Lipodystrophy
12
AT-A-GLANCE
■ Lipodystrophies are genetic or acquired disorders characterized by selective loss of body fat. The extent of fat loss determines the severity of associated metabolic complications such as diabetes mellitus, hypertriglyceridemia, hepatic steatosis, and acanthosis nigricans.
■ Four loci have been identified for the autosomal recessive congenital generalized lipodystrophy (CGL), namely, AGPAT2, BSCL2, CAV1, and CAVIN1.
■ Five loci have been identified for autosomal dominant familial partial lipodystrophy (FPL), namely, LMNA, PPARG, AKT2, PLIN1, and ADRA2A.
■ LIPE, CIDEC, PCYT1A, and RECQL2 are loci for autosomal recessive FPL, and LMNA and ZMPSTE24 are loci for mandibuloacral dysplasiaassociated lipodystrophy.
■ Molecular basis of many extremely rare forms of genetic lipodystrophies remains to be elucidated.
■ The most prevalent variety of lipodystrophy develops after prolonged duration of protease inhibitor containing highly active antiretroviral therapy in HIV-infected patients.
■ The acquired generalized lipodystrophy and acquired partial lipodystrophy are mainly autoimmune in origin.
■ Localized lipodystrophies occur as a result of drug or vaccine injections, pressure, and panniculitis, as well as other unknown reasons.
■ Current management includes cosmetic surgery and early identification and treatment of metabolic and other complications.
■ Metreleptin replacement therapy is beneficial for treating metabolic complications in hypoleptinemic patients with generalized lipodystrophies.
Lipodystrophies are a heterogeneous group of disorders characterized by selective loss of adipose tissue.1,2
The extent of fat loss varies, with some patients losing fat from small areas (localized lipodystrophy), and others having more extensive fat loss, for example, involving the extremities (partial lipodystrophy) or the entire body (generalized lipodystrophy). Depending upon the extent of fat loss, patients may be predisposed to develop complications associated with insulin resistance such as diabetes mellitus, dyslipidemia, hepatic steatosis, acanthosis nigricans, polycystic ovarian disease, and coronary heart disease.3-5 There are 2 main types of lipodystrophies: genetic and acquired. Table 74-1
provides a detailed classification of various types of lipodystrophies.
EPIDEMIOLOGY
Although the genetic lipodystrophies are rare, recent advances, such as improved definition of the phenotypes and elucidation of the molecular defects, have led to increased recognition of these syndromes. Overall, based on literature reports of approximately 1200 patients, the estimated prevalence of genetic lipodystrophies may be less than 1 in a million. Affected females are recognized easily and thus are reported more often than males. The autosomal-recessive congenital generalized lipodystrophy (CGL) has been reported in approximately 500 patients, with clustering of patients reported in Lebanon and Brazil, where there is increased prevalence of consanguinity. Recently, prevalence of CGL was reported to be approximately 1 in 2 million general population in Turkey.6 The autosomal-dominant familial partial lipodystrophy (FPL) of the Dunnigan variety is the most common with approximately 500 patients being reported; and FPL caused by PPARG mutations has been reported in approximately 40 patients. As a result of founder mutations, FPL Dunnigan type prevalence has been estimated to be as high as 1:200,000 general population from the Canadian province of Ontario7 and approximately 1:20,000 of the general population of the Reunion Island located in the Indian Ocean.8 The autosomal-recessive mandibuloacral dysplasia (MAD) that is caused by LMNA mutations has been reported in approximately 30 patients; MAD caused by ZMPSTE24 mutations has been reported in 15 patients. Acquired partial lipodystrophy was recognized approximately 125 years ago and only approximately 250 to 300 cases of various ethnicities with a maleto-female ratio of 1:4 have been reported.9 Acquired generalized lipodystrophy has been reported in fewer than 100 cases, mostly in whites with a male-to-female ratio of 1:3.10 The most common type at present is highly active antiretroviral therapy (containing protease inhibitors [PIs])-induced lipodystrophy in HIVinfected patients, which is estimated to be affecting more than 100,000 patients in the United States and many more in other countries.1,11,12
GENETIC LIPODYSTROPHIES
In the last decade or so, considerable progress has been made in elucidation of the molecular basis of many types of genetic lipodystrophies. In general, mutations
12
Genetic Lipodystrophies
- Autosomal recessive, congenital generalized lipodystrophy (CGL)
a. Type 1: AGPAT2 mutations b. Type 2: BSCL2 mutations c. Type 3: CAV1 mutation d. Type 4: CAVIN1 mutations e. Others
2. Autosomal dominant, familial partial lipodystrophy (FPL)
a. Dunnigan variety: LMNA mutations b. PPARG mutations c. AKT2 mutation d. PLIN1 mutations e. ADRA2A (adrenoceptor α2A) mutation f. Others
3. Autosomal recessive, familial partial lipodystrophy (FPL)
a. CIDEC mutation b. LIPE mutations c. PCYT1A mutations d. RECQL2 mutations
4. Autosomal recessive, mandibuloacral dysplasia (MAD)–associated lipodystrophy a. LMNA mutations b. ZMPSTE24 mutations c. Others
5. Other lipodystrophies associated with LMNA mutations
a. Atypical progeroid syndrome b. Hutchinson-Gilford progeria syndrome
6. SHORT (short stature, hyperextensibility of joints or hernia, ocular depression, Rieger anomaly, teething) syndrome a. PIK3R1 mutations
7. Neonatal progeroid (Weidemann-Rautenstrauch) syndrome
a. FBN1 mutations b. CAV1 mutations c. POLR3A mutation
8. Mandibular hypoplasia, deafness, progeroid (MDP) syndrome
a. POLD1 mutations
9. Autoinflammatory lipodystrophy (joint contractures, microcytic anemia, panniculitis-associated [JMP] lipodystrophy syndrome spectrum including chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature [CANDLE] syndrome) a. PSMB8 mutations
a. PSMB8 mutations
Acquired Lipodystrophies
- Highly active antiretroviral therapy–induced lipodystrophy in HIV-
Acquired Lipodystrophies
- Highly active antiretroviral therapy–induced lipodystrophy in HIVinfected patients
- Acquired generalized lipodystrophy
infected patients
2. Acquired generalized lipodystrophy
a. Panniculitis induced b. Autoimmune diseases associated c. Idiopathic
3. Acquired partial (Barraquer-Simons) lipodystrophy
a. Panniculitis induced b. Autoimmune diseases associated c. Idiopathic
3. Acquired partial (Barraquer-Simons) lipodystrophy
a. Autoimmune diseases associated b. Membranoproliferative glomerulonephritis associated c. Idiopathic
4. Localized lipodystrophies
a. Autoimmune diseases associated b. Membranoproliferative glomerulonephritis associated c. Idiopathic
4. Localized lipodystrophies
a. Panniculitis induced b. Pressure induced c. Drug induced d. Centrifugal e. Idiopathic
a. Panniculitis induced b. Pressure induced c. Drug induced d. Centrifugal e. Idiopathic
in genes involved in adipocyte differentiation, triglyceride synthesis, lipid droplet formation, and adipocyte survival have been reported to cause lipodystrophies.
