Relapsing Polychondritis
10
AT-A-GLANCE
■ Relapsing polychondritis is a rare multisystem autoimmune disease.
■ Different factors are implicated in the pathogenesis, including a genetic susceptibility, immunization against cartilaginous structures, and modification of cytokine and chemokine signatures.
■ More than 30% of patients have an associated disease, mainly of autoimmune or hematologic origin.
■ Recurrent episodes of chondritis lead to progressive destruction of cartilaginous structures.
■ Other proteoglycan-rich structures, such as eyes, blood vessels, or inner ear, are also affected.
■ Dermatologic manifestations occur frequently, especially in association with myelodysplasia. They are nonspecific and resemble those observed in Behçet disease and inflammatory bowel diseases.
EPIDEMIOLOGY
Relapsing polychondritis (RP) is a rare inflammatory disease with an estimated prevalence of 4.5 per million people.1 The incidence of RP has been estimated to be 0.71/million/year in the United Kingdom, being slightly higher in women than men (0.76 vs 0.66). The mean age (range) at diagnosis in men was 55 years (range: 17-81 years) and in women 51 years (range: 11-79 years).2 Development of the disease may occur in young children and in the elderly. Although most cases have been reported in whites, there is no evidence supporting the role of ethnic or geographical factors.
CLINICAL FEATURES
Disease onset is usually sudden with characteristic chondritis, and/or, less frequently, arthritis or ocular inflammation. Nonspecific initial symptoms, such as fever or weight loss, are rare.
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
Attacks of chondritis usually occur in a relapsing– remitting pattern. Inflammatory episodes generally last a few days or weeks and may subside spontaneously or upon treatment; recurrences after weeks or months occur and subsequently result in cartilage
destruction.3 Auricular chondritis is the most frequent occurrence (85% of cases), causing pain, redness, and swelling of the cartilaginous portion of the pinna, sparing the noncartilaginous lobe (Fig. 69-1). Biopsy of the auricular cartilage is unnecessary for diagnosis. After several attacks, the pinna may become soft and sloppy with a cauliflower aspect (Fig. 69-2); sometimes it is stiff from calcifications. Nasal chondritis (65% of cases) is less inflammatory, presenting with nasal pain, stuffiness, rhinorrhea, and sometimes epistaxis. The characteristic saddle-nose deformity (Fig. 69-3) may appear secondly or without previous inflammatory episodes. Other dermatologic manifestations are sometimes the presenting feature of RP (12% of cases), noticed subsequently in more than one-third of patients of a large series.4 The other dermatologic manifestations are nonspecific and include nodules on the limbs (Fig. 69-4) with an aspect of erythema nodosum, fixed annular urticarial papules on the trunk,5 purpura (Fig. 69-5), oral or complex aphthosis (Fig. 69-6), superficial phlebitis, livedo reticularis (Fig. 69-7), ulcerations on the limbs, distal necrosis. Neutrophilic dermatoses are mainly observed in association with hematologic abnormalities: sterile pustules (Fig. 69-8), Sweet syndrome and its histiocytoid subtype, erythema elevatum diutinum, and pyoderma gangrenosum. Skin lesions appear either concomitantly or not with attacks of chondritis. Pathologic features include neutrophilic or lymphocytic vasculitis, thrombotic vascular lesions, neutrophil infiltrates, and nonspecific inflammation of the dermis or subcutis. Patients with and without dermatologic manifestations have similar clinical manifestations of RP. However, the frequency of dermatologic manifestations (>90%), age at first chondritis, and male-to-female ratio seems higher when RP is associated with myelodysplasia4; so, their presence in an old person warrants repeated blood cell counts to detect a smouldering myelodysplasia.
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
Respiratory tract chondritis, although uncommon at presentation, occurs in up to 50% of RP patients, and may be lethal. Respiratory involvement is manifested by hoarseness, nonproductive persistent cough, dyspnea, and/or wheezing. Complications of respiratory tract chondritis include upper airway collapse, obstructive respiratory insufficiency, and secondary infections. Costochondritis (35% of RP patients) induces parietal pains, which may also compromise respiration. Joint pain is a common presenting feature (30% of RP patients). Large and small joints of the peripheral or
axial skeleton may all be affected. Arthritis is intermittent, migratory, asymmetric, seronegative, and usually nonerosive. Nearly 60% of RP patients developed ocular inflammation. Episcleritis and scleritis (Fig. 69-9) are the most common manifestations, followed by keratoconjunctivitis sicca, iritis, retinopathy, and keratitis. Rarely, corneal perforation, retinal vasculitis, and optic neuritis will lead to blindness. Conductive hearing loss is secondary to stenosis of the external auditory canal, eustachian tube chondritis, or serous otitis media, whereas perception hearing loss may occur as a consequence of sensorineural involvement. Symptoms of vestibular dysfunction such as dizziness, ataxia, nausea, and vomiting are usually acute and improve with time.
