川崎病 (Kawasaki Disease)
PART 22
血管疾病 (Vascular Diseases)
重點一覽 (AT-A-GLANCE)
■ 川崎病(Kawasaki disease, KD)是一種病因不明、但疑為感染性的多系統發炎過程。
■ 發生率最高者為亞洲兒童;每 80 名日本兒童中就有 1 名在 5 歲前罹患 KD。
■ KD 是已開發國家兒童後天性心臟病 (acquired heart disease) 最常見的原因。
■ KD 侵犯全身所有血管,但主要損害中型肌性動脈 (medium-sized muscular arteries),例如冠狀動脈 (coronary arteries)。
■ 主要症狀為持久高燒、結膜充血 (conjunctival injection)、口腔黏膜變化(如紅唇、咽部發紅與草莓舌 strawberry tongue)、手足發紅與腫脹、紅斑性多形性皮疹 (erythematous polymorphic rash),以及頸部淋巴結病變 (cervical lymphadenopathy)。
■ 冠狀動脈發炎可導致動脈瘤 (aneurysms),並隨後造成心肌梗塞 (myocardial infarction)、動脈瘤破裂與猝死。
■ 於發燒最初 10 天內給予靜脈注射免疫球蛋白(intravenous immunoglobulin, IVIG)與阿斯匹靈 (aspirin) 治療,可將冠狀動脈異常的盛行率從單用阿斯匹靈者的 25% 降至接受 IVIG 合併阿斯匹靈者的 5%。
■ 長期併發症局限於心臟與血管樹 (vascular tree),主要為主要冠狀動脈的血栓 (thrombosis) 與狹窄 (stenosis),並伴隨心肌缺血 (myocardial ischemia)。
川崎病(Kawasaki disease, KD)是已開發國家兒童後天性心臟病的首要原因,為一種多系統發炎性疾病,尤其侵犯血管,特別是冠狀動脈。約 25% 未治療的兒童會發展出冠狀動脈異常,包括擴張 (dilation) 與動脈瘤,可導致心肌梗塞與猝死。¹,² 病因不明,但臨床與流行病學資料支持感染性病因。在 KD 中,廣泛的器官與組織會發展出強烈的發炎細胞反應³;在中型動脈(如冠狀動脈)中,此反應可損害血管壁中的膠原蛋白 (collagen) 與彈性蛋白 (elastin) 纖維,導致其正常結構完整性的喪失,進而造成膨出 (ballooning) 或動脈瘤形成。儘管對 KD 致病機轉的理解有限,卻存在一種非常有效的療法,即靜脈注射免疫球蛋白(intravenous immunoglobulin, IVIG)合併阿斯匹靈;於發燒最初 10 天內給予時,此療法可將冠狀動脈異常的盛行率從未治療病人的 25% 降至接受治療者的 5%。⁴ 由於病因不明,並無診斷檢驗可用,診斷乃依臨床做出。典型 KD(Classic KD)的診斷見於具有持久發燒並符合 5 項其他臨床特徵中 4 項的病人。然而,不完全型 (incomplete forms) 疾病亦廣為人知,此類兒童表現持久發燒但符合少於 4 項其他臨床特徵,並隨後發展出冠狀動脈異常。這些不完全型疾病的存在,使醫師在對病因不明的持久發燒兒童準確建立診斷時面臨重大的診斷困境。
歷史觀點 (HISTORICAL PERSPECTIVE)
KD 以 Tomisaku Kawasaki(川崎富作)醫師命名,他是一位日本兒科醫師,首先辨識出此病的臨床特徵。他於 1967 年在日語文獻中描述了 50 例一種新疾病,並將其命名為黏膜皮膚淋巴結症候群(mucocutaneous lymph node syndrome)。⁵ 直到後來,部分罹患此新描述疾病的兒童發生猝死;解剖顯示因冠狀動脈瘤血栓所致的心肌梗塞。在 Kawasaki 醫師描述其臨床特徵之前,KD 僅由病理學家在解剖時辨識出來,他們稱此病為嬰兒結節性多動脈炎 (infantile periarteritis nodosa)。⁶ Kawasaki 醫師於 1974 年在英語文獻中描述了此病;緊接著這份報告之後,便有一份對同一疾病的描述,那是 Marian Melish 醫師及其同事於 1970 年代初在夏威夷獨立觀察到的。⁷,⁸
自那時起,已逐漸明朗的是,雖然 KD 的發病率在亞洲、特別是日本、韓國與中國兒童中最高,但全世界所有種族與族裔群體都會受此病影響。
流行病學 (EPIDEMIOLOGY)
KD 主要是幼兒的疾病,80% 的病例發生於 6 個月至 5 歲的兒童。⁹,¹⁰ 然而,未滿 6 個月的嬰兒也可能罹病,常表現不完全型疾病,且可能罹患特別嚴重的 KD。¹¹,¹²
同樣地,KD 也可發生於較大兒童與青少年,這些病人的診斷常被延誤,他們也可能罹患較嚴重的 KD,並有較高的冠狀動脈異常盛行率。¹¹⁻¹³ 因此,所有兒科年齡層都必須考慮此診斷。男孩比女孩更常受影響,比例為 3:2。KD 的發生率在日本兒童中約為白人兒童的 10 倍。⁹,¹⁰
