脂肪失養症 (Lipodystrophy)
PART 12
皮下組織疾病 (Subcutaneous Tissue Disorders)
重點一覽 (AT-A-GLANCE)
■ 脂肪失養症 (lipodystrophies) 是以選擇性喪失體脂肪為特徵的遺傳性或後天性疾病。脂肪喪失的程度決定相關代謝併發症的嚴重度,例如糖尿病 (diabetes mellitus)、高三酸甘油酯血症 (hypertriglyceridemia)、肝脂肪變性 (hepatic steatosis) 與黑色棘皮症 (acanthosis nigricans)。
■ 體染色體隱性遺傳的先天性全身性脂肪失養症 (congenital generalized lipodystrophy, CGL) 已辨識出四個基因座,即 AGPAT2、BSCL2、CAV1 與 CAVIN1。
■ 體染色體顯性遺傳的家族性部分脂肪失養症 (familial partial lipodystrophy, FPL) 已辨識出五個基因座,即 LMNA、PPARG、AKT2、PLIN1 與 ADRA2A。
■ LIPE、CIDEC、PCYT1A 與 RECQL2 是體染色體隱性遺傳 FPL 的基因座;LMNA 與 ZMPSTE24 則是下頜肢端發育不良 (mandibuloacral dysplasia) 相關脂肪失養症的基因座。
■ 許多極為罕見型態的遺傳性脂肪失養症,其分子基礎仍有待闡明。
■ 最常見的脂肪失養症型態,發生於 HIV 感染病人在長期使用含蛋白酶抑制劑 (protease inhibitor) 的高效抗反轉錄病毒治療 (highly active antiretroviral therapy) 之後。
■ 後天性全身性脂肪失養症 (acquired generalized lipodystrophy) 與後天性部分脂肪失養症 (acquired partial lipodystrophy) 主要源於自體免疫。
■ 局部性脂肪失養症 (localized lipodystrophies) 因藥物或疫苗注射、壓力、脂膜炎 (panniculitis),以及其他未知原因而發生。
■ 目前的處置包括美容手術,以及早期辨識與治療代謝及其他併發症。
■ 對於合併低瘦素血症 (hypoleptinemia) 的全身性脂肪失養症病人,瘦素替代療法 (metreleptin replacement therapy) 有助於治療代謝併發症。
脂肪失養症是一群以選擇性喪失脂肪組織 (adipose tissue) 為特徵的異質性疾病。¹,²
脂肪喪失的程度各異,有些病人僅從小範圍區域喪失脂肪(局部性脂肪失養症 localized lipodystrophy),另一些病人則有較廣泛的脂肪喪失,例如侵犯四肢(部分脂肪失養症 partial lipodystrophy)或全身(全身性脂肪失養症 generalized lipodystrophy)。依脂肪喪失的程度而定,病人可能傾向發展出與胰島素抗性 (insulin resistance) 相關的併發症,例如糖尿病 (diabetes mellitus)、血脂異常 (dyslipidemia)、肝脂肪變性 (hepatic steatosis)、黑色棘皮症 (acanthosis nigricans)、多囊性卵巢疾病 (polycystic ovarian disease) 與冠狀動脈心臟病 (coronary heart disease)。³⁻⁵ 脂肪失養症主要有兩大類型:遺傳性與後天性。表 74-1 提供各型脂肪失養症的詳細分類。
流行病學 (EPIDEMIOLOGY)
雖然遺傳性脂肪失養症罕見,但近期的進展——例如表型 (phenotypes) 定義的改善以及分子缺陷的闡明——已使這些症候群獲得更多的辨識。整體而言,根據文獻報告約 1200 名病人的資料,遺傳性脂肪失養症的估計盛行率可能低於百萬分之一。受影響的女性較易被辨識,因此其報告頻率高於男性。體染色體隱性遺傳的先天性全身性脂肪失養症 (CGL) 已報告約 500 名病人,病人群聚於黎巴嫩 (Lebanon) 與巴西 (Brazil),這些地區近親通婚 (consanguinity) 的盛行率較高。近期土耳其報告 CGL 的盛行率約為一般人口的 200 萬分之一。⁶ Dunnigan 型的體染色體顯性遺傳家族性部分脂肪失養症 (FPL) 最為常見,已報告約 500 名病人;由 PPARG 突變所致的 FPL 則報告約 40 名病人。由於奠基者突變 (founder mutations),FPL Dunnigan 型的盛行率估計在加拿大安大略省 (Ontario) 一般人口中高達 1:200,000⁷,而在位於印度洋的留尼旺島 (Reunion Island) 一般人口中約為 1:20,000。⁸ 由 LMNA 突變所致的體染色體隱性遺傳下頜肢端發育不良 (mandibuloacral dysplasia, MAD) 已報告約 30 名病人;由 ZMPSTE24 突變所致的 MAD 則報告 15 名病人。後天性部分脂肪失養症約於 125 年前被認識,且僅報告約 250 至 300 例各種族裔的病例,男女比為 1:4。⁹ 後天性全身性脂肪失養症已報告少於 100 例,大多為白人,男女比為 1:3。¹⁰ 目前最常見的型態是 HIV 感染病人因含蛋白酶抑制劑 (protease inhibitors, PIs) 的高效抗反轉錄病毒治療所誘發的脂肪失養症,估計在美國影響超過 100,000 名病人,在其他國家則影響更多人。¹,¹¹,¹²
遺傳性脂肪失養症 (GENETIC LIPODYSTROPHIES)
在過去約十年間,闡明許多型遺傳性脂肪失養症的分子基礎已取得相當大的進展。一般而言,已報告涉及脂肪細胞分化 (adipocyte differentiation)、三酸甘油酯合成 (triglyceride synthesis)、脂滴形成 (lipid droplet formation) 與脂肪細胞存活 (adipocyte survival) 之基因的突變會導致脂肪失養症。
遺傳性脂肪失養症分類
遺傳性脂肪失養症 (Genetic Lipodystrophies)
-
體染色體隱性遺傳,先天性全身性脂肪失養症 (congenital generalized lipodystrophy, CGL)
a. 第 1 型:AGPAT2 突變
b. 第 2 型:BSCL2 突變
c. 第 3 型:CAV1 突變
d. 第 4 型:CAVIN1 突變
e. 其他 -
體染色體顯性遺傳,家族性部分脂肪失養症 (familial partial lipodystrophy, FPL)
