分子標靶治療 (Molecular Targeted Therapies)
自 2000 年以來,由於分子遺傳學 (molecular genetics)、細胞生物學 (cell biology) 與藥理學 (pharmacology) 的進步,人類疾病的治療策略已產生戲劇性的演變。對分子病理生理學 (molecular pathophysiology) 認識的增加,使得開發出能與疾病相關之特定分子標靶相互作用的療法成為可能。透過理性藥物設計 (rational drug design)——即刻意設計化合物使其與某一生物標靶相互作用——所產生的藥物,通常被稱為「標靶治療 (targeted therapy)」,並構成「精準醫療 (precision medicine)」的基石之一。標靶治療的潛力或許在腫瘤疾病中體現得最為淋漓盡致,但理性藥物設計的前景已開始在所有醫學領域中顯現。討論醫學中所有的標靶療法已超出本章範圍。因此,我們聚焦於那些在「適應症」或「使用所造成的不良反應」上與皮膚科最有交集的分子標靶療法 (Table 194-1)。據此,我們將回顧設計用來與酪胺酸激酶 (tyrosine kinases) BCR-ABL、c-KIT、PDGFR 與 EGFR 相互作用的標靶療法;smoothened;組蛋白去乙醯酶 (histone deacetylases);以及 MAP 激酶路徑 (MAP kinase pathway) 的蛋白質。最後以黑色素瘤未來方向中針對 AKT 與 ERK 蛋白的章節作結。須注意的是,行銷適應症、禁忌症與警語是動態且持續演變的。因此,本章所涵蓋者乃截至撰寫時為最新。請參閱 FDA 產品仿單以取得最新資訊。
KIT、BCR-ABL 與 PDGFR 抑制劑 (KIT, BCR-ABL, AND PDGFR INHIBITORS)
重點一覽 (AT-A-GLANCE)
■ 小分子酪胺酸激酶抑制劑 (tyrosine kinase inhibitors) imatinib、nilotinib、dasatinib、bosutinib 與 ponatinib 適用於費城染色體陽性白血病 (Philadelphia chromosome–positive leukemia)。
■ Imatinib 亦有隆突性皮膚纖維肉瘤 (dermatofibrosarcoma protuberans) 的適應症,這是一種部分由血小板衍生生長因子受體 (platelet-derived growth factor receptor) 訊息傳遞改變所驅動的軟組織肉瘤。
■ 這些多酪胺酸激酶抑制劑的皮膚不良反應包括水腫 (edema)、麻疹樣疹 (morbilliform eruptions)、大疱性疹 (bullous eruptions)、色素異常 (dyspigmentation)、毛囊角化症樣疹 (keratosis-pilaris–like eruptions) 及嗜中性球皮膚病 (neutrophilic dermatoses)。
背景 (BACKGROUND)
酪胺酸激酶 (Tyrosine kinases) 是眾多細胞路徑的關鍵組成,參與細胞生長、增殖、遷移、血管新生、分化與存活。針對酪胺酸激酶的藥物研發始於 1980 年的最初發現:與 Abelson 鼠白血病病毒 (Abelson murine leukemia virus) 相關的致癌基因 ABL1 是一種酪胺酸激酶。¹ 這項發現帶來了以下認識:一種融合蛋白——由第 9 號染色體上的 Abl1 基因易位至第 22 號染色體斷裂點群集區 (breakpoint cluster region, BCR) 基因的一部分所產生——是大多數慢性骨髓性白血病 (chronic myeloid leukemias, CMLs) 的驅動事件。² 此融合致癌基因的蛋白產物,即所謂的費城染色體 (Philadelphia chromosome),是理性藥物設計最早的標靶之一。表現費城染色體的細胞因 BCR-ABL 激酶的組成性活化 (constitutive activation) 而發生轉化,該激酶透過多種轉導路徑——包括絲裂原活化蛋白激酶 (mitogen-activated protein kinase, MAPK)、Janus 激酶 (Janus kinase, JAK)/訊號轉導與轉錄活化因子 (signal transducer and activator of transcription, STAT),以及磷脂醯肌醇-4,5-雙磷酸 3-激酶 (phosphatidylinositol-4,5-bisphosphate 3-kinase, PI3K)——介導致癌作用 (Fig. 194-1)。「標靶治療」最早的原理驗證之一,來自於以下證明:BCR-ABL 融合致癌蛋白的小分子抑制劑——甲磺酸 imatinib (imatinib mesylate)——可抑制表現 BCR-ABL 之細胞的生長,³ 並在表現費城染色體的 CML 與急性淋巴芽細胞白血病 (acute lymphoblastic leukemia) 病人中展現了顯著的臨床活性。⁴˒⁵ 這些資料徹底改變了癌症治療的方法,並將藥物研發典範轉向辨識致病標靶。在隆突性皮膚纖維肉瘤 (dermatofibrosarcoma protuberans) 中,膠原蛋白基因 (COL1A1) 與血小板衍生生長因子 (PDGFR) β 鏈基因之間的融合,產生了一種由旁分泌 (paracrine) 與自分泌 (autocrine) 配體驅動、組成性活化的促有絲分裂原。⁶ Imatinib 抑制與 PDGFRβ 相關的酪胺酸激酶,其用於隆突性皮膚纖維肉瘤病人可帶來臨床效益。⁷
儘管 imatinib 對整體腫瘤學領域以及數種惡性腫瘤有突破性的影響,仍有近三分之一的 CML 病人需要其他療法。最常見需要替代治療的原因是 BCR-ABL1 出現抗藥性突變,影響 imatinib 與三磷酸腺苷 (adenosine triphosphate, ATP) 結合口袋相互作用的能力。因此,具有更強 BCR-ABL 親和力的第二代與第三代酪胺酸激酶抑制劑 (tyrosine kinase inhibitors, TKIs) 應運而生。這些 TKIs 包括 nilotinib、dasatinib、bosutinib 與 ponatinib。
表 194-1:分子標靶治療一覽(學名 / 商品名 / 分子標靶 / 適應症)
| 學名 (Generic Name) | 商品名 (Brand Name) | 分子標靶 (Molecular Target) | 適應症 (Indication) |
|---|---|---|---|
| Imatinib | Gleevec | BCR-ABL, c-KIT, PDGFR | Ph+-CML, GIST, ASM, DFSP, Ph+-ALL, HES/CEL |
| Nilotinib | Tasigna | BCR-ABL, c-KIT, PDGFR | Ph+-CML |
| Dasatinib | Sprycel | BCR-ABL, c-KIT, PDGFR | Ph+-CML, Ph+-ALL |
| Bosutinib | Bosulif | BCR-ABL | Ph+-CML |
| Ponatinib | Iclusig | BCR-ABL, c-KIT, PDGFR | Ph+-CML, Ph+-ALL |
| Cetuximab | Erbitux | EGFR | CRC, SCCHN |
| Panitumumab | Vectibix | EGFR | CRC |
| Gefitinib | Iressa | EGFR | NSCLC |
| Erlotinib | Tarceva | EGFR | EGFR 突變型 NSCLC, 胰臟癌 |
| Afatinib | Gilotrif | EGFR | EGFR 突變型 NSCLC |
| Osimertinib | Tagrisso | EGFR | EGFR 突變型 (T790M) NSCLC |
| Vismodegib | Erivedge | SMO | BCC |
| Sonidegib | Odomzo | SMO | BCC |
| Vorinostat | Zolinza | HDACs | CTCL |
| Romidepsin | Istodax | HDACs | CTCL, PTCL |
| Belinostat | Beleodaq | HDACs | PTCL |
| Panobinostat | Farydak | HDACs | 多發性骨髓瘤 (Multiple Myeloma) |
| Vemurafenib | Zelboraf | BRAFV600E | 黑色素瘤 (Melanoma) |
| Dabrafenib | Tafinlar | BRAFV600E | 黑色素瘤 (Melanoma) |
| Trametinib | Mekinist | MEK | 黑色素瘤 (Melanoma) |
| Cobimetinib | Cotellic | MEK | 黑色素瘤 (Melanoma) |
縮寫:ASM, aggressive systemic mastocytosis(侵襲性全身性肥大細胞增生症);BCC, basal cell carcinoma(基底細胞癌);CEL, chronic eosinophilic leukemia(慢性嗜酸性球白血病);CRC, colorectal cancer(大腸直腸癌);CTCL, cutaneous T-cell lymphoma(皮膚 T 細胞淋巴瘤);DFSP, dermatofibrosarcoma protuberans(隆突性皮膚纖維肉瘤);EGFR, epidermal growth factor receptor(表皮生長因子受體);GIST, GI stromal tumor(胃腸道間質瘤);HDAC, histone deacetylases(組蛋白去乙醯酶);HES, hypereosinophilic syndrome(高嗜酸性球症候群);NSCLC, non-small cell lung cancer(非小細胞肺癌);PDGFR, platelet-derived growth factor receptor(血小板衍生生長因子受體);Ph+-ALL, 費城染色體陽性急性淋巴芽細胞白血病;Ph+-CML, 費城染色體陽性慢性骨髓性白血病;PTCL, peripheral T-cell lymphoma(周邊 T 細胞淋巴瘤);SCCHN, squamous cell carcinoma, head and neck(頭頸部鱗狀細胞癌);SMO, smoothened。
甲磺酸 IMATINIB (GLEEVEC)
藥理學與作用機轉 (PHARMACOLOGY AND MECHANISM OF ACTION)
結構 (Structure):Imatinib,4-[(4-methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-phenyl]benzamide methanesulfonate,分子式為 C29H31N7O • CH4SO3,分子量為 589.7 daltons。Figure 194-2 顯示其結構。
代謝 (Metabolism):Imatinib 主要由細胞色素 P450 (cytochrome P450, CYP) 3A4 代謝,主要活性代謝物為一種 N-去甲基化的哌嗪 (piperazine) 衍生物。藥物及其代謝物主要經由糞便排出。原型藥物的消除半衰期約為 18 小時,N-去甲基衍生物約為 40 小時。⁸
吸收與分布 (Absorption and Distribution):Imatinib 口服吸收,生體可用率為 98%。此化合物與循環血漿蛋白(主要是白蛋白與 α1-酸性醣蛋白)有高度結合。
作用機轉 (Mechanism of Action):甲磺酸 imatinib 是一種小分子 TKI,對 BCR-ABL、c-KIT 與 PDGFR 具有活性 (見 Fig. 194-1)。Imatinib 結合於這些激酶失活、未磷酸化構型的 ATP 結合位附近,抑制該蛋白的酶活性。在具有組成性活化 BCR-ABL 的細胞中,imatinib 誘導細胞凋亡並抑制增殖。Imatinib 亦下調由異常 c-KIT 與血小板衍生生長因子 (platelet-derived growth factor, PDGF) 訊息所觸發的細胞增殖。
適應症與禁忌症 (INDICATIONS AND CONTRAINDICATIONS)
甲磺酸 imatinib 最初於 2001 年核准用於 CML。2002 年,仿單更新納入晚期或轉移性胃腸道間質瘤 (GI stromal tumors)。2006 年,美國食品藥物管理局 (U.S. Food and Drug Administration, FDA) 進一步擴大核准,納入隆突性皮膚纖維肉瘤、侵襲性全身性肥大細胞增生症 (aggressive systemic mastocytosis)、伴有 PDGFR 基因重排的骨髓增生不良/骨髓增生性疾病 (myelodysplastic/myeloproliferative diseases)、復發/難治性費城染色體陽性急性淋巴球性白血病 (Philadelphia chromosome–positive acute lymphocytic leukemia),以及伴有 FIP1L1-PDGFR 融合激酶重排的高嗜酸性球症候群/慢性嗜酸性球白血病 (hypereosinophilic syndrome/chronic eosinophilic leukemia)。Imatinib 的仿單外 (off-label) 用途已有報告,用於帶有 KIT 改變的黑色素瘤,此類改變在源自黏膜、肢端與慢性日曬受損皮膚之黑色素瘤中發生頻率較高。數項較新試驗的資料顯示,作為單一藥物在 c-KIT 突變黑色素瘤中有適度的臨床效益,且較新的美國國家綜合癌症網路 (National Comprehensive Cancer Network, NCCN) 指引將 imatinib 列為轉移性黑色素瘤的治療選項。⁹⁻¹¹ Imatinib 的臨床活性亦正被研究用於硬纖維瘤 (desmoid tumors) 以及表現 PDGFRβ 與/或 PDGFβ 的晚期或轉移性脊索瘤 (chordomas)。製造商仿單上無禁忌症。
表 194-2:Imatinib 的劑量(疾病 / 劑量)
| 疾病 (Disease) | 劑量 (Dose) |
|---|---|
| 慢性骨髓性白血病,慢性期 | 400 mg daily |
| 慢性骨髓性白血病,加速期或芽細胞危象 | 600 mg daily |
| 胃腸道間質瘤 | 400 mg daily |
| 隆突性皮膚纖維肉瘤 | 400 mg twice daily |
| 侵襲性全身性肥大細胞增生症 | 400 mg daily |
| 骨髓增生不良/骨髓增生性疾病 | 400 mg daily |
| 復發/難治性費城染色體陽性急性淋巴球性白血病 | 600 mg daily |
| 高嗜酸性球症候群/慢性嗜酸性球白血病 | 400 mg daily |
| 黑色素瘤ᵃ | 400 mg twice daily |
ᵃimatinib 用於黑色素瘤並非 FDA 核准;仿單外劑量依據 NCT00470470。¹¹
劑量療法 (DOSING REGIMEN)(見上表 194-2)
副作用與注意事項 (SIDE EFFECTS AND PRECAUTIONS)