10
The spectrum of cardiovascular manifestations is wide and includes different heart tissues (aortic and/ or mitral regurgitation, impairment of the conduction system, pericarditis) and all types of vessels (thoracic and abdominal aortitis leading to aneurysms, Takayasu-like aortic arch syndrome, medium-vessel and large-vessel vasculitis, leukocytoclastic vasculitis, and thrombophlebitis). Lesions are inflammatory and/or thrombotic. Some, but not all, thrombotic manifestations have been linked to antiphospholipid syndrome. Large-vessel vasculitis tends to occur after several years of smoldering and, often, occult disease, despite immunosuppressive therapy.
1187
10
ETIOLOGY AND PATHOGENESIS
To date, the etiology of RP is still poorly known. The inflammation is initially perichondrial, characterized by an inflammatory polymorphic infiltrate with lymphocytes, neutrophils, macrophages, and plasma cells. The T cells are primarily CD4; the antigen-presenting cells are activated with expression of human leukocyte antigen-D related (HLA-DR). Direct immunofluorescence inconsistently shows immunoglobulins and C3 deposits at the junction perichondriumcartilage. With the progression of the disease, the
1188
cartilage is invaded by inflammatory cells with release of proteolytic enzymes such as matrix proteinase (MMP)-3 and cathepsins K and L. MMP-8, MMP-9, and elastase are localized at the periphery of the cartilage that gradually deteriorated with loss of basophilia, release of glycosaminoglycans, fragmentation of collagen and elastic tissue. At a late stage, it is replaced by fibrous connective tissue that may contain gelatinous cysts and calcifications. The role of the humoral immune response was based on the presence of antibodies to collagen type II in a third of patients with RP, especially in the acute phase of RP. Antibody titers seemed to correlate with the severity of symptoms. Other antibodies also detected in patients with RP include antibodies to collagen type IX and type XI; minor collagens that represent 5% to 10% of cartilage collagens; antibodies to matrilin-1, an extracellular matrix protein, predominantly expressed in upper respiratory tract cartilage; and antibodies to cartilage oligomeric matrix protein, additional cartilage protein, expressed in auricular, tracheal, and nasal cartilage. Several animal models have been published in which immunization with these various cartilage proteins induced a variety of chondritis manifestations that mimic those seen in patients.6 Although human studies and murine models strongly support a prominent role for collagen type II, matrilin-1, and cartilage oligomeric matrix protein, as potential target antigens, the detection of antibodies toward these proteins has low sensitivity and specificity, and therefore not used in clinical practice. Triggering events of RP include mechanical stimuli such as traumas and piercing, which may expose cryptic antigens of the cartilaginous matrix and induce an autoimmune process. Influence of T cells in RP pathogenesis, although less investigated, also has been demonstrated in patients and in animal models with specificity against the same cartilage proteins. T-cell clones isolated from an RP patient were found to be specific for a peptide corresponding to residues 261 to 273 of the type II collagen and were restricted to either the DRBI∗0101 or the DRBI∗0401 allele.7 RP is likely a T-helper-1–mediated disease as serum levels of interferon-γ, interleukin (IL)-12, and IL-2 parallel changes in disease activity, while the levels of T-helper-2 cytokines do not. Other mediators are involved in the pathogenesis such as sTREM-1, MCP-1, MIP-1β, and IL-8. A complex cytokine network orchestrates the recruitment of infiltrating cells in RP lesions. The genetic background of RP seems to be different according to the different populations. HLA-DR4 was found to be associated with RP in the German but not in the Japanese population. In the Japanese population RP was associated with HLA-DRB∗16:02, HLA- DQB1∗05:02, and HLA-B∗67:01, and differs genetically from other rheumatic diseases.8
DIAGNOSIS
The different diagnostic criteria for RP are based on characteristic clinical manifestations.9 A positive histologic confirmation is rarely required (Table 69-1). Table 69-2 shows the main differential diagnoses and diseases that may be associated with RP.