約每 80 名日本兒童中就有 1 名在 5 歲前發展出 KD;在日本與美國,發病的高峰年齡皆為 9 至 11 個月。⁹,¹⁰ 日本兒童中較高的發病率在採用西方飲食與生活方式者身上依然持續存在,這很可能與亞洲兒童對 KD 的遺傳易感性有關。¹⁴ 手足發生 KD 的風險比一般族群高 10 倍,而父母曾罹患 KD 之兒童的 KD 發生率為一般族群的兩倍。¹⁵,¹⁶ 復發罕見,在日本約占 3% 的病例。¹⁰
KD 的許多流行病學特徵顯示感染性病原體為其病因。其中包括已被充分描述的疾病流行 (epidemics)¹⁴,¹⁷⁻¹⁹,以及流行期間疾病呈地理上波浪狀的擴散,這與感染性病原體的傳播相符。¹⁹ 美國的病例在冬季與春季較為常見。⁹
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
KD 常可觀察到全身性皮疹 (generalized exanthem),以軀幹與四肢最為顯著,通常呈現 3 種型態之一:麻疹樣 (morbilliform,圖 142-1A)、靶狀 (targetoid,圖 142-1B) 或猩紅熱樣 (scarlatiniform,瀰漫性紅斑,圖 142-1C)。不會觀察到大疱 (bullae)、水疱 (vesicles) 與潰瘍性病灶,但偶爾會出現細小的微膿疱疹 (micropustular rash),尤其在伸側 (extensor) 表面。皮疹可能伴有搔癢。在疾病的急性發熱期 (acute febrile phase),常可觀察到鼠蹊部 (groin) 紅斑與脫屑 (圖 142-2),可能被誤認為念珠菌性尿布皮膚炎 (candidal diaper dermatitis),甚至葡萄球菌燙傷樣皮膚症候群 (staphylococcal scalded skin syndrome)。鼠蹊部的皮膚變化可見於包尿布的兒童與已戒除尿布 (toilet-trained) 的兒童。典型的手指與腳趾甲周 (periungual) 脫屑直到發燒開始後第二至第三週才開始,並可進展至侵犯整隻手與腳 (圖 142-3);治療應在其出現之前就妥善給予。在發病後第三至第六週,常可見橫越指甲的橫向紋路(Beau 氏線 Beau lines)。這些紋路會隨指甲長出。在常規接種卡介苗 (bacille Calmette-Guérin vaccine) 的國家(如日本),KD 兒童的一項常見發現是卡介苗接種部位的紅斑與腫脹;其機轉不明,但此過程會隨 KD 的治療而消退。雖然 KD 會造成侵犯全身所有動脈與靜脈的血管炎 (vasculitis),但皮膚中的血管並未顯著受累,且皮膚切片對診斷無用,因為病理發現並無特異性。
非皮膚表現 (NONCUTANEOUS FINDINGS)
任何無其他解釋之持久發燒的兒童,都應將 KD 納入鑑別診斷。在 KD 中,所有臨床特徵可能不會同時出現。因此,重要的是向在持久發熱性疾病過程中看過病人的父母與醫師詢問是否曾出現此病的其他 5 項臨床特徵:結膜充血、口腔黏膜變化、手足變化、皮疹,以及頸部淋巴結病變。KD 兒童常有相當嚴重的手足腫脹與不適,以致他們會拒絕拿取物品或行走 (圖 142-4)。這在 KD 鑑別診斷中的大多數其他疾病兒童中並不常見,可作為診斷的重要線索。同樣地,極度易怒 (extreme irritability) 在 KD 中很常見,而在鑑別診斷中的大多數其他疾病中則不那麼常見。若無特定治療,KD 的發燒呈每日、高峰、間歇性,並持續 1 至 2 週。此病通常分為 3 個階段:急性發熱期、亞急性期(subacute phase,始於發燒消退時並持續至所有臨床特徵恢復正常),以及恢復期(convalescent phase,接在亞急性期之後,並持續至紅血球沉降率〔erythrocyte sedimentation rate, ESR〕恢復正常,通常在發燒發作後 6 至 8 週)。KD 的結膜充血為雙側性且非滲出性 (nonexudative)。可能有角膜緣豁免 (limbal sparing)(圖 142-5)。畏光 (photophobia) 是常見的伴隨特徵。口腔發現包括發紅、腫脹、乾燥、龜裂且可能出血的嘴唇 (圖 142-6)、「草莓」舌,以及口腔與咽喉的紅斑。口腔潰瘍並非 KD 的特徵。掌部與蹠部紅斑是常見的特徵,且可在手腕與腳踝處由明顯紅斑突然轉變為正常皮膚。手足可能水腫且疼痛。頸部淋巴結病變是最不常觀察到的臨床特徵,發生於約 75% 的典型 KD 兒童,但在一部分病人中可為最顯著的特徵,這些病人在做出正確診斷前常接受多個不同療程的抗生素治療而無改善。²⁰ 頸部淋巴結病變通常為單側性,可能伴隨或不伴隨表淺紅斑及/或觸診壓痛,且不具波動感 (nonfluctuant)。由於 KD 是一種多系統發炎過程,許多器官與組織都涉入發炎過程,導致各種相關的臨床特徵(表 142-1)。特別是,關節炎 (arthritis) 可發生於急性發熱期,侵犯小的指間關節 (interphalangeal joints) 與較大的關節;或可發生於疾病的亞急性期,通常侵犯較大的關節,如膝關節與踝關節。無菌性腦膜炎 (aseptic meningitis) 是接受腰椎穿刺 (lumbar puncture) 之病人的常見發現。
