Molecular Targeted Therapies
28
The treatment strategy for human disease has evolved dramatically since 2000 as a result of advances in molecular genetics, cell biology, and pharmacology. Increased understanding of molecular pathophysiology has allowed for the development of therapeutics that interact with specific molecular targets associated with disease. Agents that are the product of rational drug design, in which compounds are deliberately designed to interact with a biologic target, are often referred to as “targeted therapy” and form one of the cornerstones of “precision medicine.” The potential of targeted therapy is perhaps best exemplified with oncologic disease, but the promise of rational drug design is beginning to be seen across all fields of medicine. Discussing all targeted therapies in medicine is beyond the scope of this chapter. Consequently, we focus on those molecular targeted therapies that have the most overlap with dermatology in either their indication or the adverse effects caused by their use (Table 194-1). Thus we review targeted therapies designed to interact with the tyrosine kinases BCR- ABL, c-KIT, PDGFR and EGFR; smoothened; histone deacetylases; and proteins of the MAP kinase pathway. We finish with a section on the future directions in melanoma targeting AKT and ERK proteins. Of note, marketing indications, contra-indications and warnings are dynamic and constantly evolving. Thus, those covered in this chapter are current as of the writing. Please refer to the FDA product label for the most up-to-date information.
KIT, BCR-ABL, AND PDGFR INHIBITORS
AT-A-GLANCE
■ The small-molecule tyrosine kinase inhibitors imatinib, nilotinib, dasatinib, bosutinib, and ponatinib are indicated for Philadelphia chromosome–positive leukemia.
■ Imatinib also has indications for dermatofibrosarcoma protuberans, a soft-tissue sarcoma, driven in part by alterations in plateletderived growth factor receptor signaling.
■ Cutaneous adverse effects of these multityrosine kinase inhibitors include edema, morbilliform eruptions, bullous eruptions, dyspigmentation, keratosis-pilaris–like eruptions and neutrophilic dermatoses.
BACKGROUND
BACKGROUND
Tyrosine kinases are key components of numerous cellular pathways involved in cell growth, proliferation, migration, angiogenesis, differentiation, and survival. Drug development focusing on tyrosine kinases began after the initial discovery in 1980 that the oncogene associated with Abelson murine leukemia virus, ABL1, was a tyrosine kinase.1 This finding led to the understanding that a fusion protein, which resulted from a translocation of the Abl1 gene on chromosome 9 to a part of the breakpoint cluster region (BCR) gene on chromosome 22, was the driving event in the majority of chronic myeloid leukemias (CMLs).2 The protein product of this fusion oncogene, known as the Philadelphia chromosome, was one of the first targets for rational drug design. Cells that express the Philadelphia chromosome are transformed as a result of constitutive activation of the BCR-ABL kinase, which mediates oncogenesis through a variety of transduction pathways including the mitogen-activated protein kinase (MAPK), Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and phosphatidylinositol- 4,5-bisphosphate 3-kinase (PI3K) (Fig. 194-1). One of the earliest proof of principles of “targeted therapy” came via the demonstration that a small molecular inhibitor of the BCR-ABL fusion oncoprotein—imatinib mesylate—inhibited the growth of BCR-ABL–expressing cells3 and exhibited substantial clinical activity in patients with Philadelphia chromosome–expressing CML and acute lymphoblastic leukemia.4,5 These data revolutionized the approach to cancer therapy and shifted the drug development paradigm toward identifying diseasecausing targets. In dermatofibrosarcoma protuberans, a fusion between the collagen gene (COL1A1) and the platelet-derived growth factor (PDGFR) β-chain gene produces a constitutively active mitogen driven by paracrine and autocrine ligands.6 Imatinib inhibits the tyrosine kinase associated with PDGFRβ and its use in patients with dermatofibrosarcoma protuberans yields a clinical benefit.7
Despite the breakthrough impact that imatinib has had on the field of oncology in general and in several malignancies in particular, nearly one-third of patients with CML require other therapies. Most commonly the reason for alternate treatment is the development of resistance mutations in BCR-ABL1 that affect the ability of imatinib to interact with the adenosine triphosphate (ATP)-binding pocket. Consequently, second-generation and third-generation tyrosine
28
GENERIC NAME BRAND NAME MOLECULAR TARGET INDICATION
Imatinib Gleevec BCR-ABL, c-KIT, PDGFR Ph+-CML, GIST, ASM, DFSP, Ph+-ALL, HES/CEL
Nilotinib Tasigna BCR-ABL, c-KIT, PDGFR Ph+-CML
Dasatinib Sprycel BCR-ABL, c-KIT, PDGFR Ph+-CML, Ph+-ALL
Bosutinib Bosulif BCR-ABL Ph+-CML
Ponatinib Iclusig BCR-ABL, c-KIT, PDGFR Ph+-CML, Ph+-ALL
Cetuximab Erbitux EGFR CRC, SCCHN
Panitumumab Vectibix EGFR CRC
Gefitinib Iressa EGFR NSCLC
Erlotinib Tarceva EGFR EGFR-mutated NSCLC, Pancreatic Cancer
Afatinib Gilotrif EGFR EGFR-mutated NSCLC
Osimertinib Tagrisso EGFR EGFR-mutated (T790M) NSCLC
Vismodegib Erivedge SMO BCC
Sonidegib Odomzo SMO BCC
Vorinostat Zolinza HDACs CTCL
Romidepsin Istodax HDACs CTCL, PTCL
Belinostat Beleodaq HDACs PTCL
Panobinostat Farydak HDACs Multiple Myeloma
Vemurafenib Zelboraf BRAFV600E Melanoma
Dabrafenib Tafinlar BRAFV600E Melanoma
Trametinib Mekinist MEK Melanoma
Cobimetinib Cotellic MEK Melanoma
Cobimetinib Cotellic MEK Melanoma
ASM, aggressive systemic mastocytosis; BCC, basal cell carcinoma; CEL, chronic eosinophilic leukemia; CRC, colorectal cancer; CTCL, cutaneous T-cell lymphoma; DFSP, dermatofibrosarcoma protuberans; EGFR, epidermal growth factor receptor; GIST, GI stromal tumor; HDAC, histone deacetylases; HES, hypereosinophilic syndrome; NSCLC, non-small cell lung cancer; PDGFR, platelet-derived growth factor receptor; Ph+-ALL, Philadelphia chromosome-positive acute lymphoblastic leukemia; Ph+-CML, Philadelphia chromosome-positive chronic myeloid leukemia; PTCL, peripheral T-cell lymphoma; SCCHN, squamous cell carcinoma, head and neck; SMO, smoothened.
kinase inhibitors (TKIs) have been developed with enhanced affinity for BCR-ABL. These TKIs include nilotinib, dasatinib, bosutinib, and ponatinib.
IMATINIB MESYLATE (GLEEVEC)
IMATINIB MESYLATE
(GLEEVEC)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Imatinib, 4-[(4-methyl-1-piperazinyl) methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl] amino]-phenyl]benzamide methanesulfonate, has the molecular formula C29H31N7O • CH4SO3 and a molecular weight of 589.7 daltons. Figure 194-2 shows its structure.
Metabolism: Imatinib is predominantly metabolized by cytochrome P450 (CYP) 3A4 and the main active metabolite is a N-demethylated piperazine derivative. The drug and its metabolites are primarily excreted in the feces. The elimination half-life is
approximately 18 hours for the unchanged form and 40 hours for the N-desmethyl derivative.8
Absorption and Distribution: Imatinib is absorbed orally with bioavailability of 98%. The compound has high levels of binding to circulating plasma proteins, mainly albumin and α1-acid glycoprotein. Mechanism of Action: Imatinib mesylate is a small-molecule TKI with activity against BCR-ABL, c-KIT, and PDGFR (see Fig. 194-1). Imatinib binds near the ATP-binding site of the inactive, unphosphorylated confirmation of these kinases, inhibiting the enzyme activity of the protein. In cells with constitutively active BCR-ABL, imatinib induces apoptosis and inhibits proliferation. Imatinib also downregulates cell proliferation triggered by aberrant c-KIT and plateletderived growth factor (PDGF) signaling.
INDICATIONS AND CONTRAINDICATIONS
Imatinib mesylate was initially approved in 2001 for use in CML. In 2002, the label was updated to include
3559
28
Inhibitors of KIT/BCR-ABL/PDGFR
PDGFR c-KIT
RAS RAS RAS NF1 NF1
P
P
P
P
Shc Shc
GDP GDP GTP
P
P
P
P
Grb2 Grb2 SOS SOS
Imatinib Nilotinib Dasatinib Ponatinib
Grb2
Shc
Imatinib Nilotinib Dasatinib Ponatinib
BRAF
MEK
Bosutinib
PI3K
P P
P
BCR-ABL BCR-ABL JAK
P
Bosutinib
STAT STAT
ERK
AKT
Cell Proliferation Cell Survival
advanced or metastatic GI stromal tumors. In 2006, the U.S. Food and Drug Administration (FDA) further expanded the approval to include dermatofibrosarcoma protuberans, aggressive systemic mastocytosis, myelodysplastic/myeloproliferative diseases with PDGFR gene rearrangements, relapsed/refractory
3560
Philadelphia chromosome–positive acute lymphocytic leukemia, and hypereosinophilic syndrome/chronic eosinophilic leukemia with FIP1L1-PDGFR fusion kinase rearrangements. Off-label uses of imatinib have been reported for melanomas harboring KIT alterations, which occur at a higher frequency in melanomas arising from mucosal, acral and chronically sun-damaged skin. Data from several newer trials suggest a modest clinical benefit as a single agent in c-KIT–mutated melanoma and newer National Comprehensive Cancer Network (NCCN) guidelines include imatinib as a treatment option for metastatic melanoma.9-11 Clinical activity of imatinib is also being investigated for desmoid tumors and for advanced or metastatic chordomas expressing PDGFRβ and/or PDGFβ. There are no contraindications on the manufacturer’s label.
DISEASE DOSE
Chronic myeloid leukemia, chronic phase 400 mg daily
Chronic myeloid leukemia, accelerated phase or blast crisis 600 mg daily
GI stromal tumors 400 mg daily
Dermatofibrosarcoma protuberans 400 mg twice daily
Aggressive systemic mastocytosis 400 mg daily
Myelodysplastic/myeloproliferative diseases 400 mg daily
Relapsed/refractory Philadelphia chromosome– positive acute lymphocytic leukemia 600 mg daily
Hypereosinophilic syndrome/chronic eosinophilic leukemia 400 mg daily
Melanomaa 400 mg twice daily
Melanomaa 400 mg twice daily
aUse of imatinib for melanoma is not FDA approved; off-label dosing based on NCT00470470.11
DOSING REGIMEN
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects include edema, nausea, vomiting, muscle cramps, myalgias, diarrhea, bone pain, fatigue, and abdominal pain. Less common but serious adverse effects include severe fluid retention (eg, pericardial effusion, pulmonary edema, pleural effusions, and ascites), hematologic toxicity (eg, anemia, neutropenia, and thrombocytopenia), congestive heart failure, liver failure, and hemorrhage. A wide variety of cutaneous side effects have been reported in patients taking imatinib, including periorbital edema, dyspigmentation, morbilliform eruption, pityriasis rosea-like eruption, acute generalized exanthematous pustulosis, exacerbation of psoriasis, drug rash with eosinophilia and systemic symptoms, pseudoporphyria, mycosis fungoides–like eruption, acute neutrophilic dermatosis, erythroderma, Stevens-Johnson syndrome, perforating folliculitis, and urticaria.12
28
Women of reproductive age should be counseled that imatinib is a pregnancy category D drug as it was teratogenic when tested in rodents and there have been postmarket reports of congenital anomalies from pregnant women on imatinib. Thus, patients should be advised to use highly effective contraception and avoid pregnancy while taking imatinib. Nursing mothers should be counseled that imatinib and its metabolites have been detected in human breastmilk and nursing infants could receive up to 10% of the maternal dose. Given the risk to infants, breastfeeding is not recommended while taking imatinib.
Drug Interactions: Because of its metabolism by CYP3A4, drug concentrations of imatinib are affected by agents that inhibit or induce CYP3A4. Furthermore, studies demonstrate that imatinib is a moderate inhibitor of CYP3A4 and weakly inhibits CYP2D6; therefore, caution should be used when prescribing imatinib with CYP3A4 and CYP2D6 substrates with a narrow therapeutic window. Please see the Gleevec package insert for full drug– drug interactions and adverse effects.8
NILOTINIB (TASIGNA)
NILOTINIB (TASIGNA)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Nilotinib, 4-methyl-N-[3-(4-methyl-1Himidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4- (3-pyridinyl)-2-pyrimidinyl]amino]-benzamide, monohydrochloride, monohydrate, has the molecular formula C28H22F3N7O • HCl • H2O and molecular weight of 584 daltons. Figure 194-3 shows the structure of nilotinib.
Metabolism: Nilotinib is metabolized by CYP3A4 and metabolism is predominantly by oxidation and hydroxylation. The vast majority of the dose (93%) is eliminated in the feces. The elimination half-life is estimated at 17 hours.13
Absorption and Distribution: Nilotinib is absorbed orally and absorption is increased when given 30 minutes after a meal. Of the circulating compound, 98% is bound to serum protein.
Mechanism of Action: Similar to its forerunner imatinib, nilotinib is a multityrosine kinase inhibitor. The compound binds and stabilizes the inactive
3561
28
conformation of the kinase domain of its target proteins (BCR-ABL, KIT, PDGFR, discoidin domain receptor). It was rationally designed based on the structure of its predecessor to overcome resistance in CML to imatinib. Rational designs of the compound yield a significantly higher affinity and inhibitor activity of nilotinib against BCR-ABL compared with imatinib, while maintaining its activity against PDGFR and KIT.
INDICATIONS AND CONTRAINDICATIONS
Nilotinib is indicated for adult patients with either newly diagnosed Philadelphia chromosome–positive (Ph+) CML in chronic phase or accelerated-phase CML that is resistant or intolerant to prior treatment with imatinib. Nilotinib does not have any current FDAapproved indications for dermatologic disease. However, the results with imatinib in c-KIT–mutated melanoma have provided the rationale for early phase trials exploring the usefulness of nilotinib in these subsets. Similar to imatinib, the results have been modest. In one trial, 7 (16.7%) of 42 patients with metastatic KIT-altered melanoma achieved a response with single-agent nilotinib.14 In a second trial of patients with advanced melanoma harboring KIT mutations or amplifications who had received prior KIT inhibitor therapy, 4 (21%) of 19 patients experienced a response.15
Nilotinib is contraindicated for use in patients with long QT syndrome, hypokalemia, or hypomagnesemia.
DOSING REGIMEN
The recommended dose for newly diagnosed Ph+
chronic-phase CML is 300 mg by mouth twice daily; for resistant or intolerant Ph+ chronic-phase CML and accelerated-phase CML, 400 mg by mouth twice daily is recommended. Nilotinib should not be taken with food. It is recommended to avoid food for at least 2 hours before and 1 hour after the dose is taken. Foods that inhibit CYP3A4 should be avoided. Dose adjustments are recommended for a variety of indications, including impaired hepatic function, QT interval, and hematologic toxicity. See the Tasigna product label for full dose adjustment recommendations.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects include cutaneous toxicities (see below), thrombocytopenia, neutropenia, anemia, constipation, nausea, vomiting, hyperbilirubinemia, fatigue, elevated lipase, and elevated transaminases. Serious adverse effects include a boxed warning for QT prolongation and sudden death. Thus, nilotinib is not recommended for use in patients with hypokalemia, hypomagnesemia, or long QT syndrome. Medications known to prolong the QT interval or that strongly inhibit CYP3A4 should also be avoided. Potassium,
3562
calcium, magnesium, phosphate, or sodium electrolyte abnormalities should be corrected prior to initiation and should be periodically monitored during therapy. In the initial trial of nilotinib in CML, cutaneous toxicities were the most common adverse effects noted and included rash (specific morphologies not described), pruritus, dry skin, and alopecia. Postmarket reports have included a case of bullous Sweet syndrome.12
Nilotinib is a pregnancy category D drug, based on its mechanism of action and data in animal studies that the compound may cause fetal harm. Women with reproductive potential should be advised to use highly effective contraception during therapy. Findings in animal studies demonstrate that nilotinib may be unsafe during nursing. It is recommended to consider alternatives to breastfeeding or to weigh the risks against the benefits of use during nursing.
Drug Interactions: As nilotinib is a substrate of CYP3A4, inhibitors and inducers of CYP3A4 may affect serum concentrations. Nilotinib is an inhibitor of CYP3A4, CYP2C8, CYP2C9, and CYP2D6, and may also induce CYP2B6, CYP2C8, and CYP2C9. Thus, coadministration of nilotinib with substrates of these enzymes may affect the serum concentrations. See the Tasigna product label for full drug–drug interactions and adverse effects.13
DASATINIB (SPRYCEL)
DASATINIB (SPRYCEL)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Dasatinib, N-(2-chloro-6-methylphenyl)- 2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4- pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate, has the molecular formula C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 daltons for the monohydrate form and 488.01 daltons for the anhydrous free base.16 Figure 194-4 shows the structure of dasatinib.
