色素性紫癜性皮膚病 (Pigmented Purpuric Dermatoses)
PART 22
血管疾病 (Vascular Diseases)
重點一覽 (AT-A-GLANCE)
■ 一群以瘀點 (petechiae)、色素沉著 (pigmentation),偶爾合併毛細血管擴張 (telangiectasia) 為特徵的皮膚病。
■ 最常見於下肢;不過病灶也可能侵犯上半身,但很少泛發 (generalized)。
■ 為良性、通常無症狀的疹子,傾向呈慢性病程,並有發作與緩解 (flares and remissions) 交替。
■ 常見的組織病理學特徵包括淺層淋巴球浸潤 (superficial lymphocytic infiltrate)、紅血球外滲 (erythrocyte extravasation),以及含鐵血黃素沉積 (hemosiderin deposition)。
■ 各型疹子之間的臨床差異,使其被細分為以人名命名 (eponymic) 的數個亞群;然而彼此間常有重疊,使鑑別變得困難。
色素性紫癜性疹 (pigmented purpuric eruptions,亦稱色素性紫癜性皮膚病 [pigmented purpuric dermatoses, PPDs]) 是一群以瘀點 (petechiae)、色素沉著 (pigmentation),偶爾合併毛細血管擴張 (telangiectasia) 為特徵的皮膚病。它們最常位於下肢;不過病灶也可能侵犯軀幹與上肢,或罕見地泛發。它們被細分為數個臨床亞型,包括進行性色素性皮膚病 (progressive pigmentary dermatosis,即 Schamberg 病 [Schamberg disease])、環狀毛細血管擴張性紫癜 (purpura annularis telangiectodes,即 Majocchi 紫癜 [Majocchi purpura])、Gougerot 與 Blum 的色素性紫癜性苔癬樣皮膚病 (pigmented purpuric lichenoid dermatosis of Gougerot and Blum)、Doucas 與 Kapetanakis 的濕疹樣紫癜 (eczematid-like purpura of Doucas and Kapetanakis),以及金黃色苔癬 (lichen aureus)(表 143-1)。這些皮膚病為良性且通常無症狀,但傾向呈慢性病程,並有發作與緩解交替。儘管臨床表現各異,PPDs 的各亞型共享共同的組織病理學特徵,包括淺層淋巴球浸潤、紅血球外滲及含鐵血黃素沉積。
流行病學 (EPIDEMIOLOGY)
PPDs 相對少見。它們可於任何年齡出現,包括兒童期,而以中年最為常見(見表 143-1)。並無種族傾向,不過肉芽腫型 (granulomatous variant) 在亞裔病人中被報告較為常見。¹ 整體而言,PPDs 在男性較常發生,唯 Majocchi 亞型例外,後者在女性較常見。²
臨床特徵 (CLINICAL FEATURES)
進行性色素性皮膚病(Schamberg 病)(Progressive Pigmentary Dermatosis [Schamberg Disease])
Schamberg 於 1901 年首次描述一名 15 歲男孩腿部出現的疹子,呈不規則形狀的紅棕色斑片,伴有「針頭大小的紅色斑點,極似辣椒粉 (cayenne pepper) 顆粒」。³ Schamberg 病是 PPDs 中最常發生於兒童的型別。然而整體而言,它在成人較為常見,發生率高峰在第五個十年(50 多歲)。⁴ 病灶發展隱匿,且通常無症狀。下肢是最常受侵犯的部位。病灶亦可侵犯軀幹或上肢(圖 143-1 與 143-2)。Schamberg 病呈慢性且持續性,發作與緩解可無限期反覆。
環狀毛細血管擴張性紫癜(Majocchi 紫癜)(Purpura Annularis Telangiectodes [Majocchi Purpura])
Majocchi 於 1896 年描述了第一例環狀毛細血管擴張性紫癜,患者為一名 21 歲男性,於下肢出現由毛囊性及點狀紅棕色斑疹組成的環狀斑片,並伴有毛細血管擴張與紫癜。⁵
