脂膜炎 (Panniculitis)
PART 12
皮下組織疾病 (Subcutaneous Tissue Disorders)
皮下脂肪的發炎(脂膜炎,panniculitis)常造成診斷上的困難,因為各種脂肪組織 (adipose tissue, AT) 發炎性疾病的臨床與組織病理學表現彼此重疊(表 73-1)。一個實用的組織病理學分類法將脂膜炎分為「間隔型 (septal)」與「小葉型 (lobular)」(表 73-2),雖然有數種診斷會表現出重疊的特徵。此分類可進一步依是否合併血管炎 (vasculitis),以及發炎浸潤的細胞組成加以延伸(表 73-1)。脂肪組織具有能量儲存與消耗、食慾調節、胰島素敏感性、內分泌與生殖系統、骨代謝、發炎與免疫等功能。其起源可追溯至無脊椎動物的脂肪體 (fat body),該結構具有先天免疫系統、代謝,以及脂質、肝醣與蛋白質儲存功能。在脊椎動物的演化過程中,這些功能被分配至肝臟與脂肪組織之間,兩者皆保留了先天免疫系統的功能。
人類脂肪細胞 (adipocyte) 具有保護個體免於致病性微生物侵害的能力,其方式是透過稱為模式辨識受體 (pattern recognition receptors, PRRs) 的受體來辨識病原相關分子模式 (pathogen-associated molecular patterns)。類鐸受體 (Toll-like receptors, TLRs) 是一種跨膜型 PRR,可表現於細胞膜上(在此偵測細胞表面的微生物模式,例如革蘭氏陰性菌的脂多醣 lipopolysaccharides),或表現於內體/溶酶體 (endosomal/lysosomal) 胞器中(在此主要辨識微生物的核酸)。分泌型 PRR 會結合至微生物表面、活化補體系統 (complement system),並對微生物進行調理作用 (opsonize) 以利吞噬。一旦被活化,PRR 會啟動促發炎訊息傳導路徑,尤其是活化轉錄因子核因子 κB (nuclear factor κB)、干擾素調節因子 3 (interferon regulatory factor 3),或活化 T 細胞核因子 (nuclear factor of activated T cells),進而促進免疫反應相關基因的表現。TLRs 亦會觸發後天免疫反應的活化,包括抗體反應、第 1 型輔助 T 細胞 (T-helper type 1)、第 17 型輔助 T 細胞 CD4+ T 細胞、CD8+ T 細胞反應,以及(經由 TLR4 的)第 2 型輔助 T 細胞 (T-helper type 2) 與免疫球蛋白 E (immunoglobulin E) 反應。
脂肪細胞是白色脂肪組織中最豐富的細胞,但其他細胞類型包括周細胞 (pericytes)、纖維母細胞 (fibroblasts)、內皮細胞 (endothelial cells)、血管平滑肌細胞 (vascular smooth muscle cells) 與發炎細胞(尤其是巨噬細胞 macrophages)。
巨噬細胞衍生的細胞激素與趨化激素,包括腫瘤壞死因子 (tumor necrosis factor, TNF)-α,會誘發脂肪細胞脂解作用 (lipolysis),導致脂肪細胞釋放游離脂肪酸 (free fatty acids),進而誘發促發炎訊息傳導。此一涉及發炎細胞激素與游離脂肪酸的旁分泌迴路 (paracrine loop) 會擴大發炎。脂肪組織亦含有可促進脂肪組織發炎的淋巴球。
脂肪細胞會與血管交互作用,血管周圍脂肪組織 (perivascular AT) 與血管之間存在雙向溝通 (bidirectional crosstalk)。與其他脂肪組織儲積部位相比,血管周圍脂肪組織分泌高量的促發炎細胞激素,如介白素 (interleukin, IL)-6、IL-8 與單核球趨化蛋白-1 (monocyte chemotactic protein-1),相對於抗發炎的脂聯素 (adiponectin)。脂肪細胞的跨膜型 PRR 與 TLRs、與巨噬細胞及淋巴球交互作用的受體,以及多種細胞激素與脂肪激素 (adipokines) 的產生與分泌,皆反映了脂肪細胞在保護宿主免於感染性疾病與其他環境危害中所扮演的角色。
重點一覽(脂膜炎分類概要)
以下整理自表 73-1,列出主要脂膜炎類型之臨床、組織病理與治療要點(原文表格內容散見全章):
- 淋巴皮膚硬化症 (lipodermatosclerosis):又稱硬化性脂膜炎、硬皮樣皮下炎 (hypodermitis sclerodermiformis)、伴脂膜囊性變化之慢性脂膜炎、硬化萎縮性蜂窩組織炎、靜脈淤滯性脂膜炎。好發於 40 歲以上過重女性,與靜脈功能不全、肥胖、系統性硬化症、肺梗塞、高血壓相關。臨床為下肢硬化木質樣斑塊,最常見於前內側小腿,劇痛為最常見症狀;分為急性發炎期與慢性纖維化期(倒香檳瓶外觀)。組織病理為小葉型脂膜炎、無血管炎,脂肪小葉中心缺血性壞死、間隔增厚纖維化、皮下脂肪萎縮、脂膜囊性變化,背景有淤滯變化。治療:壓力治療(30-40 mm Hg)為主要建議治療;Stanozolol 可減輕疼痛、紅斑、硬結;Pentoxifylline、馬栗種子萃取物 (horse chestnut seed extract)、羥乙基蕓香苷 (oxerutins)、類黃酮成分 (flavonoid fraction);減重。
- 感染誘發性脂膜炎 (infectious panniculitis):由多種細菌、真菌、寄生蟲、病毒引起,可為原發接種或血行性散布。臨床為紅斑斑塊、結節、伴膿性分泌之膿瘍(波動/膿瘍型病灶),最常見於腿與足部,上肢、軀幹、臉部亦可侵犯。組織病理多為小葉型脂膜炎但呈混合型,型態視感染為接種相關或血行性而定,呈嗜中性球浸潤。治療依已知微生物而定。
- 結節性紅斑 (erythema nodosum):間隔型脂膜炎、無血管炎,早期為嗜中性球、晚期為米歇爾肉芽腫 (Miescher granulomas)。急性發作、對稱性、壓痛之結節與斑塊,侵犯下肢前側;伴發燒、疲倦、關節痛、關節炎、頭痛,無潰瘍、無萎縮、無疤痕。治療:臥床休息、阿斯匹靈、NSAIDs;SSKI 每日 3 次、每次 2-10 滴;秋水仙素 (Colchicine);皮質類固醇(極少適用)。
- 硬紅斑/結節性血管炎 (erythema induratum/nodular vasculitis):小葉型、常伴血管炎(90%),脂肪細胞早期中心壞死、嗜中性球浸潤,較舊病灶可見上皮樣組織球、多核巨細胞。臨床為下肢紅斑性皮下結節,可侵犯小腿肚、前外側小腿,伴壓痛、潰瘍、疤痕,病程遷延、反覆發作、持續 3-6 週。治療:若 MTB 檢測陽性,完整療程多藥治療;治療潛在病因;SSKI、NSAIDs、秋水仙素、抗瘧藥、皮質類固醇、金鹽 (gold);臥床休息;Pentoxifylline、壓力治療。
- α1-抗胰蛋白酶脂膜炎 (α1-antitrypsin panniculitis):小葉型脂膜炎、無血管炎;嗜中性球早期壞死,嗜中性球散布於膠原束間為特徵,液化性壞死。好發於 30-60 歲;MM 為最常見表現型(正常 AAT);ZZ 為正常值之 10%-15%,與 >60% 的病例相關。臨床為疼痛性紅斑結節與斑塊,蜂窩組織炎、波動性膿瘍型,最常見於下軀幹,亦見於臀部、近端肢體,可能危及生命。治療:輕中度為 dapsone、doxycycline;重度為蛋白質替代療法。
- 胰臟性脂膜炎 (pancreatic panniculitis):發生於 2%-3% 的胰臟疾患病人(胰臟炎、胰臟癌)。小葉型脂膜炎、無血管炎,小葉中心強烈壞死、幽靈脂肪細胞 (ghost adipocytes)、皂化 (saponification) 與鈣化。臨床為紅斑結節伴自發性潰瘍,好發於下肢(尤其關節周圍:膝、踝),成群病灶、紅棕色、萎縮性色素沉著疤痕、油性膿瘍。治療:Octreotide、血漿置換術 (plasmapheresis)。
- 狼瘡性脂膜炎 (lupus panniculitis):女性多於男性(4:1),30-60 歲,亦見於孩童;可發生於紅斑性狼瘡或盤狀紅斑性狼瘡診斷之前或之後。基底細胞層空泡變性、基底膜增厚、黏蛋白沉積、淺層與深層血管周圍浸潤;多為小葉型脂膜炎,伴淋巴濾泡、不等程度的玻璃樣脂肪壞死、硬化膠原束、淋巴球與漿細胞浸潤。治療:羥氯奎寧 (Hydroxychloroquine) 為一線治療;Quinacrine;皮質類固醇單獨或併甲胺喋呤 (methotrexate)。
- 吞噬細胞性組織球性脂膜炎 (cytophagic histiocytic panniculitis):罕見,見於成人、青少年、孩童,與 HLH、MAS 相關。小葉型脂膜炎、無血管炎,豆袋細胞 (bean bag cells):含完整或破碎紅血球、白血球或血小板之巨噬細胞;壞死。臨床為四肢、軀幹皮下紅斑至紫紅斑塊;猛爆型可有潰瘍、發燒、肝脾腫大、骨髓/淋巴結/肝/中樞神經之血球吞噬現象;可呈急性間歇性或快速致命病程。治療:皮質類固醇、環孢素 (Cyclosporine);住院多專科照護。
- 新生兒皮下脂肪壞死 (subcutaneous fat necrosis of the newborn):罕見,發生於出生最初數週,有圍產期併發症病史(胎便吸入、低體溫、低血氧、妊娠糖尿病)。小葉型脂膜炎、無血管炎,呈放射狀排列之針狀裂隙 (needle-shaped clefts),結節與斑塊自發消退;後期可有高血鈣。治療:自發消退、監測血鈣 6 個月,必要時可考慮系統性糖皮質素。
- 冷脂膜炎 (cold panniculitis,Haxthausen 病):9-14 歲男孩的陰囊冷脂膜炎,暴露於寒冷天氣、冰棒、冰敷、游泳。小葉型脂膜炎、無血管炎,血管周圍淋巴組織球浸潤。臨床為冷暴露部位的硬結紅斑斑塊或結節,侵犯臉、大腿、青春期前男孩陰囊脂肪,3 個月內消退。
- 異物/人為性脂膜炎(化妝填充物、油類、人類排泄物等):小葉型脂膜炎、無血管炎,化膿性肉芽腫侵犯脂肪小葉、可見折光性異物。臨床為侵犯臀部、背、肩、頰、大腿的紅斑至紫紅色堅實結節或斑塊,前軀幹不受侵犯,油性/白堊色物質,晚期高血鈣(監測 6 個月);偏振光可辨識巨噬細胞內折光性異物。治療:精神科治療、病灶內類固醇、手術切除。
註:以上要點濃縮自原文表 73-1(脂膜炎分類綜覽,原文以圖片表格呈現),完整表格圖片如下。
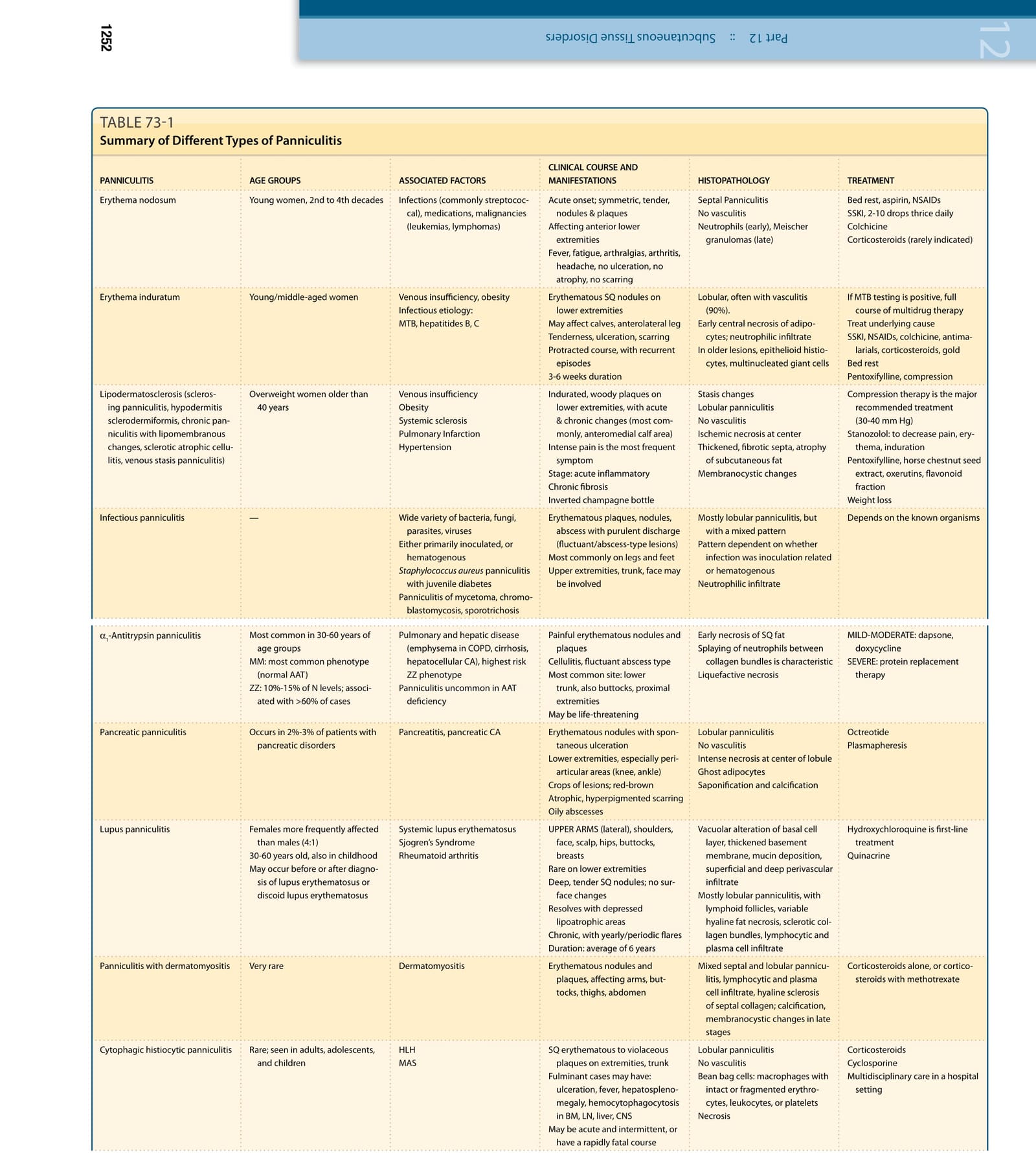
表 73-1:脂膜炎的分類(依臨床特徵、組織病理與治療整理各類型脂膜炎,含結節性紅斑、硬紅斑/結節性血管炎、淋巴皮膚硬化症、感染誘發性脂膜炎、α1-抗胰蛋白酶脂膜炎、胰臟性脂膜炎、狼瘡性脂膜炎、吞噬細胞性組織球性脂膜炎、新生兒皮下脂肪壞死、冷脂膜炎、異物/人為性脂膜炎等)。
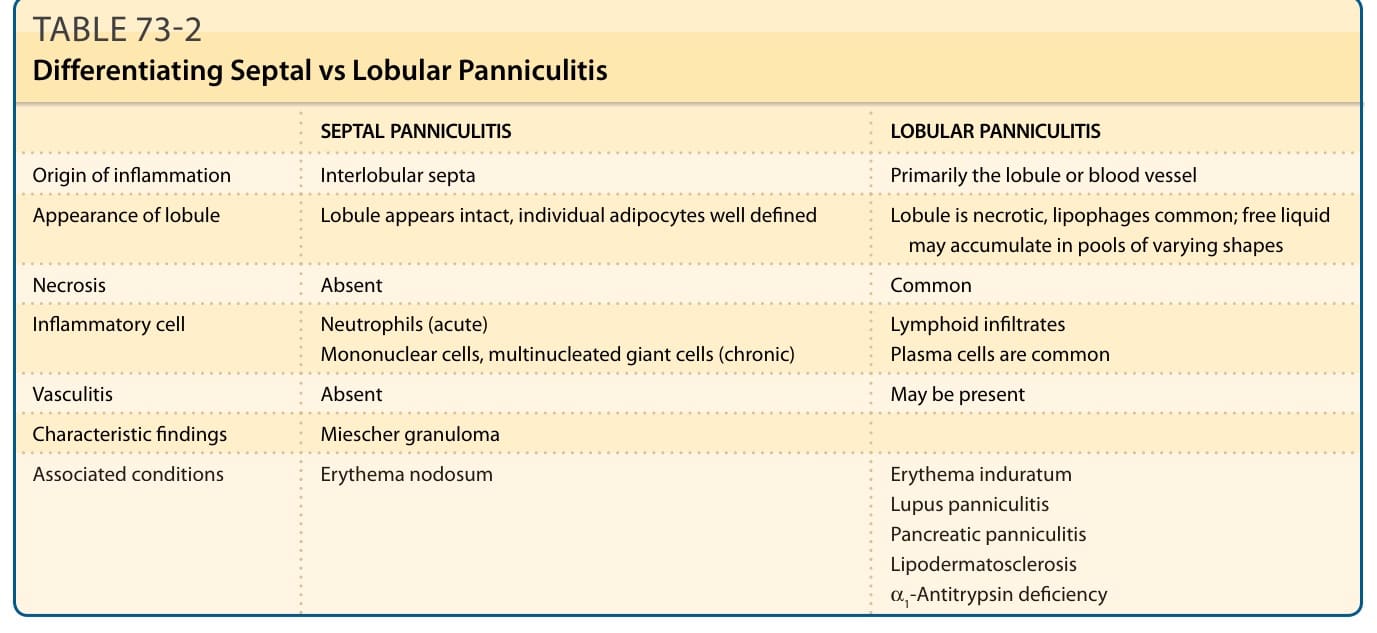
表 73-2:間隔型 (Septal) 與小葉型 (Lobular) 脂膜炎的鑑別。
間隔型脂膜炎 vs 小葉型脂膜炎(表 73-2 內容):
| 項目 | 間隔型脂膜炎 (Septal Panniculitis) | 小葉型脂膜炎 (Lobular Panniculitis) |
|---|---|---|
| 發炎起源 | 小葉間間隔 (interlobular septa) | 主要為小葉本身或血管 |
| 小葉外觀 | 小葉看似完整,個別脂肪細胞界線清楚 | 小葉壞死、噬脂細胞 (lipophages) 常見;游離液體可積聚成形狀不一的池狀 |
| 壞死 | 無 | 常見 |
| 發炎細胞 | 嗜中性球(急性);單核細胞、多核巨細胞(慢性);淋巴浸潤;漿細胞常見 | (同上,依病程而定) |
| 血管炎 | 無 | 可能存在 |
| 特徵性發現 | 米歇爾肉芽腫 (Miescher granuloma) | — |
| 相關疾病 | 結節性紅斑、硬紅斑、狼瘡性脂膜炎、胰臟性脂膜炎、淋巴皮膚硬化症、α1-抗胰蛋白酶缺乏 | 結節性紅斑、硬紅斑、狼瘡性脂膜炎、胰臟性脂膜炎、淋巴皮膚硬化症、α1-抗胰蛋白酶缺乏 |
結節性紅斑 (ERYTHEMA NODOSUM)
結節性紅斑的病因必須加以探查,因為其相關疾病對病人具有重要意義。
重點一覽 (AT-A-GLANCE)
臨床
- 下肢前側對稱性、壓痛、紅斑性結節與斑塊。
- 急性發作;無潰瘍或疤痕。
- 常見發燒、疲倦、關節痛、關節炎、頭痛。
- 女性較常見。
- 持續 3 至 6 週,新病灶可在長達 6 週內持續出現。
組織病理學
- 多為間隔型脂膜炎,不伴血管炎。
- 間隔增厚、纖維化,含發炎細胞。
- 早期病灶為嗜中性球。
- 晚期病灶可見組織球與米歇爾肉芽腫 (Miescher granulomas)。
治療
- 治療任何相關疾患。
- 臥床休息、阿斯匹靈、非類固醇抗發炎藥 (nonsteroidal antiinflammatory drugs)。
結節性紅斑(erythema nodosum, EN)是間隔型脂膜炎的典型代表。它是一種常見的脂膜炎,與多種病因相關,雖然許多病例為特發性 (idiopathic)。由單一致病因子所引起的 EN 之頻率,在過去一個世紀已有極大變化,並受地理位置與當地流行疾病所影響。
雖然診斷常為臨床診斷,但組織病理學可有助於診斷,可顯示間隔發炎以及間隔增厚與纖維化。臨床表現通常會自我緩解,但潛在的病因必須加以探查,因為相關疾病對病人有重要意義。
流行病學 (EPIDEMIOLOGY)
EN 發生於所有年齡層,多數病例侵犯生命第 2 至第 4 個十年的年輕女性。較大型研究顯示超過 80% 的 EN 病人為女性,即女男比為 5:1。然而在小兒病例中並無性別差異。在英格蘭與西班牙的盛行率為診所就診病人的 0.38% 至 0.5%。
臨床特徵 (CLINICAL FEATURES)
EN 表現為下肢出現壓痛、紅斑、溫熱的結節與斑塊(見圖 73-1)。最常侵犯下肢前側與踝部,但前臂、大腿、軀幹甚至臉部都可能受侵犯;在孩童中可見較不典型的位置。結節可融合並變成紫紅色與瘀傷樣,若有出血則稱為瘀傷樣紅斑 (erythema contusiformis)。不會出現潰瘍與疤痕。EN 可伴隨發燒、倦怠、疲倦、關節痛、關節炎、頭痛等全身症狀,較罕見者有腹痛、嘔吐、腹瀉或咳嗽。上呼吸道感染在 20% 至 30% 的病例中先於 EN 發作。實驗室異常常可指出病因;例如鏈球菌感染中咽喉培養陽性與白血球增多。在地方流行區與高風險個體中,純化蛋白衍生物 (purified protein derivative) 陽性提示結核感染。胸部 X 光異常可見於類肉瘤病 (sarcoidosis) 或肺部感染。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
病因
EN 與眾多病因相關(表 73-3)。大型回顧報告中特發性 EN 病例的高比例(37% 至 60%)反映出界定 EN 特定病因的困難。感染、藥物、惡性腫瘤、自體免疫疾病與發炎性疾患皆已被記載可引發 EN 的臨床表現。感染性病因包括細菌、病毒、真菌與原蟲,其中鏈球菌呼吸道感染是小兒 EN 病例最常見的病因。病毒性病因難以診斷,並可能混淆特發性 EN。繼發於結核的 EN 比例不一,地方流行區較高,但在美國與歐洲罕見。