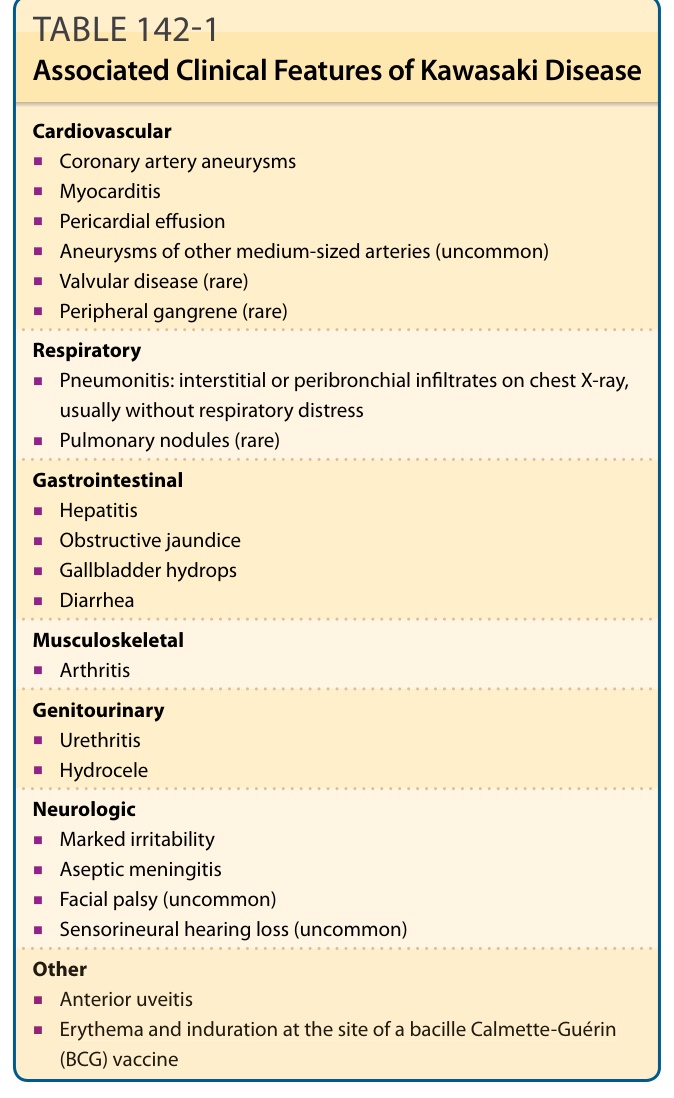
表 142-1:川崎病的相關臨床特徵 (Associated Clinical Features of Kawasaki Disease)
心血管系統 (Cardiovascular)
■ 冠狀動脈瘤 (Coronary artery aneurysms)
■ 心肌炎 (Myocarditis)
■ 心包積液 (Pericardial effusion)
■ 其他中型動脈的動脈瘤(不常見)
■ 瓣膜疾病 (Valvular disease)(罕見)
■ 周邊壞疽 (Peripheral gangrene)(罕見)
呼吸系統 (Respiratory)
■ 肺炎 (Pneumonitis):胸部 X 光上的間質性或支氣管周圍浸潤,通常無呼吸窘迫
■ 肺結節 (Pulmonary nodules)(罕見)
腸胃系統 (Gastrointestinal)
■ 肝炎 (Hepatitis)
■ 阻塞性黃疸 (Obstructive jaundice)
■ 膽囊水腫 (Gallbladder hydrops)
■ 腹瀉 (Diarrhea)
肌肉骨骼系統 (Musculoskeletal)
■ 關節炎 (Arthritis)
泌尿生殖系統 (Genitourinary)
■ 尿道炎 (Urethritis)
■ 陰囊水腫 (Hydrocele)
神經系統 (Neurologic)
■ 顯著易怒 (Marked irritability)
■ 無菌性腦膜炎 (Aseptic meningitis)
■ 顏面神經麻痺 (Facial palsy)(不常見)
■ 感音神經性聽力喪失 (Sensorineural hearing loss)(不常見)
其他 (Other)
■ 前葡萄膜炎 (Anterior uveitis)
■ 卡介苗(bacille Calmette-Guérin, BCG)接種部位的紅斑與硬結 (induration)
併發症 (COMPLICATIONS)
如上所述,約 25% 未治療的兒童會發展出冠狀動脈異常,包括擴張與動脈瘤(圖 142-7),可導致心肌梗塞、動脈瘤破裂與猝死。心肌梗塞可因動脈瘤血栓而發生,此情況在發病後最初幾個月最為常見;或因冠狀動脈狹窄而發生,此情況則在發病後數月至數年出現。動脈瘤破裂較不常見,通常發生於發病後第一個月內。超過 50% 的 KD 病人在急性發熱期有心肌炎,臨床上表現為與發燒不成比例的心搏過速 (tachycardia),此情況通常隨 IVIG 治療迅速改善。在疾病的急性期,可能出現心包積液;此情況會自行緩解。在罹患最嚴重冠狀動脈瘤的病人中,也可觀察到其他中型動脈的動脈瘤,最常見於髂動脈 (iliac)、股動脈 (femoral) 與腋動脈 (axillary)。嚴重到需要瓣膜置換的瓣膜炎 (valvulitis) 鮮有報告。雖然 KD 是一種侵犯多個器官與組織的全身性發炎疾病,但目前並無已知的心臟與血管以外的長期後果。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
KD 的病因仍然不明。最符合現有臨床、免疫學與流行病學資料的假說是:KD 源於感染一種普遍存在的病原體,此病原體通常導致無症狀感染,但在一小部分具有遺傳易感性的個體中引起 KD。遺傳易感性是傳染病的易感性與宿主反應中的一個共同主題。KD 很可能為多基因性 (polygenic)。數個國際合作團隊正在持續密集研究影響 KD 易感性的遺傳因子。ITPKC 基因(T 細胞活化的負向調節因子)的一個功能性多型性 (functional polymorphism) 與 KD 易感性及發展出冠狀動脈異常的風險有關²¹;其他免疫基因,如 CASP3、BLK、CD40、FCGR2A 與 ORAI1,也似乎與 KD 易感性有關。²²⁻²⁴
KD 血管病變 (vasculopathy) 的特徵為三個相互關聯的病理過程,其最顯著地影響冠狀動脈:嗜中性球壞死性動脈炎 (neutrophilic necrotizing arteritis),發生於發燒發作後最初 2 週;亞急性/慢性血管炎 (subacute/chronic vasculitis),始於最初 2 週但可持續數月至數年,由淋巴細胞(主要為 CD8 T 淋巴細胞²⁵)、漿細胞(特別是免疫球蛋白〔immunoglobulin, Ig〕A 漿細胞²⁶,²⁷)、嗜酸性球 (eosinophils) 與巨噬細胞 (macrophages) 組成;以及管腔肌纖維母細胞增生 (luminal myofibroblastic proliferation),其與亞急性/慢性血管炎密切相關,可導致進行性動脈狹窄。²⁸ 這 3 個過程解釋了 KD 潛在的不良結果。壞死性動脈炎可導致內膜 (intima) 與中膜 (media) 的壞死與剝落;在其最嚴重的型態中,可能僅餘一薄層外膜 (adventitia),造成偶爾破裂的巨大動脈瘤,但更常見的是累積層層血栓,這些血栓可變為阻塞性並引起心肌梗塞。