10
-
McAdam et al.10
-
McAdam et al.10
(1) Bilateral auricular chondritis (2) Nonerosive seronegative inflammatory polyarthritis (3) Nasal chondritis (4) Ocular inflammation (5) Respiratory chondritis (6) Cochlear and/or vestibular damage For the diagnosis of RP, patients must have 3 of 6 of the above criteria.
2. Damiani and Levine11
(1) Bilateral auricular chondritis (2) Nonerosive seronegative inflammatory polyarthritis (3) Nasal chondritis (4) Ocular inflammation (5) Respiratory chondritis (6) Cochlear and/or vestibular damage For the diagnosis of RP, patients must have 3 of 6 of the above criteria.
2. Damiani and Levine11
For the diagnosis of RP, patients must have (1) At least 3 of the McAdam clinical criteria (2) One or more of the McAdam clinical criteria + biopsy confirmation of cartilage inflammation (3) Chondritis at 2 or more separate anatomic locations with response to steroids and/or dapsone
3. Michet et al.12
For the diagnosis of RP, patients must have (1) At least 3 of the McAdam clinical criteria (2) One or more of the McAdam clinical criteria + biopsy confirma-
tion of cartilage inflammation (3) Chondritis at 2 or more separate anatomic locations with
response to steroids and/or dapsone
3. Michet et al.12
(1) Major criteria
(1) Major criteria
– Auricular chondritis – Nasal chondritis – Laryngotracheal chondritis (2) Minor criteria
– Auricular chondritis – Nasal chondritis – Laryngotracheal chondritis (2) Minor criteria
– Conjunctivitis, episcleritis, scleritis, or uveitis – Hearing loss – Vestibular dysfunction – Seronegative polyarthritis For the diagnosis of RP, patients must have 2 major criteria or 1 major criteria + 2 minor criteria.
– Conjunctivitis, episcleritis, scleritis, or uveitis – Hearing loss – Vestibular dysfunction – Seronegative polyarthritis For the diagnosis of RP, patients must have 2 major criteria or 1 major criteria + 2 minor criteria.
Laboratory findings in RP are nonspecific, but consistent with an acute or chronic inflammation. Urinalysis may be abnormal in case of renal involvement (mesangial expansion, immunoglobulin A nephropathy, tubulointerstitial nephritis, or necrotizing glomerulonephritis). Pulmonary function tests, including inspiratory and expiratory flow volume curves, should be performed systematically to detect occult involvement. Imaging diagnosis delivers information about the degree of disease activity that correlates better with clinical features than unspecific inflammatory laboratory markers. Additionally, clinically unapparent cartilage involvement can be detected. Indeed, most disease manifestations can be objectified by means of cross-sectional imaging (computed tomography or MRI). The complementary use of functional data like those obtained with aid of diffusion-weighted imaging and glucose metabolism analysis (eg, fluorodeoxyglucose-positron emission tomography [FDG-PET]), allow a correct evaluation of the disease activity.13
CLINICAL COURSE AND PROGNOSIS
The clinical course of RP is progressive with intermittent flares. The number of different organ manifestations, their severity, and the response to treatment
1189
10
AURICULAR CHONDRITIS NASAL CHONDRITIS
Bacterial cellulitis Sinusitis
Leishmaniasis Infectious perichondritis
Leprosy Granulomatosis with polyangiitis
Traumatisms (rugbyman, boxer) Congenital syphilis
Main Diseases Associated with Relapsing Polychondritis and/or with Clinical Similar Manifestations
Autoimmune Diseases Vascular Diseases
Rheumatoid arthritis Leukocytoclastic vasculitis
Systemic lupus erythematosus Granulomatosis with polyangiitis
Sjögren syndrome Polyarteritis nodosa