不良反應 (Adverse Effects):最常見的不良反應包括水腫、噁心、嘔吐、肌肉痙攣、肌痛、腹瀉、骨痛、疲倦與腹痛。較不常見但嚴重的不良反應包括嚴重體液滯留(如心包膜積液 pericardial effusion、肺水腫 pulmonary edema、肋膜積液 pleural effusions 與腹水 ascites)、血液學毒性(如貧血 anemia、嗜中性球低下 neutropenia 與血小板低下 thrombocytopenia)、鬱血性心衰竭 (congestive heart failure)、肝衰竭與出血。服用 imatinib 的病人已報告有多種皮膚副作用,包括眼周水腫 (periorbital edema)、色素異常 (dyspigmentation)、麻疹樣疹 (morbilliform eruption)、玫瑰糠疹樣疹 (pityriasis rosea-like eruption)、急性泛發性發疹性膿疱症 (acute generalized exanthematous pustulosis)、乾癬惡化 (exacerbation of psoriasis)、伴嗜酸性球增多及全身症狀之藥物疹 (drug rash with eosinophilia and systemic symptoms)、假性紫質症 (pseudoporphyria)、蕈狀肉芽腫樣疹 (mycosis fungoides–like eruption)、急性嗜中性球皮膚病 (acute neutrophilic dermatosis)、紅皮症 (erythroderma)、史蒂芬斯-強生症候群 (Stevens-Johnson syndrome)、穿透性毛囊炎 (perforating folliculitis) 與蕁麻疹 (urticaria)。¹²
育齡婦女應被告知 imatinib 為懷孕分級 D 類藥物,因其在囓齒類試驗中具致畸性,且上市後已有懷孕婦女服用 imatinib 後先天異常的報告。因此,應建議病人在服用 imatinib 期間使用高度有效的避孕措施並避免懷孕。哺乳母親應被告知,imatinib 及其代謝物已在人類母乳中被檢出,哺乳嬰兒可能接受到高達母體劑量 10% 的藥量。鑑於對嬰兒的風險,服用 imatinib 期間不建議哺乳。
藥物交互作用 (Drug Interactions):由於經 CYP3A4 代謝,imatinib 的藥物濃度會受到抑制或誘導 CYP3A4 之藥物影響。此外,研究顯示 imatinib 是中度的 CYP3A4 抑制劑,並弱抑制 CYP2D6;因此,當 imatinib 與治療窗狹窄的 CYP3A4 及 CYP2D6 受質併用時應謹慎。完整的藥物間交互作用與不良反應請參閱 Gleevec 仿單。⁸
NILOTINIB (TASIGNA)
藥理學與作用機轉
結構:Nilotinib,4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-benzamide, monohydrochloride, monohydrate,分子式為 C28H22F3N7O • HCl • H2O,分子量為 584 daltons。Figure 194-3 顯示 nilotinib 的結構。
代謝:Nilotinib 由 CYP3A4 代謝,代謝主要透過氧化與羥化作用。絕大部分劑量 (93%) 經糞便排出。消除半衰期估計為 17 小時。¹³
吸收與分布:Nilotinib 口服吸收,於餐後 30 分鐘給藥時吸收增加。循環化合物中有 98% 與血清蛋白結合。
作用機轉:與其前身 imatinib 類似,nilotinib 是一種多酪胺酸激酶抑制劑。此化合物結合並穩定其標靶蛋白(BCR-ABL、KIT、PDGFR、discoidin domain receptor)激酶結構域的失活構型。它是基於其前身的結構理性設計而成,以克服 CML 對 imatinib 的抗藥性。化合物的理性設計使 nilotinib 對 BCR-ABL 的親和力與抑制活性顯著高於 imatinib,同時維持其對 PDGFR 與 KIT 的活性。
適應症與禁忌症
Nilotinib 適用於成年病人,無論是新診斷的慢性期費城染色體陽性 (Ph+) CML,或是對先前 imatinib 治療具抗藥性或不耐受的加速期 CML。Nilotinib 目前無任何 FDA 核准的皮膚科疾病適應症。然而,imatinib 在 c-KIT 突變黑色素瘤的結果,為探索 nilotinib 在這些亞群用途的早期試驗提供了理論依據。與 imatinib 類似,結果為適度。在一項試驗中,42 名帶有 KIT 改變的轉移性黑色素瘤病人中,有 7 名 (16.7%) 以單一藥物 nilotinib 達到反應。¹⁴ 在第二項針對帶有 KIT 突變或擴增、且曾接受過 KIT 抑制劑治療的晚期黑色素瘤病人試驗中,19 名病人有 4 名 (21%) 出現反應。¹⁵
Nilotinib 禁用於長 QT 症候群 (long QT syndrome)、低血鉀 (hypokalemia) 或低血鎂 (hypomagnesemia) 的病人。
劑量療法
新診斷 Ph+ 慢性期 CML 的建議劑量為口服 300 mg twice daily;對於抗藥性或不耐受的 Ph+ 慢性期 CML 與加速期 CML,建議口服 400 mg twice daily。Nilotinib 不應與食物併服。建議服藥前至少 2 小時及服藥後 1 小時避免進食。應避免會抑制 CYP3A4 的食物。多種情況建議調整劑量,包括肝功能受損、QT 間期與血液學毒性。完整的劑量調整建議請參閱 Tasigna 產品仿單。
副作用與注意事項
不良反應:最常見的不良反應包括皮膚毒性(見下文)、血小板低下、嗜中性球低下、貧血、便秘、噁心、嘔吐、高膽紅素血症 (hyperbilirubinemia)、疲倦、脂肪酶升高與轉胺酶升高。嚴重不良反應包括 QT 延長與猝死的黑框警示 (boxed warning)。因此,nilotinib 不建議用於低血鉀、低血鎂或長 QT 症候群的病人。亦應避免已知會延長 QT 間期或強力抑制 CYP3A4 的藥物。鉀、鈣、鎂、磷酸鹽或鈉的電解質異常應在開始治療前矯正,並在治療期間定期監測。在 nilotinib 用於 CML 的初步試驗中,皮膚毒性是最常見的不良反應,包括皮疹(未描述特定型態)、搔癢、皮膚乾燥與脫髮。上市後報告包括一例大疱性 Sweet 症候群 (bullous Sweet syndrome)。¹²
Nilotinib 為懷孕分級 D 類藥物,根據其作用機轉及動物研究資料顯示此化合物可能造成胎兒傷害。具生育能力的婦女應被建議於治療期間使用高度有效的避孕措施。動物研究結果顯示 nilotinib 於哺乳期可能不安全。建議考慮替代哺乳,或權衡哺乳期使用的風險與效益。
藥物交互作用:由於 nilotinib 為 CYP3A4 的受質,CYP3A4 的抑制劑與誘導劑可能影響血清濃度。Nilotinib 是 CYP3A4、CYP2C8、CYP2C9 與 CYP2D6 的抑制劑,亦可能誘導 CYP2B6、CYP2C8 與 CYP2C9。因此,nilotinib 與這些酶的受質併用可能影響血清濃度。完整的藥物間交互作用與不良反應請參閱 Tasigna 產品仿單。¹³
DASATINIB (SPRYCEL)
藥理學與作用機轉
結構:Dasatinib,N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate,分子式為 C22H26ClN7O2S • H2O,對應於單水合物形式的式量 506.02 daltons 及無水游離鹼的 488.01 daltons。¹⁶ Figure 194-4 顯示 dasatinib 的結構。
代謝:Dasatinib 主要由 CYP3A4 代謝。Dasatinib 的代謝物亦由尿苷二磷酸-葡萄糖醛酸轉移酶 (uridine diphosphate-glucuronosyltransferase, UGT) 與含黃素單氧化酶 3 (flavin-containing monooxygenase 3, FMO-3) 酶產生。估計的消除半衰期為 3 至 5 小時。Dasatinib 主要經由糞便排出。
吸收與分布:口服給藥後,血漿濃度尖峰於 30 分鐘至 6 小時之間達到。表觀分布體積為 2505 L。
作用機轉:Dasatinib 是一種小分子激酶抑制劑。它抑制眾多激酶,包括 BCR-ABL、c-KIT、PDGFRβ、SRC 家族 (SRC、LCK、YES、FYN) 與 EPHA2。在臨床前的體外研究中,dasatinib 抑制了過度表現 BCR-ABL 之 CML 與急性淋巴芽細胞白血病 (acute lymphoblastic leukemia, ALL) 細胞株的生長。¹⁶ 在這些試驗中,dasatinib 對帶有賦予 imatinib 抗藥性之 BCR-ABL 激酶突變的細胞株有效。
適應症與禁忌症
Dasatinib 獲 FDA 核准用於:(a) 新診斷的 Ph+ 慢性期 CML 成人;(b) 對包含 imatinib 在內之先前治療不耐受或具抗藥性的慢性期、加速期、或骨髓性/淋巴性芽細胞期 Ph+-CML 成人;以及 (c) 對先前治療不耐受或具抗藥性的 Ph+-ALL 成人。在黑色素瘤中,dasatinib 的早期研究無論作為單一療法¹⁷ 或與全身性化療併用,¹⁸ 僅顯示出適度的臨床效益。
製造商仿單上無列出禁忌症。
劑量療法
建議口服劑量為慢性期 CML 100 mg daily,加速期 CML、骨髓性或淋巴性芽細胞期 CML 或 Ph+-ALL 為 140 mg once daily。嗜中性球低下、血小板低下及併用 CYP3A4 調節劑的劑量調整請參閱製造商仿單。未在腎功能受損病人中進行研究,因此無針對腎功能受損病人的特定劑量調整。
副作用與注意事項
不良反應:對於新診斷的慢性期 CML,最常見的不良反應為骨髓抑制 (myelosuppression)、腹瀉與體液滯留。先前曾惡化或對先前 imatinib 治療不耐受的病人,常出現骨髓抑制、體液滯留、腹瀉、頭痛、疲倦、呼吸困難、噁心、出血、肌肉骨骼疼痛與皮膚毒性。皮膚不良反應包括嗜中性球皮膚病 (neutrophilic dermatosis)、¹⁹ 毛囊角化症樣病灶與膿疱、白色角化丘疹及粟粒腫 (milia)。²⁰
藥物交互作用:當 dasatinib 與強效 CYP3A4 誘導劑(包括但不限於 phenytoin、rifampin、phenobarbital、carbamazepine、dexamethasone、rifabutin)及強效 CYP3A4 抑制劑(ketoconazole、voriconazole、itraconazole、atazanavir、nelfinavir、indinavir、ritonavir、saquinavir、nefazodone、telithromycin 與 clarithromycin)併用時應謹慎。服用 dasatinib 期間建議避免聖約翰草 (St. John’s wort) 與葡萄柚汁。建議告知具生育能力的婦女於 dasatinib 治療期間及最後一劑後至少 30 天內避免懷孕。服用 dasatinib 期間及最後一劑後至少 2 週內不建議哺乳。肝功能受損病人使用時應謹慎。完整的藥物間交互作用與不良反應請參閱 Sprycel 仿單。¹⁶
BOSUTINIB (BOSULIF)
藥理學與作用機轉
結構:Bosutinib,3-Quinolinecarbonitrile, 4-[(2,4-dichloro-5-methoxyphenyl)amino]-6-methoxy-7-[3-(4-methyl-1-piperazinyl)propoxy]-, hydrate (1:1),化學式為 C26H29Cl2N5O3 • H2O(單水合物);其分子量為 548.46(單水合物),相當於 530.46(無水)。²¹ Figure 194-5 顯示 bosutinib 的結構。
代謝:Bosutinib 主要由 CYP3A4 代謝,並主要經由糞便排出。
吸收與分布:與食物併服時口服生體可用率為 34%,口服給藥後血漿濃度尖峰於 4 至 6 小時之間達到。表觀分布體積為 6080 L ± 1230 L。
作用機轉:Bosutinib 是一種小分子量激酶抑制劑。它抑制眾多激酶,包括 BCR-ABL 與 SRC 家族激酶 SRC、LCK 與 FYN。在臨床前的體外研究中,bosutinib 抑制了 18 株對 imatinib 具抗藥性、過度表現 BCR-ABL 之鼠類骨髓細胞株中的 16 株的生長。Bosutinib 並未抑制表現 T315I 與 V299L 抗藥性突變的突變細胞株。²¹
適應症與禁忌症
Bosutinib 獲 FDA 核准用於對先前治療不耐受或具抗藥性的慢性期、加速期或芽細胞期 Ph+-CML 成人。Bosutinib 目前無任何 FDA 核准的皮膚科疾病適應症。Bosutinib 禁用於對該藥物已知過敏的病人。
劑量療法
建議口服劑量為與食物併服 500 mg once daily。嗜中性球低下、血小板低下、腎功能受損、肝功能受損及併用 CYP3A4 調節劑的劑量調整請參閱製造商仿單。
副作用與注意事項
不良反應:最常見的不良反應為腹瀉、噁心、血小板低下、嘔吐、腹痛、皮疹(未另行指明)、貧血、發熱與疲倦。Bosutinib 為懷孕分級 D 類藥物,建議告知具生育能力的婦女於治療期間避免懷孕。至於哺乳,bosutinib 可能分泌至人類乳汁中,因此須權衡對哺乳嬰兒的傷害風險與該藥物對母親的重要性。
藥物交互作用:當 bosutinib 與強效 CYP3A4 誘導劑(包括但不限於 phenytoin、rifampin、phenobarbital、carbamazepine、dexamethasone、rifabutin)及強效 CYP3A4 抑制劑(ketoconazole、voriconazole、itraconazole、atazanavir、nelfinavir、indinavir、ritonavir、saquinavir、nefazodone、telithromycin 與 clarithromycin)併用時應謹慎。服用 bosutinib 期間建議避免聖約翰草與葡萄柚汁。氫離子幫浦抑制劑 (proton pump inhibitors) 可降低 bosutinib 的暴露量,若可能應避免使用。可考慮 H2 阻斷劑或短效制酸劑。完整的藥物間交互作用與不良反應請參閱 Bosulif 仿單。²¹
PONATINIB (ICLUSIG)
藥理學與作用機轉
結構:Ponatinib,3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide hydrochloride,化學式為 C29H28ClF3N6O,對應的分子量為 569.02 daltons。²² Figure 194-6 顯示 ponatinib 的結構。
代謝:Ponatinib 由 CYP3A4 代謝,較少程度由 CYP2C8、CYP2D6 與 CYP3A5 代謝。它也由醯胺酶 (amidases) 與/或酯酶 (esterases) 代謝。Ponatinib 主要經由糞便排出。
吸收與分布:口服給藥後,血漿濃度尖峰於 6 小時內達到。表觀分布體積為 1223 L。
作用機轉:Ponatinib 是一種小分子量激酶抑制劑。它抑制多種激酶,包括 BCR-ABL、VEGFR(血管內皮生長因子受體 vascular endothelial growth factor receptor)、PDGFR、FGFR(纖維母細胞生長因子受體 fibroblast growth factor receptor)、EPH(ephrin)、KIT、RET、TIE2(TEK receptor tyrosine kinase)、FLT3(fms-related tyrosine kinase 3)與 SRC 家族激酶。Ponatinib 亦抑制賦予 imatinib 抗藥性的 T315I 突變。
