紅斑性狼瘡 (Lupus Erythematosus)
PART 10
自體免疫結締組織與風濕性疾病 (Autoimmune Connective Tissue and Rheumatologic Disorders)
重點一覽 (AT-A-GLANCE)
■ 一群異質性的疾病,其共同點為發展出對自身核酸 (self-nucleic acids) 及其相關蛋白質的免疫反應,此光譜的一端為僅侷限於皮膚的疾病,另一端則為嚴重的內臟侵犯。
■ 皮膚病灶可能為狼瘡所特有,亦可能為非特異性(在其他病況中也可見)。
■ 急性皮膚紅斑性狼瘡(acute cutaneous lupus erythematosus,顴部皮疹 malar rash)幾乎總是伴隨潛在的內臟侵犯。亞急性皮膚狼瘡 (subacute cutaneous lupus) 病人約有 50% 符合美國風濕病學會 (American College of Rheumatology) 的系統性紅斑性狼瘡 (systemic lupus erythematosus) 標準(但通常僅表現出輕微的全身性臨床表現)。慢性皮膚狼瘡 (chronic cutaneous lupus)(典型盤狀紅斑性狼瘡 classic discoid lupus erythematosus、狼瘡脂膜炎 lupus panniculitis、凍瘡狼瘡 chilblain lupus 與腫脹性紅斑性狼瘡 tumid lupus erythematosus)病人最常為僅限皮膚或以皮膚為主的疾病。
■ 典型盤狀紅斑性狼瘡會造成瘢痕,並可能導致永久性毀容。
■ 亞急性皮膚狼瘡與急性皮膚紅斑性狼瘡具高度光敏感性 (highly photosensitive),且特徵性地不形成瘢痕 (nonscarring)。
■ 紅斑性狼瘡的非特異性皮膚病灶包括非瘢痕性禿髮 (nonscarring alopecia)、口腔潰瘍 (mouth ulcers)、光敏感、雷諾現象 (Raynaud phenomenon)、血管炎/血管病變 (vasculitis/vasculopathy) 與大疱性系統性紅斑性狼瘡 (bullous systemic lupus erythematosus) 等。它們常預示著系統性紅斑性狼瘡的惡化 (flare)。
■ 治療包括物理性防曬、局部防曬乳、局部與短期全身性糖皮質素 (glucocorticoids)、抗瘧藥 (antimalarials)、類視色素 (retinoids)、沙利竇邁/來那度胺 (thalidomide/lenalidomide)、傳統免疫抑制劑 (conventional immunosuppressives),以及生物製劑療法。
■ 紅斑性狼瘡在女性中遠較常見(女男比為 9:1)。
■ 系統性紅斑性狼瘡與皮膚紅斑性狼瘡兩者都與第一型干擾素 (type 1 interferon) 訊號傳遞的上調有關。
前言
紅斑性狼瘡(lupus erythematosus, LE)是一系列多樣化臨床疾病的根本命名,這些疾病的共同連結為發展出主要針對核小體 (nucleosomes) 與核糖核蛋白 (ribonucleoproteins) 分子組成成分的自體免疫。某些病人以系統性 LE(systemic LE, SLE)的危及生命表現呈現,而另一些病人——他們所罹患的代表著相同的基本潛在疾病過程——在其整個病程中所表現的可能僅止於孤立的盤狀 LE(discoid LE, DLE)皮膚病灶。將 LE 概念化為一個臨床光譜 (clinical spectrum)(圖 61-1)是方便的,其範圍從僅有侷限性 DLE 皮膚病灶的輕微受影響病人,到那些有可能因 LE 的全身性表現(如腎炎 nephritis、中樞神經系統 (CNS) 疾病或血管炎 vasculitis)而死亡的病人。個別 LE 病人所表現的皮膚侵犯模式,可提供洞見,協助判斷該病人的疾病最適合落在此光譜上的哪個位置。James N. Gilliam 最初設計的命名與分類系統,將 LE 的皮膚表現分為兩類:顯示 LE 特徵性組織學變化的病灶(LE 特異性皮膚疾病 LE-specific skin disease,即一種介面性皮膚炎 interface dermatitis),以及那些在組織病理學上對 LE 並不獨特、且/或可能在另一種疾病過程中見到的病灶(LE 非特異性皮膚疾病 LE-nonspecific skin disease)。¹ 在此脈絡下,「LE 特異性」一詞係指那些顯示介面性皮膚炎的病灶。皮膚 LE(cutaneous LE, CLE)一詞常與「LE 特異性皮膚疾病」同義使用,作為 LE 特異性皮膚疾病三大類別的總稱:急性皮膚 LE(acute cutaneous LE, ACLE)、亞急性皮膚 LE(subacute cutaneous LE, SCLE)與慢性皮膚 LE(chronic cutaneous LE, CCLE)。這將是我們討論 LE 病人身上所發生之極為多樣化皮膚病灶集合時所使用的架構(表 61-1)。LE 的本質在於其異質性,而治療者所面臨的挑戰是在構成此千變萬化 (protean) 疾病的特徵鑲嵌中,辨識出臨床上有用的模式。LE 全身性表現的概觀可見於美國風濕病學會(American College of Rheumatology, ACR)的 SLE 分類標準²(呈現於表 61-2),以及表 61-3 所呈現之 SLE 全身性表現的概要中。
流行病學 (EPIDEMIOLOGY)
LE 整體³ 以及 CLE 特別⁴ 的流行病學與社會經濟衝擊已有相關回顧。皮膚疾病是 LE 繼關節發炎之後第二常見的臨床表現。多達 45% 的 CLE 病人經歷某種程度的職業障礙 (vocational handicap)。近期的生活品質研究顯示,SLE 病人皮膚表現所造成的衝擊,僅次於與其疾病相關的疼痛與疲勞。⁵ 皮膚科生活品質指數 (Dermatology Life Quality Index) 與 SF-36 已被用於測量 CLE 病人的生活品質。兩份問卷都顯示,有活動性皮膚病灶的病人生活品質較低,且合併禿髮的病人受到的影響特別大。⁶
顴部(或蝴蝶斑 butterfly rash,侷限性 ACLE)已在大型 LE 病人世代中被報告佔 20% 至 60%。有限的資料顯示,廣泛性 ACLE 的斑丘疹 (maculopapular) 或 SLE 皮疹存在於約 35% 至 60% 的 SLE 病人中。ACLE,如同 SLE 整體一樣,女性比男性常見 8 倍。所有種族都會受影響;然而,ACLE 的早期臨床表現在膚色深的個體中可能被忽略。以 SCLE 病灶表現的病人佔 LE 病人族群的 7% 至 27%。SCLE 主要是白人女性的疾病,發病平均年齡在第五個十年。藥物誘發的 SCLE 病人在疾病發病時年齡稍長,這或許反映了因年齡相關的醫療問題(高血壓、心血管疾病)而較多暴露於藥物。CCLE 最常見的型式——典型 DLE 皮膚病灶——存在於 15% 至 30% 以各種方式選取的 SLE 族群中。約 5% 以孤立性侷限 DLE 表現的病人隨後發展出 SLE。已發表的以族群為基礎的資料強烈主張,孤立型 CLE 的發生率與盛行率等同於 SLE。⁷
雖然 DLE 可發生於嬰兒與老年人,但它最常見於 20 至 40 歲之間的個體。DLE 的女男比為 3:2 至 3:1,遠低於 SLE。所有種族都會受影響,但研究顯示 DLE 在黑人 (blacks) 中可能較為盛行。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
LE 特異性皮膚疾病的病因與致病機轉尚未被完全了解,雖然近期研究已提供許多新的洞見。LE 特異性皮膚疾病的致病機轉與 SLE 的致病機轉密不可分地交織在一起。簡而言之,SLE 是一種疾病,其中宿主因子(susceptibility genes 易感基因、荷爾蒙環境 hormonal milieu 等)與環境因子(紫外線 [ultraviolet, UV] 輻射、病毒與藥物)之間的交互作用導致自我耐受性 (self-tolerance) 的喪失,並誘導自體免疫。隨之而來的是免疫系統的活化與擴張,最終導致對終端器官 (end organs) 的免疫損傷與疾病的臨床表現。⁸ 近期研究突顯了干擾素-α (interferon-α) 訊號傳遞在 SLE 與 LE 特異性皮膚疾病兩者致病機轉中的重要角色。
免疫學因子 (IMMUNOLOGIC FACTORS)
多項研究突顯了漿細胞樣樹突狀細胞(plasmacytoid dendritic cells, PDCs)與第一型干擾素(type 1 interferon, IFN,由干擾素-α 與干擾素-β 組成)訊號傳遞在 SLE 與 LE 特異性皮膚疾病兩者致病機轉中的重要角色。第一型 IFN 是 SLE 病人微陣列 (microarray) 研究中被辨識出上調最為顯著的基因途徑,而在 CLE 病人的血液與皮膚中也見到第一型 IFN 增加。⁹,¹⁰
大量的 PDCs 存在於 CLE 病人的皮膚病灶中。¹¹ PDCs 透過類鐸受體 (toll-like receptors) 7 與 9 對 DNA 與 RNA 刺激產生反應,製造大量的第一型 IFN。在健康狀態下,PDCs 辨識病毒 DNA 與 RNA,而非自身 DNA;然而,由自體抗體結合來自凋亡細胞 (apoptotic cells) 之 DNA 與 RNA 所生成的免疫複合體 (immune complexes) 會被 PDCs 辨識,並導致活化與第一型 IFN 的產生。慢性的第一型 IFN 反過來透過其對 B 細胞與 T 細胞的作用,促成 SLE 中耐受性的喪失。¹²,¹³
