復發性多軟骨炎 (Relapsing Polychondritis)
PART 10
自體免疫結締組織與風濕病疾患 (Autoimmune Connective Tissue and Rheumatologic Disorders)
重點一覽 (AT-A-GLANCE)
■ 復發性多軟骨炎 (relapsing polychondritis) 是一種罕見的多系統自體免疫疾病。
■ 其致病機轉牽涉多種因素,包括遺傳易感性、針對軟骨結構的免疫致敏 (immunization),以及細胞激素 (cytokine) 與趨化激素 (chemokine) 特徵的改變。
■ 超過 30% 的病人合併有相關疾病,主要為自體免疫性或血液性疾病。
■ 反覆發作的軟骨炎 (chondritis) 會導致軟骨結構進行性破壞。
■ 其他富含蛋白多醣 (proteoglycan-rich) 的結構,如眼睛、血管或內耳,也會受到侵犯。
■ 皮膚表現相當常見,尤其常與骨髓化生不良 (myelodysplasia) 相關。這些表現並無特異性,類似於貝賽特氏病 (Behçet disease) 與發炎性腸道疾病 (inflammatory bowel diseases) 中所見者。
流行病學 (EPIDEMIOLOGY)
復發性多軟骨炎 (relapsing polychondritis, RP) 是一種罕見的發炎性疾病,估計盛行率為每百萬人 4.5 例。在英國,RP 的發生率估計為 0.71/百萬/年,女性略高於男性 (0.76 對 0.66)。男性確診時的平均年齡 (範圍) 為 55 歲 (範圍:17–81 歲),女性為 51 歲 (範圍:11–79 歲)。本病可發生於幼童與老年人。雖然多數病例見於白人,但並無證據支持族裔或地理因素扮演角色。
臨床特徵 (CLINICAL FEATURES)
疾病通常以特徵性的軟骨炎 (chondritis) 突然發作,較少以關節炎 (arthritis) 或眼部發炎為起始表現。非特異性的初始症狀,如發燒或體重減輕,則少見。
皮膚表現 (CUTANEOUS FINDINGS)
