皮膚副腫瘤症候群 (Cutaneous Paraneoplastic Syndromes)
PART 21
副腫瘤症候群(paraneoplastic syndromes)係指潛在腫瘤性疾病所造成的遠端效應。這些臨床症候群可發生於多種器官系統,包括內分泌、神經肌肉、心血管、皮膚、血液、消化道 (GI) 與腎臟系統。其症狀並非轉移或腫瘤侵犯所致的直接效應,而可能源自腫瘤所產生的物質(例如荷爾蒙、胜肽或細胞激素),或源自惡性組織與正常組織之間的免疫或發炎反應。皮膚副腫瘤症候群(表 134-1)是多樣化的皮膚病學疾病實體,提示遠端惡性腫瘤的存在。在某些情況下,它們可能出現於癌症診斷之前,並協助發現潛伏 (occult) 的癌症;它們也可能提示先前已緩解的病人發生癌症復發。因此,及時辨識皮膚副腫瘤症候群,有助於在早期且高度可治療的階段偵測到潛在的惡性腫瘤。然而,副腫瘤表現可能在相關腫瘤診斷之前數月甚至數年即出現。若無法偵測到潛在癌症,仍應持續仔細追蹤數年。除了典型的副腫瘤皮膚病之外,本章亦討論腫瘤直接侵犯皮膚,以及具有惡性潛能的遺傳性皮膚病 (genodermatoses)(表 134-2)。雖然副腫瘤皮膚病在臨床上並不常見,但辨識這些與癌症相關的皮膚病灶非常重要。皮膚科醫師在較早期診斷潛伏惡性腫瘤方面,可能扮演重要角色。
過度角化性皮膚病 (Hyperkeratotic Dermatoses)
黑色棘皮症 (Acanthosis Nigricans)
第 137 章「糖尿病與其他內分泌疾病 (Diabetes and Other Endocrine Diseases)」提供關於良性黑色棘皮症的更多資訊。
重點一覽 (AT-A-GLANCE)
■ 黑色棘皮症 (acanthosis nigricans) 是一種皮膚標記,最常見為胰島素阻抗 (insulin resistance) 的表現,較少見為遺傳性疾病與惡性腫瘤的表現。
■ 特徵為對稱性、色素過度沉著、過度角化、疣狀的斑塊,質地呈天鵝絨樣,分布於間擦部位 (intertriginous) 皮膚,偶爾出現於黏膜皮膚區域。
■ 較深的膚色色素沉著、胰島素阻抗與肥胖較常與良性黑色棘皮症相關。
■ 惡性黑色棘皮症常於年長者身上迅速出現,並可侵犯非典型部位,如黏膜表面。
acanthosis nigricans(黑色棘皮症)一詞最初由 Unna 提出,雖然最早的病例為惡性腫瘤相關,並於 1890 年由 Pollitzer 與 Janovsky 各自獨立描述。Curth 在臨床上將黑色棘皮症分類為惡性、良性或症候群型黑色棘皮症,或假性黑色棘皮症(pseudoacanthosis nigricans,與肥胖相關)。今日黑色棘皮症分為兩大類:良性(家族性、肥胖相關、高胰島素血症狀態、自體免疫疾病相關)或惡性(惡性腫瘤相關)。惡性黑色棘皮症典型發生於年長病人,且常與其他副腫瘤皮膚病並存,如牛肚掌 (tripe palms) 與 Leser-Trélat 徵象 (sign of Leser-Trélat)。¹
流行病學 (EPIDEMIOLOGY)
大多數(80%)黑色棘皮症為特發性,或發生於良性情況,如內分泌病變、遺傳性疾病或藥物使用。惡性腫瘤相關的黑色棘皮症罕見,文獻中報導約有 1000 例。它通常發生於 40 歲以上個體,無性別、種族或遺傳偏好。¹
臨床特徵 (CLINICAL FEATURES)
皮膚病灶最初表現為對稱性色素過度沉著或皮膚呈髒污外觀,隨後增厚並出現皮膚紋理加深,形成色素過度沉著的天鵝絨樣斑塊。顏色可有變化,包括黃色、棕色、灰色與黑色。雖然黑色棘皮症的皮膚病灶幾乎可發生於身體任何部位,但傾向侵犯皮膚皺褶、屈側與間擦部位。最常受侵犯的位置為腋窩、頸部、外生殖器、腹股溝、顏面、大腿內側、肘前窩與膕窩、臍部與肛周區域。可能發生軟纖維瘤 (acrochordons),疊加於黑色棘皮症上或其他位置(圖 134-1)。亦有報導乳頭與乳暈的過度角化。黏膜侵犯傾向發生於惡性型,但也可見於無惡性腫瘤的黑色棘皮症,其特徵為黏膜的乳頭瘤狀增生與增厚,伴或不伴色素過度沉著,侵犯口腔(嘴唇、舌、頰黏膜與顎)(圖 134-2),罕見侵犯食道、眼、喉以及肛門與生殖器黏膜。典型上,黑色棘皮症病人無症狀,但部分病人可能有搔癢的困擾。口腔與食道乳頭瘤狀增生可造成口腔疼痛與吞嚥困難。¹
惡性與良性黑色棘皮症的病灶無法區別。然而,惡性腫瘤相關的黑色棘皮症通常突然且廣泛出現。黏膜侵犯與全身性搔癢較為常見。與旺盛皮膚乳頭瘤狀增生 (florid cutaneous papillomatosis) 及其他副腫瘤皮膚病灶(如牛肚掌(見「牛肚掌」一節)與 Leser-Trélat 徵象(見「Leser-Trélat 徵象」一節))並存,提示惡性腫瘤相關型黑色棘皮症。已報導多種惡性腫瘤與惡性黑色棘皮症相關。一項文獻回顧顯示,多達 90% 的病人有相關的腹腔內腺癌 (intraabdominal adenocarcinoma),其中約 60% 為胃癌。已有報導者包括子宮、肝、腸、卵巢、腎、乳房、肺、胰、膀胱、甲狀腺與膽囊的癌症,亦包括淋巴瘤與蕈狀肉芽腫 (mycosis fungoides)。¹
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
黑色棘皮症的發病機轉尚未充分了解。在良性黑色棘皮症中,有證據顯示胰島素透過第一型類胰島素生長因子受體 (insulin-like growth factor-1 receptor) 訊息傳遞路徑扮演重要角色(見第 137 章)。在與遺傳性症候群相關的黑色棘皮症中,第一型類胰島素生長因子受體與纖維母細胞生長因子受體 (fibroblast growth factor receptors) 可能參與其中。此外,遺傳學研究亦顯示纖維母細胞生長因子受體的活化突變。²
在惡性腫瘤相關的黑色棘皮症中,有人提出腫瘤產生轉化生長因子-α (transforming growth factor-α),其結構與表皮生長因子 (epidermal growth factor) 相似,會結合至表皮生長因子受體 (epidermal growth factor receptors) 並刺激角質形成細胞增生,進而導致黑色棘皮症的發展。此理論係基於觀察到皮膚病變在治療潛在惡性腫瘤後改善或消退。例如,在一位患有黑色棘皮症、軟纖維瘤、Leser-Trélat 徵象與黑色素瘤 (melanoma) 的病人身上,發現尿中轉化生長因子-α 升高,且病灶皮膚中表皮生長因子受體表現增加。在切除黑色素瘤後,此細胞激素及其受體的增強表現恢復正常,伴隨的皮膚病灶於術後改善。此外,部分作者認為纖維母細胞生長因子與第一型類胰島素生長因子亦可能參與惡性腫瘤相關黑色棘皮症的發病機轉。²,³
診斷 (DIAGNOSIS)
黑色棘皮症的診斷係基於臨床特徵與病灶分布。僅在有疑問的病例中需要組織病理學檢查以確認診斷。黑色棘皮症的組織病理學典型上顯示過度角化 (hyperkeratosis) 與表皮乳頭瘤狀增生 (epidermal papillomatosis)。僅有輕微的棘層肥厚 (acanthosis),且通常無色素過度沉著。部分病例可觀察到角質假性囊腫 (horn pseudocyst) 形成與黑色素色素增加。病灶的棕色係由過度角化而非黑色素所致。組織病理學發現無法與 Gougerot 與 Carteaud 的融合性網狀乳頭瘤狀增生 (confluent and reticulated papillomatosis of Gougerot and Carteaud)、脂漏性角化症 (seborrheic keratosis) 或表皮母斑 (epidermal nevus) 區別。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
惡性黑色棘皮症的臨床鑑別診斷包括在屈側區域或全身性分布呈現色素過度沉著與斑片或斑塊的情況。鑑別診斷包括乾癬樣皮膚炎 (psoriasiform dermatitis)、異位性皮膚炎 (atopic dermatitis)、糙皮病 (pellagra)、消退中的增殖性天疱瘡 (resolving pemphigus vegetans)、類乾癬 (parapsoriasis)、皮膚 T 細胞淋巴瘤 (cutaneous T-cell lymphoma)、感染(例如念珠菌病 candidiasis、紅癬 erythrasma、體癬/股癬 tinea corporis/cruris)、色素疾病(例如血色素沉著症 hemochromatosis、Addison 病、Dowling-Degos 病)以及其他(例如 Gougerot 與 Carteaud 的融合性網狀乳頭瘤狀增生、Becker 黑色素沉著 Becker melanosis、表皮母斑與堅土樣皮膚病 terra firma-forme dermatosis)。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
惡性黑色棘皮症可發生於偵測到癌症之前、期間或之後。它傾向與潛在惡性腫瘤的病程平行。皮膚病灶可能在治療潛在癌症後消退,並在惡性腫瘤復發或轉移後再次出現。不幸的是,大多數相關惡性腫瘤於進展期才表現出來,因此預後通常不佳。²,⁴
處置 (MANAGEMENT)
