Pigmented Purpuric Dermatoses
22
AT-A-GLANCE
■ A group of dermatoses characterized by petechiae, pigmentation, and occasionally, telangiectasia.
■ Found most commonly on the lower extremities; however, lesions may involve the upper body but rarely become generalized.
■ Benign, generally asymptomatic eruptions that tend to be chronic with flares and remissions.
■ Common histopathologic features include a superficial lymphocytic infiltrate, erythrocyte extravasation, and hemosiderin deposition.
■ Clinical variation between eruptions led to their subclassification into eponymic groups; however, frequent overlap may make differentiation difficult.
Pigmented purpuric eruptions (also called pigmented purpuric dermatoses [PPDs]) are a group of dermatoses that are characterized by petechiae, pigmentation, and, occasionally, telangiectasia. They are most commonly located on the lower extremities; however, lesions may involve the trunk and upper extremities, or, rarely, become generalized. They are subdivided into clinical subcategories, which include progressive pigmentary dermatosis (Schamberg disease), purpura annularis telangiectodes (Majocchi purpura), pigmented purpuric lichenoid dermatosis of Gougerot and Blum, eczematid-like purpura of Doucas and Kapetanakis, and lichen aureus (Table 143-1). These dermatoses are benign and generally asymptomatic, but tend to run a chronic course with flares and remissions. Despite differing clinical presentations, the subtypes of PPDs share common histopathologic features, including a superficial lymphocytic infiltrate, erythrocyte extravasation and hemosiderin deposition.
EPIDEMIOLOGY
PPDs are relatively uncommon. They may present at any age, including in children, and are most common in middle age (see Table 143-1). There is no ethnic predisposition, although the granulomatous variant has been reported more commonly in patients of Asian descent.1 In general, PPDs occur more frequently in males, except for the Majocchi subtype, which is seen more frequently in females.2
CLINICAL FEATURES
PROGRESSIVE PIGMENTARY DERMATOSIS (SCHAMBERG DISEASE)
PROGRESSIVE PIGMENTARY
DERMATOSIS (SCHAMBERG
DISEASE)
Schamberg first described an eruption of irregularly shaped reddish-brown patches with “pin head sized reddish puncta, closely resembling grains of cayenne pepper” over the legs of a 15-year-old boy in 1901.3 Schamberg disease is the most common of the PPDs to occur in children. It is, however, more common overall in adults with a peak incidence in the fifth decade.4 The lesions are insidious in their development and are usually asymptomatic. The lower extremities are the most common site of involvement. Lesions can involve the trunk or the upper extremities (Figs. 143-1 and 143-2). Schamberg disease is chronic and persistent with flares and remissions occurring indefinitely.
PURPURA ANNULARIS TELANGIECTODES (MAJOCCHI PURPURA)
PURPURA ANNULARIS
TELANGIECTODES
(MAJOCCHI PURPURA)
Majocchi described the first case of purpura annularis telangiectodes in 1896 in a 21-year-old male who presented with annular patches of follicular and punctate reddish-brown macules with telangiectasias and purpura on the lower extremities.5
This subtype of PPD is characterized by its distinctive annular pattern (Fig. 143-3). Individual lesions begin as punctate telangiectatic macules that extend peripherally with central hypopigmentation or slight atrophy.1 Lesions may be solitary or multiple in number. The lower extremities are the most common site of involvement and lesions can extend to involve the upper extremities and trunk. Majocchi purpura are generally asymptomatic and last several months with flares and remissions. As opposed to other subtypes of PPDs, Majocchi purpura presents most commonly in young adult females.2,6
22
SCHAMBERG MAJOCCHI GOUGEROT AND BLUM DOUCAS AND KAPETANAKIS ITCHING PURPURA LICHEN AUREUS
Average age of presentation 40s 30s 40s 40s 50s 20s-30s
Sex M > F F > M M > F M > F M > F M > F
Onset Insidious Abrupt Insidious Abrupt Abrupt Abrupt
Primary lesion Red-brown macule with pinpoint petechiae (“cayenne pepper”)
Primary lesion Red-brown macule with
Annular plaques with
Annular plaques with border of telangiectasia and purpura
pinpoint petechiae (“cayenne pepper”)
border of telangiectasia and purpura
F, female; M, male.
PIGMENTED PURPURIC LICHENOID DERMATOSIS OF GOUGEROT AND BLUM
PIGMENTED PURPURIC
LICHENOID DERMATOSIS
OF GOUGEROT AND BLUM
In 1925, Gougerot and Blum reported a pigmented eruption on the lower extremity of a 41-year-old man.7
This subtype is clinically distinguished by the presence of reddish-brown round or polygonal lichenoid papules and plaques, with a background of purpura or telangiectasias (Fig. 143-4). The term lichenoid describes this clinical appearance of lichenoid papules
Lichenoid
Red-brown
Red-brown
Orange-brown
Lichenoid papule Red-brown macule + scale Red-brown macule Orange-brown papule or plaque
papule
macule + scale
macule
papule or plaque
and plaques rather than the underlying histology. Its clinical appearance can be mistaken for Kaposi sarcoma. Similar to other subtypes of PPDs, pigmented purpuric lichenoid dermatosis of Gougerot and Blum is most commonly found on the lower extremities, occasionally involves the trunk and upper extremities, and runs a chronic course.