此 PPD 亞型以其獨特的環狀型態為特徵(圖 143-3)。個別病灶起始為點狀的毛細血管擴張性斑疹,向周邊擴展,中央則出現色素減退 (hypopigmentation) 或輕微萎縮。¹ 病灶可為單發或多發。下肢是最常受侵犯的部位,病灶可擴展至侵犯上肢與軀幹。Majocchi 紫癜通常無症狀,持續數月並伴有發作與緩解。與其他 PPD 亞型相反,Majocchi 紫癜最常出現於年輕成年女性。²,⁶
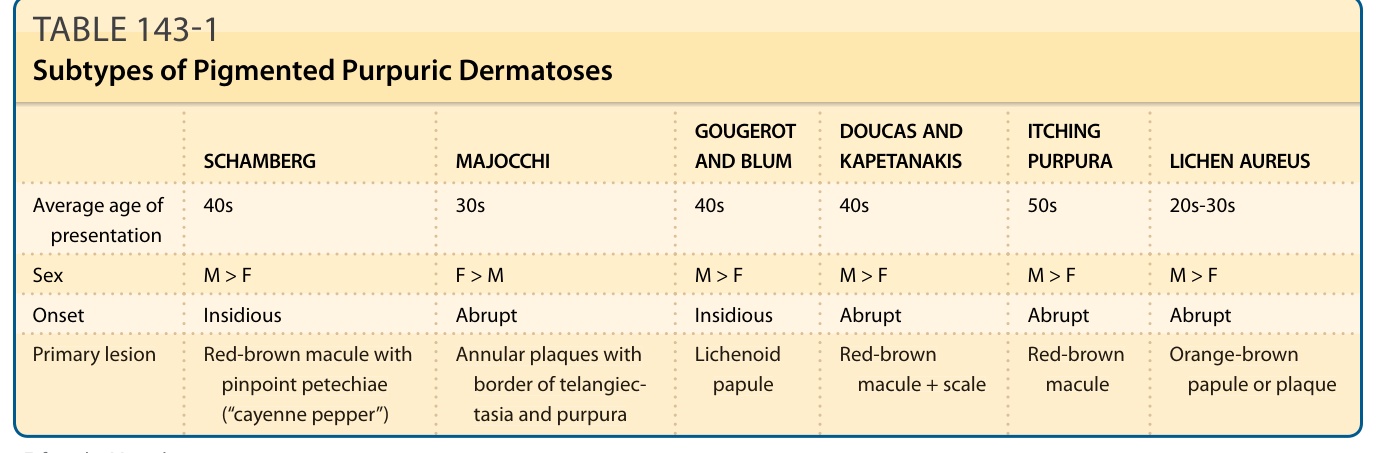
表 143-1:色素性紫癜性皮膚病的各亞型 (Subtypes of Pigmented Purpuric Dermatoses)
| 項目 | Schamberg | Majocchi | Gougerot 與 Blum | Doucas 與 Kapetanakis | 搔癢性紫癜 (Itching Purpura) | 金黃色苔癬 (Lichen Aureus) |
|---|---|---|---|---|---|---|
| 平均發病年齡 | 40 多歲 | 30 多歲 | 40 多歲 | 40 多歲 | 50 多歲 | 20–30 多歲 |
| 性別 | M > F | F > M | M > F | M > F | M > F | M > F |
| 起病 | 隱匿 | 急驟 | 隱匿 | 急驟 | 急驟 | 急驟 |
| 原發性病灶 | 帶針尖狀瘀點的紅棕色斑疹(「辣椒粉」cayenne pepper) | 帶毛細血管擴張及紫癜邊緣的環狀斑塊 | 苔癬樣丘疹 (lichenoid papule) | 紅棕色斑疹 + 脫屑 (scale) | 紅棕色斑疹 | 橘棕色丘疹或斑塊 |
F,女性 (female);M,男性 (male)。
Gougerot 與 Blum 的色素性紫癜性苔癬樣皮膚病 (Pigmented Purpuric Lichenoid Dermatosis of Gougerot and Blum)
1925 年,Gougerot 與 Blum 報告了一名 41 歲男性下肢的色素性疹子。⁷
此亞型在臨床上的特徵為出現紅棕色圓形或多角形的苔癬樣 (lichenoid) 丘疹與斑塊,背景為紫癜或毛細血管擴張(圖 143-4)。「苔癬樣 (lichenoid)」一詞描述的是苔癬樣丘疹與斑塊的這種臨床外觀,而非其底下的組織學。其臨床外觀可能被誤認為卡波西氏肉瘤 (Kaposi sarcoma)。與其他 PPD 亞型類似,Gougerot 與 Blum 的色素性紫癜性苔癬樣皮膚病最常見於下肢,偶爾侵犯軀幹與上肢,並呈慢性病程。
Doucas 與 Kapetanakis 的濕疹樣紫癜 (Eczematid-Like Purpura of Doucas and Kapetanakis)
濕疹樣紫癜由 Doucas 與 Kapetanakis 於 1953 年首次描述,當時一群病人出現無症狀的季節性疹子,發生於春季與夏季。⁸ 其臨床上的特徵為點狀紅斑性斑疹與斑片上覆有輕微脫屑,並伴隨搔癢。反覆搔抓可造成苔癬化 (lichenification)。組織病理學上,除了 PPD 的典型組織病理特徵外,還可見海綿水腫 (spongiosis)。此亞型在 15 至 30 天的期間內迅速擴散,隨後在數月至數年間未經治療而自行消退,不過仍可能復發。
金黃色苔癬 (Lichen Aureus)
1958 年,Martin 首次以「紫癜性苔癬 (lichen purpuricus)」一詞報告此 PPD 亞型,後於 1960 年由 Calnan 將其命名為金黃色苔癬 (lichen aureus),以強調其鮮明的黃橘色。⁹,¹⁰ 與 Gougerot 與 Blum 的色素性紫癜性苔癬樣皮膚病不同,後者的「苔癬樣 (lichenoid)」指的是病灶的臨床型態,而在金黃色苔癬中,「苔癬 (lichen)」同時指其臨床與組織病理學特徵。此亞型表現為較局限且持續的病灶,呈界限分明的斑疹或丘疹,帶有獨特的金色、鏽色或橘色(圖 143-5)。組織學上可見緻密的帶狀苔癬樣發炎細胞浸潤。病灶通常無症狀,但有時會劇烈搔癢。它們最常局限於一側下肢,但其他身體部位亦可受侵犯,且曾有節段性 (segmental) 分布的報告。¹¹,¹² 此疾病好發於年輕成年男性,發生率高峰在第二與第三個十年(20–30 多歲)。它呈慢性病程,病灶穩定或緩慢進展。