在可疑病例中,建議進行評估與檢查,以辨識潛在的內分泌病變或惡性腫瘤。年長病人若出現非預期的體重減輕、迅速且廣泛的黑色棘皮症併黏膜侵犯,且合併有副腫瘤皮膚病灶,應接受潛在惡性腫瘤的檢查,尤其是消化道。外觀美容問題常為病人的主要顧慮。處置任何並存的疾病或惡性腫瘤常可改善,甚至可能使黑色棘皮症消退。局部角質溶解劑 (topical keratolytics)(包括類視色素 retinoids)與口服類視色素可減少黑色棘皮症的外觀。其他報導可顯示改善的口服藥物包括膳食魚油 (dietary fish oil)、metformin 與 cyproheptadine,以 cyproheptadine 而言可能係透過抑制腫瘤分泌的生長因子。其他在病例報告中發現有益的療法包括 calcipotriol、三氯醋酸換膚 (trichloroacetic acid peeling) 與長脈衝亞歷山大雷射 (long-pulsed alexandrite laser)。³
後天性魚鱗癬 (Acquired Ichthyosis)
第 47 章「魚鱗癬 (The Ichthyoses)」提供關於後天性魚鱗癬的更多資訊。
後天性魚鱗癬 (acquired ichthyosis) 發生於成年期,可能與惡性腫瘤、內分泌與代謝疾病、HIV 與其他感染、自體免疫疾病、營養缺乏及藥物反應相關。臨床表現包括軀幹與四肢伸側 (extensor) 瀰漫、對稱、板狀脫屑,通常不侵犯屈側 (flexures)、手掌與足底。與後天性魚鱗癬相關的惡性腫瘤包括 CD30+ 淋巴增生性疾病、蕈狀肉芽腫、平滑肌肉瘤 (leiomyosarcoma)、Kaposi 肉瘤、多發性骨髓瘤 (multiple myeloma),以及卵巢、乳房、肺與子宮頸的癌症。何杰金病 (Hodgkin disease) 是最常見的腫瘤;魚鱗癬通常與淋巴瘤診斷同時或之後發生。由於成人突然發生魚鱗癬可能為潛在惡性腫瘤的表現徵象,仔細的惡性腫瘤篩檢(尤其是淋巴瘤)非常重要。在淋巴瘤中,後天性魚鱗癬可為疾病的皮膚徵象或副腫瘤現象。然而,在某些病例中這可能很難辨識,例如蕈狀肉芽腫,其魚鱗癬樣蕈狀肉芽腫 (ichthyosiform mycosis fungoides) 與蕈狀肉芽腫相關的後天性魚鱗癬在臨床與組織病理學發現上難以區別。⁵
圓形糠疹 (Pityriasis Rotunda)
圓形糠疹 (pityriasis rotunda) 是一種罕見的皮膚疾病,有些人認為它是後天性魚鱗癬的一種變異型。它曾被觀察到與慢性疾病、感染及惡性腫瘤相關。在各類型相關惡性腫瘤中,消化道(最常見為肝細胞癌 hepatocellular carcinoma)與血液惡性腫瘤經常被報導。然而,圓形糠疹也可發生於健康人。皮膚表現具特徵性,由完美的圓形或橢圓形、無症狀、邊界清楚、色素減退或色素過度沉著的魚鱗癬樣脫屑斑片組成,出現於軀幹與四肢近端。⁵ 第一型圓形糠疹與潛在的惡性或全身性疾病相關,呈現少於 30 個皮膚病灶。它較常見於黑人與東亞病人。第二型或家族性圓形糠疹發生於白人病人,呈現超過 30 個病灶(通常為色素減退),且不與任何潛在疾病相關。⁵ 治療潛在疾病可能使第一型病人的皮膚病灶消退。在第二型病人中,可能於成年期出現自發性改善。局部治療(如類視色素、水楊酸 salicyclic acid 與乳酸 lactic acid)對於此病相關的乾燥與脫屑是有用的治療。⁵
牛肚掌 (Tripe Palms)
tripe(牛肚)一詞係指牛前胃可食用內襯的皺褶表面。它於 1963 年首次應用於手掌與手指呈脊狀紋路型態的角皮症 (keratoderma) 的臨床診斷,當時一位病人向其醫師報告其雙手看起來類似牛肚。牛肚掌於 1977 年由 Clarke 首次在文獻中描述。他報導一位患有肺鱗狀細胞癌 (squamous cell carcinoma of the lung) 的病人,伴有惡性黑色棘皮症與牛肚型態的手掌增厚。自此牛肚掌被視為內臟惡性腫瘤的皮膚徵象。⁶
流行病學 (EPIDEMIOLOGY)
牛肚掌是一種罕見的副腫瘤皮膚病,文獻中報導約有 100 例。其與惡性腫瘤的關聯性很高,發生率超過 90%。它幾乎僅見於成人,且男性多於女性。⁶
臨床特徵 (CLINICAL FEATURES)
手掌粗糙、增厚、呈天鵝絨狀,並有誇張的皮紋 (dermatoglyphics)。手與手指掌面的質地可能呈苔蘚狀、鵝卵石狀或蜂巢狀(圖 134-3)。牛肚掌通常與惡性黑色棘皮症並存,約見於 70% 的病人。數位作者認為牛肚掌可能是掌部黑色棘皮症的一種型態。亦可見與其他副腫瘤皮膚病灶並存,如旺盛皮膚乳頭瘤狀增生、搔癢、手指杵狀變 (clubbing) 與 Leser-Trélat 徵象。⁶
肺癌與胃癌占與牛肚掌相關腫瘤的 50% 以上。在僅有牛肚掌的病人中,肺癌是最常相關的惡性腫瘤,尤其是鱗狀細胞型。在同時有牛肚掌與黑色棘皮症的病人中,胃癌是最常見的癌症,其次為肺癌。其他較少與牛肚掌相關的惡性腫瘤包括泌尿生殖道、乳房、頭頸部的癌症。⁶,⁷
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
具體的發展機制尚未完全闡明。與黑色棘皮症類似(見「黑色棘皮症」一節),有限的證據顯示腫瘤細胞釋放的轉化生長因子-α 在牛肚掌的發病機轉中誘發細胞增生。一般認為牛肚掌可能是惡性黑色棘皮症的一種變異型。⁷
診斷 (DIAGNOSIS)
牛肚掌的組織病理學包括過度角化、棘層肥厚與乳頭瘤狀增生。這些特徵與黑色棘皮症及脂漏性角化症所觀察到的病理發現極為相似。約 20% 的標本可有額外發現,包括真皮黏蛋白 (dermal mucin) 與肥大細胞 (mast cells)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
牛肚掌的臨床鑑別診斷包括厚皮骨膜病 (pachydermoperiostosis)、肥大性肺性骨關節病變 (hypertrophic pulmonary osteoarthropathy)、肢端肥大症 (acromegaly)、甲狀腺肢端肥厚 (thyroid acropachy)、掌蹠角皮症 (palmoplantar keratoderma) 與黑色棘皮症。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
副腫瘤性牛肚掌可發生於惡性腫瘤診斷之前 (48%)、同時 (21%) 或之後 (31%)。在三分之一的病人中,牛肚掌與相關惡性腫瘤的病程平行。在已知惡性腫瘤病例中出現牛肚掌,可能是腫瘤復發或轉移的徵象。⁶
處置 (MANAGEMENT)
由於相關惡性腫瘤的比例很高,所有牛肚掌病人都應進行完整的惡性腫瘤檢查,尤其要排除肺癌與胃癌。最低限度的檢查應包括病史、完整的理學檢查、常規實驗室檢查、胸部 X 光攝影,以及上消化道的放射線或內視鏡評估。其他檢查應依初步檢查的發現而定。牛肚掌的治療困難,主要針對潛在惡性腫瘤。牛肚掌並無特定療法。與黑色棘皮症類似,有零星報告指出單用口服類視色素,或合併 metformin,可獲改善。約 30% 的牛肚掌病例會在治療潛在腫瘤後消退。然而,也有許多病例在癌症緩解後並無效果。
Leser-Trélat 徵象 (Leser-Trélat Sign)
重點一覽 (AT-A-GLANCE)
■ 脂漏性角化症 (seborrheic keratoses) 的數量與大小迅速、爆發性增加。
■ 搔癢常見。
■ 常與惡性黑色棘皮症一起發生。
■ 消化道腺癌是最常見的相關癌症,其次為淋巴增生性惡性腫瘤。
■ 罕見於良性情況,如懷孕、HIV、心臟移植、肢端肥大症、紅皮症 (erythroderma) 與藥物反應。
Leser-Trélat 徵象的特徵為與內臟惡性腫瘤相關的脂漏性角化症 (SKs) 大小與數量突然增加。此現象歸功於 Edmund Leser 與 Ulysse Trélat 兩位歐洲外科醫師,他們於 1890 年觀察到癌症病人身上出現櫻桃血管瘤 (cherry hemangiomas)(但非 SKs)。1900 年,Hollander 是第一位將大量 SKs 的出現與內臟惡性腫瘤相關聯的人。⁷,⁸
流行病學 (EPIDEMIOLOGY)
Leser-Trélat 徵象是一種罕見的副腫瘤皮膚病,男女發生頻率相當,不同種族間亦相當。與惡性腫瘤的發生類似,此徵象較常見於年長個體。此疾病實體仍具爭議,因為 SKs 與癌症的盛行率在年長人口中皆增加。此外,爆發性 SKs 的辨識常基於病人的自我報告,具主觀性。在一項針對 1752 例連續 SKs 病例的大型族群研究中,與一般人口相比並無內臟惡性腫瘤發生率增加的統計證據。對呈現爆發性 SKs 者的亞群分析,亦未能證明內臟惡性腫瘤風險增加。在其他針對患有 SKs 且近期有實體腫瘤病人的大型研究中,與年齡及性別配對的對照組相比,在 SKs 的臨床特徵或數量上均未顯示差異。⁹
因此,大型流行病學研究尚未提供足夠證據,以明確將此徵象界定為真正的副腫瘤皮膚病。
雖然大型研究未顯示統計差異,但有零星證據顯示此徵象可能代表內臟惡性腫瘤。首先,有報導 20 多歲且患有內臟惡性腫瘤的病人出現爆發性 SKs。臨床上,Leser-Trélat 徵象常與惡性黑色棘皮症(一種較確立的副腫瘤現象)並存。此外,生長因子表現的改變與對照病人不同。綜合而言,這顯示 Leser-Trélat 徵象是一種正當但極不常見的副腫瘤皮膚病。⁷,⁸
臨床特徵 (CLINICAL FEATURES)