亞急性/慢性動脈炎與管腔肌纖維母細胞增生及進行性動脈狹窄相關,這在有或無上覆血栓的情況下都可導致心肌梗塞。²⁸ KD 中的後天性免疫反應 (adaptive immune responses) 已被證明為抗原驅動 (antigen-driven)。²⁹⁻³¹ 急性 KD 中 IgA 漿細胞與 CD8 T 細胞作為發炎浸潤的主要成分的存在,顯示對一種具呼吸道入侵門戶 (respiratory portal of entry) 的細胞內病原體 (intracellular pathogen) 的免疫反應。這些寡克隆 (oligoclonal) IgA 抗體的合成版本可在急性 KD 組織中辨識出抗原,該抗原似乎位於含有蛋白質與 RNA 的細胞質內包涵體 (intracytoplasmic inclusion bodies) 中,並高度提示病毒性病因,儘管特定病原體至今仍難以辨識,這或許是因為其與已知病毒科別缺乏同源性。³²⁻³⁵ KD 動脈炎的免疫轉錄譜 (immune transcriptional profile) 具有抗病毒免疫反應的特徵,例如活化的細胞毒性 T 淋巴細胞與第 I 型干擾素誘導 (Type I interferon-induced) 的基因上調³⁶;此資訊應有助於指引 KD 動脈炎新型免疫調節療法的發展。
危險因子 (RISK FACTORS)
亞洲兒童,特別是日本、中國與韓國兒童,有最高的 KD 發病率。¹⁰,¹⁴ 嬰兒與較大兒童在 KD 後有最高的冠狀動脈異常盛行率。¹¹⁻¹³ 日本已發展出風險評分系統 (risk-scoring systems),可用以判定哪些 KD 兒童發展出冠狀動脈疾病的風險最高,並允許對 KD 一線治療進行選擇性強化。³⁷ 然而,這些評分系統無法準確預測非日本、多族裔族群中的冠狀動脈疾病風險。³⁸
診斷 (DIAGNOSIS)
由於缺乏對 KD 病因的了解,並無診斷檢驗可用。急性 KD 的實驗室發現並無特異性,但相當具有特徵性。全血球計數 (complete blood count) 顯示正常或升高的白血球計數,並以嗜中性球為主。白血球計數低且以淋巴細胞為主在 KD 中極不尋常。可能出現正色素性、正細胞性貧血 (normochromic, normocytic anemia),並隨 KD 的緩解而自行消退。血小板計數在病程第一週為正常,儘管血小板減少 (thrombocytopenia) 已被報告與較嚴重的結果相關。血小板增多 (Thrombocytosis),血小板計數有時超過 1,000,000/mm³,是 KD 亞急性期的特徵,在發燒發作後第二至第三週達到高峰。此特徵與甲周脫屑一樣,在發燒第一週對 KD 的診斷並無幫助。有貧血與低白蛋白濃度的病人,發展出冠狀動脈疾病的風險可能較高。急性 KD 中常可觀察到肝臟轉胺酶 (liver transaminases) 的輕度升高。偶爾會發生阻塞性黃疸。膽囊水腫,伴隨右上腹疼痛,會自行緩解,無需手術介入。無菌性膿尿 (sterile pyuria) 也很常見。急性期反應物 (acute-phase reactants),如 C 反應蛋白(C-reactive protein, CRP)與 ESR,在急性 KD 病人中具特徵性地升高,而 CRP 有時用以追蹤對 IVIG 治療無反應病人的臨床反應。一旦給予 IVIG,ESR 便無法用以追蹤臨床反應,因為 IVIG 本身會短暫地升高 ESR。應於基準時進行全血球計數與 CRP 或 ESR,並於發病後 2 至 3 週與 6 至 8 週重複檢測 CRP,以監測發炎的緩解。
病理學 (PATHOLOGY)
KD 的皮膚與淋巴結切片顯示非特異性的發現,對建立診斷無用。
影像學 (IMAGING)
所有疑似 KD 的兒童都應接受心臟超音波檢查 (echocardiography),並應於診斷時、發燒發作後 2 至 3 週,以及發燒發作後 6 至 8 週進行。³⁹ 偵測冠狀動脈擴張的高峰時間為發燒發作後 2 至 3 週,即疾病的亞急性期。在這 3 次心臟超音波檢查中皆未顯現冠狀動脈異常的病人,可能不需要額外的檢查,儘管某些中心會在發病後 1 年再進行 1 次心臟超音波檢查。在發展出冠狀動脈擴張的病人中,於急性期與追蹤期將需要更頻繁的心臟超音波檢查;這些應與兒科心臟科醫師會診後安排。特別是,對於冠狀動脈管腔直徑持續擴大的病人,在最初幾週進行頻繁的心臟超音波檢查是明智的;這些病人發展出冠狀動脈血栓的風險特別高。疾病急性發熱期的心電圖 (Electrocardiogram) 最常顯示 PR 間期延長及/或非特異性的 ST 段與 T 波變化。CT 血管攝影 (CT angiography) 或磁振血管攝影 (magnetic resonance angiography) 在評估以下兩類病人的冠狀動脈時可能有用:難以藉由心臟超音波取得足夠冠狀動脈影像的青少年,以及罹患特別嚴重冠狀動脈疾病的兒童。⁴⁰,⁴¹
診斷演算法 (DIAGNOSTIC ALGORITHM)