常見的致病藥物包括抗生素、口服避孕藥與其他荷爾蒙療法,以及非類固醇抗發炎藥。較近期的病例提示 EN 與 omeprazole 使用、白三烯抑制劑 (leukotriene inhibitors) 與 vemurafenib 之間有關聯。自從引入低劑量雌激素療法後,口服避孕藥誘發 EN 的盛行率已下降。
與 EN 相關的惡性腫瘤最常包括白血病或淋巴瘤。類肉瘤病與發炎性腸道疾病亦為已知病因。類肉瘤病誘發的 EN 盛行率因地理與病人族群而異,但可佔多達三分之一的 EN 病例。顳動脈炎 (temporal arteritis) 亦曾被記載可導致 EN。
致病機轉
EN 的皮膚表現被認為是對上述病因的過敏反應 (hypersensitivity reaction)。其他理論提出免疫複合體沉積與嗜中性球的參與。整體而言,其病理生理機制尚未明瞭。早期研究顯示干擾素-γ (interferon-γ) 與 IL-2 的存在、白血球的活化、多種黏附分子 (adhesion molecules) 的上調,以及 TNF-α 啟動子、巨噬細胞移動抑制因子 (macrophage migration inhibitory factor) 或 RANTES(regulated upon activation, normal T-cell expressed and secreted)中的基因多型性。
脂肪組織可活化先天與後天免疫反應以摧毀病原。脂肪細胞過度產生與分泌促發炎脂肪激素與脂肪細胞激素 (adipocytokines),與肥胖、心血管疾病、高血壓與糖尿病相關。
相對地,EN 與較侷限的球黴菌病 (coccidiomycosis) 感染相關,並與較不嚴重、病程較短的類肉瘤病相關,尤其在帶有 HLA-DRB1∗03 陽性白血球抗原者。因此,某些與增強發炎反應相關的基因突變可能對某些病原賦予抗性。先天免疫系統功能的進一步證據是 EN 細胞激素圖譜中所見的嗜中性球參與,其中 TNF-α、IL-8、IL-6、單核球趨化蛋白-1 與顆粒球群落刺激因子 (granulocyte colony-stimulating factor) 的高度表現。
診斷 (DIAGNOSIS)
上述所有病因皆呈現相似的組織病理學特徵,且 EN 與其他脂膜炎的組織病理學特徵範圍可能多變。這需要與臨床特徵相互對照,包括病灶位置、全身症狀與實驗室發現。脂肪組織的檢查需要大型切除性切片,因為發炎浸潤可能被遺漏。脂肪組織的發炎並非靜態過程,可能需要多於 1 次的切片才能得到確定診斷。EN 一般為「間隔型」模式,發炎主要侷限於間隔。值得注意的是,「小葉型」則意指發炎主要侵犯脂肪小葉本身。早期 EN 顯示脂肪間隔水腫,伴嗜中性球與外滲的紅血球。隨著 EN 發展,間隔變寬並纖維化,可見包含淋巴球、組織球、嗜中性球與一些嗜酸性球的混合浸潤。
晚期 EN 病灶以纖維化、增寬的間隔為特徵,常伴肉芽腫,脂肪小葉可能被侵蝕並部分消失(圖 73-2A)。EN 常存在覆蓋其上的淺層與深層真皮血管周圍浸潤。
EN 具有特徵性但不具敏感性或特異性的組織球聚集,圍繞中央星狀裂隙,稱為米歇爾肉芽腫 (Miescher granuloma)(見圖 73-2B)。它被視為早期 EN 的特徵,但並非在所有 EN 都能發現,也曾在其他類型的脂膜炎中被描述。有些作者認為米歇爾肉芽腫在 EN 中持續可見。晚期 EN 病灶可顯示脂膜囊性變化 (lipomembranous change)。雖然依定義 EN 特徵性地不伴血管炎,但有些作者強調血栓性靜脈炎 (thrombophlebitis) 為早期 EN 的特徵,而中型血管動脈炎 (medium-vessel arteritis) 也可能(罕見地)出現。
結節性紅斑已報告的病因(表 73-3):
- 細菌感染:鏈球菌感染 (Streptococcal infections)、結核 (Tuberculosis)、耶氏桿菌感染 (Yersinia infections)、沙門氏菌感染 (Salmonella infections)、曲狀桿菌感染 (Campylobacter infections)、布魯氏菌病 (Brucellosis)、兔熱病 (Tularemia)、非典型分枝桿菌感染 (Atypical mycobacterial infections)、軟性下疳 (Chancroid)、腦膜炎雙球菌血症 (Meningococcemia)、白喉桿菌感染 (Corynebacterium diphtheriae infections)、貓抓病 (Cat-scratch disease)、痤瘡丙酸桿菌 (Propionibacterium acnes)、志賀氏菌感染 (Shigella infections)、淋病 (Gonorrhea)、梅毒 (Syphilis)、鉤端螺旋體病 (Leptospirosis)、Q 熱 (Q fever)、性病淋巴肉芽腫 (Lymphogranuloma venereum)、鸚鵡熱披衣菌感染 (Chlamydophila psittaci infections)、肺炎黴漿菌感染 (Mycoplasma pneumoniae infections)、幽門螺旋桿菌感染 (Helicobacter pylori infection)。
- 病毒感染:傳染性單核球增多症 (Infectious mononucleosis)、B 型肝炎 (Hepatitis B)、擠奶者結節 (Milker nodules)、羊痘瘡/接觸傳染性膿皰病 (Orf, contagious ecthyma)、單純疱疹 (Herpes simplex)、麻疹 (Measles)、巨細胞病毒感染 (Cytomegalovirus infections)。
- 真菌感染:皮癬菌 (Dermatophytes)、芽生菌病 (Blastomycosis)、組織漿菌病 (Histoplasmosis)、球黴菌病 (Coccidioidomycosis)、孢子絲菌病 (Sporotrichosis)、麴菌病 (Aspergillosis)。
- 原蟲感染:弓蟲病 (Toxoplasmosis)、鉤蟲病 (Ancylostomiasis)、阿米巴病 (Amebiasis)、梨形鞭毛蟲病 (Giardiasis)、蛔蟲病 (Ascariasis)。
- 藥物:磺胺類 (Sulfonamides)、溴化物 (Bromides)、碘化物 (Iodides)、口服避孕藥、黃體素 (Oral contraceptives, progesterone)、minocycline、金鹽 (Gold salts)、青黴素 (Penicillin)、水楊酸鹽 (Salicylates)、氯噻嗪 (Chlorothiazides)、phenytoin、aminopyrine、arsphenamine、B 型肝炎疫苗 (Hepatitis B vaccine)、nitrofurantoin、pyritinol、D-penicillamine、thalidomide、isotretinoin、介白素-2 (Interleukin-2)、omeprazole、valproate、Tdap(破傷風、白喉、百日咳)疫苗、白三烯調節劑 (Leukotriene-modifying agents)、vemurafenib。
- 雜項疾患:類肉瘤病 (Sarcoidosis)、潰瘍性結腸炎 (Ulcerative colitis)、結腸憩室病 (Colon diverticulosis)、克隆氏病 (Crohn disease)、貝賽特氏病 (Behçet disease)、反應性關節炎 (Reactive arthritis)、史威特症候群 (Sweet syndrome)、懷孕 (Pregnancy)、高安氏動脈炎 (Takayasu arteritis)、免疫球蛋白 A 腎病變 (Immunoglobulin A nephropathy)、慢性活動性肝炎 (Chronic active hepatitis)、肉芽腫性乳腺炎 (Granulomatous mastitis)、Vogt-Koyanagi 病、修格蘭氏症候群 (Sjögren syndrome)、顳動脈炎 (Temporal arteritis)、系統性紅斑性狼瘡 (Systemic lupus erythematosus)、牙齒感染 (Dental infection)。
- 惡性疾病:何杰金氏病 (Hodgkin disease)、非何杰金氏淋巴瘤(包括黏膜相關淋巴組織 MALT 淋巴瘤)、白血病 (Leukemia)、肉瘤 (Sarcoma)、腎細胞癌 (Renal carcinoma)、骨盆腔癌放射治療後 (Postradiotherapy for pelvic carcinoma)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
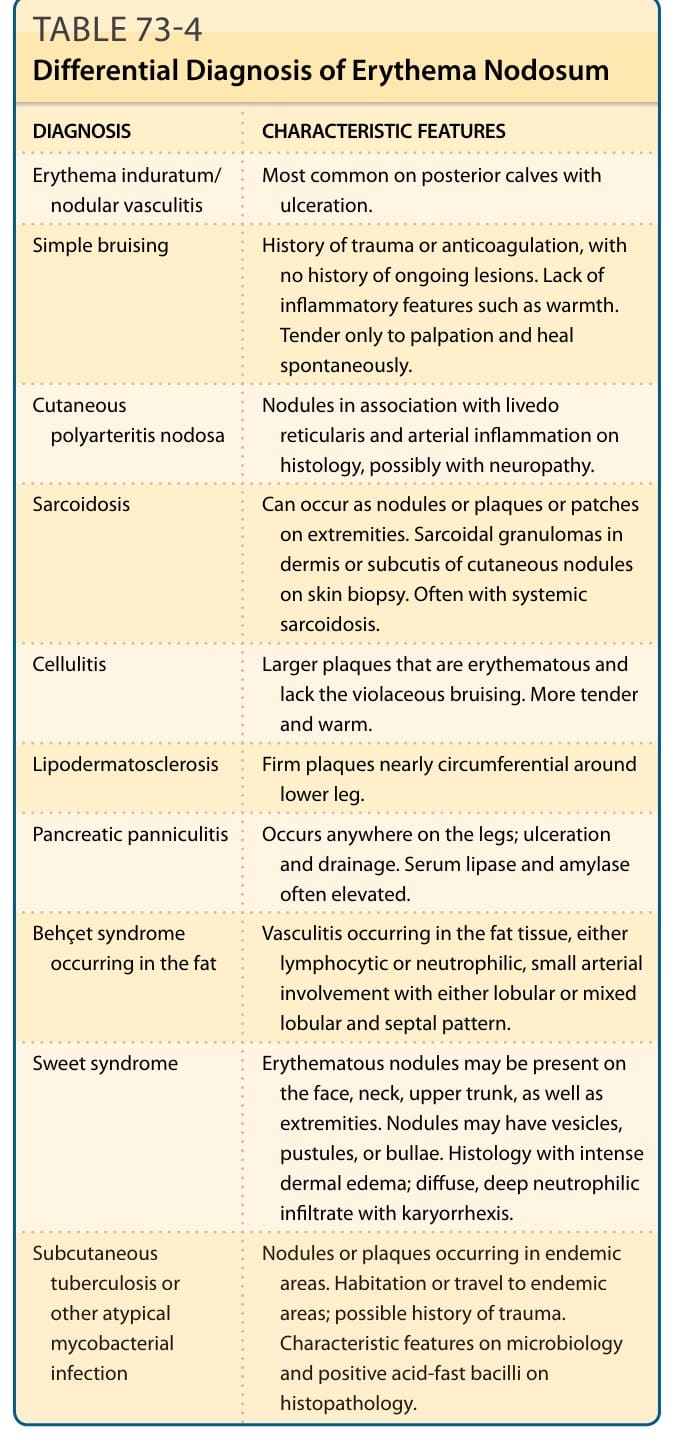
表 73-4:列出結節性紅斑 (EN) 的鑑別診斷。
結節性紅斑的鑑別診斷(表 73-4):
| 診斷 | 特徵 |
|---|---|
| 硬紅斑/結節性血管炎 (Erythema induratum/nodular vasculitis) | 最常見於後側小腿肚並伴潰瘍。 |
| 單純瘀傷 (Simple bruising) | 有外傷或抗凝血病史,無持續病灶史。缺乏溫熱等發炎特徵。僅觸診時壓痛,會自發癒合。 |
| 皮膚型結節性多發性動脈炎 (Cutaneous polyarteritis nodosa) | 結節合併網狀青斑 (livedo reticularis),組織學上有動脈發炎,可能伴神經病變。 |
| 類肉瘤病 (Sarcoidosis) | 可在四肢以結節、斑塊或斑片出現。皮膚切片中皮膚結節的真皮或皮下可見類肉瘤性肉芽腫。常伴系統性類肉瘤病。 |
| 蜂窩組織炎 (Cellulitis) | 較大的紅斑斑塊,缺乏紫紅色瘀傷。較壓痛且溫熱。 |
| 淋巴皮膚硬化症 (Lipodermatosclerosis) | 下肢近乎環周的堅實斑塊。 |
| 胰臟性脂膜炎 (Pancreatic panniculitis) | 可發生於腿部任何位置;潰瘍與引流。血清脂解酶 (lipase) 與澱粉酶 (amylase) 常升高。 |
| 發生於脂肪的貝賽特氏症候群 (Behçet syndrome occurring in the fat) | 發生於脂肪組織的血管炎,淋巴球性或嗜中性球性,小動脈受侵犯,呈小葉型或混合小葉與間隔型。 |
| 史威特症候群 (Sweet syndrome) | 紅斑結節可出現於臉、頸、上軀幹及四肢。結節可有水泡、膿皰或大皰。組織學上有強烈真皮水腫;瀰漫、深層嗜中性球浸潤伴核碎裂 (karyorrhexis)。 |
| 皮下結核或其他非典型分枝桿菌感染 (Subcutaneous tuberculosis or other atypical mycobacterial infection) | 結節或斑塊發生於地方流行區。居住或旅遊至地方流行區;可能有外傷史。微生物學上有特徵性表現,組織病理學抗酸桿菌 (acid-fast bacilli) 陽性。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
EN 是一種良性、自我緩解的皮下疾病,在初次表現後數週內消退;然而病程依病因而異。藥物誘發的 EN 在停藥後可能改善,而再次使用該藥時臨床表現會再現。EN 的新病灶在 6 週後不常見,但有持續性與復發性病例的報告。在避免致病因子後,若未找到病因,復發較為常見。繼發於惡性腫瘤、嚴重系統性疾病或感染的 EN,可能因原發疾病的預後而有較複雜的病程。繼發於類肉瘤病的 EN 病人預後較佳。患有克隆氏病與 EN 的病人,較可能有發炎性腸道疾病的結腸侵犯。雖然 EN 的復發罕見,但在與類肉瘤病、荷爾蒙療法與懷孕,以及鏈球菌感染相關時較為頻繁。多數 EN 病例癒合良好且不復發,但在報告病例中有 6% 至 34% 會復發。
處置 (MANAGEMENT)
若有可辨識的病因因子,EN 的處置著重於消除暴露或治療潛在疾病。必須尋找並治療可能的感染,可疑藥物應予停用。徹底回顧所有藥物(包括非處方與補充產品)、醫療補充劑,以及詳盡的病史(包括 EN 發作前數週的症狀、旅遊史與感染家族史),對於檢查至關重要。
在移除或治療誘發因子後,支持性照護是治療的主軸:嚴重時臥床休息並抬高下肢,並建議使用非類固醇抗發炎藥。可使用過飽和碘化鉀溶液 (supersaturated potassium iodide solution, SSKI),但給藥前必須進行甲狀腺疾患與懷孕篩檢,且有甲狀腺功能低下、甲狀腺腫、心臟與肺臟毒性的風險。SSKI 的給藥方式為滴入水或果汁中,使用 2-10 drops(1 drop = 0.03 mL = 30 mg),每日 3 次。秋水仙素 (Colchicine) 特別對貝賽特氏病相關的 EN 有效。Etanercept 與 infliximab 是發炎性腸道疾病相關 EN 的選項。其他可能的治療選項包括 oxyphenbutazone 與 hydroxychloroquine,但僅有有限的隨機對照試驗。皮質類固醇並非一線療法,因為在特發性 EN 中有感染性病因的風險。
硬紅斑與結節性血管炎 (ERYTHEMA INDURATUM AND NODULAR VASCULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 下肢的紅斑性皮下結節與斑塊;常見於小腿肚,亦見於前外側小腿、足部與大腿;罕見於上臂、前臂與臉部。
- 與靜脈功能不全相關;在中年女性較頻繁。
- 常有潰瘍與疤痕。
- 慢性、復發性病程。
- 感染性病因包括細菌(尤其是結核分枝桿菌 Mycobacterium tuberculosis)、真菌、病毒與原蟲。
組織病理學
- 多為小葉型或混合小葉與間隔型脂膜炎,90% 伴血管炎。
- 脂肪小葉中心的脂肪細胞廣泛壞死。
- 發炎浸潤不一:早期病灶為嗜中性球,發展完全的病灶為上皮樣組織球與多核巨細胞。
- 脂肪小葉小靜脈與微靜脈的血管炎。
治療
- 結核分枝桿菌檢測陽性者:完整療程的抗結核三藥療法。
- 完整治療感染性病因。
- 其他病例:碘化鉀、其他抗發炎藥、支持性繃帶與彈性襪、抬腿、臥床休息。
硬紅斑 (erythema induratum, EI) 與結節性血管炎 (nodular vasculitis, NV) 同樣表現為腿部的結節性病灶。它們最早於 1945 年被描述,與結核分枝桿菌 (Mycobacterium tuberculosis, MTB) 有關。臨床表現與組織病理學可區分 EI/NV 與 EN、皮膚型結節性多發性動脈炎,或其他見於腿部的結節。EI 最常表現為小腿肚的潰瘍性結節,並與 MTB 感染相關。一種類似的疾患後來在小腿肚與其他下肢部位被描述,但無 MTB 關聯,稱為 NV。然而在臨床上這 2 種腿部結節症候群極為相似,以致無法將它們區分開來。因此這些名詞最常被互換使用。
流行病學 (EPIDEMIOLOGY)
EI/NV 此一群組是伴血管炎之脂膜炎中最常見的診斷。EI/NV 最常見於年輕至中年女性,女男比可高達 9:1。年齡範圍從 8 個月至 66 歲,平均年齡 36.6 歲。在一份 165 名皮膚結核 (cutaneous tuberculosis, TB) 病人的回顧中,僅 2 名表現為 EI。此比例因研究時間與地點而異,香港的一份研究顯示 79.5% 的皮膚 TB 病例為 EI。小兒皮膚 TB 佔 1% 至 2% 的病例。小兒皮膚 TB 比率最高見於巴基斯坦。EI 病灶在冬季較頻繁發生,並與肥胖及靜脈功能不全相關。
