminase), aspartate aminotransferase (serum glutamic-oxaloacetic transaminase), alkaline phosphatase, γ-glutamyl transpeptidase
■Total protein, albumin, bilirubin, alanine aminotransferase (serum
glutamic-pyruvic transaminase), aspartate aminotransferase (serum glutamic-oxaloacetic transaminase), alkaline phosphatase, γ-glutamyl transpeptidase γ-γ
■Blood urea nitrogen, creatinine, electrolytes
■Blood urea nitrogen, creatinine, electrolytes
■Ferritin
■Ferritin
■Fasting glucose level
■Fasting glucose level
■Serum protein electrophoresis Coagulation studies
■Serum protein electrophoresis Coagulation studies
■International normalized ratio/prothrombin time, activated partial thromboplastin time/partial thromboplastin time, fibrinogen Early morning urine sample
■International normalized ratio/prothrombin time, activated partial
thromboplastin time/partial thromboplastin time, fibrinogen Early morning urine sample
■Specific gravity and osmolarity Abdominal ultrasound Chest radiography Bone scans, skeletal survey, or fluorodeoxyglucose positron emission tomography
■Specific gravity and osmolarity Abdominal ultrasound Chest radiography Bone scans, skeletal survey, or fluorodeoxyglucose positron emission
tomography
2025
Adapted from the recommendations of the Histiocyte Society (https:// histiocytesociety.org/).
20
Skin lesions
Skin lesions
■Skin biopsy Bicytopenia, pancytopenia, or persistent unexplained single cytopenia
■Skin biopsy Bicytopenia, pancytopenia, or persistent unexplained single
cytopenia
■Bone marrow aspirate and trephine biopsy to exclude causes other than Langerhans cell histiocytosis (LCH) Liver dysfunction
■Bone marrow aspirate and trephine biopsy to exclude causes other
than Langerhans cell histiocytosis (LCH) Liver dysfunction
■Liver biopsy only recommended if there is clinically significant liver involvement and the result will alter treatment (ie, to differentiate between active LCH and sclerosing cholangitis) Lung involvement
■Liver biopsy only recommended if there is clinically significant liver
involvement and the result will alter treatment (ie, to differentiate between active LCH and sclerosing cholangitis) Lung involvement
■Lung high-resolution computed tomography
■Lung high-resolution computed tomography
■Lung function test Abnormal lung CT and findings not characteristic for LCH or suspicious for atypical infection
■Lung function test Abnormal lung CT and findings not characteristic for LCH or
suspicious for atypical infection
■Bronchoalveolar lavage (BAL): >5% CD1a+ cells in BAL fluid is diagnostic in nonsmokers
■Bronchoalveolar lavage (BAL): >5% CD1a+ cells in BAL fluid is
diagnostic in nonsmokers
■Lung biopsy (if BAL is not diagnostic) Suspected craniofacial bone lesions, including maxilla and mandible
■Lung biopsy (if BAL is not diagnostic) Suspected craniofacial bone lesions, including maxilla and mandible
■MRI of head Suspected vertebral lesions
■MRI of head Suspected vertebral lesions
■MRI of spine Visual or neurologic abnormalities
■MRI of spine Visual or neurologic abnormalities
■MRI of head
■MRI of head
■Neurology assessment
■Neurology assessment
■Neuropsychometric assessment Suspected endocrine abnormality (eg, growth failure, polyuria, polydipsia, hypothalamic syndromes, precocious or delayed puberty)
■Neuropsychometric assessment Suspected endocrine abnormality (eg, growth failure, polyuria, poly-
dipsia, hypothalamic syndromes, precocious or delayed puberty)
■Endocrine assessment (including water deprivation test and dynamic tests of the anterior pituitary and thyroid)
■Endocrine assessment (including water deprivation test and
dynamic tests of the anterior pituitary and thyroid)
■MRI of the head Aural discharge or suspected hearing impairment/mastoid involvement
■MRI of the head Aural discharge or suspected hearing impairment/mastoid
involvement
■Formal hearing assessment
■Formal hearing assessment
■MRI of head
■MRI of head
■High-resolution CT of temporal bone Unexplained chronic diarrhea, failure to thrive, or evidence of malabsorption
■High-resolution CT of temporal bone Unexplained chronic diarrhea, failure to thrive, or evidence of
malabsorption
■Endoscopy and biopsy
■Endoscopy and biopsy
Adapted from the recommendations of the Histiocyte Society (https:// histiocytesociety.org/).
with an eosinophilic cytoplasm and typically display an irregular, vesicular, and infolded (kidney-shaped) nucleus (Fig. 117-8). In some cases, longitudinal nuclear grooves, which confer onto these cells a coffee bean-like appearance, can be seen. Conspicuously, LCH cells are 4 to 5 times larger than lymphocytes and are admixed with variable numbers of neutrophils, eosinophils, lymphocytes, plasma cells, and histiocytes, especially in early lesions. In later lesions, foamy histiocytes and a prominent fibrosis of the dermis prevail. LCH cells often show a marked epidermotropism with the formation of intraepidermal microabscesses and a periappendageal distribution (Figs. 117-7 and 117-9). The epidermotropism can be so marked that the dermoepidermal junction gets obscured and the epidermis gets thinned and even destroyed. Also, edema
2026
in the papillary dermis and multinucleated giant cells can be found on occasion. Typical immunohistochemical markers of LCH cells are CD1a (Fig. 117-9), S100B, CD207 (Langerin), and fascin. Stabilin-1 and CD34 are not expressed (Table 117-5). A typical finding in LCH cells is the presence of Birbeck granules in the cytoplasm, as identified by electron microscopy. They are organelles, which resemble a tennis racquet and have a zipper-like appearance along the “handle” (Fig. 117- 10). In the past, the detection of Birbeck granules by electron microscopy was the diagnostic gold standard for LCH. Nowadays however, immunohistochemical staining with CD207, an antibody that recognizes a C-type lectin associated with Birbeck granules, is the most sensitive marker. Electron microscopy is no longer needed for a specific diagnosis.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
LCH is a heterogeneous disease. Patients can present with lesions confined to a single-organ system with an indolent self-healing course or with multiple lesions in different organs—especially in at-risk organs—with a fatal outcome. Consequently, risk stratification is
Langerhans cell histiocytosis
Langerhans cell histiocytosis
■Positive: CD1a, CD2, CD11b, CD11c, CD13, CD66c, CD68, CD207 (Langerin), CD300LF, S100B, fascin
■Positive: CD1a, CD2, CD11b, CD11c, CD13, CD66c, CD68, CD207
(Langerin), CD300LF, S100B, fascin
■Negative: stabilin-1 Non–Langerhans cell histiocytosis
- Systemic non–Langerhans cell histiocytosis
■Negative: stabilin-1 Non–Langerhans cell histiocytosis
- Systemic non–Langerhans cell histiocytosis
■Positive: CD68
■Positive: CD68
■Negative: CD1a
2. Cutaneous non–Langerhans cell histiocytosis:
■Negative: CD1a
2. Cutaneous non–Langerhans cell histiocytosis:
■Positive: CD68, stabilin-1
■Positive: CD68, stabilin-1
■Negative: S100B, CD1a
■Negative: S100B, CD1a
20
crucial to delivering the optimal treatment regimen to each patient. The prognosis depends on a variety of factors. A recent multicenter LCH Phase III trial investigated a 12-month treatment with vinblastine and prednisolone demonstrating 5-year survival rates of 95%, 83%, and 57%, respectively, in patients who showed a complete resolution of disease, intermediate response to treatment, and progression of the disease after the 6-week induction phase.11 Therefore, one of the most important predictors of outcome in patients with multiorgan involvement is how the disease responds to systemic treatment in the first 6 weeks.11 Other predictors previously associated with a poor outcome, such as age younger than 2 years or lung involvement, are now no longer regarded as risk factors for a poor outcome. Involvement of at-risk organs such as the liver, the spleen, and the bone marrow is also associated with a poor outcome. Enlargement of the liver by more than 3 cm below the costal margins at the midclavicular line; hyperbilirubinemia greater than 3 times normal; hypoalbuminemia less than 30 g/dL; γ-glutamyl transpeptidase increased by more than 2 times normal; alanine aminotransferase and aspartate aminotransferase greater than 3 times normal; ascites; edema; or an intrahepatic nodular mass are signs for liver involvement and need to be confirmed by ultrasound and, if possible, by biopsy. Involvement of the spleen should be suspected if the organ can be palpated by more than 3 cm below the costal margin at the midclavicular line. Hematopoietic involvement can be divided into a mild form with hemoglobin between 10 and 7 g/dL, not the result of other causes, and/or a thrombocytopenia
2027
20
TYPE OF LESION
PAPULAR VESICOPUSTULAR XANTHOMATOUS NODULOULCERATIVE
Most likely Seborrheic dermatitis Scabies Papular xanthoma Juvenile xanthogranuloma
Lichen nitidus Miliaria Xanthoma disseminatum Urticaria pigmentosa
Generalized eruptive histiocytosis Varicella Juvenile xanthogranuloma Diffuse neonatal hemangiomatosis
Benign cephalic histiocytosis Intertrigo Urticaria pigmentosa Blueberry muffin baby
Darier disease Candidiasis
Hidradenitis
Lichen planus
Rosacea Folliculitis decalvans
Tuberculosis
Consider Leukemia Dermatitis Hyperlipidemic xanthomatosis Leukemia
Pityriasis lichenoides chronica Pityriasis lichenoides et varioliformis acuta Granuloma annulare disseminatum Pyoderma gangrenosum
Hidradenitis suppurativa Granuloma faciale
Cherry angioma
Psoriasis, guttate Perioral dermatitis
Tinea corporis, tinea capitis Lupus miliaris faciei
Tinea corporis, tinea capitis Lupus miliaris faciei
with platelets between 20,000 and 100,000/mm3, and a severe form with hemoglobin below 7 g/dL and platelets less than 20,000/mm3.12 To date, no data has demonstrated diabetes insipidus to be a predictor of poor outcome. Small numbers of lesions, a prompt resolution of lesions, and single-organ involvement are usually associated with a good outcome. The prognosis is, however, not only dependent on the disease itself, but also on associated findings. LCH patients tend to suffer from intercurrent infections, mostly mild ones, such as candidiasis and dermatophytosis, but they are also prone to more severe systemic infections, which can lead to sepsis and death. Also, LCH is associated with other malignancies, which may precede, occur concurrently with, or follow the diagnosis of LCH. Described associated malignant neoplasms are solid tumors (lung tumors, celiomesenteric neuroblastoma), malignant lymphomas, and leukemias, in particular, acute lymphoblastic leukemia and myeloid leukemias. Some of these malignancies may be related to alkylating chemotherapeutic regimens and radiotherapy used for LCH. These possible complications and/or associations should be considered at every clinical visit.
MANAGEMENT
MANAGEMENT
Treatment strategies depend on the extent and localization of the disease and the age of the patient. Patients with more limited disease confined to bone or skin are usually not treated systemically, except for “special site” lesions such as the odontoid peg, vertebral lesions with intraspinal soft-tissue extension, and anatomically functionally critical sites. All treatment recommendations are based on the
2028
recommendations of the Histiocyte Society. Systemic treatment also can be considered for multifocal bone lesions and “CNS-risk” lesions. In children with LCH confined to the skin, a watch-and-wait strategy is the best approach. Topical treatment can be tried with corticosteroid ointments, but topical steroids have shown little efficacy. A skin rash that does not respond to topical steroids is considered a clue for LCH. Other topical treatment options include imiquimod and tacrolimus as well as intralesional corticosteroid injections, CO2 laser therapy, or excision of single LCH nodules. Nitrogen mustard ointment can be applied to treat skin lesions in adults. Several case reports exist that show a significant improvement after narrowband ultraviolet B irradiation in adults and children. This treatment might work well with papules and eczematous lesions, but not when nodules are present. Photochemotherapy is effective in some adult patients. In case of ineffective local therapy, systemic glucocorticoids, thalidomide, or antimitotic drugs, such as low-dose methotrexate, can be tried. Thalidomide can ameliorate skin lesions, but the treatment is associated with neurologic toxicity and fatigue. Thalidomide should not be used in women with child-bearing potential if the skin is the only organ affected. There is 1 case report in which a patient went into complete remission after oral isotretinoin therapy (1.5 mg/day for 8 months). Table 117-7 outlines some treatment options. Also for single-bone lesions a watch-and-wait strategy can be used. Other options include simple curettage, complete excision of small lesions (lesions <2 cm diameter) and intralesional steroid injections of steroids (see Table 117-7). Thanks to the efforts of the Histiocyte Society, which was founded 1985, the scientific evidence for
SKIN BONE
■Observation
■Observation
■Observation
■Observation
■Nitrogen mustard
■Biopsy or curettage
■Nitrogen mustard
■Biopsy or curettage
■Tacrolimus
■Surgery
■Tacrolimus
■Surgery
■Imiquimod
■Steroid injections
■Imiquimod
■Steroid injections
■Intralesional steroids
■Vinblastine/prednisolone (multiple bone lesions or “special site” lesions)
■Intralesional steroids
■Vinblastine/prednisolone
■Narrowband ultraviolet B
■Narrowband ultraviolet B
(multiple bone lesions or “special site” lesions)
■Photochemotherapy
■Photochemotherapy
■CO2 laser
■CO2 laser
■Methotrexate
■Methotrexate
■Systemic steroids
■Systemic steroids
■Thalidomide
■Thalidomide
■Isotretinoin
■Isotretinoin
■Excision (single lesions)
■Excision (single lesions)
the treatment of LCH multisystem disease is far better than that for single-system LCH based on the reported results of multicenter trials (LCH-I, LCH-II, LCH-III). Based on the results of the most recent LCH-III trial, patients should be treated systemically for 12 months because patients treated for only 6 months have an increased frequency of early relapse. The treatment protocol consists of a 6-week induction chemotherapy phase with vinblastine (6 mg/m2
weekly intravenous bolus) combined with prednisolone (40 mg/m2/day orally for 4 weeks and then tapered over 2 weeks) and a subsequent therapy with vinblastine/prednisolone with or without mercaptopurine, depending on the treatment response after 6 weeks and the involvement of at-risk organs (Fig. 117-11). With this treatment regimen, 86% of patients without risk-organ involvement and 66% with risk-organ involvement show a response to therapy after 6 weeks—an important prognostic factor for outcome. Still, 50% of patients in this study did not respond to treatment,11 illustrating the need for additional therapeutic strategies. Other chemotherapeutic agents used as salvage strategies with proven efficacy for multisystem LCH, albeit with serious side effects, are cytarabine, cladribine, and clofarabine. Allogeneic bone marrow transplantation has been attempted in 87 patients with high-risk LCH. Among patients transplanted, 77% survived after 3 years following myeloablative and reducedintensity conditioning regimens.13 Based on recent research results, which provided evidence for a strong ERK activation arising from somatic mutations in the mitogen-activated protein kinase (MAPK) signaling pathway in LCH, novel therapeutic options include BRAF and MAPK inhibitors. Although limited data exist for these treatment regimens, first reports are promising.14,15 Also, a high programmed death ligand 1 expression in LCH has been described,6 which suggests that anti–programmed death 1 and anti–programmed death ligand 1 treatments might be successful.
