黑色素瘤 (Melanoma) 重點精華
定義與分類
- 黑色素瘤 (melanoma) 是源自黑色素細胞 (melanocytic cells) 的惡性腫瘤,可發生於任何有此類細胞之處;最常見為皮膚黑色素瘤 (cutaneous melanoma),亦可見於黏膜 (mucosal)、葡萄膜 (uveal)、腦膜。
- 百分之十的黑色素瘤以淋巴結轉移、即「原發部位不明 (unknown primary)」被偵測,可能源自結內痣 (nodal nevi)。
- 若早期診斷與手術治療可治癒,診斷太晚則具潛在致命性。
流行病學
- 全球發生率持續上升;最高為澳洲/紐西蘭(每年每 100,000 人約 35 例),其次北歐、北美(美國 21.8 例)、歐洲(13.5 例)。
- 診斷中位年齡 63 歲,15% 小於 45 歲;死亡中位年齡 69 歲。
- 黑色素瘤僅占皮膚癌診斷的 4%,卻造成 75% 的皮膚癌死亡。
臨床亞型
- 表淺擴散型黑色素瘤 (superficial spreading melanoma, SSM):最常見(約 70%),間歇曝曬部位(女性下肢、男性上背),最符合 ABCD 準則,最常與既存痣相關,病程數月至數年緩慢變化。
- 結節型黑色素瘤 (nodular melanoma, NM):第二常見(15%–30%),軀幹好發,快速演變(數週至數月),常缺乏輻射狀生長期、多為從頭新生 (de novo);典型深藍黑/藍紅隆起,5% 為無色素型。SSM 與 NM 的 BRAF 突變率最高(達 56%)。
- 惡性小痣 (lentigo maligna, LM) / 惡性小痣黑色素瘤 (LMM):LM 為原位黑色素瘤、輻射狀生長期長,侵襲後成 LMM(占 10%–15%);好發七、八十歲慢性日曬臉部;亞臨床側向生長廣、復發率高;與 c-KIT 異常相關(達 28% vs BRAF 6%)。
- 肢端小痣型黑色素瘤 (acral lentiginous melanoma, ALM):白人僅占 2%–8%,深色素族群最常見(非裔 60%–72%、亞洲 29%–46%);好發足底,其次手掌、甲下;不與日曬相關。甲下黑色素瘤源自甲基質,可見縱向黑甲 (melanonychia striata) 與 Hutchinson 徵象 (Hutchinson sign)(近端甲褶色素)。