適應症與禁忌症
Ponatinib 獲 FDA 核准用於 T315I 突變陽性的慢性期、加速期或芽細胞期 CML 成人,或 T315I 突變陽性的 Ph+-ALL。它亦核准用於無其他 TKI 治療適應的慢性期、加速期或芽細胞期 CML 或 Ph+-ALL 成人。Ponatinib 目前無任何 FDA 核准的皮膚科疾病適應症。製造商仿單上無列出禁忌症。
劑量療法
建議口服劑量為與食物併服或不併服 45 mg once daily。嗜中性球低下、血小板低下、腎功能受損、肝功能受損及併用 CYP 調節劑的劑量調整請參閱製造商仿單。
副作用與注意事項
不良反應:最常見的非血液學不良反應包括高血壓、皮疹(未另行指明)、疲倦、皮膚乾燥 (xerosis)、頭痛、腹痛、便秘、噁心、關節痛與發熱。血液學毒性包括骨髓抑制(含白血球低下 leukopenia、嗜中性球低下與血小板低下)、貧血與淋巴球低下 (lymphopenia)。Ponatinib 具有血管阻塞的黑框警示,包括動脈與靜脈血栓,發生於至少 27% 的病人、心衰竭與肝毒性。Ponatinib 為懷孕分級 D 類藥物,建議告知具生育能力的婦女於治療期間避免懷孕。關於哺乳的考量,目前未知 ponatinib 是否分泌至人類乳汁中,因此須權衡對哺乳嬰兒的傷害風險與該藥物對母親的重要性。
藥物交互作用:當 ponatinib 與強效 CYP3A 誘導劑及抑制劑併用時應謹慎。完整的藥物間交互作用與不良反應請參閱 Iclusig 仿單。²²
表皮生長因子受體抑制劑 (EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITORS)
重點一覽 (AT-A-GLANCE)
■ 表皮生長因子受體 (epidermal growth factor receptor, EGFR) 存在於多種上皮細胞的細胞表面,並在眾多惡性腫瘤中失調。
■ 阻斷表皮生長因子與 EGFR 結合的單株抗體 (monoclonal antibodies),以及 EGFR 細胞內酪胺酸激酶的小分子量抑制劑,已被核准作為數種癌症的治療。
■ 雖然在本書出版時尚無 FDA 核准的皮膚科適應症,但皮膚腫瘤學的臨床研究仍在進行中。
■ EGFR 抑制劑常有皮膚毒性,包括丘疹膿疱性疹 (papulopustular eruptions)、搔癢、皮膚乾燥 (xeroderma) 與甲溝炎 (paronychia)。
背景 (BACKGROUND)
聚焦於 EGFR(亦稱為 ErbB1 或 HER1)的藥物研發展現了標靶治療、理性藥物設計與個人化醫療的潛力。雖然抑制 EGFR 訊息的藥物目前尚無任何 FDA 核准的皮膚科適應症,但其用於皮膚惡性腫瘤的研究仍在進行中。此外,這些藥物——包括 cetuximab、panitumumab、gefitinib、erlotinib、afatinib 與 osimertinib——具有顯著的皮膚不良反應,因此瞭解其適應症、作用機轉與相關毒性與所有皮膚科醫師都相關。EGFR 是 ErbB 酪胺酸激酶受體家族的一員,該家族還包括 ErbB2 (HER2/neu)、ErbB3 (HER3) 與 ErbB4 (HER4)。這些細胞表面蛋白具有一個細胞外配體結合結構域、一個跨膜結構域與一個細胞內酪胺酸激酶結構域 (Fig. 194-7)。EGFR 存在於多種細胞類型,包括角質細胞 (keratinocytes) 與各種實體腫瘤的細胞,於表皮生長因子 (epidermal growth factor, EGF) 與其他生長因子結合後被活化。配體與 EGFR 細胞外結構域的結合導致二聚化 (dimerization),其中受體可與另一 EGFR 蛋白結合,或與 ErbB 家族的另一單體形成異二聚體 (heterodimerize)。二聚化觸發細胞內酪胺酸激酶將數個酪胺酸殘基自磷酸化 (autophosphorylate)。受體 C 端結構域上磷酸化酪胺酸殘基隨後被各種接頭蛋白 (adaptor proteins) 辨識,啟動下游訊息轉導。EGFR 的活化可觸發涉及腫瘤生長與增殖、抑制凋亡與轉移的網路,如 MAPK、STAT、PI3K 與磷脂酶路徑。EGFR 訊息的失調在數種上皮癌中很常見,可透過多種機制發生,包括基因擴增與活化突變。2004 年,EGFR 第 19 號外顯子 (exon 19) 框內活化缺失與第 21 號外顯子 (exon 21) L858R 取代的發現,深刻改變了非小細胞肺癌 (non–small-cell lung cancer, NSCLC) 的治療格局,並為腫瘤學中的精準醫療提供了另一個早期的理論依據。²³˒²⁴
已開發出兩種不同的機制來中斷 EGFR 訊息作為癌症的治療策略:(a) 透過單株抗體靶向細胞外配體結合結構域,以及 (b) 透過小分子抑制細胞內酪胺酸激酶。單株抗體 cetuximab 與 panitumumab 已在數種癌症中展現療效。Cetuximab 核准用於大腸直腸癌與頭頸癌,而 panitumumab 適用於與全身性化療併用治療大腸直腸癌。1990 年代開始了一項協同努力,以開發抑制各種酪胺酸激酶的小分子量化合物。針對 EGFR 的第一代 TKIs,gefitinib 與 erlotinib,被設計為 ATP 與細胞內激酶結構域結合的競爭性抑制劑。儘管臨床前資料前景看好,gefitinib 在肺癌的早期臨床研究令人失望,因為 ISEL(Iressa Survival Evaluation in Lung Cancer,Iressa 肺癌存活評估),一項針對未經篩選、重度預治療病人的大型試驗,並未顯示 NSCLC 病人有整體存活效益。²⁵ 有趣的是,亞組分析揭示,從不吸菸者與亞洲病人使用 gefitinib 可達到較佳的整體存活。轉譯研究隨後證明了第 19 號外顯子與第 21 號外顯子 EGFR 突變作為預測性生物標記的重要性。這些體細胞突變編碼 EGFR 的酪胺酸激酶結構域(由第 18 至 24 號外顯子編碼),在腺癌、非吸菸者與亞洲人中發生頻率較高。這些改變降低了酪胺酸激酶對 ATP 的親和力,因而賦予對 EGFR TKIs 的增強敏感性。NSCLC 中納入 EGFR 突變生物標記、針對 gefitinib 與 erlotinib 的後續研究,已證明在帶有第 19 號與第 21 號外顯子改變的病人中有更優越的臨床效益。²⁶˒²⁷ 因此,erlotinib 與第二代 TKI afatinib 僅適用於腫瘤帶有第 19 號或第 21 號外顯子改變的 NSCLC 病人。儘管對單藥 EGFR TKI 有初步反應,大多數病人在開始治療後的第 1 至 2 年內出現抗藥性。EGFR 酪胺酸激酶結構域的次發性突變與大多數病人對可逆性 TKIs 抗藥性的發展有關。
臨床上最相關的次發性突變,即第 20 號外顯子 (exon 20) 的 T790M,已在近 60% 服用 gefitinib 或 erlotinib 後產生抗藥性的病人中被檢出。²⁸ T790M 突變以較大的甲硫胺酸 (methionine) 殘基取代蘇胺酸 (threonine),同時增強 EGFR 激酶對 ATP 的親和力並在空間上阻礙藥物結合。²⁹ 展現理性藥物開發成功之處的是,osimertinib——一種對帶有 T790M 之 EGFR 具有活性的小分子抑制劑——已被證明在帶有 T790M 抗藥性突變的肺癌病人中高度有效。³⁰ 這一連串的開發成功,始於辨識一個致病蛋白、證明擾亂該標靶的臨床療效,繼而以第二代與第三代療法修改以克服抗藥性機制,正是標靶治療目標的典範。
CETUXIMAB (ERBITUX)
藥理學與作用機轉
結構:Cetuximab 是一種重組的人/鼠嵌合單株抗體 (human/mouse chimeric monoclonal antibody)。它由鼠抗 EGFR 抗體的 Fv 區與人類免疫球蛋白 G1 重鏈及 kappa 輕鏈恆定區組成。³¹ 其分子量約為 152,000 daltons。
代謝:Cetuximab 估計的消除半衰期約為 112 小時。Cetuximab 的清除似乎與其他生物製劑的代謝路徑類似,包括配體-受體複合物的內化作用,隨後從循環中移除。³²
吸收與分布:Cetuximab 經靜脈給藥,呈現非線性藥物動力學。分布體積似乎與劑量無關,約為 2 至 3 L/m²。Cetuximab 於第三次每週輸注時達到穩態血漿濃度。
作用機轉:Cetuximab 是一種對 EGFR 細胞外配體結合部分具親和力的單株抗體。Cetuximab 作為 EGF 與正常上皮及腫瘤細胞上其他配體的競爭性抑制劑。臨床前研究已證明,cetuximab 與 EGFR 的結合阻止受體相關的激酶活化,並下調與細胞生長、增殖、血管新生及轉移相關的訊息轉導路徑。³¹
適應症與禁忌症
Cetuximab 獲 FDA 核准用於大腸直腸癌與頭頸癌。2004 年初步核准用於與 irinotecan 併用治療對 irinotecan 為基礎之療法難治的轉移性大腸直腸癌 (metastatic colorectal cancer, mCRC) 病人。2006 年 3 月 1 日,FDA 擴大核准與放射治療併用,用於局部或區域晚期頭頸部鱗狀細胞癌 (squamous cell carcinoma of the head and neck, SCCHN) 病人,或作為單一療法用於腫瘤對鉑類化療難治的復發或轉移性 SCCHN 病人。2011 年,cetuximab 核准與鉑類療法加 5-fluorouracil 併用,用於復發局部區域或轉移性 SCCHN 病人。2012 年 7 月 6 日,FDA 核准 cetuximab 與 FOLFIRI(irinotecan、5-fluorouracil 與 leucovorin)併用作為表現 EGFR 之 K-ras 野生型 (wildtype) mCRC 病人的第一線療法。Erbitux 不適用於 RAS 突變型大腸直腸癌病人。
Cetuximab 已被研究用於皮膚鱗狀細胞癌 (cutaneous squamous cell carcinoma, cSCC),雖然迄今整體療效資料有限。一項針對 36 名未經化療、無法切除或轉移性 cSCC 病人的第二期非對照試驗於 2011 年發表,這些病人接受初始劑量 400 mg/m² 體表面積的 cetuximab,隨後每週給予 250 mg/m² 至少 6 週。³³ 這項小型研究報告,36 名病人中於 6 週時有 2 名 (6%) 完全反應、8 名 (22%) 部分反應、15 名 (42%) 疾病穩定。然而,反應持續時間有限,反應者的中位反應持續時間為 5 個月。需要更多更大樣本的前瞻性研究以更佳評估 cetuximab 在 cSCC 的療效。製造商仿單上無禁忌症。
劑量療法
Cetuximab 以靜脈輸注給予。初始劑量為 400 mg/m²,輸注 120 分鐘,隨後每週輸注 250 mg/m²。前置用藥建議及額外的劑量與輸注速率調整請參閱製造商仿單。
副作用與注意事項
不良反應:最常見的不良反應為皮膚毒性、頭痛、腹瀉與感染。Cetuximab 帶有嚴重、可能致命之輸注反應(發生於約 3% 的病人)以及心肺停止與/或猝死的黑框警示。強烈建議於輸注期間與輸注後密切監測血清電解質。其他嚴重不良反應包括間質性肺病 (interstitial lung disease) 與低血鎂。與其他擾亂 EGFR 訊息的藥物類似,cetuximab 的皮膚毒性常見,包括丘疹膿疱性疹、皮膚乾燥、搔癢、黏膜炎 (mucositis)、脫髮、睫毛過長 (trichomegaly)、甲溝炎與甲剝離 (onycholysis)。³⁴ 痤瘡樣疹通常於 cetuximab 治療開始後 1 至 2 週內出現,並且也與改善的臨床預後相關。³⁵
Cetuximab 為懷孕分級 C 類藥物,僅在對胎兒的潛在傷害被對母親的潛在效益所超越時,才應於懷孕期間使用。免疫球蛋白 G 抗體(如 cetuximab)會分泌至人類乳汁中;因此,若可能,應於治療期間及治療後至少 60 天內避免哺乳,以預防哺乳嬰兒的潛在不良反應。
藥物交互作用:製造商仿單上無列出藥物交互作用。
PANITUMUMAB (VECTIBIX)
藥理學與作用機轉
結構:Panitumumab 是一種重組的人類免疫球蛋白 G2 kappa 單株抗體,分子量為 147,000 daltons。³⁶ 它於中國倉鼠卵巢細胞 (Chinese hamster ovary cells) 中工程化生產。
代謝:平均估計消除半衰期為 7.5 天。
吸收與分布:給予建議劑量將於第三次輸注時達到穩態濃度。
作用機轉:Panitumumab 是一種對 EGFR 細胞外配體結合部分具親和力的單株抗體。Panitumumab 與 cetuximab 類似,作為 EGF 與正常上皮及腫瘤細胞上其他配體的競爭性抑制劑。臨床前研究已證明,panitumumab 與 EGFR 的結合降低受體相關的激酶活化,導致下調與細胞生長、增殖、血管新生及轉移相關的訊息轉導路徑。
適應症與禁忌症
Panitumumab 適用於治療經 FDA 核准檢測判定為 KRAS 野生型的 mCRC。它獲 FDA 核准作為與 FOLFOX 併用的第一線療法,以及作為含 fluoropyrimidine、oxaliplatin 與 irinotecan 之療法治療後惡化的單一療法。Panitumumab 不核准用於帶有 KRAS 突變或 KRAS 突變狀態未知的 mCRC 病人。Panitumumab 目前正在開發用於頭頸癌。Panitumumab 已有報告用於局部晚期 cSCC 並有令人鼓舞的結果。已有報告成功用於一名對 cetuximab 發生過敏反應 (anaphylactic reaction) 的老年病人,³⁷ 以及一項 16 名病人的小型第二期試驗,依 RECIST 標準產生 31% 的最佳整體反應率。³⁸ 雖然前景看好,但需要關鍵性的第三期試驗以確認這些早期研究結果。製造商仿單上無禁忌症。
劑量療法
Panitumumab 以靜脈輸注給予,建議劑量為每 14 天 6 mg/kg。腎功能或肝功能受損無特定的劑量調整。
副作用與注意事項
不良反應:最常見的不良反應為皮膚毒性、疲倦、噁心與腹瀉。嚴重不良反應包括電解質紊亂、輸注反應、肺纖維化/間質性肺病與角膜炎 (keratitis)。Panitumumab 帶有皮膚毒性的黑框警示。皮膚不良事件在註冊試驗中很常見,90% 的病人經歷任何等級的毒性,而 15% 遭受嚴重皮膚毒性。皮膚表現包括但不限於丘疹膿疱性疹、剝脫 (exfoliation)、搔癢、紅斑 (erythema)、光敏感 (photosensitivity)、皮膚乾燥與甲溝炎。已在使用 panitumumab 的病人中觀察到致命的大疱性疾病及危及生命的壞死性筋膜炎 (necrotizing fasciitis)、膿瘍與敗血症。³⁶
Panitumumab 為懷孕分級 C 類藥物。雖未在懷孕婦女中進行研究,但以建議人類劑量的 1.25 至 5 倍治療的食蟹猴 (Cynomolgus monkeys) 確實顯示出胚胎致死與流產的證據。因此,懷孕期間給藥建議極度謹慎,且對胎兒的風險須權衡對母親的潛在效益。建議於治療期間及治療完成後至少 2 個月內避免哺乳。
藥物交互作用:製造商仿單上無列出特定藥物交互作用。
GEFITINIB (IRESSA)
藥理學與作用機轉
