黑色素細胞痣 (Melanocytic Nevi) — 精華筆記
總論
- 黑色素細胞痣 (melanocytic nevi):黑色素細胞 (melanocytes) 的良性腫瘤,特徵為平庸黑色素細胞以巢 (nests,三個以上細胞直接接觸,又稱 thèque) 存在於表皮、真皮或他處。構成細胞稱痣性黑色素細胞 (nevomelanocytes)。
- 黑色素細胞增生 (melanocytic hyperplasia):黑色素細胞侷限於表皮基底層增多、無巢形成。
- 兩大來源:源自交界處黑色素細胞者多為後天性痣 (acquired nevi);源自神經嵴 (neural-crest) 前驅細胞遷移停留者為先天性黑色素細胞痣 (congenital melanocytic nevi, CMNs)。
- 發育不良痣 (dysplastic nevus, DN):具非典型構築與細胞學特徵者(曾稱 B-K 痣、Clark 痣、非典型痣等)。
先天性黑色素細胞痣 (Congenital Melanocytic Nevi, CMN)
- 流行病學:見於約百分之一至三的新生兒;多為小至中型。遲發性先天性痣 (tardive congenital nevi) 可於出生後 1 個月至 2 歲首次出現。巨大 CMN 罕見(達 99 mm 以上者約每 20,000 新生兒 1 例)。
- 分類(依預估成人尺寸):小型 <1.5 cm;中型 M1 1.5–10 cm、M2 >10–20 cm;大型 L1 >20–30 cm、L2 >30–40 cm;巨大 G1 >40–60 cm、G2 >60 cm。衛星痣計數 S1 <20、S2 >20–50、S3 >50。
- 臨床表現:多為扁平褐色斑片或斑塊,常伴多毛 (hypertrichosis);隨時間出現色素變異與表面不規則(鵝卵石樣、疣狀、分葉狀)。顏色雜亂、藍黑色調、潰瘍出血等非典型表現須警惕惡性。
- 致病機轉:胚胎黑色素細胞之合子後體細胞突變 (postzygotic somatic mutations),主要為 MAPK 路徑的 NRAS 突變(大型/巨大病灶最盛行);小至中型可帶 NRAS 或 BRAF。一研究中確認出生即存在的 32 例有百分之八十一帶 NRAS、無 BRAF。