臨床特徵 (CLINICAL FEATURES)
EI/NV 以侵犯小腿肚而為人所知,表現為復發性、紅斑至紫紅色的結節與深層斑塊,可能壓痛(圖 73-3)但常不疼痛。潰瘍常導致疤痕。表面變化包括潰瘍的結痂與周圍的鱗屑領圈 (collarette of scale)(圖 73-4)。雖然後側小腿肚是最頻繁的位置,病灶亦可出現於足部前外側、大腿,以及罕見的手臂與臉部。MTB 相關 EI 病人一致的全身性發現是結核樣皮膚試驗陽性,包括 Mantoux 試驗或結核菌素純化蛋白衍生物 (tuberculin-purified protein derivative)。與病因(TB 或其他)相關的全身性發現變異甚大。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
雖然 EI 常與 MTB 相關,但其病因具爭議性,因為切片與組織培養中並非總能鑑定出微生物。隨著聚合酶連鎖反應 (polymerase chain reaction, PCR) 技術的問世,多個培養陰性的病例被證實含有 MTB DNA。
體外研究顯示 MTB 可結合至清道夫受體 (scavenger receptors)、進入脂肪細胞,並經由 MTB 將近 20 種的脂解酶 (lipases) 挪用宿主脂質、累積胞質內脂質包涵體,並以一種對主要抗分枝桿菌藥物 isoniazid 不敏感的非複製狀態存活。然而,rifampin 與 ethambutol 可使脂肪細胞中的 MTB 細菌量減少 80% 至 90%。其他可感染脂肪細胞並作為再活化貯存庫的微生物包括巨細胞病毒、肺炎披衣菌 (Chlamydophila pneumoniae)、腺病毒、流感病毒、呼吸道融合病毒 (respiratory syncytial virus)、普氏立克次體 (Rickettsia prowazekii) 與克氏錐蟲 (Trypanosoma cruzi)。脂肪細胞的感染導致細胞激素與脂肪激素的活化,將其他先天免疫細胞(巨噬細胞、嗜中性球、肥大細胞、自然殺手細胞)以及後天免疫細胞(致敏 T 細胞)帶至感染部位以協助控制感染。
EI 被認為是一種由免疫複合體或細胞媒介過敏所媒介的過敏性疾患,表現為結核菌素皮膚試驗陽性以及對 MTB 高度陽性的干擾素-γ 釋放試驗 (interferon-γ release assay)。T 細胞、單核球與巨噬細胞以及蘭格漢細胞 (Langerhans cells) 可能提示第 IV 型過敏反應 (type IV hypersensitivity reaction)。
許多 EI 病人有皮膚外的 MTB,可能存在於肺、淋巴結、腎、腸或腎上腺,後者表現為愛迪生氏病 (Addison disease)。罕見菌種,包括 Mycobacterium monacense,亦可造成 EI/NV 的臨床與組織病理學表現。其他感染與疾患亦與 NV 相關,包括 B 型肝炎、C 型肝炎(紅指症候群 red finger syndrome 與脂膜炎)、潰瘍性結腸炎、白血病、類風濕性關節炎、甲狀腺功能低下與諾卡氏菌 (Nocardia) 感染。
克隆氏病亦為 NV 的罕見病因,當其存在時可能為轉移性疾病 (metastatic disease)。肺炎披衣菌 (Chlamydophila pneumoniae) 曾被報告可誘發 NV。藥物,包括 propylthiouracil 與 etanercept,亦與 NV 相關。卡介苗 (Bacillus Calmette-Guérin) 接種被報告可在注射後 2 至 3 個月導致 EI。
診斷 (DIAGNOSIS)
組織病理學發現與病灶持續時間相關,但共同點是多為小葉型或混合間隔與小葉型脂膜炎(圖 73-5A)。在早期病灶中,脂肪小葉含有離散的發炎細胞聚集,以嗜中性球為主,但無白血球破碎 (leukocytoclasia)。脂肪細胞壞死存在,導致泡沫狀組織球。在 EI/NV 的已建立病灶中,上皮樣組織球、多核巨細胞與淋巴球的聚集產生肉芽腫樣外觀。若有強烈血管損傷,常伴隨廣泛的乾酪樣壞死 (caseous necrosis) 區域(圖 73-5B),最終形成結核樣肉芽腫。乾酪樣壞死可能侵犯覆蓋其上的真皮至發生潰瘍的程度。嗜酸性球可能存在,且不與診斷相牴觸。
在一份 101 例符合 EI 的回顧中,90% 的病例存在血管炎(小葉微靜脈、間隔靜脈與間隔動脈)。在嚴重免疫低下病人的組織病理學中曾見缺乏血管炎。重要的是,MTB 可同時造成 EN 與 EI,因此在有適當旅遊與暴露的病人中,臨床與病理特徵可能無法排除 TB 的診斷。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
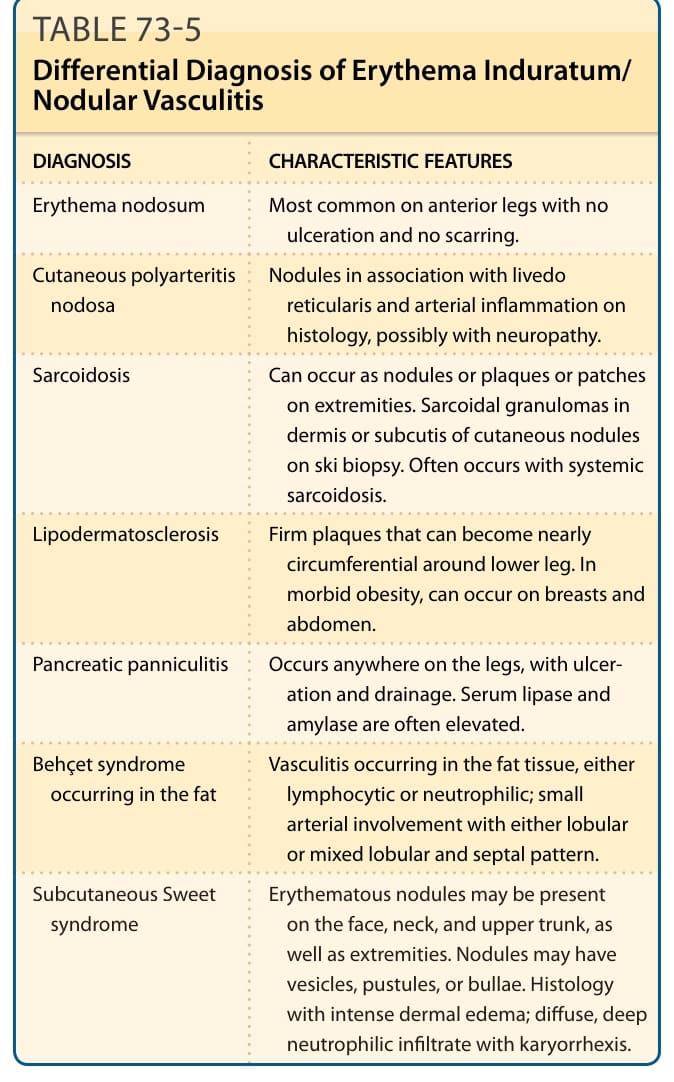
表 73-5:列出硬紅斑/結節性血管炎的鑑別診斷。
硬紅斑/結節性血管炎的鑑別診斷(表 73-5):
| 診斷 | 特徵 |
|---|---|
| 結節性紅斑 (Erythema nodosum) | 最常見於前側腿部,無潰瘍與疤痕。 |
| 皮膚型結節性多發性動脈炎 (Cutaneous polyarteritis nodosa) | 結節合併網狀青斑,組織學上有動脈發炎,可能伴神經病變。 |
| 類肉瘤病 (Sarcoidosis) | 可在四肢以結節、斑塊或斑片出現。皮膚切片中皮膚結節的真皮或皮下可見類肉瘤性肉芽腫。常伴系統性類肉瘤病。 |
| 淋巴皮膚硬化症 (Lipodermatosclerosis) | 可變成近乎環繞下肢的堅實斑塊。在病態肥胖者可發生於乳房與腹部。 |
| 胰臟性脂膜炎 (Pancreatic panniculitis) | 可發生於腿部任何位置,伴潰瘍與引流。血清脂解酶與澱粉酶常升高。 |
| 發生於脂肪的貝賽特氏症候群 (Behçet syndrome occurring in the fat) | 發生於脂肪組織的血管炎,淋巴球性或嗜中性球性;小動脈受侵犯,呈小葉型或混合小葉與間隔型。 |
| 皮下史威特症候群 (Subcutaneous Sweet syndrome) | 紅斑結節可出現於臉、頸、上軀幹及四肢。結節可有水泡、膿皰或大皰。組織學上有強烈真皮水腫;瀰漫、深層嗜中性球浸潤伴核碎裂。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
EI/NV 可有遷延的病程,數月至數年間反覆發作。EI/NV 病人除了相關疾病外通常健康。EI 的病程也常比 EN 更慢性,且 EI 的潰瘍與疤痕導致比 EN 更差的美觀與潛在的失能後果。
處置 (MANAGEMENT)
在 MTB 培養陽性、結核樣皮膚試驗陽性,或干擾素-γ 釋放試驗(如 QuantiFERON-TB Gold in-tube assay)陽性的病人中,應使用多藥抗 TB 療法。若干擾素-γ 釋放試驗陰性,但在高風險 TB 地區仍有臨床懷疑,則建議進行病灶 PCR。患有 B 型肝炎或 C 型肝炎的病人應接受適當的抗病毒介入。應尋找並治療其他感染性病因。應停用可能的致病藥物。
用於非 MTB 相關 NV 的抗發炎治療包括 SSKI、非類固醇抗發炎藥、皮質類固醇與金鹽 (gold),以及抬腿臥床休息、以壓力治療與 pentoxifylline 治療靜脈功能不全,甚至 mycophenolate mofetil。
若使用免疫抑制劑,建議監測可能的感染性病因。
淋巴皮膚硬化症 (LIPODERMATOSCLEROSIS)
重點一覽 (AT-A-GLANCE)
臨床
- 下肢硬結、堅實、木質樣質地的斑塊。
- 相關因素:慢性靜脈功能不全、身體質量指數升高、女性、動脈高血壓、動脈缺血與血栓性靜脈炎。
組織病理學
- 背景有淤滯變化;多為小葉型脂膜炎,不伴血管炎。
- 脂肪小葉中心的缺血性壞死。
- 間隔增厚與纖維化、皮下脂肪萎縮,晚期重症病例有顯著纖維化與硬化。
治療
- 壓力襪、超音波療法、pentoxifylline。
- 部分病例對同化類固醇 (anabolic steroids)、富含血小板血漿 (platelet-rich plasma) 反應良好。
淋巴皮膚硬化症 (lipodermatosclerosis, LDS) 有多個同義詞,包括硬化性脂膜炎 (sclerosing panniculitis)、硬皮樣皮下炎 (hypodermitis sclerodermiformis)、伴脂膜囊性變化之慢性脂膜炎 (chronic panniculitis with lipomembranous changes)、硬化萎縮性蜂窩組織炎 (sclerotic atrophic cellulitis) 與靜脈淤滯性脂膜炎 (venous stasis panniculitis)。它是一種侵犯下肢的硬化性脂膜炎,常與血管功能障礙有關。最早使用的名稱是 hypodermitis sclerodermiformis,早在 1950 年代即已使用。
流行病學 (EPIDEMIOLOGY)
LDS 是最常見的脂膜炎類型,臨床醫師見到的頻率遠高於 EN。LDS 隨靜脈功能不全發生,多見於 40 歲以上過重女性,年齡範圍從 31 至 74 歲及以上。女男比為 4:1,在某些研究中高達 12:1。
白種人較常與 LDS 相關。肥胖常見,85% 受侵犯病人的身體質量指數大於 30。共病包括高血壓、甲狀腺疾病、糖尿病、先前下肢蜂窩組織炎史與深部靜脈血栓。阻塞性睡眠呼吸中止症與關節炎(骨關節炎與類風濕性關節炎)亦與 LDS 相關。
臨床特徵 (CLINICAL FEATURES)
LDS 有急性發炎期與慢性纖維化期。在以急性形式表現的病人中,極為疼痛、界線不清、蜂窩組織炎樣的紅斑斑塊持續存在並演變為紫紅色、水腫或硬結的斑塊或結節,見於下肢,最常見於下前內側小腿肚(圖 73-6)。單側侵犯見於 55%,局部斑塊見於 51%,潰瘍見於 13% 的病例。病灶可極為疼痛,導致誤診為 EN、蜂窩組織炎或血栓性靜脈炎。雖然處於此急性期的病人可能缺乏明顯的靜脈疾病,但血管檢查顯示多數有靜脈功能不全。在靜脈檢查正常的病人中,多數有高身體質量指數。
LDS 的慢性形式並不總是接續於明顯的急性期之後。慢性 LDS 表現為硬結至硬化、凹陷與色素沉著的皮膚。這些發現出現於下肢的下段,主要在內側,或呈長襪狀分布。這被描述為「倒香檳瓶 (inverted champagne bottle)」或「保齡球瓶 (bowling pin)」外觀(圖 73-7)。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
少數藥物被報告可誘發 LDS,包括 gemcitabine 與 pemetrexed。額外的致病特徵可能包括:繼發於緊密連接 (tight junctions) 下調的靜水壓升高誘發血管通透性增加,伴纖維蛋白 (fibrin) 的血管外擴散;微血栓 (microthrombi);蛋白 S 與蛋白 C 的異常;缺氧 (hypoxia);發炎細胞對內皮細胞的損傷;細胞間黏附分子與血小板衍生及內皮衍生因子的上調;以及伴傷口癒合與局部膠原刺激的發炎導致纖維化與進一步的血管與淋巴損傷。此纖維化伴隨轉化生長因子-β1 (transforming growth factor-β1) 基因與蛋白表現的增加,以及第 1 型前膠原 (procollagen type 1) 基因表現的增加。
脂肪組織中的缺氧誘發伴巨噬細胞浸潤與發炎細胞激素表現的慢性發炎。脂肪細胞產生多種基質金屬蛋白酶 (matrix metalloproteinases) 以及金屬蛋白酶組織抑制劑 (tissue inhibitors of metalloproteinases),這可能促成 LDS 中所見的組織重塑。研究將脂肪組織擴張(如肥胖)與缺氧連結,導致缺氧誘導因子 1α (hypoxia-inducible factor 1α) 表現增加。這刺激細胞外因子,包括第 I 型膠原與第 III 型膠原,導致纖維化。
診斷 (DIAGNOSIS)
診斷常為臨床診斷,而 LDS 皮膚的切片可能有困難的癒合過程。值得注意的是,對切片癒合不良的顧慮與延遲切片,曾使皮膚 T 細胞淋巴瘤 (cutaneous T-cell lymphoma) 與血管肉瘤 (angiosarcoma) 的 2 個惡性病例報告未能被識別。切片最好取自侵犯的近端邊緣,以獲得最高的癒合率。較不侵入性的選項,如 MRI,曾被用於觀察疾病的範圍。
組織病理學發現反映疾病的演變。任何分期都有真皮淤滯變化,包括微血管與微靜脈的增生、小型厚壁血管、外滲的紅血球、含鐵血黃素巨噬細胞 (hemosiderin-laden macrophages)、淋巴組織球性發炎與纖維化。在皮下組織中,LDS 的早期病灶顯示間隔中稀疏的淋巴球浸潤,伴中央小葉缺血性脂肪壞死。脂肪小葉內可觀察到微血管充血,常伴內皮細胞壞死、血栓、紅血球外滲與含鐵血黃素沉積。
脂膜囊性 (lipomembranous) 或膜囊性 (membranocystic) 變化可能存在,壞死脂肪內有小型偽囊性空腔。這些空腔由玻璃樣嗜酸性物質的膜狀內襯所包覆,可被過碘酸-希夫染色 (periodic acid-Schiff staining) 突顯。脂膜囊性變化並非 LDS 所獨有。
隨著 LDS 進展,組織病理學變化的範圍涵蓋日益增加程度的脂肪壞死、間隔纖維化與增厚;淋巴球、組織球與泡沫狀巨噬細胞的發炎浸潤;以及脂肪小葉的部分至廣泛萎縮。進展期病灶顯示間隔硬化,伴繼發於噬脂性脂肪壞死 (lipophagic fat necrosis) 的脂肪小葉顯著萎縮,伴脂膜囊性變化與發炎的顯著減少。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
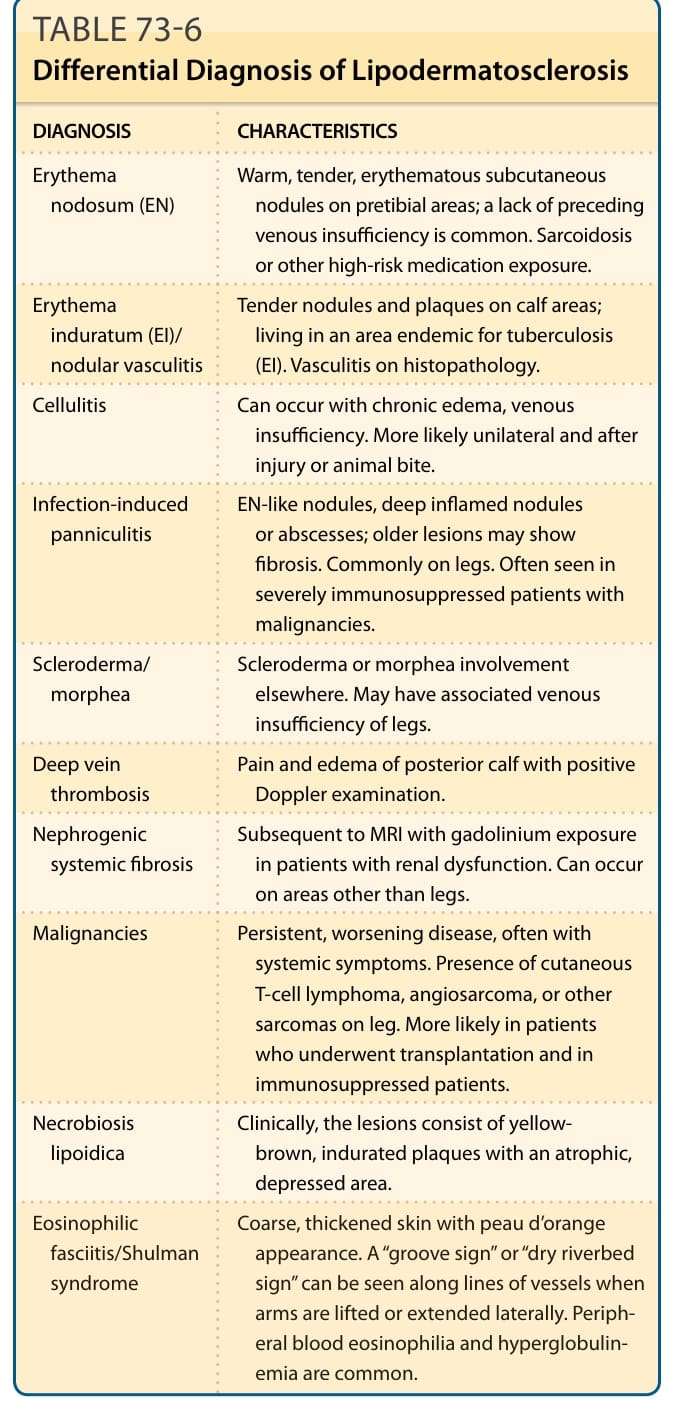
表 73-6:列出淋巴皮膚硬化症 (LDS) 的鑑別診斷。
淋巴皮膚硬化症的鑑別診斷(表 73-6):
| 診斷 | 特徵 |
|---|---|
| 結節性紅斑 (Erythema nodosum, EN) | 脛前區溫熱、壓痛、紅斑性皮下結節;常缺乏先前的靜脈功能不全。類肉瘤病或其他高風險藥物暴露。 |
| 硬紅斑 (EI)/結節性血管炎 | 小腿肚區的壓痛結節與斑塊;居住於結核地方流行區 (EI)。組織病理學有血管炎。 |
| 蜂窩組織炎 (Cellulitis) | 可隨慢性水腫、靜脈功能不全發生。較可能為單側且在受傷或動物咬傷後。 |
| 感染誘發性脂膜炎 (Infection-induced panniculitis) | EN 樣結節、深層發炎結節或膿瘍;較舊病灶可顯示纖維化。常見於腿部。常見於患惡性腫瘤的嚴重免疫抑制病人。 |
| 硬皮症/局部硬皮症 (Scleroderma/morphea) | 他處有硬皮症或局部硬皮症侵犯。可能伴腿部靜脈功能不全。 |
| 深部靜脈血栓 (Deep vein thrombosis) | 後側小腿肚疼痛與水腫,杜卜勒檢查 (Doppler) 陽性。 |
| 腎源性系統性纖維化 (Nephrogenic systemic fibrosis) | 繼發於腎功能障礙病人接受含釓 (gadolinium) MRI 暴露之後。可發生於腿部以外區域。 |