20
NON–LANGERHANS CELL HISTIOCYTOSIS
AT-A-GLANCE
■ Non–Langerhans cell histiocytosis (N-LCH) represents a group of different disorders characterized by the proliferation of histiocytes that do not meet criteria to be diagnosed as Langerhans cells
■ N-LCH are immunohistochemically positive for CD68 and negative for S100B and CD1a
■ Stabilin-1 (formerly MS-1 antigen or MS-1-HMWP) can discriminate N-LCH from LCH and other granulomatous diseases
■ Systemic manifestations, such as diabetes insipidus, or ocular involvement, as well as an association with malignancies can occur in N-LCH
■ Local skin manifestations of the disease can be treated by excision, laser therapy, or intralesional steroid injection. Radiotherapy also is used for the treatment of skin lesions and cerebral lesions. For the treatment of patients with visceral lesions, systemic glucocorticoids and chemotherapy, and in some cases targeted therapies, such as imatinib mesylate or BRAF and MEK inhibitors, are useful.
N-LCH is a group of different disorders characterized by the proliferation of histiocytes other than Langerhans cells. N-LCH can be classified as systemic or cutaneous N-LCH (see Table 117-1). Cutaneous N-LCH can be subclassified as juvenile, adult, and necrobiotic xanthogranulomas, as well as spindle-cell N-LCH.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Systemic forms of N-LCH are regarded as an accumulation of classically activated macrophages (Mφ1). In contrast, the lesions of cutaneous N-LCH are characterized by the presence of alternatively activated macrophages (Mφ2). In forms with single lesions, local trauma might play a pathogenetic role. In diffuse forms an association with malignancies and autoimmune diseases suggests an immunologic cause. Mφ1 are known to develop in response to proinflammatory stimuli such as T-helper (Th) 1 cytokines (interferon-γ or bacterial products [lipopolysaccharides]). They are characterized by secretion of proinflammatory cytokines, such as interleukin (IL)-1,
2029
20
Treatment algorithm for multisystem Langerhans cell histiocytosis
Multisystem disease (biopsy proven)
Induction 6-week treatment vinblastine plus prednisolone
Treatment response
Progression Incomplete resolution Complete resolution
Second 6-week treatment vinblastine plus prednisolone
Risk organ involved? (liver, spleen, hematopoietic system)
yes
no
no
Risk organ involved? (liver, spleen, hematopoietic system)
yes
no
Improved?
yes
Continuation therapy (to complete 12-month total treatment) vinblastine plus prednisolone
Continuation therapy (to complete 12-month total treatment) vinblastine plus prednisolone
Second-line treatment
plus mercaptopurine
IL-6, IL-1, and tumor necrosis factor-α, and possess a strong oxidative burst and a profound antimicrobial activity. In contrast to systemic N-LCHs, the lesions of cutaneous N-LCHs show alternatively activated effector macrophages (Mφ2). Mφ2 are induced by Th2 cytokines, including IL-4, IL-10, IL-13, and transforming growth factor-β, or by antiinflammatory mediators such as glucocorticoids. They express antiinflammatory cytokines such as IL-1R antagonist and IL-10,
2030
chemokine receptor antagonists such as AMAC-1, broad-spectrum receptors of innate immunity, such as macrophage mannose receptor and the haptoglobin receptor CD163. N-LCHs are positive for CD68 but negative for CD1a. Stabilin-1 (formerly MS-1 antigen or MS-1-HMWP) is expressed on some N-LCH. Because of its specific expression in N-LCH, it can be used as one marker to discriminate N-LCH from LCH or granulomatous diseases (see Table 117-5).
ROSAI-DORFMAN DISEASE (SINUS HISTIOCYTOSIS WITH MASSIVE LYMPHADENOPATHY)
ROSAI-DORFMAN DISEASE
(SINUS HISTIOCYTOSIS
WITH MASSIVE
LYMPHADENOPATHY)
PATHOPHYSIOLOGIC ASPECTS AND CLINICAL FEATURES
In 1969, sinus histiocytosis with massive lymphadenopathy was described by Juan Rosai and Ronald Dorfman. Rosai-Dorfman disease (RDD) is an idiopathic disease, but its occurrence has frequently been observed after infectious disease. A possible viral etiology, such as Epstein-Barr virus, human herpesvirus 6, parvovirus B19, and polyomavirus, has been suggested by several authors.16,17 Lymphadenopathy is the main clinical manifestation. The neck lymph nodes are the most common place of histiocyte accumulation, although accumulation outside of lymph nodes may occur. Here, the skin (Fig. 117-12) and other organs, such as the breast, kidney, thyroid, testis, and CNS, may be affected.17 Similar to other regional tumors, the symptoms of this disease vary with the site of accumulation. Despite the rarity of RDD, the co-occurrence with LCH, lymphomas, or autoimmune disorders has been reported.17 Because of the expression of S100B by histiocytes, S100B can be used as a serum marker to monitor disease progression or therapy response.
HISTOPATHOLOGIC FINDINGS
RDD cells are characteristically S100B+ (Fig. 117-13A) and CD1a−. The RDD cells are also positive for CD68, CD163, α1-antitrypsin, α1-antichymotrypsin, fascin, HAM-56 (human alveolar macrophage 56), and stabilin-1 (Fig. 117-13B). The hallmark of RDD is emperipolesis, in which different types of bone marrow cells, such as lymphocytes or neutrophils, are found in the cytoplasm of histiocytes with a background of mature lymphocytes and plasma cells.17
20
A
S100B
B
Stabilin 1
DIFFERENTIAL DIAGNOSIS
Because enlarged and massive unilateral or bilateral lymph nodes are a frequent manifestation in isolated or multiple regions, lymphoma or abscesses may be clinically suspected. Extranodal RDD can also mimic other diseases, such as meningioma.17
CLINICAL COURSE AND PROGNOSIS
Spontaneous resolution in patients with RDD was reported. There are reports that surgical resection as well as radiotherapy for affected lymph nodes or skin lesions has resulted in the complete disappearance of RDD. Oral prednisone can have a remarkably favorable response, mainly in patients with lymph node involvement. However, intralesional corticosteroid or oral prednisone does not always result in resolution of cutaneous lesions. Treatment with chemotherapeutic agents has been disappointing in general. There are reports, however, that some patients could benefit from a combination of methotrexate/ 6-mercaptopurine/vinblastine/6-thioguanine.17
2031
20
Tyrosine kinase inhibitors such as imatinib mesylate are effective in some patients with RDD.18
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
HEMOPHAGOCYTIC
LYMPHOHISTIOCYTOSIS
CLINICAL FEATURES
Hemophagocytic lymphohistiocytosis (HLH) was first described in 1952 by Farquhar and Claireaux and is a rare disease. The 70% of HLH cases are children younger than the age of 1 year. HLH is characterized by persistent fever, splenomegaly with cytopenia, hypertriglyceridemia, and hypofibrinogenemia. The infiltration of histiocytes is usually observed in reticuloendothelial systems, including the bone marrow and CNS. Various cutaneous manifestations, such as erythroderma, generalized purpuric macules and papules, and morbilliform eruptions, have been described in up to 65% of HLH patients. HLH is classified as one of the cytokine storm syndromes because high amounts of inflammatory cytokines are secreted. There are 2 major forms: primary and secondary. Primary HLH includes familial HLH and several primary immunodeficiencies, which exhibit genetic inheritance and usually occur in infancy. Several genetic defects in primary HLH—particularly familial HLH genes such as HPLH1, perforin, UNC13D, STX11, and STXBP2— have been identified. Secondary HLH is associated with infections (eg, Epstein-Barr virus), autoimmune disorders (eg, juvenile idiopathic arthritis), and malignancies (mainly non-Hodgkin lymphoma). Increased levels of various cytokines and soluble IL-2 receptor are biologic markers of HLH.19
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of HLH includes autoimmune lymphoproliferative syndrome, Griscelli syndrome, macrophage activation syndrome, and other primary immunodeficiencies that present with HLH, such as X-linked lymphoproliferative disease.19
CLINICAL COURSE AND PROGNOSIS
The prognosis is guarded with an overall mortality of 50%. It is a poor prognostic factor if HLH is associated with malignancy.20 In some individuals, secondary HLH may be self-limited. Therapeutic options include high-dose corticosteroids, cyclosporine, etoposide, methotrexate, and vincristine. Use of intravenous immunoglobulin also has been described. An aggressive therapeutic approach is warranted in most cases, including immunochemotherapy and hematopoietic stem cell transplantation.19
2032
JUVENILE XANTHOGRANULOMA
JUVENILE
XANTHOGRANULOMA
CLINICAL FEATURES
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson. It is a benign, self-healing skin disorder that primarily affects infants younger than 1 year of age, but also can be found in older children. At birth, 5% to 17% of children already have cutaneous lesions. In 40% to 70% of children who are affected by JXG, the disorder presents during the first year of life. JXG usually manifests with both solitary and multiple (oligolesional) papules or nodules that are usually located on the face, neck, and upper trunk, and on other body parts, including lungs, bone, heart, and GI tract. Early lesions show a reddish-brown color (Fig. 117-14A). Mature lesions have a reddish-yellow appearance (Fig. 117-14B). Telangiectasia can be present. Ocular lesions occur in up to 10% of children with JXG and may affect their vision. JXG is often accompanied with other disorders, such as neurofibromatosis Type 1 and juvenile chronic myelogenous leukemia.21
HISTOPATHOLOGIC FINDINGS
In early lesions, microscopic examination shows monomorphous, non–lipid-containing histiocytic infiltrates in the dermis (Fig. 117-15A). In contrast mature lesions are characterized by foam cells, Touton giant cells, and foreign-body giant cells in the superficial dermis and on the border of infiltrates (Fig. 117-15B). Also, fibrosis can be appreciated. Lesional histiocytes are positive for stabilin-1. Cells stain for macrophage markers such as CD163, CD11b, CD11c, CD36, CD68, factor XIIIa, and vimentin. S100B and CD1a are not expressed.
DIFFERENTIAL DIAGNOSIS
JXG can be distinguished from LCH by the expression of stabilin-1 and the absence of CD1a and CD207. Other differential diagnoses include, for example, molluscum contagiosum, hemangioma, and neurofibroma (Table 117-8).
CLINICAL COURSE AND PROGNOSIS
JXG usually resolves spontaneously over 1 to 5 years. Ocular lesions, however, rarely improve spontaneously and require treatment. Treatments include surgical excision, CO2 laser treatment, intralesional steroid injection, cryotherapy, and low-dose radiotherapy. In the case of a resistant or recurring lesion, chemotherapy should be considered.21
A
20
B
A
B
2033
20
■Xanthoma
■Papular mastocytosis
■Xanthoma
■Papular mastocytosis
■Neurofibroma
■Sarcoidosis
■Neurofibroma
■Sarcoidosis
■Hemangioma
■Dermatofibroma
■Hemangioma
■Dermatofibroma
■Molluscum contagiosum
■Cutaneous leukemia
■Molluscum contagiosum
■Cutaneous leukemia
■Spitz nevus
■Other forms of non– Langerhans cell histiocytosis and Langerhans cell histiocytosis
■Spitz nevus
■Other forms of non–
Langerhans cell histiocytosis and Langerhans cell histiocytosis
BENIGN CEPHALIC HISTIOCYTOSIS
BENIGN CEPHALIC
HISTIOCYTOSIS
CLINICAL FEATURES
Benign cephalic histiocytosis was reported initially by Gianotti in 1971 and was separated from JXG. Because of the ultrastructural presence of worm-like bodies, it also has been called “histiocytosis with intracytoplasmic worm-like bodies.” However, these bodies can be found in other forms of N-LCH. An overlap with other forms of N-LCH, especially with JXG, has been reported. Benign cephalic histiocytosis usually presents with small, yellow-red or yellow-brown, asymptomatic papules, located on the head and neck of young children with a tendency toward spontaneous remission.22
HISTOPATHOLOGIC FINDINGS
Histologic examination of skin samples reveals an infiltrate of histiocytes, which closely approach the epidermis, accompanied by scattered lymphocytes and eosinophils. The histiocytes express the typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and CD207 is negative.22 Ultrastructural presence of worm-like bodies is typical.
DIFFERENTIAL DIAGNOSIS
It is difficult to separate benign cephalic histiocytosis from other forms of N-LCH, especially JXG. Therefore, it can be discussed as a variant of JXG.
CLINICAL COURSE AND PROGNOSIS
Benign cephalic histiocytosis usually resolves spontaneously. Therapeutic options include CO2 laser therapies.
2034
GENERALIZED ERUPTIVE HISTIOCYTOMA OF CHILDHOOD
GENERALIZED ERUPTIVE
HISTIOCYTOMA OF
CHILDHOOD
CLINICAL FEATURES
Generalized eruptive histiocytoma (GEH) (also known as “eruptive histiocytoma” or “generalized eruptive histiocytosis”) was initially reported by Winkelmann and Muller in 1963. This rare disease is characterized by widespread, erythematous, essentially symmetrical papules, particularly involving proximal extremities as well as the trunk. Unlike in JXG, brownish papules and nodules do not develop; mucosal membranes, however, can be affected. Cutaneous lesions resolve spontaneously with remaining hyperpigmented maculae. There is also a report associating GEH with rheumatic fever. GEH can be seen as a variant of JXG.
