痲瘋病/漢生病 (Leprosy)
PART 23
細菌性疾病 (Bacterial Diseases)
重點一覽 (AT A GLANCE)
■ 定義:一種主要侵犯皮膚與神經的慢性肉芽腫性疾病,由絕對細胞內病原體痲瘋分枝桿菌 (Mycobacterium leprae) 所引起。
■ 侵犯部位:主要為皮膚與神經,但會對廣泛的組織與系統造成後遺症,包括眼睛、上呼吸道、淋巴組織、睪丸、肌肉與骨骼。
■ 診斷:根據臨床徵象與症狀,標誌性特徵包括皮膚病灶內的感覺喪失、神經腫脹或疼痛,或在皮膚抹片或切片中檢出抗酸桿菌 (acid-fast bacilli)。
■ 發生率:2016 年全球新偵測病例為 214,783 例,過去 4 年基本上維持不變。所有新病例中超過 80% 僅在 3 個國家被偵測到——印度、巴西與印尼。
■ 長期病態:儘管自 1980 年代中期以來已全球使用多重藥物治療 (multidrug therapy),仍有高達 30% 至 50% 的所有痲瘋病人會經歷某種型態的反應性發作 (reactional episode),可能導致永久性神經功能缺損或失能。
■ 臨床上的挑戰:症狀緩慢且多樣化發展前的漫長潛伏期(感染後 3-7 年)、受感染個體中極低的疾病進展率,以及誤診問題,都對於發展中斷傳播之方法構成挑戰。
■ 疾病的免疫學光譜:了解何種遺傳因子,以及宿主先天與後天免疫反應之間的交互作用如何導致對疾病的抵抗力或易感性,對於發展新型治療途徑至關重要。
前言 (INTRODUCTION)
定義 (DEFINITION)
痲瘋病是由痲瘋分枝桿菌 (Mycobacterium leprae) 引起的一種慢性肉芽腫性感染,會感染黏膜皮膚組織與周邊神經,導致皮膚感覺喪失(無論有無皮膚科病灶),並在疾病進展過程中發展出失能。WHO 表示,在流行國家中,任何呈現具有明確感覺喪失之皮膚病灶或皮膚抹片陽性的個體,皆可診斷為痲瘋病。¹
歷史觀點 (HISTORICAL PERSPECTIVE)
痲瘋病是已知折磨人類最古老的疾病之一。挪威醫師 Gerhard Armauer Hansen 於 1873 年首度描述了痲瘋桿菌 (M. leprae),辨識出第一個與人類疾病相關的細菌病原體。在某些國家(如巴西),會使用「漢生病 (Hansen disease)」一名以減少與俗名相關的汙名化。導致毀容、變形與失能的特徵性破壞性變化,是使該病在古代被汙名化的標誌之一。² 已有關於痲瘋病存在於古印度、埃及與中國的推測,最早的古病理學證據發現於印度西部拉賈斯坦邦 (Rajasthan) Balathal 墓葬遺址中一具 4000 年前的骨骸。³ 1994 年首度描述了使用聚合酶連鎖反應 (polymerase chain reaction, PCR) 分子技術,在年代追溯至西元 600 年的古代骨骸中偵測 M. leprae DNA 特異性序列。⁴ PCR 也被用於在以色列「裹屍布之墓 (Tomb of the Shroud)」墓葬遺址、年代追溯至西元 1 世紀的骨骸中辨識 M. leprae DNA,這是該地區已知痲瘋病存在的最早年代。⁵
利用古病理學與分子方法分析來自考古遺址、墓地與墓園的古代骨骸,已浮現痲瘋病傳播的證據,顯示該病自西元 4 世紀從西亞與中亞傳播至東歐與中歐,主要是由於與軍事行動、領土擴張或殖民移民相關的人口遷徙。⁶ 在 7 世紀的法國,已設立用以隔離痲瘋病及其他傳染病患者的處所,稱為 lazarets(痲瘋病院)。但直到自西元 1100 年起,十字軍從痲瘋病流行的鄂圖曼帝國 (Ottoman Empire) 諸國返回後,該病才成為日益嚴重的問題。歐洲與英國的痲瘋病在 13 與 14 世紀達到高峰,隨後開始緩慢衰退。比較取自古代歐洲、英國與斯堪地那維亞墓地的 M. leprae 全基因組顯示,過去 1000 年來僅有數十個單核苷酸多型性 (single nucleotide polymorphism, SNP) 變化,且任何與毒力或致病機轉增加相關的基因均無突變。⁷ M. leprae 從古至今基本上呈現出純系 (clonal) 的特性,顯示痲瘋病盛行率的急遽下降與病原體本身的特徵無關,而較可能與宿主抵抗力或環境的變化有關。由於患有瘤型痲瘋 (lepromatous leprosy) 的人很可能免疫狀態較弱,他們較易死於其他感染。被認為導致痲瘋病人較高死亡率的事件,包括 1325 年的一場嚴重饑荒,隨後是 1349 年爆發的瘟疫,即黑死病 (Black Death),造成歐洲與英國三分之一至三分之二的人口死亡。如此大規模的死亡,將嚴重削弱由宗教神職人員、贊助者與醫師照護病人的安養院 (hospices) 與痲瘋病院 (leprosaria) 的支持網絡,而這些人本身也很可能遭到大量毀滅。亦有提示性證據顯示,人口密度增加、過度擁擠的居住條件,以及 15 世紀後結核病的興起,促成了與多種疾病合併感染所致的死亡。⁸ 隨著那些易感個體死於疾病,倖存族群對分枝桿菌感染及該時期常見其他疾病的先天抵抗力很可能因而改善,再加上社會經濟條件、衛生與清潔狀況的改善,感染的傳播鏈於是被打斷。目前,歐洲與英國的痲瘋病地方性流行病例極為罕見。對全世界 M. leprae 分離株的比較基因組學研究揭示,該基因組的保守程度十分顯著(所有菌株型別間有 99.995% 的同一性),顯示存在 215 個多型性位點,主要由 SNP 構成。其中 4 個 SNP 代表 4 種主要菌株型別,呈現出非常強的地理關聯性,被用來根據歷史上的人類遷徙模式追溯 M. leprae 的演化與分布。⁹ SNP 第 1 型主要見於東南亞;SNP 第 2 型主要見於東非;SNP 第 3 型與歐洲/北非地區相關;而 SNP 第 4 型主要見於西非。痲瘋病在北美洲或南美洲原本並不存在,直到經由來自歐洲的殖民主義(SNP 第 3 型)以及自非洲輸入奴隸(SNP 第 4 型)而傳入。基於對來自全世界 28 個不同地區、總計 400 個樣本的現代與古代 M. leprae 基因組之 SNP 與插入/缺失事件進一步特徵分析,這 4 種主要型別被再細分為 16 種亞型,即 SNP 第 1 型 (A-D)、第 2 型 (E-H)、第 3 型 (I-M) 與第 4 型 (N-P)。¹⁰
除了人類之外,M. leprae 亦以人畜共通感染 (zoonotic infection) 的形式出現於美國南部的犰狳 (armadillos),¹¹ 以及近期在不列顛群島的紅松鼠 (red squirrels) 中被發現。¹² 有趣的是,犰狳與松鼠的 M. leprae 都具有相同的 SNP 亞型 3I,顯示其有共同的歐洲祖源。
流行病學 (EPIDEMIOLOGY)
痲瘋病仍是一種嚴重的被忽視疾病。WHO 提及在過去 10 年新病例數逐漸減少,從 2006 年的 265,661 例降至 2015 年的 210,758 例,¹³ 但不同研究團隊已證明隱藏的地方性流行可能很高,¹⁴,¹⁵ 兒童病例數正在增加¹⁶ 或維持穩定,¹⁷ 而多菌性 (multibacillary) 病人的比例正在上升,¹⁸ 第 2 級失能 (grade 2 disability) 病例似乎維持穩定,¹⁹ 但居高不下,²⁰ 顯示晚期診斷。綜合而言,這些因素表明,問題並非新病例真正下降,而是診斷的延遲²¹ 與缺乏診斷似乎才是主要問題。¹⁵,²²,²³ 事實上,數學模型顯示,到 2020 年我們全球可能有 400 萬例未被診斷的痲瘋病,亦即幾乎為目前每年診斷病例數的近 20 倍。²⁴
痲瘋病的定義隨時間而改變。目前並無實驗室檢驗可診斷痲瘋病。一旦病人完成多重藥物治療,該個體通常會自登記名冊中移除,不再被視為病例,即使失能與反應性發作在治療後長期持續亦然。數十年來,痲瘋病的治療是連續單獨使用 dapsone(單一療法 monotherapy),所有接受 dapsone 單一療法的病例都登記於系統中。由於此一做法,痲瘋病的盛行率維持在高點,1980 年代登記名冊上有超過 1000 萬名病人。隨著 1982 年實施多重藥物治療,治療被縮短至最多 2 年。活躍登記名冊隨之改為僅納入正在接受治療者,而完成療程者則隨後被「清除」。因此,在十年的過程中,數百萬名完成多重藥物治療且無症狀的病人被自登記名冊中清除。²⁵ 其淨效應是,在引入多重藥物治療後的 20 年內,痲瘋病盛行率顯著下降超過 85%,¹³ 儘管新病例偵測率近 10 年來一直相對穩定地維持在全球超過 200,000 例(圖 159-1)。目前,痲瘋病負擔最重要的流行病學指標是各國的新病例偵測率,以及兒童、多菌性疾病者與第 2 級失能者在病例中的比例,後者顯示晚期診斷。¹³ 雖然印度的痲瘋病例數居全球之冠,但巴西在所有國家中擁有最高的新病例偵測率(圖 159-2)。然而,當我們檢視另外兩個最重要的參數時,即兒童病例的百分比(圖 159-3)與第 2 級失能者的百分比(圖 159-4),就會出現明顯的轉變:大多數通報國家被歸類為高或非常高,而巴西、印度與印尼卻是中或低百分比。雖然這 3 個國家合計占全球痲瘋病負擔的 >80%,但其通報的大量病例稀釋了兒童痲瘋病人與第 2 級失能者兩者的百分比,而通報每年新病例少於 1000 例的國家,這兩項類別中通常都有高得多的比率。這兩個參數呈現高或非常高的百分比,很可能意味著兒童與成人僅在呈現出痲瘋病典型病徵性 (pathognomonic) 病灶及/或失能時才被診斷出,顯示晚期診斷。¹⁵,²¹
圖 159-1:痲瘋病全球盛行率與新病例偵測之歷史演變。WHO 於 1966 年首度正式嘗試估計全球痲瘋病負擔,當時推估病例量為 10,786,000 例,其中 60% 未登記接受治療。全球偵測數首度於 1991 年通報,1990 年全球偵測到 584,000 例新病例。目前,新病例偵測是痲瘋病負擔最重要的流行病學指標之一,連同兒童比例與第 2 級失能比例。
圖 159-2:2015 年全球每 100,000 人口之痲瘋病新病例偵測率。2015 年在 136 個國家或地區通報了超過 210,000 例新病例。印度、巴西與印尼占全球痲瘋病負擔的 81%。全球新病例偵測率為 3.2。
圖 159-3:2015 年 15 歲以下兒童中之痲瘋病例比例。雖然對此指標並無從「低」到「高」的特定分類,但它能有力地指示其居住社區內存在活躍的感染源。
圖 159-4:2015 年診斷時具第 2 級失能之痲瘋病新病例比例。它反映痲瘋病診斷的長期延遲,凸顯衛生服務體系的失敗以及疾病控制方法的缺口。
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
在 1953 年於馬德里舉行的第四屆國際痲瘋大會 (IV International Leprosy Congress) 上,痲瘋病被分為 2 種穩定型——結核樣型痲瘋 (tuberculoid leprosy) 與瘤型痲瘋 (lepromatous leprosy),以及介於這 2 個極端型之間的邊緣群 (borderline group)。1966 年,Ridley 與 Jopling 提出了一套以臨床、組織病理學與免疫學標準為基礎的 5 組分類系統,²⁶ 至今仍用於痲瘋病的分類。桿菌對皮膚的群聚與對周邊神經的侵入,繼之以宿主的先天與後天免疫反應,造就了痲瘋病的臨床光譜(圖 159-5)。整體而言對於發展痲瘋病有遺傳性抵抗力,超過 90% 的人具有天然免疫力,其中細胞媒介免疫 (cell-mediated immunity) 對於預防疾病進展最為重要。所有病人(原發神經痲瘋 (primary neural leprosy)²⁷ 病人除外)最初都呈現皮膚上 1 個或數個色素減退斑 (hypopigmented macules)。未定類痲瘋 (Indeterminate leprosy)(圖 159-6)可能持續數月或數年,之後才走向自發痊癒,或依宿主對桿菌的細胞媒介免疫力而傾向臨床光譜的某一極端型或邊緣型。在光譜的一端,具有較佳細胞媒介免疫者,是極端結核樣型痲瘋 (polar tuberculoid leprosy),其呈現界線分明的斑塊,通常僅有數個位於身體的單一節段,色素減退及/或紅斑,有時萎縮,伴有主要在病灶周邊呈環狀 (circinate) 分布的丘疹或結節 (tubercles)(圖 159-7)。一種特殊的自癒型結核樣型痲瘋,即嬰兒結節型痲瘋 (infantile nodular leprosy),可呈現為單一結節性病灶,但也可呈現為丘疹或斑塊,通常位於兒童的臉部(圖 159-8)。²⁸