a. Dunnigan 型:LMNA 突變
b. PPARG 突變
c. AKT2 突變
d. PLIN1 突變
e. ADRA2A(腎上腺素受體 α2A, adrenoceptor α2A)突變
f. 其他 -
體染色體隱性遺傳,家族性部分脂肪失養症 (familial partial lipodystrophy, FPL)
a. CIDEC 突變
b. LIPE 突變
c. PCYT1A 突變
d. RECQL2 突變 -
體染色體隱性遺傳,下頜肢端發育不良 (mandibuloacral dysplasia, MAD)–相關脂肪失養症
a. LMNA 突變
b. ZMPSTE24 突變
c. 其他 -
其他與 LMNA 突變相關的脂肪失養症
a. 非典型早老症候群 (atypical progeroid syndrome)
b. Hutchinson-Gilford 早老症 (Hutchinson-Gilford progeria syndrome) -
SHORT(矮小身材 short stature、關節過度伸展或疝氣 hyperextensibility of joints or hernia、眼窩凹陷 ocular depression、Rieger 異常 Rieger anomaly、出牙延遲 teething)症候群
a. PIK3R1 突變 -
新生兒早老 (Wiedemann-Rautenstrauch) 症候群
a. FBN1 突變
b. CAV1 突變
c. POLR3A 突變 -
下頜發育不全、耳聾、早老 (mandibular hypoplasia, deafness, progeroid, MDP) 症候群
a. POLD1 突變 -
自體發炎性脂肪失養症(關節攣縮、小球性貧血、脂膜炎相關 [JMP] 脂肪失養症候群譜系,包含合併脂肪失養與體溫升高的慢性非典型嗜中性球皮病 [CANDLE] 症候群)
a. PSMB8 突變
後天性脂肪失養症 (Acquired Lipodystrophies)
- HIV 感染病人的高效抗反轉錄病毒治療–誘發的脂肪失養症
- 後天性全身性脂肪失養症 (acquired generalized lipodystrophy)
a. 脂膜炎誘發 (panniculitis induced)
b. 自體免疫疾病相關 (autoimmune diseases associated)
c. 特發性 (idiopathic) - 後天性部分(Barraquer-Simons)脂肪失養症
a. 自體免疫疾病相關 (autoimmune diseases associated)
b. 膜增生性腎絲球腎炎相關 (membranoproliferative glomerulonephritis associated)
c. 特發性 (idiopathic) - 局部性脂肪失養症 (localized lipodystrophies)
a. 脂膜炎誘發 (panniculitis induced)
b. 壓力誘發 (pressure induced)
c. 藥物誘發 (drug induced)
d. 離心性 (centrifugal)
e. 特發性 (idiopathic)
先天性全身性脂肪失養症(Berardinelli-Seip 症候群)(CONGENITAL GENERALIZED LIPODYSTROPHY, BERARDINELLI-SEIP SYNDROME)
臨床特徵 (CLINICAL FEATURES)
此一體染色體隱性遺傳疾病有 4 種亞型,可在出生時或出生後不久因近乎完全缺乏體脂肪而被辨識出來。¹³ 病人也有明顯的肌肉發達 (muscularity)、突出的皮下靜脈 (prominent subcutaneous veins)、肢端肥大樣特徵 (acromegaloid features)、黑色棘皮症 (acanthosis nigricans)、肝腫大 (hepatomegaly) 與臍部突出 (umbilical prominence)(圖 74-1A、表 74-2)。在兒童期,他們食慾旺盛 (voracious appetite),且線性生長加速 (accelerated linear growth)。女性通常有多毛症 (hirsutism)、陰蒂肥大 (clitoromegaly)、月經稀少 (oligomenorrhea) 與多囊性卵巢 (polycystic ovaries)。僅有少數女性曾成功懷孕。某些 CGL 男性病人的生育力可能因畸形精子症 (teratozoospermia) 而受影響。部分 CGL 病人在青春期後會發展出肥厚性心肌病 (hypertrophic cardiomyopathy)、輕度智能障礙 (mild mental retardation),以及附肢骨骼 (appendicular bones) 的局部溶解性病灶 (focal lytic lesions)。¹³ 與胰島素抗性相關的代謝異常,例如糖尿病、高脂血症 (hyperlipidemia) 與肝脂肪變性,可能在年幼時即顯現,且常難以控制。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
全基因組連鎖分析 (genome-wide linkage analysis) 搭配定位選殖策略 (positional cloning strategy) 與候選基因取向 (candidate gene approach),已辨識出 CGL 的 4 個遺傳基因座:位於第 9q34 號染色體的 1-醯基甘油-3-磷酸-O-醯基轉移酶 2(1-acylglycerol-3-phosphate-O-acyltransferase 2, AGPAT2)基因¹⁴,¹⁵;位於第 11q13 號染色體的 Berardinelli-Seip 先天性脂肪失養症 2(Berardinelli-Seip congenital lipodystrophy 2, BSCL2)基因¹⁶;位於第 7q31 號染色體的小窩蛋白 1(caveolin 1, CAV1)基因¹⁷;以及位於第 17q21 號染色體的小窩相關蛋白 1(caveolae associated protein 1, CAVIN1),又稱聚合酶 I 與轉錄物釋放因子(polymerase I and transcript release factor, PTRF)基因。¹⁸ AGPAT2 是參與由甘油-3-磷酸 (glycerol-3-phosphate) 生合成三酸甘油酯與磷脂質之關鍵酵素,並在脂肪組織中高度表現。