Mixed connective tissue disease Microscopic polyangiitis
Thyroid autoimmune disease Eosinophilic granulomatosis with polyangiitis
Diabetes mellitus Behçet disease
MAGIC (mouth and genital ulcers with inflamed cartilage) syndrome
Takayasu arteritis
Antiphospholipid syndrome
Hematologic Disorders
Myelodysplastic syndromes
Immunoglobulin A myeloma
Immunoglobulin A deficiency
Others
Skin Diseases Intestinal Diseases
Vitiligo Crohn disease
Psoriasis Ulcerative colitis
Alopecia areata Others
Lichen planus Ankylosing spondylitis
Reiter syndrome
Reiter syndrome
are unpredictable.3 The Relapsing Polychondritis Disease Activity Index (RPDAI) was developed to assess disease activity thanks to an international cooperation.14 Cluster analysis of a large series of patients permitted to separate patients into 3 clinical phenotypes: hematologic, respiratory, and mild. The first group of patients with myelodysplasia was associated with death; the second group of patients with tracheobronchial involvement was associated with infections. By contrast, patients included in the mild phenotype had no severe complication and the possible occurrence of clinical remission. Factors associated with death on multivariable analysis were male sex, cardiac abnormalities, and concomitant myelodysplasia.15 The United Kingdom population-based cohort study suggested that the relative mortality in RP may be 2 to 3 times higher than in the general population.2
1190
MANAGEMENT
Because of the highly variable course of RP, individualized therapy is the key to optimum management without standardized guidelines. An algorithm for treatment cannot be established. General therapeutic guidelines are based on retrospective analyses of series of patients or isolated case reports. Nonsteroidal antiinflammatory drugs, colchicine, or dapsone may be useful for patients with mild auricular or nasal chondritis, arthralgia, or mild arthritis. More serious manifestations required oral corticosteroids in dose of 0.3 to 1 mg/kg of body weight according to their severity. Pulse intravenous steroids are prescribed for acute airway obstruction, sudden hearing loss, and/or before surgical intervention (tracheostomy, aortic aneurysm repair, cardiac valve replacement). Long-term corticosteroids decrease the frequency and severity of recurrences, although they do not prevent vital organ involvement. Whether steroid therapy should be continued during clinical remission periods remains unclear. Many kinds of immunosuppressants have been used with some success as diseasemodifying and steroid-sparing agents. Methotrexate (0.3 mg/kg/week) is often effective. Cyclophosphamide is used in severe forms of RP. Azathioprine, mycophenolate mofetil, cyclosporine, leflunomide, and chlorambucil have produced inconsistent effects. Although serum levels of IL-6 and tumor necrosis factor-α are usually not elevated in patients with active RP, infliximab has been the most frequently used biologic agent with variable results—frequently partial or complete efficacy, sometimes secondary to loss of efficacy or severe infection. Rituximab usually has no treatment effect. Other biologic agents tried in RP include tocilizumab, anakinra, etanercept, adalimumab, and certolizumab. The number of treated patients is too low to allow definitive conclusions.3
In a few patients with refractory severe RP, treatment intensification followed by autologous stem cell transplantation has been performed.
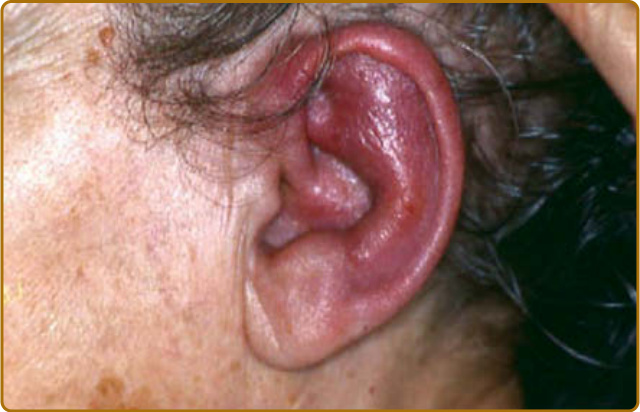
Figure 69-1 Relapsing polychondritis. Painful inflammation of the cartilaginous portion of ear.