典型 KD 的診斷見於以下情況:持久發燒持續 5 天或以上,並具有下列 5 項臨床特徵中的 4 項,且無其他解釋此病的因素:(a) 非化膿性球結膜充血 (nonpurulent bulbar conjunctival injection);(b) 發紅、腫脹、乾燥的嘴唇,可能龜裂並出血;(c) 手足的發紅與腫脹;(d) 皮疹;以及 (e) 直徑 1.5 cm 或更大的頸部淋巴結病變(表 142-2)。對於具有典型特徵的兒童,經驗豐富的醫師可在發燒第五天之前做出 KD 的診斷。具有持久發燒但符合少於 4 項其他疾病特徵的病人,若發展出冠狀動脈異常,則可診斷為 KD。不完全型(或非典型)KD 指的是持久發燒且符合少於 4 項其他疾病特徵、但具有與 KD 相符之實驗室檢查譜的兒童。此類病人應接受心臟超音波檢查並考慮以 IVIG 治療,因為 KD 病人(特別是嬰兒)在疾病急性發熱期並不總是表現出典型的診斷標準,卻仍可能發展出冠狀動脈異常。美國心臟協會的風濕熱、心內膜炎與川崎病委員會 (American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease) 已發表一套演算法,以協助臨床醫師診斷不完全型 KD。³⁹ 此演算法強調在做出診斷時結合臨床特徵、實驗室發現與心臟超音波發現的重要性(表 142-3)。在困難的病例中,應考慮諮詢 KD 專家。若病人未針對 KD 接受治療,但在發燒消退後發展出典型的甲周脫屑,且尚未確立可解釋此臨床發現的替代診斷(如猩紅熱 scarlet fever),則應重複進行心臟超音波檢查。6 個月或更年幼的嬰兒罹患 KD 時可能有輕微或不明顯的臨床發現,但有發展出冠狀動脈異常的高風險。因此,發燒 1 週或更久且無其他解釋的嬰兒,應接受實驗室檢測。若存在發炎過程的證據,即使在缺乏其他疾病臨床特徵的情況下,也應安排心臟超音波檢查並考慮 KD。

表 142-2:典型川崎病的診斷標準 (Diagnostic Criteria for Classic Kawasaki Disease)
發燒 ≥5 天ᵃ,高峰且間歇性,並具有下列 5 項臨床特徵ᵇ中至少 4 項:
- 雙側、非滲出性結膜充血。
- 口腔黏膜變化,包括發紅、乾燥、龜裂的嘴唇、咽部紅斑及/或草莓舌。
- 手足變化:急性期掌蹠紅斑及/或手足腫脹,及/或亞急性期手指與腳趾的甲周脫屑。
- 皮疹:紅斑性麻疹樣、猩紅熱樣或靶狀。
- 頸部淋巴結病變,直徑 ≥1.5 cm。
ᵃ若病人具有此病的其他臨床特徵,經驗豐富的醫師可在發燒第五天之前做出診斷。
ᵇ在無其他解釋此病之因素的情況下。
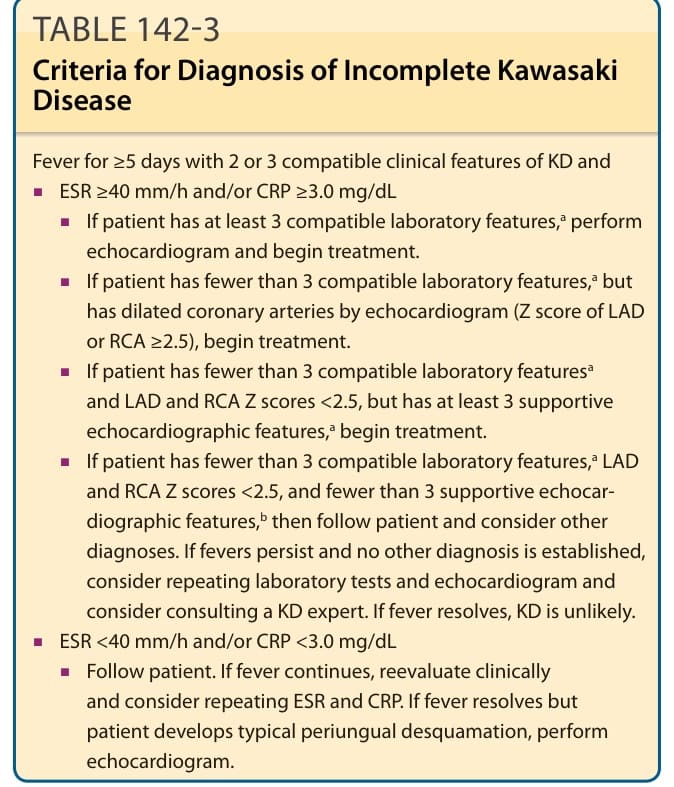
表 142-3:不完全型川崎病的診斷標準 (Criteria for Diagnosis of Incomplete Kawasaki Disease)
發燒 ≥5 天,並具有 2 或 3 項與 KD 相符的臨床特徵,且:
■ ESR ≥40 mm/h 及/或 CRP ≥3.0 mg/dL
■ 若病人具有至少 3 項相符的實驗室特徵ᵃ,進行心臟超音波檢查並開始治療。
■ 若病人具有少於 3 項相符的實驗室特徵ᵃ,但心臟超音波顯示冠狀動脈擴張(LAD 或 RCA 的 Z 分數 ≥2.5),開始治療。
■ 若病人具有少於 3 項相符的實驗室特徵ᵃ,且 LAD 與 RCA 的 Z 分數 <2.5,但具有至少 3 項支持性心臟超音波特徵ᵃ,開始治療。
■ 若病人具有少於 3 項相符的實驗室特徵ᵃ、LAD 與 RCA 的 Z 分數 <2.5,且少於 3 項支持性心臟超音波特徵ᵇ,則追蹤病人並考慮其他診斷。若發燒持續且未確立其他診斷,考慮重複實驗室檢測與心臟超音波,並考慮諮詢 KD 專家。若發燒消退,KD 的可能性不大。
■ ESR <40 mm/h 及/或 CRP <3.0 mg/dL
■ 追蹤病人。若發燒持續,臨床上重新評估並考慮重複 ESR 與 CRP。若發燒消退但病人發展出典型的甲周脫屑,進行心臟超音波檢查。
ᵃ相符的實驗室特徵:
- 白蛋白 ≤3.0 g/dL
- 與年齡相對的貧血
- 丙胺酸轉胺酶 (alanine aminotransferase) 升高
- 病程第 7 天後血小板計數 ≥450,000/mm³
- 白血球 (WBC) ≥15,000/mm³
- 尿液分析顯示每高倍視野 ≥10 個 WBC
ᵇ支持性心臟超音波特徵:
- 缺乏漸縮 (Lack of tapering)
- 左心室功能降低
- 二尖瓣逆流 (Mitral regurgitation)
- 心包積液
- LAD 或 RCA 的 Z 分數為 2.0 至 2.5
CRP, C 反應蛋白;ESR, 紅血球沉降率;LAD, 左前降支冠狀動脈 (left anterior descending coronary artery);RCA, 右冠狀動脈 (right coronary artery)。資料來源:Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110(17):2747-2771.