¹⁹ BSCL2 所編碼的蛋白質 seipin,在脂滴融合 (lipid droplet fusion) 中扮演角色,也可能參與脂肪細胞分化。²⁰⁻²² 小窩蛋白 1 是小窩 (caveolae) 的整合性成分,小窩是大量存在於脂肪細胞膜上的特化微域 (microdomains)。小窩蛋白 1 會結合脂肪酸並將其轉運至脂滴。CAVIN1 參與小窩的生成 (biogenesis),並調節小窩蛋白 1 與 3 的表現。¹⁸ 與帶有 AGPAT2、CAV1 與 PTRF 突變者(其機械性脂肪 mechanical fat 得以保留)相比,帶有 BSCL2 突變的病人缺乏位於眼眶區 (orbital region)、手掌 (palm)、腳掌 (sole) 與關節周圍區 (periarticular regions) 的機械性脂肪,也缺乏位於皮下 (subcutaneous, SQ)、腹腔內 (intraabdominal)、胸腔內 (intrathoracic) 與其他區域具代謝活性的脂肪組織 (metabolically active adipose tissue)。¹⁷,²³ 唯一報告的 CAV1 突變病人也有矮小身材 (short stature) 與推定的維生素 D 抗性 (vitamin D resistance)。¹⁷ 僅約 15 名帶有 CAVIN1 突變的病人被報告,他們有先天性肌病 (congenital myopathy)、肌酸激酶 (creatine kinase) 濃度升高、叩診誘發的肌水腫 (percussion-induced myoedema)、幽門狹窄 (pyloric stenosis)、寰樞椎不穩定 (atlantoaxial instability),以及包括 QT 間期延長 (prolonged QT interval) 與運動誘發心室頻脈 (exercise-induced ventricular tachycardia) 在內的心律障礙,這些可能導致猝死 (sudden death)。¹⁸,²⁴,²⁵ 黎巴嫩血統的病人帶有 BSCL2 基因的同型合子 c.315_319delGTATC; p.(Tyr106Cysfs∗6) 突變,而非洲血統者幾乎總是帶有 AGPAT2 基因的同型合子或複合異型合子 c.589–2A>G; p.(Val197Glufs∗32) 突變。¹⁵,¹⁶,²⁶
家族性部分脂肪失養症 (FAMILIAL PARTIAL LIPODYSTROPHY)
臨床特徵 (CLINICAL FEATURES)
FPL 在大多數病人中以體染色體顯性遺傳方式傳遞,其特徵為四肢脂肪喪失,軀幹脂肪喪失程度不一,並在非脂肪失養區域 (nonlipodystrophic regions) 有增加的皮下脂肪沉積(見圖 74-1B)。大多數 FPL 病人為 Dunnigan 型,係由核纖層蛋白 A/C(lamin A/C, LMNA)基因的異型合子突變所致。這些病人在幼兒早期有正常的體脂肪分布,但在青春期前後,四肢與軀幹的皮下脂肪逐漸喪失(見圖 74-1B)。顏面、頸部與腹腔內區域則不受影響,且該處常累積過多脂肪。²⁷,²⁸ 受影響的男性常較難以臨床診斷,因為許多正常男性也相當肌肉發達。女性在代謝方面受影響較為嚴重。²⁹
由 PPARG 基因異型合子突變所致的 FPL 病人,也有四肢脂肪喪失,尤其在遠端區域,但顏面、頸部與軀幹區域的脂肪則不受影響。³⁰
受影響者中高血壓 (hypertension) 的盛行率增加。曾報告來自單一家族、患有糖尿病與胰島素抗性的四名受試者帶有 AKT2 基因的異型合子突變。³¹ 該女性先證者 (proband) 有四肢脂肪失養,惟未進行體脂肪分布的詳細研究。近期報告 12 名 FPL 病人帶有 PLIN1 突變。³²,³³ 來自單一家族、患有非典型 FPL 且有明顯水牛肩 (buffalo hump) 的三名病人帶有異型合子 ADRA2A 突變。³⁴
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
FPL 係由下列 5 個基因之一的異型合子錯義突變 (missense mutations) 所致:位於第 1q21–22 號染色體的核纖層蛋白 A/C(lamin A/C, LMNA),³⁵⁻³⁸ 為核纖層 (nuclear lamina) 的整合性成分;位於第 3p25 號染色體的過氧化體增殖物活化受體 γ(peroxisome proliferator-activated receptor γ, PPARG),³⁰,³⁹,⁴⁰ 為參與脂肪細胞分化的關鍵轉錄因子;位於第 19q13 號染色體的 v-AKT 鼠胸腺瘤致癌基因同源物 2(v-AKT murine thymoma oncogene homolog 2, AKT2),³¹ 參與胰島素訊號傳遞的下游;位於第 15q26 號染色體的脂滴包被蛋白 1(perilipin 1, PLIN1),為脂滴的關鍵成分³²;以及腎上腺素受體 α2A(adrenoreceptor α2A, ADRA2A),為調節去甲腎上腺素 (norepinephrine) 釋放的主要突觸前抑制性回饋 G 蛋白偶聯受體。³⁴ 帶有 LMNA 突變病人的脂肪細胞喪失,可能源於核膜功能與完整性的破壞,導致過早的細胞死亡。某些在核纖層蛋白 A/C 胺基端區域帶有突變的病人,也會發展出肌病 (myopathy)、心肌病 (cardiomyopathy) 與傳導系統異常 (conduction system abnormalities),顯示為一種多系統失養症 (multisystem dystrophy)。⁴¹ 另一方面,其他在核纖層蛋白 A 極端 C 端區域帶有突變者,則可能有輕度脂肪失養。⁴²
下頜肢端發育不良–相關脂肪失養症 (MANDIBULOACRAL DYSPLASIA–ASSOCIATED LIPODYSTROPHY)
臨床特徵 (CLINICAL FEATURES)
MAD 病人有特徵性的骨骼異常,包括下頜骨 (mandible) 與鎖骨 (clavicles) 發育不全、肢端骨溶解 (acroosteolysis)、皮膚萎縮 (cutaneous atrophy)、早老樣特徵(progeroid features,例如鳥喙狀細鼻 thin beaked nose)、毛髮脫落 (hair loss)、伴有明顯淺表血管 (prominent superficial vasculature) 與斑駁性色素沉著 (mottled hyperpigmentation) 的薄皮膚、牙齒萌發 (dentition) 與顱縫閉合 (closure of cranial sutures) 延遲、關節僵硬 (joint stiffness),以及脂肪失養。⁴³,⁴⁴
