Panniculitis
PART12
Subcutaneous Tissue Disorders
Inflammation in subcutaneous fat (panniculitis) often poses a diagnostic problem as the clinical and histopathologic findings in the various inflammatory disorders of adipose tissue (AT) have overlapping features (Table 73-1). A useful histopathologic classification divides the panniculitides into “septal” and “lobular” (Table 73-2), although several diagnoses exhibit overlapping features.1 This classification is expanded by noting the presence or absence of vasculitis,2 and the composition of the inflammatory infiltrate (Table 73-1). AT functions in energy storage and expenditure, appetite modulation, insulin sensitivity, endocrine and reproductive systems, bone metabolism, inflammation, and immunity.3 Its origin is traced back to the invertebrate fat body, which has innate immune system, metabolic, and lipid, glycogen and protein storage functions. In the evolution of vertebrates these functions were divided up between the liver and AT with both retaining innate immune system functions.4,5
The human adipocyte is equipped to protect the organism from pathogenic microbes by recognition of pathogen-associated molecular patterns via receptors called pattern recognition receptors (PRRs).6 Toll-like receptors (TLRs) are a type of transmembrane PRR, expressed either on the plasma membrane where they detect cell-surface microbial patterns such as lipopolysaccharides of Gram-negative bacteria, or in endosomal/lysosomal organelles, where they recognize mainly microbial nucleic acids.4,7 Secreted PRRs bind to the surface of microbes, activate the complement system, and opsonize microbes for phagocytosis.7 Once activated, the PRRs activate proinflammatory
signaling pathways, especially activation of transcription factors nuclear factor κB, interferon regulatory factor 3, or nuclear factor of activated T cells, that promote expression of genes involved in the immune response.7,8 TLRs also trigger activation of adaptive immunity responses that include antibody responses, T-helper type 1, T-helper type 17 CD4+ T cell, CD8+ T-cell responses, and T-helper type 2 (via TLR4) and immunoglobulin E response.7
Adipocytes are the most abundant cells in white AT, but other cell types include pericytes, fibroblasts, endothelial cells, vascular smooth muscle cells and inflammatory cells, especially macrophages.3,9
Macrophage-derived cytokines and chemokines, including tumor necrosis factor (TNF)-α, induce adipocyte lipolysis, which leads to release of free fatty acids from the adipocyte, which induces proinflammatory signaling.10 This paracrine loop involving inflammatory cytokines and free fatty acids propagates inflammation.11 AT also contains lymphocytes that contribute to AT inflammation.5
Adipocytes interact with blood vessels, and bidirectional crosstalk between perivascular AT and blood vessels exists.12 Perivascular AT secretes high levels of proinflammatory cytokines such as interleukin (IL)-6, IL-8, and monocyte chemotactic protein-1 compared to antiinflammatory adiponectin, when compared to other AT depots.13 Adipocyte transmembrane PRRs and TLRs, and receptors for interaction with macrophages and lymphocytes, as well as production and secretion of multiple cytokines and adipokines reflect the role of the adipocyte in protecting the host from infectious disease and other environmental dangers.
12
Compression therapy is the major recommended treatment (30-40 mm Hg) Stanozolol: to decrease pain, erythema, induration Pentoxifylline, horse chestnut seed extract, oxerutins, flavonoid fraction Weight loss
Depends on the known organisms
Bed rest, aspirin, NSAIDs SSKI, 2-10 drops thrice daily Colchicine Corticosteroids (rarely indicated)
If MTB testing is positive, full course of multidrug therapy Treat underlying cause SSKI, NSAIDs, colchicine, antimalarials, corticosteroids, gold Bed rest Pentoxifylline, compression
Lobular, often with vasculitis (90%). Early central necrosis of adipocytes; neutrophilic infiltrate In older lesions, epithelioid histiocytes, multinucleated giant cells
Stasis changes Lobular panniculitis No vasculitis Ischemic necrosis at center Thickened, fibrotic septa, atrophy of subcutaneous fat Membranocystic changes
Septal Panniculitis No vasculitis Neutrophils (early), Meischer granulomas (late)
Acute onset; symmetric, tender, nodules & plaques Affecting anterior lower extremities Fever, fatigue, arthralgias, arthritis, headache, no ulceration, no atrophy, no scarring
Erythematous SQ nodules on lower extremities May affect calves, anterolateral leg Tenderness, ulceration, scarring Protracted course, with recurrent episodes 3-6 weeks duration
Indurated, woody plaques on lower extremities, with acute & chronic changes (most commonly, anteromedial calf area) Intense pain is the most frequent symptom Stage: acute inflammatory Chronic fibrosis Inverted champagne bottle
Overweight women older than 40 years Venous insufficiency Obesity Systemic sclerosis Pulmonary Infarction Hypertension
Lipodermatosclerosis (sclerosing panniculitis, hypodermitis sclerodermiformis, chronic panniculitis with lipomembranous changes, sclerotic atrophic cellulitis, venous stasis panniculitis)
Mostly lobular panniculitis, but with a mixed pattern Pattern dependent on whether infection was inoculation related or hematogenous Neutrophilic infiltrate
Erythematous plaques, nodules, abscess with purulent discharge (fluctuant/abscess-type lesions) Most commonly on legs and feet Upper extremities, trunk, face may be involved
Infectious panniculitis — Wide variety of bacteria, fungi, parasites, viruses Either primarily inoculated, or hematogenous Staphylococcus aureus panniculitis with juvenile diabetes Panniculitis of mycetoma, chromoblastomycosis, sporotrichosis
Corticosteroids alone, or corticosteroids with methotrexate
Hydroxychloroquine is first-line treatment Quinacrine
MILD-MODERATE: dapsone, doxycycline SEVERE: protein replacement therapy
Octreotide Plasmapheresis
Vacuolar alteration of basal cell layer, thickened basement membrane, mucin deposition, superficial and deep perivascular infiltrate Mostly lobular panniculitis, with lymphoid follicles, variable hyaline fat necrosis, sclerotic collagen bundles, lymphocytic and plasma cell infiltrate
Early necrosis of SQ fat Splaying of neutrophils between collagen bundles is characteristic Liquefactive necrosis
Mixed septal and lobular panniculitis, lymphocytic and plasma cell infiltrate, hyaline sclerosis of septal collagen; calcification, membranocystic changes in late stages
Lobular panniculitis No vasculitis Intense necrosis at center of lobule Ghost adipocytes Saponification and calcification
Pancreatic panniculitis Occurs in 2%-3% of patients with pancreatic disorders Pancreatitis, pancreatic CA Erythematous nodules with spontaneous ulceration Lower extremities, especially periarticular areas (knee, ankle) Crops of lesions; red-brown Atrophic, hyperpigmented scarring Oily abscesses
Painful erythematous nodules and plaques Cellulitis, fluctuant abscess type Most common site: lower trunk, also buttocks, proximal extremities May be life-threatening
UPPER ARMS (lateral), shoulders, face, scalp, hips, buttocks, breasts Rare on lower extremities Deep, tender SQ nodules; no surface changes Resolves with depressed lipoatrophic areas Chronic, with yearly/periodic flares Duration: average of 6 years
Pulmonary and hepatic disease (emphysema in COPD, cirrhosis, hepatocellular CA), highest risk ZZ phenotype Panniculitis uncommon in AAT deficiency
Systemic lupus erythematosus Sjogren’s Syndrome Rheumatoid arthritis
Lupus panniculitis Females more frequently affected than males (4:1) 30-60 years old, also in childhood May occur before or after diagnosis of lupus erythematosus or discoid lupus erythematosus
α1-Antitrypsin panniculitis Most common in 30-60 years of age groups MM: most common phenotype (normal AAT) ZZ: 10%-15% of N levels; associated with >60% of cases
12
(Continued)
Corticosteroids Cyclosporine Multidisciplinary care in a hospital setting
Lobular panniculitis No vasculitis Bean bag cells: macrophages with intact or fragmented erythrocytes, leukocytes, or platelets Necrosis
Cytophagic histiocytic panniculitis Rare; seen in adults, adolescents, and children HLH MAS SQ erythematous to violaceous plaques on extremities, trunk Fulminant cases may have: ulceration, fever, hepatosplenomegaly, hemocytophagocytosis in BM, LN, liver, CNS May be acute and intermittent, or have a rapidly fatal course
12
Psychiatric treatment; Intralesional steroids, surgical excision
Psychiatric treatment; Intralesional
Spontaneous resolution Monitor serum calcium for hypercalcemia Systemic glucocorticoids may be considered; nephrocalcinosis
steroids, surgical excision
Spontaneous resolution
Lobular panniculitis No vasculitis Suppurative granuloma involving fat lobule Refractile foreign material
Suppurative granuloma involving
Lobular panniculitis No vasculitis Needle-shaped clefts in radial array Nodules and plaques, resolving spontaneously
Lobular panniculitis No vasculitis Perivascular lymphohistiocytic infiltrate
Refractile foreign material
Lobular panniculitis
No vasculitis
fat lobule
Erythematous to violaceous, firm nodules or plaques affecting buttocks, back, shoulders, cheeks, thighs Anterior trunk spared Oily/chalky white material Late hypercalcemia (monitor for 6 months)
Cold panniculitis (Haxthausen disease) Scrotal cold panniculitis in 9–14-year-old males Exposure to cold weather, popsicles, ice packs, swimming Indurate, erythematous plaques or nodules at sites of cold exposure; affects face, thighs, scrotal fat of prepubertal boys Resolution within 3 months
cosmetic fillers, oils, human
waste)
Subcutaneous fat necrosis of the newborn Rare, first few weeks of life History of perinatal complications (meconium aspiration, hypothermia, hypoxemia, gestational diabetes)
12
SEPTAL PANNICULITIS LOBULAR PANNICULITIS
Origin of inflammation Interlobular septa Primarily the lobule or blood vessel
Appearance of lobule Lobule appears intact, individual adipocytes well defined Lobule is necrotic, lipophages common; free liquid may accumulate in pools of varying shapes
Necrosis Absent Common
Inflammatory cell Neutrophils (acute) Mononuclear cells, multinucleated giant cells (chronic) Lymphoid infiltrates Plasma cells are common
Vasculitis Absent May be present
Characteristic findings Miescher granuloma
Associated conditions Erythema nodosum Erythema induratum Lupus panniculitis Pancreatic panniculitis Lipodermatosclerosis α1-Antitrypsin deficiency
Associated conditions Erythema nodosum Erythema induratum Lupus panniculitis Pancreatic panniculitis Lipodermatosclerosis α1-Antitrypsin deficiency
ERYTHEMA NODOSUM etiology must be investigated as associated diseases have significance for the patient.
AT-A-GLANCE
Clinical
■ Symmetric, tender, erythematous, nodules, and plaques on the anterior aspects of the lower extremities.
■ Acute onset; no ulceration or scarring.
■ Fever, fatigue, arthralgias, arthritis, headache are common.
■ More common in women.
■ Lasts from 3 to 6 weeks, with new lesions appearing for up to 6 weeks.
Histopathology
■ Mostly septal panniculitis without vasculitis.
■ Thickened, fibrotic septae with inflammatory cells.
■ Neutrophils in early lesions.
■ Histiocytes and Miescher granulomas in late-stage lesions.
Treatment
■ Treatment of any associated disorder.
■ Bed rest, aspirin, nonsteroidal antiinflammatory drugs.
Erythema nodosum (EN) is the prototypic septal panniculitis. It is a common panniculitis and multiple etiologies are associated with EN, although many cases are idiopathic. The frequency of EN caused by a single agent has changed greatly over the past century, and is influenced by geography and endemic disease.14,15
Although the diagnosis is often clinical, the histopathology can be helpful, demonstrating septal inflammation and thickened and fibrotic septae. The clinical findings are usually self-limiting, but an underlying
EPIDEMIOLOGY
EPIDEMIOLOGY
EN occurs in person of all ages, and most cases affect young women in the second to fourth decades of life.15
Larger studies show more than 80% of EN patients are female, or a female-to-male ratio of 5:1.16 However, in pediatric cases there is no gender difference.17 Prevalence in England and Spain varies from 0.38% to 0.5% of patients seen in clinics.18,19
CLINICAL FEATURES
CLINICAL FEATURES
EN presents with tender, erythematous, warm nodules and plaques on the lower legs (see Fig. 73-1). The anterior lower legs and ankles are most commonly involved, but the forearms, upper legs, trunk, and even the face can be involved; with more atypical locations seen in children.17,18 The nodules may become confluent and violaceous and bruise-like, termed erythema contusiformis when hemorrhage is present.15 Ulceration and scarring are not seen.15 EN may be associated with systemic symptoms of fever, malaise, fatigue, arthralgia, arthritis, headache, and, more rarely, abdominal pain, vomiting, diarrhea, or cough.15,16 Upper respiratory tract infection precedes the onset of EN in 20% to 30% of cases.16,18,19 Laboratory abnormalities frequently indicate the etiology; that is, a positive throat culture and leukocytosis in streptococcal infections.20,21 A positive purified protein derivative suggests tuberculosis infection in endemic areas and high-risk individuals. An abnormal chest radiograph may be seen with either sarcoidosis or pulmonary infections.
1255
12
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
ETIOLOGY
EN is associated with numerous etiologies (Table 73-3). The abundance of idiopathic cases of EN (37% to 60%) reported in large reviews,17,22 reflects the difficulty of defining a specific causation of EN. Infections, medications, malignancies, autoimmune disease, and inflammatory disorders have all been documented to provoke the clinical findings of EN. Infectious causes include bacterial, viral, fungal, and protozoan, with streptococcal respiratory tract infections being the most common etiology in pediatric cases of EN.16,17 Viral etiologies are difficult to diagnose and may confound idiopathic EN.16 EN secondary to tuberculosis varies with higher rates seen in endemic areas but is rare in the United States and Europe.16
Common causative medications include antibiotics, oral contraceptives and other hormonal therapies, and nonsteroidal antiinflammatories.16,23
More recent cases have suggested an association between EN and omeprazole use,24 leukotriene inhibitors,20 and vemurafenib.25 The prevalence of oral contraceptive-induced EN has decreased since the introduction of low-dose estrogen therapy.23
Malignancies related to EN most often include leukemias or lymphomas.15 Sarcoidosis and inflammatory bowel disease are also known etiologies. Sarcoidosis-induced EN prevalence varies by geography and patient population but can be a cause of up to one-third of EN cases.26 Temporal arteritis also has been noted to cause EN.22
1256
PATHOGENESIS
The cutaneous findings of EN are considered to be a hypersensitivity reaction to the above etiologies. Other theories suggest immune complex deposition27 and neutrophilic involvement.28 Overall, the pathophysiologic mechanism is not understood. Early studies showed the presence of interferon-γ and IL-2, activation of leukocytes,29 upregulation of various adhesion molecules,30 and genetic polymorphisms in TNF-α promoter, macrophage migration inhibitory factor, or RANTES (regulated upon activation, normal T-cell expressed and secreted).31-33
AT can activate innate and adaptive immune responses to destroy pathogens.34 Excessive adipocyte production and secretion of proinflammatory adipokines and adipocytokines is associated with obesity, cardiovascular disease, hypertension, and diabetes.34
In contrast, EN is associated with a more limited coccidiomycosis infection35 and with a less severe and shorter duration of sarcoidosis,35,36 especially in those carrying the HLA-DRB1∗03-positive leukocyte antigen.37 Therefore, certain genetic mutations associated with enhanced inflammatory reactions may confer resistance to certain pathogens.35,37 Further evidence of the innate immune system’s function is the neutrophilic involvement seen in EN cytokine profiles, with high expression of TNF-α, IL-8, IL-6, monocyte chemoattractant protein-1, and granulocyte colonystimulating factor.28
DIAGNOSIS
DIAGNOSIS
All of the above etiologies present with similar histopathologic features, and the spectrum of histopathologic features in EN and other panniculitides may be variable.38 This necessitates correlation with clinical features, including location of lesions, systemic symptoms, and laboratory findings. Examination of AT requires large excisional biopsies, as the inflammatory infiltrate can be missed. Inflammation in AT is not a static process, and more than 1 biopsy may be necessary to come to a conclusive diagnosis. EN is generally a “septal” pattern with inflammation confined predominantly to the septa. Of note the “lobular” form implies inflammation predominantly involving the fat lobule itself. Early EN shows edema of the adipose septae with neutrophils and extravasated red blood cells. As EN develops, the septae widen and become fibrosed, and a mixed infiltrate that includes lymphocytes, histiocytes, neutrophils, and some eosinophils is seen.15
Fibrotic, wide septae characterize late EN lesions, often with granulomas, and fat lobules may be encroached upon and partially effaced (Fig. 73-2A). An overlying superficial and deep dermal perivascular infiltrate is frequently present in EN.39
EN has characteristic, but not sensitive or specific, groupings of histiocytes surrounding a central stellate cleft called Miescher granuloma (see
Reported Etiologies of Erythema Nodosum TABLE 73-3
Bacterial Infections
Bacterial Infections
■Streptococcal infections
■Streptococcal infections
■Tuberculosis
■Tuberculosis
■Yersinia infections
■Yersinia infections
■Salmonella infections
■Salmonella infections
■Campylobacter infections
■Campylobacter infections
■Brucellosis
■Brucellosis
■Tularemia
■Tularemia
■Atypical mycobacterial infections
■Atypical mycobacterial infections
■Chancroid
■Chancroid
■Meningococcemia
■Meningococcemia
■Corynebacterium diphtheriae infections
■Corynebacterium diphtheriae infections
■Cat-scratch disease
■Cat-scratch disease
■Propionibacterium acnes
■Propionibacterium acnes
■Shigella infections
■Shigella infections
■Gonorrhea
■Gonorrhea
■Syphilis
■Syphilis
■Leptospirosis
■Leptospirosis
■Q fever
■Q fever
■Lymphogranuloma venereum
■Lymphogranuloma venereum
■Chlamydophila psittaci infections
■Chlamydophila psittaci infections i
■Mycoplasma pneumoniae infections
■Mycoplasma pneumoniae infections
■Helicobacter pylori infection
■Helicobacter pylori infection i
Viral Infections
Viral Infections
■Infectious mononucleosis
■Infectious mononucleosis
■Hepatitis B
■Hepatitis B
■Milker nodules
■Milker nodules
■Orf (contagious ecthyma)
■Orf (contagious ecthyma)
■Herpes simplex
■Herpes simplex
■Measles
■Measles
■Cytomegalovirus infections
■Cytomegalovirus infections
Fungal Infections
Fungal Infections
■Dermatophytes
■Dermatophytes
■Blastomycosis
■Blastomycosis
■Histoplasmosis
■Histoplasmosis
■Coccidioidomycosis
■Coccidioidomycosis
■Sporotrichosis
■Sporotrichosis
■Aspergillosis
■Aspergillosis
Protozoal Infections
Protozoal Infections
■Toxoplasmosis
■Toxoplasmosis
■Ancylostomiasis
■Ancylostomiasis
■Amebiasis
■Amebiasis
■Giardiasis
■Giardiasis
■Ascariasis
■Ascariasis
Drugs
Drugs
■Sulfonamides
■Sulfonamides
■Bromides
■Bromides
■Iodides
■Iodides
■Oral contraceptives, progesterone
■Oral contraceptives, progesterone
12
■Minocycline
■Gold salts
■Penicillin
■Salicylates
■Chlorothiazides
■Phenytoin
■Aminopyrine
■Arsphenamine
■Hepatitis B vaccine
■Nitrofurantoin
■Pyritinol
■ D-Penicillamine
■Thalidomide
■Isotretinoin
■Interleukin-2
■Omeprazole
■Valproate
■Tdap (tetanus, diphtheria, and pertussis) vaccine
■Leukotriene-modifying agents
■Vemurafenib
Miscellaneous Conditions
■Sarcoidosis
■Ulcerative colitis
■Colon diverticulosis
■Crohn disease
■Behçet disease
■Reactive arthritis
■Sweet syndrome
■Pregnancy
■Takayasu arteritis
■Immunoglobulin A nephropathy
■Chronic active hepatitis
■Granulomatous mastitis
■Vogt-Koyanagi disease
■Sjögren syndrome
■Temporal arteritis
■Systemic lupus erythematosus
■Dental infection
Malignant Diseases
■Hodgkin disease
■Non-Hodgkin lymphoma, including mucosa-associated lymphoid tissue (MALT) lymphoma
■Leukemia
■Sarcoma
■Renal carcinoma
■Postradiotherapy for pelvic carcinoma
as a feature in early EN,41 and medium-vessel arteritis may (rarely) occur.38
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
1257
12
A
B
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
EN is a benign, self-limiting subcutaneous disease that resolves a few weeks after initial presentation; the course, however, varies based on the etiology. Medication-induced EN may improve after discontinuation of the drug with reappearance of clinical findings when the drug is reintroduced. New lesions of EN are uncommon after 6 weeks, with persistent and recurrent cases reported.18 After avoidance of causative factors, recurrence is more common if no etiology is identified.21 EN secondary to a malignancy, a severe systemic disease, or infection may have a more complicated course as a result of the prognosis of the primary disease. Patients with EN secondary to sarcoidosis have an improved prognosis.36 Patients with Crohn disease and EN are more likely to have colonic involvement of inflammatory bowel disease.42 While recurrence of EN is rare, it is more frequent when associated with sarcoidosis, hormonal therapy and pregnancy, and streptococcal infection.26 Most cases of EN heal well with no recurrence, but relapses occur in 6% to 34% of reported cases.20
1258
DIAGNOSIS CHARACTERISTIC FEATURES
Erythema induratum/ nodular vasculitis Most common on posterior calves with ulceration.
Simple bruising History of trauma or anticoagulation, with no history of ongoing lesions. Lack of inflammatory features such as warmth. Tender only to palpation and heal spontaneously.
Cutaneous polyarteritis nodosa Nodules in association with livedo reticularis and arterial inflammation on histology, possibly with neuropathy.
Sarcoidosis Can occur as nodules or plaques or patches on extremities. Sarcoidal granulomas in dermis or subcutis of cutaneous nodules on skin biopsy. Often with systemic sarcoidosis.
Cellulitis Larger plaques that are erythematous and lack the violaceous bruising. More tender and warm.