ECZEMATID-LIKE PURPURA OF DOUCAS AND KAPETANAKIS
ECZEMATID-LIKE PURPURA
OF DOUCAS AND
KAPETANAKIS
Eczematid-like purpura was first described by Doucas and Kapetanakis in 1953 in a group of patients with an asymptomatic seasonal eruption occurring in the spring and summer.8 It is distinguished
2591
22
clinically by mild scaling overlying pinpoint erythematous macules and patches with associated pruritus. Lichenification can occur from repeated scratching. Histopathologically, spongiosis is present, in addition to the classic histopathologic features of PPD. This subtype spreads rapidly over a period of 15 to 30 days and will subsequently fade without treatment over several months to years, although recurrence is possible.
LICHEN AUREUS
LICHEN AUREUS
In 1958, Martin first reported this subtype of PPD under the term lichen purpuricus, which was later named lichen aureus in 1960 by Calnan to emphasize its vivid yellow-orange color.9,10 As opposed to pigmented purpuric lichenoid dermatosis of Gougerot and Blum, where lichenoid refers to the clinical morphology of the lesions, in lichen aureus, lichen refers to both its clinical and histopathologic features. This
2592
subtype presents with more localized and persistent lesions with circumscribed macules or papules that are a distinctive gold, rust, or orange color (Fig. 143-5). Histologically a dense, band-like lichenoid infiltrate of inflammatory cells is seen. The lesions are generally asymptomatic but at times are intensely pruritic. They are most commonly localized to one lower extremity, but other body sites can be involved, and a segmental distribution has been reported.11,12 This disorder has a predilection for young adult males, with a peak incidence in the second and third decades. It runs a chronic course, with stable or slowly progressing lesions.
ITCHING PURPURA (DISSEMINATED PRURIGINOUS ANGIODERMATITIS)
ITCHING PURPURA
(DISSEMINATED
PRURIGINOUS
ANGIODERMATITIS)
Itching purpura, also known as disseminated pruriginous angiodermatitis, presents acutely with widely disseminated orange-brown to purpuric lesions associated with severe pruritus.13,14 The lesions first appear on the dorsal feet or lower extremities and then spread upward, sometimes with involvement of the trunk. Purpuric lesions are more apparent along the waistline, axilla, antecubital and popliteal fossae. Although this subtype of PPD has a chronic course, spontaneous remissions are possible.
UNILATERAL LINEAR CAPILLARITIS (SEGMENTAL PIGMENTED PURPURA)
UNILATERAL LINEAR
CAPILLARITIS (SEGMENTAL
PIGMENTED PURPURA)
In 1990, a transient PPD in a segmental distribution on the lower trunk of a middle-aged woman was reported. Later, Riordan and colleagues reported PPDs in 4 young men with linear and pseudodermatomal patterns, which they termed unilateral linear capillaritis.15
Both “segmental pigmented purpura” and “quadrantic capillaropathy” are considered subtypes of unilateral linear capillaritis.16,17 This PPD is clinically distinguished by its linear or segmental distribution. It tends to have a favorable prognosis, with spontaneous resolution occurring more commonly than in the other subtypes of PPDs.
GRANULOMATOUS PIGMENTED PURPURA
GRANULOMATOUS
PIGMENTED PURPURA
The first report of the granulomatous variant of PPDs was in 1996 by Saito and Matsuoka and since then more than 17 cases have been reported.18-20 It is most common in middle age and has been reported more commonly in patients of Asian descent.1,20 Clinically, the lesions appear similar to other PPDs with purpuric and brown macules developing most commonly on the lower extremities and dorsal feet. This subtype is distinguished by its histopathologic findings. In addition to the classic histopathologic features of a PPD, a granulomatous infiltrate is present. The granulomatous infiltrate is most commonly located in the papillary dermis, but may be in the mid to deep dermis separate from a more superficially located lichenoid infiltrate. Systemic granulomatous disorders and infectious processes (mycobacterial and deep fungal) must be ruled out in these patients. Hyperlipidemia is a relatively common association with granulomatous pigmented purpura with 9 of 17 reported cases showing elevated cholesterol levels in a review from 2014.19
PIGMENTED PURPURIC DERMATOSIS/MYCOSIS FUNGOIDES OVERLAP
PIGMENTED PURPURIC
DERMATOSIS/MYCOSIS
FUNGOIDES OVERLAP
In 1988, Barnhill and Braverman reported the first cases of pigmented purpura-like eruptions progressing to mycosis fungoides.21 Furthermore, the first patient diagnosed with lichen aureus in the United States was later diagnosed with mycosis fungoides (MF; see Chap. 119). Some evidence supports the idea that lichenoid variants of PPD may be precursors of MF, with similar histologic findings and clonal populations of lymphocytes.22-29 Although there is no clear
22
connection between these 2 diseases, and the occurrence of MF in the setting of a PPD is rare, 3 different relationships have been reported: MF mimicking pigmented purpura clinically, pigmented purpura evolving into MF, and pigmented purpura that simulates MF histologically. In a study of T-cell clonality and markers, T-cell monoclonality of PPD was most likely to predict progression to MF, whereas the absence of certain T-cell markers was a less reliable predictor.30 It is important to note that both MF and PPD can display clonality. Histologic clues of MF are subtle and include greater lymphoid atypia in the intraepidermal lymphocytes compared to dermal lymphocytes, large intraepidermal groups of lymphocytes anywhere in the epidermis, or many lymphocytes in the spinous layer.30,31 Santucci and colleagues suggested that to distinguish early MF from its inflammatory mimickers the most important feature is lymphocytes with extremely convoluted, medium to large nuclei, that are single or clustered in the epidermis and in small sheets in the dermis.32 Ackerman compared the histologic features of lichenoid purpuric eruptions with plaque stage MF and noted many similarities, concluding that it may be impossible to differentiate these two on a histologic basis alone.33
The differentiation between PPD and MF can be difficult and requires the integration of clinical, histologic and immunophenotypic information.30,33 PPDs with large areas of confluence, reticular arrangements, a superimposed violaceous hue, or pruritus that has been present or relapsing for several years are suspicious for MF. There is a predominance in adult males.21,34 For selected patients, long-term followup is needed to monitor for evolution into MF, even though the overall incidence of MF occurring in association with PPD is rare.