結構:Gefitinib,4-quinazolinamine N-(3-chloro-4-fluorophenyl)-7-methoxy-6-[3-(4-morpholinyl)propoxy],分子式為 C22H24ClFN4O3,分子質量為 446.9 daltons。³⁹ Figure 194-8 顯示 gefitinib 的結構。
代謝:Gefitinib 主要由 CYP3A4 代謝,較少程度由 CYP2D6 代謝。靜脈給藥後估計的消除半衰期為 48 小時。
吸收與分布:Gefitinib 的口服生體可用率約為 60%,血漿濃度尖峰於給藥後 3 至 7 小時達到。食物不顯著影響吸收。分布體積為 1400 L。
作用機轉:Gefitinib 是一種小分子 TKI。它可逆地結合並抑制 EGFR 的激酶結構域。與野生型 EGFR 蛋白相比,gefitinib 對帶有第 19 號外顯子缺失或第 21 號外顯子點突變 L858R 的 EGFR 具有更高的親和力。
適應症與禁忌症
Gefitinib 在美國的適應症有限,僅限於目前正在接受並從 gefitinib 受益、或先前曾從 gefitinib 治療受益的 NSCLC 病人。Gefitinib 最初於 2003 年 5 月由 FDA 加速核准,用於腫瘤對鉑類療法及 docetaxel 難治的 NSCLC 病人。然而,在兩項研究證明缺乏療效後,FDA 於 2005 年變更仿單,撤回對新病人的核准。後續研究已證明 gefitinib 在帶有 EGFR 突變之腫瘤病人中的臨床有效性,且該藥在歐洲適用於晚期 NSCLC。Gefitinib 目前無任何 FDA 核准的皮膚科疾病適應症。製造商仿單上無禁忌症。
劑量療法
建議劑量為口服 250 mg once daily,與食物併服或不併服皆可。
副作用與注意事項
不良反應:皮膚毒性與腹瀉是最常見的不良反應。皮膚毒性常見,並與其他靶向 EGFR 之藥物有共同特徵。丘疹膿疱性疹、皮膚乾燥與搔癢皆常見。丘疹膿疱性疹通常於治療開始後 1 至 2 週出現,並與改善的整體存活相關。⁴⁰ 其他皮膚不良表現包括光敏感、黏膜炎以及指甲與毛髮變化。指甲影響包括甲剝離、甲溝炎與甲褶的化膿性肉芽腫樣 (pyogenic granuloma-like) 病灶。毛髮變化包括脫髮、睫毛過長與多毛症 (hirsutism)。¹²
根據臨床前動物研究,gefitinib 給予懷孕婦女時可能造成胎兒傷害。因此,建議強烈勸告於懷孕期間避免使用 gefitinib。Gefitinib 已在大鼠乳汁中被檢出,因此應使哺乳婦女瞭解哺乳期間對嬰兒的潛在傷害。
藥物交互作用:當 gefitinib 與 CYP3A4 的抑制劑及誘導劑併用時應謹慎。亦建議避免會影響胃部 pH 值的化合物,如氫離子幫浦抑制劑。與 warfarin 併用時,應密切監測凝血酶原時間 (prothrombin time) 與/或國際標準化比值 (international normalized ratio)。完整的藥物間交互作用與不良反應請參閱 Iressa 仿單。³⁹
鹽酸 ERLOTINIB (TARCEVA)
藥理學與作用機轉
結構:Erlotinib,N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine,分子式為 C22H23N3O4 • HCl,分子量為 429.90 daltons。⁴¹ Figure 194-9 顯示 erlotinib 的結構。
代謝:Erlotinib 主要由 CYP3A4 代謝,較少程度由 CYP1A2 與 CYP1A1 代謝。消除半衰期估計為 36.2 小時。血漿達到穩態的時間為 7 至 8 天。
吸收與分布:口服劑量於 4 小時後達到血漿濃度尖峰。口服吸收為 60%,若與食物併服則增加至約 100%。分布體積為 232 L。
作用機轉:Erlotinib 是一種可逆性的 EGFR 小分子抑制劑。它與 ATP 競爭結合 EGFR 的細胞內結構域,從而阻止酪胺酸殘基的自磷酸化並阻斷下游訊息轉導。與野生型 EGFR 蛋白相比,erlotinib 對帶有第 19 號外顯子缺失或第 21 號外顯子 (L858R) 突變的 EGFR 蛋白展現優先結合。
適應症與禁忌症
Erlotinib 獲 FDA 核准用於 NSCLC 與胰臟癌 (pancreatic carcinoma)。在 NSCLC 中,其核准範圍涵蓋第一線、維持治療,或在經至少 1 個化療療程後惡化、且轉移性腫瘤帶有 EGFR 第 19 號外顯子缺失或第 21 號外顯子 (L858R) 取代突變(經 FDA 核准檢測偵測)之病人的第二線或更後線療法。Erlotinib 亦適用於局部晚期、無法切除或轉移性胰臟癌病人,作為與 gemcitabine 併用的第一線療法。Erlotinib 正被研究與放射治療及其他全身性藥物併用於 cSCC。在黑色素瘤中,一項針對轉移性疾病病人之 erlotinib 與 bevacizumab 的第二期試驗顯示令人失望的結果,無惡化存活期 (progression-free survival, PFS) 僅 2 個月。⁴²
製造商仿單上無列出禁忌症。
劑量療法
對於 NSCLC,erlotinib 的建議劑量為口服 150 mg once daily,直到疾病惡化或不可接受的毒性。胰臟癌的劑量為 100 mg once daily,與 gemcitabine 併用,直到疾病惡化或不可接受的毒性。建議空腹服用 erlotinib。製造商仿單未提供腎功能受損病人初始治療期間的劑量調整。服用 erlotinib 期間建議監測腎功能,且發生腎功能受損的病人應停藥,直到毒性緩解。肝功能受損的特定劑量尚未定義。基線肝臟異常病人的調整請參閱製造商仿單。
副作用與注意事項
不良反應:最常見的不良反應包括皮膚毒性、腹瀉、疲倦、厭食、呼吸困難、咳嗽、噁心與嘔吐。嚴重的藥物毒性包括間質性肺病(發生於 1.1% 服用 erlotinib 的病人)、肝毒性、胃腸道穿孔、心肌缺血/梗塞、微血管病性溶血性貧血 (microangiopathic hemolytic anemia)、腦血管意外、角膜穿孔/潰瘍與持續性嚴重角膜炎。皮膚毒性非常常見,大多數接受 erlotinib 的病人都會出現某種程度的皮膚毒性。最常見者包括丘疹膿疱性疹、皮膚乾燥與搔癢。丘疹膿疱性疹常於治療開始後 1 至 2 週出現,並與改善的整體存活相關。⁴⁰ 其他皮膚不良表現包括大疱性疹、光敏感與黏膜炎,以及指甲與毛髮變化。指甲影響包括甲剝離、甲溝炎與甲褶的化膿性肉芽腫樣病灶。毛髮變化包括脫髮、睫毛過長與多毛症。¹²
Erlotinib 可造成胎兒傷害,具生育能力的婦女應被建議使用高度有效的避孕措施。關於哺乳,目前未知 erlotinib 是否存在於母乳中,但基於潛在的嚴重傷害,建議仔細權衡服用 Tarceva 的風險與效益。
藥物交互作用:當 erlotinib 與 CYP3A4 及 CYP1A2 的抑制劑或誘導劑併用時應謹慎,因其可能影響血漿濃度。吸菸可加速 erlotinib 的清除,可能降低其抗腫瘤效果。與會增加胃部 pH 值並可能降低 erlotinib 血漿濃度的化合物(如氫離子幫浦抑制劑、H2 受體拮抗劑與制酸劑)併用時亦應謹慎。完整的藥物間交互作用與不良反應請參閱 Tarceva 仿單。
二馬來酸 AFATINIB (GILOTRIF)
藥理學與作用機轉
結構:Afatinib dimaleate,2-butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2E)-,(2Z) 2-butenedioate (1:2),分子式為 C32H33ClFN5O11,分子量為 718.1 daltons。⁴³ Figure 194-10 顯示 afatinib 的結構。
代謝:Afatinib 的酶促代謝極少,該化合物在與循環蛋白共價加成後主要經由糞便排出。消除半衰期估計為 37 小時。血漿達到穩態的時間約為 8 天。
吸收與分布:口服劑量於 2 至 5 小時後達到血漿濃度尖峰。口服吸收約為 92%,高脂餐使最高血漿濃度降低 50%。
作用機轉:Afatinib 是一種第二代小分子激酶抑制劑,對全部 4 個 ErbB 家族成員具有活性。與可逆性抑制 EGFR 的第一代 EGFR TKIs gefitinib 與 erlotinib 不同,afatinib 共價結合受體的激酶結構域。因此,afatinib 不可逆地抑制相關酪胺酸殘基的自磷酸化,導致下調細胞生長與存活的訊息轉導路徑。
適應症與禁忌症
Afatinib 獲 FDA 核准作為第一線藥物,用於帶有 EGFR 第 19 號外顯子缺失或第 21 號外顯子 (L858R) 取代突變(經 FDA 核准檢測偵測)的轉移性 NSCLC。Afatinib 目前無任何 FDA 核准的皮膚科疾病適應症。製造商仿單上無列出禁忌症。
劑量療法
Afatinib 的建議劑量為口服 40 mg once daily,直到疾病惡化或不可接受的毒性。建議於餐前至少 1 小時或餐後 2 小時服用 afatinib。對於腎功能受損病人(估計腎絲球過濾率:15 至 29 mL/min),建議減量至口服 30 mg daily。Afatinib 未在腎絲球過濾率 <15 mL/min 的病人中進行研究;因此,無建議特定的劑量調整。Afatinib 未在重度(Child-Pugh C 級)肝功能受損病人中進行研究。對於輕度至中度肝功能受損病人,無提供特定的劑量調整。
副作用與注意事項
不良反應:afatinib 最常見的不良反應包括腹瀉、皮膚毒性、厭食、噁心與嘔吐。嚴重的藥物毒性包括間質性肺病(發生於 1.6% 服用 afatinib 的病人)、肝毒性、角膜炎與胚胎胎兒毒性。⁴³
皮膚毒性常見,包括丘疹膿疱性疹、皮膚乾燥、搔癢、口腔炎 (stomatitis) 與指甲變化。相關研究顯示,丘疹膿疱性疹的出現可能可預測腫瘤反應,與其他 EGFR 抑制劑所見類似。⁴⁴ 與 gefitinib 與 erlotinib 相比,afatinib 已被證明以更高的頻率及更快的發作誘發甲溝炎。⁴⁵ 雖然此差異的機制尚未闡明,但 afatinib 是不可逆性抑制劑而 gefitinib 與 erlotinib 皆為可逆性抑制劑這一事實,已被引述為一種可能的解釋。⁴⁵ 有一例 afatinib 相關史蒂芬斯-強生症候群的報告。⁴⁶ 其他皮膚效應包括睫毛與眉毛的多毛症 (hypertrichosis)。⁴⁷
Afatinib 可造成胎兒傷害,具生育能力的婦女應被建議於治療期間及最後一劑後至少 2 週內使用高度有效的避孕措施。關於哺乳,目前未知 afatinib 是否存在於人類母乳中,但基於潛在的嚴重傷害,建議服用 Gilotrif 的婦女於治療期間及最後一劑後 2 週內避免哺乳。
藥物交互作用:當 afatinib 與會調節 P-醣蛋白 (P-glycoprotein) 的藥物併用時應謹慎。併用 P-醣蛋白抑制劑——包括但不限於 ketoconazole、itraconazole、ritonavir、cyclosporine、tacrolimus、erythromycin、verapamil、quinidine、nelfinavir、saquinavir 與 amiodarone——可增加 afatinib 濃度。反之,與 P-醣蛋白誘導劑(如 phenytoin、rifampicin、phenobarbital、carbamazepine 與聖約翰草)併用時,afatinib 的暴露量可能降低。⁴³
完整的藥物間交互作用與不良反應請參閱 Gilotrif 仿單。
OSIMERTINIB (TAGRISSO)
藥理學與作用機轉
結構:Osimertinib (AZD9291),N-(2-{2-dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino}phenyl)prop-2-enamide mesylate salt,分子式為 C28H33N7O2 • CH4O3S,分子量為 596 daltons。⁴⁸ Figure 194-11 顯示 osimertinib 的結構。
代謝:Osimertinib 主要由 CYP3A 經氧化與脫烷基化代謝。它主要經由糞便排出,估計的消除半衰期為 48 小時。
吸收與分布:給藥後,達到最高濃度的中位時間為 6 小時,範圍為 3 至 24 小時。穩態時的平均分布體積為 986 L。
作用機轉:Osimertinib 是一種 EGFR 的小分子激酶抑制劑。它不可逆地結合於某些突變型 EGFR,如 T790M、L858R 與第 19 號外顯子缺失,且與野生型 EGFR 蛋白相比有更高的親和力。在體外,osimertinib 已被證明可抑制 ACK1、BLK 與 HER2、HER3、HER4,即其他 3 個 ErbB 成員。
適應症與禁忌症
Osimertinib 於 2015 年 11 月獲加速核准,用於帶有 EGFR T790M 突變且在先前 EGFR TKI 治療下惡化的轉移性 NSCLC 病人。核准基於兩項 AURA 第二期研究(AURA extension 與 AURA2)的資料。這些是針對 411 名帶有 T790M 突變 EGFR NSCLC、且在先前 EGFR TKI 治療下惡化之病人的多中心單臂試驗,整體而言,osimertinib 治療的病人有 59% 的整體客觀反應率。³⁰ Osimertinib 目前無任何 FDA 核准的皮膚科疾病適應症。2018 年,FDA 核准用於帶有第 19 號外顯子缺失或第 21 號外顯子 L858R 突變之轉移性 NSCLC 病人的第一線治療。製造商仿單上無禁忌症。
劑量療法
對於確認帶有 T790M EGFR 突變的病人,建議劑量為口服 80 mg once daily,與食物併服或不併服皆可。
副作用與注意事項
不良反應:與其他 EGFR 激酶抑制劑類似,osimertinib 常造成腹瀉、皮疹、皮膚乾燥、指甲變化、噁心與厭食。嚴重毒性包括間質性肺病/肺炎、QTc 間期延長、心肌病變與胚胎胎兒毒性。根據其作用機轉與動物研究,osimertinib 給予懷孕婦女時可能對發育中的胎兒造成傷害。建議告知育齡女性於治療期間及最後一劑 osimertinib 後 6 週內使用有效的避孕措施。此外,建議告知與具生育能力女性有性行為的男性,於最後一劑後 4 個月內使用高度有效的避孕措施。服用 osimertinib 期間及最後一劑後 2 週內不建議哺乳。
藥物交互作用:當 osimertinib 與強效 CYP3A 誘導劑(如 phenytoin、rifampin、carbamazepine、聖約翰草)併用時應謹慎。Osimertinib 與 rosuvastatin(一種乳癌抗藥蛋白 [breast cancer resistance protein, BRCP] 受質)併用會增加 rosuvastatin 的血漿濃度,而與 simvastatin(一種 CYP3A4 受質)併用對 simvastatin 濃度則無臨床顯著影響。因此,當 osimertinib 與 BRCP 受質(如 rosuvastatin、sulfasalazine、topotecan)併用時,建議密切監測不良反應。完整的藥物間交互作用與不良反應請參閱 Tagrisso 仿單。
SMOOTHENED 抑制劑 (SMOOTHENED INHIBITORS)
重點一覽 (AT-A-GLANCE)
■ Hedgehog 路徑在絕大多數基底細胞癌 (basal cell carcinomas) 中受到擾亂。
■ 腫瘤抑制因子 PTCH1 的喪失或 smoothened 的活化突變,導致不受調控的細胞生長與致癌。
■ 小分子 vismodegib 與 sonidegib 是 smoothened 的抑制劑,核准用於治療無法切除及晚期基底細胞癌。
■ Vismodegib 與 sonidegib 帶有胚胎胎兒毒性的黑框警示。
背景 (BACKGROUND)