光譜的另一端是極端瘤型痲瘋 (polar lepromatous leprosy),其特徵為完全缺乏細胞媒介免疫,通常呈現遍布全身的大量結節性病灶,伴有瀰漫性浸潤(圖 159-9),包括耳部(圖 159-10)與臉部,其臉部特徵可能如此顯著,以致呈現出獅子臉的外觀,稱為獅面 (leonine facies)(圖 159-11)。²⁹ 一種特殊型的瘤型痲瘋是組織樣型痲瘋 (histoid leprosy),其桿菌負荷量甚至高於一般的瘤型痲瘋,含有稱為球體 (globi) 的桿菌叢集,呈現瀰漫性發亮的結節與丘疹,皮膚浸潤程度不一(圖 159-12)。³⁰ 某些瘤型痲瘋病例,當主要皮膚表現為浸潤時(圖 159-13),對經驗較不足的醫療專業人員而言是診斷上的挑戰。此一特殊型稱為 Lucio 痲瘋 (Lucio leprosy),由 Lucio 與 Alvarado 於 1852 年在墨西哥首度描述。³¹ 自 Lucio 病人分離並特徵分析出此一新菌種,稱為痲瘋瘤分枝桿菌 (Mycobacterium lepromatosis),³² 連同全基因組定序,³³ 現已穩固確立此一密切相關的分枝桿菌會引起這種主要見於墨西哥與加勒比海地區的痲瘋病型。介於其間的 3 種邊緣型(邊緣結核樣型 borderline-tuberculoid、邊緣邊緣型 borderline-borderline 與邊緣瘤型 borderline-lepromatous)在免疫學上都不穩定。所有邊緣型病人都有皮膚浸潤,病灶從少數到許多不等,位於身體的一個或多個區域。雖然結核樣型痲瘋病人只有丘疹或結節而無浸潤,但邊緣結核樣型痲瘋在病灶周邊呈現出清楚的浸潤帶,從結核樣型痲瘋中非常銳利的邊界,變化為邊緣結核樣型痲瘋中較瀰漫的浸潤外層(圖 159-14)。隨著病型朝瘤型端進展,在邊緣邊緣型(圖 159-15)及其典型的小凹狀 (foveolar) 病灶,以及邊緣瘤型痲瘋(圖 159-16)中,有效的細胞媒介免疫遞減,使桿菌得以漸進性擴散與數量增加,病灶浸潤增加,並演變形成更多結節性病灶,常侵犯臉部與耳部。痲瘋病的診斷是根據在病灶中偵測到感覺減退或完全麻木 (hypo- or total anesthesia),此可能伴隨少汗 (hypohidrosis) 與脫髮 (alopecia)。結核樣型痲瘋病人可呈現皮膚乾燥與限於病灶範圍內的脫髮。相對地,瘤型痲瘋病人可能呈現大面積乾燥,尤其在腿部,在晚期病例可能導致眉毛脫落 (madarosis) 與皮膚不同部位的毛髮脫落。邊緣型病人遵循相同模式,邊緣結核樣型痲瘋特徵較侷限,邊緣瘤型痲瘋則較瀰漫。感覺異常 (Paresthesia) 是痲瘋病例常伴隨的症狀。灼熱、麻木、刺癢與其他感覺,可能出現在病灶之內,或沿著受侵犯區域神經幹所支配的範圍出現。病人可能在急性發作時感受到這些感覺,尤其在寒冷天氣的夜間,且可能頻繁復發,並隨疾病進展而日益常見。所有型別的痲瘋病灶都必須接受徹底的感覺評估,包括血管運動反射 (vasomotor reflex)、出汗功能、溫度、疼痛與觸覺敏感性。血管微血管擴張作為一種次級軸突反射性紅斑 (axon reflex erythema),其取決於神經完整性,可使用 1:1000 組織胺溶液,於正常與病灶皮膚內皮下注射來測試。在 5 至 10 秒內,紅斑會因組織胺對微血管的直接作用而產生,於正常與病灶兩區域均造成血管擴張。在此之後 2 分鐘,由微血管擴張引起的次級紅斑將僅在正常皮膚上發生。三聯路易士反應 (triple Lewis response) 的最後一相是液體滲出至真皮,於兩區域均形成風疹塊 (wheal)。因此,三聯路易士反應僅在病灶皮膚上不完全,缺乏次級紅斑。
碘澱粉 (Iodine-starch) 或茜素紅 (alizarin red)³⁴ 可用於評估痲瘋病灶的出汗功能。在塗碘繼以澱粉後,或在塗茜素紅後,可能需要運動以誘發出汗。當自主神經功能受影響時,出汗會受損或完全缺乏,皮膚保持乾燥。在正常皮膚上會出現藍色或深棕色(碘澱粉)或紫色(茜素紅),而在痲瘋病灶上則無反應(無汗 anhidrosis)或不規則出汗(少汗 hypohidrosis)。溫度、疼痛與觸覺,以及 Semmes-Weinstein 單絲 (Monofilaments) 測試,全都直接依賴病人正確的口頭回應。因此,向病人解釋每項測試的目的、如何對引發的感覺給予適當回饋,並在測試病灶區域前先於正常非病灶皮膚部位進行測試以使個體熟悉該感覺,以便測量任何變化,是至關重要的。溫度測試是根據用裝有熱水(±45°C [113°F])或冷水的 2 支試管觸碰皮膚時辨別冷熱敏感性的能力。將試管表面隨機觸碰病灶與正常皮膚,隨後記錄病人的回答。專業人員必須小心避免同時觸碰病灶與正常皮膚,尤其對於較小的病灶。疼痛敏感性是根據區分針尖或針底的能力,因為其一會產生疼痛而另一不會。同樣地,須隨機以針尖或針底觸碰病灶與非病灶皮膚,隨後記錄回答。此方法的一個明顯限制是使用穿刺器械,可能使某些病人(尤其是兒童)感到恐懼。觸覺敏感性以棉絮測試,病人應回答是否感覺到棉絮輕觸皮膚,無論正常或病灶皮膚。Semmes-Weinstein 單絲或感覺計 (esthesiometer) 套組是不同顏色、粗細分級的單絲線,附著於塑膠柱上,以對皮膚施加不同量的目標壓力。其顏色與目標力量範圍從綠色(範圍 0.008-0.07 g,正常皮膚感覺,其最細者如同蚊子停在皮膚上的感覺),到最粗的紅色(高達 300 g)。³⁵ 這些裝置簡單而便宜,可輕易測量糖尿病神經病變或痲瘋神經損傷所致神經病變造成的感覺減退或喪失。使用藍色 Semmes-Weinstein 單絲檢測到感覺喪失,表示輕觸覺感覺減退(0.16-0.4 g);紫色表示保護性感覺減退(0.6-2 g);紅色表示保護性感覺有更深度的喪失(4-300 g)。較近期,Semmes-Weinstein 單絲的簡便與易用性已取代所有其他觸覺、溫度與疼痛測試。³⁶ 在向病人示範單絲如何運作,並在其感覺到觸碰時得到「有」的回應後,可使用各種粗細的 Semmes-Weinstein 單絲,隨機測試可疑病灶內外的皮膚區域,如 mhprofessional.com/fitzderm9evideos 上的 Video 159-1 所示。即使年僅 6 至 7 歲的兒童也能對此類測試反應良好,而那些無法良好溝通的兒童,若感覺到裝置觸碰,仍可指出皮膚上的位置(見 mhprofessional.com/fitzderm9evideos 上的 Video 159-1)。總之,依 Ridley-Jopling 分類所定義的「典型」病例具有界線分明的病灶,伴隨一系列徵象與症狀,有助於痲瘋病的診斷。在 35 年的多重藥物治療之後,現今的挑戰是早期診斷病例,目標是消除失能。
非皮膚表現 (NONCUTANEOUS FINDINGS)
雖然痲瘋病的診斷主要根據皮膚病灶的存在,但通常當偵測到皮膚科徵象時,此時周邊神經已被 M. leprae 本身及/或我們免疫系統的反應所侵入並損傷。事實上,神經可能是 M. leprae 的第一個標靶,而感染本身連同免疫細胞浸潤與發炎,可在臨床上經由觸診偵測到。觸診周邊神經幹可確立神經的粗細與壓痛。然而,即使對於受過高度訓練的醫療專業人員而言,要偵測兩側粗細的差異,或判定神經是否柔軟(因而正常)或纖維化,也非易事。此外,唯有當這些差異伴隨某種功能性損害時,才應加以考量,如 (1) 由神經支配範圍上的感覺減退或完全麻木所定義的感覺喪失;(2) 運動功能障礙,如骨間肌萎縮 (interosseous muscle hypotrophy) 的情形;或 (3) 自主神經改變,如皮膚出汗缺損。雖然結核樣型痲瘋病人可能僅在一條特定周邊神經幹有明顯改變,通常與皮膚病灶位於相同節段,但瘤型痲瘋病人常呈現多條神經的粗細與壓痛變異,於身體不同節段伴隨或不伴隨功能性改變。邊緣結核樣型、邊緣邊緣型與邊緣瘤型痲瘋病人通常呈現神經改變,從邊緣結核樣型痲瘋中少數神經幹受侵犯,到邊緣瘤型痲瘋中許多神經幹受侵犯不等。許多病例會有某種程度的疼痛,由病人自發報告,或在觸診時提及。除了上下肢之外,當顏面神經或三叉神經受損時,臉部也可能受侵犯,可導致感覺減退或麻木(包括角膜),以及肌肉萎縮,尤其當眼瞼肌受侵犯時,導致兔眼 (lagophthalmos)(圖 159-17)。經觸診偵測到周邊神經幹有任何這些改變或功能喪失,即排除未定類痲瘋的診斷。另一方面,在所有痲瘋病人中,有 5% 至 17% 僅有神經發炎或功能缺損的徵象而無任何皮膚病灶,此時診斷可能為純神經炎型 (pure neuritic),或更常用的詞——原發神經痲瘋 (primary neural leprosy),³⁷ 因為高達 35% 的此類病例可能在原發神經痲瘋診斷後發展出皮膚病灶。³⁸
原發神經痲瘋約占所有痲瘋病例的 4% 至 8%,不過在印度可能高達 17%。³⁷ 裂隙皮膚抹片 (slit-skin smear) 上抗酸桿菌陽性結果可排除原發神經痲瘋,但神經切片在 16% 的此類病例中可顯示抗酸桿菌的存在,而 PCR 在其中近半數呈陽性。³⁹ 原發神經痲瘋的確定診斷並非易事,可能需要臨床徵象、神經組織病理學、電生理學與超音波,³⁷ 不過這些技術中的大多數在高度流行國家並不普及。內分泌功能障礙在病人中是次於神經與皮膚病灶最為顯著者,但不易被偵測到,可達 25% 的病例,⁴⁰ 導致甲狀腺功能低下、正常甲狀腺病態症候群 (euthyroid sick syndrome)、性腺功能低下、不孕與骨質疏鬆等問題。⁴¹ 睪固酮 (testosterone) 濃度與皮膚病灶數目呈反向相關,⁴⁰ 而腎上腺雄激素硫酸脫氫表雄酮 (dehydroepiandrosterone sulphate) 濃度與介白素 (interleukin, IL)-6 及腫瘤壞死因子 (tumor necrosis factor, TNF)-α 呈反向相關,而促性腺激素——黃體生成素 (luteinizing hormone) 與濾泡刺激素 (follicle-stimulating hormone)——則與促發炎細胞激素呈正向相關,⁴² 提示痲瘋病中可能存在神經—免疫—內分泌的相關性。
併發症 (COMPLICATIONS)
痲瘋病的自然病史是演變為損害,尤其在眼睛、手與足,包括軟組織與骨骼,導致毀容與變形,這是所有痲瘋病相關汙名的根源。即使在實施多重藥物治療達到細菌學治癒,並有參與失能者教育、訓練與重返社會等社會網絡的協助,全世界因 M. leprae 而生活在不同程度失能中的人數,包括第 1 級(部分失能)到第 2 級(可能完全失能且無法工作),估計可能介於 100 萬至 400 萬之間。⁴³