Lipodermatosclerosis Firm plaques nearly circumferential around lower leg.
Pancreatic panniculitis Occurs anywhere on the legs; ulceration and drainage. Serum lipase and amylase often elevated.
Behçet syndrome occurring in the fat Vasculitis occurring in the fat tissue, either lymphocytic or neutrophilic, small arterial involvement with either lobular or mixed lobular and septal pattern.
Sweet syndrome Erythematous nodules may be present on the face, neck, upper trunk, as well as extremities. Nodules may have vesicles, pustules, or bullae. Histology with intense dermal edema; diffuse, deep neutrophilic infiltrate with karyorrhexis.
Subcutaneous
Nodules or plaques occurring in endemic
Subcutaneous tuberculosis or other atypical mycobacterial infection
Nodules or plaques occurring in endemic areas. Habitation or travel to endemic areas; possible history of trauma. Characteristic features on microbiology and positive acid-fast bacilli on histopathology.
tuberculosis or other atypical mycobacterial infection
areas. Habitation or travel to endemic areas; possible history of trauma. Characteristic features on microbiology and positive acid-fast bacilli on histopathology.
MANAGEMENT
MANAGEMENT
If there is an identifiable etiologic factor, management of EN focuses on eliminating the exposure, or treating the underlying diseases. Potential infection must be sought and treated and suspected medications should be discontinued. An extensive review of all medications (including nonprescription and supplemental products), medical supplements and a thorough medical history, including symptoms several weeks prior to the EN onset, travel history, and family history of infections are critical to the workup.18
Supportive care is the mainstay of treatment after removing or treating the provoking factors: bed rest if severe, with lower-extremity elevation, and
nonsteroidal antiinflammatory agents are recommended. Supersaturated potassium iodide solution (SSKI) can be used, but thyroid disorder and pregnancy screening must be done prior to administration, and there are risks of hypothyroidism, goiter, and heart and lung toxicity.43 Dosing SSKI is via drops in water or juice, using 2-10 drops (1 drop = 0.03 mL = 30 mg) 3 times per day.43 Colchicine is effective in Behçet-related EN specifically.44 Etanercept45 and infliximab46 are options for inflammatory bowel disease–associated EN. Other potential treatment options include oxyphenbutazone47 and hydroxychloroquine,48
with limited, randomized, controlled trials. Corticosteroids are not first-line therapy as there is a risk of infectious etiology in idiopathic EN.
ERYTHEMA INDURATUM AND NODULAR VASCULITIS
AT-A-GLANCE
Clinical
■ Erythematous subcutaneous nodules and plaques of lower legs; common on calves, but also on anterolateral legs, feet, and thighs; rarely also on arms, forearms, and face.
■ Associated with venous insufficiency; more frequent in middle-aged women.
■ Often, ulceration and scarring present.
■ Chronic, relapsing course.
■ Infectious etiologies include bacterial (especially Mycobacterium tuberculosis), fungal, viral, and protozoal.
Histopathology
■ Mostly lobular or mixed lobular and septal panniculitis with vasculitis in 90%.
■ Extensive necrosis of the adipocytes in the center of the adipose lobule.
■ Variable inflammatory infiltrate: neutrophils in early lesions and epithelioid histiocytes and multinucleated giant cells in fully developed lesions.
■ Vasculitis of the small veins and venules of the fat lobule.
Treatment
■ With positive Mycobacterium tuberculosis studies: a full course of antituberculosis triple-agent therapy.
■ Complete treatment of infectious etiologies.
■ In other cases: potassium iodide, other antiinflammatory drugs, supporting bandages and hose, leg elevation, bed rest.
12
Erythema induratum (EI) and nodular vasculitis (NV) also present as nodular lesions on the legs. They were first described as early as 1945, with a relationship with Mycobacterium tuberculosis (MTB).49 The clinical presentation and histopathology differentiate EI/NV from EN, cutaneous polyarteritis nodosa, or other nodules seen on the legs. EI most commonly presents with ulcerated nodules on the calves, and is associated with MTB infection. A similar disorder, appearing in calves and other lower-extremity sites was subsequently described without MTB association and was called NV. However, clinically the 2 nodular leg syndromes are so similar that it is impossible to separate them.50-52 Consequently, the terms are most often used interchangeably.
EPIDEMIOLOGY
EPIDEMIOLOGY
The EI/NV grouping is the most common diagnosis of a panniculitis with vasculitis.53 EI/NV is seen most commonly in young to middle-aged women,51,54 as dominant as a 9:1 ratio.55 Ages range from 8 months to 66 years with a mean age of 36.6 years.56,57 In a review of 165 patients with cutaneous tuberculosis (TB), only 2 presented with EI.58 This varies by time of study and location, as a study in Hong Kong showed 79.5% of cutaneous TB cases to be EI.59 Pediatric cutaneous TB accounts for 1% to 2% of cases. The highest rates of pediatric cutaneous TB are seen in Pakistan.60 EI lesions develop more frequently during winter, and are associated with obesity and venous insufficiency.51
CLINICAL FEATURES
CLINICAL FEATURES
EI/NV is known for affecting the calves as recurrent, erythematous to violaceous nodules and deep plaques that may be tender51 (Fig. 73-3) but often are not painful.50 Ulceration often leads to scarring.51 Surface changes include crusting of the ulcers and a surrounding collarette of scale (Fig. 73-4).50 Although the posterior calf is the most frequent location, lesions may also appear in the anterolateral areas of the feet,61 thighs, and, rarely, the arms and face.50 A consistent systemic finding in patients with MTB-associated EI is a positive tuberculoid skin test, including the Mantoux test or tuberculinpurified protein derivative. Systemic findings related to the etiology, TB or otherwise, vary greatly.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Although EI was frequently associated with MTB, the etiology was controversial because organisms are not always identified in the biopsies and tissue cultures. With the advent of polymerase chain reaction (PCR) techniques, multiple culture-negative cases were shown to contain MTB DNA.62
1259
12
In vitro studies show that MTB can bind to scavenger receptors, enter adipocytes, and, via the nearly 20 lipases of MTB, can appropriate the host lipids, accumulate intracytoplasmic lipid inclusions, and survive in a nonreplicating state that is insensitive to the major antimycobacterial medication isoniazid.63 However, rifampin and ethambutol can reduce MTB bacterial load in adipocytes by 80% to 90%.63 Other organisms that can infect adipocytes and serve as a reservoir of reactivation include cytomegalovirus, Chlamydophila pneumoniae, adenovirus, influenza virus, respiratory syncytial virus, Rickettsia prowazekii, and Trypanosoma cruzi.64-67 Infection of adipocytes leads to activation of cytokines and adipokines that bring other innate immune cells (macrophages, neutrophils, mast cells, natural killer cells) as well as adaptive immune cells (sensitized T cells) to the infection site to help contain the infection.57
EI is considered to be a hypersensitivity disorder mediated by immune complexes or cell-mediated hypersensitivity, manifested by the presence of tuberculin skin tests and highly positive interferon-γ release assay tests to MTB.68,69 The T cells, monocytes, and macrophages as well as Langerhans cells may suggest a type IV hypersensitivity reaction.70
Extracutaneous MTB is found in many EI patients, and may be present in the lung, lymph nodes, kidney, bowel,51 or adrenal glands, presenting as Addison disease.55 Rare species, including Mycobacterium monacense, can also cause the EI/NV clinical and histopathologic presentation.71 Other infections and disorders also have been associated with NV, including hepatitis B, hepatitis C (red finger syndrome and panniculitis), ulcerative colitis, leukemia, rheumatoid arthritis, hypothyroidism, and Nocardia infection.51,72-74
Crohn disease is also a rare cause of NV, and when present it may be metastatic disese.75 Chlamydophila pneumoniae has been reported to induce NV.76 Medications,
1260
A
B
including propylthiouracil77 and etanercept,78 also are associated with NV. Bacillus Calmette-Guérin vaccination is reported to cause EI, 2 and 3 months after injection.56,57
DIAGNOSIS
DIAGNOSIS
Histopathologic findings correlate with lesion duration, but the common denominator is a mostly lobular or mixed septal and lobular panniculitis (Fig. 73-5A). In early lesions, fat lobules contain discrete aggregates of inflammatory cells, with neutrophils predominating
A
B
without leukocytoclasia.53 Adipocyte necrosis is present, which leads to foamy histiocytes. In established lesions of EI/NV, collections of epithelioid histiocytes, multinucleated giant cells, and lymphocytes produce a granulomatous appearance.52,53 Intense vascular damage, when present, is accompanied by extensive areas of caseous necrosis (Fig. 73-5B), eventuating in tuberculoid granuloma formation.52 The caseous necrosis may involve the overlying dermis to such an extent that ulceration occurs.52 Eosinophils may be present and do not conflict the diagnosis.79
In a review of 101 cases consistent with EI, vasculitis (of lobular venules, septal veins, and septal arteries) was present in 90% of cases.51 Lack of vasculitis has been seen on the histopathology in severely immunocompromised patients.80 Importantly, MTB can cause both EN and EI, and thus in a patient with appropriate travel and exposures, clinical and pathologic characteristics may not rule out a diagnosis of TB.81
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
12
DIAGNOSIS CHARACTERISTIC FEATURES
Erythema nodosum Most common on anterior legs with no ulceration and no scarring.
Cutaneous polyarteritis nodosa Nodules in association with livedo reticularis and arterial inflammation on histology, possibly with neuropathy.
Sarcoidosis Can occur as nodules or plaques or patches on extremities. Sarcoidal granulomas in dermis or subcutis of cutaneous nodules on ski biopsy. Often occurs with systemic sarcoidosis.
Lipodermatosclerosis Firm plaques that can become nearly circumferential around lower leg. In morbid obesity, can occur on breasts and abdomen.
Pancreatic panniculitis Occurs anywhere on the legs, with ulceration and drainage. Serum lipase and amylase are often elevated.
Behçet syndrome occurring in the fat Vasculitis occurring in the fat tissue, either lymphocytic or neutrophilic; small arterial involvement with either lobular or mixed lobular and septal pattern.
Subcutaneous Sweet
Erythematous nodules may be present
Subcutaneous Sweet syndrome Erythematous nodules may be present on the face, neck, and upper trunk, as well as extremities. Nodules may have vesicles, pustules, or bullae. Histology with intense dermal edema; diffuse, deep neutrophilic infiltrate with karyorrhexis.
syndrome
on the face, neck, and upper trunk, as well as extremities. Nodules may have vesicles, pustules, or bullae. Histology with intense dermal edema; diffuse, deep neutrophilic infiltrate with karyorrhexis.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
EI/NV can have a protracted course with recurrent episodes over months50 to years.51,52 Patients with EI/NV are generally healthy, except for the associated disease. The course of EI is also often more chronic than EN, and the ulceration and scarring of EI leads to a worse cosmetic and potentially debilitating outcome than EN.70
MANAGEMENT
MANAGEMENT
In patients with positive MTB cultures, positive tuberculoid skin test or positive interferon-γ release assay such as the QuantiFERON-TB Gold in-tube assay, treatment with multiple agent anti-TB therapy is indicated.70 If an interferon-γ release assay is negative but clinical suspicion in a high-risk TB area persists, lesional PCR is recommended.82 Patients with hepatitis B or hepatitis C should receive appropriate antiviral intervention. Other infectious etiologies should be sought and treated if present. Potentially causative medications should be discontinued.
1261
12
Antiinflammatory treatments that have been used in NV not associated with MTB include SSKI, nonsteroidal antiinflammatory agents, corticosteroids, and gold, as well as bed rest with leg elevation, and treatment of venous insufficiency with compression and pentoxifylline, and even mycophenolate mofetil.54,83,84
If immunosuppressive agents are used, monitoring for possible infectious etiology is recommended.
LIPODERMATOSCLEROSIS
AT-A-GLANCE
Clinical
■ Indurated, firm plaques of wood-like consistency on the lower legs.
■ Associations: chronic venous insufficiency, elevated body mass index, female gender, arterial hypertension, arterial ischemia, and thrombophlebitis.
Histopathology
■ Background of stasis changes; mostly lobular panniculitis without vasculitis.
■ Ischemic necrosis at the center of fat lobule.
■ Thickened and fibrotic septae and atrophy of the subcutaneous fat, with marked fibrosis and sclerosis in late-stage severe cases.
Treatment
■ Compression stockings, ultrasound therapy, pentoxifylline.
■ Successful response to anabolic steroids, plateletrich plasma, in cases.
Lipodermatosclerosis (LDS) has multiple synonyms, including sclerosing panniculitis, hypodermitis sclerodermiformis, chronic panniculitis with lipomembranous changes, sclerotic atrophic cellulitis, and venous stasis panniculitis. It is a form of sclerosing panniculitis involving the lower legs, often related to vascular malfunction. The first name used was hypodermitis sclerodermiformis, which was used as early as the 1950s.85
EPIDEMIOLOGY
EPIDEMIOLOGY
LDS is the most common form of panniculitis, seen by clinicians far more frequently than EN. LDS occurs with venous insufficiency, mostly in overweight women older than 40 years,86,87 with age ranges varying from 31 to 74 years and older.88 The female-to-male ratio is 4:1,88 and in some studies it is as high as 12:1.89
White race has been associated more often with LDS.89
Obesity is common, with 85% of affected patients having a body mass index greater than 30.90 Comorbidities include hypertension, thyroid disease, diabetes
1262
mellitus, prior history of lower-extremity cellulitis, and deep vein thrombosis.90 Obstructive sleep apnea and arthritis (both osteoarthritic and rheumatoid) also are associated with LDS.88
CLINICAL FEATURES
CLINICAL FEATURES
LDS has an acute inflammatory stage and a chronic fibrotic stage. In patients presenting with the acute form, very painful, poorly demarcated, cellulitis-like erythematous plaques persist and evolve to violaceous, edematous, or indurated plaques or nodules, which are seen on the lower legs, most commonly on the lower anteromedial calf (Fig. 73-6).87,91 Unilateral
A
B
involvement is seen in 55%, a localized plaque in 51%, and ulceration in 13% of cases.90 Lesions can be very painful, leading to misdiagnoses of EN, cellulitis, or thrombophlebitis.87,91 Although patients in this acute phase may lack obvious venous disease,87 vascular studies show venous insufficiency in the majority.91 In those patients with normal venous studies, most have a high body mass index.90
The chronic form of LDS does not always follow an obvious acute phase.87 Chronic LDS presents as indurated92 to sclerotic, depressed and hyperpigmented skin. These findings occur on the lower portion of the lower leg, predominantly on the medial aspect, or in a stocking distribution. This is described as an “inverted champagne bottle” or a “bowling pin” appearance (Fig. 73-7).86,87,91
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
A few medications are reported to induce LDS, including gemcitabine93 and pemetrexed.94 Additional pathogenic features may include elevated hydrostatic pressureinduced vascular permeability secondary to downregulation of tight junctions95 with extravascular diffusion of fibrin90; microthrombi96; abnormalities in protein S and protein C97; hypoxia98; damage to endothelial cells by inflammatory cells99; upregulation of intercellular adhesion molecules and platelet-derived and endothelialderived factors100; and inflammation with wound healing and local collagen stimulation leading to fibrosis and further vascular and lymphatic damage.90 The fibrosis is accompanied by increased transforming growth factor-β1 gene and protein expression,101 as well as an increase in procollagen type 1 gene expression.102
Hypoxia in AT induces chronic inflammation with macrophage infiltration and inflammatory cytokine expression.103 The adipocyte produces multiple matrix metalloproteinases as well as tissue inhibitors of metalloproteinases,104 which may contribute to tissue remodeling seen in LDS. Studies have linked expansion of AT (as in obesity) to hypoxia, causing an increase in hypoxia-inducible factor 1α expression.105 This stimulates extracellular factors, including collagen I and collagen III, leading to fibrosis.106
12
DIAGNOSIS
DIAGNOSIS
The diagnosis is often clinical, and biopsy of LDS skin can have a difficult course of healing.92 Of note, concern for poor healing and delay of biopsy would have failed to identify 2 cases of malignancy in case reports of cutaneous T-cell lymphoma107 and angiosarcoma.108 Biopsies are best taken from the proximal edge of involvement for highest rates of healing. Less-invasive options, such as MRI, have been used to view the extent of disease.109
Histopathologic findings reflect the evolution of disease. Dermal stasis changes are present at any stage, including a proliferation of capillaries and venules, small thick-walled blood vessels, extravasated erythrocytes, hemosiderin-laden macrophages, lymphohistiocytic inflammation, and fibrosis.52,110 In the subcutis, early lesions of LDS show a sparse infiltrate of lymphocytes in the septa, with central lobular ischemic fat necrosis. Capillary congestion is observed within fat lobules, often with endothelial cell necrosis, thrombosis, red cell extravasation, and hemosiderin deposition.52,110
Lipomembranous or membranocystic changes may be present with small pseudocystic spaces within necrotic fat. The spaces are lined by a membranous lining of hyaline eosinophilic material, which is highlighted by periodic acid-Schiff staining. Lipomembranous changes are not exclusive to LDS.52,111
With progression of LDS, the spectrum of histopathologic changes encompasses increasing degrees of fat necrosis, septal fibrosis, and thickening; an inflammatory infiltrate of lymphocytes, histiocytes, and foamy macrophages; and partial to extensive atrophy of fat lobules.52,87,110 Advanced lesions show septal sclerosis with marked atrophy of fat lobules secondary to lipophagic fat necrosis, accompanied by lipomembranous change and a marked reduction in inflammation.52,110
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
The course of LDS is variable and depends on the patient’s comorbidities and compliance with the treatment regimen, particularly compression, which can be difficult to adhere to daily. The acute form may last months or even a year.91 The chronic form can last years and can be refractory to standard treatment regimens. The ulcerations and resulting scarring are permanent and can be cosmetically distressful. Secondary infection of ulcers must be avoided. Dermatosclerosis in patients with systemic sclerosis has been associated with pulmonary infarction and hypertension secondary to leg thrombi.112 Additionally, untreated LDS can result in venolymphedema.92
1263
12
DIAGNOSIS CHARACTERISTICS
Erythema nodosum (EN) Warm, tender, erythematous subcutaneous nodules on pretibial areas; a lack of preceding venous insufficiency is common. Sarcoidosis or other high-risk medication exposure.
Erythema induratum (EI)/ nodular vasculitis
Tender nodules and plaques on calf areas; living in an area endemic for tuberculosis (EI). Vasculitis on histopathology.
Cellulitis Can occur with chronic edema, venous insufficiency. More likely unilateral and after injury or animal bite.
Infection-induced panniculitis EN-like nodules, deep inflamed nodules or abscesses; older lesions may show fibrosis. Commonly on legs. Often seen in severely immunosuppressed patients with malignancies.
Scleroderma/ morphea Scleroderma or morphea involvement elsewhere. May have associated venous insufficiency of legs.
Deep vein thrombosis Pain and edema of posterior calf with positive Doppler examination.
Nephrogenic systemic fibrosis Subsequent to MRI with gadolinium exposure in patients with renal dysfunction. Can occur on areas other than legs.
Malignancies Persistent, worsening disease, often with systemic symptoms. Presence of cutaneous T-cell lymphoma, angiosarcoma, or other sarcomas on leg. More likely in patients who underwent transplantation and in immunosuppressed patients.
Necrobiosis lipoidica Clinically, the lesions consist of yellowbrown, indurated plaques with an atrophic, depressed area.
Eosinophilic
Coarse, thickened skin with peau d’orange
Eosinophilic fasciitis/Shulman syndrome
Coarse, thickened skin with peau d’orange appearance. A “groove sign” or “dry riverbed sign” can be seen along lines of vessels when arms are lifted or extended laterally. Peripheral blood eosinophilia and hyperglobulinemia are common.
fasciitis/Shulman syndrome
appearance. A “groove sign” or “dry riverbed sign” can be seen along lines of vessels when arms are lifted or extended laterally. Peripheral blood eosinophilia and hyperglobulinemia are common.
MANAGEMENT
MANAGEMENT
Diagnostic tests to evaluate peripheral vascular disease include the ankle brachial index for arterial evaluation, as well as venous Doppler examinations to detect thrombi and duplex sonography to detect direction of flow and severity of venous reflux.91 A patient with arterial compromise should not undergo compression therapy, which is a first-line treatment for LDS.87,90 Higher compression gradient (30 to 40 mm Hg) may be more effective, but lower pressure (15 to 20 mm Hg or 20 to 30 mm Hg) encourages compliance, especially in the elderly, and effectively decreases edema.113 Compression tightens vascular tight junctions, inhibiting permeability of fluid into the perivascular tissue.95,114 Stockings must be worn all day; a few days without compression
1264
may lead to recurrence of edema and inflammation.113 Because of the difficulty of wearing stockings, adaptive compression modalities have been developed, which use sustained or intermittent pneumatic compression. Stanozolol is effective in LDS; it decreases pain, erythema, and induration.115 Patients tolerate the treatment well, and the risk of hepatotoxicity has been evaluated as an asymptomatic increase in liver transaminases that resolves with medication cessation.116 This drug is not available in the United States. Other anabolic steroids, such as oxandrolone and danazol, also have been used.117,118
Pentoxifylline has been successfully used in LDS cases with and without associated ulceration and is a useful adjunct to compression for treating venous ulcers and may be effective as monotherapy.119 In fact, in one study, hydroxychloroquine and pentoxifylline combined therapy, without compression, led to a 50% reduction in pain from baseline.120
Ultrasound therapy can reduce and even resolve induration, tenderness, and erythema.121,122 Available through physical therapy departments, it is a simple and safe treatment and may be used as adjunctive therapy.121,122 Because obesity is a common condition among affected patients, weight loss is critical. Capsaicin may alleviate pain associated with LDS.123 Finally, refractory disease has responded to intralesional, autologous, platelet-rich plasma, injected in a grid-like pattern, and repeated every 2 weeks.124
One author (IKA) has noted improvement in patients treated with rutin and vitamin C, particularly adjunctive to compression therapy.
INFECTION-INDUCED PANNICULITIS
AT-A-GLANCE
Clinical
■ Caused by multiple infectious agents: bacteria, fungi, parasites, and viruses.