搔癢性紫癜(瀰漫性癢疹性血管皮膚炎)(Itching Purpura [Disseminated Pruriginous Angiodermatitis])
搔癢性紫癜,亦稱瀰漫性癢疹性血管皮膚炎 (disseminated pruriginous angiodermatitis),急性發作,表現為廣泛瀰漫的橘棕色至紫癜性病灶,並伴隨嚴重搔癢。¹³,¹⁴ 病灶首先出現於足背或下肢,然後向上擴散,有時侵犯軀幹。紫癜性病灶在腰線、腋窩、肘前窩 (antecubital fossa) 與膕窩 (popliteal fossa) 處較為明顯。雖然此 PPD 亞型呈慢性病程,但仍可能自發緩解。
單側線狀毛細血管炎(節段性色素性紫癜)(Unilateral Linear Capillaritis [Segmental Pigmented Purpura])
1990 年,曾報告一名中年女性下軀幹出現呈節段性分布的短暫性 PPD。其後,Riordan 與同僚報告了 4 名年輕男性出現呈線狀及偽皮節 (pseudodermatomal) 型態的 PPDs,他們將之命名為單側線狀毛細血管炎 (unilateral linear capillaritis)。¹⁵
「節段性色素性紫癜 (segmental pigmented purpura)」與「象限性毛細血管病變 (quadrantic capillaropathy)」皆被視為單側線狀毛細血管炎的亞型。¹⁶,¹⁷ 此 PPD 在臨床上的特徵為其線狀或節段性分布。它傾向有良好的預後,自發消退較其他 PPD 亞型更為常見。
肉芽腫性色素性紫癜 (Granulomatous Pigmented Purpura)
PPDs 肉芽腫型的首次報告於 1996 年由 Saito 與 Matsuoka 提出,自此已有超過 17 例被報告。¹⁸⁻²⁰ 它在中年最常見,並在亞裔病人中被報告較為常見。¹,²⁰ 臨床上,病灶外觀與其他 PPDs 相似,紫癜性與棕色斑疹最常發生於下肢與足背。此亞型以其組織病理學發現為特徵。除了 PPD 的典型組織病理特徵外,還可見肉芽腫性浸潤 (granulomatous infiltrate)。肉芽腫性浸潤最常位於乳頭層真皮 (papillary dermis),但也可能位於中至深層真皮,與位置較淺的苔癬樣浸潤分隔開來。對這些病人必須排除全身性肉芽腫性疾病及感染性病程(分枝桿菌與深部真菌)。高血脂 (hyperlipidemia) 與肉芽腫性色素性紫癜的關聯相對常見,在一篇 2014 年的回顧中,17 例報告中有 9 例顯示膽固醇值升高。¹⁹
色素性紫癜性皮膚病/蕈狀肉芽腫重疊 (Pigmented Purpuric Dermatosis/Mycosis Fungoides Overlap)
1988 年,Barnhill 與 Braverman 報告了首批類色素性紫癜疹進展為蕈狀肉芽腫 (mycosis fungoides) 的病例。²¹ 此外,美國首位被診斷為金黃色苔癬的病人後來被診斷為蕈狀肉芽腫 (mycosis fungoides, MF;見第 119 章)。有些證據支持 PPD 的苔癬樣亞型可能為 MF 的前驅病變這一觀點,因兩者有相似的組織學發現與淋巴球的單株 (clonal) 族群。²²⁻²⁹ 雖然這兩種疾病之間並無明確關聯,且 MF 在 PPD 背景下發生屬罕見,但已有 3 種不同的關係被報告:MF 在臨床上模擬色素性紫癜、色素性紫癜演變為 MF,以及色素性紫癜在組織學上模擬 MF。在一項關於 T 細胞單株性 (clonality) 與標記的研究中,PPD 的 T 細胞單株性 (T-cell monoclonality) 最有可能預測進展為 MF,而某些 T 細胞標記的缺失則為較不可靠的預測因子。³⁰ 值得注意的是,MF 與 PPD 皆可呈現單株性。MF 的組織學線索相當細微,包括相較於真皮淋巴球,表皮內淋巴球有更大的淋巴樣異型性 (lymphoid atypia)、表皮內任何位置出現大群淋巴球,或棘層 (spinous layer) 中出現許多淋巴球。³⁰,³¹ Santucci 與同僚指出,要將早期 MF 與其發炎性模擬病區分開來,最重要的特徵是具有極度扭曲、中至大型核的淋巴球,這些淋巴球單個或成簇出現於表皮中,並在真皮中以小片狀 (small sheets) 出現。³² Ackerman 將苔癬樣紫癜性疹與斑塊期 (plaque stage) MF 的組織學特徵進行比較,注意到許多相似之處,並結論認為僅憑組織學基礎可能無法區分這兩者。³³