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
具有持久高燒、明顯結膜充血、發紅腫脹的手足、紅斑性皮疹,以及發紅、龜裂、出血嘴唇的 KD 兒童,若有明顯升高的 ESR 及/或 CRP,以及升高且以嗜中性球為主的周邊白血球計數,則為一種非常具特色的疾病,通常在診斷上不構成任何困難。在其他僅具有部分 KD 臨床特徵的病人,或臨床發現不那麼戲劇化的病人中,則必須仔細考慮其他診斷(表 142-4)。在麻疹 (measles) 仍盛行的地區,要區分這兩種疾病可能很困難。典型上,麻疹病人的皮疹始於耳後的臉部,並在口腔內有柯氏斑 (Koplik spots);這兩項特徵在 KD 中都不會觀察到。在麻疹感染的較晚期,皮疹變得瀰漫,柯氏斑也不再可見。結膜充血與手足水腫在這兩種疾病中都可觀察到。在單純的麻疹中,周邊白血球計數與 ESR 通常較低。麻疹 IgM 抗體在麻疹皮疹出現時幾乎總是陽性,是區分這兩種病況的最佳單一檢驗。對於具有猩紅熱樣皮疹的 KD 病人,鑑別診斷應考慮 A 群鏈球菌 (group A streptococcal) 感染,可藉由陰性的咽喉培養排除。在身為 A 群鏈球菌帶原者的 KD 病人中,可能產生診斷上的不確定性。給予抗生素治療後於 24 至 48 小時重新評估,通常可釐清診斷;A 群鏈球菌性咽炎的兒童對治療有迅速反應,而抗生素對 KD 則無效。腺病毒 (adenovirus) 感染可能類似 KD。滲出性結膜炎與滲出性咽炎的存在顯示腺病毒為最可能的診斷。其他病毒偶爾也可引起持久發燒,如腸病毒 (enterovirus);在區分 KD 病人與單純病毒感染病人時,ESR、CRP 與尿液分析等實驗室檢驗可能有幫助,因為膿尿與明顯升高的急性期反應物更典型於 KD。藥物過敏反應 (Drug hypersensitivity reactions) 可能類似 KD。史蒂芬斯—強生症候群 (Stevens–Johnson syndrome) 中所見的口腔與黏膜潰瘍在 KD 中不存在。臉部水腫,特別是眼睛周圍的水腫,較提示藥物反應而非 KD。一般而言,藥物過敏反應中的 ESR 與 CRP 為正常或僅輕度升高。葡萄球菌燙傷樣皮膚症候群很容易區分,其典型發現為疼痛性皮膚(這在 KD 中不會觀察到),以及鬆弛性水疱、脫屑與陽性的尼氏徵象 (Nikolsky sign),這些都存在於葡萄球菌燙傷樣皮膚症候群中,而在 KD 中不存在。雖然低血壓在 KD 中不尋常,但偶爾會發生 KD 休克症候群 (KD shock syndrome)⁴²,⁴³,而毒性休克症候群 (toxic shock syndrome) 在此類病人的鑑別診斷之列。腎臟侵犯與肌酸磷酸激酶 (creatinine phosphokinase) 升高較可能見於毒性休克症候群而非 KD。為可能的 KD 診斷而給予的 IVIG 可作為毒性休克症候群的輔助療法。幼年型類風濕性關節炎 (Juvenile rheumatoid arthritis) 是一種排除性診斷,偶爾有此疾病的病人最初可能被診斷並治療為不完全型 KD。當減少高劑量阿斯匹靈治療後發燒復發時,正確診斷可能變得明顯,這意味著原本的臨床反應是來自阿斯匹靈而非 IVIG。

表 142-4:川崎病的鑑別診斷 (Differential Diagnosis of Kawasaki Disease)
■ 麻疹 (Measles)
■ 腺病毒感染 (Adenovirus infection)
■ 腸病毒感染 (Enterovirus infection)
■ 猩紅熱 (Scarlet fever)