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
位於第 1p34 號染色體的 LMNA 與鋅金屬蛋白酶(zinc metalloproteinase, ZMPSTE24)突變會導致體染色體隱性遺傳的 MAD–相關脂肪失養症。⁴⁵,⁴⁶ ZMPSTE24 參與前核纖層蛋白 A(prelamin A)轉譯後蛋白水解處理成成熟核纖層蛋白 A(mature lamin A)的過程,其缺乏會導致前核纖層蛋白 A 在細胞內累積,被認為會造成毒性。帶有 ZMPSTE24 突變者較早在生命中發展出臨床表現,出生時即早產,並可能發展出局部節段性腎絲球硬化 (focal segmental glomerulosclerosis)、鈣化的皮膚結節 (calcified skin nodules),以及薄而有光澤的皮膚。⁴⁷,⁴⁸
其他型態 (OTHER TYPES)
臨床特徵 (CLINICAL FEATURES)
筆者與同事曾報告一種新型症候群,即下頜發育不全、耳聾、早老樣特徵–相關脂肪失養症。⁴⁹ 所有患有下頜發育不全、耳聾、早老樣特徵的男性都有隱睪症 (undescended testes) 且為性腺功能低下 (hypogonadal)。一名成年女性顯示缺乏乳房發育。患有自體發炎性脂肪失養症 (autoinflammatory lipodystrophy) 的病人表現為一系列臨床表現譜,發病時間從出生後最初幾個月⁵⁰ 到兒童期後期不等。⁵¹ 早期特徵包括反覆發燒 (recurrent fevers)、環狀紫紅色斑塊 (annular violaceous plaques)、體重與身高增長不良、持續性紫紅色眼瞼腫脹 (persistent violaceous eyelid swelling)、肝腫大 (hepatomegaly)、關節痛 (arthralgias)、程度不一的肌肉萎縮 (muscle atrophy),以及進行性脂肪失養。⁵²⁻⁵⁴ 病灶皮膚的組織病理學檢查顯示髓系 (myeloid lineage) 的非典型單核細胞浸潤與成熟嗜中性球 (mature neutrophils),實驗室異常包括慢性貧血 (chronic anemia)、急性期反應物 (acute-phase reactants) 升高,以及肝臟酵素升高,其細胞激素譜顯示干擾素誘導蛋白–1(interferon-inducible protein–1)、單核細胞趨化蛋白–1(monocyte chemotactic protein–1)、白介素-6(interleukin-6)與白介素-1 受體拮抗劑(interleukin-1 receptor antagonist)的高濃度,符合干擾素訊號傳遞特徵 (interferon signaling signature)。⁵²,⁵³
此一表現被稱為 CANDLE 症候群(chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature,合併脂肪失養與體溫升高的慢性非典型嗜中性球皮病)。⁵⁰ 年齡較大且帶有 PSMB8 突變的個體表現出關節攣縮 (joint contractures)、肌肉萎縮 (muscle atrophy)、小球性貧血 (microcytic anemia),以及脂膜炎誘發 (panniculitis-induced) 的兒童期發病脂肪失養 (JMP)(圖 74-2)。⁵¹ JMP 症候群的其他特徵包括高γ球蛋白血症 (hypergammaglobulinemia)、紅血球沉降速率 (erythrocyte sedimentation rate) 升高、肝脾腫大 (hepatosplenomegaly),以及基底核鈣化 (calcification of basal ganglia)。⁵¹ 其他罕見的脂肪失養症候群包括體染色體隱性遺傳的 FPL 與 SHORT(short stature 矮小身材、hyperextensibility or inguinal hernia 關節過度伸展或腹股溝疝氣、ocular depression 眼窩凹陷、Rieger anomaly Rieger 異常、teething delay 出牙延遲)症候群。⁵⁵,⁵⁶ 脂肪喪失通常局限於顏面、上肢與軀幹,有時也包括臀部。⁵⁵,⁵⁶ 患有新生兒早老 (Wiedemann-Rautenstrauch) 症候群的病人,在出生時即表現出全身性的體脂肪與肌肉量喪失以及早老外觀。⁵⁷ Wiedemann-Rautenstrauch 症候群病人的表型有顯著的異質性。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
患有下頜發育不全、耳聾、早老樣特徵症候群的病人,已被報告有聚合酶(DNA)δ 1 催化次單元(polymerase [DNA] delta 1, catalytic subunit, POLD1)基因的新生 (de novo) 反覆出現異型合子突變。⁵⁸
大多數 SHORT 症候群病人有磷酸肌醇-3-激酶調節次單元 1(phosphoinositide-3-kinase regulatory subunit 1, PIK3R1)基因的新生反覆出現異型合子錯義突變。⁵⁹ Wiedemann-Rautenstrauch 症候群病人已被報告有原纖維蛋白 1(fibrillin 1, FBN1)⁶⁰ 與小窩蛋白 1(caveolin 1, CAV1)⁶¹ 的新生異型合子突變,以及 RNA 聚合酶 III 次單元 A(RNA polymerase III subunit A, POLR3A)的雙等位基因突變 (biallelic mutations)。⁶² 近期,一名具有體染色體隱性遺傳 FPL 表型的病人被發現帶有位於第 3p25 號染色體、參與脂滴形成之細胞死亡誘導 DNA 片段化因子 a 樣效應子 c(cell death-inducing DNA fragmentation factor a-like effector c, CIDEC)基因的同型合子錯義突變。⁶³ 該病人皮下脂肪組織的組織病理學顯示脂肪細胞內有多房性、小型的脂滴 (multilocular, small lipid droplets)。另外兩名具有體染色體隱性遺傳部分與全身性脂肪失養表型的病人,帶有磷酸胞苷轉移酶 1α(phosphate cytidylyltransferase 1α, PCYT1A)基因突變,該基因編碼磷脂醯膽鹼 (phosphatidyl choline) 生合成路徑中的限速酵素。⁶⁴ 激素敏感性脂解酶(hormone sensitive lipase, LIPE)基因的功能喪失突變已被報告會導致一種伴有多發對稱性脂肪瘤病 (multiple symmetric lipomatosis) 與肌病的體染色體隱性遺傳 FPL 表型。⁶⁵⁻⁶⁷ 兩名患有 FPL 並具 Werner 症候群特徵的女性,已被報告帶有 Werner 症候群 RecQ 樣解旋酶(Werner syndrome RecQ like helicase, WRN)基因的無效突變 (null mutations)。⁶⁸