軟骨炎的發作通常呈現復發–緩解 (relapsing–remitting) 的型態。發炎發作期一般持續數天至數週,可自發性消退或於治療後消退;數週或數月後可再次復發,最終導致軟骨破壞。耳廓軟骨炎 (auricular chondritis) 最為常見 (佔 85% 病例),造成耳廓 (pinna) 軟骨部位疼痛、發紅與腫脹,而不侵犯不含軟骨的耳垂 (lobe)(圖 69-1)。診斷上不需做耳廓軟骨的切片。經過數次發作後,耳廓可能變得鬆軟且下垂,呈現「花椰菜樣 (cauliflower)」外觀(圖 69-2);有時則因鈣化 (calcifications) 而變得僵硬。鼻軟骨炎 (nasal chondritis)(佔 65% 病例)的發炎程度較輕,表現為鼻痛、鼻塞、流鼻水,有時伴隨鼻出血 (epistaxis)。特徵性的鞍鼻畸形 (saddle-nose deformity)(圖 69-3)可能在後續出現,或在沒有先前發炎發作的情況下出現。其他皮膚表現有時是 RP 的起始表現 (佔 12% 病例),在一個大型系列中超過三分之一的病人後續被察覺到。其他皮膚表現並無特異性,包括四肢上類似結節性紅斑 (erythema nodosum) 外觀的結節(圖 69-4)、軀幹上固定的環狀蕁麻疹樣丘疹 (fixed annular urticarial papules)、紫斑 (purpura)(圖 69-5)、口腔或複雜性阿弗他潰瘍 (oral or complex aphthosis)(圖 69-6)、淺表性靜脈炎 (superficial phlebitis)、網狀青斑 (livedo reticularis)(圖 69-7)、四肢潰瘍、肢端壞死 (distal necrosis)。嗜中性球皮膚病 (neutrophilic dermatoses) 主要在合併血液學異常時觀察到:無菌性膿疱 (sterile pustules)(圖 69-8)、史威特氏症候群 (Sweet syndrome) 及其組織球樣 (histiocytoid) 亞型、持久性隆起性紅斑 (erythema elevatum diutinum),以及壞疽性膿皮症 (pyoderma gangrenosum)。皮膚病灶與軟骨炎發作可能同時出現或不同時出現。病理特徵包括嗜中性球性或淋巴球性血管炎 (neutrophilic or lymphocytic vasculitis)、血栓性血管病灶、嗜中性球浸潤,以及真皮或皮下組織的非特異性發炎。有與沒有皮膚表現的病人,其 RP 的臨床表現相似。然而,當 RP 與骨髓化生不良 (myelodysplasia) 相關時,皮膚表現的頻率 (>90%)、首次軟骨炎的年齡,以及男女比例似乎都較高;因此,老年人若出現這些表現,應重複進行血球計數,以偵測隱匿性的骨髓化生不良。