■ 葡萄球菌燙傷樣皮膚症候群 (Staphylococcal scalded skin syndrome)
■ 毒性休克症候群 (Toxic shock syndrome)
■ 藥物過敏反應 (Drug hypersensitivity reaction)
■ 幼年型類風濕性關節炎 (Juvenile rheumatoid arthritis)
若病人有流行病學危險因子:
■ 落磯山斑疹熱 (Rocky Mountain spotted fever)
■ 鉤端螺旋體病 (Leptospirosis)
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
約 85% 在病程最初 10 天內以 IVIG 與阿斯匹靈治療的 KD 兒童會有反應,發燒與其他臨床徵象迅速緩解。絕大多數及時診斷與治療的 KD 病人預後良好,不會發展出心臟併發症。然而,約 15% 在病程最初 10 天內接受治療的 KD 兒童,在單次 IVIG 合併阿斯匹靈輸注後仍持續發燒,需要額外治療;這些病人發展出冠狀動脈異常的風險較高。⁴⁴ 一項評估在病程最初 10 天內接受治療之 KD 病人的研究指出,18% 在發病後第 5 週時冠狀動脈 Z 分數高於 2。⁴⁵ 動脈瘤越大,其管腔直徑隨時間恢復正常的可能性就越小。非常大或「巨大 (giant)」的冠狀動脈瘤與最嚴重的結果相關。⁴⁶ 動脈瘤管腔直徑的減少可能源於血栓或管腔肌纖維母細胞增生,這可導致冠狀動脈狹窄與心肌梗塞。²⁸ 在年幼嬰兒與兒童中,心肌梗塞通常表現為休克、嘔吐及/或腹痛。⁴⁷ 具有顯著冠狀動脈疾病的病人可能需要導管介入處置⁴⁸、冠狀動脈繞道手術 (coronary artery bypass surgery)⁴⁹,或罕見地,需要心臟移植。⁵⁰ 應依冠狀動脈異常的嚴重度,間隔進行心血管風險評估。³⁹
治療 (MANAGEMENT)
介入處置 (INTERVENTIONS)
藥物 (MEDICATIONS)
應在診斷後盡快對急性 KD 兒童給予單次 IVIG 2 g/kg 合併阿斯匹靈(80-100 mg/kg/day,每 6 小時口服一次)輸注(表 142-5),如 Newburger 及其同事的一項研究所證明。⁵¹ 此治療方案在發燒最初 10 天內給予 KD 兒童時,已被證明可將冠狀動脈異常的盛行率從未治療病人的 25% 降至接受治療者的 5%。然而,此研究是在使用冠狀動脈 Z 分數來辨識冠狀動脈擴張之前進行的,而較近期的研究顯示 KD 病人整體冠狀動脈擴張的盛行率高於先前的認知。⁴⁵,⁵²
大多數兒童在治療後會經歷發燒與其他臨床徵象的迅速緩解,以及急性期反應物的改善。IVIG 在 KD 中的作用機轉不明。阿斯匹靈在急性 KD 期間以高劑量給予,以達抗發炎效果。其通常持續以 80 至 100 mg/kg/day 給予,直到病程第 14 天,或直到病人退燒至少 2 天。然後將阿斯匹靈減至 3 至 5 mg/kg/day,以單次每日劑量給予,以達其抗血栓 (antithrombotic) 效果。若所有心臟超音波檢查皆正常且急性期反應物已恢復正常,則於發病後 6 至 8 週停用阿斯匹靈。若發展出冠狀動脈異常,則持續使用低劑量阿斯匹靈。依冠狀動脈疾病的嚴重度,可能有指徵使用其他療法,如氯吡格雷 (clopidogrel) 與華法林 (warfarin)³⁹;此類決定應與兒科心臟科醫師會診後做出。對於在病程第 10 天之後才就診、且存在發燒及/或持續發炎之臨床與實驗室徵象的 KD 病人,也會給予 IVIG 與阿斯匹靈,儘管此臨床情況下該療法的療效並不確定。在日本,已發展出風險評分系統,能以良好的準確度預測哪些日本兒童有發展出冠狀動脈異常的最高風險,而「RAISE」研究顯示,以 IVIG 並合併在 2 至 3 週內漸減的潑尼松龍 (prednisolone) 療程作為一線治療,可改善高風險病人的結果。³⁷