Hedgehog (HH) 路徑是胚胎發育所必需的訊號級聯,現已知對基底細胞癌 (basal cell carcinoma, BCC) 的分子致病機轉至關重要。在 1990 年代,腫瘤抑制因子 patched 1 (PTCH1) 基因的突變在源自基底細胞母斑症候群 (basal cell nevus syndrome)⁴⁹ 與偶發病例⁵⁰ 的 BCC 中皆被發現。PTCH1 是下游蛋白 smoothened (SMO) 的抑制因子,其喪失導致 HH 訊息的組成性上調以及負責細胞存活、生長、增殖、血管化與癒合之基因的過度表現 (Fig. 194-12)。幾乎所有偶發性 BCC 都帶有促進疾病的 HH 訊息突變,其中約 90% 帶有至少 1 個 PTCH1 等位基因的喪失,10% 帶有 SMO 的活化突變。⁵¹ BCC 的分子標靶治療於 2012 年隨著同類首創 (first-in-class) 的 SMO 抑制劑 vismodegib (Erivedge) 問世。2015 年,同類第二個 (second-in-class) 抑制劑 sonidegib (Odomzo) 獲核准。
VISMODEGIB (ERIVEDGE)
藥理學與作用機轉
結構:Vismodegib,2-Chloro-N-[4-chloro-3-(2-pyridinyl)phenyl]-4-(methylsulfonyl)benzamide,分子式為 C19H14Cl2N2O3S,分子量為 421.3 g/mol。⁵² Figure 194-13 顯示 vismodegib 的結構。
代謝:Vismodegib 主要由 CYP2C9 與 CYP3A4/5 經氧化與葡萄糖醛酸化代謝,雖然它主要以未變化的原型排出。⁵² 估計的半衰期為單次劑量後 12 天,連續每日給藥則為 4 天。
吸收與分布:Vismodegib 口服吸收,生體可用率為 31.8%。分布體積為 16.4 L 至 26.6 L。Vismodegib 與血清白蛋白及 α1-酸性醣蛋白結合。
作用機轉:Vismodegib 是 7 次跨膜蛋白 SMO(HH 訊息轉導路徑的關鍵成員)的同類首創小分子抑制劑。抑制 SMO 阻止轉錄因子 GLI(glioma-associated oncogene homolog,神經膠瘤相關致癌基因同源物)的活化與轉位,從而減少涉及細胞增殖與存活之基因的誘導。
適應症與禁忌症
Vismodegib 於 2012 年 1 月 30 日獲 FDA 核准,用於治療轉移性或無法切除 BCC 的成年病人。核准源自 ERIVANCE BCC 試驗的資料,這是一項針對 104 名局部晚期 (n = 63) 或轉移性 BCC (n = 33) 病人的多中心、第二期、單臂、雙世代、開放標籤 II 試驗。⁵³ 口服 vismodegib 在轉移性 BCC 病人中產生 10 名 (30.3%) 的客觀反應,在局部晚期 BCC 病人中產生 27 名 (42.8%) 的客觀反應。中位反應持續時間為 7.6 個月。製造商仿單上無禁忌症。
劑量療法
Vismodegib 的建議劑量為轉移性與局部晚期 BCC 口服 150 mg daily。通常持續至疾病惡化或出現不可接受的不良反應。Vismodegib 在肝功能或腎功能受損病人中的安全性與有效性尚未建立。
副作用與注意事項
不良反應:使用 vismodegib 常經歷治療相關不良反應。在 STEVIE(Safety Events in Vismodegib,Vismodegib 安全事件)研究中,這是一項針對 499 名轉移性 BCC 或局部晚期 BCC 病人的多中心開放標籤試驗,491 名病人至少有 1 項不良反應。⁵⁴ 常見不良反應包括肌肉痙攣、味覺障礙 (dysgeusia)、無力 (asthenia)、體重減輕、疲倦、噁心、食慾減退與腹瀉。皮膚不良反應常見。脫髮發生於大多數病人 (58% 至 63%)。⁵⁵ 治療期間發生的角化棘皮瘤 (keratoacanthomas) 與分化良好的鱗狀細胞癌亦曾被描述。⁵⁵ STEVIE 中約三分之一的病人因不可接受的毒性而中止治療。已有 vismodegib 病人發生第 5 級不良反應的報告,雖然根據試驗研究者,這些事件被認為與 vismodegib 無關。⁵³˒⁵⁴ 暴露於 Erivedge 的兒科病人已有報告骨骺早期融合 (premature fusion of the epiphyses),且在某些情況下,融合在停藥後仍持續進展。⁵²
動物生殖研究證明 vismodegib 具致畸性、胚胎毒性與胎兒毒性。在大鼠中,約為人類建議劑量曲線下面積 0.2 倍的劑量導致骨骼與內臟結構的畸形、發育遲緩或變異。因此,Erivedge 產品仿單包含胚胎胎兒毒性的黑框警示。婦女應於開始治療前 7 天篩檢懷孕,且建議女性於治療期間及最後一劑後最多 24 個月內避免懷孕。建議男性病人於治療期間及停藥後 3 個月內使用含殺精劑的保險套。應勸告婦女於治療期間及最後一劑後 24 個月內不要哺乳。
病人被建議於治療期間及完成治療後 24 個月內不要捐血。
藥物交互作用:目前對 vismodegib 的藥物交互作用尚無明確證據。資料顯示 vismodegib 是 P-醣蛋白外排轉運體的受質;因此,與 P-醣蛋白抑制劑(如巨環內酯類抗生素 macrolide antibiotics)併用可能增加藥物濃度與全身毒性。影響胃部 pH 值的藥物可能降低該藥的生體可用率,但尚無正式研究評估改變 pH 值藥物對 vismodegib 的影響。完整的藥物間交互作用與不良反應請參閱 Erivedge 仿單。⁵²
SONIDEGIB (ODOMZO)
藥理學與作用機轉
結構:Sonidegib,N-[6-(cis-2,6-dimethylmorpholin-4-yl)pyridine-3-yl]-2-methyl-4′-(trifluoromethoxy)[1,1′-biphenyl]-3-carboxamide diphosphate,分子式為 C26H26F3N3O3 • 2H3PO4,分子量為 681.49 daltons。Figure 194-14 顯示 sonidegib 的結構。
代謝:Sonidegib 由 CYP3A 代謝,該化合物及其代謝物主要經由腸肝循環 (enterohepatic circulation) 排出。消除半衰期約為 28 天,吸收劑量的 70% 經糞便排出,30% 經尿液排出。⁵⁶
吸收與分布:口服給藥時,劑量的不到 10% 被吸收。食用高脂餐會增加該藥的全身暴露量。穩態分布體積為 9166 L,該化合物與血漿蛋白高度結合。
作用機轉:Sonidegib 與 vismodegib 類似,是 7 次跨膜蛋白 SMO 的小分子抑制劑。因此,暴露於該化合物被認為可減弱涉及細胞增殖與存活之 HH 訊息基因的表現。
適應症與禁忌症
Sonidegib 於 2015 年 7 月 24 日獲 FDA 核准,用於治療在手術切除或放射治療後復發的局部晚期 BCC 成年病人,以及不適合手術或放射治療的病人。核准基於 BOLT(Basal Cell Carcinoma Outcomes With LDE225 Treatment)試驗的資料,這是一項針對 230 名病人的國際多中心、隨機、雙臂、非比較性試驗。⁵⁷ 每日給予 200 mg sonidegib 在 43% 的病人中產生客觀反應,800 mg daily 達到 38% 的客觀反應。該藥產品仿單上無禁忌症。
劑量療法
建議劑量為口服 200 mg daily,於餐前至少 1 小時或餐後 2 小時服用,直到疾病惡化或不可接受的不良反應。開始治療前,建議取得具生育能力婦女的懷孕狀態驗證,以及所有病人的血清肌酸激酶 (creatinine kinase) 與腎功能檢測。輕度至中度腎功能受損 (CrCl 30 至 59 mL/min) 或輕度肝功能受損的病人無建議劑量調整。更嚴重肝功能受損的病人缺乏資料。
副作用與注意事項
不良反應:最常見的不良事件包括肌肉痙攣、脫髮、味覺障礙、噁心、肌酸激酶升高、疲倦、體重減輕、厭食、肌痛、頭痛與搔癢。不良反應限制了約三分之一病人的治療持續時間。研究者報告了一例橫紋肌溶解症 (rhabdomyolysis),雖然經獨立審查後,該病例被判定不符合橫紋肌溶解症。儘管如此,建議測量基線肌酸激酶值,且對於首次血清肌酸激酶升高至正常上限 (ULN) 2.5 至 10 倍之間,建議中斷劑量。雖然無懷孕婦女的資料,但動物毒性研究證明 sonidegib 與 vismodegib 類似,具致畸性、胚胎毒性與胎兒毒性。因此,Odomzo 產品仿單上有胚胎胎兒毒性的黑框警示。據此,具生育能力的婦女應被警告潛在的胚胎胎兒死亡與嚴重的出生缺陷,並應被建議於治療期間及最後一劑後至少 20 個月內避免懷孕。雖然 sonidegib 在精液中的濃度尚未評估,但建議男性病人於治療期間及停藥後至少 8 個月內使用含殺精劑的保險套。對於哺乳期評估 sonidegib 安全性的文獻不足;因此,建議婦女於 sonidegib 治療期間及最後一劑後至少 20 個月內避免哺乳。無關於 sonidegib 對人類生殖影響的資料;然而,該化合物在 1 至 2 倍人類建議劑量下確實降低了雌性囓齒類的生育力。病人被建議於治療期間及完成治療後 20 個月內不要捐血。
藥物交互作用:鑑於 sonidegib 由 CYP3A 代謝,CYP3A 的抑制劑與誘導劑可影響藥物濃度,使用此類藥物應謹慎。臨床前研究顯示 sonidegib 抑制 CYP2B6 與 CYP2C9,因此可影響這些受質的藥物濃度。完整的藥物間交互作用與不良反應請參閱 Odomzo 仿單。⁵⁶
組蛋白去乙醯酶抑制劑 (HISTONE DEACETYLASE INHIBITORS)
重點一覽 (AT-A-GLANCE)
■ 組蛋白修飾 (histone modification) 是調控基因表現的關鍵表觀遺傳 (epigenetic) 現象。
■ 組蛋白 (histone) 蛋白乙醯化的異常是數種惡性腫瘤中常見的情形。
■ 組蛋白乙醯化的調節由組蛋白乙醯轉移酶 (histone acetyltransferases) 與組蛋白去乙醯酶 (histone deacetylases, HDACs) 控制。
■ HDACs 的抑制劑已展現作為癌症治療的療效,其給藥被認為透過恢復組蛋白與非組蛋白蛋白的正常乙醯化,誘導分化、細胞週期停滯與凋亡。
■ 在目前核准的 4 種 HDAC 抑制劑中,有 2 種——vorinostat 與 romidepsin——獲 FDA 核准用於治療皮膚 T 細胞淋巴瘤 (cutaneous T-cell lymphoma)。
背景 (BACKGROUND)
基因表現失調是致癌的標誌。基因的異常表現可源自 DNA 核苷酸序列的突變,或透過表觀遺傳的調節,如染色質修飾 (chromatin modification)。DNA 與組蛋白複合物形成染色質的基本結構單位,即核小體 (nucleosome)。核小體與染色質的構型結構部分由組蛋白蛋白的乙醯化所調控。在其濃縮形式中,帶負電的 DNA 緊密纏繞在帶正電的組蛋白蛋白周圍,限制了轉錄因子接近 DNA 啟動子區域的能力。組蛋白乙醯轉移酶將組蛋白尾部的離胺酸 (lysine) 殘基乙醯化,中和了組蛋白上的正電荷 (Fig. 194-15)。這導致染色質結構更為鬆弛,並允許轉錄因子更大程度地接近基因啟動子,促進基因表現。相反地,組蛋白去乙醯酶 (HDACs) 的活性促進一種乙醯化不足的狀態,透過損害核小體的可及性而促成轉錄沉默。乙醯化與去乙醯化之間的微妙平衡在數種人類疾病中受到擾亂。在癌細胞中,組蛋白去乙醯化與促凋亡基因以及對分化至關重要之基因的下調有關。⁵⁸ 除了染色質調節之外,組蛋白乙醯轉移酶與 HDACs 已被證明參與涉及致癌之非組蛋白蛋白的調節,如 p53、核因子 κB (nuclear factor κB)、E2F 與低氧誘導因子 1α (hypoxia-inducible factor 1α)。⁵⁸˒⁵⁹
因此,HDACs 的抑制劑已被開發用於治療用途,並在數種惡性腫瘤中展現前景,包括皮膚 T 細胞淋巴瘤。雖然確切的治療機制未知,但抑制 HDACs 的藥物被認為透過恢復正常乙醯化來誘導分化、細胞週期停滯與凋亡。⁵⁸ FDA 已核准 4 種 HDAC 抑制劑用於癌症治療:vorinostat、romidepsin、belinostat 與 panobinostat。Vorinostat 與 romidepsin 核准用於皮膚 T 細胞淋巴瘤,而 romidepsin 與 belinostat 適用於治療周邊 T 細胞淋巴瘤 (peripheral T-cell lymphoma)。Panobinostat 核准用於多發性骨髓瘤 (multiple myeloma)。
VORINOSTAT (ZOLINZA)
藥理學與作用機轉
結構:Vorinostat,suberanilohydroxamic acid 或 N-hydroxy-N′-phenyloctanediamide,是一種異羥肟酸 (hydroxamic acid) 衍生物,經驗式為 C14H20N2O3,分子量為 264.32 daltons。Figure 194-16 顯示 vorinostat 的結構。
代謝:葡萄糖醛酸化 (glucuronidation) 與水解是 vorinostat 的主要代謝途徑。由 CYP 進行的生物轉化可忽略不計。平均末端半衰期約為 2 小時。
吸收與分布:與高脂餐口服併服時,達到最高濃度的中位時間為 4 小時。進食狀態下多次給藥後的穩態濃度於 4 小時內達到(範圍:0.5 至 14 小時)。
作用機轉:Vorinostat 是數種 HDACs 的抑制劑,包括 HDAC1、HDAC2、HDAC3 與 HDAC6。在體外,vorinostat 導致乙醯化組蛋白的累積,並在某些轉化細胞中誘導凋亡與/或細胞週期停滯。⁶⁰
適應症與禁忌症
Vorinostat 於 2006 年核准用於治療在 2 種全身性療法治療期間或之後仍有持續性、復發性或進行性疾病的皮膚 T 細胞淋巴瘤病人。核准基於 2 項第二期試驗,這些試驗在重度預治療的病人中展現了 24% 至 30% 的整體客觀反應率。⁶¹˒⁶² 基線搔癢的改善亦在 32% 至 45% 的病人中被觀察到。⁶¹˒⁶² 雖然 vorinostat 在難治性疾病病人中展現療效,但通常被視為第二線療法之後的選擇。藥物仿單上無特定禁忌症。
劑量療法
Vorinostat 的建議劑量為與食物併服口服 400 mg once daily。Vorinostat 在腎功能受損病人中尚未充分研究。雖然腎臟排泄在 vorinostat 的消除中不扮演角色,但對於既有腎功能受損的病人仍建議謹慎。與腎功能正常的病人相比,輕度(膽紅素 >1 至 1.5 倍 ULN 或天門冬胺酸轉胺酶 > ULN 但膽紅素 ≤ ULN)與中度(膽紅素 1.5 至 ≤3 倍 ULN)肝功能受損的病人,其平均曲線下面積值增加了 50%。因此,建議在輕度至中度肝功能受損的病人中減量,雖然因資料不足,產品仿單上未做出特定建議。
副作用與注意事項
不良反應:最常見的不良反應(發生率 ≥20%)為疲倦、腹瀉、噁心、味覺障礙、血小板低下、厭食與體重減輕。在導致該藥核准的臨床試驗中,18.6% 的病人觀察到脫髮。⁶⁰ 導致核准的臨床試驗中,最常見的嚴重不良反應為肺栓塞 (pulmonary embolism)、鱗狀細胞癌與貧血。Vorinostat 為懷孕分級 D 類藥物。雖然在懷孕婦女中無足夠的研究,但臨床前動物研究證明 vorinostat 可能造成胎兒傷害。因此,建議勸告於懷孕期間避免使用 vorinostat。Vorinostat 於哺乳期的安全性未知。然而,由於它可能不安全,應使哺乳婦女瞭解哺乳期間對嬰兒的潛在傷害。