非皮膚表現 (NONCUTANEOUS FINDINGS)
呼吸道軟骨炎 (respiratory tract chondritis) 雖然在發病初期不常見,但會發生於高達 50% 的 RP 病人,且可能致命。呼吸道侵犯表現為聲音沙啞、非生產性的持續咳嗽、呼吸困難,以及/或喘鳴 (wheezing)。呼吸道軟骨炎的併發症包括上呼吸道塌陷、阻塞性呼吸功能不全,以及續發性感染。肋軟骨炎 (costochondritis)(佔 35% 的 RP 病人)會引起胸壁疼痛,亦可能影響呼吸。關節痛是常見的起始表現 (佔 30% 的 RP 病人)。周邊或中軸骨骼的大小關節皆可能受侵犯。其關節炎呈間歇性、遊走性、不對稱、血清陰性 (seronegative),且通常無侵蝕性 (nonerosive)。將近 60% 的 RP 病人會出現眼部發炎。表層鞏膜炎 (episcleritis) 與鞏膜炎 (scleritis)(圖 69-9)是最常見的表現,其次為乾性角結膜炎 (keratoconjunctivitis sicca)、虹膜炎 (iritis)、視網膜病變 (retinopathy) 與角膜炎 (keratitis)。罕見情況下,角膜穿孔、視網膜血管炎 (retinal vasculitis) 與視神經炎 (optic neuritis) 會導致失明。傳導性聽力喪失 (conductive hearing loss) 續發於外耳道狹窄、耳咽管 (eustachian tube) 軟骨炎,或漿液性中耳炎;而感知性聽力喪失 (perception hearing loss) 則可能因感覺神經性侵犯所致。前庭功能障礙的症狀,如頭暈、運動失調 (ataxia)、噁心與嘔吐,通常為急性發作,並隨時間改善。
心血管表現的範圍很廣,涵蓋不同的心臟組織(主動脈瓣與/或二尖瓣逆流、傳導系統受損、心包炎)以及各種類型的血管(胸主動脈與腹主動脈炎導致動脈瘤、類高安氏病 (Takayasu-like) 的主動脈弓症候群、中血管與大血管血管炎、白血球破碎性血管炎 (leukocytoclastic vasculitis) 與血栓性靜脈炎 (thrombophlebitis))。病灶可為發炎性與/或血栓性。部分(但非全部)血栓性表現與抗磷脂質症候群 (antiphospholipid syndrome) 有關。大血管血管炎傾向於在多年的隱匿性、且往往是不顯性 (occult) 疾病後發生,即使在免疫抑制治療下仍可能出現。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
