Lupus Erythematosus
PART10
Autoimmune Connective Tissue and Rheumatologic Disorders
AT-A-GLANCE
■ A group of heterogeneous illnesses that have in common the development of immunity to selfnucleic acids and their associated proteins, with skin-only disease at one end of the spectrum and severe visceral involvement at the other.
■ Skin lesions may be specific to lupus or nonspecific, being seen in other conditions as well.
■ Acute cutaneous lupus erythematosus (malar rash) is almost always associated with underlying visceral involvement. Subacute cutaneous lupus patients meet American College of Rheumatology systemic lupus erythematosus criteria approximately 50% of the time (but typically express only mild systemic clinical manifestations). Chronic cutaneous lupus (classic discoid lupus erythematosus, lupus panniculitis, chilblain lupus, and tumid lupus erythematosus) patients most often have skin-only or skin-predominant disease.
■ Classical discoid lupus erythematosus causes scarring and can be permanently disfiguring.
Lupus erythematosus (LE) is the root designation for a diverse array of clinical illnesses that are linked together by the development of autoimmunity directed predominantly at the molecular constituents of nucleosomes and ribonucleoproteins. Some patients present with life-threatening manifestations of systemic LE (SLE), whereas others, who are affected with what represents the same basic underlying disease process, express little more than isolated discoid LE (DLE) skin lesions throughout their illness. It is convenient to conceptualize LE as a clinical spectrum (Fig. 61-1) ranging from mildly affected patients with only localized DLE
Subacute cutaneous lupus and acute cutaneous lupus erythematosus are highly photosensitive and are characteristically nonscarring.
■ Lupus erythematosus–nonspecific skin lesions include nonscarring alopecia, mouth ulcers, photosensitivity, Raynaud phenomenon, vasculitis/ vasculopathy, and bullous systemic lupus erythematosus, among others. They often herald a systemic lupus erythematosus flare.
■ Treatment consists of physical sun protection, topical sunscreens, local and short-term systemic glucocorticoids, antimalarials, retinoids, thalidomide/lenalidomide, conventional immunosuppressives, and biologic therapies.
■ Lupus erythematosus occurs much more commonly in women (9:1 female-to-male ratio).
■ Both systemic lupus erythematosus and cutaneous lupus erythematosus are associated with upregulation of type 1 interferon signaling.
skin lesions to those at risk of dying from the systemic manifestations of LE such as nephritis, CNS disease, or vasculitis. The pattern of skin involvement expressed by an individual patient with LE can provide insight about the position on the spectrum where the patient’s illness might best be placed. The nomenclature and classification system originally devised by James N. Gilliam divides the cutaneous manifestations of LE into those lesions that show characteristic histologic changes of LE (LE-specific skin disease; ie, an interface dermatitis) and those that are not histopathologically distinct for LE and/or may
10
Lupus erythematosus spectrum
SCLE
Generalized DLE LE profundus
ACLE
Localized DLE Hypertrophic DLE
Skin disease only
Systemic disease
be seen as a feature of another disease processes (LEnonspecific skin disease).1 Within this context, the term LE-specific relates to those lesions displaying an interface dermatitis. The term cutaneous LE (CLE) is often used synonymously with “LE-specific skin disease” as an umbrella designation for the 3 major categories of LE-specific skin disease: acute cutaneous LE (ACLE), subacute cutaneous LE (SCLE), and chronic cutaneous LE (CCLE). This will be the framework used in our discussion of the extraordinarily diverse set of cutaneous lesions that occur in patients with LE (Table 61-1). The essence of LE is in its heterogeneity, and the challenge for those who treat it is to recognize clinically useful patterns within the mosaic of features that constitute this protean illness. An overview of the systemic manifestations of LE can be seen in the American College of Rheumatology’s (ACR) classification criteria for SLE,2 which are presented in Table 61-2, and from the outline of the systemic manifestations of SLE presented in Table 61-3.
EPIDEMIOLOGY
The epidemiology and socioeconomic impact of LE in general,3 and CLE specifically,4 have been reviewed. Skin disease is the second most frequent clinical manifestation of LE after joint inflammation. As many as 45% of patients with CLE experience some degree of vocational handicap. Recent quality-of-life studies suggested that the impact of skin manifestations in patients with SLE was preceded only by pain and fatigue related to their disease.5 The Dermatology Life Quality Index and SF-36 have been used to measure quality of life in patients with CLE. Both questionnaires showed that patients with active skin lesions had lower quality of life and that patients with associated alopecia were particularly impacted.6
1038
LE-SPECIFIC SKIN DISEASE (CUTANEOUS LE/CLE) LE-NONSPECIFIC SKIN DISEASE
A. ACLE
A. ACLE
A. Cutaneous vascular disease
A. Cutaneous vascular disease
-
Localized ACLE (malar rash,
-
Vasculitis
-
Localized ACLE (malar rash, butterfly rash)
-
Generalized ACLE (lupus maculopapular lupus rash, SLE rash, rash, photosensitive lupus dermatitis) a. TEN-like ACLE B. SCLE
-
Vasculitis
butterfly rash)
2. Generalized ACLE (lupus
a. Leukocytoclastic
a. Leukocytoclastic
i. Palpable purpura ii. Urticarial vasculitis b. Polyarteritis nodosa-like
i. Palpable purpura ii. Urticarial vasculitis b. Polyarteritis nodosa-like cutaneous lesions
2. Vasculopathy
maculopapular lupus rash, SLE rash, rash, photosensitive lupus dermatitis) a. TEN-like ACLE B. SCLE
cutaneous lesions
2. Vasculopathy
a. Degos disease-like
a. Degos disease-like lesions b. Secondary atrophie blanche (syn. livedoid vasculitis, livedo vasculitis)
3. Periungual telangiectasia
4. Livedo reticularis
5. Thrombophlebitis
6. Raynaud phenomenon
7. Erythromelalgia B. Nonscarring alopecia
- Annular SCLE (syn. Lupus
lesions b. Secondary atrophie
- Annular SCLE (syn. Lupus marginatus, symmetric erythema centrifugum, autoimmune annular erythema, lupus erythematosus gyrates repens)
- Papulosquamous SCLE (syn. Disseminated DLE, subacute disseminated LE, superficial disseminated LE, psoriasiform LE, pityriasiform LE, maculopapular photosensitive LE) C. CCLE
marginatus, symmetric erythema centrifugum, autoimmune annular erythema, lupus erythematosus gyrates repens)
2. Papulosquamous SCLE
blanche (syn. livedoid vasculitis, livedo vasculitis)
3. Periungual telangiectasia
4. Livedo reticularis
5. Thrombophlebitis
6. Raynaud phenomenon
7. Erythromelalgia B. Nonscarring alopecia
(syn. Disseminated DLE, subacute disseminated LE, superficial disseminated LE, psoriasiform LE, pityriasiform LE, maculopapular photosensitive LE) C. CCLE
-
“Lupus hair”
-
Telogen effluvium
-
Alopecia areata C. Sclerodactyly D. Rheumatoid nodules E. Calcinosis cutis F. LE-nonspecific bullous lesions G. Urticaria H. Papulonodular mucinosis I. Cutis laxa/anetoderma J. Acanthosis nigricans K. Erythema multiforme (syn.
-
“Lupus hair”
-
Telogen effluvium
-
Alopecia areata C. Sclerodactyly D. Rheumatoid nodules E. Calcinosis cutis F. LE-nonspecific bullous lesions G. Urticaria H. Papulonodular mucinosis I. Cutis laxa/anetoderma J. Acanthosis nigricans K. Erythema multiforme (syn. Rowell syndrome) L. Leg ulcers M. Lichen planus
-
Classic DLE
-
Classic DLE
a. Localized DLE b. Generalized DLE
2. Hypertrophic/verrucous
a. Localized DLE b. Generalized DLE
2. Hypertrophic/verrucous DLE
3. Lupus profundus/lupus panniculitis
4. Mucosal DLE
5. Oral DLE
6. Conjunctival DLE
7. Lupus tumidus (urticarial plaque phase of LE)
8. Chilblain LE (chilblain lupus, perniotic lupus)
9. Lichenoid DLE (LE/lichen planus overlap, lupus planus)
DLE
3. Lupus profundus/lupus
panniculitis
4. Mucosal DLE
5. Oral DLE
6. Conjunctival DLE
7. Lupus tumidus (urticarial
Rowell syndrome) L. Leg ulcers M. Lichen planus
plaque phase of LE)
8. Chilblain LE (chilblain
lupus, perniotic lupus)
9. Lichenoid DLE (LE/lichen
planus overlap, lupus planus)
ACLE, acute cutaneous lupus erythematosus; CCLE, chronic cutaneous lupus erythematosus; DLE, discoid lupus erythematosus; LE, lupus erythematosus; SCLE, subacute cutaneous lupus erythematosus; SLE, systemic lupus erythematosus; TEN, toxic epidermal necrolysis. Adapted from Sontheimer RD. The lexicon of cutaneous lupus erythematosus—a review and personal perspective on the nomenclature and classification of the cutaneous manifestations of lupus erythematosus. Lupus. 1997;6(2):84-95. Copyright © Stockton Press. Reprinted with permission of SAGE Publications.
Malar, or butterfly rash (localized ACLE), has been reported in 20% to 60% of large cohorts of patients with LE. Limited data suggest that the maculopapular or SLE rash of generalized ACLE is present in approximately 35% to 60% of patients with SLE. ACLE, like SLE in general, is 8 times more common in women than men. All races are affected; however, the early
10
CRITERION DEFINITION
-
Malar rash Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds
-
Discoid rash Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions
-
Photosensitivity Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation
-
Oral ulcers Oral or nasopharyngeal ulceration, usually painless, observed by a physician
-
Arthritis Nonerosive arthritis involving 2 or more peripheral joints, characterized by tenderness, swelling, or effusion
-
Serositis a. Pleuritis—convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion
or
b. Pericarditis—documented by electrocardiogram or rub or evidence of pericardial effusion
- Renal disorder a. Persistent proteinuria— >0.5 g/day or greater than 3+ if quantitation not performed
or
b. Cellular casts—may be red cell, hemoglobin, granular, tubular, or mixed
- Neurologic disorder a. Seizures—in the absence of offending drugs or known metabolic derangements (eg, uremia, ketoacidosis, or electrolyte imbalance)
or
b. Psychosis—in the absence of offending drugs or known metabolic derangements (eg, uremia, ketoacidosis, or electrolyte imbalance)
- Hematologic disorder a. Hemolytic anemia—with reticulocytosis
or
b. Leukopenia— <4000 µL total on 2 or more occasions
or
c. Lymphopenia— <1500/µL on 2 or more occasions
or
d. Thrombocytopenia— <100,000 µL in the absence of offending drugs
- Immunologic disorder a. Anti-DNA—antibody to native DNA in abnormal titer
or
b. Anti–Smith antigen—presence of antibody to Smith nuclear antigen
or
c. Positive finding of antiphospholipid antibodies based on (1) an abnormal serum level of immunoglobulin G or immunoglobulin M anticardiolipin antibodies, (2) a positive test result for lupus anticoagulant using a standard method, or (3) a false-positive serologic test for syphilis known to be positive for at least 6 months and confirmed by Treponema pallidum immobilization or fluorescent treponemal antibody absorption test
-
Antinuclear antibody An abnormal titer of antinuclear antibody by immunofluorescence of an equivalent assay at any point in time and in
-
Antinuclear antibody An abnormal titer of antinuclear antibody by immunofluorescence of an equivalent assay at any point in time and in the absence of drugs known to be associated with “drug-induced lupus” syndrome
the absence of drugs known to be associated with “drug-induced lupus” syndrome
aThe proposed classification is based on 11 criteria. For the purpose of identifying patients in clinical studies, a person shall be said to have systemic lupus erythematosus if any 4 or more of the 11 criteria are present, serially or simultaneously, during any interval or observation. From Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11): 1271-1277, copyright 1982, with permission of the American College of Rheumatology.
clinical manifestations of ACLE can be overlooked in a dark-skinned individual. Patients presenting with SCLE lesions constitute 7% to 27% of LE patient populations. SCLE is primarily a disease of white females, with the mean age of onset being in the fifth decade. Drug-induced SCLE patients are somewhat older at disease onset, perhaps reflecting greater exposure to drugs for age-related medical problems (hypertension, cardiovascular disease). The most common form of CCLE, a classic DLE skin lesion, is present in 15% to 30% of SLE populations
selected in various ways. Approximately 5% of patients presenting with isolated localized DLE subsequently develop SLE. Published population-based data have argued strongly that the incidence and prevalence of isolated forms of CLE are equivalent to those of SLE.7
Although DLE can occur in infants and the elderly, it is most common in individuals between 20 and 40 years of age. DLE has a female-to-male ratio of 3:2 to 3:1, which is much lower than that of SLE. All races are affected, but investigations suggest that DLE might be more prevalent in blacks.
1039
10
■General
■General
■Fever, fatigue, malaise, weight loss
■Fever, fatigue, malaise, weight loss
■Alopecia
■Alopecia
■Musculoskeletal
■Musculoskeletal
■Arthralgia, arthritis, morning stiffness
■Arthralgia, arthritis, morning stiffness
■Myalgia, myositis
■Myalgia, myositis
■Avascular necrosis
■Avascular necrosis
■Hematologic
■Hematologic
■Anemia
■Anemia
■Anemia of chronic disease (normocytic)
■Anemia of chronic disease (normocytic)
■Autoimmune hemolytic anemia (Coombs positive)
■Autoimmune hemolytic anemia (Coombs positive)
■Leukopenia, lymphopenia, neutropenia
■Leukopenia, lymphopenia, neutropenia
■Thrombocytopenia
■Thrombocytopenia
■Antiphospholipid syndrome, thrombosis (thrombophlebitis, deep vein thrombosis)
■Antiphospholipid syndrome, thrombosis (thrombophlebitis,
deep vein thrombosis)
■Cardiopulmonary
■Cardiopulmonary
■Pleurisy, pleural effusion, aseptic pneumonitis, pulmonary hemorrhage, Interstitial lung disease, pulmonary embolism, coronary artery disease
■Pleurisy, pleural effusion, aseptic pneumonitis, pulmonary
hemorrhage, Interstitial lung disease, pulmonary embolism, coronary artery disease
■Chest pain, pericarditis, myocarditis Libman-Sacks endocarditis
■Chest pain, pericarditis, myocarditis Libman-Sacks endocarditis
■Renal
■Renal
■Membranoproliferative glomerulonephritis (World Health Organization [WHO] classes II-IV)
■Membranoproliferative glomerulonephritis (World Health
Organization [WHO] classes II-IV)
■Proteinuria, hematuria, red blood cell casts
■Proteinuria, hematuria, red blood cell casts
■Membranous glomerulonephritis (WHO class V)
■Membranous glomerulonephritis (WHO class V)
■Severe proteinuria, nephrotic syndrome
■Severe proteinuria, nephrotic syndrome
■Neuropsychiatric
■Neuropsychiatric
■Peripheral neuropathy, transverse myelitis, Guillain-Barré syndrome
■Peripheral neuropathy, transverse myelitis, Guillain-Barré
syndrome
■Chorea, choreoathetosis
■Chorea, choreoathetosis
■Seizures
■Seizures
■Headaches
■Headaches
■Acute confusional state, psychosis
■Acute confusional state, psychosis
■Cognitive dysfunction
■Cognitive dysfunction
■Aseptic meningitis
■Aseptic meningitis
■GI
■GI
■Anorexia, nausea, vomiting, abdominal pain
■Anorexia, nausea, vomiting, abdominal pain
■Hepatitis, hepatomegaly
■Hepatitis, hepatomegaly
■Pancreatitis
■Pancreatitis
■Mesenteric vasculitis
■Mesenteric vasculitis
■Protein-losing enteropathy
■Protein-losing enteropathy
■Ocular
■Ocular
■Retinal vascular disease, optic neuropathy
■Retinal vascular disease, optic neuropathy
■Conjunctivitis, episcleritis
■Conjunctivitis, episcleritis
■Dry eyes, keratitis
■Dry eyes, keratitis
■Nasooral
■Nasooral
■Palatal ulceration
■Palatal ulceration
■Sicca syndrome, secondary Sjögren Syndrome
■Sicca syndrome, secondary Sjögren Syndrome
■Lymphatic system
■Lymphatic system
■Lymphadenopathy
■Lymphadenopathy
■Splenomegaly
■Splenomegaly
ETIOLOGY AND PATHOGENESIS
The cause(s) of and pathogenic mechanisms responsible for LE-specific skin disease are not fully understood, although recent work has provided many new insights. The pathogenesis of LE-specific skin disease
1040
is inextricably intertwined with SLE pathogenesis. Simply put, SLE is a disorder in which the interplay between host factors (susceptibility genes, hormonal milieu, etc) and environmental factors (ultraviolet [UV] radiation, viruses, and drugs) leads to loss of self-tolerance, and induction of autoimmunity. This is followed by activation and expansion of the immune system, and eventuates in immunologic injury to end organs and clinical expression of disease.8 Recent work highlights the important role of interferon-α signaling in the pathogenesis of both SLE and LE-specific skin disease.