近期研究已辨識出一條獨立的 DNA 感知與 RNA 感知途徑,稱為免疫刺激性 DNA 途徑 (immunostimulatory DNA pathway),它與內體 (endosomal) 類鐸受體不同,於細胞質內具有活性。由免疫刺激性 DNA 途徑基因突變所引起的單基因症候群 (Monogenetic syndrome),如見於 Aicardi-Goutières 症候群與 SAVI 中者,會導致免疫刺激性 DNA 途徑活性上調與慢性 IFN-β 的產生,並與自發性 LE 共有許多特徵。¹⁴ 免疫刺激性 DNA 途徑基因中的單核苷酸多型性 (single-nucleotide polymorphisms) 與 CLE 及 SLE 有關。¹⁵ 來自這些被稱為干擾素病變 (interferonopathies) 之疾病的洞見,進一步支持不當的第一型 IFN 活性(無論是 IFN-α 或 IFN-β)作為狼瘡致病機轉中近端 (proximal) 且驅動性事件的角色。
環境因子 (ENVIRONMENTAL FACTORS)
對於狼瘡體質 (lupus diathesis) 的遺傳易感性,其本身並不會產生疾病。確切而言,此類病人中自體免疫的誘導,似乎是由某種誘發事件所觸發,很可能是一種環境暴露。
藥物、病毒、UV 光,以及可能的菸草,都會誘發 SLE 的發展。UV 輻射很可能是 SLE 誘導期中最重要的環境因子,尤其是 LE 特異性皮膚疾病。UV 光很可能導致自我免疫與耐受性的喪失,因為它造成角質細胞 (keratinocytes) 的凋亡,這反過來使先前隱蔽的胜肽 (cryptic peptides) 得以被免疫監視 (immunosurveillance)。UVB 輻射使自體抗原(如 Ro/SS-A 及相關自體抗原 La/SS-B 與鈣網蛋白 calreticulin)從其於表皮角質細胞內部的正常位置移位至細胞表面。¹⁶ UVB 照射誘導 CCL27(皮膚 T 細胞吸引趨化激素 cutaneous T cell-attracting chemokine)的釋放,而 CCL27 上調趨化激素的表現,這些趨化激素會活化自體反應性 T 細胞與第一型 IFN,並產生樹突狀細胞 (dendritic cells),後者很可能在狼瘡致病機轉中扮演核心角色。¹⁷,¹⁸
近期一項大型病例對照研究報告,吸菸者比不吸菸者與曾吸菸者有較高的發展 SLE 風險。Werth 及其同事建立的一個合作式網路資料庫的橫斷面分析記錄了,治療抗拒型 (treatment-resistant) CLE 病人更可能吸菸。¹⁹ 數位作者已顯示,吸菸的 LE 特異性皮膚疾病病人對抗瘧藥治療的反應較差。²⁰⁻²²
許多藥物被認為涉及誘導 SLE 的各種特徵(表 61-4)。誘導 CLE 的藥物可由其光敏感 (photosensitizing) 特性連結起來。有人提出這些藥物造成角質細胞凋亡的增加、先前位於細胞內的胜肽暴露於表皮細胞表面,並增強促發炎細胞激素 (proinflammatory cytokines),如腫瘤壞死因子 (tumor necrosis factor, TNF)-α 與第一型 IFN。²³,²⁴
關於傳染性病原體(尤其是病毒)在 SLE 與 CLE 誘導中的角色有許多臆測。SLE 病人對艾司坦-巴爾病毒 (Epstein-Barr virus) 的血清轉換 (seroconversion) 幾乎是普遍的,而近期資料證明 SLE 病人對潛伏性艾司坦-巴爾病毒感染的控制有缺陷,這很可能源自針對艾司坦-巴爾病毒的 T 細胞反應改變。
臨床表現 (CLINICAL FINDINGS)
區分 LE 特異性皮膚疾病的各亞型是重要的,因為 LE 中皮膚侵犯的型式可反映潛在的 SLE 活性模式。事實上,就 CLE 而言,急性、亞急性與慢性這些命名指的是任何相關 SLE 的進展速度與嚴重度,而不一定與個別病灶已存在多久有關。例如,ACLE 幾乎總是發生在急性惡化的 SLE 背景下,而 CCLE 則常發生在沒有 SLE,或存在輕微、悶燒型 (smoldering) SLE 的情況下。SCLE 在此臨床光譜中佔據中間位置。亞分類雖然對於評估風險很重要,但有時很困難,因為在同一病人身上見到一種以上亞型的 LE 特異性皮膚疾病並不罕見,尤其在 SLE 病人中。
急性皮膚紅斑性狼瘡 (ACUTE CUTANEOUS LUPUS ERYTHEMATOSUS)
雖然侷限於顏面的 ACLE 是通常的表現型式,但 ACLE 可呈現廣泛性分布。侷限性 ACLE 一般被稱為 SLE 的典型蝴蝶斑或顴部皮疹(圖 61-2)。在侷限性 ACLE 中,融合性對稱性紅斑與水腫集中於顴部隆起 (malar eminences) 並橫跨鼻樑(已有 ACLE 單側侵犯的描述)。鼻唇溝 (nasolabial folds) 特徵性地不受侵犯。前額、下巴與頸部的 V 形區域可能受侵犯,並可能發生嚴重的顏面腫脹。偶爾,ACLE 起始為顏面上的小斑點 (macules) 及/或丘疹 (papules),之後可能變得融合並過度角化 (hyperkeratotic)。廣泛性 ACLE 表現為一種廣布的麻疹樣 (morbilliform) 或發疹性 (exanthematous) 皮疹,常集中於手臂與手部的伸側,並特徵性地不侵犯指關節(圖 61-3A)。雖然可能發生血管周圍甲褶紅斑 (perivascular nail fold erythema) 與毛細血管擴張 (telangiectasia)(圖 61-3B),但它們在皮肌炎 (dermatomyositis) 與系統性硬化症 (systemic sclerosis) 中相當更為常見且以更誇張的型式出現。廣泛性 ACLE 已被不加區別地稱為 SLE 的斑丘疹皮疹、光敏感性狼瘡皮膚炎 (photosensitive lupus dermatitis) 與 SLE 皮疹。一種極為急性型式的 ACLE 罕見可見,它可模擬毒性表皮壞死溶解症(toxic epidermal necrolysis, TEN;圖 61-4)。此型式的 LE 特異性水疱大疱性疾病 (vesiculobullous disease) 源於表皮角質細胞的廣泛凋亡,並最終導致全層表皮皮膚壞死 (full-thickness epidermal skin necrosis) 的區域,隨後剝脫。ACLE 可與真正的 TEN 區分,因為它主要發生在曝曬陽光的皮膚上,且發病較為隱匿。黏膜可能受侵犯也可能不受侵犯,如同 TEN 中一樣。ACLE 通常由暴露於 UV 光所誘發或加重。此型式的 CLE 可能相當短暫,僅持續數小時、數日或數週;然而,某些病人經歷較長期的活動期。發炎後色素變化 (Postinflammatory pigmentary change) 在膚色深重的病人中最為顯著。萎縮性瘢痕 (Atrophic scarring) 不會發生於 ACLE,除非該過程因繼發性細菌感染而複雜化。
亞急性皮膚紅斑性狼瘡 (SUBACUTE CUTANEOUS LUPUS ERYTHEMATOSUS)
SCLE 皮膚病灶的存在於 1979 年首次被描述為 LE 的一個獨特免疫遺傳亞群。²⁵ 以 SCLE 病灶為主的疾病表現,標誌著一個具有特徵性臨床、血清學與遺傳特徵之獨特 LE 亞群的存在。雖然發現循環中對 Ro/SS-A 核糖核蛋白顆粒的自體抗體強烈支持 SCLE 的診斷,但要做出 SCLE 的診斷並不需要此種自體抗體特異性的存在。SCLE 最初表現為紅斑性斑點及/或丘疹,演變為過度角化的丘疹鱗屑性 (papulosquamous) 或環狀/多環狀 (annular/polycyclic) 斑塊(圖 61-5)。雖然大多數病人具有環狀或丘疹鱗屑性 SCLE,但少數病人發展出兩種型態變異的元素。SCLE 病灶特徵性地具光敏感性,並主要發生於曝曬陽光的區域(即上背部、肩部、手臂伸側、頸部 V 形區域,較少見於顏面)。SCLE 病灶通常癒合而不留瘢痕,但可能消退並留下持久(若非永久)的白斑樣 (vitiligo-like) 白皮症 (leukoderma) 與毛細血管擴張。已有數種 SCLE 的變異型被描述。有時,SCLE 病灶最初以多形性紅斑 (erythema multiforme) 的外觀表現。此類病例類似 Rowell 症候群(在存在 La/SS-B 自體抗體的 SLE 病人身上發生之多形性紅斑樣病灶)。由於對表皮基底細胞 (basal cells) 的強烈損傷,環狀 SCLE 病灶的活動性邊緣偶爾會發生水疱大疱性變化,隨後可能產生顯著的結痂外觀。此類病灶可模擬史蒂芬斯-強生症候群(Stevens-Johnson syndrome)/TEN。其致病機轉與前述 TEN 樣 ACLE 所描述者相似。罕見地,SCLE 以剝脫性紅皮症 (exfoliative erythroderma) 表現,或顯示一種特殊的環狀病灶肢端分布 (acral distribution)。已有 SCLE 的糠疹樣 (pityriasiform) 與發疹性變異型的報告。新生兒 LE(neonatal LE,發生於經胎盤接受 IgG 抗 Ro/SS-A 及偶爾其他自體抗體特異性之新生兒身上的短暫性、光敏感性、非瘢痕性 LE 特異性皮膚病灶)的皮膚病灶與 SCLE 共有許多特徵。與 ACLE 皮膚病灶不同,SCLE 病灶傾向較 ACLE 病灶不那麼短暫,且癒合時伴隨較多色素變化。它們也較 ACLE 病灶不那麼水腫,並更為過度角化。SCLE 較常侵犯頸部、肩部、上肢與軀幹,而 ACLE 較常侵犯顏面的顴部區域。當顏面受 SCLE 侵犯時,最常為顏面外側,並不侵犯中央的顴部區域。與 SCLE 病灶相比,DLE 病灶一般伴隨較大程度的過度色素沉著 (hyperpigmentation) 與色素脫失 (hypopigmentation)、萎縮性真皮瘢痕 (atrophic dermal scarring)、毛囊角栓 (follicular plugging) 與附著性鱗屑 (adherent scale)。一個一致的臨床差異是,DLE 病灶特徵性地有硬結 (indurated),而 SCLE 病灶則無;此差異反映了 DLE 病灶在組織病理學上所見較深的發炎程度。約有一半的 SCLE 病人符合 ACR 修訂的 SLE 分類標準。