Metabolism: Dasatinib is predominantly metabolized by CYP3A4. Dasatinib metabolites are also produced by uridine diphosphate-glucuronosyltransferase (UGT) and flavin-containing monooxygenase 3 (FMO-
3) enzymes. The estimated elimination half-life is 3 to 5 hours. Dasatinib is primarily excreted through the feces.
Absorption and Distribution: Following oral administration, peak plasma concentrations are reached between 30 minutes and 6 hours. The apparent volume of distribution is 2505 L.
Mechanism of Action: Dasatinib is a small molecule kinase inhibitor. It inhibits numerous kinases, including BCR-ABL, c-KIT, PDGFRβ, SRC family (SRC, LCK, YES, FYN), and EPHA2. In preclinical in vitro studies, dasatinib inhibited the growth of BCR- ABL overexpressing CML and acute lymphoblastic leukemia (ALL) cell lines.16 In these assays, dasatinib was effective in cell lines that possess BCR-ABL kinase mutations that conferred resistance to imatinib.
INDICATIONS AND CONTRAINDICATIONS
Dasatinib has FDA approval for adults with (a) newly diagnosed Ph+ chronic-phase CML, (b) chronic, accelerated, or myeloid or lymphoid blast-phase Ph+-CML with intolerance or resistance to previous therapy including imatinib, and (c) adults with Ph+-ALL with intolerance or resistance to previous therapy. In melanoma, early-phase investigations with dasatinib have shown only a modest clinical benefit when used a monotherapy17 or in combination with systemic chemotherapy.18
There are no contraindications listed on the manufacturer’s label.
DOSING REGIMEN
The recommended oral dose is 100 mg daily for chronic-phase CML and 140 mg once daily for accelerated-phase CML, myeloid or lymphoid blast-phase CML, or Ph+-ALL. Please see the manufacturer’s label for dose modification for neutropenia, thrombocytopenia, and concomitant use of CYP3A4 modifiers. Studies were not performed in patients with impaired renal function, thus there are no specific dose modifications for patients with renal impairment.
SIDE EFFECTS AND PRECAUTIONS
Adverse Effects: For newly diagnosed chronicphase CML, the most common adverse effects are myelosuppression, diarrhea, and fluid retention. Patients who have previously progressed, or were intolerant to prior imatinib therapy, commonly experience myelosuppression, fluid retention, diarrhea, headache, fatigue, dyspnea, nausea, hemorrhage, musculoskeletal pain, and skin toxicity. Cutaneous adverse effects include neutrophilic dermatosis,19 keratosis pilaris–like lesions and pustules, white keratotic papules, and milia.20
Drug Interactions: Caution is advised when dasatinib is used concomitantly with strong CYP3A4 inducers (including, but not limited to phenytoin, rifampin, phenobarbital, carbamazepine, dexamethasone, rifabutin) and strong CYP3A4 inhibitors (ketoconazole, voriconazole, itraconazole, atazanavir, nelfinavir, indinavir, ritonavir, saquinavir, nefazodone, telithromycin, and
28
clarithromycin). It is recommended to avoid St. John’s wort and grapefruit juice while taking dasatinib. It is recommended to advise women of reproductive potential to avoid pregnancy during treatment with dasatinib and for at least 30 days following the final dose. Breastfeeding is not recommended while taking dasatinib and for at least 2 weeks after the final dose. Use with caution in patients with hepatic impairment. See the Sprycel package insert for full drug–drug interactions and adverse effects.16
BOSUTINIB (BOSULIF)
BOSUTINIB (BOSULIF)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Bosutinib, 3-Quinolinecarbonitrile, 4-[(2,4-dichloro-5-methoxyphenyl)amino]-6-methoxy- 7-[3-(4-methyl-1-piperazinyl) propoxy]-, hydrate (1:1), has the chemical formula C26H29Cl2N5O3 • H2O (monohydrate); its molecular weight is 548.46 (monohydrate), equivalent to 530.46 (anhydrous).21 Figure 194-5 shows the structure of bosutinib.
Metabolism: Bosutinib is predominantly metabolized by CYP3A4 and primarily excreted through the feces.
Absorption and Distribution: Oral bioavailability is 34% when taken with food and following oral administration; peak plasma concentrations are reached between 4 and 6 hours. The apparent volume of distribution is 6080 L ± 1230 L.
Mechanism of Action: Bosutinib is a smallmolecular-weight kinase inhibitor. It inhibits numerous kinases, including BCR-ABL and the SRC family kinases SRC, LCK, and FYN. In preclinical in vitro studies, bosutinib inhibited the growth of 16 of 18 BCR- ABL overexpressing murine myeloid cell lines that were resistant to imatinib. Bosutinib did not inhibit the mutant cell lines that expressed the T315I and V299L resistance mutations.21
INDICATIONS AND CONTRAINDICATIONS
Bosutinib has FDA approval for adults with chronicphase, accelerated-phase, or blast-phase Ph+-CML with
3563
28
intolerance or resistance to previous therapy. Bosutinib does not have any current FDA-approved indications for dermatologic disease. Bosutinib is contraindicated in patients with a known hypersensitivity to the drug.
DOSING REGIMEN
The recommended oral dose is 500 mg once daily with food. See the manufacturer’s label for dose modifications for neutropenia, thrombocytopenia, renal impairment, hepatic impairment, and concomitant use of CYP3A4 modifiers.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects are diarrhea, nausea, thrombocytopenia, vomiting, abdominal pain, rash (not otherwise specified), anemia, pyrexia, and fatigue. Bosutinib is a pregnancy category D drug, and it is recommended to advise women of reproductive potential to avoid pregnancy during treatment. As to breastfeeding, bosutinib may be excreted in human milk and thus the risk of harm to a nursing infant must be weighed with the importance of the drug to the mother.
Drug Interactions: Caution is advised when bosutinib is used concomitantly with strong CYP3A4 inducers (including, but not limited to, phenytoin, rifampin, phenobarbital, carbamazepine, dexamethasone, rifabutin) and strong CYP3A4 inhibitors (ketoconazole, voriconazole, itraconazole, atazanavir, nelfinavir, indinavir, ritonavir, saquinavir, nefazodone, telithromycin, and clarithromycin). It is recommended to avoid St. John’s wort and grapefruit juice while taking bosutinib. Proton pump inhibitors can reduce exposure to bosutinib and should be avoided if possible. Consider H2 blockers or short-acting antacids. See the Bosulif package insert for full drug–drug interactions and adverse effects.21
PONATINIB (ICLUSIG)
PONATINIB (ICLUSIG)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Ponatinib, 3-(imidazo[1,2-b]pyridazin- 3-ylethynyl)-4-methyl-N-{4-[(4-methylpiperazin-1-yl) methyl]-3-(trifluoromethyl)phenyl}benzamide hydrochloride, has the chemical formula C29H28ClF3N6O, which corresponds to a molecular weight of 569.02 daltons.22 Figure 194-6 shows the structure of ponatinib.
3564
Metabolism: Ponatinib is metabolized by CYP3A4, and to a lesser degree by CYP2C8, CYP2D6, and CYP3A5. It is also metabolized by amidases and/or esterases. Ponatinib is primarily excreted through the feces.
Absorption and Distribution: Following oral administration, peak plasma concentrations are reached within 6 hours. The apparent volume of distribution is 1223 L.
Mechanism of Action: Ponatinib is a smallmolecular-weight kinase inhibitor. It inhibits a variety of kinases, including BCR-ABL, VEGFR (vascular endothelial growth factor receptor), PDGFR, FGFR (fibroblast growth factor receptor), EPH (ephrin), KIT, RET, TIE2 (TEK receptor tyrosine kinase), FLT3 (fmsrelated tyrosine kinase 3), and the SRC family kinases. Ponatinib also inhibits the T315I mutation that confers resistance to imatinib.
INDICATIONS AND CONTRAINDICATIONS
Ponatinib has FDA approval for adults with T315Imutant-positive chronic-phase, accelerated-phase, or blast-phase CML or T315I-mutant-positive Ph+-ALL. It is also approved for adults with chronic-phase, accelerated-phase, or blast-phase CML or Ph+-ALL in which no other TKI treatment is indicated. Ponatinib does not have any current FDA-approved indications for dermatologic disease. There are no contraindications listed on the manufacturer’s label.
DOSING REGIMEN
The recommended oral dose is 45 mg once daily taken with or without food. Please see the manufacturer’s label for dose modification for neutropenia, thrombocytopenia, renal impairment, hepatic impairment, and concomitant use of CYP modifiers.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common nonhematologic adverse effects include hypertension, rash (not otherwise specified), fatigue, xerosis, headache, abdominal pain, constipation, nausea, arthralgia, and pyrexia. Hematologic toxicities include
myelosuppression including leukopenia, neutropenia and thrombocytopenia, anemia, and lymphopenia. Ponatinib has a boxed warning for vascular occlusion, including arterial and venous thrombosis, which occurred in at least 27% of patients, heart failure and hepatotoxicity. Ponatinib is a pregnancy category D drug and it is recommended to advise women of reproductive potential to avoid pregnancy during treatment. With considerations to breastfeeding, it is unknown if ponatinib is excreted in human milk and thus the risk of harm to a nursing infant must be weighed with the importance of the drug to the mother.
Drug Interactions: Caution is advised when ponatinib is used concomitantly with strong CYP3A inducers and inhibitors. See the Iclusig package insert for full drug–drug interactions and adverse effects.22
EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITORS
AT-A-GLANCE
■ The epidermal growth factor receptor (EGFR) is found on the cell surface of a variety of epithelial cells and is dysregulated in numerous malignancies.
■ Monoclonal antibodies that block epidermal growth factor binding to EGFR and smallmolecular-weight inhibitors of the EGFR intracellular tyrosine kinase have been approved as therapy for several types of cancer.
■ Although there are no FDA-approved dermatologic indications at the time of publication of this book, clinical investigations in cutaneous oncology are ongoing.
■ EGFR inhibitors commonly have skin toxicities that include papulopustular eruptions, pruritus, xeroderma, and paronychia.
BACKGROUND
BACKGROUND
Drug development focused on the EGFR (also known as ErbB1 or HER1) showcases the potential of targeted therapy, rational drug design and personalized medicine. Although agents that inhibit EGFR signaling do not currently have any FDA-approved dermatologic indications, investigations into their use in cutaneous malignancy are ongoing. In addition these agents, which include cetuximab, panitumumab, gefitinib, erlotinib, afatinib, and osimertinib have significant
28
cutaneous adverse effects and thus knowledge of their indication, mechanism of action, and associated toxicities is relevant to all dermatologists. EGFR is a member of the ErbB family of tyrosine kinase receptors, which also includes ErbB2 (HER2/ neu), ErbB3 (HER3), and ErbB4 (HER4). These cellsurface proteins possess an extracellular ligandbinding domain, a transmembrane domain and an intracellular tyrosine kinase domain (Fig. 194-7). EGFR, which is found on a variety of cell types, including keratinocytes and cells of various solid tumors, is activated following binding of epidermal growth factor (EGF) and other growth factors. Ligand binding to the extracellular domain of EGFR results in dimerization in which the receptor either binds with another EGFR protein or heterodimerizes with an additional monomer of the ErbB family. Dimerization triggers the intracellular tyrosine kinase to autophosphorylate several tyrosine residues. Subsequent recognition of the phosphorylated tyrosine residues on the C-terminal domain of the receptor by various adaptor proteins initiates downstream signal transduction signaling. EGFR activation can trigger networks involved in tumor growth and proliferation, inhibition of apoptosis and metastases such as the MAPK, STAT, PI3K, and phospholipase pathways. Dysregulation of EGFR signaling is common in several epithelial cancers and can occur through multiple mechanisms including gene amplification and activating mutations. In 2004, the discovery of in-frame activating deletions in exon 19 and the L858R substitution in exon 21 of EGFR profoundly changed the therapeutic landscape in non–small-cell lung cancer (NSCLC) and provided an additional early rationale for precision medicine in oncology.23,24
Two distinct mechanisms have been developed to interrupt EGFR signaling as a therapeutic strategy in cancer: (a) targeting of the extracellular ligand binding domain via monoclonal antibodies and (b) inhibition of intracellular tyrosine kinase via small molecules. The monoclonal antibodies cetuximab and panitumumab have demonstrated efficacy in several carcinomas. Cetuximab holds approval for colorectal and head and neck cancer and panitumumab is indicated in combination with systemic chemotherapy for colorectal cancer. During the 1990s a concerted effort began to develop small-molecular-weight compounds that inhibit various tyrosine kinases. The first-generation TKIs against EGFR, gefitinib and erlotinib, were designed as competitive inhibitors of ATP binding to the intracellular kinase domain. Despite promising preclinical data, early clinical studies in lung cancer with gefitinib were discouraging as ISEL (Iressa Survival Evaluation in Lung Cancer), a large trial of unselected, heavily pretreated patients, did not show an overall survival benefit in patients with NSCLC.25 Interestingly, subgroup analysis revealed that never-smokers and Asian patients achieved better overall survival with gefitinib. Translational studies subsequently demonstrated the importance of the exon 19 and exon 21 EGFR mutations as predictive biomarkers. These somatic mutations,
3565
28
Epidermal growth factor receptor pathway
Panitumumab Cetuximab
RAS RAS
NF1
PIP2 PIP3
Shc PLC
GTP GDP P
P
Grb2 SOS
P
P
PI3K
AKT
DAG BRAF
JAK 1/2
MEK
STAT1 STAT3
ERK
IP3
Gefitinib
Erlotinib Afatinib Osimertinib
mTOR
Ca2+
PKC
CM PKC
Cell Proliferation Cell Survival
which encode the tyrosine kinase domain of EGFR (encoded by exons 18 to 24), occur at higher frequency in adenocarcinomas, nonsmokers, and Asians. These alterations reduce the affinity of the tyrosine kinase to ATP and thus confer enhanced sensitivity to EGFR TKIs. Followup studies in NSCLC with gefitinib and erlotinib incorporating EGFR-mutation biomarkers have demonstrated superior clinical benefit in patients with exon 19 and exon 21 alterations.26,27 As a result, erlotinib and the second-generation TKI afatinib are indicated only for NSCLC patients with tumors that possess exon 19 or exon 21 alterations. Despite initial response to monotherapy EGFR TKI, resistance emerges for the majority of patients within the first 1 to 2 years following initiation of treatment. Secondary mutations in the tyrosine kinase domain of EGFR are associated with the development of resistance to reversible TKIs in the majority of patients.
3566
The most clinically relevant secondary mutation, the T790M of exon 20, has been detected in nearly 60% of patients who have become resistant while taking gefitinib or erlotinib.28 The T790M mutation replaces threonine with a larger methionine residue, simultaneously enhancing the affinity of the EGFR kinase for ATP and sterically impeding drug binding.29 Showcasing the success of rational drug development, osimertinib, a small molecule inhibitor with activity against EGFR possessing T790M, was shown to be highly active in lung cancer patients with the T790M resistance mutation.30 This sequence of developmental successes, beginning with the identification a disease-causing protein, demonstrating the clinical efficacy with disruption of that target, followed by second-generation and third-generation modifications of therapies to overcome mechanisms of resistance, exemplifies the goal of targeted therapy.
CETUXIMAB (ERBITUX)
CETUXIMAB (ERBITUX)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Cetuximab is a recombinant, human/ mouse chimeric monoclonal antibody. It is a composite of the Fv region of a murine anti-EGFR antibody with a human immunoglobulin G1 heavy-chain and kappa light-chain constant regions.31 It has an approximate molecular weight of 152,000 daltons.
Metabolism: The estimated elimination half-life of cetuximab is approximately 112 hours. Clearance of cetuximab appears to be similar to pathways of metabolism for other biologics, including internalization of the ligand-receptor complex with subsequent removal from the circulation.32
Absorption and Distribution: Cetuximab is administered intravenously and exhibits nonlinear pharmacokinetics. The volume of distribution appeared to be independent of dose and approximated 2 to 3 L/m2. Cetuximab reaches steady-state plasma concentrations by the third weekly infusion.