就眼睛而言,角膜感覺喪失可導致傷口,繼之以感染與失明,而眼瞼肌萎縮可導致兔眼,這也可能促成角膜感染。晚期病例,尤其在多菌性痲瘋、多偏向瘤型痲瘋端者,可能呈現特徵性的顏面骨畸形、吸收與凹蝕,特別是前鼻棘 (anterior nasal spine) 的破壞、上頜齒槽突 (alveolar process of the maxilla) 的吸收,有時伴隨牙齒脫落,統稱為鼻上頜症候群 (rhinomaxillary syndrome)。⁴⁴
就手與足而言,併發症始於感覺喪失,可能在燒燙傷、外傷,或病人未察覺的反覆中度壓力所致皮膚破損後導致傷口形成(圖 159-18),並可能演變為裂隙與潰瘍、軟組織發炎性自體分解 (autolysis)、肌肉萎縮、骨骼脫鈣、骨炎與吸收(圖 159-19)、融合與關節脫位、骨關節炎與破壞。⁴⁵ 同時,骨間肌萎縮或肌肉萎縮 (amyotrophy) 可能導致輕癱或癱瘓,導致形成爪狀手(足)或垂手(足)(見 mhprofessional.com/fitzderm9evideos 上的 Video 159-2),起初可能仍可活動,但對痲瘋病人構成嚴重損害,⁴⁶ 例如行走(見 mhprofessional.com/fitzderm9evideos 上的 Video 159-3)。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
危險因子 (RISK FACTORS)
痲瘋分枝桿菌 (Mycobacterium leprae) 是一種無法培養的絕對細胞內病原體,主要損傷皮膚與周邊神經,是痲瘋病的致病因子,造成範圍廣泛、伴隨麻木的皮膚病灶、經由神經損傷的周邊神經病變,以及肌肉無力與萎縮,並因吸收導致骨質流失,伴隨相關的變形、毀容與失能,連同折磨人類數千年來與此病相關的社會汙名化。雖然 M. leprae 與結核分枝桿菌 (M. tuberculosis) 共有大約 1,439 個基因直系同源物 (orthologs) 與同源物 (homologs),但發生在 1000 萬至 2000 萬年前的一場退化性演化事件,導致大規模的基因缺失與衰減,使得近半數的所有編碼基因轉變為無功能的截短基因殘餘或假基因 (pseudogenes)。⁴⁷ 此一退化性演化過程已發生在數種絕對細胞內病原體中,包括立克次體 (Rickettsia) 與披衣菌 (Chlamydia),並被認為是對生態棲位或生活型態劇烈變化的一種存活反應。許多曾為自由生活物種存活所需的基因與路徑,在細胞內棲地中將屬多餘,因而被精簡並消除,導致基因組大部分被刪除或去活化。因此,M. tuberculosis 有 4.41 Mb,其中 >90% 的基因組編碼 3,998 個蛋白質編碼序列,而 M. leprae 有 3.27 Mb,其中僅不到 50% 的基因組編碼 1614 個功能性基因,其餘編碼 1306 個假基因與基因殘餘,其完整對應物可在 M. tuberculosis 中找到。基因縮減的綜合效應造就了一組最精簡的基因集,減少了參與所有功能性代謝路徑的基因數目,包括參與基因調控、解毒、DNA 修復、代謝物與小分子之轉運或外排的關鍵路徑,同時普遍降低降解性路徑相對於合成性路徑的基因頻率,以及呼吸酵素的匱乏。由於這些缺陷,M. leprae 擁有所有細菌中最長的倍增時間之一,約 13 天,這可能解釋了從感染到發展出臨床疾病之間異常漫長的潛伏期,通常介於 3 至 7 年之間,不過在某些病例中可達 20 年。儘管數十年來嘗試不斷,仍無法在無菌培養基 (axenic medium) 中培養 M. leprae,這很可能是基因縮減與關鍵代謝路徑突變綜合效應的結果。痲瘋桿菌的標誌之一是能夠附著並侵入與周邊神經系統相關的許旺細胞 (Schwann cells),導致神經內的群聚與發炎,造成神經損傷、去髓鞘 (demyelination) 與神經病變。與許旺細胞基底層 (basal lamina) 中層黏連蛋白-2 (laminin-2) 分子的結合,是由 2 種細菌細胞壁成分所媒介,即層黏連蛋白結合蛋白(由 ML1683c 編碼)與 M. leprae 特異性酚醣脂 I (phenolic glycolipid I, PGL-I) 的末端三醣。⁴⁸ 傳播被認為是經由氣溶膠 (aerosol) 途徑發生,桿菌進入並群聚於鼻黏膜與鼻甲 (turbinate) 中常駐的巨噬細胞,⁴⁹ 然後經由血流播散至組織與神經。在細胞層級宿主—病原體交互作用所涉的早期事件,很可能由參與模式辨識受體 (pattern recognition receptors) 與分枝桿菌攝取的宿主基因所媒介(類鐸受體 [Toll-like receptors, TLR]、含核苷酸結合寡聚化結構域 2 [nucleotide-binding oligomerization domain containing 2, NOD2],以及甘露糖受體 C 型 1 凝集素 [mannose receptor C-Type 1 lectin, MRC1]),這些基因調控自噬作用 (autophagy)。例如,辨識並結合宿主細胞表面 TLR 1/2 異二聚體配體的細菌細胞壁脂蛋白,會觸發先天免疫反應,其結果將決定桿菌是被包圍或殺死於肉芽腫內,抑或不受控制地生長。整體而言對於發展痲瘋病有遺傳性抵抗力,全世界超過 90% 的人具有天然免疫力。⁵⁰ 分枝桿菌結合至這些模式辨識受體並進入細胞的早期先天免疫反應,會調控細胞代謝以活化 NF-κB 與維生素 D 受體路徑,上調細胞激素產生以及對於形成並維持包圍桿菌所需肉芽腫至關重要的基因,包括 TNF、干擾素 γ (interferon gamma, IFN-γ) 與淋巴毒素 α (lymphotoxin alpha)。接下來發生的事,很可能取決於後天免疫反應(涉及細胞媒介與體液免疫反應)的複雜交互作用:T 輔助細胞 1 (T helper 1, Th1) 細胞激素與促發炎反應導致細胞媒介反應增強,控制細菌生長並防止播散;而轉向 T 輔助細胞 2 (T helper 2, Th2) 細胞激素產生,則下調發炎反應,導致不受控制的生長、對細菌抗原無效的高量抗體反應,以及漸進惡化的疾病症狀。
流行病學研究,包括雙胞胎研究、複雜分離分析 (complex segregation analyses),以及在來自不同痲瘋病流行國家、遺傳多樣化的各族群中進行的全基因組分析,已指出宿主遺傳學在此病易感性或抵抗力中可能的重要性。1960 與 1970 年代在印度進行的雙胞胎研究顯示,同卵雙胞胎對發展痲瘋病有壓倒性的一致性(60%-80%)。⁵¹,⁵² 檢驗多個候選基因連鎖與關聯的全基因組分析研究顯示,發展痲瘋病的易感性以及發展特定疾病型的傾向均受遺傳控制。後者的證據可見於墨西哥與菲律賓,當地 90% 的病例發展為瘤型疾病,而在許多非洲國家與巴西,發展為結核樣型或瘤型疾病的病人數目則大致相當。廣泛的研究已牽連出與人類白血球抗原 (human leukocyte antigen, HLA) 複合體基因(第一類與第二類)的關聯,因其在後天宿主免疫反應中扮演主要角色;當檢驗不同國家的特定族裔人口時,有許多對偶基因 (alleles) 在痲瘋病易感性或結核樣型或瘤型亞型的發展中過度呈現。痲瘋病與若干發炎性疾病之間也有共同的遺傳背景,包括克隆氏症 (Crohn disease)(含核苷酸結合寡聚化結構域 2 [NOD2])、心肌梗塞(淋巴毒素 α [LTA])、第 1 型糖尿病與乾癬(維生素 D 受體 [VDR]),以及帕金森氏病 (Parkinson disease)(E3 泛素蛋白連接酶 [PARK2])。⁵⁰ 較近期,檢驗中國痲瘋病例與對照之間 SNP 差異的全基因組關聯研究,已增加了可能參與調控與痲瘋病易感性或抵抗力相關之先天與後天免疫反應路徑的基因清單。⁵³ 在 6 個基因中偵測到的 15 個 SNP 與痲瘋病相關(HLA-DR-DQ、RIPK2、TNFSF15、CCDC122、C13orf31 與 NOD2),而路徑分析辨識出總計 35 個參與痲瘋病易感性或抵抗力單一網絡的基因。除了遺傳危險因子之外,許多研究顯示尚有若干其他因子(操作性或社會經濟性)會增加風險或使個體易於發展疾病。與一名未治療之多菌性痲瘋指標病例 (index case) 同住一屋簷下的家戶接觸者,最終罹患疾病的風險最高,尤其當該家戶接觸者是指標病例的血親時。⁵⁴ 抗 PGL-I 效價陽性的個體,發展痲瘋病的風險可高達 8 倍。其他危險因子包括居住於痲瘋病的流行或高度流行地區、貧窮、居住於 >2 人同房共眠的高密度家戶、營養狀況不良、衛生不良或缺乏潔淨水源,以及缺乏醫療照護的可近性。⁵⁵ 改善這些根本問題將大幅降低痲瘋病高風險者最終發展此病的可能性。
診斷 (DIAGNOSIS)
輔助檢查 (SUPPORTIVE STUDIES)
病理學 (PATHOLOGY) 痲瘋病的病理生理學 (Physiopathology of Leprosy): M. leprae 用以進入標靶細胞(主要為許旺細胞)的途徑,一直是討論的議題。基本上已提出 4 種不同的路徑:(1) 表皮中裸露的神經絲;(2) M. leprae 進入表皮,並由此進入其他許旺細胞;(3) 真皮巨噬細胞對 M. leprae 的吞噬作用,這些巨噬細胞隨後侵入神經外膜 (perineurium),釋放桿菌進入許旺細胞;以及 (4) 經由血液,亦即 M. leprae 可經由神經內微血管 (intraneural capillaries) 進入神經。腫大的內皮細胞可能促進桿菌進入神經系統,最終進入許旺細胞。⁵⁶
M. leprae 與內皮細胞的交互作用及其被內皮細胞吞噬,早已被辨識。⁵⁷ 犰狳研究顯示,在 M. leprae 感染淋巴與血管內皮細胞之巨噬細胞後出現神經外膜增厚。⁵⁸ 雖然此一感染模式已廣為人知,但 M. leprae 隨後如何從那些細胞轉移至許旺細胞,仍不清楚。一旦 M. leprae 到達細胞外基質 (extracellular matrix),PGL-I⁴⁸ 或一種類組蛋白的 21-kDa 層黏連蛋白結合蛋白⁵⁹ 會結合至層黏連蛋白 α2 鏈 (laminin α2 chain),以侵入許旺細胞。α-肌縮蛋白聚醣 (α-dystroglycan, α-DG) 之 G 結構域的存在,可能是 M. leprae 黏附至許旺細胞所必需(圖 159-20)。基質細胞骨架連結(α-DG、α2-層黏連蛋白、β-DG)可能是 M. leprae 用以進入宿主細胞的路徑。M. leprae 繞過神經調節蛋白受體 (neuregulin receptor)(一種表皮生長因子家族成分),直接與 ErbB2 進行細菌性連結,在無 ErbB3 異二聚化的情況下傳訊,並放大 Erk1/2 傳訊,這可能誘導髓鞘 (myelin sheath) 降解。⁶¹ 此外,M. leprae 利用非經典路徑與不依賴 MEK 的傳訊,可經由淋巴細胞激酶 (p56LcK) 直接活化 Erk1/2,誘導細胞增生並維持其增生棲位。⁶² 感染時,9-O-乙醯基 GD3 神經節苷脂 (9-O-acetyl GD3 ganglioside) 的表現增加,這是一種參與抗凋亡傳訊與神經再生的分子(圖 159-20)。對許旺細胞上 9-O-乙醯基 GD3 神經節苷脂進行免疫阻斷,會減少 Erk1/2 與細胞增生。⁶³