■ AT may serve as reservoir for various infections.
■ Patients may be immunosuppressed with primary inoculation or hematogenous spread.
■ Erythematous plaques, nodules, abscesses, ulcers with purulent discharge.
Histopathology
■ Suppurative granulomas within fat lobule.
■ In primary cutaneous infections, epicenter of inflammation is superficial dermis; in secondary infections, epicenter is deep reticular dermis and subcutaneous fat.
■ Special stains, cultures, and serologic studies for detection of microorganisms.
Treatment
■ Appropriate antimicrobial therapy selected according to susceptibility tests.
Infection-induced panniculitis (IIP), also termed infectious panniculitis and infective panniculitis, is panniculitis directly caused by an infectious agent.74 AT infection can be caused by bacteria, mycobacteria, fungi, protozoa, or viruses.52,74,125 Primary infections produced by direct inoculation at a wound site (injury, surgical procedure, catheter, injection, acupuncture) usually result in a single lesion which may spread locally.52,74,125 Secondary infections caused by sepsis and hematogenous spread may manifest as single or multiple lesions.52,74,125 In immunosuppressed patients, microorganisms may be numerous and identified on routine histopathology, or with special stains. In immunocompetent patients, microorganisms may be sparse, requiring positive cultures, lesional PCR, or serologic studies for identification.52,74,125
EPIDEMIOLOGY
EPIDEMIOLOGY
IIP is most frequently seen in immunocompromised hosts, including those with diabetes mellitus.126 The epidemiology depends on the infectious etiology with geographic and host susceptibility. Recent reports of infectious etiologies in association with various autoimmune disorders include Staphylococcus aureus panniculitis with juvenile dermatomyositis,127 mucormycosis in systemic lupus erythematosus (SLE),126 Mycobacterium-associated and Histoplasma-associated panniculitis with rheumatoid arthritis,128,129 as well as Histoplasma causing panniculitis in 2 patients with polymyositis.130 Complicating these associations is the fact that autoimmune disorders are often treated with immunosuppressive therapies.
CLINICAL FEATURES
CLINICAL FEATURES
The clinical appearance of IIP varies from fluctuant or abscess-like lesions with purulence and ulcerations to nonspecific erythematous, firm subcutaneous plaques, purpuric plaques, and EN-like lesions.74,125,131 Deep nodules or plaques may not always appear fluctuant, and pustules, fluctuant papules, and ulcers can be superimposed on top of firm, deep nodules.125 The most common sites of infection are the legs and feet, but upper extremities, trunk, and face also may be involved.74,125 Immunosuppression of varying etiology is the most frequent association.52,74,125 Immunosuppression is associated with more widespread and abscess-type lesions. In immunocompetent patients, granulomas are common.125,132 Clinical features vary with the setting, the infective organism and patient’s immunocompetence.125,131
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Innumerable infectious agents can cause IIP via direct inoculation or hematologic spread. Diffuse fusariosis has presented with panniculitis in a patient with
12
acute lymphoblastic leukemia.133 One case reported cutaneous aspergillosis panniculitis in a patient with acute lymphoblastic leukemia, and gouty panniculitis was suspected with the presence of urate crystals. This patient also had the characteristic “ghost cell” changes of pancreatic panniculitis, which along with the urate crystals may be secondary to fungal enzymatic changes.134 These findings have been appreciated in mucormycoses as well.126 Transplantation patients are at risk for disseminated infections, as seen in a review of 15 solid-organ transplantation patients (mainly renal transplantations) with cryptococcal panniculitis. Duration between transplantation and infection ranged from 3 months to 31 years.135 Cryptococcal infection in the transplantation group is highest in heart transplant recipients.136 Pseudomonas aeruginosa may cause panniculitis, most often as a disseminated infection, but it has presented without septicemia.137
Epstein-Barr virus also has been reported to cause IIP localized to the trunk in a patient with methotrexateassociated lymphoproliferative disorder, with resolution after discontinuation of methotrexate.138
Immunocompetent patients with IIP also have been reported. Panniculitis caused by syphilis has presented with cutaneous findings of secondary syphilis, with an infiltrate containing many plasma cells and positive Treponema pallidum immunohistochemical staining.139 Cutaneous leishmaniasis has also presented as inflammation of the AT, noted in 2 Iraqi cases with both septal and lobular histopathology, improving with local treatment standard for cutaneous leishmaniasis.140 Borrelia burgdorferi is also associated with panniculitis, in an immunocompetent host, mimicking a subcutaneous panniculitis-like T-cell lymphoma (SPTCL), which responded promptly to doxycycline.141
The adipocyte is an innate immune cell that tailors specific and distinct receptor-mediated transcriptional responses to the various microbial infections.142 Adipocytes can be infected in vitro with infectious agents, including Chlamydophila pneumoniae, cytomegalovirus, adenovirus, respiratory syncytial virus, influenza,64
MTB,63 R. prowazekii,65 T. cruzi,143 Coxiella burnetii,65 and HIV.144
In vitro infection of adipocytes with T. cruzi (the cause of Chagas disease) results in increased expression of multiple proinflammatory cytokines and chemokines, increased expression of TLR2 and TLR9 and acute-phase reactants, but decreased expression of adiponectin and peroxisome-proliferator-activated receptor γ, the negative regulators of inflammation.9,66 In mice, real-time PCR has shown that at 300 days postinfection with T. cruzi, a comparable number of parasites are present in both AT and heart tissue, indicating that AT is a reservoir for the parasites.9 Interestingly, the T. cruzi amastigotes are more numerous in brown AT than in white α1-antitrypsin.9
In another murine study, Mycobacterium leprae had very limited growth potential in AT, as preadipocytes and adipocytes were not permissive for M. leprae multiplication and logarithmic growth.145 Because adipocytes have high expression of TLR4, adipocytes are responsive to endotoxin, producing proinflammatory
1265
12
cytokines at comparable or higher levels than macrophages.9 TLR2 induction in adipocytes confers a high degree of sensitivity to fungal cell-wall components.9
Mutations coding TLRs or downstream signaling proteins can increase the risk of infections.146 The genetic variants in TLRs, and other innate immune signaling components and their effect on adipocyte function and development of panniculitis are not yet known.
DIAGNOSIS
DIAGNOSIS
Evaluation includes histopathologic studies with stains for organisms as well as tissue culture, with sensitivities. Immunosuppression may lead to the presence of numerous microorganisms but diagnosis is more difficult in immunocompetent patients.52,74,125 Molecular PCR techniques have increased detection of mycobacterium infections.62,79 Histopathologic features vary with the organism and its virulence, the host immune status, and the duration of the lesion.74 A mostly lobular panniculitis,52 IIP often reveals a mixed septal and lobular pattern.74 The superficial dermis has more inflammation in infections acquired by direct inoculation, while infections secondary to hematogenous spread involve the deep reticular dermis and subcutaneous fat.52
Additional features of IIP include hemorrhage, vascular proliferation, foci of basophilic necrosis, and sweat gland necrosis.74 Overlying epidermal changes, such as parakeratosis, acanthosis, and spongiosis are seen in nonulcerated specimens. Dermal findings include edema, a diffuse perivascular inflammatory infiltrate, proliferation of thick-walled vessels, and focal or diffuse hemorrhage.74 Observation of these features warrants stains for bacteria, mycobacteria, and fungi, and additional immunohistochemistry or PCR amplification techniques may be necessary. Occasionally histopathologic changes suggest a particular etiology. Suppurative granulomas formed by epithelioid histiocytes surrounding aggregated neutrophils are common with atypical mycobacteria.52 Viral inclusions within endothelial cells may be seen in cytomegalovirus-associated panniculitis.147
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
The clinical course and prognosis of IIP are affected by the infective organism and whether the organism was introduced locally or hematologically. Organisms treated with antibiotics, and a prompt diagnosis, portend a good prognosis. Immunocompromised patients are at risk for a more complicated course.
1266
DIAGNOSIS CHARACTERISTICS
α1-Antitrypsin panniculitis Nodules, plaques, and cellulitis-like lesions to fluctuant abscess-like lesions that tend to ulcerate and drain, and appear on lower trunk and proximal extremities.
Pancreatic panniculitis Occurs anywhere on the legs, with ulceration and drainage. Serum lipase and amylase often elevated. Fungal strains should be used to rule out mucormycosis or aspergillosis which can cause “ghost cell” adipocytes, especially in immunocompromised patients.
Traumatic/factitial panniculitis Variable nodules, plaques, ulceration. Need high index of suspicion by caring physicians. Trauma can predispose to infection-induced panniculitis.
Erythema induratum/ nodular vasculitis
Crops of tender nodules and plaques on posterior lower legs, often develop ulceration and drainage.
Pyoderma gangrenosum Can present at any location. Highly inflammatory with characteristic rolled, elevated edges. Related to trauma. Not classically confined to the lower legs.
Cutaneous/ subcutaneous lymphomas
Nodules and plaques, some with ulceration, predominantly on legs. Can be mimicked by Borrelia burgdorferi–induced panniculitis in an immunocompetent host.141
Lymphoproliferative disorders Immunocompromised or posttransplantation patient (but not restricted to such). Must be differentiated into benign and malignant behavior/characteristics both clinically and with histopathology.
Lymphoproliferative
Immunocompromised or posttransplantation
disorders
patient (but not restricted to such). Must be differentiated into benign and malignant behavior/characteristics both clinically and with histopathology.
MANAGEMENT
MANAGEMENT
Treatment varies depending on the suspected or known organisms and their cultures and sensitivities. In cases involving bacteria such as MTB and parasites such as T. cruzi, the capability to remain dormant in AT necessitates the use of appropriate antibiotics, often for extended treatment courses.9,63
`1-ANTITRYPSIN PANNICULITIS
AT-A-GLANCE
Clinical
■ ZZ-, MZ-, MS-, and SS-phenotype-associated panniculitis. Rare, with >60% occurring in ZZ cases.
■ Low levels of α1-antitrypsin are associated with emphysema, hepatitis, cirrhosis, vasculitis, and angioedema.
(Continued)
AT-A-GLANCE (Continued)
Continued
AT A GLANCE (
)
■ Subcutaneous nodules mostly located on the lower abdomen, buttocks, and proximal extremities.
■ Frequent ulceration, draining lesions, poor healing after surgery, and isomorphic phenomenon.
Histopathology
■ Mostly lobular panniculitis without vasculitis.
■ Dense inflammatory infiltrate of neutrophils and neutrophils between collagen bundles of deep reticular dermis.
■ Normal fat adjacent to necrotic adipocytes.
Treatment
■ Dapsone, doxycycline.
■ Supplemental intravenous infusion of α1- antitrypsin or liver transplantation for severe forms of homozygous ZZ disease.
α1-Antitrypsin (α1AT) is a glycoprotein made in the liver. It is a serine protease inhibitor that has widespread function as well as an acute phase reactant, increased in times of stress. α1AT deficiency is inherited as a codominant disorder, and more than 100 alleles have been identified.148,149 The α1AT phenotypes are classified according to gel electrophoresis migration as F (fast), M (medium), S (slow), and Z (very slow), and null variants that do not produce any α1AT and patients may have dysfunctional α1AT with normal levels.150 Deficiency in α1AT was first seen in the 1960s with protein electrophoresis isolation.151 The first description of panniculitis secondary to glycoprotein deficiency was in 1972.152
EPIDEMIOLOGY
EPIDEMIOLOGY
Panniculitis uncommonly occurs in α1AT deficiency, fewer than 50 cases having been reported153 in ZZ, MZ, MS, and SS phenotypes. More than 60% involve ZZ phenotypes and 65% affect women.153 Homozygous MM represents 90% to 97% of the population.154 Presenting in all ages, panniculitis is most often seen between ages 30 and 60 years.153,155 The estimated prevalence of α1AT deficiency in whites is 1 per 3000 to 5000 in the United States, with incidence in white newborns similar to that of cystic fibrosis.150 Cases also have been noted to initially present after pregnancy as acute-phase reactants (including α1AT) rise and fall.156 Up to 35% of cases have preceding trauma.154
CLINICAL FEATURES
CLINICAL FEATURES
Patients present with painful erythematous nodules and plaques, but early lesions may have a cellulitic or fluctuant abscess-type appearance (Fig. 73-8A).
12
A
B
Lesions may ulcerate with oily or serosanguinous discharge (Fig. 73-8B) and resolve with atrophic scars.52
The lesions appear most commonly on the lower trunk (buttocks) (Fig. 73-9) and proximal extremities, but the lower legs and other sites may be affected.52,153,157 α1AT panniculitis may coexist with autoimmune disease, cancer, or infection,158,159 thus the presence of α1AT deficiency in association with panniculitis should not preclude a search for infection or other underlying medical problems such as autoimmune disorders or malignancies.
1267
12
α1AT deficiency is associated with pulmonary and hepatic disease, leading to chronic obstructive pulmonary disease, hepatic cirrhosis, or hepatocellular carcinoma150; the ZZ phenotype is at highest risk. There is no association of the null variant with hepatic disease, as it is the accumulation of polymerized α1AT in the liver that induces damage, and accumulation does not occur in this variant.150
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
α1AT is a glycoprotein that is produced and secreted mainly by hepatocytes, but also in small amounts by monocytes/macrophages and neutrophils,160 and is known to inhibit numerous proteases. α1AT may also help regulate complement activation.161,162
Homozygous MM is associated with normal levels of α1AT (90 to 220 mg/dL),163 whereas those homozygous for ZZ have low levels at 10% to 15% of normal, and those heterozygous for S or Z allele have levels in between.150 ZZ is the most common phenotype to induce panniculitis. MS heterozygous individuals may have low-normal serum levels causing panniculitis clinically164,165 and the F variant may increase lung disease166 and is associated with panniculitis only as the FZ phenotype.,154,167
Possible mechanisms leading to the development of α1AT panniculitis include lack of interference with the various proteases that lead to activation of lymphocytes, macrophages, complement, activation of the autoinflammatory cascade mediated by activation of IL-1 and IL-1β,168 and lysis and destruction of connective tissue at sites of inflammation. Trauma to adipocytes may result in release of adipokines and cytokines that are chemotactic to inflammatory cells, whose released proteases are unopposed because of the absence of the α1AT, leading to severe damage in involved tissue.154,169,170 Animal models of soft-tissue injury show elevated levels of IL-6 and monocyte chemotactic protein-1, and increased systemic inflammatory mediators.169 Interestingly, a case has been
1268
reported of a patient with MM phenotype acquiring a ZZ phenotype from a donor liver; after the transplantation patient developed panniculitis.171
DIAGNOSIS
DIAGNOSIS
Histopathologic findings vary with the age and type of lesion biopsied. Early in development, nodular lesions reveal edema and degeneration of adipocytes, with ruptured and collapsed cell membranes and a perivascular mononuclear infiltrate.172 Also reported at this stage is a mild infiltrate of neutrophils and macrophages in septa and lobules, with foci of early necrosis of subcutaneous fat. This may be accompanied by splaying of neutrophils between collagen bundles throughout the overlying reticular dermis, considered an early and distinctive diagnostic clue.173 More advanced lesions have masses of neutrophils and histiocytes associated with necrosis and replacement of fat lobules (Fig. 73-10). Involvement
A
B
also may be focal, manifested by large areas of normal fat in immediate proximity to necrotic septa and fat lobules.174 Liquefactive necrosis and dissolution of dermal collagen may be accompanied by ulceration, and degeneration of elastic tissue may lead to septal destruction and the appearance of “floating” necrotic fat lobules.159,175 Neutrophils and necrotic adipocytes are less prevalent in late-stage lesions, with replacement by lymphocytes, foamy histiocytes, and variable amount of fibrosis within fat lobules.159,176
At times the histopathology fails to demonstrate the characteristic features.177 These rare cases suggest that this disease should be considered in a patient with ulcerative lesions if pathology is not suggestive of a diagnosis with serum protein levels and phenotype testing in the correct clinical setting. Because of the complex nature of these proteins, it is best to obtain serum protein levels as well as phenotyping.150
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
DIAGNOSIS CHARACTERISTICS
Infection-induced panniculitis Erythema nodosum–like nodules, deep inflamed nodules or abscesses; older lesions may show fibrosis. Commonly on legs. Often seen in severely immunosuppressed patients with malignancies.
Factitial panniculitis Geometric morphology. Patient often has an underlying psychiatric disturbance. Identifying foreign material is helpful. Histopathology varies based on mechanism.
Erythema induratum/ nodular vasculitis
Tender nodules and plaques on calf areas, living in an area endemic for tuberculosis (erythema induratum). Vasculitis on histopathology.
Pancreatic panniculitis Occurs anywhere on the legs, ulceration and drainage. Serum lipase and amylase often elevated.
Subcutaneous Sweet syndrome Erythematous nodules may be present on the face, neck, upper trunk, as well as extremities. Nodules may have vesicles, pustules or bullae. Histology with intense dermal edema; diffuse, deep neutrophilic infiltrate with karyorrhexis.
Pyoderma
Violaceous, undermined borders with isomor-
Pyoderma gangrenosum Violaceous, undermined borders with isomorphic response.
gangrenosum
phic response.
12
panniculitis, pancreatic panniculitis, medicationassociated panniculitis (vemurafenib, ponatinib); subcutaneous sweets syndrome or neutrophilic panniculitis associated with myelodysplasia. Additionally cutaneous Crohn disease, familial Mediterranean fever, rheumatoid arthritis and cases of idiopathic neutrophilic panniculitis can cause a neutrophilic panniculitis.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
Cutaneous and subcutaneous necrosis can develop rapidly. Fatal cases have been reported, mainly the ZZ variant.178,179 A rare fatal case with MZ phenotype that mimicked pyoderma gangrenosum also has been reported.180 Prompt diagnosis and treatment of severe disease with α1AT augmentation may significantly improve the panniculitis and may induce clinical remission, requiring augmentation therapy infusions only with recurrence. Although the panniculitis may lead to significant morbidity, the lung and liver disease associated with α1AT deficiency are a much higher priority and comanagement with pulmonologists and hepatologists is ideal.
MANAGEMENT
MANAGEMENT
Many medications have been used in the treatment of α1AT-deficiency panniculitis including colchicine, antimalarials, dapsone, doxycycline, plasma infusion and plasma exchange, intravenous augmentation therapy of α1AT protein, and liver transplantation.157,181,182 Steroids, immunosuppressives, and cytotoxic agents are less effective. Doxycycline, and especially dapsone, may be useful in mild to moderate disease, especially in heterozygous cases such as MZ. Severe panniculitis unresponsive to standard treatment requires protein-replacement therapy (augmentation therapy).183 For severe panniculitis, α1AT infusions are recommended at 60 mg/kg, often with a few weekly infusions, with repeat infusions for recurrence.183 Long-term supplementation may be required in severe cases.149
Panniculitis has also resolved with liver transplantation,184 and panniculitis acquired after liver transplantation has successfully been treated with retransplantation.171 It is critical to note the tendency of α1AT-deficiency panniculitis for poor wound healing, as a case of an untreated α1AT patient resulted in poor healing after intraabdominal surgery.185 Not initiating augmentation therapy in a timely fashion may cause significant complications and must be considered in the appropriate clinical setting.185 As trauma may induce lesions in one-third of patients, debridement is discouraged.52
1269
12
PANCREATIC PANNICULITIS
AT-A-GLANCE
Clinical
■ Erythematous subcutaneous nodules that often ulcerate spontaneously.
■ Lower extremities (around ankles and knees) are the most frequent sites of involvement.
■ Regression of pancreatitis-associated cutaneous lesions occurs as pancreas improves, but those associated with pancreatic carcinoma tend to persist; may be fatal.
Histopathology
■ Mostly lobular panniculitis without vasculitis.
■ Intense necrosis of adipocytes at center of fat lobule.
■ “Ghost cell” adipocytes with finely granular and basophilic intracytoplasmic material.
Treatment
■ Treatment of the underlying pancreatic disease.
■ Additional treatment options: octreotide, plasmapheresis.
■ Stop precipitating medications.
Panniculitis associated with and secondary to underlying pancreatic disease was first noted in 1883.186
Variable pathologies of the pancreas can induce panniculitis, including acute pancreatitis, even following endoscopic retrograde cholangiopancreatography procedures.187 The panniculitis resolves as the pancreatic disease improves, which may be difficult or impossible in cases of malignancy.