Mechanism of Action: Cetuximab is a monoclonal antibody with affinity for the extracellular ligand-binding portion of EGFR. Cetuximab acts as a competitive inhibitor for EGF and other ligands on normal epithelium as well as tumor cells. Preclinical studies have demonstrated that cetuximab binding to EGFR precludes receptor-associated kinase activation and downregulates signal transduction pathways associated with cell growth, proliferation, angiogenesis, and metastases.31
INDICATIONS AND CONTRAINDICATIONS
Cetuximab is FDA approved for use in colorectal and head and neck cancer. Initial approval in 2004 was granted for use in metastatic colorectal cancer (mCRC) in combination with irinotecan in patients who are refractory to irinotecan-based therapy. On March 1, 2006, the FDA expanded approval for use in combination with radiotherapy for patients with local or regionally advanced squamous cell carcinoma of the head and neck (SCCHN) or as monotherapy for patients with recurrent or metastatic SCCHN whose tumors were refractory to platinum-based chemotherapy. In 2011, cetuximab was approved for patients with recurrent locoregional or metastatic SCCHN in combination with platinum-based therapy plus 5-fluorouracil. On July 6, 2012, the FDA granted approval to cetuximab for first-line therapy in combination with FOLFIRI (irinotecan, 5-fluorouracil, and leucovorin) in patients with wildtype K-ras mCRC that expresses EGFR. Erbitux is not indicated for use in patients with RAS-mutated colorectal cancer.
28
Cetuximab has been investigated for use in cutaneous squamous cell carcinoma (cSCC), although the totality of the efficacy data to date are limited. A Phase II, uncontrolled trial of 36 chemotherapy-naïve patients with unresectable or metastatic cSCC who were treated with an initial dose of 400 mg/m2 body surface area of cetuximab, followed by weekly doses of 250 mg/m2 for at least 6 weeks, was published in 2011.33 This small study reported a complete response in 2 (6%), a partial response in 8 (22%), and stable disease in 15 (42%) of the 36 patients at 6 weeks. Nevertheless, responses were limited in duration, with the median duration of response for the responders of 5 months. Additional prospective studies with larger number of patients are needed to better assess the efficacy of cetuximab in cSCC. There are no contraindications on the manufacturer’s label.
DOSING REGIMEN
Cetuximab is given as an IV infusion. The initial dose is 400 mg/m2 infused over 120 minutes, followed by weekly infusions at 250 mg/m2. See the manufacturer’s label for premedication recommendations and additional dose and infusion rate modifications.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse reactions are cutaneous toxicity, headache, diarrhea, and infection. Cetuximab carries a boxed warning for serious, potentially fatal, infusion reactions that occurred in approximately 3% of patients and for cardiopulmonary arrest and/or sudden death. Close monitoring of serum electrolytes during and after infusion is strongly recommended. Other serious adverse effects include interstitial lung disease and hypomagnesemia. Similar to other agents that disrupt EGFR signaling, skin toxicities are common with cetuximab and include a papulopustular eruption, xerosis, pruritus, mucositis, alopecia, trichomegaly, paronychia, and onycholysis.34 The development of an acne-like eruption occurs typically within 1 to 2 weeks of initiation of cetuximab therapy and also is associated with improved clinical outcomes.35
Cetuximab is a pregnancy category C drug, and should be used during pregnancy only if the potential harms to the fetus are outweighed by the potential benefit to the mother. Immunoglobulin G antibodies, such as cetuximab, are secreted in human milk; consequently, breastfeeding should be avoided during and at least up to 60 days after treatment, if possible, to prevent potential adverse reactions to nursing infants.
Drug Interactions: No drug interactions are listed on the manufacturer’s label.
3567
28
PANITUMUMAB (VECTIBIX)
PANITUMUMAB (VECTIBIX)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Panitumumab is a recombinant, human immunoglobulin G2 kappa monoclonal antibody with a molecular weight of 147,000 daltons.36 It is engineered in Chinese hamster ovary cells.
Metabolism: The mean estimated elimination half-life is 7.5 days.
Absorption and Distribution: Administration of the recommended dose will result in reaching steady-state concentrations by the third infusion.
Mechanism of Action: Panitumumab is a monoclonal antibody with affinity for the extracellular ligand-binding portion of EGFR. Panitumumab, like cetuximab, functions as a competitive inhibitor for EGF and other ligands on both normal epithelium and tumor cells. Preclinical studies have demonstrated that binding to EGFR by panitumumab decreases receptor-associated kinase activation, resulting in downregulated signal transduction pathways that are associated with cell growth, proliferation, angiogenesis, and metastases.
INDICATIONS AND CONTRAINDICATIONS
Panitumumab is indicated for the treatment of mCRC that is wildtype for KRAS as determined by an FDAapproved test. It has FDA approval as first-line therapy in combination with FOLFOX and as monotherapy following progression after treatment with a regimen incorporating fluoropyrimidine, oxaliplatin, and irinotecan. Panitumumab is not approved for use in mCRC patients who possess a KRAS mutation or for whom the mutation status of KRAS is unknown. Panitumumab is currently in development for head and neck cancer. Use of panitumumab has been reported in locally advanced cSCC with promising results. The successful use of panitumumab in an elderly patient who had an anaphylactic reaction to cetuximab37 and in a small Phase II trial of 16 patients who produced a best overall response rate by RECIST criteria of 31% have been reported.38 Although promising, pivotal Phase III trials are needed to confirm these early investigational results. There are no contraindications on the manufacturer’s label.
DOSING REGIMEN
Panitumumab is administered as an IV infusion and the recommended dose is 6 mg/kg every 14 days. There are no specific dosing adjustments for renal or hepatic impairment.
3568
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects are skin toxicity, fatigue, nausea, and diarrhea. Serious adverse effects include electrolyte disturbance, infusion reactions, pulmonary fibrosis/interstitial lung disease, and keratitis. Panitumumab carries a box warning for dermatologic toxicity. Skin adverse events were common in the registration trial, with 90% of patients experiencing toxicity of any grade, while 15% suffered from severe skin toxicity. The cutaneous manifestations include, but are not limited to, papulopustular eruptions, exfoliation, pruritus, erythema, photosensitivity, xeroderma, and paronychia. Fatal bullous disease and life-threatening necrotizing fasciitis, abscesses and sepsis have been observed in patients on panitumumab.36
Panitumumab is a pregnancy category C drug. No studies have been performed in pregnant women, although Cynomolgus monkeys treated with 1.25 to 5 times the recommended human dose did demonstrate evidence of embryo lethality and abortions. Consequently, extreme caution is recommended with administration during pregnancy and the risks to the fetus must be weighed against the potential benefit to the mother. It is recommended that breastfeeding by avoided during treatment of at least 2 months following completion of treatment.
Drug Interactions: No specific drug interactions are listed on the manufacturer’s label.
GEFITINIB (IRESSA)
GEFITINIB (IRESSA)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Gefitinib, 4-quinazolinamine N-(3- chloro-4-uorophenyl)-7-methoxy-6-[3-(4-morpholinyl) propoxy], has the molecular formula C22H24ClFN4O3 and a molecular mass of 446.9 daltons.39 Figure 194-8 shows the structure of gefitinib.
Metabolism: Gefitinib is metabolized predominantly by CYP3A4 and to a lesser degree by CYP2D6. The estimated elimination half-life is 48 hours after intravenous administration.
Absorption and Distribution: The oral bioavailability of gefitinib is approximately 60% and peak plasma levels are reached 3 to 7 hours after dosing.
Food does not significantly affect absorption. The volume of distribution is 1400 L.
Mechanism of Action: Gefitinib is a smallmolecule TKI. It reversibly binds and inhibits the kinase domain of EGFR. Gefitinib has a higher affinity for EGFR harboring exon 19 deletions or the exon 21 point mutation L858R, as compared with the wildtype EGFR protein.
INDICATIONS AND CONTRAINDICATIONS
Gefitinib has limited indication in the United States to patients with NSCLC who are currently receiving and benefitting from gefitinib or have previously benefited from therapy with gefitinib. Gefitinib was initially granted accelerated approval by the FDA in May 2003 for patients with NSCLC tumors that were refractory to a platinum-based regimen and docetaxel. However, after 2 studies demonstrated a lack of efficacy, the FDA changed the label in 2005, withdrawing approval for new patients. Subsequent studies have demonstrated clinical effectiveness of gefitinib in patients with tumors harboring EGFR mutations and the drug is indicated for use in advanced NSCLC in Europe. Gefitinib does not have any current FDA-approved indications for dermatologic disease. There are no contraindications on the manufacturer’s label.
DOSING REGIMEN
Recommended dosing is 250 mg once daily by mouth, with or without food.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: Skin toxicities and diarrhea are the most common adverse effects. Cutaneous toxicities are common and share the characteristics of other agents targeting EGFR. A papulopustular eruption, xeroderma, and pruritus are all common. The papulopustular eruption typically develops 1 to 2 weeks following treatment initiation and has been associated with improved overall survival.40 Other adverse cutaneous manifestations include photosensitivity, mucositis as well as nail and hair changes. Nail effects include onycholysis, paronychia, and pyogenic granulomalike lesions of the nailfold. Hair changes include alopecia, trichomegaly and hirsutism.12
Based on preclinical animal studies, gefitinib may produce fetal harm when administered to pregnant women. Thus, it is recommended to strongly advise avoidance of gefitinib during pregnancy. Gefitinib has been detected in rat milk and thus lactating women should be made aware of the potential harm to infants during nursing.
Drug Interactions: Caution is advised when gefitinib is administered concomitantly with inhibitors and inducers of CYP3A4. It is also recommended
28
to avoid compounds that can affect gastric pH such as proton pump inhibitors. Close monitoring of the prothrombin time and/or international normalized ratio when used with warfarin is warranted. See the Iressa package insert for full drug–drug interactions and adverse effects.39
ERLOTINIB HYDROCHLORIDE (TARCEVA)
ERLOTINIB
HYDROCHLORIDE
(TARCEVA)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Erlotinib, N-(3-ethynylphenyl)-6,7-bis(2- methoxyethoxy)-4-quinazolinamine, has the molecular formula C22H23N3O4 • HCl and a molecular weight of 429.90 daltons.41 Figure 194-9 shows the structure of erlotinib.
Metabolism: Erlotinib is predominantly metabolized by CYP3A4 and to a lesser degree by CYP1A2 and CYP1A1. The elimination half-life is estimated to be 36.2 hours. Time to steady-state in the plasma is 7 to 8 days.
Absorption and Distribution: Peak plasma levels are reached after 4 hours of an oral dose. Oral absorption is 60%, which is increased to approximately 100% if administered with food. The volume of distribution is 232 L.
Mechanism of Action: Erlotinib is a reversible small-molecule inhibitor of EGFR. It competes with the binding of ATP to the intracellular domain of EGFR, thus preventing autophosphorylation of the tyrosine residues and precluding downstream signal transduction. Erlotinib exhibits preferential binding to EGFR proteins that possess the exon 19 deletion or exon 21 (L858R) mutations as compared to the wildtype EGFR protein.
INDICATIONS AND CONTRAINDICATIONS
Erlotinib has FDA approval for use in NSCLC and pancreatic carcinoma. In NSCLC its approval extends to first-line, maintenance, or second-line or greater therapy after progression following at least 1 regimen of chemotherapy in patients with metastatic tumors that
3569
28
possess either exon 19 deletions or exon 21 (L858R) substitution mutations in EGFR, as detected by an FDA-approved test. Erlotinib is also indicated for patients with locally advanced, unresectable or metastatic pancreatic carcinoma as frontline therapy in combination with gemcitabine. Erlotinib is being investigated in combination with radiotherapy and other systemic agents in cSCC. In melanoma, a Phase II trial of erlotinib and bevacizumab for patients with metastatic disease demonstrated disappointing results, with a progression-free survival (PFS) of only 2 months.42
There are no contraindications listed on manufacturer’s label.
DOSING REGIMEN
For NSCLC, the recommended dose of erlotinib is 150 mg by mouth administered once daily until disease progression or unacceptable toxicity. The dose for pancreatic cancer is 100 mg once daily, in combination with gemcitabine, until disease progression or unacceptable toxicity. It is recommended that erlotinib be taken on an empty stomach. No dose adjustments are provided in the manufacturer’s labeling during initial treatment in patients with renal impairment. Monitoring of renal function is recommended while taking erlotinib and the drug should be stopped in patients developing renal impairment until toxicity has resolved. Specific dosing for hepatic impairment has not been defined. See the manufacturer’s label for adjustments in patients with baseline liver abnormalities.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects include cutaneous toxicity, diarrhea, fatigue, anorexia, dyspnea, cough, nausea, and vomiting. Serious drug toxicities include interstitial lung disease, which occurs in 1.1% of patients who are taking erlotinib, hepatotoxicity, GI perforations, myocardial ischemia/infarction, microangiopathic hemolytic anemia, cerebrovascular accident, corneal perforations/ulcerations, and persistent severe keratitis. Skin toxicities are very common and occur to some degree in most patients receiving erlotinib. The most common include the development of a papulopustular eruption, xeroderma, and pruritus. The papulopustular eruption often develops 1 to 2 weeks following treatment initiation and has been associated with improved overall survival.40 Other adverse cutaneous manifestations include bullous eruptions, photosensitivity, and mucositis, as well as nail and hair changes. Nail effects include onycholysis, paronychia, and pyogenic granuloma-like lesions of the nailfold. Hair changes include alopecia, trichomegaly, and hirsutism.12
Erlotinib can cause fetal harm and women of reproductive potential should be advised to use highly effective contraception. In regards to breastfeeding,
3570
it is not known if erlotinib is found in breastmilk, but based on the potential for serious harm, it is recommended that the risks and benefits of taking Tarceva be weighed carefully.
Drug Interactions: Caution should be advised when erlotinib is administered with inhibitors or inducers of CYP3A4 and CYP1A2 as they can affect the plasma concentrations. Cigarette smoking can accelerate clearance of erlotinib, potentially decreasing its antitumor effects. Caution is also advised with concomitant use of compounds such as proton pump inhibitors, H2-receptor antagonists, and antacids, which increase gastric pH and can decrease erlotinib plasma concentrations. See the Tarceva package insert for full drug–drug interactions and adverse effects.
AFATINIB DIMALEATE (GILOTRIF)
AFATINIB DIMALEATE
(GILOTRIF)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Afatinib dimaleate, 2- butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)- tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4- (dimethylamino)-,(2E)-,(2Z) 2-butenedioate (1:2), has the molecular formula C32H33ClFN5O11 and a molecular weight of 718.1 daltons.43 Figure 194-10 shows the structure of afatinib.
Metabolism: Enzymatic metabolism of afatinib is minimal and the compound is principally excreted in the feces following covalent adducts to circulating proteins. The elimination half-life is estimated to be 37 hours. Time to steady-state in the plasma is approximately 8 days.
Absorption and Distribution: Peak plasma levels are reached 2 to 5 hours after an oral dose. Oral absorption is approximately 92%, and a high-fat meal decreased maximal plasma concentrations by 50%.
Mechanism of Action: Afatinib is a secondgeneration small-molecule kinase inhibitor with activity against all 4 ErbB family members. Unlike
the first-generation EGFR TKIs gefitinib and erlotinib, which reversibly inhibit EGFR, afatinib covalently binds the kinase domain of the receptor. Consequently, afatinib irreversibly inhibits autophosphorylation of associated tyrosine residues, resulting in downregulation of cell growth and survival signal transduction pathways.
INDICATIONS AND CONTRAINDICATIONS
Afatinib has FDA approval for use as a first-line agent in metastatic NSCLC that possesses either exon 19 deletions or exon 21 (L858R) substitution mutations in EGFR, as detected by an FDA-approved test. Afatinib does not have any current FDA-approved indications for dermatologic disease. There are no contraindications listed on manufacturer’s label.
DOSING REGIMEN
The recommended dose for afatinib is 40 mg by mouth administered once daily until disease progression or unacceptable toxicity. It is recommended that afatinib be taken at least 1 hour before or 2 hours after a meal. For patients with renal impairment (estimated glomerular filtration rate: 15 to 29 mL/min), a reduction to 30 mg by mouth daily is recommended. Afatinib has not been studied in patients with glomerular filtration rates of <15 mL/min; consequently, no specific dose adjustments are recommended. Afatinib has not been studied in patients with severe (Child-Pugh grade C) hepatic impairment. For patients with mild-to-moderate hepatic impairment, no specific dose modifications are given.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects of afatinib include diarrhea, cutaneous toxicity, anorexia, nausea, and vomiting. Serious drug toxicities include interstitial lung disease, which occurs in 1.6% of patients who are taking afatinib, hepatotoxicity, keratitis, and embryofetal toxicity.43
Skin toxicities are common and include the development of a papulopustular eruption, xeroderma, pruritus, stomatitis, and nail changes. Correlative studies suggest that the development of the papulopustular eruption might be predictive of tumor response, similar to that seen in other EGFR inhibitors.44 When compared to gefitinib and erlotinib, afatinib has been shown to induce paronychia at a higher frequency and with accelerated onset.45 Although the mechanism for this difference has not been elucidated, the fact that afatinib is an irreversible inhibitor while both gefitinib and erlotinib are both reversible inhibitors, has been cited as a possible explanation.45 There is a report of afatinib-associated Stevens-Johnson syndrome.46
28
Other cutaneous effects include hypertrichosis of the eyelashes and eyebrows.47
Afatinib can cause fetal harm and women of reproductive potential should be recommended to use highly effective contraception during treatment and for at least 2 weeks after the last dose. In regards to breastfeeding, it is not known if afatinib is found in human breastmilk, but based on the potential for serious harm, it is recommended that breastfeeding be avoided in women taking Gilotrif and for 2 weeks following the final dose.