M. leprae 可使成熟的許旺細胞去分化並重編程為類幹細胞 (stem cell–like cells),可能藉此促進感染的播散。⁶⁴ 桿菌的存活可能透過不同機制維持。在侵入後(圖 159-20),M. leprae 干擾 (1) 內吞成熟,抑制吞噬體 (phagosomes) 囊泡的酸化⁶⁵;(2) 宿主細胞脂質恆定,透過細胞骨架重組與 PI3K 傳訊(不依賴 TLR-2)誘導並累積脂滴 (lipid droplets)⁶⁶;以及 (3) 氧化路徑,經由強化葡萄糖攝取與增加葡萄糖-6-磷酸去氫酶 (glucose-6-phosphate dehydrogenase),一旦此酶被抑制,可使 M. leprae 存活率降低達 70%。⁶⁷
圖 159-20:M. leprae 的宿主細胞交互作用。此圖顯示細菌進入並維持於許旺細胞內的主要機制。
免疫反應可能在宿主—細菌交互作用的任何階段啟動。巴西高度流行地區的流行病學調查顯示,學齡兒童間循環的抗 PGL-I IgM 抗體比例很高(達 50% 或更高),顯示曾感染 M. leprae 並繼之以對桿菌的早期抗體反應。⁶⁸ 雖然神經去髓鞘可能發生於 T 與 B 淋巴球被剔除的小鼠,⁶⁹ 但感染過程中會發生多樣的免疫反應。痲瘋病的人類免疫反應有一明確的二分性 (dichotomy):位於光譜結核樣端者有強烈的 Th1 細胞媒介反應,而位於瘤型端者則有偏斜的 Th2 反應並伴隨 T 細胞無反應性 (anergy)。結核樣型痲瘋病例有強烈的 Th1 反應,產生 IL-2、TNF-α、IFN-γ 與 IL-12 細胞激素,而瘤型痲瘋病人有 Th2 反應,產生 IL-4、IL-5、IL-10 與高量抗體。這些特徵也見於皮膚,如病灶組織化學染色組織切片中浸潤皮膚之免疫細胞的漸進性失序所示,這 2 種狀況一般可顯現出來。⁷⁰ 結核樣型痲瘋病人呈現組織良好的肉芽腫,含類上皮細胞 (epithelioid cells)、CD4+ T 細胞、良好的細胞媒介免疫、幾乎無抗體產生,且裂隙皮膚抹片菌檢 (bacilloscopy) 未發現桿菌。另一方面,瘤型痲瘋病人呈現大量充滿大量桿菌的泡沫狀巨噬細胞浸潤,淋巴球少(多為 CD8+ T 細胞),細胞媒介免疫有缺陷,並有對 M. leprae 抗原(包括 PGL-I)的高效價抗體。⁷¹
類鐸受體,如 TLR-1、TLR-2 與 TLR-4,連同 DC-SIGN (CD209) 與 CD163,可能參與巨噬細胞/樹突細胞與 M. leprae 的交互作用。TLR-1 與 TLR-2 在結核樣型痲瘋的表現高於瘤型痲瘋病灶,而 TLR-1/TLR-2 異二聚體媒介單核球與樹突細胞的活化,刺激 TNF-α 與 IL-12 產生⁷²(圖 159-19)。M. leprae 亦可經由巨噬細胞中的 TLR-4 傳訊刺激 TNF-α、IL-6 與 CXCL10 (IP-10) 的產生。⁷³ 另一方面,許旺細胞中的 TLR-2 傳訊與細胞凋亡相關。⁷⁴
M. leprae 經由 DC-SIGN 傳訊增加樹突細胞中 IL-10 的表現,活化 Raf-1,在 TLR 誘導 NFκB 活化後造成 NFκB p65 次單元的乙醯化。⁷⁵ IL-10 經由 DC-SIGN 誘導巨噬細胞的吞噬作用,並透過上調氧化型低密度脂蛋白 (oxidized low-density lipoprotein) 的攝取,使單核球分化為泡沫狀巨噬細胞,而 IL-15 則誘導維生素 D 抗菌路徑,呈現較少的吞噬作用。⁷⁶ 連同 DC-SIGN 與吲哚胺 2,3-雙加氧酶 (indoleamine 2,3-dioxygenase) 的上調,CD163 也增加,並促成鐵的攝取,以及在瘤型痲瘋巨噬細胞中為 M. leprae 進入與存活創造有利環境⁷⁷(圖 159-20)。M. leprae 可調節許旺細胞中的 NFκB 活化,此一功能可被 thalidomide 抑制。⁷⁸ 此外,金屬蛋白酶 2 與 9 及 TNF-α 在原發神經痲瘋神經的許旺細胞、巨噬細胞與內皮細胞中上調,⁷⁹ 與非痲瘋周邊神經病變相比,造成顯著的神經內膜 (endoneurial) 浸潤,伴隨神經外膜纖維化與腫大。⁸⁰
除了 Th1/Th2 細胞激素譜之外,其他因子如 IL-17、抗菌肽 LL-37 (cathelicidin LL-37) 與類胰島素生長因子 I (insulin-like growth factor I),似乎在痲瘋病的病理生理學中也很重要。VCAM-1 在痲瘋病人的血清中增加,⁸¹ 而血管內皮生長因子 (vascular endothelial growth factor) 與凝血致活酶 (thromboplastin) 的表現則在痲瘋病人的內皮細胞中增加。⁸²
IL-17 在所有痲瘋病人中相較於非痲瘋對照組均較低,但在瘤型痲瘋中更低。⁸³ 雖然產生 IL-17 的 CD4+CD45RO+ Th17 細胞在結核樣型痲瘋病人的周邊血液單核細胞中增加,但產生 IL-10 的 Foxp3+ Treg 細胞在瘤型痲瘋中的盛行率為結核樣型痲瘋病人的 5 倍,顯示 Treg 在多菌性痲瘋發展中扮演角色。⁸⁴ 抗菌肽 LL-37(cathelicidin LL-37,是人類中發現的一種獨特抗菌肽家族宿主防禦肽,已知會調節對 M. tuberculosis 的免疫反應)在所有痲瘋病人中均偏低⁸⁵(圖 159-21)。類胰島素生長因子 I(已知會降低巨噬細胞的抗菌能力,抑制 M. leprae 的殺滅⁸⁶)在瘤型痲瘋病人中也被發現偏低,尤其是那些未發展出第 2 型反應或痲瘋性結節性紅斑 (erythema nodosum leprosum, ENL) 者。⁸⁷
ENL 是一種免疫學第三型過敏反應 (Type III hypersensitivity response),伴隨免疫複合體沉積⁸⁸ 以及抗 PGL-I 與抗單核球趨化蛋白-I (monocyte chemoattractant protein-I) 抗體,⁸⁹ Th17、IL-6、IL-1β、sIL2R 與 sIL6R 上調、Treg 反應減少,以及嗜中性球 (neutrophils) 湧入病灶⁹⁰(圖 159-21)。此外,ENL 可在瘤型痲瘋病人皮內注射 IFN-γ 後被啟動,⁹¹ CD4+/CD8+ 比值增加,並發現高血清濃度的 TNF-α,⁹² 使用 TLR-9 致效劑會增加 TNF-α、IL-6 與 IL-1β。⁹³ E-選擇素 (E-selectin) 以血管型態表現,在 ENL 中高於非反應性瘤型痲瘋病人,⁹⁴ 而 FcγRI 在 ENL 病人的循環嗜中性球中增加⁹⁵(圖 159-21)。基因表現分析顯示一個「細胞移動」生物學群組的表現增加,包括 P-選擇素、E-選擇素,以及嗜中性球對內皮細胞的黏附,伴隨遷移與發炎。體外刺激 TLR-2 會誘導 IL-1β 與 FcR 表現,連同 IFN-γ 與顆粒球巨噬細胞群落刺激因子 (granulocyte macrophage colony–stimulating factor),增加 E-選擇素表現,並增加嗜中性球對內皮細胞的黏附。⁹⁴ Thalidomide 抑制此一嗜中性球募集路徑,減少嗜中性球湧入、FcγRI 表現與 TNF-α 產生。經典補體路徑的 C1qA、B 與 C 成分,以及受體 C3AR1 與 C5AR1,在瘤型痲瘋病人中也增加。⁵⁹
圖 159-21:痲瘋病的免疫學。圖中呈現結核樣型痲瘋與瘤型痲瘋兩極的主要差異,連同逆轉反應與痲瘋性結節性紅斑之間的關鍵變異。
第 1 型反應或逆轉反應 (reversal reaction),一種第四型過敏免疫反應 (Type IV hypersensitivity immune response),是由針對 M. leprae 之細胞媒介免疫特異性增加所引起,可能迅速演變為神經損傷。連同 CD4+ T 細胞浸潤增加並伴隨 IL-1β、IL-2、TNF-α 與 IFN-γ 上調⁹⁶(圖 159-21),亦觀察到 CC 趨化因子單核球趨化蛋白-I 與 RANTES⁹⁷ 的增加。此外,已證明血管內皮生長因子、IL-10、CXCL-9 與 IL-17A 在逆轉反應發作時上調,連同 IL-10 與顆粒球群落刺激因子 (granulocyte colony-stimulating factor) 的下調。此一表現譜與 CD39+CCL4+CD25++ 調節性 T 細胞亞群的減少,以及與細胞毒性 T 細胞相關之 GNLY、GZMA/B 與 PRF1 基因的增加有關。⁹⁸
實驗室檢測 (LABORATORY TESTING)
進一步減少痲瘋病需要對處於疾病早期階段者進行診斷,以便治療來預防神經損傷與損害,但估計大多數病人的診斷會延遲達 2 年。⁹⁹ 其原因錯綜複雜,但包括全世界受過訓練的痲瘋病臨床醫師與實驗室技術員數目減少,以及將痲瘋病診斷併入一般家庭健康服務體系,導致誤診程度增加或開始治療的延遲。當對臨床診斷有疑慮時,可使用實驗室檢驗來協助或確認痲瘋病的推定病例。痲瘋病的臨床光譜呈現皮膚病灶與神經損傷的一系列病理表現,這與宿主免疫反應的能力相符,並取決於細胞媒介與抗體反應兩者的強度與交互作用。¹⁰⁰ 皮膚切片的實驗室診斷標準,是以石碳酸品紅技術 (carbol fuchsin technique) 之 Fite-Faraco 修改法偵測抗酸桿菌,以及以蘇木精—伊紅 (hematoxylin–eosin) 染色偵測構成痲瘋病免疫病理型別的特徵性細胞組織學模式。廣泛使用的 Ridley-Jopling 分類系統²⁶ 根據抗酸桿菌的數目以及淋巴球浸潤與組織化的程度,將疾病分為 5 型,如病理學(上文)所述。多處皮膚切片或裂隙皮膚抹片的菌檢可確立對數刻度上的細菌指數 (bacillary index),其範圍可從 0(結核樣型痲瘋病灶中未偵測到抗酸桿菌)到 6+(瘤型痲瘋中每視野 >1000 個抗酸桿菌)(圖 159-22)。就治療目的而言,在皮膚病灶或裂隙皮膚抹片中偵測到抗酸桿菌,會自動將病人歸入多菌性類別,接受 12 個月的多重藥物治療。目前並無實驗室檢驗能夠診斷痲瘋病,或辨識正朝向發展早期症狀進展的無症狀個體,因此發展一種簡單而便宜、能協助醫療專業人員根據免疫學或代謝體學感染生物標記正確診斷此病的檢驗,是值得期望的。有許多研究團隊已發展出多種部分達成此目標的檢驗,以快速側流裝置 (lateral flow devices) 測量對分枝桿菌抗原的抗體效價;細胞媒介細胞激素釋放試驗(如偵測 IFN-γ,類似用於偵測 M. tuberculosis 感染的商用全血試驗);以 PCR 擴增分枝桿菌 DNA;或使用代謝體學偵測血液或尿液中 M. leprae 感染特有的分子特徵。經調整以評估痲瘋病人對 M. leprae 特異性抗原 PGL-I 抗體效價的實驗室試驗,已使用超過 30 年,¹⁰¹,¹⁰² 包括酵素連結免疫吸附試驗 (enzyme-linked immunosorbent assays) 與側流裝置,後者使用偶聯至牛或人類白蛋白的 PGL-I 可溶性合成雙醣,分別稱為 ND-O-BSA 或 ND-O-HSA。另一種是蛋白質抗原 LID-1,由 2 種公認的 M. leprae 蛋白質 ML0405 與 ML2331 融合而成,可被 >95% 的瘤型病人辨識。¹⁰³ 將 PGL-I 的合成雙醣偶聯至 LID-1,並將此 NDO-LID 醣蛋白納入側流檢驗,可增強偵測痲瘋病人的敏感度,即使對於光譜寡菌性端 (paucibacillary end) 者亦然。¹⁰⁴ 另一團隊將側流試驗與上轉換磷光體報告技術 (up-converting phosphor reporter technology) 結合,於單一樣本中同時評估抗 PGL-I 效價與 T 細胞媒介細胞激素反應(IFN-γ、IL-10 等)。能夠定量發炎性 Th1 細胞激素相對於下調性 Th2 反應的比值,可提供更多有關個體內細胞與體液免疫複雜交互作用的資訊,可用來更佳地預測無症狀感染者或正進展至疾病者。¹⁰⁵,¹⁰⁶ 另一種用於診斷困難病例的方法,是以 PCR 分子偵測 M. leprae 特異性重複性 RLEP 序列。由於基因組中有 37 個 RLEP 序列的拷貝,這使得能在樣本中偵測到少至 3 個細菌基因組。近期證據顯示,在皮膚病灶部位切片、耳垂裂隙皮膚抹片或鼻甲中 RLEP PCR 陽性,同時抗 PGL-I 效價也呈陽性的個體,很可能有無症狀感染,且發展疾病的風險最高。⁴⁹ 我們自己在巴西帕拉 (Pará) 高度流行地區的家戶接觸者研究支持這些發現。對居住於「熱區 (hot zones)」之多個家庭的初步結果顯示,在許多情況下 >80% 的家戶接觸者抗 PGL-I 效價呈陽性,>70% 的裂隙皮膚抹片 RLEP PCR 呈陽性,達 65% 兩種生物標記皆陽性,顯示極高的感染率,每個家戶中均有 1 名或多名個體根據臨床徵象被診斷為痲瘋病(Salgado 等人,未發表觀察)。最後,代謝體學已被用於辨識痲瘋病人血清中發現的分子特徵,顯示瘤型疾病個體中循環多元不飽和脂肪酸與磷脂質增加。¹⁰⁷ 近期一份報告甚至顯示,僅藉由將矽膠板壓在病人皮膚病灶上,即可用質譜法 (mass spectrometry) 辨識感染的分子特徵。¹⁰⁸ 此外,痲瘋病的微小 RNA 組 (miRNome) 定序近期已發表,揭示了參與痲瘋病病理生理學的新標記。¹⁰⁹