EPIDEMIOLOGY
EPIDEMIOLOGY
Panniculitis in association with pancreatic disease is an uncommon occurrence, developing in 0.3% to 3% of patients with pancreatic disorders such as acute or chronic pancreatitis, pancreatic carcinoma, or pancreatic pseudocysts.188-190 Pancreatic disease of any kind may be associated with pancreatic panniculitis. Although pancreatitis is most commonly a result of alcohol abuse, cholelithiasis, medications, trauma, and viral infections are also known etiologic factors.190,191 Congenital pancreatic abnormalities may also cause panniculitis.190
CLINICAL FEATURES
CLINICAL FEATURES
The cutaneous lesions appear as crops on the lower legs, especially the periarticular areas, but are also on the arms, wrists,192 thighs, and trunk.188 The lesions are ill-defined erythematous to red-brown edematous and tender nodules, which may involute and resolve with atrophic
1270
hyperpigmented scars.188 They may have central “softer” areas or may become fluctuant, abscess-like, and drain an oily material similar to lesions of α1AT-deficiency panniculitis (Fig. 73-11).188 The cutaneous panniculitis may precede the diagnosis of the associated pancreatic disease by weeks to months in up to 45% of patients.188,191
Extracutaneous manifestations include periarticular fat necrosis with concomitant arthritis,188,193 and painful medullary fat necrosis in bone.194,195 Monoarticular or oligoarticular arthritis secondary to periarticular fat necrosis may be present in more than half of patients.188 This triad of pancreatic disease, panniculitis, and polyarthritis (PPP syndrome) is very rare and is associated with both pancreatitis and pancreatic carcinoma.194,196 Osteonecrosis may also develop as enzymes extend to the joint capsules and cause purulent joints.195
Pleural effusions and serositis also may be seen with pancreatic panniculitis,194,197 and pleural effusions are associated with a high mortality rate.197 Eosinophilia may be seen in pancreatic panniculitis resulting from either pancreatitis or pancreatic malignancies.190,197 A pancreatic tumor (or pancreatitis) in association with panniculitis, polyarthritis, and eosinophilia (the Schmid triad) imparts a poor prognosis.198,199 An important complication of pancreatic panniculitis is secondary infection.200
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Pancreatic panniculitis has generally been attributed to release of pancreatic enzymes such as lipase, amylase, and trypsin into the circulation, promoting vascular permeability and damage, leading to release of fatty
A
B
12
acids from adipocytes and subsequent fat necrosis.194,201 However, there are reports of pancreatic panniculitis in the setting of normal serum levels of pancreatic enzymes.188 Additionally, incubation of normal AT with amylase, lipase and a pancreatitis patient’s serum failed to induce fat necrosis in vitro.202 Resistin and leptin are potential markers of extrapancreatic fat necrosis.203
Other than the more common causes of pancreatitis, pancreatic panniculitis may be seen in association with pancreatitis following renal or combined renal and pancreatic transplantation,204,205 liver transplantation,206 with SLE with hemophagocytic syndrome,207 as well as HIV with hemophagocytic syndrome.208 Pancreatic cancer and metastatic disease can cause a panniculitis that improves with resection of the malignant tissue.209 Panniculitis also may be associated with acute fatty liver of pregnancy and HELLP syndrome (hemolysis, elevated liver enzymes, low platelet count).210
Rare drug-induced cases have been reported, including to l-asparaginase in the setting of treating acute lymphoblastic leukemia,211 and treatment of hepatitis C.212 Identical histopathology has been seen at the site of subcutaneous interferon β injections.213
DIAGNOSIS
DIAGNOSIS
Developed lesions of pancreatic panniculitis demonstrate lobular fat necrosis (Fig. 73-12A). Adipocytes lose their nuclei but maintain peripheral outlines, forming characteristic “ghost cells” (Fig. 73-12B). Saponification causes calcification, producing fine, granular basophilic deposits within and around necrotic adipocytes. The ghost cells are frequently aggregated in small clusters at the center of fat lobules, with a peripheral inflammatory infiltrate of neutrophils.52 In older lesions, necrosis and ghost cells are replaced by foamy histiocytes, multinucleated giant cells, lymphocytes, and, eventually, fibrosis.52,214
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
1271
12
DIAGNOSIS CHARACTERISTICS
α1-Antitrypsin deficiency Tender, erythematous nodules, often with a suppurative exudate. Located on the abdomen, buttocks, and proximal extremities.
Erythema induratum/nodular vasculitis
Tender nodules and plaques on calf areas, living in an area endemic for tuberculosis (erythema induratum). Vasculitis present on histopathology.
Cutaneous polyarteritis nodosa
Nodules in association with livedo reticularis and arterial inflammation on histology, possibly with neuropathy.
Infectioninduced panniculitis
Erythema nodosum–like nodules, deep inflamed nodules or abscesses; older lesions may show fibrosis. Most commonly on legs. Often seen in severely immunosuppressed patients. “Ghost cell” changes and urate crystals secondary to fungal enzymatic changes can be seen in mucormycosis and aspergillosis associated pancreatitis.126,134
Pyoderma gangrenosum Violaceous, undermined borders with isomorphic response.
Malignancies Persistent, worsening disease, often with systemic symptoms. Presence of cutaneous T-cell lymphoma, angiosarcoma, or other sarcomas on leg with/without venous hypertension. More likely in patients with solid-organ transplants or other immunosuppressed states.
Behçet syndrome
Vasculitis occurring in the fat tissue, either
Behçet syndrome occurring in the fat
Vasculitis occurring in the fat tissue, either lymphocytic or neutrophilic, small arterial involvement with either lobular or mixed lobular and septal pattern.
occurring in the fat
lymphocytic or neutrophilic, small arterial involvement with either lobular or mixed lobular and septal pattern.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
Overall, when the pancreatic pathology resolves, the cutaneous symptoms subsequently improve. Panniculitis associated with pancreatic disease may be fatal, with a mortality rate of 24%196 to 42%,197 and mortality rates near 100% in those with pancreatic carcinoma. Acute pancreatitis may be treated and may resolve promptly with supportive care, however pancreatic carcinoma and associated panniculitis is far more difficult to treat.
MANAGEMENT
MANAGEMENT
The focus of treatment of pancreatic panniculitis is on the underlying pancreatic disease. Care is often supportive. Because patients may lack abdominal symptoms at presentation, pancreatic panniculitis should be considered in any patient with panniculitis. Octreotide,215 a somatostatin analog, and plasmapheresis are associated with resolution of pancreatic panniculitis.216 Treating pancreatic
1272
cancer with surgical resection and combination chemotherapy may ameliorate the cutaneous findings.192
LUPUS PANNICULITIS
AT-A-GLANCE
Clinical
■ Erythematous nodules on face, shoulders, upper arms, scalp, chest, buttocks; rarely on lower extremities.
■ Persistent areas of lipoatrophy in regressed lesions.
Histopathology
■ Mostly lobular panniculitis (with or without lymphocytic vasculitis).
■ Lobular lymphocytic infiltrate; may include plasma cells and eosinophils.
■ Lymphoid follicles and sclerotic collagen bundles within septae.
■ Hyaline necrosis and atrophy of entire fat lobule in late-stage lesions.
■ Interface changes of discoid lupus erythematosus in 20% to 30% of cases.
Treatment
■ Antimalarials, thalidomide, topical steroids.
■ If active and severely inflamed, short oral courses of corticosteroids.
■ Dapsone, cyclosporine, methotrexate, intravenous immunoglobulin, rituximab, azathioprine, tacrolimus.
Lupus erythematosus panniculitis (LEP), also termed lupus profundus, subcutaneous lupus erythematosus, and Irrgang-Kaposi syndrome, was first described in 1883 by Kaposi and named lupus erythematous profundus by Irrgang in 1940.217 Overall, it is very rare, even among the lupus erythematosus cohort.
EPIDEMIOLOGY
EPIDEMIOLOGY
LEP is a rare variant of lupus erythematosus, presenting as inflammation in the AT. LEP may appear as the sole manifestation of lupus erythematosus or may occur prior to or after the onset of discoid lupus erythematosus (DLE) or SLE.218,219 The incidence of SLE in patients with LEP is reported to range from 10% to 41%.220 DLE in patients with LEP has a higher incidence, ranging from 21% to 60%.218 Among those who have SLE, only 2% to 5% will have LEP.218 Although rare, LEP occurs worldwide, more frequently among women than men, with a female-to-male ratio of approximately 4:1.221 LEP is most common between the ages of 30 and 60 years, but may rarely be seen in childhood or even as neonatal lupus.222,223 When present in
association with SLE, LEP tends to occur with lesssevere SLE.221,224 Patients with LEP may have other autoimmune disorders such as Sjögren syndrome and rheumatoid arthritis.218 Raynaud phenomenon is present in 10% of LEP patients.221
Other associations with LEP include TNF inhibitors, which caused SLE-like reactions as well as LEP in a patient with rheumatoid arthritis.225 Treatment of a female patient with hairy cell leukemia with interferon has been associated with emergence of disseminated ulcerating lupus panniculitis.226
CLINICAL FEATURES
CLINICAL FEATURES
LEP lesions may be tender and painful and usually appear on the lateral upper arms, shoulders, face, scalp, hips, buttocks, breasts, and, rarely, on the lower extremities (Fig. 73-13).218,224 Orbital involvement may present with periorbital edema,227 and Blaschko linear patterns have been reported.228 The lesions are deep subcutaneous nodules either with or without surface changes, including erythema and DLE features such as atrophy, hyperkeratosis, hyperpigmentation or hypopigmentation, telangiectasia, follicular plugging, and focal necrosis.218 Ulceration can be seen in up to 28% of cases.221 Areas of ongoing indurated panniculitis may coexist with DLE and lead to subcutaneous atrophy and scar formation on the cutaneous surface. Atrophy of facial lesions leads to severe cosmetic alterations. Lesions may be induced by trauma, including injections and surgical procedures.218,224
Serology may be normal or abnormal.218,224 Patients with associated SLE tend to have higher positive antinuclear antibody (ANA) titers224 and C4 deficiency may be found.218,229 Other laboratory findings may include rheumatoid factor, false-positive Venereal Disease Research Laboratory, ANA, leukopenia, anemia, or thrombocytopenia.230 Antiphospholipid antibodies may be positive, and if thrombi are noted on histopathology, testing for antiphospholipid antibodies should be performed.231
Lupus mastitis is a rare form of lupus panniculitis involving the breast. It may clinically mimic Paget disease. On mammography there is calcification paralleling the course of fat necrosis, and early deposition is concerning for malignancy. MRI shows the extent of lupus mastitis and nonspecific fat necrosis; adding contrast may significantly aid in diagnosis.232
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
The innate immune system plays a significant role in the development of LEP. Adipocytes are important cells of the innate immune system expressing TLR1, TLR2, TLR3, TLR4, and TLR6.233 The stromovascular fraction of AT expresses TLR5, TLR7, TLR8, TLR9, and TLR10.233 In SLE, TLR7 and TLR9 recognize RNA and
12
A
B
DNA patterns, respectively, and appear to provide a mechanism for recognition of self-DNA or self-RNA, with subsequent activation of the adaptive immune system and production of autoantibodies to nucleic acids and proteins bound to nucleic acids.234,235 Inhibitors of TLR7 and TLR9 can prevent disease in mouse models of autoimmunity.234 Genetic variations in TLR9 receptor predispose to SLE236 and regression of SLE was seen in a patient who acquired TLR7 and TLR9 defects and antibody deficiency.237 Both receptors have
1273
12
been suggested as therapeutic targets.234,235 Interestingly, hydroxychloroquine blocks intracellular TLRs in vitro.238
DIAGNOSIS
DIAGNOSIS
Histopathologic features of DLE are seen in approximately 20% to 30% of LEP cases. These features include vacuolization of the basal cell layer, thickened basement membrane, mucin deposition between dermal collagen bundles, and a superficial and deep perivascular inflammatory infiltrate of lymphocytes.52,224 The AT shows a lobular or mixed lobular and septal panniculitis, with lymphocytes, formation of lymphoid follicles (20% germinal centers),218 variable hyaline fat necrosis,52,224 and hyalinized and sclerotic septal collagen bundles associated with an interstitial infiltrate of lymphocytes and plasma cells.52 Karyorrhexis, lymphocytic vasculitis, and membranocystic changes may be seen.218 Calcification and/or fibrin thrombi also may be noted in 10% of cases.224 Eosinophils may be seen in 22% to 41% of cases.218
Direct immunofluorescence of blood vessels and basement membrane demonstrates positive findings in almost all SLE-associated LEP, and a high percentage of positive findings in LEP without SLE.224 Immunohistochemical studies of LEP show a predominance of lymphocytes, with both αβ T-helper (CD4+) and cytotoxic (CD8+) lymphocytes mixed with (CD20+) B cells and plasma cells.220
In cases with only AT involvement and without other features specific for DLE or SLE, differentiation from subcutaneous T-cell lymphoma is essential and difficult.239 Anaplastic large-cell lymphoma may also have overlapping features with LEP.240 There are reports of a spectrum of subcutaneous lymphoid dyscrasias, with lesions originally diagnosed as LEP, progressing to indeterminate lymphocytic lobular panniculitis and eventuating in a diagnosis of SPTCL.241 Lymphocytic atypia and lymphocytic rimming is seen in SPTCL.242 SPTCL may coexist with LEP, and must be distinguished from atypical lymphocytic panniculitis in which clonal T-cell infiltrates are associated with indolent behavior.243-245 There may be epidermal involvement in SPTCL,246-248 making the distinction more difficult from LEP. Lack of adipocyte membrane disruption suggests LEP.242 Hyaline lipomembranous change is common in LEP, but seen infrequently in SPTCL.242
In 1 study, high levels of the interferon-α protein marker human myxovirus resistance protein 1 (MxA) were found in the infiltrate of LEP lesions.249 Minimal, less than 20% staining, with MxA suggests a lymphoma.249 CCL5 (the ligand of the receptor often expressed in adipocytes CCR5) expression is much higher in lymphomas compared to LEP.250 Ki-67 staining of greater than 20% and focal areas of increased uptake are more suggestive of SPTCL.242,251 Presence of CD123-positive staining also suggests LEP242 and is an important prognostic indicator.
1274
Even though serology is not specific it should be added to the workup, because SLE will require further treatment. ANA serology has a high false-positive rate, and may be positive in patients with SPTCL.242
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
LEP is a chronic inflammatory disorder with periodic flares or long remissions. Average disease duration is 6 years, with a range of less than 1 year to 38 years.221 CD123 immunohistochemical staining can be a helpful prognostic indicator, as a higher percentage of CD123 positive cells has a greater response to steroid therapy.252 Prompt treatment aids in reducing permanent and potentially disfiguring scarring.
DIAGNOSIS CHARACTERISTICS
Tumid lupus Erythematous, edematous plaques mainly in photoexposed areas with extensive mucin deposition on histopathology without epidermal change.
Cutaneous lymphoma (including subcutaneous panniculitis-like T-cell lymphoma [SPTCL])
Persistent, worsening disease, often with systemic symptoms. Histopathology is critical if no surface changes or additional signs or symptoms suggest lupus erythematosus panniculitis (LEP). LEP more often involves the face, whereas SPTCL more often involves the scalp, lower extremities, and buttocks.242 Lymphocytic atypia and lymphocytic rimming is seen in SPTCL. Up to 19% of SPTCL cases with an αβ phenotype can have an associated autoimmune disorder with lupus erythematosus being the most common.247
Erythema induratum/ nodular vasculitis Tender nodules and plaques on calf areas, living in an area endemic for tuberculosis (erythema induratum). Vasculitis on histopathology.
Morphea/systemic sclerosis Early lymphocytic panniculitis with subsequent subcutaneous septal and dermal sclerosis. In association with clinical features of morphea and systemic sclerosis.
Pancreatic panniculitis Occurs most commonly on the legs with
Pancreatic panniculitis Occurs most commonly on the legs with ulceration and drainage. Serum lipase and amylase often elevated.
ulceration and drainage. Serum lipase and amylase often elevated.
MANAGEMENT
MANAGEMENT
Treatment is challenging, as is assessment of response to therapy, other than clinical assessment of erythema, induration, and tenderness. Just as in other forms of cutaneous lupus, sun protection and avoidance are often recommended.253 Antimalarials are the first-line treatment of LEP218,254 and may be the only medications needed.254 When monotherapy with hydroxychloroquine is ineffective, combination with quinacrine has been used successfully but is difficult to obtain.218,255
Antimalarials interfere with both inflammatory cytokines256 and TLRs.238 Antimalarials have a diminished effect in smokers.257,258
Topical steroids also may be successful, particularly under occlusion.253 Systemic corticosteroids are effective and useful if the disease is active and severe,224 and used as short courses or during antimalarial initiation. Long-term use of corticosteroids is avoided because of the chronic nature of LEP. Intralesional steroids are not recommended as the trauma may induce further activation as well as atrophy. Other treatments include dapsone,259 thalidomide,260 cyclosporine,261 methotrexate,262 azathioprine,253 intravenous immunoglobulin (IVIG),262 and tacrolimus.263 Severe, recalcitrant cases may require rituximab264 or infliximab, although caution must be advised with this modality because of its association with known activation of lupus erythematosus in some instances.265 Considering the common calcification seen in LEP histopathology, calcium-channel blockers such as diltiazem are effective adjuncts, improving the calcinosis.266
CYTOPHAGIC HISTIOCYTIC PANNICULITIS
AT-A-GLANCE
Clinical
■ Subcutaneous erythematous to violaceous plaques and nodules on extremities, trunk, and less frequently elsewhere; lesions may ulcerate.
■ Fever, hepatosplenomegaly, 2 or more cytopenias; hemocytophagocytosis in bone marrow, lymph nodes, liver, or CNS.
■ Hypertriglyceridemia, ferritin >500 mg/L; increased soluble CD25, CD163 levels.
■ Low or absent natural killer cell activity, fibrinogen levels.
■ Potentially rapidly fatal disease course if associated with hemophagocytic lymphohistiocytosis and macrophage activation syndrome; intermittent remissions and exacerbations prior to death; or nonfatal acute or intermittent course.
Histopathologic
■ Mostly lobular panniculitis without vasculitis.
12
■ Histiocytes and mature lymphocytes within fat lobules with necrosis of adipocytes.
■ “Bean-bag” cells: macrophages that contain intact or fragmented erythrocytes, leukocytes or platelets within their cytoplasm; may be focal.
Treatment
■ First-line treatment is immunosuppression with cyclosporine and corticosteroids. Other options include tacrolimus, azathioprine, anakinra, cyclophosphamide with IVIG.
■ Cytophagic histiocytic panniculitis with hemophagocytic lymphohistiocytosis or macrophage activation syndrome requires combined chemotherapy and aggressive supportive care.
■ Diagnose and treat associated malignancies or infections.
Cytophagic histiocytic panniculitis (CHP) is a rare panniculitis that varies in duration from 3 months to 27 years and may be fatal.267-269 It may be an aggressive disease requiring chemotherapy if related to hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome, but a more indolent course requiring immunosuppressant therapy with an excellent remission rate is also reported. Of note, HLH may be primary (genetic) or secondary (to infections, autoimmune disease), but CHP is only secondary in etiology. CHP itself may represent a “smoldering” lymphoma rather than a purely inflammatory disease.270
EPIDEMIOLOGY
EPIDEMIOLOGY
CHP is rare, but is seen in adults, adolescents, and children. The oldest patient reported was 80271 years old at time of diagnosis. Children affected include one with a perforin gene mutation (likely suffering from familial HLH).272 One adult with familial multiple lipomatosis syndrome developed CHP at age 43 years.273 As a consequence of the variable etiologies, epidemiology is largely based on the cause of CHP.
CLINICAL FEATURES
CLINICAL FEATURES
Lesions are subcutaneous plaques and nodules of variable sizes that may coalesce on the extremities and trunk and less frequently on the head and neck (Fig. 73-14).267,274,275 Mucosal involvement is rare.275 Features vary from skin colored to erythematous, purpuric, ecchymotic, or hyperpigmented; flat or raised; well circumscribed or diffuse; and indurated to fluctuant or ulcerated.269,274 Individual lesions can reach 20 cm in diameter.276
Fulminant forms of the disease may be associated with fever, hepatosplenomegaly, lymphadenopathy, serosal effusions, pancytopenia, intravascular coagulation,
1275
12
liver failure, hemorrhagic diatheses, and death.268,269,274,277 Laboratory tests should include a complete blood count with differential, liver function, ferritin, erythrocyte sedimentation rate, and lipid profile. Interestingly, erythrocyte sedimentation rate is often normal or low.275 Soluble hemoglobin-haptoglobin scavenger receptor CD163 serum levels are associated with hemophagocytic disease activity and correlate with serum ferritin levels.278 Serum CD163 level in CHP is much higher than levels seen in sepsis and in normal controls.278 Other tests are determined by history and physical findings and the clinical course. A positive ANA may be detected and does not alone prove LEP.279,280
As CHP may cause hypertriglyceridemia, highly elevated serum lipid levels cause pancreatitis, which may improve with plasmapheresis.281 Elevated cholesterol levels may persist for years after improvement of the panniculitis.279 The elevated triglycerides during the hemophagocytic disease are secondary to inhibition of lipoprotein lipase, leading to IL-6, IL-1, and TNF secretion from macrophages.281,282
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
CHP may belong to a spectrum of panniculitides with benign lymphocytic infiltrate that
1276
develop reactive HLH/macrophage activation syndrome.269,271,275 Secondary HLH is associated with infections, connective tissue diseases, and malignancies, or precipitated by medication.283-285 Infections associated with HLH/macrophage activation syndrome include Epstein-Barr virus, cytomegalovirus, parvovirus, varicella, human herpesviruses 6 and 8, avian influenza, rubella, adenovirus, hepatitis B, HIV, bacterial, acid-fast bacterial, parasitic, and fungal infections.285,286 CHP has presented after H1N1 influenza vaccine287 and visceral leishmaniasis.288 Traumainduced CHP also has been reported.280
HLH is considered an autoinflammatory disease, with impairment of cytolytic T-cell and natural killer cell functions leading to a compensatory cascade of potentially fatal reactions.289 Underlying gene mutations affect vesicular transport, granule shedding, and pore-forming cytolytic proteins involved in granulemediated cytotoxicity.251,286,289,290 Inability of the cytotoxic cells to eliminate infected cells leads to increased activation and proliferation of T cells, which produce high levels of cytokines that stimulate and activate macrophages without the ability to terminate the reaction via apoptosis.251,289
Both LEP and CHP may, in some cases, fit into a spectrum of subcuticular lymphoid dyscrasia, ranging from benign reactive changes to SPTCL, including atypical indeterminate lobular panniculitis.241-244,270 Immunophenotyping and genotyping studies are important for differentiating malignant SPTCL, which may have αβ or γδ rearranged T cells, from CHP with benign T cells. Monitoring biopsies is prudent as transformation has been reported.277 Both SPTCL types can be associated with hemophagocytic syndrome/HLH and present with CHP; the incidence of HLH is much higher among γδ cases than among αβ cases.247
DIAGNOSIS
DIAGNOSIS
Multiple biopsies may be needed to establish the diagnosis of CHP as cytophagocytosis on histopathology may be focal. The histopathology is principally a lobular panniculitis (Fig. 73-15A) with fat lobules containing an infiltrate of histiocytes and mature lymphocytes with variable plasma cells, neutrophils, and eosinophils.52 Phagocytic histiocytes contain intact or fragmented cells and nuclear debris within their cytoplasm; this represents the characteristic “beanbag cell” (Fig. 73-15B).52 This is unlike emperipolesis (the presence of an intact cell within the cytoplasm of another cell) in which inflammatory cells pass through a histiocyte and no nuclear debris is seen.291
Immunohistochemical studies demonstrate that cytophagic histiocytes express histiocytic markers such as CD68, whereas most lymphocytes show a T-cell immunophenotype. The presence of atypical lymphocytes, lymphocytic rimming of adipocytes, and clonality are concerning for lymphoma.242,247 In rapidly progressive cases of CHP with HLH, cytophagic features can also be seen in internal organs including
A
B
lymph nodes, spleen, liver, and bone marrow, and in cerebrospinal fluid fluid.267-269,274,277
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
Patients may have a rapidly fatal disease course, a longer disease course with intermittent remissions and exacerbations for many years prior to death, or a nonfatal acute or intermittent course responsive to treatment.268,274,277,292 Patients with the benign, nonfatal course were found to have less-severe laboratory abnormalities.292 Prompt treatment with corticosteroids
12
DIAGNOSIS CHARACTERISTICS
α1-Antitrypsin deficiency Tender, erythematous nodules that often have a suppurative exudate. Located on the lower abdomen, buttocks, and proximal extremities. Serology for enzymatic levels and phenotype helpful.