Drug Interactions: Caution is advised when afatinib is used with agents that modify P-glycoprotein. Coadministration of inhibitors of P-glycoprotein— including, but not limited to, ketoconazole, itraconazole, ritonavir, cyclosporine, tacrolimus, erythromycin, verapamil, quinidine, nelfinavir, saquinavir, and amiodarone—can increase afatinib concentrations. Conversely, afatinib exposure can be decreased when coadministered with inducers of P-glycoprotein, such as phenytoin, rifampicin, phenobarbital, carbamazepine, and St. John’s wort.43
See the Gilotrif package insert for full drug–drug interactions and adverse effects.
OSIMERTINIB (TAGRISSO)
OSIMERTINIB (TAGRISSO)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Osimertinib (AZD9291), N-(2-{2- dimethylaminoethyl-methylamino}-4-methoxy-5-{[4- (1-methylindol-3-yl)pyrimidin-2-yl]amino}phenyl) prop-2-enamide mesylate salt, has the molecular formula C28H33N7O2 • CH4O3S, and the molecular weight of 596 daltons.48 Figure 194-11 shows the structure of osimertinib.
Metabolism: Osimertinib is metabolized primarily by oxidation via CYP3A and dealkylation. It is excreted primarily in the feces and the estimated elimination half-life is 48 hours.
Absorption and Distribution: Following administration, the median time to maximum concentration is 6 hours, with a range of 3 to 24 hours. The mean volume of distribution at steady-state is 986 L.
3571
28
Mechanism of Action: Osimertinib is a smallmolecule kinase inhibitor of the EGFR. It binds irreversibly to certain mutant forms of EGFR, such as T790M, L858R, and exon 19 deletion, and with a higher affinity as compared with the wildtype EGFR protein. In vitro, osimertinib has been shown to inhibit ACK1, BLK, and HER2, HER3, and HER4, the other 3 ERbB members.
INDICATIONS AND CONTRAINDICATIONS
Osimertinib received accelerated approval in November 2015 for patients with metastatic NSCLC that possess a T790M mutation in EGFR and have progressed on prior EGFR TKI therapy. Approval was based on data from the 2 AURA Phase II studies (AURA extension and AURA2). These were multicentered single-arm trials of 411 patients with T790M-mutant EGFR NSCLC who had progressed on prior EGFR TKI therapy and in aggregate osimertinib-treated patients had a 59% overall objective response rate.30 Osimertinib does not have any current FDA-approved indications for dermatologic disease. In 2018, the FDA granted an approval for first-line treatment in patients with metastatic NSCLC with exon 19 deletions or exon 21 L858R mutations. There are no contraindications on the manufacturer’s label.
DOSING REGIMEN
The recommended dosing in patients with a confirmed T790M EGFR mutation is 80 mg once daily by mouth, with or without food.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: Similar to other EGFR kinase inhibitors, osimertinib commonly causes diarrhea, rash, xerosis, nail changes, nausea, and anorexia. Serious toxicities include interstitial lung disease/pneumonitis, QTc interval prolongation, cardiomyopathy, and embryofetal toxicity. Based on the mechanism of action and animal studies, osimertinib can cause harm to a developing fetus when administered to a pregnant woman. It is recommended to advise females of reproductive age to use effective contraception during treatment and for 6 weeks after the final dose of osimertinib. Additionally, it is recommended to advise males to use highly effective contraception for 4 months after the final dose if they are sexually active with females of reproductive potential. Breastfeeding while taking osimertinib and for the 2 weeks following the final dose is not recommended.
Drug Interactions: Caution is advised when osimertinib is administered concomitantly with strong inducers of CYP3A (eg, phenytoin, rifampin, carbamazepine, St. John’s wort). Coadministering osimertinib with rosuvastatin (a breast cancer resistance protein [BRCP] substrate) increased plasma concentrations
3572
of rosuvastatin, whereas coadministration with simvastatin (a CYP3A4 substrate) had no clinically significant effect on simvastatin concentrations. Thus, close monitoring for adverse effects is recommended when osimertinib is used with BRCP substrates (eg, rosuvastatin, sulfasalazine, topotecan). See the Tagrisso package insert for full drug–drug interactions and adverse effects.
SMOOTHENED INHIBITORS
AT-A-GLANCE
■ The hedgehog pathway is disrupted in the vast majority of basal cell carcinomas.
■ Loss of the tumor-suppressor PTCH1 or activating mutations in smoothened leads to unregulated cell growth and oncogenesis.
■ The small molecules vismodegib and sonidegib are inhibitors of smoothened and are approved for the treatment of unresectable and advanced basal cell carcinoma.
■ Vismodegib and sonidegib carry boxed warnings for embryofetal toxicity.
BACKGROUND
The hedgehog (HH) pathway, a cascade vital for embryonic development, is now known to be critical in the molecular pathogenesis of basal cell carcinoma (BCC). In the 1990s, mutations in the tumor-suppressor patched 1 (PTCH1) gene were found in both BCCs arising in basal cell nevus syndrome49 and in sporadic cases.50 Loss of PTCH1, an inhibitor of the downstream protein smoothened (SMO), results in constitutive upregulation of HH signaling and overexpression of genes responsible for cell survival, growth, proliferation, vascularization and healing (Fig. 194-12). Nearly all sporadic BCCs have a disease-promoting mutation in HH signaling, with approximately 90% possessing a loss of at least 1 allele PTCH1 and 10% having activating mutations in SMO.51 Molecularly targeted therapy for BCC emerged in 2012 with the first-in-class SMO inhibitor vismodegib (Erivedge). In 2015, the secondin-class inhibitor sonidegib (Odomzo) was approved.
VISMODEGIB (ERIVEDGE)
VISMODEGIB (ERIVEDGE)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Vismodegib, 2-Chloro-N-[4-chloro-3-(2- pyridinyl)phenyl]-4-(methylsulfonyl)benzamide, has the molecular formula C19H14Cl2N2O3S and molecular weight of 421.3 g/mol.52 Figure 194-13 shows the structure of vismodegib.
28
Hedgehog signaling
Vismodegib Sonidegib
SHH
PTCH1 SMO
SUFU
GLI1 GLI2
GLI3
Cell Proliferation Cell Survival
Metabolism: Vismodegib is metabolized via oxidation and glucuronidation primarily by CYP2C9 and CYP3A4/5, although it is excreted predominantly in its unchanged form.52 The estimated half-life is 12 days following a single dose and 4 days with continuous daily dosing.
Absorption and Distribution: Vismodegib is absorbed orally and has a bioavailability of 31.8%.
The volume of distribution is 16.4 L to 26.6 L. Vismodegib binds to serum albumin and α1-acid glycoprotein.
Mechanism of Action: Vismodegib is a firstin-class small-molecule inhibitor of the 7-pass transmembrane protein SMO, a key member of the HH signal-transduction pathway. Inhibition of SMO precludes activation and translocation of the transcription factor GLI (glioma-associated oncogene homolog), thereby decreasing the induction of genes involved with cell proliferation and survival.
INDICATIONS AND CONTRAINDICATIONS
Vismodegib was approved by the FDA on January 30, 2012, for treatment of adult patients with metastatic or
3573
28
unresectable BCC. Approval stemmed from data from the ERIVANCE BCC trial, a multicenter, Phase II, single-arm, two-cohort, open-label II trial of 104 patients with locally advanced (n = 63) or metastatic BCC (n = 33).53 Vismodegib taken orally produced objective responses in 10 (30.3%) patients with metastatic BCC and 27 (42.8%) patients with locally advanced BCC. Median duration of response was 7.6 months. There are no contraindications on the manufacturer’s label.
DOSING REGIMEN
Recommended dosing for vismodegib is 150 mg daily by mouth for metastatic and locally advanced BCC. It is usually continued until disease progression or the development of unacceptable adverse effects. The safety and effectiveness of vismodegib has not been established in patients with hepatic or renal impairment.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: Treatment-emergent adverse effects are commonly experienced with vismodegib. In the STEVIE (Safety Events in Vismodegib) study, a multicenter, open-label trial of 499 patients with metastatic BCC or locally advanced BCC, 491 patients had at least 1 adverse effect.54 Common adverse effects include muscle spasms, dysgeusia, asthenia, decrease in weight, fatigue, nausea, decrease in appetite, and diarrhea. Cutaneous adverse effects are common. Alopecia occurs in the majority of patients (58% to 63%).55 Keratoacanthomas and well-differentiated squamous cell carcinomas occurring during treatment also have been described.55 Roughly onethird of patients in STEVIE discontinued treatment because of unacceptable toxicity. Grade 5 adverse effects have been reported in patients using vismodegib, although according to the trial investigators, those events were thought to be unrelated to vismodegib.53,54 Premature fusion of the epiphyses has been reported in pediatric patients exposed to Erivedge, and in some cases, the fusion progressed after discontinuation of the drug.52
Animal reproductive studies demonstrated that vismodegib is teratogenic, embryotoxic, and fetotoxic. In rats, doses approximately 0.2 times the area under the curve of the recommended dose in humans resulted in malformation, retardations, or variations in skeletal and visceral structures. As a result, the Erivedge product label contains a boxed warning of embryofetal toxicity. Women should be screened for pregnancy 7 days prior to starting and it is recommended that females avoid pregnancy during treatment and for up to 24 months after the last dose. It is recommended that male patients use condoms with spermicide during treatment and for 3 months after discontinuation. Women should be counseled not to breastfeed during treatment and for 24 months after the last dose.
3574
Patients are advised not to donate blood products during treatment and for 24 months after completing treatment.
Drug Interactions: There is little definitive evidence at this time of drug interactions with vismodegib. Data indicates that vismodegib is a substrate of the P-glycoprotein efflux transporter; consequently, coadministration with inhibitors of P-glycoprotein, such as macrolide antibiotics, may increase drug levels and systemic toxicity. Medications that affect gastric pH may reduce the drug’s bioavailability, but there are no formal studies evaluating the effect of pH-altering drugs on vismodegib. See the Erivedge package insert for full drug–drug interactions and adverse effects.52
SONIDEGIB (ODOMZO)
SONIDEGIB (ODOMZO)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Sonidegib, N-[6-(cis-2,6-dimethylmorpholin-4-yl)pyridine-3-yl]-2-methyl-4′-(trifluoromethoxy) [1,1′-biphenyl]-3-carboxamide diphosphate, has the molecular formula C26H26F3N3O3 • 2H3PO4 and molecular weight of 681.49 daltons. Figure 194-14 shows the structure of sonidegib.
Metabolism: Sonidegib is metabolized by CYP3A, and the compound and its metabolites are primarily excreted via the enterohepatic circulation. The elimination half-life is approximately 28 days and 70% of the absorbed dose is eliminated in the feces and 30% is excreted in the urine.56
Absorption and Distribution: When given orally, less than 10% of the dose is absorbed. Consumption of a high-fat meal results in an increased systemic exposure of the drug. Steady-state volume of distribution is 9166 L with the compound exhibiting high binding to plasma proteins.
Mechanism of Action: Sonidegib, like vismodegib, is a small-molecule inhibitor of the 7-pass, transmembrane protein SMO. Consequently, exposure to the compound is thought to attenuate the expression of HH signaling genes involved with cell proliferation and survival.
INDICATIONS AND CONTRAINDICATIONS
Sonidegib was approved by the FDA on July 24, 2015, for treatment of adults patients with locally advanced BCC that recurs following surgical excision or radiotherapy, and for those patients who are not candidates for surgery or radiotherapy. The approval was based on data from the BOLT (Basal Cell Carcinoma Outcomes With LDE225 Treatment) trial, an international multicenter, randomized, 2-arm, noncomparative trial of 230 patients.57 Daily administration of 200 mg of sonidegib produced objective responses in 43% of patients, and 800 mg daily achieved objective responses of 38%. The drug has no contraindications on the product label.
DOSING REGIMEN
Recommending dosing is 200 mg by mouth daily, taken at least 1 hour prior to or 2 hours after a meal, until disease progression or unacceptable adverse effects. Prior to starting treatment, it is recommended to obtain verification of the pregnancy status of women with reproductive potential, and serum creatinine kinase and kidney function tests for all patients. There are no recommended dose adjustments for patients with mild-to-moderate renal impairment (CrCl 30 to 59 mL/min) or for patients with mild hepatic impairment. Data is lacking in patients with more significant liver impairment.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse events include muscle spasms, alopecia, dysgeusia, nausea, elevated creatinine kinase, fatigue, weight loss, anorexia, myalgia, headache, and pruritus. Adverse effects limit the duration of treatment in roughly one-third of patients. One case of rhabdomyolysis was reported by the study investigators, although upon independent review, the case was deemed not to be consistent with rhabdomyolysis. Nevertheless, baseline creatinine kinase levels are suggested and dose interruptions are recommended for the first occurrence of serum creatinine kinase elevation between 2.5 and 10 times the ULN. Although there are no data from pregnant women, animal toxicity studies demonstrated that sonidegib, like vismodegib, is teratogenic, embryotoxic, and fetotoxic. As a result, there is a boxed warning on the Odomzo package label for embryofetal toxicity. Consequently, women with childbearing potential should be warned of the potential embryofetal death and severe birth defects and should be advised to avoid pregnancy during treatment and for at least 20 months after the last dose. Although the concentration of sonidegib in semen has not been evaluated, it is recommended that male patients use condoms with spermicide during treatment and for at least 8 months after discontinuation. There is inadequate literature to assess the safety
28
of sonidegib in regards to lactation; consequently, it is recommended that women avoid breastfeeding during treatment with sonidegib for at least 20 months after the last dose. There are no data regarding the effects of sonidegib on human reproduction; however, the compound did decrease fertility in female rodents at doses 1 to 2 times the recommended human dose. Patients are advised not to donate blood products during treatment and for 20 months after completing treatment.
Drug Interactions: Given that sonidegib is metabolized by CYP3A, inhibitors and inducers of CYP3A can affect drug concentrations and such agents should be used with caution. Preclinical studies suggest that sonidegib inhibits CYP2B6 and CYP2C9 and thus can affect drug levels of those substrates. See the Odomzo package insert for full drug–drug interactions and adverse effects.56
HISTONE DEACETYLASE INHIBITORS
AT-A-GLANCE
■ Histone modification is a key epigenetic phenomena that regulates gene expression.
■ Abnormalities in the acetylation of histone proteins is a common occurrence in several malignancies.
■ Regulation of histone acetylation is controlled by histone acetyltransferases and histone deacetylases (HDACs).
■ Inhibitors of HDACs have demonstrated efficacy as cancer therapy and their administration is thought to induce differentiation, cell-cycle arrest and apoptosis through restoration of normal acetylation of histone and nonhistone proteins.
■ Of the 4 HDAC inhibitors currently approved, 2— vorinostat and romidepsin—have FDA approval for the treatment of cutaneous T-cell lymphoma.
BACKGROUND
BACKGROUND
Dysregulated gene expression is a hallmark of oncogenesis. Aberrant expression of genes can result from mutations of the DNA nucleotide sequence or via epigenetic modulations, such as chromatin modification. DNA and histone complexes form the basic structural unit of chromatin, the nucleosome. The conformational structure of nucleosomes and chromatin is regulated, in part, by acetylation of histone proteins. In its condensed form, negatively charged DNA is wrapped tightly around positively charged histone proteins, limiting the ability of transcription factors to access DNA promoter
3575
28
Mechanism of histone deacetylase inhibitors
regions. The acetylation of lysine residues in the tails of histones by histone acetyltransferases neutralizes the positive charge on histones (Fig. 194-15). This results in a more relaxed structure for chromatin and permits greater access to gene promoters by transcription factors, facilitating gene expression. In contrast, the activity of histone deacetylases (HDACs) promotes an underacetylated state, contributing to transcriptional silencing by impairing nucleosome accessibility. The fine balance between acetylation and deacetylation is disrupted in several human diseases. In cancer cells, histone deacetylation is associated with the downregulation of proapoptotic genes as well as of genes critical for differentiation.58 In addition to chromatin modulation, histone acetyltransferases and HDACs have been shown to be involved in the regulation of nonhistone proteins involved in oncogenesis, such as p53, nuclear factor κB, E2F, and hypoxia-inducible factor 1α.58,59
Consequently, inhibitors of HDACs have been developed for therapeutic use and have shown promise in several malignancies, including cutaneous T-cell lymphoma. Although the exact therapeutic mechanism is unknown, drugs that inhibit HDACs are thought to induce differentiation, cell-cycle arrest and apoptosis through restoration of normal acetylation.58 The FDA has approved 4 HDAC inhibitors for cancer therapy: vorinostat, romidepsin, belinostat, and panobinostat.