雖然許多研究團隊在辨識感染生物標記方面已有進展,但要將此轉化為一種快速、便宜的即時照護 (point-of-care) 檢驗,以協助診斷整個痲瘋病臨床光譜的所有病人(包括無症狀個體),仍有很長的路要走。目前,痲瘋病的診斷必須仍仰賴受過良好訓練的痲瘋病臨床醫師與醫療工作者的稱職之手。我們已證明,在以學校為基礎的監測中針對學齡兒童,以及追蹤這些被診斷兒童的家戶接觸者,以在疾病過程早期偵測病例的重要性。使用地理資訊系統 (Geographic Information System)、空間分析工具與實驗室檢驗(抗 PGL-I 酵素連結免疫吸附試驗與 RLEP PCR),已增強辨識高度流行城市內「熱區」的能力,¹¹⁰,¹¹¹ 進而可讓在當地工作的社區醫療人員聚焦針對感染病灶。儘管如此,鑑於檢查、診斷並確保對居住於高度流行地區個體之治療的複雜性,¹⁵ 發展一種簡單的實驗室檢驗以早期診斷痲瘋病是高度值得期望的,並將有助於打斷傳播鏈,最終達到消除痲瘋病的目標。
組織病理學 (HISTOPATHOLOGY)
皮膚切片應包括病灶的真皮,若可能也應包括皮下組織 (subcutis)。蘇木精—伊紅染色以 Fite-Faraco 染色法或其他偵測抗酸桿菌的方法加以輔助。
組織病理學發現依 Ridley 與 Jopling 量表分級。²⁶ 未定類痲瘋的組織反應為非特異性。正常表皮或皮膚基底層顯示黑色素減少。少數巨噬細胞與淋巴球的血管周圍或神經周圍、淺層與深層真皮浸潤是常見發現。有時浸潤環繞皮膚附屬器,神經中偶有桿菌(圖 159-23)。結核樣型痲瘋的表皮通常正常,表皮下透明帶 (subepidermal clear zone) 缺如。有一種非乾酪樣 (noncaseating) 的真皮肉芽腫過程,主要由活化的巨噬細胞(類上皮細胞)構成,類上皮細胞中央有 CD4+ T 細胞,環繞肉芽腫的外圍 (mantle) 有 CD8+ T 細胞,並有 Langhans 型巨細胞。肉芽腫可能接觸表皮,且常環繞神經與血管排列。周邊神經受侵犯、汗腺的細胞浸潤,以及肉芽腫浸潤侵入立毛肌 (arrectores pilorum muscle) 是常見的。沒有抗酸桿菌,或當其出現時,較常見於周邊神經、立毛肌或甚至肉芽腫內¹¹²,¹¹³(圖 159-24)。邊緣結核樣型的組織病理學可藉由表皮下無細胞帶 (subepidermal grenz zone) 的存在與結核樣型痲瘋相區別。一般而言,沒有由組織良好的類上皮細胞聚集所構成的明確肉芽腫,淋巴球頻率降低,Langhans 細胞稀少,抗酸桿菌罕見(圖 159-25)。
在邊緣邊緣型中,有類上皮細胞的聚集、零散稀疏的淋巴球、無 Langhans 多核巨細胞,以及數目漸增的抗酸桿菌(圖 159-26)。邊緣瘤型痲瘋顯示表皮下無細胞帶、巨噬細胞聚集、偶見胞質豐富的類上皮細胞,以及一些泡沫細胞 (foamy cells),淋巴球少。可發現大量桿菌與一些球體(圖 159-27)。¹¹⁴
在瘤型痲瘋中,表皮正常至扁平、有表皮下無細胞帶、泡沫狀巨噬細胞的聚集與成片,混雜著遍布真皮並延伸至皮下脂肪的主要為 CD8+ 淋巴球與漿細胞。在泡沫狀巨噬細胞(Virchow 細胞)、神經、立毛肌、毛囊上皮與汗腺內,可發現大量的抗酸桿菌與球體(圖 159-28)。組織樣型痲瘋的特徵為表皮萎縮、表皮下無細胞帶,以及真皮中呈現成片的主要為紡錘狀細胞,伴核固縮 (nuclear pyknosis) 與泡沫狀、空泡化的胞質,並以漩渦狀 (storiform) 模式排列。有一些多角形細胞、巨噬細胞與發炎細胞。有些病例可能呈現假性包膜 (pseudocapsule)。此病灶類似纖維組織細胞瘤 (fibrohistiocytic tumor)(圖 159-29)。Fite-Faraco 染色揭示大量抗酸桿菌,多為桿菌叢集 (rafts) 或球體(圖 159-30)。桿菌可位於神經、許旺細胞、外分泌腺 (eccrine glands) 與血管內皮。組織樣型痲瘋中存在 CD68 陽性的巨噬細胞與紡錘細胞。¹¹⁵
在痲瘋逆轉反應中,組織病理學特徵為水腫(細胞外與類上皮細胞肉芽腫內均有)、浸潤中淋巴球與 Langhans 巨細胞數目增加、小型的類上皮細胞聚集,以及組織不良的肉芽腫。嚴重的逆轉反應病例會出現類纖維蛋白壞死 (fibrinoid necrosis)。桿菌見於巨噬細胞與神經中(圖 159-31)。降級反應 (downgrading reaction) 則顯示泡沫狀巨噬細胞的聚集、淋巴球數目顯著減少或完全消失,以及數目較多的抗酸桿菌。¹¹⁶,¹¹⁷
ENL 或第 2 型反應。其特徵性組織學特徵為水腫,以及真皮與皮下組織中的混合性發炎浸潤,主要為嗜中性球,伴嗜酸性球、淋巴球、泡沫狀巨噬細胞聚集、漿細胞與肥大細胞。大多數病例會出現血管炎,以及混合性的小葉性與中隔性脂層炎 (lobular and septal panniculitis)(圖 159-32)。大量桿菌(通常呈顆粒狀外觀)易於發現。ENL 中主要存在的淋巴球是 T 輔助細胞,而瘤型痲瘋中則以 T 抑制細胞為主。¹¹⁷,¹¹⁸
Lucio 現象 (Lucio phenomenon) 或壞死性紅斑 (erythema necrotisans)。其主要顯微特徵為皮膚或皮下的壞死性血管炎 (necrotizing vasculitis)。有小型與中型血管的類纖維蛋白壞死。Lucio 現象中報告的其他組織學表現有:表皮與淺層真皮的壞死、微膿瘍 (micro-abscess) 形成、血管新生 (angiogenesis)、內皮腫脹、管腔血栓所致的血管阻塞,以及真皮與皮下小血管壁的纖維蛋白沉積。有混合性的真皮及/或皮下浸潤,含嗜中性球、嗜酸性球、淋巴球與核塵 (nuclear dust)。桿菌見於內皮細胞、血管、神經、立毛肌、毛囊上皮、皮脂腺與汗腺。¹¹⁹⁻¹²²
神經傳導檢查——電神經肌電圖 (NERVE CONDUCTION STUDY—ELECTRONEUROMYOGRAPHY)
痲瘋病神經病變表現為不對稱的局灶性或多灶性病灶、單神經病變 (mononeuropathy) 或多發性單神經炎 (mononeuritis multiplex),由 M. leprae 對神經的直接損傷以及宿主的發炎性免疫反應所引起。有慢性神經病變,伴隨逆轉反應或 ENL 所致的急性惡化。除了局灶性病灶之外,腫大神經的卡壓 (entrapment) 也是一個顧慮,可能需要外科介入以減壓神經。評估包括神經觸診、疼痛評估、感覺評估、肌力測量與自主神經檢查。電神經肌電圖 (Electroneuromyography) 是一種精密的工具,用於在診斷時評估神經功能,以及用於病人追蹤以偵測並特徵化新病灶,尤其在逆轉反應或 ENL 期間,或用於評估卡壓症候群與神經病變性疼痛。痲瘋病神經病變始於許旺細胞去髓鞘,可能演變為軸突喪失。常受侵犯的部位包括尺神經的肘部、正中神經的腕隧道、腓神經的腓骨頭,以及脛後神經的跗隧道,這些都是評估神經病變的關鍵區域。¹²³
影像 (IMAGING)
使用高解析度超音波進行周邊神經評估是一項既定的程序。然而,直到近期它才較常被用於痲瘋病神經病變中神經功能損害的評估。神經腫大的臨床檢查可能難以測量,即使對於經驗豐富的痲瘋學家亦然,且並無穩健的參數可供記錄以追蹤痲瘋病人在多重藥物治療期間與之後的情形。高解析度超音波可用於評估回音性 (echogenicity)、血管性 (vascularity) 與神經增厚,對包括痲瘋病在內的多種神經病變使用客觀的參數與數值。¹²⁴ 此外,神經膿瘍與卡壓也可早期偵測並評估。¹²³ 其用於痲瘋病神經病變評估的應用仍在演進中,但極具前景。回音性異常、神經內都卜勒 (intraneural Doppler),以及多重藥物治療後橫切面面積高於正常上限且降低不到 30%,均與不良預後相關。多重藥物治療後神經異常的惡化,被發現與痲瘋病分類或反應的存在無關。¹²⁵
診斷流程 (DIAGNOSTIC ALGORITHM)
見圖 159-33。
圖 159-33:痲瘋病診斷流程。
痲瘋病診斷流程(圖 159-33 內容):
請病人脫去衣物,檢查所有皮膚表面並觸診神經幹。
- 具有改變之敏感性的皮膚病灶?或 僅有增厚的神經?或 神經增厚且伴隨敏感性或運動功能損害?
- 否 → 非痲瘋病
- 是 →
- 若僅有增厚的神經:
- 測試神經支配範圍的敏感性
- 測試運動功能
- 正常 → 自主神經改變?
- 1 或 2 項異常 → 痲瘋病
- 瘤型痲瘋 (LL):瀰漫性浸潤、結節與自主神經功能障礙;許多增厚的神經
- 邊緣瘤型 (BL):一個或多個浸潤性病灶,可能呈小凹狀;通常超過一條增厚的神經 → 痲瘋病
- 邊緣邊緣型 (BB):一個或數個浸潤性病灶 → 痲瘋病
- 邊緣結核樣型 (BT):一個或數個結核樣型病灶;不超過一條增厚的神經 → 痲瘋病
- 結核樣型 (TT):一個或數個色素減退病灶;無增厚的神經 → 痲瘋病
- 未定類痲瘋 (Indeterminate):一個或數個色素減退病灶;無自主神經改變
- 若僅有增厚的神經:
(其他病型對應:許多浸潤性病灶,伴隨或不伴隨結節+許多增厚的神經 → 痲瘋病。)
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 159-1。
原發病灶 (Primary Lesions)
■ 斑與片 (Macules and patches)。白色糠疹 (pityriasis alba) 與未定類痲瘋的色素減退彼此相似。若病人出生於或曾居住於流行地區,則兩者間的區別可藉由神經學或組織學檢查來做出。色素減退的 BL 斑塊可能只有極輕微的硬結,以致類似斑片。微血管擴張 (Telangiectasias) 可能呈爆發性,或在臉部與上軀幹呈墊狀 (mats)。