ADULT XANTHOGRANULOMA
ADULT
XANTHOGRANULOMA
CLINICAL FEATURES
Adult xanthogranuloma was originally described by Gartmann and Tritsch in 1963. Both solitary and oligolesional yellow-orange papules usually appear on the face, neck, and lower arms. In contrast to JXG, adult xanthogranuloma does not show a spontaneous remission. No association with systemic diseases such as neurofibromatosis or leukemia has been reported. Histologic findings of adult and juvenile forms of the disease are almost identical. Usually in adult xanthogranuloma there are more giant cells. Therapeutic options include excisions of lesions or CO2 laser therapies.
PAPULAR XANTHOMA
PAPULAR XANTHOMA
CLINICAL FEATURES
Papular xanthoma was reported for the first time in 1981 by Winkelmann. It is a rare disease in normolipemic patients who present clinically. It appears mainly as a solitary yellowish papule, and seems to appear more often in males. It shows a biphasic occurrence in young adolescents and in persons of middle age. A congenital form has been reported. Mucous membranes were affected in some cases but there is no systemic involvement. A plaquelike form of papular xanthoma can be seen as a variant.23,24
HISTOPATHOLOGIC FINDINGS
Histology shows a normal epidermis and a dense infiltration of xanthomatized macrophages interspersed by numerous Touton-type giant cells. Immunohistochemically mononucleated and multinucleated macrophages are positive for KiM1p. Only giant cells are positive for CD68. Up to 50% of the xanthomatized cells are positive for the lectin peanut agglutinin. Stainings for factor XIIIa and CD1a are negative.
DIFFERENTIAL DIAGNOSIS
Differential diagnoses include xanthoma, atheroma, keloid, histiocytoma, Spitz nevus, clear cell acanthoma, and other benign and malignant cutaneous tumors.
CLINICAL COURSE AND PROGNOSIS
Papular xanthoma usually resolves spontaneously. Therapeutic options include surgical excision and CO2 laser therapy.22,23
GENERALIZED ERUPTIVE HISTIOCYTOMA
GENERALIZED ERUPTIVE
HISTIOCYTOMA
CLINICAL FEATURES
GEH was first described by Winkelmann and Muller in
1963. There are fewer than 50 case reports worldwide and it affects mainly adults.25 GEH is characterized by multiple asymptomatic and symmetrically distributed brownish erythematous papules that involve the axial regions—trunk, face, and proximal extremities—that frequently evolve to flares. The big flexures are spared (Fig. 117-16). Mucous membranes were reported to be affected in some cases.26 Although the disorder usually follows a benign clinical course, there are 2 reports of an atypical form associated with acute monocytic leukemia.
HISTOPATHOLOGIC FINDINGS
Skin biopsies show a normal epidermis. The papillary dermis is infiltrated by mostly small, nonlipidized cells and few lymphocytes underneath a Grenz zone, which is characteristic for GEH (Fig. 117-17). Giant cells are missing. Macrophages are positive for CD68 and stabilin-1, and negative for CD1a, CD34, and S100B.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis includes LCH, other forms of N-LCH, and urticaria pigmentosa. Other N-LCHs are easily distinguished by histology.
CLINICAL COURSE AND PROGNOSIS
GEH usually resolves spontaneously. Therapeutic options include surgical excision and CO2 laser
20
A
B
therapy. Systemic psoralen and ultraviolet A therapy was reported to produce rapid regression of the skin lesions. Given the potential development of acute monocytic leukemia, close follow-up of patients with GEH is recommended.25,26
XANTHOMA DISSEMINATUM
XANTHOMA DISSEMINATUM
CLINICAL FEATURES
Xanthoma disseminatum was described by Montgomery in 1938. It preferentially affects male children. The disease is characterized by the sudden appearance of small, yellow-red to brown papules and nodules that are discrete and disseminated with a predilection for the flexural and intertriginous areas as well as mucous
2035
20
membranes (Fig. 117-18). Involvement of mucosae and a variety of internal organs can result in significant morbidity and mortality. Systemic associations include central diabetes insipidus owing to involvement of the pituitary stalk and paraproteinemias, such as multiple myeloma.27
Variants of xanthoma disseminatum include disseminated xanthosiderohistiocytosis, which was described by Halprin and Lorincz in 1960 and is associated with multiple myeloma. In 1998, Ferrando described a case
2036
of systemic xanthohistiocytoma that can be also seen as a variant of xanthoma disseminatum.
HISTOPATHOLOGIC FINDINGS
Histopathologic examination of early lesions of xanthoma disseminatum shows a predominance of scalloped histiocytes, whereas more established lesions consist mainly of foamy histiocytes with few scalloped cells. In the most established or mature lesions, a mixture of scalloped cells, foam cells, lymphocytes, and Touton giant cells is seen. These cells are positive for stabilin-1, HAM-56, HHF35, KP1, KiM1P, factor XIIIa, and vimentin, and negative for S100B and CD1a (Fig. 117-19).27
DIFFERENTIAL DIAGNOSIS
The main differential diagnosis includes eruptive xanthomas, other N-LCH, and LCH.
CLINICAL COURSE AND PROGNOSIS
Xanthoma disseminatum is a slowly progressing disease. Cutaneous lesions can be treated, for example, by CO2 laser therapy. Lesions are not very radiosensitive. Several medications, including statins, fibrates, glitazones, prednisolone, azathioprine, cyclophosphamide, and thalidomide, have been tried with variable results. A successful therapeutic approach seems to be the administration of 2-chlorodeoxyadenosine.27,28
MULTICENTRIC RETICULOHISTIOCYTOSIS
MULTICENTRIC
RETICULOHISTIOCYTOSIS
CLINICAL FEATURES
Multicentric reticulohistiocytosis (MRH) was first reported by Weber and Freudenthal in 1937. The term was coined by Goltz and Laymon in 1954. It is a rare, multisystem inflammatory disease that usually appears around the age of 50 years, but there are a few reports of its occurrence during childhood. It is twice as common in females. Cutaneous lesions are firm, yellow-brownish papules or nodules that reach a size of several centimeters and progress slowly in size. The occurrence of lesions over the joints of fingers and wrist is typical (Figs. 117-20 and 117-21). Leonine facies can occur via confluence of facial lesions. Involvement of mucosae and conjunctivae can be appreciated in every second patient. MRH can affect any organ. However, the most common clinical manifestations are cutaneous eruptions and symmetric inflammatory polyarthritis. Internal organs, such as the lungs (resulting in pleural effusion) and heart (pericardial effusion and congestive heart failure), have been described as affected by MRH. In addition, mesenteric lymphadenopathy and
A
20
B
2037
20
urogenital lesions have been reported.29 Constitutional symptoms such as fever, weight loss, and malaise can be observed.30 A malignancy rate of 33 (25%) in 133 cases of MRH has been reported in the literature. The malignancies were most commonly hematologic, breast, or stomach carcinomas.31
HISTOPATHOLOGIC FINDINGS
Histology reveals mid-dermal infiltrates of mononuclear histiocytes and multinucleated histiocytes with a ground-glass appearance and a variable number of vacuolated, spindle-shaped, and xanthomatized mononuclear histiocytes. In the case of MRH, immunohistochemical analyses are usually positive for CD45, CD68 and HLA-DR (human leukocyte antigen-D related), but are negative for S100B, CD1a, or HHF-35 actin.29,30
DIFFERENTIAL DIAGNOSIS
Differential diagnosis includes François syndrome (dermochondrocorneal dystrophy), Morbus Farber (lipogranulomatosis disseminata) as well as different metabolic diseases such as gout.
CLINICAL COURSE AND PROGNOSIS
The disease can be self-limited. However, severe joint destruction usually results. Surgical excision, pulseddye laser, oral corticosteroids, methotrexate, cyclophosphamide, and tocilizumab have shown success in treating extensive MRH lesions. A systematic review of 17 cases of MRH treated with tumor necrosis factor antagonists showed an efficacy of anti–tumor necrosis factor treatment in MRH.30
ERDHEIM-CHESTER DISEASE
ERDHEIM-CHESTER
DISEASE
CLINICAL FEATURES
The first report of the disease was by Chester who cooperated with Erdheim in 1930. In 1972, Jaffe reported on a similar case and named the disease Erdheim-Chester disease. Erdheim-Chester disease is a rare disease with onset in middle age. Long bone involvement is almost universal in Erdheim-Chester disease patients, and is bilateral and symmetrical in nature. It encompasses a spectrum of disorders, ranging from asymptomatic bone lesions to multisystem, life-threatening variants. More than 50% of cases have some sort of extraskeletal involvement. This can include kidney, skin, brain, and lung involvement; less frequently, retro-orbital tissue, pituitary gland, and heart involvement is observed.32,33
On the skin, xanthelasma and xanthoma are present in one-sixth of cases. Yellow-brown papular and
2038
widespread infiltrated lesions have been described in some patients. Newer studies demonstrate that approximately 50% of Erdheim-Chester disease patients have BRAF mutations in early multipotent myelomonocytic precursors or in tissue-resident histiocytes. Rarely, other genes involved in the MAPK signaling pathway also mutate.32
HISTOPATHOLOGIC FINDINGS
Tissue samples show a xanthomatous or xanthogranulomatous infiltration by lipid-laden or foamy histiocytes that are usually surrounded by fibrosis, especially in longer-standing lesions. Bone biopsies show infiltration of lipid-laden macrophages, multinucleated giant cells, inflammatory infiltrates of lymphocytes, and histiocytes as well as a generalized sclerosis of the long bones.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis mainly includes LCH.
CLINICAL COURSE AND PROGNOSIS
Interferon-α is the first-line treatment in Erdheim- Chester disease, as it has been clearly demonstrated to increase overall survival. Anakinra and infliximab have also led to encouraging results and should be considered when treatment with interferon-α fails. More recently, BRAF inhibitors have been shown to be efficient in all treated cases.33
NECROBIOTIC XANTHOGRANULOMA
NECROBIOTIC
XANTHOGRANULOMA
CLINICAL FEATURES
Necrobiotic xanthogranuloma was first reported by Kossard and Winkelmann in 1980. However, a necrobiosis with xanthogranulomas associated with paraproteinemia was already observed in the 1960s. It is a multisystem disease that affects older adults and it manifests as yellowish plaques and nodules that can ulcerate. Necrobiotic xanthogranuloma is predominantly located on the trunk, the extremities, and the face (periorbital lesions). Extracutaneous involvement also has been described, such as of the eyes, heart, skeletal muscle, larynx, spleen, and ovaries. A systemic association in the form of serum monoclonal gammopathy usually of immunoglobulin G κ and λ type is seen in 80% of necrobiotic xanthogranuloma patients. There is an increased risk for concomitant hematologic and lymphoproliferative malignancies.34
HISTOPATHOLOGIC FINDINGS
Necrobiotic xanthogranuloma is characterized by typical necrobiotic areas surrounded by granulomas
composed of lymphocytes, Touton giant cells, foamy histiocytes and foreign body-type multinucleated giant cells in the dermis as well as subcutis. As with other N-LCHs, there is no expression of CD1a antigen or CD207. There are no Birbeck granules.
DIFFERENTIAL DIAGNOSIS
Necrobiosis lipoidica, subcutaneous granuloma annulare, and noduli rheumatica can be considered as differential diagnoses. In cases with only a modest necrobiosis, xanthoma that is associated with paraproteinemia and lymphomas should be considered. In cases with periorbital skin lesions, xanthelasma can be a differential diagnosis.
CLINICAL COURSE AND PROGNOSIS
The prognosis is poor with several treatments showing variable results. Neither treatment nor nontreatment of monoclonal gammopathy with alkylating agents necessarily influences the activity of the skin disease. There are no randomized controlled trials and studies on long-term outcomes do not exist. Treatment options are mainly described in case reports and include topical and systemic corticosteroids, thalidomide, high-dose intravenous immunoglobulin, chlorambucil, cyclophosphamide, fludarabine, rituximab, melphalan, infliximab, interferon-α, cladribine, hydroxychloroquine, azathioprine, and methotrexate. In addition, laser therapy, radiotherapy, surgery, psoralen and ultraviolet A, plasmapheresis, and extracorporeal photopheresis are described (systematically reviewed in Miguel et al35).
HEREDITARY PROGRESSIVE MUCINOUS HISTIOCYTOSIS
HEREDITARY PROGRESSIVE
MUCINOUS HISTIOCYTOSIS
CLINICAL FEATURES
Hereditary progressive mucinous histiocytosis was described by Bork and Hoede in 1988. It is a rare, potentially autosomal dominant inherited disease. Progressive eruptions of self-resolving skin-colored to red-brown papules usually develop in the first decade on the nose, hands, forearms, and thighs (Fig. 117-22). These lesions can later develop into persistent and progressive erythematous papules. Visceral involvement has not yet been reported.
HISTOPATHOLOGIC FINDINGS
Collections of epithelioid S100B/CD1a− and CD68 as well as weak stabilin-1+ histiocytes and telangiectatic vessels in the upper dermis of early lesions can be seen. In the mid-dermis of early and well-developed lesions, nodular aggregates of tightly packed spindle-shaped cells are present. Moderate to extensive mucin production was demonstrated in epithelioid histiocytes and spindle-shaped cells (Fig. 117-23).36
20
DIFFERENTIAL DIAGNOSIS
Acral persistent papular mucinosis, scleromyxedema, granuloma annulare, dermatofibroma, and GEH can be distinguished by histology.
2039
20
CLINICAL COURSE AND PROGNOSIS
Ablative laser therapy for single lesions is an option.