IMMUNOLOGIC FACTORS
IMMUNOLOGIC FACTORS
Multiple studies highlight the important role of plasmacytoid dendritic cells (PDCs) and type 1 interferon (IFN) signaling, comprised of interferon-α and interferon-β, in the pathogenesis of both SLE and LEspecific skin disease. type 1 IFN is the most upregulated gene pathway identified in microarray studies in SLE patients, and increased type 1 IFN has been seen in the blood and skin of patients with CLE.9,10
Large numbers of PDCs are present in skin lesions of patients with CLE.11 PDCs produce large amounts of type 1 IFN in response to DNA and RNA stimulation through toll-like receptors 7 and 9. In healthy state conditions, PDCs recognize viral DNA and RNA and not self-DNA; however, immune complexes generated by autoantibody binding of DNA and RNA from apoptotic cells are recognized by PDCs and lead to activation and production of type 1 IFN. Chronic type 1 IFN, in turn, promotes loss of tolerance in SLE through its actions of B and T cells.12,13
Recent work has identified a separate DNA- sensing and RNA-sensing pathway, termed the immunostimulatory DNA pathway, that is active within the cytoplasm in contrast to endosomal toll-like receptors. Monogenetic syndrome caused by mutations in immunostimulatory DNA pathway genes, as seen in Aicardi-Goutières syndrome and SAVI, result in upregulated immunostimulatory DNA pathway activity and chronic IFN-β production, and share many features with spontaneous LE.14 Single- nucleotide polymorphisms in immunostimulatory DNA pathway genes are associated with CLE and SLE.15 Insights from these diseases, termed interferonopathies, further support the role for inappropriate type 1 IFN activity, whether IFN-α or IFN-β, as a proximal and driving event in lupus pathogenesis.
ENVIRONMENTAL FACTORS
ENVIRONMENTAL FACTORS
Genetic predisposition for a lupus diathesis does not, in itself, produce disease. Rather, it appears that induction of autoimmunity in such patients is triggered by some inciting event, likely an environmental exposure.
Drugs, viruses, UV light, and, possibly, tobacco induce development of SLE. UV radiation is probably the most important environmental factor in the induction phase of SLE, especially of LE-specific skin disease. UV light likely leads to self-immunity and loss of tolerance because it causes apoptosis of keratinocytes, which in turn, makes previously cryptic peptides available for immunosurveillance. UVB radiation displaces autoantigens such as Ro/SS-A and related autoantigens, La/SS-B, and calreticulin, from their normal locations inside epidermal keratinocytes to the cell surface.16 UVB irradiation induces the release of CCL27 (cutaneous T cell- attracting chemokine), which upregulates the expression of chemokines that activate autoreactive T cells and type 1 IFN, producing dendritic cells, which likely play a central role in lupus pathogenesis.17,18
A recent, large, case-control study reported that smokers are at a greater risk of developing SLE than are nonsmokers and former smokers. A cross-sectional analysis of a collaborative Web-based database established by Werth and colleagues documented that patients with treatment-resistant CLE were much more likely to smoke.19 Several authors have shown that patients with LE-specific skin disease who smoke are less responsive to antimalarial treatment.20-22
Numerous drugs are implicated in inducing various features of SLE (Table 61-4). The drugs that induce CLE can be linked by their photosensitizing properties. It is suggested that these drugs cause an increase in keratinocyte apoptosis, exposure of previously intracellular peptides on epidermal cell surfaces, and enhance proinflammatory cytokines such as tumor necrosis factor (TNF)-α and type 1 IFN.23,24
There is much speculation about the role of infectious agents, particularly viruses, in the induction of SLE and CLE. Seroconversion to Epstein-Barr virus among patients with SLE is nearly universal, and recent data demonstrate that patients with SLE have defective control of latent Epstein-Barr virus infection that probably stems from altered T-cell responses against Epstein-Barr virus.
CLINICAL FINDINGS
It is important to distinguish among the subtypes of LE-specific skin disease, because the type of skin involvement in LE can reflect the underlying pattern of SLE activity. In fact, the designations acute, subacute, and chronic, in regard to CLE, refer to the pace and severity of any associated SLE and are not necessarily related to how long individual lesions have been present. For example, ACLE almost always occurs in the setting of acutely flaring SLE, whereas CCLE often occurs in the absence of SLE or in the presence of mild, smoldering SLE. SCLE occupies an intermediate position in this clinical spectrum. Subclassification, although important for assigning risk, is sometimes difficult, as it is not uncommon to see more than 1 subtype of LE-specific skin disease in the same patient, especially in patients with SLE.
10
Drug-Induced Subacute Cutaneous Lupus Erythematosus
Antacidsa
Protein pump inhibitors (esomeprazole, lansoprazole, omeprazole, pantoprazole) Antibiotics (Amoxicillin/clavulanic acid, doxycycline, minocycline, nitrofurantoin, norfloxacin) Anticonvulsants/antiepileptics (carbamazepine, phenytoin, lamotrigine) Antifungalsa (griseofulvin, terbinafine) Antihistamines (brompheniramine, ranitidine) Antiinflammatory Nonsteroidal antiinflammatories (naproxen, piroxicam) Immunomodulators (hydroxychloroquine, imiquimod, interferon-α, interferon-β, leflunomide) Biologicsa (adalimumab, bevacizumab, efalizumab, etanercept, golimumab, infliximab, ranibizumab, rituximab) Cardiovascular Antihypertensives Angiotensin-converting enzyme (ACE) inhibitors (captopril, cilazapril, enalapril, lisinopril, ramipril) ACEII receptor antagonists (losartan) Beta blockers (acebutolol, oxypurinol) Calcium channel blockers (amlodipine, diltiazem, nifedipine, nitrendipine, verapamil) Diuretics (chlorothiazide, hydrochlorothiazide, hydrochlorothiazide/triamterene) Statins (pravastatin, simvastatin) Chemotherapeutica (capecitabine, docetaxel, doxorubicin, doxorubicin/cyclophosphamide, fluorouracil, gemcitabine, mitotane, paclitaxel, pazopanib, pemetrexed/carboplatin, tamoxifen) Hormone-altering (anastrozole, leuprorelin) Ultraviolet therapy (psoralen and ultraviolet A [PUVA]) Others (allopurinol, amiodarone, bupropion, citalopram, iodine-131, torsemide, tiotropium, ticlopidine)
Drug-Induced Systemic Lupus Erythematosus (Typically without Skin Involvement)
Antihyperlipidemic agents Anti–tumor necrosis factor biologics Hydralazine Isoniazid Minocycline Procainamide
Antihyperlipidemic agents Anti–tumor necrosis factor biologics Hydralazine Isoniazid Minocycline Procainamide
aIndicates most commonly imputed drug classes. Imputed drugs are listed alphabetically by class. Data summarized from Michaelis TC, Sontheimer RD, Lowe GC. An update in drug-induced subacute cutaneous lupus erythematosus. Dermatol Online J. 2017;23(3).
ACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
ACUTE CUTANEOUS LUPUS
ERYTHEMATOSUS
Although ACLE localized to the face is the usual pattern of presentation, ACLE can assume a generalized distribution. Localized ACLE has commonly been referred to as the classic butterfly rash or malar rash of SLE (Fig. 61-2). In localized ACLE, confluent symmetric erythema and edema are centered over the malar eminences and bridges over the nose (unilateral involvement with ACLE has been described). The nasolabial folds are
1041
10
characteristically spared. The forehead, chin, and V area of the neck can be involved, and severe facial swelling may occur. Occasionally, ACLE begins as small macules and/or papules on the face that later may become confluent and hyperkeratotic. Generalized ACLE presents as a widespread morbilliform or exanthematous eruption often focused over the extensor aspects of the arms and hands and characteristically sparing the knuckles (Fig. 61-3A). Although perivascular nail fold erythema and telangiectasia can occur (Fig. 61-3B), they are considerably more common and occur in more exaggerated forms in dermatomyositis and systemic sclerosis. Generalized ACLE has been indiscriminately referred to as
the maculopapular rash of SLE, photosensitive lupus dermatitis, and SLE rash. An extremely acute form of ACLE is rarely seen that can simulate toxic epidermal necrolysis (TEN; Fig. 61-4). This form of LE-specific vesiculobullous disease results from widespread apoptosis of epidermal keratinocytes, and eventuates in areas of full-thickness epidermal skin necrosis, which is subsequently denuded. ACLE can be differentiated from true TEN because it occurs on predominantly sun-exposed skin and has a more insidious onset. The mucosa may or may not be involved, as in TEN. ACLE is typically precipitated or exacerbated by exposure to UV light. This form of CLE can be quite ephemeral, lasting only hours, days, or weeks; however, some patients experience more prolonged periods of activity. Postinflammatory pigmentary change is most prominent in patients with heavily pigmented skin. Atrophic scarring does not occur in ACLE unless the process is complicated by secondary bacterial infection.
SUBACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
SUBACUTE CUTANEOUS
LUPUS ERYTHEMATOSUS
The presence of SCLE skin lesions was first characterized as a distinctive immunogenetic subset of LE in 1979.25 A disease presentation dominated by SCLE lesions marks the presence of a distinct subset of LE having characteristic clinical, serologic, and genetic features. Although a finding of circulating autoantibodies to the Ro/SS-A ribonucleoprotein particle strongly supports a diagnosis of SCLE, the presence of this autoantibody specificity is not required to make a diagnosis of SCLE. SCLE initially presents as erythematous macules and/or papules that evolve into hyperkeratotic papulosquamous or annular/polycyclic plaques (Fig. 61-5). Whereas most patients have either annular
A
B
1042
A
C
10
B
D
A
B
1043
10
or papulosquamous SCLE, a few develop elements of both morphologic varieties. SCLE lesions are characteristically photosensitive and occur in predominantly sun-exposed areas (ie, upper back, shoulders, extensor aspects of the arms, V area of the neck, and, less commonly, the face). SCLE lesions typically heal without scarring but can resolve with long-lasting, if not permanent, vitiligo-like leukoderma, and telangiectasias. Several variants of SCLE have been described. On occasion, SCLE lesions present initially with an appearance of erythema multiforme. Such cases are similar to Rowell syndrome (erythema multiforme–like lesions occurring in patients with SLE in the presence of La/ SS-B autoantibodies). As a result of intense injury to epidermal basal cells, the active edge of an annular SCLE lesion occasionally undergoes a vesiculobullous change that can subsequently produce a strikingly crusted appearance. Such lesions can mimic Stevens- Johnson syndrome/TEN. Pathogenesis is similar to that described above for TEN-like ACLE. Rarely, SCLE presents with exfoliative erythroderma or displays a curious acral distribution of annular lesions. Pityriasiform and exanthematous variants of SCLE have been reported. The skin lesions of neonatal LE (transient, photosensitive, nonscarring LE-specific skin lesions in neonates who have received immunoglobulin (Ig) G anti-Ro/ SS-A, and, occasionally, other autoantibody specificities transplacentally) share many features with SCLE. Unlike ACLE skin lesions, SCLE lesions tend to be less transient than ACLE lesions and heal with more pigmentary change. They are also less edematous and more hyperkeratotic than ACLE lesions. SCLE more commonly involves the neck, shoulders, upper extremities, and trunk, whereas ACLE more commonly affects the malar areas of the face. When the face is involved with SCLE, it is most often the lateral face, with sparing of the central, malar regions. In comparison to SCLE lesions, DLE lesions are generally associated with a greater degree of hyperpigmentation and hypopigmentation, atrophic dermal scarring, follicular plugging, and adherent scale. A consistent clinical difference is that DLE lesions are characteristically indurated, whereas SCLE lesions are not; this difference reflects the greater depth of inflammation seen histopathologically in DLE lesions. Approximately one-half of patients with SCLE meet the ACR’s revised criteria for the classification of SLE. However, manifestations of severe SLE, such as nephritis, CNS disease, and systemic vasculitis, develop in only 10% to 15% of patients with SCLE. It has been suggested that the papulosquamous type of SCLE, leukopenia, high titer of antinuclear antibody (ANA) (>1:640), and anti–double-stranded DNA (dsDNA) antibodies are risk factors for the development of SLE in a patient presenting with SCLE lesions. SCLE can overlap with other autoimmune diseases that share the 8.1 ancestral haplotype (human leukocyte antigen A1-B8-DR3-DQ2), including Sjögren’s syndrome, dermatitis herpetiformis, sarcoidosis, and Hashimoto thyroiditis. Other disorders that have been anecdotally related to SCLE are rheumatoid arthritis, Sweet syndrome, porphyria cutanea
1044
tarda, gluten-sensitive enteropathy, and Crohn disease. There has also been the suggestion anecdotally that SCLE may be associated with internal malignancy (breast, lung, gastric, uterine, hepatocellular, and laryngeal carcinomas, as well as with Hodgkin lymphoma).26 Several different types of chemotherapeutic agents are known to be capable of triggering drug-induced SCLE. This could perhaps confound the possibility that SCLE has a significant association with internal malignancy.