然而,嚴重 SLE 的表現(如腎炎、CNS 疾病與全身性血管炎)僅在 10% 至 15% 的 SCLE 病人中發展。有人提出,丘疹鱗屑型的 SCLE、白血球減少 (leukopenia)、抗核抗體 (antinuclear antibody, ANA) 高效價(>1:640)與抗雙股 DNA(anti–double-stranded DNA, dsDNA)抗體,是以 SCLE 病灶表現之病人發展出 SLE 的危險因子。SCLE 可與其他共有 8.1 祖先單倍型(human leukocyte antigen A1-B8-DR3-DQ2)的自體免疫疾病重疊,包括修格蘭氏症候群 (Sjögren’s syndrome)、疱疹樣皮膚炎 (dermatitis herpetiformis)、類肉瘤病 (sarcoidosis) 與橋本甲狀腺炎 (Hashimoto thyroiditis)。其他軼事性地與 SCLE 相關的疾病有類風濕性關節炎 (rheumatoid arthritis)、Sweet 症候群、遲發性皮膚紫質症 (porphyria cutanea tarda)、麩質敏感性腸病變 (gluten-sensitive enteropathy) 與克隆氏症 (Crohn disease)。也有軼事性地提出 SCLE 可能與內臟惡性腫瘤相關(乳房、肺、胃、子宮、肝細胞與喉部癌,以及霍奇金淋巴瘤 Hodgkin lymphoma)。²⁶ 已知數種不同類型的化療藥物能夠觸發藥物誘發的 SCLE。這或許會混淆 SCLE 與內臟惡性腫瘤有顯著相關的可能性。
慢性皮膚紅斑性狼瘡 (CHRONIC CUTANEOUS LUPUS ERYTHEMATOSUS)
典型盤狀紅斑性狼瘡 (CLASSIC DISCOID LUPUS ERYTHEMATOSUS)
典型 DLE 病灶——CCLE 最常見的型式——起始為紅紫色斑點、丘疹或小斑塊,並迅速發展出過度角化的表面。早期的典型 DLE 病灶通常演變為界線分明、錢幣狀(即盤狀 discoid)的紅斑性斑塊,覆有顯著的附著性鱗屑,並延伸進入擴張之毛囊的開口(圖 61-6)。DLE 病灶通常以周邊的紅斑與過度色素沉著向外擴張,留下標誌性的中央萎縮性瘢痕、毛細血管擴張與色素脫失(圖 61-7)。此階段的 DLE 病灶可融合形成大片、融合性、毀容的斑塊。某些族裔背景(如亞洲印度人 Asian Indians)的人身上的 DLE,臨床上可表現為孤立的斑狀過度色素沉著區域。當出現於有毛髮的皮膚(頭皮、眼瞼緣與眉毛)時,DLE 造成瘢痕性禿髮 (scarring alopecia),可能導致毀容並顯著衝擊生活品質。DLE 中的毛囊侵犯是一個顯著特徵。角栓 (Keratotic plugs) 堆積於擴張的毛囊中,這些毛囊很快變得無毛。當附著性鱗屑從較進展的病灶上掀起時,可見到類似地毯釘 (carpet tacks) 外觀的角化棘突從鱗屑的下表面突出(即「地毯釘 (carpet tack)」徵象)。DLE 病灶在白人病人中可能難以診斷,因為特徵性的周邊過度色素沉著常付之闕如。肥厚型 DLE 病灶可能與肥厚型光化性角化症 (hypertrophic actinic keratosis)、角化棘皮瘤 (keratoacanthoma) 與鱗狀細胞癌 (squamous cell carcinoma) 混淆。DLE 病灶最常見於顏面、頭皮、耳朵、頸部 V 形區域與手臂伸側。顏面的任何區域,包括眉毛、眼瞼、鼻與唇,都可能受侵犯。偶爾可在顏面的顴部區域與鼻樑上發現對稱、過度角化、蝴蝶形的 DLE 斑塊。此類病灶不應與發生於相同區域之較短暫、水腫、最低限度脫屑的 ACLE 紅斑反應混淆。顏面 DLE,如同 ACLE 與 SCLE 一樣,通常不侵犯鼻唇溝。要區分顴部 DLE 與 ACLE 的早期病灶可能很困難,但硬結與對局部類固醇/鈣調神經磷酸酶抑制劑 (calcineurin inhibitors) 的抗拒性傾向支持前者的診斷。當 DLE 病灶發生於口周時,它們可消退並留下顯著的痤瘡樣 (acneiform) 凹陷性瘢痕型態。DLE 特徵性地侵犯外耳,包括耳甲腔 (conchal bowl) 與外耳道的外部(圖 61-8A)。此類病灶常最初表現為擴張、過度色素沉著的毛囊。頭皮在 60% 的 DLE 病人中受侵犯;由此種侵犯所致的不可逆瘢痕性禿髮已被報告發生於三分之一的病人中(圖 61-8B)。DLE 所致的不可逆瘢痕性禿髮,與 SLE 病人在全身疾病活動期間常發展的可逆、非瘢痕性禿髮不同。此種落髮,即所謂的狼瘡髮 (lupus hair),可能代表因疾病惡化而發生的休止期脫髮 (telogen effluvium)。侷限性 DLE 病灶僅發生於頭部或頸部,而廣泛性 DLE 病灶則同時發生於頸部以上與以下。廣泛性 DLE 較常與潛在的 SLE 相關,並常對標準治療較為抗拒,頻繁需要疊加抗瘧藥與免疫抑制藥物。頸部以下的 DLE 病灶最常發生於手臂、前臂與手部的伸側,雖然它們幾乎可發生於身體的任何部位。手掌與腳掌可能是疼痛性、有時令人失能的糜爛性 (erosive) DLE 病灶的部位。偶爾,僅發生於毛囊開口周圍的小 DLE 病灶出現於肘部與他處(毛囊型 DLE follicular DLE)。我們觀察到肘部/伸側手臂病灶似乎與 DLE 的肢端手指病灶一同發生,而具有此種徵象組合的病人較常有活動性的全身疾病。DLE 活動可侷限於甲單位 (nail unit)。指甲可能受其他型式的 CLE 以及 SLE 影響,產生甲褶紅斑與毛細血管擴張、紅色甲半月 (red lunulae)、杵狀指 (clubbing)、甲溝炎 (paronychia)、點狀凹陷 (pitting)、條狀白甲 (leukonychia striata) 與甲剝離 (onycholysis)。DLE 病灶可能被陽光曝曬增強,但程度低於 ACLE 與 SCLE 病灶。DLE 以及其他型式的 LE 皮膚疾病活動,可由任何型式的皮膚創傷誘發(即柯本現象 Koebner phenomenon 或同型反應 isomorphic effect)。典型 DLE 與 SLE 之間的關係一直是許多爭論的主題。²⁷ 可做出以下總結要點:(a) 5% 以典型 DLE 病灶表現的病人隨後發展出明確的 SLE 證據;(b) 廣泛性 DLE(即頸部以上與以下均有病灶)的病人比侷限性 DLE 病人有稍高的免疫學異常率、進展為 SLE 的較高風險,以及發展出更嚴重 SLE 表現的較高風險。約四分之一的 SLE 病人在其病程的某個時點發展出 DLE 病灶,而此類病人傾向有較不嚴重型式的 SLE。除了典型 DLE 之外,還有數種其他較不常見的 CCLE 變異型,它們之所以被如此亞分類,是因為其組織學重疊,以及傾向以低頻率與潛在的 SLE 相關發生。
肥厚型盤狀紅斑性狼瘡 (HYPERTROPHIC DISCOID LUPUS ERYTHEMATOSUS)
肥厚型 DLE,也被稱為過度角化型 (hyperkeratotic) 或疣狀 (verrucous) DLE,是 CCLE 的一種罕見變異型,其中典型 DLE 病灶中正常所見的過度角化被大幅誇大。手臂伸側、上背部與顏面是最常受影響的區域。肥厚型 LE 與扁平苔癬 (lichen planus) 的重疊特徵已在「狼瘡扁平苔癬 (lupus planus)」的標題下被描述。「肥厚與深部紅斑性狼瘡 (lupus erythematosus hypertrophicus et profundus)」此一實體似乎代表肥厚型 DLE 的一種罕見型式。肥厚型 DLE 病人發展出 SLE 的風險可能並不比典型 DLE 病灶病人來得高。
黏膜盤狀紅斑性狼瘡 (MUCOSAL DISCOID LUPUS ERYTHEMATOSUS)
黏膜 DLE 發生於約 25% 的 CCLE 病人。口腔黏膜最常受影響;然而,鼻、結膜與生殖器黏膜表面都可能成為標的。在口腔中,頰黏膜表面最常受侵犯,而顎、齒槽突 (alveolar processes) 與舌則為較少受侵犯的部位。病灶起始為無痛、紅斑性的斑片,演變為可能與扁平苔癬混淆的慢性斑塊。慢性頰黏膜斑塊界線分明,並有不規則扇貝狀、白色的邊界,伴有放射狀白色條紋 (white striae) 與毛細血管擴張。覆蓋於顎黏膜上之此類斑塊的表面常有蜂窩狀 (honeycomb) 外觀。中央凹陷常發生於較舊的病灶中,並可能發展出疼痛性潰瘍。罕見地,口腔黏膜 DLE 病灶可退變為鱗狀細胞癌,類似長期存在的皮膚 DLE 病灶。黏膜 DLE 病灶內任何程度的結節性不對稱都應評估惡性退變的可能性。慢性 DLE 斑塊也出現於唇的紅唇緣 (vermilion border) 上。有時,唇部的 DLE 侵犯可表現為瀰漫性唇炎 (diffuse cheilitis),尤其在較曝曬陽光的下唇。DLE 病灶可能出現於鼻、結膜與肛門生殖器黏膜上。鼻中隔 (nasal septum) 穿孔較常與 SLE 而非 DLE 相關。結膜 DLE 病灶侵犯下眼瞼較上眼瞼為多。病灶起始為局灶性的非特異性發炎區域,最常侵犯瞼結膜 (palpebral conjunctivae) 或眼瞼緣。隨著病灶成熟,瘢痕變得明顯,並可能發展出睫毛的永久喪失與眼瞼外翻 (ectropion),產生相當大的失能。
深部紅斑性狼瘡/紅斑性狼瘡脂膜炎 (LUPUS ERYTHEMATOSUS PROFUNDUS/LUPUS ERYTHEMATOSUS PANNICULITIS)