PPD 與 MF 之間的鑑別可能困難,需要整合臨床、組織學及免疫表型 (immunophenotypic) 資訊。³⁰,³³ 若 PPDs 出現大範圍融合、網狀排列、疊加的紫紅色調 (violaceous hue),或已存在或反覆數年的搔癢,則應懷疑 MF。其以成年男性為主。²¹,³⁴ 對於部分病人,即使 MF 與 PPD 合併發生的整體發生率屬罕見,仍需長期追蹤以監測是否演變為 MF。
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
關於 PPDs 的發病機轉有 3 種不同的觀點。第一種是皮膚血管存在紊亂或脆弱,導致毛細血管脆性 (capillary fragility) 與紅血球外滲。然而,此提出的機轉無法解釋這些疹子中也可見到的發炎性浸潤。第二種理論是 PPDs 源自體液免疫反應 (humoral immune response)。此理論獲得直接免疫螢光 (direct immunofluorescence) 研究的支持,後者顯示血管壁有 C3、C1q、免疫球蛋白 M (immunoglobulin M) 或免疫球蛋白 A (immunoglobulin A) 的沉積。³⁵,³⁶ 最後一種理論是 PPDs 源自細胞免疫反應 (cellular immune response)。³⁷,³⁸
此理論認為,由淋巴球、巨噬細胞 (macrophages) 與蘭格漢氏細胞 (Langerhans cells) 組成的發炎性浸潤,導致血管脆弱及隨後的紅血球外滲。Aiba 與 Tagami 在 8 例 Schamberg 病中運用免疫組織學研究,證實真皮浸潤主要由輔助—誘導性 T 細胞 (helper-inducer T-cells) 與 OKT6 反應性細胞組成,而表皮則顯示以人類白血球抗原-DR 抗體 (human leukocyte antigen-DR antibody) 與 OKT6 抗體染色的細胞間染色。³⁹ 根據此研究,他們結論認為細胞免疫反應,特別是蘭格漢氏細胞,可能在發病機轉中扮演重要角色。此外,Ghersetich 與同僚顯示細胞媒介機轉 (cell-mediated mechanisms) 在 Schamberg 病的發展中可能很重要,因為 CD4+ T 細胞與 CD1a 樹突細胞 (dendritic cells) 在早期病灶中佔優勢,且此浸潤在強效局部類固醇治療後清除。³⁸
PPD 可能由多種藥物誘發(表 143-2)。¹,⁴⁰⁻⁵³ 這些藥物誘發的疹子較可能泛發,但仍會出現下肢侵犯。它們通常為短暫性,平均持續 3 至 4 個月,許多病例在數週內消退。⁵³ 時序關係可能難以建立,因為病人可能在疹子發作前數月至數年即已服用該致病藥物。再激發試驗 (rechallenge) 已被用於確認診斷。除了藥物之外,亦有其他原因被指涉(表 143-3)。⁵⁴⁻⁶¹ 少數病例顯示有家族性受侵犯,其中一篇報告為體染色體顯性 (autosomal dominant) 遺傳模式。⁶² 重力與靜脈壓增加可能解釋了病灶局限於下肢的原因。整體而言,大多數 PPDs 病例為原發性 (idiopathic)。
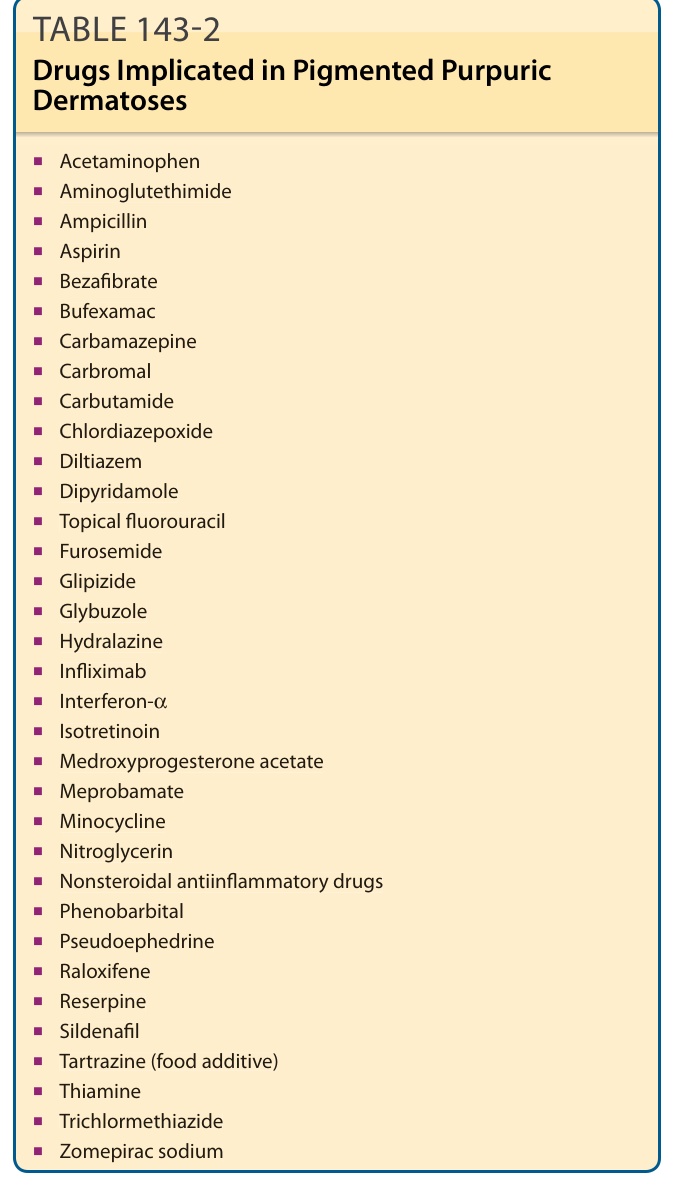
表 143-2:與色素性紫癜性皮膚病相關的藥物 (Drugs Implicated in Pigmented Purpuric Dermatoses)
- 乙醯胺酚 (Acetaminophen)
- 胺基麩希醯亞胺 (Aminoglutethimide)
- 安比西林 (Ampicillin)
- 阿斯匹靈 (Aspirin)
- 苯紮貝特 (Bezafibrate)
- Bufexamac
- 卡馬西平 (Carbamazepine)
- Carbromal
- Carbutamide
- 氯二氮平 (Chlordiazepoxide)
- 地爾硫卓 (Diltiazem)
- 雙嘧達莫 (Dipyridamole)
- 局部氟尿嘧啶 (Topical fluorouracil)
- 呋塞米 (Furosemide)
- 格列吡嗪 (Glipizide)
- Glybuzole
- 肼屈嗪 (Hydralazine)