ETIOLOGY AND PATHOGENESIS
There are 3 different views on the pathogenesis of PPDs. The first is that there is a disturbance or weakness of cutaneous blood vessels, leading to capillary fragility and erythrocyte extravasation. This proposed mechanism, however, does not account for the inflammatory infiltrate that is also seen in these eruptions. The second theory is that PPDs develop from a humoral immune response. This theory is supported by direct immunofluorescence studies showing vascular deposition of C3, C1q, immunoglobulin M, or immunoglobulin A.35,36 The final theory is that PPDs develop as a result of a cellular immune response.37,38
This theory suggests that the inflammatory infiltrate, consisting of lymphocytes, macrophages, and Langerhans cells, leads to vascular fragility and subsequent extravasation of erythrocytes. Aiba and Tagami used immunohistologic studies in 8 cases of Schamberg disease to demonstrate that the dermal infiltrate was predominantly composed of helper-inducer T-cells and
2593
22
OKT6-reactive cells, whereas the epidermis showed intercellular staining with human leukocyte antigen- DR antibody and OKT6 antibody.39 Based on this study, they concluded that a cellular immune reaction, specifically with Langerhans cell, likely plays an important role in the pathogenesis. Additionally, Ghersetich and colleagues showed that cell-mediated mechanisms may be important in the development of Schamberg disease as CD4+ T cells and CD1a dendritic cells predominate in early lesions and this infiltrate clears with potent topical steroid therapy.38
PPD may be induced by multiple drugs (Table 143-2).1,40-53 These drug-induced eruptions are more likely to be generalized, but still present with lower-extremity involvement. They are often transient, with a mean duration of 3 to 4 months, with many cases resolving within a few weeks.53 A temporal relationship may be difficult to establish as patients can be taking the culprit medication for months to years prior to the onset of the eruption. Rechallenge has been used to confirm diagnosis. In addition to medications, other causes also have been implicated (Table 143-3).54-61 A handful of cases show familial involvement, with one report of an
■Acetaminophen
■Acetaminophen
■Aminoglutethimide
■Aminoglutethimide
■Ampicillin
■Ampicillin
■Aspirin
■Aspirin
■Bezafibrate
■Bezafibrate
■Bufexamac
■Bufexamac
■Carbamazepine
■Carbamazepine
■Carbromal
■Carbromal
■Carbutamide
■Carbutamide
■Chlordiazepoxide
■Chlordiazepoxide
■Diltiazem
■Diltiazem
■Dipyridamole
■Dipyridamole
■Topical fluorouracil
■Topical fluorouracil
■Furosemide
■Furosemide
■Glipizide
■Glipizide
■Glybuzole
■Glybuzole
■Hydralazine
■Hydralazine
■Infliximab
■Infliximab
■Interferon-α
■Interferon-α
■Isotretinoin
■Isotretinoin
■Medroxyprogesterone acetate
■Medroxyprogesterone acetate
■Meprobamate
■Meprobamate
■Minocycline
■Minocycline
■Nitroglycerin
■Nitroglycerin
■Nonsteroidal antiinflammatory drugs
■Nonsteroidal antiinflammatory drugs
■Phenobarbital
■Phenobarbital
■Pseudoephedrine
■Pseudoephedrine
■Raloxifene
■Raloxifene
■Reserpine
■Reserpine
■Sildenafil
■Sildenafil
■Tartrazine (food additive)
■Tartrazine (food additive)
■Thiamine
■Thiamine
■Trichlormethiazide
■Trichlormethiazide
2594
■Zomepirac sodium
■Zomepirac sodium
■Contact dermatitis
■Contact dermatitis
■Disperse dyes
■Disperse dyes
■Paraphenylenediamine
■Paraphenylenediamine
■Black rubber
■Black rubber
■Cobalt
■Cobalt
■Benzoyl peroxide
■Benzoyl peroxide
■Epoxy resin
■Epoxy resin
■Methylmethacrylate
■Methylmethacrylate
■Eutectic mixture of local anesthetics
■Eutectic mixture of local anesthetics
■Venous stasis/acroangiodermatitis
■Venous stasis/acroangiodermatitis
■Step aerobics
■Step aerobics
■Strenuous exercise
■Strenuous exercise
■Rheumatoid arthritis
■Rheumatoid arthritis
■Systemic lupus erythematosus
■Systemic lupus erythematosus
■Diabetes mellitus
■Diabetes mellitus
■Thyroid dysfunction
■Thyroid dysfunction
■Hyperlipidemia
■Hyperlipidemia
■Benign hyperglobulinemic purpura (of Waldenstrom)/ dysproteinemic purpura)
■Benign hyperglobulinemic purpura (of Waldenstrom)/
dysproteinemic purpura)
■Hodgkin disease
■Hodgkin disease
■Hepatitides B and C
■Hepatitides B and C
■Odontogenic infection
■Odontogenic infection
■Polycythemia
■Polycythemia
■Hereditary spherocytosis
■Hereditary spherocytosis
■Porphyrias
■Porphyrias
■Purpuric mycosis fungoides
■Purpuric mycosis fungoides
autosomal dominant inheritance pattern.62 Gravity and increased venous pressure may account for the lesions localizing to the lower extremities. Overall, the majority of cases of PPDs are idiopathic.