Erythema induratum/ nodular vasculitis
Tender nodules and plaques on calf areas, living in an area endemic for tuberculosis (erythema induratum). Vasculitis present on histopathology.
Cutaneous polyarteritis nodosa
Nodules in association with livedo reticularis and arterial inflammation on histology, possibly with neuropathy.
Infection-induced panniculitis Erythema nodosum–like nodules, deep and inflamed nodules, or abscesses; older lesions may show fibrosis. Most commonly on legs. Systemic symptoms possible with fever. Often seen in severely immunosuppressed patients with malignancies. “Ghost cell” changes and urate crystals secondary to fungal enzymatic changes can be seen in mucormycosis and aspergillosis associated pancreatitis.126,134
Pyoderma gangrenosum Violaceous, undermined borders with isomorphic response. Rare to mild systemic symptoms.
Malignancies Persistent, worsening disease, often with sys-
Malignancies Persistent, worsening disease, often with systemic symptoms. Subcutaneous panniculitislike T-cell lymphoma must be distinguished from atypical lymphocytic panniculitis. Presence of cutaneous T-cell lymphoma, angiosarcoma, or other sarcomas on leg with or without venous hypertension. More likely, but not exclusively, in patients with solid-organ transplants or other immunosuppressed states. Systemic symptoms possible.
temic symptoms. Subcutaneous panniculitislike T-cell lymphoma must be distinguished from atypical lymphocytic panniculitis. Presence of cutaneous T-cell lymphoma, angiosarcoma, or other sarcomas on leg with or without venous hypertension. More likely, but not exclusively, in patients with solid-organ transplants or other immunosuppressed states. Systemic symptoms possible.
and cyclosporine in the appropriate patient may reduce a 70% mortality rate to near 0%.267,279 The panniculitis by itself is not always a marker of disease severity as 1 patient with CHP had resolution of cutaneous lesions with high-dose corticosteroid and cyclosporine therapy followed by recurrence 5 months later, progression, and death with generalized HLH.293 This 1998 case was the first report of the failure of cyclosporine, and highlights the need for careful followup for systemic involvement even after an apparent therapeutic response.293
MANAGEMENT
MANAGEMENT
Multidisciplinary care is critical in CHP management owing to its potentially rapidly fatal course. Hospitalization may be required for a rapid workup and institution of treatment. In HLH associated with an
1277
12
infection, treating the pathogen may result in recovery, but this is unpredictable.294
Treatment of CHP includes immunosuppressive, immunomodulating, or cytotoxic agents that target activated macrophages/histiocytes (steroids, etoposide, high-dose IVIG) and T cells (steroids, cyclosporine, antithymocyte globulin).275 Among the published cases, cyclosporine and corticosteroids are often used and are often successful. In patients with both SLE and CHP, the combination of corticosteroids with cyclosporine may induce remission of both diseases.276
Cyclosporine-resistant disease also has been reported to respond to tacrolimus.295 Tacrolimus may be an option for patients who have adverse effects with cyclosporine, and it may be a more realistic long-term treatment option for cases of CHP that are not able to taper off therapy.296 Resistant cases may warrant a trial with cyclophosphamide and IVIG.297 Addition of combined chemotherapy may be helpful.275,298 Multiple rounds of combination chemotherapy or autologous stem cell transplantation may be required to induce remission.299
Cyclosporine, and cyclosporine with etoposide,280 are effective regimens for CHP in both children and adults. In 2 pediatric patients, who developed CHP after viral infections, focal areas within the histopathology suggested SPTCL versus a reactive T-cell proliferation with in situ T-cell clonality; however, cyclosporine with corticosteroids induced remission for 69 and 29 months, respectively.300 This report highlights the need to discriminate between CHP associated with a nonmalignant condition or with SPTCL. This report also suggests that when the diagnosis suggests a reactive T-cell proliferation rather than true lymphoma, cyclosporine may be used because nonmalignant CHP often improves with cyclosporine and prednisone whereas CHP with SPTCL is best treated aggressively.300 One adolescent with lifelong CHP and severe HLH experienced improvement signs with anakinra, an IL-1 receptor antagonist, in addition to prednisone and cyclosporine.301
SUBCUTANEOUS FAT NECROSIS OF THE NEWBORN
AT-A-GLANCE
Clinical
■ Circumscribed, red to violaceous, subcutaneous nodules, or plaques with predilection for buttocks, shoulders, cheeks, and thighs.
■ Hypercalcemia in some cases, even presenting after the acute episode. Rarely, hypertriglyceridemia, hypoglycemia, thrombocytopenia, anemia.
Histopathology
1278
■ Mostly lobular panniculitis without vasculitis.
■ Dense inflammatory infiltrate of lymphocytes, histiocytes, lipophages, and multinucleated giant cells.
■ Needle-shaped clefts, often in radial array, within cytoplasm of histiocytes and multinucleated giant cells.
Treatment
■ Nodules and plaques usually resolve spontaneously.
■ Monitor for hypercalcemia for 6 months following onset, treat if hypercalcemia develops.
Subcutaneous fat necrosis of the newborn (SCFN) is a rare panniculitis that occurs in the first few days to weeks of life. Nearly all cases resolve spontaneously. The most common complication is hypercalcemia. Additional secondary effects include hypertriglyceridemia, hypoglycemia, thrombocytopenia, and anemia.302-304
EPIDEMIOLOGY
EPIDEMIOLOGY
SCFN presents in full-term or postterm newborns (preterm cases also have been noted305) with a preceding history of perinatal complications, including meconium aspiration, asphyxia, hypothermia (eg, for cardiac surgery or ice pack application for supraventricular tachycardia, or for hypoxic ischemic encephalopathy complicating lack of respiratory effort306), hypoxemia, seizures, sepsis, gestational diabetes, preeclampsia, factors requiring cesarean section, forceps delivery, severe neonatal anemia, maternal cocaine use, and/or failure to thrive.302-304,307,308 Within the population requiring hypothermic treatment of asphyxia, macrosomia and hemodynamic instability represent additional risk factors.309
CLINICAL FEATURES
CLINICAL FEATURES
Lesions are sharply demarcated, erythematous to violaceous, firm, indurated nodules or plaques located on the back, shoulders, arms, buttocks, thighs, or face, but rarely on the anterior trunk or the anterolateral shins (Fig. 73-16A).303,306,307 Visceral involvement has been reported with MRI findings in one case consistent with abdominal wall, perihepatic, perisplenic, and perirenal fat necrosis.310 The subcutaneous nodules range in size from several millimeters to up to 11 cm in diameter,311,312 may be single or multiple, and are usually well defined.311 Rarely, fluctuant nodules may drain an oily or chalky white material.312 Larger blisters may form and may need drainage.306 Lesions are not warm to touch311 and are commonly painless; rarely, morphine is needed for pain control.311 While often asymptomatic, the location and the size of the
A
12
B
C
lesions can have local mass effect, such as in a case of radial nerve palsy secondary to SCFN that improved spontaneously within 10 weeks.313
There are several metabolic complications that may occur during and even after resolution of the panniculitis.303 These include hypercalcemia (which affects 36% to 56% of patients with SCFN),314 thrombocytopenia,302,303 hypertriglyceridemia (likely related to the fat necrosis), hypoglycemia,303,304 and anemia.303 It is unclear if the glucose and hematologic abnormalities are secondary to the SCFN or to the initial insult that provokes the SCFN, as they may precede onset of cutaneous findings.305,315 Hypercalcemia may be asymptomatic,304 or may progress to symptomatic failure to thrive, irritability, fever, vomiting, hypotonia, seizures, polyuria and polydipsia, and even death.311,312 Soft tissue and organ calcification may occur with variable outcomes.302,311
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
The ultimate cause of SCFN is not known, but hypothermia and hypoxia are presumed to be involved. Therapeutic hypothermia is used in pediatric intensive care in cases of asphyxia. It is unclear whether the hypothermia assists with the process or is a direct
cause of the SCFN.316 Cold exposure during surgical procedures may also induce SCFN.317 Possible explanations have included a biochemical defect in the composition or metabolism of neonatal fat, leading to crystallization, fat necrosis, and subsequent inflammation after cold stress.303,318 The hypercalcemia may be related to increased 25-hydroxyvitamin D3–1α hydroxylase within the granulomatous infiltrates.319
Brown adipose tissue (BAT) in neonates rapidly converts fat stores to heat under conditions of cold stress.320 SCFN affecting BAT has been confirmed with immunohistochemical staining, as well as uncoupling protein isoform 1, found specifically in BAT.321 The BAT cells generate heat. The mechanism is complex, and uses uncoupling protein isoform 1, cytosolic fatty acids, and Ca2+ ions.322
DIAGNOSIS
DIAGNOSIS
Characteristically, SCFN is a mostly lobular panniculitis (see Fig. 73-16B), with focal necrosis of the fat lobule and a dense inflammatory infiltrate of lymphocytes, histiocytes, and foreign-body giant cells; a few eosinophils are possible.52,303,323
A primarily neutrophilic infiltrate may mimic an infection, especially if lesions are biopsied when 1 day
1279
12
old.324 Many adipocytes retain their cellular outlines, but contain fine eosinophilic strands and granules, as well as needle-shaped clefts in radial array (see Fig. 73-16C).52,303,323 On frozen section, these clefts are occupied by doubly refractile crystals, representing triglycerides. Similar clefts and crystals also may be seen within the cytoplasm of the multinucleated giant cells.52,303,323 Late-stage lesions may demonstrate fibrosis and calcified areas within fat lobules.52,311 Fine-needle aspiration may be less painful, more cost-effective, and a more rapid diagnosis.325,326 Ultrasonography also may be an adjunctive noninvasive diagnostic tool. AT is hyperechoic with high blood flow on Doppler and calcifications are seen.327
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
SCFN is a benign process with an excellent prognosis. Lesions regress in a few weeks to 6 months, with some eventuating in atrophy.303,304,310 Hypercalcemia is the most severe potential adverse outcome and may cause nephrocalcinosis.329 Even severe hypercalcemia may be asymptomatic.314 The hypercalcemia may have a delayed onset up to 6 months after appearance of the skin lesions; therefore, serial monitoring of serum calcium levels is necessary.303,304
DIAGNOSIS CHARACTERISTICS
Cellulitis Larger plaques that are erythematous, usually lacking the violaceous bruising. Tender and warm.
Cold panniculitis With any exposure to cold in both children and adults, not specific to the setting of hypoxia. Histopathology lacks needle-shaped clefts within the subcutaneous adipose tissue. Resolves with rewarming.
Sclerema neonatorum Diffuse skin hardening, particularly on buttocks and thighs, in stressed infants in the first week of life, who are often low weight and premature.52,328 Histopathology also shows needle-shaped clefts, with minimal inflammation. Poor prognosis with systemic illness.52
Poststeroid panniculitis Occurs in children, usually 10 days after rapid cessation of high-dose systemic corticosteroid therapy. Similar histopathology to subcutaneous fat necrosis of the newborn.
Poststeroid
Occurs in children, usually 10 days after rapid
panniculitis
cessation of high-dose systemic corticosteroid therapy. Similar histopathology to subcutaneous fat necrosis of the newborn.
1280
MANAGEMENT
MANAGEMENT
SCFN nodules resolve spontaneously, and treatment should be conservative in most cases, except for lesions that may benefit from aspiration to prevent rupture, infections, necrosis, and scarring.302,303
Surgical management has been used in severe cases that persist for months with incision and curettage of small calcifications.330
Serum calcium should be monitored serially, for several months after presentation in case of delayed elevations. If hypercalcemia is present, management includes hydration, use of the calcium-wasting diuretic furosemide, and a low calcium formula.303,314
Systemic glucocorticoids may be used to inhibit vitamin D production by the macrophages in the inflamed AT.303,307,312 Bisphosphonates, including alendronate,331 etidronate,332 and pamidronate,333 treat hypercalcemia in SCFN. Importantly, nephrocalcinosis may still develop in spite of cutaneous response.301 The degree and duration of hypercalcemia and hypercalciuria may be critical in development of nephrocalcinosis, encouraging early monitoring and recognition of hypercalcemia, as rapid therapeutic intervention may prevent dystrophic calcification.334 Renal ultrasound detects nephrocalcinosis.314 Persistent nephrocalcinosis does not result consistently in renal dysfunction.314
For prevention of SCFN, a specific nursing protocol has reduced the risk of SCFN while cooling. It includes rotation of the neonate every 3 hours and a gentler cooling surface–cooling device.335
COLD PANNICULITIS
AT-A-GLANCE
Clinical
■ Circumscribed, red to violaceous, subcutaneous plaques or nodules on the face and thighs, and rarely of the scrotal fat in prepubertal boys.
■ Follows exposure to cold weather, water, popsicles, and ice packs.
Histopathology
■ Mostly lobular panniculitis with a lymphohistiocytic or mixed infiltrate, without vasculitis.
■ Perivascular lymphohistiocytic infiltrate involving blood vessels at the dermal–subcutaneous junction and within overlying dermis.
■ Findings may be similar to those seen in perniosis.
Treatment
■ Avoid direct ice placement on skin and cold exposure.
■ Spontaneous resolution within 3 months.
Cold panniculitis, also known as Haxthausen disease,336 was first described in 1902 with hardening of a child’s chin after cold weather exposure.337 When occurring in equestrians, the terms horse rider’s pernio and equestrian panniculitis are also used.338 Cold panniculitis is an inflammatory reaction of AT that occurs after skin is exposed to cold weather and ice application (as in cold pack application for supraventricular tachycardia),336 and can occur on mucosae as well (popsicle panniculitis).337,339
EPIDEMIOLOGY
EPIDEMIOLOGY
Cold panniculitis occurs in children and in young female horseback riders as well as other adults with appropriate exposures. Symptoms may be secondary to intraoral frozen teething rings,340 or after ice packs are applied to treat febrile episodes.341 Scrotal cold panniculitis presents in 9- to 14-year-old overweight boys after cold exposure (including swimming in cold ocean water).342 Symptoms of cold panniculitis in horseback riders have been reported in both males and females. A survey in Finland suggested 25% of riders have symptoms in the winter months.338 Moderate smoking is statistically significant for an increased risk of cold panniculitis in equestrians, as is wearing tight riding clothes throughout the day, riding for long periods, and being younger than 35 years old.338 Other risk factors for equestrian-like panniculitis include motorbike or open vehicle riding, wading through cold rivers, and young women wearing tight jeans.343,344
CLINICAL FEATURES
CLINICAL FEATURES
Clinical findings of indurated, erythematous plaques or nodules occur at sites of cold exposure and may include mucosa. The onset ranges from 6 to 72 hours after exposure,345 appearing directly at sites that have contact with cold.336 In equestrians with cold panniculitis, the presentation is on the lateral thighs.338
Scrotal cold panniculitis, also known as scrotal fat necrosis, manifests as unilateral or bilateral tender masses below the testes. Recognizing these changes is important to avoid unnecessary biopsy or intervention, as spontaneous resolution over days to several weeks will occur.342 Equestrian panniculitis appears hours after long outdoor cold exposure while wearing tight-fitting, noninsulated pants. The lesions are pruritic, erythematous nodules and plaques that may focally ulcerate, crust, and drain.337,338
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
In affected infants, the mechanism of inflammation has been attributed to a higher proportion of saturated fat to
12
unsaturated fat in infants, with the composition becoming more unsaturated with age.337,346 Other mechanisms include the effects of cold on BAT, which is more prevalent in infants than adults, and its activation to generate heat.347 Cold exposure leads to changes in BAT including increased vascular endothelial growth factor, enhanced free fatty acid degradation, and metabolic changes.348,349 Scrotal cold panniculitis may be related to the greater concentration of scrotal fat and the increased cold sensitivity of the scrotal AT of younger males.350
Equestrian panniculitis may represent perniosis/ chilblains, rather than a true panniculitis.343,344,351 Although high levels of cold agglutinins were detected in 2 Scottish women with equestrian panniculitis,351 other series do not show this abnormality338,346 and cryoglobulins and cold agglutinins are not thought to play a role.352
DIAGNOSIS
DIAGNOSIS
The diagnosis is often clinical given the history of the cold exposure to the affected areas. In general, the histopathologic picture of cold panniculitis is a mostly lobular panniculitis, with a lymphohistiocytic or mixed inflammatory infiltrate present within fat lobules.52,337
There is a superficial and deep perivascular, as well as periadnexal, lymphocytic infiltrate with prominent inflammation of veins most notable at the dermal–subcutaneous fat junction; these histopathologic findings closely resemble those seen in perniosis.343,346 The title “cold-associated perniosis of the thighs” has been suggested.343 In scrotal cases, ultrasound findings may help confirm the diagnosis.342
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
Cold panniculitis is self-limiting and resolves upon rewarming without continued exposure to cold. Full improvement may be seen over days341 to 3 months.340
Postinflammatory hyperpigmentation may remain.352
MANAGEMENT
MANAGEMENT
There is no additional treatment needed of cold panniculitis other than avoidance of further cold exposure; the best prevention is avoidance of cold exposure.340
Equestrians with cold panniculitis may benefit from warm clothing and local topical corticosteroid application. Prevention in this population includes warm and
1281
12
DIAGNOSIS CHARACTERISTICS
Cellulitis Large plaques that are often unilateral, erythematous, tender and warm. Systemic findings may include fever and leukocytosis.
Subcutaneous fat necrosis of the newborn
Cold exposure in infants in the setting of hypoxia. Histopathology with needle-shaped clefts within the adipose tissue. Complications such as hypercalcemia are possible.
Sclerema neonatorum Diffuse skin hardening, particularly on buttocks and thighs, in stressed infants in the first week of life, who are often low weight and premature.52,328 Histopathology also shows needleshaped clefts with minimal inflammation.52 Poor prognosis with systemic illness.52
Poststeroid panniculitis Occurs in children, usually within 10 days after rapid cessation of high-dose systemic corticosteroid therapy. Similar histopathology to subcutaneous fat necrosis of the newborn.
Erythema
Violaceous or red plaques on the anterior legs
Erythema nodosum Violaceous or red plaques on the anterior legs with no ulceration.
nodosum
with no ulceration.
loose-fitting clothing both while riding and during the day, as well as smoking cessation.338 Nifedipine is not an effective treatment.338,345
FACTITIAL AND TRAUMATIC PANNICULITIS
AT-A-GLANCE
Clinical
■ In factitial panniculitis (a variant of Munchausen syndrome),353 self-induced trauma (mechanical, physical, or chemical) causes subcutaneous inflammation. In trauma-induced panniculitis the injury is not induced by the patient, such as in a postsurgical panniculitis or panniculitis following a sport injury.354,355
■ Erythematous geometric papules and nodules with erosions or ulcerations (Fig. 73-17),356 with disparate location or appearance.
■ Factitial panniculitis is often seen in patients with personality aberrations and subcutaneous implantation may include medications, cosmetic fillers, oils, and food or human waste.356
■ Traumatic panniculitis may be secondary to cupping, acupuncture, and electropuncture,357,358 or may be iatrogenic from immunizations or therapeutic injections.356
■ Secondary hypertrichosis at affected sites is rare.359,360
1282
Histopathology
■ Mostly a nonspecific lobular panniculitis without vasculitis, varying with the cause.
■ Suppurative granuloma involving the fat lobule; requires cultures for various organisms. Necrosis is often present.361
■ Polarization of the slide may identify refractile foreign material within macrophages.356
■ Late stages may show foamy histocytes with fibrotic changes and dystrophic calcific deposits.356,362 Foamy lipophagic histiocytes, adipocyte anisocytosis with pseudocystic degeneration, single adipocyte necrosis, erythrocyte extravasation, and foreignbody granuloma with multinucleated giant cells are all potential features. Frank scarring, deep phlebitis, and traumatic neuromas may be present in postsurgical panniculitis.354
Treatment
■ Psychiatric treatment for self-inflicted factitial panniculitis.52
■ Supportive care and interference with injection of responsible agents.
■ Often the implanted material must be surgically excised.356,357
■ Prevention of ongoing trauma and supportive care is needed for traumatic panniculitis as it is selflimiting without further trauma.361
CONNECTIVE TISSUE DISEASE-ASSOCIATED PANNICULITIS
AT-A-GLANCE
Clinical
■ Most often associated with dermatomyositis, and may precede, occur with, or appear late in the disease course, with severity correlating with disease flares.363
■ Erythematous, tender to painful nodules and plaques on the arms, buttocks, thighs, and abdomen. Complications include ulceration and lipoatrophy.364,365
■ Rarely, panniculitis is the primary presentation of dermatomyositis,52,366 and juvenile dermatomyositis-associated panniculitis is uncommon but has been reported.367 Panniculitis in a child should prompt workup for connective tissue disease.368
■ Infection may occur concurrently and must be treated prior to immunosuppression.367
■ Panniculitis is also associated with morphea and systemic sclerosis, but the association is rare.