- 英夫利昔單抗 (Infliximab)
- 干擾素-α (Interferon-α)
- 異維 A 酸 (Isotretinoin)
- 醋酸甲羥孕酮 (Medroxyprogesterone acetate)
- Meprobamate
- 米諾環素 (Minocycline)
- 硝化甘油 (Nitroglycerin)
- 非類固醇抗發炎藥 (Nonsteroidal antiinflammatory drugs)
- 苯巴比妥 (Phenobarbital)
- 偽麻黃鹼 (Pseudoephedrine)
- 雷洛昔芬 (Raloxifene)
- 利血平 (Reserpine)
- 西地那非 (Sildenafil)
- 檸檬黃 (Tartrazine,食品添加物)
- 硫胺 (Thiamine)
- 三氯甲噻嗪 (Trichlormethiazide)
- 佐美酸鈉 (Zomepirac sodium)

表 143-3:色素性紫癜性皮膚病其他被指涉的原因 (Other Implicated Causes of Pigmented Purpuric Dermatoses)
- 接觸性皮膚炎 (Contact dermatitis)
- 分散染料 (Disperse dyes)
- 對苯二胺 (Paraphenylenediamine)
- 黑色橡膠 (Black rubber)
- 鈷 (Cobalt)
- 過氧化苯甲醯 (Benzoyl peroxide)
- 環氧樹脂 (Epoxy resin)
- 甲基丙烯酸甲酯 (Methylmethacrylate)
- 局部麻醉劑共熔混合物 (Eutectic mixture of local anesthetics)
- 靜脈鬱滯/肢端血管皮膚炎 (Venous stasis/acroangiodermatitis)
- 階梯有氧運動 (Step aerobics)
- 劇烈運動 (Strenuous exercise)
- 類風濕性關節炎 (Rheumatoid arthritis)
- 全身性紅斑性狼瘡 (Systemic lupus erythematosus)
- 糖尿病 (Diabetes mellitus)
- 甲狀腺功能障礙 (Thyroid dysfunction)
- 高血脂 (Hyperlipidemia)
- 良性高球蛋白血症性紫癜(Waldenström 氏)/異常蛋白血症性紫癜 (Benign hyperglobulinemic purpura [of Waldenstrom]/dysproteinemic purpura)
- 何杰金氏病 (Hodgkin disease)
- B 型與 C 型肝炎 (Hepatitides B and C)
- 牙源性感染 (Odontogenic infection)
- 多血症 (Polycythemia)
- 遺傳性球形紅血球症 (Hereditary spherocytosis)
- 紫質症 (Porphyrias)
- 紫癜性蕈狀肉芽腫 (Purpuric mycosis fungoides)
診斷 (DIAGNOSIS)
診斷通常以臨床方式做出,並可由組織病理學檢查支持。以血壓計 (sphygmomanometer) 應用測量毛細血管脆性的臨床檢查(Hess 試驗 [Hess test])似乎並不可靠,因為 PPD 中並非一致地見到毛細血管脆性增加。應檢視病人的病史、用藥及潛在的接觸過敏原。若病灶呈慢性且不緩解,應考慮 MF。
支持性檢查 (Supportive Studies)
實驗室檢測 (Laboratory Testing)
診斷不需要實驗室檢測。在部分病例中,實驗室檢測可包括全血球計數合併周邊血液抹片(complete blood cell count with peripheral smear,以排除血小板低下 [thrombocytopenia] 或其他血液疾病)、凝血功能檢查(以排除其他造成紫癜的原因),以及抗核抗體 (antinuclear antibody)、類風濕因子 (rheumatoid factor) 與肝炎血清學檢查。¹
病理學 (PATHOLOGY)