PROGRESSIVE NODULAR HISTIOCYTOSIS
PROGRESSIVE NODULAR
HISTIOCYTOSIS
CLINICAL FEATURES
Progressive nodular histiocytosis was described by Taunton and colleagues in 1978. The disease is characterized by generalized, discrete yellow to red-brown papules and nodules measuring a few centimeters in size. A prominent facial involvement has been described. Other organs are not affected. There is an association with chronic myeloid leukemia as well as with tumors of the hypothalamus.37
HISTOPATHOLOGIC FINDINGS
The dermis contains abundant spindle-shaped histiocytes, some with foamy cytoplasm. Cells are positive for CD68, CD163, vimentin, and fascin, while negative for CD1a and S100B.
DIFFERENTIAL DIAGNOSIS
MRH, which usually shows an involvement of other organ sites, is a differential diagnosis.
CLINICAL COURSE AND PROGNOSIS
Surgical ablation, for example, with a CO2 laser, of all visible lesions is a therapeutic option.
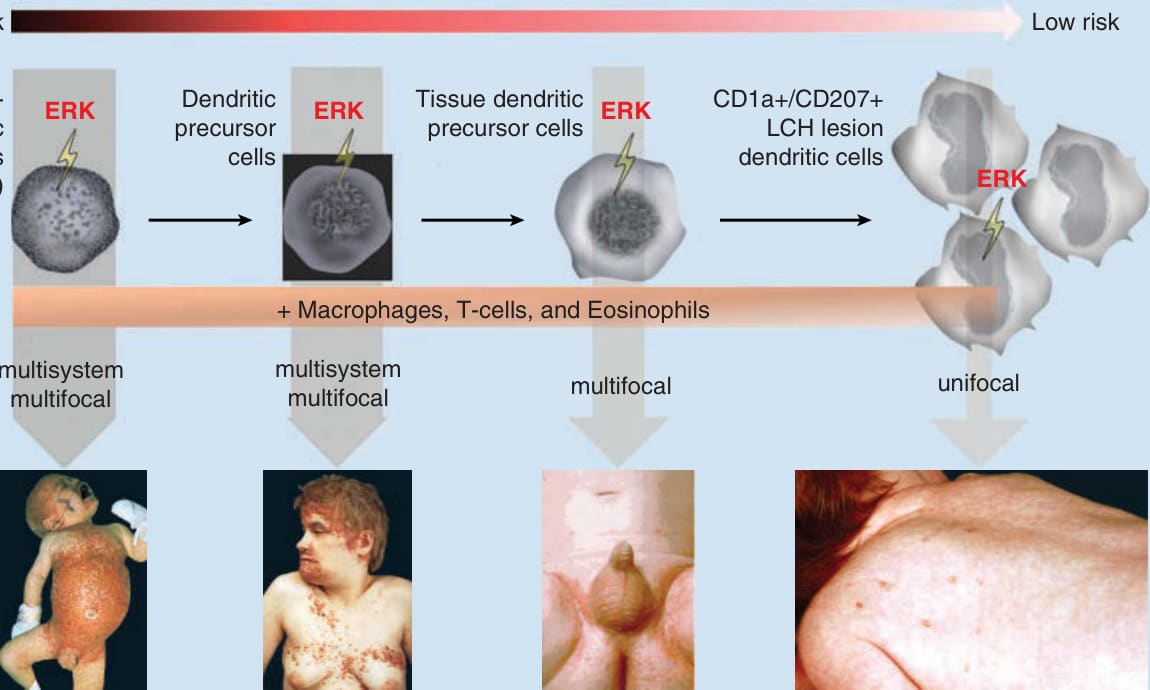
Figure 117-1 A, Reported mutations in Langerhans cell histiocytosis (LCH). This figure illustrates the frequency of identified mutations in LCH, leading to a downstream activation of the transcription factor ERK (extracellular signal-regulated kinase). In 50% to 65% of LCH cases, a mutation in the BRAF (v-Raf murine sarcoma viral oncogene homolog B) gene was found. The BRAF-V600E mutation was by far the most common mutation found, but BRAF-V600D, BRAF-600DLAT, and BRAF-T599A mutations also have been described. MAP2K1, mitogen-activated protein kinase kinase 1; MAP3K, mitogenactivated protein kinase kinase kinase; MEK, mitogen-activated protein/extracellular signal-related kinase kinase; RTK, receptor tyrosine kinase. B, The “Misguided Myeloid Dendritic Cell Model.” Based on the “Misguided Myeloid Dendritic Cell Model,” the clinical manifestation depends on the stage of differentiation of the hematopoietic cell in which the ERK activation takes place. ERK activation in the hematopoietic stem cell and the dendritic precursor cell leads to an extensive LCH, whereas ERK activation in the tissue dendritic precursor cell and the CD1a+/CD207+ LCH lesion dendritic cell leads to a single-system LCH.
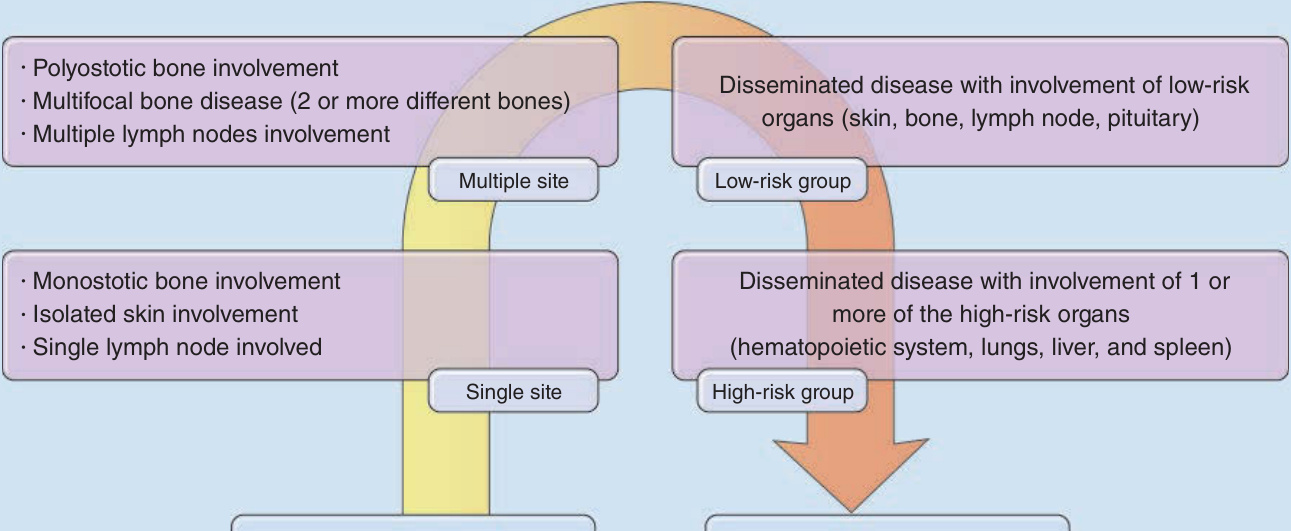
Figure 117-2 Clinical spectrum of Langerhans cell histiocytosis (LCH). Any single patient affected by LCH can be situated in a given point on the arrow.
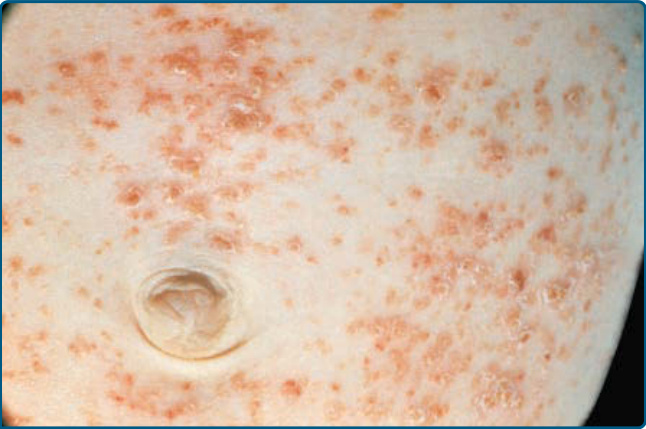
Figure 117-3 Rose-yellowish papules on the abdominal area of an infant suffering from extensive Langerhans cell histiocytosis.
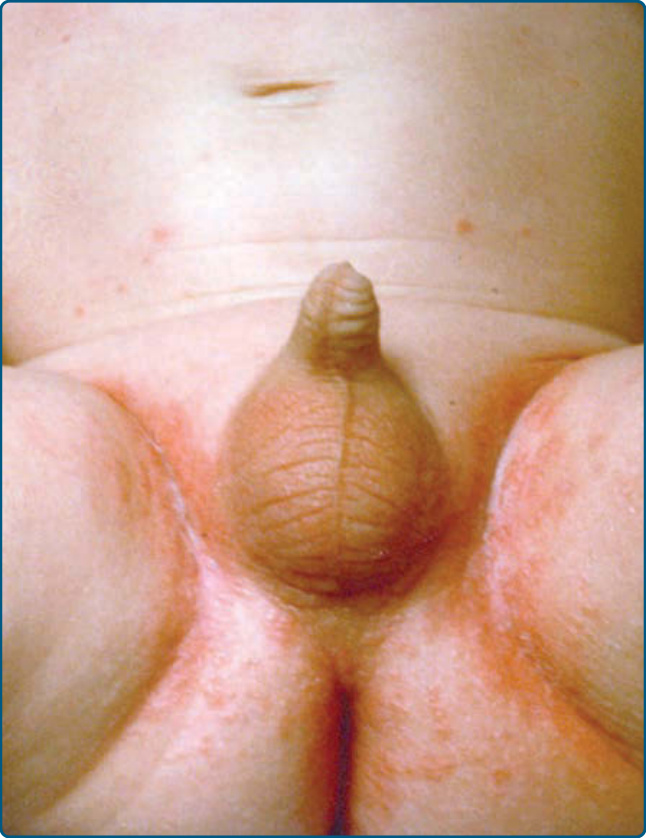
Figure 117-4 “Candida intertrigo”–like eruptions in the genital area of an infant with Langerhans cell histiocytosis.
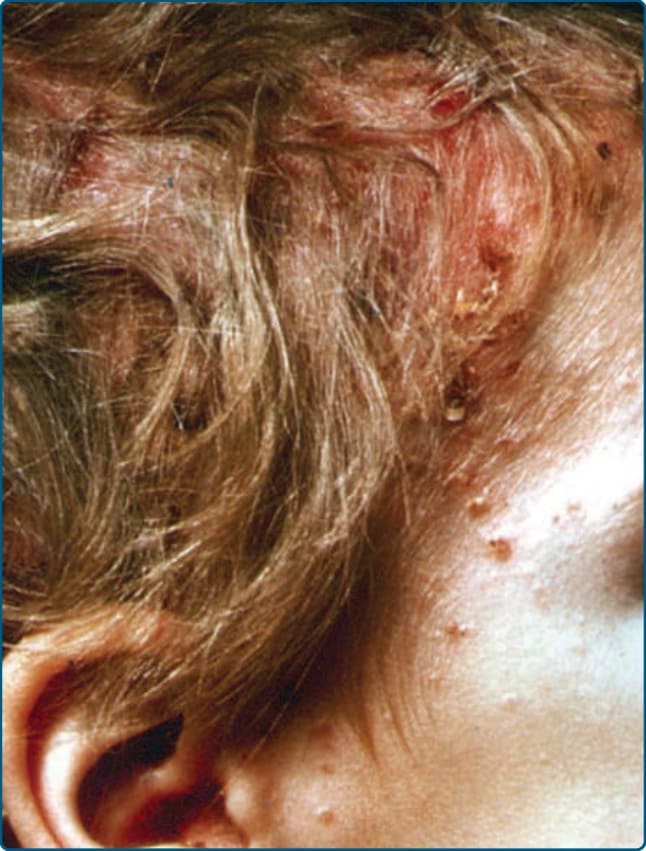
Figure 117-5 “Seborrheic dermatitis”–like eruptions on the scalp of an infant with Langerhans cell histiocytosis.
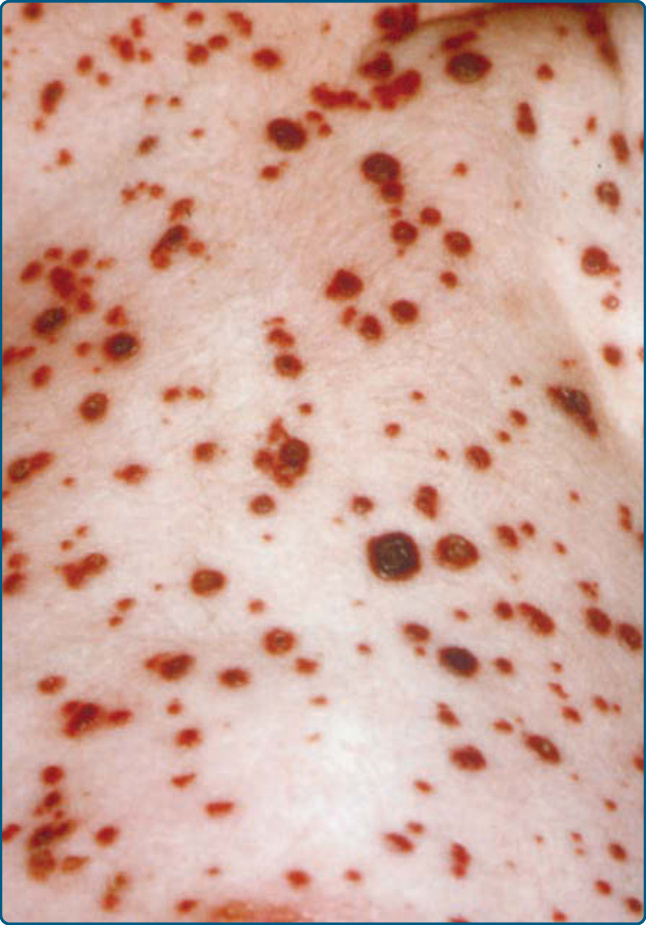
Figure 117-6 Hemorrhagic papules and nodules on the abdominal area of a patient who has Langerhans cell histiocytosis.