深部 LE/LE 脂膜炎(Kaposi-Irrgang 病)是 CCLE 的一種罕見型式,特徵為下真皮與皮下組織的發炎性病灶。約 70% 患有此型 CCLE 的病人也有典型的 DLE 病灶,常覆蓋於脂膜炎病灶之上。有些人使用「深部 LE」一詞來指稱同時具有 LE 脂膜炎與 DLE 病灶的病人,而以「LE 脂膜炎」來指稱僅有皮下侵犯者。典型的皮下病灶表現為堅實的結節,直徑 1 至 3 cm。覆蓋的皮膚常附著於皮下結節並向內拉,產生深陷、碟狀的凹陷(圖 61-9)。頭部、上臂近端、胸部、背部、乳房、臀部與大腿是常受影響的部位。LE 脂膜炎在沒有覆蓋性 DLE 的情況下,可能產生乳房結節,在臨床上與影像學上模擬癌症(狼瘡乳腺炎 lupus mastitis)。對藥物無反應的廣泛乳房侵犯可能嚴重到需要根除性乳房切除術 (radical mastectomy)。融合性的顏面侵犯可模擬脂肪萎縮 (lipoatrophy) 的外觀。萎縮性鈣化 (Dystrophic calcification) 常發生於深部 LE/LE 脂膜炎的較舊病灶中,而與此種鈣化相關的疼痛有時可能是主要的臨床問題。約 50% 的深部 LE/脂膜炎病人有 SLE 的證據。然而,LE 脂膜炎/深部 LE 病人的全身性特徵傾向較不嚴重,類似於有 DLE 皮膚病灶之 SLE 病人者。
凍瘡紅斑性狼瘡 (CHILBLAIN LUPUS ERYTHEMATOSUS)
凍瘡 LE 病灶最初發展為腳趾、手指與顏面上的紫紅色斑片、丘疹與斑塊,由寒冷、潮濕的氣候所誘發,在臨床與組織學上類似特發性凍瘡(idiopathic chilblains, pernio)(圖 61-10)。隨著它們的演變,這些病灶通常呈現為伴有相關毛細血管擴張的瘢痕性萎縮斑塊的外觀。它們可能類似 DLE 的舊病灶,或可能模擬小血管血管炎 (small vessel vasculitis) 的肢端病灶。組織學發現包括表淺與深層的淋巴球性血管反應 (lymphocytic vascular reaction),以及網狀真皮 (reticular dermal) 中真皮基底血管的纖維蛋白沉積 (fibrin deposition)。凍瘡 LE 病人常在顏面與頭部有典型的 DLE 病灶。凍瘡 LE 有可能起始為一種典型的肢端、寒冷誘導的病灶,然後柯本化 (Koebnerizes) 出 DLE 病灶,從而解釋了臨床組織學發現的光譜,這些發現似乎依病灶病程中採取切片樣本的時間點而異。凍瘡 LE 似乎與抗 Ro/SS-A 抗體相關²⁸,並在許多情況下與雷諾現象連結。²⁹ 病灶持續至寒冷月份之後、ANA 陽性,或在診斷凍瘡病灶時存在其他 ACR 之 SLE 標準之一,有助於將凍瘡 LE 與特發性凍瘡區分。³⁰ 約 20% 以凍瘡 LE 表現的病人之後發展出 SLE。凍瘡 LE 是一個未被充分認識的實體,然而它很可能是 LE 病人指(趾)端病灶最常見的原因之一。它有時被誤診為血管炎,並可能如前所述與肢端 DLE 重疊。已有一種體染色體顯性、家族性型式的凍瘡 LE 被描述,由 TREX 1(核酸內切酶修復 endonuclease repair)基因的錯義突變 (missense mutation) 所引起。³¹
腫脹性紅斑性狼瘡 (LUPUS ERYTHEMATOSUS TUMIDUS)
腫脹性紅斑性狼瘡(lupus erythematosus tumidus, LET;tumid LE)是 CCLE 的一種變異型,其組織學評估發現 DLE 的真皮表現,即過量的黏蛋白沉積 (mucin deposition) 與表淺的血管周圍及附屬器周圍 (periadnexal) 發炎。LE 特異性皮膚疾病的特徵性表皮組織學變化僅以最低限度表現(若有的話)。這導致多汁、水腫、蕁麻疹樣 (urticaria-like) 的斑塊,表面變化甚少(圖 61-11)。也可見到環狀的蕁麻疹樣斑塊。表皮變化的稀少常造成將 LET 作為一種 CCLE 型式的診斷混淆。³²,³³
數篇近期報告支持此種亞分類,並進一步描述此 CCLE 亞型的特徵。³⁴⁻³⁹ 雖然據描述發生於某些 SLE 病人中,但大多數 LET 病人有陰性的 ANA 與良性的疾病病程。LET 似乎是皮膚狼瘡中最具光敏感性的亞型,並通常對抗瘧藥顯示良好反應。此外,LET 病灶傾向完全消退而不留下瘢痕或萎縮。關於 LET 作為 LE 皮膚疾病真實型式的有效性仍持續有爭論。有些人主張 LET 病灶事實上可能並非 CLE 的一種型式³²,而另一些人則認為 LET 值得被認可為一種獨特的 CLE 類型(間歇性 CLE intermittent CLE),其重要性等同於 ACLE、SCLE 與 CCLE。³⁵ 在 UV 光誘導的 LE 腫脹病灶皮膚中第一型干擾素上調的證據支持後一論點。⁴⁰
其他變異型 (OTHER VARIANTS)
其他罕見型式的 CCLE/CLE 已被描述。這些包括肥厚與深部 LE(LE hypertrophicus et profundus)、苔癬樣 DLE(lichenoid DLE)、蟲蝕狀 LE(LE vermiculatus)、毛細血管擴張性 LE(LE telangiectaticus)、線狀 CLE(linear CLE)與水腫性 LE(LE edematous,可能是 DLE 之蕁麻疹樣斑塊變異型及/或腫脹性 LE 的一個歷史性命名)。關於這些臨床實體的進一步資訊可另行取得。⁴¹
紅斑性狼瘡非特異性皮膚疾病 (LUPUS ERYTHEMATOSUS NONSPECIFIC SKIN DISEASE)
在狼瘡病人(尤其是 SLE)身上已發現許多其他皮膚徵象;它們列於表 61-1。值得注意的是,LE 非特異性皮膚變化的存在,尤其當與 LE 特異性皮疹一同見到時,對應於較高的全身疾病活性。⁴² 特定徵象(如網狀青斑 livedo reticularis、血栓性靜脈炎 thrombophlebitis 或皮膚梗塞 cutaneous infarct)可能提示繼發性抗磷脂症候群 (secondary antiphospholipid syndrome) 的存在。篇幅限制不允許在此對這些實體進行特定討論。
非皮膚表現 (NONCUTANEOUS FINDINGS)
系統性狼瘡可造成無數的皮膚外表現,這些表現概述於表 61-3。
診斷 (DIAGNOSIS)
實驗室檢查 (LABORATORY TESTING)
由於 ACLE 與 SLE 之間的強烈關聯,ACLE 的免疫學實驗室特徵即為與 SLE 相關者(高效價 ANA、抗 dsDNA、抗 Smith 抗原、低補體血症 hypocomplementemia、高伽瑪球蛋白血症 hypergammaglobinemia 等)。其他實驗室發現,如血球減少 (cytopenias)、腎功能下降與尿液變化(血尿 hematuria、蛋白尿 proteinuria),反映疾病活性,並可能因人而異,取決於終端器官侵犯的程度。SCLE 的實驗室標記為抗 Ro/SS-A(70% 至 90%)以及較少見的抗 La/SS-B(30% 至 50%)自體抗體的存在。ANA 存在於 60% 至 80% 的 SCLE 病人中,類風濕因子 (rheumatoid factor) 存在於約 33%。SCLE 病人的其他自體抗體包括梅毒血清學試驗偽陽性(性病研究實驗室 [Venereal Disease Research Laboratory, VDRL] 快速血漿反應素試驗 rapid plasma reagin;7% 至 33%)、抗心磷脂 (anticardiolipin)(10% 至 16%)、抗甲狀腺 (antithyroid)(18% 至 44%)、抗 Smith 抗原(10%)、抗 dsDNA(10%)與抗 U1 核糖核蛋白(10%)。SCLE 病人(尤其是有全身侵犯者)可能有許多實驗室異常,包括貧血 (anemia)、白血球減少、血小板低下 (thrombocytopenia)、紅血球沉降速率 (erythrocyte sedimentation rate) 升高、高伽瑪球蛋白血症、蛋白尿、血尿、尿圓柱 (urine casts)、血清肌酸酐 (serum creatine) 與血中尿素氮 (blood urea nitrogen) 升高,以及補體濃度降低(源自遺傳性缺乏或補體消耗增加)。ANA 以低效價存在於 30% 至 40% 的 DLE 病人中;然而,少於 5% 的病人具有明顯 SLE 病人特徵性的較高 ANA 濃度(間接免疫螢光試驗 indirect immunofluorescence assay 效價 >1:320)。針對單股 DNA (single-stranded DNA) 的抗體在 DLE 中並不少見,但針對 dsDNA 的抗體則顯著少見。針對 U1 核糖核蛋白的沉澱抗體 (Precipitating antibodies) 有時可在病程以 DLE 病灶為主的病人中發現;然而,此類病人通常僅有輕微的 SLE 表現,或重疊的結締組織疾病,如混合性結締組織病 (mixed connective tissue disease)。沉澱性的 Ro/SS-A 與 La/SS-B 自體抗體在 DLE 病人中罕見;以酵素連結免疫測定法 (enzyme-linked immunoassay) 偵測到的低濃度抗 Ro/SS-A 抗體則較常見。一小部分 DLE 病人有輕度貧血、生物性梅毒血清學試驗偽陽性(VDRL 快速血漿反應素)、類風濕因子試驗陽性、血清補體濃度輕微降低、γ-球蛋白輕度升高與輕度白血球減少。有人提出此類發現是發展 SLE 的危險因子。ANA 存在於 70% 至 75% 的深部 LE/脂膜炎病人中,但抗 dsDNA 抗體罕見。與 SLE 以及 CLE 相關的實驗室發現,在成人與兒童中,已於他處回顧。⁴³,⁴⁴ 應注意的是,上述討論之自體抗體偵測的測定方法隨時間持續演進。本書出版時,商業參考實驗室大多已改用固相免疫測定 (solid-phase immunoassay) 技術,尤其是多重流式免疫測定 (multiplex flow immunoassay)。上節所討論之自體抗體的疾病發生率/盛行率,可能與今日從商業實驗室所收到的自體抗體測定結果略有不同。
組織病理學 (HISTOPATHOLOGY)
LE 特異性皮膚疾病的組織病理學是一組獨特的組合:過度角化、表皮萎縮、基底細胞空泡變性 (vacuolar basal cell degeneration)、真皮—表皮交界 (dermal–epidermal junction) 基底膜增厚、真皮水腫、真皮黏蛋白沉積,以及真皮—表皮交界與真皮的單核細胞浸潤(集中於血管周圍與附屬器周圍分布)。在不同型式的 LE 特異性皮膚疾病中,遭遇到這些特徵的程度不一。對於 ACLE、SCLE 與 DLE 病灶能否僅基於其組織病理學外觀就可靠地區分,存在不同的意見。⁴⁴⁻⁴⁶