所有 PPDs 共享相似的組織病理學。乳頭層真皮 (papillary dermis) 中可見血管周圍淋巴球浸潤,伴有內皮腫脹 (endothelial swelling)、外滲的紅血球,以及巨噬細胞內的含鐵血黃素沉積(圖 143-6)。在較舊的病灶中,發炎較少,外滲的紅血球可能不再存在。雖然對於是否存在毛細血管炎 (capillaritis) 尚無共識,但 Ackerman 主張並無真正的毛細血管炎,因為管腔壁缺乏纖維蛋白 (fibrin),且無血栓。¹
表皮可顯示海綿水腫 (spongiosis) 與角化不全 (parakeratosis),特別是在 Gougerot 與 Blum 的色素性紫癜性苔癬樣皮膚炎與 Doucas 與 Kapetanakis 的濕疹樣紫癜中。在金黃色苔癬中,上層真皮可見帶狀單核細胞浸潤,與表皮間隔著一薄層未受侵犯的膠原蛋白。在肉芽腫性色素性紫癜中,肉芽腫性浸潤位於乳頭層真皮,或位於中至深層真皮,與位置較淺的苔癬樣浸潤分隔開來。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
PPD 的鑑別診斷詳列於表 143-4。

表 143-4:色素性紫癜性皮膚病的鑑別診斷 (Differential Diagnosis of Pigmented Purpuric Dermatoses)
- 接觸性皮膚炎 (Contact dermatitis)
- 白血球破裂性血管炎 (Leukocytoclastic vasculitis)
- 鬱滯性皮膚炎與紫癜 (Stasis dermatitis and purpura)
- 蛇行性血管瘤 (Angioma serpiginosum)
- 卡波西氏肉瘤(Gougerot-Blum)(Kaposi sarcoma)
- 蕈狀肉芽腫 (Mycosis fungoides)
- 類肉瘤病 (Sarcoidosis)
- 壞血病 (Scurvy)
- 高球蛋白血症性紫癜 (Hypergammaglobinemic purpura)
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
PPDs 為良性且通常無症狀。一般而言,PPDs 呈慢性,並有發作與緩解交替。此慢性病程的例外包括單側線狀毛細血管炎與藥物誘發的 PPDs,後兩者傾向有較短的臨床病程與較佳的整體預後。
處置 (MANAGEMENT)
PPD 的處置具有挑戰性,因為治療往往無效。應檢視病人的病史、用藥及潛在的接觸過敏原。若疹子由藥物或接觸過敏原誘發,停用致病因子可導致完全消退。然而,殘餘的色素沉著可能持續數年。
介入治療 (Interventions)
藥物治療 (Medications)
常見的初始療法包括局部類固醇與抗組織胺,後兩者對治療伴隨的搔癢特別有幫助,以及使用彈性襪 (compression stockings) 以治療伴隨的靜脈功能不全。除局部類固醇外,亦有使用局部 pimecrolimus。以局部 pimecrolimus 每日兩次治療 10 週,清除了一名 10 歲男孩的金黃色苔癬病灶。該病人在停藥後有輕微復發,經 2 週的重複治療後改善。⁶³ 病灶內注射皮質類固醇 (intralesional corticosteroids) 亦曾使病情改善,不過可能發生組織萎縮,且無法防止新病灶的發生。⁶
口服生物類黃酮(bioflavonoid,蘆丁苷 [rutoside],50 mg 每日兩次)與抗壞血酸(ascorbic acid,500 mg 每日兩次)在一項開放試驗中給予 3 名慢性 PPD 病人,使全部 3 名病人於 4 週內清除,並在治療後 3 個月維持緩解。⁶⁴ 在一項初步研究中,鈣劑多貝西酸鹽(calcium dobesilate,500 mg 每日兩次)給予 9 名病人為期 3 個月。⁶⁵ 7 名病人有輕度至中度的改善,並在停藥後 1 年仍維持。灰黃黴素(griseofulvin,500 mg 至 750 mg 每日)在一項 6 名病人的開放試驗中,於一週內改善既有病灶,並在平均 33 天內阻止新病灶。⁶⁶ 秋水仙素(colchicine,0.5 mg 每日兩次)清除了一名患有頑固性 Schamberg 病的 28 歲女性。⁶⁷ 米諾環素 (Minocycline) 亦曾被報告有益。己酮可可鹼(pentoxifylline,可抑制 T 細胞黏附於內皮細胞與角質細胞)可能有幫助。一項隨機、研究者盲性試驗,比較了己酮可可鹼(400 mg 每日 3 次)與局部二丙酸倍他米松乳膏(betamethasone dipropionate cream,0.05% 每日兩次)為期 2 個月。以己酮可可鹼治療的病人改善顯著優於以局部類固醇治療者。然而,停藥後療效未能維持。⁶⁸