■ SPTCL must be considered in the differential diagnosis of a connective tissue disease panniculitis. Up to 19% of SPTCL with an αβ phenotype in one series had an associated autoimmune disorder.247 SPTCL with a γδ phenotype was reported as developing in a patient with rheumatoid arthritis after 3 years of anti-TNF therapy.247 One patient with SPTCL presented with clinical features of dermatomyositis.369
Histopathology
■ Mostly lobular panniculitis with a lymphocytic and plasma cell infiltrate, with dermal mucin and vacuolar interface change, similar to lupus panniculitis.52,370
■ Calcification may be present.363
■ Membranocystic change may be present in late-stage lesions,52,230 and indicate a worse prognosis.371
Treatment
■ Same as for dermatomyositis: corticosteroids, immunosuppressants (including azathioprine, mycophenolate mofetil, and methotrexate370) and IVIG.364,366,371,372
■ Calcification within lesions may benefit from IVIG.373
■ Antimalarials are not effective in dermatomyositisassociated panniculitis.370
12
GOUTY PANNICULITIS
AT-A-GLANCE
Clinical
■ Subcutaneous urate crystals are a rare manifestation of gout.374
■ Most commonly firm, indurated nodules or plaques, with variable tenderness on the lower extremities,375 with possible urate crystal deposition in bone marrow375 and otherwise diffuse involvement.376
■ A chalky substance may overly ulceration.376,377
■ Massive AT hypertrophy may be seen, requiring surgical resection.378
■ Hyperuricemia is associated,379 as well as arthralgias (gouty arthritis).380
■ The lesions may precede a diagnosis of gout,376 but they are often a late consequence of chronic gout.377
■ Differential diagnosis includes foreign-body panniculitis, calcium, and other crystal deposition diseases.375
Histopathology
■ Fine, radially oriented, needle-shaped urate crystals within AT are surrounded by granulomatous inflammation. The monosodium urate crystals are refractile and negatively birefringent.375,378,379
■ Alcohol fixation allows for enhanced crystal visualization.375
■ There is no evidence of vasculitis.379
Treatment
■ Standard treatments for gout include allopurinol (high doses),380 probenecid, and antiinflammatory medications, including colchicine, which can be used to reduce uric acid levels.375
■ Acute panniculitis may benefit from corticosteroids.379
■ Surgery may be necessary for ulcerated lesions that do not heal.377
POSTSTEROID PANNICULITIS
AT-A-GLANCE
Clinical
■ Poststeroid panniculitis is rare, most often seen in children after high doses of systemic corticosteroid treatment, days to weeks after abrupt corticosteroid withdrawal.303,381 Cases in adults are very rare.382,383
■ Erythematous, tender nodules and plaques on the cheeks, trunk, arms,382 and legs.381
1283
(Continued)
12
AT-A-GLANCE (Continued)
Continued
AT A GLANCE (
)
■ Resolves in weeks or months without scarring; can leave residual hyperpigmentation. Most common complication is ulceration; systemic symptoms are rare.382
Histopathology
■ Lobular panniculitis with a lymphocytic and granulomatous infiltrate, without vasculitis. Lipophages and crystals can be present.382
■ Needle-shaped clefts in a radial orientation are seen, similar to those seen in SCFN and sclerema neonatorum; sclerema neonatorum lacks or has sparse inflammation.382
■ Multinucleated giant cells may contain the needleshaped clefts with foamy histiocytes.382,383
Treatment
■ Lesions resolve within weeks or months without scarring even without treatment, leaving residual hyperpigmentation.382,383
■ It has been postulated that gradual tapering of steroids can eliminate the risk of poststeroid panniculitis.381
ACKNOWLEDGMENTS
The authors wish to acknowledge Dr. Patricia Fishman for her work on the prior edition of this chapter.
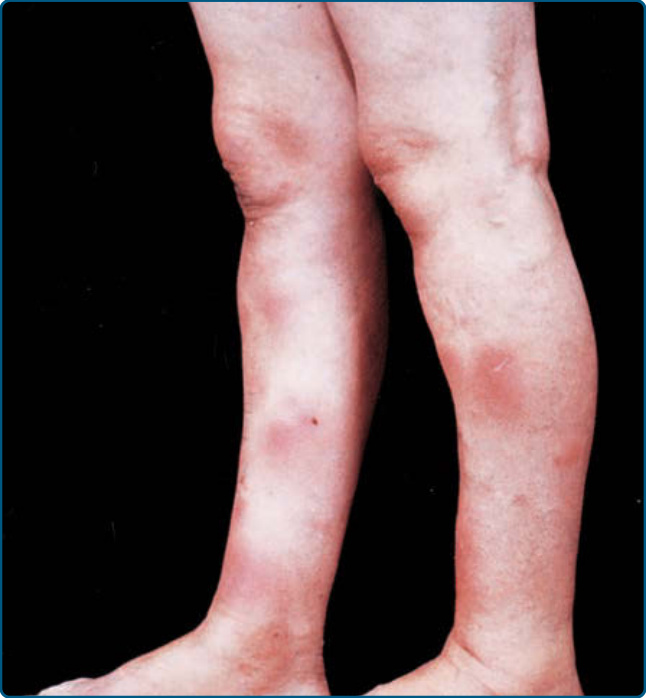
Figure 73-1 Erythema nodosum. Erythematous nodules located mainly on the anterior lower legs.
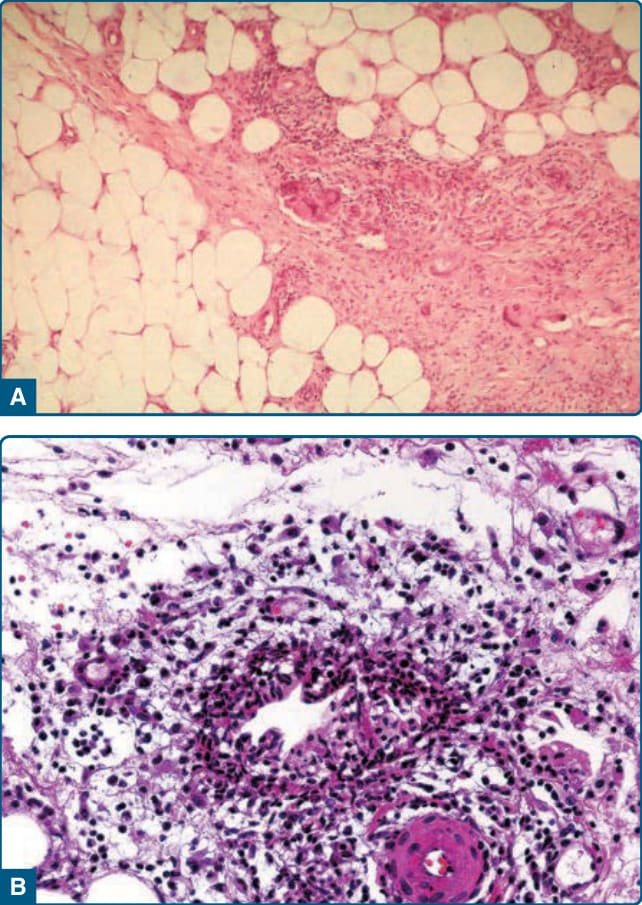
Fig. 73-2B).15 It is considered characteristic of early EN, but is not universally found in EN,39 and has been described in other types of panniculitis.39 Some authors believe the Miescher granulomas are consistently found in EN.15 Late EN lesions may demonstrate lipomembranous change.15,40 Although by definition, vasculitis is characteristically absent in EN, thrombophlebitis has been emphasized by some

Fig. 73-2B).15 It is considered characteristic of early EN, but is not universally found in EN,39 and has been described in other types of panniculitis.39 Some authors believe the Miescher granulomas are consistently found in EN.15 Late EN lesions may demonstrate lipomembranous change.15,40 Although by definition, vasculitis is characteristically absent in EN, thrombophlebitis has been emphasized by some
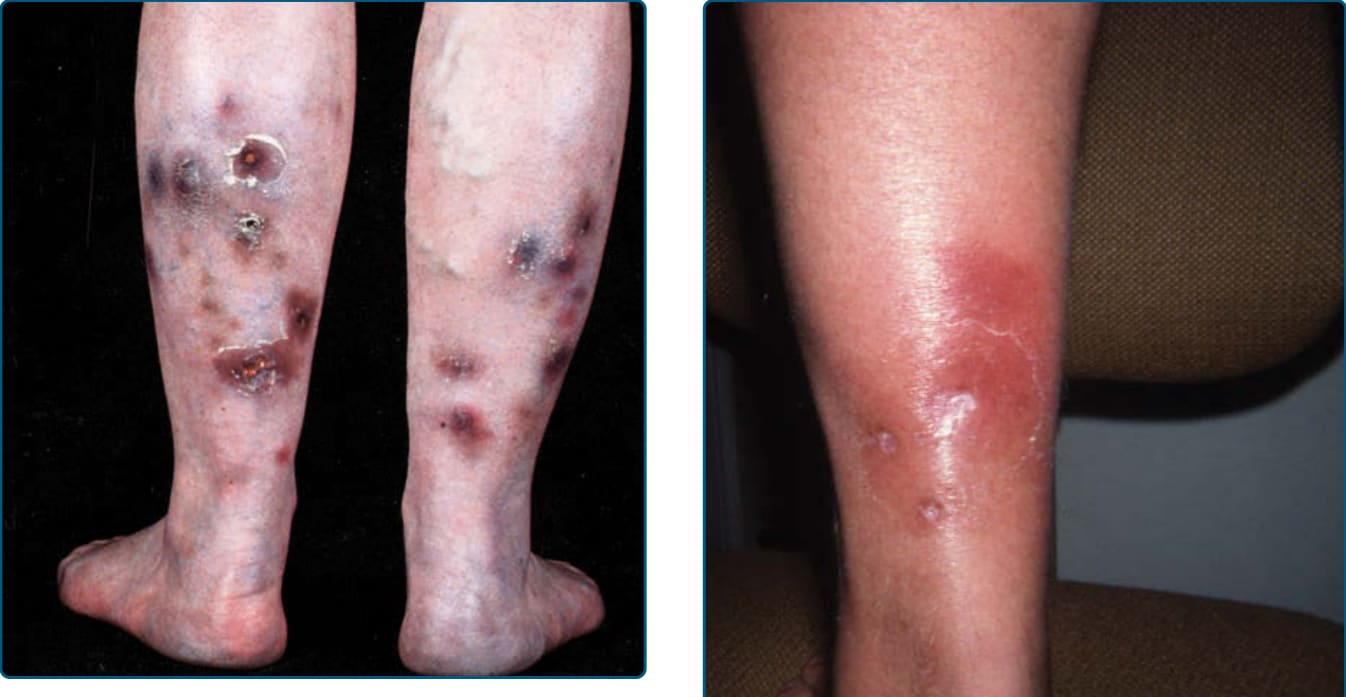
Figure 73-3 Erythema induratum. Erythematous to brown and bluish nodules with ulceration on calves.
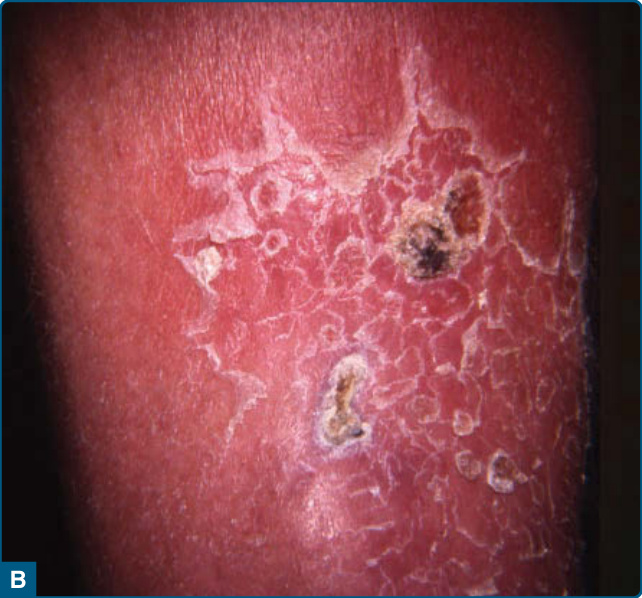
Figure 73-4 A and B, Erythema induratum with surrounding collarette of scale.
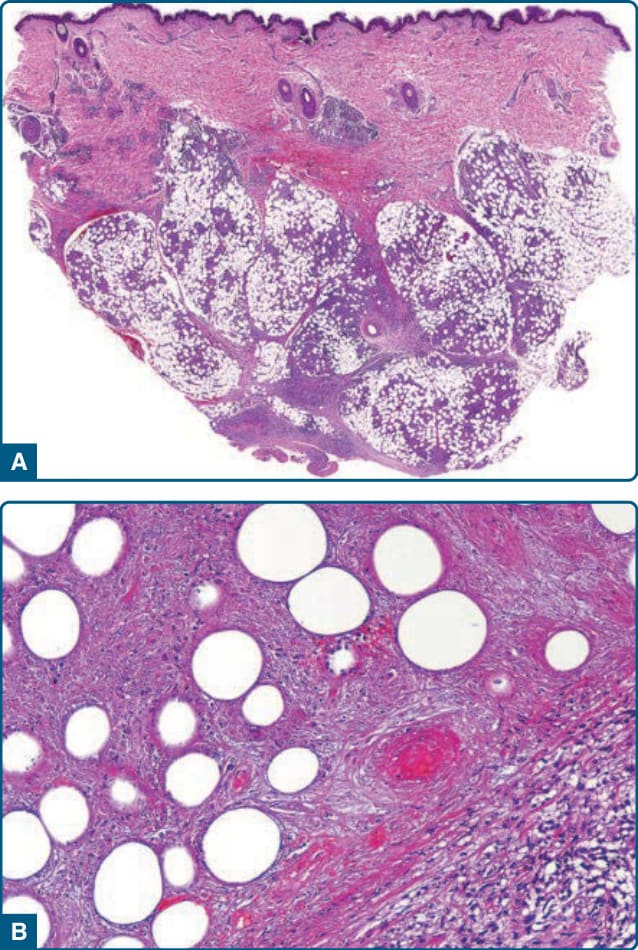
Figure 73-5 Erythema induratum nodular vasculitis. A, Low-power magnification shows a mostly lobular panniculitis. B, Higher-power magnification shows extensive adipocyte necrosis and vascular damage—necrotizing vasculitis of small venules in the fat lobule.
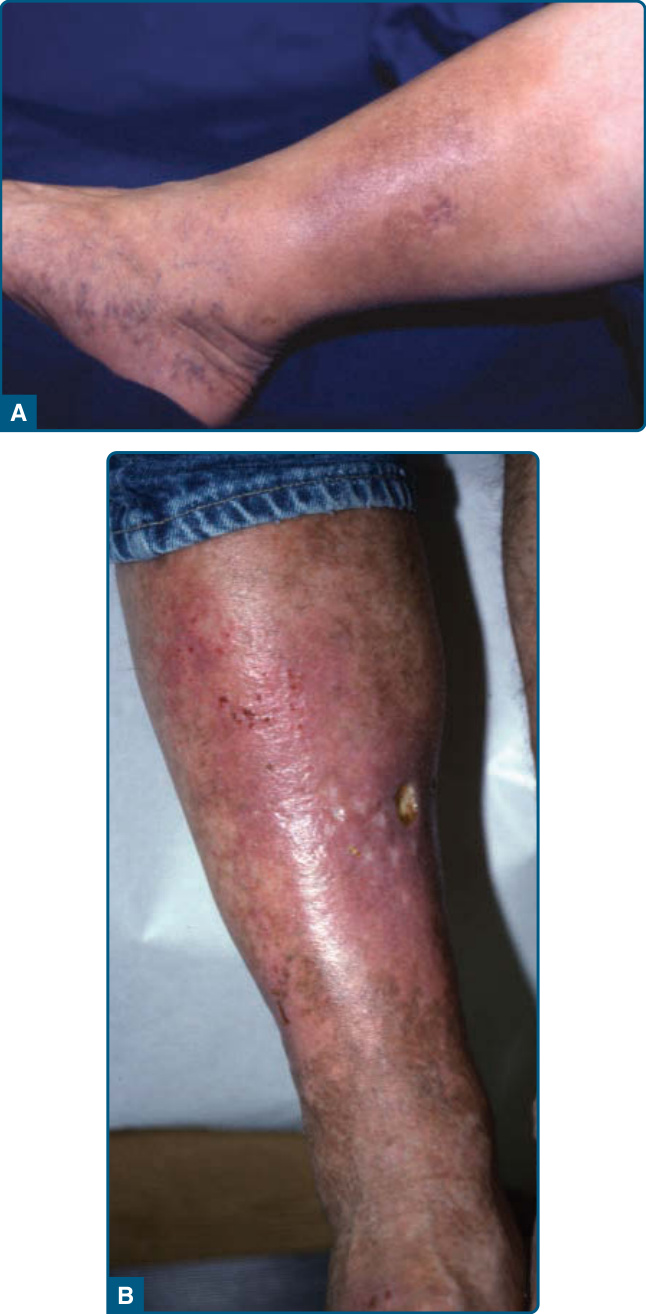
Figure 73-6 A, Chronic lipodermatosclerosis (LDS) with mild erythema of the medial lower leg, indicating presence of active inflammation. Cellulitis-like plaques on the medial lower leg. B, Severe acute inflammation with ulcerations superimposed on chronic LDS.
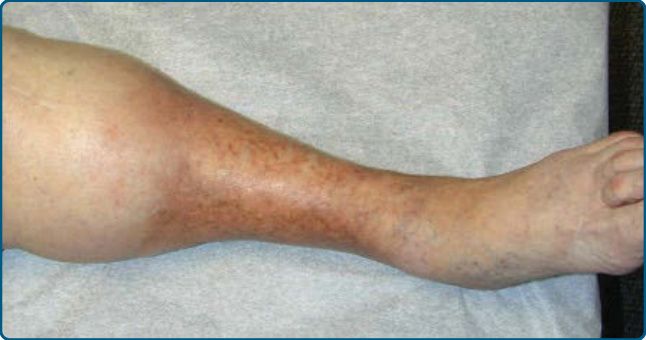
Figure 73-7 Chronic lipodermatosclerosis with champagne bottle/bowling pin deformity.
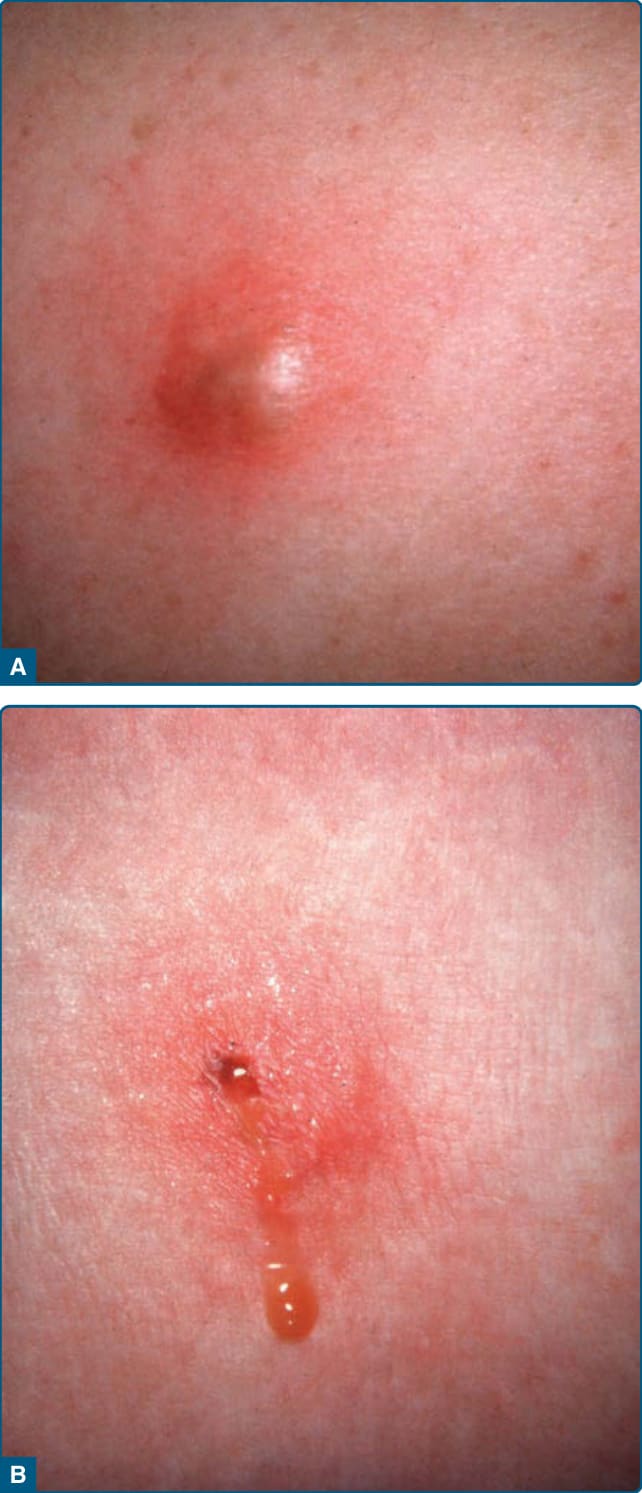
Figure 73-8 α1-Antitrypsin deficiency–associated panniculitis. A, Fluctuant abscess appearance. B, Discharge of oily material from lesion.
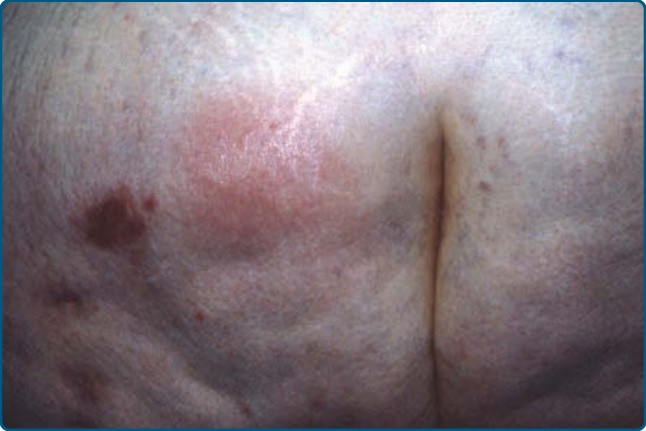
Figure 73-9 α1-Antitrypsin deficiency–associated panniculitis. Nodular lesion on the buttock.