- 促結締組織增生性黑色素瘤 (desmoplastic melanoma, DM):第六、七十年日曬頭頸部,堅實硬化、半數無色素,傾向神經周圍生長;>90% 帶 NF1 突變,無 BRAF/NRAS 突變。
- 黏膜黑色素瘤 (mucosal melanoma):占 1.3%,頭頸、生殖器、肛門直腸黏膜;女性較常見(生殖道為主),鼻腔為兩性最常見部位;分子上以 RAS、c-kit 突變較多,<10% 帶 BRAF。
- 痣樣黑色素瘤 (nevoid melanoma)、Spitz 樣黑色素瘤 (spitzoid melanoma):組織學易誤診;Spitz 樣可達 >1 cm、Breslow >2 mm、有絲分裂多者較支持惡性。
- 葡萄膜黑色素瘤 (uveal melanoma):約占 5%,最常見原發性眼內惡性腫瘤;第 3 號染色體單體 (monosomy 3) 預後差(5 年疾病特異性死亡率 75.1% vs 雙體 13.2%),主要轉移至肝。
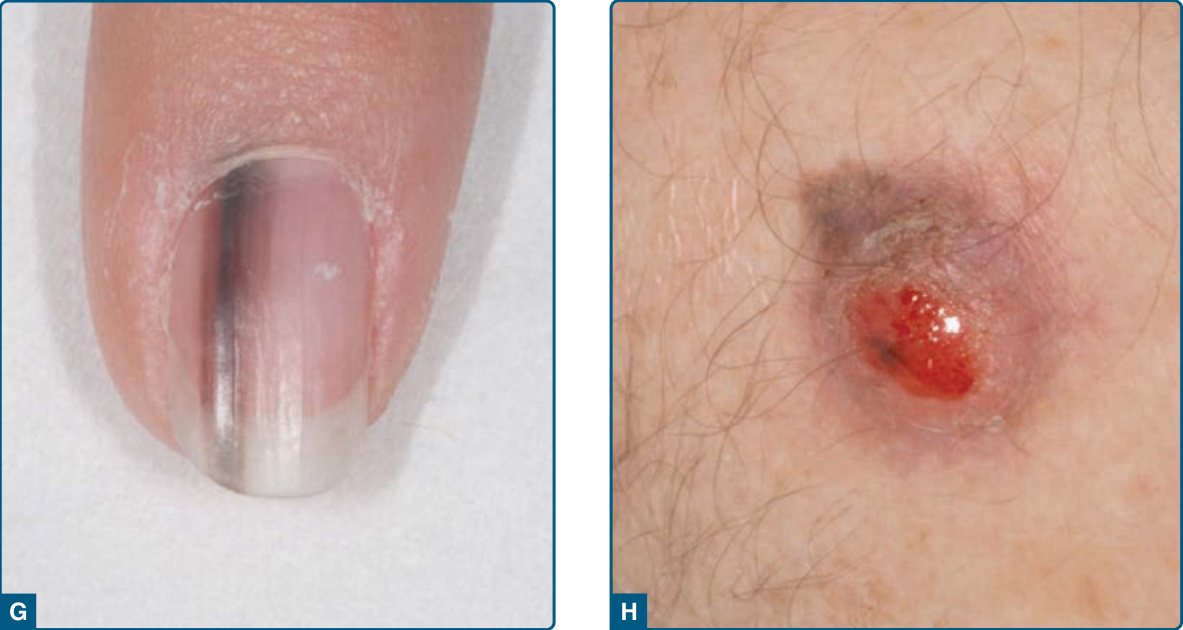
圖 116-2:黑色素瘤臨床亞型(A、B 表淺擴散型,C 無色素結節型,D 色素結節型,E 惡性小痣黑色素瘤,F 肢端小痣型,G 甲下,H 促結締組織增生性)。
病因與發病機轉
- 兩大最重要因子:日曬與遺傳。
- 日曬 (UV):間歇性強烈曝曬(尤其童年青春期)較長期持續曝曬更重要(間歇性曝曬假說);童年一次起水泡曬傷使日後黑色素瘤風險增加超過一倍。PUVA、UVB、助曬床均增加風險(助曬床勝算比 1.69)。澳洲試驗顯示每日防曬乳使原發黑色素瘤發生率減半(11 vs 22 例)。
- 皮膚表型:淺膚色、金/紅髮、藍/綠眼、雀斑、Fitzpatrick I–II 型風險增加 2–3 倍;V–VI 型少見。
- 黑色素細胞痣:>100 顆典型痣成人、>50 顆典型痣兒童、任何非典型痣者皆有風險;單一發育不良痣使風險加倍,≥10 顆非典型痣風險升 12 倍。巨大先天性痣(成年 >20 cm)一生黑色素瘤風險 2%–10%。
- 家族史:占 5%–12%;一位一等親加倍風險,≥3 位升 35–70 倍。
- 遺傳基因:CDKN2A(編碼 p16 與 p14ARF)生殖細胞系突變約占遺傳性病例 40%,帶因者一生風險 76%(美國)、91%(澳洲)、58%(英國),並增胰臟癌風險(約 15%);CDKN2A、CDK4、POT1、TERT 突變賦予 60%–90% 一生風險。