| 惡性腫瘤 (Malignancies) | 持續、惡化的疾病,常伴全身症狀。腿部出現皮膚 T 細胞淋巴瘤、血管肉瘤或其他肉瘤。較可能見於接受移植與免疫抑制的病人。 |
| 類脂質漸進性壞死 (Necrobiosis lipoidica) | 臨床上病灶由黃棕色、硬結斑塊組成,伴萎縮、凹陷區域。 |
| 嗜酸性筋膜炎/Shulman 症候群 (Eosinophilic fasciitis/Shulman syndrome) | 粗糙、增厚的皮膚伴橘皮 (peau d’orange) 外觀。當手臂抬起或向外側伸展時,沿血管走向可見「溝槽徵 (groove sign)」或「乾河床徵 (dry riverbed sign)」。週邊血液嗜酸性球增多與高球蛋白血症常見。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
LDS 的病程多變,取決於病人的共病與對治療方案(尤其是壓力治療,每日遵從可能有困難)的順從性。急性形式可能持續數月甚至一年。慢性形式可持續數年並可能對標準治療方案頑固。潰瘍及其產生的疤痕是永久性的,可能在美觀上造成困擾。必須避免潰瘍的繼發感染。系統性硬化症病人的皮膚硬化曾與繼發於腿部血栓的肺梗塞與高血壓相關。此外,未治療的 LDS 可導致靜脈淋巴水腫 (venolymphedema)。
處置 (MANAGEMENT)
評估週邊血管疾病的診斷檢查包括用於動脈評估的踝肱指數 (ankle brachial index),以及偵測血栓的靜脈杜卜勒檢查與偵測血流方向及靜脈逆流嚴重度的雙功超音波 (duplex sonography)。有動脈損害的病人不應接受壓力治療,而壓力治療是 LDS 的一線治療。較高的壓力梯度(30 to 40 mm Hg)可能更有效,但較低壓力(15 to 20 mm Hg 或 20 to 30 mm Hg)可鼓勵順從性,尤其在老年人,並有效減輕水腫。壓力可緊縮血管緊密連接,抑制液體滲入血管周圍組織。長襪必須整天穿著;幾天不使用壓力治療可能導致水腫與發炎復發。由於穿著長襪的困難,已發展出使用持續或間歇性氣動壓力 (pneumatic compression) 的適應性壓力模式。Stanozolol 對 LDS 有效;它可減輕疼痛、紅斑與硬結。病人對此治療耐受良好,肝毒性風險被評估為無症狀的肝轉胺酶升高,停藥後可恢復。此藥在美國無法取得。其他同化類固醇,如 oxandrolone 與 danazol,亦曾被使用。
Pentoxifylline 已成功用於伴與不伴潰瘍的 LDS 病例,是治療靜脈潰瘍時壓力治療的有用輔助,並可能作為單一療法有效。事實上,在一項研究中,hydroxychloroquine 與 pentoxifylline 合併療法(不使用壓力治療)使疼痛較基準減少 50%。
超音波療法可減輕甚至消除硬結、壓痛與紅斑。可透過物理治療部門使用,是簡單而安全的治療,可作為輔助療法。由於肥胖是受侵犯病人的常見狀況,減重至關重要。辣椒素 (Capsaicin) 可緩解與 LDS 相關的疼痛。最後,頑固性疾病曾對病灶內、自體、富含血小板血漿 (platelet-rich plasma) 有反應,以網格狀模式注射,每 2 週重複一次。
一位作者 (IKA) 注意到以芸香苷 (rutin) 與維生素 C 治療的病人有改善,尤其作為壓力治療的輔助。
感染誘發性脂膜炎 (INFECTION-INDUCED PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 由多種感染性病原引起:細菌、真菌、寄生蟲與病毒。
- 脂肪組織可作為各種感染的貯存庫。
- 病人可能為免疫抑制,伴原發接種或血行性散布。
- 紅斑斑塊、結節、膿瘍、伴膿性分泌的潰瘍。
組織病理學
- 脂肪小葉內的化膿性肉芽腫 (suppurative granulomas)。
- 在原發性皮膚感染中,發炎中心位於淺層真皮;在繼發性感染中,中心位於深層網狀真皮與皮下脂肪。
- 以特殊染色、培養與血清學研究偵測微生物。
治療
- 依藥敏試驗選擇適當的抗微生物療法。
感染誘發性脂膜炎 (infection-induced panniculitis, IIP),亦稱感染性脂膜炎 (infectious panniculitis) 與感染脂膜炎 (infective panniculitis),是由感染性病原直接引起的脂膜炎。脂肪組織感染可由細菌、分枝桿菌、真菌、原蟲或病毒引起。在傷口部位(受傷、手術操作、導管、注射、針灸)直接接種所產生的原發性感染,通常導致單一病灶,可能局部擴散。由敗血症與血行性散布引起的繼發性感染,可能表現為單一或多個病灶。在免疫抑制病人中,微生物可能數量眾多,可在常規組織病理學或特殊染色上鑑定。在免疫健全病人中,微生物可能稀疏,需要陽性培養、病灶 PCR 或血清學研究來鑑定。
流行病學 (EPIDEMIOLOGY)
IIP 最常見於免疫低下的宿主,包括患有糖尿病者。流行病學取決於感染性病因,伴地理與宿主易感性。近期與各種自體免疫疾患相關的感染性病因報告,包括伴幼年型皮肌炎 (juvenile dermatomyositis) 的金黃色葡萄球菌 (Staphylococcus aureus) 脂膜炎、系統性紅斑性狼瘡 (systemic lupus erythematosus, SLE) 中的毛黴菌病 (mucormycosis)、伴類風濕性關節炎的分枝桿菌相關與組織漿菌 (Histoplasma) 相關脂膜炎,以及組織漿菌在 2 名多發性肌炎 (polymyositis) 病人中引起脂膜炎。使這些關聯更複雜的是,自體免疫疾患常以免疫抑制療法治療。
臨床特徵 (CLINICAL FEATURES)
IIP 的臨床外觀從伴膿液與潰瘍的波動性或膿瘍樣病灶,到非特異性紅斑、堅實皮下斑塊、紫斑斑塊與 EN 樣病灶。深層結節或斑塊不一定都呈波動性,膿皰、波動性丘疹與潰瘍可疊加在堅實、深層的結節之上。最常見的感染部位是腿與足部,但上肢、軀幹與臉部亦可受侵犯。各種病因的免疫抑制是最頻繁的關聯。免疫抑制與較廣泛及膿瘍型病灶相關。在免疫健全病人中,肉芽腫常見。臨床特徵隨情境、感染微生物與病人的免疫健全程度而異。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
無數感染性病原可經由直接接種或血行性散布造成 IIP。瀰漫性鐮孢菌病 (fusariosis) 曾在一名急性淋巴芽細胞白血病 (acute lymphoblastic leukemia) 病人中以脂膜炎表現。一例報告了一名急性淋巴芽細胞白血病病人的皮膚麴菌病 (aspergillosis) 脂膜炎,並因尿酸鹽結晶 (urate crystals) 的存在而懷疑痛風性脂膜炎 (gouty panniculitis)。此病人亦有胰臟性脂膜炎特徵性的「幽靈細胞 (ghost cell)」變化,這與尿酸鹽結晶一同可能繼發於真菌酵素變化。這些發現在毛黴菌病中亦曾被觀察到。移植病人有散播性感染的風險,如一份 15 名實質器官移植病人(主要為腎移植)伴隱球菌 (cryptococcal) 脂膜炎的回顧所示。移植與感染之間的間隔從 3 個月至 31 年不等。移植族群中隱球菌感染以心臟移植受者最高。綠膿桿菌 (Pseudomonas aeruginosa) 可造成脂膜炎,最常為散播性感染,但也曾在無敗血症的情況下表現。
Epstein-Barr 病毒亦曾被報告在一名 methotrexate 相關淋巴增生性疾患病人中造成侷限於軀幹的 IIP,停用 methotrexate 後緩解。
免疫健全病人的 IIP 亦有報告。由梅毒引起的脂膜炎曾以續發性梅毒的皮膚表現呈現,浸潤中含許多漿細胞,梅毒螺旋體 (Treponema pallidum) 免疫組織化學染色陽性。皮膚利什曼原蟲病 (leishmaniasis) 亦曾表現為脂肪組織的發炎,見於 2 個伊拉克病例,呈間隔與小葉型組織病理學,以皮膚利什曼原蟲病標準的局部治療而改善。伯氏疏螺旋體 (Borrelia burgdorferi) 亦與脂膜炎相關,在一名免疫健全宿主中模擬皮下脂膜炎樣 T 細胞淋巴瘤 (subcutaneous panniculitis-like T-cell lymphoma, SPTCL),對 doxycycline 反應迅速。
脂肪細胞是一種先天免疫細胞,會對各種微生物感染量身打造特定且不同的受體媒介轉錄反應。脂肪細胞可在體外被感染性病原感染,包括肺炎披衣菌 (Chlamydophila pneumoniae)、巨細胞病毒、腺病毒、呼吸道融合病毒、流感、MTB、普氏立克次體 (R. prowazekii)、克氏錐蟲 (T. cruzi)、貝氏考克斯菌 (Coxiella burnetii) 與 HIV。
以克氏錐蟲(恰加斯病 Chagas disease 的病因)在體外感染脂肪細胞,導致多種促發炎細胞激素與趨化激素表現增加、TLR2 與 TLR9 及急性期反應物 (acute-phase reactants) 表現增加,但脂聯素 (adiponectin) 與過氧化體增殖物活化受體 γ (peroxisome-proliferator-activated receptor γ)——發炎的負調節因子——的表現減少。在小鼠中,即時 PCR 顯示在以克氏錐蟲感染後 300 天,脂肪組織與心臟組織中存在相當數量的寄生蟲,顯示脂肪組織是寄生蟲的貯存庫。有趣的是,克氏錐蟲的無鞭毛體 (amastigotes) 在棕色脂肪組織中比在白色脂肪組織中更多。
在另一項小鼠研究中,痲瘋分枝桿菌 (Mycobacterium leprae) 在脂肪組織中的生長潛能非常有限,因為前脂肪細胞與脂肪細胞不允許 M. leprae 增殖與對數生長。由於脂肪細胞具有高 TLR4 表現,脂肪細胞對內毒素 (endotoxin) 有反應,產生與巨噬細胞相當或更高量的促發炎細胞激素。脂肪細胞中的 TLR2 誘導賦予對真菌細胞壁成分的高度敏感性。
編碼 TLRs 或下游訊息傳導蛋白的突變可增加感染風險。TLRs 與其他先天免疫訊息傳導成分的基因變異,以及它們對脂肪細胞功能與脂膜炎發展的影響,尚未明瞭。
診斷 (DIAGNOSIS)
評估包括含微生物染色的組織病理學研究以及帶藥敏的組織培養。免疫抑制可能導致眾多微生物的存在,但在免疫健全病人中診斷較困難。分子 PCR 技術已增加分枝桿菌感染的偵測。組織病理學特徵隨微生物及其毒力、宿主免疫狀態與病灶持續時間而異。IIP 多為小葉型脂膜炎,但常顯示混合間隔與小葉型模式。由直接接種獲得的感染中淺層真皮有較多發炎,而繼發於血行性散布的感染則侵犯深層網狀真皮與皮下脂肪。
IIP 的其他特徵包括出血、血管增生、嗜鹼性壞死灶與汗腺壞死。覆蓋其上的表皮變化,如角化不全 (parakeratosis)、棘層肥厚 (acanthosis) 與海綿水腫 (spongiosis),見於未潰瘍的檢體。真皮發現包括水腫、瀰漫血管周圍發炎浸潤、厚壁血管增生與局灶或瀰漫出血。觀察到這些特徵應進行細菌、分枝桿菌與真菌的染色,並可能需要額外的免疫組織化學或 PCR 擴增技術。組織病理學變化偶爾提示特定病因。由上皮樣組織球圍繞聚集嗜中性球所形成的化膿性肉芽腫常見於非典型分枝桿菌。在巨細胞病毒相關脂膜炎中可見內皮細胞內的病毒包涵體。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
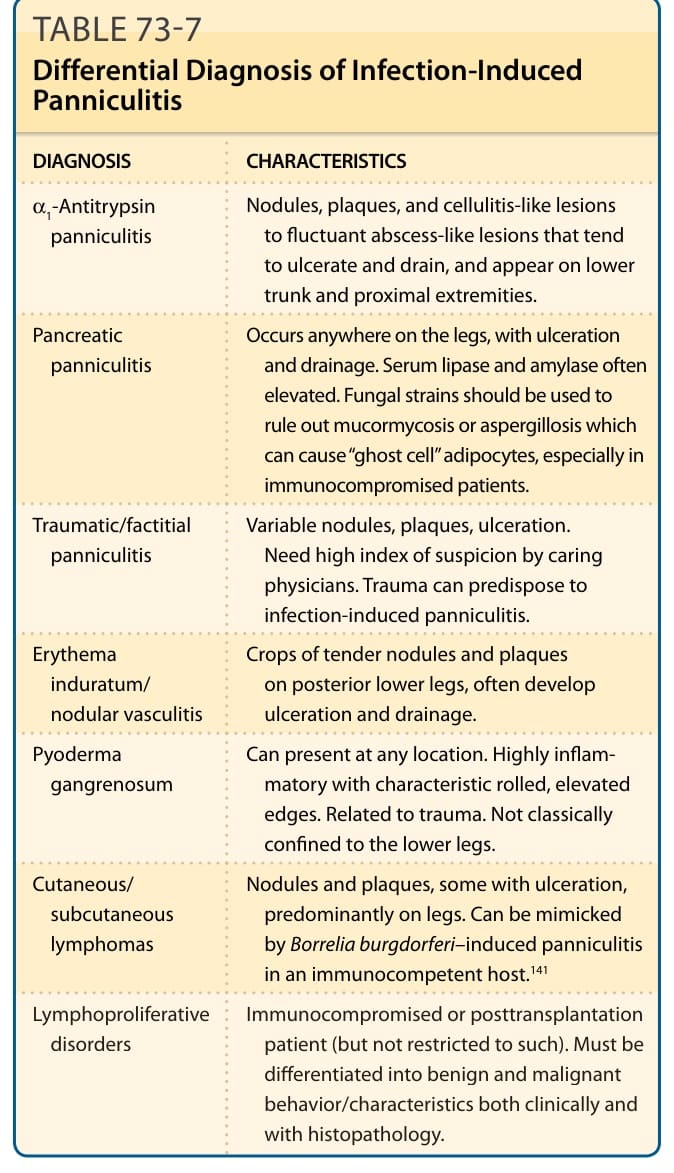
表 73-7:列出感染誘發性脂膜炎 (IIP) 的鑑別診斷。
感染誘發性脂膜炎的鑑別診斷(表 73-7):
| 診斷 | 特徵 |
|---|---|
| α1-抗胰蛋白酶脂膜炎 (α1-Antitrypsin panniculitis) | 結節、斑塊與蜂窩組織炎樣病灶至波動性膿瘍樣病灶,傾向潰瘍與引流,出現於下軀幹與近端肢體。 |
| 胰臟性脂膜炎 (Pancreatic panniculitis) | 可發生於腿部任何位置,伴潰瘍與引流。血清脂解酶與澱粉酶常升高。應使用真菌染色以排除毛黴菌病或麴菌病,後二者可造成「幽靈細胞」脂肪細胞,尤其在免疫低下病人。 |
| 外傷性/人為性脂膜炎 (Traumatic/factitial panniculitis) | 多變的結節、斑塊、潰瘍。需照護醫師高度懷疑。外傷可誘發感染誘發性脂膜炎。 |
| 硬紅斑/結節性血管炎 (Erythema induratum/nodular vasculitis) | 後側下肢成群的壓痛結節與斑塊,常發展出潰瘍與引流。 |
| 壞疽性膿皮症 (Pyoderma gangrenosum) | 可在任何位置出現。高度發炎,伴特徵性捲曲、隆起的邊緣。與外傷有關。不典型地侷限於下肢。 |
| 皮膚/皮下淋巴瘤 (Cutaneous/subcutaneous lymphomas) | 結節與斑塊,部分伴潰瘍,主要在腿部。可被免疫健全宿主中伯氏疏螺旋體誘發的脂膜炎所模擬。 |
| 淋巴增生性疾患 (Lymphoproliferative disorders) | 免疫低下或移植後病人(但不限於此)。必須在臨床與組織病理學上區分良性與惡性的行為/特徵。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
IIP 的臨床病程與預後受感染微生物以及微生物為局部或血行性引入所影響。以抗生素治療的微生物,加上及時診斷,預示良好預後。免疫低下病人有較複雜病程的風險。
處置 (MANAGEMENT)
治療依可疑或已知的微生物及其培養與藥敏而異。在涉及細菌(如 MTB)與寄生蟲(如克氏錐蟲)的病例中,其能在脂肪組織中保持休眠的能力,使得需要使用適當的抗生素,常需延長的治療療程。
α1-抗胰蛋白酶脂膜炎 (α1-ANTITRYPSIN PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- ZZ-、MZ-、MS- 與 SS- 表現型相關的脂膜炎。罕見,>60% 發生於 ZZ 病例。
- 低 α1-抗胰蛋白酶水平與肺氣腫、肝炎、肝硬化、血管炎與血管性水腫 (angioedema) 相關。
- 皮下結節多位於下腹部、臀部與近端肢體。
- 常見潰瘍、引流性病灶、術後癒合不良與同形現象 (isomorphic phenomenon)。
組織病理學
- 多為小葉型脂膜炎,不伴血管炎。
- 嗜中性球的濃密發炎浸潤,以及深層網狀真皮膠原束間的嗜中性球。
- 壞死脂肪細胞旁有正常脂肪。
治療
- Dapsone、doxycycline。
- 對於嚴重型同合子 ZZ 疾病,補充靜脈輸注 α1-抗胰蛋白酶或肝臟移植。
α1-抗胰蛋白酶 (α1-antitrypsin, α1AT) 是肝臟製造的醣蛋白。它是一種絲胺酸蛋白酶抑制劑 (serine protease inhibitor),具有廣泛功能,也是一種急性期反應物,在壓力時升高。α1AT 缺乏為共顯性遺傳 (codominant disorder),已鑑定出超過 100 種等位基因。α1AT 表現型依凝膠電泳遷移 (gel electrophoresis migration) 分類為 F(fast,快)、M(medium,中)、S(slow,慢)與 Z(very slow,極慢),以及不產生任何 α1AT 的無效變異 (null variants),且病人可能有功能異常的 α1AT 但水平正常。α1AT 缺乏最早於 1960 年代以蛋白質電泳分離被發現。繼發於醣蛋白缺乏的脂膜炎之首次描述在 1972 年。