不幸的是,風險評分系統至今在北美等多族裔族群中表現不佳,使得在非日本族群中選擇病人進行強化一線治療更具問題性。³⁸
約 15% 的急性 KD 病人對初始治療無反應;這些「難治性 (refractory)」KD 病人的最佳療法尚不清楚。這些病人中的大多數會對第二次 2 g/kg IVIG 輸注有反應。⁴⁴ 對於對初始治療無反應、且因冠狀動脈擴張的存在而已屬高風險類別的病人,應考慮給予第二劑 IVIG 並合併在 2 至 3 週內漸減方案的潑尼松龍。⁵³
IVIG 無反應者的其他選項包括每日一次、連續 3 天的高劑量靜脈注射甲基潑尼松龍 (methylprednisolone),或英夫利昔單抗 (infliximab)。⁵⁴,⁵⁵ 更具特異性的療法則有待 KD 病因的辨識。

表 142-5:急性川崎病的治療 (Treatment of Acute Kawasaki Disease)
■ 靜脈注射丙種球蛋白(intravenous gammaglobulin, IVIG)2 g/kg,於 10 至 12 小時內輸注;輸注期間仔細監測生命徵象,並合併阿斯匹靈 80 至 100 mg/kg/day,每 6 小時分次口服,直到病程第 14 天或直到病人退燒至少 2 天,然後將劑量減至 3 至 5 mg/kg/day,以單次每日劑量給予。
■ 若 2 至 3 週與 6 至 8 週的心臟超音波檢查正常,且急性期反應物已恢復正常,則停用阿斯匹靈。
諮詢衛教 (COUNSELING)
未發展出冠狀動脈異常的病人,目前並無已知會發展出任何長期後遺症。具有冠狀動脈異常但管腔直徑隨時間恢復正常的病人,應由兒科心臟科醫師追蹤;這些病人未來可能有發展出動脈狹窄的風險。具有顯著持續冠狀動脈異常的病人,應由兒科心臟科醫師追蹤,後者可依據美國心臟協會所建立的指引,就長期治療與活動限制提出建議。³⁹
治療演算法 (TREATMENT ALGORITHM)
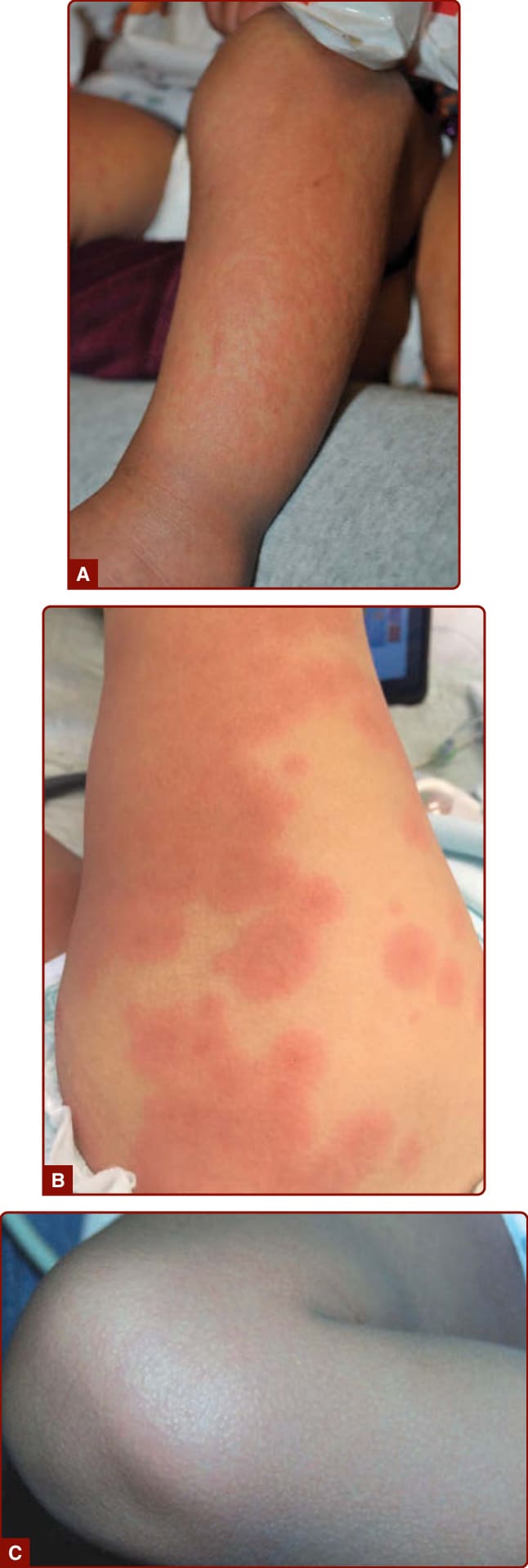
圖 142-1:急性川崎病所觀察到的皮疹 (exanthems)。A,紅斑性麻疹樣皮疹;B,靶狀或蕁麻疹樣變化;C,瀰漫性紅斑。
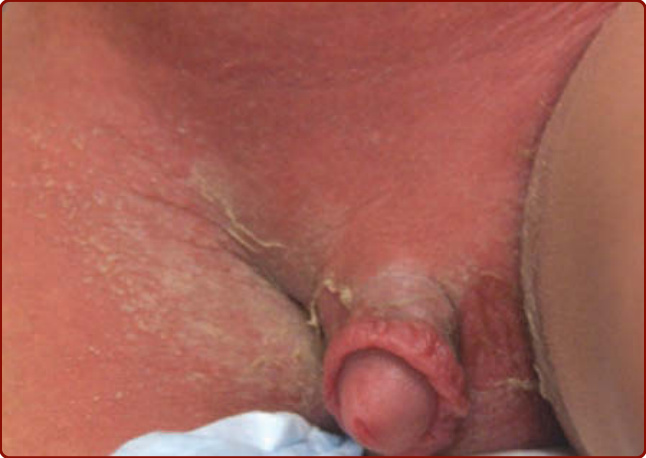
圖 142-2:急性川崎病的紅斑性脫屑鼠蹊部皮疹。
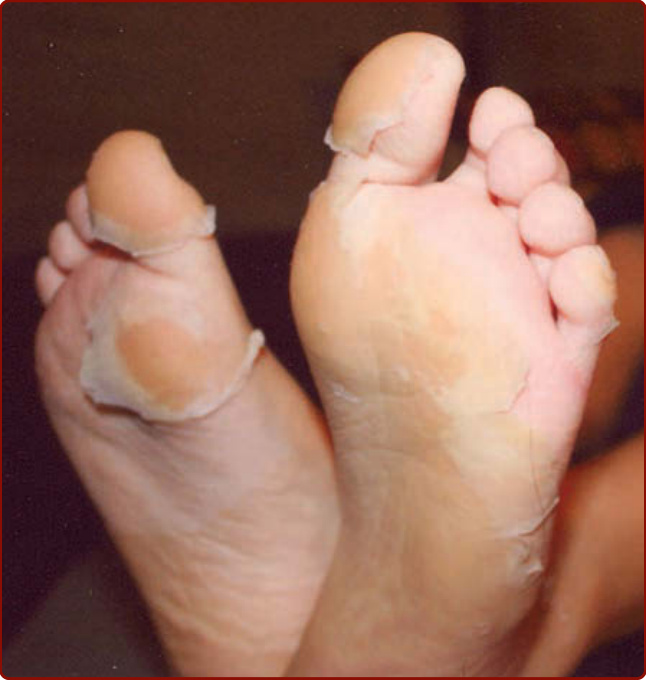
圖 142-3:川崎病亞急性期足部的脫屑;此過程始於甲周並進展至侵犯整個腳掌。
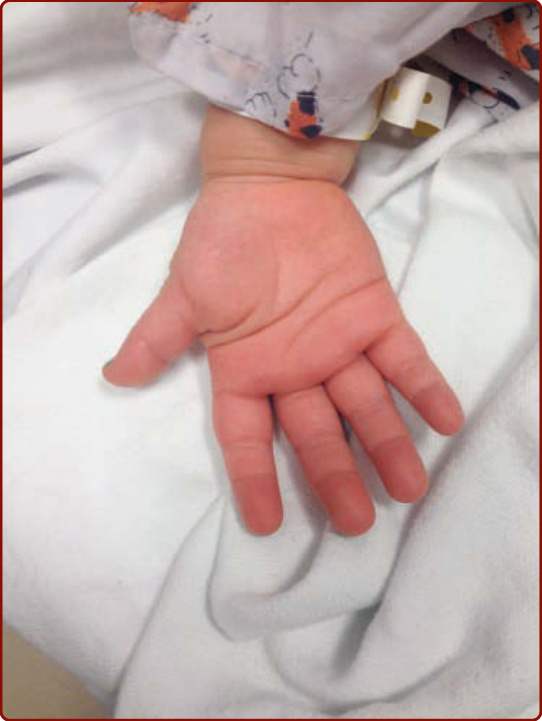
圖 142-4:川崎病中手部的紅斑與水腫。
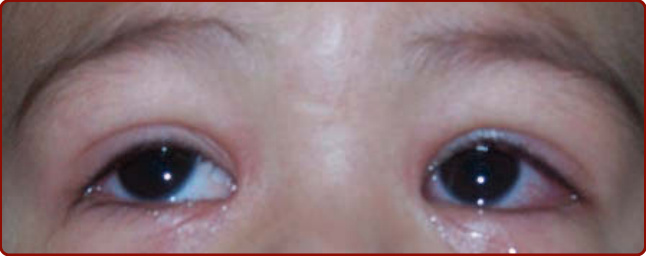
圖 142-5:急性川崎病中伴有角膜緣豁免的結膜充血。
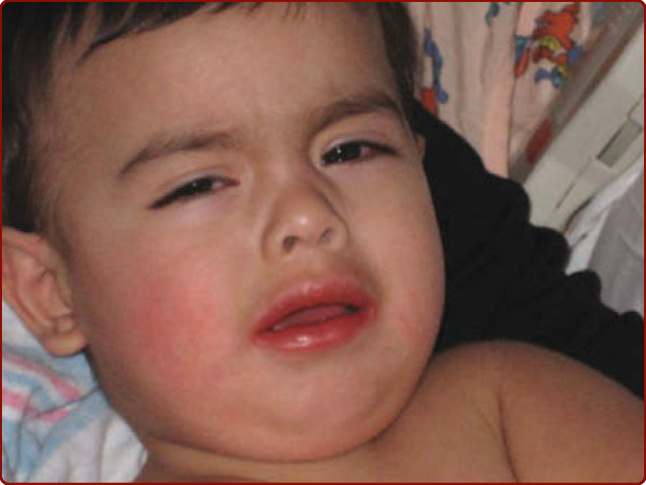
圖 142-6:急性川崎病的典型顏面特徵,顯示結膜充血與發紅乾燥的嘴唇。
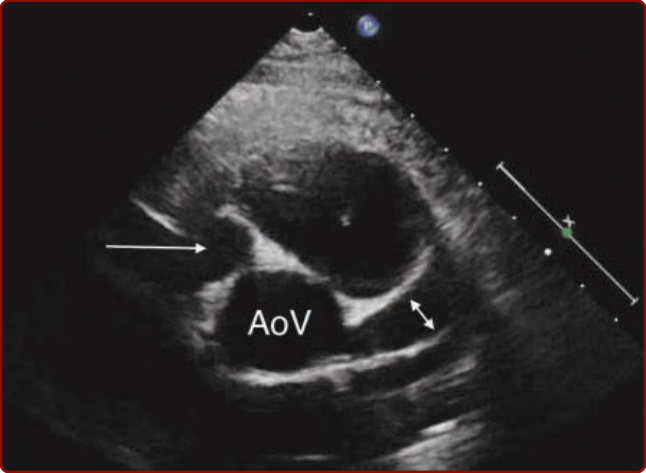
圖 142-7:藉由心臟超音波所示之川崎病冠狀動脈瘤。AoV,主動脈瓣 (aortic valve);雙箭頭,左前降支冠狀動脈近端的動脈瘤;單箭頭,右冠狀動脈近端的大型動脈瘤。