迄今為止,RP 的病因仍不甚明瞭。發炎最初位於軟骨膜周圍 (perichondrial),特徵為含有淋巴球、嗜中性球、巨噬細胞與漿細胞的多形性發炎浸潤。T 細胞主要為 CD4;抗原呈現細胞 (antigen-presenting cells) 被活化並表現人類白血球抗原-D 相關抗原 (human leukocyte antigen-D related, HLA-DR)。直接免疫螢光 (direct immunofluorescence) 在軟骨膜–軟骨交界處不一致地顯示免疫球蛋白與 C3 沉積。隨著疾病進展,軟骨被發炎細胞侵入,並釋放出蛋白水解酶,如基質蛋白酶 (matrix proteinase, MMP)-3 以及組織蛋白酶 (cathepsins) K 與 L。MMP-8、MMP-9 與彈性蛋白酶 (elastase) 定位於軟骨周邊,軟骨逐漸劣化,伴隨嗜鹼性 (basophilia) 喪失、糖胺聚醣 (glycosaminoglycans) 釋出、膠原蛋白與彈性組織的碎裂。到了晚期,軟骨被纖維結締組織取代,其中可能含有膠狀囊腫 (gelatinous cysts) 與鈣化。體液性免疫反應扮演角色的依據,源自三分之一 RP 病人(尤其在 RP 急性期)體內存在抗第 II 型膠原蛋白 (collagen type II) 的抗體。抗體效價似乎與症狀嚴重度相關。RP 病人中亦偵測到的其他抗體包括:抗第 IX 型與第 XI 型膠原蛋白的抗體(這些次要膠原蛋白佔軟骨膠原蛋白的 5% 至 10%);抗 matrilin-1(一種主要表現於上呼吸道軟骨的細胞外基質蛋白)的抗體;以及抗軟骨寡聚基質蛋白 (cartilage oligomeric matrix protein) 的抗體(這是另一種表現於耳廓、氣管與鼻軟骨的軟骨蛋白)。已有數種動物模型發表,以這些不同的軟骨蛋白進行免疫致敏,可誘發各種類似病人所見的軟骨炎表現。雖然人體研究與小鼠模型強力支持第 II 型膠原蛋白、matrilin-1 與軟骨寡聚基質蛋白作為潛在標靶抗原扮演重要角色,但偵測針對這些蛋白的抗體其敏感度與特異度均低,因此並未用於臨床實務。