3576
Vorinostat and romidepsin have approval for use in cutaneous T-cell lymphoma, while romidepsin and belinostat are indicated to treat peripheral T-cell lymphoma. Panobinostat is approved for use in multiple myeloma.
VORINOSTAT (ZOLINZA)
VORINOSTAT (ZOLINZA)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Vorinostat, suberanilohydroxamic acid or N-hydroxy-N′-phenyloctanediamide, is a hydroxamic acid derivative with the empirical formula C14H20N2O3 and molecular weight of 264.32 daltons. Figure 194-16 shows the structure of vorinostat.
Metabolism: Glucuronidation and hydrolysis are the major metabolic pathways of vorinostat. There is negligible biotransformation by CYP. The mean terminal half-life is approximately 2 hours.
Absorption and Distribution: When taken orally with a high-fat meal, the median time to maximum concentration is 4 hours. Steady-state concentrations after multiple doses in the fed-state are achieved in 4 hours (range: 0.5 to 14 hours).
Mechanism of Action: Vorinostat is an inhibitor of the several HDACs including HDAC1, HDAC2, HDAC3, and HDAC6. In vitro, vorinostat results in the accumulation of acetylated histones, and in some transformed cells, induces apoptosis and/or arrest of the cell cycle.60
INDICATIONS AND CONTRAINDICATIONS
Vorinostat was approved in 2006 for treatment of cutaneous T-cell lymphoma in patients with persistent, recurrent, or progressive disease on or following 2 systemic therapies. Approval was based on 2 Phase II trials that demonstrated overall objective response rates ranging from 24% to 30% in heavily pretreated patients.61,62 Improvement in baseline pruritus was also noted in 32% to 45% of patients.61,62 Although vorinostat demonstrates efficacy in patients with refractory disease, it is typically regarded as beyond second-line therapy. There are no specific contraindications on the drug label.
DOSING REGIMEN
The recommended dose of vorinostat is 400 mg by mouth once daily with food. Vorinostat has not been adequately studied in patients with renal impairment. Although renal excretion does not play a role in the elimination of vorinostat, caution is advised in patients with preexisting renal impairment. Compared to patients with normal renal function, patients with mild (bilirubin >1 to 1.5 times ULN or aspartate aminotransferase > ULN but bilirubin ≤ ULN) and moderate (bilirubin 1.5 to ≤3 times ULN) hepatic impairment has mean area-under-the-curve levels that were increased by 50%. Therefore, it is recommended to reduce the dose in patients with mildto-moderate hepatic impairment, although no specific recommendations are made on the package label because of insufficient data.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse reactions (incidence ≥20%) are fatigue, diarrhea, nausea, dysgeusia, thrombocytopenia, anorexia, and weight loss. Alopecia was observed in 18.6% of patients in
28
the clinical trials leading to the drug’s approval.60 The most common serious adverse reactions in the clinical trials that led to approval were pulmonary embolism, squamous cell carcinoma, and anemia. Vorinostat is a pregnancy category D medication. Although there are no adequate studies in pregnant women, preclinical animal studies demonstrated that vorinostat may cause fetal harm. Thus, it is recommended to advise avoidance of vorinostat during pregnancy. The safety of vorinostat during breastfeeding is unknown. However, because it is possibly unsafe for use, lactating women should be made aware of the potential harm to infants during nursing.
Drug Interactions: GI bleeding and severe thrombocytopenia have been observed when vorinostat is used concurrently with other HDAC inhibitors such as valproic acid. Prolongation of prothrombin time and international normalized ratio were observed when vorinostat was coadministered with coumarin-derivative anticoagulants. Close monitoring of prothrombin time and international normalized ratio is recommended with concurrently administered vorinostat and coumarin derivatives such as warfarin. See the Zolinza package insert for full drug–drug interactions and adverse effects.60
ROMIDEPSIN (ISTODAX)
ROMIDEPSIN (ISTODAX)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Romidepsin, (1S,4S,7Z,10S,16E,21R)- 7-ethylidene-4,21-bis(1-methylethyl)-2-oxa-12,13- dithia-5,8,20,23-tetraazabicyclo[8.7.6]tricos-16- ene-3,6,9,19,22-pentone, has the empirical formula C24H36N4O6S2 and a molecular weight of 540.71 daltons.63 Figure 194-17 shows the structure of romidepsin.
Metabolism: Romidepsin is principally metabolized by CYP3A4 with partial contribution by CYP3A5, CYP2B6, CYP2C19, and CYP1A1. The elimination halflife is approximately 3 hours.
Absorption and Distribution: When given intravenously in concentrations ranging from 1 to 24 mg/m2 romidepsin exhibits linear pharmacokinetics.
3577
28
The drug is a substrate for the P-glycoprotein ABCB1 efflux transporter.
Mechanism of Action: Romidepsin acts as a HDAC inhibitor. In preclinical studies, romidepsin was shown to induce cell-cycle arrest and apoptosis in some cancer cell lines.63 The full mechanism of action, however, has not been fully elucidated.
INDICATIONS AND CONTRAINDICATIONS
Romidepsin was approved by the FDA in 2009 for treatment of cutaneous T-cell lymphoma in patients who have received at least 1 previous systemic therapy. Approval was based on 2 Phase II clinical trials. Clinical benefit was noted across all stages of disease, with overall response rates of 34%, including 38% in patients with advanced disease (Stage IIB or greater). Complete responses were noted in 5.6% to 7% of patients.64,65
In 2011, the FDA expanded approval for treatment of peripheral T-cell lymphoma in patients who have received at least 1 prior therapy. There are no specific contraindications on the drug label.
DOSING REGIMEN
Romidepsin is administered as an IV infusion. The recommended dose is 14 mg/m2 given over 4 hours on days 1, 8, and 15 of a 28-day cycle. Cycles are repeated for as long as the patient tolerates the treatment and continues to demonstrate a clinical benefit. Although there have been no dedicated hepatic or renal impairment studies with romidepsin, caution is advised in patients with moderate-to-severe hepatic impairment and endstage renal impairment.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects are nausea, asthenia/fatigue, thrombocytopenia, vomiting, diarrhea, diarrhea, pyrexia, constipation, neutropenia, and electrocardiogram T-wave changes. The most common serious adverse effect observed in trials leading to romidepsin’s approval was infection. Additional serious adverse effects that occurred in at least 2% of patients included pyrexia, vomiting, cellulitis, deep vein thrombosis, febrile neutropenia, abdominal pain, chest pain, pulmonary embolism, dyspnea, and dehydration. One case of grade IV “dermatitis medicamentosa” and a single case of oral candidiasis were noted in the pivotal Phase II trial.65 Several treatment-emergent changes in the morphology of electrocardiograms were observed in the clinical studies. Thus, the package label recommends considering cardiovascular monitoring in patients with congenital long QT syndrome, patients taking QT-prolonging medications, and in patients with a history of significant cardiovascular disease.
3578
Based on the mechanism of action and preclinical animal studies, romidepsin may produce fetal harm when administered to pregnant women. Thus, it is recommended to advise avoidance of romidepsin during pregnancy. Romidepsin is possibly unsafe for use during breastfeeding, and lactating women should be made aware of the potential harm to infants during nursing.
Drug Interactions: Concomitant use of strong CYP3A4 inducers such as rifampin is not recommended. Close monitoring of toxicities is recommended when coadministering strong CYP3A4 inhibitors and warfarin and coumarin derivatives. See the Istodax package insert for full drug–drug interactions and adverse effects.63
BELINOSTAT (BELEODAQ)
BELINOSTAT (BELEODAQ)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Belinostat, (2E)-N-hydroxy-3-[3- (phenylsulfamoyl)phenyl]prop-2-enamide, has the molecular formula C15H14N2O4S and the molecular weight is 318.35 g/mol.66 Figure 194-18 shows the structure of belinostat.
Metabolism: Belinostat is primarily metabolized by UGT1A1, but also undergoes hepatic metabolism by CYP3A4, CYP2A6, and CYP2C9. The elimination half-life is 1.1 hours.
Absorption and Distribution: After intravenous injection, the mean volume of distribution of belinostat approaches total body water.
Mechanism of Action: Belinostat inhibits the activity of HDACs. In preclinical studies, belinostat was shown to induce cell-cycle arrest and apoptosis in some cancer cells. Compared to normal cells, belinostat demonstrates preferential cytotoxicity to transformed cells.66
INDICATIONS AND CONTRAINDICATIONS
Beleodaq was granted FDA approval in 2014 for treatment of relapsed or refractory peripheral T-cell lymphoma. Approval was based on a pivotal Phase II trial of 120 patients with relapsed or refractory disease. Overall response rates were 25.8%, including a complete response in 13 patients (10.8%).67
There are no specific contraindications on the drug label.
DOSING REGIMEN
The recommended dosing and schedule of Beleodaq is 1000 mg/m2 administered IV on days 1 to 5 of a 21-day cycle. Patients with moderate-to-severe hepatic impairment were not included in the pivotal clinical trials; as a result, there is insufficient data to recommend a dose of Beleodaq in patients with hepatic impairment. The exposure to Beleodaq was not altered in patients with a CrCl of greater than 39 mL/min. There is insufficient data to recommend a dose of Beleodaq in patients with a CrCl of less than 39 mL/min.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects of belinostat are nausea, fatigue, pyrexia, anemia, vomiting, constipation, diarrhea, dyspnea, rash, and peripheral edema. The most common serious adverse effects include pneumonia, pyrexia, infection, anemia, increased creatinine, thrombocytopenia, and multiorgan failure. Belinostat is a pregnancy category D medication. It may cause embryofetal lethality as it targets actively dividing cells and is genotoxic.66 Thus, it is recommended to advise avoidance of belinostat during pregnancy. The safety of belinostat during breastfeeding is unknown. However, because it is possibly unsafe for use, nursing women should be made aware of the potential harm to infants during breastfeeding.
Drug Interactions: It is recommended to avoid concomitant use of belinostat with strong UGT1A1 inhibitors. See the Beleodaq package insert for full drug–drug interactions and adverse effects.66
PANOBINOSTAT (FARYDAK)
PANOBINOSTAT (FARYDAK)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Panobinostat, (2E)-N-hydroxy-3-[4- [[[2- (2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]- 2-propenamide, has the molecular formula C21H23N3O2 • C3H6O3 and molecular weight of 439.51 daltons (as a lactate), which is equivalent to 349.43 of free base.68 Figure 194-19 shows the structure of panobinostat.
28
Metabolism: Panobinostat is metabolized via oxidation, reduction, hydrolysis, and glucuronidation. Approximately 40% of the compound is metabolized by CYP3A. Minor contributions come via CYP2D6 and CYP2C19. Glucuronidation occurs via UGT1A1, UGT1A3, UGT1A7, UGT1A8, UGT1A9, and UGT2B4. The elimination half-life is approximately 37 hours.
Absorption and Distribution: The oral bioavailability of panobinostat is approximately 21% and peak plasma concentrations are achieved within 2 hours of oral administration. In vitro, panobinostat is approximately 90% bound to plasma proteins.
Mechanism of Action: Panobinostat is an HDAC inhibitor. In vitro, panobinostat was shown to induce cell-cycle arrest and apoptosis in some transformed cells. Panobinostat demonstrates preferential cytotoxicity to transformed cells compared to normal cells.
INDICATIONS AND CONTRAINDICATIONS
Farydak was approved by the FDA in 2015 for use in combination with bortezomib and dexamethasone as treatment of multiple myeloma in patients who have received at least 2 prior regimens. Panobinostat does not have any current FDA-approved indications for dermatologic disease, although there is an ongoing Phase Ib/II trial exploring the treatment of resistant metastatic melanoma with panobinostat and the epigenetic modifier decitabine along with temozolomide (NCT00925132). There are no specific contraindications on the drug label.
DOSING REGIMEN
The recommended dose of panobinostat is 20 mg, taken by mouth once every other day for 3 doses per week (on days 1, 3, 5, 8, 10, 12) of weeks 1 and 2 of each 21-day cycle for 8 cycles. It is recommended to reduce the starting dose of panobinostat to 15 mg for patients with mild hepatic impairment and to 10 mg in patients with moderate hepatic impairment. Use in patients with severe hepatic impairment is not recommended. The exposure to Farydak was not altered in patients with mild or severe renal impairment. It was not studied in patients with end stage renal disease or those on dialysis. Dose and/or schedule modifications of Farydak may be required based on toxicity. See the Farydak package insert for the appropriate dose reduction.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects (incidence of at least 20%) in clinical studies are
3579
28
diarrhea, fatigue, nausea, peripheral edema, anorexia, pyrexia, and vomiting. The most common hematologic adverse effects (≥60% incidence) include thrombocytopenia, lymphopenia, leukopenia, neutropenia, and anemia. Electrolyte abnormalities, such as hypophosphatemia, hypokalemia, and hyponatremia, occur in 40% or more of patients. Farydak possesses a boxed warning for severe diarrhea and severe and fatal cardiac ischemic events, severe arrhythmias, and electrocardiogram changes. Severe diarrhea occurred in 25% of patients receiving Farydak. Fatal and serious cases of GI and pulmonary hemorrhage have been reported. Farydak may cause fetal harm if administered during pregnancy. In preclinical studies, panobinostat was teratogenic in rabbits and rats. Patients should be advised to avoid getting pregnant while taking Farydak. It is also recommended that women use highly effective contraception for at least 1 month after the last dose of Farydak. Men should be advised to use condoms while on treatment and for 3 months after the last dose of Farydak. The safety of panobinostat during breastfeeding is unknown. However, because it is possibly unsafe for use, nursing women should be made aware of the potential harm to infants during breastfeeding.
Drug Interactions: It is recommended to avoid concomitant use of Farydak with strong inducers of CYP3A4, CYP2D6 substrates, and antiarrhythmic/ QT-prolonging drugs. The dose of Farydak should be reduced with concomitant use of strong inhibitors of CYP3A4. See the Farydak package insert for full drug–drug interactions and adverse effects.68
MITOGEN-ACTIVATED PROTEIN KINASE INHIBITORS
AT-A-GLANCE
■ Between 75% and 85% of melanomas harbor oncogenic mutations in the mitogen-activated protein kinase (MAPK) pathway, including BRAF (v-raf murine sarcoma viral oncogene homolog B), RAS, and neurofibromin 1 (NF1).
■ Activating mutations in BRAF, such as V600E and V600K, can trigger unregulated cell growth and promote melanocyte transformation.
■ Vemurafenib and dabrafenib are inhibitors of certain mutated forms of BRAF, including V600E and V600K.
■ Trametinib and cobimetinib are inhibitors of mitogen-activated extracellular signal-regulated kinase (MEK).
3580
■ Combination BRAF and MEK inhibition can lead to clinical benefit, including improved overall survival, in patients with unresectable or metastatic melanoma in patients harboring BRAF V600E or V600K mutations.
BACKGROUND
BACKGROUND
Melanomagenesis results from the dysregulation of a number of cellular circuits including the PI3K pathway, telomerase promoter, the retinoblastoma pathway, and the MAPK pathway. Targeted therapy in melanoma became a possibility following the 2002 discovery that activating mutations of the protooncogene v-raf murine sarcoma viral oncogene homolog B (BRAF) drive nearly half of melanomas.69 BRAF, a serine-threonine kinase in the MAPK pathway, is normally activated by RAS, resulting in signal transduction via the phosphorylation of mitogen-activated extracellular signal-regulated kinase (MEK) (Fig. 194-20). Mutations in BRAF—usually V600E, but occasionally V600K and other substitutions—result in constitutive activation of the kinase domain, leading to RAS-independence and hyperactivation of downstream mediators such as MEK and extracellular signal-regulated kinase (ERK). Aberrant activation of the MAPK pathway can lead to cell-cycle dysregulation, resistance to apoptosis, immune evasion, and increased invasion and metastases. The discovery that gain-of-function alterations in BRAF were a key determinant in melanomagenesis drove the development of therapeutic strategies to intervene at multiple nodes in the MAPK pathway. Precision medicine in melanoma became a possibility with the demonstration that the small-molecule inhibitors of mutant BRAF, vemurafenib and dabrafenib, could prolong disease-free and overall survival when used as monotherapy.70-72 In addition, inhibition of MEK with trametinib monotherapy improves PFS in patients with BRAF-mutant melanoma.73 Although initially promising, it became evident that the acquisition of resistance to single-agent therapy precludes durable responses in advanced disease. Building off of preclinical data that showed dual-targeting of BRAF and MEK cooperate to reduce MAPK output, clinical trials have subsequently validated the safety and superior efficacy of combined BRAF/MEK inhibition in BRAF-mutant melanoma.74-77 Indeed, many of the dermatologic toxicities seen with single-agent BRAF inhibition, such as cSCC and keratoacanthomas, are attenuated with dual BRAF inhibition/MEK inhibition. However, despite reducing many of the adverse effects and delaying the emergence of therapeutic resistance, combined BRAF inhibition/MEK inhibition does not entirely circumvent mechanisms of resistance and the vast majority of patients progress on treatment. In addition, 20% to 25% of patients have mutations in MAPK signaling
28
Mitogen-activated protein kinase pathway
Vemurafenib
BRAF
Trametinib
MEK
Growth Factor Receptor
RAS RAS NF1
P
P
Shc
GDP GTP
P
P
Grb2
SOS
Dabrafenib
Cobimetinib
ERK
Cell Proliferation and Survival
proteins that are not currently druggable, such as RAS and neurofibromin 1. Consequently, additional targets and approaches are being explored such as AKT and ERK, as well as immune checkpoint blockade.