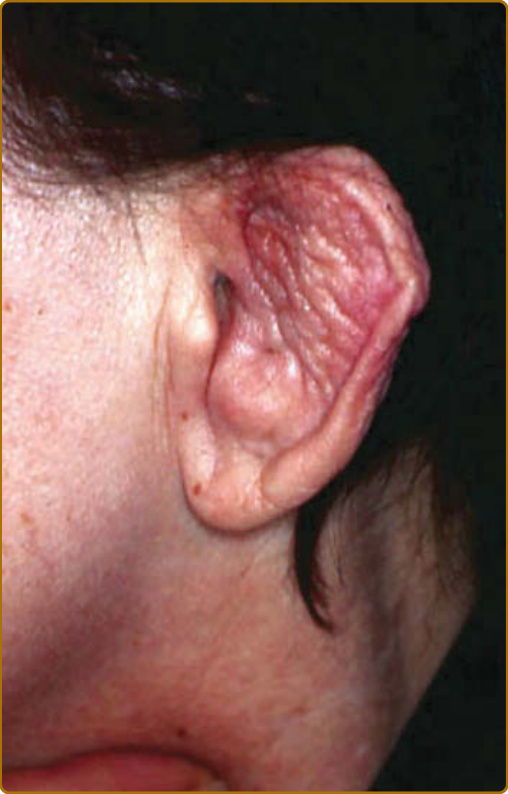
Figure 69-2 Relapsing polychondritis. Soft and sloppy ear with a “cauliflower” aspect.
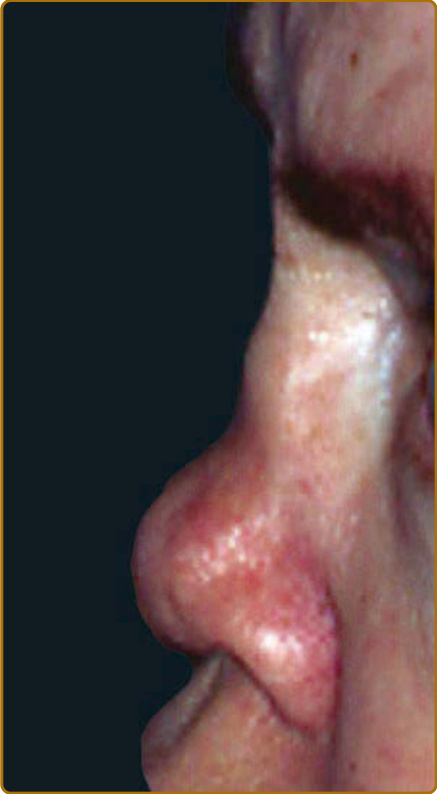
Figure 69-3 Relapsing polychondritis. Saddle-nose deformity.
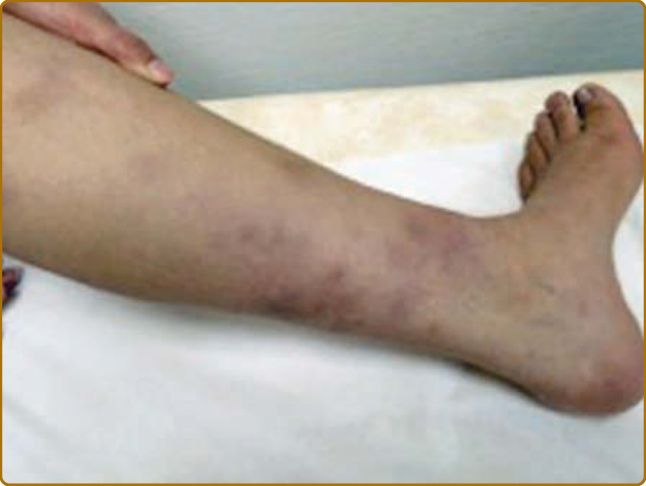
Figure 69-4 Relapsing polychondritis. Nodules of the limbs that may be secondary to septal panniculitis, vasculitis, thrombosis, or deep neutrophils infiltrate.
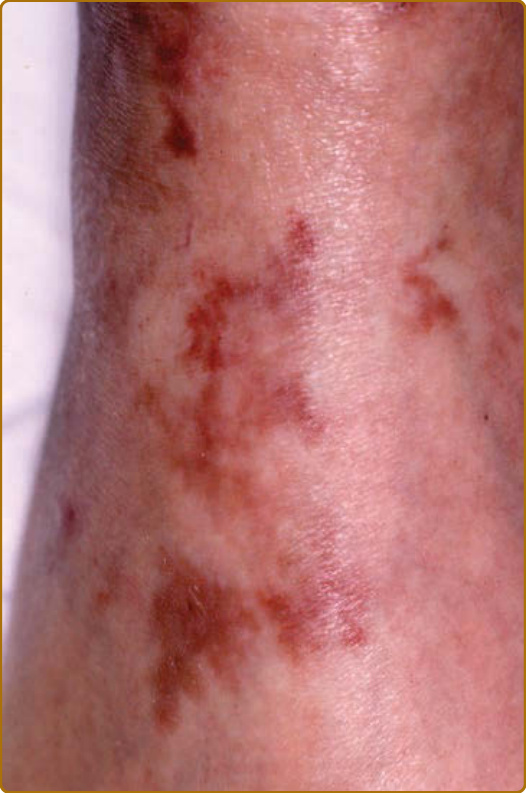
Figure 69-5 Relapsing polychondritis. Purpura secondary to leukocytoclastic vasculitis.