提示副腫瘤現象的臨床特徵包括大量、廣泛、爆發性的 SKs,主要分布於軀幹與四肢。然而,就爆發期長度,以及診斷所需的 SKs 數量與大小而言,爆發性 SKs 的量化仍付之闕如。Leser-Trélat 徵象中所見的個別 SKs 在臨床與組織學上皆與正常 SKs 相似。搔癢是一項顯著特徵,發生於約半數病人。與其他過度角化性副腫瘤皮膚病並存很常見,約三分之一的病人有黑色棘皮症。亦有一些病例報告 Leser-Trélat 徵象與牛肚掌並存。⁸ 搔癢性、爆發性 SKs 的迅速出現,尤其在黑色棘皮症的情境下,應提醒臨床醫師潛在內臟惡性腫瘤的可能性(圖 134-4)。腺癌占 Leser-Trélat 徵象所描述惡性腫瘤的大多數,其中 32% 的 Leser-Trélat 徵象與消化道惡性腫瘤相關,尤其是胃癌。淋巴增生性疾病,包括 Sézary 症候群、蕈狀肉芽腫、其他淋巴瘤與白血病,是第二常見的相關惡性腫瘤,發生於約 20% 的病人。其他腫瘤,如肺、膀胱、腎與卵巢癌,以及黑色素瘤,亦曾被報導。⁷
類 Leser-Trélat 爆發或爆發性 SKs 罕有報導於良性情況,如懷孕、HIV、心臟移植、肢端肥大症與紅皮症,以及與藥物相關,如 cytarabine 與腫瘤壞死因子-α 抑制劑 (tumor necrosis factor-α inhibitors)。⁷,¹⁰ 假性 Leser-Trélat 徵象 (pseudosign of Leser-Trélat) 一詞曾被用來指稱非惡性腫瘤相關的爆發性 SKs。不幸的是,這些例子無助於進一步界定此一已然令人困惑且具爭議的臨床實體。
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
確切的發病機轉尚待闡明。然而,與黑色棘皮症及牛肚掌類似,Leser-Trélat 徵象亦有證據顯示生長因子恆定性的改變具貢獻。在數個案例中,曾觀察到生長因子表現增加的狀態,在患有潛在惡性腫瘤與爆發性 SKs 的病人中偵測到尿中表皮生長因子與轉化生長因子-α 升高。隨後,生長因子濃度在原發腫瘤切除後下降。⁷
除了生長因子表現增加之外,副腫瘤性黑色棘皮症與 SKs 的病灶皮膚在細胞外基質 (extracellular matrix) 上有所改變。生長因子訊息傳遞對皮膚的直接影響尚不清楚。提出的想法包括主要誘發過度增生狀態,或藉由改變周圍環境(如透過細胞外基質)而達成。這些相似的機制進一步支持黑色棘皮症與 Leser-Trélat 徵象之間存在連續譜 (continuum) 的想法。
診斷 (DIAGNOSIS)
可進行皮膚切片以確認診斷。此症候群中 SK 的組織病理學發現與一般 SK 相同,包括表皮過度角化、乳頭瘤狀增生與棘層肥厚,並有角質物質的囊性內含物(角質假性囊腫 horn pseudocysts)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
此症候群中個別皮膚病灶的鑑別診斷與 SKs 相同,包括纖維上皮性息肉 (fibroepithelial polyp)、表皮母斑、黑色素細胞母斑 (melanocytic nevus)、色素性基底細胞癌 (pigmented basal cell carcinoma)、鱗狀細胞癌、惡性黑色素瘤 (malignant melanoma)、疣狀角化異常瘤 (warty dyskeratoma)、尋常疣 (verruca vulgaris) 與尖形濕疣 (condyloma acuminatum)。在廣泛且全身性的 SKs(可出現於年長健康個體)中,重要的是區分良性表現與 Leser-Trélat 徵象。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
Leser-Trélat 徵象可於惡性腫瘤診斷之前約 5 個月至之後 10 個月發生。超過半數病人於診斷時已有轉移性腫瘤。預後不佳,診斷後估計存活時間為 1 年。此爆發在某些病例中與潛在惡性腫瘤的病程平行,但並非通則。
處置 (MANAGEMENT)
治療應針對潛在腫瘤。若病灶有症狀或病人有美容上的顧慮,可進行 SK 的局部治療,如 α-羥基酸 (α-hydroxy acids)、類視色素、三氯醋酸、合併或不合併刮除術的冷凍手術 (cryosurgery)、皮膚磨削術 (dermabrasion)、雷射與削除術 (shave removal)。
Bazex 副腫瘤性肢端角化症 (Acrokeratosis Paraneoplastica of Bazex)
重點一覽 (AT-A-GLANCE)
■ 特徵性皮膚病灶與分布,對稱侵犯耳輪 (helices)、鼻、頰、手指與指甲。
■ 病灶由非特異性皮膚炎與甲溝炎 (paronychia) 演變為發炎性斑塊、肢端角皮症 (acral keratoderma) 與甲板變化。
■ 皮膚病灶常早於內臟惡性腫瘤的偵測。
■ 上呼吸消化道 (upper aerodigestive tract) 惡性腫瘤最為常見。
Bazex 副腫瘤性肢端角化症於 1965 年首次在法國皮膚科學會發表。Bazex 與其同事描述一位四肢有鱗屑性紅斑病灶的病人,患有梨狀窩 (pyriform fossa) 的癌症,其皮膚病灶在治療癌症後清除。這些病灶被提出為內臟惡性腫瘤的皮膚標記。¹¹
此病亦稱 Bazex 症候群 (Bazex syndrome),此一名稱被用來描述兩種不同的臨床實體。第一種指本章所討論的 Bazex 副腫瘤性肢端角化症,第二種為 Bazex-Dupré-Christol 症候群,是一種非常罕見的遺傳性皮膚病,特徵為先天性毛髮稀少 (congenital hypotrichosis)、毛囊性萎縮性皮膚 (follicular atrophoderma),以及早年即出現的基底細胞癌 (basal cell carcinomas)。
流行病學 (EPIDEMIOLOGY)
醫學文獻中已報導超過 100 例 Bazex 症候群。此病與潛在惡性腫瘤高度相關。它幾乎僅發生於 40 歲以上的男性。約 60% 的相關惡性腫瘤為上呼吸消化道的鱗狀細胞癌。第二常見的是肺癌。其他較不常見的腫瘤位置為泌尿生殖道與下消化道。¹²
臨床特徵 (CLINICAL FEATURES)
特徵性的皮膚發現為對稱性的紅斑至紫紅色鱗屑性斑片或斑塊,分布於肢端四肢、耳朵與鼻樑。色素過度沉著傾向出現於膚色較深的個體。手與足偶可觀察到小水疱 (vesicles) 與大疱 (bullae)。曾有描述遠端指骨的球狀膨大。最常受侵犯的部位為指甲 (77%)、耳朵 (76%)、手指 (65%)、鼻 (62%)、手掌 (56%)/手 (51%),以及足底 (49%)/足 (44%)。數位病人有額外的皮膚副腫瘤症候群,包括後天性魚鱗癬、Leser-Trélat 徵象與杵狀變。¹² Bazex 與 Griffiths 描述了與潛在腫瘤生長及散播平行的三個皮膚病灶階段。在第一階段,腫瘤常未被偵測到。耳輪、鼻、手指與足趾通常以對稱方式受侵犯。早期病灶為邊界不清的鱗屑性丘疹,類似非特異性皮膚炎。此爆發典型上無症狀,但搔癢可能是困擾。甲溝炎是甲侵犯的第一個徵象(圖 134-5),可能有壓痛。曾有報導甲營養不良 (nail dystrophy)、甲下過度角化 (subungual hyperkeratosis) 與甲剝離 (onycholysis),進展至甲板完全破壞。在第二階段,腫瘤因局部擴展或轉移性散播而出現症狀,皮膚爆發變得更廣泛。典型的紅至紫色鱗屑性斑塊可能侵犯整個耳廓、頰部或上唇。手掌與足底發展出角皮症,常不侵犯中央掌面,但可能導致疼痛性皸裂(圖 134-6)。甲板變化包括變黃、增厚、甲剝離,以及水平與垂直的脊狀紋路。最終階段見於腫瘤未治療或治療無反應時。上述所有徵象與症狀持續存在,丘疹鱗屑性病灶開始出現於其他部位,如軀幹、肘、膝、頭皮,以及手足背側。罕見有小水疱與大疱,最常見於手指、手與足。甲變化可相當多變,從典型的增厚到甲萎縮與甲板脫落。
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
副腫瘤性肢端角化症的病理生理學仍不清楚。許多作者提出免疫學機制,即針對腫瘤的抗體與角質形成細胞及基底膜抗原 (basement membrane antigen) 交叉反應,或對表皮中類腫瘤抗原的 T 細胞媒介免疫反應。在某些病人中曾觀察到免疫球蛋白 (IgG、IgA、IgM) 與補體 (C3) 沿基底膜帶 (basement membrane zone) 沉積的發現。部分作者亦認為腫瘤分泌的表皮生長因子可能在表皮增生與過度角化中扮演角色,如本章其他過度角化性皮膚病所述。其他假說包括血清維生素 A 與鋅 (zinc) 濃度偏低。¹²
診斷 (DIAGNOSIS)
組織病理學略呈非特異性。乾癬樣皮膚炎是最常見的型態。過度角化、角化不全 (parakeratosis) 與表淺淋巴組織球性浸潤 (superficial lymphohistiocytic infiltrate) 是主要的組織病理學特徵。介面變化,包括空泡變性 (vacuolar degeneration)、角化異常角質形成細胞 (dyskeratotic keratinocytes)、苔蘚樣浸潤 (lichenoid infiltrate) 與真皮中含黑色素的巨噬細胞,報導較不常見。¹²