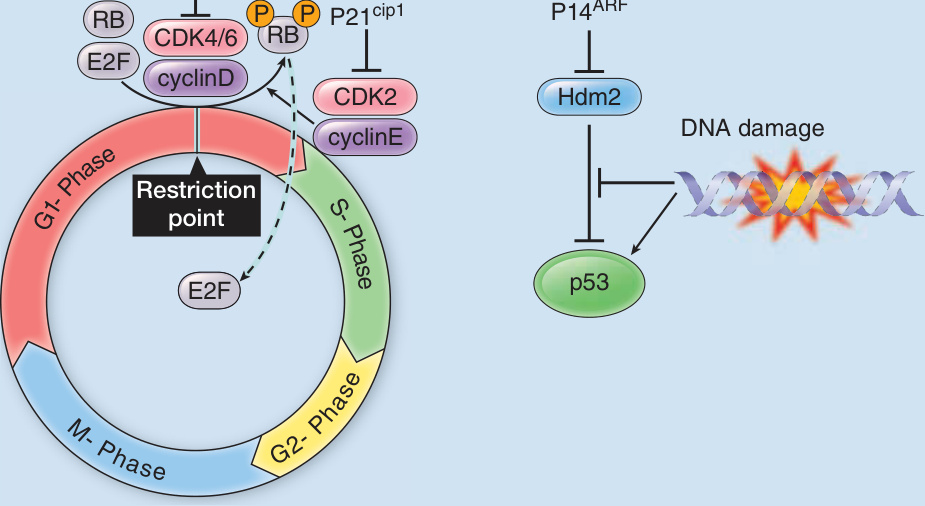
圖 116-5:腫瘤抑制基因 CDKN2A 突變導致細胞週期控制喪失與抗細胞凋亡。
診斷
- 早期偵測為改善預後關鍵;無單一臨床特徵可確診或排除。
- ABCDE 準則:A 不對稱、B 邊緣、C 顏色、D 直徑(>5 mm)、E 演變;敏感度高、特異度低;不適用於結節型與促結締組織增生性。另有「醜小鴨」徵象 (“ugly duckling” sign)。
- 臨床診斷可在約 80%–90% 病例做出。
- 皮膚鏡 (dermoscopy):改善敏感度與特異度;型態分析 (pattern analysis) 最廣泛使用;序列性數位皮膚鏡改善早期診斷。
- 組織學:首選窄切緣切除性切片 (excisional biopsy with narrow margins),避免寬切緣以利後續 SLNB;主要特徵為不對稱、界限不良、體積大(>5–6 mm)、巢缺乏成熟、佩吉特樣擴散 (pagetoid spread)。免疫組化:S100、Sox10 幾乎全表現;HMB-45 特異度高;Melan-A 比 HMB-45 敏感、比 S100 特異;DM 僅表現 S100、Sox10。組織學另提供 Breslow 厚度與潰瘍狀態(AJCC 分期所需)。
- 實驗室:LDH 為遠端轉移病人 AJCC 分類所需;S100B 較特異但敏感度低,用於追蹤。
- 影像:皮膚與淋巴結超音波最敏感;CT/MRI/PET 僅用於高風險(>4 mm Breslow)與已知轉移者,原發階段不建議(偽陽性 8%–15%)。
- 前哨淋巴結切片 (SLNB):強大分期與預後工具,建議用於 Breslow ≥1 mm;較薄者(0.8–1 mm 或 <0.8 mm)若有高有絲分裂指數、淋巴血管侵犯或年輕亦可考慮。鎝-99 標記放射性膠體加藍染劑可 >98% 偵測 SLN。MSLT-I:SLN 陰性 5 年存活 90.2% vs 陽性 72.3%。DECOG-SLT 與最終分析顯示僅顯微結節疾病者立即 CLND 無存活益處,已不再建議。
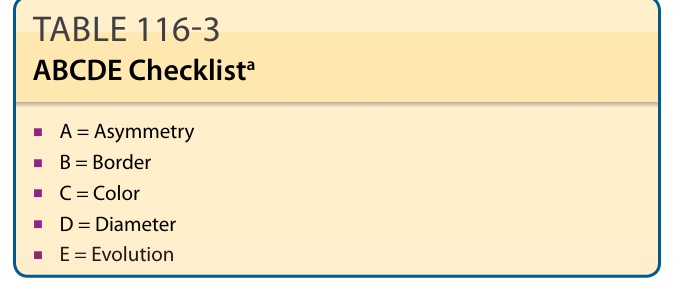
表 116-3:ABCDE 檢核表。
圖 116-7:診斷演算法。
臨床病程與預後
- 初次表現:約 85% 局限性、10% 區域轉移、5% 遠端轉移。