急性皮膚紅斑性狼瘡
ACLE 病灶的組織病理學變化一般不如 SCLE 與 DLE 病灶來得顯著,主要為一種細胞稀少的介面性皮膚炎(cell-poor interface dermatitis)(圖 61-13A)。淋巴組織細胞性 (lymphohistiocytic) 細胞浸潤相對稀疏。某些作者注意到浸潤中嗜中性球 (neutrophils) 數量的增加,尤其當對新近發病的病灶進行切片時。除了毛細血管擴張與紅血球滲出 (extravasation of erythrocytes) 之外,還可見到輕度局灶性的基底角質細胞空泡改變。可能見到個別壞死的角質細胞,而在其最嚴重的型式中,ACLE 可顯示類似 TEN 的廣泛表皮壞死。上真皮通常顯示明顯的黏蛋白沉積 (mucinosis),這在區分 ACLE 與其他細胞稀少介面性皮膚炎原因時可能非常有幫助。在 ACLE 中見到基底膜帶 (basement membrane zone) 增厚、毛囊角栓或表皮厚度改變並不常見,雖然有時存在表皮萎縮。⁴⁵,⁴⁶
亞急性皮膚紅斑性狼瘡
SCLE 也通常表現為一種介面性皮膚炎,伴有基底角質細胞空泡改變的病灶與苔癬樣皮膚炎 (lichenoid dermatitis) 區域交替(見圖 61-13B)。常存在明顯的表皮萎縮。SCLE 與萎縮性扁平苔癬 (atrophic lichen planus) 及苔癬樣藥物疹 (lichenoid drug eruptions) 同列於萎縮性苔癬樣皮膚炎 (atrophic lichenoid dermatitis) 的鑑別診斷中。真皮變化包括水腫、明顯的黏蛋白沉積,以及通常侷限於真皮上三分之一血管周圍與附屬器周圍結構之區域的稀疏單核細胞浸潤。較輕程度的過度角化、毛囊角栓、附屬器結構的單核細胞浸潤與真皮噬黑色素細胞 (melanophages),可能有助於將 SCLE 病灶與 DLE 病灶區分。僅憑組織病理學標準,尚無法可靠地區分丘疹鱗屑型與環狀型 SCLE。⁴³
慢性皮膚紅斑性狼瘡
在典型 DLE 病灶中,表皮變化包括過度角化、程度不一的萎縮,以及類似 SCLE 所述之介面變化(見圖 61-13C)。表皮基底膜明顯增厚。真皮變化包括主要由 CD4 T 淋巴球與巨噬細胞組成的緻密單核細胞浸潤(主要位於附屬器周圍與血管周圍區域)、噬黑色素細胞與真皮黏蛋白沉積。該浸潤常相當緻密,並通常深入較深的網狀真皮及/或皮下,這可能有助於將其與 ACLE 或 SCLE 區分。在慢性瘢痕性 DLE 病灶中,緻密的發炎細胞浸潤消退,並被真皮纖維增生 (dermal fibroplasia) 取代。已有 DLE 的一種親毛囊變異型 (folliculotropic variant) 被描述(其中發炎浸潤主要圍繞毛囊),以及淋巴瘤樣變異型 (lymphomatoid variants)(其中有極為緻密的浸潤,可能含有非典型淋巴細胞)。⁴⁵
免疫組織學 (IMMUNOHISTOLOGY)
免疫組織學常有助於確認 LE 特異性皮膚疾病的診斷,並已被顯示能提升診斷的敏感度與特異度。⁴⁵
由於在急性、亞急性與慢性 LE 病人中見到陰性免疫螢光研究結果,以及在健康個體中見到偽陽性結果並不罕見,免疫組織學必須在特定病人的臨床與組織學發現的脈絡下解讀。自 1960 年代初以來,已觀察到 IgG、IgA、IgM 與補體成分(C3、C4、Clq、備解素 properdin、因子 B 與膜攻擊複合體 membrane attack complex C5b-C9)以連續顆粒狀或線狀帶狀排列沉積於 LE 病人之病灶與非病灶皮膚的真皮—表皮交界處(圖 61-14)。然而,此領域術語上的爭論持續使其混淆不清。有些人將「狼瘡帶試驗 (lupus band test)」一詞的使用限定為檢查非病灶皮膚切片是否存在此種帶狀排列的免疫反應物於真皮—表皮交界處。另一些人則將狼瘡帶試驗限定為「病灶性 (lesional)」或「非病灶性 (nonlesional)」。若統一採用「病灶性狼瘡帶試驗」與「非病灶性狼瘡帶試驗」這些術語,混淆可能會較少。
急性皮膚紅斑性狼瘡
現有的稀少資料顯示,60% 至 100% 的 ACLE 病灶顯示病灶性狼瘡帶。然而,認識到來自其他方面健康個體的陽光受損皮膚可顯示類似的免疫病理,已稀釋了此發現的臨床價值。
亞急性皮膚紅斑性狼瘡
初步研究指出,約 60% 的 SCLE 病人有病灶性狼瘡帶。一種圍繞表皮基底角質細胞之 IgG 沉積的「塵狀顆粒 (dustlike particle)」型態,已被認為對 SCLE 更具特異性,因為它反映了體內結合的 Ro/SS-A 自體抗體的存在。
慢性皮膚紅斑性狼瘡
早期報告指出,超過 90% 的典型 DLE 病灶在真皮—表皮交界處有病灶性免疫反應物,常沿著毛囊基底膜延伸,但後續研究報告的比率稍低。頭、頸與手臂的病灶比軀幹的病灶(20%)更常呈陽性(80%)。病灶性狼瘡帶似乎也是所檢查病灶年齡的函數,較舊的病灶(>3 個月)比較年輕者更常呈陽性。免疫球蛋白在真皮—表皮交界處的超微結構定位確認,這些蛋白質沉積於上真皮膠原纖維上,並沿著表皮基底膜帶的緻密板 (lamina densa)。在深部 LE 中,免疫球蛋白與補體沉積通常見於深真皮與皮下的血管壁。真皮—表皮交界處的免疫球蛋白沉積可能存在也可能不存在,取決於切片的部位、伴隨 SLE 的有無,以及真皮—表皮交界處有無覆蓋性 DLE 變化。
非病灶性狼瘡帶試驗 (NONLESIONAL LUPUS BAND TEST)
過去三十年來,關於取自 LE 病人之非病灶皮膚的真皮—表皮交界處免疫球蛋白/補體帶的診斷與預後意義,一直有許多爭論。⁴⁴ 當取樣完全受陽光保護的非病灶皮膚(如臀部)時,若真皮—表皮交界處存在 3 種或更多的免疫反應物,對 SLE 的診斷特異度似乎非常高。前瞻性確定的追蹤資料也顯示,非病灶性狼瘡帶試驗的存在與發展出 LE 腎炎的風險呈正相關。然而,非病灶性狼瘡帶試驗作為一種臨床工具已不再受青睞,主要是因為所獲得的資訊未被證明比更容易取得的血清學測定(如抗 dsDNA 抗體)的結果具有顯著更高的價值。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
CLE 的鑑別診斷概述於表 61-5。此外,網狀紅斑性黏蛋白沉積症 (reticulated erythematosus mucinosis) 已被某些人認為是光敏感性 CLE 的一種型式,或許與 LE 腫脹有關。網狀紅斑性黏蛋白沉積症表現為上胸部與背部呈網狀排列的斑點及/或丘疹。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
急性皮膚紅斑性狼瘡
侷限性與廣泛性兩種型式的 ACLE 病灶都與潛在的 SLE 疾病活性平行地惡化與緩解。因此,任何特定 ACLE 病人的預後取決於潛在 SLE 的模式。過去四十年來,SLE 的 5 年(80% 至 95%)與 10 年(70% 至 90%)存活率因更敏感的實驗室檢測所促成的早期診斷以及改良的免疫抑制治療方案而逐步改善。SLE 中不祥的預後徵象為高血壓、腎炎、全身性血管炎與 CNS 疾病。
亞急性皮膚紅斑性狼瘡
由於 SCLE 被認可為一個獨立的疾病實體僅二十年,與 SCLE 病灶相關的長期結果尚待確定。根據作者的經驗,大多數 SCLE 病人在長期間內有間歇性的皮膚疾病活動復發,而沒有全身侵犯的顯著進展(在約 150 名 SCLE 病人中,我們僅知有 1 例死亡可直接歸因於 SLE)。其他病人享有長期(若非永久)的皮膚疾病活動緩解。少數病人經歷持續不退的皮膚疾病。根據作者的經驗,約 10% 的 SCLE 病人發展出活動性 SLE,包括狼瘡腎炎。此亞群病人的特徵為丘疹鱗屑型 SCLE、侷限性 ACLE、高效價 ANA、白血球減少,及/或針對 dsDNA 的抗體的存在。需要對 SCLE 進行長期追蹤研究,以確定以 SCLE 皮膚病灶表現之病人發生嚴重全身疾病進展的真實風險。CCLE 病灶(通常為典型 DLE)也曾出現於最初以 SCLE 表現的病人中。證據顯示 SCLE 與修格蘭氏症候群之間發生重疊。發展出修格蘭氏症候群的 SCLE 病人有發展出與修格蘭氏症候群相關之腺體外全身性併發症的風險,包括血管炎、周邊神經病變、自體免疫甲狀腺炎、腎小管酸中毒、肌炎、慢性肝炎、原發性膽汁性肝硬化、精神病、淋巴結病變、脾腫大與 B 細胞淋巴瘤。
慢性皮膚紅斑性狼瘡
大多數未經治療的典型 DLE 病灶病人遭受惰性進展,發展為大面積的皮膚萎縮與瘢痕性禿髮,這在社會心理上可能是毀滅性的,並可造成職業失能。然而,透過治療,皮膚疾病可大致受到控制。自發緩解偶爾發生,且疾病活動可在較舊、不活動病灶的部位再度發作。停止治療後的反彈 (Rebound) 是典型的,因此建議在不活動期間較緩慢地遞減藥物。鱗狀細胞癌偶爾發展於慢性悶燒型 DLE 病灶中。最初以侷限性 DLE 表現的病人死於 SLE 顯著少見。如前述「流行病學」所討論,以侷限性 DLE 表現的病人僅有 5% 的機會隨後發展出臨床上顯著的 SLE 疾病活性。廣泛性 DLE 與持續性、低度的實驗室異常似乎是此種疾病進展的危險因子。在長期存在之 DLE 皮膚病灶內發展出的未被認出的鱗狀細胞癌,可能是病態與死亡的原因。
結果測量 (OUTCOME MEASURES)
近期開發出一種測量 CLE 活性的經驗證工具——皮膚紅斑性狼瘡疾病面積與嚴重度指數(Cutaneous Lupus Erythematosus Disease Area and Severity Index, CLASI),使得客觀追蹤病人的疾病病程與對治療的反應成為可能。此工具對損傷(瘢痕)與活性有各自獨立的評分,這很重要,因為我們不會期望一個「燒盡 (burned-out)」的瘢痕區域會因旨在減輕 LE 活性的藥物而正常化。⁴⁷ 它已被驗證為測量臨床反應的有用工具。⁴⁸