RP 的觸發事件包括機械性刺激,如創傷與穿洞 (piercing),這些可能暴露軟骨基質的隱蔽抗原 (cryptic antigens) 並誘發自體免疫過程。T 細胞在 RP 致病機轉中的影響雖然較少被研究,但已在病人與動物模型中得到證實,其專一性針對相同的軟骨蛋白。自一名 RP 病人分離出的 T 細胞純株 (T-cell clones),被發現對應於第 II 型膠原蛋白第 261 至 273 位殘基的胜肽具有專一性,且受限於 DRBI∗0101 或 DRBI∗0401 等位基因。RP 可能是一種第一型輔助 T 細胞 (T-helper-1) 介導的疾病,因為血清中干擾素-γ (interferon-γ)、介白素 (interleukin, IL)-12 與 IL-2 的濃度與疾病活動度的變化呈平行,而第二型輔助 T 細胞 (T-helper-2) 細胞激素的濃度則否。其他參與致病機轉的介質包括 sTREM-1、MCP-1、MIP-1β 與 IL-8。一個複雜的細胞激素網絡協調著 RP 病灶中浸潤細胞的招募。RP 的遺傳背景在不同族群間似乎有所不同。HLA-DR4 在德國族群中被發現與 RP 相關,但在日本族群中則否。在日本族群中,RP 與 HLA-DRB∗16:02、HLA-DQB1∗05:02 與 HLA-B∗67:01 相關,且在遺傳上與其他風濕病不同。
診斷 (DIAGNOSIS)
RP 的各種診斷標準是以特徵性的臨床表現為基礎。陽性的組織學確認則鮮少需要(表 69-1)。表 69-2 列出主要的鑑別診斷以及可能與 RP 相關的疾病。
1. McAdam 等人
(1) 雙側耳廓軟骨炎 (2) 無侵蝕性血清陰性發炎性多關節炎 (3) 鼻軟骨炎 (4) 眼部發炎 (5) 呼吸道軟骨炎 (6) 耳蝸與/或前庭損傷。診斷 RP 時,病人必須符合上述 6 項標準中的 3 項。
2. Damiani 與 Levine
診斷 RP 時,病人必須具備 (1) 至少符合 3 項 McAdam 臨床標準 (2) 符合一項或多項 McAdam 臨床標準+軟骨發炎的切片確認 (3) 在 2 個或以上不同解剖部位出現軟骨炎,且對類固醇與/或 dapsone 有反應。
3. Michet 等人
(1) 主要標準
– 耳廓軟骨炎 – 鼻軟骨炎 – 喉氣管軟骨炎 (laryngotracheal chondritis)
(2) 次要標準