在組織病理學與特徵性皮膚發現的背景下,Bazex 副腫瘤性肢端角化症可能是一個可考慮的診斷。若病人無惡性腫瘤病史,應迅速進行適當的檢查。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括乾癬 (psoriasis)、毛髮紅糠疹 (pityriasis rubra pilaris)、脂漏性或接觸性皮膚炎、濕疹性藥物疹 (eczematous drug eruption)、感染(例如皮癬菌病 dermatophytosis、甲癬 onychomycosis)、紅斑性狼瘡 (lupus erythematosus)、掌蹠角皮症與蕈狀肉芽腫。在副腫瘤性肢端角化症中幾乎總是存在的特徵性臨床表現是耳輪(圖 134-7)與鼻尖的侵犯。典型上,Bazex 症候群對傳統治療具抗性。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
皮膚爆發可發生於腫瘤診斷之前 (67%)、同時 (18%) 或之後 (15%)。皮膚病灶平均較腫瘤診斷提早 12 個月出現。副腫瘤性肢端角化症預期在成功治療潛在惡性腫瘤後消退,並可隨腫瘤復發而再次出現。然而,儘管潛在惡性腫瘤與其他皮膚病灶顯著改善,甲營養不良與色素性皮膚變化仍可能持續存在。¹²
處置 (MANAGEMENT)
治療原發腫瘤是最有效的療法。皮膚病灶通常對過度角化性疾病的標準療法具抗性,且針對副腫瘤性肢端角化症皮膚成分的特定治療鮮有報導。全身性類視色素 (systemic retinoids) 曾被使用,成效不一。零星報告顯示口服補骨脂素與紫外線 A 光療 (oral psoralen and ultraviolet A phototherapy)、局部水楊酸與皮質類固醇有益。
膠原-血管疾病 (Collagen–Vascular Disease)
皮肌炎 (Dermatomyositis)
第 62 章「皮肌炎 (Dermatomyositis)」提供關於皮肌炎的更多資訊。成人發病型皮肌炎 (adult-onset dermatomyositis, DM) 顯著與惡性腫瘤相關。DM 的癌症風險高於單純的多發性肌炎 (polymyositis)。約 18% 至 25% 的成人發病型 DM 病人可能有惡性腫瘤,並與存活率下降相關。惡性腫瘤可發生於 DM 診斷之前、同時或之後。DM 的病程並不總是與惡性腫瘤的病程一致。相對地,青少年發病型 DM 並無強烈增加的惡性腫瘤風險。有或無相關惡性腫瘤的 DM 臨床表現相似。先前研究報導了伴隨惡性腫瘤的預測徵象,如皮膚壞死 (cutaneous necrosis)、皮膚血管炎 (cutaneous vasculitis)、缺乏間質性肺病 (interstitial lung disease)、吞嚥困難、男性、紅血球沉降率 (erythrocyte sedimentation rate) 升高與發病年齡較大。然而,其中部分發現不一致且仍具爭議。近期,抗 p155 抗體 (anti-p155 antibodies) 被報導為成人 DM 病人有前景的癌症標記。相關惡性腫瘤的類型在各研究間有所不同,可能反映不同族群間惡性腫瘤風險的差異。肺、乳房、卵巢、子宮頸、胰、胃、結直腸與攝護腺惡性腫瘤經常被報導;血液惡性腫瘤亦可能與 DM 相關。已注意到鼻咽癌 (nasopharyngeal carcinoma) 在亞洲族群中較常見。¹³
在成人 DM 病人中,惡性腫瘤風險在診斷後第一年最高,隨後穩定下降,但即使在 5 年後仍略為升高。因此,所有成人 DM 病人都應在診斷時接受惡性腫瘤評估,隨後進行長期監測。DM 病人的癌症篩檢並無明確指引。多數作者建議初步篩檢應包括完整的病史與理學檢查(包括女性的骨盆檢查)、標準實驗室檢查(全血球計數、完整代謝功能組合、尿液分析、糞便潛血檢查)與胸部 X 光攝影。進一步檢查依任何異常發現而定。部分臨床醫師亦建議大腸鏡檢查與胸、腹、骨盆的電腦斷層掃描。應考慮額外的依年齡、性別與族群適當的惡性腫瘤篩檢,包括女性病人的經陰道骨盆超音波 (transvaginal pelvic sonography)、子宮頸抹片 (Papanicolaou smear) 與乳房 X 光攝影 (mammography),或對亞裔病人由耳鼻喉科醫師評估以排除鼻咽癌。¹³
進行性全身性硬化症 (Progressive Systemic Sclerosis)
第 63 章「全身性硬化症 (Systemic Sclerosis)」提供關於進行性全身性硬化症的更多資訊。大多數流行病學研究顯示,與一般人口相比,進行性全身性硬化症 (progressive systemic sclerosis, PSS) 病人的癌症風險增加,並證明男性風險高於女性。惡性腫瘤的頻率約為 3% 至 11%。¹⁴ 癌症風險在 PSS 診斷後的前 12 個月內較高,可能為副腫瘤現象。然而,納入較新統合分析的其他研究並未顯示癌症風險增加。¹⁵
肺、膀胱、乳房、肝、食道與口咽癌、非黑色素瘤皮膚癌 (nonmelanoma skin cancer) 與血液惡性腫瘤經常被報導與 PSS 相關。肺癌在大多數研究中是最常見的癌症類型,可能與間質性肺病的存在以及吸菸史相關。有趣的是,在一項美國研究中,舌癌的發生率增加了 25 倍。¹⁴,¹⁶
此癌症風險增加可能源自慢性發炎與纖維化造成的損傷、PSS 的免疫抑制治療、環境暴露,以及/或對發展癌症與自體免疫的遺傳易感性。雖然資料具爭議,但部分研究顯示瀰漫型皮膚疾病 (diffuse cutaneous disease) 病人的癌症風險可能較大。數項較新研究確認,具有 RNA 聚合酶 III 自體抗體 (RNA polymerase III autoantibodies) 或 PSS 發病年齡較大的病人,PSS 相關癌症風險增加。這些族群的病人可能受益於診斷時的積極惡性腫瘤篩檢與長期癌症監測。¹⁶
反應性紅斑 (Reactive Erythemas)
匍行性迴狀紅斑 (Erythema Gyratum Repens)
第 46 章「離心性環狀紅斑與其他圖形紅斑 (Erythema Annulare Centrifugum and Other Figurate Erythemas)」提供關於匍行性迴狀紅斑的更多資訊。此特殊的環狀(gyratum 為拉丁文「圓圈」之意)紅斑曾被認為是與潛在腫瘤相關最具特異性的皮膚病之一。然而,較新的資料顯示相關良性情況的比例高於先前報導。匍行性迴狀紅斑 (erythema gyratum repens) 是一種罕見疾病,呈現戲劇性的外觀與演變。文獻中報導近 100 例。它主要影響 60 多歲的成人,男性多於女性(比例 2:1)。眾多蛇行帶狀 (serpiginous bands) 以平行排列方式構成同心圓的紅色漩渦,遍布身體大部分。此表現偶被稱為「木紋 (wood-grain)」外觀(圖 134-8)。更引人注目的是病灶相對迅速的遷移速率(repens 為拉丁文「爬行」之意),估計每天 1 cm。沿紅斑的後緣可能有細微脫屑(圖 134-9)。手、足與顏面常不受侵犯,偶有掌面過度角化例外。許多病例存在魚鱗癬。搔癢為普遍現象,可能很嚴重。¹⁷
潛在惡性腫瘤約於 70% 的時候與匍行性迴狀紅斑相關。在超過 80% 的病例中,皮膚爆發出現於惡性腫瘤診斷之前,平均期間為 7 個月(範圍:1 至 72 個月),但也可與惡性腫瘤診斷同時或之後發生。在有相關癌症的病人中,幾乎半數病例有肺癌,而 8% 有胃癌,7% 有食道癌,5% 有乳癌。也有許多其他類型相關腫瘤的個別病例報告發表,以及 5% 的病例原發部位不明。在患有匍行性迴狀紅斑但無可偵測潛在惡性腫瘤的個體中,有些病例為特發性;有些則呈現並存情況,包括並存的皮膚疾病(毛髮紅糠疹、乾癬、魚鱗癬)、結締組織疾病(CREST [鈣質皮膚沉著 calcinosis cutis、雷諾現象 Raynaud phenomenon、食道動力障礙 esophageal motility disorder、指端硬化 sclerodactyly 與微血管擴張 telangiectasia] 症候群、類風濕性關節炎)、感染(結核 tuberculosis)、高嗜伊紅性白血球症候群 (hypereosinophilic syndrome) 或藥物反應 (azathioprine)。¹⁸
匍行性迴狀紅斑的確切病因不明,但有人認為腫瘤可能誘發周圍組織正常成分的化學改變。隨後發生分子模擬 (molecular mimicry),針對腫瘤的發炎反應與良性皮膚蛋白交叉反應。此理論受到下列證據支持:在一例與肺癌相關的病例中,發現受侵犯皮膚與支氣管基底膜有 IgG 與 C3 沉積。遷移是所有圖形紅斑的特徵,但在匍行性迴狀紅斑中尤為迅速。其機制尚不清楚,雖然部分研究著重於纖維母細胞活性。發炎細胞與/或纖維母細胞可能媒介基質 (ground substance) 改變,這可能侷限發炎,進而以圖形模式協調浸潤的移動。¹⁷
匍行性迴狀紅斑的治療涉及找出並治療原發惡性腫瘤。在充分控制癌症後,皮疹通常會消退,但在診斷時已廣泛轉移的病例中,這可能無法達成。否則,此爆發常具治療抗性,雖然全身性類固醇的結果不一。局部類固醇、維生素 A 與 azathioprine 並無助益。已知此爆發會在死亡前立即消退,可能係因死前全身性免疫抑制所致。¹⁷
壞死性遊走性紅斑 (Necrolytic Migratory Erythema)
重點一覽 (AT-A-GLANCE)
■ 疼痛、糜爛、結痂的間擦部位與顏面皮膚爆發。
■ 高度提示胰臟惡性腫瘤(升糖素瘤 glucagonoma)。
■ 半數腫瘤於診斷時已轉移。