1296
CONGENITAL GENERALIZED LIPODYSTROPHY (BERARDINELLI-SEIP SYNDROME)
CONGENITAL GENERALIZED
LIPODYSTROPHY
(BERARDINELLI-SEIP
SYNDROME)
CLINICAL FEATURES
This autosomal recessive disorder has 4 subtypes and can be recognized at birth or soon thereafter from the near total lack of body fat.13 Patients also have marked muscularity, prominent subcutaneous veins, acromegaloid features, acanthosis nigricans, hepatomegaly, and umbilical prominence (Fig. 74-1A, Table 74-2). During childhood, they have a voracious appetite, and accelerated linear growth. Females usually have hirsutism, clitoromegaly, oligomenorrhea, and polycystic ovaries. Only a few women have had successful pregnancies. Fertility may be affected in some men with CGL because of teratozoospermia. Some CGL patients develop hypertrophic cardiomyopathy, mild mental retardation, and focal lytic lesions in the appendicular bones after puberty.13 Metabolic abnormalities related to insulin resistance, such as diabetes mellitus, hyperlipidemia, and hepatic steatosis, may manifest at a young age and are often difficult to control.
ETIOLOGY AND PATHOGENESIS
Genome-wide linkage analysis with positional cloning strategy and candidate gene approach have led to the identification of 4 genetic loci for CGL: 1-acylglycerol- 3-phosphate-O-acyltransferase 2 (AGPAT2) gene on chromosome 9q3414,15; Berardinelli-Seip congenital lipodystrophy 2 (BSCL2) gene on chromosome 11q1316; caveolin 1 (CAV1) gene on chromosome 7q3117; and caveolae associated protein 1 (CAVIN1), also known as polymerase I and transcript release factor (PTRF) gene on chromosome 17q21.18 AGPAT2 is a critical enzyme involved in the biosynthesis of triglycerides and phospholipids from glycerol-3-phosphate and is expressed highly in the adipose tissue.19 The BSCL2-encoded protein, seipin, plays a role in lipid droplet fusion and also may be involved in adipocyte differentiation.20-22 Caveolin 1 is an integral component of caveolae, specialized microdomains seen in abundance on adipocyte membranes. Caveolin 1 binds fatty acids and translocates them to lipid droplets. CAVIN1 is involved in biogenesis of caveolae and regulates expression of caveolins 1 and 3.18 Patients with BSCL2 mutations lack mechanical fat located in the orbital region, palm, sole, and in periarticular regions, as well as metabolically active adipose tissue located in the subcutaneous (SQ), intraabdominal, intrathoracic, and other areas as compared to those with AGPAT2, CAV1, and PTRF mutations where mechanical fat is preserved.17,23 The only reported patient with CAV1 mutation also had short stature and presumed vitamin D resistance.17 Only approximately 15 patients with CAVIN1 mutations have been reported
12
A B C D E
and they have congenital myopathy, increased creatine kinase levels, percussion-induced myoedema, pyloric stenosis, atlantoaxial instability, and cardiac rhythm disturbances including prolonged QT interval and exercise-induced ventricular tachycardia, which can result in sudden death.18,24,25 Patients of Lebanese origin harbor homozygous c.315_319delGTATC; p.(Tyr106Cysfs∗6) mutation in BSCL2, whereas those of African origin nearly always have either homozygous or compound heterozygous c.589–2A>G; p.(Val197Glufs∗32) mutation in AGPAT2 gene.15,16,26
FAMILIAL PARTIAL LIPODYSTROPHY
FAMILIAL PARTIAL
LIPODYSTROPHY
CLINICAL FEATURES
FPL in most patients is transmitted in an autosomal dominant fashion and is characterized by fat loss
from the limbs with variable fat loss from the trunk and increased SQ fat deposition in nonlipodystrophic regions (see Fig. 74-1B). Most FPL patients have the Dunnigan variety as a result of heterozygous mutations in lamin A/C (LMNA) gene. These patients have normal body fat distribution during early childhood, but around the time of puberty, SQ fat from the extremities and trunk is progressively lost (see Fig. 74-1B). The face, neck, and intraabdominal region are spared, and often excess fat accumulates there.27,28 Affected men are often more difficult to diagnose clinically, as many normal men are also quite muscular. Women are more severely affected metabolically.29
Patients with FPL caused by heterozygous mutations in PPARG gene also have fat loss from the extremities, especially from distal regions, but the fat from the face, neck, and truncal areas is spared.30
There is increased prevalence of hypertension among the affected subjects. Four subjects from a single family with diabetes and insulin resistance were reported to harbor a heterozygous mutation in AKT2 gene.31 The female proband had lipodystrophy of the
1297
12
SUBTYPE OF CONGENITAL GENERALIZED LIPODYSTROPHY (CGL)
CHARACTERISTIC CGL1 CGL2 CGL3 CGL4
Gene AGPAT2 BSCL2 CAV1 CAVIN1
Loss of metabolically active adipose tissue +++ +++ ++ ++
Loss of mechanical adipose tissue − + − −
Bone marrow fat − − + +
Lytic bone lesions ++ + − −
Mild mental retardation − + − −
Cardiomyopathy − + − +
Echocardiogram Normal Abnormal Normal Normal
Catecholaminergic polymorphic ventricular tachycardia − − − +
Prolonged QT interval − − − +
Sudden death − − − +
Congenital pyloric stenosis − − − +
Atlantoaxial instability − − − +
Acanthosis nigricans +++ +++ ++ +/−
Hepatomegaly + + + +
Congenital myopathy − − − +
Diabetes mellitus + + + −
Hypertriglyceridemia + + + +
Hypocalcemia − − + −
Hyperinsulinemia + + + +/−
Cirrhosis + ++ − −
Cirrhosis + ++ − −
−, absent; +/−, absent/present; +, mild; ++, moderate; +++, severe.