免疫抑制劑,例如全身性皮質類固醇、環孢素 (cyclosporine) 與甲胺喋呤 (methotrexate),通常有效,但因本疾病性質良性且停藥後會復發,故甚少有適應症使用。⁶,⁶⁹ 甲胺喋呤曾被報告以每週僅 15 mg 的劑量治療 4 週,即清除了一名 Majocchi 紫癜病人,不過她在停藥時復發。再次強調,除非發生泛發性疹子或嚴重症狀,否則此良性疾病通常不需要積極治療。
光療與雷射治療 (Phototherapy and Laser Therapy)
補骨脂素併紫外線 A 光(psoralen and ultraviolet A light, PUVA)與窄波段紫外線 B(narrowband ultraviolet B, nbUVB)皆曾被報告可清除病灶。⁷⁰⁻⁷⁹ 提出的機轉是光療誘發的細胞媒介反應免疫抑制。⁶
Gudi 與 White 首先報告了 nbUVB 在治療一名 Schamberg 病病人的有效性。⁷⁰ 在 Lasocki 與 Kelly 的一篇報告中,nbUVB 被用於在逐步減量口服 prednisolone 的同時維持疾病抑制。該病人在每 2 週一次的劑量下維持清除,但在完全停止 UVB 後復發。⁷¹ 另有其他病例報告了 nbUVB 在治療 PPD 的有效性。⁷²⁻⁷⁵
PUVA 抑制 T 細胞介白素-2 (interleukin-2) 的產生,並導致血管周圍淋巴球浸潤的消退。⁷⁶ Krisa 與同僚以 PUVA 治療 7 名病人,於 7 至 20 次治療後清除。⁷⁷ 其中 5 名病人維持緩解,2 名病人復發,但對第二療程的 PUVA 有反應。在另一篇報告中,一群 11 名 PPDs 病人對 PUVA 有反應。⁷⁸
光動力療法 (photodynamic therapy) 曾被報告有益,因為它同時影響免疫調節與血管破壞。⁸⁰ 此外,曾有報告以 595-nm 脈衝染料雷射 (pulsed-dye laser) 經 5 次每月一次的治療後,成功清除 Schamberg 病。⁸¹
諮詢衛教 (Counseling)
應向病人說明這些疹子良性但慢性且易復發的本質。可能需要持續追蹤以監測疾病進展。將治療目標設定為治療症狀,而非以治癒疾病為目標,可能可提升病人的滿意度,因為旨在清除疹子的治療往往無效。
致謝 (ACKNOWLEDGMENTS)
作者感謝本章前任作者 Theresa Schroeder Devere 與 Anisha B. Patel 的貢獻。
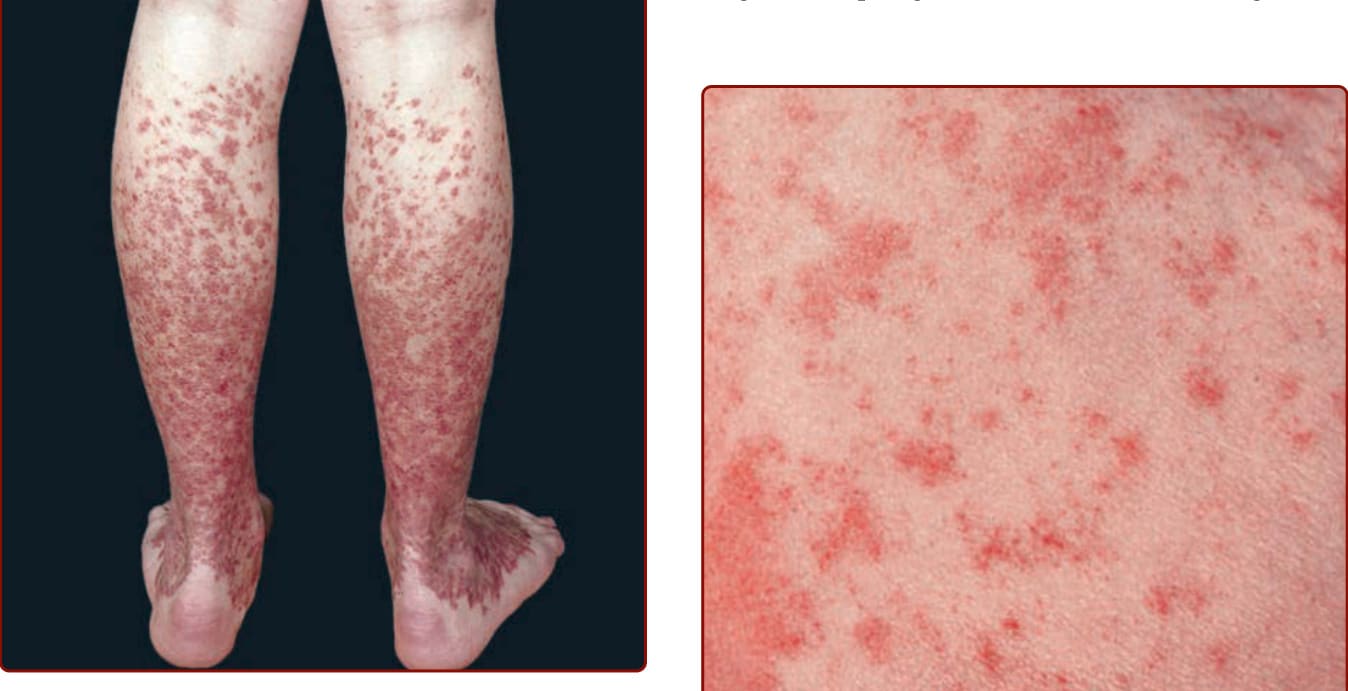
圖 143-1:色素性紫癜性皮膚病:Schamberg 病。腿部多發、散在且融合的不可觸及 (nonpalpable)、不褪色 (nonblanching) 的紫癜性病灶,已持續數月。急性微出血 (microhemorrhages) 隨含鐵血黃素沉積而消退,形成毀容性的深棕色胡椒狀染色。
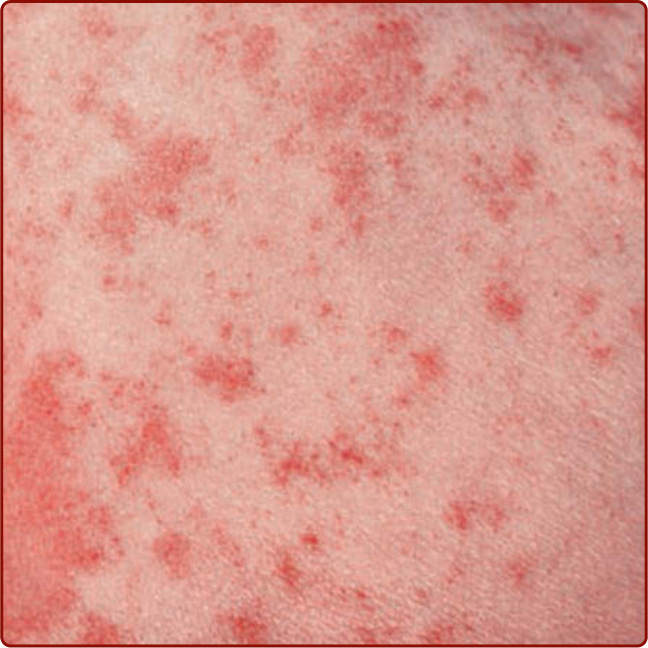
圖 143-2:Schamberg 病的「辣椒粉 (cayenne pepper)」外觀。(From Wolff K, Johnson R, Saavedra AP, et al. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 8th ed. New York, NY: McGraw-Hill; 2017, with permission.)
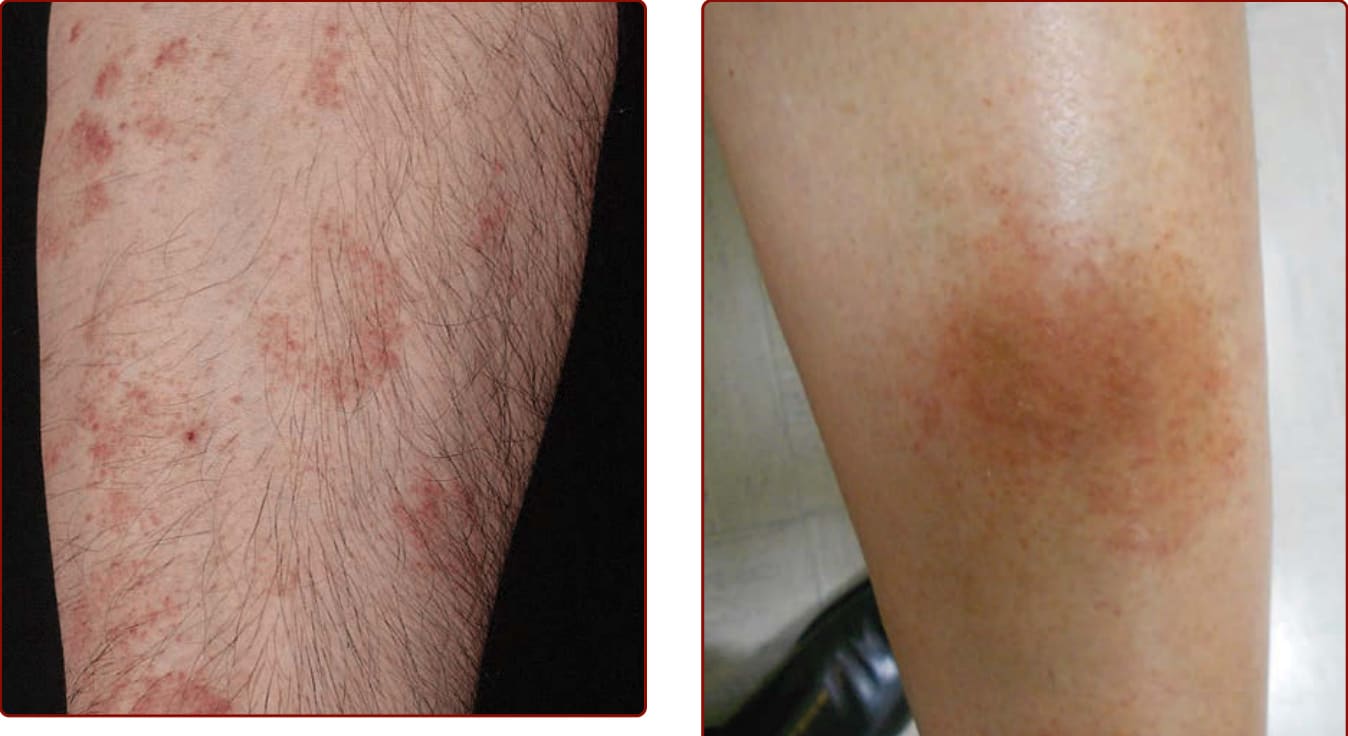
圖 143-3:色素性紫癜性皮膚病:Majocchi 病。多發、不可觸及、不褪色的紫癜性病灶,排列成環狀構型,並伴有細小的毛細血管擴張。注意較舊病灶的棕色變色。
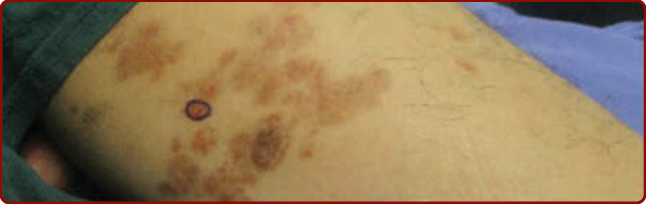
圖 143-4:Gougerot 與 Blum 的苔癬樣皮膚病。(Used with permission from Dr. April Armstrong.)