異型合子 LMNA 突變也可在非典型早老症候群 (atypical progeroid syndrome) 病人中導致部分或全身性脂肪失養⁶⁹,並在 Hutchinson-Gilford 早老症 (Hutchinson-Gilford progeria syndrome) 中導致皮下脂肪的全身性喪失。⁷⁰ 自體發炎性脂肪失養症候群係由蛋白酶體次單元 β 型 8(proteasome subunit, beta-type, 8, PSMB8)的突變所致。⁵²,⁵³ PSMB8 編碼免疫蛋白酶體 (immunoproteasome) 的 β5i 次單元,參與主要組織相容性複合體第 I 類分子 (major histocompatibility complex Class I molecules) 所呈現之免疫原性表位 (immunogenic epitopes) 的蛋白水解切割。⁵² 許多其他遺傳性脂肪失養症候群的分子遺傳基礎仍不明確(見表 74-1)。
後天性脂肪失養症 (ACQUIRED LIPODYSTROPHIES)
儘管後天性脂肪失養症被認識已超過一個世紀,但在理解其潛在致病機轉方面的進展一直緩慢。
後天性部分脂肪失養症(Barraquer-Simons 症候群)(ACQUIRED PARTIAL LIPODYSTROPHY, BARRAQUER-SIMONS SYNDROME)
臨床特徵 (CLINICAL FEATURES)
後天性部分脂肪失養症在大多數病人中於 15 歲前發病。病人以對稱方式逐漸喪失皮下脂肪,從顏面開始,然後向下擴展。大多數病人表現為顏面、頸部、上肢與軀幹的脂肪喪失,而皮下腹部脂肪 (SQ abdominal fat) 與下肢則不受影響(見圖 74-1C)。約 20% 的病人會發展出膜性微血管(膜增生性)腎絲球腎炎(mesangiocapillary [membranoproliferative] glomerulonephritis),有些則發展出玻璃膜疣 (drusen)。⁹ 病人通常不會發展出代謝併發症。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
皮下脂肪喪失的確切致病機轉仍不明確,但有強烈證據顯示自體免疫介導的脂肪細胞喪失,因為超過 80% 的病人有低濃度的補體 3(complement 3, C3),並存在一種循環免疫球蛋白 G(immunoglobulin G)——一種阻斷酵素 C3 轉化酶 (C3 convertase) 降解的 C3 腎炎因子(C3-nephritic factor)。⁹ 脂肪喪失可能是 C3 腎炎因子誘導表現因子 D(factor D)之脂肪細胞溶解的結果。
後天性全身性脂肪失養症(Lawrence 症候群)(ACQUIRED GENERALIZED LIPODYSTROPHY, LAWRENCE SYNDROME)
臨床特徵 (CLINICAL FEATURES)
後天性全身性脂肪失養症通常在兒童期表現出程度不一的皮下脂肪喪失。雖然許多病人有全身性的脂肪喪失,但某些區域不受影響(見圖 74-1D)。通常腹腔內或骨髓脂肪 (bone marrow fat) 不受影響。¹⁰ 然而,病人會發展出極為嚴重的肝脂肪變性與纖維化 (fibrosis)、糖尿病與高三酸甘油酯血症,這些都難以處置。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
脂肪喪失的確切機轉尚不明瞭。在約 25% 的病人中,皮下脂肪喪失發生於皮下發炎性結節 (SQ inflammatory nodules) 出現之後,這些結節在切片上顯示脂膜炎 (panniculitis)。¹⁰ 這些病灶起初導致局部性脂肪喪失,隨後發展為全身性脂肪喪失。另外 25% 的病人有相關的自體免疫疾病,尤其是幼年型皮肌炎 (juvenile dermatomyositis)。¹⁰,⁷¹ 在其餘的特發性型病人中,可能涉及多種未知機轉。¹⁰ 脂膜炎相關型 (panniculitis-associated variety) 的病人,其脂肪喪失與代謝併發症較其他型態為輕。有些病人被報告有低血清補體 4(complement 4)濃度。⁷²
HIV 感染病人的高效抗反轉錄病毒治療–誘發脂肪失養症 (HIGHLY ACTIVE ANTIRETROVIRAL THERAPY–INDUCED LIPODYSTROPHY IN HIV-INFECTED PATIENTS)
臨床特徵 (CLINICAL FEATURES)
接受含蛋白酶抑制劑 (PI-containing) 高效抗反轉錄病毒治療的 HIV 感染病人,通常在開始治療 2 年以上後,從顏面、軀幹與四肢喪失皮下脂肪(見圖 74-1E、74-3A 與 74-3B)。顏面的脂肪喪失可能嚴重到造成消瘦憔悴的外觀。其中有些人發展出水牛肩 (buffalo hump)、雙下巴 (double chin),且腹腔內脂肪 (intraabdominal fat) 增加。脂肪喪失隨著持續進行的高效抗反轉錄病毒治療而逐漸惡化,且在停用 PIs 後不會逆轉。有些病例發展出糖尿病,許多病例發展出合併型高脂血症 (combined hyperlipidemia),可能使病人傾向發展冠狀動脈心臟病。¹²
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
HIV-1 PIs 與核苷類似物 (nucleoside analogs) 等藥物被認為會在 HIV 感染病人中導致脂肪失養症。許多(但非全部)PIs 可能透過抑制 ZMPSTE24,導致前核纖層蛋白 A 的累積,而誘發脂肪失養。⁷³ 其他機轉可能包括 PI 誘導改變參與脂質生成 (lipogenesis) 與脂肪細胞分化之關鍵轉錄因子的表現,例如固醇調節元件結合蛋白 1c(sterol regulatory element-binding protein 1c)與 PPARG。⁷⁴ PIs 也會降低葡萄糖轉運蛋白 4(glucose transporter 4)的表現,這可能是誘發胰島素抗性的機轉之一。⁷⁵ 核苷類似物,尤其是齊多夫定 (zidovudine) 與司他夫定 (stavudine),可能透過抑制聚合酶-γ(polymerase-γ,一種參與粒線體 DNA 複製的粒線體酵素)而誘發脂肪喪失。⁷⁶ 由於大多數病人同時接受多種抗反轉錄病毒藥物,PIs 或核苷反轉錄酶抑制劑 (nucleoside reverse transcriptase inhibitors) 對表型的個別影響尚不清楚。