CHRONIC CUTANEOUS LUPUS ERYTHEMATOSUS
CHRONIC CUTANEOUS
LUPUS ERYTHEMATOSUS
CLASSIC DISCOID LUPUS ERYTHEMATOSUS
Classic DLE lesions, the most common form of CCLE, begin as red-purple macules, papules, or small plaques and rapidly develop a hyperkeratotic surface. Early classic DLE lesions typically evolve into sharply demarcated, coin-shaped (ie, discoid) erythematous plaques covered by a prominent, adherent scale that extends into the orifices of dilated hair follicles (Fig. 61-6). DLE lesions typically expand with erythema and hyperpigmentation at the periphery, leaving hallmark atrophic central scarring, telangiectasia, and hypopigmentation (Fig. 61-7). DLE lesions at this stage can merge to form large, confluent, disfiguring plaques. DLE in persons of certain ethnic backgrounds, such as Asian Indians, can present clinically as isolated
areas of macular hyperpigmentation. When present on hair-bearing skin (scalp, eyelid margins, and eyebrows), DLE causes scarring alopecia, which can lead to disfigurement and markedly impact quality of life. Follicular involvement in DLE is a prominent feature. Keratotic plugs accumulate in dilated follicles that soon become devoid of hair. When the adherent scale is lifted from more advanced lesions, keratotic spikes similar in appearance to carpet tacks can be seen to project from the undersurface of the scale (ie, the “carpet tack” sign). DLE lesions can be difficult to diagnose in white patients because the characteristic peripheral hyperpigmentation is often absent. Hypertrophic DLE lesions can be confused with hypertrophic actinic keratosis, keratoacanthoma, and squamous cell carcinoma. DLE lesions are most frequently encountered on the face, scalp, ears, V area of the neck, and extensor aspects of the arms. Any area of the face, including the eyebrows, eyelids, nose, and lips, can be affected. A symmetric, hyperkeratotic, butterfly-shaped DLE plaque is occasionally found over the malar areas of the face and bridge of the nose. Such lesions should not be confused with the more transient, edematous, minimally scaling ACLE erythema reactions that occur in the same areas. Facial DLE, like ACLE and SCLE, usually spares the nasolabial folds. It may be difficult to distinguish early lesions of malar DLE from ACLE, but induration and recalcitrance to topical steroids/calcineurin inhibitors favor the former diagnosis. When DLE lesions occur periorally, they can resolve with a striking acneiform pattern of pitted
10
scarring. DLE characteristically affects the external ear, including the conchal bowl and outer portion of the external auditory canal (Fig. 61-8A). Such lesions often present initially as dilated, hyperpigmented follicles. The scalp is involved in 60% of patients with DLE; irreversible, scarring alopecia resulting from such involvement has been reported in one-third of patients (Fig. 61-8B). The irreversible, scarring alopecia resulting from DLE differs from the reversible, nonscarring alopecia that patients with SLE often develop during periods of systemic disease activity. This type of hair loss, so-called lupus hair, may represent telogen effluvium occurring as the result of flaring systemic disease. Localized DLE lesions occur only on the head or neck, whereas generalized DLE lesions occur both above and below the neck. Generalized DLE is more commonly associated with underlying SLE and is often more recalcitrant to standard therapy, frequently requiring layering of antimalarial and immunosuppressive medications. DLE lesions below the neck most commonly occur on the extensor aspects of the arms, forearms, and hands, although they can occur at virtually any site on the body. The palms and soles can be the sites of painful, and at times disabling, erosive DLE lesions. On occasion, small DLE lesions occurring only around follicular orifices appear at the elbow and elsewhere (follicular DLE). We have observed that elbow/ extensor arm lesions seem to occur with acral finger lesions of DLE, and that patients with this combination of findings more frequently have active systemic disease. DLE activity can localize to the nail unit. The nail can be impacted by other forms of CLE as well as SLE, producing nail fold erythema and telangiectasia, red lunulae, clubbing, paronychia, pitting, leukonychia striata, and onycholysis. DLE lesions can be potentiated by sunlight exposure but to a lesser extent than ACLE and SCLE lesions. DLE, as well as other forms of LE skin disease activity, can be precipitated by any form of cutaneous trauma (ie, the Koebner phenomenon or isomorphic effect). The relationship between classic DLE and SLE has been the subject of much debate.27 The following summary points can be made: (a) 5% of patients presenting with classic DLE lesions subsequently develop unequivocal evidence of SLE and (b) patients with generalized DLE (ie, lesions both above and below the neck) have somewhat higher rates of immunologic abnormalities, a higher risk for progressing to SLE, and a higher risk for developing more severe manifestations of SLE than patients with localized DLE. Roughly one-fourth of patients with SLE develop DLE lesions at some point in the course of their disease, and such patients tend to have less-severe forms of SLE. Aside from classic DLE, there are several other lesscommon variants of CCLE, which are subclassified as such because of their overlapping histologies and tendency to occur in a low frequency in association with underlying SLE.
1045
10
A
C
HYPERTROPHIC DISCOID LUPUS ERYTHEMATOSUS
Hypertrophic DLE, also referred to as hyperkeratotic or verrucous DLE, is a rare variant of CCLE in which the hyperkeratosis normally found in classic DLE lesions is greatly exaggerated. The extensor aspects of the arms, the upper back, and the face are the areas most frequently affected. Overlapping features of hypertrophic LE and lichen planus have been described under the rubric lupus planus. The entity lupus erythematosus hypertrophicus et profundus appears to represent a rare form of hypertrophic DLE. Patients with hypertrophic DLE probably do not have a greater risk for developing SLE than do patients with classic DLE lesions.
MUCOSAL DISCOID LUPUS ERYTHEMATOSUS
Mucosal DLE occurs in approximately 25% of patients with CCLE. The oral mucosa is most frequently affected; however, nasal, conjunctival, and
1046
B
genital mucosal surfaces can be targeted. In the mouth, the buccal mucosal surfaces are most commonly involved, with the palate, alveolar processes, and tongue being sites of less frequent involvement. Lesions begin as painless, erythematous patches that evolve to chronic plaques that can be confused with lichen planus. Chronic buccal mucosal plaques are sharply marginated and have irregularly scalloped, white borders with radiating white striae and telangiectasia. The surfaces of these plaques overlying the palatal mucosa often have a honeycomb appearance. Central depression often occurs in older lesions, and painful ulceration can develop. Rarely, oral mucosal DLE lesions can degenerate into squamous cell carcinoma, similar to longstanding cutaneous DLE lesions. Any degree of nodular asymmetry within a mucosal DLE lesion should be evaluated for the possibility of malignant degeneration. Chronic DLE plaques also appear on the vermilion border of the lips. At times, DLE involvement of the lips can present as a diffuse cheilitis, especially on the more sunexposed lower lip. DLE lesions may present on the nasal, conjunctival, and anogenital mucosa. Perforation of the nasal
septum is more often associated with SLE than DLE. Conjunctival DLE lesions affect the lower lid more often than the upper lid. Lesions begin as focal areas of nondescript inflammation most commonly affecting the palpebral conjunctivae or the lid margin. Scarring becomes evident as lesions mature, and the permanent loss of eyelashes and ectropion can develop, producing considerable disability.
LUPUS ERYTHEMATOSUS PROFUNDUS/LUPUS ERYTHEMATOSUS PANNICULITIS
LE profundus/LE panniculitis (Kaposi-Irrgang disease) is a rare form of CCLE typified by inflammatory lesions in the lower dermis and subcutaneous tissue. Approximately 70% of patients with this type of CCLE also have typical DLE lesions, often overlying the panniculitis lesions. Some have used the term LE profundus to designate those patients who have both LE panniculitis and DLE lesions, and LE panniculitis to refer to those having only subcutaneous involvement. Typical subcutaneous lesions present as firm nodules, 1 to 3 cm in diameter. The overlying skin often becomes attached to the subcutaneous nodules and is drawn inward to produce deep, saucerized depressions (Fig. 61-9). The head, proximal upper arms, chest, back, breasts, buttocks, and thighs are the sites frequently affected. LE panniculitis, in the absence of overlying DLE, may produce breast nodules that can mimic carcinoma clinically and radiologically (lupus mastitis). Medically unresponsive extensive breast involvement can be so severe as to require radical mastectomy. Confluent facial involvement can simulate the appearance of lipoatrophy. Dystrophic calcification frequently occurs in older lesions of LE profundus/LE panniculitis, and pain associated with such calcification can, at times, be the dominant clinical
10
problem. Roughly 50% of patients with LE profundus/ panniculitis have evidence of SLE. However, the systemic features of patients with LE panniculitis/ profundus tend to be less severe, similar to those of patients with SLE who have DLE skin lesions.
CHILBLAIN LUPUS ERYTHEMATOSUS
Chilblain LE lesions initially develop as purple-red patches, papules, and plaques on the toes, fingers, and face, which are precipitated by cold, damp climates and are clinically and histologically similar to idiopathic chilblains (pernio) (Fig. 61-10). As they evolve, these lesions usually assume the appearance of scarred atrophic plaques with associated telangiectases. They may resemble old lesions of DLE or may mimic acral lesions of small vessel vasculitis. Histologic findings include a superficial and deep lymphocytic vascular reaction in addition to fibrin deposition in reticular, dermal-based blood vessels. Patients with chilblain LE often have typical DLE lesions on the face and head. It is possible that chilblain LE begins as a classic acral, cold-induced lesion that then Koebnerizes DLE lesions, thus explaining the spectrum of clinicohistologic findings, which seem to vary based on when, in the course of the lesion, the biopsy sample is taken. Chilblain LE appears to be associated with anti-Ro/ SS-A antibodies,28 and is linked to Raynaud phenomenon in many cases.29 Persistence of lesions beyond the cold months, a positive ANA, or presence of one of the other ACR criteria for SLE at the time of diagnosis of chilblain lesions helps to distinguish chilblain LE from idiopathic chilblains.30 Approximately 20% of patients presenting with chilblain LE later develop SLE. Chilblain LE is an underrecognized entity, yet it is likely that it is one of the most common causes of digital lesions in patients with LE. It is sometimes misdiagnosed as vasculitis and may overlap with acral DLE as mentioned above. An autosomal dominant, familial form of Chilblain LE has been described, and is caused
1047
10
by a missense mutation in the TREX 1 (endonuclease repair) gene.31
LUPUS ERYTHEMATOSUS TUMIDUS
Lupus erythematosus tumidus (LET; tumid LE) is a variant of CCLE in which the dermal findings of DLE, namely, excessive mucin deposition and superficial perivascular and periadnexal inflammation, are found on histologic evaluation. The characteristic epidermal histologic changes of LE-specific skin disease are only minimally expressed, if at all. This results in succulent, edematous, urticaria-like plaques with little surface change (Fig. 61-11). Annular urticaria-like plaques can also be seen. The paucity of epidermal change often produces confusion concerning the diagnosis of LET as a form of CCLE.32,33
Several recent reports support this subclassification and further characterize this subtype of CCLE.34-39 Although described to occur in some patients with SLE, most patients with LET have a negative ANA and a benign disease course. LET appears to be the most photosensitive subtype of cutaneous lupus, and typically demonstrates a good response to antimalarials. Additionally, LET lesions tend to resolve completely without either scarring or atrophy. There continues to be debate about the validity of LET as an authentic form of LE-skin disease. Some argue that LET lesions may in fact not be a form of CLE32 while others feel that LET deserves to be recognized as a distinct type of CLE (intermittent CLE) equivalent in importance to ACLE, SCLE, and CCLE.35 Evidence of type I interferon upregulation in skin lesions of UV light-induced LE tumidus support the latter argument.40
OTHER VARIANTS
Other rare forms of CCLE CLE have been described. These include, LE hypertrophicus et profundus, lichenoid DLE, LE vermiculatus, LE telangiectaticus, linear CLE, and LE edematous (probably a historical designation for urticaria-like plaque variant of DLE
1048
and/or tumid LE). Further information on these clinical entities is available.41
LUPUS ERYTHEMATOSUS NONSPECIFIC SKIN DISEASE
LUPUS ERYTHEMATOSUS
NONSPECIFIC SKIN DISEASE
Many other cutaneous findings have been found in patients with lupus patients, particularly SLE; they are listed in Table 61-1. Of note, the presence of LE- nonspecific skin changes, especially when seen in conjunction with LE-specific rashes, corresponds with higher systemic disease activity.42 Specific findings such as livedo reticularis, thrombophlebitis, or cutaneous infarct may suggest the presence of secondary antiphospholipid syndrome. Space constraints do not allow specific discussions of these entities here.
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
Systemic lupus can cause a myriad of extracutaneous manifestations, which manifestations are outlined in Table 61-3.
DIAGNOSIS
LABORATORY TESTING
LABORATORY TESTING
Because of the strong association between ACLE and SLE, the immunologic laboratory features of ACLE are those associated with SLE (high-titer ANA, antidsDNA, anti-Smith antigen, hypocomplementemia, hypergammaglobinemia, etc). Other laboratory findings, such as cytopenias, decreased kidney function, and urine changes (hematuria, proteinuria), reflect disease activity and can vary from patient to patient depending on the extent of end-organ involvement. The laboratory markers for SCLE are the presence of anti-Ro/SS-A (70% to 90%) and, less commonly, anti-La/SS-B (30% to 50%) autoantibodies. ANA are present in 60% to 80% of patients with SCLE, and rheumatoid factor is present in approximately 33%. Other autoantibodies in patients with SCLE include falsepositive serologic tests for syphilis (Venereal Disease Research Laboratory [VDRL] rapid plasma reagin; 7% to 33%), anticardiolipin (10% to 16%), antithyroid (18% to 44%), anti-Smith antigen (10%), anti-dsDNA (10%), and anti-U1 ribonucleoprotein (10%). Patients with SCLE, particularly those with systemic involvement, may have a number of laboratory abnormalities, including anemia, leukopenia, thrombocytopenia, elevated erythrocyte sedimentation rate, hypergammaglobulinemia, proteinuria, hematuria, urine casts,
10
Approach to the patient with skin lesions suspicious for cutaneous lupus erythematosus
Photosensitive facial eruption
Annular or psoriasiform plaques
Scarring alopecia
Triangular truncal pattern Dorsal arm involvement
NO
Consider photoallergic drug eruption GA EAC Tinea Psoriasis
Hardening
Hallmark hand changes of dermatomyositis Involvement of knuckles
Prominent centrofacial telangiectasia Pustules Nasolabial fold nvolvement
YES NO
Nonscarring malar erythematosus macules/papules
Rosacea
YES
YES
Dermatomyositis
Annular plaques with lateral facial predominance
Scarring lesions Conchal lesions Scarring alopecia
Confirm with biopsy
ACLE DLE
SCLE
Consider druginduced SCLE
Comprehensive medical history and examination (include rheumatologic review of systems) CBC, chemistry panel, urinalysis, CRP ANA, ENA, dsDNA, C3, C4, antiphospholipid panel
Meets SLE criteria
Refer to rheumatologist
SLE criteria not met
Begin stepwise approach to treatment of CLE
elevated serum creatine and blood urea nitrogen, and depressed complement levels (resulting from genetic deficiency or increased complement consumption). ANA are present in low titer in 30% to 40% of patients with DLE; however, fewer than 5% have the higher ANA
levels that are characteristic of patients with overt SLE (>1:320 titer by indirect immunofluorescence assay). Antibodies to single-stranded DNA are not uncommon in DLE, but antibodies to dsDNA are distinctly uncommon. Precipitating antibodies to U1 ribonucleoprotein
1049
10
are sometimes found in patients whose disease course is dominated by DLE lesions; however, such patients usually have only mild manifestations of SLE or overlapping connective tissue disorders such as mixed connective tissue disease. Precipitating Ro/SS-A and La/ SS-B autoantibodies are rare in patients with DLE; low levels of anti-Ro/SS-A antibody detected by enzymelinked immunoassay are more common. A small percentage of patients with DLE have low-grade anemia, biologic false-positive serologic tests for syphilis (VDRL rapid plasma reagent), positive rheumatoid factor tests, slight depressions in serum complement levels, modest elevations in γ-globulin, and modest leukopenia. It has been suggested that such findings are risk factors for the development of SLE. ANA are present in 70% to 75% of patients with LE profundus/panniculitis, but anti-dsDNA antibodies are rare. The laboratory findings associated with SLE, as well as with CLE, in both adults and children, are reviewed elsewhere.43,44 It should be noted that the assay methodologies for the detection of the autoantibodies discussed above continue to evolve over time. Commercial reference laboratoriesat the time of this publication have largely moved to the use of solid-phase immunoassay techniques, especially multiplex flow immunoassay. The disease incidence/prevalence of the autoantibodies discussed in the section above might vary somewhat
A
B
from the results of the autoantibody assays that one might receive from commercial laboratories today.