limbs, although detailed studies of body fat distribution were not performed. Recently, 12 FPL patients were reported to harbor PLIN1 mutations.32,33 Three patients from a single family with atypical FPL and marked buffalo hump had a heterozygous ADRA2A mutation.34
ETIOLOGY AND PATHOGENESIS
FPL results from heterozygous missense mutations in 1 of the 5 genes: lamin A/C (LMNA) on chromosome 1q21–22,35-38 an integral component of nuclear lamina; peroxisome proliferator-activated receptor γ (PPARG) on chromosome 3p25,30,39,40 a key transcription factor involved in adipocyte differentiation; v-AKT murine thymoma oncogene homolog 2 (AKT2) on chromosome 19q13,31 which is involved in downstream insulin signaling; perilipin 1 (PLIN1) on chromosome 15q26, a key component of lipid droplets32; and adrenoreceptor α2A (ADRA2A), the main presynaptic inhibitory feedback G-protein–coupled receptor regulating norepinephrine release.34 Adipocyte loss in patients with LMNA mutations may be a result of disruption of nuclear envelope function and integrity
1298
resulting in premature cell death. Some patients with mutations in the aminoterminal region of lamin A/C also develop myopathy, cardiomyopathy, and conduction system abnormalities indicative of a multisystem dystrophy.41 On the other hand, others with mutations in the extreme C-terminal region of lamin A may have mild lipodystrophy.42
MANDIBULOACRAL DYSPLASIA–ASSOCIATED LIPODYSTROPHY
MANDIBULOACRAL
DYSPLASIA–ASSOCIATED
LIPODYSTROPHY
CLINICAL FEATURES
Patients with MAD have characteristic skeletal abnormalities including hypoplasia of the mandible and clavicles, acroosteolysis, cutaneous atrophy, progeroid features such as thin beaked nose, hair loss, thin skin with prominent superficial vasculature and mottled hyperpigmentation, delayed dentition
and closure of cranial sutures, joint stiffness, and lipodystrophy.43,44
ETIOLOGY AND PATHOGENESIS
Mutations in LMNA and zinc metalloproteinase (ZMP- STE24) on chromosome 1p34 cause autosomal-recessive MAD-associated lipodystrophies.45,46 ZMPSTE24 is involved in posttranslational proteolytic processing of prelamin A to mature lamin A and its deficiency can result in accumulation of prelamin A in cells which is supposed to cause toxicity. Those persons with ZMP- STE24 mutations develop clinical manifestations earlier in life, are premature at birth, and can develop focal segmental glomerulosclerosis, calcified skin nodules, and thin shiny skin.47,48
OTHER TYPES
OTHER TYPES
CLINICAL FEATURES
My colleagues and I reported a novel syndrome with mandibular hypoplasia, deafness, progeroid features–associated lipodystrophy.49 All males with mandibular hypoplasia, deafness, progeroid features had undescended testes and were hypogonadal. One adult female showed lack of breast development. Patients with autoinflammatory lipodystrophy present as a spectrum of clinical manifestations, with onset ranging from during the first months of life50 to later during childhood.51 Early features are recurrent fevers, annular violaceous plaques, poor weight and height gain, persistent violaceous eyelid swelling, hepatomegaly, arthralgias, variable muscle atrophy, and progressive lipodystrophy.52-54 Histopathologic examination of lesional skin shows atypical mononuclear infiltrates of myeloid lineage and mature neutrophils, and laboratory abnormalities include chronic anemia, elevated acute-phase reactants, and raised liver enzymes with a cytokine profile showing high levels of interferoninducible protein–1, monocyte chemotactic protein–1, interleukin-6, and interleukin-1 receptor antagonist, consistent with an interferon signaling signature.52,53
This presentation has been called CANDLE syndrome (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature).50 Older individuals with PSMB8 mutations show joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced (JMP) childhood-onset lipodystrophy (Fig. 74-2).51 Other features of JMP syndrome include hypergammaglobulinemia, elevated erythrocyte sedimentation rate, hepatosplenomegaly, and calcification of basal ganglia.51 Other rare syndromes of lipodystrophy include autosomal-recessive FPL and SHORT (short stature, hyperextensibility or inguinal hernia, ocular depression, Rieger anomaly, and teething delay) syndrome.55,56 The fat loss is usually confined to the face, upper extremities, and trunk, and sometimes the buttocks.55,56 Patients with neonatal
12
progeroid (Wiedemann-Rautenstrauch) syndrome present with generalized loss of body fat and muscle mass and progeroid appearance at birth.57 There is a marked heterogeneity in the phenotype of patients with Wiedemann-Rautenstrauch syndrome.
ETIOLOGY AND PATHOGENESIS
De novo recurrent heterozygous mutation in polymerase (DNA) delta 1, catalytic subunit (POLD1) gene has been reported in patients with mandibular hypoplasia, deafness, progeroid features syndrome.58
Most patients with SHORT syndrome have a de novo recurrent heterozygous missense mutation in phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) gene.59 De novo heterozygous mutations in fibrillin 1 (FBN1)60 and caveolin 1 (CAV1)61 and biallelic mutations in RNA polymerase III subunit A (POLR3A)62
have been reported in Wiedemann-Rautenstrauch syndrome patients. Recently, a single patient with autosomal recessive, FPL phenotype was found to harbor a homozygous missense mutation in cell deathinducing DNA fragmentation factor a-like effector c (CIDEC) gene on chromosome 3p25, involved in lipid droplet formation.63 The histopathology of the SQ adipose tissue of the patient revealed multilocular, small lipid droplets in adipocytes. Two additional patients with autosomal recessive partial and generalized lipodystrophy phenotype had mutations in phosphate cytidylyltransferase 1α (PCYT1A) gene that encodes the rate-limiting enzyme in phosphatidyl choline biosynthetic pathway.64 Loss-of-function mutations in hormone sensitive lipase (LIPE) gene have been reported to cause an autosomal recessive FPL phenotype with multiple symmetric lipomatosis and myopathy.65-67 Two women with FPL and features of Werner syndrome have been reported to harbor null mutations in Werner syndrome RecQ like helicase (WRN) gene.68
Heterozygous LMNA mutations can also cause partial or generalized lipodystrophy in patients with atypical progeroid syndrome69 and generalized loss of SQ fat in Hutchinson-Gilford progeria syndrome.70 Autoinflammatory lipodystrophy syndrome results from mutations in proteasome subunit, beta-type, 8 (PSMB8).52,53 PSMB8 encodes the β5i subunit of the immunoproteasome involved in proteolytic cleavage of immunogenic epitopes presented by major histocompatibility complex Class I molecules.52 The molecular genetic basis of many other genetic lipodystrophy syndromes remains unclear (see Table 74-1).
ACQUIRED LIPODYSTROPHIES
Despite recognition of acquired lipodystrophies for more than a century, progress in understanding underlying pathogenic mechanisms has been slow.
1299
12
A
B
C
D
E
1300
ACQUIRED PARTIAL LIPODYSTROPHY (BARRAQUER-SIMONS SYNDROME)
ACQUIRED PARTIAL
LIPODYSTROPHY
(BARRAQUER-SIMONS
SYNDROME)
CLINICAL FEATURES
Acquired partial lipodystrophy develops in most patients before age 15 years. Patients lose SQ fat gradually in a symmetric fashion starting with the face and then spreading downward. Most patients present with fat loss from the face, neck, upper extremities, and trunk, with sparing of SQ abdominal fat and lower extremities (see Fig. 74-1C). Approximately, 20% of the patients develop mesangiocapillary (membranoproliferative) glomerulonephritis, and some develop drusen.9 Usually, patients do not develop metabolic complications.
ETIOLOGY AND PATHOGENESIS
The exact pathogenesis of SQ fat loss remains unclear but there is strong evidence of autoimmune-mediated adipocyte loss as more than 80% of the patients have low levels of complement 3 (C3) and presence of a circulating immunoglobulin G, a C3-nephritic factor that blocks degradation of the enzyme C3 convertase.9 Loss of fat could be the result of C3-nephritic factor–induced lysis of adipocytes expressing factor D.
ACQUIRED GENERALIZED LIPODYSTROPHY (LAWRENCE SYNDROME)
ACQUIRED GENERALIZED
LIPODYSTROPHY
(LAWRENCE SYNDROME)
CLINICAL FEATURES
Acquired generalized lipodystrophies present with variable amount of SQ fat loss usually during childhood. Although many patients have generalized loss of fat, some areas are spared (see Fig. 74-1D). Usually, intraabdominal or bone marrow fat is spared.10 However, patients develop extremely severe hepatic steatosis and fibrosis, diabetes, and hypertriglyceridemia, which are difficult to manage.