DIAGNOSIS
Diagnosis is generally made clinically and may be supported by histopathologic examination. The clinical measurement of capillary fragility by application of a sphygmomanometer (Hess test) does not appear to be reliable, as increased capillary fragility is not consistently seen in PPD. The patient’s medical history, medications, and potential contact allergens should be reviewed. MF should be considered if the lesions are chronic and unremitting.
SUPPORTIVE STUDIES
SUPPORTIVE STUDIES
LABORATORY TESTING
Laboratory testing is not required for diagnosis. In selected cases, laboratory testing may include a complete blood cell count with peripheral smear (to rule out thrombocytopenia or other hematologic disorders), coagulation studies (to rule out other causes of purpura), as well as an antinuclear antibody, rheumatoid factor, and hepatitis serologies.1
A
22
B
PATHOLOGY
All PPDs share similar histopathology. A lymphocytic perivascular infiltrate is present in the papillary dermis with endothelial swelling and extravasated erythrocytes and hemosiderin deposition within macrophages (Fig. 143-6). In older lesions, there is less inflammation, and extravasated red cells may no longer be present. Although there is no consensus on whether a capillaritis is present, Ackerman asserts that there is no true capillaritis because of the lack of fibrin in the luminal walls and absence of thrombi.1
The epidermis may show spongiosis and parakeratosis, particularly in pigmented purpuric lichenoid dermatitis of Gougerot and Blum and eczematidlike purpura of Doucas and Kapetanakis. In lichen aureus, there is a band-like mononuclear infiltrate in the upper dermis separated from the epidermis by a thin rim of uninvolved collagen. In granulomatous pigmented purpura, a granulomatous infiltrate is present either in the papillary dermis, or in the mid to deep dermis, which separated from a more superficially located lichenoid infiltrate.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
PPDs are benign and commonly asymptomatic. In general, PPDs are chronic with flares and remissions. Exceptions to this chronic course include unilateral linear capillaritis and drug-induced PPDs, which tend to have shorter clinical courses and more favorable overall prognoses.
MANAGEMENT
Management of PPD is challenging, as treatments are often ineffective. The patient’s medical history, medications, and potential contact allergens should be reviewed. If the eruption is induced by a drug or contact allergen, discontinuation of the causative agent can lead to complete resolution. However, residual pigmentation may persist for years.
INTERVENTIONS
INTERVENTIONS
MEDICATIONS
Common initial therapies include topical steroids and antihistamines, which are particularly helpful in treating associated pruritus, as well as the use of compression stockings to treat associated venous insufficiency. In addition to topical steroids, topical pimecrolimus has been used. Treatment with topical pimecrolimus twice daily for 10 weeks cleared lichen aureus lesions in a 10-year-old boy. The patient had a minor flare after discontinuation that responded to 2 weeks of repeat treatment.63 Intralesional corticosteroids have
■Contact dermatitis
■Contact dermatitis
■Leukocytoclastic vasculitis
■Leukocytoclastic vasculitis
■Stasis dermatitis and purpura
■Stasis dermatitis and purpura
■Angioma serpiginosum
■Angioma serpiginosum
■Kaposi sarcoma (Gougerot-Blum)
■Kaposi sarcoma (Gougerot-Blum)
■Mycosis fungoides
■Mycosis fungoides
■Sarcoidosis
■Sarcoidosis
■Scurvy
■Scurvy
2595
■Hypergammaglobinemic purpura
■Hypergammaglobinemic purpura
22
also led to improvement, although tissue atrophy may develop and the development of new lesions is not prevented.6
Oral bioflavonoid (rutoside, 50 mg twice daily) and ascorbic acid (500 mg twice daily) given to 3 patients with chronic PPD in an open trial led to clearance in all 3 patients within 4 weeks with maintenance of remission 3 months after treatment.64 In a pilot study, calcium dobesilate, 500 mg twice daily, was given to 9 patients for 3 months.65 Seven patients had mild-to-moderate improvement that was sustained at 1 year after cessation of therapy. Griseofulvin, 500 mg to 750 mg daily, improved existing lesions within a week and stopped new lesions within a mean of 33 days in an open trial of 6 patients.66 Colchicine, 0.5 mg twice daily, cleared a 28-year-old woman with recalcitrant Schamberg disease.67 Minocycline also has been reported to be beneficial. Pentoxifylline, which inhibits T-cell adherence to endothelial cells and keratinocytes, may be helpful. A randomized, investigator-blinded trial compared pentoxifylline, 400 mg 3 times daily, with topical betamethasone dipropionate cream, 0.05% twice daily, for 2 months. Patients treated with pentoxifylline had significantly greater improvement than those treated with the topical steroid. However, the response was not sustained after discontinuation of the medication.68
Immunosuppressants, such as systemic corticosteroids, cyclosporine, and methotrexate are often effective but are rarely indicated because of the benign nature of the disorder as well as recurrence upon discontinuation of these medications.6,69 Methotrexate was reported to clear a patient with Majocchi purpura with just 15 mg weekly dosing for 4 weeks, although she had reoccurrence when the medication was discontinued. Again, aggressive therapy is not often warranted for this benign condition unless a generalized eruption or severe symptoms occur.