HISTOPATHOLOGY
HISTOPATHOLOGY
The LE-specific skin disease histopathology is a distinctive constellation of hyperkeratosis, epidermal atrophy, vacuolar basal cell degeneration, dermal–epidermal junction basement membrane thickening, dermal edema, dermal mucin deposition, and mononuclear cell infiltration of the dermal–epidermal junction and dermis, focused in a perivascular and periappendageal distribution. Variable degrees of these features are encountered in the different forms of LE-specific skin disease. Differences of opinion exist as to whether ACLE, SCLE, and DLE lesions can be distinguished reliably on the basis of their histopathologic appearances alone.44-46
ACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
The histopathologic changes in ACLE lesions are generally less impressive than those in SCLE and DLE lesions, and are mainly those of a cell-poor interface dermatitis (Fig. 61-13A). The lymphohistiocytic
C
1050
cellular infiltrate is relatively sparse. Some authors have noted an increase in the number of neutrophils in the infiltrate, especially when recent-onset lesions are biopsied. A mild degree of focal vacuolar alteration of basal keratinocytes can be seen, in addition to telangiectases and extravasation of erythrocytes. One may see individually necrotic keratinocytes, and in its most severe form, ACLE can display extensive epidermal necrosis similar to TEN. The upper dermis usually shows pronounced mucinosis and may be very helpful in distinguishing ACLE from other causes of a cell-poor interface dermatitis. It is uncommon to see basement membrane zone thickening, follicular plugging, or alteration of epidermal thickness in ACLE, although epidermal atrophy is sometimes present.45,46
SUBACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
SCLE also typically presents as an interface dermatitis, with foci of vacuolar alteration of basal keratinocytes alternating with areas of lichenoid dermatitis (see Fig. 61-13B). Pronounced epidermal atrophy is often present. SCLE is in the differential diagnosis of atrophic lichenoid dermatitis, along with atrophic lichen planus and lichenoid drug eruptions. Dermal changes include edema, prominent mucin deposition, and sparse mononuclear cell infiltration usually limited to areas around blood vessels and periadnexal structures in the upper one-third of the dermis. Lesser degrees of hyperkeratosis, follicular plugging, mononuclear cell infiltration of adnexal structures, and dermal melanophages might help distinguish SCLE lesions from DLE lesions. It has not been possible to reliably differentiate papulosquamous from annular SCLE by histopathologic criteria alone.43
CHRONIC CUTANEOUS LUPUS ERYTHEMATOSUS
In classic DLE lesions, epidermal changes include hyperkeratosis, variable atrophy, and interface changes similar to those described for SCLE (see Fig. 61-13C). The epidermal basement membrane is markedly thickened. Dermal changes include a dense mononuclear cell infiltrate composed primarily of CD4 T lymphocytes and macrophages predominantly in the periappendageal and perivascular areas, melanophages, and dermal mucin deposition. The infiltrate is often quite dense and typically extends well into the deeper reticular dermis and/ or subcutis, which may help to distinguish it from ACLE or SCLE. In chronic scarring DLE lesions, the dense inflammatory cell infiltrate subsides and is replaced by dermal fibroplasia. A folliculotropic variant of DLE, in which the inflammatory infiltrate is predominantly around hair follicles, has been described, as have lymphomatoid variants, in which there are extremely dense infiltrates that may contain atypical lymphoid cells.45
10
IMMUNOHISTOLOGY
IMMUNOHISTOLOGY
Immunohistology is often helpful in confirming a diagnosis of LE-specific skin disease and has been shown to boost the sensitivity and specificity of diagnosis.45
Because it is not uncommon to see negative immunofluorescence studies in patients with acute, subacute, and chronic LE, and false-positive studies in healthy individuals, immunohistology must be interpreted in the context of clinical and histologic findings in a given patient. IgG, IgA, IgM, and complement components (C3, C4, Clq, properdin, factor B, and the membrane attack complex C5b-C9) deposited in a continuous granular or linear band-like array at the dermal–epidermal junction have been observed in the lesional and nonlesional skin of patients with LE since the early 1960s (Fig. 61-14). However, debate about terminology in this area continues to cloud the field. Some restrict the use of the term lupus band test to refer to the examination of nonlesional skin biopsies for the presence of this band-like array of immunoreactants at the dermal-epidermal junction. Others qualify the lupus band test as being either “lesional” or “nonlesional.” Less confusion might exist if the terms lesional lupus band test and nonlesional lupus band test were uniformly adopted.
ACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
The sparse data that exist suggest that 60% to 100% of ACLE lesions display a lesional lupus band. However, the realization that sun-damaged skin from otherwise healthy individuals can display similar immunopathology has diluted the clinical value of this finding.
1051
10
SUBACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
Initial studies indicated that approximately 60% of patients with SCLE had lesional lupus bands. A “dustlike particle” pattern of IgG deposition focused around epidermal basal keratinocytes has been suggested to be more specific for SCLE by reflecting the presence of in vivo bound Ro/SS-A autoantibody.
CHRONIC CUTANEOUS LUPUS ERYTHEMATOSUS
Early reports suggested that more than 90% of classic DLE lesions had lesional immunoreactants at the dermal– epidermal junction, often extending along the basement membrane of the hair follicle, but subsequent studies report somewhat lower rates. Lesions on the head, neck, and arms are positive more frequently (80%) than those on the trunk (20%). The lesional lupus band also appears to be a function of the age of the lesion being examined, with older lesions (>3 months) being positive more often than younger ones. Ultrastructural localization of immunoglobulin at the dermal–epidermal junction confirms that these proteins are deposited on the upper dermal collagen fibers and along the lamina densa of the epidermal basement membrane zone. In LE profundus, immunoglobulin and complement deposits are usually found in blood vessel walls of the deep dermis and subcutis. Immunoglobulin deposits at the dermal–epidermal junction may or may not be present, depending on the site biopsied, the presence or absence of accompanying SLE, and the presence or absence of overlying changes of DLE at the dermal– epidermal junction.
NONLESIONAL LUPUS BAND TEST
There has been much debate over the past 3 decades regarding the diagnostic and prognostic significance of an immunoglobulin/complement band at the dermal– epidermal junction of nonlesional skin taken from patients with LE.44 When totally sun-protected nonlesional skin (eg, buttocks) is sampled, the diagnostic specificity for SLE appears to be very high when 3 or more immunoreactants are present at the dermal–epidermal junction. Prospectively ascertained followup data also suggest that the presence of a nonlesional lupus band test correlates positively with risk for developing LE nephritis. However, the nonlesional lupus band test has fallen out of favor as a clinical tool largely because the information gained has not been proven to be of significantly greater value than the results of more readily available serologic assays such as antibody to dsDNA.
DIFFERENTIAL DIAGNOSIS
1052
perhaps related to LE tumidus. Reticulated erythematosus mucinosis presents as a reticulated array of macules and/or papules on the upper chest and back.
CLINICAL COURSE AND PROGNOSIS
ACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
ACUTE CUTANEOUS LUPUS
ERYTHEMATOSUS
Both localized and generalized forms of ACLE lesions flare and abate in parallel with underlying SLE disease activity. Therefore, the prognosis for any given patient with ACLE is dictated by the pattern of the underlying SLE. Both 5-year (80% to 95%) and 10-year (70% to 90%) survival rates for SLE have progressively improved over the past 4 decades as a result of earlier diagnosis made possible by more sensitive laboratory testing and improved immunosuppressive treatment regimens. Ominous prognostic signs in SLE are hypertension, nephritis, systemic vasculitis, and CNS disease.
SUBACUTE CUTANEOUS LUPUS ERYTHEMATOSUS
SUBACUTE CUTANEOUS
LUPUS ERYTHEMATOSUS
Because SCLE has been recognized as a separate disease entity for only 2 decades, the long-term outcome associated with SCLE lesions has yet to be determined. It is the authors’ experience that most patients with SCLE have intermittent recurrences of skin disease activity over long periods of time without significant progression of systemic involvement (we are aware of only 1 death directly attributable to SLE in approximately 150 patients with SCLE). Other patients enjoy long-term if not permanent remissions of their skin disease activity. A few patients have experienced unremitting cutaneous disease. It has also been the authors’ experience that approximately 10% of the patients with SCLE develop active SLE, including lupus nephritis. This subgroup of patients is marked by the presence of papulosquamous SCLE, localized ACLE, high-titer ANA, leukopenia, and/or antibodies to dsDNA. Long-term followup studies of SCLE are required to determine the true risk of severe systemic disease progression in patients presenting with SCLE skin lesions. CCLE lesions, typically classic DLE, have also arisen in patients initially presenting with SCLE. Evidence suggests that overlap occurs between SCLE and Sjögren syndrome. Patients with SCLE who develop Sjögren syndrome are at risk for developing the extraglandular systemic complications associated with Sjögren syndrome, including vasculitis, peripheral neuropathy, autoimmune thyroiditis, renal tubular acidosis, myositis, chronic hepatitis, primary biliary cirrhosis, psychosis, lymphadenopathy, splenomegaly, and B-cell lymphoma.
10
MOST LIKELY CONSIDER ALWAYS RULE OUT
■ACLE
■ACLE
■ACLE
■ACLE
■Localized
■Localized
■Localized
■Localized
■Acne rosacea
■Seborrheic dermatitis
■Acne rosacea
■Seborrheic dermatitis
■Dermatomyositis
■ACLE
■ACLE
■Generalized
■Generalized
■Toxic epidermal necrolysis
■Toxic epidermal necrolysis
■Polymorphous light eruption
■Dermatomyositis
■CCLE
■Polymorphous light eruption
■Generalized
■CCLE
■Photoallergic contact dermatitis
■Generalized
■DLE
■Photoallergic contact dermatitis
■Drug hypersensitivity reaction
■Generalized
■Drug hypersensitivity reaction
■Generalized
■Photoallergic/phototoxic drug reactions
■Dermatomyositis
■Photoallergic/phototoxic drug
■Dermatomyositis
■SCLE
■SCLE
reactions
■Viral exanthems
■Papulosquamous
■Viral exanthems
■Papulosquamous
■SCLE
■DLE
■Tinea incognito
■Tinea incognito
■Cutaneous T-cell lymphoma
■Cutaneous T-cell lymphoma
■Lupus panniculitis
■Lupus panniculitis
■Infectious panniculitis (deep fungal/ atypical mycobacterial organisms)
■Infectious panniculitis (deep fungal/
■Photoallergic/photo lichenoid drug reaction
■SCLE
■Photoallergic/photo lichenoid drug reaction
■Papulosquamous
■Annular
■Papulosquamous
■Annular
■Photosensitive psoriasis
atypical mycobacterial organisms)
■Calciphylaxis
■Calciphylaxis
■Erythema gyratum repens
■Photosensitive psoriasis
■Erythema gyratum repens
■Annular
■DLE
■Annular
■DLE
■Erythema annulare centrifugum
■Early DLE/LET
■Erythema annulare centrifugum
■Early DLE/LET
■Granuloma annulare
■Granuloma faciale
■Granuloma annulare
■Granuloma faciale
■CCLE
■Sarcoidosis
■CCLE
■Sarcoidosis
■Early DLE/LET
■Jessner benign lymphocytic infiltration of the skin
■Early DLE/LET
■Jessner benign lymphocytic infiltration of
■Polymorphous light eruption
■Polymorphous light eruption
the skin
■Fully evolved DLE/hypertrophic DLE
■Pseudolymphoma
■Fully evolved DLE/hypertrophic DLE
■Pseudolymphoma
■Squamous cell carcinoma
■Lymphoma cutis
■Squamous cell carcinoma
■Lymphoma cutis
■Hypertrophic actinic keratosis
■Lupus vulgaris/cutaneous tuberculosis
■Hypertrophic actinic keratosis
■Lupus vulgaris/cutaneous tuberculosis
■Keratoacanthoma
■Urticaria
■Keratoacanthoma
■Urticaria
■Lupus panniculitis
■Urticarial vasculitis
■Lupus panniculitis
■Urticarial vasculitis
■Morphea profundus
■Fully evolved DLE/hypertrophic DLE
■Morphea profundus
■Fully evolved DLE/hypertrophic DLE
■Prurigo nodularis
■Prurigo nodularis
■Hypertrophic lichen planus
■Hypertrophic lichen planus
■Lupus panniculitis
■Lupus panniculitis
■Subcutaneous sarcoidosis
■Subcutaneous sarcoidosis
■Traumatic panniculitis
■Traumatic panniculitis
■Eosinophilic fasciitis
■Eosinophilic fasciitis
ACLE, acute cutaneous lupus erythematosus; CCLE, chronic cutaneous lupus erythematosus; DLE, discoid lupus erythematosus; LET, lupus erythematosus tumidus; SCLE, subacute cutaneous lupus erythematosus.
CHRONIC CUTANEOUS LUPUS ERYTHEMATOSUS
CHRONIC CUTANEOUS
LUPUS ERYTHEMATOSUS
Most patients with untreated classic DLE lesions suffer indolent progression to large areas of cutaneous dystrophy and scarring alopecia that can be psychosocially devastating and occupationally disabling. However, with treatment, skin disease can be largely controlled. Spontaneous remission occurs occasionally, and the disease activity can recrudesce at the sites of older, inactive lesions. Rebound after discontinuation of treatment is typical, and slower taper of medications during periods of inactivity is recommended. Squamous cell carcinoma occasionally develops in chronic smoldering DLE lesions. Death from SLE is distinctly uncommon in patients who present initially with localized DLE. As discussed in “Epidemiology” above, patients presenting with localized DLE have only a 5% chance of subsequently developing clinically significant SLE disease activity. Generalized DLE and persistent, low-grade laboratory abnormalities appear to be risk factors for such disease progression. Unrecognized squamous cell carcinoma
developing within a longstanding DLE skin lesion could be a cause of morbidity and mortality.