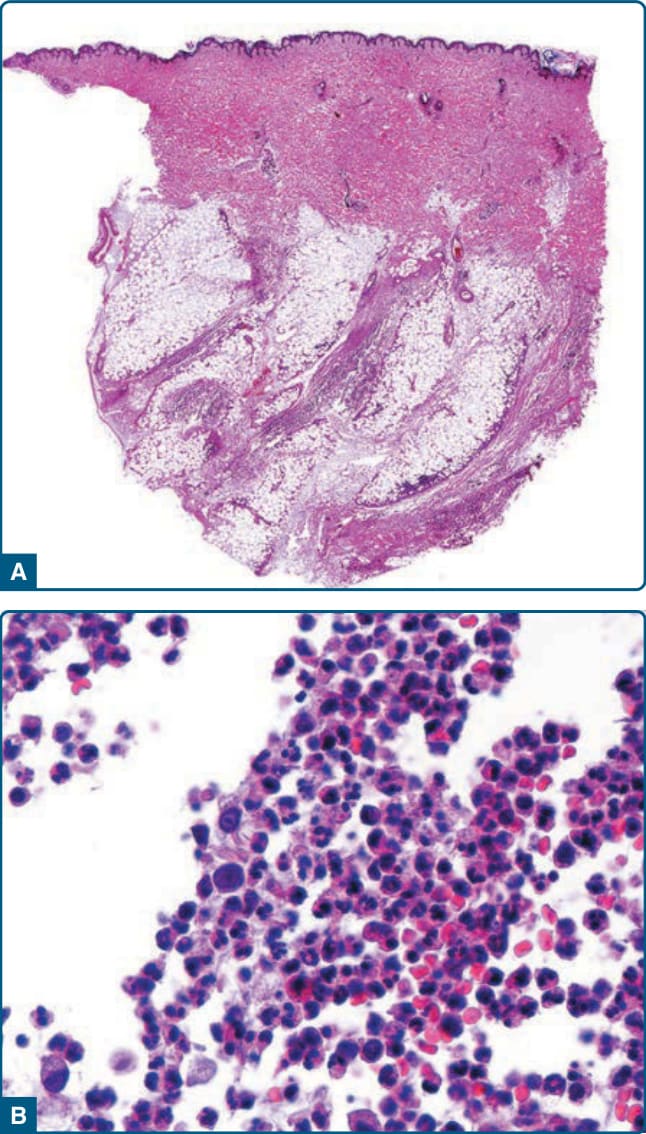
Figure 73-10 α1-Antitrypsin deficiency–associated panniculitis. A,. Low-power magnification shows a mostly lobular panniculitis. B, High-power magnification shows dense inflammatory infiltrate of neutrophils in the fat lobule.
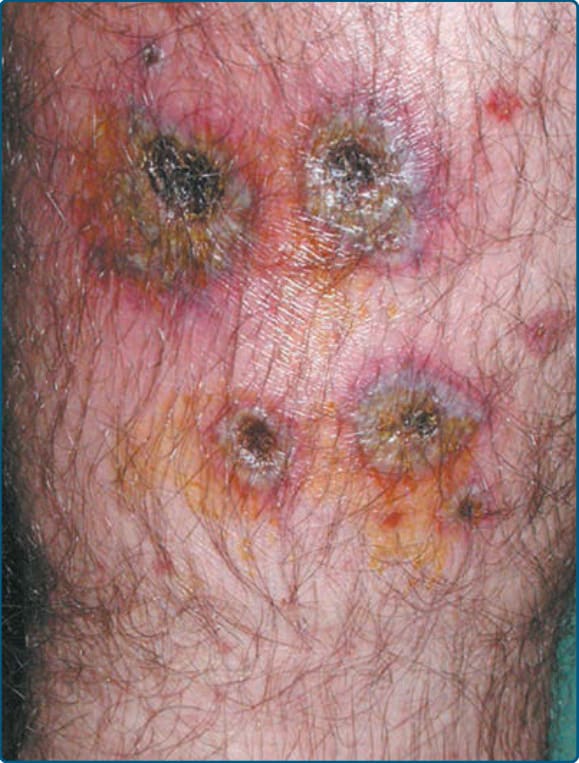
Figure 73-11 Pancreatic panniculitis. Erythematous subcutaneous nodules that ulcerate and exude an oily material.
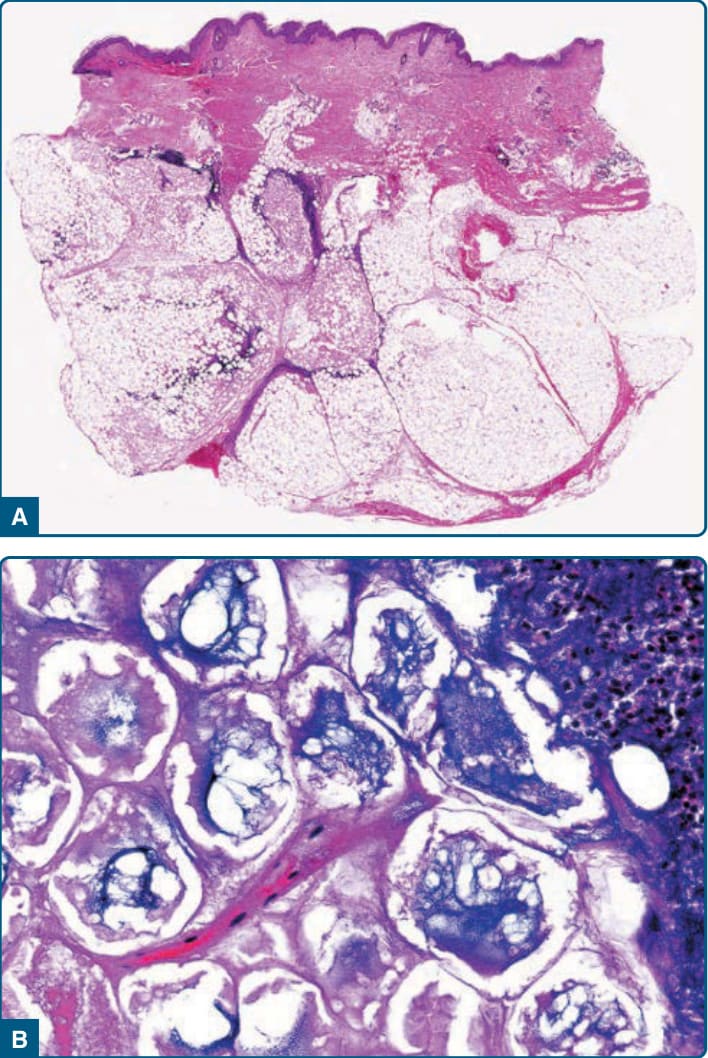
Figure 73-12 Pancreatic panniculitis. A, Low-power magnification shows a mostly lobular panniculitis with adipocyte necrosis at the center of the fat lobule. B, Higher-power magnification shows ghost adipocytes, necrotic adipocytes without nuclei and with cytoplasmic fine, granular basophilic material resulting from calcification.
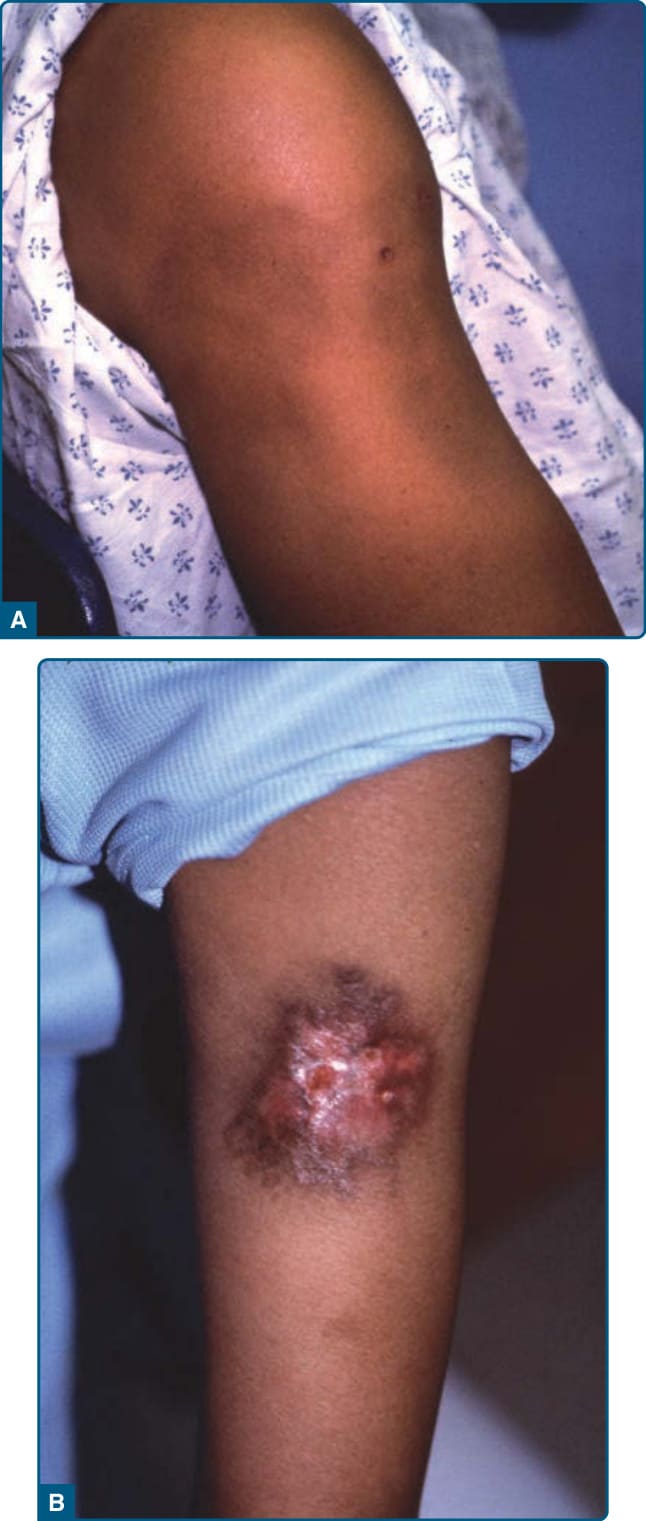
Figure 73-13 Lupus panniculitis. A, Atrophic upper arm lesion with superficial hyperpigmentation. B, With superimposed discoid lupus erythematosus and ulceration.
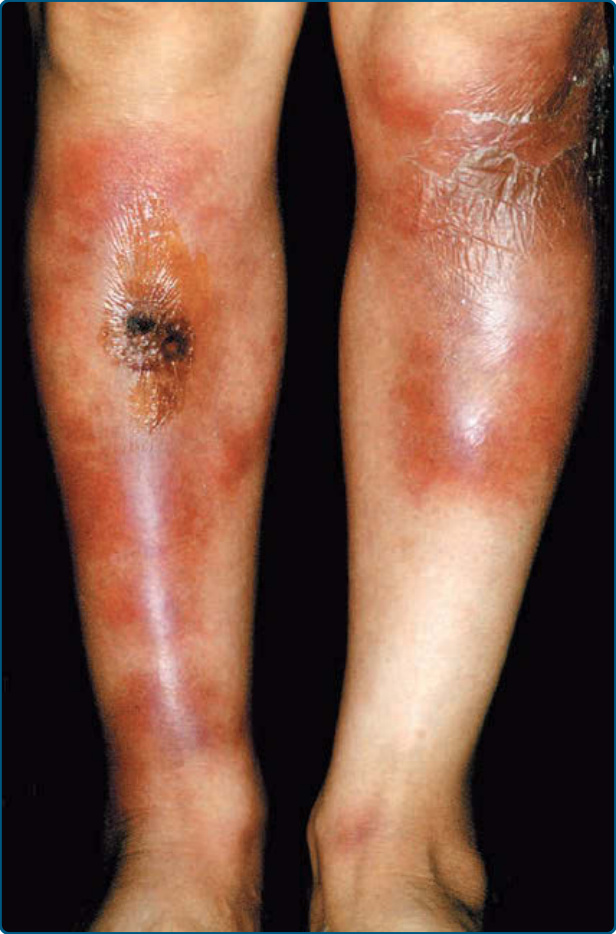
Figure 73-14 Cytophagic histiocytic panniculitis. Multiple erythematous plaques and nodules on the lower extremity.
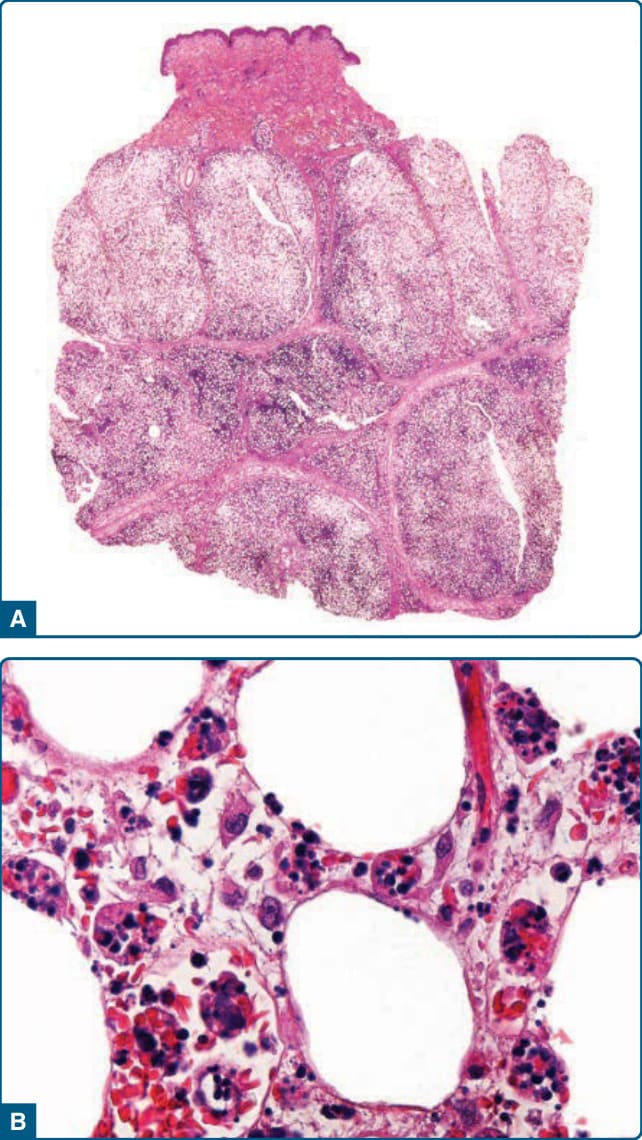
Figure 73-15 Cytophagic histiocytic panniculitis. A, Lowpower magnification histologic image shows lobular panniculitis. B. Cytophagocytic “bean bag” cells in the adipose tissue.
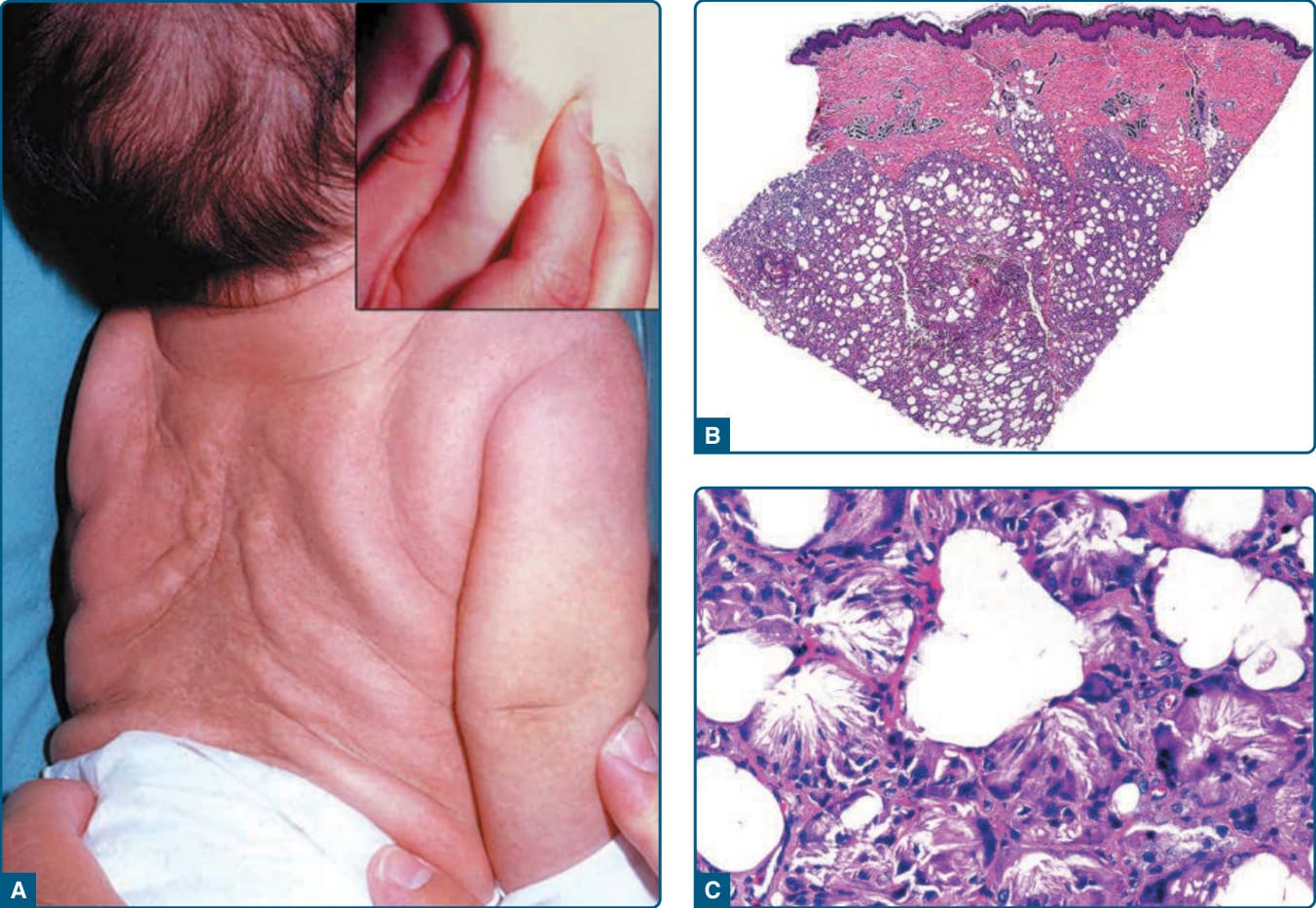
Figure 73-16 Subcutaneous fat necrosis of the newborn. A, Circumscribed, indurated subcutaneous nodules on the back. B, Low-power magnification histology shows mostly lobular panniculitis. C, Higher-power magnification shows needleshaped clefts of adipocytes, histiocytes, and multinucleated giant cells.
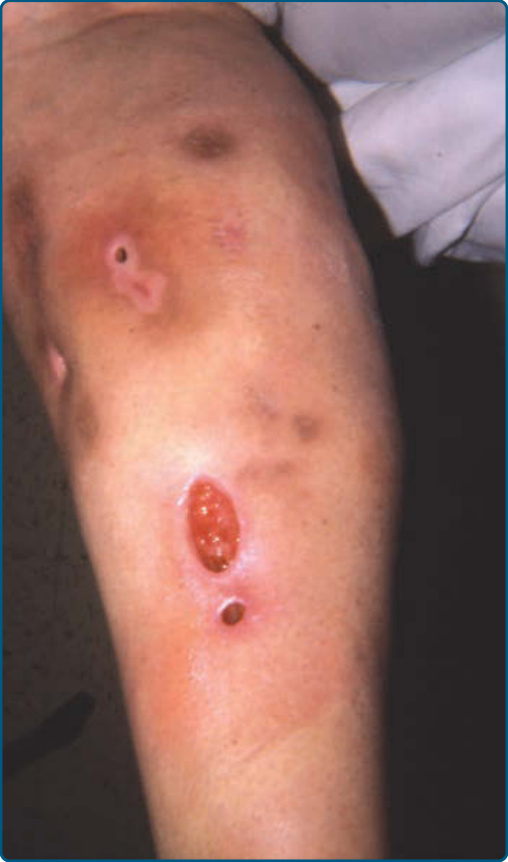
Figure 73-17 Factitial panniculitis. Self-induced round and angulated ulceration on the leg at sites of injections and trauma.