處置 (MANAGEMENT)
任何型式 CLE 病人的初始處置,都應包括在診斷時進行評估以排除潛在的 SLE 疾病活性。所有 CLE 病人都應接受關於防護陽光與人工 UV 輻射來源的指導,並應被建議避免使用潛在的光敏感藥物,如氫氯噻嗪 (hydrochlorothiazide)、四環素 (tetracycline)、灰黃黴素 (griseofulvin) 與吡羅昔康 (piroxicam)。關於特定的藥物治療,應將局部措施最大化,並在顯著的局部疾病活性持續存在或全身活性疊加時使用全身性藥物。ACLE 病灶通常對治療潛在 SLE 疾病活性所需的全身性免疫抑制措施有反應,而此種潛在 SLE 活性如此頻繁地伴隨此型式的 CLE(如全身性糖皮質素、azathioprine 與 cyclophosphamide)。越來越多的證據顯示,氨基喹啉類抗瘧藥(aminoquinoline antimalarial agents,如 hydroxychloroquine)可對 SLE 有節省類固醇 (steroid-sparing) 的效應,且這些藥物在 ACLE 中可能有價值。下文「局部治療」中所討論的局部措施也可對治療 ACLE 有價值。由於 SCLE 與 CCLE 的病灶常見於少有或沒有潛在全身疾病活性證據的病人中(與 ACLE 的病灶不同),對 SCLE 與 CCLE 較偏好非免疫抑制治療型式(表 61-6)。大致上,SCLE 與 CCLE 病灶對此類藥物的反應相同。準確的診斷始終是最佳藥物處置的關鍵。在臨床上因人而異地表現之遺傳上複雜的臨床疾病(如 LE)中,一旦確立了 LE 的診斷,便有將新的臨床發現過度歸因於 LE 的傾向。為了喚起對此重要臨床公理的注意,風濕病學家 Robert Greenwald 制定了他的「狼瘡定律 (Law of Lupus)」。它指出:「SLE 常被錯誤地指控為造成在 SLE 診斷之後病人身上可能發生的任何與所有事情的原因。」⁴⁹ 在急性醫療情境中未能認識此原則,可能導致誤診與潛在嚴重的處置失當。常見的發炎性皮膚病灶(如乾癬與濕疹性皮膚炎)可發生於 LE 個體中。這些皮膚病灶與 LE 及其治療完全無關。同樣地,混淆可能因治療 LE 病人所需之全身性免疫抑制治療所致的皮膚變化而產生(如帶狀疱疹 herpes zoster、類固醇誘發的痤瘡、藥物疹、伺機性感染 opportunistic infections)。
局部治療 (LOCAL THERAPY)
防曬 (SUN PROTECTION)
建議病人避免直接曝曬陽光、穿著緊密編織的衣物與寬邊帽,並規律使用廣效、抗水的防曬乳(SPF [防曬係數 sun protection factor] ≥30,含有效的 UVA 阻斷劑,如光穩定型 (photostabilized) 的 avobenzone [Parsol 1789]、微粒化二氧化鈦 micronized titanium dioxide、微粒化氧化鋅 micronized zinc oxide 或 ecamsule [Mexoryl SX])。應將阻 UV 膜貼於住家與汽車的窗戶上,並將壓克力擴散屏 (acrylic diffusion shields) 置於螢光燈上。矯正性偽裝化妝品(corrective camouflage cosmetics,如 Dermablend 與 Covermark)提供雙重益處:既是高效的物理性防曬,又是美觀上令人愉悅的化妝遮蓋劑。Ting 與 Sontheimer 對自體免疫結締組織皮膚疾病的實務與理論性光保護及局部治療提供了深入的討論。⁵⁰
由於需要避免陽光以及或許其他因素,CLE 與 SLE 病人常被發現維生素 D 不足或缺乏。⁵¹,⁵² 將維生素 D 代謝正常化對人體健康有明確的益處。矯正維生素 D 不足/缺乏是否對皮膚與 SLE 疾病活性有治療性衝擊正在研究中。
局部糖皮質素 (TOPICAL GLUCOCORTICOIDS)
雖然某些人偏好對敏感區域(如顏面)使用中等強度的製劑(如 triamcinolone acetonide 0.1%),但超強效局部第一級藥物(superpotent topical class I agents,如 clobetasol propionate 0.05% 或 betamethasone dipropionate 0.05%)在 CLE 中產生最大的益處。將超強效製劑每日兩次塗抹於病灶皮膚 2 週,之後接著 2 週的休息期,可將局部併發症(如類固醇萎縮 steroid atrophy 與毛細血管擴張)的風險降至最低。或者,可在停用局部皮質類固醇的 2 週休息期間每日使用局部鈣調神經磷酸酶抑制劑。對於更為過度角化的病灶(如肥厚型 DLE),軟膏比乳膏更為有效。以浸漬糖皮質素的膠帶(glucocorticoid-impregnated tape,如 flurandrenolide)或以糖皮質素加塑膠食物保鮮膜(如 Saran 或 Glad Press-N-Seal)進行的封閉治療,可增強局部糖皮質素的有益效應,但也帶有較高的局部副作用風險。第一級或第二級局部糖皮質素溶液與凝膠最適於治療頭皮。不幸的是,即使是最積極的局部糖皮質素方案,其本身對大多數 SCLE 與 CCLE 病人也無法提供足夠的改善。
局部鈣調神經磷酸酶抑制劑 (TOPICAL CALCINEURIN INHIBITORS)
Pimecrolimus 1% 乳膏與 tacrolimus 0.1% 軟膏已在 ACLE、DLE 與 SCLE 的治療中展現療效。⁵³⁻⁵⁵ 一項雙盲、安慰劑對照的試驗性研究顯示,pimecrolimus 1% 乳膏在治療顏面 DLE 上與 betamethasone valerate 0.1% 乳膏有相同的療效⁵⁶,而另一項不同的研究證明了 tacrolimus 0.3% 配於 clobetasol propionate 0.05% 中的複方對抗拒型 CLE 的療效。⁵⁷
針對其他臨床適應症正在開發之新型靶向非類固醇抗發炎藥的局部給藥(如局部 apremilast、局部 tofacitinib)是否可能對 CLE 有益,仍有待證明。
病灶內糖皮質素 (INTRALESIONAL GLUCOCORTICOIDS)
病灶內糖皮質素(如 triamcinolone acetonide 懸浮液,顏面用 2.5 至 5 mg/mL,較不敏感部位可使用較高濃度)在 DLE 的處置上比 SCLE 更為有用。病灶內糖皮質素本身可產生皮膚與皮下萎縮(深入皮下組織的注射會增強此風險)。偏好使用 30 號針頭,因為它在穿透時僅產生輕微的不適,尤其當垂直於皮膚注射時。應徹底浸潤病灶的活動性邊緣。病灶內治療適用於特別過度角化的病灶或對局部糖皮質素無反應的病灶,但大多數 CLE 病人的病灶過多,無法僅以病灶內糖皮質素注射來處置。
全身性治療 (SYSTEMIC THERAPY)
抗瘧藥 (ANTIMALARIALS)
對於未能從上述「局部治療」所描述之局部措施中獲得足夠益處的 CLE 病人,約 75% 可對一種或合併使用氨基喹啉類抗瘧藥有效。應與病人討論視網膜毒性 (retinal toxicity) 的風險,並應進行治療前的眼科檢查。然而,抗瘧藥視網膜病變 (antimalarial retinopathy) 的風險極為罕見,尤其在治療的前 10 年內,前提是不超過這些藥物建議的每日最大劑量(hydroxychloroquine,6.5 mg/kg/day,根據理想體重;chloroquine,3 至 4 mg/kg/day)。病人在治療期間應每 6 至 12 個月進行追蹤眼科評估。硫酸 hydroxychloroquine(Plaquenil),按瘦體重 6 至 6.5 mg/kg,應每日給予,可每日一次或分兩次以預防腸胃副作用。約需 6 週才能達到 hydroxychloroquine 的平衡血中濃度。應告知病人治療益處有 2 至 3 個月的延遲發作期。若在 8 至 12 週後未見反應,可將鹽酸 quinacrine,100 mg/day(目前在美國僅透過複方藥局取得),加至 hydroxychloroquine 中而不增加視網膜病變的風險(quinacrine 不造成視網膜病變)。若在此合併方案 4 至 6 週後仍未達到足夠的臨床控制,應考慮以二磷酸 chloroquine(Aralen),3 至 4 mg/kg/day,取代此合併方案中的 hydroxychloroquine。對於腎或肝功能下降的病人,劑量可能需要調整。在歐洲,chloroquine 一般被認為在治療 CLE 上比 hydroxychloroquine 更有效,這或許是因為 chloroquine 與 hydroxychloroquine 相比,達到穩態血中濃度所需的時間較短,因而可能發生較早的治療反應。Hydroxychloroquine 與 chloroquine 不應同時使用,因為會增強視網膜毒性的風險。有一些證據顯示 chloroquine 可能比 hydroxychloroquine 更具視網膜毒性。現在已可測定 hydroxychloroquine 的血清濃度。然而,此種測定尚未由美國的商業檢測實驗室廣泛提供。檢視此一終點的近期研究發現,高達 10% 的 CLE 與 SLE 病人未依處方順從地服用其 hydroxychloroquine 治療。⁵⁸,⁵⁹ 此外,這些及其他研究顯示,在接受 hydroxychloroquine 治療的皮膚與 SLE 病人中,hydroxychloroquine 血清濃度與疾病活性測量呈負相關。⁶⁰