– 結膜炎、表層鞏膜炎、鞏膜炎或葡萄膜炎 (uveitis) – 聽力喪失 – 前庭功能障礙 – 血清陰性多關節炎。診斷 RP 時,病人必須符合 2 項主要標準,或 1 項主要標準+2 項次要標準。
RP 的實驗室檢查結果並無特異性,但與急性或慢性發炎一致。在腎臟受侵犯的情況下,尿液分析可能異常(繫膜增生 mesangial expansion、免疫球蛋白 A 腎病變、腎小管間質腎炎 tubulointerstitial nephritis,或壞死性腎絲球腎炎 necrotizing glomerulonephritis)。肺功能檢查,包括吸氣與呼氣的流量–容積曲線,應例行性地執行,以偵測隱匿性的侵犯。影像診斷可提供疾病活動度的資訊,且與臨床特徵的相關性優於非特異性的發炎實驗室標記。此外,臨床上不顯著的軟骨侵犯亦可被偵測到。事實上,大多數疾病表現都可藉由斷層影像(電腦斷層或 MRI)加以客觀化。輔以功能性資料,例如擴散加權影像 (diffusion-weighted imaging) 與葡萄糖代謝分析(如 fluorodeoxyglucose-positron emission tomography [FDG-PET])所得者,可正確評估疾病活動度。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
RP 的臨床病程呈進行性,並伴隨間歇性發作。受侵犯器官表現的數目、其嚴重度以及對治療的反應皆無法預測。復發性多軟骨炎疾病活動度指數 (Relapsing Polychondritis Disease Activity Index, RPDAI) 是透過國際合作所開發,用以評估疾病活動度。對一大型病人系列進行群集分析 (cluster analysis),可將病人分為 3 種臨床表現型:血液型、呼吸型與輕症型。第一組合併骨髓化生不良的病人與死亡相關;第二組合併氣管支氣管侵犯的病人與感染相關。相對地,歸入輕症表現型的病人沒有嚴重併發症,且可能出現臨床緩解。在多變項分析中,與死亡相關的因素為男性、心臟異常與合併骨髓化生不良。英國的族群基礎世代研究指出,RP 的相對死亡率可能比一般族群高出 2 至 3 倍。
治療處置 (MANAGEMENT)