PHOTOTHERAPY AND LASER THERAPY
Psoralen and ultraviolet A light (PUVA) and narrowband ultraviolet B (nbUVB) have both been reported to clear lesions.70-79 The phototherapy-induced immunosuppression of the cell-mediated response is the proposed mechanism.6
Gudi and White were the first to report the effectiveness of nbUVB in treating a patient with Schamberg disease.70 In a report by Lasocki and Kelly, nbUVB was used to maintain disease suppression while tapering oral prednisolone. The patient remained clear on a once-every-2-week dosing, but had recurrence when UVB was completely stopped.71 Additional cases have reported effectiveness of nbUVB in the treatment of PPD.72-75
PUVA inhibits T-cell interleukin-2 production and leads to resolution of the perivascular lymphocytic infiltrate.76 Krisa and colleagues treated 7 patients with PUVA with clearance after 7 to 20 treatments.77 Five of
2596
these patients remained in remission and 2 patients relapsed, but responded to a second course of PUVA. In another report, a cohort of 11 patients with PPDs responded to PUVA.78
Photodynamic therapy has been reported as beneficial as it affects both immunomodulation and vascular destruction.80 In addition, successful clearance of Schamberg disease with the use of a 595-nm pulsed-dye laser after 5 monthly treatments has been reported.81
COUNSELING
Patients should be counseled about the benign but chronic and relapsing nature of these eruptions. Ongoing follow up may be warranted to monitor disease progression. Setting the expectation of treating symptoms, rather than aiming to cure the condition, may lead to improved patient satisfaction, as treatments to clear the eruption are often ineffective.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Theresa Schroeder Devere and Anisha B. Patel, the former authors of this chapter.
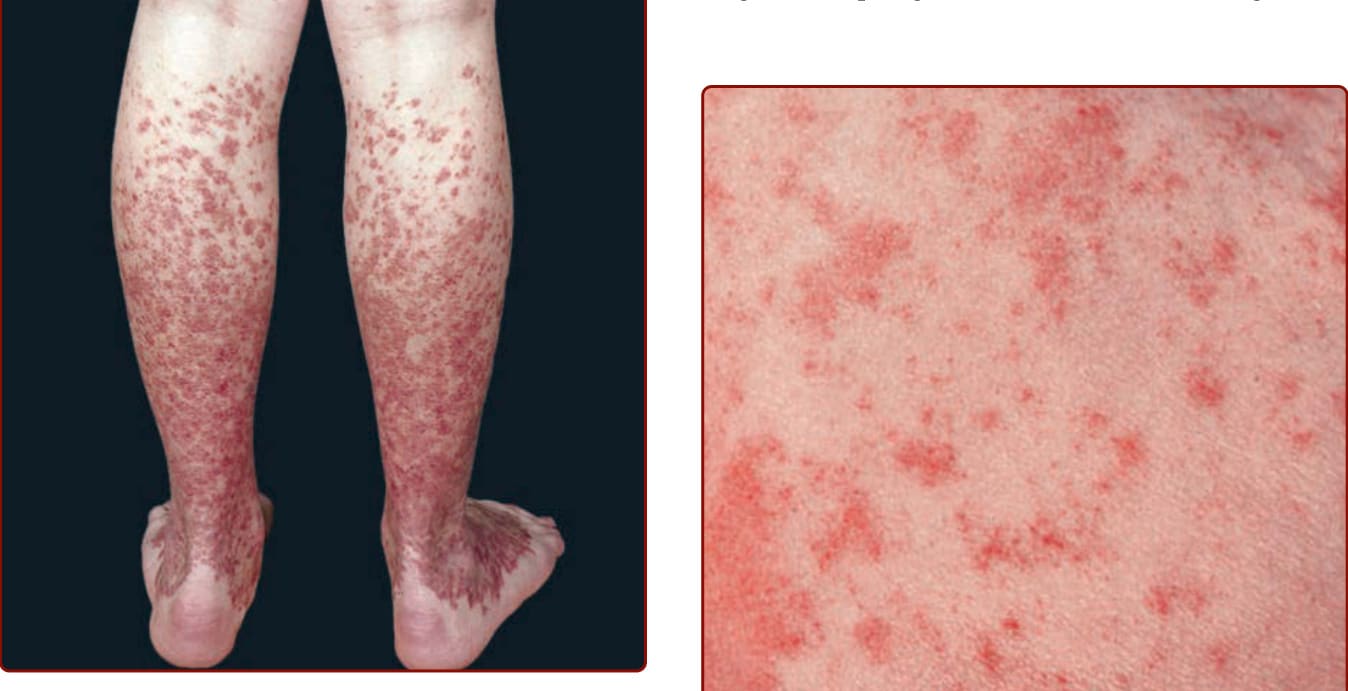
Figure 143-1 Pigmented purpuric dermatosis: Schamberg disease. Multiple, discrete, and confluent nonpalpable, nonblanching purpuric lesions of many months’ duration on the legs. Acute microhemorrhages resolve with deposition of hemosiderin, creating a disfiguring dark-brown peppered stain.