■ 丘疹至結節性病灶 (Papular to nodular lesions)。在真皮中,痲瘋病可能模仿或被以下疾病所模仿:皮膚纖維瘤 (dermatofibromas)、組織細胞瘤 (histiocytomas)、淋巴瘤、類肉瘤病 (sarcoidosis)、神經纖維瘤病 (neurofibromatosis)、梅毒、無反應性利什曼病 (anergic leishmaniasis)、副球黴菌病 (paracoccidioidomycosis)、著色芽生菌病 (chromoblastomycosis)、孢子絲菌病 (sporotrichosis)、Lobo 黴菌病 (lobomycosis)、結核,以及其他肉芽腫。爆發性與復發性發炎性皮下結節可能為 ENL、結節性紅斑 (erythema nodosum)、硬結性紅斑 (erythema induratum) 與血管炎。Lucio 痲瘋中可觸及但不可見的皮下結節可能模仿脂肪瘤。
■ 斑塊 (Plaques)。紅斑性斑塊可能模仿蕈狀肉芽腫 (mycosis fungoides)。無色素改變的斑塊可能外觀如風疹塊,造成與蕁麻疹 (urticaria) 的混淆。色素減退斑塊可能模仿丘疹鱗屑性疹 (papulosquamous eruptions)。斑塊內的正常皮膚島可能令人聯想到乾癬 (psoriasis)。
■ 多形性水疱大疱性疹/真皮表皮分離 (Polymorphous vesiculobullous eruption/Dermoepidermal separation)。它們可能發生於 ENL。在 LL 中,IgM 沉積於表皮基底膜並不少見。這些抗體不一定具致病性,但可能混淆診斷。
■ 環狀病灶 (Annular lesions)。痲瘋病可能模仿或被環狀紅斑 (annular erythemas)、類肉瘤病、梅毒或癬 (tinea) 所模仿。
次發病灶 (Secondary Lesions)
■ 梗塞 (Infarcts)。Lucio 現象病灶與壞死性 ENL 模仿敗血性梗塞。
■ 潰瘍 (Ulcers)。潰瘍發生於 Lucio 現象,以及繼發於血管阻塞的 ENL。在有神經破壞的病人,神經營養性潰瘍 (neurotrophic ulcers) 發生於蹠面;Lucio 痲瘋中可見繼發於靜脈功能不全的腿部潰瘍。
臨床綜合徵 (Clinical Constellations)
■ 類全身性紅斑性狼瘡之變化 (Systemic lupus erythematosus–like changes)。梭形手指 (fusiform fingers)、天鵝頸畸形 (swan neck deformity)、偽陽性梅毒試驗、抗磷脂質抗體 (antiphospholipid antibodies)、狼瘡抗凝物 (lupus anticoagulant)、高球蛋白血症 (hyperglobulinemia) 與貧血。
■ 血管炎 (Vasculitis)。真正的血管炎可能發生於 ENL、Lucio 現象與 Lucio 痲瘋。在臨床上,呈結節性質的痲瘋病灶可能被誤診為「血管炎」。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
痲瘋病臨床病程中最重要的問題之一,是急性或亞急性發炎發作的出現,定義為反應 (reactions)。痲瘋病反應由針對 M. leprae 抗原的免疫反應所引起,分為第 1 型或逆轉反應(主要涉及周邊神經與皮膚)與第 2 型或 ENL(可能有局部或全身症狀)。急性神經炎也可被視為一種反應。反應從不發生於未定類病人。所有接受多重藥物治療的病人中,多達 50% 可能在治療期間出現反應,但這些反應也可能發生於治療前與治療後。診斷時即存在的神經病變、多菌性痲瘋、疾病範圍,以及覆蓋在周邊神經幹上之病灶的存在,是增加反應與神經功能損害風險的因子。¹²⁶
逆轉反應與 ENL 在某些病人中可能一起發生(表 159-2)。逆轉反應可發生於多達 30% 的病人,而結核樣型痲瘋病例則鮮少受影響,¹²⁷ 大多數的反應性發作發生於邊緣型,主要是邊緣瘤型與邊緣邊緣型,其次是瘤型痲瘋。¹²⁸ 它始於皮膚病灶與神經功能損害的突然惡化,無明顯的全身侵犯。除了反應前病灶呈現更多浸潤與脫屑外,先前與新發生的病灶可能呈現鮮紅、灼熱與觸痛(圖 159-34),有時潰瘍,常伴隨周邊神經腫大,且通常伴隨疼痛。逆轉反應需要立即介入,因為它可能導致神經損害與永久性失能。ENL 是一種伴隨免疫複合體沉積、侵犯不同器官的侵襲性血管炎,導致的後遺症包括神經炎、脂層炎、絲球體腎炎 (glomerulonephritis)、關節痛、副睪炎 (epididymitis)、睪丸炎 (orchitis)、眼睛發炎、骨炎與淋巴腺炎,並伴隨發燒、水腫與倦怠等全身症狀。¹²⁹
表 159-2 痲瘋反應與處置 (Lepra Reactions and Management)
| 項目 | 逆轉反應(第 1 型)Reversal Reaction (Type I) | 痲瘋性結節性紅斑(第 2 型)Erythema Nodosum Leprosum (Type II) |
|---|---|---|
| 主要痲瘋亞型 | 邊緣結核樣型、邊緣邊緣型、邊緣瘤型、瘤型痲瘋 | 瘤型痲瘋 |
| 徵象與症狀 | 皮膚病灶急性惡化;神經功能損害急性惡化;無明顯全身症狀 | 結節性紅斑;多形性紅斑;嚴重皮膚壞死性血管炎;發燒、水腫、倦怠 |
| 皮膚外侵犯 | 神經炎 | 神經炎、脂層炎、絲球體腎炎、關節痛、副睪炎、睪丸炎、眼睛發炎、骨炎、淋巴腺炎 |
| 治療 | Prednisone(1-2 mg/kg/d,緩慢遞減約 3 個月) | Thalidomide 100-400 mg/d 併用 prednisone(1-2 mg/kg/d);或 Pentoxifylline 400 mg 一日三次 |
高細菌指數與瀰漫性皮膚浸潤是重要的危險因子,¹³⁰ 而 65% 的病例有超過一次的 ENL 發作。¹³¹ 主要皮膚表現為結節性紅斑,較可觸及而不易見(圖 159-35),可能伴隨多形性紅斑或嚴重皮膚壞死性血管炎(Lucio 現象)(圖 159-36)。免疫學疾患,如 HIV 與 HTLV 感染、免疫抑制藥物或免疫生物製劑,可能干擾痲瘋病的緩解。2003 年描述了第一例因 HIV 感染個體免疫重建發炎症候群 (immune reconstitution inflammatory syndrome, IRIS) 所致的痲瘋病例。¹³² AIDS 病人因抗病毒治療所致細胞媒介免疫反應的提升,現已在 IRIS 中廣為認可,¹³³ 而痲瘋病人中 HIV 與其他病毒感染的百分比很高,顯示所有痲瘋病人都應接受 HIV、HBV、HCV 與 HTLV-1 檢測。¹³⁴ 病毒共同感染會降低痲瘋病人的存活,¹³⁵ 同時增加神經炎、神經功能損害與痲瘋病復發的比率。¹³⁴ 抗 TNF 治療可在受感染個體於停用該免疫生物製劑後導致臨床痲瘋病的發生,¹³⁶⁻¹³⁸ 無論是否涉及反應性發作。區分反應與復發 (relapses) 是重要的。反應可能發生於多重藥物治療之前、期間與之後數年。通常,它們是急性的,新病灶快速出現、舊病灶浸潤、神經功能惡化及/或全身侵犯,並對抗發炎藥物治療反應良好。另一方面,復發一般是緩慢進展的,幾乎總是伴隨原發病灶的再現,繼之以新病灶的逐漸出現,連同神經侵犯,且相對地,對抗發炎藥物無反應。真正的復發必須在確認病人完成第一次治療後才能定義。若確認完成完整療程,則有必要檢測藥物抗藥性,不過無法排除再感染。若病人被錯誤分類,則可能是療程長度不足。組織病理學有助於區分反應與復發病例。¹³⁹
雖然痲瘋病以皮膚感覺喪失而聞名,但神經病變性疼痛可能在多重藥物治療期間或之後出現,原因為組織發炎或神經系統功能障礙。¹⁴⁰ 神經幹疼痛的發作可能為自發性,或在觸診後出現,可能突然或隱伏,但許多病人會經歷反覆發作。超過半數的病人在多重藥物治療期間或之後有某種疼痛發作,瘤型痲瘋的盛行率較高,朝邊緣型與結核樣型遞減。最常受影響的神經是尺神經與脛神經。¹⁴⁰
若疼痛在治療期間持續,或在完成多重藥物治療後變得長期持續,則可能定義為慢性神經炎或神經病變。¹⁴¹
處置 (MANAGEMENT)
介入 (INTERVENTIONS)
藥物 (MEDICATIONS)
用於治療痲瘋病的藥物基本上有 3 大類:抗生素、抗發炎或免疫抑制劑,以及止痛藥物。第一類抗生素有明確的治療標準,即 WHO 多重藥物治療方案,其含有 rifampicin 與 dapsone,加或不加 clofazimine,以每月泡殼包裝 (blister packs) 提供。抗發炎藥物(通常為 prednisone 與 thalidomide)用於藉由減少發炎來控制痲瘋病反應,而止痛藥則用於控制神經病變性疼痛。在 1941 年 Guy Faget 發現磺胺類 (sulfones) 改善痲瘋病徵象的效力之前,¹⁴² 大風子油 (chaulmoogra oil)——一種在印度使用數十年的藥物——是唯一常用的痲瘋病治療方法,雖然其療效令人質疑,因為它一般僅在皮膚誘發局部發炎反應。Dapsone 是一種簡單、低成本、高效的痲瘋病藥物,使用每日劑量 100 mg 或 1 至 2 mg/kg。該藥由胃腸道吸收,經腎臟排除。它通常耐受良好,雖然其作用依賴葡萄糖 6-磷酸去氫酶 (glucose 6-phosphate dehydrogenase, G6PD) 的存在,這是一種 X 染色體遺傳的酵素,全世界有 4 億人缺乏此酵素,大多在瘧疾存在的熱帶地區,¹⁴³ 也是痲瘋病盛行的地區。G6PD 缺乏會因氧化壓力導致嚴重的溶血事件,包括變性血紅素 (methemoglobin) 的形成,臨床上偵測為鞏膜、嘴唇與手指末端呈紫色,伴隨倦怠、頭痛與呼吸困難。除了高度溶血之外,使用 dapsone 的 G6PD 缺乏病人發展出嚴重危及生命之溶血性貧血的風險較高,必須更換藥物。¹⁴⁴ Dapsone 過敏症候群是一種罕見但可能致命的事件,呈現發燒與皮膚紅疹,最終侵犯內臟器官(尤其是肺部),伴嗜酸性球浸潤與肺炎。它可能在治療期間任何時間發生,並可能與伴嗜酸性球增多及全身症狀之藥物疹(DRESS 症候群)相關。¹⁴⁵
Clofazimine 是一種色素,除其未知的抗生素機制外,也具有抗發炎特性。就病人而言,主要的反對意見是它對脂肪組織與巨噬細胞沉積物的親和性,導致皮膚色素沉著,尤其在病灶處。另一個副作用是皮膚乾燥,連同色素沉著,使皮膚呈現高度乾皮病 (xerodermic) 的外觀(圖 159-37)。它的使用劑量為每月一次 300 mg,以及每日 50 mg,僅用於多菌性病人。Clofazimine 為痲瘋病人耐受良好。¹⁴⁶
Rifampicin 具有高度殺菌性,與 dapsone 及 clofazimine 的每日投藥不同,它僅在監督下每月投予一次,兒童 450 mg、成人 600 mg。不良反應包括臉部與頸部潮紅、搔癢與皮膚紅疹、食慾不振、噁心、嘔吐與腹瀉、倦怠(可能需要停藥)、紫斑與鼻出血。類流感症候群是使用間歇劑量 rifampicin 時發生的一種未充分理解的免疫學副作用,其特徵為發燒、虛弱、肌肉痛與頭痛,有時伴隨骨痛。最終可能發展出嗜酸性球增多、腎炎、血小板低下與休克。雖然被認為罕見,但它是一項涵蓋 20,667 名接受多重藥物治療之痲瘋病人的巴西研究中所報告的主要副作用。¹⁴⁷
現今使用的多重藥物治療方案與 1982 年首度實施時相同(表 159-3),對多菌性病例處方 dapsone、clofazimine 與 rifampicin 達 24 個月,或對寡菌性病人處方 dapsone 與 rifampicin 達 6 個月。懷孕與哺乳並不禁忌使用多重藥物治療。¹⁴⁴