OUTCOME MEASURES
OUTCOME MEASURES
The recent development of a validated instrument to measure activity of CLE, the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI), has made it possible to objectively follow patients’ disease course and response to therapy. The instrument has separate scores for damage (scarring) and activity which is important, because one would not expect a “burned-out” scarred area to normalize with drugs that were meant to abate LE activity.47 It has been validated as a useful tool to measure clinical response.48
MANAGEMENT
The initial management of patients with any form of CLE should include an evaluation to rule out underlying SLE disease activity at the time of diagnosis. All patients with CLE should receive instruction about protection
1053
10
from sunlight and artificial sources of UV radiation, and should be advised to avoid the use of potentially photosensitizing drugs such as hydrochlorothiazide, tetracycline, griseofulvin, and piroxicam. With regard to specific medical therapy, local measures should be maximized and systemic agents used if significant local disease activity persists or systemic activity is superimposed. ACLE lesions usually respond to the systemic immunosuppressive measures required to treat the underlying SLE disease activity that so frequently accompanies this form of CLE (eg, systemic glucocorticoids, azathioprine, and cyclophosphamide). Increasing evidence suggests that aminoquinoline antimalarial agents such as hydroxychloroquine can have a steroid-sparing effect on SLE, and these drugs can be of value in ACLE. The local measures discussed in “Local Therapy” below can also be of value in treating ACLE. Because the lesions of SCLE and CCLE are often found in patients who have little or no evidence of underlying systemic disease activity, unlike the lesions of ACLE, nonimmunosuppressive treatment modalities are preferred for SCLE and CCLE
(Table 61-6). For the most part, SCLE and CCLE lesions respond equally to such agents. Accurate diagnosis is always the key to optimal medical management. In genetically complex clinical disorders that are variably expressed clinically from one person to another, such as LE, there is a tendency to over ascribe new clinical findings to LE once a diagnosis of LE has been established. To call attention to this important clinical axiom, Robert Greenwald, a rheumatologist, formulated his “Law of Lupus.” It states that “SLE is often falsely accused of causing anything and everything that might happen to a patient subsequent to the diagnosis of SLE.”49 Failure to recognize this principle in an acute medical setting can result in misdiagnosis and potentially serious mismanagement. Common inflammatory skin lesions, such as psoriasis and eczematous dermatitis, can occur in individuals with LE. These skin lesions are completely independent of LE and its treatment. Likewise, confusion can arise as a result of skin changes that occur as a result of systemic immunosuppressive therapy that might be needed to treat LE patients (eg, herpes zoster,
DRUG DOSE
First line Topical glucocorticoids, topical calcineurin inhibitor Class I topical steroid daily to twice daily for 2 weeks alternating with pimecrolimus 1% or tacrolimus 0.1% twice daily for 2 weeks
Intralesional triamcinolone acetonide 2.5-10 mg/cc
Second line Hydroxychloroquine 6.5 mg/kg/day based on ideal body weight
Chloroquine 3-3.5 mg/kg/day based on ideal body weight
Quinacrine 100 mg/day (available at compounding pharmacies
If monotherapy fails, add quinacrine to either hydroxychloroquine or chloroquine
Short course only (2-16 weeks) Prednisone 5-60 mg/day
Thalidomide 50-200 mg/day; taper to 50 mg every other day on response
Third line (safer immunosuppressives) Azathioprine 1.5-2.5 mg/kg/day
Mycophenolate mofetil 1-1.5 g/dose, twice daily
Methotrexate 7.5-25 mg by mouth or subcutaneously, once weekly
Worth considering Dapsone 50-200 mg/day
Accutane 0.5-2 mg/kg/day
Acitretin 10-50 mg/day
Gold Titrate to 50 mg intramuscularly weekly, taper after 1 g
Investigational (some currently
Leflunomide
Investigational (some currently available for other indications) Leflunomide
available for other indications)
Anti–tumor necrosis factor biologics
Anti–tumor necrosis factor biologics
Rituximab
Rituximab
Belimumab
Belimumab
Abatacept
Abatacept
Janus kinase (JAK) inhibitors (tofacitinib,
Janus kinase (JAK) inhibitors (tofacitinib, ruxolitinib)
ruxolitinib)
Antiinterferon antibodies
Antiinterferon antibodies
1054
CXCR3 receptor inhibitors
CXCR3 receptor inhibitors
steroid-induced acne, drug eruptions, opportunistic infections).
LOCAL THERAPY
LOCAL THERAPY
SUN PROTECTION
Advise patients to avoid direct sun exposure, wear tightly woven clothing and broad-brimmed hats, and regularly use broad-spectrum, water-resistant sunscreens (SPF [sun protection factor] ≥30 with an efficient ultraviolet A blocking agent such as a photostabilized form of avobenzone [Parsol 1789], micronized titanium dioxide, micronized zinc oxide, or ecamsule [Mexoryl SX]). UV-blocking films should be applied to home and automobile windows, and acrylic diffusion shields should be placed over fluorescent lighting. Corrective camouflage cosmetics such as Dermablend and Covermark offer the dual benefit of being highly effective physical sunscreens as well as aesthetically pleasing cosmetic masking agents. Ting and Sontheimer provide an in-depth discussion of practical and theoretical photoprotection and local therapy for autoimmune connective tissue skin disease.50
As a result of the need for sun avoidance, and perhaps of other factors as well, both CLE and SLE patients are often found to be vitamin D insufficient or deficient.51,52 There are clear human health benefits to normalizing vitamin D metabolism. Whether correcting vitamin D insufficiency/deficiency has a therapeutic impact on the cutaneous and SLE disease activity is under study.
TOPICAL GLUCOCORTICOIDS
Although some prefer intermediate-strength preparations, such as triamcinolone acetonide 0.1%, for sensitive areas such as the face, superpotent topical class I agents, such as clobetasol propionate 0.05% or betamethasone dipropionate 0.05%, produce the greatest benefit in CLE. Twice-daily application of the superpotent preparations to lesional skin for 2 weeks followed by a 2-week rest period can minimize the risk of local complications such as steroid atrophy and telangiectasia. Alternatively, a topical calcineurin inhibitor can be used daily during the 2-week rest period from topical corticosteroids. Ointments are more effective than creams for more hyperkeratotic lesions such as hypertrophic DLE. Occlusive therapy with glucocorticoid-impregnated tape (eg, flurandrenolide) or glucocorticoids with plastic food wrap (eg, Saran or Glad Press-N-Seal) can potentiate the beneficial effects of topical glucocorticoids but also carries a higher risk of local side effects. Class I or class II topical glucocorticoid solutions and gels are best for treating the scalp. Unfortunately, even the most aggressive regimen of topical glucocorticoids by itself does not provide adequate improvement for most patients with SCLE and CCLE.
TOPICAL CALCINEURIN INHIBITORS
Pimecrolimus 1% cream and tacrolimus 0.1% ointment have demonstrated efficacy in the treatment of ACLE,
10
DLE, and SCLE.53-55 A double-blind, placebo-controlled pilot study showed that pimecrolimus 1% cream had equal efficacy with betamethasone valerate 0.1% cream in treating facial DLE,56 and a different study demonstrated the efficacy of topical tacrolimus 0.3% compound in clobetasol propionate 0.05% for recalcitrant CLE.57
It remains to be demonstrated whether topical delivery of newer classes of targeted nonsteroidal antiinflammatory agents that are being developed for other clinical indications might be of benefit for CLE (eg, topical apremilast, topical tofacitinib).
INTRALESIONAL GLUCOCORTICOIDS
Intralesional glucocorticoids (eg, triamcinolone acetonide suspension, 2.5 to 5 mg/mL for the face with higher concentrations allowable in less-sensitive sites) are more useful in the management of DLE than SCLE. Intralesional glucocorticoids themselves can produce cutaneous and subcutaneous atrophy (deep injections into the subcutaneous tissue enhances this risk). A 30-gauge needle is preferred because it produces only mild discomfort on penetration, especially when injected perpendicularly to the skin. The active borders of lesions should be thoroughly infiltrated. Intralesional therapy is indicated for particularly hyperkeratotic lesions or lesions that are unresponsive to topical glucocorticoids, but most patients with CLE have too many lesions to be managed exclusively by intralesional glucocorticoid injections.
SYSTEMIC THERAPY
SYSTEMIC THERAPY
ANTIMALARIALS
One or a combination of the aminoquinoline antimalarials can be effective for approximately 75% of patients with CLE who have failed to benefit adequately from the local measures described in “Local Therapy” above. The risks of retinal toxicity should be discussed with the patient, and a pretreatment ophthalmologic examination should be performed. However, the risk of antimalarial retinopathy is extremely rare, particularly in the first 10 years of therapy, if the recommended daily dose maximum levels of these agents are not exceeded (hydroxychloroquine, 6.5 mg/kg/day based on ideal body weight; chloroquine, 3 to 4 mg/kg/day). Patients should have followup ophthalmologic evaluations every 6 to 12 months while on therapy. Hydroxychloroquine sulfate (Plaquenil), 6 to 6.5 mg/kg of lean body mass, should be given daily, either once daily or in 2 divided doses to prevent GI side effects. Approximately 6 weeks are required to reach equilibrium blood levels of hydroxychloroquine. Patients should be informed about a 2- to 3-month delayed onset of therapeutic benefit. If no response is seen after 8 to 12 weeks, quinacrine hydrochloride, 100 mg/day (currently available in the United States only through compounding pharmacies), can be added
1055
10
to the hydroxychloroquine without enhancing the risk of retinopathy (quinacrine does not cause retinopathy). If, after 4 to 6 weeks of this combined regimen adequate clinical control has not been achieved, consideration should be given to replacing the hydroxychloroquine in this combined regimen with chloroquine diphosphate (Aralen), 3 to 4 mg/kg/day. Doses may need to be adjusted for patients with decreased renal or hepatic function. In Europe, chloroquine is generally felt to be more efficacious than hydroxychloroquine in treating CLE, perhaps because of the earlier therapeutic responses that might occur as a result of the shorter time period required to reach steady-state blood levels with chloroquine as compared to hydroxychloroquine. Hydroxychloroquine and chloroquine should not be used simultaneously because of enhanced risk for retinal toxicity. There is some evidence that chloroquine may be more retinotoxic than hydroxychloroquine. It is now possible to assay serum levels of hydroxychloroquine. However, this assay has not yet been made available widely by commercial testing laboratories in the United States. Recent studies examining this end point have found that up to 10% of CLE and SLE patients are not compliant in taking their hydroxychloroquine therapy as prescribed.58,59 In addition, these and other studies have suggested that hydroxychloroquine serum levels and disease activity measures are inversely correlated in both cutaneous and SLE patients undergoing hydroxychloroquine therapy.60
It is now generally agreed that the modern hydroxychloroquine regimen can be used safely in children (with the appropriate dose adjustment based on lean body mass) and in women who are pregnant. However, the therapeutic index in children is quite narrow. Therefore, extra caution should be taken to prevent accidental overdosing of children. Multiple side effects other than retinal toxicity are associated with the use of antimalarials. Quinacrine is associated with a higher incidence of side effects, such as headache, GI intolerance, hematologic toxicity, pruritus, lichenoid drug eruptions, and mucosal or cutaneous pigmentary deposition, than is either hydroxychloroquine or chloroquine. However, at the currently recommended dose of no more than 100 mg/day, such adverse effects are extremely rare. Quinacrine commonly produces a yellow discoloration of the entire skin and sclera in fair-skinned individuals, which is completely reversible when the dose of the drug is reduced or discontinued altogether. Quinacrine can produce significant hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency (this adverse effect has also been reported to occur rarely with hydroxychloroquine and chloroquine). Each of the aminoquinoline antimalarials can produce bone marrow suppression, including aplastic anemia, although this effect is exceedingly rare with the current dosage regimens. Toxic psychosis, grand mal seizures, neuromyopathy, and cardiac arrhythmias occurred with the use of high doses of these drugs in the past; these reactions are uncommon with the lower daily dose regimens used today.
1056
Before therapy with hydroxychloroquine and chloroquine is begun, complete blood cell counts, as well as liver and renal function tests, should be performed; these tests should be repeated 4 to 6 weeks after therapy is initiated, and every 4 to 6 months thereafter. A screen for hematologic toxicity when quinacrine is used is recommended more often. Patients with overt or subclinical porphyria cutanea tarda are at particularly high risk for developing acute hepatotoxicity, which often simulates an acute surgical abdomen, when treated with therapeutic doses of antimalarials for CLE.
NONIMMUNOSUPPRESSIVE OPTIONS FOR ANTIMALARIAL– REFRACTORY DISEASE
Some patients with refractory CLE (SCLE more than DLE) respond to diaminodiphenylsulfone (dapsone).61
An initial dose of 25 mg by mouth twice daily can be increased up to 200 to 400 mg/day, if necessary. Significant dose-related hemolysis and/or methemoglobinemia can result from the use of dapsone, especially in individuals deficient in G6PD activity. Consequently, complete blood counts and liver function tests should be performed regularly, and testing for G6PD status should be considered prior to starting treatment, especially in high-risk populations. Isotretinoin, 0.5 to 2.0 mg/kg/day, and acitretin, 10 to 50 mg/day, also have been used in this setting, but their efficacy is limited by their side effects (teratogenicity, mucocutaneous dryness, and hyperlipidemia). In addition, breakthrough of CLE activity has been a problem with the long-term use of retinoids. Thalidomide (50 to 200 mg/day) is strikingly effective for CLE that is refractory to other medications. Numerous studies cite response rates between 85% and 100%, with many patients experiencing complete remission.62 However, strict prescribing regulations put in place in the United States in 1998 because of severe teratogenicity, make thalidomide challenging to dispense to women of childbearing potential. Sensory neuropathy is another toxicity associated with thalidomide, and 25% to 75% of patients with CLE develop peripheral neuropathy while taking the drug. Most cases are reversible when therapy is stopped. Neuropathy seems to correlate with total treatment times so that short courses are preferred. Relapses after the drug is stopped are common. Excess somnolence as well as constipation and other minor side effects sometimes limit its use, although these effects usually abate with lower daily doses.63 Thromboembolism is a serious adverse event that may occur in patients with a preexisting hypercoagulable state (eg, presence of antiphospholipid antibodies). Oncologists who use thalidomide for multiple myeloma frequently initiate concomitant anticoagulation therapy to prevent this side effect. Lenalidomide (Revlimid, Celgene), a thalidomide analog, is U.S. Food and Drug Administration (FDA) approved for the treatment of multiple
myeloma. In preliminary studies, its efficacy rate in CLE appears to be similar to that of thalidomide. Safety data to date indicates a much lower rate of peripheral neuropathy compared to thalidomide; however, it has a similar rate of thromboembolism (especially when combined with glucocorticoids) and leukopenia compared to thalidomide. Its potential teratogenicity in humans has yet to be determined. Other drugs reported to be of value in the treatment of refractory CLE are gold and clofazimine; however, the benefit varies from case to case and both of these agents are associated with the risk of significant side effects. Vitamin E, phenytoin, sulfasalazine, danazol, DHEA, and phototherapy (UVA1 phototherapy, photopheresis) also have been reported clinical trials to be of potential value in CLE.
IMMUNOSUPPRESSIVE OPTIONS FOR ANTIMALARIAL–REFRACTORY DISEASE Systemic Glucocorticoids: Every effort should be made to avoid the use of systemic glucocorticoids in patients with LE limited to the skin. However, in occasional patients who have especially severe and symptomatic skin disease, intravenous pulse methylprednisolone has been used. In less-acute cases, moderate daily doses of oral glucocorticoids (prednisone, 20 to 40 mg/day, given as a single morning dose) can be used as supplemental therapy during the loading phase of therapy with an antimalarial agent. The dose should be reduced at the earliest possible time because of the complications of long-term glucocorticoid therapy, especially avascular (aseptic) bone necrosis, a side effect to which patients with LE are particularly susceptible. Because steroid-induced bone loss occurs most rapidly in the first 6 months of use, all patients who do not have contraindications should begin agents to prevent osteoporosis with the initiation of steroid therapy. An excellent review describing current recommendations for prevention of bone loss and other side effects of systemic glucocorticoids has been published.64 When the disease activity is controlled, the daily dosage should be reduced by 5- to 10-mg decrements until activity flares again or until a daily dosage of 20 mg/day is achieved. The daily dose should then be lowered by 2.5-mg decrements (some physicians prefer to use 1-mg dose decrements below 10 mg/day). Alternateday glucocorticoid therapy has not been successful in suppressing disease activity in most patients with CLE or SLE. Prednisolone rather than prednisone should be used in patients who have significant underlying liver disease, because prednisone requires hydroxylation in the liver to become biologically active. Any amount of prednisone given as a single oral dose in the morning has less adrenal-suppressing activity than the same amount given in divided doses throughout the day. However, any given amount of this drug, taken in divided doses, has a greater LE-suppressing activity than does the same amount of drug given as a single
10
morning dose. Daily doses of prednisone less than or equal to 7.5 mg/day eliminate glucocorticoid-induced damage accrual in SLE patients.65
Traditional Immunosuppressives: Azathioprine (Imuran) (1.5 to 2 mg/kg per day orally) can play a glucocorticoid-sparing role in the severely affected patient with CLE. Mycophenolate mofetil (CellCept) (2.5 to 3 g divided twice daily orally) is a purine analog similar to azathioprine, but with more specific inhibition of the de novo pathway in lymphocytes. This characteristic may allow for more efficacy and less toxicity in treating severe, recalcitrant CLE, and such results have been reported in several studies.66,67 Methotrexate (7.5 to 25 mg orally 1 day per week) is effective for severe refractory CLE. A double-blind, randomized, placebo-controlled trial in patients with SLE showed that moderate doses of methotrexate (15 to 20 mg weekly) effectively controlled cutaneous and articular activity and permitted a reduction in prednisone dose.68 Other immunosuppressives that have been used anecdotally include cytosine arabinoside (Cytarabine) and cyclosporine. Immunotherapy with high-dose IV gammaglobulin has also been used.