■ 假性升糖素瘤症候群 (Pseudoglucagonoma syndrome) 發生於無升糖素瘤的情況下。
■ 皮膚隨潛在營養異常的治療而改善。
1942 年,Becker 首次描述一位皮膚爆發與 α 細胞胰臟腫瘤相關的病人。1966 年,McGavran 發現另一位有類似臨床表現並有高升糖素血症 (hyperglucagonemia) 的病人。後來,壞死性遊走性紅斑 (necrolytic migratory erythema, NME) 一詞由 Wilkinson 於 1973 年所創,用以描述胰臟癌病人特殊的皮疹。¹⁹
流行病學 (EPIDEMIOLOGY)
NME 是一種非常罕見的副腫瘤皮膚疾病,被視為升糖素瘤的標誌,且於腫瘤診斷時存在於超過三分之二的病人。¹⁹ 升糖素瘤是一種極為罕見、生長緩慢的胰臟 α 細胞神經內分泌腫瘤 (neuroendocrine tumor)。全球發生率約為每 2000 萬人 1 例。它典型上以升糖素瘤症候群 (glucagonoma syndrome) 表現。² 無性別或種族偏好,最常影響 60 多歲的人。
臨床特徵 (CLINICAL FEATURES)
NME 的皮膚病灶呈多形性,但通常可見糜爛與結痂。原發病灶為紅斑性斑片,演變為斑塊並發展出中央大疱。水疱迅速糜爛、形成結痂,並最終消退。紅斑性與糜爛性環狀斑片與斑塊融合成大片地圖狀區域。它們典型上疼痛且搔癢。此爆發在數週的病程中自發性消失與再現。NME 的分布具特徵性,包括間擦部位(例如腹股溝、會陰、臀部與下腹部)、中央顏面(尤其是口周)與遠端四肢(圖 134-10)。黏膜侵犯表現為口角炎 (angular cheilitis)、萎縮性舌炎 (atrophic glossitis) 與口炎 (stomatitis)。營養不良性指甲 (dystrophic nails) 可能伴隨此症候群。¹⁹,²⁰
升糖素瘤症候群的特徵為 NME、體重減輕、口腔疼痛、腹瀉、糖尿病、深部靜脈栓塞 (deep vein thrombosis)、正色素性正球性貧血 (normochromic normocytic anemia) 與神經精神疾病 (neuropsychiatric disorders)。體重減輕是最常見的表現徵象。升糖素瘤傾向緩慢生長。轉移至肝、區域淋巴結與骨骼很常見,但出現於疾病晚期。假性升糖素瘤症候群表現相同,但無 α 細胞胰臟腫瘤,這可能解釋為何血清升糖素濃度未升高。在假症候群病人中發現的潛在疾病包括肝病、胰臟炎、乳糜瀉 (celiac sprue)、發炎性腸道疾病、腸病性肢端皮膚炎 (acrodermatitis enteropathica)、糙皮病與非胰臟惡性腫瘤。¹⁹,²⁰
由於 NME 可為這些症候群的首發表現,及早辨識很重要,並可能提供較佳的結果。
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
NME 的確切病因仍不清楚。皮疹可歸因於過量升糖素的代謝效應,因為皮疹可在手術切除升糖素瘤或以藥物使升糖素濃度正常化後消退。高升糖素血症刺激肝臟糖質新生 (gluconeogenesis),導致血糖濃度增加。糖質新生路徑中的胺基酸消耗導致血清胺基酸減少。低胺基酸血症 (hypoaminoacidemia) 導致表皮蛋白缺乏(組胺酸 histidine 與色胺酸 tryptophan),可能誘發表皮壞死。升糖素亦增加皮膚的花生四烯酸 (arachidonic acid)、前列腺素 (prostaglandins) 與白三烯素 (leukotrienes) 濃度,可能誘發皮膚發炎反應。然而,NME 無法完全歸因於高升糖素血症,因為它可能在無升糖素濃度升高或胰臟腫瘤的情況下出現,如假性升糖素瘤症候群所報導。鋅、蛋白質、必需脂肪酸與維生素 B 的缺乏也可能在 NME 的發展中扮演角色。²⁰
診斷 (DIAGNOSIS)
NME 的組織病理學具特徵性,但非病理徵象性 (pathognomonic)。急性病灶在棘層 (stratum spinosum) 上層呈現顯著程度的表皮壞死,可能與下方存活的表皮分離。可觀察到壞死層中的嗜中性球浸潤導致角質層下膿疱 (subcorneal pustules)。慢性病灶顯示乾癬樣皮膚炎。可顯現角化不全、顆粒層消失、基底空泡化 (basal vacuolization) 與散在的壞死角質形成細胞。這些發現也可見於其他營養缺乏、移植物對抗宿主疾病 (graft-versus-host disease)、結締組織疾病與光毒性藥物疹 (phototoxic drug eruptions)。¹⁹,²⁰
實驗室異常包括血清升糖素顯著升高,通常大於 1000 pg/mL(參考範圍:50 至 150 pg/mL)。大多數病人有高血糖與正色素性正球性貧血。存在肝功能異常,且血清胺基酸、總蛋白、白蛋白與膽固醇濃度偏低。偶爾鋅濃度亦降低。應進行影像學檢查以偵測胰臟腫瘤。已使用各種方法來辨識胰臟腫瘤,如血管攝影、超音波、CT、MRI、正子斷層掃描 (positron emission tomography)、octreotide 閃爍掃描 (octreotide scintigraphy) 與體抑素受體閃爍掃描 (somatostatin receptor scintigraphy)。²⁰
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
其他與 NME 皮膚表現相似的疾病包括腸病性肢端皮膚炎、營養缺乏、乾癬、濕疹、脂漏性皮膚炎、念珠菌病、表淺性天疱瘡 (superficial pemphigus) 與某些化療的副作用。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
在與升糖素瘤症候群相關的 NME 中,疾病病程依腫瘤診斷時的分期而異。若升糖素瘤未轉移且可完全切除,症候群的症狀將消退。不幸的是,於診斷時,腫瘤在大多數病例中常已巨大且轉移。所幸腫瘤生長緩慢,病人可能透過手術減少腫瘤負荷而獲得症狀改善,雖然存活可能不受此手術影響。
處置 (MANAGEMENT)
必須處理高升糖素血症的潛在病因,以根除此疼痛的皮膚疾病。對於升糖素瘤病人,切除腫瘤對於症狀緩解很重要。若腫瘤侷限於胰臟,完整的手術切除可治癒。此腫瘤常對化療具抗性。體抑素類似物 (somatostatin analog)(octreotide、pasireotide 與 lanreotide)可改善皮膚症狀,並可能延緩腫瘤進展。其他治療方式,如干擾素 (interferon)、everolimus(哺乳類雷帕黴素標的抑制劑 mammalian target of rapamycin inhibitor)、sunitinib(酪胺酸激酶抑制劑 tyrosine kinase inhibitor)與胜肽受體放射性核素治療 (peptide receptor radionuclide therapy),皆有良好結果的報導。在某些病例中,補充以矯正鋅、胺基酸或脂肪酸缺乏可改善皮膚病灶。¹⁹
嗜中性球皮膚病 (Neutrophilic Dermatoses)
Sweet 症候群 (Sweet Syndrome)
第 36 章「Sweet 症候群 (Sweet Syndrome)」提供關於 Sweet 症候群的更多資訊。Sweet 症候群,又稱急性發熱性嗜中性球皮膚病 (acute febrile neutrophilic dermatosis),特徵為發燒、白血球增多 (leukocytosis),以及由真皮嗜中性球所造成的皮膚丘疹或斑塊。約 21% 被診斷為 Sweet 症候群的病人報導有惡性腫瘤,可為血液 (15%) 或實體 (6%) 惡性腫瘤。最常相關的血液惡性腫瘤為急性骨髓母細胞性白血病 (acute myeloblastic leukemia)。其他血液腫瘤包括骨髓增生性腫瘤 (myeloproliferative neoplasms)、瀰漫性大 B 細胞淋巴瘤 (diffuse large B-cell lymphoma)、何杰金淋巴瘤、骨髓化生不良症候群 (myelodysplastic syndrome) 與骨髓纖維化 (myelofibrosis)。與 Sweet 症候群相關最常見的實體惡性腫瘤為泌尿生殖器官、乳房與消化道的癌症,最常為腺癌 (57%)。²¹
數位作者提出區分惡性腫瘤相關 Sweet 症候群與本病典型型的特徵。惡性腫瘤相關 Sweet 症候群較少先有上呼吸道感染,且發病在時間上與癌症的新發現或復發相關。此外,無性別優勢,而典型型則以女性為主。²²
臨床上,大疱性與潰瘍性皮膚病灶、皮下結節與口腔黏膜侵犯,在惡性腫瘤相關 Sweet 症候群中可能比典型型更常觀察到。實驗室異常,包括貧血 (82% 至 83%) 與血小板計數異常 (68%),在惡性腫瘤相關 Sweet 症候群中有報導。雖然周邊白血球增多是 Sweet 症候群的診斷標準之一,但部分惡性腫瘤相關 Sweet 症候群病人可觀察到嗜中性球減少 (neutropenia)。少數患有血液惡性腫瘤的病人在病灶皮膚切片中顯示同時存在白血病皮膚浸潤 (leukemia cutis) 與 Sweet 症候群。²¹,²²