ETIOLOGY AND PATHOGENESIS
The exact mechanisms of fat loss are not known. In approximately 25% of patients, SQ fat loss occurs following development of SQ inflammatory nodules that on biopsy reveal panniculitis.10 These lesions initially
12
result in localized fat loss followed by generalized loss of fat. Another 25% of the patients have associated autoimmune diseases, especially juvenile dermatomyositis.10,71 In the remaining patients with the idiopathic variety, multiple unknown mechanisms are likely involved.10 Patients with the panniculitis- associated variety have less-severe fat loss and metabolic complications than seen in other types. Some patients have been reported to have low serum levels of complement 4.72
HIGHLY ACTIVE ANTIRETROVIRAL THERAPY–INDUCED LIPODYSTROPHY IN HIV-INFECTED PATIENTS
HIGHLY ACTIVE
ANTIRETROVIRAL
THERAPY–INDUCED
LIPODYSTROPHY IN
HIV-INFECTED PATIENTS
CLINICAL FEATURES
Patients infected with HIV who are receiving PIcontaining highly active antiretroviral therapy usually lose SQ fat from the face, trunk, and extremities 2 years or more after initiation of therapy (see Figs. 74-1E, 74-3A, and 74-3B). Fat loss from the face can be so severe as to result in an emaciated appearance. Some of them develop buffalo hump, double chin, and also gain intraabdominal fat. The fat loss progressively gets worse with ongoing highly active antiretroviral therapy and does not reverse on discontinuation of PIs. Some cases develop diabetes mellitus and many develop combined hyperlipidemia that can predispose the patients to coronary heart disease.12
ETIOLOGY AND PATHOGENESIS
Drugs such as HIV-1 PIs and nucleoside analogs are implicated in causing lipodystrophy in HIV-infected patients. Many, but not all, PIs may induce lipodystrophy by inhibiting ZMPSTE24, resulting in accumulation of prelamin A.73 Other mechanisms may include PI-induced alteration of expression of key transcription factors involved in lipogenesis and adipocyte differentiation, such as sterol regulatory element- binding protein 1c and PPARG.74 PIs also reduce glucose transporter 4 expression, which may be a mechanism for inducing insulin resistance.75 Nucleoside analogs, especially zidovudine and stavudine, may induce fat loss by inhibiting polymerase-γ, a mitochondrial enzyme involved in replication of mitochondrial DNA.76 As most patients receive multiple antiretroviral drugs together, the individual effects of PIs or nucleoside reverse transcriptase inhibitors on the phenotype are not clear.
1301
12
A
B
LOCALIZED LIPODYSTROPHY
LOCALIZED
LIPODYSTROPHY
CLINICAL FEATURES
Localized lipodystrophies present with SQ fat loss from a focal region resulting in a dimple or a crater with overlying skin usually unaffected. In some patients, large contiguous or anatomically distinct areas on any region of the body may be involved.3
ETIOLOGY AND PATHOGENESIS
This can occur from SQ injection of various drugs, panniculitis, pressure, and other mechanisms.3
DIAGNOSIS
Lipodystrophies should be strongly suspected in “lean or nonobese” patients who present with premature onset of diabetes, hypertriglyceridemia, hepatic steatosis, acanthosis nigricans, and polycystic ovarian syndrome. These patients should be examined carefully for evidence of loss of SQ fat especially from the hips and thighs, as well as excess SQ fat deposition in various anatomic regions. For those presenting with generalized lipodystrophy during childhood, pictures
1302
at birth should be evaluated for evidence of fat loss. If lipodystrophy phenotype is discovered at or shortly after birth, CGL should be considered; otherwise, the patient may have acquired lipodystrophy.
HISTORY
HISTORY
Patients should be asked about their age at time of onset and the progression of the lipodystrophy and other associated manifestations. Taking a detailed family history, including the history of consanguinity, is very important to understand the mode of inheritance of genetic lipodystrophies. Associated autoimmune diseases, especially juvenile dermatomyositis, should be considered in patients with acquired lipodystrophies. Those with localized lipodystrophies should be asked about local injections, trauma, pressure, or other insults. A detailed history of duration and type of antiretroviral therapy should be obtained from HIV-infected patients with lipodystrophy.
CUTANEOUS LESIONS
CUTANEOUS LESIONS
The most common cutaneous lesion seen in patients with lipodystrophies is acanthosis nigricans in the axillae,
12
A B C
D E F G
groins, neck, and sometimes even on the knuckles, Achilles tendons, and trunk (Figs. 74-4A and B). Many patients develop clitoromegaly and hirsutism as a result of associated polycystic ovarian syndrome. Freckles were noted in a patient with atypical progeroid syndrome (Fig. 74-4C). A thin, beaked nose with loss of scalp, eyebrow, and axillary hair with cutaneous atrophy and mottled hyperpigmentation can be seen in patients with progeroid syndromes and MAD (Fig. 74-4D), along with acroosteolysis (Fig. 74-4E).43,69,70 Rare patients with MAD develop shiny, taut, atrophic skin with a tendency to breakdown. Eruptive, tuberous, and planar xanthomas are also commonly seen in patients with extreme hypertriglyceridemia (Figs. 74-4F and G). Loss of SQ fat from the soles can result in plantar calluses. SQ nodules with overlying erythema may be seen in patients with panniculitis.
LABORATORY TESTING
LABORATORY TESTING
Laboratory testing depends upon the type of lipodystrophy. Except for patients with localized lipodystrophies, a serum chemistry profile for glucose, lipids, liver enzymes, and uric acid should be obtained. Measurement of fasting and postprandial serum glucose and insulin during an oral glucose tolerance test can provide some estimate of insulin resistance. Serum leptin measurements are not diagnostic, but can help guide treatment decisions as far as investigational human recombinant leptin (metreleptin) replacement therapy is concerned. Serum leptin and adiponectin levels are very low in patients with generalized lipodystrophies.77 Patients with acquired partial
1303
12
lipodystrophy should be tested for serum C3 and C3-nephritic factor and annually checked for proteinuria. Radiographs can show presence of lytic lesions in appendicular bones in patients with CGL and skeletal defects in those with MAD. Skin biopsy is useful for patients with localized lipodystrophy or panniculitisassociated varieties.
SPECIAL TESTS (INCLUDING IMAGING STUDIES)
SPECIAL TESTS (INCLUDING
IMAGING STUDIES)
Distinction between various types of lipodystrophies can be made by physical examination and supported by anthropometry, including measurement of skinfold thickness with calipers at various sites. For in-depth phenotyping of body fat distribution, whole-body dual-energy X-ray absorptiometry, and T1-weighted MRI can be conducted. For those genetic lipodystrophies whose molecular basis is known, various commercial and research laboratories offer genetic testing. Prenatal genetic testing is also feasible. FPL patients, atypical progeroid syndrome patients and CGL, Type 4 patients who are predisposed to cardiomyopathy should undergo electrocardiography and Holter monitoring to detect arrhythmias and echocardiography to assess cardiac function.
DIFFERENTIAL DIAGNOSIS
The most important differential diagnosis of generalized lipodystrophies is with conditions presenting with severe weight loss, such as malnutrition, famine, starvation, anorexia nervosa, uncontrolled diabetes mellitus, thyrotoxicosis, adrenocortical insufficiency, cancer cachexia, HIV-associated wasting, diencephalic syndrome, and chronic infections. For partial lipodystrophies, distinction should be made with Cushing syndrome, generalized and truncal obesity, and multiple symmetric lipomatosis (Madelung disease). Patients with MAD and progeroid syndromes-associated lipodystrophies should be differentiated from those with Werner syndrome and leprechaunism (Donahue syndrome).