由於 RP 的病程高度多變,在缺乏標準化指引的情況下,個別化治療是達到最佳處置的關鍵。無法建立一套治療演算法。一般性的治療指引乃基於對病人系列的回溯性分析或個別病例報告。對於輕度耳廓或鼻軟骨炎、關節痛或輕度關節炎的病人,非類固醇抗發炎藥 (nonsteroidal antiinflammatory drugs)、秋水仙素 (colchicine) 或 dapsone 可能有用。較嚴重的表現則需依其嚴重度給予口服皮質類固醇,劑量為每公斤體重 0.3 至 1 mg/kg。對於急性呼吸道阻塞、突發性聽力喪失,以及/或手術介入前(氣管造口術、主動脈瘤修補、心臟瓣膜置換)會給予脈衝式靜脈類固醇 (pulse intravenous steroids)。長期使用皮質類固醇可降低復發的頻率與嚴重度,但無法預防重要器官的侵犯。在臨床緩解期間是否應持續類固醇治療,仍不明確。許多種免疫抑制劑已被使用,作為疾病修飾與類固醇節制 (steroid-sparing) 藥物,並取得一定成效。Methotrexate (0.3 mg/kg/week) 往往有效。Cyclophosphamide 用於 RP 的嚴重型。Azathioprine、mycophenolate mofetil、cyclosporine、leflunomide 與 chlorambucil 的效果則不一致。雖然活動性 RP 病人血清中的 IL-6 與腫瘤壞死因子-α (tumor necrosis factor-α) 濃度通常並未升高,infliximab 仍是最常使用的生物製劑,但結果不一——常為部分或完全有效,有時則續發於療效喪失或嚴重感染。Rituximab 通常無治療效果。其他曾試用於 RP 的生物製劑包括 tocilizumab、anakinra、etanercept、adalimumab 與 certolizumab。接受治療的病人數目過少,無法做出明確結論。
在少數難治型嚴重 RP 的病人中,曾施行治療強化 (treatment intensification) 後接續自體幹細胞移植 (autologous stem cell transplantation)。
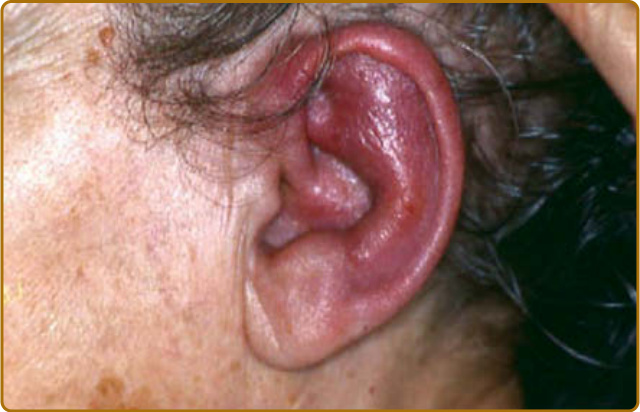
圖 69-1:復發性多軟骨炎 (Relapsing polychondritis)。耳朵軟骨部位的疼痛性發炎。
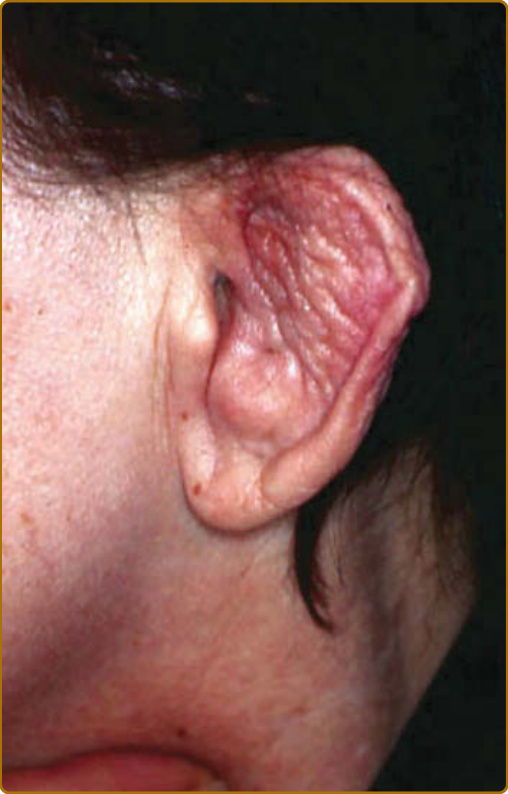
圖 69-2:復發性多軟骨炎 (Relapsing polychondritis)。鬆軟下垂、呈「花椰菜樣 (cauliflower)」外觀的耳朵。