現在一般同意,現代的 hydroxychloroquine 方案可安全用於兒童(根據瘦體重進行適當的劑量調整)與孕婦。然而,兒童的治療指數 (therapeutic index) 相當狹窄。因此,應採取額外的謹慎以預防兒童的意外過量。除視網膜毒性外,抗瘧藥的使用還伴隨多種副作用。Quinacrine 比 hydroxychloroquine 或 chloroquine 伴隨較高的副作用發生率,如頭痛、腸胃不耐、血液學毒性、搔癢、苔癬樣藥物疹,以及黏膜或皮膚色素沉積。然而,在目前建議不超過 100 mg/day 的劑量下,此類不良反應極為罕見。Quinacrine 在膚色白皙的個體中常產生整個皮膚與鞏膜的黃色變色,當藥物劑量減少或完全停用時可完全逆轉。Quinacrine 可在葡萄糖-6-磷酸去氫酶(glucose-6-phosphate dehydrogenase, G6PD)缺乏的病人中產生顯著溶血(此不良反應也曾被報告罕見發生於 hydroxychloroquine 與 chloroquine)。每一種氨基喹啉類抗瘧藥都可產生骨髓抑制,包括再生不良性貧血 (aplastic anemia),雖然此效應在目前的劑量方案下極為罕見。中毒性精神病 (Toxic psychosis)、大發作癲癇 (grand mal seizures)、神經肌肉病變 (neuromyopathy) 與心律不整 (cardiac arrhythmias) 過去曾在高劑量使用這些藥物時發生;這些反應在今日使用的較低每日劑量方案下並不常見。
在以 hydroxychloroquine 與 chloroquine 開始治療之前,應進行全血球計數以及肝腎功能檢測;這些檢測應在治療開始後 4 至 6 週重複,之後每 4 至 6 個月一次。使用 quinacrine 時建議更頻繁地篩檢血液學毒性。有明顯或亞臨床遲發性皮膚紫質症的病人,在以治療劑量的抗瘧藥治療 CLE 時,發展出急性肝毒性的風險特別高,而此種肝毒性常模擬急性外科腹症。
對抗瘧藥難治性疾病的非免疫抑制選項 (NONIMMUNOSUPPRESSIVE OPTIONS FOR ANTIMALARIAL–REFRACTORY DISEASE)
某些難治性 CLE 病人(SCLE 多於 DLE)對二氨基二苯碸(diaminodiphenylsulfone, dapsone)有反應。⁶¹
初始劑量為每日兩次口服 25 mg,必要時可增加至 200 至 400 mg/day。使用 dapsone 可能導致顯著的劑量相關溶血及/或變性血紅素血症 (methemoglobinemia),尤其在 G6PD 活性缺乏的個體中。因此,應定期進行全血球計數與肝功能檢測,並應考慮在開始治療前檢測 G6PD 狀態,尤其在高風險族群中。異維 A 酸(Isotretinoin),0.5 至 2.0 mg/kg/day,與 acitretin,10 至 50 mg/day,也已用於此情境,但其療效受其副作用(致畸性 teratogenicity、黏膜皮膚乾燥與高血脂)所限。此外,CLE 活性的突破 (breakthrough) 一直是類視色素長期使用的問題。沙利竇邁(Thalidomide,50 至 200 mg/day)對於難治於其他藥物的 CLE 顯著有效。眾多研究引述介於 85% 與 100% 之間的反應率,許多病人經歷完全緩解。⁶² 然而,1998 年在美國因嚴重致畸性而實施的嚴格處方規範,使得 salidomide 難以開立給有生育潛力的女性。感覺神經病變 (Sensory neuropathy) 是另一種與 salidomide 相關的毒性,而 25% 至 75% 的 CLE 病人在服藥期間發展出周邊神經病變。大多數病例在治療停止時可逆。神經病變似乎與總治療時間相關,因此偏好短療程。藥物停用後的復發很常見。過度嗜睡以及便秘與其他輕微副作用有時限制其使用,雖然這些效應通常隨較低的每日劑量而減輕。⁶³ 血栓栓塞 (Thromboembolism) 是一種嚴重的不良事件,可能發生於先前已存在高凝血狀態 (hypercoagulable state) 的病人中(如存在抗磷脂抗體)。使用 salidomide 治療多發性骨髓瘤的腫瘤科醫師常啟動同時的抗凝血治療以預防此副作用。來那度胺(Lenalidomide [Revlimid, Celgene])是一種 salidomide 類似物,已獲美國食品藥物管理局(U.S. Food and Drug Administration, FDA)核准用於治療多發性骨髓瘤。在初步研究中,其在 CLE 的療效率似乎與 salidomide 相似。迄今的安全性資料顯示,與 salidomide 相比,周邊神經病變的發生率低得多;然而,它與 salidomide 相比有相似的血栓栓塞率(尤其當與糖皮質素合併使用時)與白血球減少率。其在人類的潛在致畸性尚待確定。其他被報告在治療難治性 CLE 上有價值的藥物為金製劑 (gold) 與 clofazimine;然而,益處因病例而異,且這兩種藥物都伴隨顯著副作用的風險。維生素 E、phenytoin、sulfasalazine、danazol、DHEA 與光照治療(UVA1 光照治療、光分離術 photopheresis)也已在臨床試驗中被報告對 CLE 有潛在價值。
對抗瘧藥難治性疾病的免疫抑制選項 (IMMUNOSUPPRESSIVE OPTIONS FOR ANTIMALARIAL–REFRACTORY DISEASE)
全身性糖皮質素(Systemic Glucocorticoids): 對於 LE 侷限於皮膚的病人,應盡一切努力避免使用全身性糖皮質素。然而,在偶爾有特別嚴重且症狀明顯之皮膚疾病的病人中,已使用靜脈脈衝 methylprednisolone (intravenous pulse methylprednisolone)。在較不急性的病例中,中等每日劑量的口服糖皮質素(prednisone,20 至 40 mg/day,以單次晨間劑量給予)可在以抗瘧藥治療的負荷期 (loading phase) 中作為補充治療使用。由於長期糖皮質素治療的併發症,尤其是無血管性(無菌性)骨壞死(avascular [aseptic] bone necrosis,一種 LE 病人特別易感的副作用),劑量應在儘可能早的時間減少。由於類固醇誘發的骨質流失在使用的前 6 個月內發生最快,所有沒有禁忌症的病人都應在啟動類固醇治療時開始使用預防骨質疏鬆的藥物。一篇描述全身性糖皮質素之骨質流失與其他副作用之現行預防建議的優秀回顧已被發表。⁶⁴ 當疾病活性受控時,每日劑量應以 5 至 10 mg 的遞減量減少,直到活性再次惡化或達到每日劑量 20 mg/day。每日劑量接著應以 2.5 mg 的遞減量降低(某些醫師偏好在低於 10 mg/day 時使用 1 mg 的劑量遞減量)。隔日 (Alternate-day) 糖皮質素治療在大多數 CLE 或 SLE 病人中未能成功抑制疾病活性。對於有顯著潛在肝病的病人,應使用 prednisolone 而非 prednisone,因為 prednisone 需要在肝臟中羥基化 (hydroxylation) 才能具生物活性。任何量的 prednisone 以單次晨間口服劑量給予時,其腎上腺抑制活性低於相同量分次給予整天的劑量。然而,任何特定量的此藥物,以分次劑量服用時,其抑制 LE 的活性大於相同量以單次晨間劑量給予者。每日劑量小於或等於 7.5 mg/day 的 prednisone 可消除 SLE 病人中糖皮質素誘發的損傷累積。⁶⁵
傳統免疫抑制劑(Traditional Immunosuppressives): Azathioprine(Imuran)(每日口服 1.5 至 2 mg/kg)可在嚴重受影響的 CLE 病人中扮演節省糖皮質素的角色。Mycophenolate mofetil(CellCept)(口服 2.5 至 3 g,分兩次給予)是一種類似 azathioprine 的嘌呤類似物 (purine analog),但對淋巴球中的從頭合成途徑 (de novo pathway) 有更特異的抑制。此特性可能允許在治療嚴重、抗拒型 CLE 上有更高的療效與較少的毒性,而此類結果已在數項研究中被報告。⁶⁶,⁶⁷ 甲胺喋呤(Methotrexate,每週 1 天口服 7.5 至 25 mg)對嚴重難治性 CLE 有效。一項在 SLE 病人中的雙盲、隨機、安慰劑對照試驗顯示,中等劑量的 methotrexate(每週 15 至 20 mg)有效控制皮膚與關節活性,並允許減少 prednisone 劑量。⁶⁸ 其他曾被軼事性地使用的免疫抑制劑包括胞嘧啶阿拉伯糖苷(cytosine arabinoside, Cytarabine)與環孢素 (cyclosporine)。以高劑量靜脈丙種球蛋白 (IV gammaglobulin) 進行的免疫治療也曾被使用。
生物製劑療法(Biologic Therapies): 已有報告記錄抗 TNF 藥物(etanercept、adalimumab、infliximab)在治療抗拒型 CLE(尤其是 SCLE)上的療效。⁶⁹⁻⁷¹ 然而,這些藥物也廣為人知會誘發 SLE 與 CLE,突顯了 TNF 恆定 (homeostasis) 在易發生 LE 之病人中的重要性。⁷²,⁷³
B 淋巴球刺激因子(B-lymphocyte stimulator, BLyS [同義詞:B 細胞活化因子 B-cell activating factor (BAFF)])是 TNF 細胞激素家族近期被發現的一個成員。BLyS 由單核球與巨噬細胞製造,並在這些細胞被活化時隨後釋放。BLyS 結合於僅見於 B 細胞上的一種受體,刺激其成熟為分泌抗體的漿細胞 (plasma cells)。一種針對 BLyS 的完全人源化單株抗體(belimumab [Benlysta])抑制 BLyS 的生物活性。Belimumab 現已獲 FDA 核准用於 SLE。在 belimumab 治療 SLE 的臨床試驗中,除了全身性終點之外,SLE 病人的皮膚終點也被達到。然而,迄今關於 belimumab 在孤立性 CLE 的臨床療效少有報告。如前所述,干擾素-α 的異常調節似乎透過上調促發炎細胞激素與趨化激素,在 LE 之皮膚與全身表現兩者的致病機轉中扮演核心角色。三種靶向第一型 IFN 的生物製劑目前正在活動性 SLE 病人中測試其療效與安全性(兩種抗干擾素-α 單株抗體 [rontalizumab 與 sifalimumab],以及一種結合於第一型 IFN 受體的單株抗體 [anifrolumab])。⁷⁴ 針對第一型 IFN 與干擾素受體的重組人源化抗體正在開發中。若這些新生物製劑證明對 SLE 疾病活性有益,便可推斷它們可能對 CLE 疾病活性有幫助。