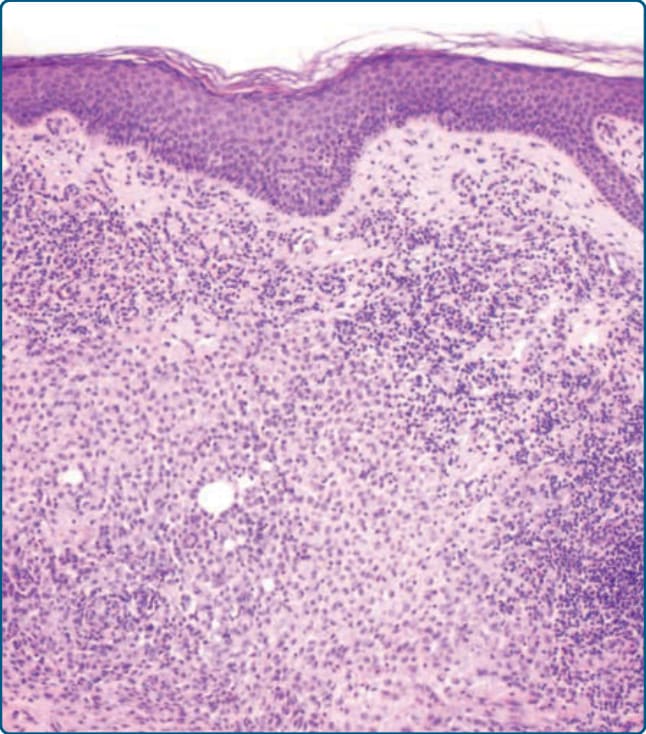
Figure 117-7 Histology of Langerhans cell histiocytosis (LCH) showing a dense mononuclear infiltrate of the dermis with oval to rounded LCH cell and lymphocytes as well as some eosinophils with epidermal involvement.
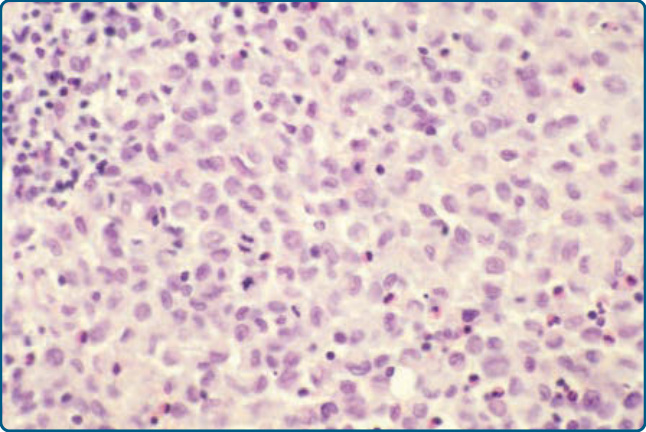
Figure 117-8 Histology of Langerhans cell histiocytosis (LCH). LCH cells have a kidney-shaped nucleus.
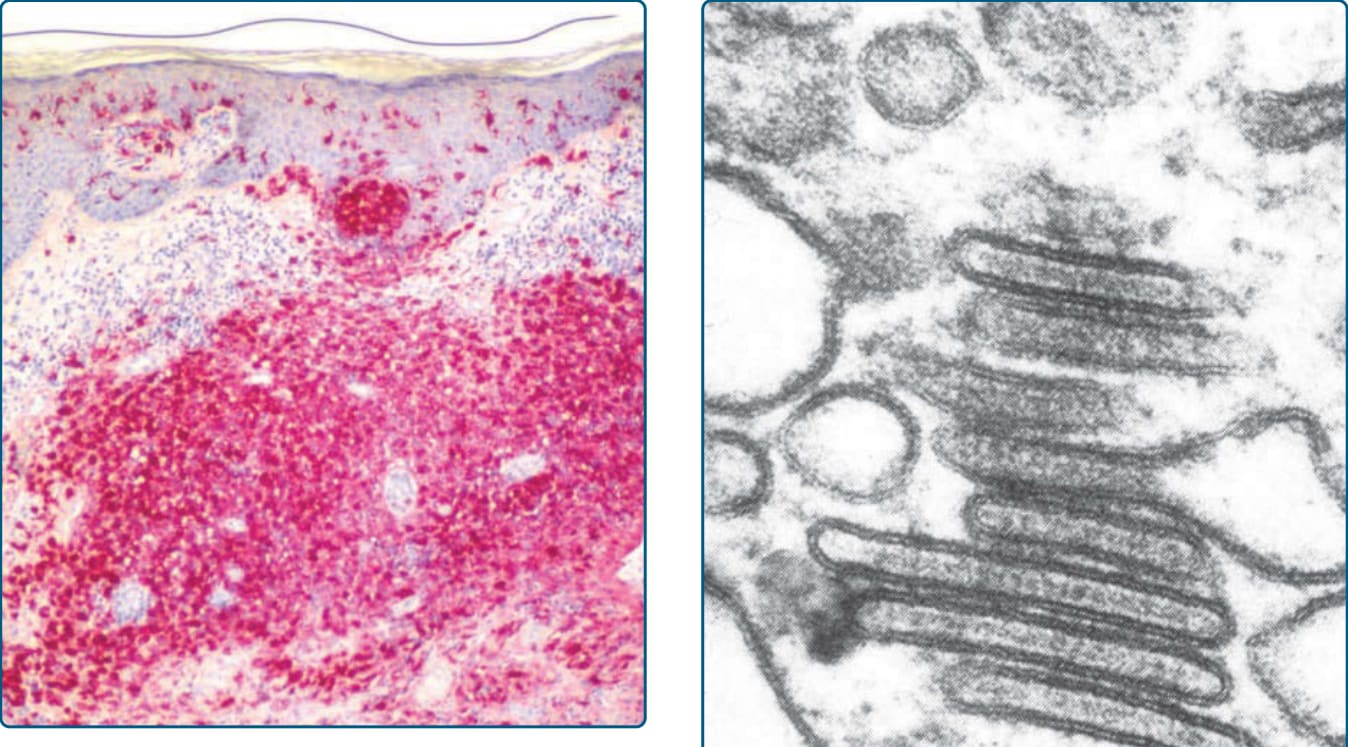
Figure 117-9 Histology of Langerhans cell histiocytosis (LCH). The LCH cells stain for CD1a (red) and show a marked epidermotropism.
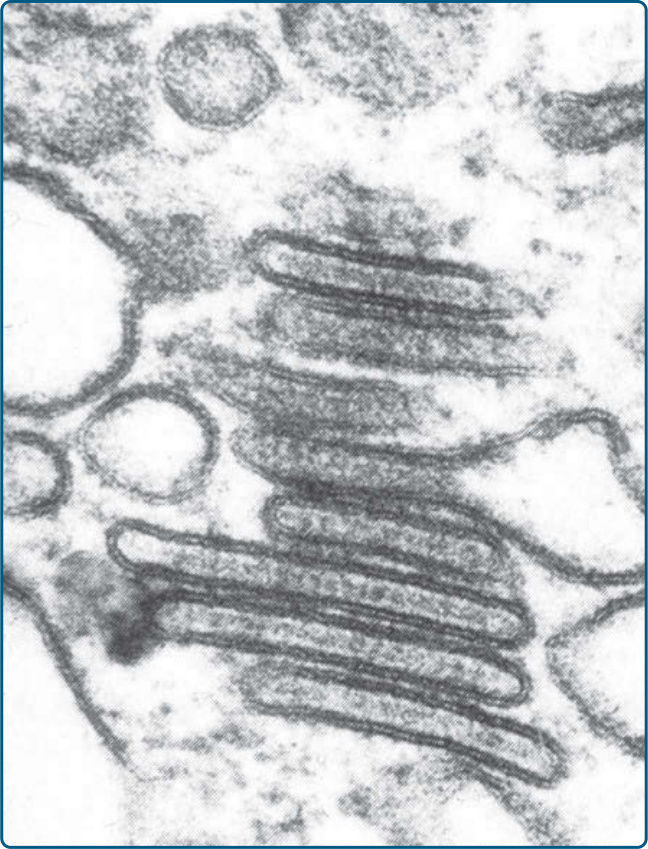
Figure 117-10 Electron microscope picture of a Langerhans cell histiocytosis cell shows typical Birbeck granules.
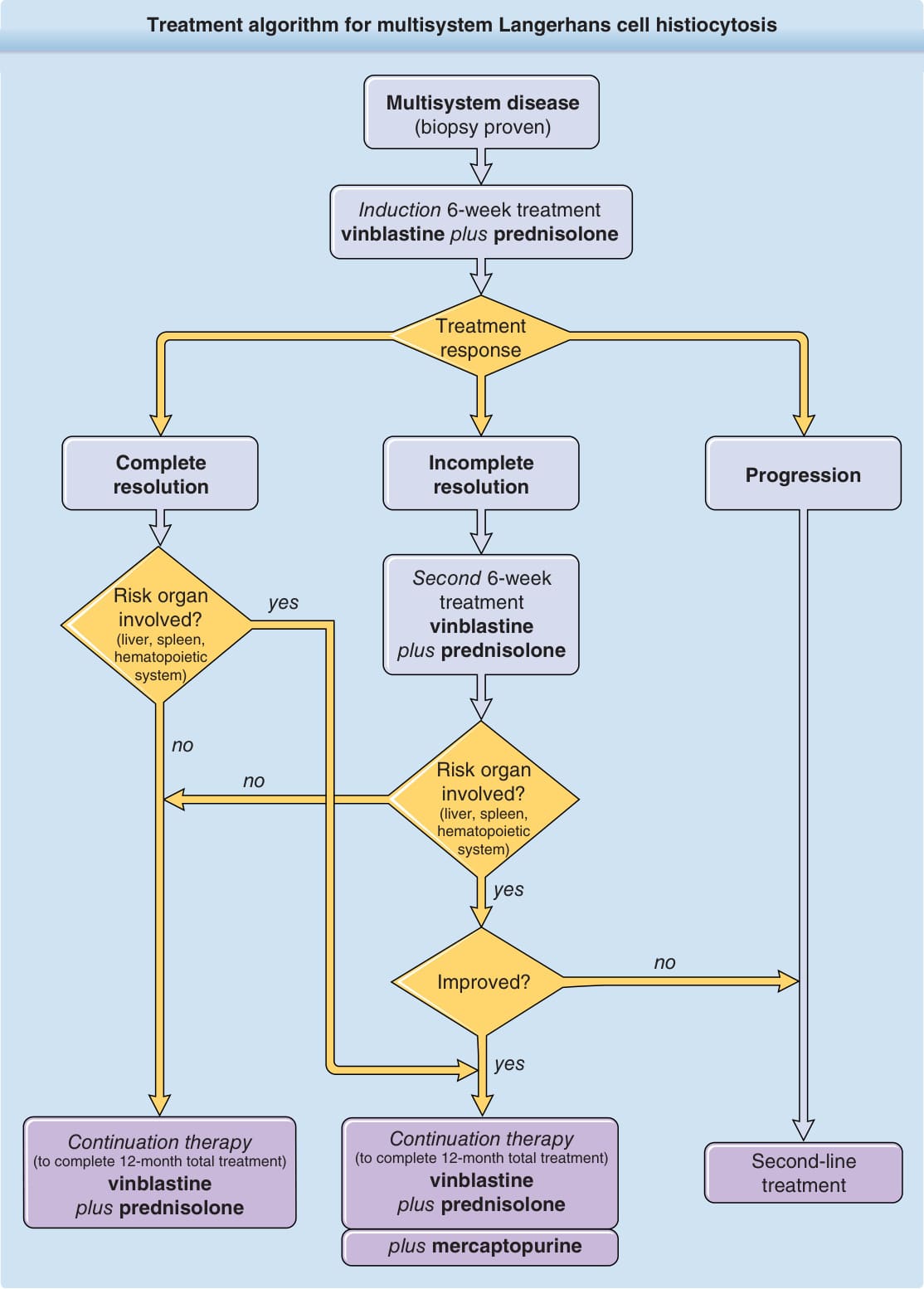
Figure 117-11 Treatment algorithm for multisystem Langerhans cell histiocytosis.
Figure 117-12 Rosai-Dorfman disease.
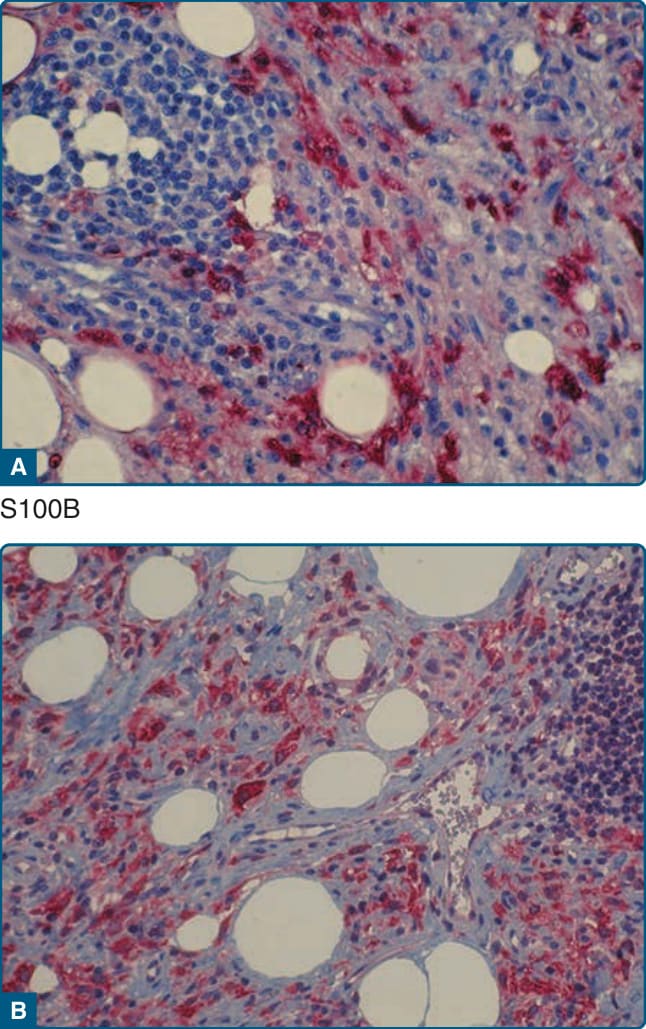
Figure 117-13 Photomicrographs of a lesion of Rosai- Dorfman disease. Histiocytes are positive for S100B (A) and stabilin-1 (B).