流行病學 (EPIDEMIOLOGY)
脂膜炎在 α1AT 缺乏中不常見,在 ZZ、MZ、MS 與 SS 表現型中已報告少於 50 例。超過 60% 涉及 ZZ 表現型,65% 影響女性。同合子 MM 佔人口的 90% 至 97%。可在所有年齡出現,脂膜炎最常見於 30 至 60 歲之間。白人 α1AT 缺乏的估計盛行率在美國為每 3000 至 5000 人中 1 人,白人新生兒的發生率與囊狀纖維化 (cystic fibrosis) 相似。亦有病例被注意到隨著急性期反應物(包括 α1AT)的升降而在懷孕後初次出現。多達 35% 的病例有先前外傷。
臨床特徵 (CLINICAL FEATURES)
病人表現為疼痛性紅斑結節與斑塊,但早期病灶可能有蜂窩組織炎或波動性膿瘍型外觀(圖 73-8A)。病灶可能潰瘍並伴油性或血清血液性分泌(圖 73-8B),並以萎縮性疤痕消退。
病灶最常出現於下軀幹(臀部)(圖 73-9)與近端肢體,但下肢與其他部位亦可受侵犯。α1AT 脂膜炎可與自體免疫疾病、癌症或感染共存,因此 α1AT 缺乏與脂膜炎的關聯並不排除尋找感染或其他潛在醫療問題(如自體免疫疾患或惡性腫瘤)。
α1AT 缺乏與肺部及肝臟疾病相關,導致慢性阻塞性肺病、肝硬化或肝細胞癌;ZZ 表現型風險最高。無效變異與肝臟疾病無關聯,因為是聚合化 α1AT 在肝臟中的累積誘發損傷,而此變異不發生累積。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
α1AT 是一種主要由肝細胞產生與分泌的醣蛋白,但也由單核球/巨噬細胞與嗜中性球少量產生,已知可抑制眾多蛋白酶。α1AT 亦可能協助調節補體活化。
同合子 MM 與正常 α1AT 水平相關(90 to 220 mg/dL),而 ZZ 同合子者水平低,為正常的 10% 至 15%,S 或 Z 等位基因異合子者水平介於兩者之間。ZZ 是最常誘發脂膜炎的表現型。MS 異合子個體可能有低正常血清水平,臨床上造成脂膜炎,而 F 變異可能增加肺部疾病,僅在 FZ 表現型時與脂膜炎相關。
導致 α1AT 脂膜炎發展的可能機制包括:缺乏對各種蛋白酶的干擾,導致淋巴球、巨噬細胞、補體的活化;由 IL-1 與 IL-1β 活化所媒介的自體發炎級聯反應 (autoinflammatory cascade) 的活化;以及發炎部位結締組織的溶解與破壞。脂肪細胞的外傷可能導致釋放對發炎細胞具趨化性的脂肪激素與細胞激素,其釋放的蛋白酶因缺乏 α1AT 而無對抗,導致受侵犯組織的嚴重損傷。軟組織損傷的動物模型顯示 IL-6 與單核球趨化蛋白-1 水平升高,以及系統性發炎媒介物增加。有趣的是,曾有一例報告,一名 MM 表現型病人從捐贈肝臟獲得 ZZ 表現型;移植後病人發展出脂膜炎。
診斷 (DIAGNOSIS)
組織病理學發現隨切片的病灶年齡與類型而異。在發展早期,結節病灶顯示脂肪細胞的水腫與退化,伴破裂與塌陷的細胞膜,以及血管周圍單核浸潤。此階段也報告了間隔與小葉中嗜中性球與巨噬細胞的輕微浸潤,伴皮下脂肪的早期壞死灶。這可能伴隨嗜中性球散布於覆蓋其上的網狀真皮各處的膠原束間,被視為早期且獨特的診斷線索。較進展的病灶有與脂肪小葉壞死及取代相關的大量嗜中性球與組織球(圖 73-10)。侵犯亦可能為局灶性,表現為大片正常脂肪緊鄰壞死的間隔與脂肪小葉。液化性壞死與真皮膠原的溶解可能伴隨潰瘍,彈性組織的退化可能導致間隔破壞與「漂浮 (floating)」壞死脂肪小葉的外觀。嗜中性球與壞死脂肪細胞在晚期病灶中較不普遍,被淋巴球、泡沫狀組織球與脂肪小葉內不等量的纖維化所取代。
組織病理學有時無法呈現特徵性表現。這些罕見病例提示,在有潰瘍性病灶的病人中,若病理學在正確的臨床情境下不提示診斷,應以血清蛋白水平與表現型檢測來考慮此疾病。由於這些蛋白質性質複雜,最好同時取得血清蛋白水平與表現型分析。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
值得注意的是,可造成嗜中性球性脂膜炎的疾患包括 α1AT 脂膜炎、胰臟性脂膜炎、藥物相關脂膜炎(vemurafenib、ponatinib);與骨髓增生不良 (myelodysplasia) 相關的皮下史威特症候群或嗜中性球性脂膜炎。此外,皮膚克隆氏病、家族性地中海熱 (familial Mediterranean fever)、類風濕性關節炎與特發性嗜中性球性脂膜炎的病例亦可造成嗜中性球性脂膜炎。
α1-抗胰蛋白酶脂膜炎的鑑別診斷:
| 診斷 | 特徵 |
|---|---|
| 感染誘發性脂膜炎 (Infection-induced panniculitis) | 結節性紅斑樣結節、深層發炎結節或膿瘍;較舊病灶可顯示纖維化。常見於腿部。常見於患惡性腫瘤的嚴重免疫抑制病人。 |
| 人為性脂膜炎 (Factitial panniculitis) | 幾何形態。病人常有潛在精神障礙。辨識異物有幫助。組織病理學依機制而異。 |
| 硬紅斑/結節性血管炎 (Erythema induratum/nodular vasculitis) | 小腿肚區的壓痛結節與斑塊,居住於結核地方流行區(硬紅斑)。組織病理學有血管炎。 |
| 胰臟性脂膜炎 (Pancreatic panniculitis) | 可發生於腿部任何位置,伴潰瘍與引流。血清脂解酶與澱粉酶常升高。 |
| 皮下史威特症候群 (Subcutaneous Sweet syndrome) | 紅斑結節可出現於臉、頸、上軀幹及四肢。結節可有水泡、膿皰或大皰。組織學上有強烈真皮水腫;瀰漫、深層嗜中性球浸潤伴核碎裂。 |
| 壞疽性膿皮症 (Pyoderma gangrenosum) | 紫紅色、潛蝕性 (undermined) 邊緣,伴同形反應 (isomorphic response)。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
皮膚與皮下壞死可迅速發展。曾報告致命病例,主要為 ZZ 變異。亦曾報告一罕見的 MZ 表現型致命病例,模擬壞疽性膿皮症。以 α1AT 增補 (augmentation) 對嚴重疾病的及時診斷與治療,可顯著改善脂膜炎並可能誘發臨床緩解,僅在復發時需要增補療法輸注。雖然脂膜炎可能導致顯著的罹病率,但與 α1AT 缺乏相關的肺部與肝臟疾病優先順序高得多,與肺科醫師及肝臟科醫師共同管理最為理想。
處置 (MANAGEMENT)
許多藥物曾被用於治療 α1AT 缺乏脂膜炎,包括秋水仙素、抗瘧藥、dapsone、doxycycline、血漿輸注與血漿置換、α1AT 蛋白的靜脈增補療法,以及肝臟移植。類固醇、免疫抑制劑與細胞毒性藥物效果較差。Doxycycline,尤其是 dapsone,可能在輕至中度疾病中有用,特別是在異合子病例如 MZ。對標準治療無反應的嚴重脂膜炎需要蛋白質替代療法(增補療法)。對於嚴重脂膜炎,建議 α1AT 輸注 60 mg/kg,常以每週數次輸注,並在復發時重複輸注。嚴重病例可能需要長期補充。
脂膜炎亦曾以肝臟移植緩解,肝臟移植後獲得的脂膜炎已成功以再移植治療。重要的是要注意 α1AT 缺乏脂膜炎傾向傷口癒合不良,曾有一例未治療的 α1AT 病人在腹腔內手術後癒合不良。未及時開始增補療法可能造成顯著併發症,必須在適當的臨床情境中加以考量。由於外傷可能在三分之一的病人中誘發病灶,不鼓勵清創 (debridement)。
胰臟性脂膜炎 (PANCREATIC PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 常自發潰瘍的紅斑性皮下結節。
- 下肢(踝部與膝部周圍)是最頻繁的侵犯部位。
- 隨胰臟改善,胰臟炎相關皮膚病灶會消退,但與胰臟癌相關者傾向持續;可能致命。
組織病理學
- 多為小葉型脂膜炎,不伴血管炎。
- 脂肪小葉中心脂肪細胞的強烈壞死。
- 帶有細顆粒狀嗜鹼性胞質內物質的「幽靈細胞 (ghost cell)」脂肪細胞。
治療
- 治療潛在的胰臟疾病。
- 額外治療選項:octreotide、血漿置換術。
- 停用誘發藥物。
與潛在胰臟疾病相關且繼發於其的脂膜炎首次於 1883 年被注意到。
胰臟的多種病理可誘發脂膜炎,包括急性胰臟炎,甚至在內視鏡逆行性膽胰管攝影 (endoscopic retrograde cholangiopancreatography) 操作後。脂膜炎隨胰臟疾病改善而消退,但在惡性腫瘤病例中可能困難或不可能。
流行病學 (EPIDEMIOLOGY)
與胰臟疾病相關的脂膜炎不常發生,在 0.3% 至 3% 患有胰臟疾患(如急性或慢性胰臟炎、胰臟癌或胰臟假性囊腫)的病人中發展。任何種類的胰臟疾病皆可能與胰臟性脂膜炎相關。雖然胰臟炎最常為酒精濫用的結果,但膽石症、藥物、外傷與病毒感染也是已知的病因因子。先天性胰臟異常亦可能造成脂膜炎。
臨床特徵 (CLINICAL FEATURES)
皮膚病灶以成群方式出現於下肢,尤其關節周圍區域,但也見於手臂、手腕、大腿與軀幹。病灶為界線不清的紅斑至紅棕色水腫與壓痛結節,可能退化並以萎縮性色素沉著疤痕消退。它們可能有中央「較軟」區域,或可能變成波動性、膿瘍樣,並引流出類似 α1AT 缺乏脂膜炎病灶的油性物質(圖 73-11)。在多達 45% 的病人中,皮膚脂膜炎可能比相關胰臟疾病的診斷早數週至數月。
皮膚外表現包括伴隨關節炎的關節周圍脂肪壞死,以及骨內疼痛性髓質脂肪壞死。繼發於關節周圍脂肪壞死的單關節或寡關節關節炎可能存在於超過半數的病人。胰臟疾病、脂膜炎與多關節炎的這個三聯徵(PPP 症候群)非常罕見,並與胰臟炎及胰臟癌皆相關。當酵素延伸至關節囊並造成化膿性關節時,也可能發展出骨壞死 (osteonecrosis)。
胰臟性脂膜炎亦可見胸腔積液與漿膜炎 (serositis),且胸腔積液與高死亡率相關。胰臟性脂膜炎可見嗜酸性球增多,無論其源於胰臟炎或胰臟惡性腫瘤。胰臟腫瘤(或胰臟炎)合併脂膜炎、多關節炎與嗜酸性球增多(Schmid 三聯徵)預示不良預後。胰臟性脂膜炎的一個重要併發症是繼發感染。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
胰臟性脂膜炎一般歸因於胰臟酵素(如脂解酶、澱粉酶與胰蛋白酶 trypsin)釋放至循環中,促進血管通透性與損傷,導致脂肪細胞釋放脂肪酸與後續的脂肪壞死。然而,有胰臟酵素血清水平正常情況下發生胰臟性脂膜炎的報告。此外,將正常脂肪組織與澱粉酶、脂解酶及一名胰臟炎病人的血清一同培養,在體外並未誘發脂肪壞死。抵抗素 (Resistin) 與瘦素 (leptin) 是胰臟外脂肪壞死的潛在標記。
除了較常見的胰臟炎病因外,胰臟性脂膜炎可能與下列情況相關:腎臟或合併腎臟與胰臟移植後的胰臟炎、肝臟移植後、伴噬血症候群 (hemophagocytic syndrome) 的 SLE,以及伴噬血症候群的 HIV。胰臟癌與轉移性疾病可造成脂膜炎,隨惡性組織切除而改善。脂膜炎亦可能與妊娠急性脂肪肝 (acute fatty liver of pregnancy) 與 HELLP 症候群(溶血、肝酵素升高、血小板低下)相關。
曾報告罕見的藥物誘發病例,包括在治療急性淋巴芽細胞白血病情境中的 l-asparaginase,以及 C 型肝炎的治療。在皮下注射干擾素 β (interferon β) 部位曾見相同的組織病理學。
診斷 (DIAGNOSIS)
胰臟性脂膜炎的發展期病灶顯示小葉脂肪壞死(圖 73-12A)。脂肪細胞失去細胞核但維持週邊輪廓,形成特徵性的「幽靈細胞 (ghost cells)」(圖 73-12B)。皂化作用造成鈣化,在壞死脂肪細胞內與周圍產生細緻、顆粒狀的嗜鹼性沉積物。幽靈細胞常聚集成小簇於脂肪小葉中心,週邊有嗜中性球的發炎浸潤。在較舊病灶中,壞死與幽靈細胞被泡沫狀組織球、多核巨細胞、淋巴球,以及最終的纖維化所取代。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
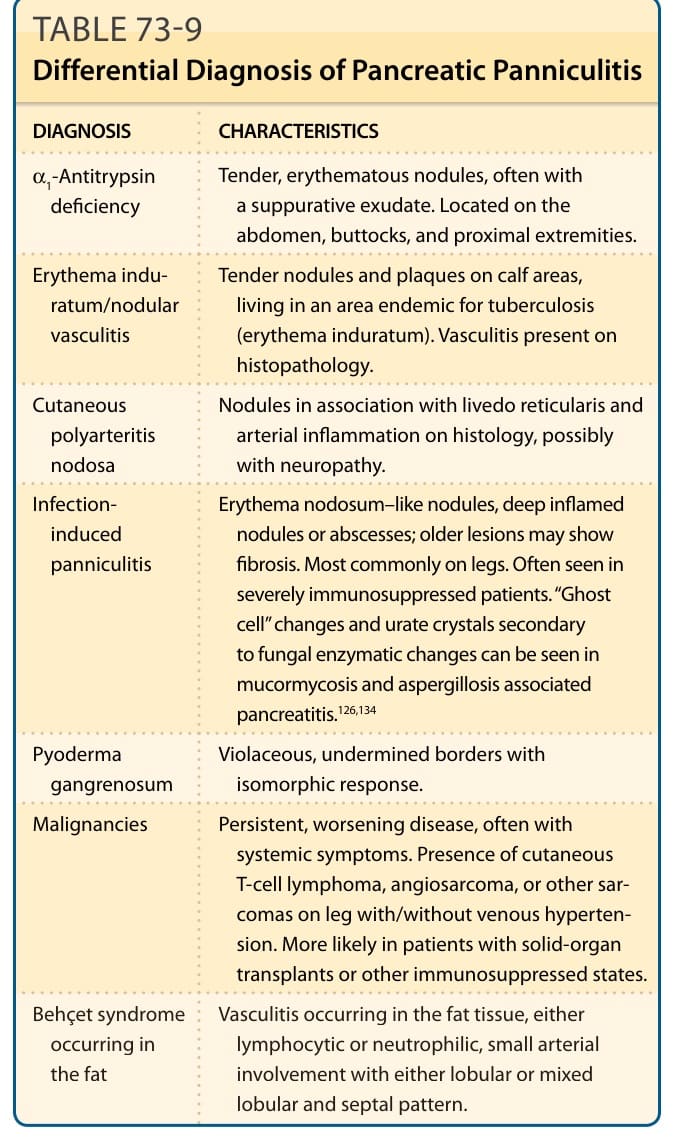
表 73-9:列出胰臟性脂膜炎的鑑別診斷。
胰臟性脂膜炎的鑑別診斷(表 73-9):
| 診斷 | 特徵 |
|---|---|
| α1-抗胰蛋白酶缺乏 (α1-Antitrypsin deficiency) | 壓痛、紅斑結節,常伴化膿性滲出物。位於腹部、臀部與近端肢體。 |
| 硬紅斑/結節性血管炎 (Erythema induratum/nodular vasculitis) | 小腿肚區的壓痛結節與斑塊,居住於結核地方流行區(硬紅斑)。組織病理學有血管炎。 |
| 皮膚型結節性多發性動脈炎 (Cutaneous polyarteritis nodosa) | 結節合併網狀青斑,組織學上有動脈發炎,可能伴神經病變。 |
| 感染誘發性脂膜炎 (Infection-induced panniculitis) | 結節性紅斑樣結節、深層發炎結節或膿瘍;較舊病灶可顯示纖維化。最常見於腿部。常見於嚴重免疫抑制病人。在毛黴菌病與麴菌病相關胰臟炎中可見繼發於真菌酵素變化的「幽靈細胞」變化與尿酸鹽結晶。 |
| 壞疽性膿皮症 (Pyoderma gangrenosum) | 紫紅色、潛蝕性邊緣,伴同形反應。 |
| 惡性腫瘤 (Malignancies) | 持續、惡化的疾病,常伴全身症狀。腿部出現皮膚 T 細胞淋巴瘤、血管肉瘤或其他肉瘤,伴或不伴靜脈高血壓。較可能見於實質器官移植或其他免疫抑制狀態的病人。 |
| 發生於脂肪的貝賽特氏症候群 (Behçet syndrome occurring in the fat) | 發生於脂肪組織的血管炎,淋巴球性或嗜中性球性,小動脈受侵犯,呈小葉型或混合小葉與間隔型。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
整體而言,當胰臟病理消退時,皮膚症狀隨後改善。與胰臟疾病相關的脂膜炎可能致命,死亡率為 24% 至 42%,在患胰臟癌者中死亡率接近 100%。急性胰臟炎可被治療並可能以支持性照護迅速消退,然而胰臟癌及相關脂膜炎則難以治療得多。
處置 (MANAGEMENT)
胰臟性脂膜炎的治療重點在於潛在的胰臟疾病。照護常為支持性。由於病人在表現時可能缺乏腹部症狀,任何有脂膜炎的病人都應考慮胰臟性脂膜炎。Octreotide(一種體抑素 somatostatin 類似物)與血漿置換術與胰臟性脂膜炎的消退相關。以手術切除與合併化療治療胰臟癌可能改善皮膚表現。
狼瘡性脂膜炎 (LUPUS PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 臉、肩、上臂、頭皮、胸、臀部的紅斑結節;罕見於下肢。
- 消退病灶中持續的脂肪萎縮 (lipoatrophy) 區域。
組織病理學
- 多為小葉型脂膜炎(伴或不伴淋巴球性血管炎)。
- 小葉淋巴球浸潤;可包含漿細胞與嗜酸性球。
- 間隔內的淋巴濾泡與硬化膠原束。
- 晚期病灶整個脂肪小葉的玻璃樣壞死與萎縮。
- 20% 至 30% 的病例有盤狀紅斑性狼瘡 (discoid lupus erythematosus) 的介面變化。
治療
- 抗瘧藥、thalidomide、外用類固醇。
- 若活躍且嚴重發炎,短療程口服皮質類固醇。
- Dapsone、cyclosporine、methotrexate、靜脈注射免疫球蛋白、rituximab、azathioprine、tacrolimus。