第一型干擾素的作用透過 Janus 激酶(Janus kinase, JAK)—訊號轉導子與轉錄活化子(signal transducer and activator of transcription, STAT)細胞內訊號傳遞途徑進行轉導。破壞此細胞內訊號傳遞途徑可能提供另一種下調 SLE 與 CLE 疾病活性及損傷的方法。兩種口服給藥的 JAK 同功酶 (isoenzyme) 抑制劑已被 FDA 核准用於其他臨床疾病:tofacitinib(JAK 1/3)用於類風濕性關節炎,ruxolitinib(JAK 1/2)用於骨髓化生不良疾病 (myelodysplastic disorders)。有病例報告顯示這兩種藥物對 CLE 與皮膚皮肌炎兩者似乎都有臨床益處。⁷⁵,⁷⁶
更具特異性的 JAK1 同功酶抑制劑目前正在開發中(如 filgotinib),有望減少此類新型靶向小分子治療的副作用。
外科與美容治療 (SURGICAL AND COSMETIC THERAPY)
DLE 病灶可產生永久性瘢痕性禿髮、美容上令人困擾的真皮萎縮,以及持久的色素變化。受此影響的病人常詢問美容矯正這些變化的可能性。外科介入(如毛髮移植 hair transplantation 與磨皮術 dermabrasion)帶有一定的風險,因為 CLE 的特徵為非特異性機械創傷(包括外科切開或雷射消融)有加重疾病活性的傾向(即柯本現象/同型現象)。某些病人若正在接受維持性全身治療(如抗瘧藥),可耐受瘢痕修復技術(包括磨皮術)。已有以氬雷射 (argon) 與脈衝染料雷射 (pulsed-dye laser) 治療成功處置活動性 CLE 的軼事報告。⁷⁷ 以鉺:YAG(Erbium:YAG)或 Fraxel 二氧化碳磨皮雷射對萎縮性瘢痕進行表面重建 (resurfacing) 據報告有益。⁷⁸,⁷⁹ 自體脂肪移植 (Autologous fat transplantation) 對於有燒盡型狼瘡脂膜炎/深部 LE 之萎縮性瘢痕的病人可有臨床價值。應避免以膠原蛋白或其他類似的外來美容材料注射萎縮性病灶。
治療演算法 (TREATMENT ALGORITHM)
以前述藥物治療 CLE 通常以逐步 (stepwise) 的方式進行,以將副作用的風險降至最低,並概述於表 61-6。
預防 (PREVENTION)
目前尚無法預測與預防 LE 的初始臨床表現,無論是皮膚疾病或全身性。然而,由於許多 LE 病人會因 UV 光暴露而出現皮膚疾病活性惡化,應鼓勵物理性防護陽光與人工 UV 光來源,以及規律使用 SPF 30 或更高的廣效防曬乳。應鼓勵 CLE 病人戒菸。吸菸的 CLE 病人與不吸菸者相比,對抗瘧治療的反應較差。已觀察到吸菸且對抗瘧治療無反應的 CLE 病人,在戒菸後(其治療無其他改變)其 CLE 活性經歷完全緩解。吸菸不會改變抗瘧藥的代謝。有人提出,吸菸(無論抗瘧治療與否)與 CLE 強烈相關,而與 SLE 的相關性則稍弱。⁸⁰,⁸¹
圖表 (FIGURES AND TABLES)
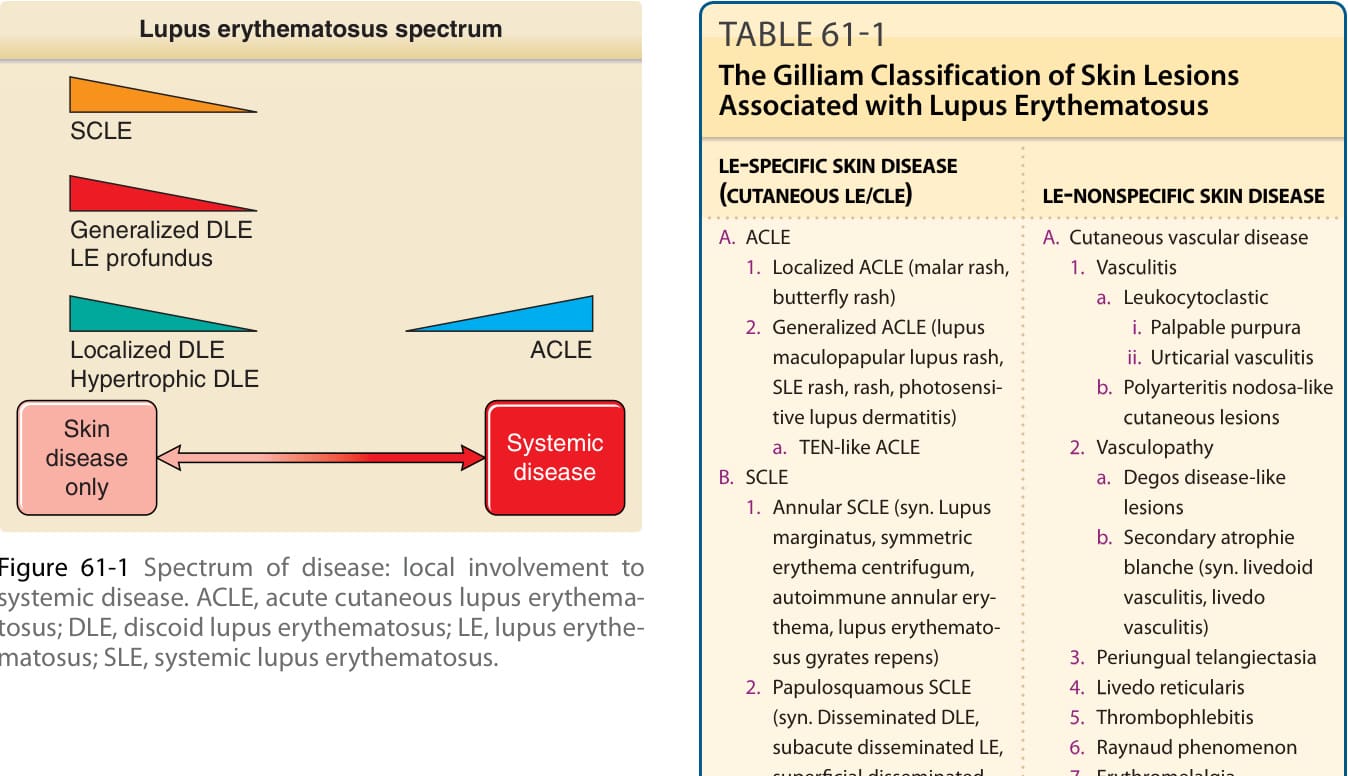
圖 61-1:疾病光譜:從局部侵犯到全身性疾病。ACLE, acute cutaneous lupus erythematosus(急性皮膚紅斑性狼瘡);DLE, discoid lupus erythematosus(盤狀紅斑性狼瘡);LE, lupus erythematosus(紅斑性狼瘡);SLE, systemic lupus erythematosus(系統性紅斑性狼瘡)。
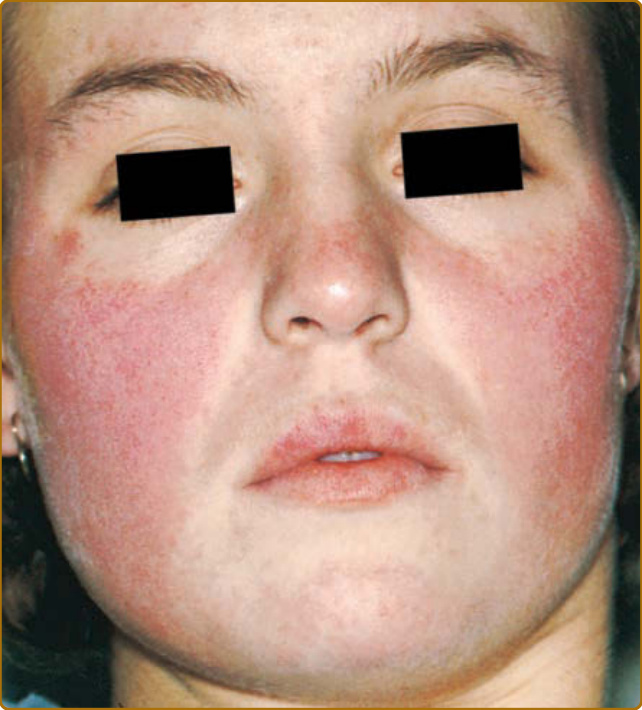
圖 61-2:侷限性急性皮膚紅斑性狼瘡。可見於顴部區域呈「蝴蝶 (butterfly)」分布之紅斑性、輕微水腫、界線分明的紅斑。
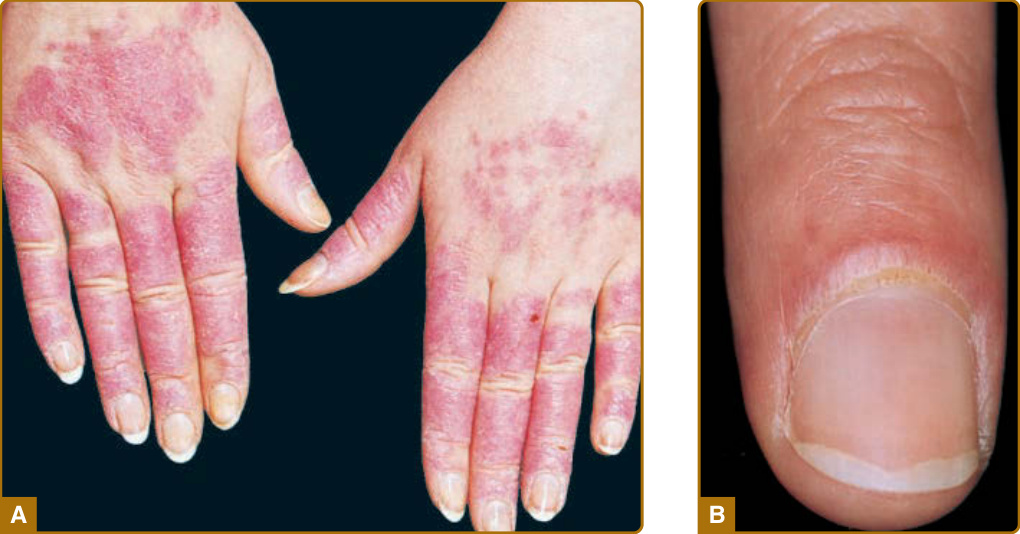
圖 61-3:廣泛性急性皮膚紅斑性狼瘡。A,手背、手指與甲周區域上界線分明的紅斑斑片,覆有細微鱗屑。注意特徵性地不侵犯指關節,而指關節在皮肌炎中為優先受侵犯之處。B,甲周紅斑與肉眼可見之毛細血管擴張的特寫。雖然這些病灶可見於紅斑性狼瘡,但它們更為皮肌炎所典型。
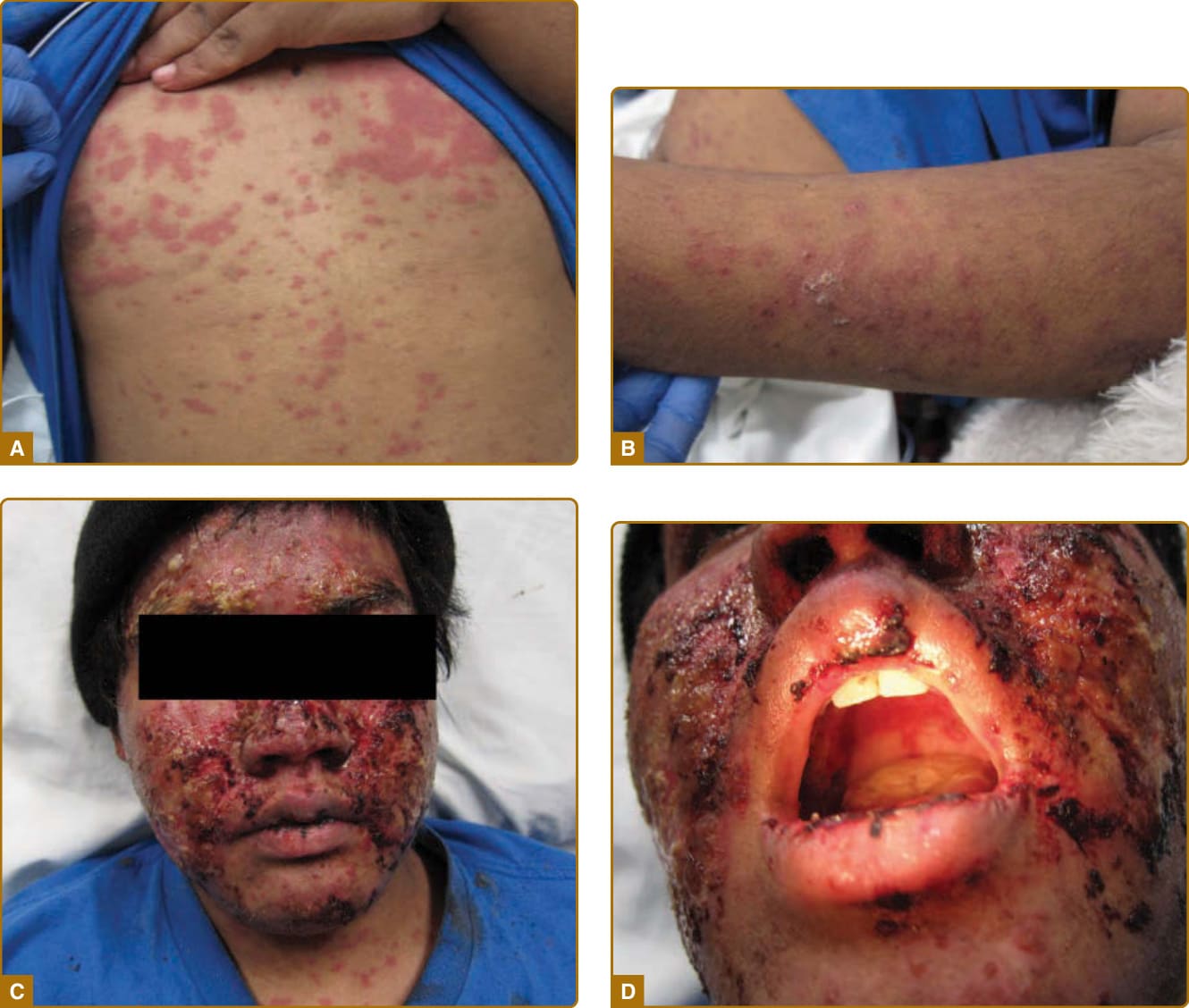
圖 61-4:一名 16 歲男孩的毒性表皮壞死溶解症 (TEN) 樣急性皮膚狼瘡。A,軀幹上廣布的紅斑性丘疹伴水疱大疱性變化。B,手臂上的類似變化。C,一名 16 歲男孩顏面的嚴重表皮喪失與剝脫。D,存在系統性紅斑性狼瘡特徵性之無痛顎部潰瘍,相對於 TEN 的嚴重黏膜侵犯。
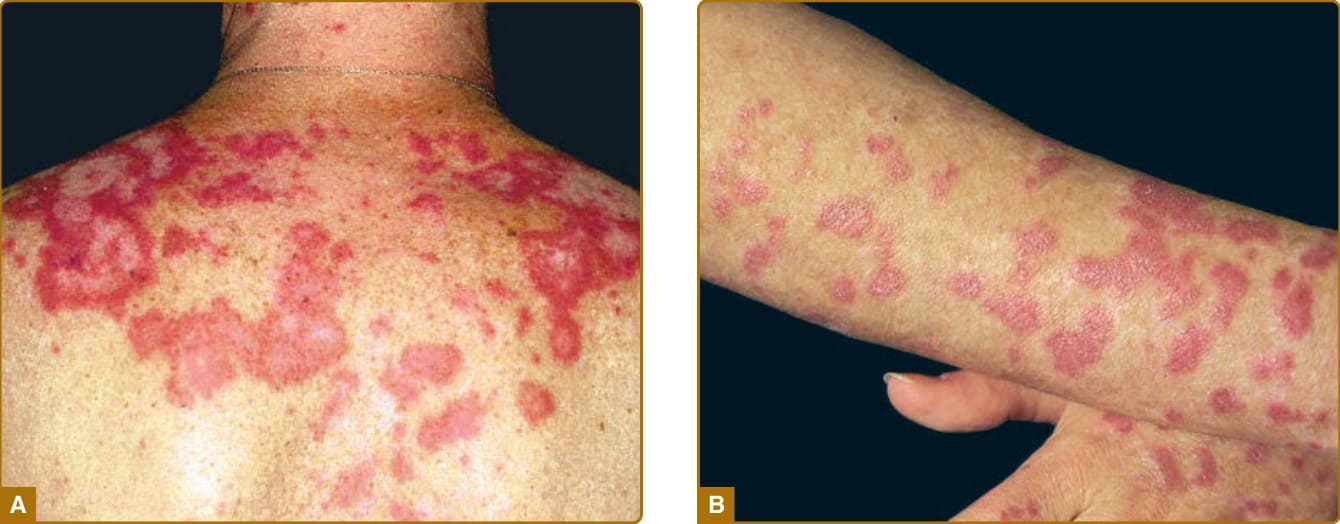
圖 61-5:亞急性皮膚紅斑性狼瘡 (SCLE)。A,一名 38 歲女性上背部的環狀 SCLE。注意中央色素脫失區域,其中不存在真皮萎縮。B,一名 26 歲女性前臂伸側的丘疹鱗屑型 SCLE。
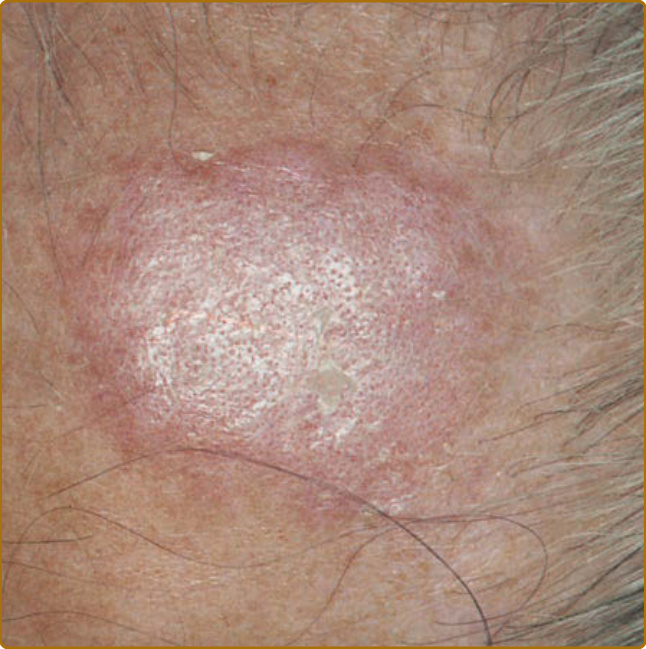
圖 61-6:典型盤狀紅斑性狼瘡。一名有 25 年皮膚紅斑性狼瘡病史的 60 歲男性,其前額上典型的早期紅斑性斑塊,顯示過度角化與毛囊開口的強化。該病灶已存在 3 個月;此階段不存在真皮萎縮。
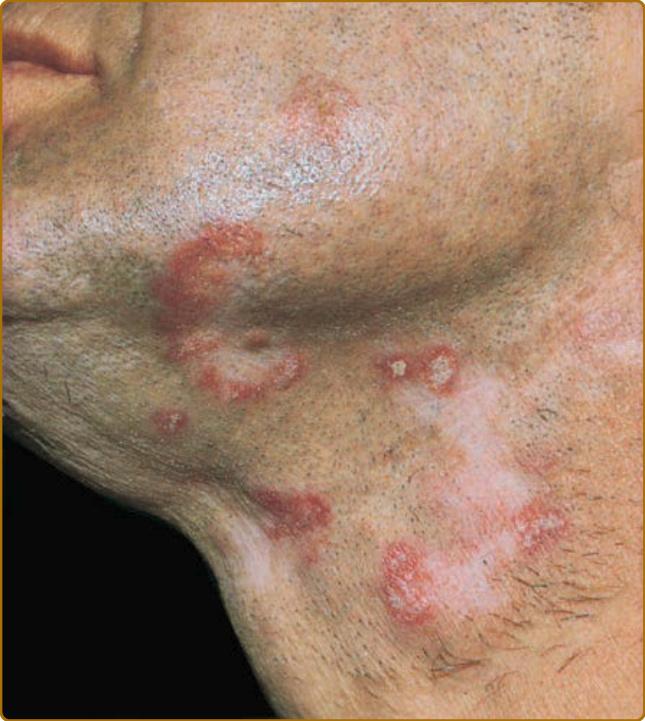
圖 61-7:典型盤狀紅斑性狼瘡。頸部與顏面上界線分明、圓形至卵圓形、輕微硬結的紅斑性斑塊。大多數斑塊顯示輕度的過度角化,某些顯示真皮萎縮。非發炎的色素脫失與瘢痕區域標誌著先前已消退病灶的部位。
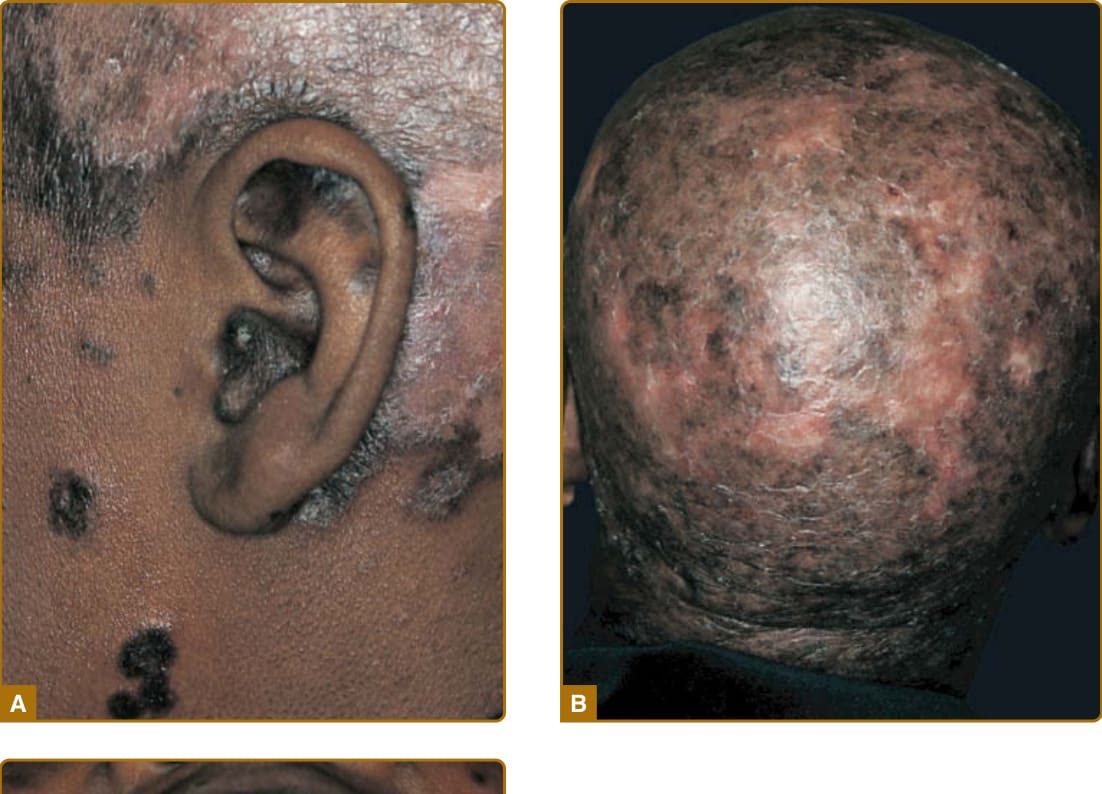
圖 61-8:一名有 20 年未經治療皮膚紅斑性狼瘡病史的 45 歲非裔美國女性的典型盤狀紅斑性狼瘡 (DLE) 與黏膜 DLE。A,耳朵的特徵性侵犯顯示伴有萎縮與發炎後過度色素沉著的病灶,以及頭皮上伴有發炎後色素脫失的發炎性紅色斑塊。B,頭皮上的融合性病灶已導致廣泛的瘢痕性禿髮。C,顎黏膜上的 DLE 斑塊顯示與皮膚病灶相似的型態特徵。
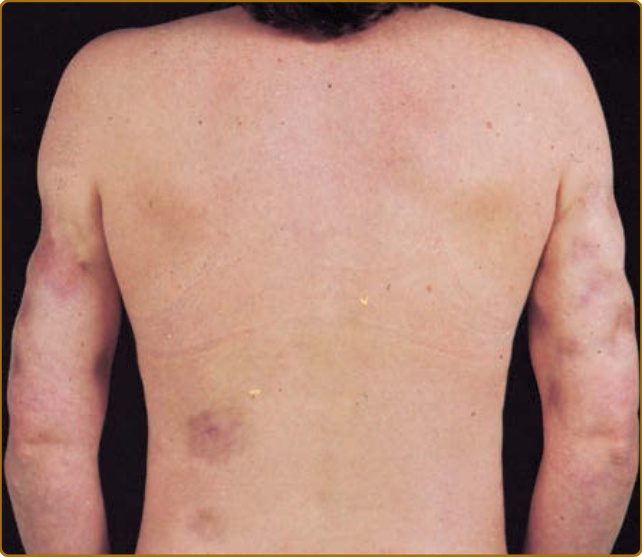
圖 61-9:紅斑性狼瘡脂膜炎。狼瘡脂膜炎已導致大片、凹陷的覆蓋皮膚區域;存在皮膚的紅斑與萎縮。
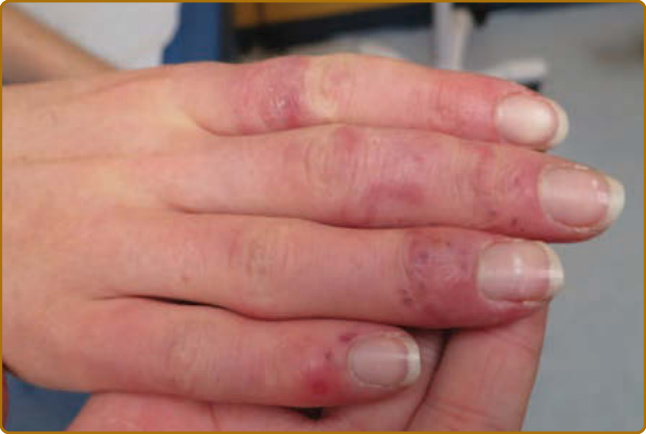
圖 61-10:凍瘡狼瘡。一名長期狼瘡與狼瘡腎炎病人侵犯手指的凍瘡病灶。這些皮膚病灶為慢性,且在沒有寒冷暴露的情況下發生。
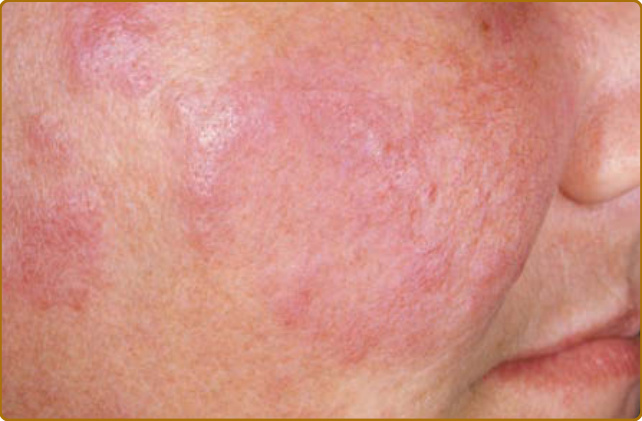
圖 61-11:腫脹性紅斑性狼瘡。注意多汁、硬結的斑塊。
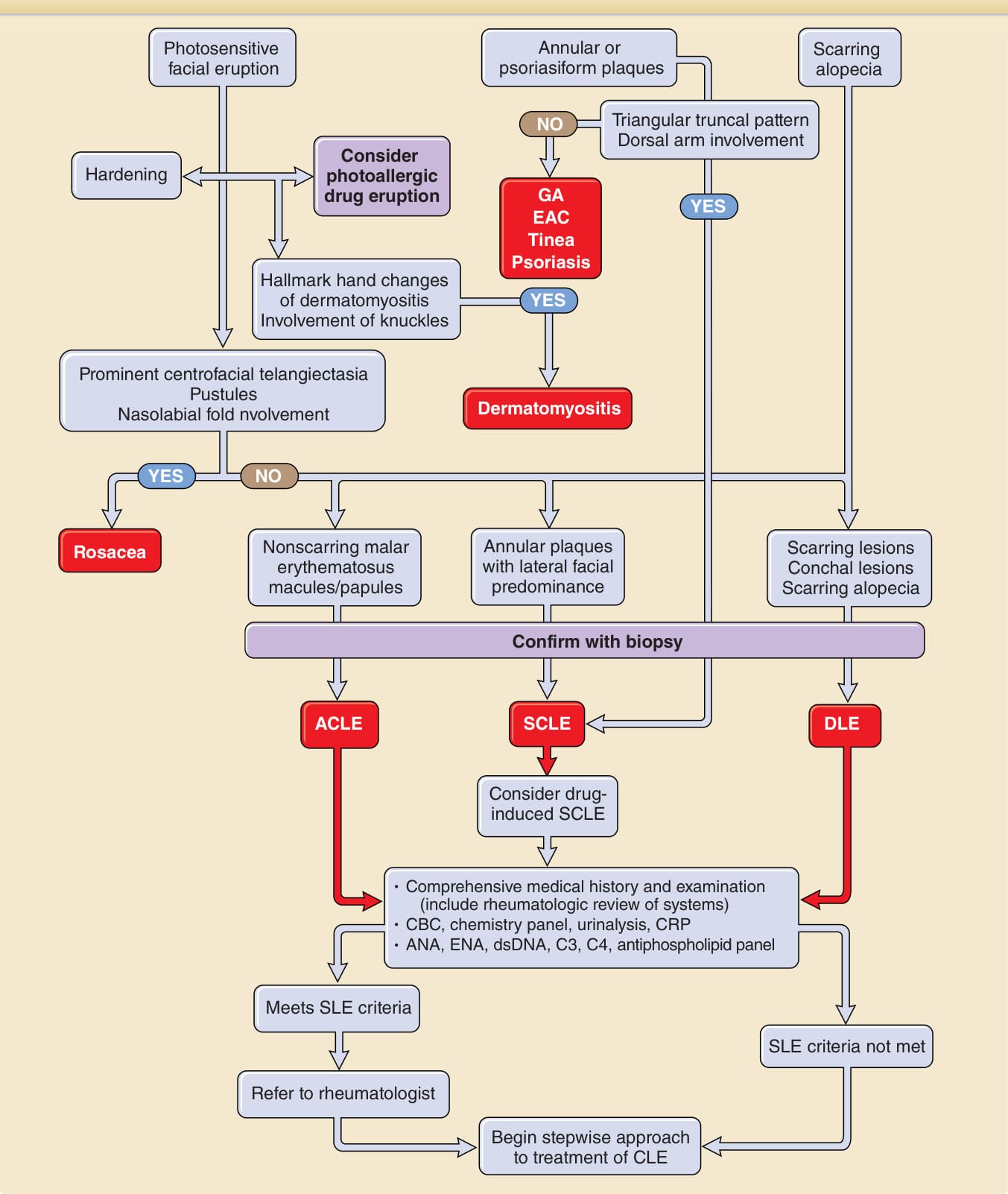
圖 61-12:概述對疑似 CLE 之皮膚病灶病人的處理方式。
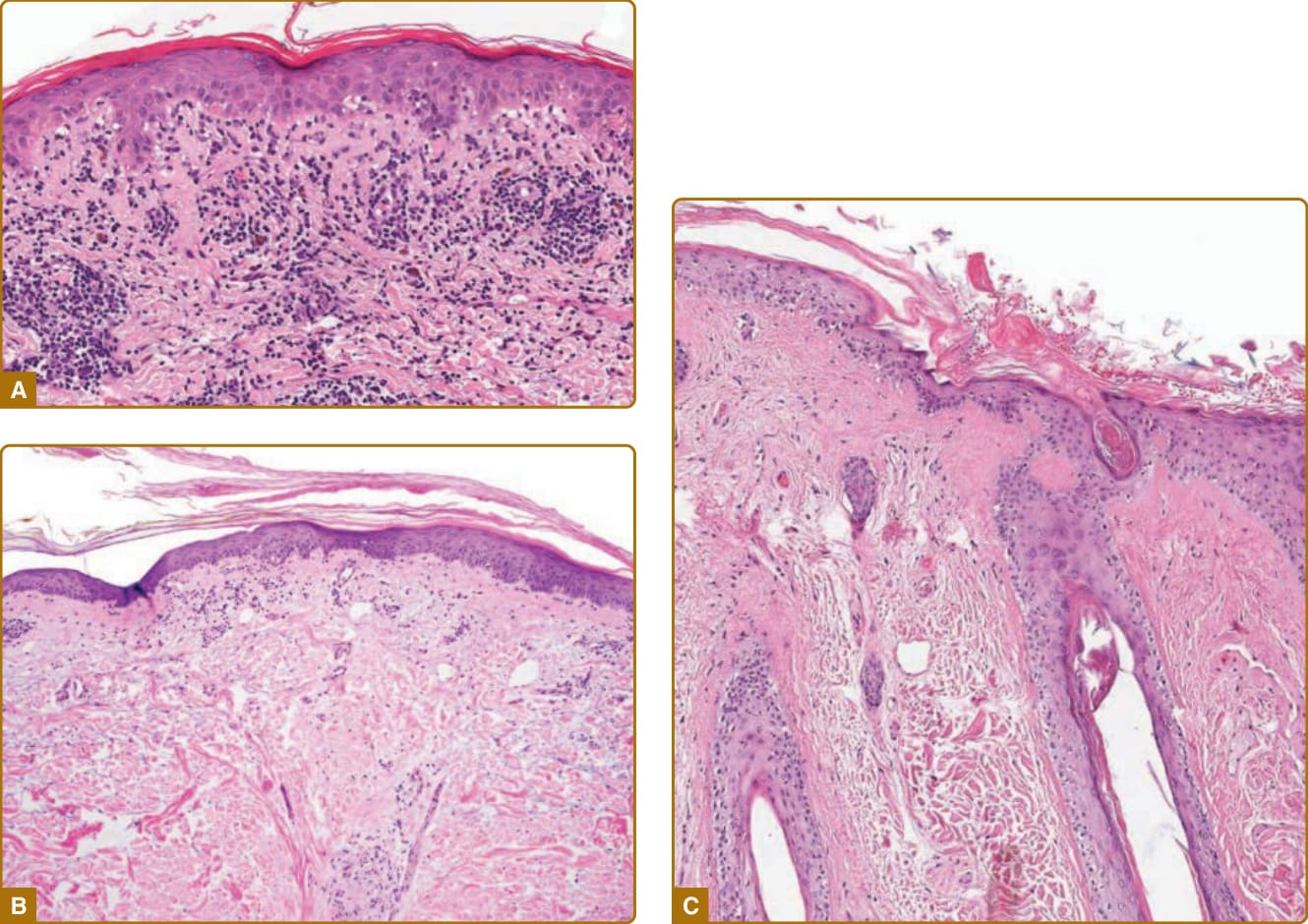
圖 61-13:紅斑性狼瘡特異性皮膚疾病的組織病理學。A,急性皮膚狼瘡病灶(蘇木精與伊紅 [hematoxylin and eosin, H&E] 染色)顯示介面性皮膚炎與空泡變化,以及血管周圍發炎細胞浸潤(×128)。B,亞急性皮膚狼瘡病灶(H&E 染色)伴有輕度介面性皮膚炎、空泡變化與輕度過度角化。也可見到輕度表皮萎縮與上真皮明顯的黏蛋白沉積(×100)。C,盤狀狼瘡病灶顯示典型的介面變化以及過度角化、毛囊角栓。也可見到輕度真皮纖維增生(×84)。
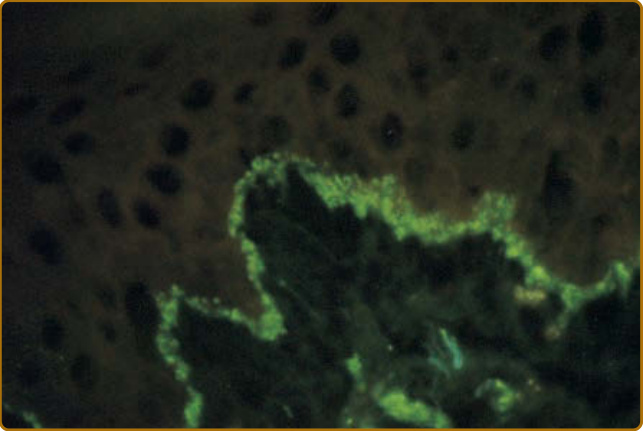
圖 61-14:紅斑性狼瘡特異性皮膚疾病的免疫病理學。一個盤狀紅斑性狼瘡病灶皮膚切片的直接免疫螢光 (direct immunofluorescence) 檢查,因以異硫氰酸螢光素 (fluorescein isothiocyanate) 結合的山羊抗免疫球蛋白 G 染色,而顯示真皮—表皮交界處連續的顆粒狀螢光帶。
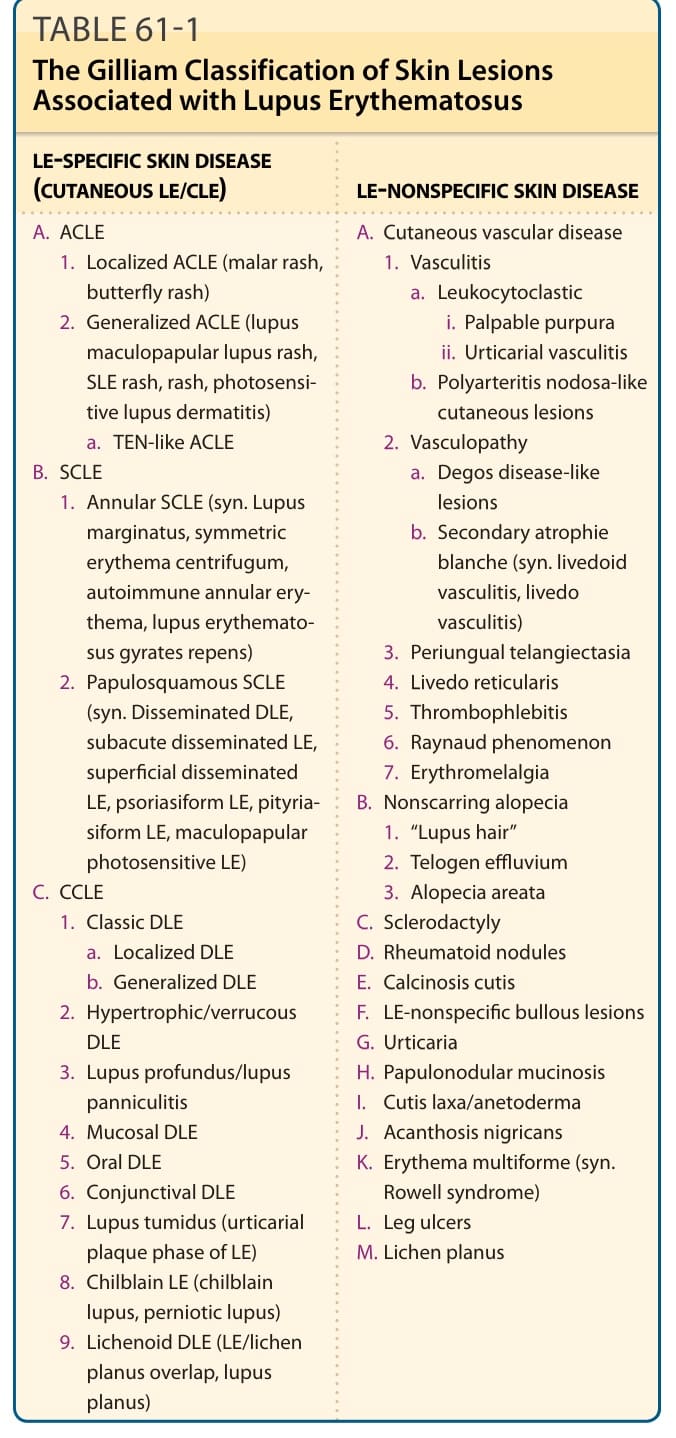
表 61-1:與紅斑性狼瘡相關之皮膚病灶的 Gilliam 分類 (The Gilliam Classification of Skin Lesions Associated with Lupus Erythematosus)
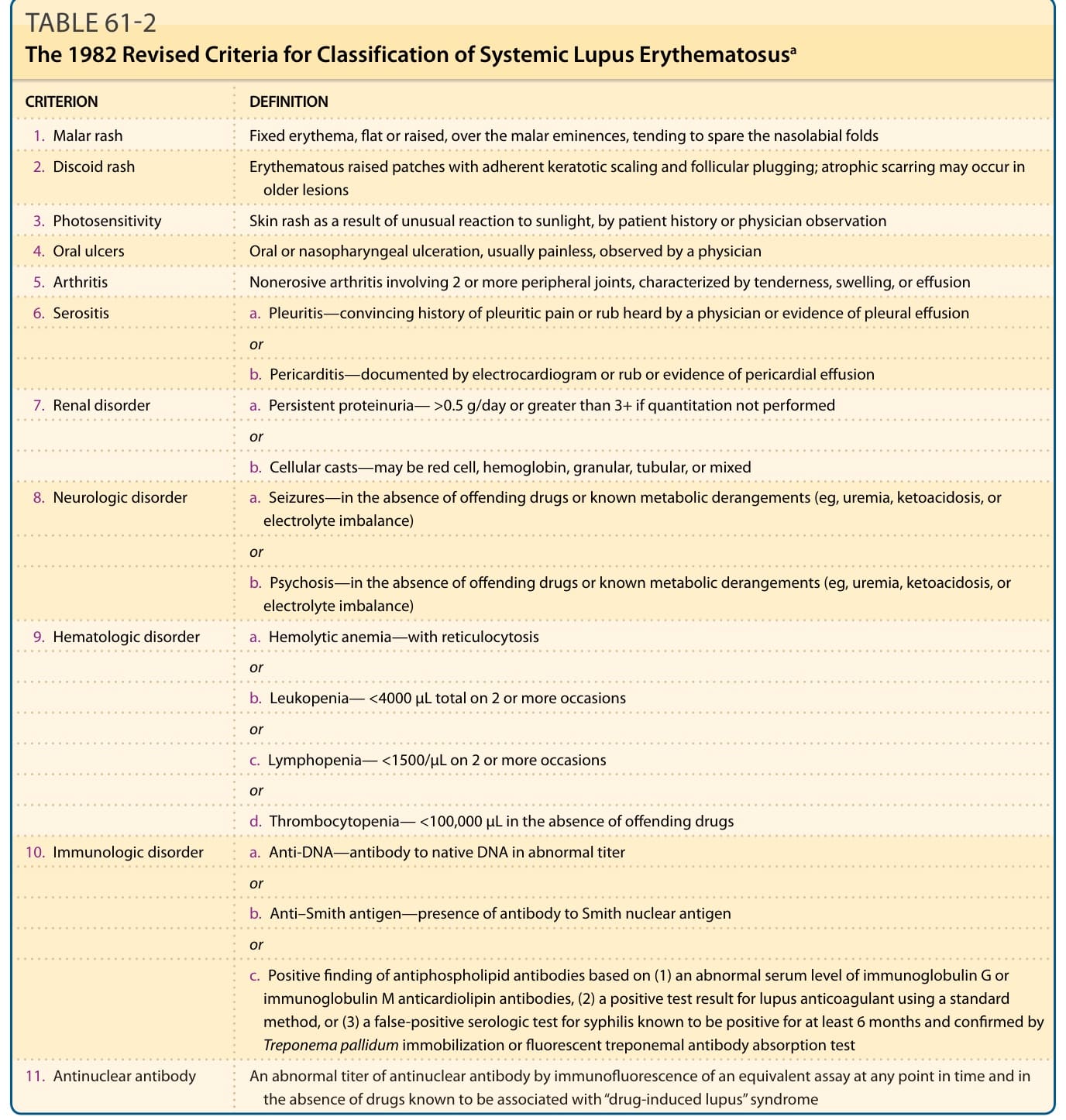
表 61-2:1982 年修訂的系統性紅斑性狼瘡分類標準 (The 1982 Revised Criteria for Classification of Systemic Lupus Erythematosus)

表 61-3:系統性紅斑性狼瘡皮膚外表現概觀 (Overview of the Extracutaneous Manifestations of Systemic Lupus Erythematosus)

表 61-4:藥物誘發狼瘡的原因 (Causes of Drug-Induced Lupus)

表 61-5:概述 CLE 的鑑別診斷。此外,網狀紅斑性黏蛋白沉積症已被某些人認為是光敏感性 CLE 的一種型式。
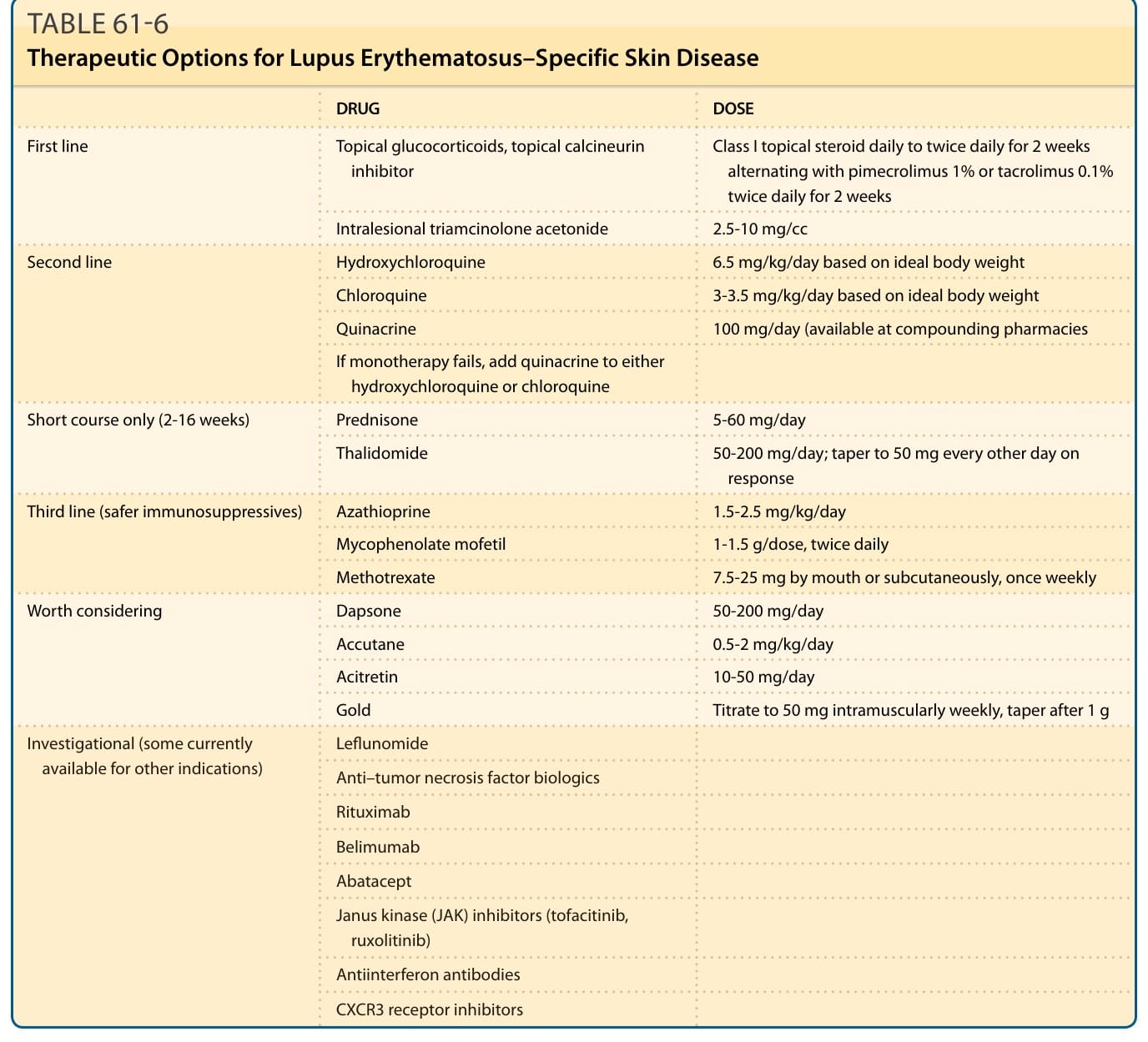
表 61-6:紅斑性狼瘡特異性皮膚疾病的治療選項 (Therapeutic Options for Lupus Erythematosus–Specific Skin Disease)