藥物交互作用:當 vorinostat 與其他 HDAC 抑制劑(如 valproic acid)併用時,已觀察到胃腸道出血與嚴重的血小板低下。當 vorinostat 與香豆素衍生物抗凝血劑 (coumarin-derivative anticoagulants) 併用時,觀察到凝血酶原時間與國際標準化比值延長。當 vorinostat 與香豆素衍生物(如 warfarin)併用時,建議密切監測凝血酶原時間與國際標準化比值。完整的藥物間交互作用與不良反應請參閱 Zolinza 仿單。⁶⁰
ROMIDEPSIN (ISTODAX)
藥理學與作用機轉
結構:Romidepsin,(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-bis(1-methylethyl)-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone,經驗式為 C24H36N4O6S2,分子量為 540.71 daltons。⁶³ Figure 194-17 顯示 romidepsin 的結構。
代謝:Romidepsin 主要由 CYP3A4 代謝,CYP3A5、CYP2B6、CYP2C19 與 CYP1A1 有部分貢獻。消除半衰期約為 3 小時。
吸收與分布:當以 1 至 24 mg/m² 的濃度靜脈給予時,romidepsin 展現線性藥物動力學。該藥是 P-醣蛋白 ABCB1 外排轉運體的受質。
作用機轉:Romidepsin 作為 HDAC 抑制劑。在臨床前研究中,romidepsin 已被證明在某些癌細胞株中誘導細胞週期停滯與凋亡。⁶³ 然而,完整的作用機轉尚未完全闡明。
適應症與禁忌症
Romidepsin 於 2009 年獲 FDA 核准,用於治療至少接受過 1 種先前全身性療法的皮膚 T 細胞淋巴瘤病人。核准基於 2 項第二期臨床試驗。臨床效益在所有疾病分期皆有觀察到,整體反應率為 34%,包括晚期疾病(IIB 期或更高)病人的 38%。完全反應在 5.6% 至 7% 的病人中觀察到。⁶⁴˒⁶⁵
2011 年,FDA 擴大核准用於治療至少接受過 1 種先前療法的周邊 T 細胞淋巴瘤病人。藥物仿單上無特定禁忌症。
劑量療法
Romidepsin 以靜脈輸注給予。建議劑量為 14 mg/m²,於 28 天週期的第 1、8、15 天輸注 4 小時。週期重複進行,只要病人能耐受治療並持續展現臨床效益。雖然 romidepsin 尚無專門的肝功能或腎功能受損研究,但對於中度至重度肝功能受損及末期腎功能受損的病人,建議謹慎。
副作用與注意事項
不良反應:最常見的不良反應為噁心、無力/疲倦、血小板低下、嘔吐、腹瀉、發熱、便秘、嗜中性球低下與心電圖 T 波變化。導致 romidepsin 核准的試驗中觀察到的最常見嚴重不良反應為感染。發生於至少 2% 病人的額外嚴重不良反應包括發熱、嘔吐、蜂窩性組織炎 (cellulitis)、深層靜脈血栓 (deep vein thrombosis)、發熱性嗜中性球低下、腹痛、胸痛、肺栓塞、呼吸困難與脫水。在關鍵性的第二期試驗中,注意到一例第 IV 級「藥物性皮膚炎 (dermatitis medicamentosa)」及一例口腔念珠菌感染 (oral candidiasis)。⁶⁵ 在臨床研究中觀察到數種治療相關的心電圖型態變化。因此,產品仿單建議考慮對先天性長 QT 症候群病人、服用 QT 延長藥物的病人,以及有顯著心血管疾病病史的病人進行心血管監測。
根據作用機轉與臨床前動物研究,romidepsin 給予懷孕婦女時可能造成胎兒傷害。因此,建議勸告於懷孕期間避免使用 romidepsin。Romidepsin 於哺乳期可能不安全,應使哺乳婦女瞭解哺乳期間對嬰兒的潛在傷害。
藥物交互作用:不建議併用強效 CYP3A4 誘導劑(如 rifampin)。當併用強效 CYP3A4 抑制劑與 warfarin 及香豆素衍生物時,建議密切監測毒性。完整的藥物間交互作用與不良反應請參閱 Istodax 仿單。⁶³
BELINOSTAT (BELEODAQ)
藥理學與作用機轉
結構:Belinostat,(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]prop-2-enamide,分子式為 C15H14N2O4S,分子量為 318.35 g/mol。⁶⁶ Figure 194-18 顯示 belinostat 的結構。
代謝:Belinostat 主要由 UGT1A1 代謝,但亦經由 CYP3A4、CYP2A6 與 CYP2C9 進行肝臟代謝。消除半衰期為 1.1 小時。
吸收與分布:靜脈注射後,belinostat 的平均分布體積接近全身水分。
作用機轉:Belinostat 抑制 HDACs 的活性。在臨床前研究中,belinostat 已被證明在某些癌細胞中誘導細胞週期停滯與凋亡。與正常細胞相比,belinostat 對轉化細胞展現優先的細胞毒性。⁶⁶
適應症與禁忌症
Beleodaq 於 2014 年獲 FDA 核准,用於治療復發或難治性周邊 T 細胞淋巴瘤。核准基於一項針對 120 名復發或難治性疾病病人的關鍵性第二期試驗。整體反應率為 25.8%,包括 13 名病人 (10.8%) 的完全反應。⁶⁷
藥物仿單上無特定禁忌症。
劑量療法
Beleodaq 的建議劑量與排程為 1000 mg/m²,於 21 天週期的第 1 至 5 天靜脈給予。中度至重度肝功能受損的病人未納入關鍵性臨床試驗;因此,資料不足以建議肝功能受損病人的 Beleodaq 劑量。在 CrCl 大於 39 mL/min 的病人中,Beleodaq 的暴露量未改變。資料不足以建議 CrCl 小於 39 mL/min 病人的 Beleodaq 劑量。
副作用與注意事項
不良反應:belinostat 最常見的不良反應為噁心、疲倦、發熱、貧血、嘔吐、便秘、腹瀉、呼吸困難、皮疹與周邊水腫。最常見的嚴重不良反應包括肺炎、發熱、感染、貧血、肌酸酐升高、血小板低下與多重器官衰竭。Belinostat 為懷孕分級 D 類藥物。它可能造成胚胎胎兒致死,因為它靶向活躍分裂的細胞且具基因毒性。⁶⁶ 因此,建議勸告於懷孕期間避免使用 belinostat。Belinostat 於哺乳期的安全性未知。然而,由於它可能不安全,應使哺乳婦女瞭解哺乳期間對嬰兒的潛在傷害。
藥物交互作用:建議避免將 belinostat 與強效 UGT1A1 抑制劑併用。完整的藥物間交互作用與不良反應請參閱 Beleodaq 仿單。⁶⁶
PANOBINOSTAT (FARYDAK)
藥理學與作用機轉
結構:Panobinostat,(2E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide,分子式為 C21H23N3O2 • C3H6O3,分子量為 439.51 daltons(以乳酸鹽計),相當於游離鹼的 349.43。⁶⁸ Figure 194-19 顯示 panobinostat 的結構。
代謝:Panobinostat 經由氧化、還原、水解與葡萄糖醛酸化代謝。約 40% 的化合物由 CYP3A 代謝。CYP2D6 與 CYP2C19 有少量貢獻。葡萄糖醛酸化經由 UGT1A1、UGT1A3、UGT1A7、UGT1A8、UGT1A9 與 UGT2B4 進行。消除半衰期約為 37 小時。
吸收與分布:Panobinostat 的口服生體可用率約為 21%,血漿濃度尖峰於口服給藥後 2 小時內達到。在體外,panobinostat 約 90% 與血漿蛋白結合。
作用機轉:Panobinostat 是一種 HDAC 抑制劑。在體外,panobinostat 已被證明在某些轉化細胞中誘導細胞週期停滯與凋亡。與正常細胞相比,panobinostat 對轉化細胞展現優先的細胞毒性。
適應症與禁忌症
Farydak 於 2015 年獲 FDA 核准,用於與 bortezomib 及 dexamethasone 併用治療至少接受過 2 種先前療法的多發性骨髓瘤病人。Panobinostat 目前無任何 FDA 核准的皮膚科疾病適應症,雖然有一項正在進行的第 Ib/II 期試驗,探索以 panobinostat 與表觀遺傳修飾劑 decitabine 加上 temozolomide 治療難治性轉移性黑色素瘤 (NCT00925132)。藥物仿單上無特定禁忌症。
劑量療法
Panobinostat 的建議劑量為口服 20 mg,每隔一天一次,每週 3 劑(於第 1、3、5、8、10、12 天),於每個 21 天週期的第 1 與第 2 週服用,共 8 個週期。建議將 panobinostat 的起始劑量減至輕度肝功能受損病人 15 mg,中度肝功能受損病人 10 mg。重度肝功能受損病人不建議使用。在輕度或重度腎功能受損的病人中,Farydak 的暴露量未改變。它未在末期腎臟病或透析病人中進行研究。Farydak 可能需要根據毒性調整劑量與/或排程。適當的減量請參閱 Farydak 仿單。
副作用與注意事項
不良反應:臨床研究中最常見的不良反應(發生率至少 20%)為腹瀉、疲倦、噁心、周邊水腫、厭食、發熱與嘔吐。最常見的血液學不良反應(≥60% 發生率)包括血小板低下、淋巴球低下、白血球低下、嗜中性球低下與貧血。電解質異常,如低磷酸鹽血症 (hypophosphatemia)、低血鉀與低血鈉 (hyponatremia),發生於 40% 或更多的病人。Farydak 具有嚴重腹瀉以及嚴重和致命之心臟缺血事件、嚴重心律不整與心電圖變化的黑框警示。嚴重腹瀉發生於 25% 接受 Farydak 的病人。已有致命與嚴重之胃腸道與肺出血病例的報告。Farydak 若於懷孕期間給藥可能造成胎兒傷害。在臨床前研究中,panobinostat 在兔與大鼠中具致畸性。應建議病人於服用 Farydak 期間避免懷孕。亦建議婦女於最後一劑 Farydak 後至少 1 個月內使用高度有效的避孕措施。應建議男性於治療期間及最後一劑 Farydak 後 3 個月內使用保險套。Panobinostat 於哺乳期的安全性未知。然而,由於它可能不安全,應使哺乳婦女瞭解哺乳期間對嬰兒的潛在傷害。
藥物交互作用:建議避免將 Farydak 與強效 CYP3A4 誘導劑、CYP2D6 受質及抗心律不整/QT 延長藥物併用。併用強效 CYP3A4 抑制劑時應減少 Farydak 的劑量。完整的藥物間交互作用與不良反應請參閱 Farydak 仿單。⁶⁸
絲裂原活化蛋白激酶抑制劑 (MITOGEN-ACTIVATED PROTEIN KINASE INHIBITORS)
重點一覽 (AT-A-GLANCE)
■ 75% 至 85% 的黑色素瘤帶有絲裂原活化蛋白激酶 (mitogen-activated protein kinase, MAPK) 路徑的致癌突變,包括 BRAF (v-raf murine sarcoma viral oncogene homolog B)、RAS 與神經纖維瘤蛋白 1 (neurofibromin 1, NF1)。
■ BRAF 的活化突變,如 V600E 與 V600K,可觸發不受調控的細胞生長並促進黑色素細胞轉化。
■ Vemurafenib 與 dabrafenib 是某些突變型 BRAF(包括 V600E 與 V600K)的抑制劑。
■ Trametinib 與 cobimetinib 是絲裂原活化細胞外訊號調節激酶 (mitogen-activated extracellular signal-regulated kinase, MEK) 的抑制劑。
■ 在帶有 BRAF V600E 或 V600K 突變的無法切除或轉移性黑色素瘤病人中,合併 BRAF 與 MEK 抑制可帶來臨床效益,包括改善的整體存活。
背景 (BACKGROUND)
黑色素瘤生成 (melanomagenesis) 源自多個細胞迴路的失調,包括 PI3K 路徑、端粒酶啟動子 (telomerase promoter)、視網膜母細胞瘤路徑 (retinoblastoma pathway) 與 MAPK 路徑。黑色素瘤的標靶治療在 2002 年發現原癌基因 v-raf murine sarcoma viral oncogene homolog B (BRAF) 的活化突變驅動近半數黑色素瘤之後成為可能。⁶⁹ BRAF 是 MAPK 路徑中的一種絲胺酸-蘇胺酸激酶,通常由 RAS 活化,透過絲裂原活化細胞外訊號調節激酶 (MEK) 的磷酸化進行訊息轉導 (Fig. 194-20)。BRAF 的突變——通常為 V600E,偶爾為 V600K 與其他取代——導致激酶結構域的組成性活化,造成 RAS 非依賴性以及下游介導者(如 MEK 與細胞外訊號調節激酶 extracellular signal-regulated kinase, ERK)的過度活化。MAPK 路徑的異常活化可導致細胞週期失調、對凋亡的抵抗、免疫逃逸,以及增加的侵犯與轉移。BRAF 功能獲得性改變是黑色素瘤生成關鍵決定因素的發現,推動了在 MAPK 路徑多個節點介入之治療策略的開發。當證明突變型 BRAF 的小分子抑制劑 vemurafenib 與 dabrafenib 作為單一療法可延長無病存活與整體存活時,黑色素瘤的精準醫療成為可能。⁷⁰⁻⁷² 此外,以 trametinib 單一療法抑制 MEK 可改善 BRAF 突變黑色素瘤病人的 PFS。⁷³ 雖然初期前景看好,但顯然單藥治療抗藥性的獲得使晚期疾病無法達到持久反應。基於顯示雙重靶向 BRAF 與 MEK 協同降低 MAPK 輸出的臨床前資料,臨床試驗隨後驗證了 BRAF/MEK 合併抑制在 BRAF 突變黑色素瘤中的安全性與更優越的療效。⁷⁴⁻⁷⁷ 確實,許多單藥 BRAF 抑制所見的皮膚毒性,如 cSCC 與角化棘皮瘤,在 BRAF/MEK 雙重抑制下會減輕。然而,儘管減少了許多不良反應並延遲了治療抗藥性的出現,合併 BRAF 抑制/MEK 抑制並未完全規避抗藥性機制,絕大多數病人仍在治療中惡化。此外,20% 至 25% 的病人帶有目前無法藥物靶向之 MAPK 訊息蛋白的突變,如 RAS 與神經纖維瘤蛋白 1。因此,正在探索額外的標靶與方法,如 AKT 與 ERK,以及免疫檢查點阻斷 (immune checkpoint blockade)。
VEMURAFENIB (ZELBORAF)
藥理學與作用機轉
結構:Vemurafenib,propane-1-sulfonic acid {3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl}-amide,分子式為 C23H18ClF2N3O3S,分子量為 489.92 daltons。Figure 194-21 顯示 vemurafenib 的結構。
代謝:體外研究證明 vemurafenib 是 CYP3A4 的受質。口服給藥後,94% 的 vemurafenib 劑量於糞便中回收,約 1% 於尿液中發現。中位消除半衰期為 57 小時。⁷⁸
吸收與分布:vemurafenib 的生體可用率尚未確定;然而,多次給藥後,達到最高血漿濃度的中位時間約為 3 小時。在導致其核准的臨床試驗中,Zelboraf 的服用不論進食與否。
作用機轉:Vemurafenib 是某些突變型絲胺酸-蘇胺酸激酶 BRAF(包括 V600E)的小分子量抑制劑。此外,vemurafenib 抑制野生型 BRAF、CRAF、ARAF、SRMS、ACK1、MAP4K5 與 FGR。在帶有 V600E 突變 BRAF 的細胞中,vemurafenib 具有抗腫瘤效果。
適應症與禁忌症