紅斑性狼瘡脂膜炎 (lupus erythematosus panniculitis, LEP),亦稱深部狼瘡 (lupus profundus)、皮下紅斑性狼瘡 (subcutaneous lupus erythematosus) 與 Irrgang-Kaposi 症候群,首次於 1883 年由 Kaposi 描述,並於 1940 年由 Irrgang 命名為深部紅斑性狼瘡 (lupus erythematosus profundus)。整體而言它非常罕見,即使在紅斑性狼瘡的世代中亦然。
流行病學 (EPIDEMIOLOGY)
LEP 是紅斑性狼瘡的罕見變異型,表現為脂肪組織的發炎。LEP 可作為紅斑性狼瘡的唯一表現,或可發生於盤狀紅斑性狼瘡 (discoid lupus erythematosus, DLE) 或 SLE 發作之前或之後。LEP 病人中 SLE 的發生率報告為 10% 至 41%。LEP 病人中 DLE 的發生率較高,為 21% 至 60%。在患有 SLE 者中,僅 2% 至 5% 會有 LEP。雖然罕見,LEP 在全球皆有發生,女性比男性更頻繁,女男比約 4:1。LEP 最常見於 30 至 60 歲之間,但罕見可見於孩童甚或新生兒狼瘡 (neonatal lupus)。當與 SLE 相關出現時,LEP 傾向與較不嚴重的 SLE 一同發生。LEP 病人可能有其他自體免疫疾患,如修格蘭氏症候群與類風濕性關節炎。10% 的 LEP 病人有雷諾現象 (Raynaud phenomenon)。
LEP 的其他關聯包括 TNF 抑制劑,曾在一名類風濕性關節炎病人中造成 SLE 樣反應以及 LEP。以干擾素治療一名毛細胞白血病 (hairy cell leukemia) 女性病人,曾與散播性潰瘍性狼瘡性脂膜炎的出現相關。
臨床特徵 (CLINICAL FEATURES)
LEP 病灶可能壓痛與疼痛,通常出現於外側上臂、肩、臉、頭皮、髖、臀部、乳房,以及罕見的下肢(圖 73-13)。眼眶侵犯可能以眼周水腫表現,並曾報告布拉斯科線狀 (Blaschko linear) 模式。病灶為深層皮下結節,伴或不伴表面變化,包括紅斑與 DLE 特徵如萎縮、過度角化、色素沉著或色素減退、毛細血管擴張、毛囊角栓 (follicular plugging) 與局灶壞死。多達 28% 的病例可見潰瘍。持續硬結的脂膜炎區域可能與 DLE 共存,並導致皮下萎縮與皮膚表面疤痕形成。臉部病灶的萎縮導致嚴重的美觀改變。病灶可能由外傷誘發,包括注射與手術操作。
血清學可能正常或異常。相關 SLE 的病人傾向有較高的抗核抗體 (antinuclear antibody, ANA) 陽性效價,並可能發現 C4 缺乏。其他實驗室發現可能包括類風濕因子、偽陽性的性病研究實驗室試驗 (Venereal Disease Research Laboratory)、ANA、白血球減少、貧血或血小板減少。抗磷脂抗體 (Antiphospholipid antibodies) 可能陽性,若組織病理學上注意到血栓,則應進行抗磷脂抗體檢測。
狼瘡性乳腺炎 (Lupus mastitis) 是侵犯乳房的罕見狼瘡性脂膜炎形式。它臨床上可能模擬佩吉特氏病 (Paget disease)。乳房攝影上有與脂肪壞死走向平行的鈣化,早期沉積令人擔憂惡性。MRI 顯示狼瘡性乳腺炎與非特異性脂肪壞死的範圍;加上對比劑可能顯著助於診斷。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
先天免疫系統在 LEP 的發展中扮演重要角色。脂肪細胞是先天免疫系統的重要細胞,表現 TLR1、TLR2、TLR3、TLR4 與 TLR6。脂肪組織的基質血管部分 (stromovascular fraction) 表現 TLR5、TLR7、TLR8、TLR9 與 TLR10。在 SLE 中,TLR7 與 TLR9 分別辨識 RNA 與 DNA 模式,似乎提供辨識自體 DNA 或自體 RNA 的機制,隨後活化後天免疫系統並產生對核酸及與核酸結合的蛋白質的自體抗體。TLR7 與 TLR9 的抑制劑可在自體免疫小鼠模型中預防疾病。TLR9 受體的基因變異使人易患 SLE,且曾在一名獲得 TLR7 與 TLR9 缺陷及抗體缺乏的病人中見到 SLE 消退。兩種受體都被提議作為治療標的。有趣的是,hydroxychloroquine 在體外可阻斷細胞內 TLRs。
診斷 (DIAGNOSIS)
約 20% 至 30% 的 LEP 病例可見 DLE 的組織病理學特徵。這些特徵包括基底細胞層的空泡化、基底膜增厚、真皮膠原束間的黏蛋白沉積,以及淋巴球的淺層與深層血管周圍發炎浸潤。脂肪組織顯示小葉型或混合小葉與間隔型脂膜炎,伴淋巴球、淋巴濾泡形成(20% 有生發中心 germinal centers)、不等程度的玻璃樣脂肪壞死,以及與淋巴球及漿細胞的間質浸潤相關的玻璃樣化與硬化的間隔膠原束。可見核碎裂、淋巴球性血管炎與膜囊性變化。10% 的病例亦可見鈣化及/或纖維蛋白血栓。22% 至 41% 的病例可見嗜酸性球。
血管與基底膜的直接免疫螢光 (Direct immunofluorescence) 在幾乎所有 SLE 相關 LEP 中顯示陽性發現,在無 SLE 的 LEP 中也有高比例的陽性發現。LEP 的免疫組織化學研究顯示淋巴球佔優勢,αβ 輔助 (CD4+) 與細胞毒性 (CD8+) 淋巴球與 (CD20+) B 細胞及漿細胞混合。
在僅有脂肪組織侵犯而無 DLE 或 SLE 其他特異性特徵的病例中,與皮下 T 細胞淋巴瘤的區分至關重要且困難。退行性大細胞淋巴瘤 (Anaplastic large-cell lymphoma) 亦可能與 LEP 有重疊特徵。曾有皮下淋巴樣血液惡質 (lymphoid dyscrasias) 系譜的報告,病灶原先診斷為 LEP,進展為不確定的淋巴球性小葉脂膜炎,最終診斷為 SPTCL。SPTCL 可見淋巴球異型性與淋巴球鑲邊 (lymphocytic rimming)。SPTCL 可與 LEP 共存,且必須與非典型淋巴球性脂膜炎區分,後者的克隆性 T 細胞浸潤與惰性 (indolent) 行為相關。SPTCL 可能有表皮侵犯,使其與 LEP 的區分更困難。缺乏脂肪細胞膜破壞提示 LEP。玻璃樣脂膜囊性變化在 LEP 中常見,但在 SPTCL 中不常見。
在一項研究中,LEP 病灶的浸潤中發現高水平的干擾素-α 蛋白標記人類黏液病毒抗性蛋白 1 (human myxovirus resistance protein 1, MxA)。MxA 染色極少(少於 20%)提示淋巴瘤。CCL5(脂肪細胞常表現的受體 CCR5 的配體)在淋巴瘤中的表現比 LEP 高得多。Ki-67 染色大於 20% 與局灶性增加攝取區域,更提示 SPTCL。CD123 陽性染色的存在亦提示 LEP,並是重要的預後指標。
即使血清學不具特異性,仍應加入檢查,因為 SLE 將需要進一步治療。ANA 血清學有高偽陽性率,並可能在 SPTCL 病人中呈陽性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)

表 73-10:列出狼瘡性脂膜炎 (LEP) 的鑑別診斷。
狼瘡性脂膜炎的鑑別診斷(表 73-10):
| 診斷 | 特徵 |
|---|---|
| 腫脹性狼瘡 (Tumid lupus) | 紅斑、水腫斑塊,主要在光暴露區域,組織病理學有廣泛黏蛋白沉積但無表皮變化。 |
| 皮膚淋巴瘤(包括皮下脂膜炎樣 T 細胞淋巴瘤 [SPTCL]) | 持續、惡化的疾病,常伴全身症狀。若無表面變化或無其他徵象/症狀提示 LEP,組織病理學至關重要。LEP 較常侵犯臉部,而 SPTCL 較常侵犯頭皮、下肢與臀部。SPTCL 可見淋巴球異型性與淋巴球鑲邊。多達 19% 帶 αβ 表現型的 SPTCL 病例可有相關自體免疫疾患,以紅斑性狼瘡最常見。 |
| 硬紅斑/結節性血管炎 (Erythema induratum/nodular vasculitis) | 小腿肚區的壓痛結節與斑塊,居住於結核地方流行區(硬紅斑)。組織病理學有血管炎。 |
| 局部硬皮症/系統性硬化症 (Morphea/systemic sclerosis) | 早期淋巴球性脂膜炎,隨後皮下間隔與真皮硬化。伴局部硬皮症與系統性硬化症的臨床特徵。 |
| 胰臟性脂膜炎 (Pancreatic panniculitis) | 最常見於腿部,伴潰瘍與引流。血清脂解酶與澱粉酶常升高。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
LEP 是一種慢性發炎疾患,伴週期性發作或長期緩解。平均疾病持續時間為 6 年,範圍從少於 1 年至 38 年。CD123 免疫組織化學染色可作為有用的預後指標,因為較高百分比的 CD123 陽性細胞對類固醇療法有較大的反應。及時治療有助於減少永久且可能毀容的疤痕。
處置 (MANAGEMENT)
治療具挑戰性,除了臨床評估紅斑、硬結與壓痛外,治療反應的評估也是如此。正如其他形式的皮膚狼瘡,常建議防曬與避免日曬。抗瘧藥是 LEP 的一線治療,並可能是唯一所需的藥物。當 hydroxychloroquine 單一療法無效時,與 quinacrine 合併已成功使用,但取得困難。
抗瘧藥同時干擾發炎細胞激素與 TLRs。抗瘧藥在吸菸者中效果減弱。
外用類固醇亦可能成功,尤其在封閉 (occlusion) 下。系統性皮質類固醇有效且有用,若疾病活躍且嚴重,並以短療程或在抗瘧藥起始期間使用。由於 LEP 的慢性本質,避免長期使用皮質類固醇。不建議病灶內類固醇,因為外傷可能誘發進一步活化以及萎縮。其他治療包括 dapsone、thalidomide、cyclosporine、methotrexate、azathioprine、靜脈注射免疫球蛋白 (IVIG) 與 tacrolimus。嚴重、頑固的病例可能需要 rituximab 或 infliximab,但使用此方式必須謹慎,因為它在某些情況下與已知的紅斑性狼瘡活化相關。考量到 LEP 組織病理學中常見的鈣化,鈣離子通道阻斷劑如 diltiazem 是有效的輔助,可改善鈣質沉著 (calcinosis)。
吞噬細胞性組織球性脂膜炎 (CYTOPHAGIC HISTIOCYTIC PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 四肢、軀幹及較少其他部位的皮下紅斑至紫紅色斑塊與結節;病灶可能潰瘍。
- 發燒、肝脾腫大、2 種或以上的血球減少;骨髓、淋巴結、肝或中樞神經的血球吞噬現象 (hemocytophagocytosis)。
- 高三酸甘油酯血症、鐵蛋白 (ferritin) >500 mg/L;可溶性 CD25、CD163 水平增加。
- 自然殺手細胞活性低或缺失、纖維蛋白原 (fibrinogen) 水平。
- 若與噬血性淋巴組織球增多症 (hemophagocytic lymphohistiocytosis) 及巨噬細胞活化症候群 (macrophage activation syndrome) 相關,可能有快速致命的病程;死亡前有間歇性緩解與惡化;或非致命的急性或間歇性病程。
組織病理學
- 多為小葉型脂膜炎,不伴血管炎。
- 脂肪小葉內的組織球與成熟淋巴球,伴脂肪細胞壞死。
- 「豆袋 (bean-bag)」細胞:胞質內含有完整或破碎紅血球、白血球或血小板的巨噬細胞;可為局灶性。
治療
- 一線治療為以 cyclosporine 與皮質類固醇進行免疫抑制。其他選項包括 tacrolimus、azathioprine、anakinra、cyclophosphamide 加 IVIG。
- 伴噬血性淋巴組織球增多症或巨噬細胞活化症候群的吞噬細胞性組織球性脂膜炎需要合併化療與積極的支持性照護。
- 診斷並治療相關的惡性腫瘤或感染。
吞噬細胞性組織球性脂膜炎 (cytophagic histiocytic panniculitis, CHP) 是一種罕見的脂膜炎,持續時間從 3 個月至 27 年不等,可能致命。若與噬血性淋巴組織球增多症 (hemophagocytic lymphohistiocytosis, HLH) 或巨噬細胞活化症候群相關,它可能是需要化療的侵襲性疾病,但也有報告為需要免疫抑制療法且緩解率極佳的較惰性病程。值得注意的是,HLH 可能為原發性(遺傳)或繼發性(繼發於感染、自體免疫疾病),但 CHP 在病因上僅為繼發性。CHP 本身可能代表「悶燒型 (smoldering)」淋巴瘤,而非純粹的發炎性疾病。
流行病學 (EPIDEMIOLOGY)
CHP 罕見,但見於成人、青少年與孩童。報告中最年長的病人在診斷時為 80 歲。受侵犯的孩童包括一名帶有穿孔素 (perforin) 基因突變者(可能患有家族性 HLH)。一名患有家族性多發性脂肪瘤症候群 (familial multiple lipomatosis syndrome) 的成人在 43 歲時發展出 CHP。由於病因多變,流行病學主要依 CHP 的病因而定。
臨床特徵 (CLINICAL FEATURES)
病灶為大小不等的皮下斑塊與結節,可在四肢與軀幹融合,較少於頭頸部(圖 73-14)。黏膜侵犯罕見。特徵從膚色至紅斑、紫斑、瘀斑或色素沉著;平坦或隆起;界線清楚或瀰漫;硬結至波動或潰瘍。個別病灶可達直徑 20 cm。
疾病的猛爆型可能與發燒、肝脾腫大、淋巴結病、漿膜積液、全血球減少、血管內凝血、肝衰竭、出血性體質與死亡相關。實驗室檢查應包括含分類的全血球計數、肝功能、鐵蛋白、紅血球沉降率與血脂圖譜。有趣的是,紅血球沉降率常正常或偏低。可溶性血紅素-結合珠蛋白清道夫受體 CD163 (soluble hemoglobin-haptoglobin scavenger receptor CD163) 血清水平與噬血疾病活性相關,並與血清鐵蛋白水平相關。CHP 中的血清 CD163 水平遠高於敗血症與正常對照所見的水平。其他檢查由病史、身體發現與臨床病程決定。ANA 可能陽性,但其本身不單獨證明 LEP。
由於 CHP 可造成高三酸甘油酯血症,極高的血脂水平造成胰臟炎,可能隨血漿置換而改善。膽固醇水平升高可能在脂膜炎改善後持續數年。噬血疾病期間的三酸甘油酯升高繼發於脂蛋白脂解酶 (lipoprotein lipase) 的抑制,導致巨噬細胞分泌 IL-6、IL-1 與 TNF。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
CHP 可能屬於一個帶有良性淋巴球浸潤、會發展出反應性 HLH/巨噬細胞活化症候群的脂膜炎系譜。繼發性 HLH 與感染、結締組織疾病與惡性腫瘤相關,或由藥物誘發。與 HLH/巨噬細胞活化症候群相關的感染包括 Epstein-Barr 病毒、巨細胞病毒、小病毒 (parvovirus)、水痘、人類疱疹病毒 6 與 8、禽流感、德國麻疹、腺病毒、B 型肝炎、HIV、細菌、抗酸桿菌、寄生蟲與真菌感染。CHP 曾在 H1N1 流感疫苗與內臟利什曼原蟲病 (visceral leishmaniasis) 後出現。亦曾報告外傷誘發的 CHP。