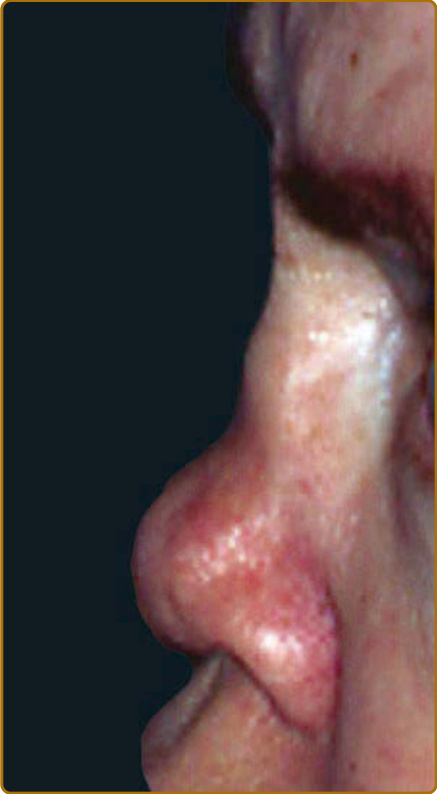
圖 69-3:復發性多軟骨炎 (Relapsing polychondritis)。鞍鼻畸形 (Saddle-nose deformity)。
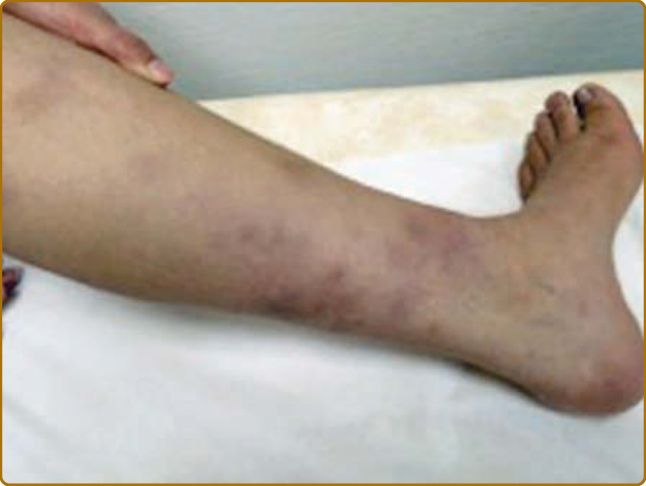
圖 69-4:復發性多軟骨炎 (Relapsing polychondritis)。四肢的結節,可能續發於間隔性脂層炎 (septal panniculitis)、血管炎、血栓,或深層嗜中性球浸潤。
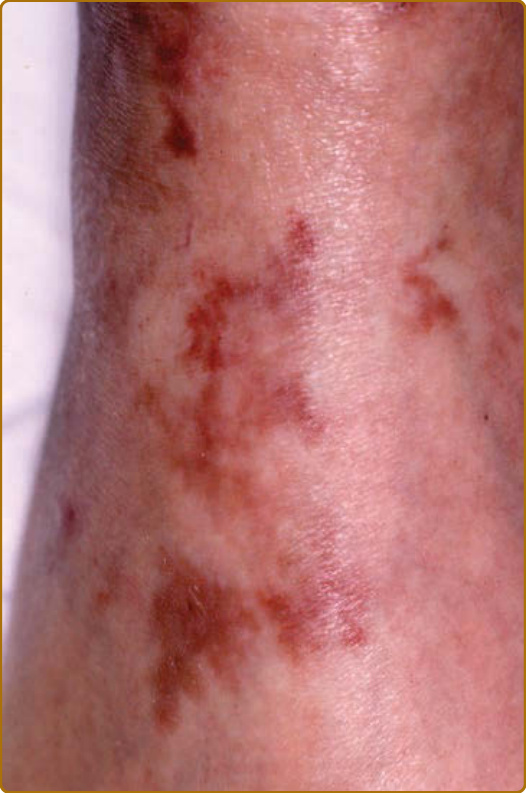
圖 69-5:復發性多軟骨炎 (Relapsing polychondritis)。續發於白血球破碎性血管炎 (leukocytoclastic vasculitis) 的紫斑 (Purpura)。
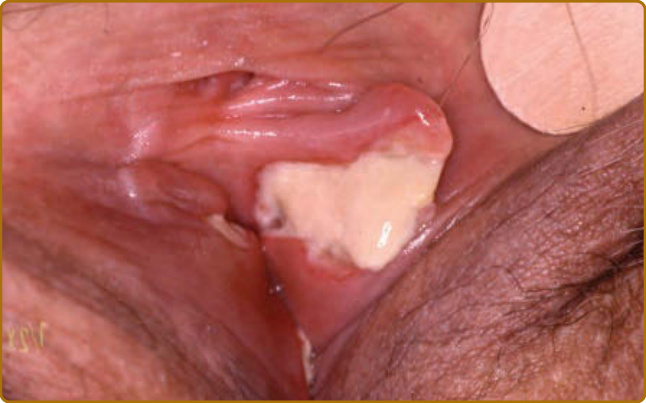
圖 69-6:復發性多軟骨炎 (Relapsing polychondritis)。與急性耳廓軟骨炎同時發生的巨大外陰阿弗他潰瘍 (vulvar aphtha)。
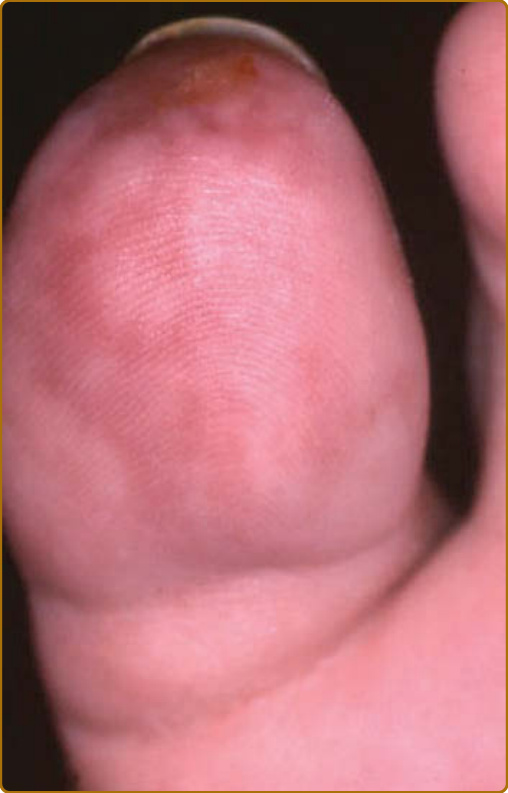
圖 69-7:復發性多軟骨炎 (Relapsing polychondritis)。續發於白血球破碎性血管炎 (leukocytoclastic vasculitis) 的壞死性網狀青斑 (necrotic livedo)。
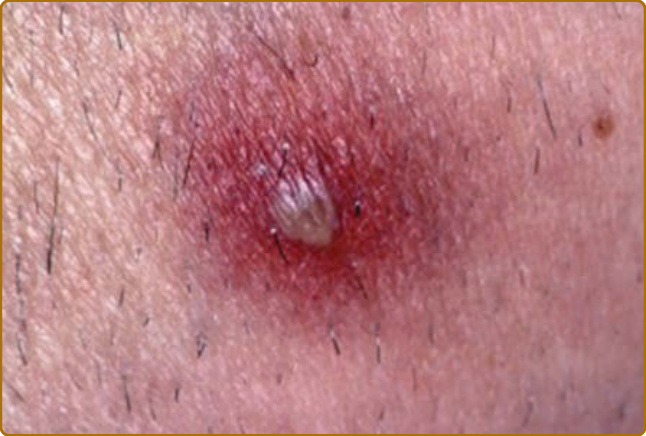
圖 69-8:復發性多軟骨炎 (Relapsing polychondritis)。如貝賽特氏病 (Behçet disease) 中所見的無菌性膿疱 (sterile pustule)。
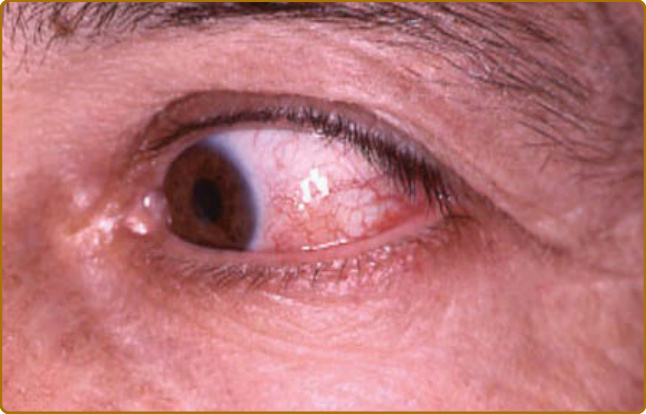
圖 69-9:復發性多軟骨炎 (Relapsing polychondritis)。提示鞏膜炎 (scleritis) 的紅眼,必須由眼科醫師確認。

表 69-1:各種診斷標準 (Different Diagnostic Criteria)
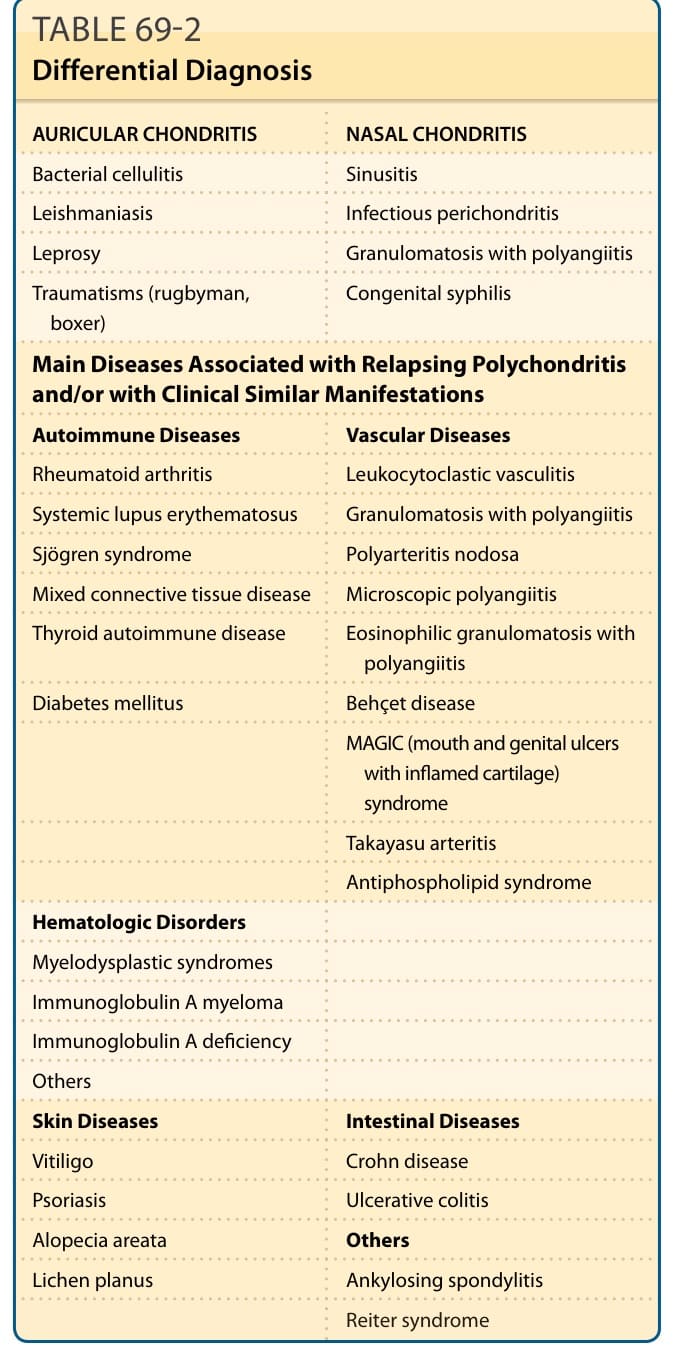
表 69-2:鑑別診斷 (Differential Diagnosis)