Biologic Therapies: There have been reports documenting efficacy of anti-TNF medications (etanercept, adalimumab, infliximab) in the treatment of recalcitrant CLE, particularly SCLE.69-71 However, these agents are also well-known to induce both SLE and CLE, highlighting the importance of TNF homeostasis in patients predisposed to LE.72,73
B-lymphocyte stimulator (BLyS [synonym: B-cell activating factor (BAFF)]), is a recently discovered member of the TNF cytokine family. BLyS is made by monocytes and macrophages with subsequent release when these cells are activated. BLyS binds to a receptor found only on B cells which stimulates maturation into antibodysecreting plasma cells. A fully human monoclonal antibody directed against BLyS (belimumab [Benlysta]) inhibits the biologic activity of BLyS. Belimumab is now FDA approved for SLE. In the clinical trials of belimumab in SLE, the cutaneous end points in SLE patients were met in addition to the systemic end points. However, little has been reported to date concerning the clinical efficacy of belimumab in isolated CLE. As previously discussed abnormal regulation of interferon-α appears to play a central role in the pathogenesis of both cutaneous and systemic manifestations of LE by upregulating proinflammatory cytokines and chemokines. Three biologics that target type 1 IFN are currently being tested for their efficacy and safety in active SLE patients (2 anti–interferon-α monoclonal antibodies [rontalizumab and sifalimumab] and one monoclonal antibody that binds to a type 1 IFN receptor [anifrolumab]).74 Recombinant humanized antibodies to type 1 IFN and to the interferon receptor are under development. If these new biologics prove to be of benefit in SLE disease activity, one could extrapolate that they might be helpful in CLE disease activity.
1057
10
Class I interferon actions are transduced through the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) intracellular signaling pathway. Disrupting this intracellular signaling pathway might provide another approach to downregulating SLE and CLE disease activity and damage. Two orally administered JAK isoenzyme inhibitors have been approved by the FDA for other clinical disorders: tofacitinib (JAK 1/3) for rheumatoid arthritis and ruxolitinib (JAK 1/2) for myelodysplastic disorders. There are case reports in which these 2 drugs appeared to have clinical benefit in both CLE and cutaneous dermatomyositis.75,76
More specific JAK1 isoenzyme inhibitors are currently under development (eg, filgotinib) which promise to lessen the side effects of this new class of targeted, small-molecule therapy.
SURGICAL AND COSMETIC THERAPY
SURGICAL AND COSMETIC
THERAPY
DLE lesions can produce permanent scarring alopecia, cosmetically disturbing dermal atrophy, and long-lasting pigmentary changes. Patients so affected often ask about the possibility of cosmetic correction of these changes. Surgical interventions such as hair transplantation and dermabrasion carry finite risks because CLE is characterized by a tendency for nonspecific mechanical trauma, including surgical incision or laser ablation, to exacerbate disease activity (ie, Koebner phenomenon/isomorphic phenomenon). Some patients tolerate scar revision techniques, including dermabrasion, if they are on maintenance systemic therapy (eg, antimalarials). There are anecdotal reports of the successful management of active CLE with argon and pulsed-dye laser therapy.77 Resurfacing of atrophic scars with the Erbium:YAG or Fraxel carbon dioxide resurfacing laser is reported to be beneficial.78,79 Autologous fat transplantation can be of clinical value in patients with atrophic scarring from burned out lupus panniculitis/profundus. The injection of atrophic lesions with collagen or other similar foreign cosmetic materials should be avoided.
TREATMENT ALGORITHM
TREATMENT ALGORITHM
Treatment of CLE with the aforementioned agents is typically done in a stepwise manner to minimize the risk of side effects and is outlined in Table 61-6.
PREVENTION
PREVENTION
Predicting and preventing the initial clinical manifestation of LE, whether it is skin disease or systemic, is not feasible at this time. However, as many LE patients exhibit worsening of their skin disease activity with
1058
UV light exposure, physical protection from sunlight and artificial sources of UV light as well as the regular use of broad-spectrum sunscreens having a SPF of 30 or greater should be encouraged. CLE patients should be encouraged to discontinue cigarette smoking. CLE patients who smoke cigarettes are not as responsive to antimalarial therapy compared to those who do not smoke. CLE patients who smoke and are not responding to antimalarial therapy have been observed to experience complete remission in their CLE activity after they discontinue smoking with no other change in their therapy. Cigarette smoking does not alter antimalarial metabolism. It has been suggested that cigarette smoking, irrespective of antimalarial therapy, is strongly associated with CLE and somewhat less so with SLE.80,81
Figure 61-1 Spectrum of disease: local involvement to systemic disease. ACLE, acute cutaneous lupus erythematosus; DLE, discoid lupus erythematosus; LE, lupus erythematosus; SLE, systemic lupus erythematosus.
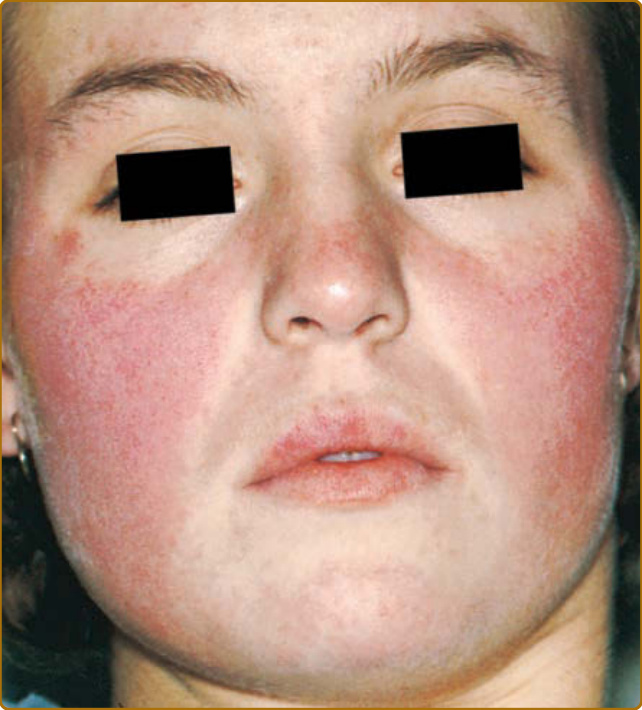
Figure 61-2 Localized acute cutaneous lupus erythematosus. Erythematous, slightly edematous, sharply demarcated erythema is seen on the malar areas in a “butterfly” distribution.
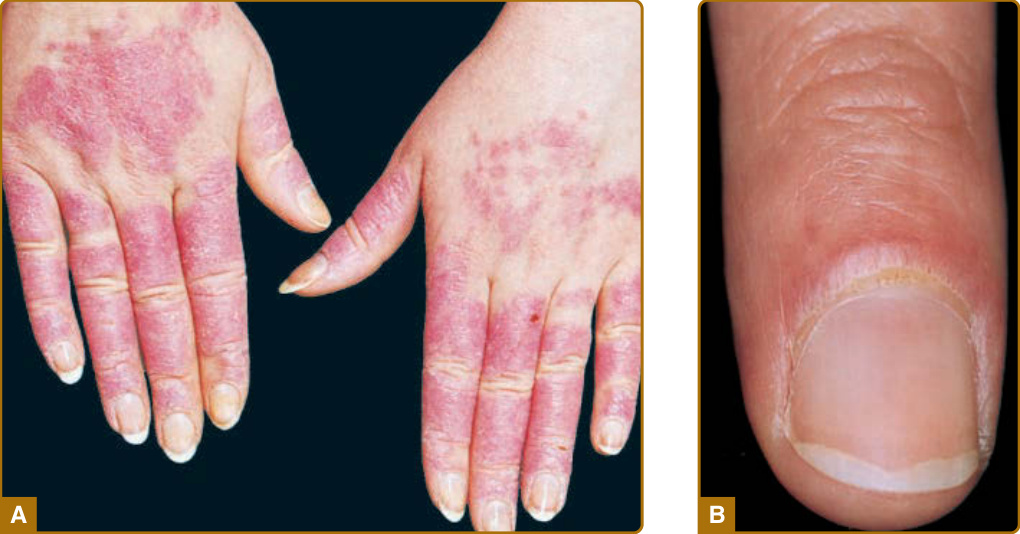
Figure 61-3 Generalized acute cutaneous lupus erythematosus. A, Well-demarcated patches of erythema with fine overlying scale on the dorsal aspect of the hands, fingers, and periungual areas. Note the characteristic sparing of the knuckles, which are preferentially involved in dermatomyositis. B, Closeup view of periungual erythema and grossly visible telangiectasia. Although these lesions can be seen in lupus erythematosus, they are much more typical of dermatomyositis.
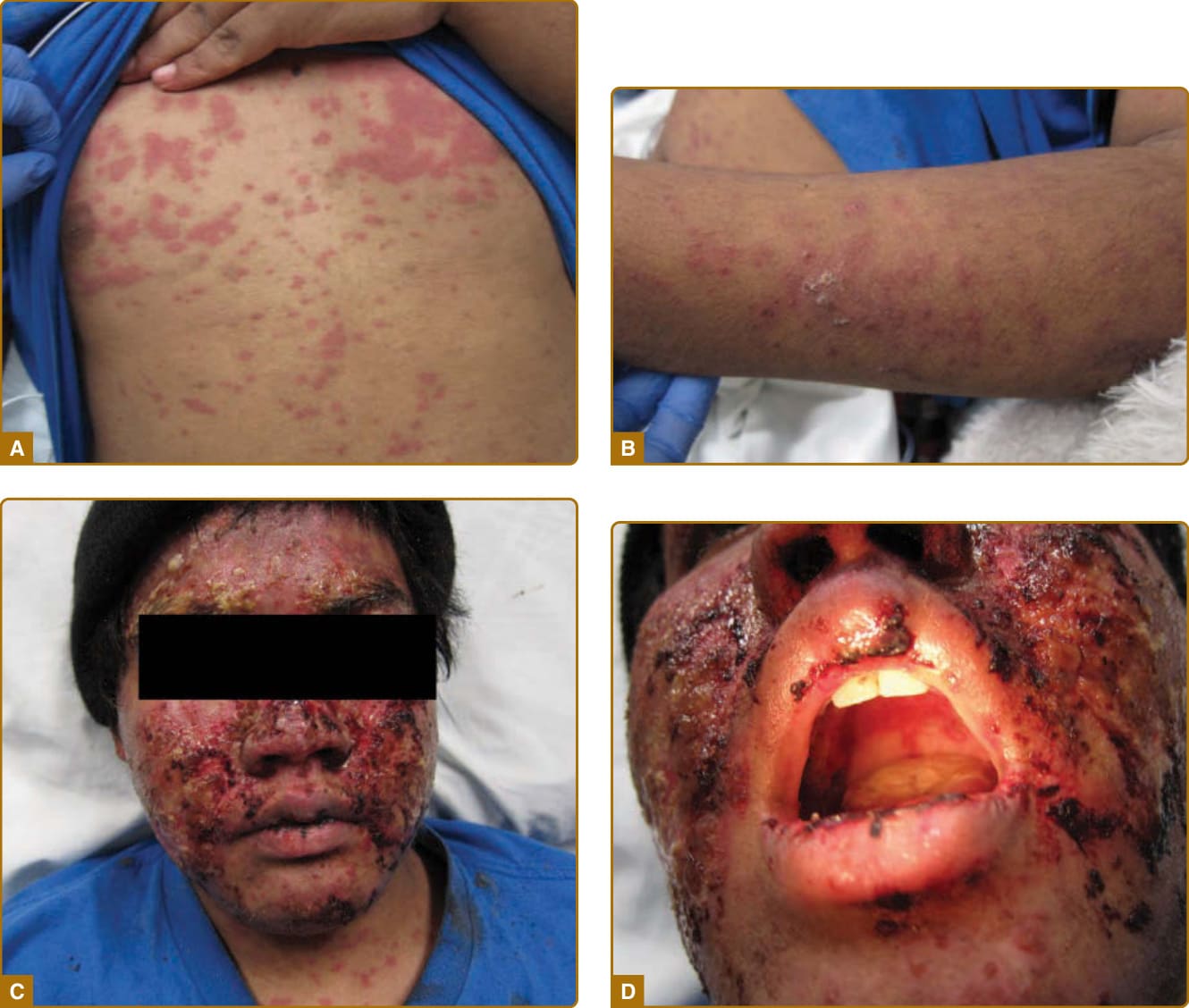
Figure 61-4 Toxic epidermal necrolysis (TEN)-like acute cutaneous lupus in a 16-year-old boy. A, Widespread erythematous papules with vesiculobullous changes over the trunk. B, Similar changes over the arm. C, Severe epidermal loss and denuding over the face in a 16-year-old boy. D, Painless palatal ulceration characteristic of systemic lupus erythematosus was present as opposed to severe mucosal involvement of TEN.
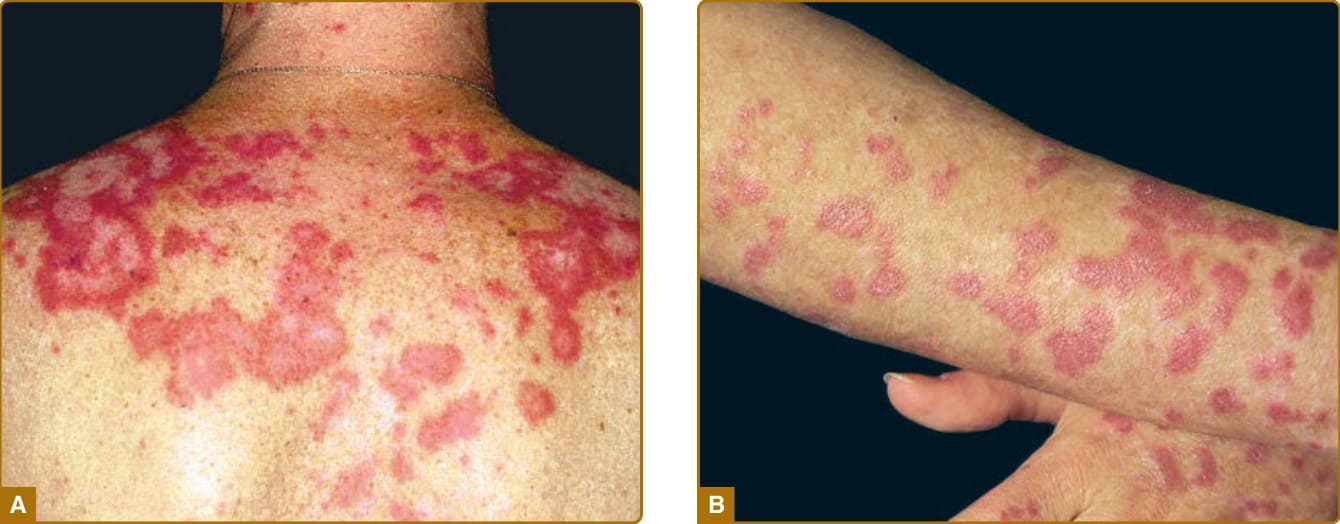
Figure 61-5 Subacute cutaneous lupus erythematosus (SCLE). A, Annular SCLE on the upper back of a 38-year-old woman. Note the central areas of hypopigmentation in which no dermal atrophy is present. B, Papulosquamous SCLE over the extensor aspect of the forearm of a 26-year-old woman.