- 相關狀況:頭頸或後正中線 CMN 須注意神經皮膚黑色素沉著症 (neurocutaneous melanosis, NCM)——痣細胞侵犯腦膜/腦實質,嬰幼期以癲癇、水腦症表現,有症狀者預後差,常於症狀後 3 年內死亡。
- 併發症(黑色素瘤):整體發生率約百分之一至二,與痣尺寸/細胞數成比例。>40 cm 且帶衛星痣者終生風險約百分之十至十五。傾向發生於較深真皮/皮下,侵襲性高。增生性結節 (proliferative nodules) 為良性但須切除,組織學上有時難與黑色素瘤區分。
- 診斷:通常直接;頭部/軸向中線者以 MRI 評估有無 NCM。
- 病理:痣性黑色素細胞自交界區侵犯至皮下,沿神經血管束、附屬器與毛囊皮脂腺單位強化,膠原束間呈索狀/單列排列;可神經化 (neurotization) 形成假梅斯納小體 (pseudomeissnerian corpuscles)。
- 治療:依黑色素瘤風險加美觀/功能考量個別化。可行時對大型痣減積 (debulking)、小至中型切除,大型可分階段;建議延至出生 6 個月後以降低麻醉風險。破壞性治療(磨皮、雷射)不能消除惡性潛能。臨床良性者可選擇仔細監測。所有 CMN 出生時應以高品質照片記錄。
- 預後:除非併發 NCM 或黑色素瘤,否則極佳。UV 在出生即存在病灶之初始發展中不扮演角色。
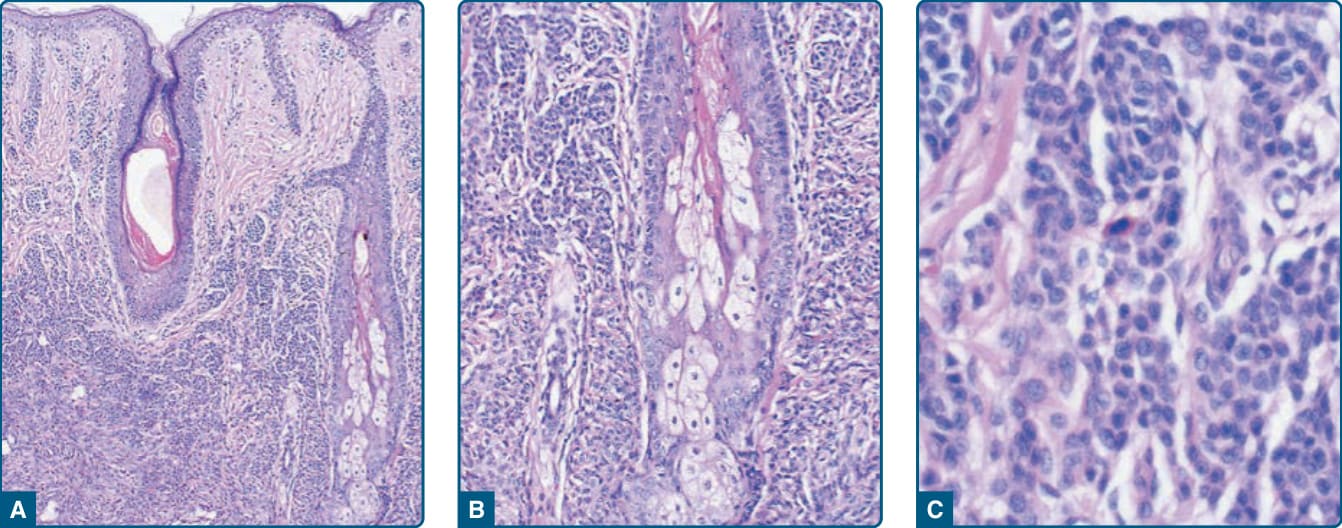
圖 115-4:先天性痣性黑色素細胞痣的組織病理特徵——真皮下三分之二密集累積、侵入附屬器結構。
斑痣 (Nevus Spilus)
- 又稱斑點性雀斑樣痣 (speckled lentiginous nevus),視為先天性痣變異型,見於約百分之一至二人口。
- 臨床:淡褐色(咖啡牛奶色)背景斑片,上有散布的深色斑點或丘疹;常見於軀幹與四肢,可呈節段性。