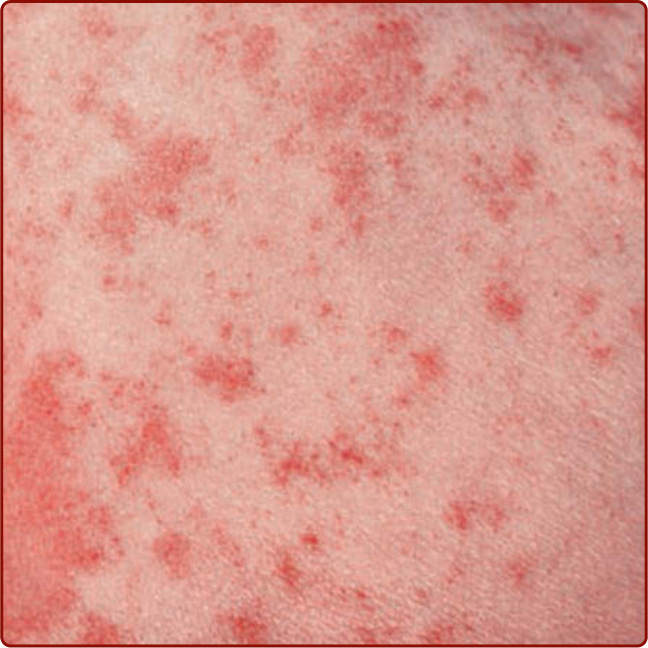
Figure 143-2 “Cayenne pepper” appearance of Schamberg disease. (From Wolff K, Johnson R, Saavedra AP, et al. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 8th ed. New York, NY: McGraw-Hill; 2017, with permission.)
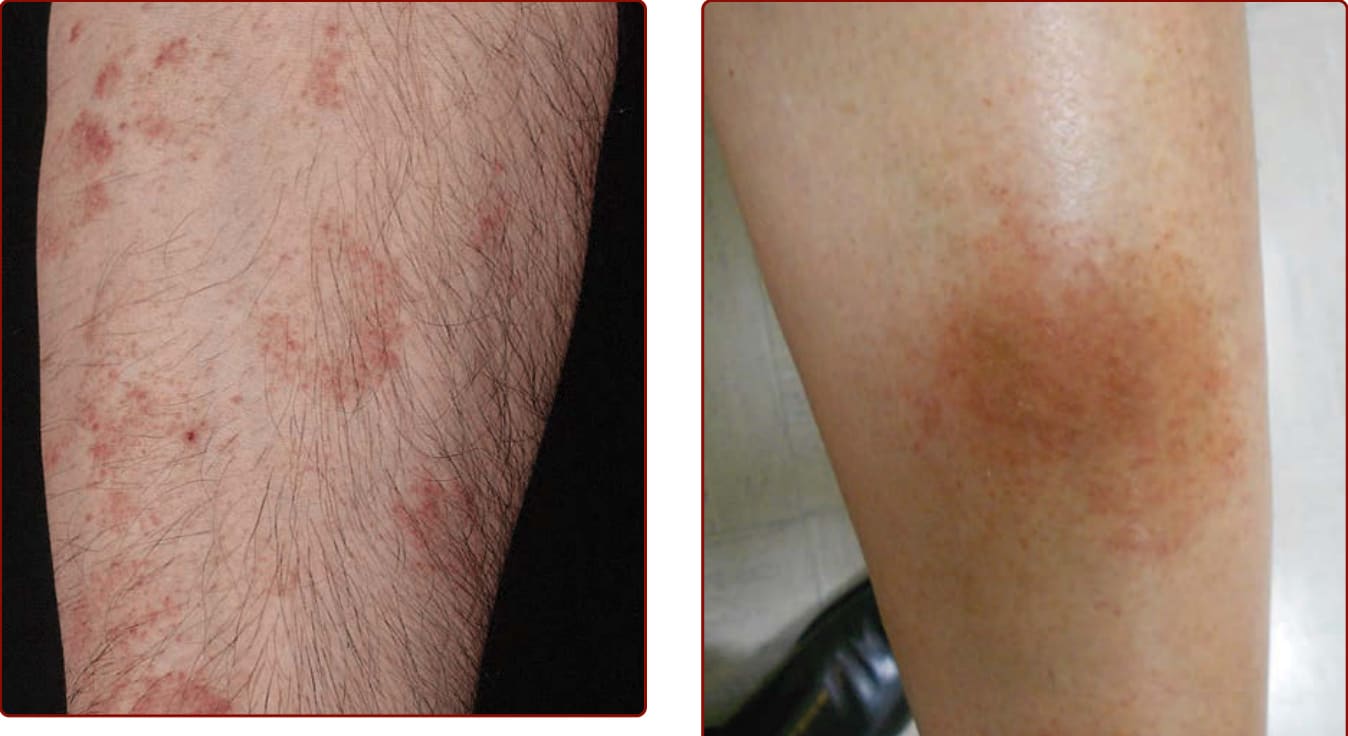
Figure 143-3 Pigmented purpuric dermatosis: Majocchi disease. Multiple nonpalpable, nonblanching purpuric lesions arranged in annular configurations and associated with tiny telangiectasias. Note brownish discoloration of older lesions.