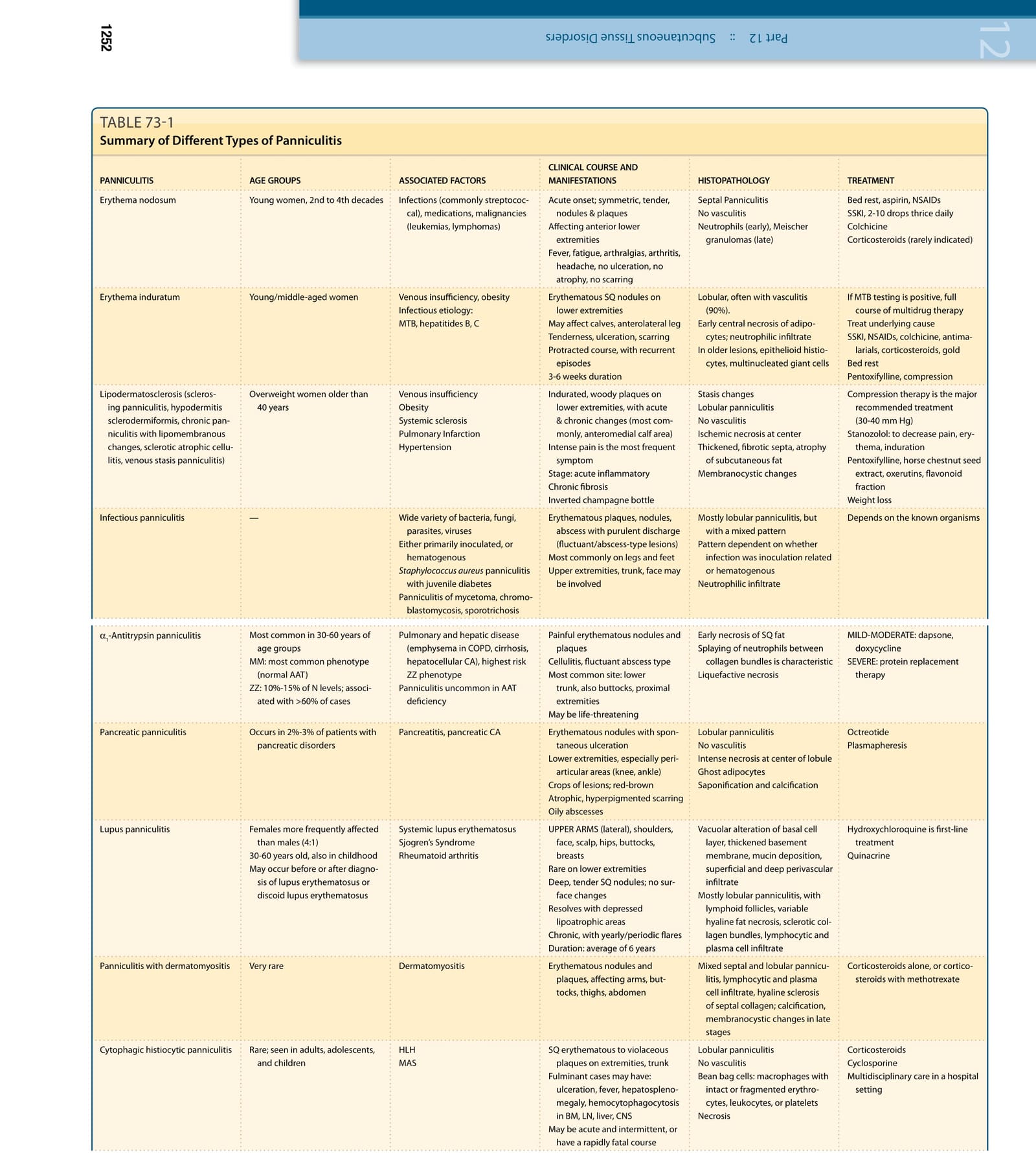
TABLE 73-1
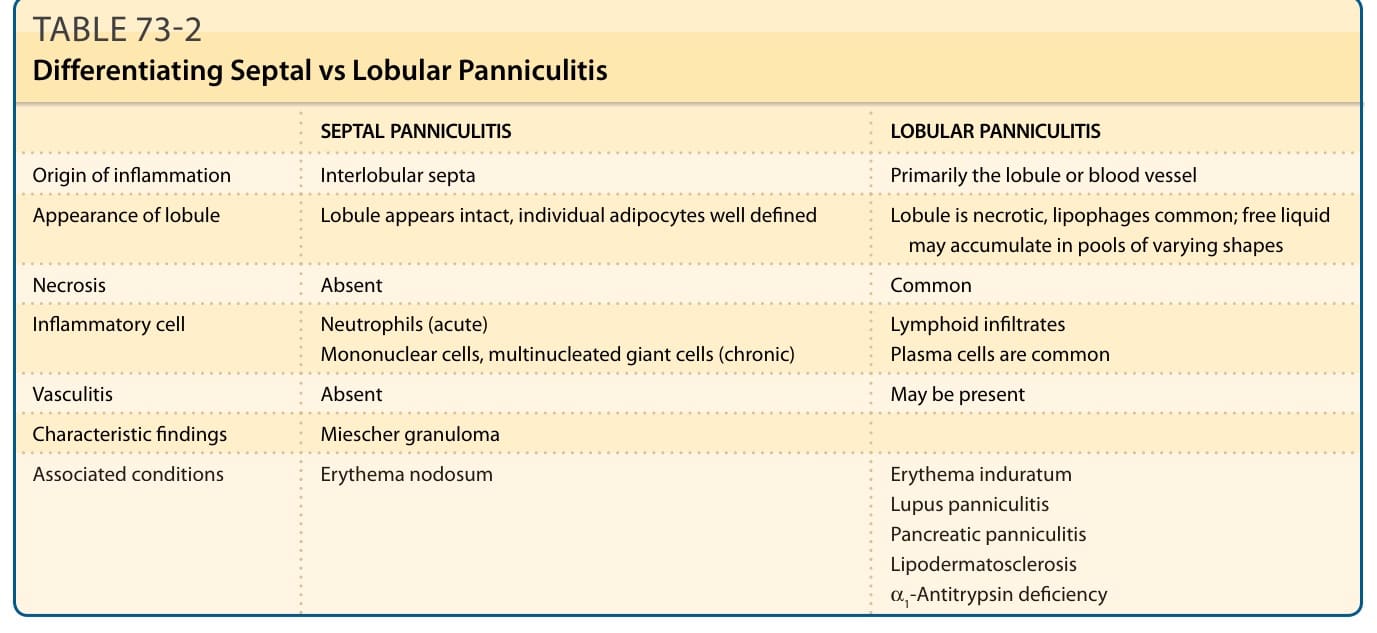
TABLE 73-2 Differentiating Septal vs Lobular Panniculitis
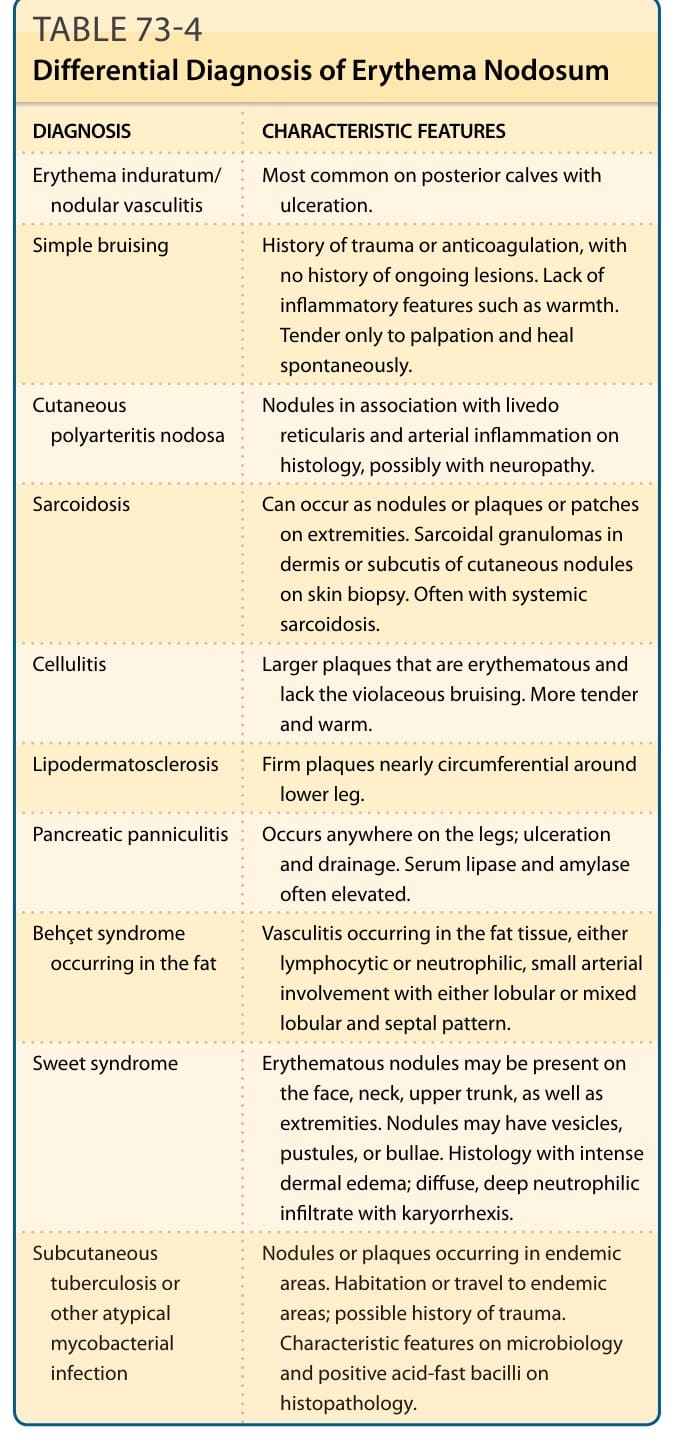
Table 73-4 outlines the differential diagnosis of EN.
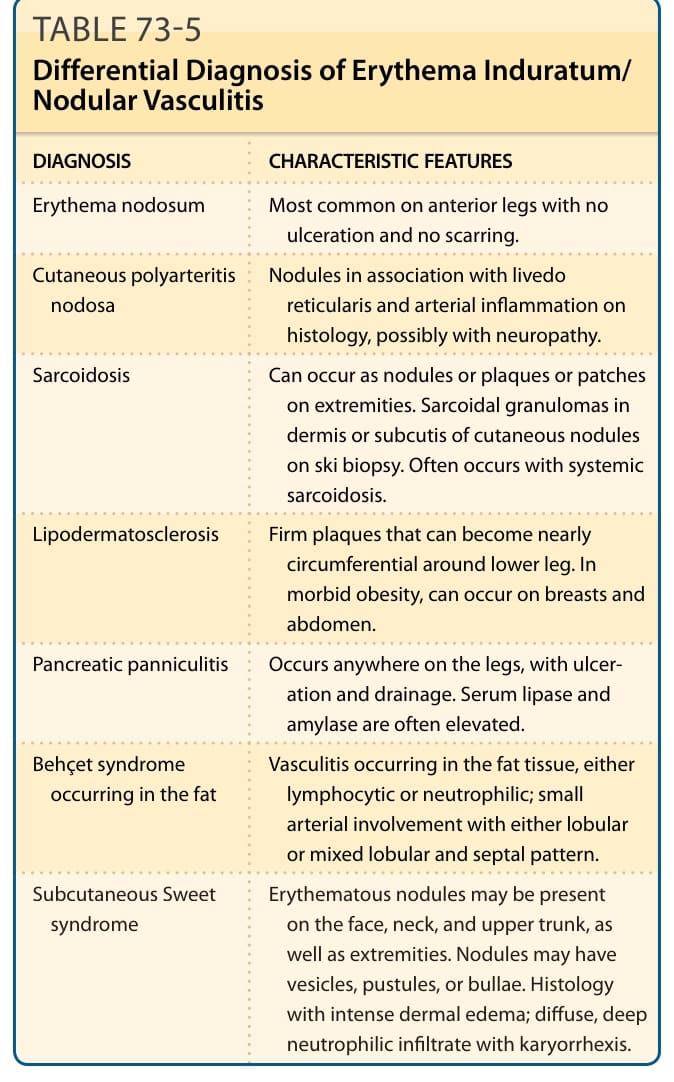
Table 73-5 outlines the differential diagnosis of erythema induratum/nodular vasculitis.
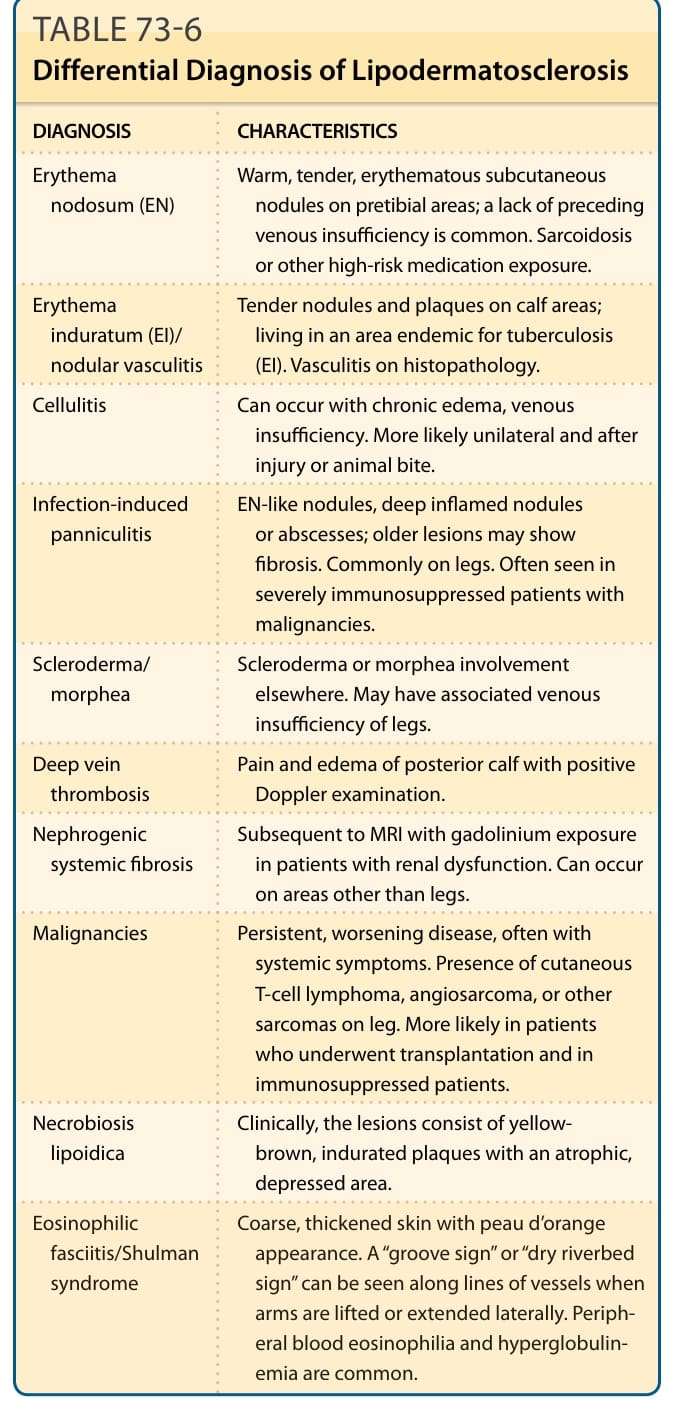
Table 73-6 outlines the differential diagnosis of LDS.
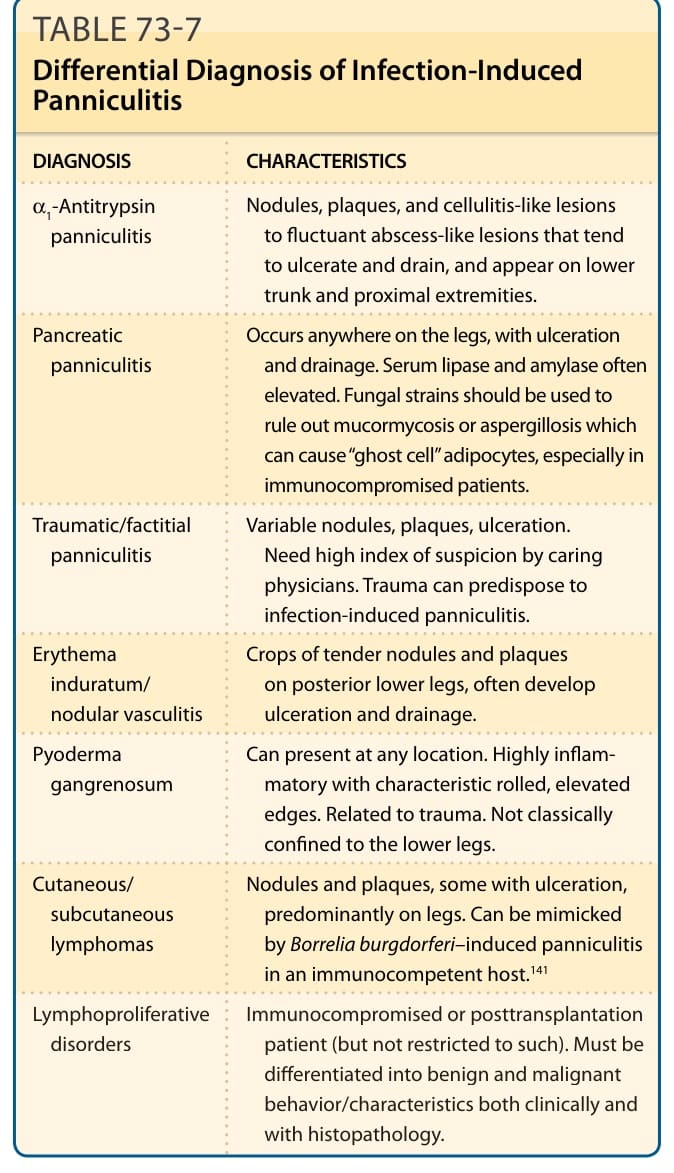
Table 73-7 outlines the differential diagnosis of IIP.
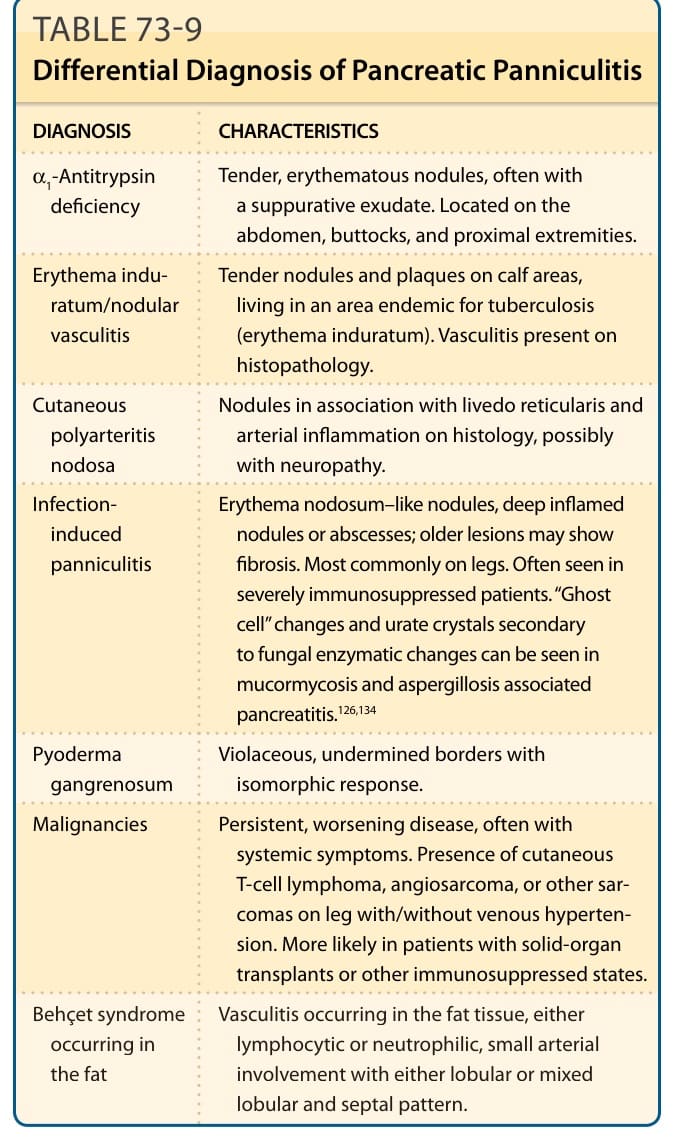
Table 73-9 outlines the differential diagnosis of pancreatic panniculitis.

Table 73-10 outlines the differential diagnosis of LEP.
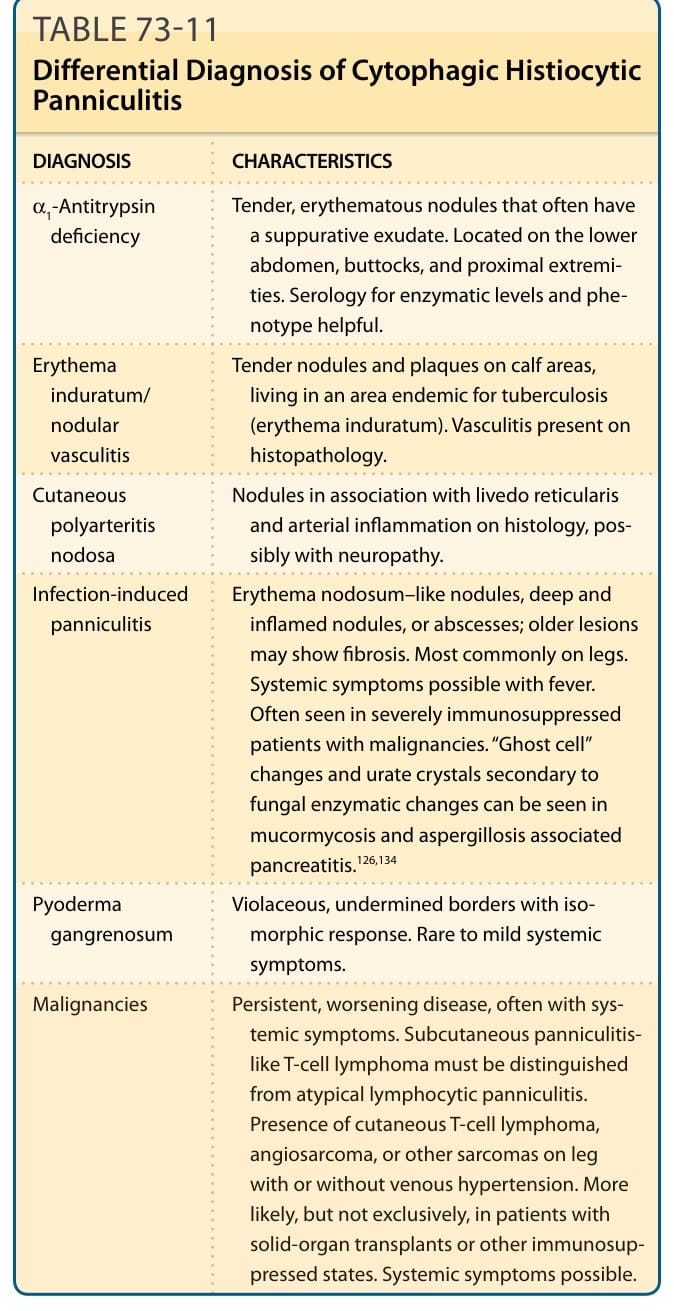
Table 73-11 outlines the differential diagnosis of CHP.

Table 73-12 outlines the differential diagnosis of SCFN.
Table 73-13 outlines the differential diagnosis of cold panniculitis.