- 致病機轉:合子後突變啟動黑色素細胞克隆場。散發小型斑痣帶活化性 HRAS 突變 (c.37G->C, p.Gly13Arg);斑痣型先天性黑色素細胞痣 (nevus spilus-type CMN) 則帶 NRAS 活化性突變。
- 相關:可與斑點玫瑰斑痣症 (phakomatosis spilorosea,斑狀型) 或色素角化母斑病 (phakomatosis pigmentokeratotica,丘疹型) 並存。
- 併發症:發育不良斑痣或罕見黑色素瘤;風險隨尺寸增加,尤其 >40 cm 之大型節段性病灶。
- 治療:無標準指引;非典型或不穩定成分應切片排除黑色素瘤。預後極佳。
常見後天性黑色素細胞痣 (Common Acquired Melanocytic Nevus)
- 又稱痣細胞痣 (nevocellular nevus);依細胞位置分交界型 (junctional)、皮內型 (intradermal)、複合型 (compound)。
- 流行病學:出生後發展,數目於第三個十年達高峰後減少;淺膚色、易曬傷者較多。
- 臨床:多 <6 mm,表面與色素均質、圓至橢圓、邊緣清楚。淺膚色者出現很深褐/黑、或藍灰紅白色須警惕黑色素瘤。甲器痣呈縱向條紋,色素延伸至近端甲褶 (proximal nail fold) 或超出遠端甲溝須懷疑黑色素瘤(Hutchinson 徵概念)。
- 暈痣 (Halo nevi):多無症狀、多發於青少年軀幹,代表既有痣的自體免疫退化(CD8+ 細胞毒性 T 淋巴細胞媒介);中央痣周圍對稱色素脫失暈。又稱 Sutton 痣。
- Meyerson 痣:中央痣周圍紅色濕疹樣暈,皮膚不脫色、痣不退化,多自行消退。
- 爆發性痣 (eruptive nevi):多發痣同時發展,見於水疱性疾病、免疫抑制、腫瘤、化療/免疫調節劑。
- 致病機轉:克隆性腫瘤,多帶活化性 BRAF 突變(早期啟動事件)。
- 併發症:痣數目增多增加黑色素瘤風險——高痣計數者(101–120 顆)相對風險 6.89;五顆非典型痣者相對風險 6.52。約百分之二十黑色素瘤有相關痣成分。