Sweet 症候群的發病可早於、晚於病人腫瘤的診斷,或與之同時出現。Sweet 症候群的持續期間多變。病灶復發不僅可發生於受惡性腫瘤影響的病人,也可發生於特發性或疾病相關 Sweet 症候群的個體。惡性腫瘤相關 Sweet 症候群的治療並無特定指引。全身性皮質類固醇與其他用於典型或特發性 Sweet 症候群的標準治療為治療的主軸。成功治療潛在惡性腫瘤可能使惡性腫瘤相關 Sweet 症候群完全消退,而皮膚病學發現的再現可能代表癌症復發。²¹,²²
壞疽性膿皮症 (Pyoderma Gangrenosum)
第 37 章「壞疽性膿皮症 (Pyoderma Gangrenosum)」提供關於壞疽性膿皮症的更多資訊。壞疽性膿皮症 (pyoderma gangrenosum, PG) 是一種罕見、疼痛、潰瘍性的嗜中性球皮膚病,與多種內臟疾病相關。PG 病人中惡性腫瘤的發生率在各研究間有所不同,發生於約 4% 至 20% 的 PG 病人。²³
血液惡性腫瘤是最常見的相關腫瘤;其他相關惡性腫瘤包括急性與慢性骨髓性白血病 (acute and chronic myelogenous leukemia)、慢性淋巴球性白血病 (chronic lymphocytic leukemia)、多發性骨髓瘤、骨髓化生不良症候群、真性紅血球增多症 (polycythemia vera)、何杰金淋巴瘤與皮膚 T 細胞淋巴瘤。PG 的非典型特徵,如突然發病、表淺病灶、出血性大疱與上肢侵犯,曾於 27% 患有潛在血液疾病或惡性腫瘤的 PG 病人中描述。²⁴ 已記載大疱性 PG 隨潛在惡性腫瘤的治療而改善。數位作者描述意義未明的單株免疫球蛋白病變 (monoclonal gammopathy of undetermined significance) 為 PG 的相關情況,其中 IgA 是最常相關的副蛋白血症 (paraproteinemia)。²³ 這些免疫球蛋白病變臨床上為良性,但偶爾進展為骨髓瘤。因此,建議長期監測。PG 病人也可能發生潛在的實體腫瘤。偶有報導各種器官的癌症,如乳房、攝護腺、肺、膀胱、結腸、肝、卵巢、喉、多形性膠質母細胞瘤 (glioblastoma multiforme) 與黑色素瘤。²³
真皮增生性疾病 (Dermal Proliferative Disorders)
多中心網狀組織球增生症 (Multicentric Reticulohistiocytosis)
第 117 章「組織球增生症 (Histiocytosis)」提供關於多中心網狀組織球增生症的更多資訊。多中心網狀組織球增生症 (multicentric reticulohistiocytosis, MRH) 是一種罕見的全身性疾病,呈現破壞性多關節炎 (destructive polyarthritis) 與典型的皮膚病灶。孤立或成群的紅棕色至膚色丘疹與結節主要見於顏面與手部,甲周丘疹呈特徵性的「珊瑚珠 (coral bead)」外觀。多達 25% 的病人有相關惡性腫瘤,且在某些病例中,多中心網狀組織球增生症的診斷早於癌症診斷。²⁵
此病無主要的相關癌症類型。已報導各種腫瘤,包括乳房、肺、甲狀腺、消化道、泌尿生殖、肉瘤、淋巴瘤、白血病與黑色素瘤。疾病病程可能為自限性而無關節變形;起伏不定;或具侵襲性而有殘毀性關節炎 (mutilating arthritis)。自發性緩解通常於多年後發生。在大多數病人中,它並不與相關惡性腫瘤的病程平行。在某些報導的病例中,治療伴隨的癌症僅可解決關節與皮膚疾病。數位作者間仍在爭論是否將多中心網狀組織球增生症標記為副腫瘤疾病。無論如何,每位被診斷為多中心網狀組織球增生症的病人都應進行潛在惡性腫瘤的完整檢查。²⁶
壞死生物性黃色肉芽腫 (Necrobiotic Xanthogranuloma)
第 57 章「細胞間 IgA 皮膚病(IgA 天疱瘡)(Intercellular IgA Dermatosis [IgA Pemphigus])」提供關於壞死生物性黃色肉芽腫的更多資訊。壞死生物性黃色肉芽腫 (necrobiotic xanthogranuloma) 是一種罕見的非朗格漢斯細胞組織球增生症 (non–Langerhans cell histiocytosis),與血液疾病有強烈關聯。皮膚是最常受侵犯的部位,超過 80% 的病人有眼周病灶。皮膚外侵犯罕見。病灶表現為黃色至紅橙色或紫紅色的丘疹、斑塊或結節,伴有潰瘍、微血管擴張或萎縮區域。大多數病例無症狀,病程緩慢。多達 80% 的病人有 IgG 型單株免疫球蛋白病變,伴有 κ 或 λ 輕鏈。²⁷ 最常相關的漿細胞惡液質 (plasma cell dyscrasias) 為意義未明的單株免疫球蛋白病變、無症狀多發性骨髓瘤 (smoldering multiple myeloma) 與多發性骨髓瘤。其他血液異常,包括非何杰金淋巴瘤、慢性淋巴球性白血病、何杰金淋巴瘤與淋巴漿細胞性淋巴瘤 (lymphoplasmacytic lymphoma),可能與壞死生物性黃色肉芽腫相關。²⁷
患有與單株免疫球蛋白病變相關的壞死生物性黃色肉芽腫的病人,有轉變為多發性骨髓瘤的風險。因此,應持續仔細監測以早期偵測疾病進展。²⁷
真皮沉積疾病 (Dermal Deposition)
硬化性黏液水腫 (Scleromyxedema)
第 67 章「硬腫病與硬化性黏液水腫 (Scleredema and Scleromyxedema)」提供關於硬化性黏液水腫的更多資訊。硬化性黏液水腫 (scleromyxedema) 是丘疹性黏液病 (papular mucinosis;苔蘚樣黏液水腫 lichen myxedematosus) 的全身型,是一種與單株免疫球蛋白病變及全身性器官侵犯相關的不常見疾病。典型的皮膚表現好發於顏面、手臂與手。心肺、風濕、消化道與神經侵犯很常見。大多數(80%)硬化性黏液水腫病人有 IgGλ 輕鏈的意義未明單株免疫球蛋白病變。應進行血清蛋白電泳 (serum protein electrophoresis) 並合併甲狀腺檢查,以排除甲狀腺功能障礙與黏液水腫 (myxedema)。所幸副蛋白血症僅罕見轉變為多發性骨髓瘤,但一旦發生,則預示不良預後。有數則病例報告硬化性黏液水腫病人中出現其他血液與非血液惡性腫瘤,包括白血病、何杰金或非何杰金淋巴瘤與某些實體腫瘤。然而,尚不確定某些相關惡性腫瘤是偶發的,抑或與以 melphalan 等藥物治療硬化性黏液水腫所造成的續發性惡性腫瘤相關。²⁸ 疾病病程通常為進行性,主要全身性器官侵犯可能造成不良結果。
全身性類澱粉沉著症 (Systemic Amyloidosis)
第 124 章「紫質症 (The Porphyrias)」提供關於全身性類澱粉沉著症的更多資訊。原發性全身性類澱粉沉著症被稱為 AL 類澱粉沉著症 (AL amyloidosis),其中有單株免疫球蛋白輕鏈纖維的細胞外沉積,典型上由一小群漿細胞株所產生。續發性 (AA) 類澱粉沉著症發生於自體免疫或發炎疾病、惡性腫瘤與慢性感染。全身性類澱粉沉著症的皮膚表現包括「捏夾性紫斑 (pinch purpura)」(眼周瘀血)、巨舌症 (macroglossia),以及較少見的蠟樣皮膚增厚與皮下結節。AL 類澱粉沉著症是與惡性腫瘤相關最常見的型態。預後一般不佳,若未治療,中位存活時間為 12 個月。在 AL 類澱粉沉著症中,多個器官與組織(腎、心、肝、周邊/自主神經、軟組織)通常受侵犯。重要的是將全身性 AL 類澱粉沉著症的皮膚病灶,與較不常見、侷限性的皮膚變異型區別,後者不會進展至多系統侵犯。多發性骨髓瘤是最常見的相關惡性腫瘤,見於約 20% 的 AL 類澱粉沉著症病人。²⁹ 其他腫瘤,如非何杰金淋巴瘤、黏膜相關淋巴組織淋巴瘤 (mucosa-associated lymphoid tissue lymphoma)、淋巴漿細胞性淋巴瘤,以及其他實體器官腫瘤的個別病例報告,鮮少被報導。對於所有疑似 AL 類澱粉沉著症的病人,應在基線時進行血清與尿液蛋白電泳/免疫固定電泳 (immunofixation electrophoresis)、血清游離輕鏈分析 (serum free light chain assay)、骨髓切片與骨骼影像學檢查,以排除多發性骨髓瘤的存在。³⁰
部分研究者描述了轉甲狀腺素類澱粉沉著症 (transthyretin amyloidosis, ATTR) 中非何杰金淋巴瘤風險增加。³¹ 此外,某些腫瘤,如肝細胞癌、腎細胞癌、Castleman 病、何杰金病與成人毛細胞白血病 (adult hairy cell leukemia),可造成反應性或續發性 (AA) 類澱粉沉著症。³⁰
大疱性疾病 (Bullous Disorders)
副腫瘤性天疱瘡 (Paraneoplastic Pemphigus)
第 53 章「副腫瘤性天疱瘡 (Paraneoplastic Pemphigus)」提供關於副腫瘤性天疱瘡的更多資訊。副腫瘤性天疱瘡 (paraneoplastic pemphigus) 是一種危及生命的疾病,死亡率高。疼痛、出血性的口腔糜爛是最早的特徵性臨床發現。多形性皮膚爆發由類天疱瘡 (pemphigus-like)、類大疱性類天疱瘡 (bullous pemphigoid–like)、類多形性紅斑 (erythema multiforme–like)、類移植物對抗宿主疾病 (graft-versus-host disease–like) 與類扁平苔蘚 (lichen planus–like) 的皮膚病灶組成,可侵犯任何部位。已記載多器官侵犯,如肺、甲狀腺、腎、平滑肌與消化道。副腫瘤性天疱瘡的發病機轉涉及細胞與體液免疫。幾乎所有病人都有潛在腫瘤,最常為淋巴增生性疾病。最常報導的相關腫瘤為非何杰金淋巴瘤、慢性淋巴球性白血病、Castleman 病與胸腺瘤 (thymoma)。惡性腫瘤相關副腫瘤性天疱瘡病人的預後不佳,但良性腫瘤病人的預後可能較佳。³²