CLINICAL COURSE AND PROGNOSIS
Some patients develop extreme hypertriglyceridemia and chylomicronemia, which result in acute pancreatitis and even death. Long-term complications of diabetes such as nephropathy, neuropathy, and retinopathy are frequently seen. Many patients develop coronary heart disease and other atherosclerotic vascular complications.29,78 Hepatic steatosis can lead to cirrhosis, necessitating liver transplantation. Sudden death has been reported during childhood in CGL, Type 4, likely from arrhythmias.24 Some patients with acquired partial lipodystrophy and membranoproliferative glomerulonephritis may require kidney transplantation.9 Patients
1304
with Hutchinson-Gilford progeria syndrome die of acute myocardial infarction or cerebrovascular accidents during their teenage years.79 Some patients with atypical progeroid syndrome and FPL-Dunnigan develop cardiomyopathy with valvular dysfunction, congestive heart failure, and arrhythmias requiring pacemaker implantation.41,69 Two adult patients with MAD caused by ZMPSTE24 mutations died of renal failure resulting from focal segmental glomerulosclerosis.47 Some patients with acquired generalized lipodystrophy have been reported to develop peripheral T-cell lymphomas.80
The prognosis is dependent upon the type of lipodystrophy. In most of the published cases of CGL the patients have been children, resulting in a lack of data about their prognosis. In my experience, some patients have died of complications of acute pancreatitis or cirrhosis, or developed end-stage renal disease, requiring renal transplantation, and blindness as a consequence of diabetic retinopathy. Patients with FPL are also predisposed to metabolic complications and die of atherosclerotic vascular and coronary heart disease or cardiomyopathy and rhythm disturbances. Some patients with MAD have reportedly died during childhood and some died later in their third and fourth decades from complications of renal failure.47,81 Patients with acquired generalized lipodystrophy suffer severe metabolic complications. Patients with acquired partial lipodystrophy and membranoproliferative glomerulonephritis develop renal failure, but others have a normal life span as do those with localized lipodystrophy. HIV-infected patients with lipodystrophy are predisposed to developing coronary heart disease.
MANAGEMENT
Treatment of various types of lipodystrophies is quite challenging. There is no specific treatment available to reverse the loss of body fat. The mainstay of treatment includes cosmetic surgery and management of complications. Patients with partial lipodystrophies can undergo autologous adipose tissue transplantation or implantation of dermal fillers such as hyaluronic acid, calcium hydroxylapatite, silicone, polyacrylamide gels, or poly-l-lactic acid.82 Unwanted excess adipose tissue can be surgically excised or removed by liposuction. Those with CGL can undergo reconstructive facial surgery, including facial grafts from thighs, free flaps from anterolateral thigh, anterior abdomen, or temporalis muscle.1 Support of the parents is critical for preventing unwanted stress and psychological sequelae in children affected with lipodystrophies. All patients are advised to consume low-fat diets. These diets can improve chylomicronemia in patients with extreme hypertriglyceridemia. However, high carbohydrate intake may also raise very low-density lipoprotein triglyceride concentrations. Increased physical activity should be encouraged to mitigate insulin resistance and its complications except in those who have cardiomyopathy. Lytic bone lesions in appendicular bones in patients with CGL usually do not pose an increased risk of fractures.
There are no well-controlled trials available to guide treatment decisions about how to manage metabolic complications. For severe hypertriglyceridemia, an extremely low-fat diet along with fibrates and n-3 polyunsaturated fatty acids from fish oils should be used.5 Statins can be added if required. Any form of estrogen therapy should be avoided, as it can pose the risk of severe hypertriglyceridemia-induced acute pancreatitis. Diabetes should be managed initially with metformin. Thiazolidinediones should be used with caution in patients with partial lipodystrophies as they can potentially increase unwanted fat deposition in nonlipodystrophic regions.83 Although thiazolidinediones can be useful in FPL patients with PPARG mutations, the data on their efficacy are equivocal.84 If hyperglycemia persists despite using various combinations of oral antidiabetic drugs, insulin therapy should be initiated. For those with extreme insulin resistance, highly concentrated U-200 or U-500 insulin should be used. Although SQ metreleptin replacement therapy improves diabetes control, hepatic steatosis, and hypertriglyceridemia in markedly hypoleptinemic patients with generalized lipodystrophies,85,86 its effects in patients with FPL so far have been equivocal.87,88 Metreleptin may be effective in selected FPL patients with severe metabolic derangements or low serum leptin levels.89 Metreleptin therapy is approved by the U.S. Food and Drug Administration for patients with generalized lipodystrophies to treat metabolic complications and in Japan for both generalized and partial lipodystrophies. Frequent side effects of metreleptin include injection-site reactions and hypoglycemia (in patients on concomitant insulin therapy). The significance of reported neutralizing antibodies to leptin remains unclear. There is also a risk of developing T-cell lymphoma in acquired generalized lipodystrophy patients. Switching from PIs and nucleoside reverse transcriptase inhibitors strongly associated with lipodystrophy to other regimens may improve dyslipidemia and insulin resistance in HIV-infected patients with lipodystrophy; however, loss of SQ fat may not improve.90
With the discovery of the molecular genetic basis of many types of inherited lipodystrophies, prenatal diagnosis can be offered for those families with an affected child. Premarital genetic counseling can be provided to those with a high prevalence of consanguinity and CGL, such as those from Lebanon and certain regions of Brazil. If the newer highly active antiretroviral therapy regimens (not including PIs) are proven not to be associated with lipodystrophy and are deemed to be efficacious and safe, we may be able to prevent development of lipodystrophy in HIV-infected patients.