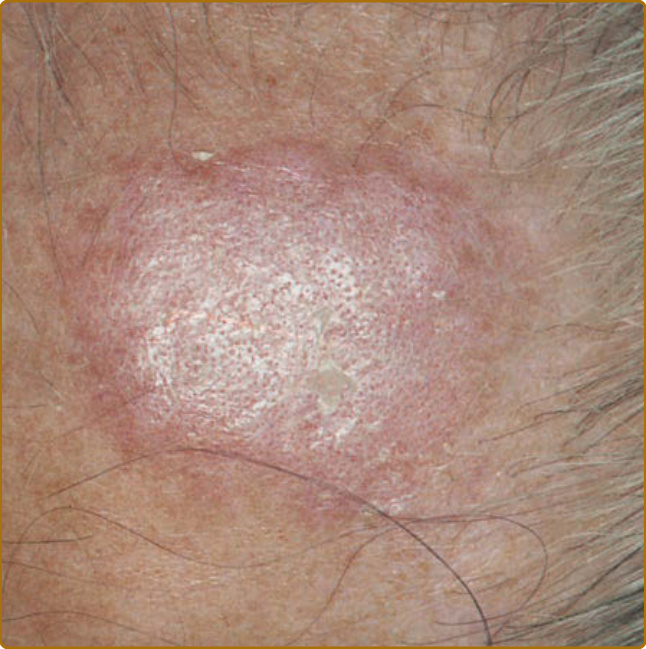
Figure 61-6 Classic discoid lupus erythematosus. Typical early erythematous plaque on the forehead demonstrating hyperkeratosis and accentuation of follicle orifices in a 60-year-old man with a 25-year history of cutaneous lupus erythematosus. The lesion had been present for 3 months; no dermal atrophy was present at this stage.
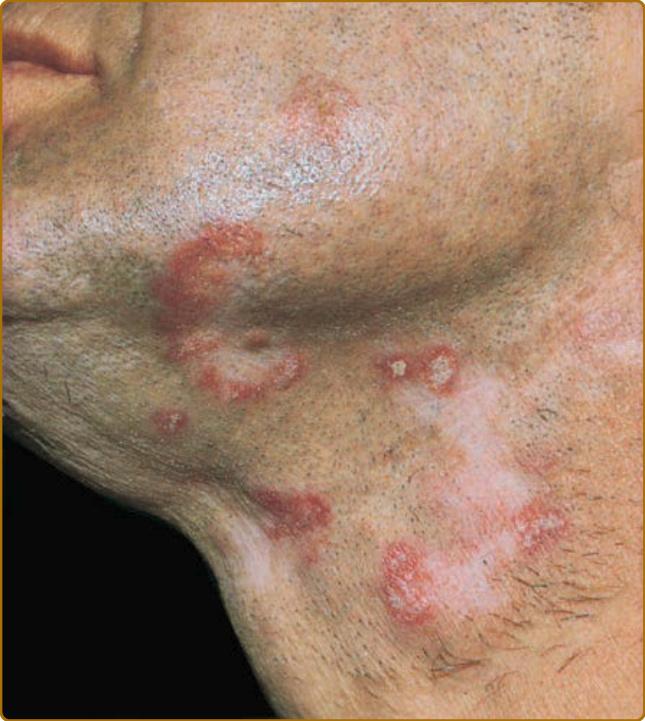
Figure 61-7 Classic discoid lupus erythematosus. Sharply demarcated, round-to-ovoid slightly indurated, erythematous plaques on the neck and face. Most plaques show a mild degree of hyperkeratosis, and some show dermal atrophy. Noninflamed areas of hypopigmentation and scarring mark the sites of prior lesions that have resolved.
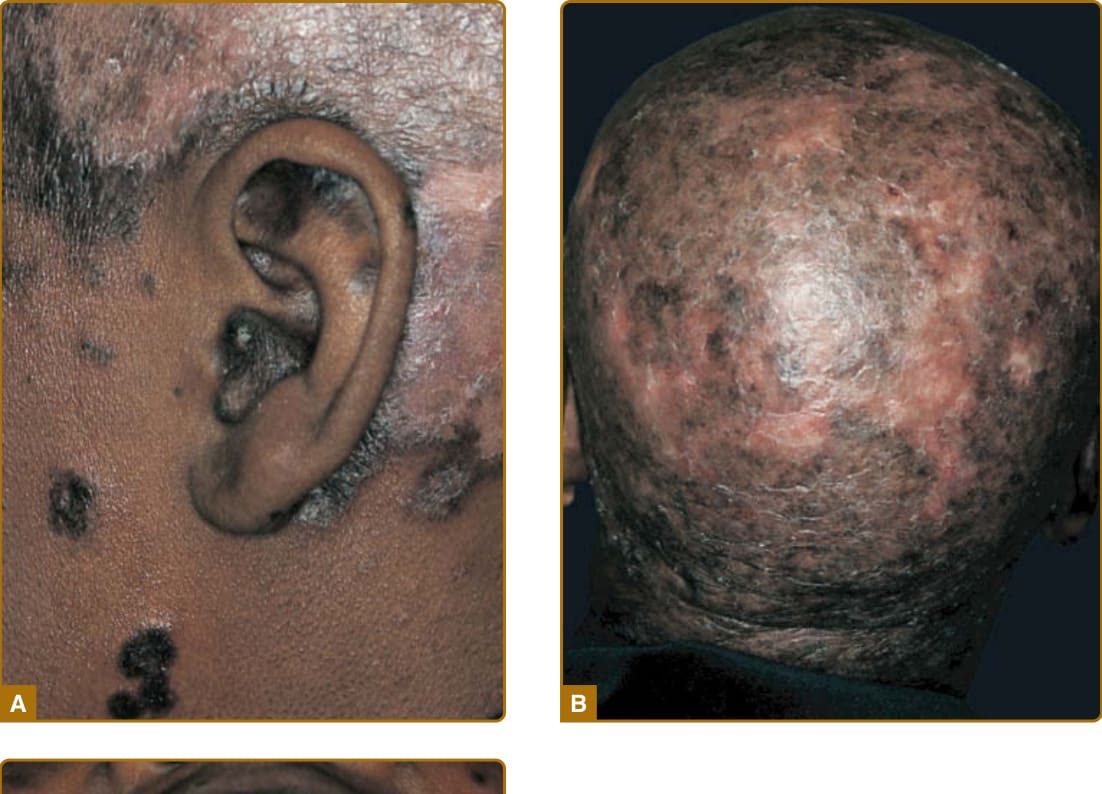
Figure 61-8 Classic discoid lupus erythematosus (DLE) and mucosal DLE in a 45-year-old African American woman with a 20-year history of untreated cutaneous lupus erythematosus. A, Characteristic involvement of the ear shows lesions with atrophy and postinflammatory hyperpigmentation as well as inflammatory red plaques on the scalp with postinflammatory hypopigmentation. B, Confluent lesions on the scalp have resulted in extensive scarring alopecia. C, Plaques of DLE on the palatal mucosa showing morphologic features similar to those of cutaneous lesions.
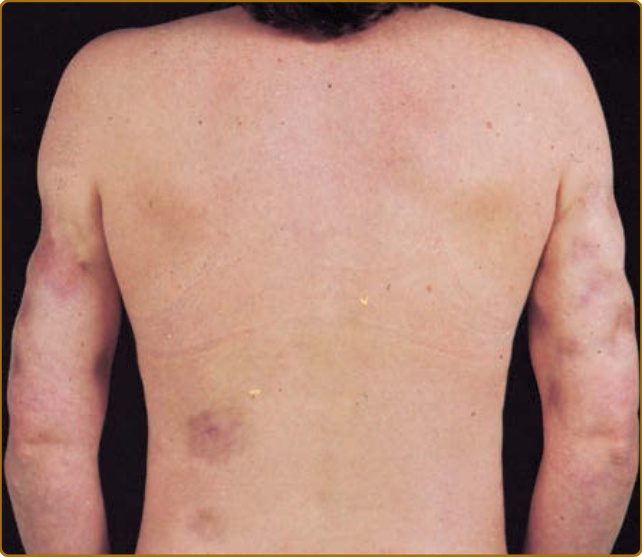
Figure 61-9 Lupus erythematosus panniculitis. Lupus panniculitis has resulted in large, sunken areas of overlying skin; erythema and atrophy of the skin are present.
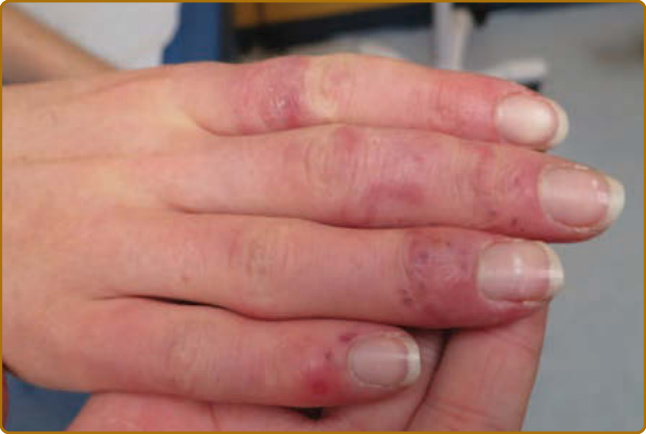
Figure 61-10 Chilblain lupus. Chilblain lesions affecting the fingers in a patient with longstanding lupus and lupus nephritis. The skin lesions were chronic and occurred without cold exposure.
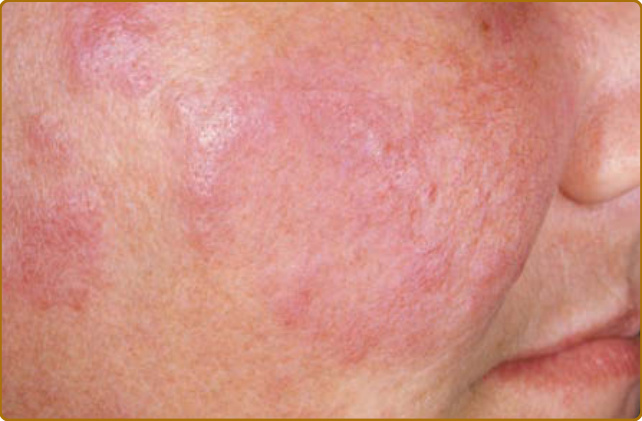
Figure 61-11 Lupus erythematosus tumidus. Note succulent, indurated plaques.
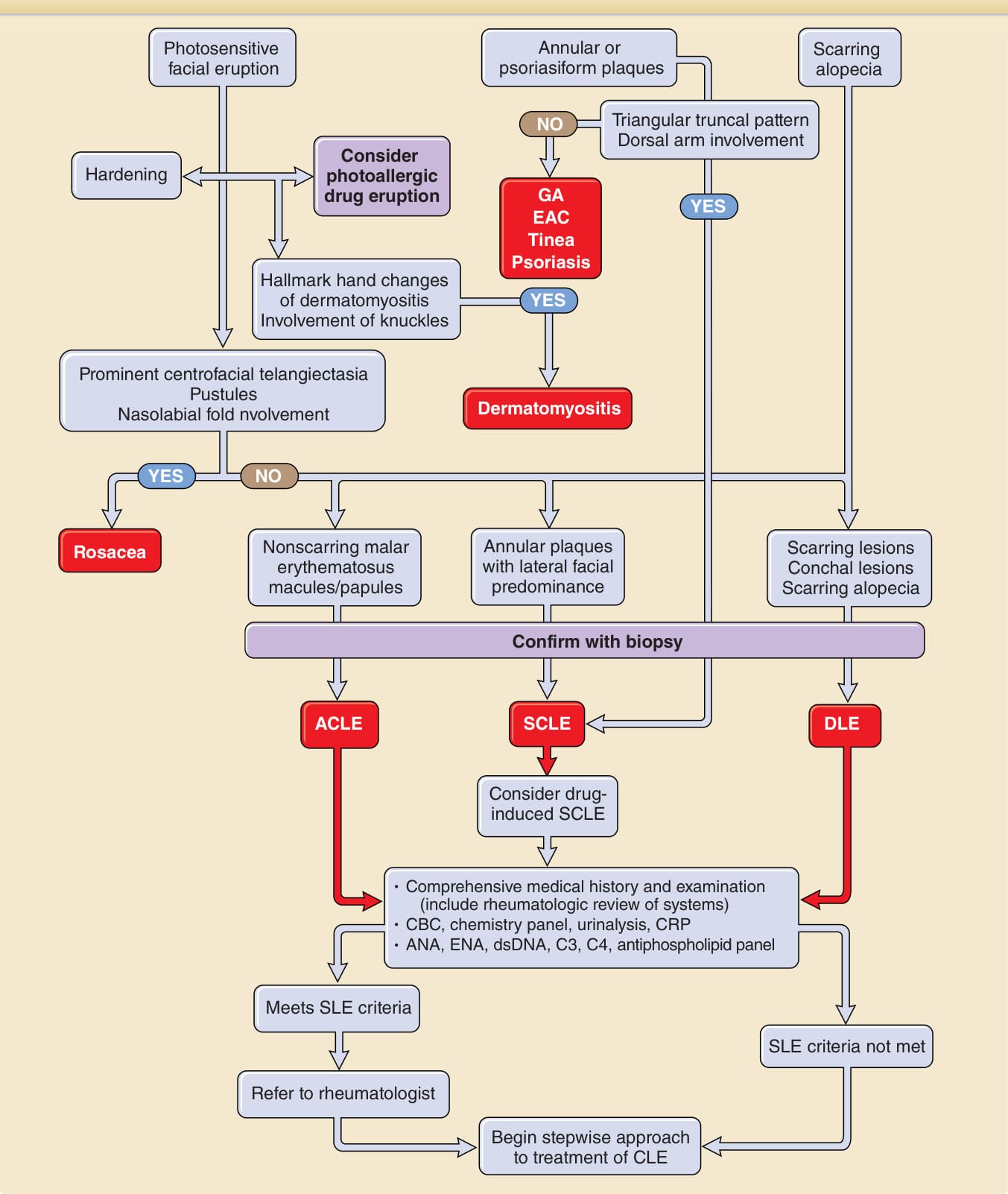
Figure 61-12 outlines approaches to the patient with skin lesions suspicious for CLE.