VEMURAFENIB (ZELBORAF)
VEMURAFENIB (ZELBORAF)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Vemurafenib, propane-1-sulfonic acid {3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine- 3-carbonyl]-2,4-difluoro-phenyl}-amide, has the molecular formula C23H18ClF2N3O3S and molecular weight of 489.92 daltons. Figure 194-21 shows the structure of vemurafenib.
Metabolism: In vitro studies demonstrate that vemurafenib is a substrate of CYP3A4. Following oral administration, 94% of the dose of vemurafenib was recovered in the feces, while approximately 1% was found in the urine. The median elimination half-life is 57 hours.78
3581
28
Absorption and Distribution: The bioavailability of vemurafenib has not been determined; however, following multiple doses, the median time to maximum plasma concentration was approximately 3 hours. In the clinical trials leading to its approval, Zelboraf was taken without regard to food.
Mechanism of Action: Vemurafenib is a smallmolecular-weight inhibitor of certain mutated forms of the serine-threonine kinase BRAF, including V600E. In addition, vemurafenib inhibits wildtype BRAF, CRAF, ARAF, SRMS, ACK1, MAP4K5, and FGR. In cells with V600E-mutated BRAF, vemurafenib has antitumor effects.
INDICATIONS AND CONTRAINDICATIONS
The FDA approved Zelboraf in 2011 for use as monotherapy in the treatment of unresectable or metastatic melanoma with the BRAFV600E mutation, as detected by an FDA-approved test. Approval was based on 2 trials that demonstrated improved response rates and survival rates as compared to dacarbazine.70,71 The median PFS was 5.3 months and 1.6 months, and the objective response rates were 48% and 5% in the vemurafenibtreated and dacarbazine-treated groups, respectively.71
The median overall survival in patients treated with vemurafenib was 15.9 months in the single-arm Phase II trial.70
When used in combination with the MEK inhibitor cobimetinib (Cotellic), vemurafenib demonstrated improved efficacy, achieving an overall response rate of 68% and a PFS of 9.9 months.77
Zelboraf is also indicated to treat patients with Erdhiem-Chester disease harboring V600 mutations. There are no contraindications listed on the drug label.
DOSING REGIMEN
The recommended dose of vemurafenib is 960 mg, taken by mouth with or without a meal, twice daily administered approximately 12 hours apart. No formal studies have been conducted to evaluate the effects of renal or hepatic impairment on the pharmacokinetics of Zelboraf. No dose adjustments are recommended for patients with mild and moderate renal or hepatic impairment. Caution is advised for severe renal or hepatic impairment.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common noncutaneous adverse effects of Zelboraf include arthralgia, fatigue, and nausea. Zelboraf is associated with a variety of skin toxicities, including rash, alopecia, photosensitivity, pruritus, and new primary malignancies. Skin eruptions occurred in up to 68% of the patients taking vemurafenib.70,71,79 The rashes have been described as folliculocentric, toxic erythema– like, or exaggerated keratosis pilaris–like.80 The toxic
3582
erythema–like rash has been described as having a morphology similar to a classic maculopapular drug hypersensitivity rash with histopathologic features suggestive of an exanthematous drug eruption.80
Squamous proliferations ranging from benign papillomas and verrucous keratoses to keratoacanthomas and squamous cell carcinomas are commonly seen in patients taking single-agent vemurafenib. Papillomas were reported in 29% of patients and cSCCs and keratoacanthomas were described in 26% of patients in the Phase II trial.70 In a newer prospective study of the cutaneous adverse effects of vemurafenib, verrucous papillomas occurred in 79% of patients, with nearly half of the patients developing between 4 and 20 efflorescent and verruciform lesions.81 A hand–foot skin reaction characterized by hyperkeratotic plaques located on pressure points of the sole was observed in 60% of patients.81 Additional skin toxicities that have been described with monotherapy include hair growth modification, hyperkeratotic follicular rash, xerosis, cystic lesions, facial erythema, cheilitis, nipple hyperkeratosis, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms syndrome, nevi efflorescence, and radiodermatitis.81 Several of the cutaneous adverse effects appear to be dependent on MEK output via paradoxical activation of MAPK signaling, as concomitant use of the MEK inhibitor cobimetinib was associated with lower rates of keratoacanthomas, cSCCs, and alopecia.77
Additional adverse effects of Zelboraf include hepatoxicity, QT prolongation, uveitis, photophobia, and renal injury, including interstitial nephritis and acute tubular necrosis. Based on its mechanism of action, Zelboraf may cause fetal harm and women should be made aware of the potential risk to a fetus. It is recommended that women of reproductive age be advised to use highly effective contraception during treatment and for 2 weeks after the final dose of Zelboraf. Although there is no information available regarding the presence of vemurafenib in human milk, the potential for harm to nursing infants exists; therefore, women should be advised to avoid breastfeeding during treatment and for at least 2 weeks following the last dose of Zelboraf.
Drug Interactions: It is recommended to avoid concomitant use of strong CYP3A4 inhibitors or inducers and CYP1A2 substrates with a narrow therapeutic window. See the Zelboraf package insert for full drug–drug interactions and adverse effects.78
DABRAFENIB (TAFINLAR)
DABRAFENIB (TAFINLAR)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Dabrafenib mesylate, N-{3-[5-(2-amino-4- pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-
2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt, has the molecular formula C23H20F3N5O2S2 CH4O3S and a molecular weight of 615.68 daltons.82 Figure 194-22 shows the structure of dabrafenib mesylate.
Metabolism: Dabrafenib is primarily metabolized by CYP2C8 and CYP3A4. Excretion through the feces is the main route of elimination (71%) of dabrafenib, while urinary excretion accounts for 23% of its elimination. After oral administration, the half-life of dabrafenib is 8 hours.
Absorption and Distribution: Dabrafenib exhibits 95% bioavailability and peak plasma concentrations are reached in 2 hours after oral administration; 99.7% of dabrafenib is bound to plasma proteins and its volume of distribution is 70.3 L.
Mechanism of Action: Dabrafenib is a smallmolecular-weight inhibitor of certain mutated forms of the BRAF, including V600E, V600K, and V600D. It is a reversible ATP-competitive inhibitor, with a concentration required for 50% inhibition that is 5 times lower for BRAFV600E than for wildtype BRAF.72
In addition, dabrafenib inhibits wildtype BRAF, CRAF, SIK1, NEK11, and LIMK1. In melanoma cells with V600-mutated BRAF, dabrafenib inhibits cell growth.
INDICATIONS AND CONTRAINDICATIONS
Tafinlar was given FDA approval in 2013 for treatment of unresectable or metastatic melanoma with BRAF V600E mutation, as detected by an FDA-approved test. Approval was based on data from a multicenter, open-label, randomized trial of 250 patients that demonstrated improved PFS as compared to dacarbazine (5.1 months vs. 2.7 months).72 Tafinlar was also given accelerated approval in 2014 and full approval in 2015 for use in combination with the MEK inhibitor trametinib (Mekinist) in unresectable or metastatic BRAF V600E or V600K mutated melanoma. Approval was given after the combination demonstrated objective response rates of 64% to 76% and PFS of 9.3 to 11.4 months.74-76 Long-term followup of patients from that cohort demonstrated on overall survival of 25 months and a 3-year overall survival of 38%.83
Tafinlar is also indicated in combination with Mekinist as: 1) adjuvant treatment for patients with V600
28
mutant melanoma involving lymph nodes following complete resection;2) treatment for metastatic V600 mutant NSCLC; and 3) metastatic or locally advanced V600 mutant anaplastic thyroid cancer with no satisfactory locoregional treatment options. There are no specific contraindications on the drug label.
DOSING REGIMEN
The recommend dose of Tafinlar is 150 mg taken by mouth twice daily at least 1 hour before or at least 2 hours after a meal. An FDA-approved test must be performed that confirms either the presence of a BRAF V600E mutation prior to monotherapy with Tafinlar, or the presence of a BRAF V600E or V600K mutation prior to combined therapy with trametinib. No formal studies have been conducted to evaluate the effects of renal or hepatic impairment on the pharmacokinetics of Tafinlar. No specific dose adjustments are recommended for patients with mild hepatic or mild or moderate renal impairment. An appropriate dose has not been determined for moderate to severe hepatic or severe renal impairment. Dose modifications of Tafinlar may be required based on toxicity. See the Tafinlar product label for the appropriate dose reduction.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common noncutaneous adverse effects include headache, pyrexia, and arthralgia. Cutaneous toxicities are similar to those seen with vemurafenib and include verruciform keratotic squamoproliferative lesions, papillomas, alopecia, rash, Grover disease, plantar hyperkeratosis, cSCC, keratoacanthoma, increased incidence of melanoma, seborrheic dermatitis–like eruption and palmar-plantar erythrodysesthesia syndrome.82,84 New primary malignancies, cutaneous and noncutaneous, have been reported during treatment with Tafinlar. Additional toxicities include hemorrhage, cardiomyopathy, uveitis (including iritis and iridocyclitis), hyperglycemia, and hemolytic anemia in patients with glucose-6-phosphate dehydrogenase deficiency. Based on data from animal studies and its mechanism of action, Tafinlar may cause fetal harm and women should be made aware of the potential risk to a fetus. It is recommended that women of reproductive age be advised to use highly effective contraception during treatment and for 2 weeks after the final dose of Tafinlar. Although there is no information available regarding the presence of dabrafenib in human milk, there is a potential for serious adverse reactions to nursing infants; consequently, women should be advised to avoid breastfeeding during treatment and for at least 2 weeks following the last dose of Tafinlar.
Drug Interactions: It is recommended to avoid concurrent use of strong inhibitors or inducers of CYP3A4 or CYP2C8. Coadministration of compounds that are sensitive substrates of CYP3A4, CYP2B6,
3583
28
CYP2C8, CYP2C9, or CYP2C19 may result in decreased efficacy of these agents. See the Tafinlar package insert for full drug–drug interactions and adverse effects.82
TRAMETINIB (MEKINIST)
TRAMETINIB (MEKINIST)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Trametinib dimethyl sulfoxide, or acetamide, N-[3-[3-cyclopropyl-5-[(2-fluoro-4-iodophenyl) amino]-3,4,6,7-tetrahydro-6,8-dimethyl-2,4,7- trioxopyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl]-, compound with 1,1′-sulfinylbis[methane] (1:1), has the molecular formula C26H23FIN5O4 • C2H6OS and molecular mass of 693.53 daltons. Figure 194-23 shows the structure of trametinib dimethyl sulfoxide.
Metabolism: Trametinib is primarily metabolized by non–CYP-mediated deacetylation alone or in combination with monooxygenation or glucuronidation biotransformation pathways. The estimated elimination half-life is 3.9 to 4.8 hours. Greater than 80% is excreted through the feces, while less than 20% is eliminated through the urine.
Absorption and Distribution: Trametinib is 72% bioavailable and the volume of distribution is 214 L.
Mechanism of Action: Trametinib is a reversible, small-molecule inhibitor of activation and kinase activity of MEK1 and MEK2.85 Trametinib inhibits the in vitro and in vivo cell growth of BRAF V600 mutation-positive melanoma.
INDICATIONS AND CONTRAINDICATIONS
Mekinist is indicated for use as a single agent or in combination with dabrafenib for the treatment of unresectable or metastatic BRAF V600E–mutated or BRAF V600K–mutated melanoma, as determined by an FDA-approved test. Approval as monotherapy was given in 2013 based on the Phase III METRIC (MEK Versus Dacarbazine [DTIC] or Paclitaxel [Taxol] in Metastatic Melanoma) study which demonstrated improved PFS in patients receiving trametinib 2 mg
3584
once daily (4.8 months) as compared to those receiving either dacarbazine or paclitaxel (1.5 months).73
As stated above, Mekinist is synergistic when used in combination with Tafinlar resulting in superior objective response rates (64% to 76%) and PFS (9.3 to 11.4 months) compared to monotherapy.74-76
Mekinist is also indicated in combination with Tafinlar as: 1) adjuvant treatment for patients with V600 mutant melanoma involving lymph nodes following complete resection;2) treatment for metastatic V600 mutant NSCLC; and 3) metastatic or locally advanced V600 mutant anaplastic thyroid cancer with no satisfactory locoregional treatment options. There are no specific contraindications on the product label.
DOSING REGIMEN
The recommend dose of trametinib is 2 mg taken by mouth once daily at least 1 hour before or at least 2 hours after a meal. No dedicated studies have been conducted to evaluate the effects of renal or hepatic impairment on the pharmacokinetics of Mekinist. No specific dose adjustments are recommended for patients with mild hepatic or mild or moderate renal impairment. An appropriate dose of Mekinist has not been determined for moderate to severe hepatic or severe renal impairment. Dose modifications of Mekinist may be required based on toxicity. See the Mekinist package insert for the appropriate dose reduction.
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse effects include rash, diarrhea, and lymphedema. When used in combination with Tafinlar, the most common toxicities include pyrexia, nausea, rash, diarrhea, vomiting, chills, hypertension, and peripheral edema. The most common cutaneous toxicity is a papulopustular eruption similar to those seen with inhibitors of EGFR, occurring in 40% to 93% of patients.86 Other skin changes include pruritus, xerosis, alopecia, paronychia, and eruptions described as maculopapular and urticarial-to-targetoid with central duskiness.86 Other adverse effects include venous thromboembolism, cardiomyopathy, retinal vein occlusion, retinal pigment epithelial detachment, interstitial lung disease, and hyperglycemia. Based on data from animal studies and its mechanism of action, Mekinist may cause fetal harm and women should be made aware of the potential risk to a fetus. It is recommended that women of reproductive age be advised to use highly effective contraception during treatment and for 4 months after the final dose of Mekinist. Increased follicular cysts and decreased corpora lutea were observed in animal studies; thus, women should be advised that Mekinist may impair fertility. Although there is no information available regarding the presence of trametinib in human milk,
there is a potential for serious adverse reactions to nursing infants; consequently, women should be advised to avoid breastfeeding during treatment and for at least 4 months following the last dose of Mekinist.
Drug Interactions: Trametinib is not a substrate of CYP enzymes and at the clinically relevant systemic concentration of 0.04 µM, it is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, or CYP3A4. See the Mekinist package insert for full drug–drug interactions and adverse effects.82
COBIMETINIB (COTELLIC)
COBIMETINIB (COTELLIC)
PHARMACOLOGY AND MECHANISM OF ACTION Structure: Cobimetinib fumarate, (S)-[3,4- difluoro- 2-(2-fluoro-4-iodophenylamino)phenyl] [3-hydroxy-3- (piperidin-2-yl)azetidin-1-yl]methanone hemifumarate, has the molecular formula C46H46F6I2N6O8 and molecular mass of 1178.71 daltons. Figure 194-24 shows the structure of cobimetinib fumarate.
Metabolism: Cobimetinib is primary metabolized by oxidization via CYP3A and glucuronidation by way of UGT2B7. The elimination half-life following oral administration is 44 hours. Excretion is primarily via the fecal route (76%).
Absorption and Distribution: The bioavailability of cobimetinib is 46% and its volume of distribution is 806 L.
Mechanism of Action: Cobimetinib fumarate is a reversible inhibitor of MEK1 and MEK2. The compound inhibits BRAF V600E–mutant murine cells transplanted into mice.87
INDICATIONS AND CONTRAINDICATIONS
Cotellic is indicated for the treatment of unresectable or metastatic BRAF V600E–mutant or BRAF V600K–mutant melanoma, in combination with vemurafenib. Approval was based on results from the coBRIM (Cobimetinib Combined with Vemurafenib in Advanced BRAF V600–mutant Melanoma) trial which showed that the combination of vemurafenib
28
and cobimetinib produced improved PFS and response rates compared to single-agent vemurafenib in patients with previously untreated, unresectable or metastatic BRAF V600 mutation-positive melanoma (PFS: 9.9 months vs. 6.2 months; objective response rate 68% vs. 45%).77
There are no specific contraindications on the product label.