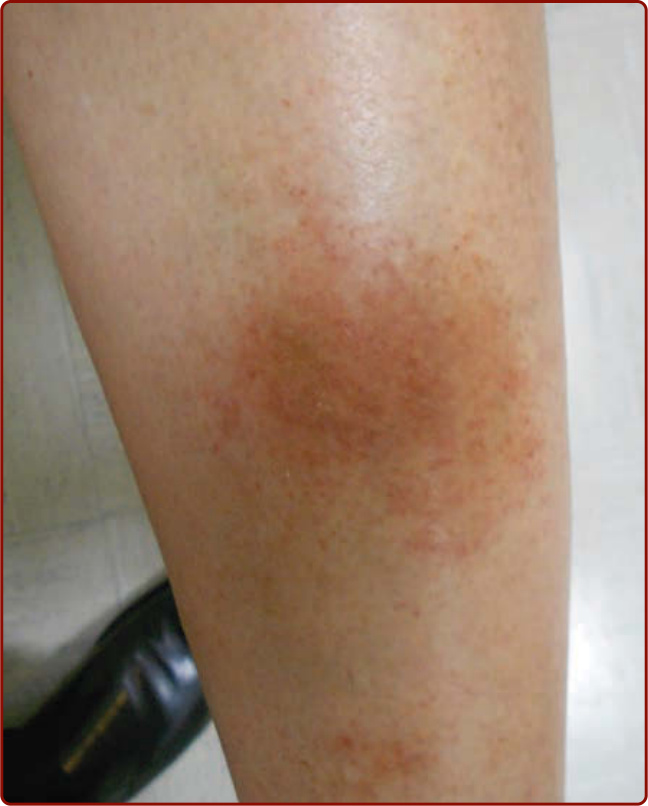
圖 143-5:金黃色苔癬 (Lichen aureus)。(Used with permission from Dr. Ashley Crew.)
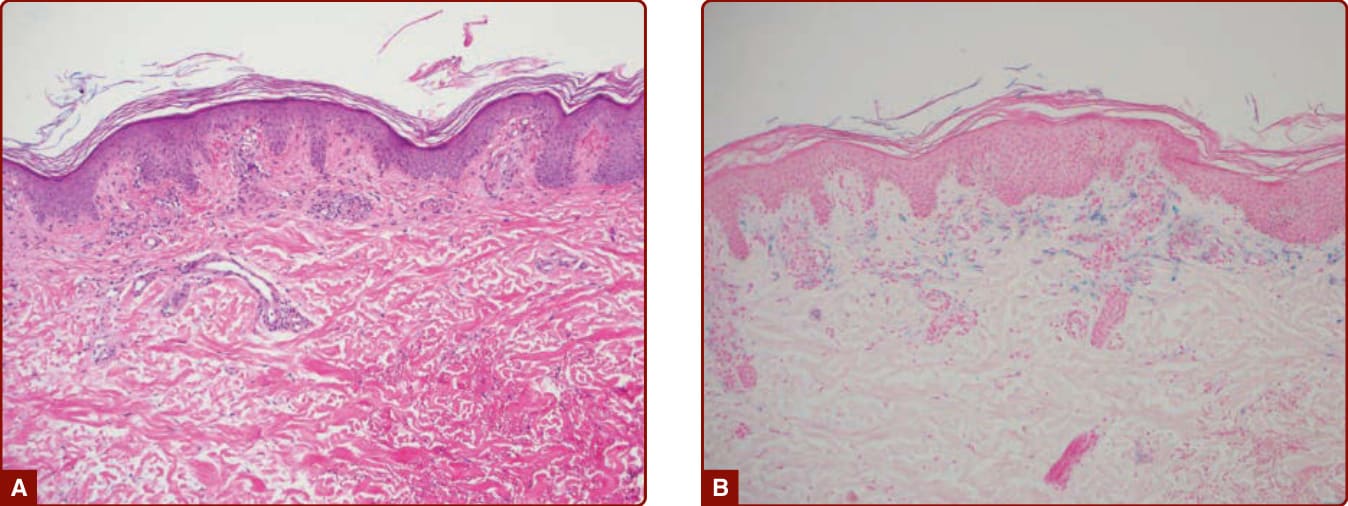
圖 143-6:A,色素性紫癜性皮膚病,顯示淺層血管周圍淋巴球與外滲的紅血球。B,以普魯士藍染色 (Prussian Blue staining) 凸顯的含鐵血黃素沉積。(Used with permission from Gene H. Kim, MD.)