- 危險因子:UVR 暴露為最關鍵因子,間歇性強烈日曬風險最大;遺傳因素亦相關。
- 病理:交界痣巢狀位於基底層;複合/皮內痣自上而下「成熟 (maturation)」——淺層較大上皮樣細胞、深層較小細胞並喪失色素,深層真皮 HMB-45 表現降低。變異型含氣球細胞痣 (balloon cell nevus)、複合痣 (combined nevus)、復發性痣 (recurrent nevus,限於疤痕內、外觀可非典型)。特殊部位痣 (special site nevi:頭皮、耳、乳頭、臍、肛門生殖器、肢端) 構築變異較大。
- 特殊檢查:低 Ki67、深層 HMB45 喪失支持良性;FISH/CGH 可助區分痣樣黑色素瘤。
- 治療:絕大多數不需治療;可疑病灶須切除做組織病理,避免破壞性治療(電乾燥、冷凍、磨皮、雷射)以保留組織。完全移除以切除為佳。
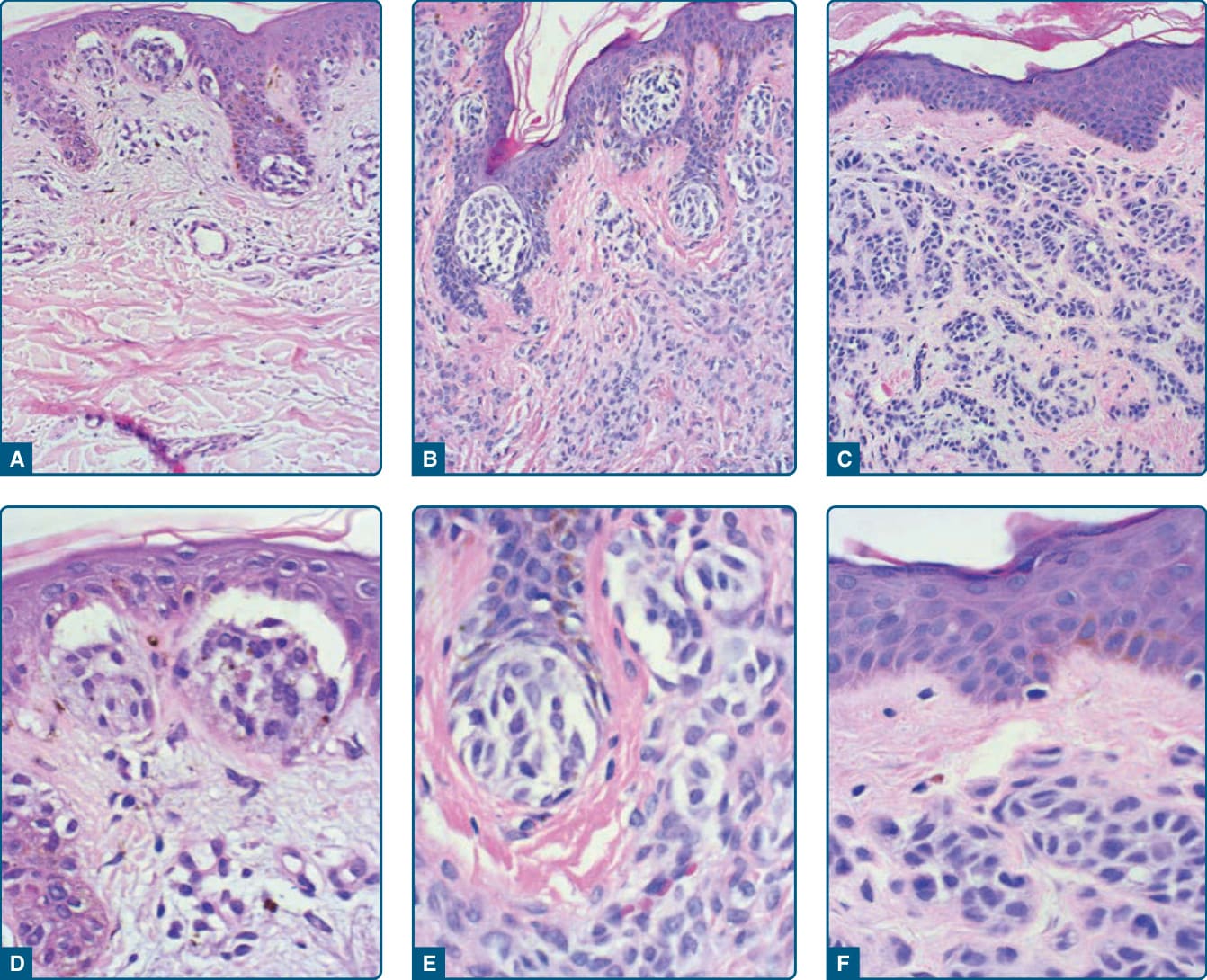
圖 115-8:後天性痣組織病理——交界痣 (A)、複合痣 (B)、皮內痣 (C) 及其高倍率。
藍痣 (Blue Nevus)
- 真皮內深度色素性紡錘形或上皮樣黑色素細胞組成;含常見、細胞型、複合型、非典型細胞型。藍色調來自廷得耳效應 (Tyndall effect)。
- 流行病學:約百分之一至二白人成人、百分之三至五日本成人;女性較多,多於第二個十年出現。
- 臨床:藍/藍灰/藍黑色平滑丘疹或結節,質地堅實。常見藍痣 <1 cm;細胞型藍痣常 >1 cm。相關病灶含太田/伊藤母斑、蒙古斑(多於幼兒早期退化)、真皮黑色素細胞錯構瘤。
- 相關:多發/上皮樣藍痣可見於 Carney 複合症/LAMB 症候群(雀斑樣痣、心房黏液瘤、黏膜皮膚黏液瘤、藍痣)。
- 致病機轉:多帶 GNAQ(高達百分之八十七,幾乎全在 codon 209)、較少 GNA11 啟動性突變,活化 MAPK 路徑。深部穿透痣 (deep penetrating nevi) 不帶 GNAQ/GNA11,部分帶 HRAS、並帶 MAPK 與 β-catenin 路徑突變。