HLH 被認為是一種自體發炎疾病 (autoinflammatory disease),伴細胞溶解性 T 細胞與自然殺手細胞功能的損害,導致一連串可能致命反應的代償性級聯。潛在的基因突變影響囊泡運輸、顆粒釋放,以及參與顆粒媒介細胞毒性的成孔細胞溶解蛋白。細胞毒性細胞無法清除受感染細胞,導致 T 細胞活化與增殖增加,產生高水平的細胞激素,刺激並活化巨噬細胞,而無法經由細胞凋亡 (apoptosis) 終止反應。
LEP 與 CHP 在某些病例中,可能都符合皮下淋巴樣血液惡質 (subcuticular lymphoid dyscrasia) 的系譜,範圍從良性反應性變化到 SPTCL,包括非典型不確定小葉脂膜炎。免疫表現型 (Immunophenotyping) 與基因型分析研究對於區分可能有 αβ 或 γδ 重排 T 細胞的惡性 SPTCL 與帶良性 T 細胞的 CHP 很重要。監測切片是審慎的做法,因為曾報告轉化 (transformation)。兩種 SPTCL 類型都可與噬血症候群/HLH 相關並以 CHP 表現;γδ 病例中 HLH 的發生率遠高於 αβ 病例。
診斷 (DIAGNOSIS)
可能需要多次切片以確立 CHP 的診斷,因為組織病理學上的細胞吞噬作用 (cytophagocytosis) 可能為局灶性。組織病理學主要為小葉型脂膜炎(圖 73-15A),脂肪小葉含有組織球與成熟淋巴球的浸潤,伴不等量的漿細胞、嗜中性球與嗜酸性球。吞噬性組織球在其胞質內含有完整或破碎的細胞與核碎片;這代表特徵性的「豆袋細胞 (bean-bag cell)」(圖 73-15B)。這不同於伸入運動 (emperipolesis)(一個完整細胞存在於另一細胞的胞質內),後者中發炎細胞通過組織球而無核碎片可見。
免疫組織化學研究顯示吞噬性組織球表現組織球標記如 CD68,而多數淋巴球顯示 T 細胞免疫表現型。非典型淋巴球、脂肪細胞的淋巴球鑲邊與克隆性的存在令人擔憂淋巴瘤。在伴 HLH 的快速進展性 CHP 病例中,內臟器官包括淋巴結、脾、肝與骨髓,以及腦脊髓液中亦可見細胞吞噬特徵。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
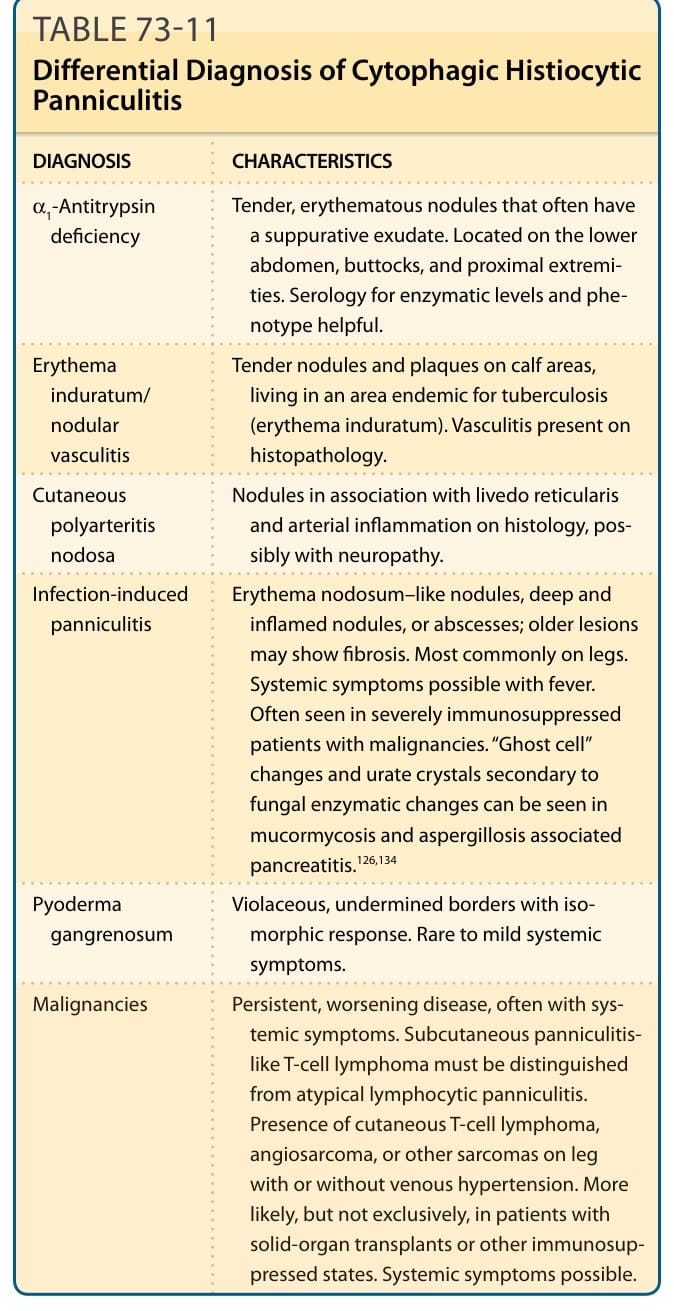
表 73-11:列出吞噬細胞性組織球性脂膜炎 (CHP) 的鑑別診斷。
吞噬細胞性組織球性脂膜炎的鑑別診斷(表 73-11):
| 診斷 | 特徵 |
|---|---|
| α1-抗胰蛋白酶缺乏 (α1-Antitrypsin deficiency) | 壓痛、紅斑結節,常有化膿性滲出物。位於下腹部、臀部與近端肢體。酵素水平與表現型的血清學有幫助。 |
| 硬紅斑/結節性血管炎 (Erythema induratum/nodular vasculitis) | 小腿肚區的壓痛結節與斑塊,居住於結核地方流行區(硬紅斑)。組織病理學有血管炎。 |
| 皮膚型結節性多發性動脈炎 (Cutaneous polyarteritis nodosa) | 結節合併網狀青斑,組織學上有動脈發炎,可能伴神經病變。 |
| 感染誘發性脂膜炎 (Infection-induced panniculitis) | 結節性紅斑樣結節、深層發炎結節或膿瘍;較舊病灶可顯示纖維化。最常見於腿部。可能有發燒等全身症狀。常見於患惡性腫瘤的嚴重免疫抑制病人。在毛黴菌病與麴菌病相關胰臟炎中可見繼發於真菌酵素變化的「幽靈細胞」變化與尿酸鹽結晶。 |
| 壞疽性膿皮症 (Pyoderma gangrenosum) | 紫紅色、潛蝕性邊緣,伴同形反應。罕見至輕微的全身症狀。 |
| 惡性腫瘤 (Malignancies) | 持續、惡化的疾病,常伴全身症狀。皮下脂膜炎樣 T 細胞淋巴瘤必須與非典型淋巴球性脂膜炎區分。腿部出現皮膚 T 細胞淋巴瘤、血管肉瘤或其他肉瘤,伴或不伴靜脈高血壓。較可能(但不限於)見於實質器官移植或其他免疫抑制狀態的病人。可能有全身症狀。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
病人可能有快速致命的病程、較長的病程(在死亡前許多年伴間歇性緩解與惡化),或對治療有反應的非致命急性或間歇性病程。具有良性、非致命病程的病人被發現有較不嚴重的實驗室異常。在適當的病人中以皮質類固醇與 cyclosporine 及時治療,可將 70% 的死亡率降至接近 0%。脂膜炎本身並不總是疾病嚴重度的標記,因為一名 CHP 病人以高劑量皮質類固醇與 cyclosporine 療法使皮膚病灶消退,隨後在 5 個月後復發、進展,並死於全身性 HLH。這個 1998 年的病例是 cyclosporine 失敗的首次報告,並強調即使在明顯的治療反應後仍需謹慎追蹤全身侵犯。
處置 (MANAGEMENT)
由於 CHP 可能有快速致命的病程,多專科照護在 CHP 管理中至關重要。可能需要住院以快速檢查並開始治療。在與感染相關的 HLH 中,治療病原可能導致恢復,但這不可預測。
CHP 的治療包括針對活化巨噬細胞/組織球(類固醇、etoposide、高劑量 IVIG)與 T 細胞(類固醇、cyclosporine、抗胸腺細胞球蛋白 antithymocyte globulin)的免疫抑制、免疫調節或細胞毒性藥物。在已發表的病例中,cyclosporine 與皮質類固醇常被使用且常成功。在同時患有 SLE 與 CHP 的病人中,皮質類固醇與 cyclosporine 的合併可能誘發兩種疾病的緩解。
對 cyclosporine 有抗性的疾病亦曾被報告對 tacrolimus 有反應。對於對 cyclosporine 有不良反應的病人,tacrolimus 可能是一個選項,並可能是無法停止療法的 CHP 病例較實際的長期治療選項。頑固病例可能需嘗試 cyclophosphamide 與 IVIG。加入合併化療可能有幫助。可能需要多輪合併化療或自體幹細胞移植以誘發緩解。
Cyclosporine,以及 cyclosporine 加 etoposide,是兒童與成人 CHP 的有效方案。在 2 名病毒感染後發展出 CHP 的小兒病人中,組織病理學內的局灶區域提示 SPTCL 而非反應性 T 細胞增生,伴原位 (in situ) T 細胞克隆性;然而 cyclosporine 加皮質類固醇分別誘發 69 與 29 個月的緩解。這份報告強調需區分與非惡性狀況相關的 CHP 與 SPTCL 相關的 CHP。這份報告也提示,當診斷提示反應性 T 細胞增生而非真正淋巴瘤時,可使用 cyclosporine,因為非惡性 CHP 常以 cyclosporine 與 prednisone 改善,而伴 SPTCL 的 CHP 最好積極治療。一名終身患有 CHP 與嚴重 HLH 的青少年,在 prednisone 與 cyclosporine 之外加上 anakinra(一種 IL-1 受體拮抗劑)後,經歷改善的徵象。
新生兒皮下脂肪壞死 (SUBCUTANEOUS FAT NECROSIS OF THE NEWBORN)
重點一覽 (AT-A-GLANCE)
臨床
- 界線清楚、紅至紫紅色的皮下結節或斑塊,好發於臀部、肩、頰與大腿。
- 部分病例有高血鈣,甚至在急性發作後出現。罕見有高三酸甘油酯血症、低血糖、血小板減少、貧血。
組織病理學
- 多為小葉型脂膜炎,不伴血管炎。
- 淋巴球、組織球、噬脂細胞 (lipophages) 與多核巨細胞的濃密發炎浸潤。
- 組織球與多核巨細胞胞質內呈針狀裂隙 (needle-shaped clefts),常呈放射狀排列。
治療
- 結節與斑塊通常自發消退。
- 發作後監測高血鈣達 6 個月,若發生高血鈣則治療。
新生兒皮下脂肪壞死 (subcutaneous fat necrosis of the newborn, SCFN) 是一種罕見的脂膜炎,發生於生命最初數天至數週。幾乎所有病例都自發消退。最常見的併發症是高血鈣。其他繼發效應包括高三酸甘油酯血症、低血糖、血小板減少與貧血。
流行病學 (EPIDEMIOLOGY)
SCFN 出現於足月或過期產的新生兒(亦有早產病例的注意),有先前的圍產期併發症病史,包括胎便吸入、窒息、低體溫(如心臟手術或室上性心搏過速的冰敷,或缺氧缺血性腦病合併呼吸努力不足)、低血氧、抽搐、敗血症、妊娠糖尿病、子癇前症、需要剖腹產的因素、產鉗分娩、嚴重新生兒貧血、母親使用古柯鹼及/或生長遲滯。在需要低體溫治療窒息的族群中,巨嬰症 (macrosomia) 與血流動力不穩定是額外的風險因子。
臨床特徵 (CLINICAL FEATURES)
病灶為界線分明、紅斑至紫紅色、堅實、硬結的結節或斑塊,位於背、肩、手臂、臀部、大腿或臉,但罕見於前軀幹或前外側脛部(圖 73-16A)。曾報告內臟侵犯,一例的 MRI 發現符合腹壁、肝周、脾周與腎周的脂肪壞死。皮下結節大小從數毫米至直徑達 11 cm 不等,可能單一或多發,且通常界線清楚。罕見地,波動性結節可能引流出油性或白堊色物質。可能形成較大的水泡並可能需要引流。病灶觸之不溫熱且常無痛;罕見地需要嗎啡 (morphine) 控制疼痛。雖然常無症狀,但病灶的位置與大小可能有局部腫塊效應,例如一例繼發於 SCFN 的橈神經麻痺 (radial nerve palsy),於 10 週內自發改善。
有數種代謝併發症可能在脂膜炎期間甚至消退後發生。這些包括高血鈣(影響 36% 至 56% 的 SCFN 病人)、血小板減少、高三酸甘油酯血症(可能與脂肪壞死相關)、低血糖與貧血。尚不清楚血糖與血液學異常是繼發於 SCFN 或繼發於引發 SCFN 的初始損傷,因為它們可能先於皮膚發現出現。高血鈣可能無症狀,或可能進展為有症狀的生長遲滯、易怒、發燒、嘔吐、肌張力低下、抽搐、多尿與多渴,甚至死亡。軟組織與器官鈣化可能發生,結果不一。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
SCFN 的最終病因不明,但低體溫與缺氧被推測涉及其中。治療性低體溫用於小兒加護中的窒息病例。尚不清楚低體溫是協助此過程或是 SCFN 的直接病因。手術操作期間的冷暴露亦可能誘發 SCFN。可能的解釋包括新生兒脂肪組成或代謝的生化缺陷,導致冷壓力後的結晶化、脂肪壞死與後續發炎。高血鈣可能與肉芽腫浸潤內 25-羥維生素 D3-1α 羥化酶 (25-hydroxyvitamin D3–1α hydroxylase) 增加有關。
新生兒的棕色脂肪組織 (brown adipose tissue, BAT) 在冷壓力條件下迅速將脂肪儲存轉化為熱能。侵犯 BAT 的 SCFN 已以免疫組織化學染色確認,以及解偶聯蛋白同型 1 (uncoupling protein isoform 1)(特異存在於 BAT)。BAT 細胞產生熱能。其機制複雜,使用解偶聯蛋白同型 1、胞質脂肪酸與 Ca2+ 離子。
診斷 (DIAGNOSIS)
特徵性地,SCFN 為多為小葉型脂膜炎(見圖 73-16B),伴脂肪小葉的局灶壞死,以及淋巴球、組織球與異物巨細胞 (foreign-body giant cells) 的濃密發炎浸潤;可能有少數嗜酸性球。
以嗜中性球為主的浸潤可能模擬感染,尤其若病灶在 1 天大時切片。許多脂肪細胞保留其細胞輪廓,但含有細緻的嗜酸性條索與顆粒,以及呈放射狀排列的針狀裂隙(見圖 73-16C)。在冷凍切片上,這些裂隙被代表三酸甘油酯的雙折光性結晶 (doubly refractile crystals) 所佔據。類似的裂隙與結晶亦可見於多核巨細胞的胞質內。晚期病灶可能顯示脂肪小葉內的纖維化與鈣化區域。細針抽吸 (Fine-needle aspiration) 可能較不疼痛、更具成本效益且診斷更快速。超音波檢查亦可能是輔助性的非侵入診斷工具。脂肪組織在杜卜勒上為高回音 (hyperechoic) 伴高血流,並可見鈣化。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)

表 73-12:列出新生兒皮下脂肪壞死 (SCFN) 的鑑別診斷。
新生兒皮下脂肪壞死的鑑別診斷(表 73-12):
| 診斷 | 特徵 |
|---|---|
| 蜂窩組織炎 (Cellulitis) | 較大的紅斑斑塊,通常缺乏紫紅色瘀傷。壓痛且溫熱。 |
| 冷脂膜炎 (Cold panniculitis) | 兒童與成人任何冷暴露皆可發生,不特定於缺氧情境。組織病理學在皮下脂肪組織內缺乏針狀裂隙。隨復溫消退。 |
| 新生兒硬化症 (Sclerema neonatorum) | 瀰漫性皮膚硬化,特別在臀部與大腿,發生於生命第一週承受壓力的嬰兒,常為低體重與早產。組織病理學亦顯示針狀裂隙,伴極少發炎。伴系統性疾病者預後不佳。 |
| 類固醇後脂膜炎 (Poststeroid panniculitis) | 發生於兒童,通常在快速停用高劑量系統性皮質類固醇療法後 10 天。組織病理學與新生兒皮下脂肪壞死相似。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
SCFN 是預後極佳的良性過程。病灶在數週至 6 個月內消退,部分以萎縮告終。高血鈣是最嚴重的潛在不良結果,並可能造成腎鈣質沉著症 (nephrocalcinosis)。即使嚴重的高血鈣也可能無症狀。高血鈣可能在皮膚病灶出現後延遲長達 6 個月發作;因此,連續監測血清鈣水平是必要的。
處置 (MANAGEMENT)
SCFN 結節自發消退,多數病例的治療應為保守,除了可能受益於抽吸以預防破裂、感染、壞死與疤痕的病灶。
外科處置曾用於持續數月的嚴重病例,以切開與刮除小型鈣化。
血清鈣應連續監測,在表現後數月內以防延遲升高。若有高血鈣,處置包括水化、使用排鈣利尿劑 furosemide 與低鈣配方。
可使用系統性糖皮質素以抑制發炎脂肪組織中巨噬細胞的維生素 D 製造。雙磷酸鹽 (Bisphosphonates),包括 alendronate、etidronate 與 pamidronate,可治療 SCFN 中的高血鈣。重要的是,儘管皮膚有反應,腎鈣質沉著症仍可能發展。高血鈣與高尿鈣的程度與持續時間可能對腎鈣質沉著症的發展至關重要,鼓勵早期監測與識別高血鈣,因為迅速的治療介入可能預防營養不良性鈣化 (dystrophic calcification)。腎臟超音波可偵測腎鈣質沉著症。持續的腎鈣質沉著症並不一致地導致腎功能障礙。
為預防 SCFN,一個特定的護理流程已降低降溫期間 SCFN 的風險。它包括每 3 小時旋轉新生兒,以及更溫和的降溫表面-降溫裝置。
冷脂膜炎 (COLD PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 界線清楚、紅至紫紅色的皮下斑塊或結節,位於臉與大腿,罕見於青春期前男孩的陰囊脂肪。
- 接續於暴露於寒冷天氣、水、冰棒與冰敷。
組織病理學
- 多為小葉型脂膜炎,伴淋巴組織球性或混合浸潤,不伴血管炎。
- 血管周圍淋巴組織球浸潤,侵犯真皮-皮下交界處的血管以及覆蓋其上的真皮內血管。
- 發現可能類似凍瘡 (perniosis) 所見。
治療
- 避免將冰直接放置於皮膚與冷暴露。
- 3 個月內自發消退。
冷脂膜炎,又稱 Haxthausen 病,首次於 1902 年在一名孩童冷天氣暴露後下巴硬化中被描述。當發生於騎馬者時,亦使用騎馬者凍瘡 (horse rider’s pernio) 與馬術脂膜炎 (equestrian panniculitis) 等名詞。冷脂膜炎是脂肪組織的發炎反應,發生於皮膚暴露於寒冷天氣與冰敷(如室上性心搏過速的冷敷應用)之後,亦可發生於黏膜(冰棒脂膜炎 popsicle panniculitis)。