疱疹樣皮膚炎 (Dermatitis Herpetiformis)
第 59 章「疱疹樣皮膚炎 (Dermatitis Herpetiformis)」提供關於疱疹樣皮膚炎的更多資訊。疱疹樣皮膚炎 (dermatitis herpetiformis) 是一種自體免疫皮膚疾病,呈現嚴重搔癢的皮膚爆發,伴多形性病灶,並與麩質敏感性 (gluten sensitivity) 相關。大多數研究已證明疱疹樣皮膚炎病人的非何杰金淋巴瘤風險顯著增加,儘管整體惡性腫瘤風險與一般人口相似。³³,³⁴ 已注意到病程久遠的疱疹樣皮膚炎病人風險特別高。疱疹樣皮膚炎病人相關惡性腫瘤的報導頻率高達 4.3%,以男性為主。³³ 然而,儘管與非何杰金淋巴瘤有此關聯,疱疹樣皮膚炎病人的死亡率似乎並未增加。³³,³⁴
與疱疹樣皮膚炎相關的淋巴瘤可為 B 細胞或 T 細胞淋巴瘤,並可發生於消化道內外,作為淋巴結性或淋巴結外疾病。此外,典型上與乳糜瀉相關的腸病相關 T 細胞淋巴瘤 (enteropathy-associated T-cell lymphoma),曾於疱疹樣皮膚炎病人中報導。³⁴ 無麩質飲食 (gluten-free diet) 可能預防癌症的發展,但需要更多研究來證實此發現。³³,³⁴
大疱性類天疱瘡 (Bullous Pemphigoid)
第 54 章「大疱性類天疱瘡 (Bullous Pemphigoid)」提供關於大疱性類天疱瘡的更多資訊。大疱性類天疱瘡 (bullous pemphigoid, BP) 是一種自體免疫水疱性疾病,呈現於老年病人,有完整皮膚的水疱與搔癢。除了與病人年齡相關的風險外,大疱性類天疱瘡是否有惡性腫瘤風險增加仍具爭議。數項研究,包括較新的大型流行病學研究,並未發現大疱性類天疱瘡與癌症之間的關聯。然而,其他研究,尤其來自日本者,證明大疱性類天疱瘡病人的惡性腫瘤風險高於正常人口。許多作者不建議對大疱性類天疱瘡病人進行常規癌症篩檢,除非為早發型類天疱瘡、有惡性腫瘤病史或標準治療失敗者。³⁵
其他 (Miscellaneous)
後天性胎毛性多毛症 (Hypertrichosis Lanuginosa Acquisita)
後天性胎毛性多毛症 (acquired hypertrichosis lanuginosa,「惡性絨毛 malignant down」) 是一種罕見疾病,特徵為長、細、無色素的胎毛 (lanugo;拉丁文「絨毛」之意) 相對突然出現。胎毛最常於病程早期出現於顏面與耳朵(圖 134-11)。毛髮可能長至驚人的長度;眉毛與睫毛可能長達數英寸。長而細的毛髮也可見於軀幹與四肢,包括腋窩,但手掌、足底、恥骨上與生殖器區域通常不受侵犯。後天性胎毛性多毛症常以頭尾方向 (cephalocaudal direction) 發展,並可能伴隨舌炎、舌乳頭肥大、口腔色素過度沉著、味覺與嗅覺障礙、黑色棘皮症、腹瀉、淋巴結病變與體重減輕。³⁶
此病常為副腫瘤性,可為潛在惡性腫瘤的標記。它通常出現於進展期或轉移性癌症,因此預後不佳。最常相關的惡性腫瘤在男性為肺癌與結直腸癌,在女性為結直腸癌,其次為肺癌與乳癌。卵巢、子宮與膀胱的癌症、淋巴瘤與白血病,以及其他惡性腫瘤,亦於後天性胎毛性多毛症病人中發現。³⁷
後天性胎毛性多毛症必須與非惡性病因相關的多毛症 (hirsutism) 與多毛 (hypertrichosis) 區別,這些病因包括代謝與內分泌疾病,如神經性厭食症 (anorexia nervosa)、甲狀腺毒症 (thyrotoxicosis) 與遲發性皮膚紫質症 (porphyria cutanea tarda)(見第 124 章),或藥物,如 cyclosporine、phenytoin、penicillamine、spironolactone、補骨脂素 (psoralens)、皮質類固醇、干擾素、diazoxide 或 minoxidil。³⁶,³⁷
呈現後天性胎毛性多毛症而無藥物使用史或潛在代謝、內分泌或惡性疾病的病人,應懷疑潛在癌症。應考慮對潛伏惡性腫瘤進行適當的診斷評估,尤其是肺、結腸、直腸區域與乳房。
Trousseau 症候群 (Trousseau Syndrome)
Trousseau 症候群的定義隨時間演變。它最初描述與潛在胃癌相關的遊走性血栓性靜脈炎 (migratory thrombophlebitis) 的發生。後來,數位作者將其擴展為包括癌症相關的高凝血狀態,伴隨廣泛的臨床表現,包括無臨床症狀的異常凝血檢查、表淺遊走性血栓性靜脈炎、深部靜脈栓塞、消瘦性心內膜炎 (marantic endocarditis)、肺栓塞,以及與瀰漫性血管內凝血 (disseminated intravascular coagulation) 相關的大量血栓栓塞現象。靜脈血栓栓塞 (venous thromboembolism, VTE) 似乎是最常見的臨床表現。³⁸
臨床上,惡性腫瘤病人的 VTE 可能比無惡性腫瘤者更嚴重且更廣泛。不尋常部位的深部靜脈栓塞,如上肢、內臟器官與腦的血栓,以及動脈血栓栓塞,皆曾報導發生於癌症相關病例。Trousseau 症候群約占所有 VTE 病人的 20% 至 30%,並發生於 1% 至 8% 的潛在惡性腫瘤病人。其發生率已增加,可能係因更頻繁使用較新的致血栓性化療與免疫調節劑(例如 thalidomide 或 lenalidomide),以及癌症病人存活期延長所致。不幸的是,VTE 是癌症病人第二常見的死因。患有活動性癌症的病人血栓風險比正常人口高出 4 至 7 倍,且某些癌症的風險最高,如胰、腦、肺、胃與卵巢癌。³⁸⁻⁴⁰
雖然癌症病人 Trousseau 症候群的發病機轉不清楚,但致血栓過程可能涉及多重因素。異常血流或淤滯、血管壁損傷與血液高凝血狀態的組合,稱為 Virchow 三要素 (Virchow triad),促成血栓形成。數項研究描述腫瘤細胞組織因子 (tissue factor) 的過度表現,以及參與凝血路徑的血漿微粒 (plasma microparticles) 的增加。化療與腫瘤壞死可能誘發發炎細胞激素,導致內皮細胞損傷。嗜中性球細胞外捕捉網 (neutrophil extracellular traps, NETs),因嗜中性球程序性細胞死亡 (NETosis) 而釋放,對內在凝血路徑有影響。此外,數位研究者已記載促成癌細胞促凝血表型 (procoagulant phenotype) 的特定訊息傳遞路徑,係源自致癌基因(例如 K-ras、EGFR、PML/RAR-α、MET)與腫瘤抑制基因(例如 p53、PTEN)的突變。³⁸,⁴⁰
由於 Trousseau 症候群可為潛伏癌症的表現表徵,數位作者建議對特發性 VTE 病人進行依年齡與性別特定的惡性腫瘤篩檢檢查。建議治療潛在惡性腫瘤並合併抗凝血治療。低分子量肝素 (low-molecular-weight heparin) 是第一線治療。
涉及皮膚與/或黏膜並與惡性腫瘤相關的遺傳症候群 (Genetic Syndromes Involving Skin and/or Mucous Membranes and Associated with Malignancy)
臨床醫師辨識易導致內臟惡性腫瘤的遺傳性疾病的皮膚特徵很重要。在數個病例中,皮膚特徵早於惡性腫瘤的發展,且加強監測可能改善這些病人的整體存活。對於與遺傳性皮膚病相關的惡性腫瘤,家庭成員發展癌症的風險可能增加,應考慮適當的遺傳諮詢與檢查。
腫瘤直接侵犯皮膚 (Direct Tumor Involvement of the Skin)
皮膚轉移 (Cutaneous Metastases)
皮膚轉移 (cutaneous metastases) 代表遠端原發腫瘤透過轉移性散播侵犯皮膚。皮膚侵犯的整體發生率約為 5%。在女性中,最常轉移至皮膚的惡性腫瘤為乳癌,其次為結腸癌與黑色素瘤。在男性中,最常見的是肺癌,其次為結腸癌與黑色素瘤。⁴⁷
皮膚轉移最常表現為迅速出現的單發或多發、無症狀、膚色、可移動、質硬、圓形或橢圓形結節。轉移性乳癌可表現為各種型態的皮膚病灶。例如,丹毒樣癌 (carcinoma erysipelatoides) 呈現溫熱、壓痛的紅斑性斑片或斑塊,類似丹毒 (erysipelas) 或蜂窩性組織炎 (cellulitis)(圖 134-12A),但無發燒或白血球增多。另一臨床變異型為硬化性轉移性乳癌的皮革樣皮膚變化,稱為鎧甲狀癌 (carcinoma en cuirasse),後續可能表現為結節與潰瘍(圖 134-12B、C)。雖然鎧甲狀癌曾被報導為乳癌的表現徵象,但它更常作為潛在乳癌治療後的局部復發而發生。丹毒樣癌與鎧甲狀癌皆不侷限於乳癌;它們也可見於肺、腎、消化道與其他轉移性惡性腫瘤。⁴⁸ 來自惡性黑色素瘤的轉移通常有色素(圖 134-13)。病灶常帶有藍色調,即使深在皮膚中亦然。然而,即使原發腫瘤有色素,轉移可能為無色素 (amelanotic),反之亦然。胸部是皮膚轉移最常見的部位,係因轉移性乳癌與肺癌的高頻率所致。頭皮是另一個常見部位,為來自肺(圖 134-14)、腎與乳房腫瘤的轉移所在。在頭皮上,轉移性腫瘤典型上表現為單發或多發的硬結節,但偶可見瘢痕性禿髮 (scarring alopecia),稱為腫瘤性禿髮 (alopecia neoplastica),尤其來自轉移性乳癌。顏面與頸部可能受口咽癌轉移侵犯。來自腎與甲狀腺癌的轉移可能有搏動 (pulsatile) 且可能有雜音 (bruit)。⁴⁸