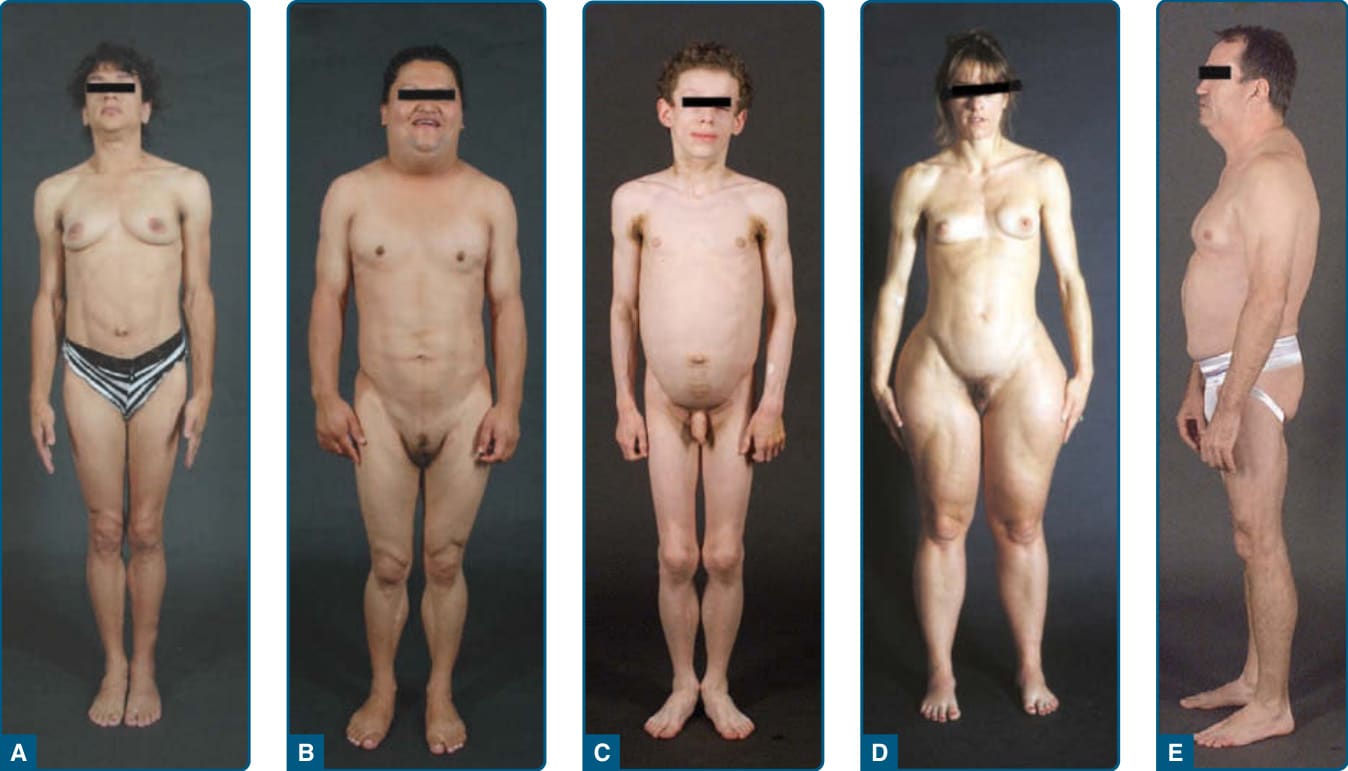
Figure 74-1 Clinical features of patients with various types of lipodystrophies. A, Anterior view of a 33-year-old Hispanic female with congenital generalized lipodystrophy (also known as Berardinelli-Seip congenital lipodystrophy), Type 1 caused by homozygous c.589–2A>G; p.(Val197Glufs∗32) mutation in the AGPAT2 gene. The patient had generalized loss of subcutaneous fat with acanthosis nigricans in the axillae and neck. She has umbilical prominence and acromegaloid features (enlarged mandible, hands, and feet). B, Anterior view of a 27-year-old Native American Hispanic female with familial partial lipodystrophy of the Dunnigan variety caused by heterozygous p.Arg482Trp mutation in LMNA gene. She had marked loss of subcutaneous fat from the limbs and anterior truncal region. The breasts were atrophic. She had increased subcutaneous fat deposits in the face, anterior neck, and vulvar regions. C, Anterior view of an 8-year-old German boy with acquired generalized lipodystrophy. He had severe generalized loss of subcutaneous fat with marked acanthosis nigricans in the neck, axillae, and groin. D, Anterior view of a 39-year-old white female with acquired partial lipodystrophy (Barraquer-Simons syndrome). She had marked loss of subcutaneous fat from the face, neck, upper extremities, and chest, but had lipodystrophy on localized regions on anterior thighs. She had increased subcutaneous fat deposition in the lower extremities. E, Lateral view of a 53-year-old white male infected with HIV with highly active antiretroviral therapy– induced lipodystrophy. He had marked loss of subcutaneous fat from the face and limbs, but had increased subcutaneous fat deposition in the neck region anteriorly and posteriorly showing buffalo hump. Abdomen was protuberant because of excess intraabdominal fat. He had been on protease inhibitor–containing antiretroviral therapy for more than 8 years.
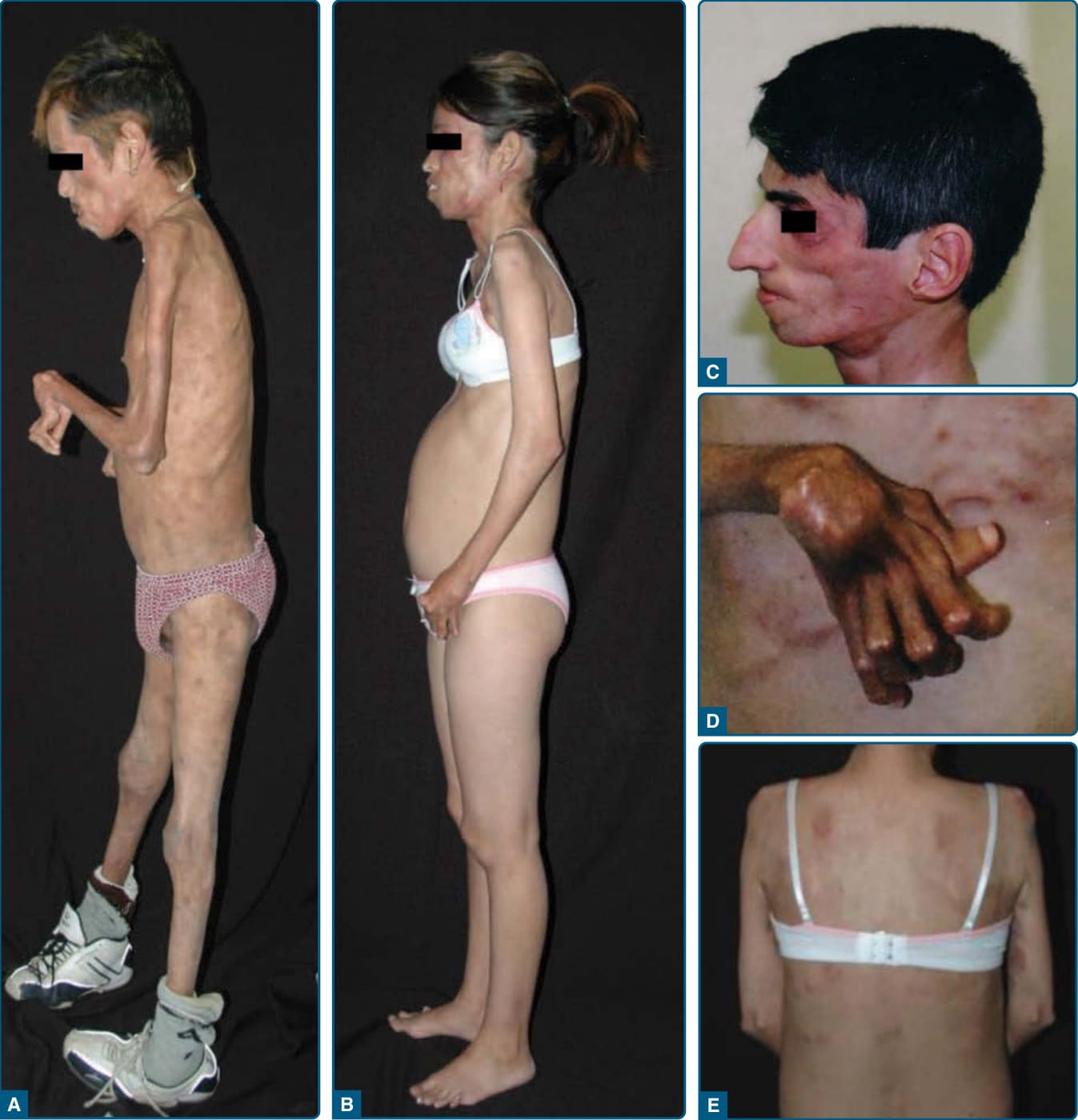
Figure 74-2 Phenotype of patients with joint contractures, muscle atrophy, microcytic anemia, and panniculitisinduced lipodystrophy (JMP) syndrome. A, Left lateral view of a 30-year-old Mexican male with JMP syndrome showing marked generalized loss of subcutaneous fat and muscle mass from the face, neck, chest, and extremities. Flexion contracture at the elbow, wrist, and hand are seen. B, Left lateral view of a 26-year-old Mexican female with JMP syndrome showing lipodystrophy affecting mostly the upper body, that is, the face, neck, thorax, and upper extremities. C, Left lateral view of the face of a 35-year-old Portuguese male with JMP syndrome showing marked loss of subcutaneous fat. D, Dorsal view of the hand from a 35-year-old Portuguese male showing flexion contractures of the wrist, proximal and distal interphalangeal joints and hyperextension of the metacarpophalangeal joints. E, Posterior view of the chest of a 26-year-old Mexican female with JMP syndrome showing many discrete, small, erythematous maculopapular and nodular skin lesions on the chest and medial side of right arm. (Reproduced with permission from Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87:866-872. Copyright © Elsevier.)