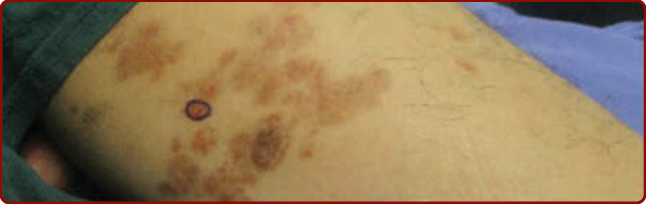
Figure 143-4 Lichenoid dermatosis of Gougerot and Blum. (Used with permission from Dr. April Armstrong.)
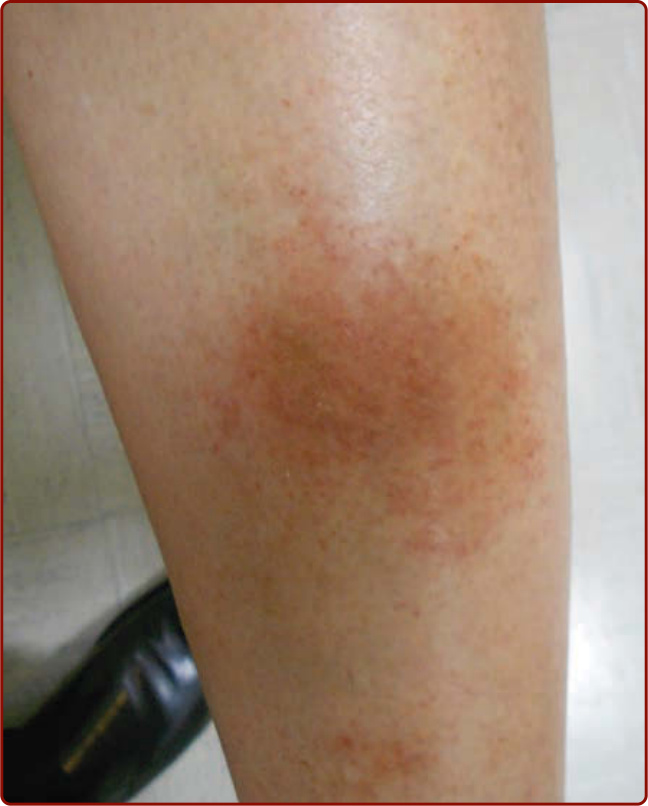
Figure 143-5 Lichen aureus. (Used with permission from Dr. Ashley Crew.)
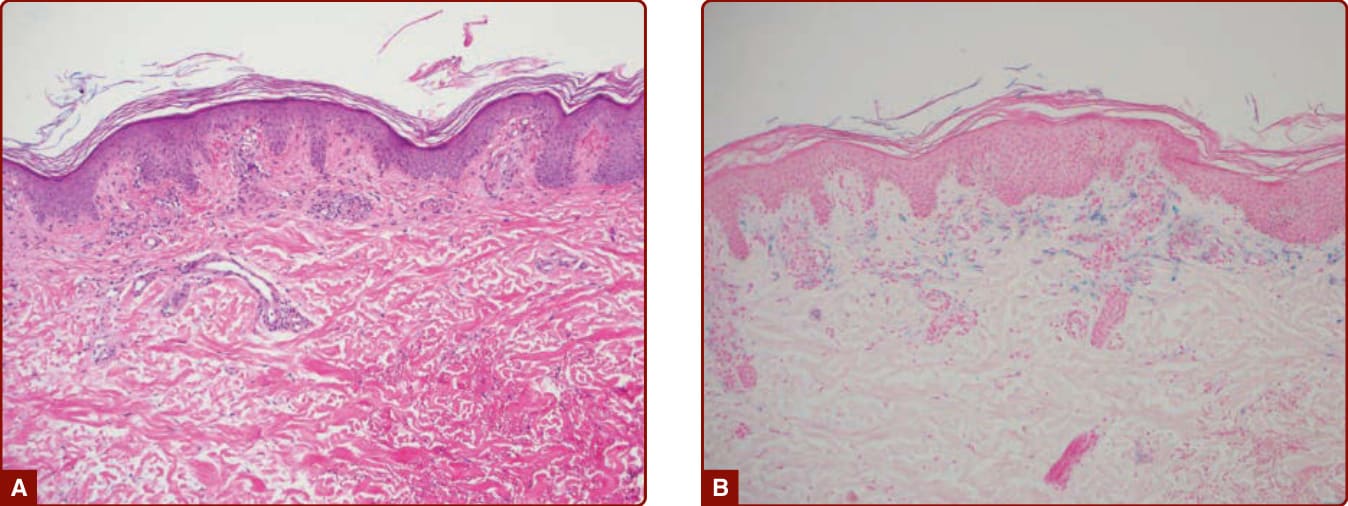
Figure 143-6 A, Pigmented purpuric dermatosis showing superficial perivascular lymphocytes and extravasated erythrocytes. B, Hemosiderin deposition highlighted by Prussian Blue staining. (Used with permission from Gene H. Kim, MD.)