在 1960 年代,連同確認 clofazimine 與 rifampicin¹⁴⁸ 對 M. leprae 的療效,第一批 dapsone 抗藥性痲瘋病例出現。¹⁴⁹ 在 1970 年代,WHO 決定以 3 藥策略取代 dapsone 單一療法,將 dapsone、rifampicin 與 clofazimine 結合於一種新的藥物方案中以治療痲瘋病,稱為多重藥物治療。雖然痲瘋病的抗藥性似乎維持在低點,¹⁵⁰ 但文獻中多重抗藥性 (MDR) 痲瘋的報告正在增加,¹⁵¹⁻¹⁵³ 在不久的將來可能成為痲瘋病治療的一項顧慮。¹⁵⁴ 可供使用的替代藥物,無論是對抗藥性菌株或對多重藥物治療有副作用的病人,即 ofloxacin、minocycline 或 clarithromycin(表 159-3),對痲瘋病治療似乎安全有效,¹⁴⁶ 但仍需要新的替代藥物。雖然有少數結構良好、中等品質證據的臨床試驗用於治療痲瘋病的神經損傷,包括一項靜脈注射 methylprednisolone 脈衝療法的試驗,¹⁵⁵ WHO 建議痲瘋病反應必須立即以抗發炎或免疫抑制藥物治療。¹⁴⁴ 最廣泛使用的是皮質類固醇與 thalidomide。逆轉反應可用 prednisone 以 1 至 2 mg/kg/d 的劑量、採遞減方案治療,每 15 天減少 10% 至 15% 的劑量,完整的治療週期最長持續 3 個月。若臨床情況惡化,可能有必要回到先前較高的劑量,將此類固醇治療水準延長 30 至 45 天,隨後再次逐漸減量。使用皮質類固醇期間必須控制血糖值與血壓。長期使用可能發生青光眼、白內障、月亮臉、紋路 (striae)、腎上腺萎縮與骨質疏鬆,且如同其他免疫抑制劑,可能出現其他感染性疾病,如全身性黴菌感染與結核。此外,糞小桿線蟲 (Strongyloides stercoralis) 過度感染也是一項顧慮。可使用 Ivermectin 200 µg/kg/d 共 2 天、2 週後重複,作為預防。¹⁵⁶ 經由等效換算計算後,prednisone 可由其他類固醇取代,包括其代謝物 prednisolone、dexamethasone 或 deflazacort。對任何使用糖皮質素的病人,無論劑量與療程長短,均建議鈣攝取 1200 至 1500 mg/d 與維生素 D 補充。¹⁵⁷
ENL 可用 100 至 400 mg/d 的 thalidomide 治療。由於 thalidomide 是一種致畸胎藥物,對育齡婦女在開始治療前,必須檢測懷孕並處方 2 種避孕方法。通常,ENL 伴隨神經損傷出現,需要併用類固醇治療。較高劑量的 prednisone(高達 2 mg/kg/d)與改善的神經功能結果相關,但若在反應的第一個徵象出現後立即開始治療,較低劑量 1 mg/kg/d 可能有相同效果。¹⁵⁸ 若 ENL 伴隨任何其他組織發炎,如睪丸炎或虹膜炎,或伴隨手足反應(如骨關節炎與軟組織發炎),也需要類固醇。在必須避免使用 thalidomide 的病例中,pentoxifylline 400 mg、一日三次,是一種替代藥物,對控制肢體水腫與全身症狀相當有用。¹⁵⁹
類固醇與 thalidomide 可藉由減少水腫與針對神經的免疫反應來幫助有神經病變性疼痛的病人,尤其在急性發作時。然而,其治療仍是一項挑戰,尤其對於有慢性神經炎/神經病變者。中樞神經系統藥物,如三環抗憂鬱劑 amitriptyline 與 imipramine,或抗痙攣劑 carbamazepine 與 gabapentin,已被嘗試用於控制這些病人的慢性神經炎/神經病變,但這些藥物不會干預神經損傷過程,因此無法保護痲瘋病人免於神經惡化。雖然一些軼事性報告顯示,免疫抑制劑如 cyclosporine 與 azathioprine 可能有助於治療慢性神經炎/神經病變病人,但近期一項使用 azathioprine 的試驗並未顯示逆轉反應病人有改善。¹⁶⁰ 在病理生理學以及治療神經病變性疼痛的新藥方面,仍需要更多研究。
程序 (PROCEDURES)
病人可使用簡單的預防技巧來防止損害的進展。手與足皮膚的按摩與保濕對於避免裂隙、潰瘍與固定爪狀形成是必要的。除了對失去感覺與有失能的足部使用特殊靴底或客製化鞋子外,自我檢查對於外傷的早期偵測與立即治療是必須的。對於手,內收、外展與對掌運動對於維持營養良好的肌肉與健康的關節是必要的。工作器具與家庭用品的改造有助於預防外傷與燒燙傷。對於眼睛,在兔眼的病例中使用潤滑眼藥水可預防角膜炎。在較複雜的病例中,病人可能被轉介至專科中心,由多專業團隊進行更複雜的治療,以進行身體、心理與社會復健。在痲瘋病的不同階段,可能需要手術。神經膿瘍不常見,較多出現於原發神經痲瘋與結核樣型痲瘋,朝瘤型痲瘋端則較少,但它可能是疾病的第一個臨床表現。¹⁶¹ 在此類病例中,膿瘍引流是必須的,其內容物必須送實驗室檢查。神經減壓 (Nerve decompression) 可用於改善痲瘋病神經病變與肌肉功能,儘管並無可靠的臨床試驗能確切證明其用處。¹⁶²
最後,重建手術可恢復手與足的某些功能面向,如握住玻璃杯的能力或抬起足部的能力;矯正眼睛問題,如兔眼,預防角膜炎、感染與失明;並改善美觀,如鼻部塌陷的手術矯正(現今已罕見)¹⁶³ 或手部骨間與第一指蹼間隙萎縮的矯正。¹⁶⁴
諮商 (COUNSELING)
諮商是痲瘋病人處置中涉及的關鍵過程。神經損傷及其處置包括諮商與傷害減少。¹⁶⁵ 雖然近數十年來因為對抗歧視、協助人們重返社會並廢除歧視性法律的活躍社會運動之存在而有所改善,但汙名在現代社會中仍然存在。痲瘋病雖然可治癒,但仍是一種導致嚴重失能的疾病,即使在疾病初期階段被診斷的病人也害怕經由失能而惡化。因此,諮商應對每位被診斷為痲瘋病的人及其家屬提供。¹⁶⁶ 自我照護團體是必要的,但它們不應是排他性的。痲瘋病相關的失能照護與處置,必須與一般衛生服務中對其他疾病所致失能的照護一併納入。¹⁶⁷
治療流程 (TREATMENT ALGORITHM)
見圖 159-38。
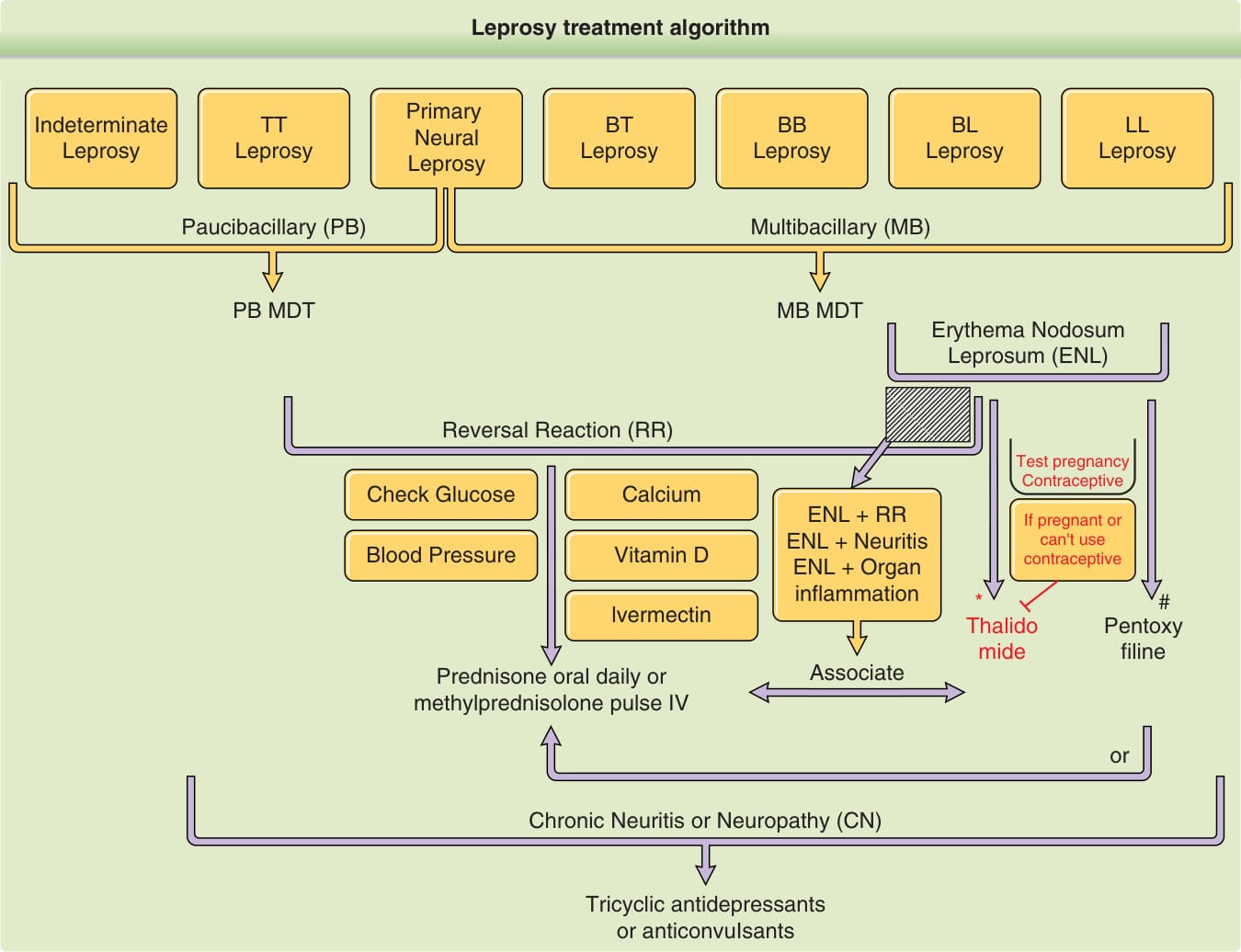
*圖 159-38:痲瘋病治療。多重藥物治療是痲瘋病治療的選擇。然而,當對任一藥物不耐受時,可使用可供替代之藥物,如 ofloxacin、minocycline 或 clarithromycin。此外,若對多重藥物治療無反應,可檢測藥物抗藥性,並以相同的替代藥物取代 1 種或多種多重藥物治療藥物。對於反應,thalidomide 並非所有國家都有,甚至可能被禁止。不允許對孕婦或育齡婦女處方 thalidomide。若有必要使用,有些國家有嚴格的規定或法律,可能要求做懷孕測試並使用避孕方法才能處方 thalidomide。#Pentoxifylline 對懷孕而言屬「C 類」藥物,因此唯有當潛在益處足以證明對胎兒的潛在風險為合理時才應使用。
痲瘋病治療流程(圖 159-38 內容):
- 原發神經痲瘋、未定類痲瘋、TT 痲瘋 → 寡菌性 (PB) → PB 多重藥物治療 (PB MDT)
- BT、BB、BL、LL 痲瘋 → 多菌性 (MB) → MB 多重藥物治療 (MB MDT)
- 逆轉反應 (RR):檢查血糖、血壓;給予 prednisone 口服每日或 methylprednisolone 脈衝靜脈注射
- 痲瘋性結節性紅斑 (ENL):測試懷孕、避孕、補充鈣與維生素 D;給予 thalidomide;ENL+RR、ENL+神經炎、ENL+器官發炎時可合併;若懷孕或無法使用避孕,則使用 pentoxifylline
- 慢性神經炎或神經病變 (CN):三環抗憂鬱劑或抗痙攣劑
預防/篩檢 (PREVENTION/SCREENING)
痲瘋病的預防有一項基本要求,即接觸者檢查。檢查所有家戶接觸者,應是流行國家任何痲瘋病防治計畫的強制部分,有些計畫將此概念擴及社會接觸者,即與指標病例一起工作或有較近距離接近者。家戶接觸者罹患痲瘋病的風險,比未與病例接觸者高 5 至 8 倍(圖 159-39)。然而,即使有持續的警戒,社區中也僅有多達 30% 的病例會在家戶接觸者中被偵測到;因此,可能還有其他遺傳或環境因子涉及感染的維持。¹⁶⁸
早期偵測病例是另一項有效預防痲瘋病的工具。除了接觸者檢查之外,在一般人口或特殊社區(如學齡兒童¹¹⁰)中的痲瘋病活動,可用於提高警覺、減少汙名,並增加痲瘋病早期病例的偵測。雖然以 rifampicin 進行化學預防 (chemoprophylaxis) 在最初 2 年顯示出某種程度的保護力 (57%),但 4 年後在 rifampicin 與安慰劑組之間並未觀察到差異,¹⁶⁹ 因此目前並無官方建議對痲瘋病接觸者使用化學預防。就免疫預防 (immunoprophylaxis) 而言,巴西長期以來一直對接觸者使用卡介苗 (Bacillus Calmette-Guerin, BCG) 再接種。一項為期 18 年的追蹤研究發現,相較於未接種疫苗者,接種 BCG 的接觸者有 56% 的保護力,¹⁷⁰ 因此,巴西的痲瘋病指引持續對所有家戶接觸者納入 BCG 接種。
圖表 (FIGURES AND TABLES)
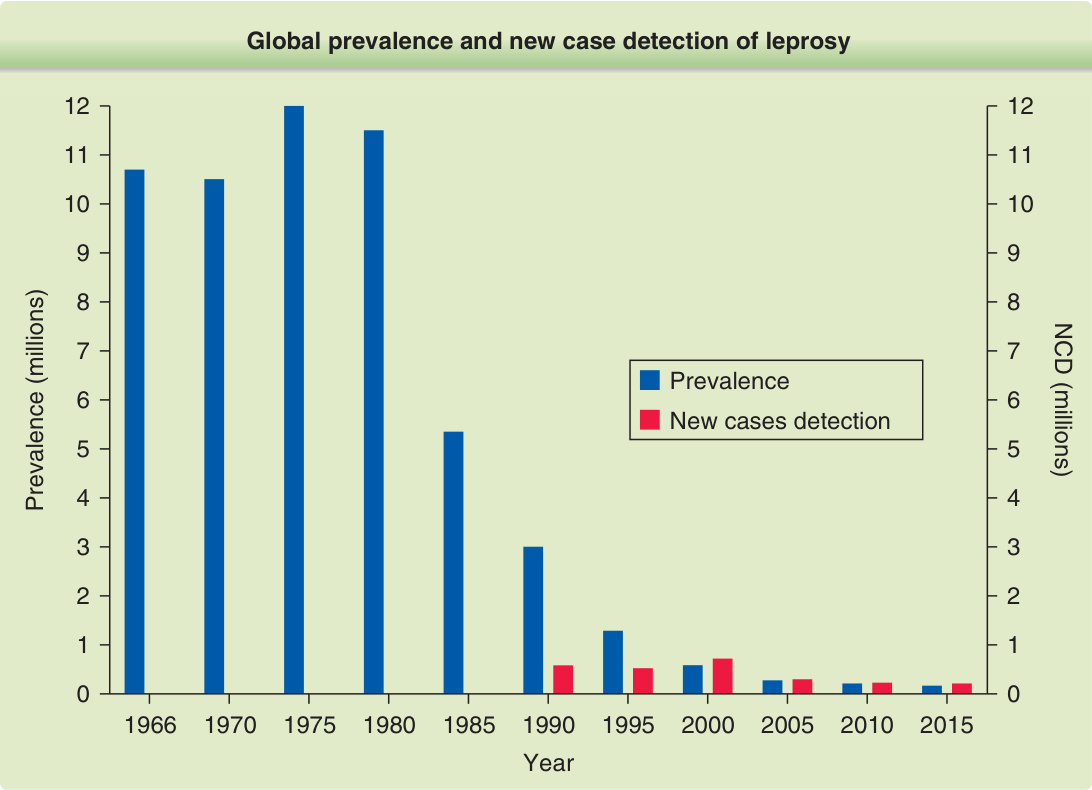
圖 159-1:痲瘋病全球盛行率與新病例偵測之歷史演變。WHO 於 1966 年首度正式嘗試估計全球痲瘋病負擔,當時推估病例量為 10,786,000 例,其中 60% 未登記接受治療。全球偵測數首度於 1991 年通報,1990 年全球偵測到 584,000 例新病例。目前,新病例偵測是痲瘋病負擔最重要的流行病學指標之一,連同兒童比例與第 2 級失能比例。(經巴西帕拉聯邦大學 Josafá Barreto 教授許可使用。)
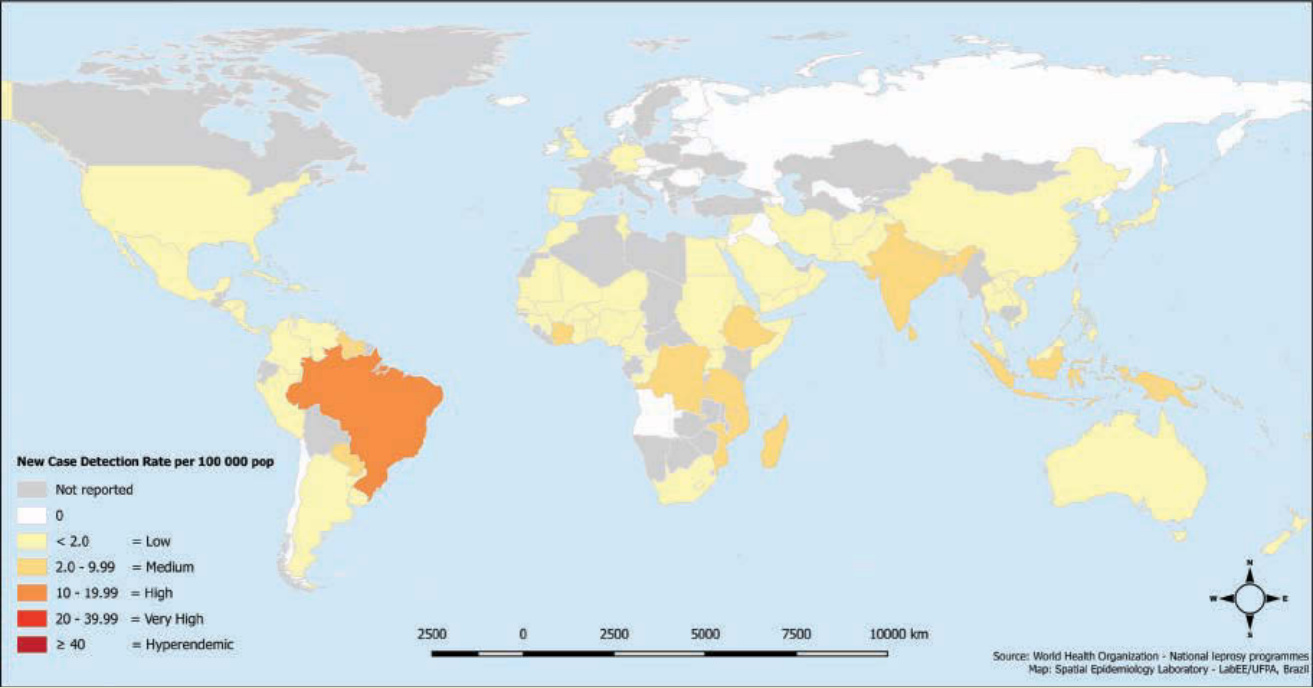
圖 159-2:2015 年全球每 100,000 人口之痲瘋病新病例偵測率。2015 年在 136 個國家或地區通報了超過 210,000 例新病例。印度、巴西與印尼占全球痲瘋病負擔的 81%。全球新病例偵測率為 3.2。(經巴西帕拉聯邦大學 Josafá Barreto 教授許可使用。)
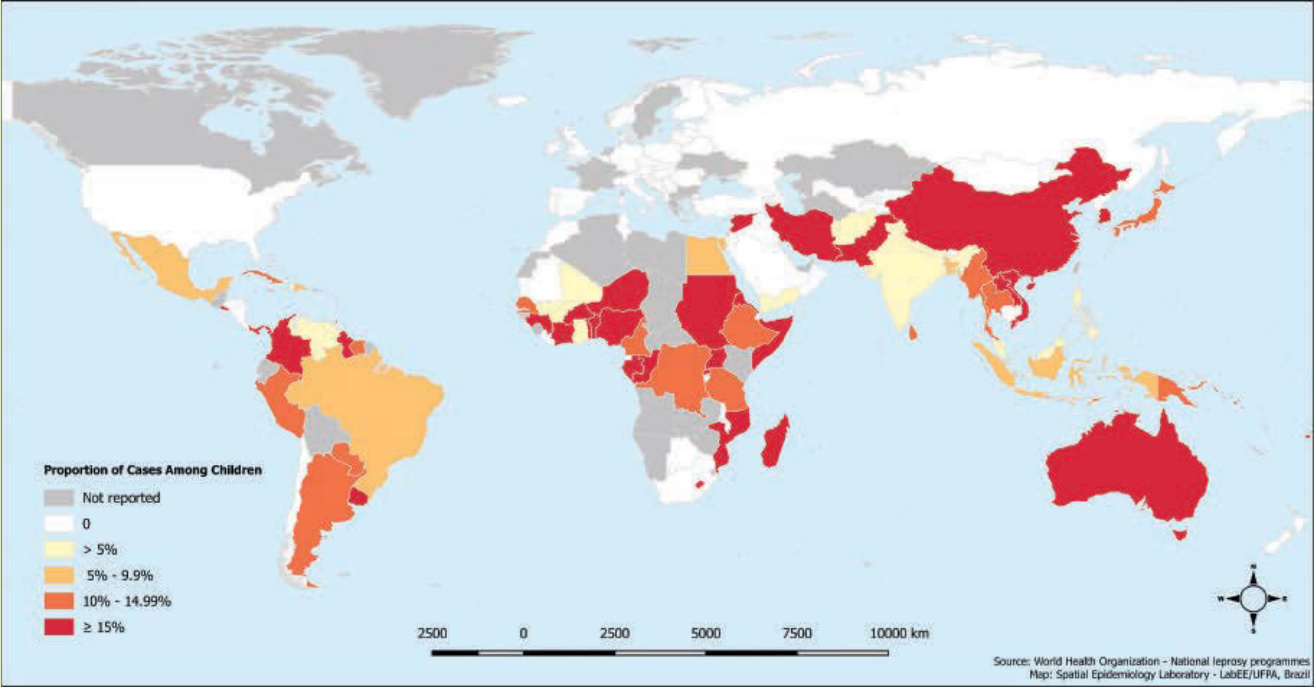
圖 159-3:2015 年 15 歲以下兒童中之痲瘋病例比例。雖然對此指標並無從「低」到「高」的特定分類,但它能有力地指示其居住社區內存在活躍的感染源。(經巴西帕拉聯邦大學 Josafá Barreto 教授許可使用。)
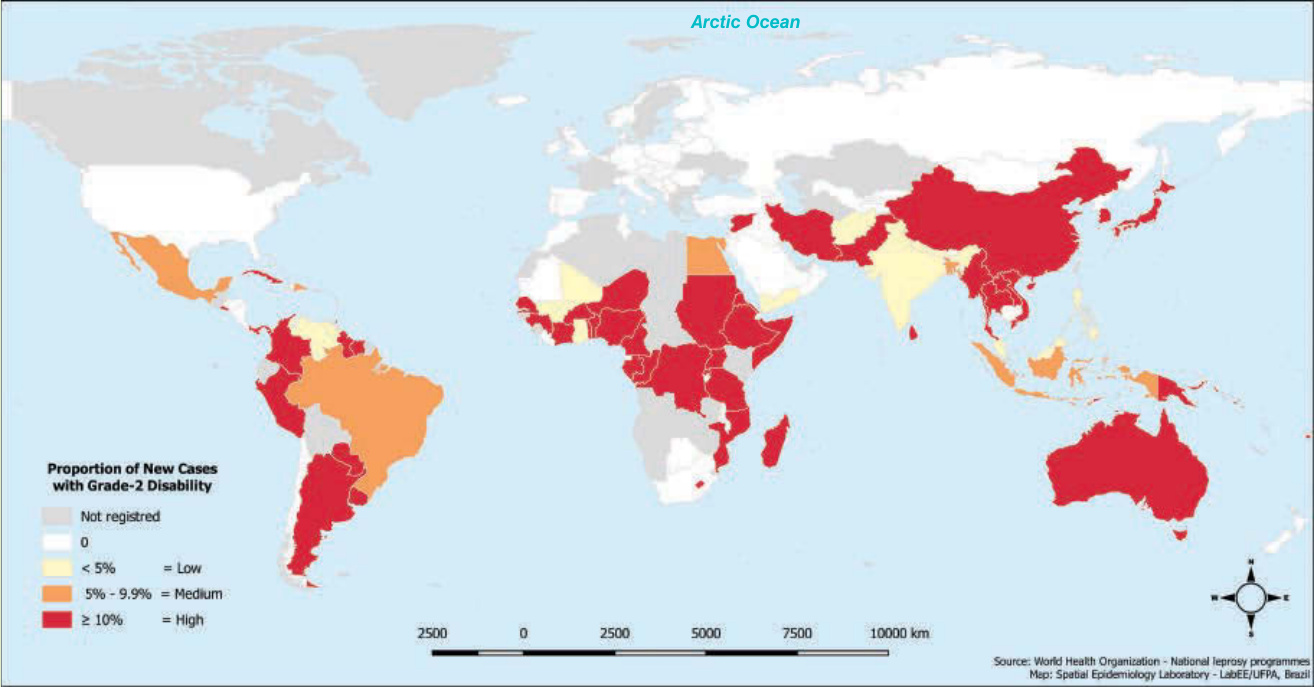
圖 159-4:2015 年診斷時具第 2 級失能之痲瘋病新病例比例。它反映痲瘋病診斷的長期延遲,凸顯衛生服務體系的失敗以及疾病控制方法的缺口。(經巴西帕拉聯邦大學 Josafá Barreto 教授許可使用。)
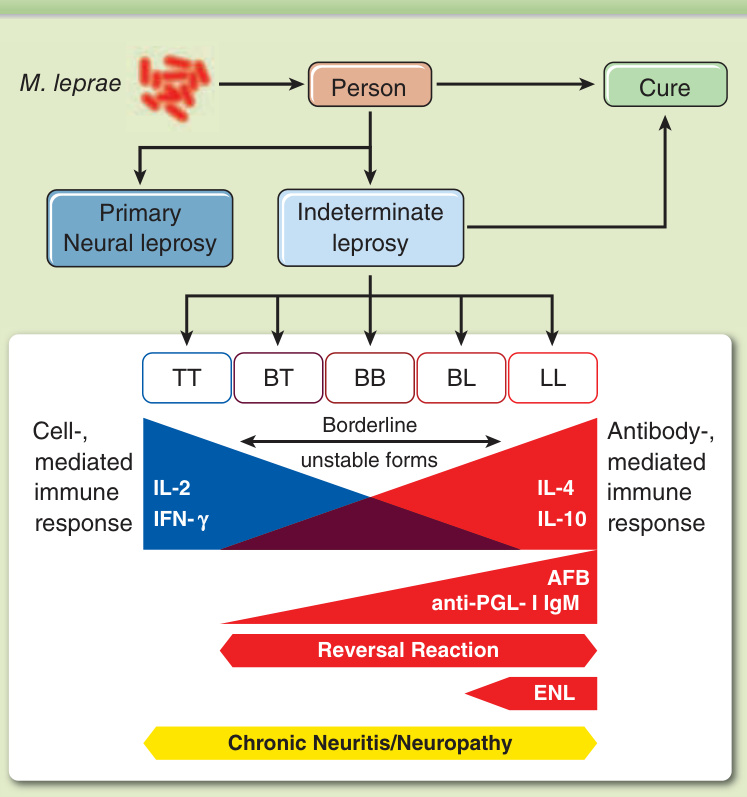
圖 159-5:痲瘋病臨床光譜。多達 80% 暴露於 M. leprae 的人可能在症狀出現前或亞臨床痲瘋後解決問題並清除桿菌。有些病人會發展出原發神經痲瘋,無皮膚病灶。所有有皮膚病灶者都會經過一個未定類型,然後演變為極端結核樣型痲瘋 (TT) 或瘤型痲瘋 (LL),或演變為不穩定的邊緣型痲瘋。朝 TT 的寡菌性 (PB) 端有良好的細胞免疫反應 (CIR),存在 Th1 細胞激素,而朝 LL 的多菌性端則呈現受損的 CIR 與高抗體反應,含 Th2 細胞激素。抗酸桿菌與抗 PGL-I IgM 兩者在 PB 端均低或陰性,並朝多菌性端增加。逆轉反應可能特別發生於邊緣型痲瘋,而痲瘋性結節性紅斑則發生於邊緣瘤型痲瘋 (BL) 與 LL 病人。慢性神經炎或神經病變可能發生於原發神經痲瘋以及除未定類痲瘋外的所有臨床型。