DOSING REGIMEN
The recommended dose of Cotellic is 60 mg by mouth once daily, with or without food, for the first 21 days of a 28-day cycle. No dedicated studies have been conducted to evaluate the effects of renal or hepatic impairment on the pharmacokinetics of Cotellic. No specific dose adjustments are recommended for patients with mild hepatic or mild or moderate renal impairment. An appropriate dose of Cotellic has not been determined for moderate to severe hepatic or severe renal impairment. Dose modifications of Cotellic may be required based on toxicity. See the Cotellic package insert for the appropriate dose reduction.87
SIDE EFFECTS AND PRECAUTIONS Adverse Effects: The most common adverse reactions for Cotellic are diarrhea, photosensitivity reaction, nausea, pyrexia, and vomiting. The most common grade 4 event (4%) was elevated creatinine kinase, a known class effect of MEK blockade.77 Other serious laboratory abnormalities include increased γ-glutamyl transferase, hypophosphatemia, increased alanine aminotransferase and aspartate aminotransferase, lymphopenia, increased alkaline phosphatase, and hyponatremia. Although squamoproliferative events are seen with combination therapy, cSCC and keratoacanthomas appear to be less common with the dual therapy cobimetinib and vemurafenib compared with single-agent vemurafenib.77 A grade 3 to grade 4 rash was seen in 16% of patients receiving dual therapy compared with 17% receiving single agent vemurafenib. Other noncutaneous adverse effects include cardiomyopathy, hemorrhage, noncutaneous malignancies, retinopathy, and rhabdomyolysis. Based on data from animal studies and its mechanism of action, Cotellic may cause fetal harm and women should be made aware of the potential risk to a fetus. It is recommended that women of reproductive age be advised to use highly effective contraception during treatment and for 2 weeks following the final dose of Cotellic. Patients should be advised that Cotellic may impair fertility in both females and males. Although there is no information available regarding the presence of cobimetinib in human milk, there is a potential for serious adverse reactions to nursing infants; therefore, women should be advised to avoid breastfeeding during treatment and for at least 2 weeks following the last dose of Cotellic.
3585
28
Drug Interactions: Moderate or strong inhibitor or inducers of CYP3A should be avoided when taking Cotellic. See the Cotellic package insert for full drug–drug interactions and adverse effects.
FUTURE DIRECTIONS IN MELANOMA: AKT AND ERK INHIBITION
AKT INHIBITORS
AKT INHIBITORS
PI3K–AKT–mTOR (mammalian target of rapamycin) signaling is an important oncogenic pathway in melanoma development and progression. The hyperactivation of this pathway is associated with activation of receptor tyrosine kinases, somatic mutations in key signaling genes such as PIK3CA, or inactivation of the tumor-suppressor gene PTEN.88
Preclinical studies with AKT inhibitors have demonstrated the usefulness of AKT inhibitors in melanoma treatment. Conjunctival melanoma is a rare, but deadly, variant of melanoma with limited effective treatment options. Preclinical in vitro studies demonstrate that the AKT inhibitor, MK2206, suppresses growth in conjunctival melanoma cell lines, and furthermore, a synergistic effect was observed when combined with the MEK inhibitor MEK162.89 These preliminary findings have supported the rationale for a recently initiated Phase II clinical trial studying the efficacy of the MEK inhibitor trametinib, with or without the AKT inhibitor GSK2141795, for treatment of patients with stage IV uveal melanoma (NCT01979523). Also, in a Phase I trial investigating the oral AKT inhibitor MK2206 combined with chemotherapy for solid tumors, 2 patients with stage IV BRAF WT melanoma tolerated and experienced disease-free progression with MK2206 combined with carboplatin and paclitaxel.90
More recently, another Phase I study revealed that the AKT inhibitor ipatasertib (GDC-0068) is capable of safe and robust targeting of AKT in patients with solid tumors.91 These preliminary studies are paving the way for larger randomized, controlled clinical trials, which may result in the addition of AKT inhibitors to the armamentarium of therapeutics for advanced melanoma.
EXTRACELLULAR SIGNAL- REGULATED KINASE INHIBITORS
EXTRACELLULAR SIGNAL-
REGULATED KINASE
INHIBITORS
The MAPK (RAS-RAF-MEK-ERK) pathway is hyperactivated in more than 90% of melanomas.92 RAF and MEK inhibitors were the first clinically available therapeutics aimed at targeting the MAPK pathway for advanced melanoma patients. However, acquired
3586
resistance to BRAF and MEK inhibitors proves to be a major limitation to sustainable outcomes.93 One wellcharacterized mechanism of resistance is the reactivation of the downstream MAPK pathway target ERK; hence, investigators are focusing on the development of ERK inhibitors.93 Preclinical studies show that ERK inhibition is more effective, compared to MEK inhibition, in suppressing MAPK activity and tumor growth in multiple BRAF inhibitor–resistant melanoma cell lines.94 Also, in vitro data demonstrates that MEK inhibitor–resistant melanoma cell lines are sensitive to ERK inhibition, and that combined treatment with MEK and ERK inhibitors is synergistic and overcomes acquired resistance to MEK inhibition.95 Although the preclinical data is promising, ERK inhibitors are capable of targeting a wide range of kinases, hence a potential narrow therapeutic index is a major concern. Currently, there are Phase I clinical trials underway exploring safety and tolerability of the ERK inhibitors, BVD-523 and LTT462, in patients with advanced solid malignancies (NCT01781429, NCT02711345).
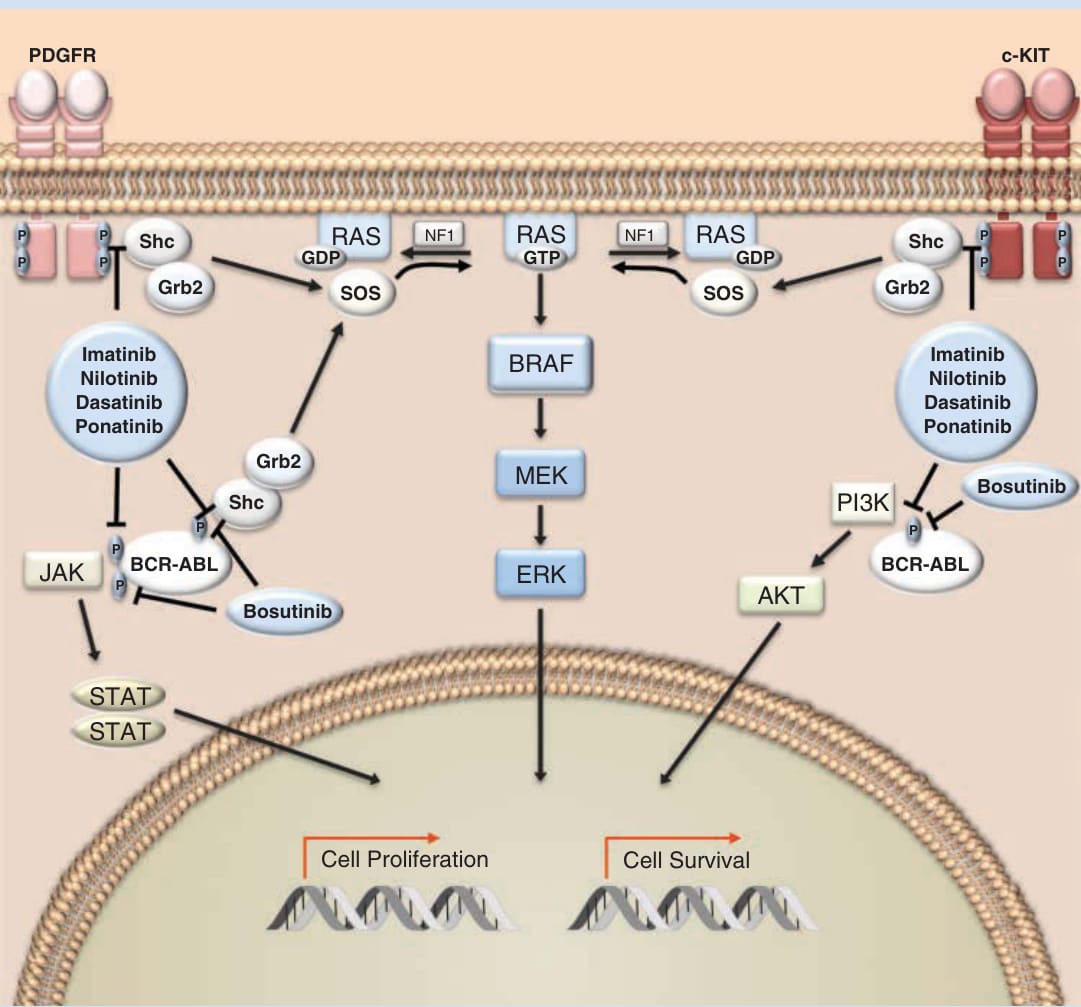
Figure 194-1 Inhibitors of KIT/BCR-ABL/PDGFR. Imatinib, nilotinib, dasatinib, and ponatinib are multi–tyrosine kinase inhibitors that modulate autophosphorylation of tyrosine residues associated with BCR-ABL (breakpoint cluster region–Abelson murine leukemia virus), c-KIT, and PDGFR (platelet-derived growth factor receptor). c-KIT and PDGFR are receptor tyrosine kinases and BCR-ABL is a nonreceptor tyrosine kinase. Bosutinib is an inhibitor of BCR-ABL, but not of c-KIT or PDGFR. The signal transduction pathways downstream of these tyrosine kinases, such as MAPK (mitogen-activated protein kinase) cascade, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) regulate the expression of genes involved in cell growth, proliferation, and cell survival.
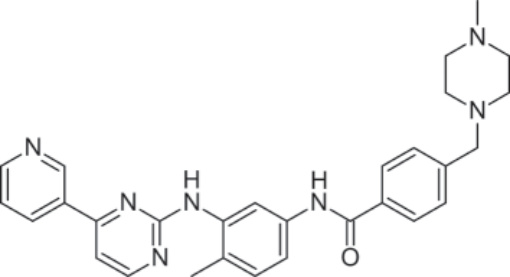
Figure 194-2 Structure of Imatinib.
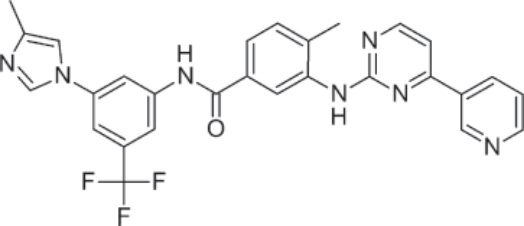
Figure 194-3 Structure of Nilotinib.
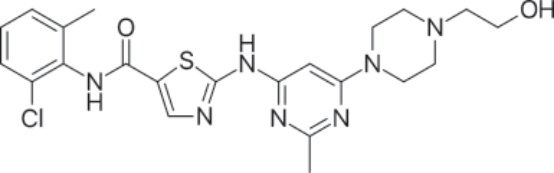
Figure 194-4 Structure of Dasatinib.
Figure 194-5 Structure of Bosutinib.
Figure 194-6 Structure of Ponatinib.
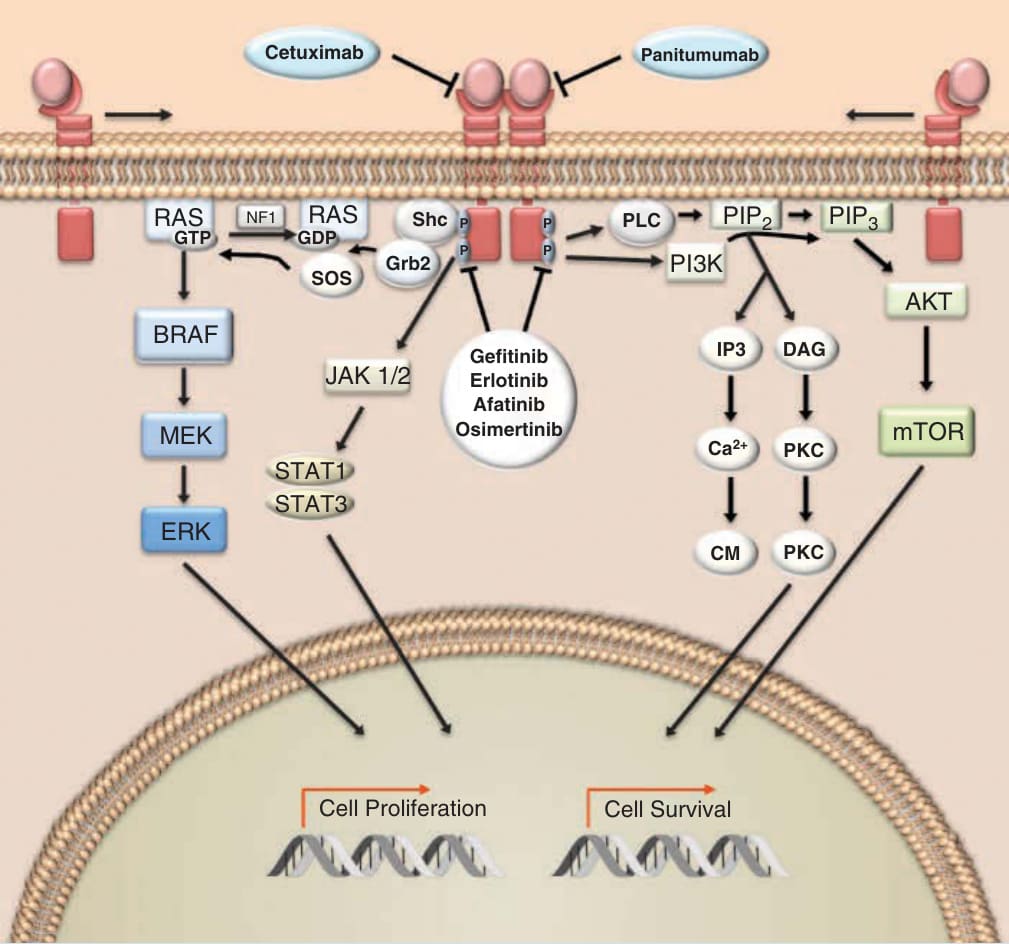
Figure 194-7 Epidermal growth factor receptor (EGFR) pathway. The human epidermal growth receptor is a cell-surface protein comprised of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain. Binding of the epidermal growth factor and other ligands to the extracellular domain results in dimerization in which the receptor either binds with another EGFR protein (seen above) or heterodimerizes with an additional monomer of the ErbB family. Dimerization triggers the intracellular tyrosine kinase to autophosphorylate several tyrosine residues. Subsequent recognition of the phosphorylated tyrosine residues on the C-terminal domain of the receptor by various adaptor proteins initiates downstream signal transduction signaling. EGFR activation can trigger networks involved in cell proliferation, cell survival and metastases such as the mitogen-activated protein kinase (MAPK), Janus kinase(JAK)/signal transducer and activator of transcription (STAT), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), and phospholipase C (PLC) pathways.
Figure 194-8 Structure of gefitinib.
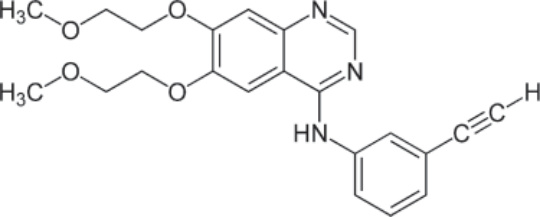
Figure 194-9 Structure of erlotinib.
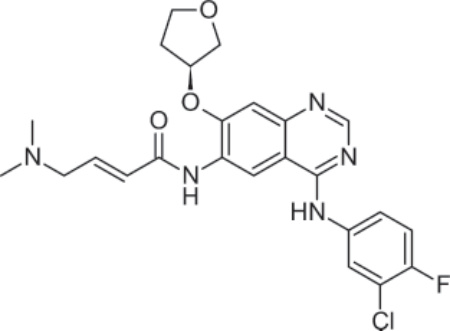
Figure 194-10 Structure of afatinib.
Figure 194-11 Structure of osimertinib.