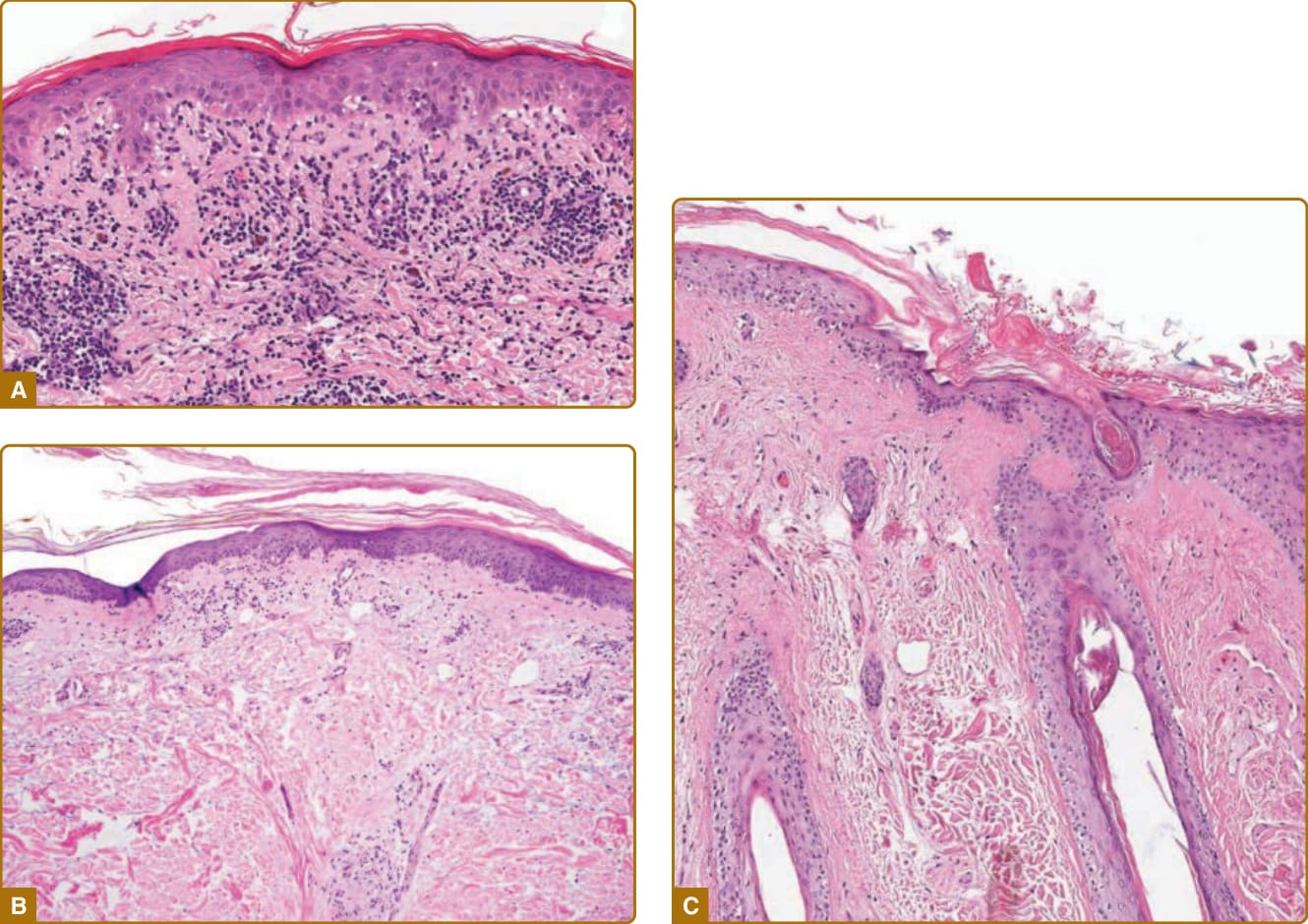
Figure 61-13 Histopathology of lupus erythematosus-specific skin disease. A, Acute cutaneous lupus lesion (hematoxylin and eosin [H&E] stain) showing interface dermatitis and vacuolar changes as well as perivascular inflammatory cell infiltrate (×128). B, Subacute cutaneous lupus lesion (H&E stain) with mild interface dermatitis, vacuolar changes, and mild hyperkeratosis. Mild epidermal atrophy and prominent mucinosis in upper dermis are also seen (×100). C, Discoid lupus lesion showing typical interface changes as well as hyperkeratosis, follicular plugging. Mild dermal fibroplasia is also seen (×84).
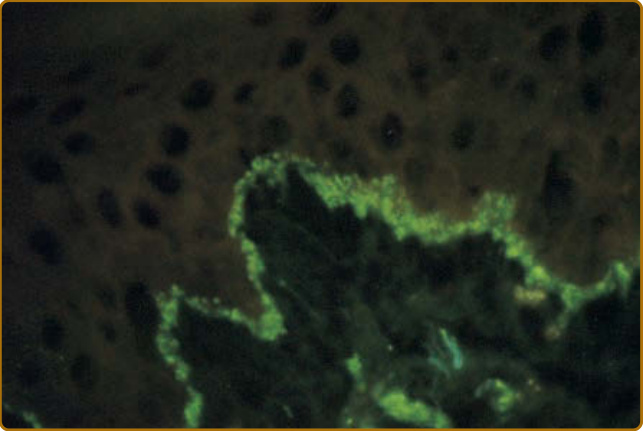
Figure 61-14 Immunopathology of lupus erythematosus– specific skin disease. Direct immunofluorescence examination of a discoid lupus erythematosus lesional skin biopsy showing a continuous band of granular fluorescence at the dermal–epidermal junction as a result of staining with fluorescein isothiocyanate–conjugated goat anti–immunoglobulin G.
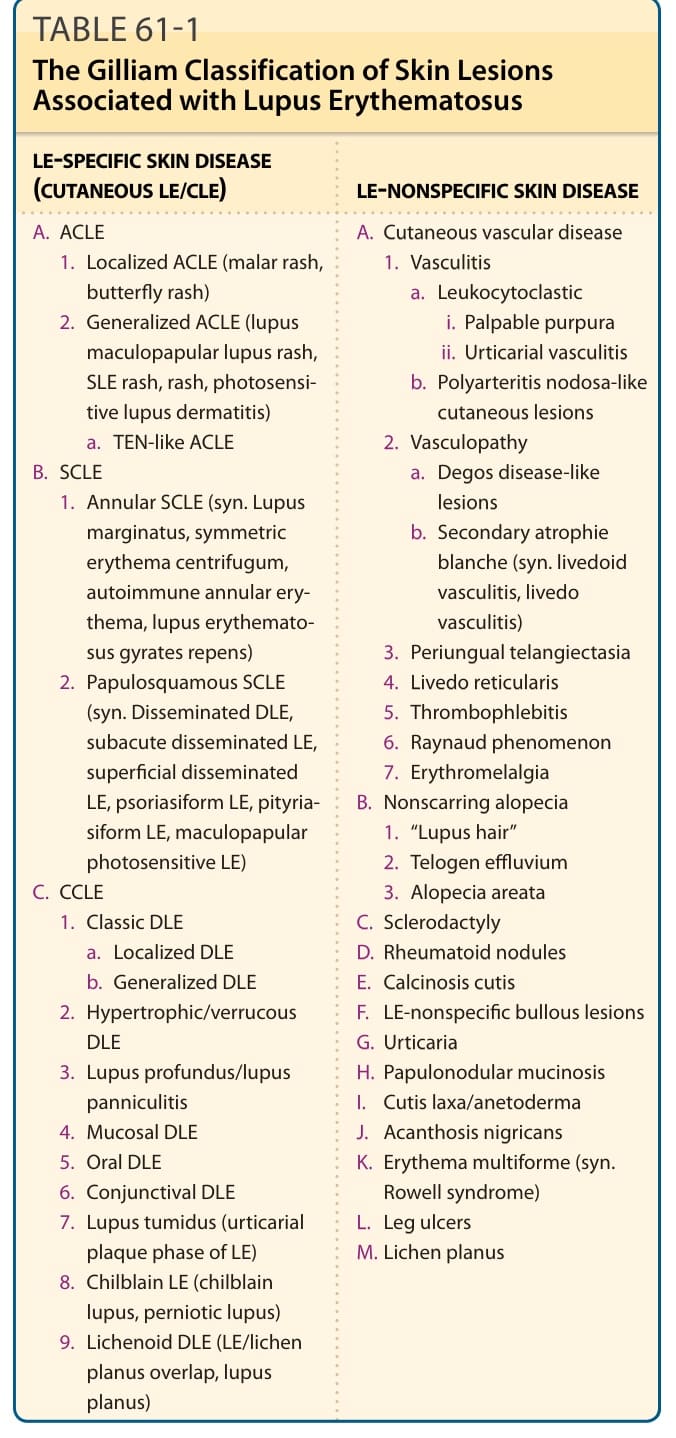
TABLE 61-1 The Gilliam Classification of Skin Lesions Associated with Lupus Erythematosus
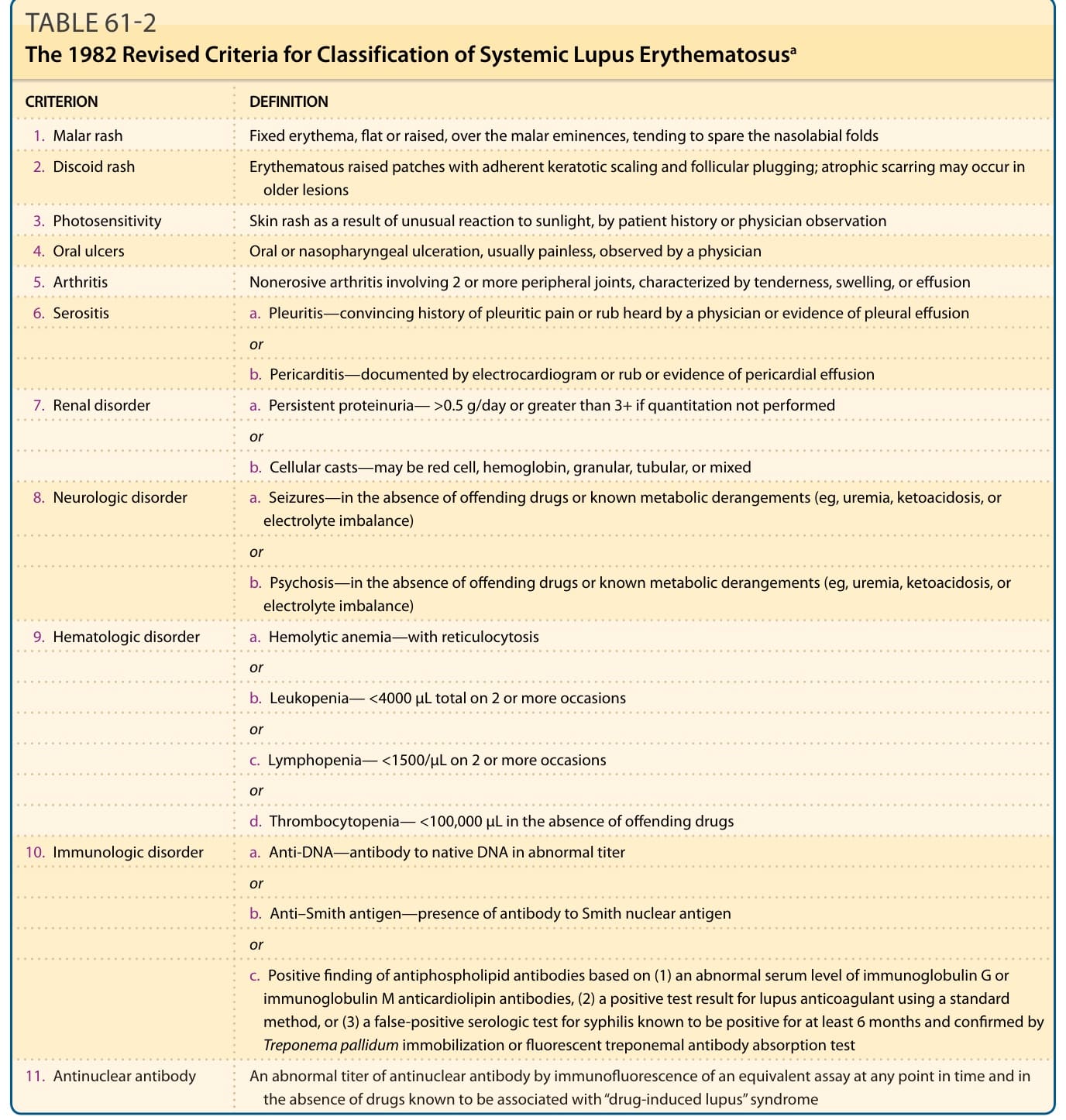
TABLE 61-2 The 1982 Revised Criteria for Classification of Systemic Lupus Erythematosusa

TABLE 61-3 Overview of the Extracutaneous Manifestations of Systemic Lupus Erythematosus

TABLE 61-4 Causes of Drug-Induced Lupus

Table 61-5 outlines the differential diagnosis of CLE. In addition, reticulated erythematosus mucinosis has been suggested by some to be a form of photosensitive CLE
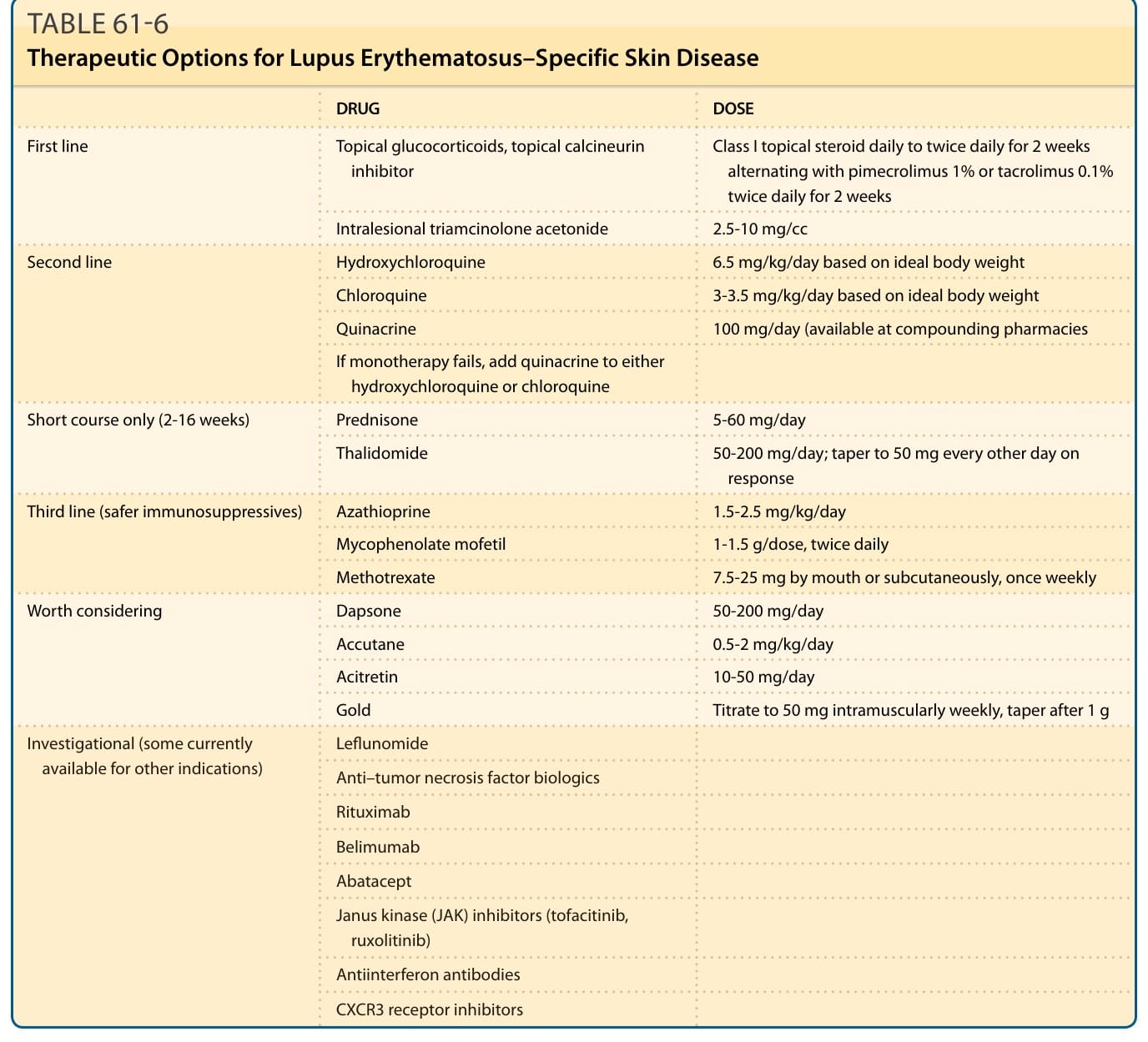
TABLE 61-6 Therapeutic Options for Lupus Erythematosus–Specific Skin Disease