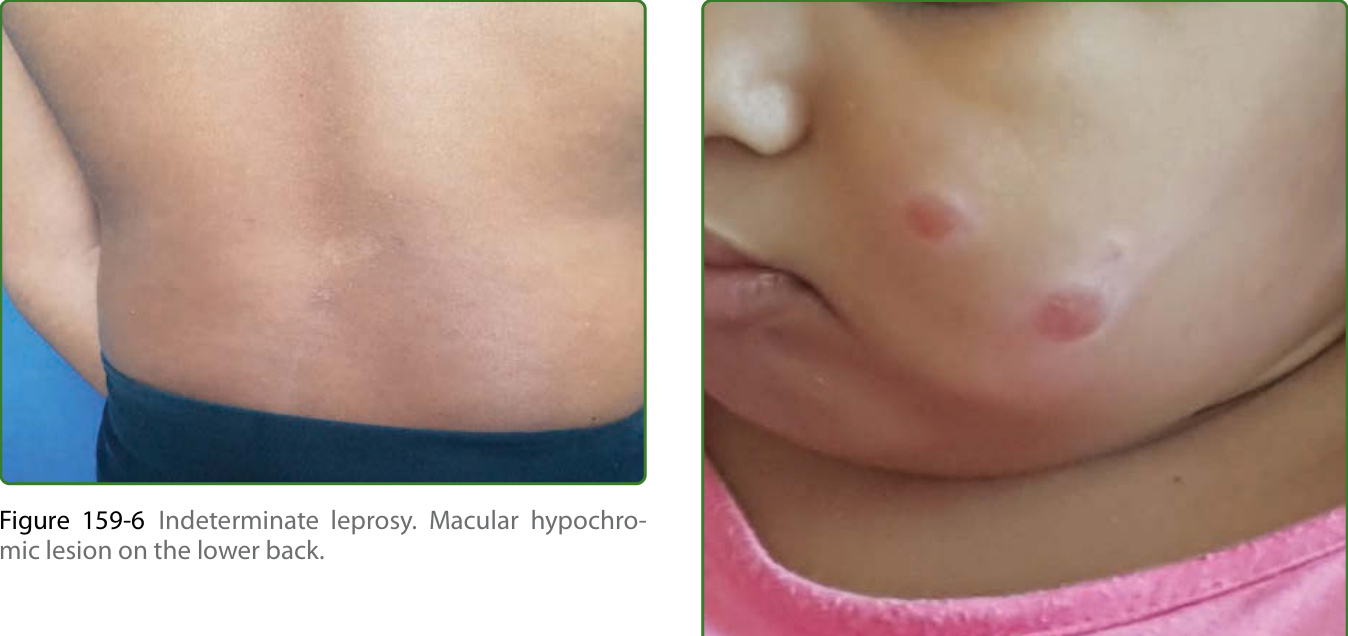
圖 159-6:未定類痲瘋。下背部的斑狀色素減退病灶。
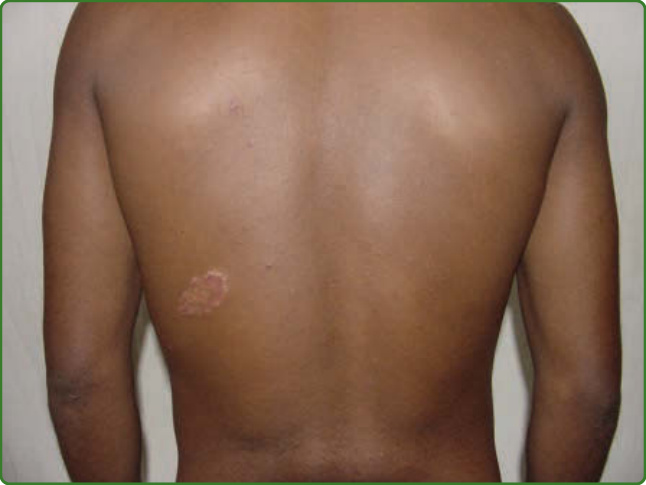
圖 159-7:結核樣型痲瘋。界線分明的病灶,中央呈斑狀色素減退與萎縮外觀,周邊有一群以環狀模式分布的丘疹。
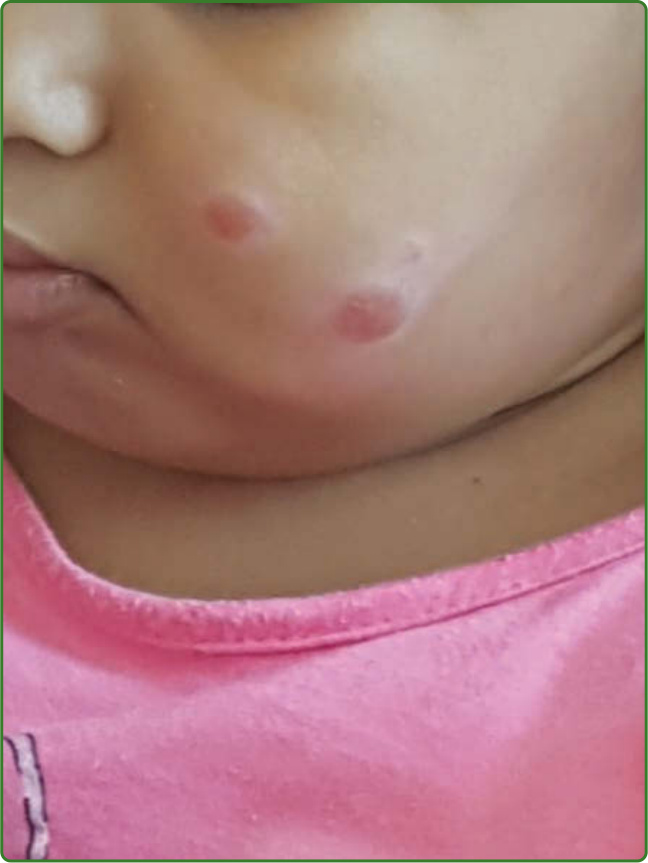
圖 159-8:嬰兒結節型痲瘋。一名兒童臉部出現 2 個結節性病灶,其家庭中有 2 名成人被診斷為多菌性痲瘋。
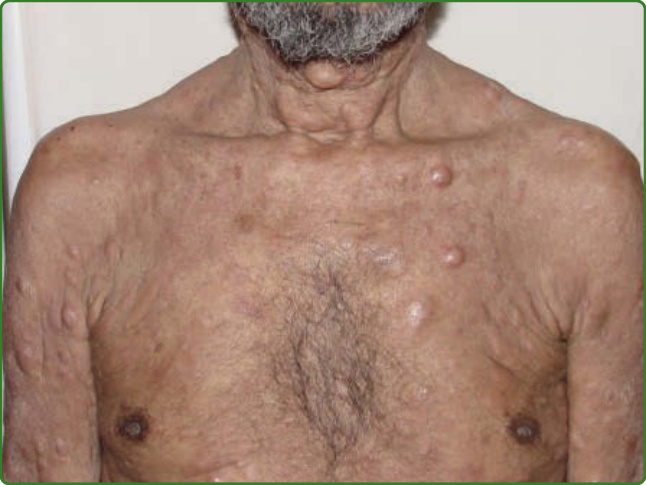
圖 159-9:瘤型痲瘋。多發性結節(hansenomes 或 lepromes)遍布全身皮膚,伴隨瀰漫性浸潤。
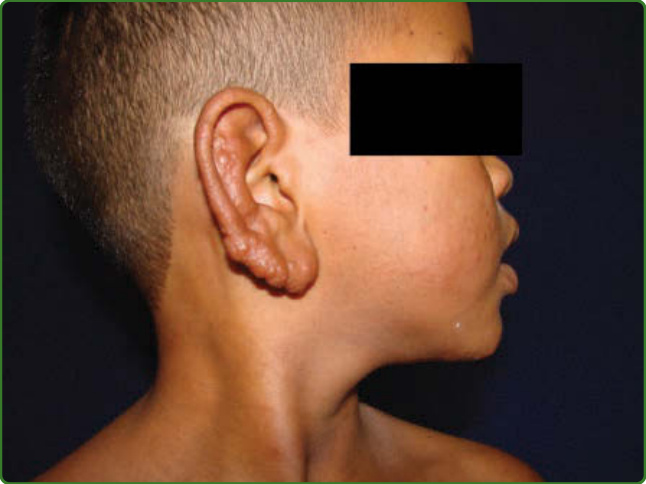
圖 159-10:瘤型痲瘋。一名兒童耳部的丘疹、結節與浸潤。他在皮膚其他部位也有許多病灶。
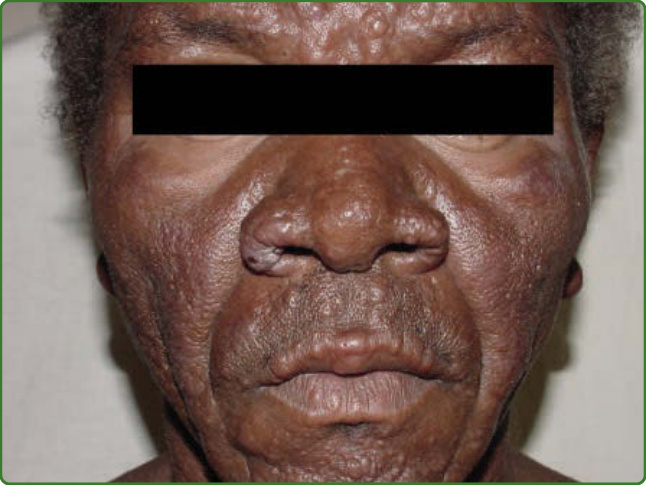
圖 159-11:獅面 (Leonine facies)。瘤型病人臉部有瀰漫性結節與浸潤,造成獅子臉外觀。
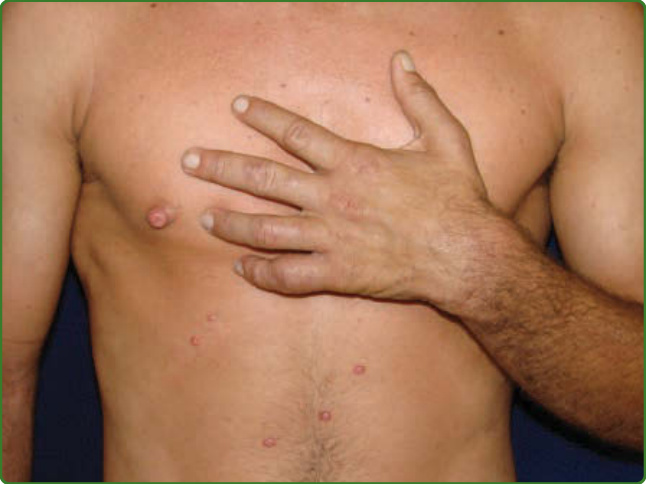
圖 159-12:瘤型痲瘋組織樣型或 Wade 痲瘋。出現結節,可能瀰漫或散在,有些類似傳染性軟疣 (molluscum contagiosum) 病灶(如圖所示),但請注意右乳頭上有一結節,左手也有一些浸潤伴爪狀形成。
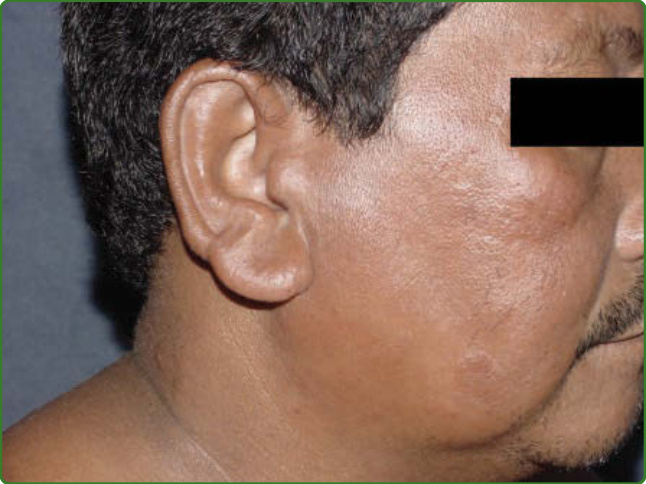
圖 159-13:瘤型痲瘋。皮膚的瀰漫性浸潤。
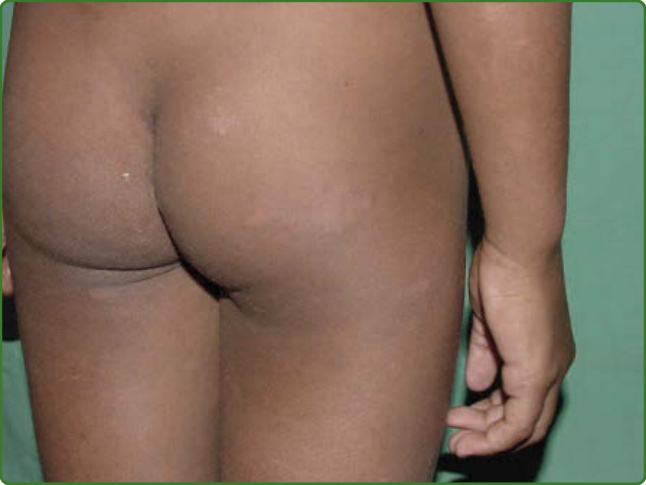
圖 159-14:邊緣結核樣型痲瘋。一名兒童臀部與大腿出現多個色素減退環狀病灶,周邊有丘疹與浸潤。
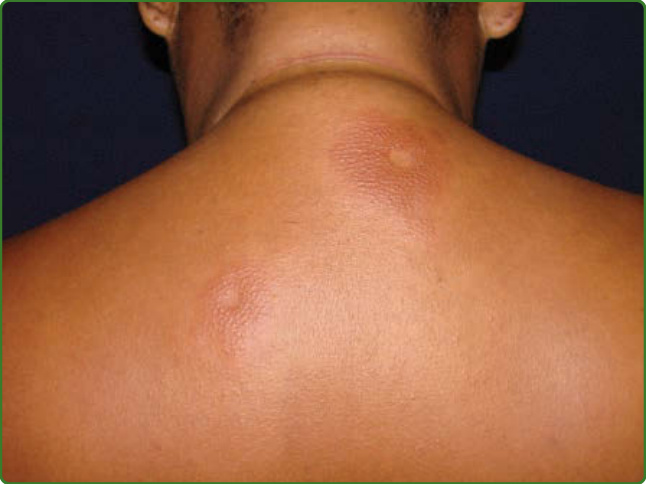
圖 159-15:邊緣邊緣型痲瘋。上背部胸廓的 2 個小凹狀病灶。
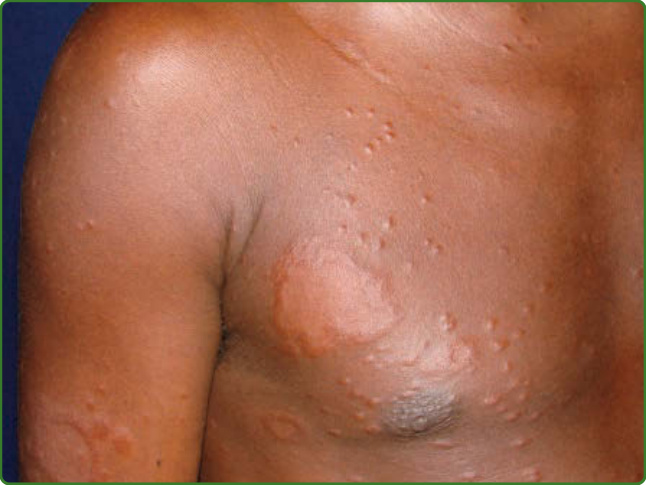
圖 159-16:邊緣瘤型痲瘋。小凹狀與結節性浸潤病灶。
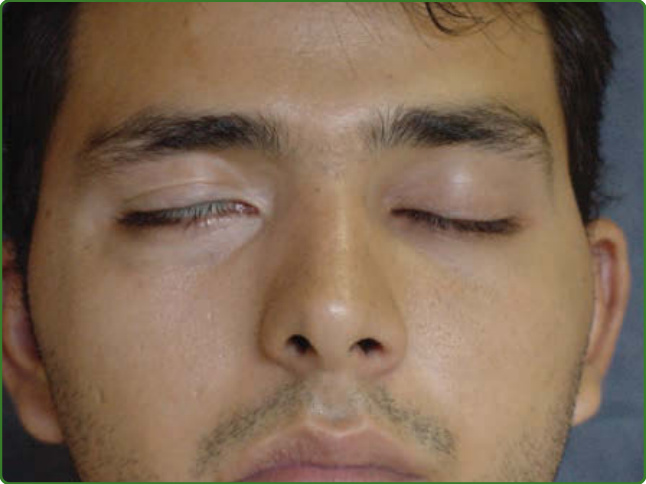
圖 159-17:右眼兔眼 (Lagophthalmos)。
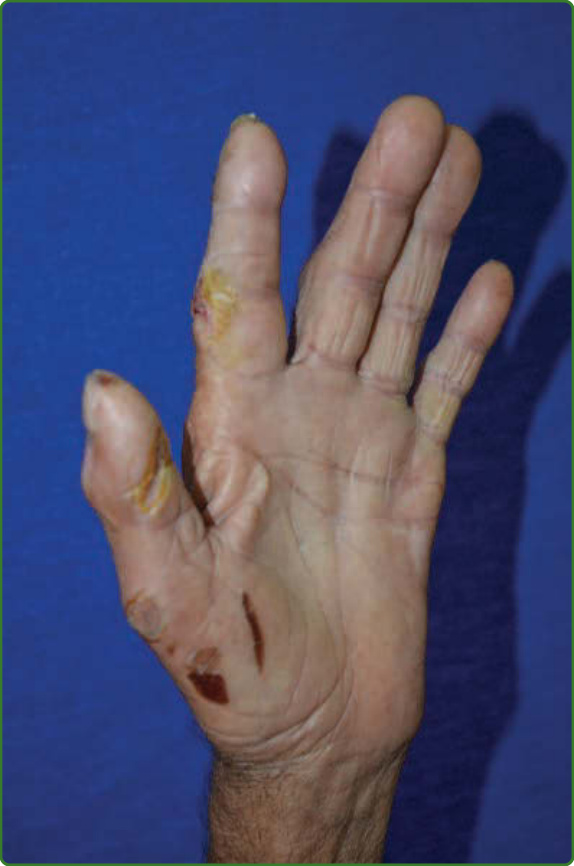
圖 159-18:一名 LL 病人萎縮且無汗之手部的潰瘍。
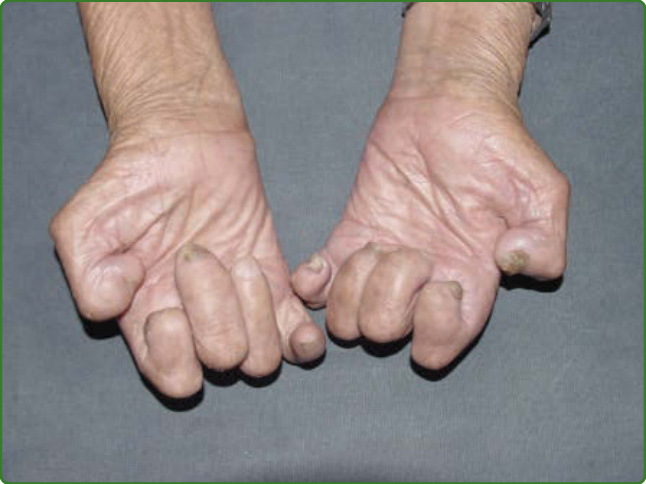
圖 159-19:一名邊緣瘤型痲瘋病人手部的固定爪狀與骨吸收後遺症,伴隨無汗與萎縮。
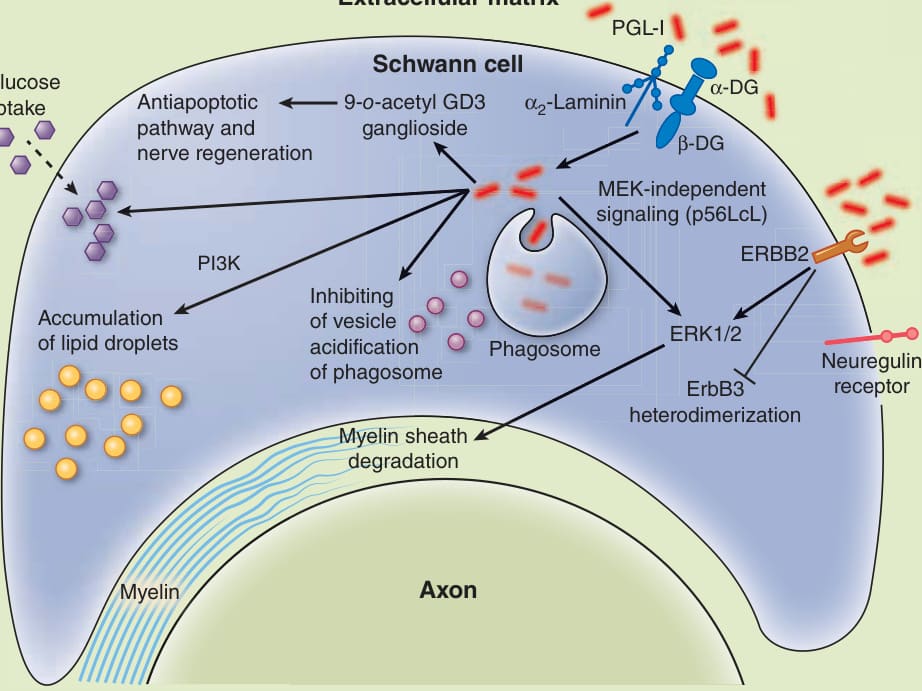
圖 159-20:M. leprae 的宿主細胞交互作用。此圖顯示細菌進入並維持於許旺細胞內的主要機制。
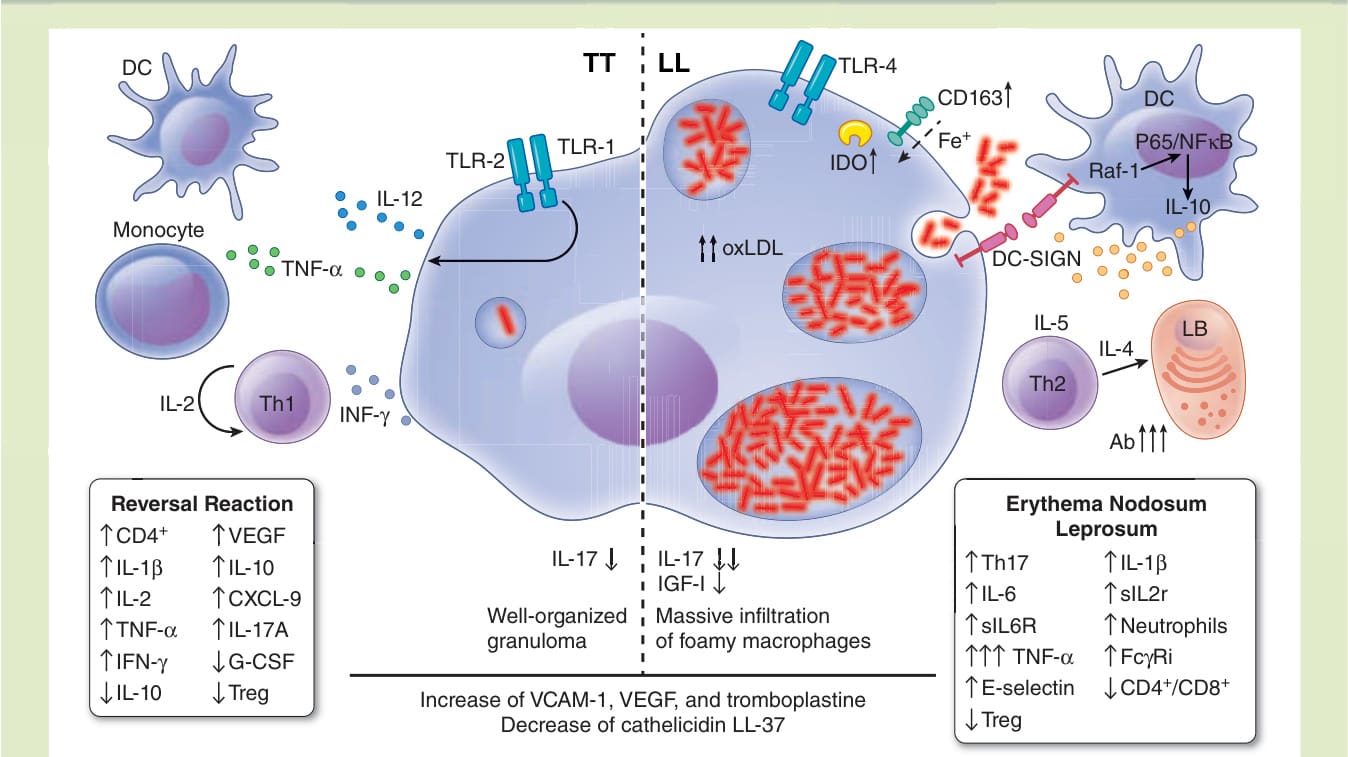
圖 159-21:痲瘋病的免疫學。圖中呈現結核樣型痲瘋與瘤型痲瘋兩極的主要差異,連同逆轉反應與痲瘋性結節性紅斑之間的關鍵變異。
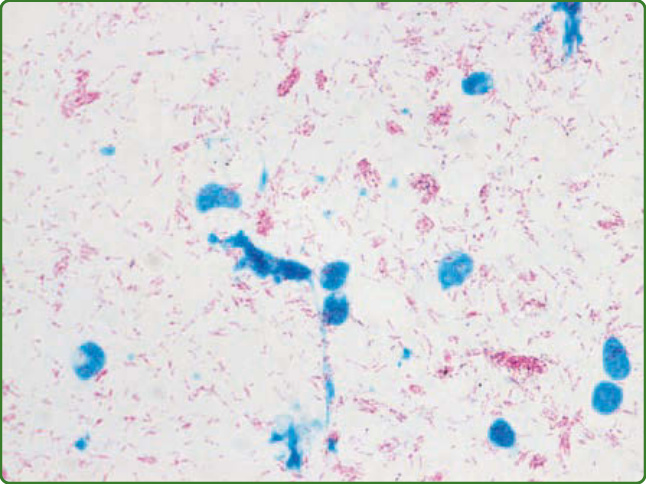
圖 159-22:一名瘤型痲瘋病人裂隙皮膚抹片的抗酸桿菌。細胞呈藍色,細菌呈紅色,形成球體。
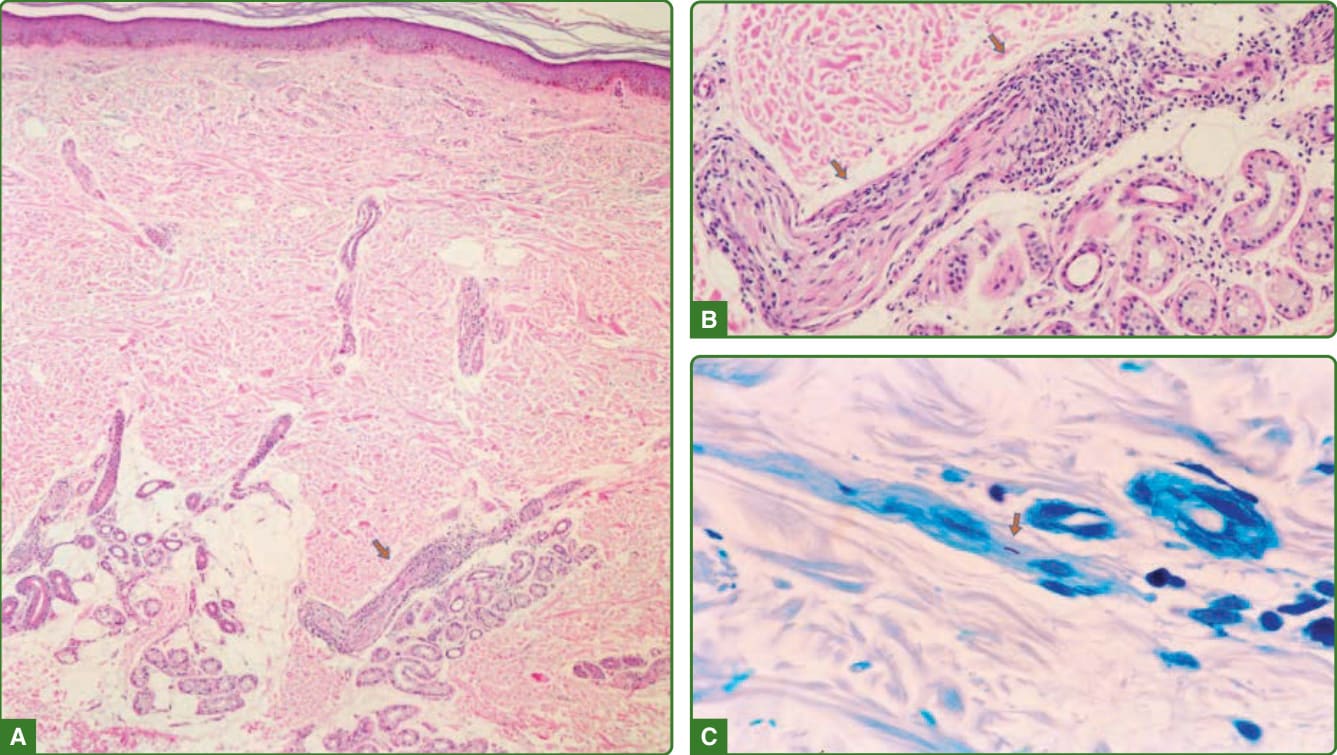
圖 159-23:未定類痲瘋組織病理學。多達 70% 的未定類病例可能有非特異性組織病理學。在 30% 中,可能觀察到伴神經分層 (delamination) 的神經周圍浸潤(A 與 B,箭頭),如此處所示,若廣泛搜尋,有時可能找到一個抗酸桿菌(C,箭頭)。(經巴西 Lauro de Souza Lima 研究所 Jaison Barreto 醫師許可使用。)
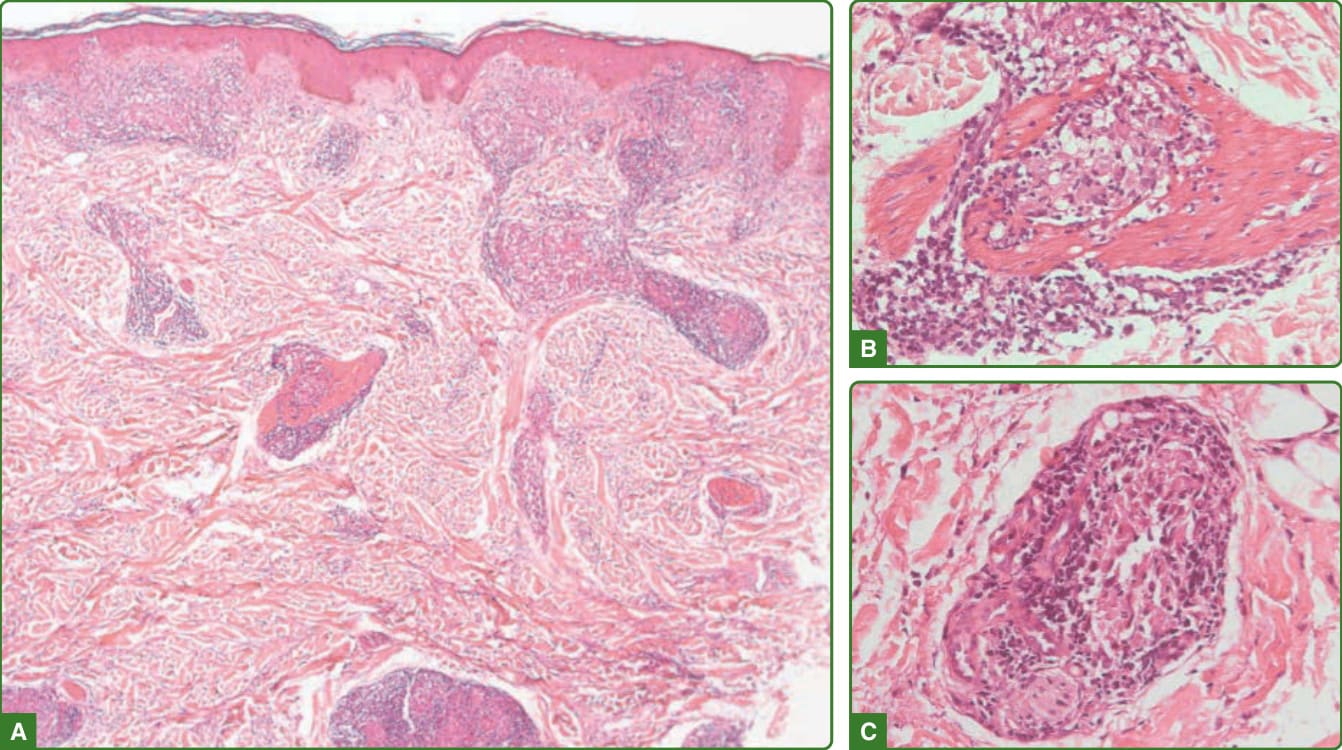
圖 159-24:結核樣型痲瘋組織病理學。出現接觸表皮的深層與淺層發育良好之肉芽腫(A),伴隨環繞或侵入並破壞皮膚附屬器(如神經、立毛肌(B)或汗腺(C))的淋巴球浸潤。
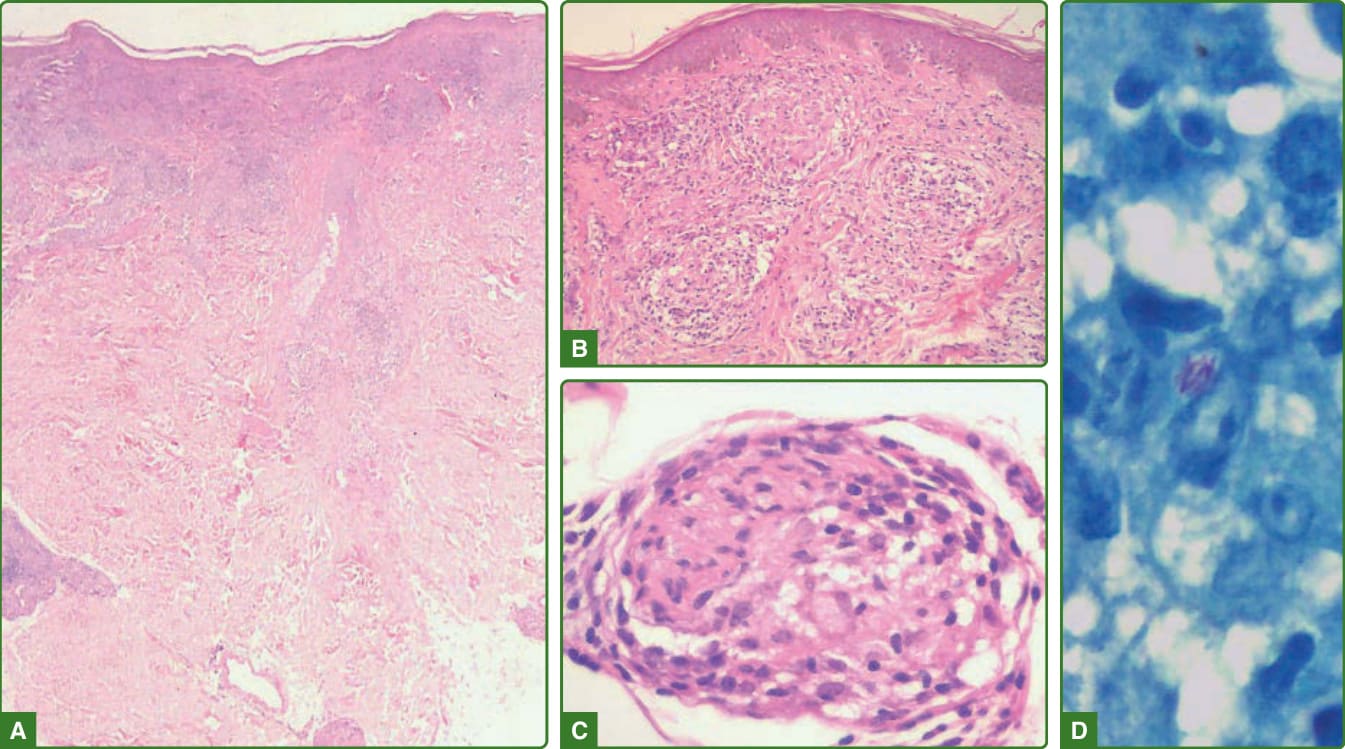
圖 159-25:邊緣結核樣型痲瘋組織病理學。不接觸表皮(B)的深層與淺層結核樣型肉芽腫(A)。可見結核樣型肉芽腫侵入神經(C),並可找到抗酸桿菌(D,100×)。(經巴西 Lauro de Souza Lima 研究所 Jaison Barreto 醫師許可使用。)
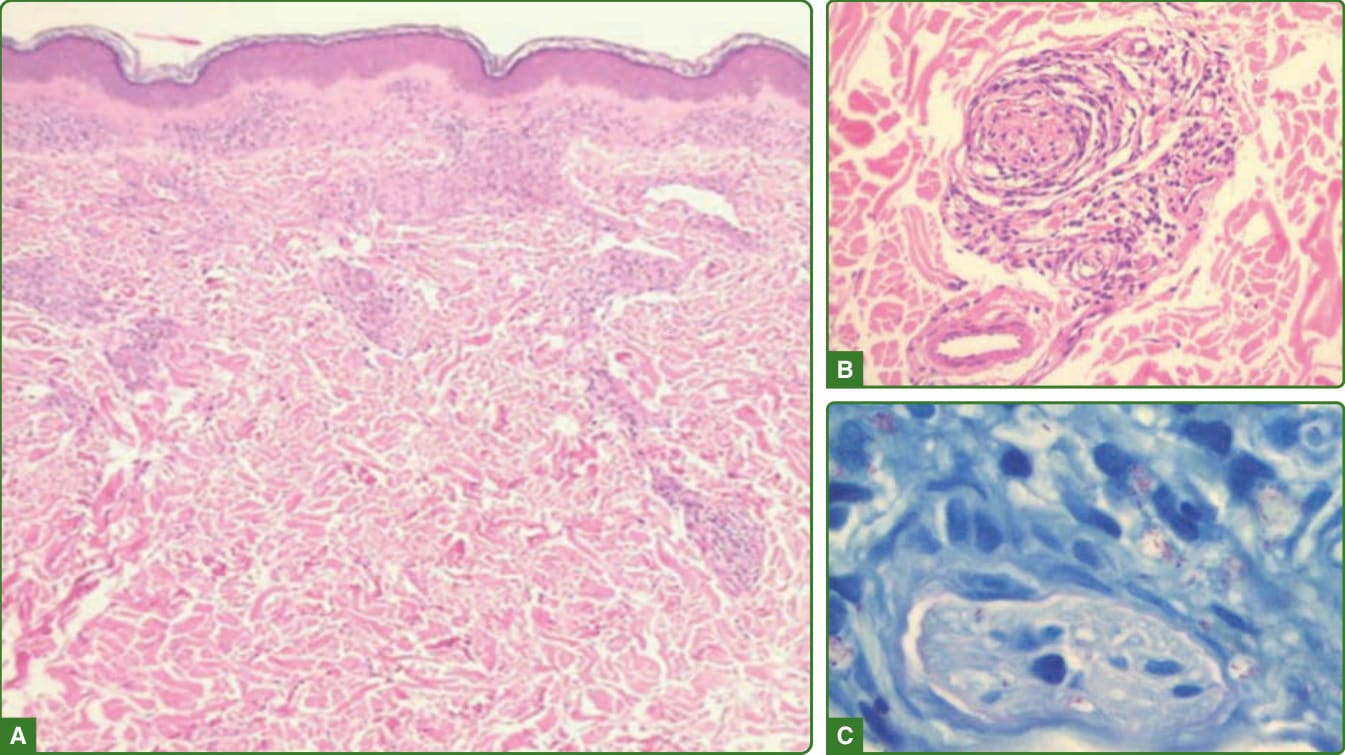
圖 159-26:邊緣邊緣型痲瘋組織病理學。不接觸表皮、開始形成無細胞帶的深層與淺層結核樣型肉芽腫(A)。發炎細胞正侵入皮膚附屬器與已退化的神經(B),抗酸桿菌可較易找到,有些形成球體(C)。(經巴西 Lauro de Souza Lima 研究所 Cleverson Soares 醫師許可使用。)
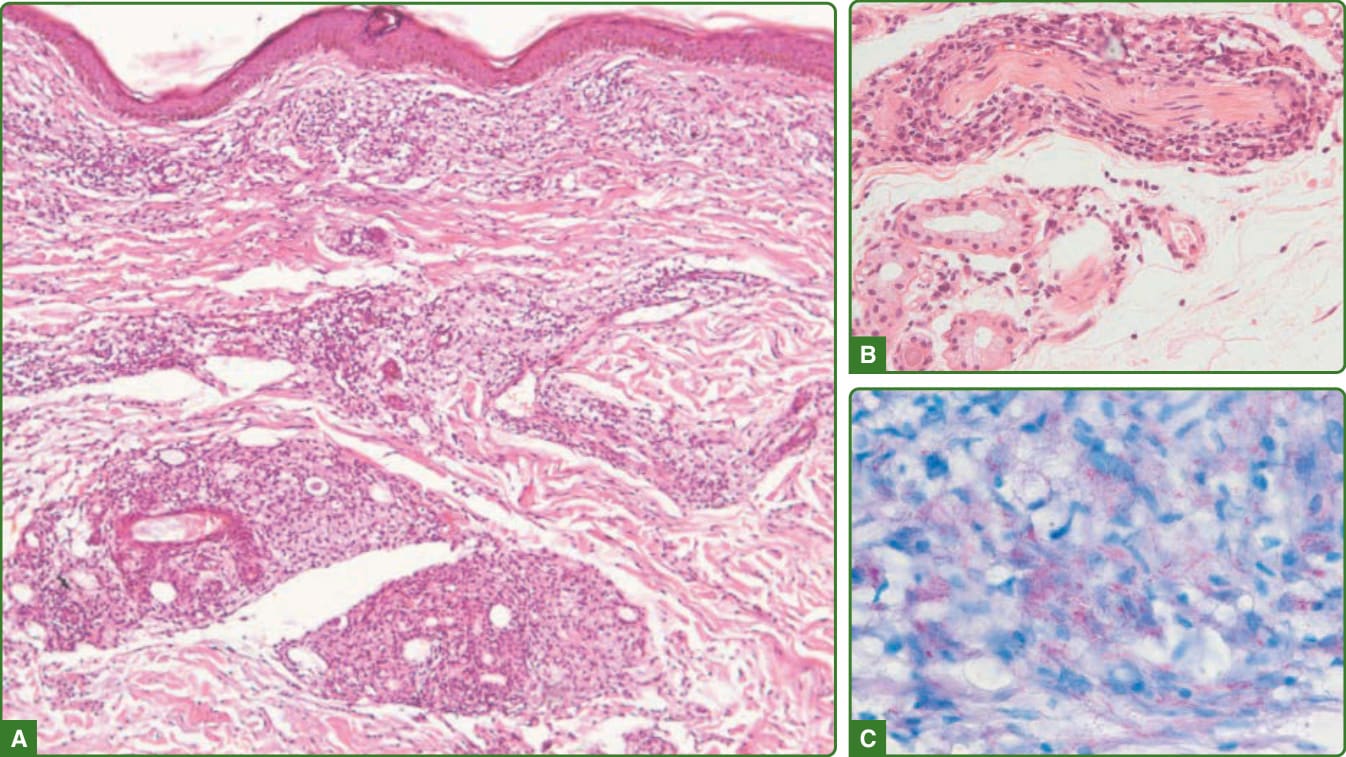
圖 159-27:邊緣瘤型痲瘋組織病理學。淺層與深層真皮有混合性巨噬細胞淋巴球發炎浸潤(巨噬細胞肉芽腫)(A)。混合性浸潤不接觸表皮,可見環繞或侵入神經束(B)。可見大量抗酸桿菌(C)。
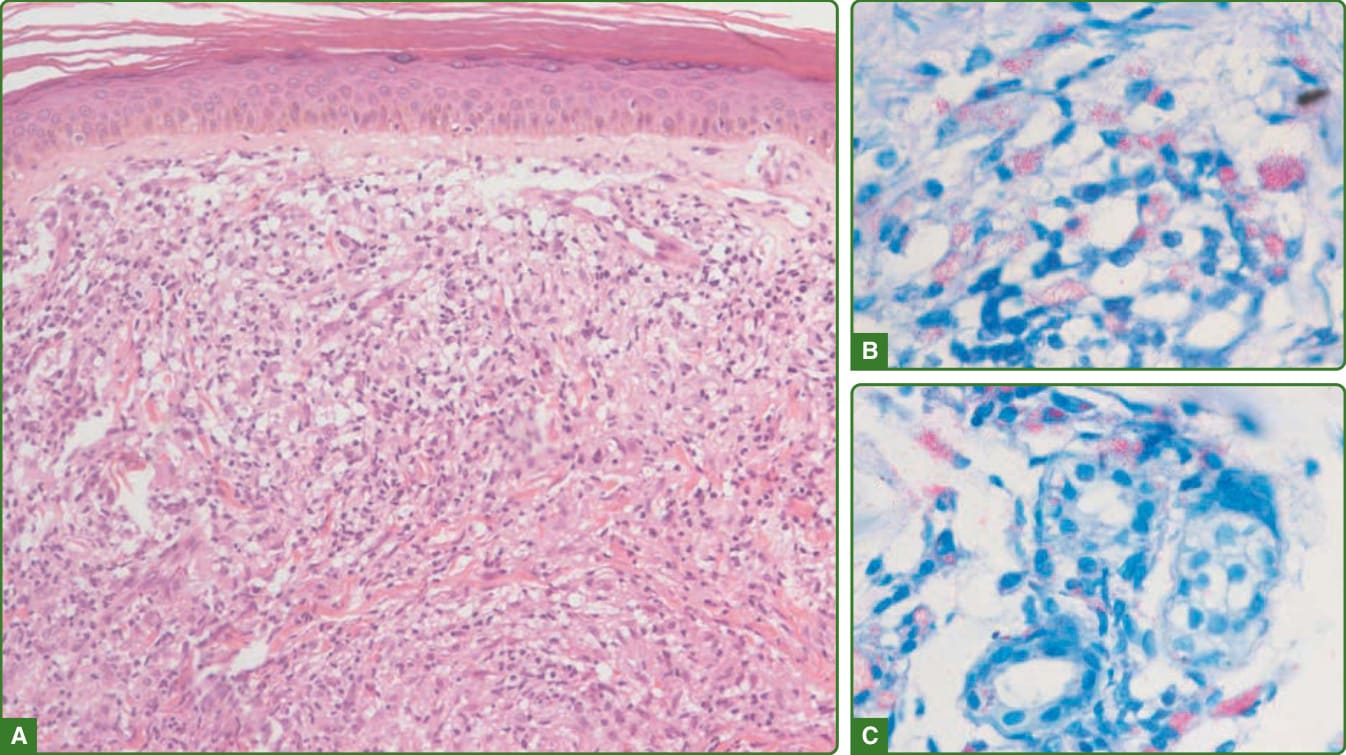
圖 159-28:瘤型痲瘋組織病理學。真皮可見主要由泡沫狀巨噬細胞構成的發炎浸潤。表皮扁平,真皮—表皮交界處有清楚的無細胞帶(A)。Fite-Faraco 顯示巨噬細胞內(B)與汗腺細胞上(C)的抗酸桿菌球體。
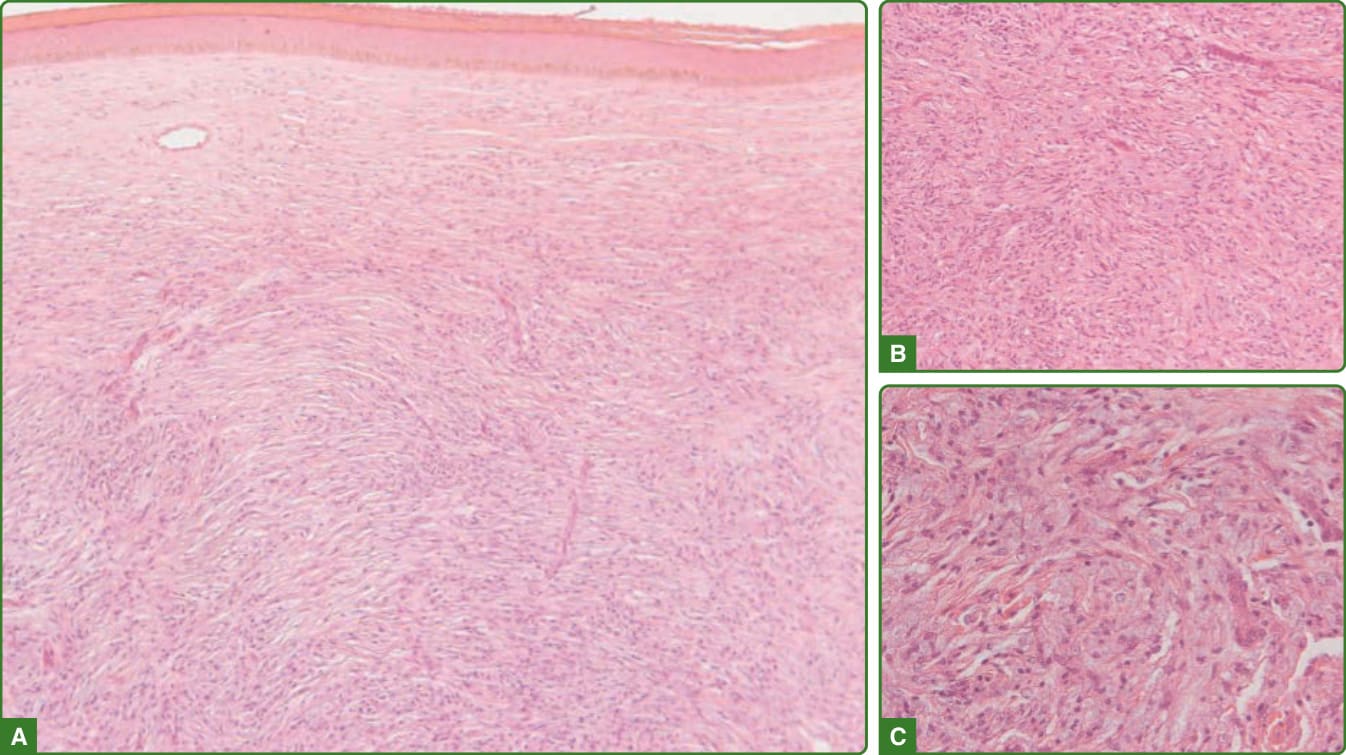
圖 159-29:瘤型痲瘋組織樣型 Wade 痲瘋組織病理學。在密集的紡錘狀與類上皮細胞(以漩渦狀或束狀模式排列)發炎浸潤(A、B)之上,可見扁平的表皮,近觀可能呈泡沫狀外觀(C)。
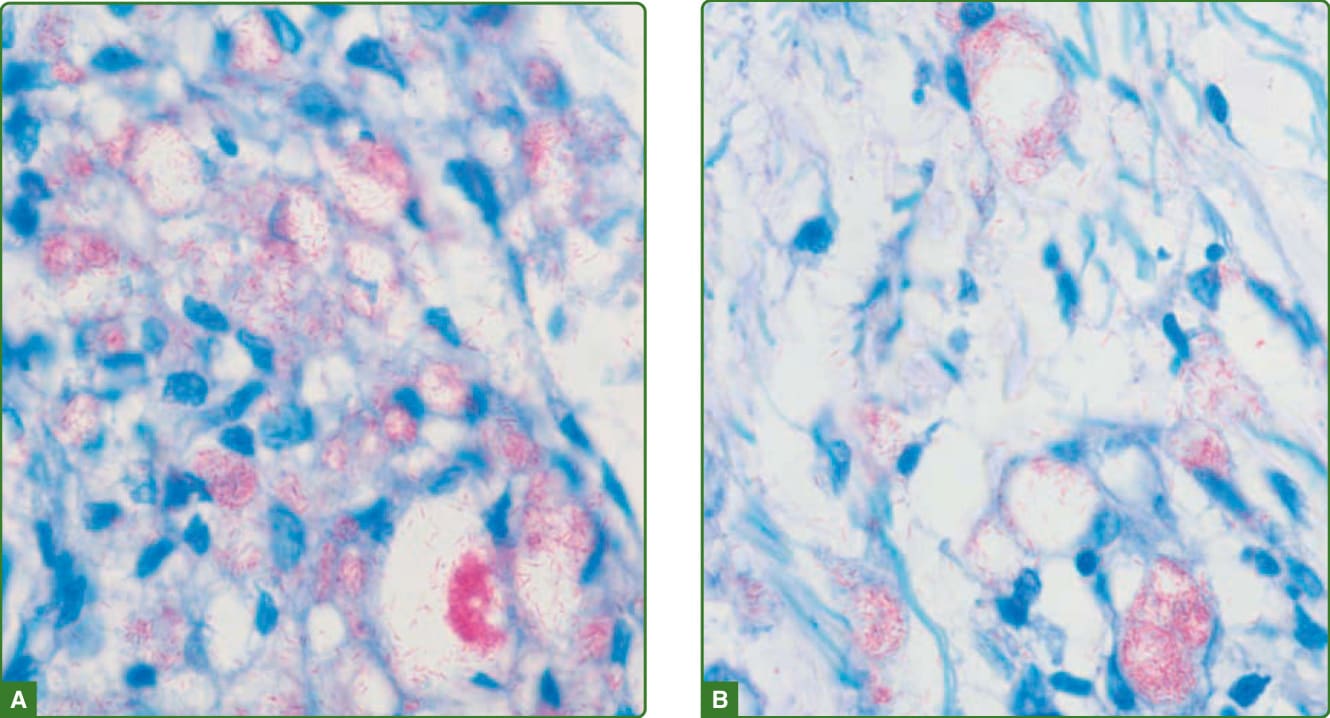
圖 159-30:球體 (Globi)。宿主細胞內的桿菌聚集(球體),如此處巨噬細胞內所見(A 與 B),是 M. leprae 的特徵。
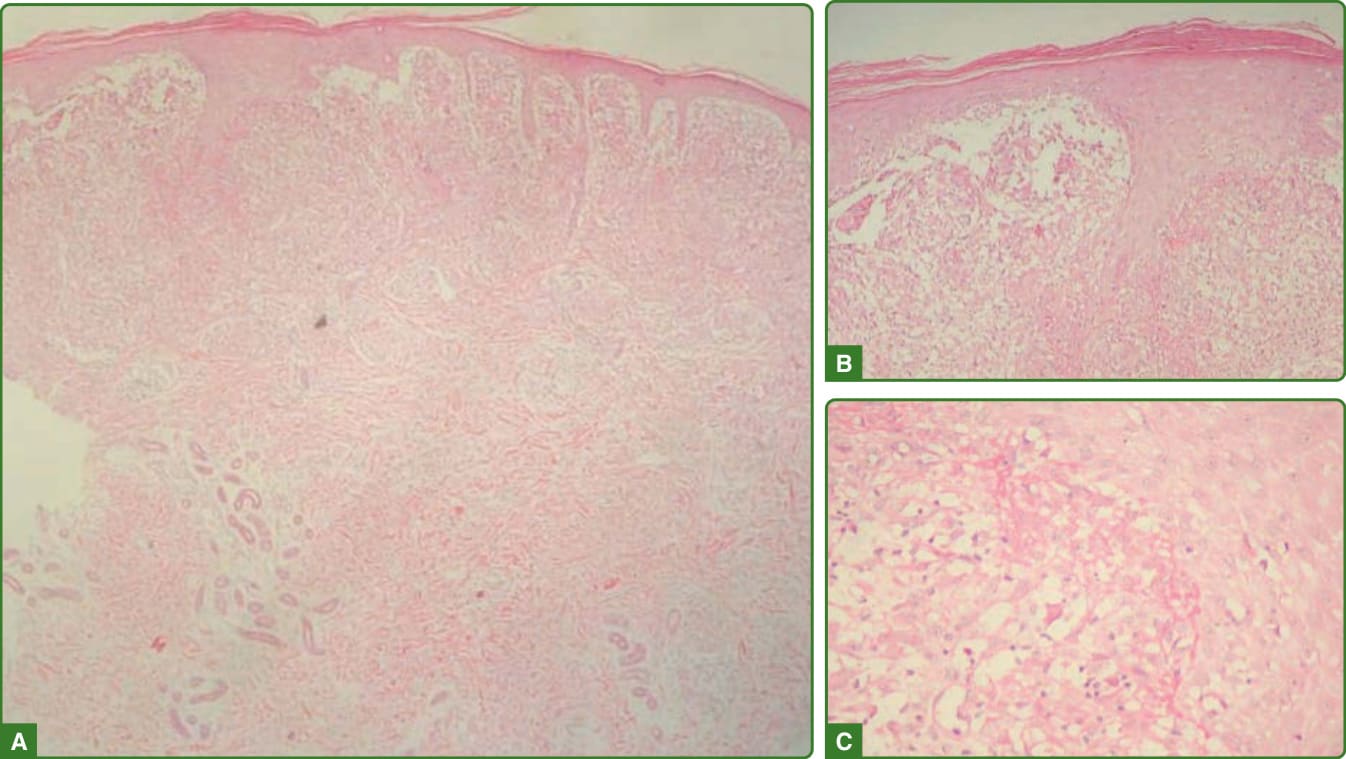
圖 159-31:逆轉反應組織病理學。出現發炎浸潤,伴隨上皮增生(A)以及表皮侵犯灶並伴真皮水腫(B)與纖維蛋白沉積(C)。(經巴西 Lauro de Souza Lima 研究所 Jaison Barreto 醫師許可使用。)
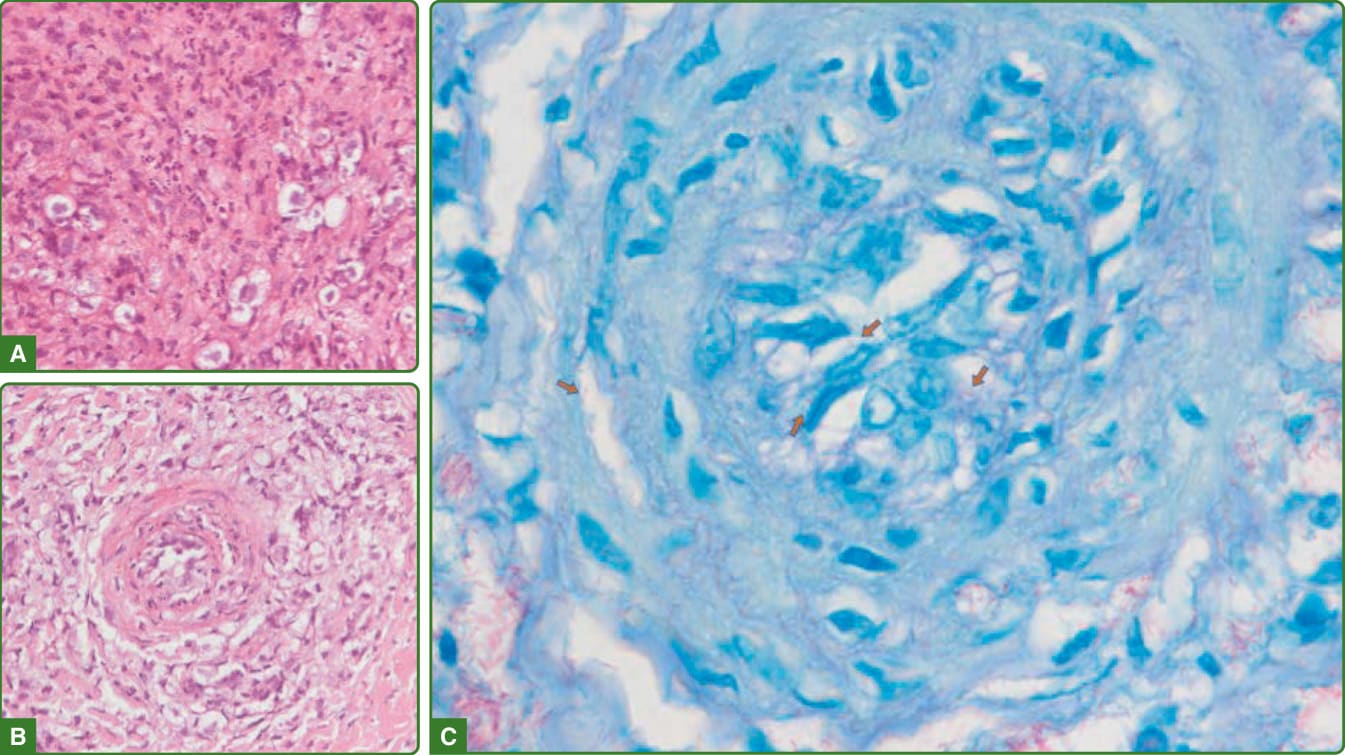
圖 159-32:痲瘋性結節性紅斑組織病理學。顯示由具大空泡的泡沫狀巨噬細胞與嗜中性球構成的混合性發炎浸潤(A)。泡沫狀巨噬細胞也見於環繞一條正進行類纖維蛋白壞死的血管,其管腔與管壁內有發炎細胞聚集(B),Fite-Faraco 在此處揭示大量球體的存在,以及血管內與管壁中的單一桿菌(C,箭頭)。
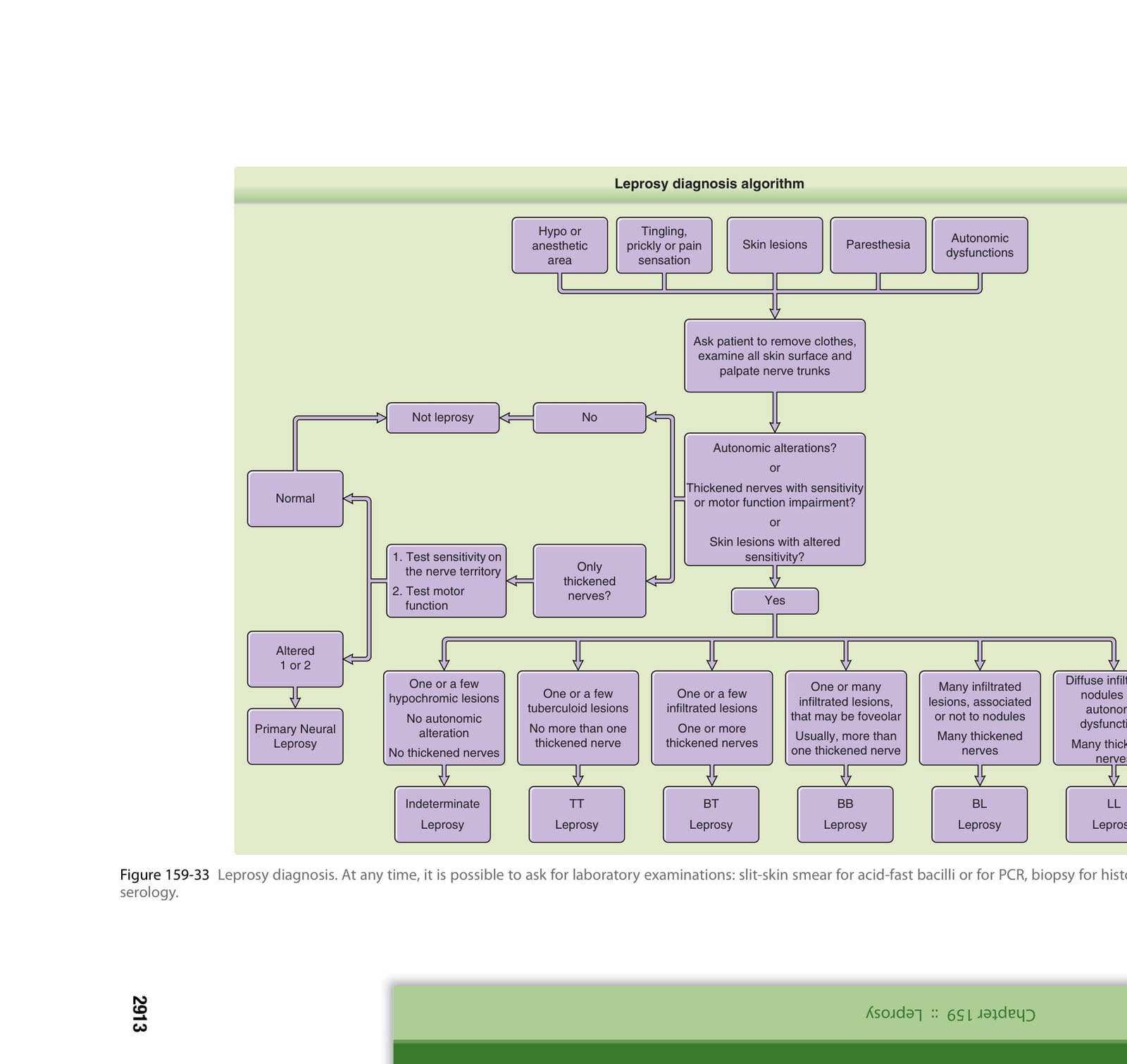
圖 159-33:痲瘋病診斷流程。
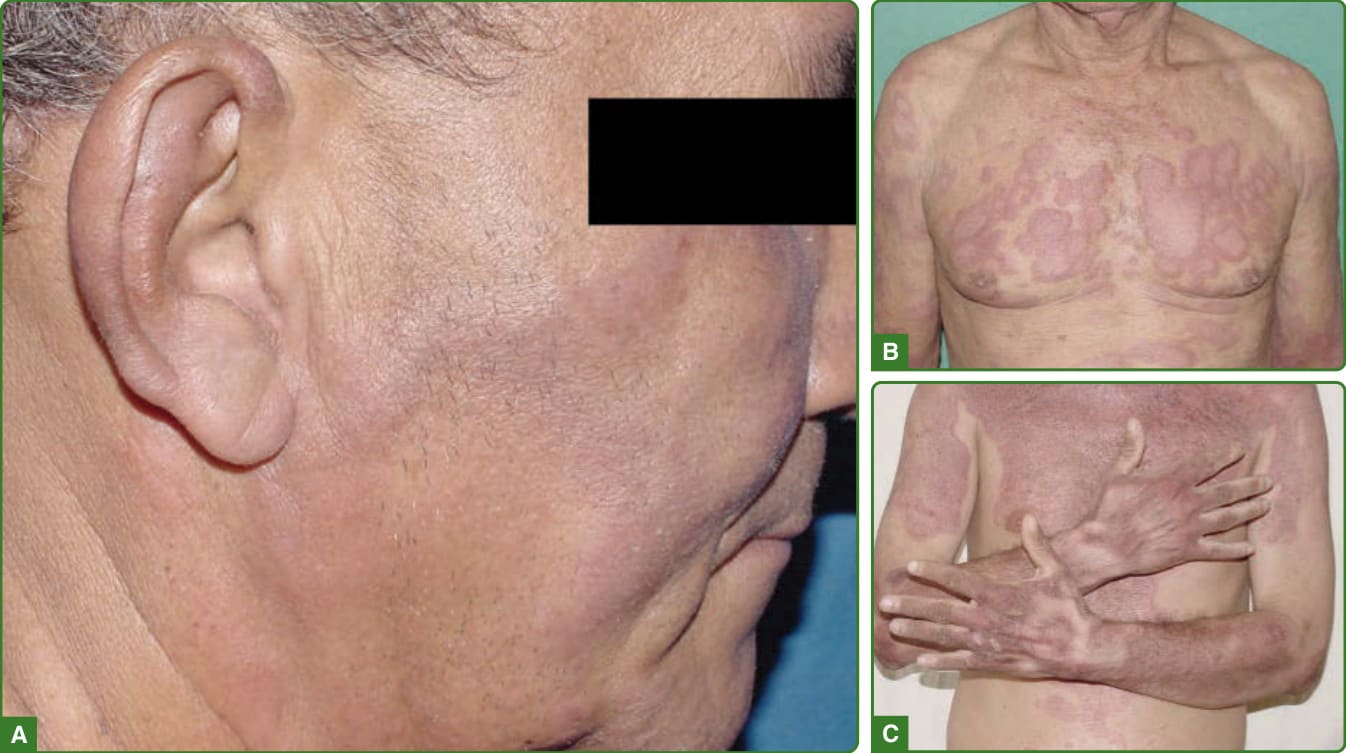
圖 159-34:逆轉反應。多重藥物治療前病灶在治療期間或之後再現或惡化(A),通常呈現嚴重的浸潤性病灶,可能融合成小型小凹狀(B)或大型鱗屑性(C)斑塊。
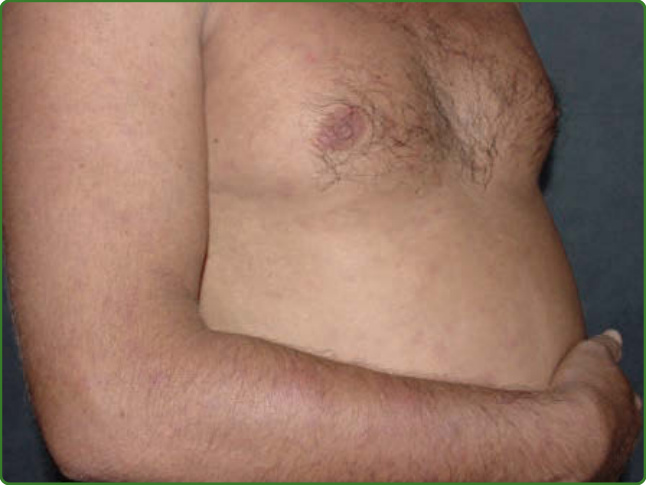
圖 159-35:痲瘋性結節性紅斑。出現發燒與倦怠,伴有易於觸診但遠不如逆轉反應病灶明顯的疼痛性皮下結節。
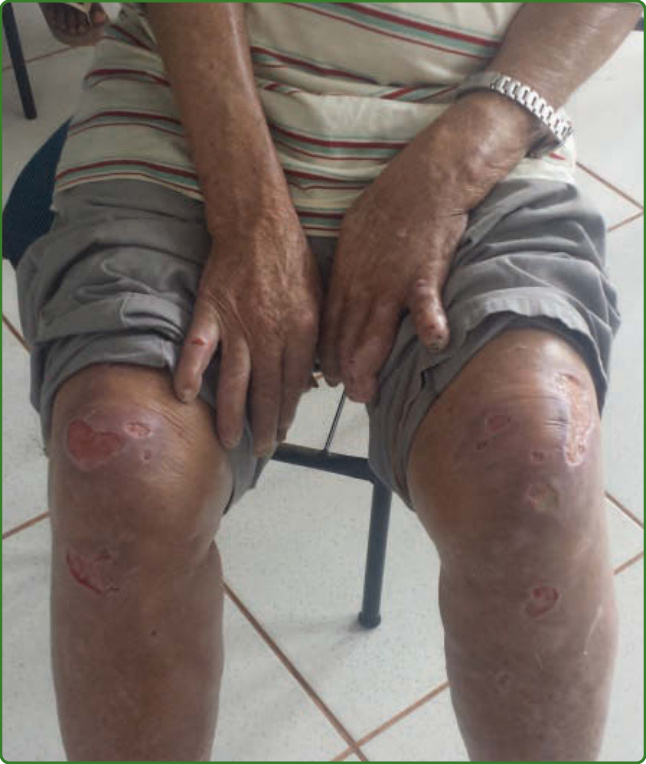
圖 159-36:Lucio 現象。瘤型痲瘋病人的小型與大型潰瘍伴浸潤。這些病人的裂隙皮膚抹片載有大量抗酸桿菌。
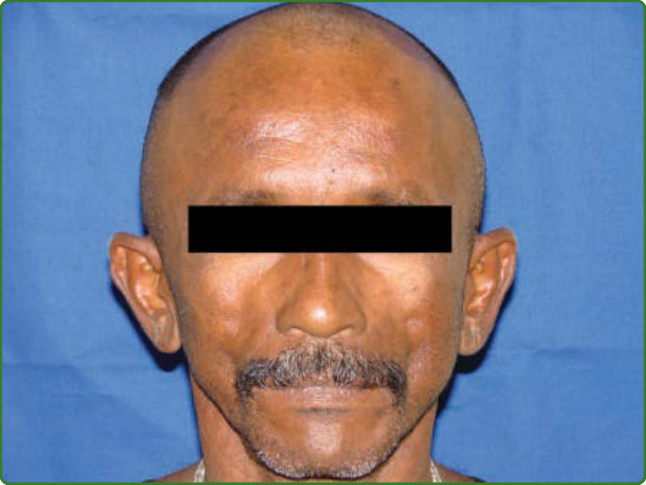
圖 159-37:Clofazimine 色素沉著。一名接受多菌性多重藥物治療之病人的棕色與乾燥皮膚。
*圖 159-38:痲瘋病治療。多重藥物治療是痲瘋病治療的選擇。然而,當對任一藥物不耐受時,可使用可供替代之藥物,如 ofloxacin、minocycline 或 clarithromycin。此外,若對多重藥物治療無反應,可檢測藥物抗藥性,並以相同的替代藥物取代 1 種或多種多重藥物治療藥物。對於反應,thalidomide 並非所有國家都有,甚至可能被禁止。不允許對孕婦或育齡婦女處方 thalidomide。若有必要使用,有些國家有嚴格的規定或法律,可能要求做懷孕測試並使用避孕方法才能處方 thalidomide。#Pentoxifylline 對懷孕而言屬「C 類」藥物,因此唯有當潛在益處足以證明對胎兒的潛在風險為合理時才應使用。
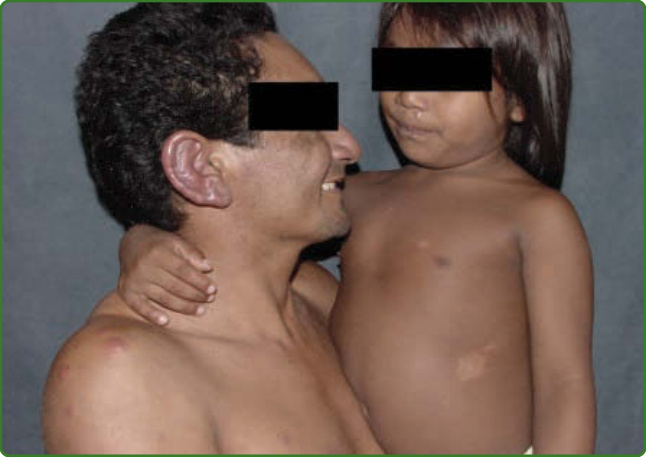
圖 159-39:接觸者檢查在痲瘋病計畫中為強制性。請注意一名父親被診斷為瘤型痲瘋之兒童身上的斑狀色素減退麻木病灶。

表 159-1:鑑別診斷 (Differential Diagnosis)
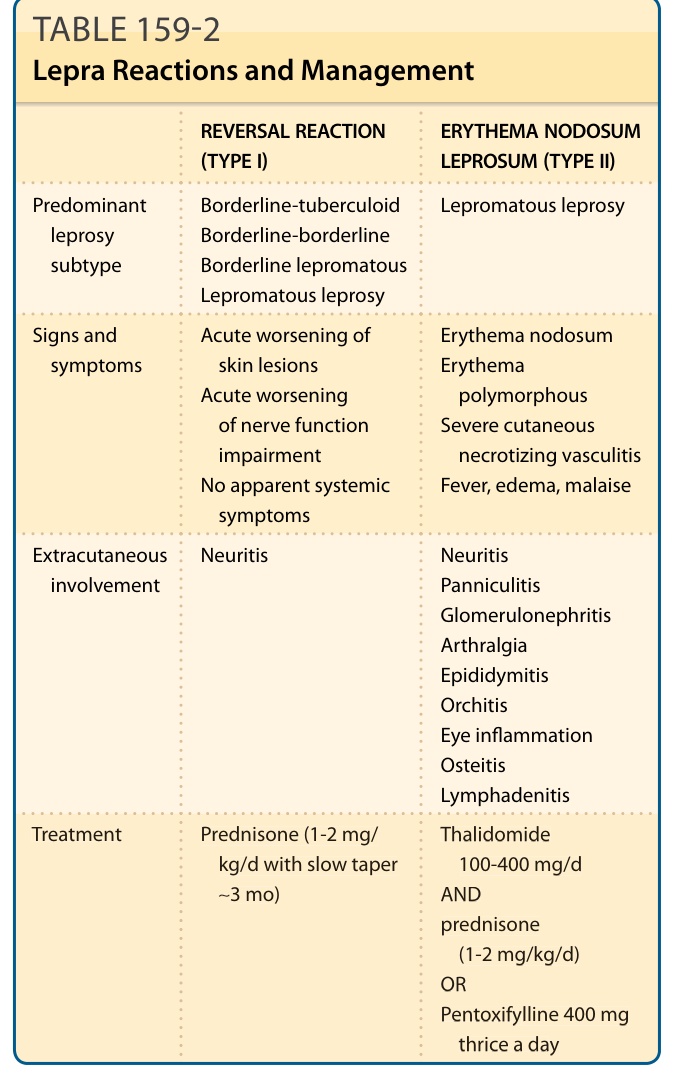
表 159-2:痲瘋反應與處置 (Lepra Reactions and Management)
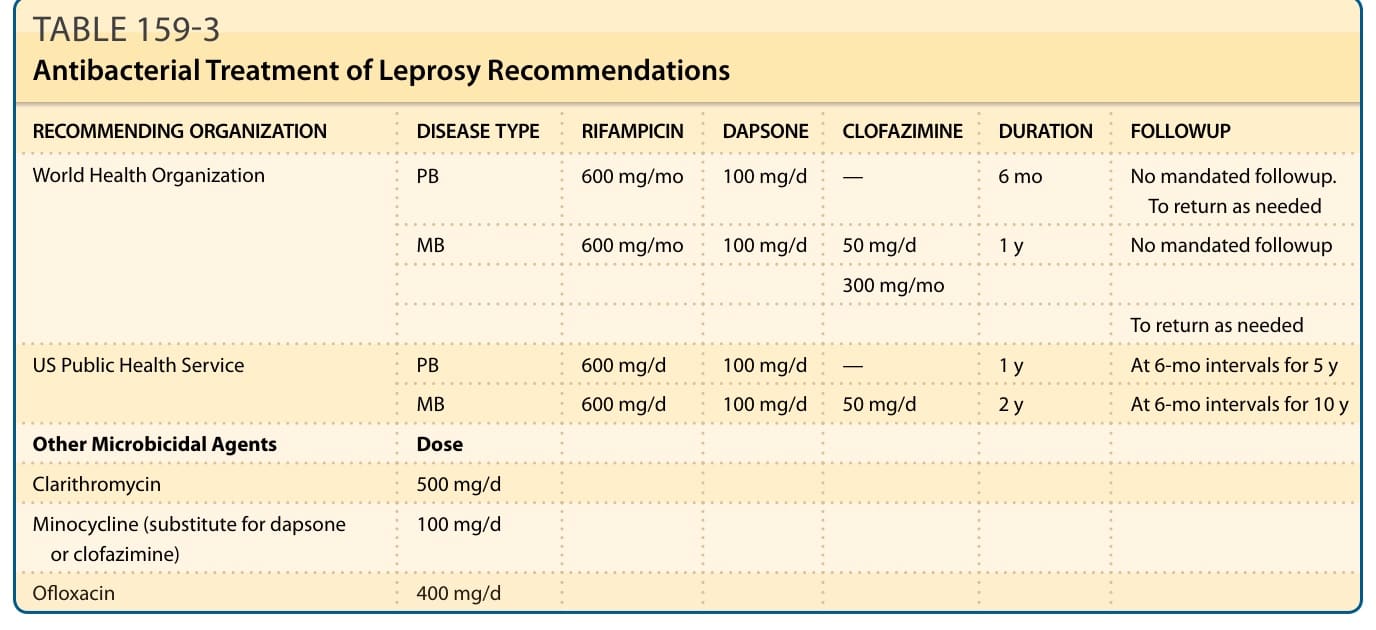
表 159-3:痲瘋病抗菌治療建議 (Antibacterial Treatment of Leprosy Recommendations)
表 159-3 痲瘋病抗菌治療建議(圖內容重排):
| 建議組織 | 疾病型別 | RIFAMPICIN | DAPSONE | CLOFAZIMINE | 療程 | 追蹤 |
|---|---|---|---|---|---|---|
| 世界衛生組織 (WHO) | PB | 600 mg/mo | 100 mg/d | — | 6 mo | 無強制追蹤,視需要回診 |
| MB | 600 mg/mo | 100 mg/d | 50 mg/d;300 mg/mo | 1 y | 無強制追蹤,視需要回診 | |
| 美國公共衛生署 (US Public Health Service) | PB | 600 mg/d | 100 mg/d | — | 1 y | 每 6 個月一次,持續 5 年 |
| MB | 600 mg/d | 100 mg/d | 50 mg/d | 2 y | 每 6 個月一次,持續 10 年 |
其他殺菌劑與劑量:Clarithromycin 500 mg/d;Minocycline(替代 dapsone 或 clofazimine)100 mg/d;Ofloxacin 400 mg/d。
PB,寡菌性 (paucibacillary);MB,多菌性 (multibacillary)。