局部性脂肪失養症 (LOCALIZED LIPODYSTROPHY)
臨床特徵 (CLINICAL FEATURES)
局部性脂肪失養症表現為從局灶區域喪失皮下脂肪,造成凹陷 (dimple) 或凹坑 (crater),而其上方的皮膚通常不受影響。在某些病人中,身體任何區域的大片連續或解剖學上獨立的區域都可能受侵犯。³
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
這可能由各種藥物的皮下注射、脂膜炎、壓力及其他機轉所致。³
診斷 (DIAGNOSIS)
對於表現出提早發病之糖尿病、高三酸甘油酯血症、肝脂肪變性、黑色棘皮症與多囊性卵巢症候群的「瘦弱或非肥胖 (lean or nonobese)」病人,應強烈懷疑脂肪失養症。應仔細檢查這些病人是否有皮下脂肪喪失的證據(尤其是臀部與大腿),以及各種解剖區域是否有過多皮下脂肪沉積。對於兒童期表現出全身性脂肪失養者,應評估其出生時的照片以尋找脂肪喪失的證據。若脂肪失養表型在出生時或出生後不久即被發現,應考慮 CGL;否則,病人可能患有後天性脂肪失養症。
病史 (HISTORY)
應詢問病人脂肪失養症的發病年齡與進展情形,以及其他相關表現。詳細詢問家族史(包括近親通婚史)對於理解遺傳性脂肪失養症的遺傳模式非常重要。對於後天性脂肪失養症病人,應考慮相關的自體免疫疾病,尤其是幼年型皮肌炎。對於局部性脂肪失養症病人,應詢問局部注射、外傷、壓力或其他損傷。對於患有脂肪失養症的 HIV 感染病人,應取得抗反轉錄病毒治療的持續時間與類型的詳細病史。
皮膚病灶 (CUTANEOUS LESIONS)
脂肪失養症病人最常見的皮膚病灶是腋窩 (axillae)、腹股溝 (groins)、頸部,有時甚至指關節 (knuckles)、阿基里斯腱 (Achilles tendons) 與軀幹的黑色棘皮症 (acanthosis nigricans)(圖 74-4A 與 B)。許多病人因相關的多囊性卵巢症候群而發展出陰蒂肥大 (clitoromegaly) 與多毛症 (hirsutism)。在一名非典型早老症候群病人身上注意到雀斑 (freckles)(圖 74-4C)。在早老症候群與 MAD 病人身上可見鳥喙狀細鼻 (thin, beaked nose)、頭皮、眉毛與腋下毛髮脫落、皮膚萎縮與斑駁性色素沉著(圖 74-4D),並伴有肢端骨溶解 (acroosteolysis)(圖 74-4E)。⁴³,⁶⁹,⁷⁰ 罕見的 MAD 病人會發展出有光澤、緊繃、萎縮且容易破損的皮膚。在極度高三酸甘油酯血症病人身上也常見發疹性 (eruptive)、結節性 (tuberous) 與扁平 (planar) 黃色瘤 (xanthomas)(圖 74-4F 與 G)。腳掌皮下脂肪喪失可造成足底胼胝 (plantar calluses)。脂膜炎病人可能出現伴有上方紅斑 (overlying erythema) 的皮下結節 (SQ nodules)。
實驗室檢查 (LABORATORY TESTING)
實驗室檢查取決於脂肪失養症的類型。除了局部性脂肪失養症病人外,應取得包含葡萄糖 (glucose)、血脂 (lipids)、肝臟酵素 (liver enzymes) 與尿酸 (uric acid) 的血清生化檢查。在口服葡萄糖耐量試驗 (oral glucose tolerance test) 期間測量空腹與餐後血清葡萄糖與胰島素,可對胰島素抗性提供某種程度的估計。血清瘦素 (serum leptin) 測量並非診斷性的,但有助於指引關於試驗性人類重組瘦素(metreleptin)替代療法的治療決策。全身性脂肪失養症病人的血清瘦素與脂聯素 (adiponectin) 濃度非常低。⁷⁷ 後天性部分脂肪失養症病人應檢測血清 C3 與 C3 腎炎因子,並每年檢查蛋白尿 (proteinuria)。X 光攝影可顯示 CGL 病人附肢骨中的溶解性病灶,以及 MAD 病人的骨骼缺陷。皮膚切片對於局部性脂肪失養症或脂膜炎相關型病人很有用。
特殊檢查(包括影像學檢查)(SPECIAL TESTS, INCLUDING IMAGING STUDIES)
各型脂肪失養症之間的區別可透過身體檢查來進行,並以人體測量學 (anthropometry) 加以佐證,包括以測徑器 (calipers) 在各部位測量皮褶厚度 (skinfold thickness)。為深入進行體脂肪分布的表型分析,可進行全身雙能量 X 光吸收測量 (whole-body dual-energy X-ray absorptiometry) 與 T1 加權 MRI (T1-weighted MRI)。對於分子基礎已知的遺傳性脂肪失養症,各種商業與研究實驗室提供基因檢測。產前基因檢測 (prenatal genetic testing) 也是可行的。傾向發展心肌病的 FPL 病人、非典型早老症候群病人,以及 CGL 第 4 型病人,應接受心電圖 (electrocardiography) 與霍特氏監測 (Holter monitoring) 以偵測心律不整,並接受心臟超音波 (echocardiography) 以評估心臟功能。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
全身性脂肪失養症最重要的鑑別診斷是與表現出嚴重體重減輕的病況,例如營養不良 (malnutrition)、饑荒 (famine)、飢餓 (starvation)、神經性厭食症 (anorexia nervosa)、未控制的糖尿病、甲狀腺毒症 (thyrotoxicosis)、腎上腺皮質功能不全 (adrenocortical insufficiency)、癌症惡病質 (cancer cachexia)、HIV 相關消耗症 (HIV-associated wasting)、間腦症候群 (diencephalic syndrome) 與慢性感染。對於部分脂肪失養症,應與庫欣症候群 (Cushing syndrome)、全身性與軀幹性肥胖 (truncal obesity),以及多發對稱性脂肪瘤病(multiple symmetric lipomatosis, Madelung 病)相區分。患有 MAD 與早老症候群相關脂肪失養症的病人,應與患有 Werner 症候群與妖精症(leprechaunism, Donahue 症候群)者相區分。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