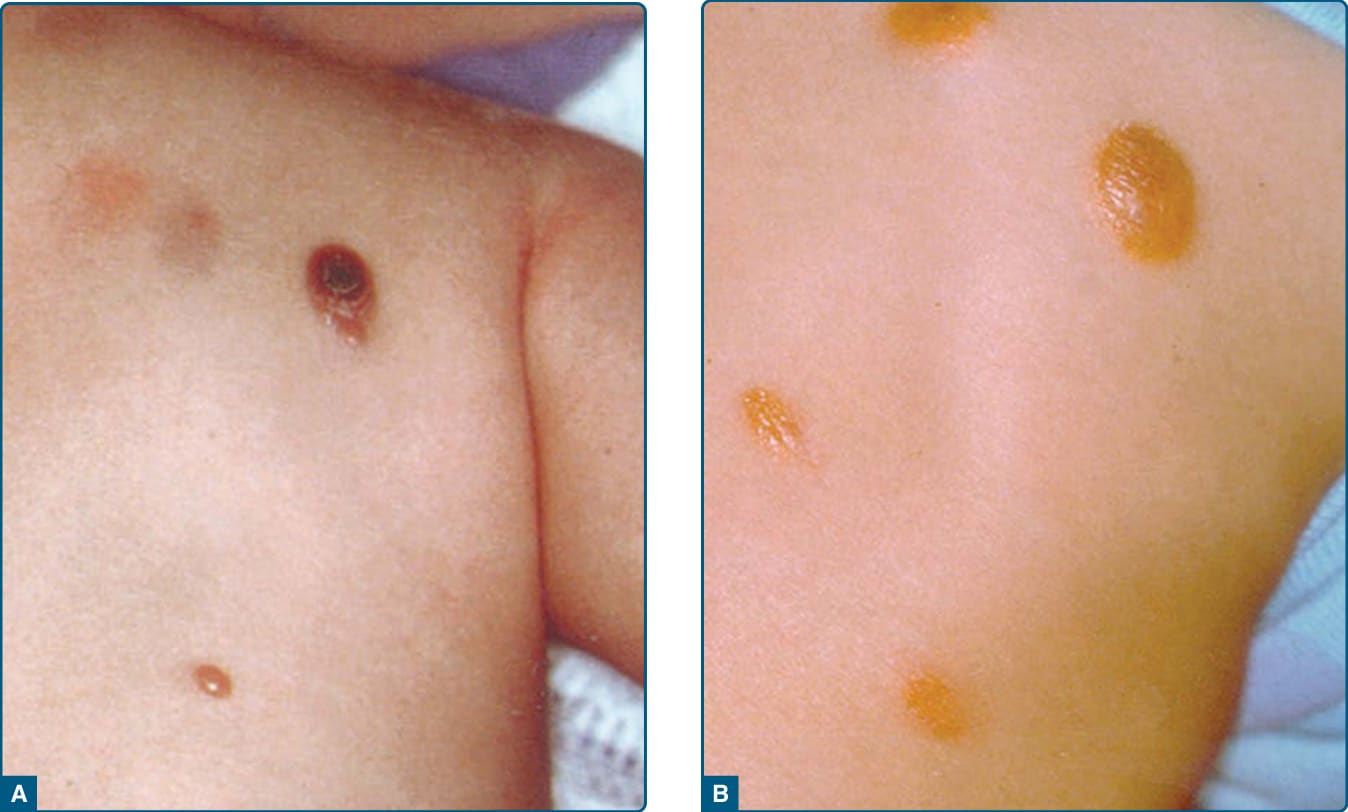
Figure 117-14 Juvenile xanthogranuloma. A, Early stage presents with reddish papules. B, Mature stage shows a reddishyellow shade of color.
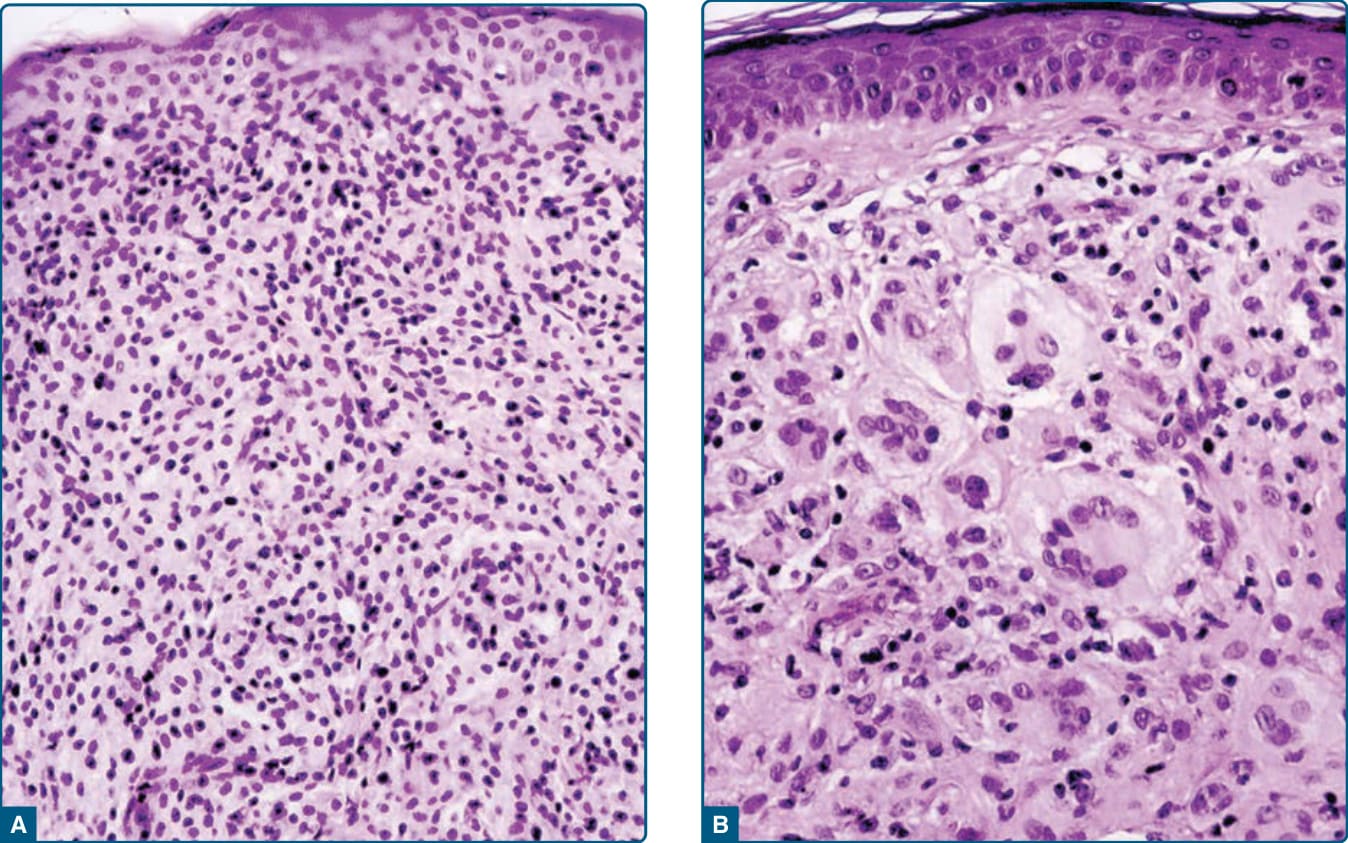
Figure 117-15 Photomicrographs of lesions of juvenile xanthogranuloma. A, The early stage is characterized by small, mostly mononuclear histiocytes with a small cytoplasm and less lipid reaching the epidermis without any Grenz zone. B, The mature stage shows big, cytoplasm-rich, lipidized histiocytes, big foam cells, and typical Touton giant cells.
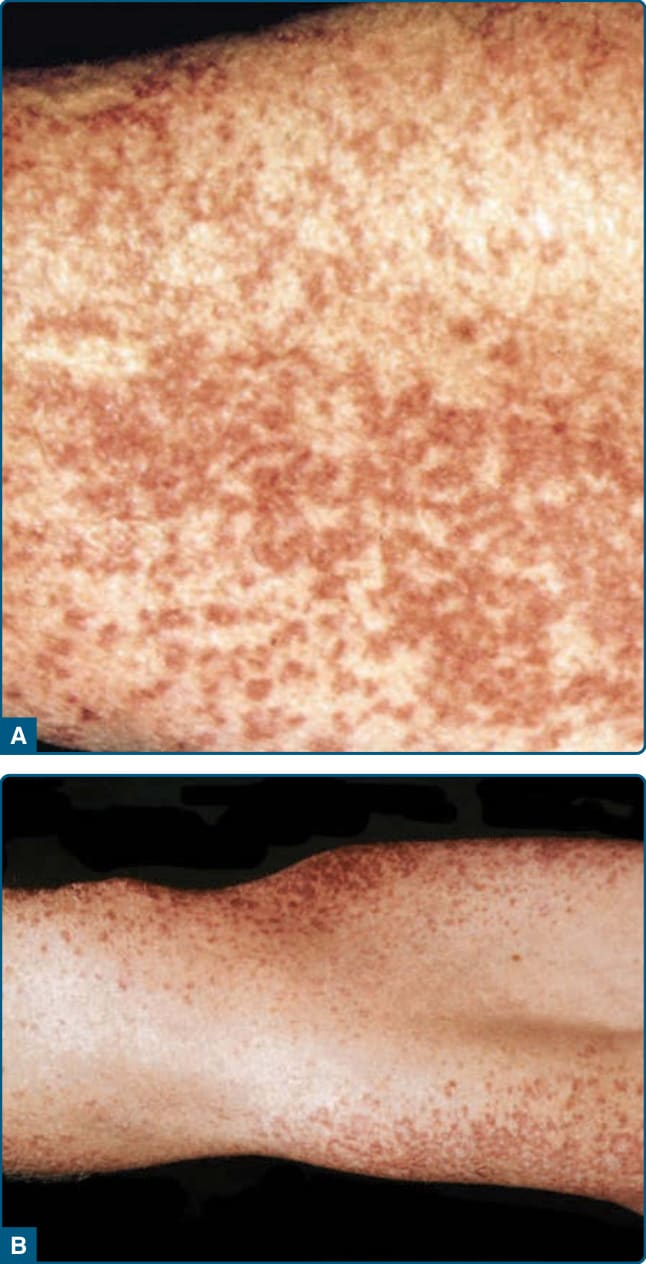
Figure 117-16 Generalized eruptive histiocytoma. Note that the big flexures are spared, as seen in part B of the figure.
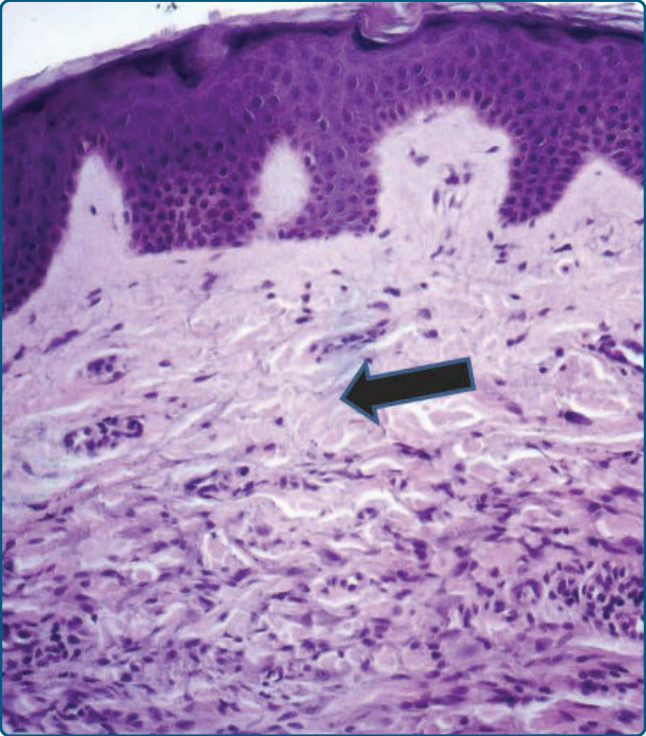
Figure 117-17 Photomicrographs of a lesion of generalized eruptive histiocytoma. In the papillary dermis an infiltration of mostly small, nonlipidized cells and of few lymphocytes underneath a Grenz zone (arrow) can be seen. This is a typical sign for generalized eruptive histiocytoma. Giant cells are missing.
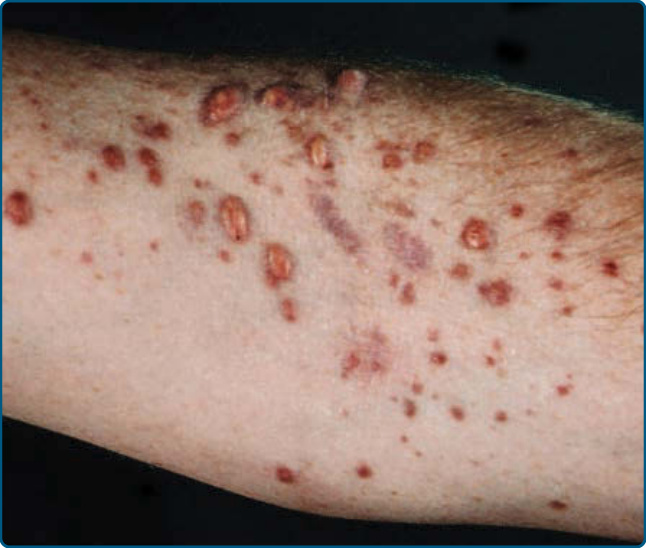
Figure 117-18 Xanthoma disseminatum.