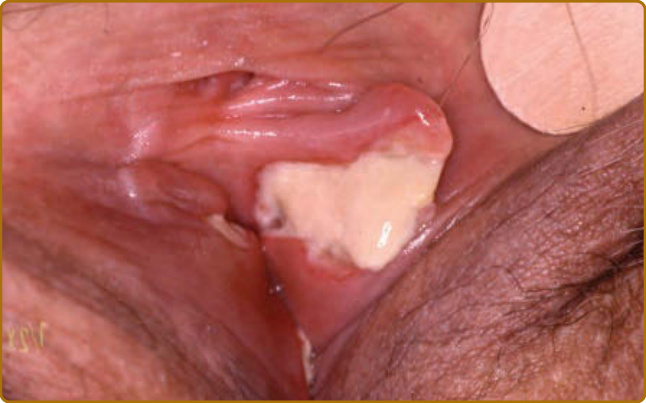
Figure 69-6 Relapsing polychondritis. Giant vulvar aphtha contemporary to acute auricular chondritis.
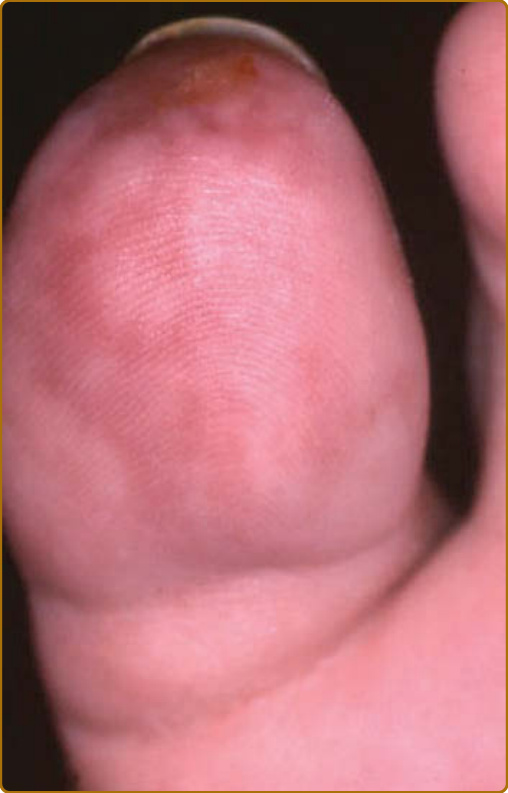
Figure 69-7 Relapsing polychondritis. Necrotic livedo secondary to leukocytoclastic vasculitis.
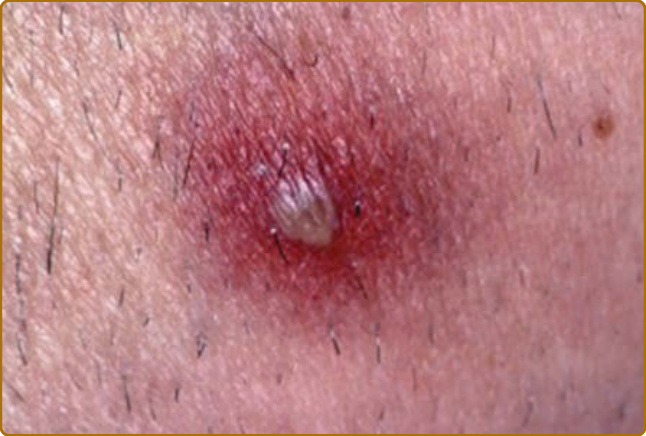
Figure 69-8 Relapsing polychondritis. Sterile pustule as observed in Behçet disease.