有些病人發展出極度高三酸甘油酯血症與乳糜微粒血症 (chylomicronemia),導致急性胰臟炎 (acute pancreatitis) 甚至死亡。糖尿病的長期併發症,例如腎病變 (nephropathy)、神經病變 (neuropathy) 與視網膜病變 (retinopathy),頻繁可見。許多病人發展出冠狀動脈心臟病與其他動脈粥樣硬化性血管併發症 (atherosclerotic vascular complications)。²⁹,⁷⁸ 肝脂肪變性可導致肝硬化 (cirrhosis),需要肝臟移植。CGL 第 4 型在兒童期曾報告猝死,可能源於心律不整。²⁴ 某些患有後天性部分脂肪失養症與膜增生性腎絲球腎炎的病人可能需要腎臟移植。⁹ 患有 Hutchinson-Gilford 早老症的病人在青少年期死於急性心肌梗塞 (acute myocardial infarction) 或腦血管意外 (cerebrovascular accidents)。⁷⁹ 某些患有非典型早老症候群與 FPL-Dunnigan 的病人發展出伴有瓣膜功能障礙 (valvular dysfunction)、充血性心衰竭 (congestive heart failure) 與心律不整的心肌病,需要植入心律調節器 (pacemaker)。⁴¹,⁶⁹ 兩名由 ZMPSTE24 突變所致 MAD 的成年病人死於局部節段性腎絲球硬化導致的腎衰竭。⁴⁷ 某些患有後天性全身性脂肪失養症的病人已被報告發展出周邊 T 細胞淋巴瘤 (peripheral T-cell lymphomas)。⁸⁰
預後取決於脂肪失養症的類型。在大多數已發表的 CGL 病例中,病人都是兒童,導致缺乏關於其預後的資料。依筆者經驗,有些病人死於急性胰臟炎或肝硬化的併發症,或發展出末期腎病 (end-stage renal disease) 而需要腎臟移植,並因糖尿病視網膜病變而失明。FPL 病人也傾向發生代謝併發症,並死於動脈粥樣硬化性血管與冠狀動脈心臟病,或死於心肌病與心律障礙。某些 MAD 病人據報於兒童期死亡,有些則在第三、四個十年因腎衰竭併發症而較晚死亡。⁴⁷,⁸¹ 後天性全身性脂肪失養症病人遭受嚴重的代謝併發症。患有後天性部分脂肪失養症與膜增生性腎絲球腎炎的病人會發展出腎衰竭,但其他病人則與局部性脂肪失養症病人一樣有正常的壽命。患有脂肪失養症的 HIV 感染病人傾向發展冠狀動脈心臟病。
處置 (MANAGEMENT)
各型脂肪失養症的治療相當具有挑戰性。目前沒有可逆轉體脂肪喪失的特定治療。治療的主軸包括美容手術與併發症的處置。部分脂肪失養症病人可接受自體脂肪組織移植 (autologous adipose tissue transplantation),或植入真皮填充劑 (dermal fillers),例如玻尿酸 (hyaluronic acid)、氫氧基磷灰石鈣 (calcium hydroxylapatite)、矽膠 (silicone)、聚丙烯醯胺凝膠 (polyacrylamide gels) 或聚左旋乳酸 (poly-l-lactic acid)。⁸² 不想要的過多脂肪組織可以手術切除或以抽脂 (liposuction) 移除。CGL 病人可接受重建性顏面手術,包括取自大腿的顏面移植片 (facial grafts)、取自前外側大腿、前腹部或顳肌 (temporalis muscle) 的游離皮瓣 (free flaps)。¹ 父母的支持對於預防罹患脂肪失養症兒童的不必要壓力與心理後遺症至關重要。建議所有病人攝取低脂飲食 (low-fat diets)。這些飲食可改善極度高三酸甘油酯血症病人的乳糜微粒血症。然而,高碳水化合物攝取也可能提高極低密度脂蛋白 (very low-density lipoprotein) 三酸甘油酯濃度。應鼓勵增加身體活動以減輕胰島素抗性及其併發症,但患有心肌病者除外。CGL 病人附肢骨中的溶解性骨病灶通常不會增加骨折風險。
目前沒有設計良好的對照試驗可供指引關於如何處置代謝併發症的治療決策。對於嚴重高三酸甘油酯血症,應使用極低脂飲食,並搭配纖維酸鹽類 (fibrates) 與來自魚油的 n-3 多元不飽和脂肪酸 (n-3 polyunsaturated fatty acids)。⁵ 必要時可加入史他汀類藥物 (statins)。應避免任何形式的雌激素治療 (estrogen therapy),因為它可能造成嚴重高三酸甘油酯血症誘發急性胰臟炎的風險。糖尿病起初應以二甲雙胍 (metformin) 處置。在部分脂肪失養症病人中應謹慎使用噻唑烷二酮類 (thiazolidinediones),因為它們可能增加非脂肪失養區域不想要的脂肪沉積。⁸³ 雖然噻唑烷二酮類在帶有 PPARG 突變的 FPL 病人中可能有用,但關於其療效的資料尚無定論。⁸⁴ 若儘管使用各種口服降血糖藥物組合,高血糖仍持續存在,則應開始胰島素治療。對於有極度胰島素抗性者,應使用高濃度的 U-200 或 U-500 胰島素。雖然皮下 metreleptin 替代療法能改善明顯低瘦素血症之全身性脂肪失養症病人的糖尿病控制、肝脂肪變性與高三酸甘油酯血症,⁸⁵,⁸⁶ 但其在 FPL 病人中的效果至今尚無定論。⁸⁷,⁸⁸ Metreleptin 可能在某些具有嚴重代謝紊亂或低血清瘦素濃度的特定 FPL 病人中有效。⁸⁹ Metreleptin 療法已獲美國食品藥物管理局 (U.S. Food and Drug Administration) 核准用於全身性脂肪失養症病人以治療代謝併發症,並在日本核准用於全身性與部分脂肪失養症兩者。Metreleptin 常見的副作用包括注射部位反應 (injection-site reactions) 與低血糖 (hypoglycemia)(發生於同時接受胰島素治療的病人)。所報告的瘦素中和抗體 (neutralizing antibodies to leptin) 之意義仍不明確。後天性全身性脂肪失養症病人也有發展 T 細胞淋巴瘤的風險。對於患有脂肪失養症的 HIV 感染病人,將與脂肪失養症強烈相關的 PIs 與核苷反轉錄酶抑制劑換成其他治療方案,可能改善血脂異常與胰島素抗性;然而,皮下脂肪喪失可能不會改善。⁹⁰