- 併發症:惡性藍痣(黑色素瘤)較常與細胞型藍痣連續發生;頭皮、高頻 GNA11 突變伴 BAP1 表現喪失。
- 治療:穩定常見藍痣不需治療;臨床變化或大型/非典型須切片。細胞型藍痣(尤其成人)應評估切除,並含皮下脂肪以移除深層成分。
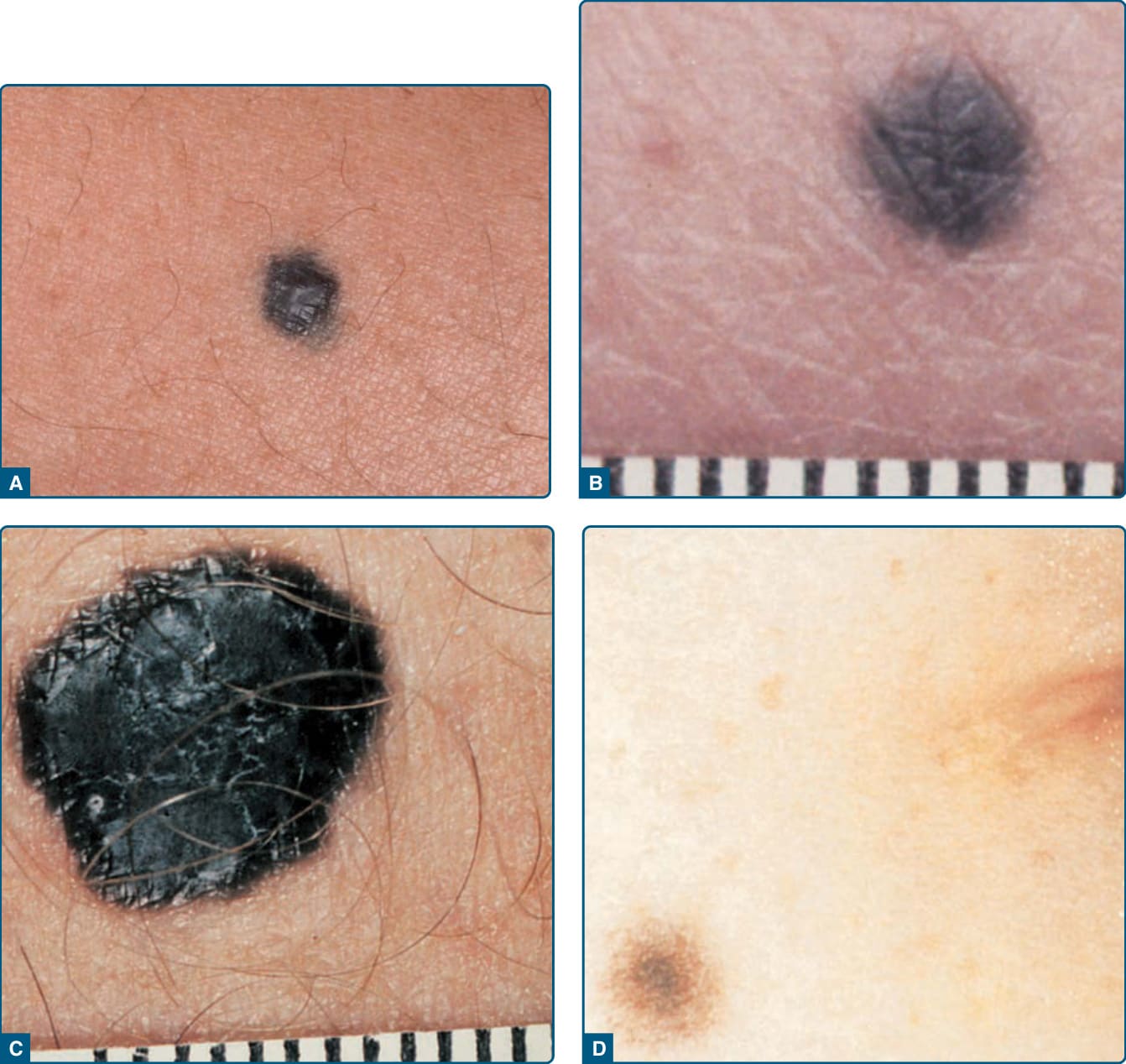
圖 115-10:藍痣——常見藍痣、細胞型藍痣與複合型藍痣的臨床表現。
色素性紡錘細胞痣 (Pigmented Spindle Cell Nevus, PSCN)
- 又稱 Reed 痣 (Reed nevus),由重度色素性紡錘形黑色素細胞以巢狀主要侷限於表皮組成;多被視為 Spitz 痣變異型。
- 流行病學:多於第三個十年出現(平均約 25 歲),女多於男(達 2:1),偏好四肢尤其大腿。
- 臨床:界限分明、墨黑色丘疹通常 <7 mm;皮膚鏡常見條紋/假足構成「星芒狀 (starburst)」外觀。
- 病理:均一合成色素之紡錘形黑色素細胞束沿交界處;可有 Kamino 樣特徵與佩吉特樣細胞(多限於中央)。非典型變異型須與黑色素瘤鑑別。
- 治療:因與黑色素瘤鑑別困難,應切除取得乾淨邊緣。預後良性。
Spitz 痣 (Spitz Nevus)
- 又稱紡錘與上皮樣細胞痣 (spindle and epithelioid cell nevus);上皮樣與紡錘形黑色素細胞,豐富嗜伊紅細胞質、大核、顯著核仁。
- 流行病學:年發生率約每十萬人 1.4–1.6 例;約百分之七十發生於 20 歲以下;白人較多。
- 臨床:典型單發、無症狀、粉紅/紅色、無毛、堅實、圓頂狀丘疹。可呈群集 (agminated),常於斑痣背景內或大型 CMN 內發生。偏好頭頸與四肢,不發生於掌蹠黏膜。
- 致病機轉:常帶 HRAS 突變或激酶基因 (ALK, ROS1, NTRK1, BRAF, RET, MET) 基因組重排。BAP1 喪失合併 BRAF 突變為一非典型亞群特徵;生殖系 BAP1 突變致 BAP1 相關癌症易感症候群(皮膚/葡萄膜黑色素瘤、間皮瘤、腎細胞癌等風險增)。