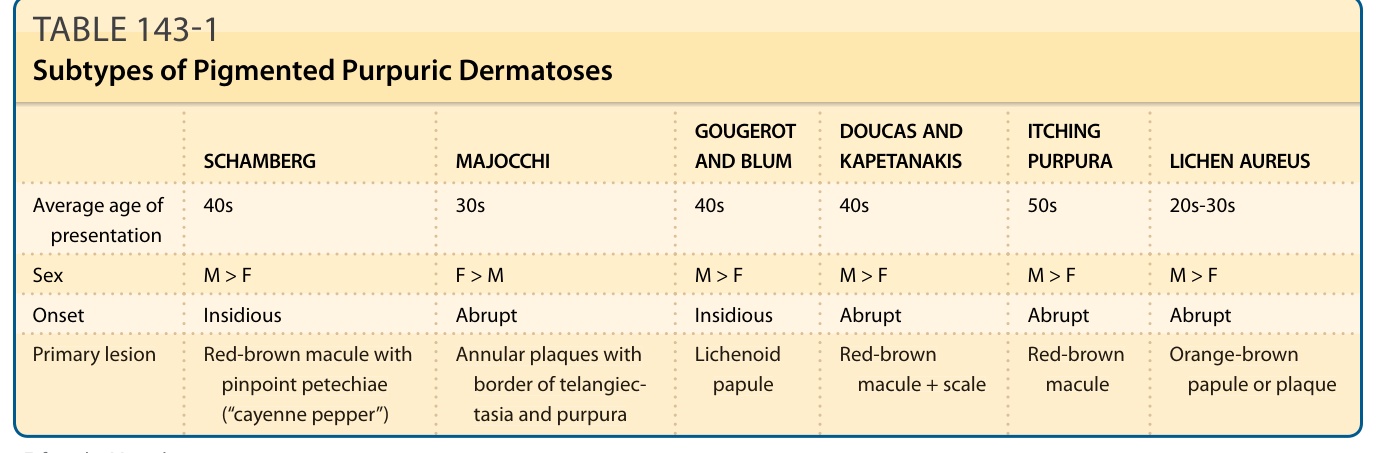
TABLE 143-1 Subtypes of Pigmented Purpuric Dermatoses
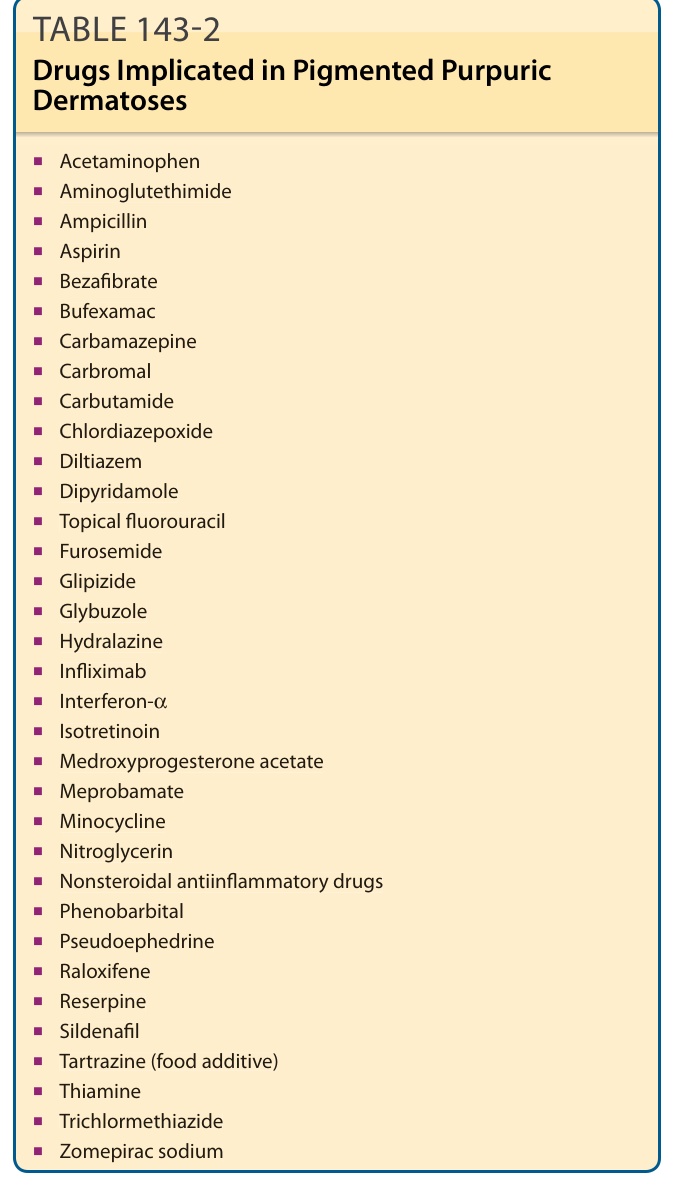
TABLE 143-2 Drugs Implicated in Pigmented Purpuric Dermatoses

TABLE 143-3 Other Implicated Causes of Pigmented Purpuric Dermatoses

Table 143-4 outlines the differential diagnosis of PPD.