- 第 I/II 期:Breslow <1 mm 局限性薄原發者 5–10 年存活 >90%;多數復發在 5 年內,晚期復發(≥10 年)約 1%–5%。
- 第 III 期:整體 5 年存活 38%–78%,主要取決於陽性淋巴結數量(最重要)、腫瘤負荷、年齡、潰瘍、Breslow 厚度。
- 第 IV 期:可轉移至任何器官;常見內臟部位為肺(18%–36%)、肝(14%–20%)、腦(12%–20%)、骨(11%–17%)、胃腸道(1%–7%)。未治療中位存活約 6 至 9 個月,標靶與免疫療法可增至 2 年。
- 原發部位不明轉移 (MUP):占 2%–5%,約 60% 牽涉淋巴結(可能源自結內痣);轉移至淋巴結 5/10 年存活 46%/41%,轉移至內臟中位存活 6 個月。
- AJCC 2017 分期:取自 >46,000 病人;TNM 系統為骨幹。
- 預後因子:女性優於男性;年齡增加預後差(>60 歲男性死亡率最高)。原發病灶最重要因子為腫瘤厚度 (Breslow index)(從顆粒層頂端量至最深侵襲,mm);Clark 分級已不用於常規分期。潰瘍 (ulceration) 為獨立預後因子(<1 mm 為 6%、>4 mm 為 63%),有潰瘍者預後較差並使分期上升。有絲分裂率在新分類已非獨立因子。顯微衛星病灶 (N1c) 即屬第 III 期。腫瘤浸潤淋巴球 (TILs) 分級越高(brisk)預後越好。區域:淋巴結狀態最強,淋巴結數量最顯著;中途/衛星轉移 (N1c/N2c/N3c) 5 年存活 <50%。遠端:內臟轉移較非內臟差,升高 LDH 預後差。
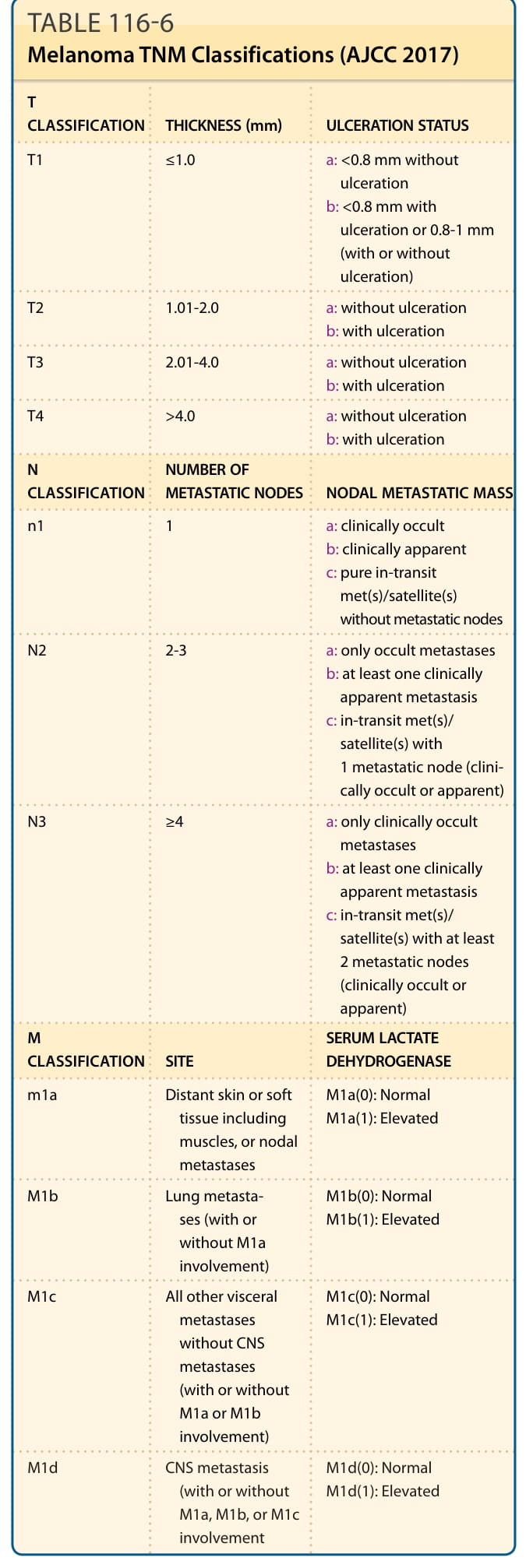
表 116-6:黑色素瘤 TNM 分類(AJCC 2017)。
治療
原發黑色素瘤手術
- 標準為廣泛局部切除 (wide local excision, WLE)。切緣依 Breslow 厚度:原位黑色素瘤 0.5–1 cm;<1 mm 為 1 cm;1–2 mm 為 1–2 cm;>2 mm 為 2 cm。高達 5 cm 的較寬切除無益處。原則為腫瘤清除優先、重建其次。
區域轉移手術
- 選擇性淋巴結廓清 (ELND) 今日已無作用。顯微結節疾病者立即 CLND 無存活益處(DECOG-SLT:3 年 OS 81.2% vs 81.7%;僅降低區域復發 8.3% vs 14.6%),不再建議。巨觀(IIIB/C)結節疾病標準為對受累盆 CLND。