流行病學 (EPIDEMIOLOGY)
冷脂膜炎發生於兒童、年輕女性騎馬者,以及其他有適當暴露的成人。症狀可能繼發於口腔內冷凍固齒器 (frozen teething rings),或在施用冰敷以治療發燒發作後。陰囊冷脂膜炎出現於 9 至 14 歲過重男孩冷暴露(包括在寒冷海水中游泳)後。騎馬者的冷脂膜炎症狀在男女皆有報告。芬蘭的一項調查提示 25% 的騎馬者在冬季月份有症狀。中度吸菸對騎馬者冷脂膜炎風險增加有統計顯著性,整天穿著緊身騎馬服、長時間騎馬與年齡小於 35 歲亦然。馬術樣脂膜炎的其他風險因子包括騎摩托車或開放式車輛、涉過寒冷河流,以及年輕女性穿著緊身牛仔褲。
臨床特徵 (CLINICAL FEATURES)
硬結、紅斑斑塊或結節的臨床發現出現於冷暴露部位,可能包括黏膜。發作範圍為暴露後 6 至 72 小時,直接出現於與冷接觸的部位。在患冷脂膜炎的騎馬者中,表現於大腿外側。
陰囊冷脂膜炎,又稱陰囊脂肪壞死,表現為睪丸下方的單側或雙側壓痛腫塊。識別這些變化很重要,以避免不必要的切片或介入,因為會在數天至數週內自發消退。馬術脂膜炎在穿著緊身、無隔熱的褲子長時間戶外冷暴露後數小時出現。病灶為搔癢、紅斑的結節與斑塊,可能局灶潰瘍、結痂與引流。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在受侵犯的嬰兒中,發炎機制歸因於嬰兒飽和脂肪對不飽和脂肪比例較高,其組成隨年齡變得更不飽和。其他機制包括冷對 BAT 的效應(BAT 在嬰兒中比成人更普遍)及其活化以產生熱能。冷暴露導致 BAT 的變化,包括血管內皮生長因子 (vascular endothelial growth factor) 增加、游離脂肪酸降解增強與代謝變化。陰囊冷脂膜炎可能與陰囊脂肪較高的濃度以及年輕男性陰囊脂肪組織對冷敏感性增加有關。
馬術脂膜炎可能代表凍瘡/凍傷 (perniosis/chilblains),而非真正的脂膜炎。雖然在 2 名患馬術脂膜炎的蘇格蘭女性中偵測到高水平的冷凝集素 (cold agglutinins),但其他系列並未顯示此異常,且冷球蛋白 (cryoglobulins) 與冷凝集素被認為不扮演角色。
診斷 (DIAGNOSIS)
鑑於受侵犯區域冷暴露的病史,診斷常為臨床診斷。一般而言,冷脂膜炎的組織病理學圖像為多為小葉型脂膜炎,伴脂肪小葉內的淋巴組織球性或混合發炎浸潤。
有淺層與深層血管周圍以及附屬器周圍的淋巴球浸潤,伴最明顯於真皮-皮下脂肪交界處的靜脈顯著發炎;這些組織病理學發現與凍瘡所見密切相似。曾建議「大腿冷相關凍瘡 (cold-associated perniosis of the thighs)」的名稱。在陰囊病例中,超音波發現可能有助於確認診斷。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
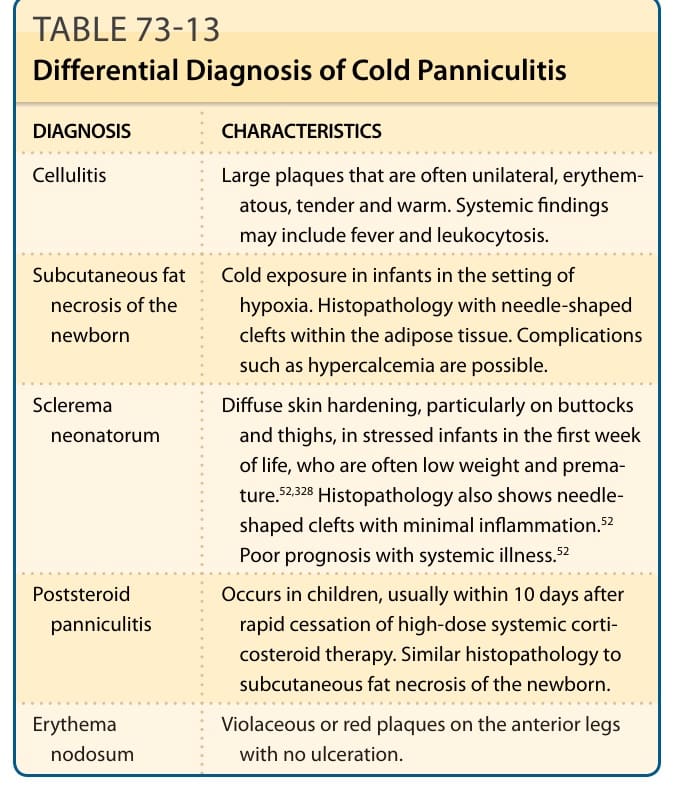
表 73-13:列出冷脂膜炎的鑑別診斷。
冷脂膜炎的鑑別診斷(表 73-13):
| 診斷 | 特徵 |
|---|---|
| 蜂窩組織炎 (Cellulitis) | 常為單側、紅斑、壓痛且溫熱的大斑塊。全身發現可能包括發燒與白血球增多。 |
| 新生兒皮下脂肪壞死 (Subcutaneous fat necrosis of the newborn) | 嬰兒在缺氧情境下的冷暴露。組織病理學在脂肪組織內有針狀裂隙。可能有高血鈣等併發症。 |
| 新生兒硬化症 (Sclerema neonatorum) | 瀰漫性皮膚硬化,特別在臀部與大腿,發生於生命第一週承受壓力的嬰兒,常為低體重與早產。組織病理學亦顯示針狀裂隙,伴極少發炎。伴系統性疾病者預後不佳。 |
| 類固醇後脂膜炎 (Poststeroid panniculitis) | 發生於兒童,通常在快速停用高劑量系統性皮質類固醇療法後 10 天內。組織病理學與新生兒皮下脂肪壞死相似。 |
| 結節性紅斑 (Erythema nodosum) | 前側腿部的紫紅色或紅色斑塊,無潰瘍。 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
冷脂膜炎為自我緩解,在復溫且不持續冷暴露後消退。可在數天至 3 個月內見到完全改善。
可能留有發炎後色素沉著 (postinflammatory hyperpigmentation)。
處置 (MANAGEMENT)
冷脂膜炎除了避免進一步冷暴露外不需額外治療;最佳預防是避免冷暴露。
患冷脂膜炎的騎馬者可能受益於保暖衣物與局部外用皮質類固醇。此族群的預防包括騎馬時與整天穿著溫暖、寬鬆的衣物,以及戒菸。Nifedipine 不是有效的治療。
人為性與外傷性脂膜炎 (FACTITIAL AND TRAUMATIC PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 在人為性脂膜炎 (factitial panniculitis)(孟喬森症候群 Munchausen syndrome 的一種變異型)中,自我誘發的外傷(機械性、物理性或化學性)造成皮下發炎。在外傷誘發性脂膜炎中,損傷並非病人自行誘發,例如術後脂膜炎或運動傷害後的脂膜炎。
- 紅斑、幾何形丘疹與結節,伴糜爛或潰瘍(圖 73-17),位置或外觀不一致。
- 人為性脂膜炎常見於有人格偏差的病人,皮下植入物可能包括藥物、化妝填充物、油類,以及食物或人類排泄物。
- 外傷性脂膜炎可能繼發於拔罐、針灸與電針,或可能為免疫接種或治療性注射的醫源性。
- 受侵犯部位的繼發性多毛症 (hypertrichosis) 罕見。
組織病理學
- 多為非特異性小葉型脂膜炎,不伴血管炎,依病因而異。
- 侵犯脂肪小葉的化膿性肉芽腫;需要對各種微生物進行培養。常存在壞死。
- 玻片的偏振光 (polarization) 可辨識巨噬細胞內的折光性異物。
- 晚期可能顯示泡沫狀組織球伴纖維化變化與營養不良性鈣質沉積。泡沫狀噬脂性組織球、脂肪細胞大小不一伴偽囊性退化、單一脂肪細胞壞死、紅血球外滲,以及伴多核巨細胞的異物肉芽腫,皆為潛在特徵。術後脂膜炎可能有明顯疤痕、深部靜脈炎與外傷性神經瘤 (traumatic neuromas)。
治療
- 自我造成的人為性脂膜炎需精神科治療。
- 支持性照護與干預責任藥劑的注射。
- 植入物質常需手術切除。
- 外傷性脂膜炎需預防持續外傷與支持性照護,因為在無進一步外傷下為自我緩解。
結締組織疾病相關脂膜炎 (CONNECTIVE TISSUE DISEASE-ASSOCIATED PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 最常與皮肌炎 (dermatomyositis) 相關,可能先於、伴隨或晚於疾病病程出現,嚴重度與疾病發作相關。
- 手臂、臀部、大腿與腹部的紅斑、壓痛至疼痛的結節與斑塊。併發症包括潰瘍與脂肪萎縮。
- 罕見地,脂膜炎是皮肌炎的初始表現,而幼年型皮肌炎相關脂膜炎不常見但已有報告。孩童的脂膜炎應促使對結締組織疾病進行檢查。
- 感染可能同時發生,必須在免疫抑制前治療。
- 脂膜炎亦與局部硬皮症 (morphea) 與系統性硬化症相關,但此關聯罕見。
- 結締組織疾病脂膜炎的鑑別診斷必須考慮 SPTCL。在一個系列中,多達 19% 帶 αβ 表現型的 SPTCL 有相關自體免疫疾患。帶 γδ 表現型的 SPTCL 曾報告在一名類風濕性關節炎病人接受 3 年抗 TNF 療法後發展。一名 SPTCL 病人曾以皮肌炎的臨床特徵表現。
組織病理學
- 多為小葉型脂膜炎,伴淋巴球與漿細胞浸潤,伴真皮黏蛋白與空泡介面變化,類似狼瘡性脂膜炎。
- 可能存在鈣化。
- 晚期病灶可能存在膜囊性變化,並指出較差的預後。
治療
- 與皮肌炎相同:皮質類固醇、免疫抑制劑(包括 azathioprine、mycophenolate mofetil 與 methotrexate)與 IVIG。
- 病灶內的鈣化可能受益於 IVIG。
- 抗瘧藥對皮肌炎相關脂膜炎無效。
痛風性脂膜炎 (GOUTY PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 皮下尿酸鹽結晶是痛風的罕見表現。
- 最常為堅實、硬結的結節或斑塊,下肢有不等程度的壓痛,可能有骨髓的尿酸鹽結晶沉積,以及其他瀰漫侵犯。
- 白堊樣物質可能覆蓋潰瘍。
- 可能見到大量脂肪組織肥大,需要手術切除。
- 與高尿酸血症 (hyperuricemia) 相關,以及關節痛(痛風性關節炎)。
- 病灶可能先於痛風診斷,但常為慢性痛風的晚期後果。
- 鑑別診斷包括異物性脂膜炎、鈣質與其他結晶沉積疾病。
組織病理學
- 脂肪組織內細緻、放射狀排列、針狀的尿酸鹽結晶被肉芽腫性發炎所包圍。單鈉尿酸鹽結晶 (monosodium urate crystals) 具折光性且為負雙折射 (negatively birefringent)。
- 酒精固定 (Alcohol fixation) 可增強結晶的可視化。
- 無血管炎的證據。
治療
- 痛風的標準治療包括 allopurinol(高劑量)、probenecid 與抗發炎藥物,包括可用於降低尿酸水平的秋水仙素。
- 急性脂膜炎可能受益於皮質類固醇。
- 不癒合的潰瘍性病灶可能需要手術。
類固醇後脂膜炎 (POSTSTEROID PANNICULITIS)
重點一覽 (AT-A-GLANCE)
臨床
- 類固醇後脂膜炎罕見,最常見於兒童在高劑量系統性皮質類固醇治療後,於突然停用皮質類固醇後數天至數週。成人病例非常罕見。
- 頰、軀幹、手臂與腿部的紅斑、壓痛結節與斑塊。
- 在數週或數月內消退而無疤痕;可留下殘餘色素沉著。最常見的併發症是潰瘍;全身症狀罕見。
組織病理學
- 小葉型脂膜炎,伴淋巴球與肉芽腫浸潤,不伴血管炎。可存在噬脂細胞與結晶。
- 可見放射狀排列的針狀裂隙,類似 SCFN 與新生兒硬化症所見;新生兒硬化症缺乏或僅有稀疏的發炎。
- 多核巨細胞可能含有針狀裂隙與泡沫狀組織球。
治療
- 病灶在數週或數月內消退而無疤痕,即使不治療亦然,留下殘餘色素沉著。
- 曾有人推測逐漸減量類固醇可消除類固醇後脂膜炎的風險。
致謝 (ACKNOWLEDGMENTS)
作者們希望感謝 Patricia Fishman 醫師對本章前一版的貢獻。
圖片 (FIGURES)
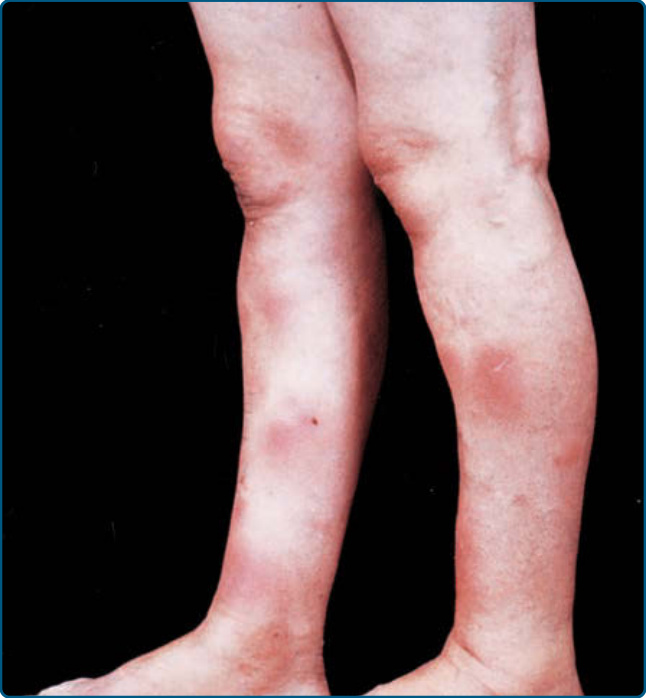
圖 73-1:結節性紅斑 (Erythema nodosum)。紅斑結節主要位於下肢前側。
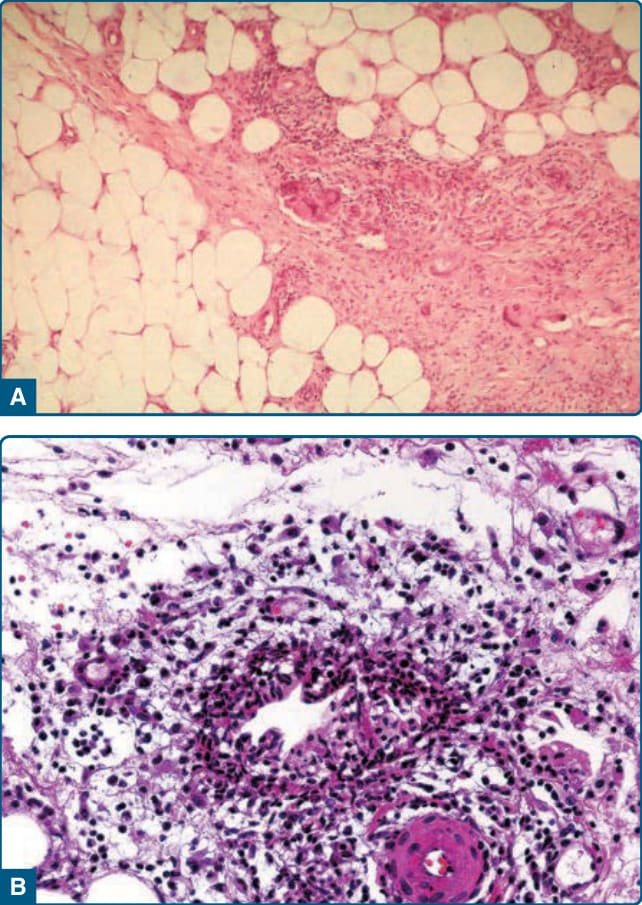
圖 73-2A:結節性紅斑。低倍放大顯示晚期 EN 病灶的纖維化、增寬間隔;脂肪小葉被侵蝕並部分消失。

圖 73-2B:結節性紅斑。米歇爾肉芽腫 (Miescher granuloma)——圍繞中央星狀裂隙的組織球聚集,被視為早期 EN 的特徵,但非在所有 EN 都能發現,也曾於其他類型脂膜炎中被描述;晚期 EN 病灶可顯示脂膜囊性變化。
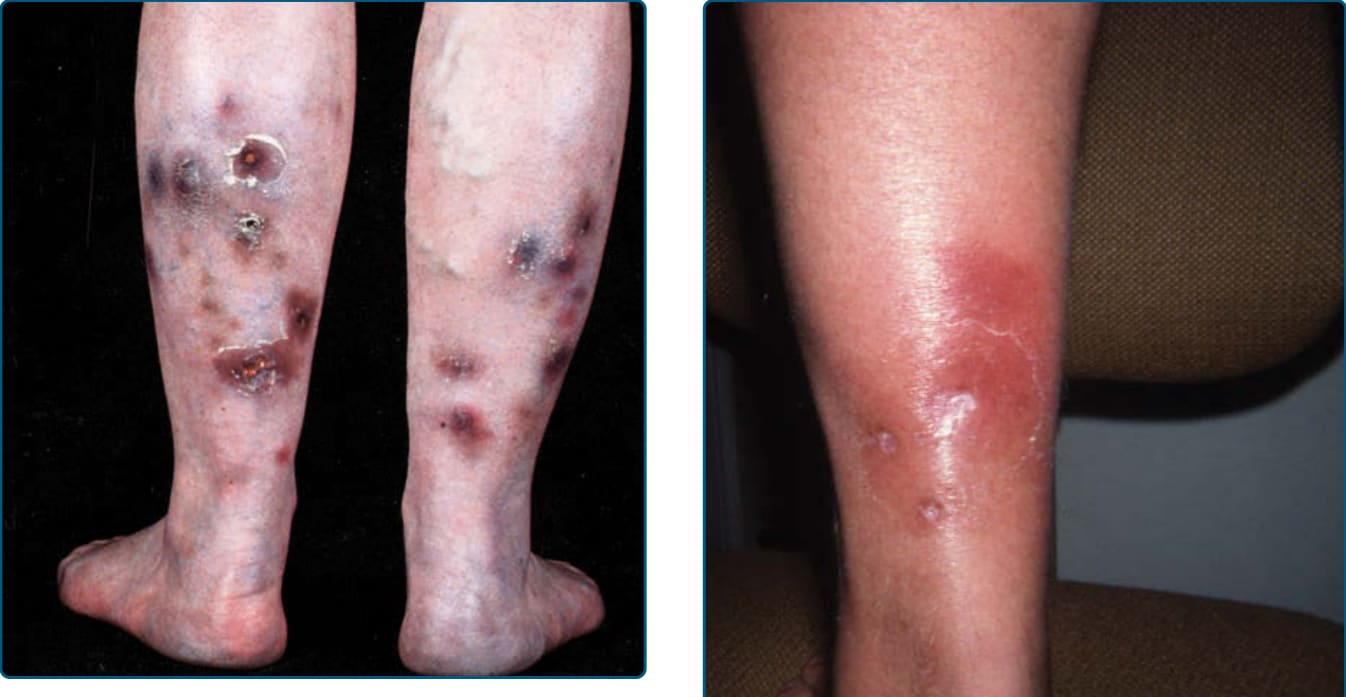
圖 73-3:硬紅斑 (Erythema induratum)。小腿肚上紅斑至棕色與藍色的結節伴潰瘍。
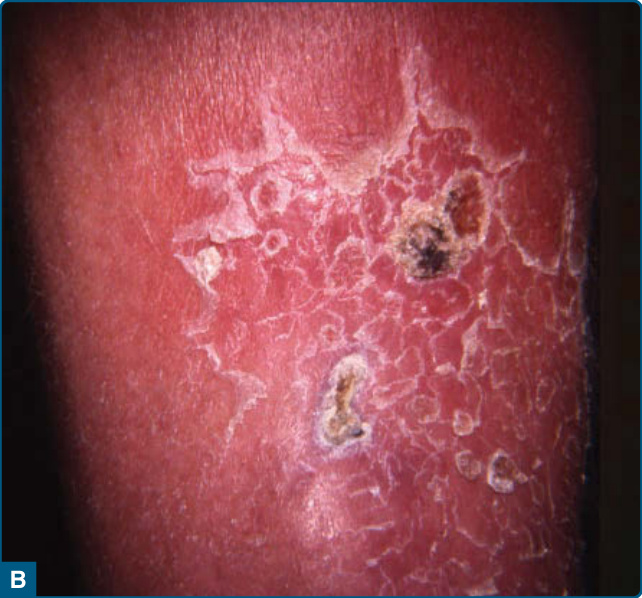
圖 73-4:A 與 B,硬紅斑伴周圍的鱗屑領圈 (collarette of scale)。
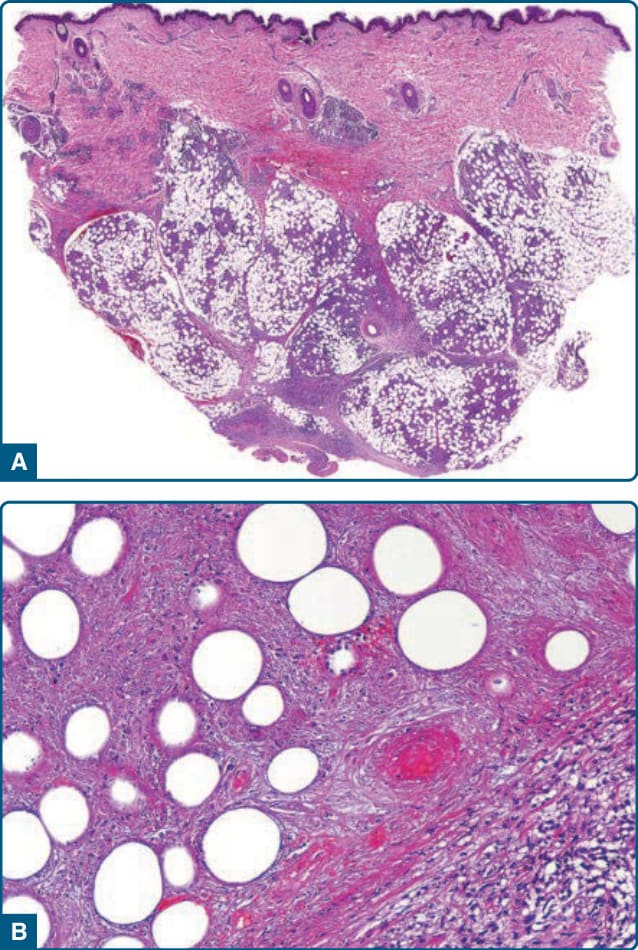
圖 73-5:硬紅斑結節性血管炎 (Erythema induratum nodular vasculitis)。A,低倍放大顯示多為小葉型脂膜炎。B,高倍放大顯示廣泛的脂肪細胞壞死與血管損傷——脂肪小葉中小靜脈的壞死性血管炎 (necrotizing vasculitis)。
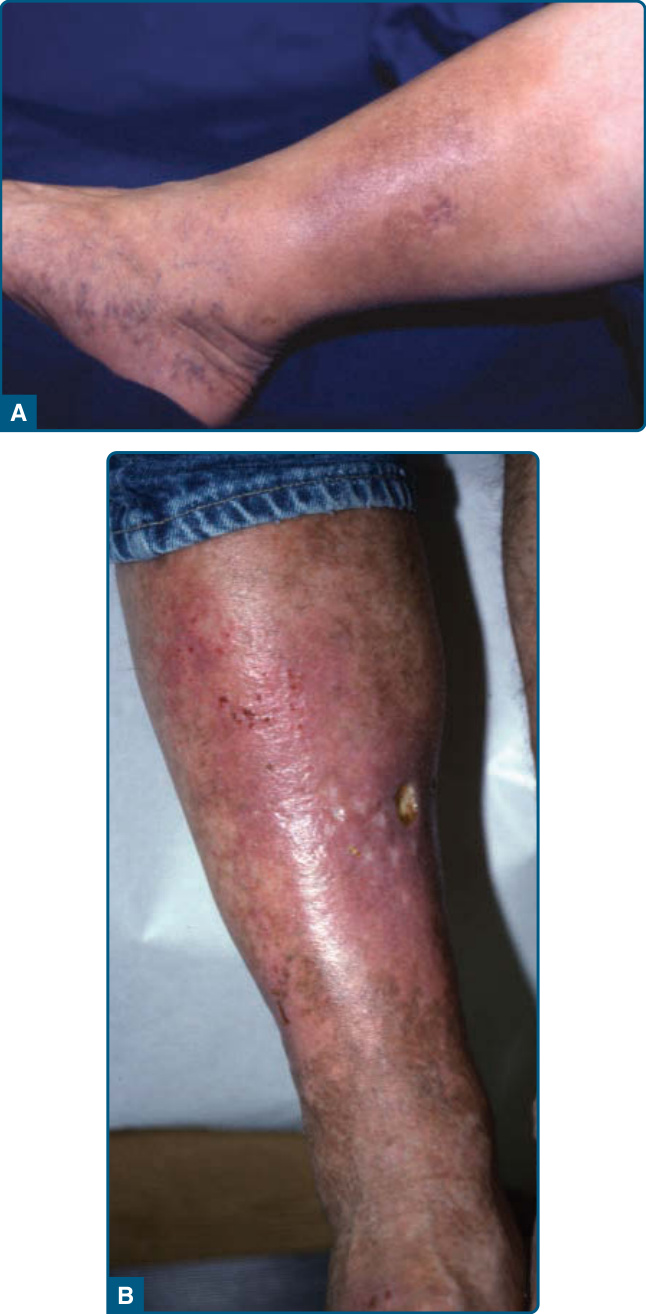
圖 73-6:A,慢性淋巴皮膚硬化症 (lipodermatosclerosis, LDS) 伴內側下肢輕度紅斑,指出活躍發炎的存在。內側下肢的蜂窩組織炎樣斑塊。B,疊加於慢性 LDS 之上的嚴重急性發炎伴潰瘍。
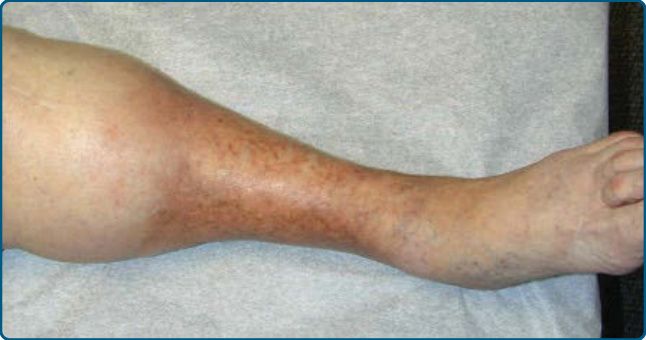
圖 73-7:慢性淋巴皮膚硬化症伴香檳瓶/保齡球瓶 (champagne bottle/bowling pin) 變形。
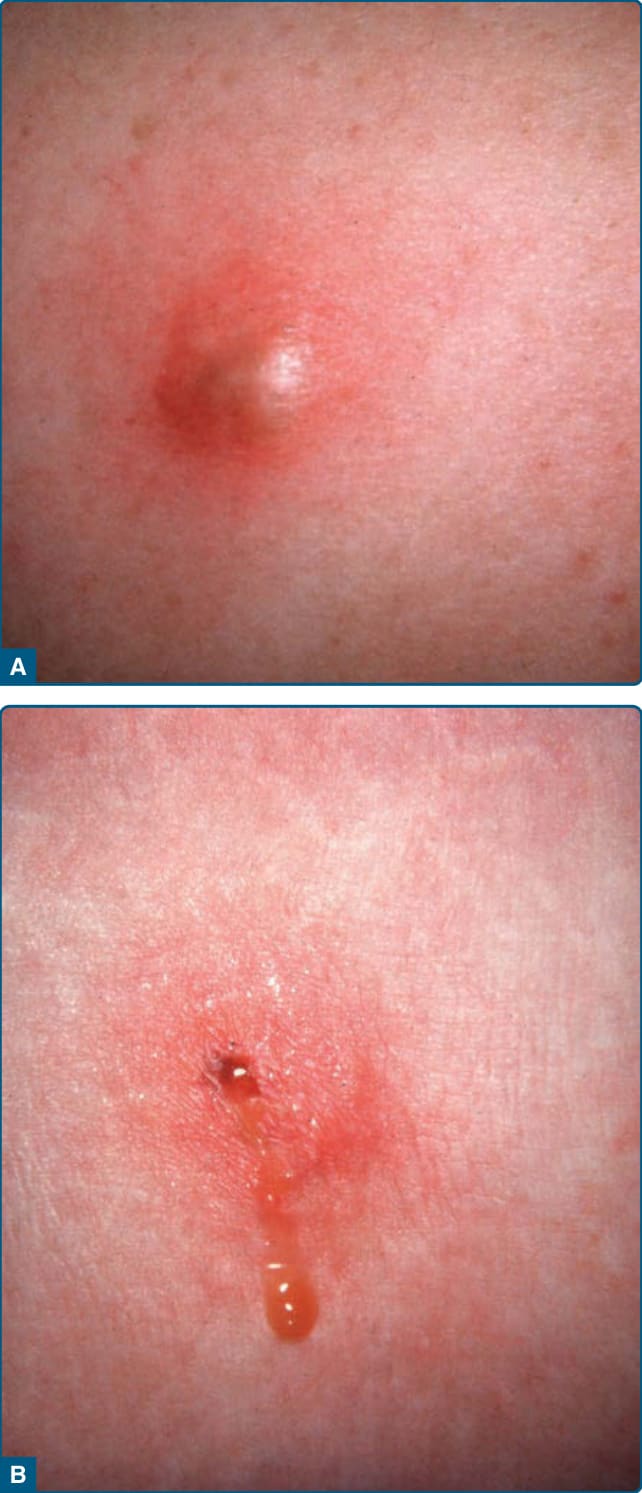
圖 73-8:α1-抗胰蛋白酶缺乏相關脂膜炎 (α1-Antitrypsin deficiency–associated panniculitis)。A,波動性膿瘍外觀。B,病灶排出油性物質。
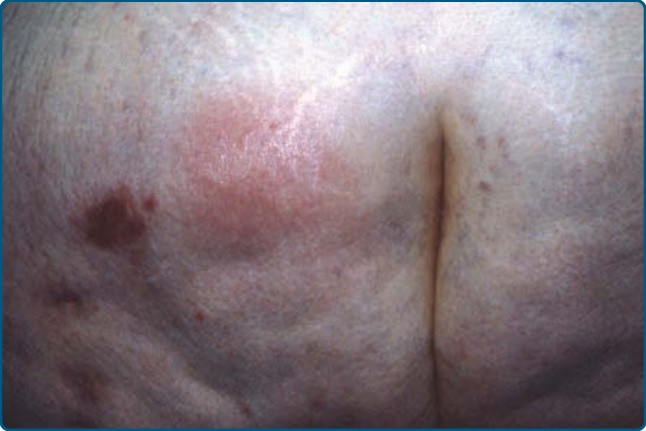
圖 73-9:α1-抗胰蛋白酶缺乏相關脂膜炎。臀部的結節性病灶。
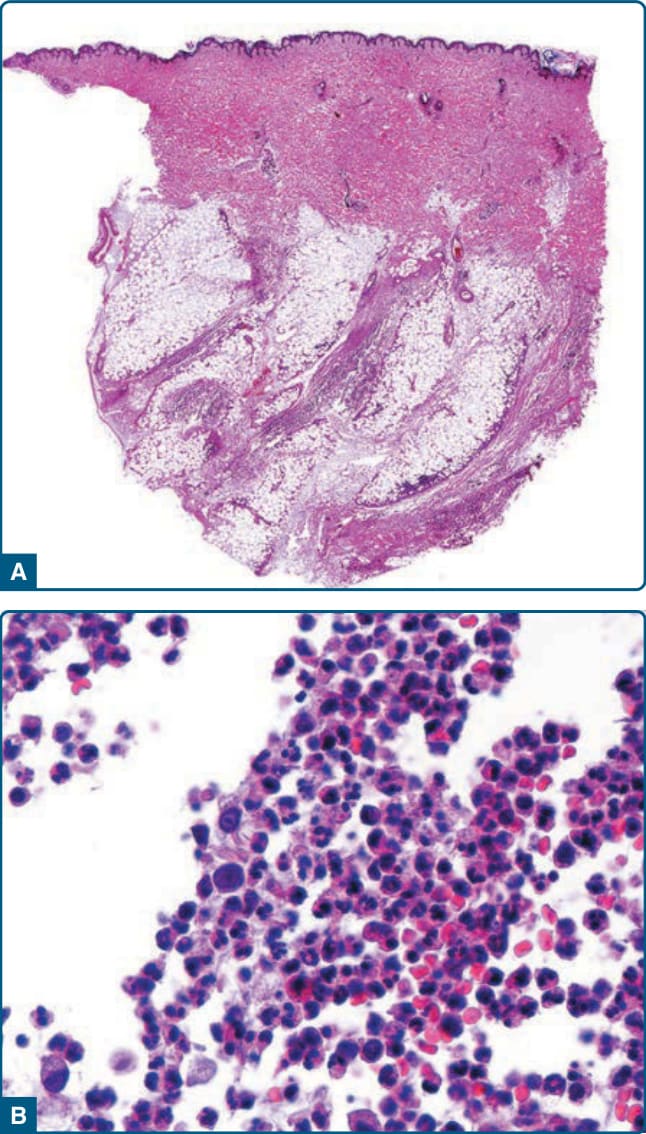
圖 73-10:α1-抗胰蛋白酶缺乏相關脂膜炎。A,低倍放大顯示多為小葉型脂膜炎。B,高倍放大顯示脂肪小葉中嗜中性球的濃密發炎浸潤。
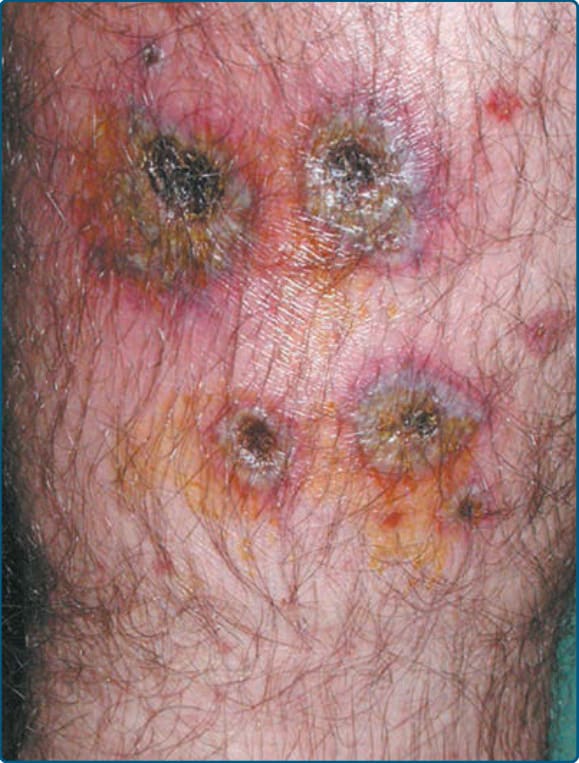
圖 73-11:胰臟性脂膜炎 (Pancreatic panniculitis)。潰瘍並滲出油性物質的紅斑性皮下結節。
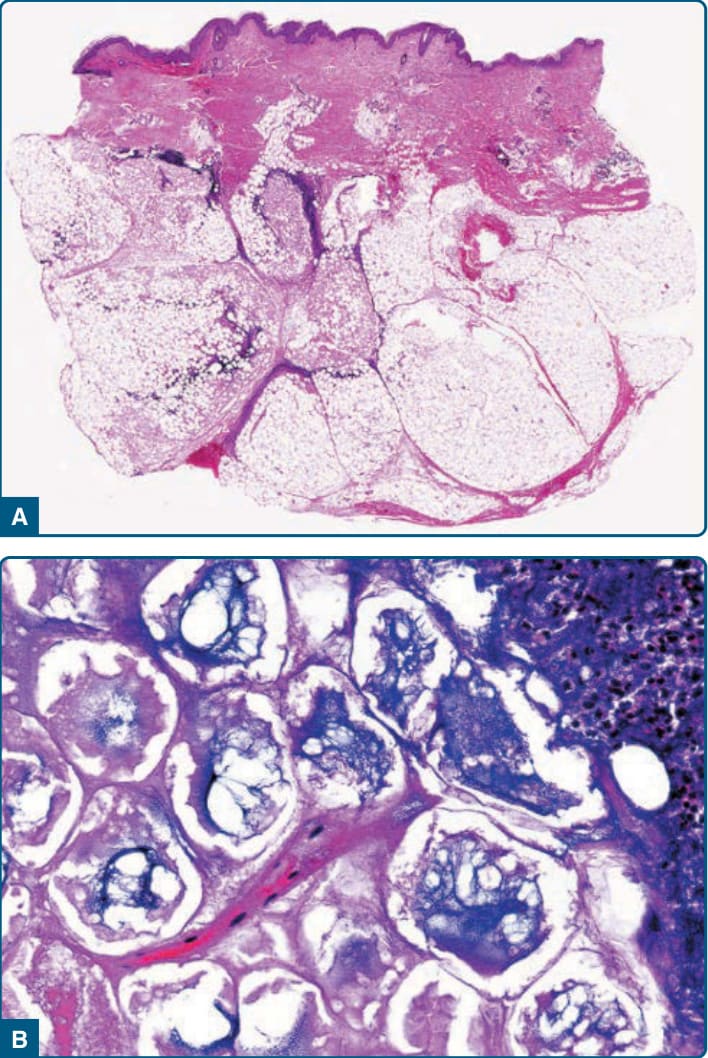
圖 73-12:胰臟性脂膜炎。A,低倍放大顯示多為小葉型脂膜炎,伴脂肪小葉中心的脂肪細胞壞死。B,高倍放大顯示幽靈脂肪細胞 (ghost adipocytes),即無細胞核、胞質內因鈣化而有細緻顆粒狀嗜鹼性物質的壞死脂肪細胞。
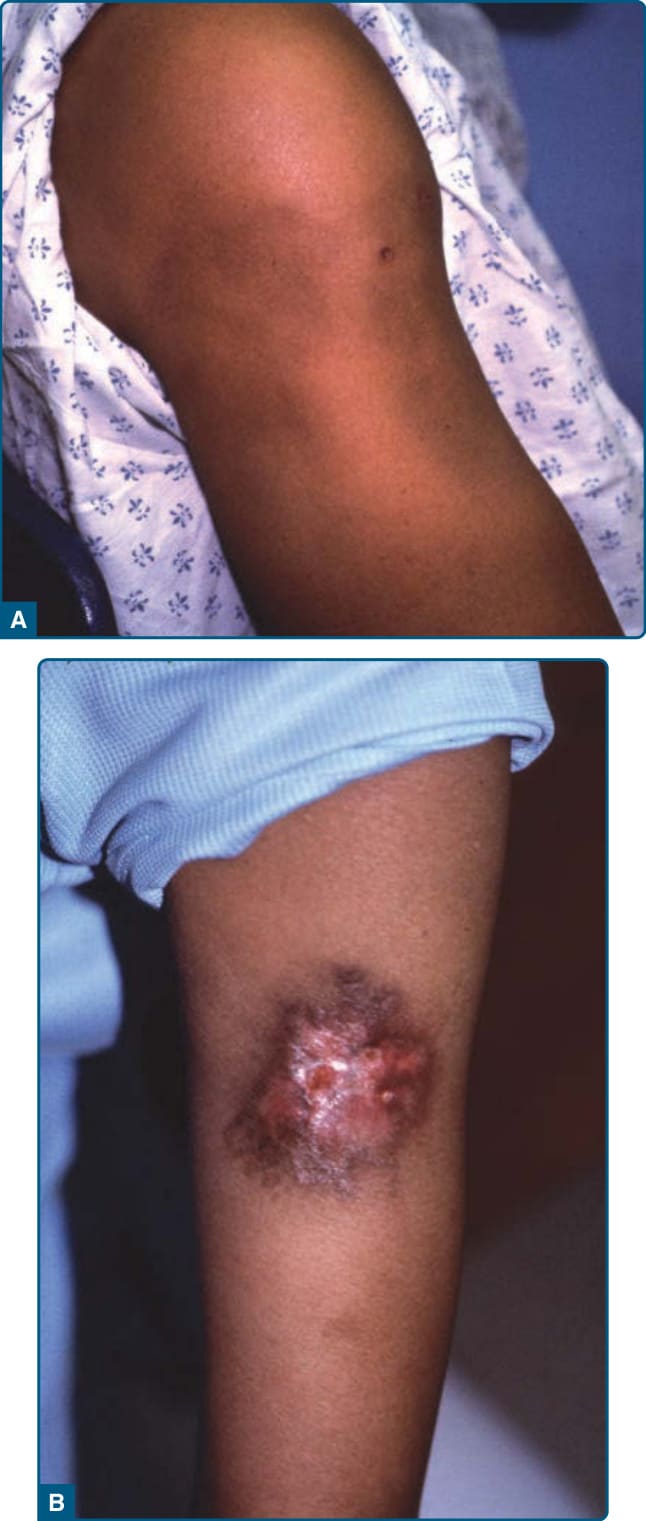
圖 73-13:狼瘡性脂膜炎 (Lupus panniculitis)。A,上臂萎縮性病灶伴淺層色素沉著。B,伴疊加的盤狀紅斑性狼瘡與潰瘍。
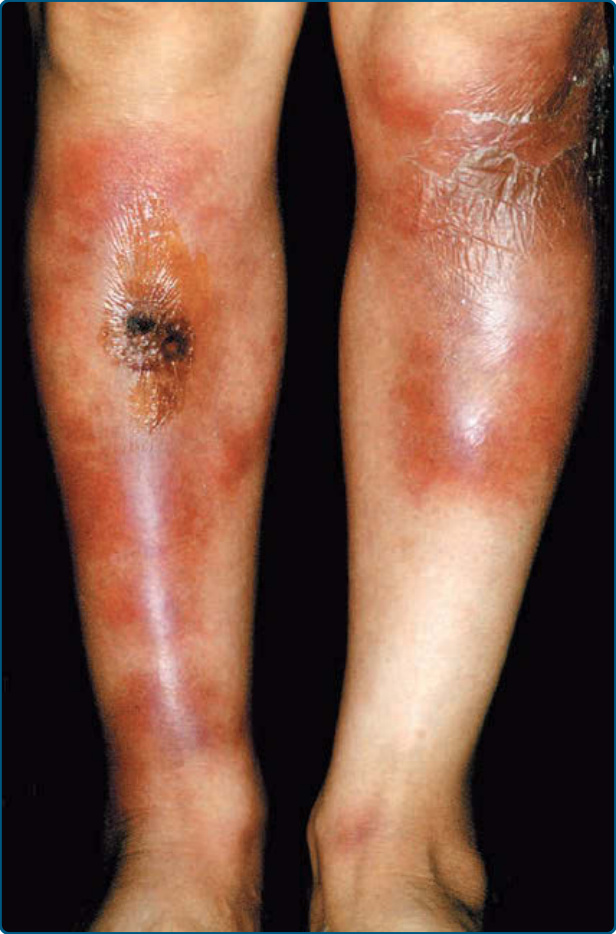
圖 73-14:吞噬細胞性組織球性脂膜炎 (Cytophagic histiocytic panniculitis)。下肢多個紅斑斑塊與結節。
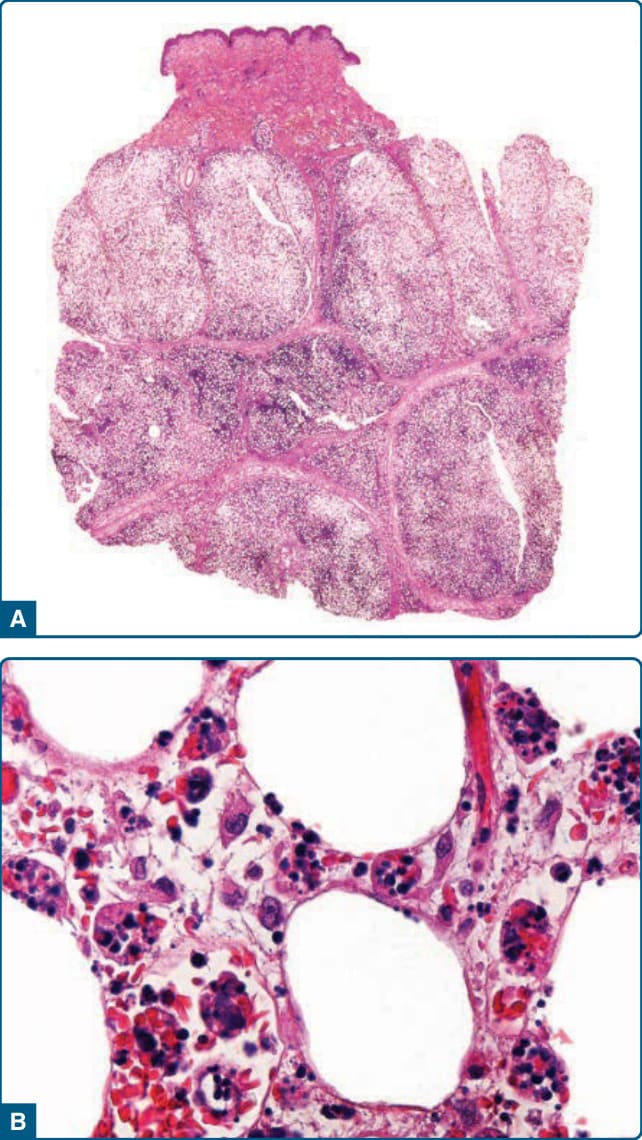
圖 73-15:吞噬細胞性組織球性脂膜炎。A,低倍放大組織學影像顯示小葉型脂膜炎。B,脂肪組織中的細胞吞噬性「豆袋 (bean bag)」細胞。
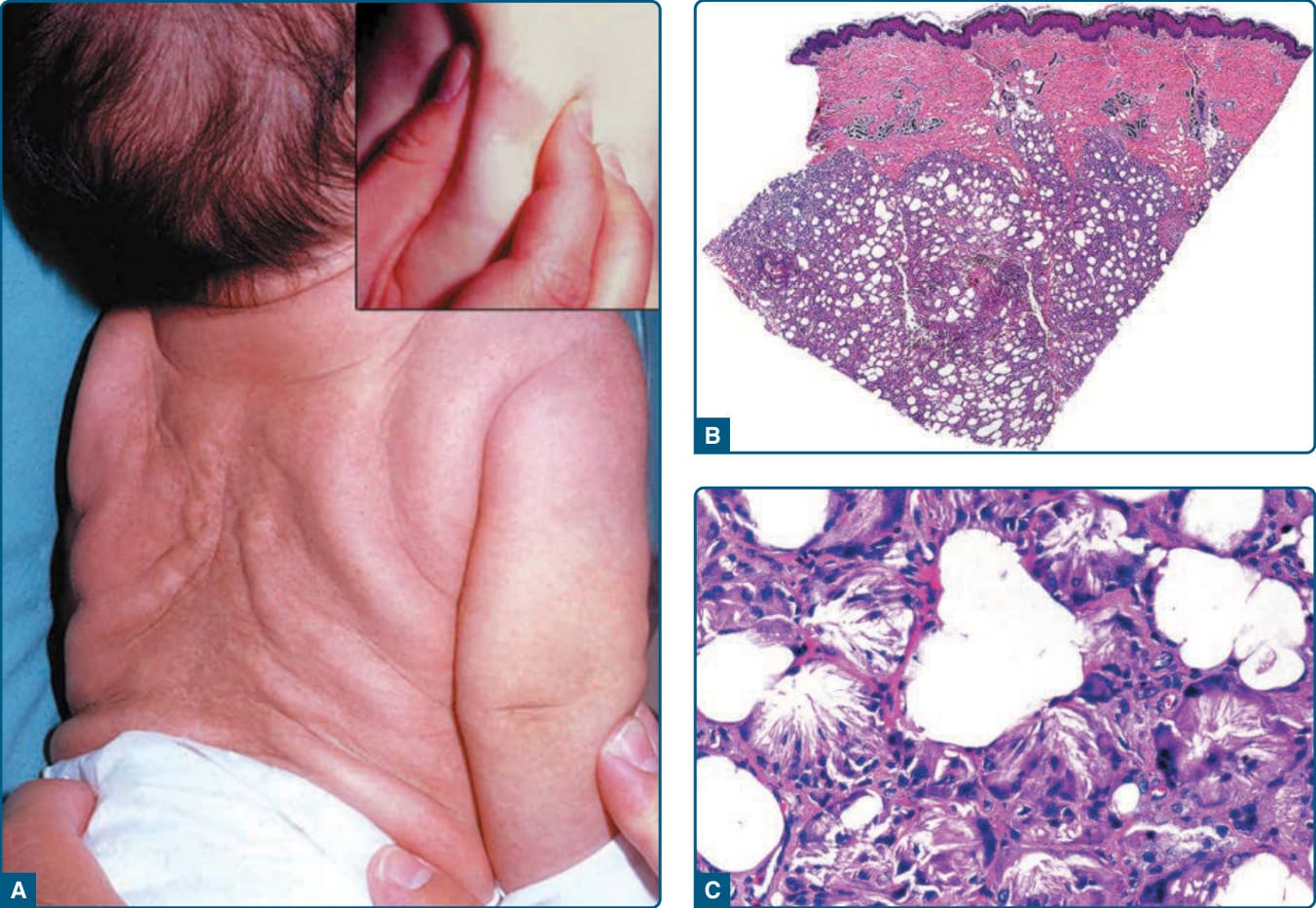
圖 73-16:新生兒皮下脂肪壞死 (Subcutaneous fat necrosis of the newborn)。A,背部界線清楚、硬結的皮下結節。B,低倍放大組織學顯示多為小葉型脂膜炎。C,高倍放大顯示脂肪細胞、組織球與多核巨細胞的針狀裂隙 (needle-shaped clefts)。
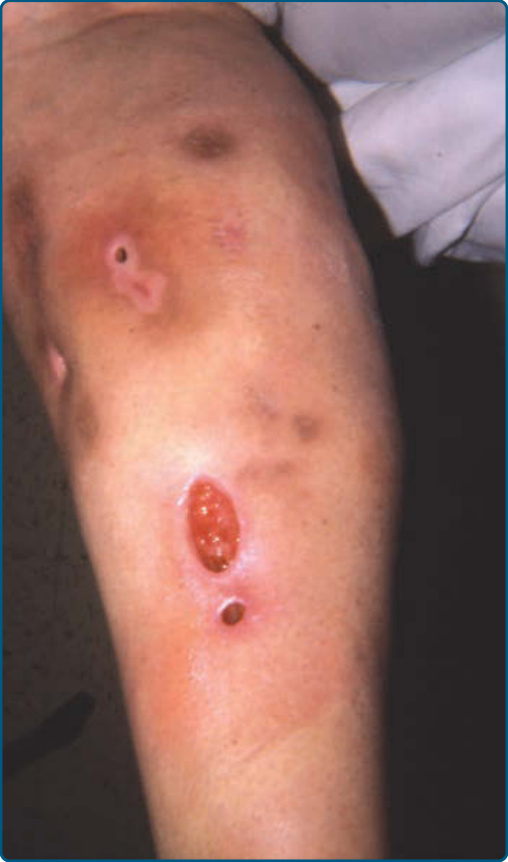
圖 73-17:人為性脂膜炎 (Factitial panniculitis)。腿部注射與外傷部位自我誘發的圓形與成角潰瘍。