FDA 於 2011 年核准 Zelboraf 作為單一療法,用於治療帶有 BRAFV600E 突變(經 FDA 核准檢測偵測)的無法切除或轉移性黑色素瘤。核准基於 2 項試驗,這些試驗證明與 dacarbazine 相比,反應率與存活率有所改善。⁷⁰˒⁷¹ vemurafenib 治療組與 dacarbazine 治療組的中位 PFS 分別為 5.3 個月與 1.6 個月,客觀反應率分別為 48% 與 5%。⁷¹
在單臂第二期試驗中,以 vemurafenib 治療的病人之中位整體存活為 15.9 個月。⁷⁰
當與 MEK 抑制劑 cobimetinib (Cotellic) 併用時,vemurafenib 展現改善的療效,達到 68% 的整體反應率與 9.9 個月的 PFS。⁷⁷
Zelboraf 亦適用於治療帶有 V600 突變的 Erdheim-Chester 病 (Erdheim-Chester disease) 病人。藥物仿單上無列出禁忌症。
劑量療法
vemurafenib 的建議劑量為口服 960 mg,與餐併服或不併服皆可,twice daily,間隔約 12 小時給予。尚無正式研究評估腎功能或肝功能受損對 Zelboraf 藥物動力學的影響。輕度與中度腎功能或肝功能受損的病人無建議劑量調整。重度腎功能或肝功能受損建議謹慎。
副作用與注意事項
不良反應:Zelboraf 最常見的非皮膚不良反應包括關節痛、疲倦與噁心。Zelboraf 與多種皮膚毒性相關,包括皮疹、脫髮、光敏感、搔癢與新發原發性惡性腫瘤。皮膚疹發生於高達 68% 服用 vemurafenib 的病人。⁷⁰˒⁷¹˒⁷⁹ 這些皮疹被描述為以毛囊為中心 (folliculocentric)、毒性紅斑樣 (toxic erythema–like) 或誇大的毛囊角化症樣 (exaggerated keratosis pilaris–like)。⁸⁰ 毒性紅斑樣疹被描述為具有類似典型斑丘疹型藥物過敏疹的型態,組織病理學特徵提示為發疹性藥物疹 (exanthematous drug eruption)。⁸⁰
從良性乳突瘤 (papillomas) 與疣狀角化 (verrucous keratoses) 到角化棘皮瘤與鱗狀細胞癌的鱗狀增生,在服用單藥 vemurafenib 的病人中常見。在第二期試驗中,29% 的病人報告有乳突瘤,26% 的病人描述有 cSCCs 與角化棘皮瘤。⁷⁰ 在一項較新的 vemurafenib 皮膚不良反應前瞻性研究中,疣狀乳突瘤發生於 79% 的病人,近半數病人發展出 4 至 20 個盛放性 (efflorescent) 與疣狀 (verruciform) 病灶。⁸¹ 一種以位於足底受壓點之過度角化斑塊為特徵的手足皮膚反應 (hand–foot skin reaction) 在 60% 的病人中被觀察到。⁸¹ 單藥治療所描述的額外皮膚毒性包括毛髮生長改變、過度角化性毛囊疹、皮膚乾燥、囊腫性病灶、顏面紅斑、唇炎 (cheilitis)、乳頭過度角化、史蒂芬斯-強生症候群、毒性表皮壞死溶解 (toxic epidermal necrolysis)、伴嗜酸性球增多及全身症狀之藥物疹症候群、痣盛放 (nevi efflorescence) 與放射性皮膚炎 (radiodermatitis)。⁸¹ 數種皮膚不良反應似乎透過 MAPK 訊息的弔詭性活化 (paradoxical activation) 而依賴 MEK 輸出,因為併用 MEK 抑制劑 cobimetinib 與較低的角化棘皮瘤、cSCCs 與脫髮發生率相關。⁷⁷
Zelboraf 的額外不良反應包括肝毒性、QT 延長、葡萄膜炎 (uveitis)、畏光 (photophobia) 與腎損傷,包括間質性腎炎 (interstitial nephritis) 與急性腎小管壞死 (acute tubular necrosis)。根據其作用機轉,Zelboraf 可能造成胎兒傷害,應使婦女瞭解對胎兒的潛在風險。建議育齡婦女於治療期間及最後一劑 Zelboraf 後 2 週內使用高度有效的避孕措施。雖然無關於 vemurafenib 存在於人類乳汁的資訊,但對哺乳嬰兒存在潛在傷害;因此,應建議婦女於治療期間及最後一劑 Zelboraf 後至少 2 週內避免哺乳。
藥物交互作用:建議避免併用強效 CYP3A4 抑制劑或誘導劑,以及治療窗狹窄的 CYP1A2 受質。完整的藥物間交互作用與不良反應請參閱 Zelboraf 仿單。⁷⁸
DABRAFENIB (TAFINLAR)
藥理學與作用機轉
結構:Dabrafenib mesylate,N-{3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt,分子式為 C23H20F3N5O2S2 CH4O3S,分子量為 615.68 daltons。⁸² Figure 194-22 顯示 dabrafenib mesylate 的結構。
代謝:Dabrafenib 主要由 CYP2C8 與 CYP3A4 代謝。經糞便排泄是 dabrafenib 的主要消除途徑 (71%),而尿液排泄佔其消除的 23%。口服給藥後,dabrafenib 的半衰期為 8 小時。
吸收與分布:Dabrafenib 展現 95% 的生體可用率,血漿濃度尖峰於口服給藥後 2 小時達到;99.7% 的 dabrafenib 與血漿蛋白結合,其分布體積為 70.3 L。
作用機轉:Dabrafenib 是某些突變型 BRAF(包括 V600E、V600K 與 V600D)的小分子量抑制劑。它是一種可逆性的 ATP 競爭性抑制劑,對 BRAFV600E 達到 50% 抑制所需的濃度比野生型 BRAF 低 5 倍。⁷² 此外,dabrafenib 抑制野生型 BRAF、CRAF、SIK1、NEK11 與 LIMK1。在帶有 V600 突變 BRAF 的黑色素瘤細胞中,dabrafenib 抑制細胞生長。
適應症與禁忌症
Tafinlar 於 2013 年獲 FDA 核准,用於治療帶有 BRAF V600E 突變(經 FDA 核准檢測偵測)的無法切除或轉移性黑色素瘤。核准基於一項針對 250 名病人的多中心、開放標籤、隨機試驗的資料,該試驗證明與 dacarbazine 相比,PFS 有所改善(5.1 個月 vs. 2.7 個月)。⁷² Tafinlar 亦於 2014 年獲加速核准、2015 年獲完全核准,用於與 MEK 抑制劑 trametinib (Mekinist) 併用治療帶有 BRAF V600E 或 V600K 突變的無法切除或轉移性黑色素瘤。核准是在該合併療法展現 64% 至 76% 的客觀反應率與 9.3 至 11.4 個月的 PFS 之後給予。⁷⁴⁻⁷⁶ 該世代病人的長期追蹤證明整體存活為 25 個月,3 年整體存活為 38%。⁸³
Tafinlar 亦適用於與 Mekinist 併用作為:1) 完全切除後侵犯淋巴結之 V600 突變黑色素瘤病人的輔助治療;2) 轉移性 V600 突變 NSCLC 的治療;以及 3) 無滿意局部區域治療選項之轉移性或局部晚期 V600 突變未分化甲狀腺癌 (anaplastic thyroid cancer)。藥物仿單上無特定禁忌症。
劑量療法
Tafinlar 的建議劑量為口服 150 mg twice daily,於餐前至少 1 小時或餐後至少 2 小時服用。在以 Tafinlar 單藥治療前,必須進行 FDA 核准檢測以確認 BRAF V600E 突變的存在;或在與 trametinib 合併治療前確認 BRAF V600E 或 V600K 突變的存在。尚無正式研究評估腎功能或肝功能受損對 Tafinlar 藥物動力學的影響。輕度肝功能或輕度或中度腎功能受損的病人無建議特定的劑量調整。中度至重度肝功能或重度腎功能受損的適當劑量尚未確定。Tafinlar 可能需要根據毒性調整劑量。適當的減量請參閱 Tafinlar 產品仿單。
副作用與注意事項
不良反應:最常見的非皮膚不良反應包括頭痛、發熱與關節痛。皮膚毒性與 vemurafenib 所見類似,包括疣狀角化性鱗狀增生病灶、乳突瘤、脫髮、皮疹、Grover 病 (Grover disease)、足底過度角化、cSCC、角化棘皮瘤、黑色素瘤發生率增加、脂漏性皮膚炎樣疹 (seborrheic dermatitis–like eruption) 與掌蹠紅腫症候群 (palmar-plantar erythrodysesthesia syndrome)。⁸²˒⁸⁴ 在 Tafinlar 治療期間已報告新發原發性惡性腫瘤(皮膚與非皮膚)。額外的毒性包括出血、心肌病變、葡萄膜炎(包括虹膜炎 iritis 與虹膜睫狀體炎 iridocyclitis)、高血糖,以及葡萄糖-6-磷酸去氫酶缺乏症 (glucose-6-phosphate dehydrogenase deficiency) 病人的溶血性貧血。根據動物研究資料與其作用機轉,Tafinlar 可能造成胎兒傷害,應使婦女瞭解對胎兒的潛在風險。建議育齡婦女於治療期間及最後一劑 Tafinlar 後 2 週內使用高度有效的避孕措施。雖然無關於 dabrafenib 存在於人類乳汁的資訊,但對哺乳嬰兒存在嚴重不良反應的可能性;因此,應建議婦女於治療期間及最後一劑 Tafinlar 後至少 2 週內避免哺乳。
藥物交互作用:建議避免併用強效 CYP3A4 或 CYP2C8 的抑制劑或誘導劑。併用對 CYP3A4、CYP2B6、CYP2C8、CYP2C9 或 CYP2C19 敏感的受質可能導致這些藥物療效降低。完整的藥物間交互作用與不良反應請參閱 Tafinlar 仿單。⁸²
TRAMETINIB (MEKINIST)
藥理學與作用機轉
結構:Trametinib dimethyl sulfoxide,或 acetamide, N-[3-[3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-3,4,6,7-tetrahydro-6,8-dimethyl-2,4,7-trioxopyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl]-, compound with 1,1′-sulfinylbis[methane] (1:1),分子式為 C26H23FIN5O4 • C2H6OS,分子質量為 693.53 daltons。Figure 194-23 顯示 trametinib dimethyl sulfoxide 的結構。
代謝:Trametinib 主要由非 CYP 介導的去乙醯化作用單獨進行,或與單氧化或葡萄糖醛酸化生物轉化途徑合併進行。估計的消除半衰期為 3.9 至 4.8 小時。超過 80% 經糞便排出,不到 20% 經尿液消除。
吸收與分布:Trametinib 的生體可用率為 72%,分布體積為 214 L。
作用機轉:Trametinib 是 MEK1 與 MEK2 活化及激酶活性的可逆性小分子抑制劑。⁸⁵ Trametinib 抑制 BRAF V600 突變陽性黑色素瘤的體外與體內細胞生長。
適應症與禁忌症
Mekinist 適用於作為單一藥物或與 dabrafenib 併用,治療帶有 BRAF V600E 突變或 BRAF V600K 突變(經 FDA 核准檢測判定)的無法切除或轉移性黑色素瘤。作為單一療法的核准於 2013 年給予,基於第三期 METRIC(MEK Versus Dacarbazine [DTIC] or Paclitaxel [Taxol] in Metastatic Melanoma)研究,該研究證明接受 trametinib 2 mg once daily 的病人(4.8 個月)與接受 dacarbazine 或 paclitaxel 的病人(1.5 個月)相比,PFS 有所改善。⁷³
如上所述,Mekinist 與 Tafinlar 併用時具協同作用,與單藥治療相比,產生更優越的客觀反應率(64% 至 76%)與 PFS(9.3 至 11.4 個月)。⁷⁴⁻⁷⁶
Mekinist 亦適用於與 Tafinlar 併用作為:1) 完全切除後侵犯淋巴結之 V600 突變黑色素瘤病人的輔助治療;2) 轉移性 V600 突變 NSCLC 的治療;以及 3) 無滿意局部區域治療選項之轉移性或局部晚期 V600 突變未分化甲狀腺癌。產品仿單上無特定禁忌症。
劑量療法
trametinib 的建議劑量為口服 2 mg once daily,於餐前至少 1 小時或餐後至少 2 小時服用。尚無專門研究評估腎功能或肝功能受損對 Mekinist 藥物動力學的影響。輕度肝功能或輕度或中度腎功能受損的病人無建議特定的劑量調整。中度至重度肝功能或重度腎功能受損的 Mekinist 適當劑量尚未確定。Mekinist 可能需要根據毒性調整劑量。適當的減量請參閱 Mekinist 仿單。
副作用與注意事項
不良反應:最常見的不良反應包括皮疹、腹瀉與淋巴水腫 (lymphedema)。當與 Tafinlar 併用時,最常見的毒性包括發熱、噁心、皮疹、腹瀉、嘔吐、寒顫、高血壓與周邊水腫。最常見的皮膚毒性是與 EGFR 抑制劑所見類似的丘疹膿疱性疹,發生於 40% 至 93% 的病人。⁸⁶ 其他皮膚變化包括搔癢、皮膚乾燥、脫髮、甲溝炎,以及被描述為斑丘疹樣與蕁麻疹至標靶樣 (targetoid) 伴中央暗沉的疹子。⁸⁶ 其他不良反應包括靜脈血栓栓塞 (venous thromboembolism)、心肌病變、視網膜靜脈阻塞 (retinal vein occlusion)、視網膜色素上皮剝離 (retinal pigment epithelial detachment)、間質性肺病與高血糖。根據動物研究資料與其作用機轉,Mekinist 可能造成胎兒傷害,應使婦女瞭解對胎兒的潛在風險。建議育齡婦女於治療期間及最後一劑 Mekinist 後 4 個月內使用高度有效的避孕措施。動物研究中觀察到濾泡囊腫增加與黃體減少;因此,應告知婦女 Mekinist 可能損害生育力。雖然無關於 trametinib 存在於人類乳汁的資訊,但對哺乳嬰兒存在嚴重不良反應的可能性;因此,應建議婦女於治療期間及最後一劑 Mekinist 後至少 4 個月內避免哺乳。
藥物交互作用:Trametinib 不是 CYP 酶的受質,且在臨床相關的全身濃度 0.04 µM 下,它不是 CYP1A2、CYP2A6、CYP2B6、CYP2C9、CYP2C19、CYP2D6 或 CYP3A4 的抑制劑。完整的藥物間交互作用與不良反應請參閱 Mekinist 仿單。⁸²
COBIMETINIB (COTELLIC)
藥理學與作用機轉
結構:Cobimetinib fumarate,(S)-[3,4-difluoro-2-(2-fluoro-4-iodophenylamino)phenyl][3-hydroxy-3-(piperidin-2-yl)azetidin-1-yl]methanone hemifumarate,分子式為 C46H46F6I2N6O8,分子質量為 1178.71 daltons。Figure 194-24 顯示 cobimetinib fumarate 的結構。
代謝:Cobimetinib 主要由 CYP3A 經氧化及由 UGT2B7 經葡萄糖醛酸化代謝。口服給藥後的消除半衰期為 44 小時。排泄主要經由糞便途徑 (76%)。
吸收與分布:cobimetinib 的生體可用率為 46%,其分布體積為 806 L。
作用機轉:Cobimetinib fumarate 是 MEK1 與 MEK2 的可逆性抑制劑。該化合物抑制移植入小鼠之 BRAF V600E 突變的鼠類細胞。⁸⁷
適應症與禁忌症