輔助治療
- 干擾素-α (interferon-α):高劑量 (HDI) 與低劑量 (LDI)。HDI 誘導期 20 million units/m²/day 靜脈注射、每週 5 天、4 週,維持期 10 million units/m²/day 皮下、每週 3 次、48 週;LDI 為 3 million units、每週 3 次皮下、1.5 年。Cochrane 統合分析顯示改善無病存活(HR 0.83)與整體存活(HR 0.91);HDI 因有更有效藥物已不再建議。
- 免疫檢查點阻斷劑:第 III 期 ipilimumab(10 mg/kg 每 3 週共 4 劑,後每 3 個月最多 3 年)使中位無復發存活從 17.1 延至 26.1 個月。nivolumab、pembrolizumab(抗 PD-1)更有效、耐受性更佳,已 FDA 核准(Checkmate 238、Keynote-054)。
- 輔助放射治療:CLND 後控制結節盆轉移;風險因子為 ≥3 個淋巴結轉移、包膜外擴散、淋巴結 >3 cm;提升局部區域控制但無存活益處。
衛星/中途轉移
- 盡量手術切除。其他選擇:放射(加高溫療法)、病灶內介白素-2(小型表淺轉移,每週 2–3 次反應率 >80%)、溶瘤病毒 T-VEC(不可切除 IIIB/C 或 M1a)、電化學療法 (ECT,bleomycin 或 cisplatin 加電脈衝,單一轉移反應率 80.6%)、隔離肢體灌流 (ILP,melphalan,反應達 80%)。
不可切除轉移性疾病
- 手術/放射:可考慮轉移切除術、腦轉移立體定位放射、緩和性放射。
- 免疫療法:ipilimumab(抗 CTLA-4)為首個對第 IV 期 OS 有顯著益處者,反應率約 10% 但 >20% 達 >3 年長期控制。抗 PD-1(nivolumab、pembrolizumab)30%–40% 顯著縮小、2 年 OS 約 60%。ipilimumab + nivolumab 組合(Checkmate 067)反應率 58%、中位 PFS 11.5 個月(單藥 ipilimumab 19.0%、2.9 個月);3 年 OS 組合 58%、nivolumab 52%、ipilimumab 34%。副作用為免疫相關不良事件 (irAEs):皮膚、肝、胃腸道、內分泌,組合治療嚴重副作用達 55%。眼部黑色素瘤免疫療法成效有限(中位 OS 6.8 個月)。