- 病理:界限分明、對稱,隨深度漸進成熟,呈楔形向皮下變窄;表皮內嗜伊紅小球 (Kamino 小體) PAS 陽性且抗澱粉酶;交界處巢有垂直流動 (vertical streaming)。非典型特徵(核多形性、有絲分裂增多/深層、不對稱、缺乏成熟)須警惕非典型 Spitz 腫瘤或黑色素瘤。
- 特殊檢查:Spitz 痣多保留 p16,Spitzoid 黑色素瘤傾向喪失 p16;Ki-67 在痣中低、黑色素瘤升高;CGH/FISH 助判生物學潛能。
- 治療:良性 Spitz 痣完全切除至乾淨邊緣即足;令人擔憂者留較寬邊緣。前哨淋巴結切片對非典型 Spitz 腫瘤角色具爭議。
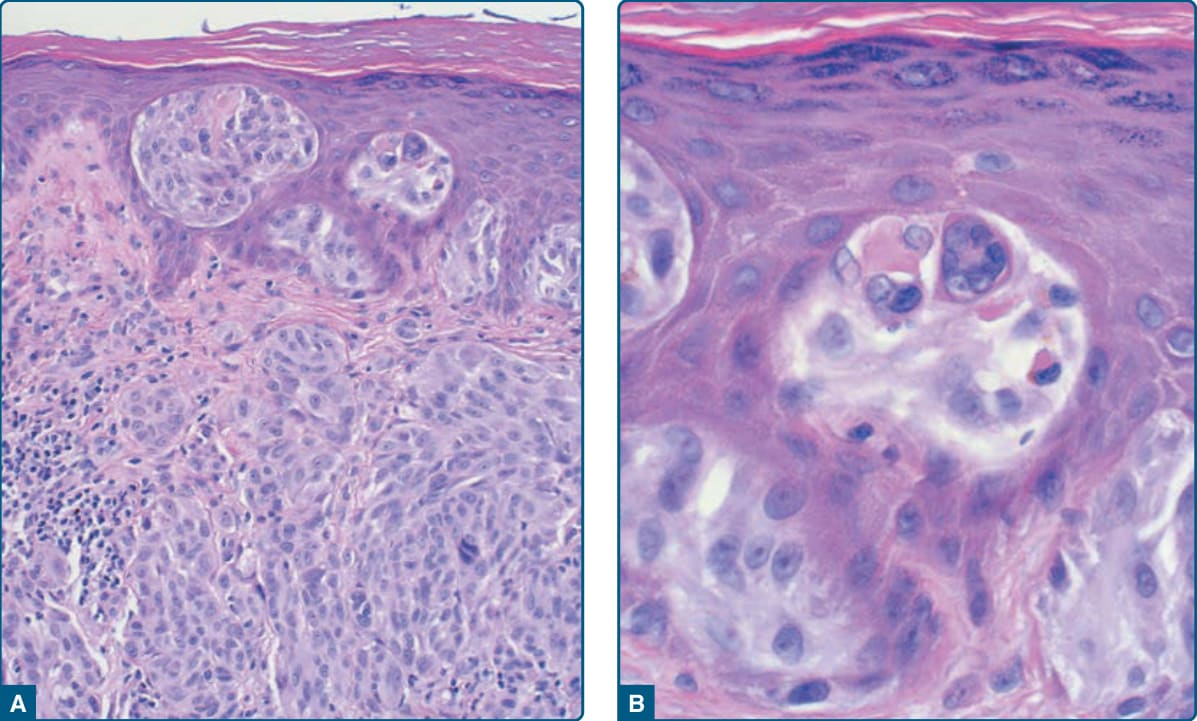
圖 115-15:Spitz 腫瘤組織病理——表皮內嗜伊紅小球(Kamino 小體)。
淋巴結痣 (Nodal Nevi)
- 存在於淋巴結(特徵性位於被膜 capsule,亦可在實質)的良性黑色素細胞腫瘤,通常無症狀偶然發現。
- 頻率:與黑色素瘤無關之淋巴結切除中 0.3%–7.3%;因黑色素瘤移除之區域淋巴結中高達百分之二十二。與引流區皮膚痣(尤以先天性痣)顯著相關。
- 機轉(兩理論):發育期黑色素母細胞被困於淋巴結;或自皮膚病灶被動轉移(如同碳、刺青墨水、放射性膠體)。
- 病理:缺乏細胞異型性與有絲分裂活動。HMB-45 弱/陰性、缺乏 Ki67、強 p16 表現支持良性;脂肪酸合成酶與乙醯輔酶 A 羧化酶在淋巴結痣不表現但在轉移性黑色素瘤表現。
- 臨床意義:誤判為惡性可致不必要淋巴結廓清與全身治療,誤判惡性為良性則治療不足。良性者不需治療。
單純性雀斑樣痣 (Lentigo Simplex)
- 色素增多斑點,可單發、群集 (agminated) 或廣泛分布;發生於皮膚、結膜與黏膜,常於幼兒早期發展。良性、無直接併發症。
- 臨床:界限分明 1–5 mm 斑點,均勻淡褐至黑色。多發雀斑樣痣可與遺傳症候群相關:LEOPARD 症候群、Carney 症候群、Peutz-Jeghers 症候群、Laugier-Hunziker 症候群、顏面中央雀斑樣痣病、LAMB 症候群。亦可與愛迪生氏病、tacrolimus 等鈣調神經磷酸酶抑制劑相關。