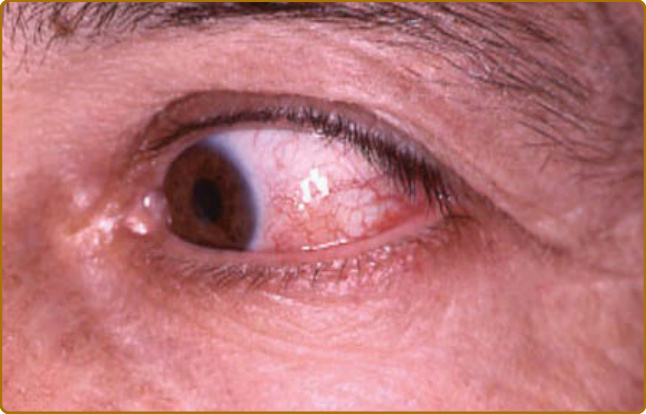
Figure 69-9 Relapsing polychondritis. Red eye suggestive of scleritis, which must be confirmed by an ophthalmologist.

TABLE 69-1 Different Diagnostic Criteria9
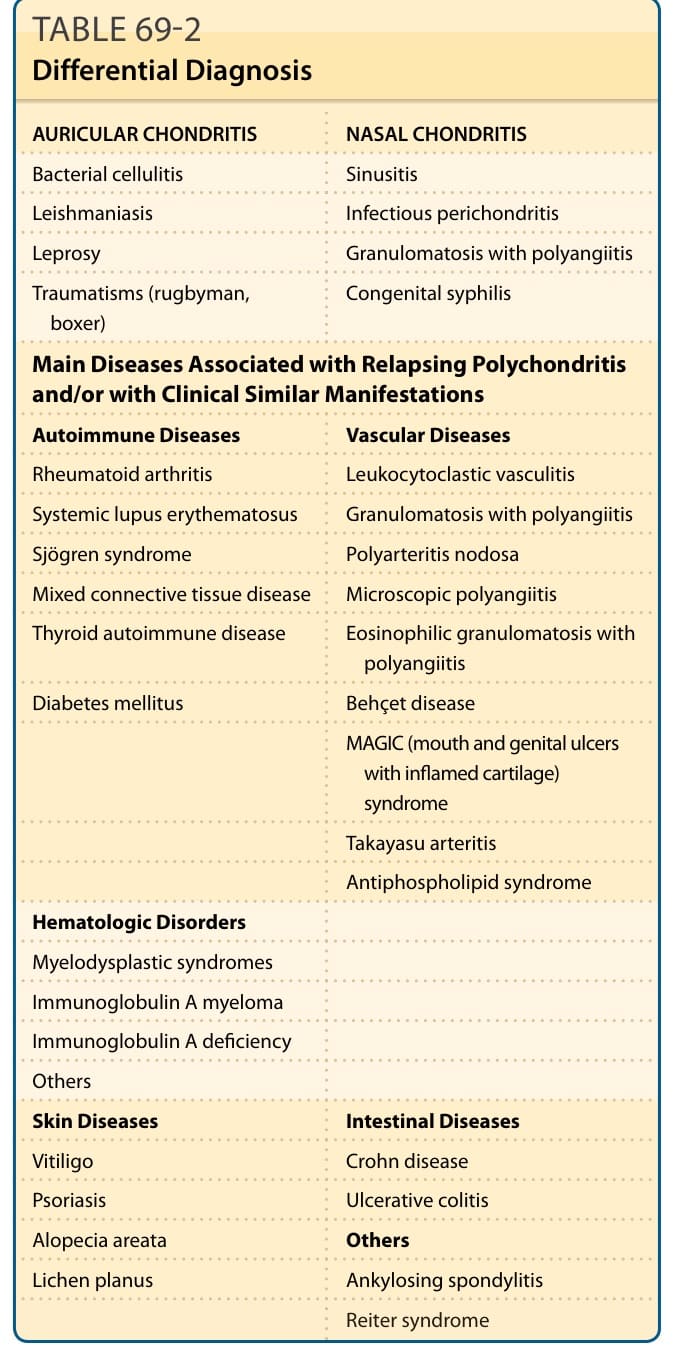
TABLE 69-2 Differential Diagnosis