- irAEs 處置:第 1 級症狀治療;第 2 級口服 1 mg/kg prednisolone 並中斷治療;第 3/4 級住院、IV 2 mg/kg methylprednisolone,2 天無改善加 mycophenolate mofetil 3 g 口服/日(肝炎)或 infliximab 5 mg/kg IV(結腸炎)。
- 過繼性 T 細胞療法 (ACT/TIL)、高劑量 IL-2(單藥反應率 16%,持久反應 5%–8%;現一般不再全身性使用)。
標靶治療
- BRAF V600E 突變可見於約 50% 黑色素瘤。選擇性 BRAF 抑制劑 vemurafenib、dabrafenib(FDA 核准),約 50% BRAF V600 突變者反應且極快速,中位 PFS 7–8 個月、中位 OS 16–18 個月;多數日後出現抗藥性。BRAF + MEK 抑制劑(trametinib、cobimetinib)組合反應率達 70%、中位 PFS 10–11 個月、中位 OS 約 2 年,為 BRAF 突變病人照護標準。皮膚副作用含皮疹、過度角化、鱗狀細胞癌(矛盾性 MAPK 活化);vemurafenib 光敏感、dabrafenib 發燒;MEK 抑制劑可致心臟與眼部副作用。NRAS 突變者 binimetinib 療效有限(反應率 15%、PFS 2.8 個月)。
- c-KIT 抑制:肢端與黏膜黑色素瘤帶 KIT 突變者,imatinib、nilotinib 反應率 16%–23%(exon 11 之 L576P 反應最高)。
化療
- dacarbazine (DTIC) 為唯一 FDA 核准化療,反應率約 10%、中位反應 4–6 個月;temozolomide (TMZ) 口服、療效與 DTIC 相當。組合化療無存活益處且毒性高。免疫與標靶治療核准後化療退居次要。
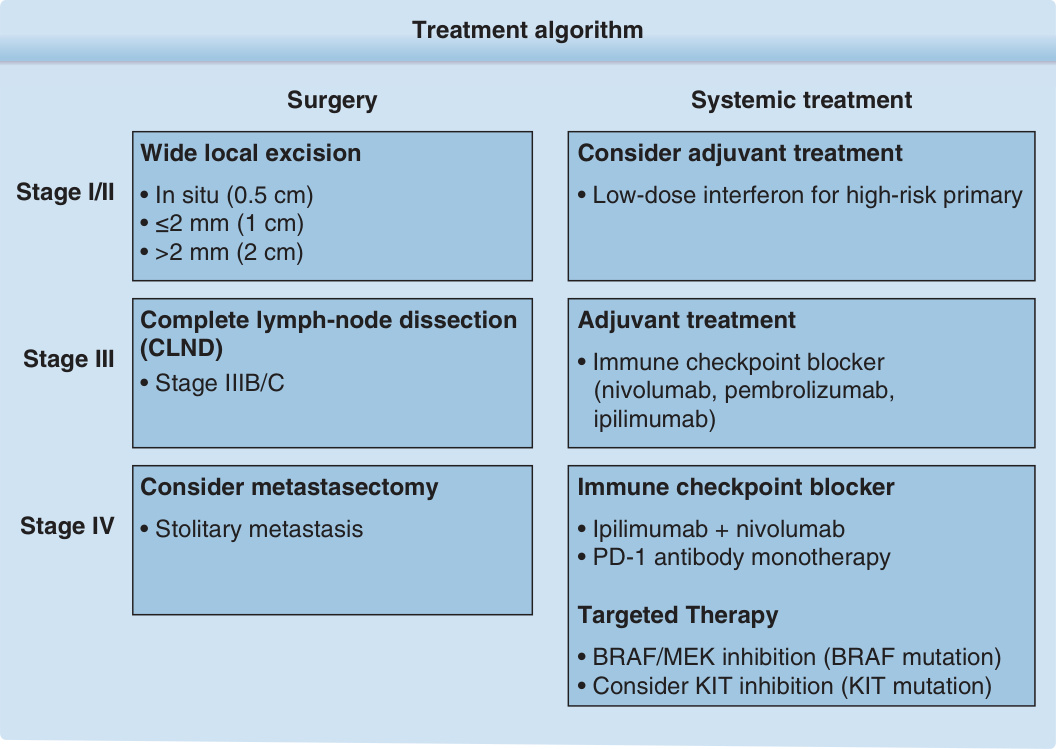
圖 116-14:治療演算法。
預防與追蹤
- 初級預防:安全日曬、限制 UV、防曬(兒童青春期最重要);次級預防:早期診斷。
- 規律追蹤;NCCN 建議終身至少每年一次,診斷後 1–3 年每 3–6 個月,之後依分期每年。強家族史或胰臟癌家族史應轉介遺傳諮詢。