隨著許多型遺傳性脂肪失養症之分子遺傳基礎的發現,可為有患病子女的家庭提供產前診斷 (prenatal diagnosis)。可為近親通婚與 CGL 盛行率高的族群(例如來自黎巴嫩與巴西某些地區者)提供婚前遺傳諮詢 (premarital genetic counseling)。若較新的高效抗反轉錄病毒治療方案(不含 PIs)被證明與脂肪失養症無關,且被認為具療效與安全性,我們或許就能預防 HIV 感染病人發展脂肪失養症。
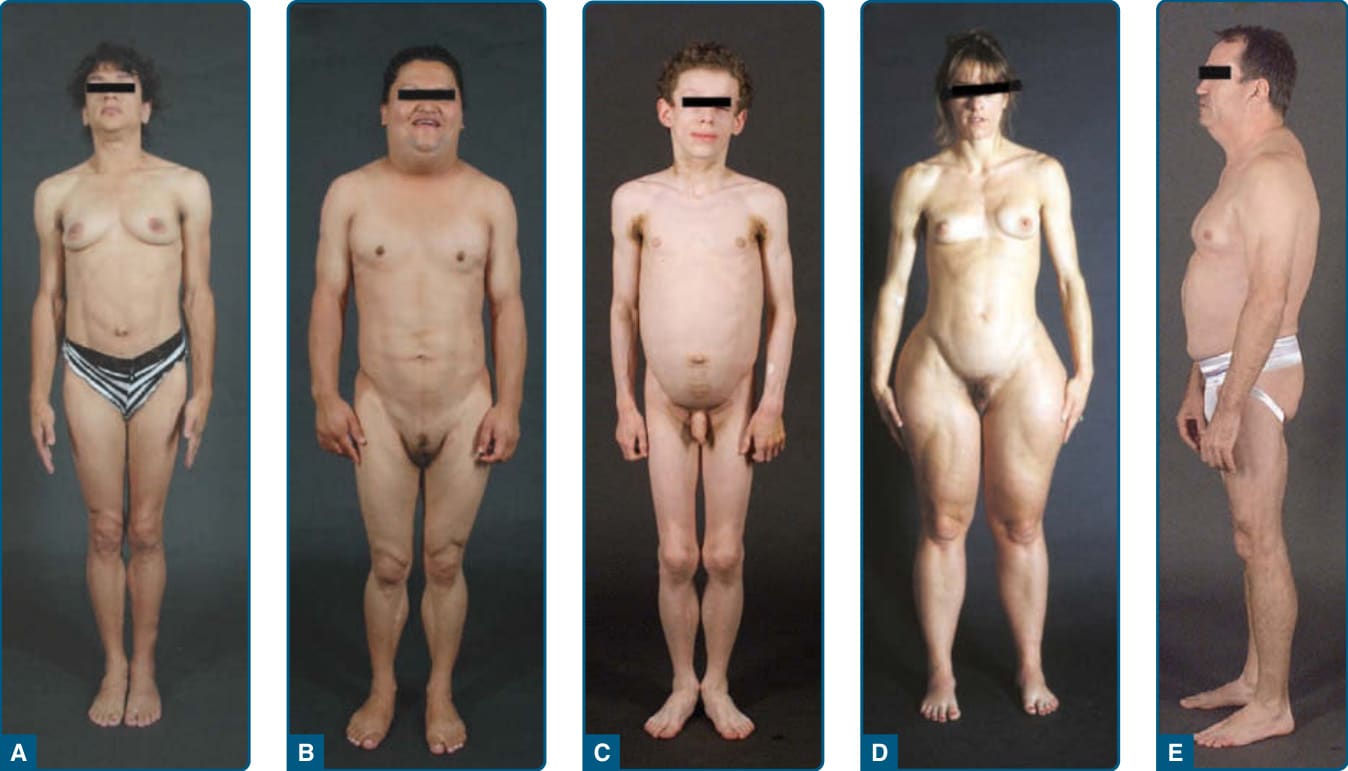
圖 74-1:各型脂肪失養症病人的臨床特徵。A,一名 33 歲西班牙裔女性的前視圖,患有先天性全身性脂肪失養症(亦稱 Berardinelli-Seip 先天性脂肪失養症),第 1 型,由 AGPAT2 基因的同型合子 c.589–2A>G; p.(Val197Glufs∗32) 突變所致。病人有全身性皮下脂肪喪失,腋窩與頸部有黑色棘皮症 (acanthosis nigricans)。她有臍部突出與肢端肥大樣特徵(下頜、手、足增大)。B,一名 27 歲美洲原住民西班牙裔女性的前視圖,患有 Dunnigan 型家族性部分脂肪失養症,由 LMNA 基因的異型合子 p.Arg482Trp 突變所致。她的四肢與前軀幹區域有明顯的皮下脂肪喪失。乳房萎縮。她在顏面、前頸部與外陰區域有增加的皮下脂肪沉積。C,一名 8 歲德國男孩的前視圖,患有後天性全身性脂肪失養症。他有嚴重的全身性皮下脂肪喪失,頸部、腋窩與腹股溝有明顯的黑色棘皮症。D,一名 39 歲白人女性的前視圖,患有後天性部分脂肪失養症(Barraquer-Simons 症候群)。她的顏面、頸部、上肢與胸部有明顯的皮下脂肪喪失,但在前大腿的局部區域有脂肪失養。她在下肢有增加的皮下脂肪沉積。E,一名感染 HIV 的 53 歲白人男性的側視圖,患有高效抗反轉錄病毒治療–誘發的脂肪失養症。他的顏面與四肢有明顯的皮下脂肪喪失,但在頸部前後區域有增加的皮下脂肪沉積,呈現水牛肩 (buffalo hump)。腹部因腹腔內脂肪過多而突出。他接受含蛋白酶抑制劑的抗反轉錄病毒治療已超過 8 年。
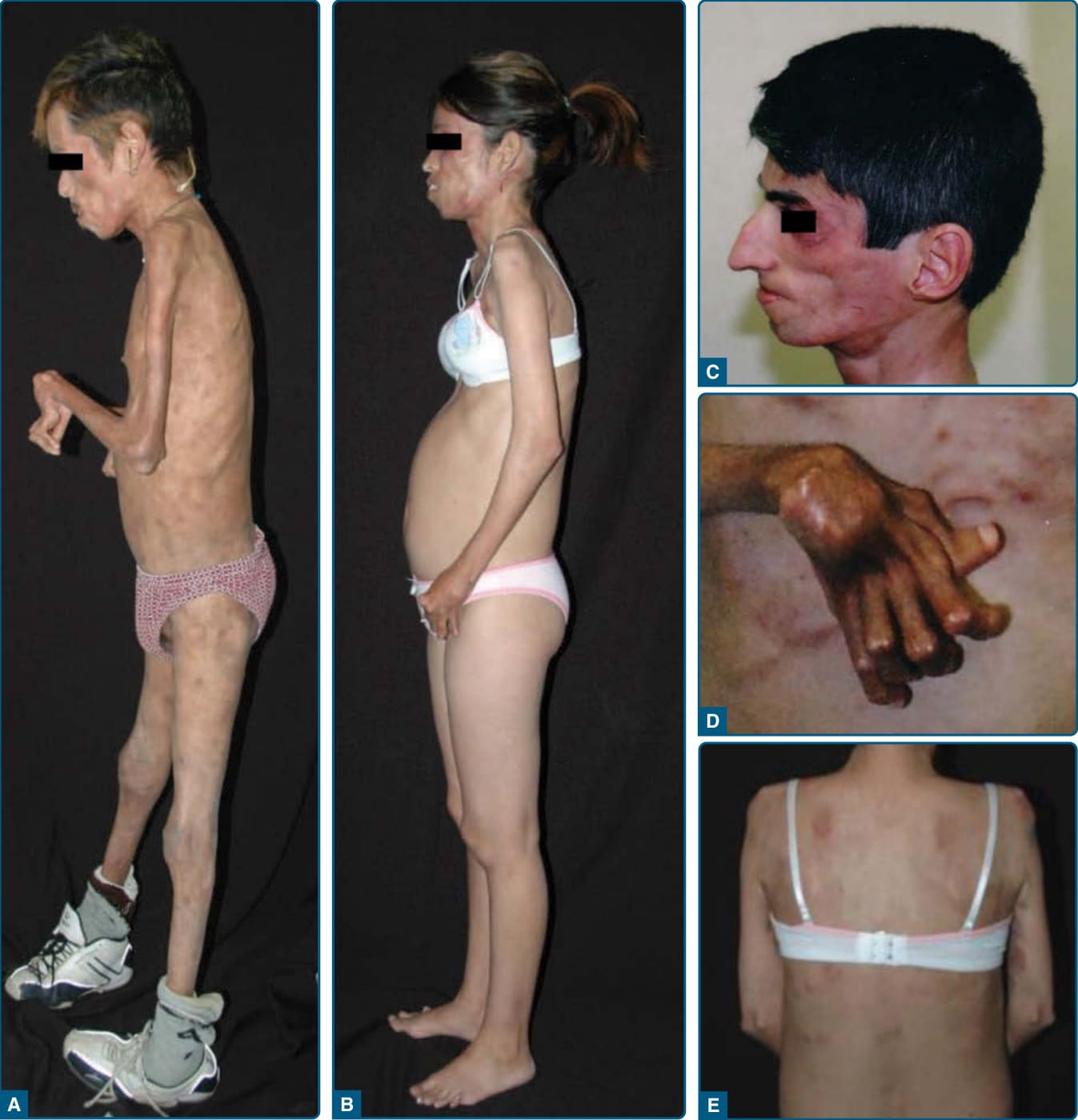
圖 74-2:患有關節攣縮、肌肉萎縮、小球性貧血與脂膜炎誘發脂肪失養 (JMP) 症候群病人的表型。A,一名 30 歲墨西哥男性 JMP 症候群病人的左側視圖,顯示顏面、頸部、胸部與四肢有明顯的全身性皮下脂肪與肌肉量喪失。可見肘部、腕部與手部的屈曲攣縮 (flexion contracture)。B,一名 26 歲墨西哥女性 JMP 症候群病人的左側視圖,顯示脂肪失養主要影響上半身,即顏面、頸部、胸廓與上肢。C,一名 35 歲葡萄牙男性 JMP 症候群病人顏面的左側視圖,顯示明顯的皮下脂肪喪失。D,一名 35 歲葡萄牙男性手部的背側視圖,顯示腕部、近端與遠端指間關節 (interphalangeal joints) 的屈曲攣縮,以及掌指關節 (metacarpophalangeal joints) 的過度伸展。E,一名 26 歲墨西哥女性 JMP 症候群病人胸部的後視圖,顯示胸部與右臂內側有許多離散、小型、紅斑性斑丘疹與結節性皮膚病灶。(Reproduced with permission from Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87:866-872. Copyright © Elsevier.)
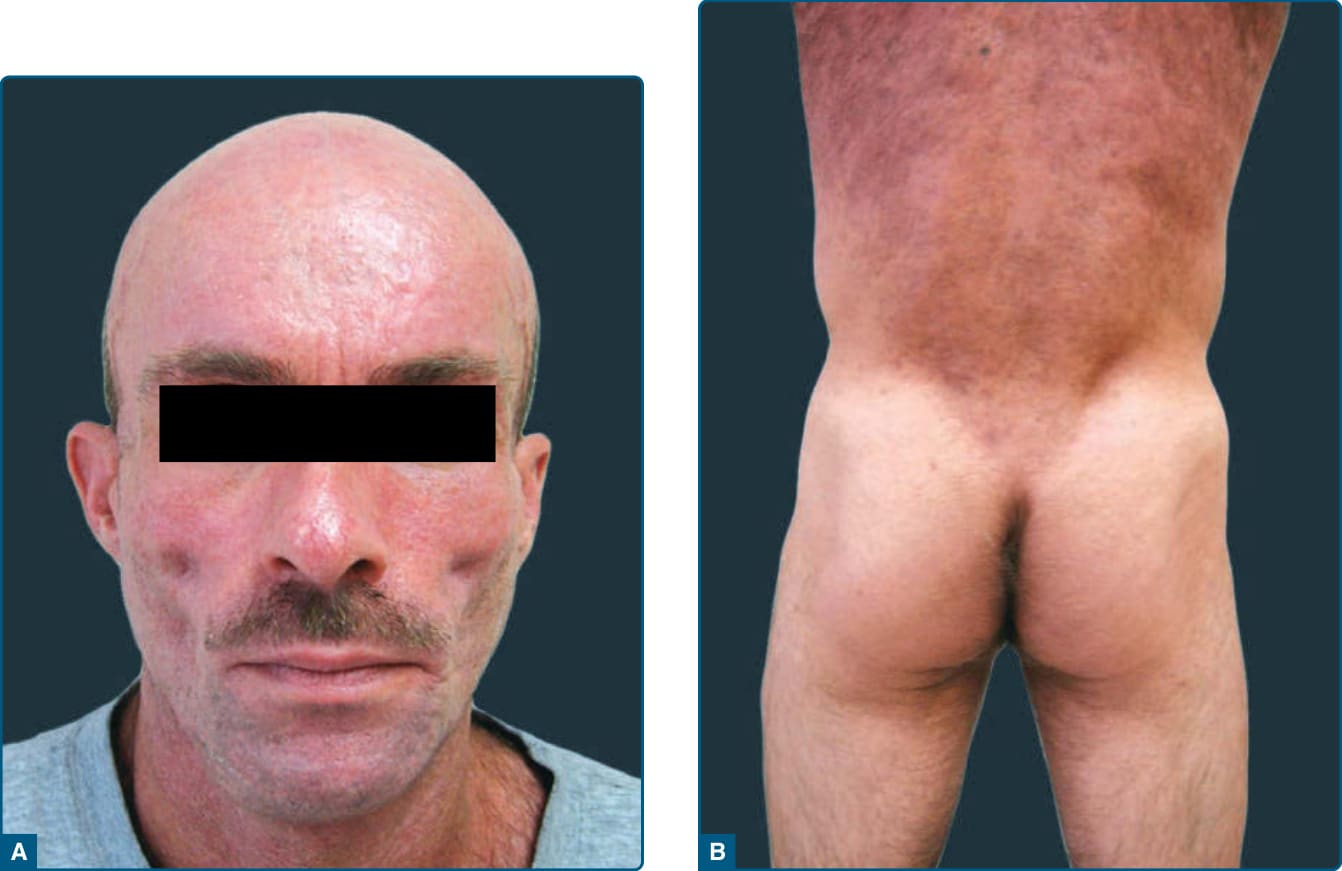
圖 74-3:A,一名 HIV 感染相關脂肪失養症病人的高效抗反轉錄病毒治療–誘發脂肪失養症。頰脂肪 (buccal fat) 喪失導致顴弓 (zygomatic arch) 突出。B,高效抗反轉錄病毒治療–誘發脂肪失養症,伴有外側臀部皮下脂肪喪失與軀幹脂肪沉積,造成腰臀比 (waist-to-hip ratio) 增加。
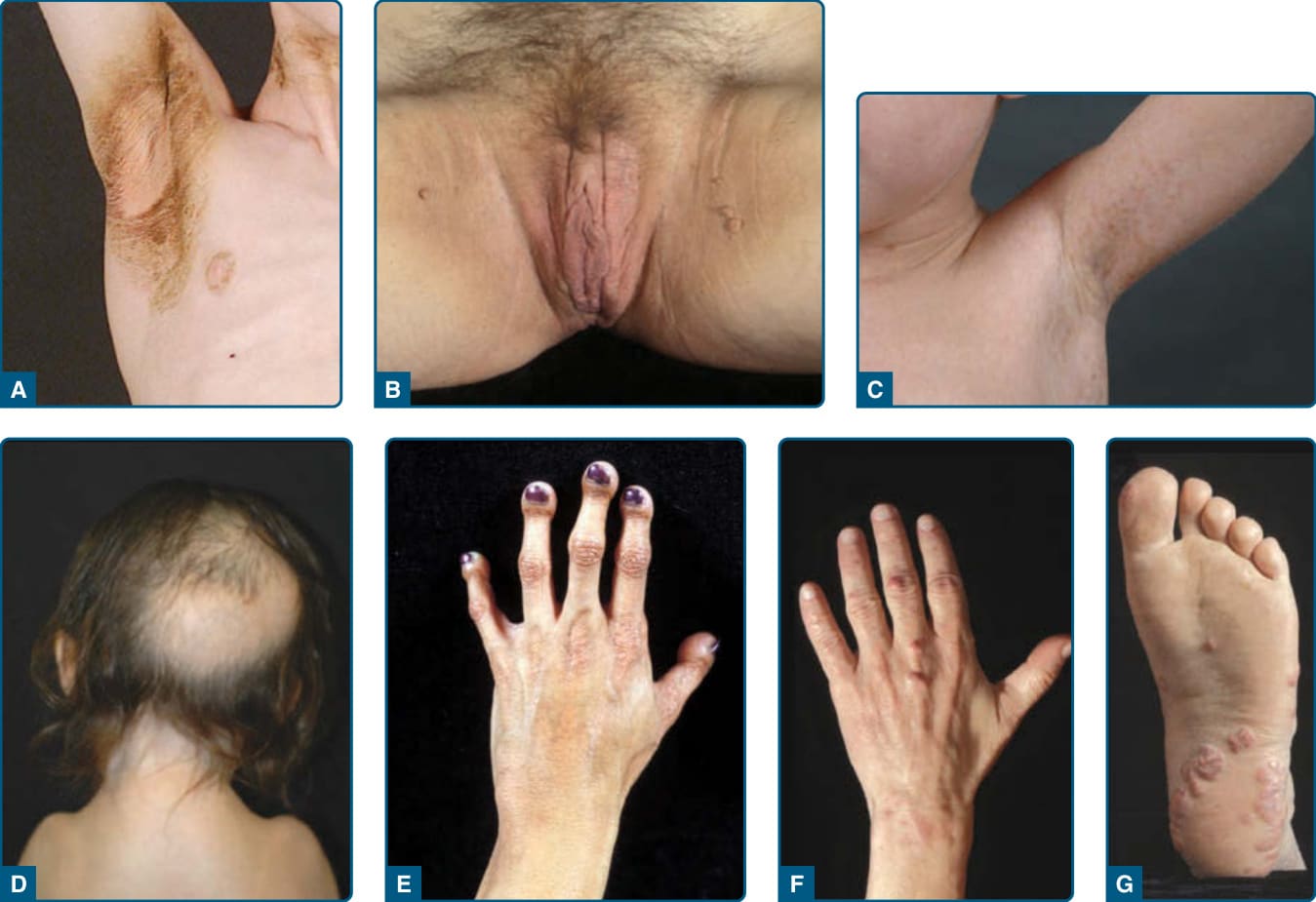
圖 74-4:脂肪失養症病人所見的皮膚科表現。A,一名 8 歲白人男孩(患有後天性全身性脂肪失養症)腋窩與前頸部的黑色棘皮症 (acanthosis nigricans,伴有皮膚增厚的棕色變色)。B,一名 37 歲女性(患有家族性部分脂肪失養症 FPL)會陰與近端大腿內側的黑色棘皮症。多發、小型的皮膚贅瘤 (skin tags) 伴隨色素增加與皮膚增厚。C,一名 7 歲男孩(患有由 LMNA 基因異型合子 p.(Cys588Arg) 突變所致的非典型早老症候群)的多發、輕微色素沉著扁平斑塊(雀斑 freckles)。D,一名 5 歲女孩(患有由 LMNA 基因同型合子 p.(Arg527Cys) 突變所致的重度下頜肢端發育不良 MAD)後頭皮區域的毛髮脫落。她因鎖骨發育不全而有狹窄的肩膀。E,一名 20 歲西班牙裔女性(患有由 LMNA 基因同型合子 p.(Arg527His) 突變所致的 MAD)的肢端骨溶解 (acroosteolysis)。末端指節因末端指骨 (terminal phalanges) 吸收而顯得短而呈球狀。手背皮膚萎縮,尤其在近端指間關節與掌指關節之上。F,一名 45 歲白人病人(患有由 LMNA 基因異型合子 p.(Arg482Gln) 突變所致 Dunnigan 型 FPL 相關之重度高脂血症)中指上的結節性黃色瘤 (tuberous xanthomas)。G,F 所述病人腳掌上的扁平黃色瘤 (planar xanthomas)。(Panel C reproduced with permission from Garg A, Subramanyam L, Agarwal AK, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. 2009;94:4971-4983. Copyright 2009, The Endocrine Society. Panel G reproduced with permission from Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol. 2006;17:162-169. Wolters Kluwer/Lippincott Williams & Wilkins.)

表 74-1:脂肪失養症的分類 (Classification of Lipodystrophies)
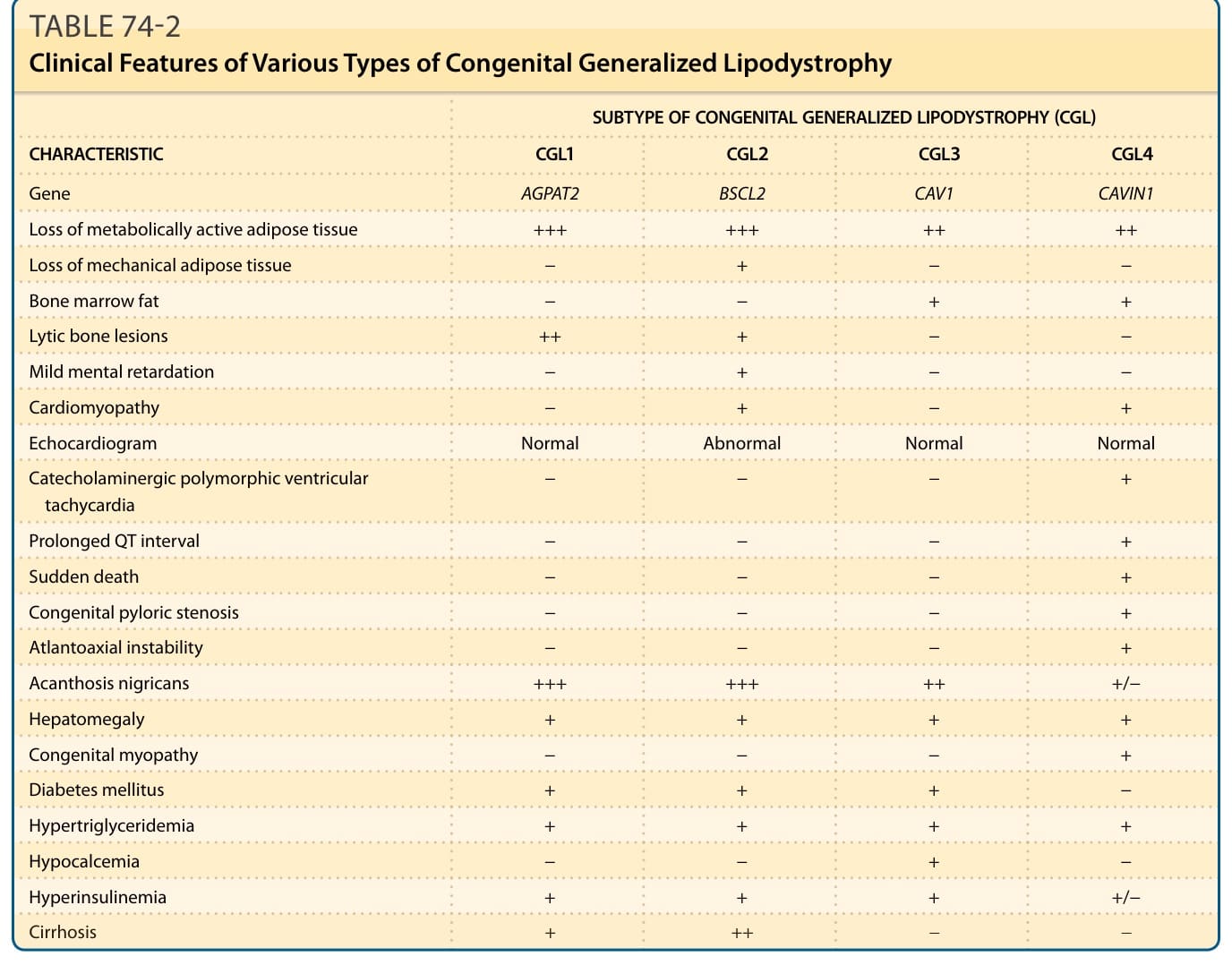
表 74-2:各型先天性全身性脂肪失養症的臨床特徵 (Clinical Features of Various Types of Congenital Generalized Lipodystrophy)