Cotellic 適用於與 vemurafenib 併用,治療帶有 BRAF V600E 突變或 BRAF V600K 突變的無法切除或轉移性黑色素瘤。核准基於 coBRIM(Cobimetinib Combined with Vemurafenib in Advanced BRAF V600–mutant Melanoma)試驗的結果,該試驗顯示,在先前未治療、無法切除或轉移性 BRAF V600 突變陽性黑色素瘤病人中,vemurafenib 與 cobimetinib 的合併療法與單藥 vemurafenib 相比,產生改善的 PFS 與反應率(PFS:9.9 個月 vs. 6.2 個月;客觀反應率 68% vs. 45%)。⁷⁷
產品仿單上無特定禁忌症。
劑量療法
Cotellic 的建議劑量為口服 60 mg once daily,與食物併服或不併服皆可,於 28 天週期的前 21 天服用。尚無專門研究評估腎功能或肝功能受損對 Cotellic 藥物動力學的影響。輕度肝功能或輕度或中度腎功能受損的病人無建議特定的劑量調整。中度至重度肝功能或重度腎功能受損的 Cotellic 適當劑量尚未確定。Cotellic 可能需要根據毒性調整劑量。適當的減量請參閱 Cotellic 仿單。⁸⁷
副作用與注意事項
不良反應:Cotellic 最常見的不良反應為腹瀉、光敏感反應、噁心、發熱與嘔吐。最常見的第 4 級事件 (4%) 為肌酸激酶升高,這是 MEK 阻斷已知的類效應 (class effect)。⁷⁷ 其他嚴重的實驗室異常包括 γ-麩胺醯轉移酶 (γ-glutamyl transferase) 升高、低磷酸鹽血症、丙胺酸轉胺酶 (alanine aminotransferase) 與天門冬胺酸轉胺酶升高、淋巴球低下、鹼性磷酸酶 (alkaline phosphatase) 升高與低血鈉。雖然合併療法可見鱗狀增生事件,但與單藥 vemurafenib 相比,cobimetinib 與 vemurafenib 的雙重療法 cSCC 與角化棘皮瘤似乎較不常見。⁷⁷ 接受雙重療法的病人有 16% 出現第 3 至 4 級皮疹,相比之下接受單藥 vemurafenib 者為 17%。其他非皮膚不良反應包括心肌病變、出血、非皮膚惡性腫瘤、視網膜病變 (retinopathy) 與橫紋肌溶解症。根據動物研究資料與其作用機轉,Cotellic 可能造成胎兒傷害,應使婦女瞭解對胎兒的潛在風險。建議育齡婦女於治療期間及最後一劑 Cotellic 後 2 週內使用高度有效的避孕措施。應告知病人 Cotellic 可能損害男女雙方的生育力。雖然無關於 cobimetinib 存在於人類乳汁的資訊,但對哺乳嬰兒存在嚴重不良反應的可能性;因此,應建議婦女於治療期間及最後一劑 Cotellic 後至少 2 週內避免哺乳。
藥物交互作用:服用 Cotellic 時應避免中度或強效 CYP3A 的抑制劑或誘導劑。完整的藥物間交互作用與不良反應請參閱 Cotellic 仿單。
黑色素瘤的未來方向:AKT 與 ERK 抑制 (FUTURE DIRECTIONS IN MELANOMA: AKT AND ERK INHIBITION)
AKT 抑制劑 (AKT INHIBITORS)
PI3K–AKT–mTOR(mammalian target of rapamycin,雷帕黴素哺乳動物標靶)訊息是黑色素瘤發展與惡化中一條重要的致癌路徑。此路徑的過度活化與受體酪胺酸激酶的活化、關鍵訊息基因(如 PIK3CA)的體細胞突變,或腫瘤抑制基因 PTEN 的失活有關。⁸⁸
以 AKT 抑制劑進行的臨床前研究已證明 AKT 抑制劑在黑色素瘤治療中的用處。結膜黑色素瘤 (conjunctival melanoma) 是一種罕見但致命的黑色素瘤變異型,有效治療選項有限。臨床前體外研究證明,AKT 抑制劑 MK2206 抑制結膜黑色素瘤細胞株的生長,此外,當與 MEK 抑制劑 MEK162 併用時觀察到協同效應。⁸⁹ 這些初步發現支持了近期啟動之第二期臨床試驗的理論依據,該試驗研究 MEK 抑制劑 trametinib 加或不加 AKT 抑制劑 GSK2141795 治療第 IV 期葡萄膜黑色素瘤 (uveal melanoma) 病人的療效 (NCT01979523)。此外,在一項研究口服 AKT 抑制劑 MK2206 與化療併用治療實體腫瘤的第一期試驗中,2 名第 IV 期 BRAF 野生型 (WT) 黑色素瘤病人耐受 MK2206 與 carboplatin 及 paclitaxel 併用,並經歷無病惡化。⁹⁰
更近期,另一項第一期研究揭示,AKT 抑制劑 ipatasertib (GDC-0068) 能夠在實體腫瘤病人中安全且穩健地靶向 AKT。⁹¹ 這些初步研究正為更大型的隨機對照臨床試驗鋪路,這些試驗可能使 AKT 抑制劑加入晚期黑色素瘤的治療武器庫。
細胞外訊號調節激酶抑制劑 (EXTRACELLULAR SIGNAL-REGULATED KINASE INHIBITORS)
MAPK (RAS-RAF-MEK-ERK) 路徑在超過 90% 的黑色素瘤中過度活化。⁹² RAF 與 MEK 抑制劑是首批臨床可用、針對晚期黑色素瘤病人靶向 MAPK 路徑的療法。然而,對 BRAF 與 MEK 抑制劑獲得性抗藥性的出現,被證明是可持續預後的主要限制。⁹³ 一種已充分闡明的抗藥性機制是下游 MAPK 路徑標靶 ERK 的再活化;因此,研究者正聚焦於 ERK 抑制劑的開發。⁹³ 臨床前研究顯示,與 MEK 抑制相比,ERK 抑制在多種 BRAF 抑制劑抗藥性黑色素瘤細胞株中更有效地抑制 MAPK 活性與腫瘤生長。⁹⁴ 此外,體外資料證明 MEK 抑制劑抗藥性黑色素瘤細胞株對 ERK 抑制敏感,且 MEK 與 ERK 抑制劑的合併治療具協同作用並克服對 MEK 抑制的獲得性抗藥性。⁹⁵ 雖然臨床前資料前景看好,但 ERK 抑制劑能夠靶向廣泛的激酶,因此潛在的狹窄治療指數 (narrow therapeutic index) 是一大隱憂。目前有第一期臨床試驗正在進行,探索 ERK 抑制劑 BVD-523 與 LTT462 在晚期實體惡性腫瘤病人中的安全性與耐受性 (NCT01781429, NCT02711345)。
圖表 (FIGURES AND TABLES)
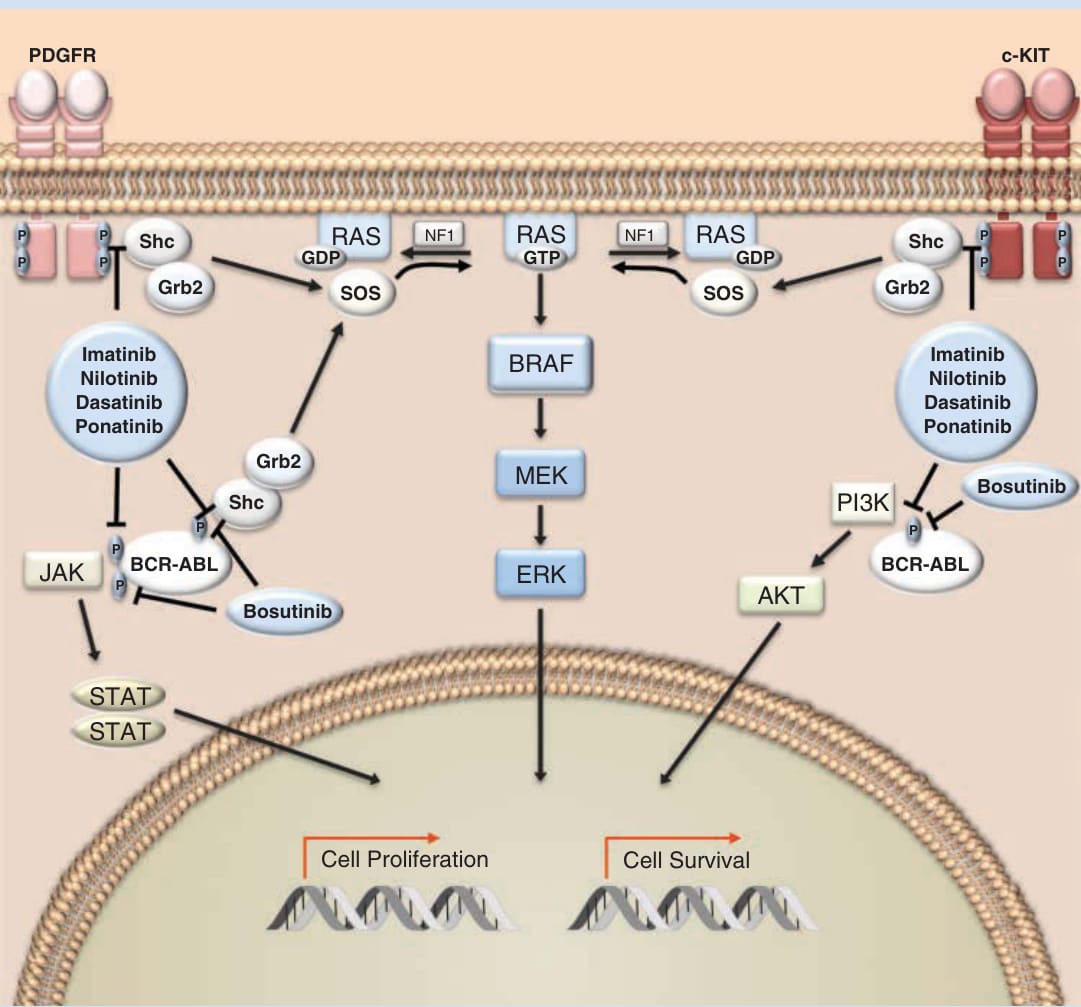
圖 194-1:KIT/BCR-ABL/PDGFR 的抑制劑。Imatinib、nilotinib、dasatinib 與 ponatinib 是多酪胺酸激酶抑制劑,調節與 BCR-ABL(breakpoint cluster region–Abelson murine leukemia virus,斷裂點群集區–Abelson 鼠白血病病毒)、c-KIT 與 PDGFR(platelet-derived growth factor receptor,血小板衍生生長因子受體)相關之酪胺酸殘基的自磷酸化。c-KIT 與 PDGFR 是受體酪胺酸激酶 (receptor tyrosine kinases),而 BCR-ABL 是非受體酪胺酸激酶 (nonreceptor tyrosine kinase)。Bosutinib 是 BCR-ABL 的抑制劑,但不抑制 c-KIT 或 PDGFR。這些酪胺酸激酶下游的訊息轉導路徑,如 MAPK(mitogen-activated protein kinase,絲裂原活化蛋白激酶)級聯、Janus kinase (JAK)/signal transducer and activator of transcription (STAT) 與 phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K),調控涉及細胞生長、增殖與細胞存活之基因的表現。
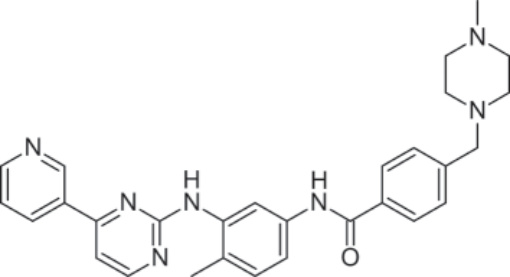
圖 194-2:Imatinib 的結構。
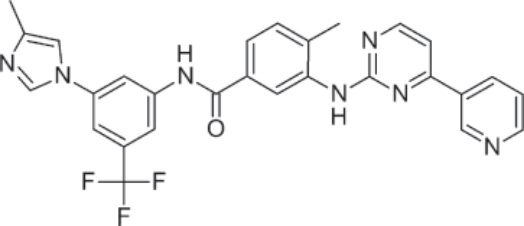
圖 194-3:Nilotinib 的結構。
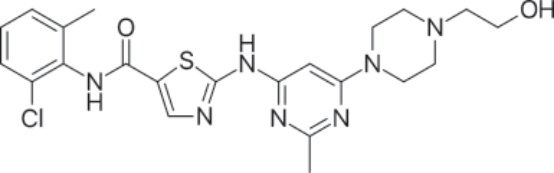
圖 194-4:Dasatinib 的結構。
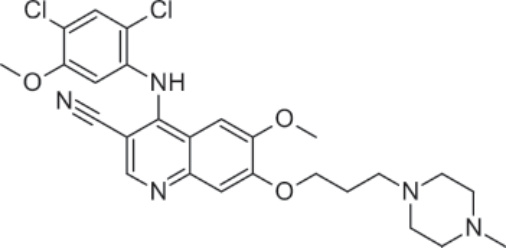
圖 194-5:Bosutinib 的結構。
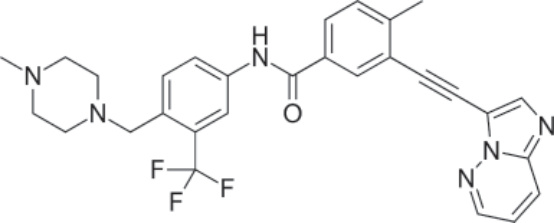
圖 194-6:Ponatinib 的結構。
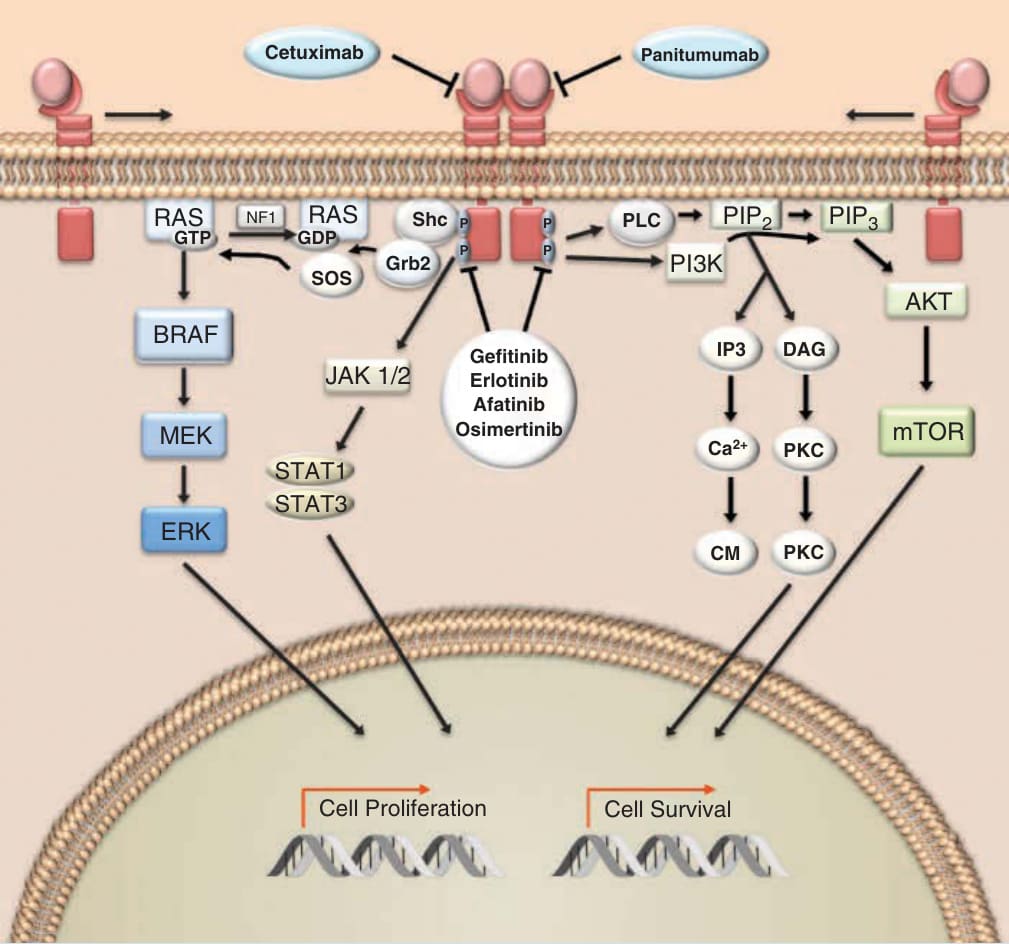
圖 194-7:表皮生長因子受體 (epidermal growth factor receptor, EGFR) 路徑。人類表皮生長受體是一種細胞表面蛋白,由細胞外配體結合結構域、跨膜結構域與細胞內酪胺酸激酶結構域組成。表皮生長因子與其他配體與細胞外結構域的結合導致二聚化,其中受體可與另一 EGFR 蛋白結合(如上圖所示)或與 ErbB 家族的另一單體形成異二聚體。二聚化觸發細胞內酪胺酸激酶將數個酪胺酸殘基自磷酸化。受體 C 端結構域上磷酸化酪胺酸殘基隨後被各種接頭蛋白辨識,啟動下游訊息轉導。EGFR 的活化可觸發涉及細胞增殖、細胞存活與轉移的網路,如 MAPK(mitogen-activated protein kinase,絲裂原活化蛋白激酶)、Janus kinase (JAK)/signal transducer and activator of transcription (STAT)、phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) 與 phospholipase C (PLC) 路徑。
圖 194-8:Gefitinib 的結構。
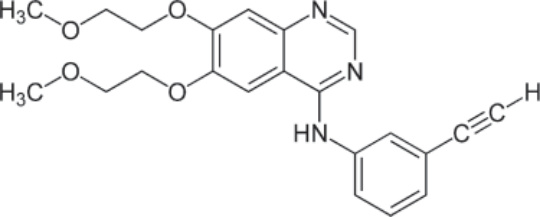
圖 194-9:Erlotinib 的結構。
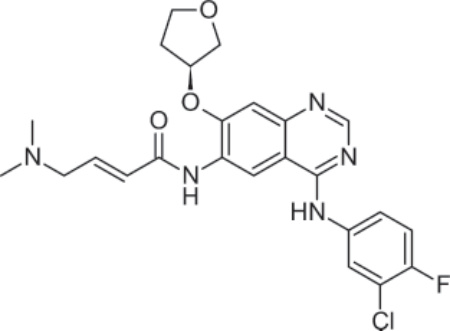
圖 194-10:Afatinib 的結構。
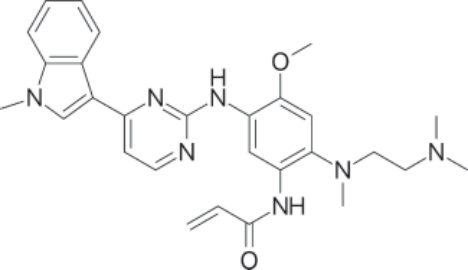
圖 194-11:Osimertinib 的結構。
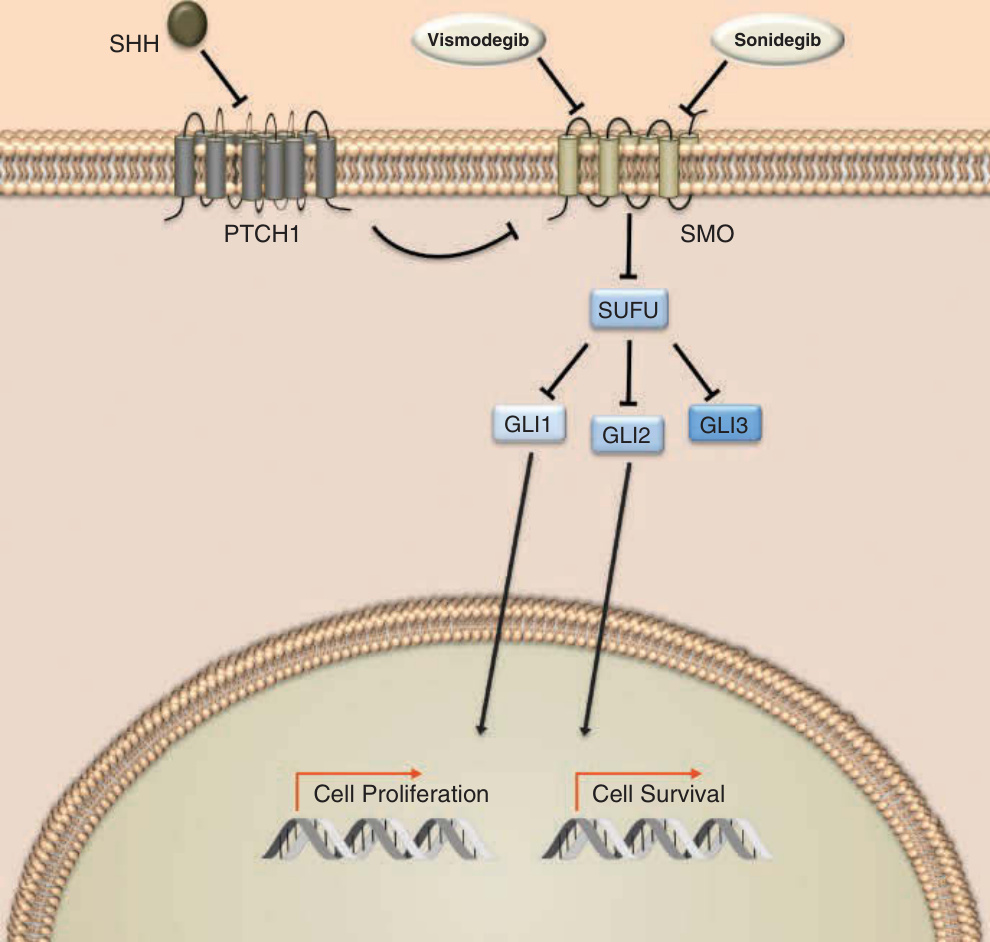
圖 194-12:Hedgehog 訊息傳遞。Hedgehog (HH) 訊息由 3 種細胞外 HH 配體之一——sonic hedgehog (SHH)、Indian hedgehog (IHH) 與 desert hedgehog (DHH)——的結合所啟動。這些配體與 12 次跨膜蛋白受體 Patched 1 (PTCH1) 與 Patched 2 (PTCH2) 結合。當未結合時,Patched 受體相互作用並抑制 7 次跨膜轉導蛋白 Smoothened (SMO)。配體與 Patched 結合後,SMO 的抑制被解除,透過一系列相互作用的蛋白(包括 suppressor of fused, SUFU)進行轉導,導致 glioma-associated oncogene homolog (GLI) 轉錄因子 GLI1、GLI2 與 GLI3 的轉位。GLI1 專一作為轉錄活化因子,而 GLI2 與 GLI3 的作用則取決於訊息傳遞情境。在缺乏 HH 配體時,GLI3 作為轉導級聯的主要抑制因子。核轉位後,GLI 轉錄因子調節眾多細胞程式的表現,包括涉及細胞生長、增殖、上皮-間質轉化 (epithelial–mesenchymal transition)、血管新生、抑制凋亡與幹細胞維持者。小分子 vismodegib 與 sonidegib 結合 SMO 並損害 HH 路徑的訊息傳遞。
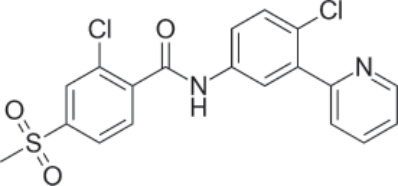
圖 194-13:Vismodegib 的結構。
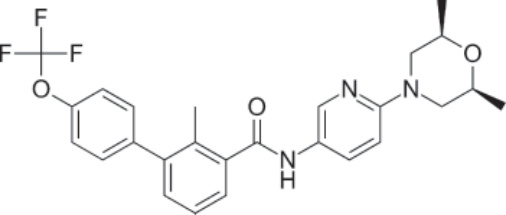
圖 194-14:Sonidegib 的結構。
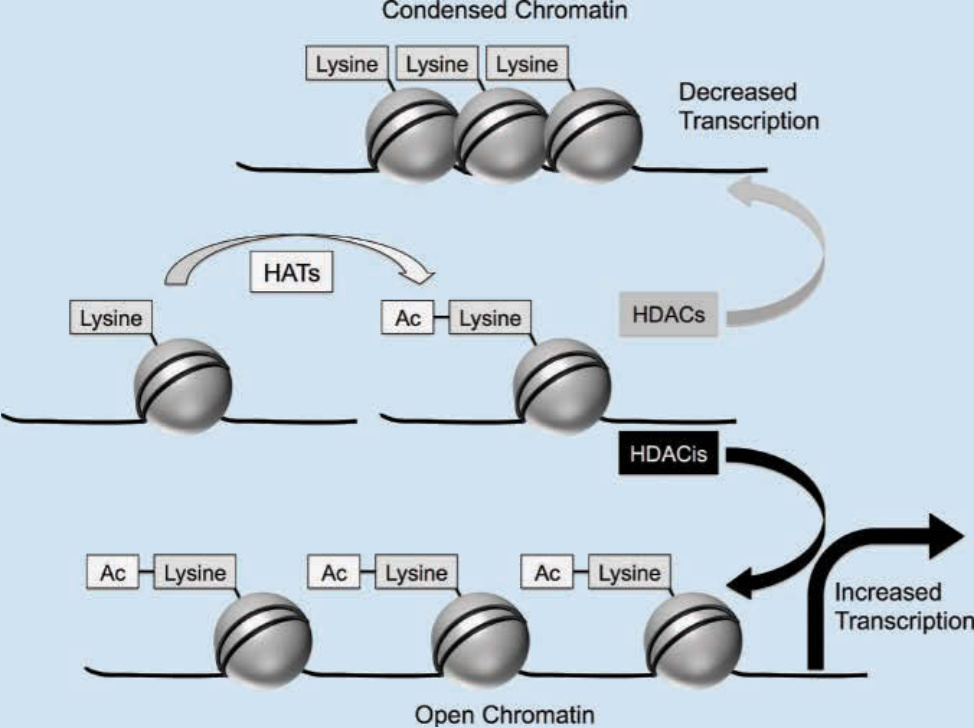
圖 194-15:組蛋白去乙醯酶抑制劑的機制。在其濃縮形式中,帶負電的 DNA(黑線)緊密纏繞在帶正電的組蛋白蛋白(灰色球體)周圍,限制了轉錄因子接近 DNA 啟動子區域的能力。組蛋白乙醯轉移酶 (histone acetyltransferases, HATs) 透過將乙醯基添加至組蛋白尾部的離胺酸殘基來中和組蛋白上的正電荷。這些殘基的乙醯化導致染色質結構更為鬆弛,並允許轉錄因子更大程度地接近基因啟動子,促進基因表現。相反地,組蛋白去乙醯酶 (histone deacetylases, HDACs) 的活性促進一種乙醯化不足的狀態,透過損害核小體的可及性而促成轉錄沉默(上圖)。抑制 HDACs 的藥物 (HDACis) 被認為可恢復正常乙醯化,導致調控分化、細胞週期停滯與凋亡之基因的轉錄增加(下圖)。
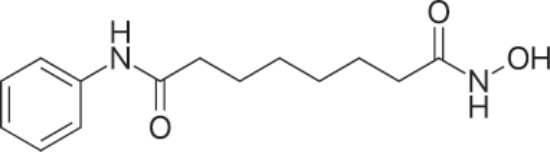
圖 194-16:Vorinostat 的結構。
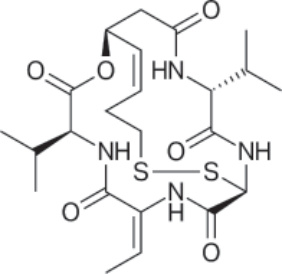
圖 194-17:Romidepsin 的結構。
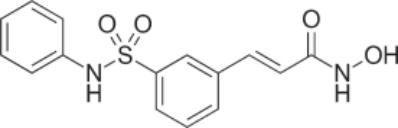
圖 194-18:Belinostat 的結構。
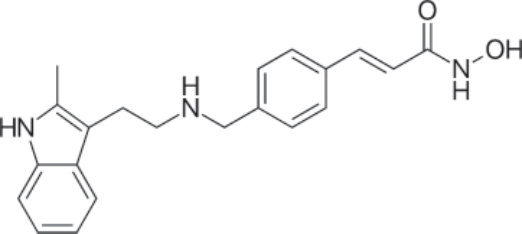
圖 194-19:Panobinostat 的結構。
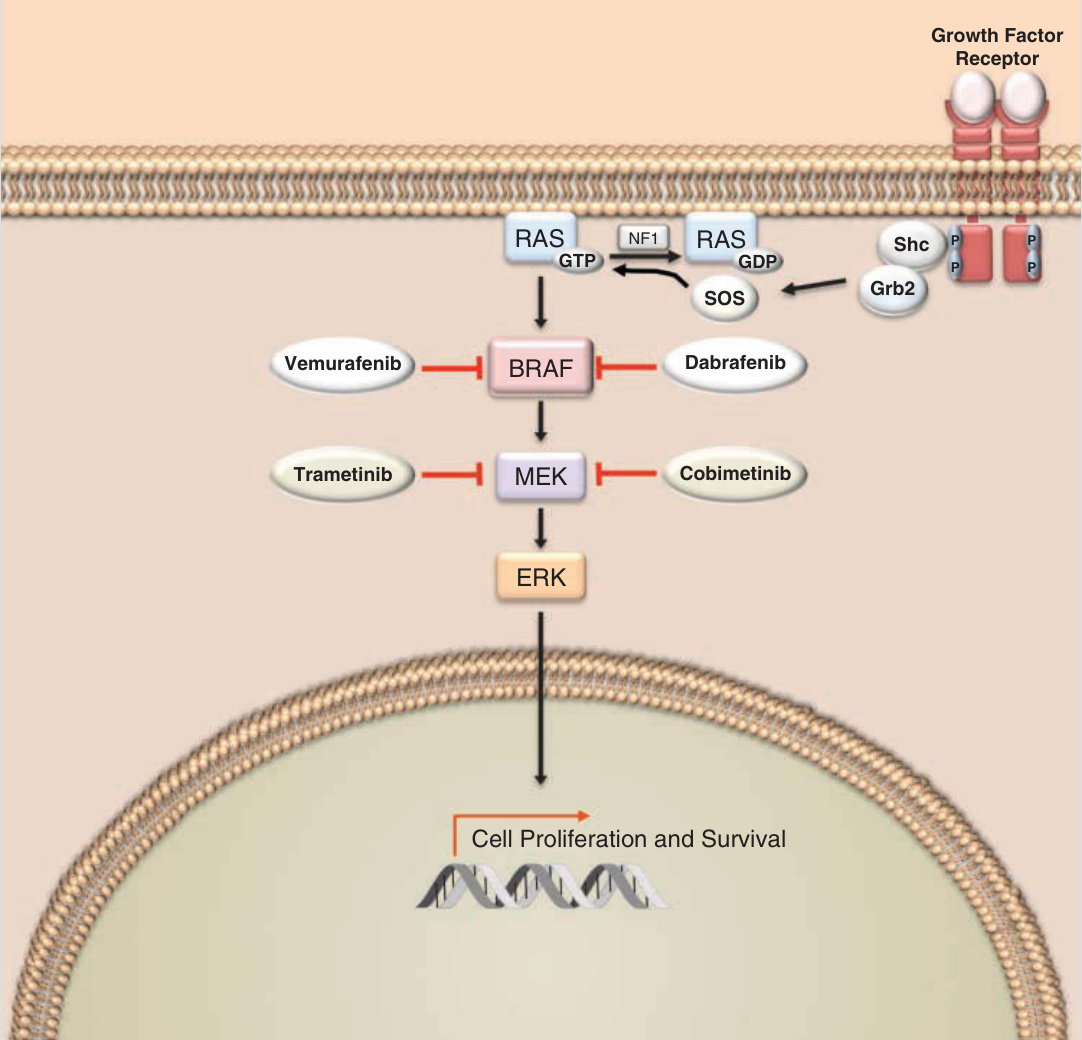
圖 194-20:絲裂原活化蛋白激酶 (mitogen-activated protein kinase, MAPK) 路徑。受體酪胺酸激酶(如 c-KIT)的活化啟動透過 MAPK 路徑的訊息傳遞。鳥嘌呤核苷酸交換因子 (guanine nucleotide exchange factors, GEFs)(如 son of sevenless, SOS)的作用加速 RAS 將鳥苷二磷酸 (guanosine diphosphate, GDP) 交換為鳥苷三磷酸 (guanosine triphosphate, GTP)。活化的 RAS 觸發 MAPK 級聯(包括 BRAF、MEK 與 ERK),導致細胞增殖與存活。RAS 的活性由鳥苷三磷酸酶活化蛋白(如神經纖維瘤蛋白 1, NF1)減弱。小分子 vemurafenib 與 dabrafenib 抑制 BRAF 的活性,而 trametinib 與 cobimetinib 抑制 MEK。
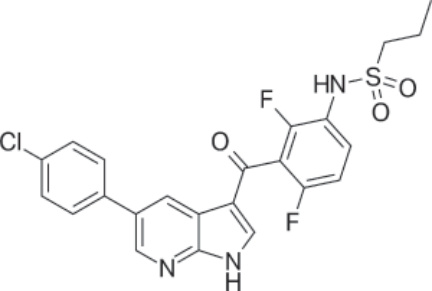
圖 194-21:Vemurafenib 的結構。
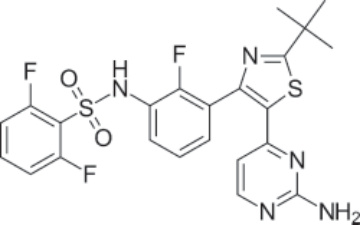
圖 194-22:Dabrafenib 的結構。
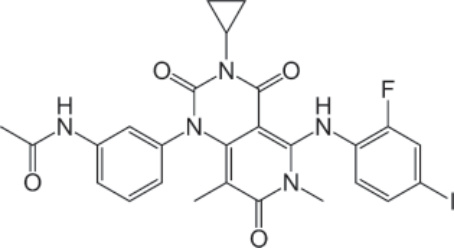
圖 194-23:Trametinib 的結構。
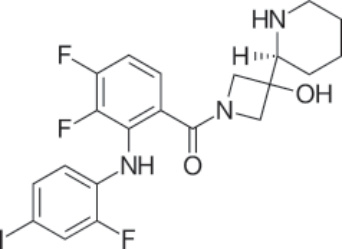
圖 194-24:Cobimetinib 的結構。
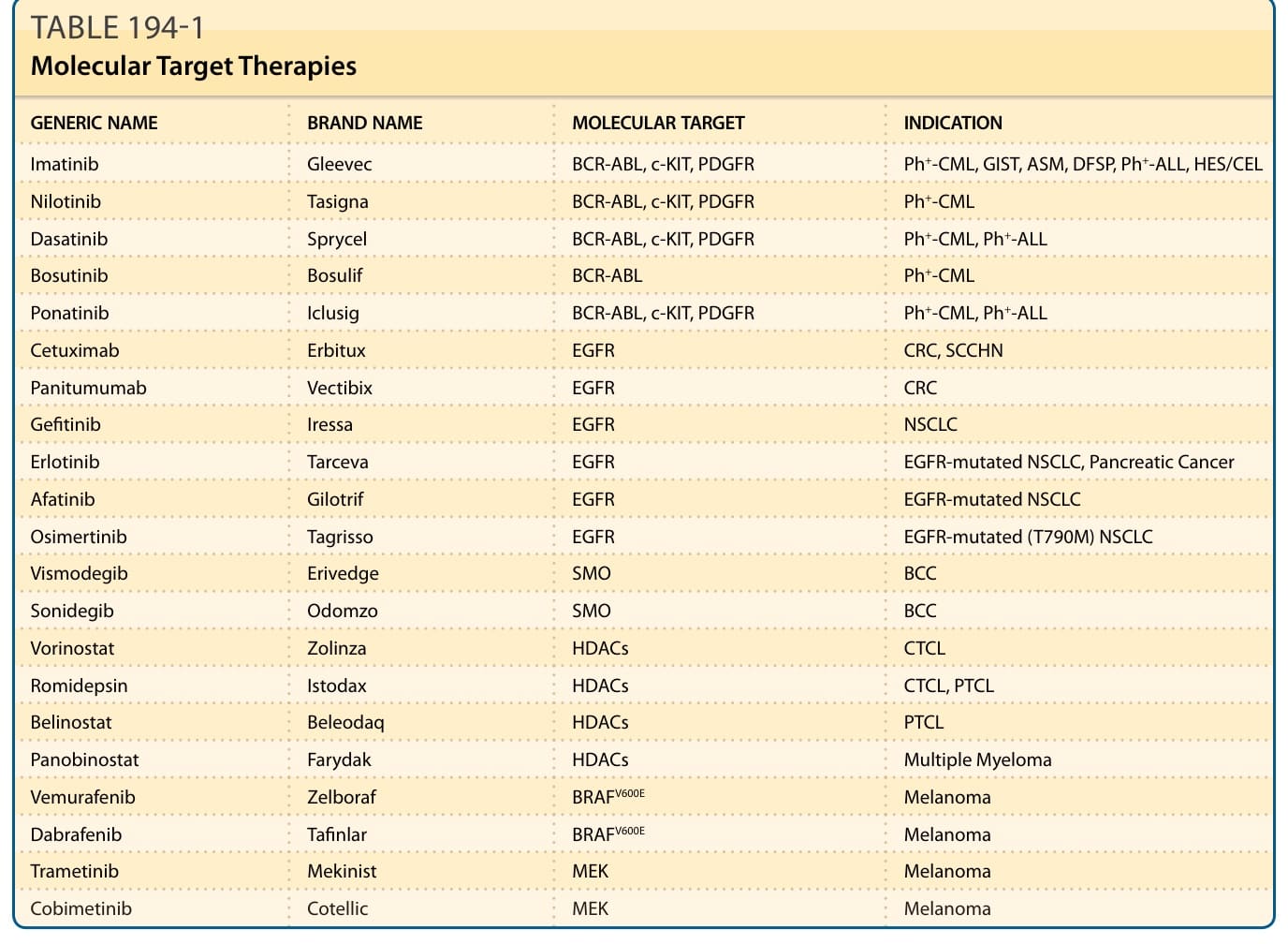
表 194-1:分子標靶治療 (Molecular Target Therapies)。(內容已於上文重排為 Markdown 表格呈現)
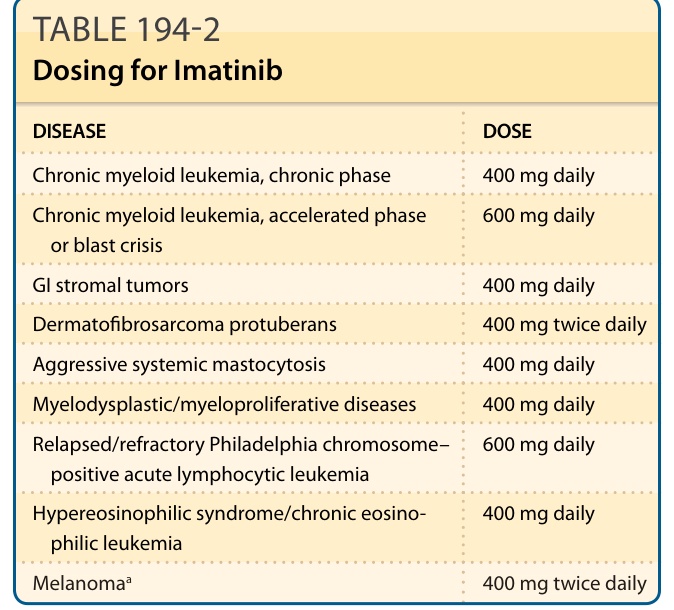
表 194-2:Imatinib 的劑量 (Dosing for Imatinib)。(內容已於上文重排為 Markdown 表格呈現)