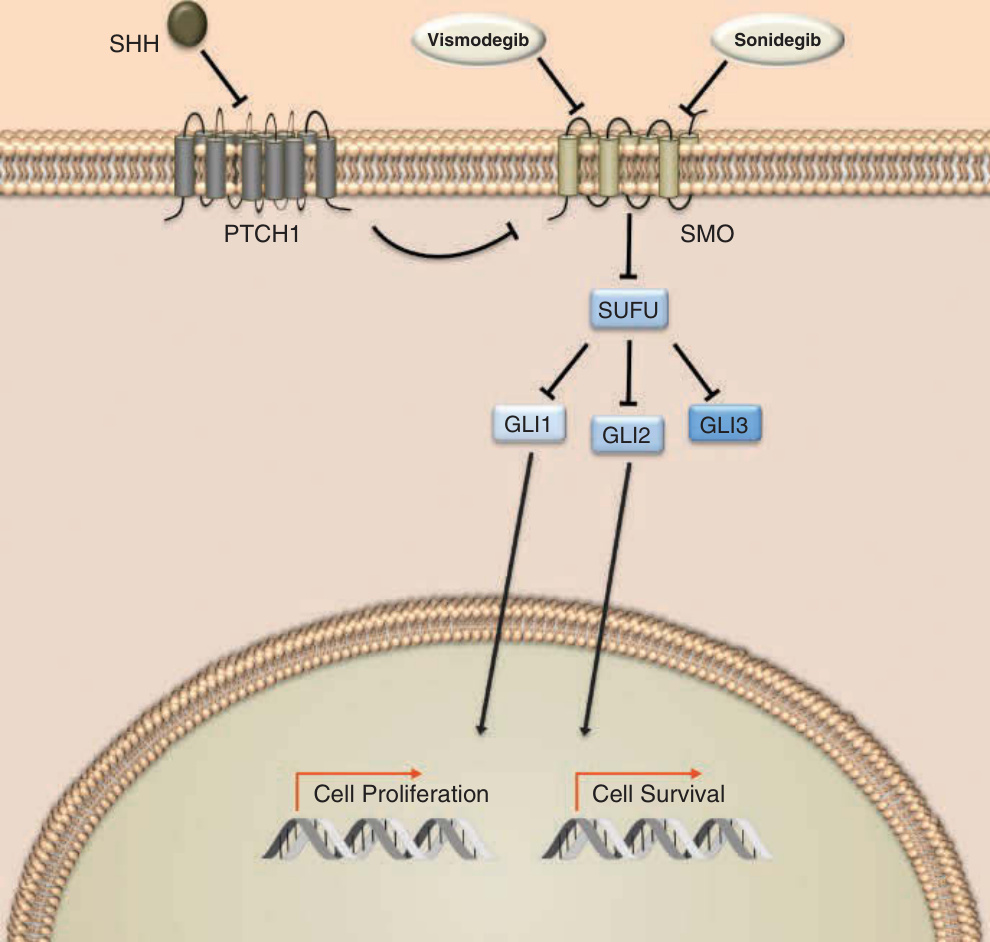
Figure 194-12 Hedgehog signaling. Hedgehog (HH) signaling is initiated by binding of 1 of 3 extracellular HH ligands, sonic hedgehog (SHH), Indian hedgehog (IHH) and desert hedgehog (DHH). These ligands bind to 12-pass transmembrane protein receptors Patched 1 (PTCH1) and Patched 2 (PTCH2). When unbound, Patched receptors interact and inhibit the 7-pass transmembrane transduction protein Smoothened (SMO). Following ligand binding to Patched, the inhibition of SMO is relieved and transduction through a series of interacting proteins, including suppressor of fused (SUFU), results in translocation of the glioma-associated oncogene homolog (GLI) transcription factors: GLI1, GLI2, and GLI3. GLI1 functions exclusively as an activator of transcription, while the actions of GLI2 and GLI3 are dependent on the signaling context. In the absence of HH ligands, GLI3 functions as the primary repressor of the transduction cascade. Following nuclear translocation, GLI transcription factors modulate the expression on numerous cell programs including those involved in cell growth, proliferation, epithelial–mesenchymal transition, angiogenesis, inhibition of apoptosis, and stem cell maintenance. The small molecules vismodegib and sonidegib bind to SMO and impair signaling through the HH pathway.
Figure 194-13 Structure of vismodegib.
Figure 194-14 Structure of sonidegib.
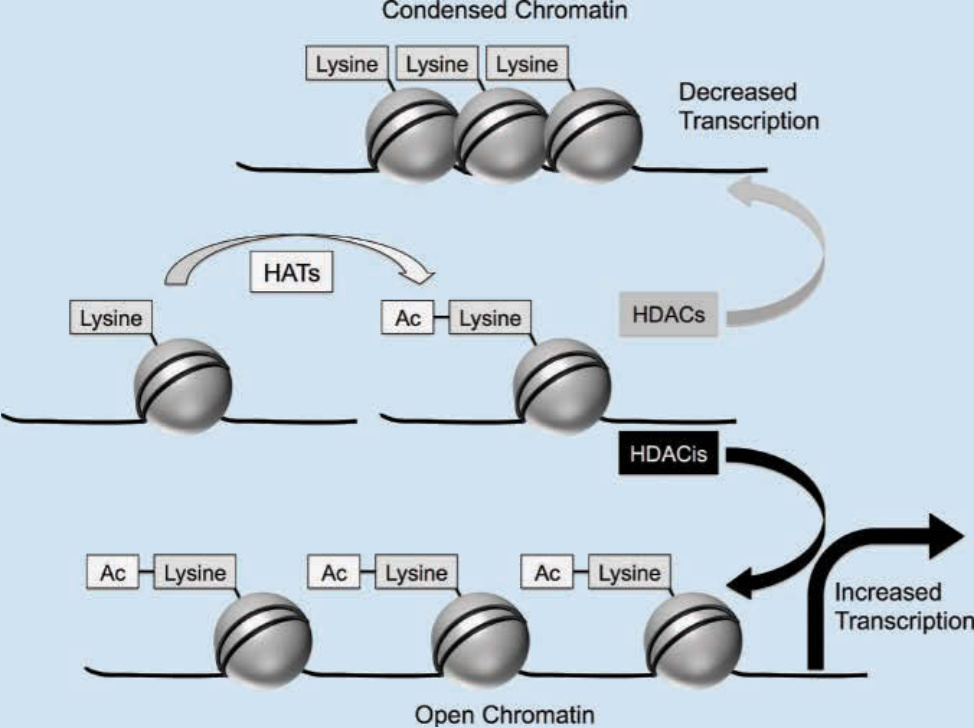
Figure 194-15 Mechanism of histone deacetylase inhibitors. In its condensed form, negatively charged DNA (black lines) is wrapped tightly around positively charged histone proteins (gray spheres), limiting the ability of transcription factors to access DNA promoter regions. Histone acetyltransferases (HATs) neutralize the positive charge on histones by the addition of acetyl groups to the lysine residues in the tails of histones. Acetylation of these residues results in a more relaxed structure for chromatin and permits greater access to gene promoters by transcription factors, facilitating gene expression. In contrast, the activity of histone deacetylases (HDACs) promotes an underacetylated state, contributing to transcriptional silencing by impairing nucleosome accessibility (upper panel). Agents that inhibit HDACs (HDACis) are thought to restore normal acetylation, resulting in increased transcription of genes that regulate differentiation, cell-cycle arrest, and apoptosis (bottom panel).
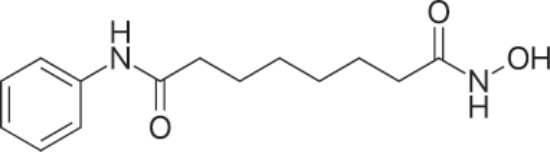
Figure 194-16 Structure of vorinostat.
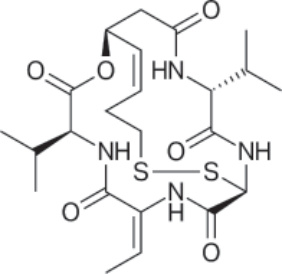
Figure 194-17 Structure of romidepsin.
Figure 194-18 Structure of belinostat.
Figure 194-19 Structure of panobinostat.
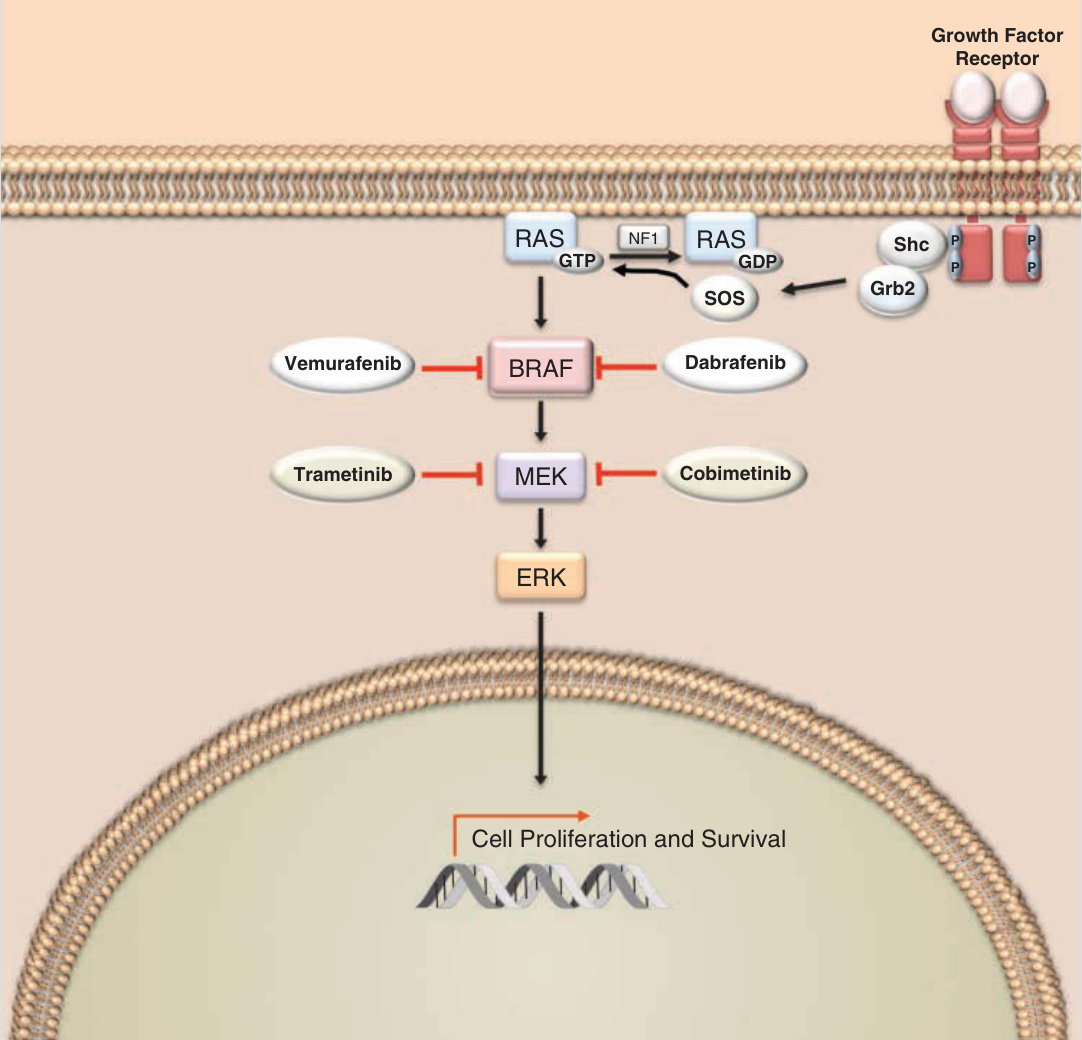
Figure 194-20 Mitogen-activated protein kinase (MAPK) pathway. Activation of receptor tyrosine kinases, such as c-KIT initiates signaling through the MAPK pathway. The action of guanine nucleotide exchange factors (GEFs), such as son of sevenless (SOS), accelerates the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) by RAS. Activated RAS triggers the MAPK cascade, which includes BRAF, MEK, and ERK, leading to cell proliferation and survival. The activity of RAS is attenuated by guanosine triphosphatase–activating proteins such as neurofibromin 1 (NF1). The small molecules vemurafenib and dabrafenib inhibit the activity of BRAF, whereas trametinib and cobimetinib inhibit MEK.
Figure 194-21 Structure of vemurafenib.
Figure 194-22 Structure of dabrafenib.
Figure 194-23 Structure of trametinib.
Figure 194-24 Structure of cobimetinib.
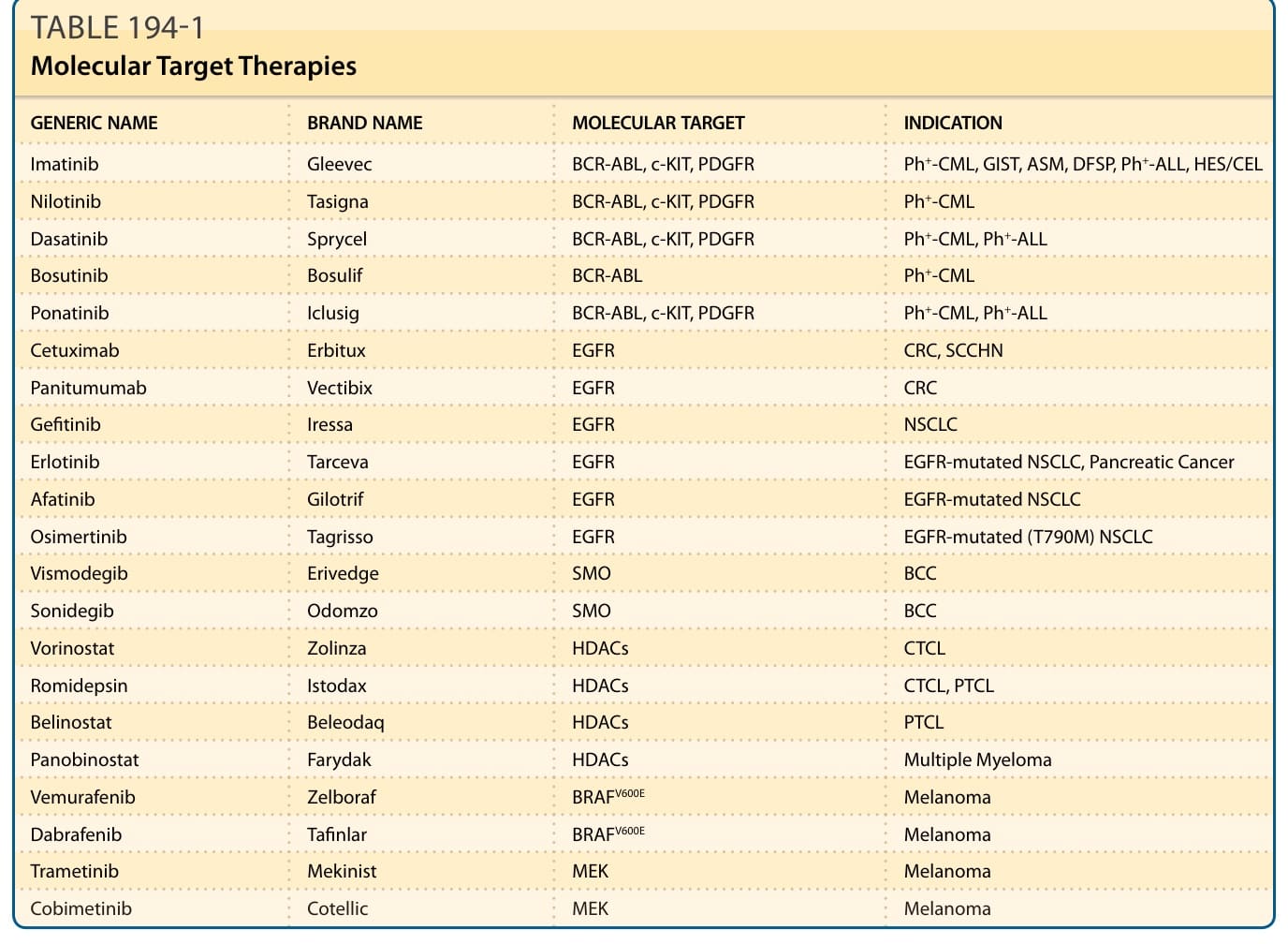
TABLE 194-1 Molecular Target Therapies
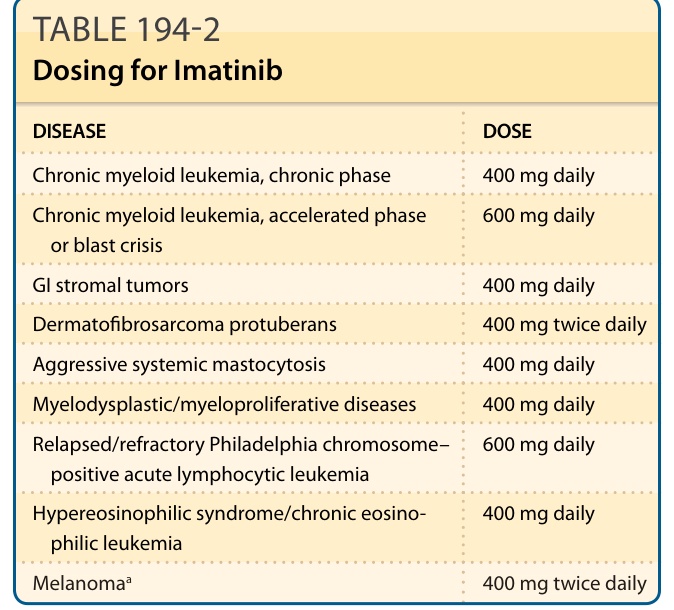
TABLE 194-2 Dosing for Imatinib