- 致病機轉:機轉未明;與日光性雀斑樣痣不同,缺乏 BRAFV600E、FGFR3、PIK3CA 突變。
- 病理:網脊輕至中度伸長、基底層黑色素細胞增生、表皮內色素增加、乳頭層真皮噬黑色素細胞;黑色素細胞無異型性。
- 治療:不需治療;可冷凍或 Q 開關雷射 (Q-switched laser) 美容性移除。無惡性轉化風險增加。
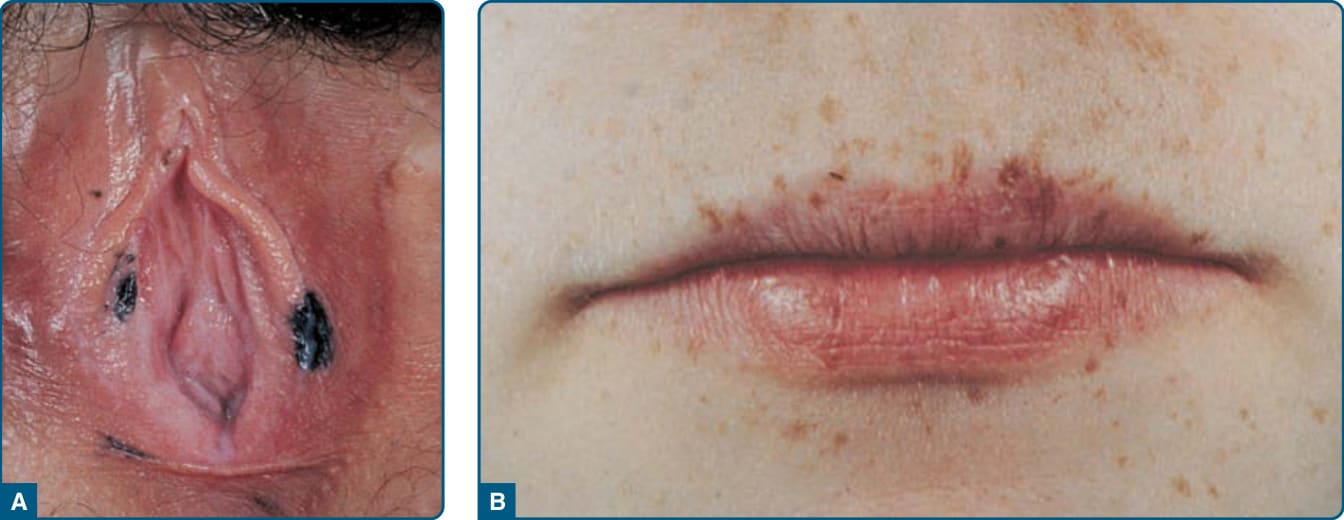
圖 115-16:單純性雀斑樣痣——LAMB(Carney 複合症)患者外陰與唇部多發雀斑樣痣。
日光性雀斑樣痣 (Solar Lentigo)
- 又稱光化性雀斑樣痣、肝斑、老年性雀斑樣痣;發生於光損傷皮膚的色素增多斑點,主要於較年長者。
- 流行病學:超過百分之九十白皮膚者 50 歲前發展。慢性 PUVA(補骨脂素加 UVA)可誘發 PUVA 雀斑樣痣(一研究百分之五十三患者出現臀部雀斑樣痣,平均治療後 5.7 年)。
- 臨床:界限清楚、不規則邊緣,好發臉部與前臂手背;淡至深褐色。墨點雀斑樣痣 (ink spot lentigo) 為黑色變異型。PUVA 雀斑樣痣亦見於臀部/生殖器等防曬部位。
- 致病機轉:基底黑色素細胞增殖與黑色素產生增加,UVR 透過角質細胞 ROS、MC1R-MITF 路徑活化。帶 FGFR3 (約 17%)、PIK3CA (約 7%) 突變;PUVA 雀斑樣痣百分之三十三帶 T1799A BRAF 突變。
- 病理:網脊伸長伴芽狀/棒槌狀突起、色素增多、黑色素細胞正常或略增多、真皮日光性彈力組織變性 (solar elastosis)。
- 臨床病程:可演變為苔癬樣角化 (lichenoid keratoses) 而消退,或演變為大細胞棘皮瘤。與 UV 相關皮膚惡性腫瘤(鱗狀細胞癌、基底細胞癌、黑色素瘤)風險增加相關。
- 治療:良性不需治療;可冷凍或 Q 開關雷射美容移除。避免 UVR 可降低新發風險。