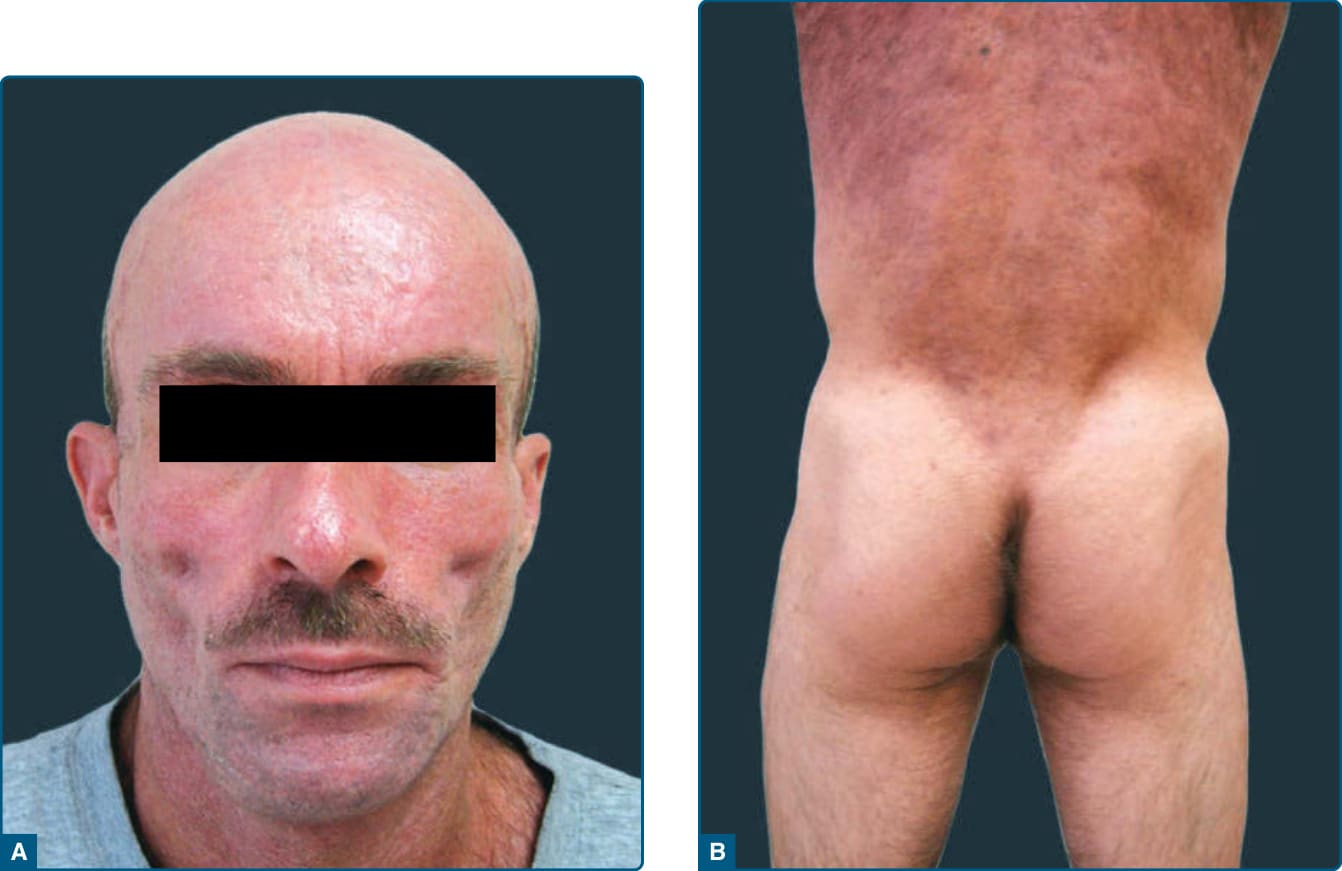
Figure 74-3 A, Highly active antiretroviral therapy–induced lipodystrophy in a patient with HIV infection–associated lipodystrophy. Loss of buccal fat results in prominence of the zygomatic arch B, Highly active antiretroviral therapy-induced lipodystrophy with loss of subcutaneous fat from the lateral buttock and deposit in the trunk, causing an increased waistto-hip ratio.
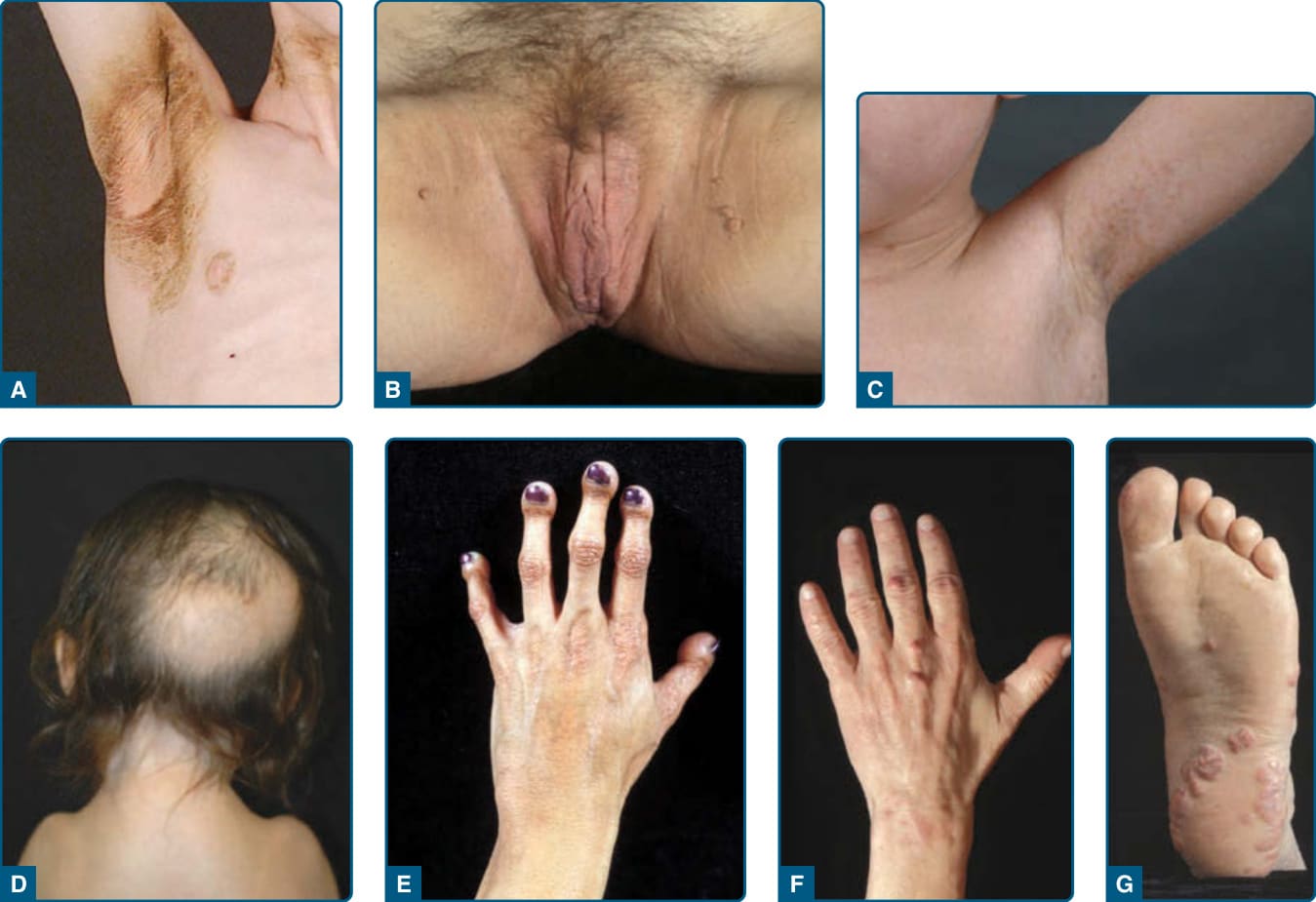
Figure 74-4 Dermatologic manifestations seen in patients with lipodystrophies. A, Acanthosis nigricans (brownish discoloration with thickening of the skin) in the axilla and anterior neck in an 8-year-old white boy with acquired generalized lipodystrophy. B, Acanthosis nigricans in the perineum and medial parts of the proximal thighs in a 37-year-old female with familial partial lipodystrophy (FPL). Multiple, small skin tags accompany increased pigmentation and thick skin. C, Multiple, slightly hyperpigmented flat plaques (freckles) in a 7-year-old boy with atypical progeroid syndrome caused by a heterozygous p.(Cys588Arg) mutation in the LMNA gene. D, Loss of hair from the posterior scalp region in a 5-year-old girl with severe mandibuloacral dysplasia (MAD) caused by a homozygous p.