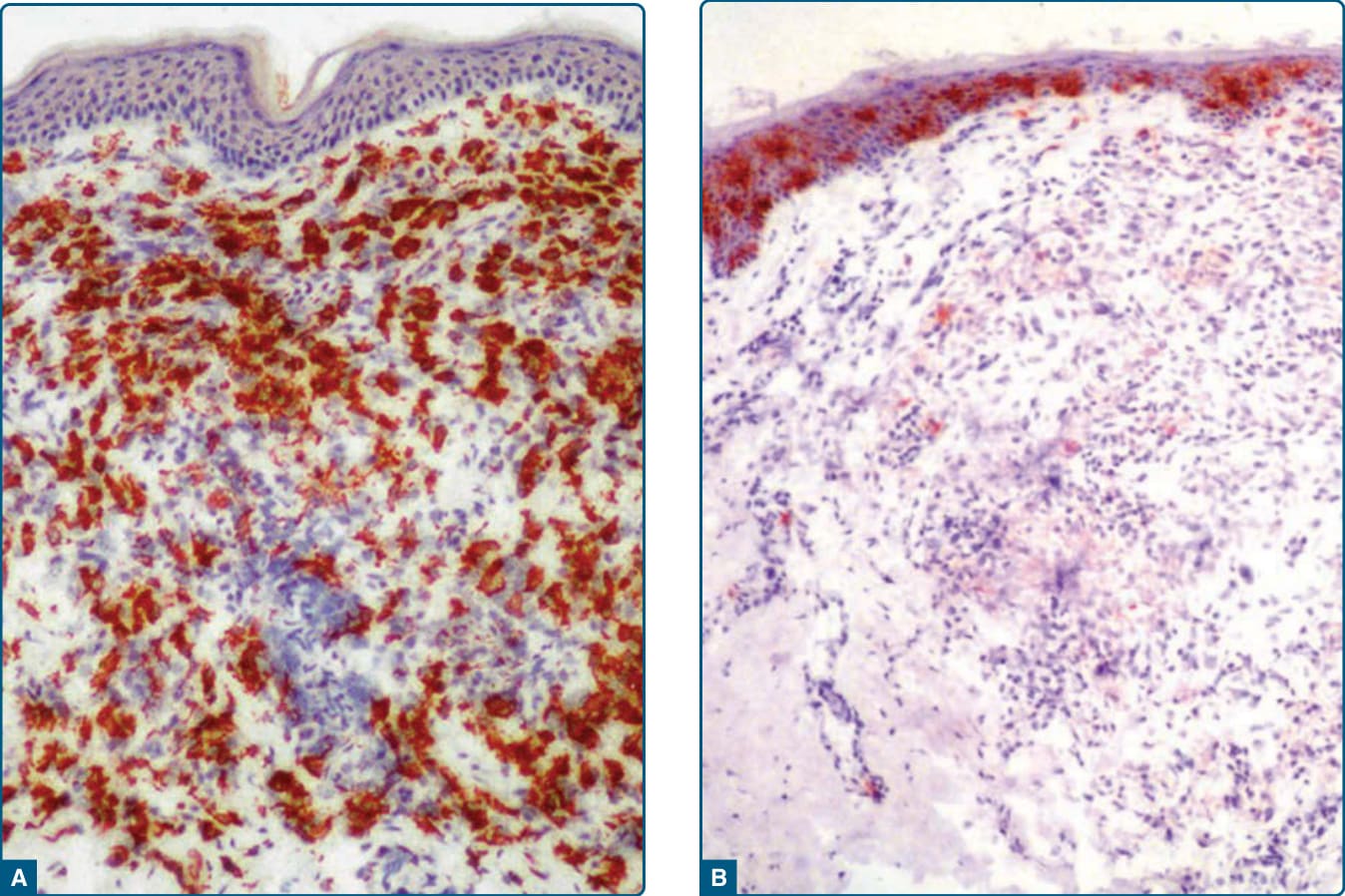
Figure 117-19 Photomicrographs of an early-stage lesion of xanthoma disseminatum. A, Stabilin-1 shows strong expression in lesional histiocytes. B, CD1a staining is limited mainly to the epidermal Langerhans cells.
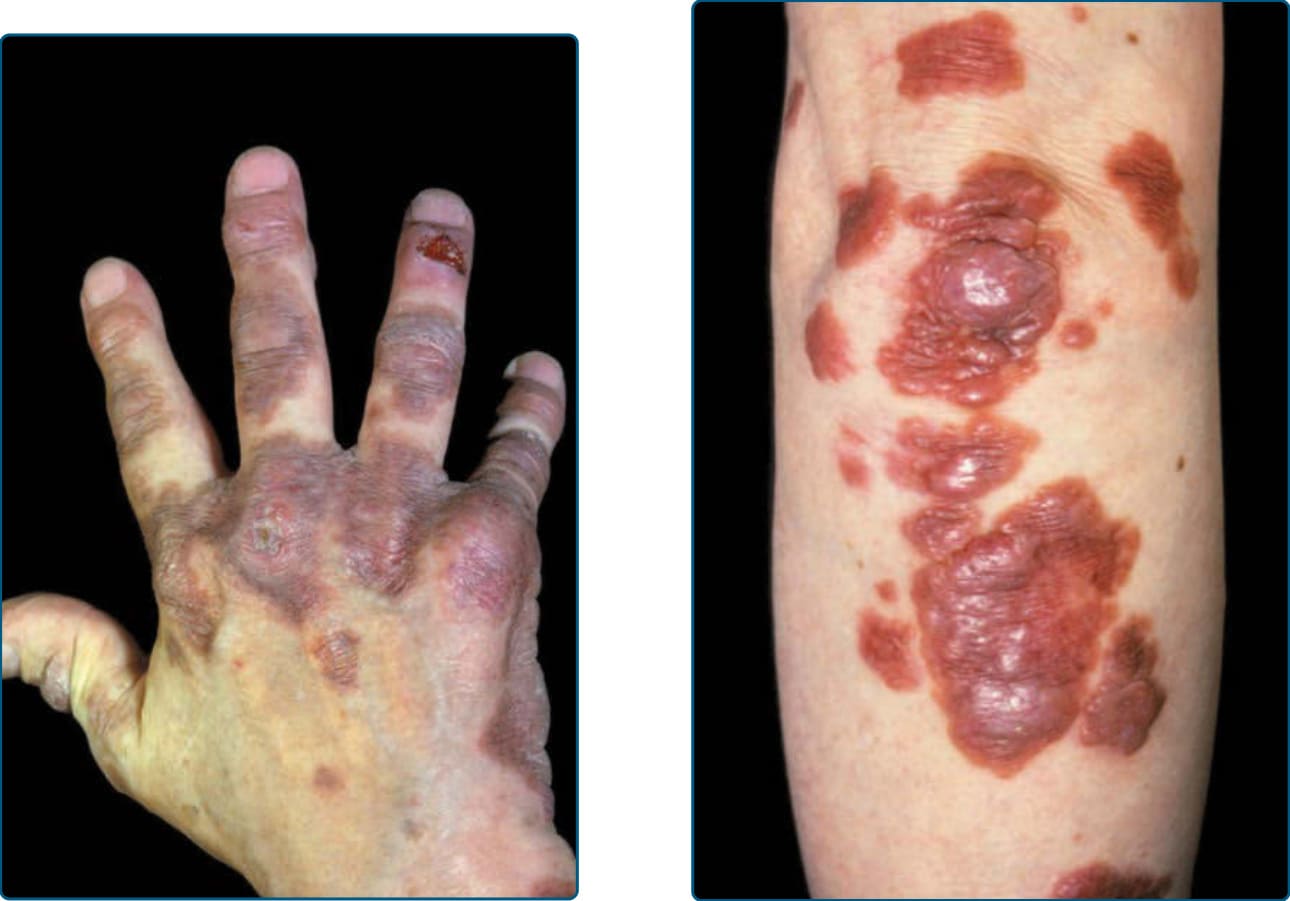
Figure 117-20 Multicentric reticulohistiocytosis.
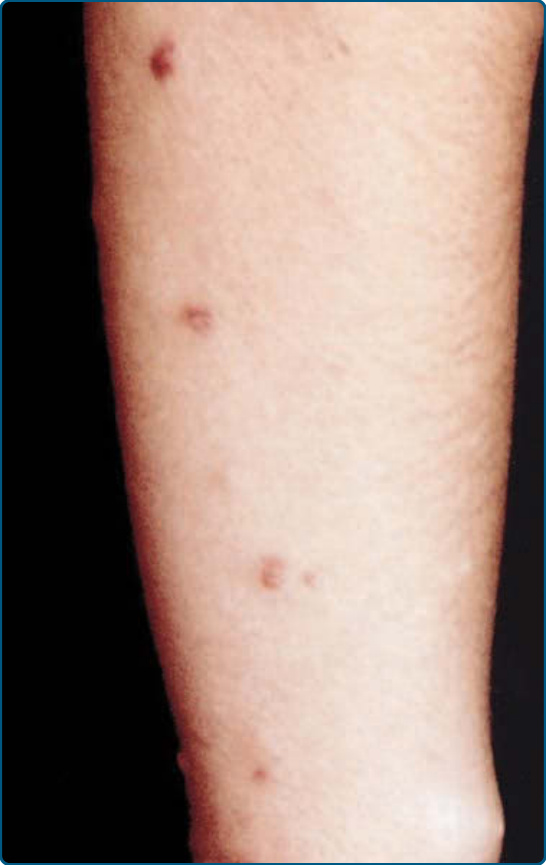
Figure 117-22 Hereditary progressive mucinous histiocytosis.
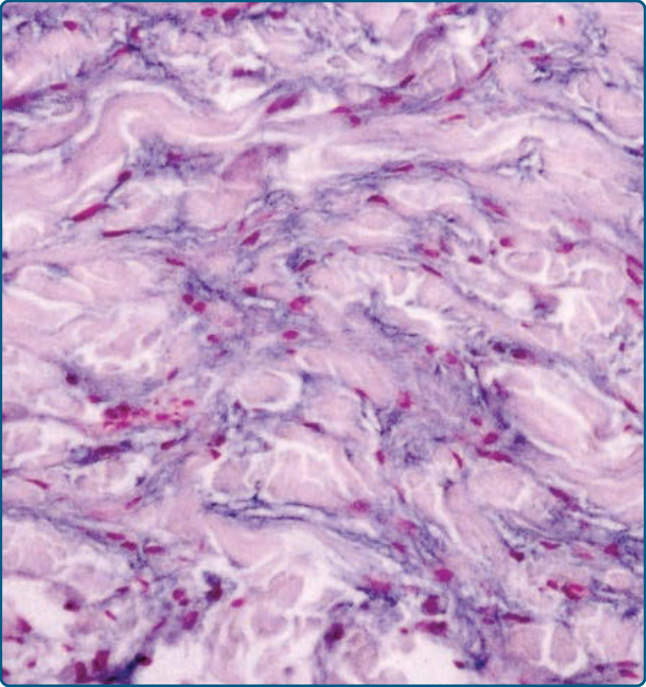
Figure 117-23 Photomicrographs of a lesion of hereditary progressive mucinous histiocytosis. Production of mucin by histiocytes in the dermis is a typical feature of the disease.