發育不良黑色素細胞痣 (Dysplastic Melanocytic Nevi, DN)
- 常見於高加索人;辨識出黑色素瘤的風險族群。好發中度日曬區(尤其上背部),與大量常見後天性痣相關。
- 流行病學:黑色素瘤患者中存在於百分之三十點四(對照組百分之一點八)。發育不良痣症候群 (DNS) 者青春期前可發展超過 100 個臨床非典型痣。
- 臨床:常 ≥5 mm,不規則不清邊緣、多變色素(淡褐+褐色,可帶粉紅;黑色較關聯黑色素瘤),常有扁平斑狀「肩部」成分;略隆起、鵝卵石樣。ABCD 規則(不對稱、邊緣不規則、顏色變異、直徑 >6 mm)。
- 致病機轉:機轉未明;DNS 可散發或體染色體顯性(變異表現度、不完全外顯率)。CDKN2A (9p21-22) 突變約見於百分之四十 DNS。與常見痣相似 BRAF 突變率,可有 PTEN 喪失、p53 改變。UVR 與曬傷(尤其 20 歲前)重要。
- 併發症:黑色素瘤——散發性 DN 約 10 倍風險;若至少兩位親屬有黑色素瘤家族史則達 200 倍。
- 病理:表皮內雀斑樣黑色素細胞增生伴細胞學與構築異型性、真皮基質反應(圖 115-22);網脊尖端巢橋接、肩化交界成分延伸超出真皮成分至少三個網脊;乳頭層真皮纖維增生。發育不良程度應評分(觀察者間一致性可變)。
- 特殊檢查:皮膚鏡 (dermatoscopy) 提高黑色素瘤偵測;多發病灶或 DNS 建議全身攝影 (total-body photography / mole mapping)。
- 臨床病程:多數不進展為黑色素瘤;僅約百分之二十黑色素瘤源自 DN。高風險者初始每 4–6 個月密集監測;高危家族監測自 10 歲起。
- 治療:可疑/變化/有症狀病灶切片;疑黑色素瘤應切除。重度異型性 DN 可能須再切除取得 5-mm 邊緣。對 DNS 患者大規模移除 DN 不顯著降低終生風險。教育 ABCD 與防曬。
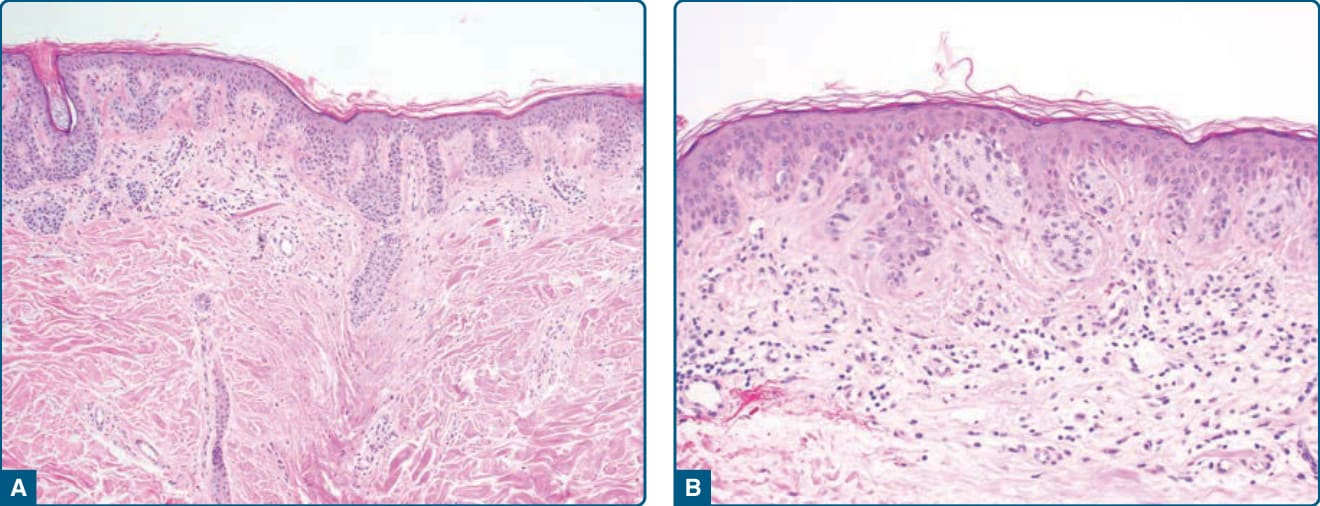
圖 115-22:發育不良痣組織病理——交界處巢延伸超出真皮成分、隨機細胞學異型性、乳頭層纖維增生與色素失禁。