(Arg527Cys) mutation in the LMNA gene. She had narrow shoulders as a result of clavicular hypoplasia. E, Acroosteolysis in a 20-year-old Hispanic woman with MAD caused by a homozygous p.(Arg527His) mutation in the LMNA gene. The terminal digits appear short and bulbous because of resorption of the terminal phalanges. The skin on the dorsum of the hand is atrophic, especially over the proximal interphalangeal and metacarpophalangeal joints. F, Tuberous xanthomas over the middle finger of a 45-year-old white patient with severe hyperlipidemia associated with FPL of the Dunnigan variety caused by a heterozygous p.(Arg482Gln) mutation in the LMNA gene. G, Planar xanthomas on the sole of the patient described in F. (Panel C reproduced with permission from Garg A, Subramanyam L, Agarwal AK, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. 2009;94:4971-4983. Copyright 2009, The Endocrine Society. Panel G reproduced with permission from Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol. 2006;17: 162-169. Wolters Kluwer/Lippincott Williams & Wilkins.)

TABLE 74-1 Classification of Lipodystrophies
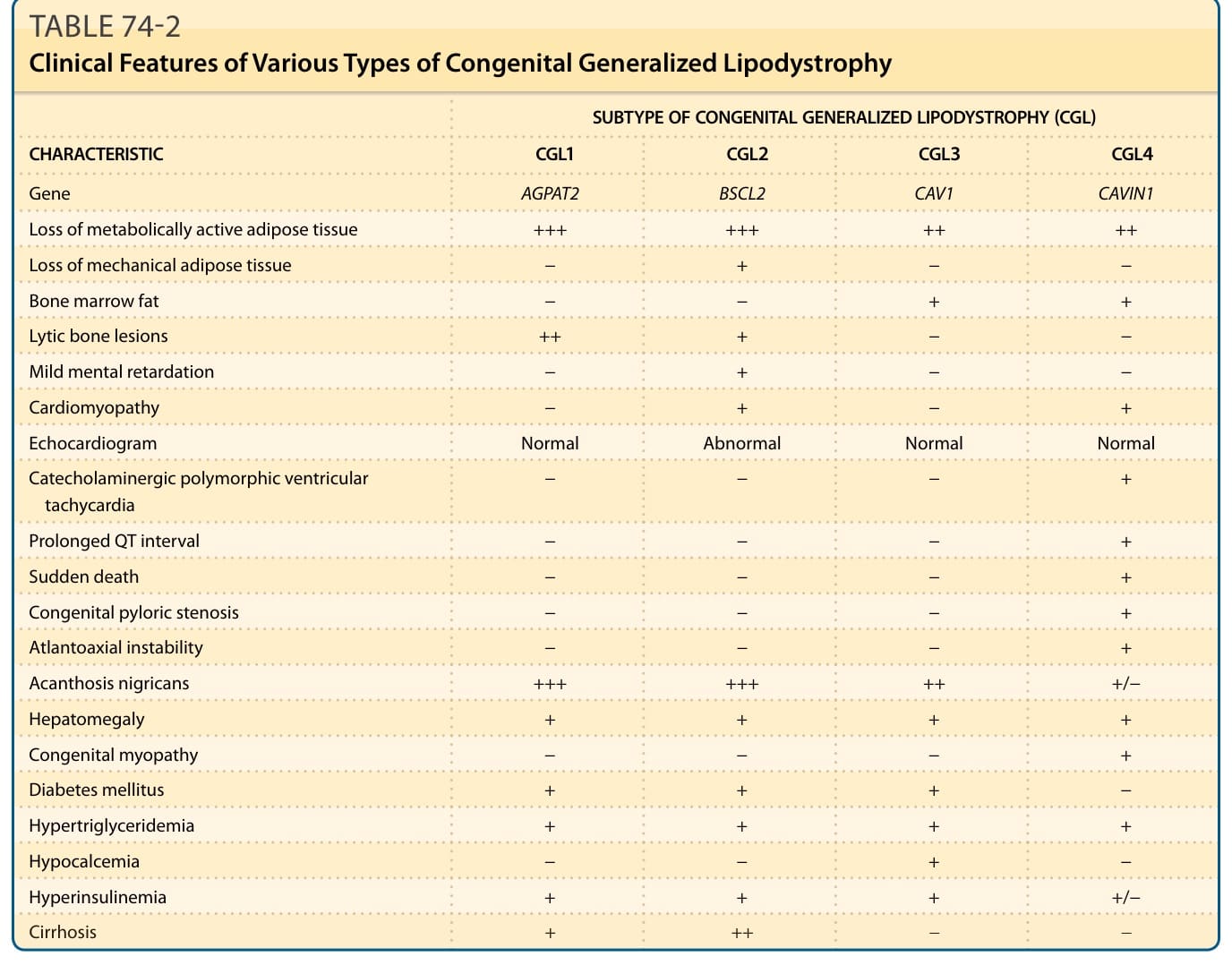
TABLE 74-2 Clinical Features of Various Types of Congenital Generalized Lipodystrophy