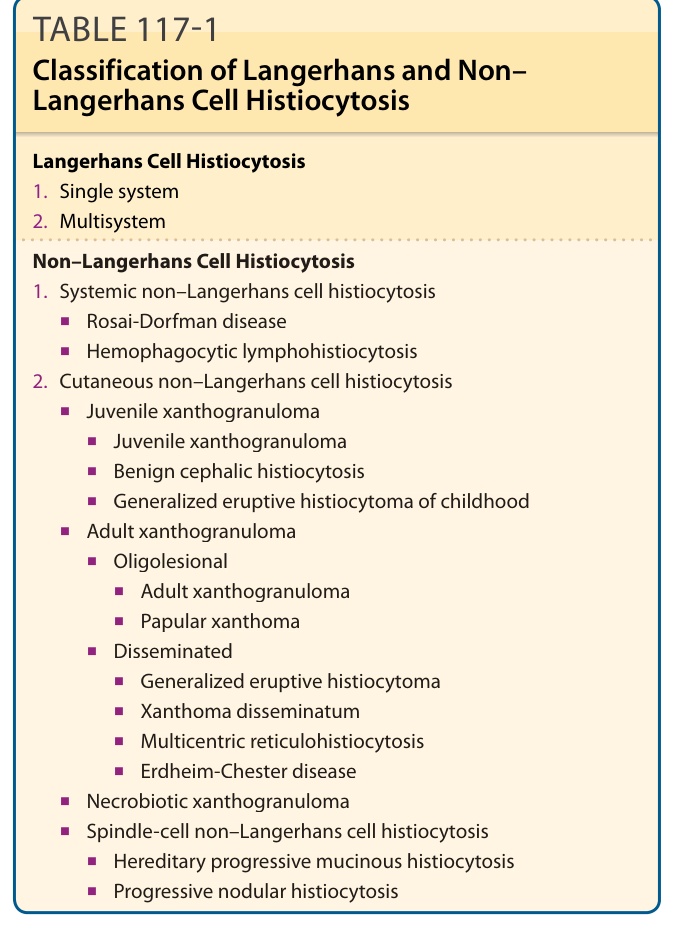
TABLE 117-1 Classification of Langerhans and Non– Langerhans Cell Histiocytosis
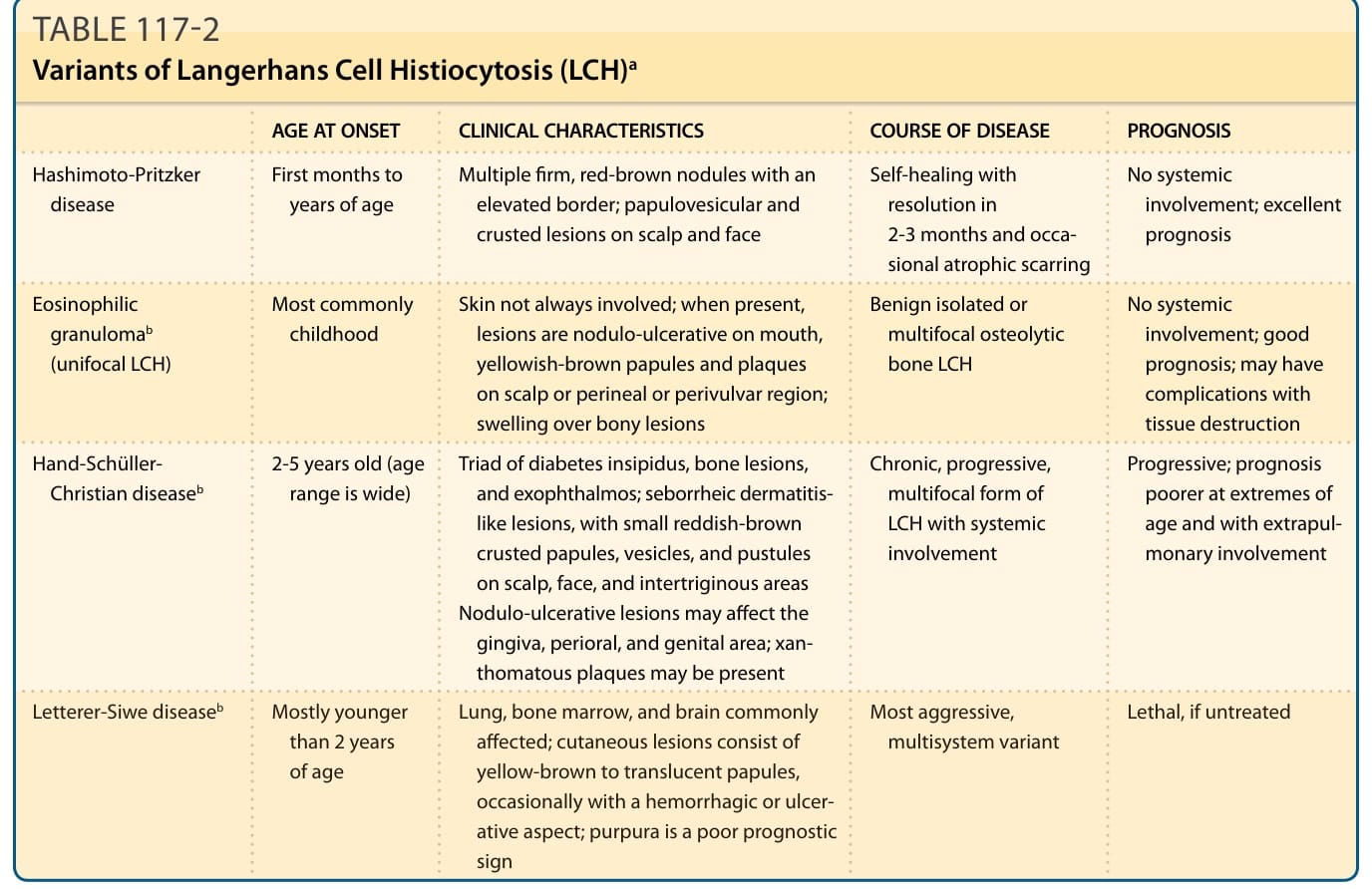
TABLE 117-2 Variants of Langerhans Cell Histiocytosis (LCH)a
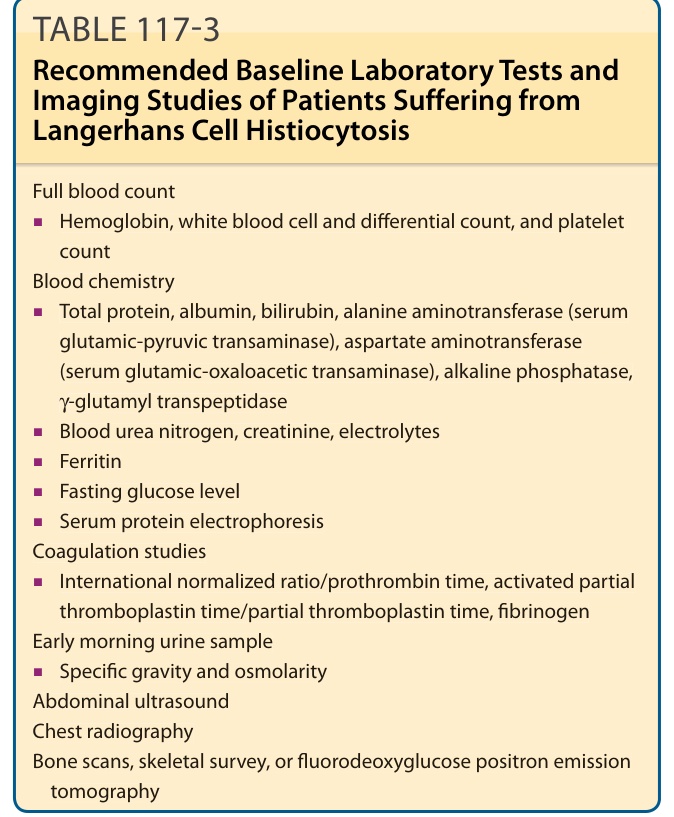
Table 117-3 lists the recommended laboratory investigations and imaging studies for patients suffering from LCH. Table 117-4 identifies the recommendations for specific clinical scenarios.
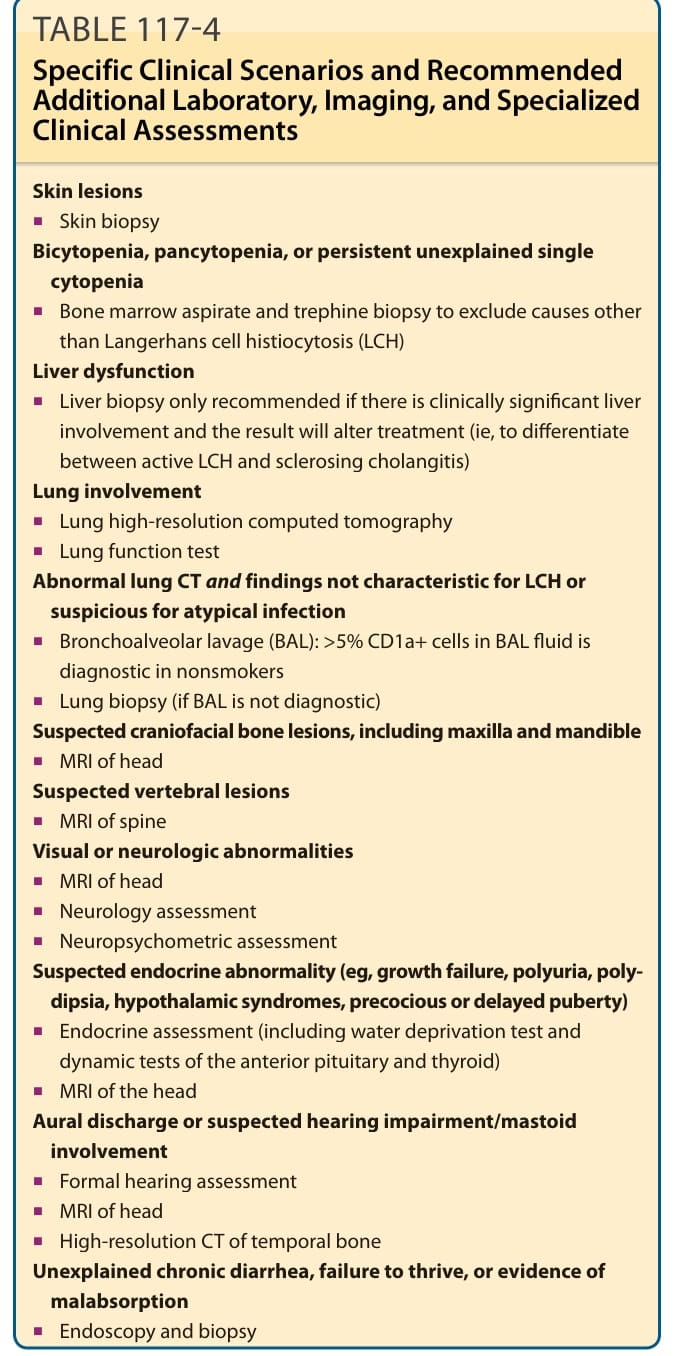
TABLE 117-4 Specific Clinical Scenarios and Recommended Additional Laboratory, Imaging, and Specialized Clinical Assessments
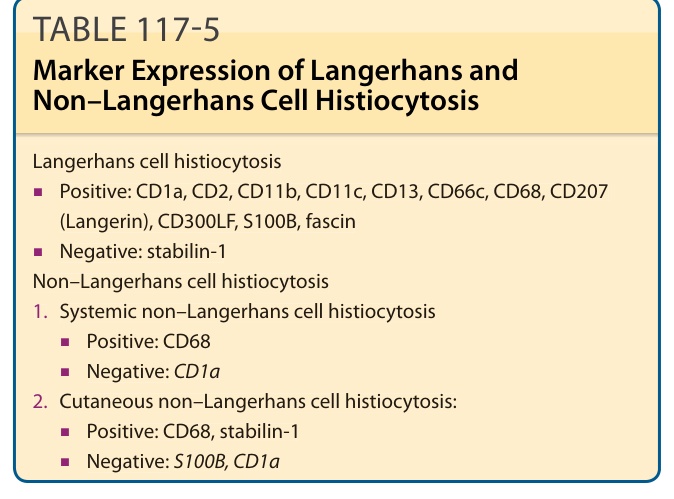
TABLE 117-5 Marker Expression of Langerhans and Non–Langerhans Cell Histiocytosis
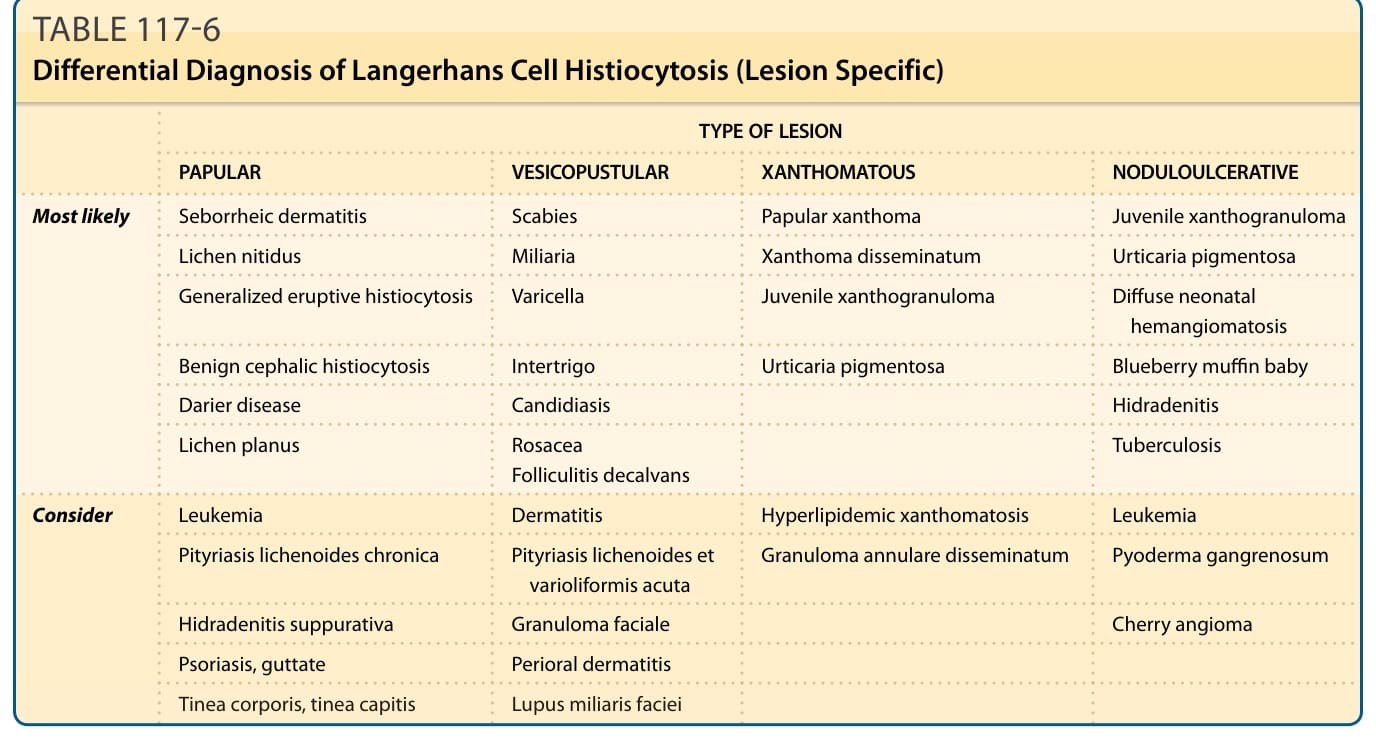
Table 117-6 lists possible differential diagnoses for different clinical manifestations of LCH in the skin.
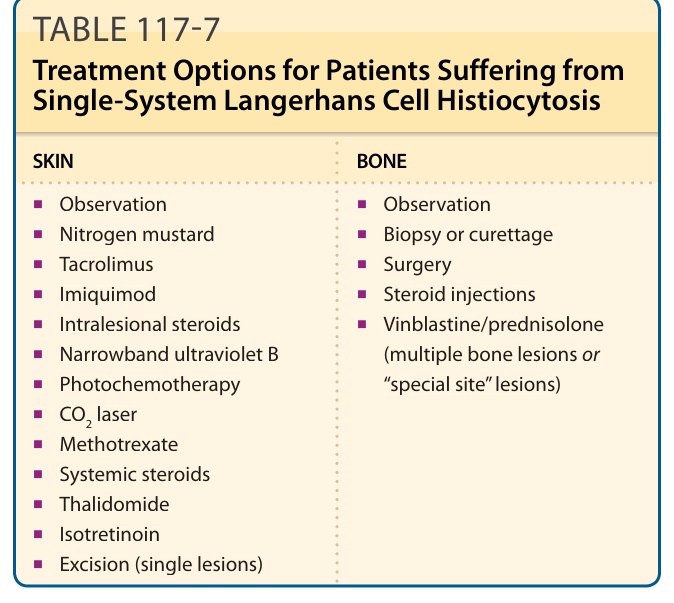
TABLE 117-7 Treatment Options for Patients Suffering from Single-System Langerhans Cell Histiocytosis

TABLE 117-8 Differential Diagnosis of Juvenile Xanthogranuloma