數項組織病理學特徵可能有助於辨識原發腫瘤的來源。顯微鏡下,腫瘤細胞的聚集(通常類似其原發惡性腫瘤)一般見於真皮與/或皮下組織。其他特徵,如腫瘤細胞呈「印度列隊 (Indian filing)」型態、淋巴血管侵犯、壞死與無腫瘤的「grenz 帶 (grenz zone)」,亦有助於轉移性皮膚病灶的診斷。在某些類型的癌症或分化不良的腫瘤中,常需要免疫組織化學染色 (immunohistochemical staining) 以達成正確診斷。例如,CK7、CK20 與 S-100 的免疫組織化學組合是診斷乳房、肺與消化道癌症及黑色素瘤的有用工具。其他建議的標記包括 TTF-1(甲狀腺轉錄因子-1 thyroid transcription factor-1;肺)、HMB-45(homatropine methylbromide;黑色素瘤)、PSA(攝護腺特異性抗原 prostate-specific antigen;攝護腺)、雌激素/黃體素受體 (estrogen/progesterone receptors)(乳房)與嗜鉻顆粒蛋白 (chromogranin)(神經內分泌腫瘤)。⁴⁷,⁴⁸
皮膚轉移可能代表進展期疾病與不良預後。皮膚轉移診斷後的估計平均存活在 6 個月時為 50%。然而,根據近期研究,乳癌病人的預後可能優於其他癌症病人。⁴⁸
白血病與淋巴瘤皮膚浸潤 (Leukemia and Lymphoma Cutis)
第 119 章「皮膚淋巴瘤 (Cutaneous Lymphoma)」提供關於白血病與淋巴瘤皮膚浸潤的更多資訊。來自白血病與淋巴瘤的皮膚轉移分別稱為白血病皮膚浸潤 (leukemia cutis) 與淋巴瘤皮膚浸潤 (lymphoma cutis)。兩種疾病的皮膚表現可能有相似的外觀。病灶一般表現為粉紅色、紅色與/或棕色至紫色的斑、丘疹、斑塊與結節,通常質硬且無痛。白血病皮膚浸潤是白血病的髓外表現,特徵為白血病細胞侵入表皮、真皮或皮下。皮膚爆發可出現於軀幹、四肢與頭部,無好發部位。偶爾可見紅斑、斑、水疱、潰瘍與紅皮症。黏膜侵犯(尤其是牙齦肥大)很常見。顏面結節狀或斑塊狀浸潤可導致獅面 (leonine facies) 的發展。皮膚病灶迅速且全身性的爆發可能提示急性型白血病。白血病皮膚浸潤最常發生於患有急性骨髓性白血病 (acute myeloid leukemia) 的病人,尤其是單核球性 (M5) 與骨髓單核球性 (M4) 亞型,但也可發生於患有其他白血病亞型的病人,如慢性淋巴球性白血病、慢性骨髓性白血病、慢性骨髓單核球性白血病 (chronic myelomonocytic leukemia) 與急性淋巴球性白血病。大多數白血病皮膚浸潤病人在已知白血病診斷後發展出此症,但在罕見病例中,「非白血病性 (aleukemic)」白血病皮膚浸潤或無全身性侵犯的白血病皮膚浸潤可作為表現表徵,於發展出全身性侵犯之前數月或數年出現。皮膚切片對於確認診斷至關重要。免疫組織化學與分子遺傳學方法對於確定白血病亞型至關重要。治療針對潛在白血病的特定類型。部分作者認為白血病皮膚浸潤的出現可能代表侵襲性疾病與不良預後。⁴⁹
淋巴瘤皮膚浸潤發生於全身性淋巴瘤續發性散播至皮膚時。有時,可能難以區分原發性皮膚淋巴瘤 (primary cutaneous lymphoma) 與淋巴瘤皮膚浸潤。需要進一步的分期評估以記載皮膚淋巴瘤的來源。皮膚播散可發生於 T 細胞與 B 細胞淋巴瘤。全身性間變性大細胞淋巴瘤 (systemic anaplastic large-cell lymphoma) 是一種 T 細胞淋巴瘤,其皮膚侵犯在間變性淋巴瘤激酶陰性 (anaplastic lymphoma kinase–negative) 亞型中更常發生。在成人 T 細胞白血病-淋巴瘤 (adult T-cell leukemia-lymphoma) 中,超過半數病人於診斷時呈現皮膚病灶,其慢性與無症狀 (smoldering) 亞型類似蕈狀肉芽腫的皮膚病灶。何杰金淋巴瘤的皮膚播散遠較非何杰金淋巴瘤少見。許多類型的全身性 B 細胞淋巴瘤也可有淋巴結外散播至皮膚(全身性濾泡性淋巴瘤 systemic follicular lymphoma、邊緣帶淋巴瘤 marginal zone lymphoma,或瀰漫性大 B 細胞淋巴瘤),雖然原發性皮膚 B 細胞淋巴瘤可能比全身性 B 細胞淋巴瘤的續發性皮膚侵犯更常見。在淋巴瘤樣肉芽腫病 (lymphomatoid granulomatosis) 中,皮膚是最常見的肺外部位,淋巴瘤樣肉芽腫病在 50% 的病例中以進行性型態發生。其他罕見有皮膚侵犯的 B 細胞淋巴瘤類型包括血管內 B 細胞淋巴瘤 (intravascular B-cell lymphoma) 與前驅 B 細胞淋巴母細胞性淋巴瘤 (precursor B-cell lymphoblastic lymphoma)。⁵⁰
Paget 病與乳房外 Paget 病 (Paget and Extramammary Paget Diseases)
第 114 章「Paget 病 (Paget Disease)」提供關於 Paget 病與乳房外 Paget 病的更多資訊。乳頭 Paget 病是乳頭與乳暈的慢性紅斑性脫屑爆發,提示潛在乳房的導管癌 (ductal carcinoma)。乳房外 Paget 病 (extramammary Paget disease) 最常發生於肛門生殖器皮膚,其次為腋窩與陰莖。它分為 3 型。最常見的型態表現為原發性表皮內腺癌 (intraepidermal adenocarcinoma)。第二型 (25%) 與內臟惡性腫瘤相關,最常為消化道或泌尿生殖系統。第三型最不常見,與鄰近的汗腺癌 (sweat gland carcinoma) 相關。
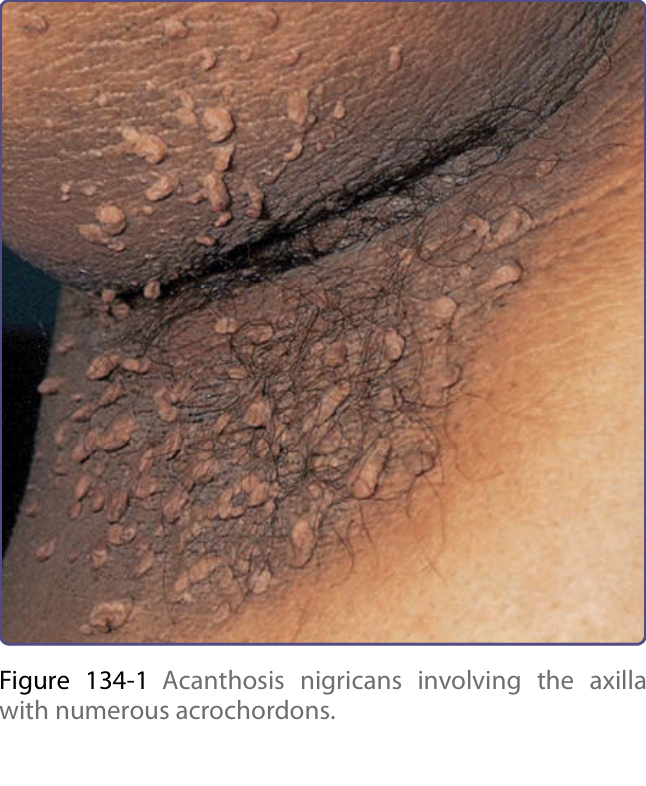
圖 134-1:黑色棘皮症 (acanthosis nigricans) 侵犯腋窩,伴有眾多軟纖維瘤 (acrochordons)。
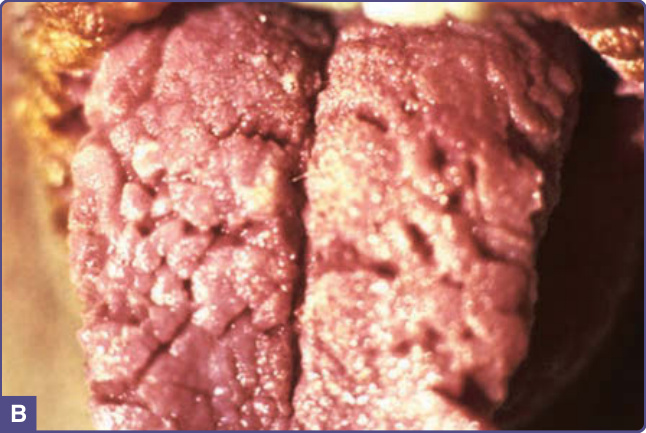
圖 134-2:黑色棘皮症 (acanthosis nigricans)。A,唇紅緣的疣狀與乳頭瘤狀增生。B,舌的天鵝絨樣增厚。(A 與 B 並非同一病人。)(A 與 B,取自 Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, NY: McGraw-Hill; 2009,經許可使用。)
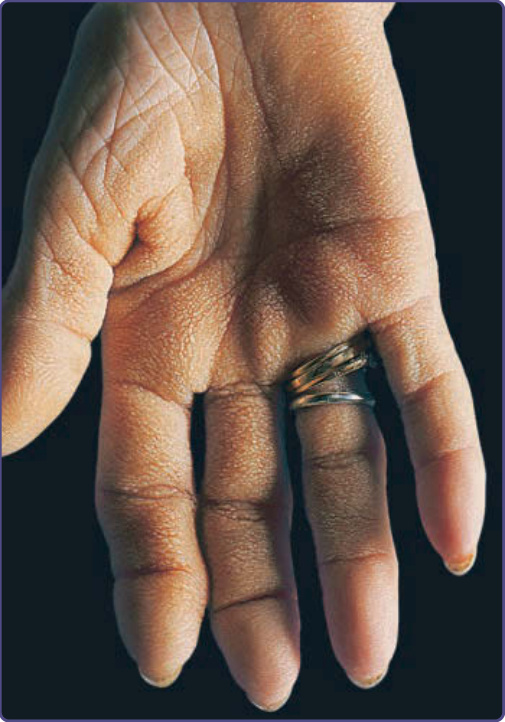
圖 134-3:牛肚掌 (tripe palm)。掌部脊紋呈最大程度的加深,因而模擬反芻動物胃部的黏膜。
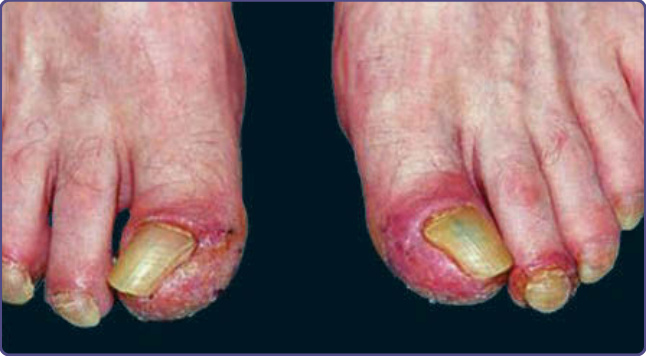
圖 134-5:早期副腫瘤性肢端角化症中,足趾遠端水腫、疼痛性全身性甲溝炎與遠端甲下過度角化。
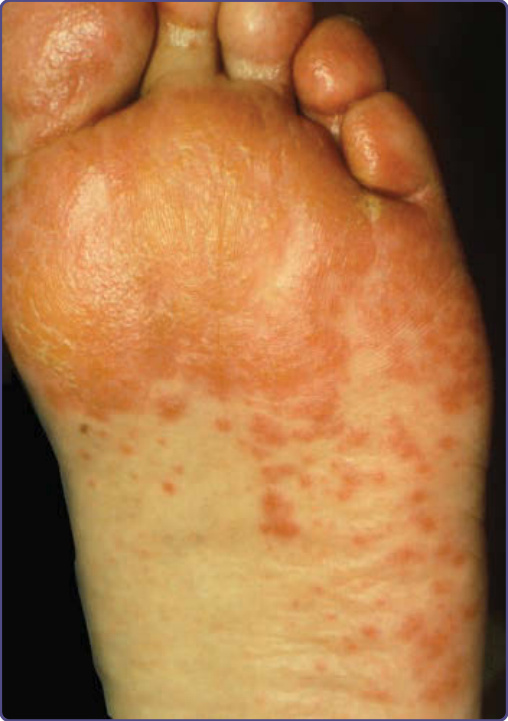
圖 134-6:副腫瘤性肢端角化症中,角皮症 (keratoderma) 特徵性地不侵犯足底(與手掌)表面的中央部分。
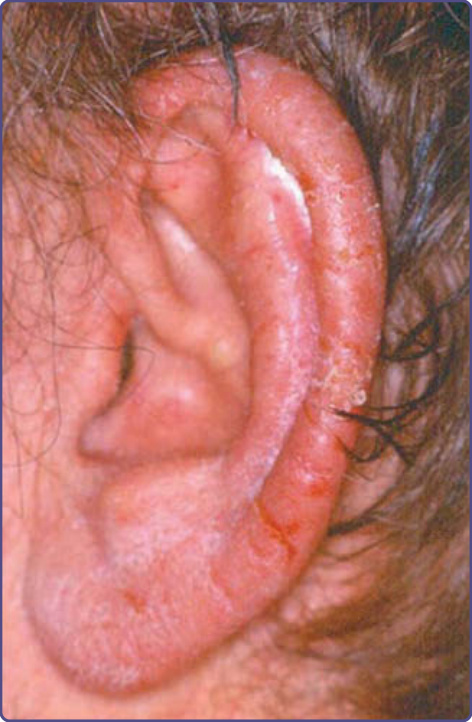
圖 134-7:副腫瘤性肢端角化症中特徵性的耳輪 (helical) 發炎伴脫屑。
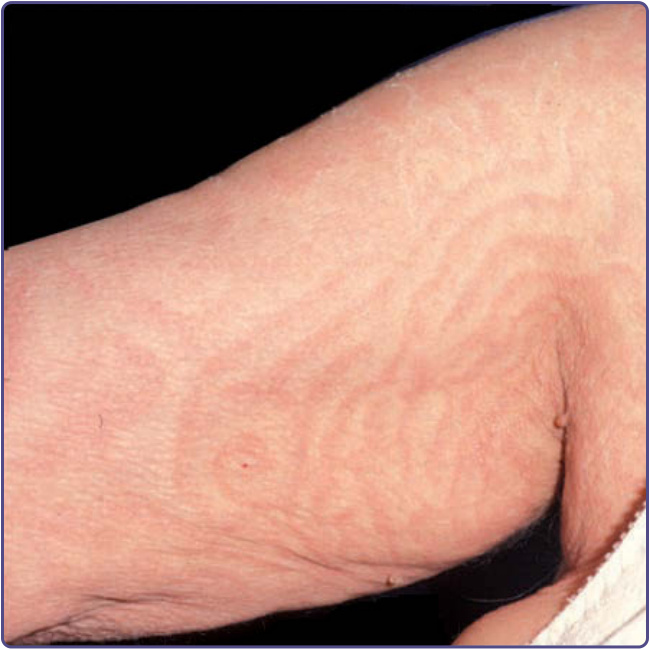
圖 134-8:匍行性迴狀紅斑 (erythema gyratum repens)。腋窩與手臂上的蛇行平行帶狀病灶。(經 Michael Adler 許可使用。)
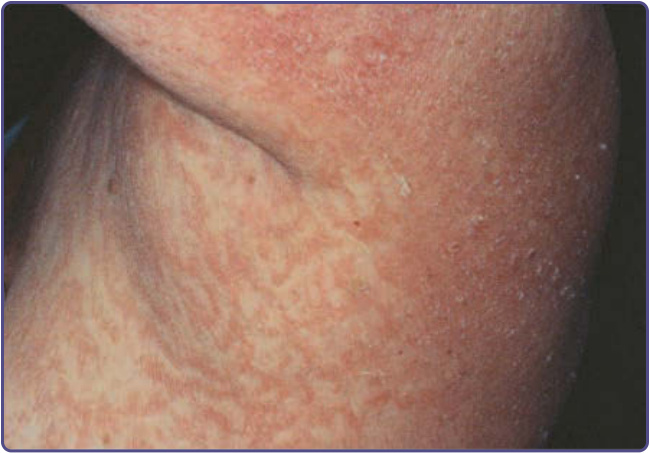
圖 134-9:匍行性迴狀紅斑 (erythema gyratum repens)。一位乳癌女性病人特徵性的紅斑「木紋 (wood-grain)」型態與輕微脫屑。(經 Jill McKenzie, MD 許可使用。)
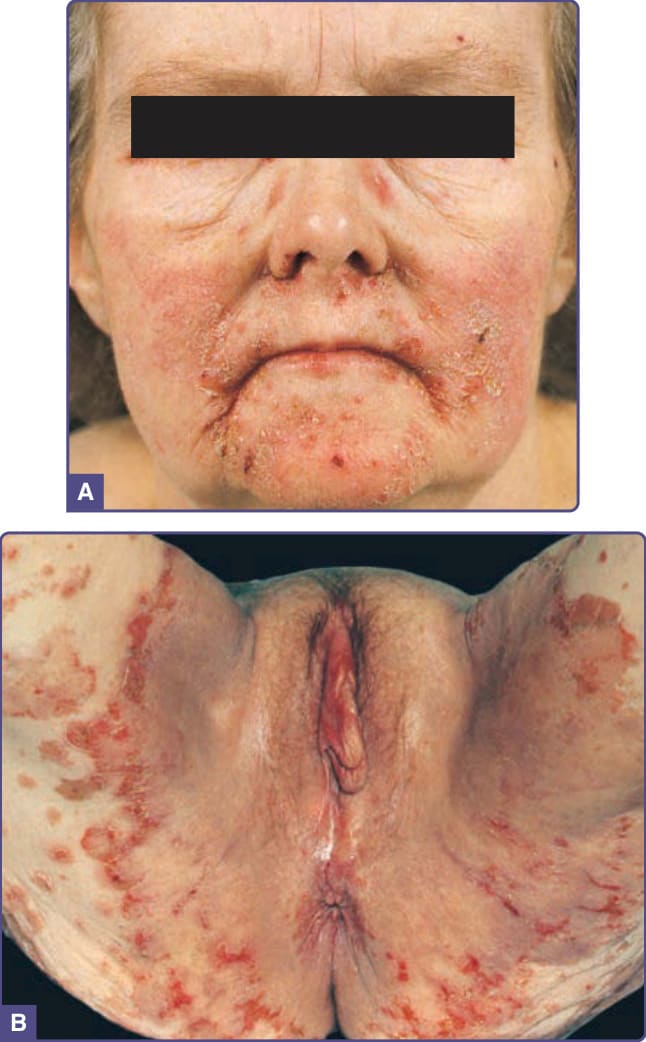
圖 134-10:升糖素瘤症候群,壞死性遊走性紅斑 (necrolytic migratory erythema)。(A) 鬆弛性與丘疹水疱性病灶,伴孔口周圍的糜爛、結痂與皸裂,以及 (B) 腹股溝中呈現地圖狀、環形的「壞死性遊走性紅斑」。(A 與 B 並非同一病人。)
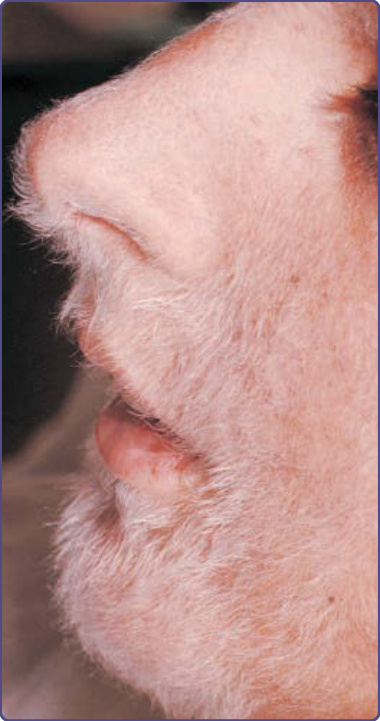
圖 134-11:一位 19 歲患有胰臟癌的女性病人的後天性胎毛性多毛症 (hypertrichosis lanuginosa acquisita)。
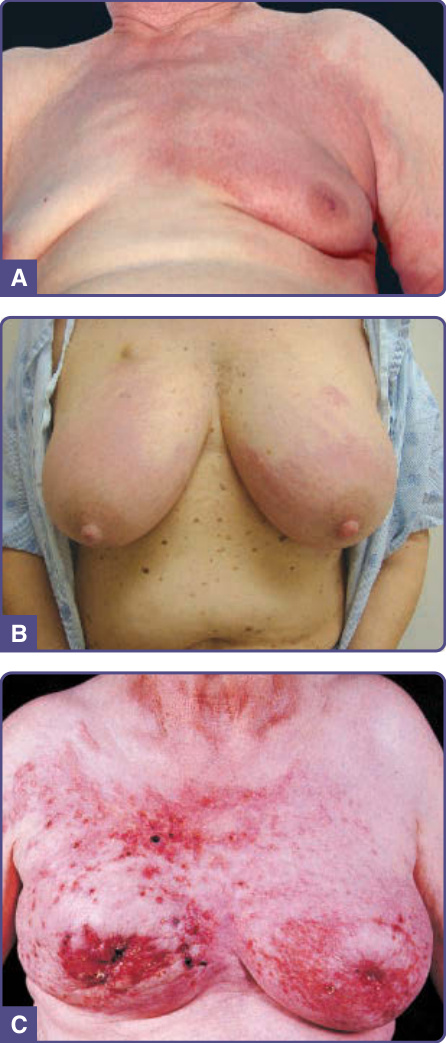
圖 134-12:A,丹毒樣癌 (carcinoma erysipelatoides)。乳癌的淋巴管內散播,表現為類似丹毒的紅斑。B,來自潛在乳癌的雙側皮膚轉移。C,鎧甲狀癌 (carcinoma en cuirasse) 侵犯雙側乳房與胸壁。
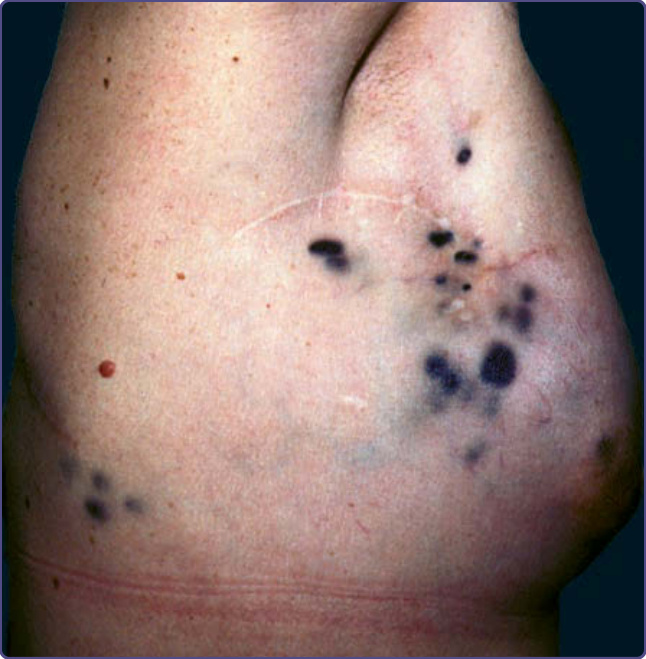
圖 134-13:來自黑色素瘤的深色素皮膚轉移。
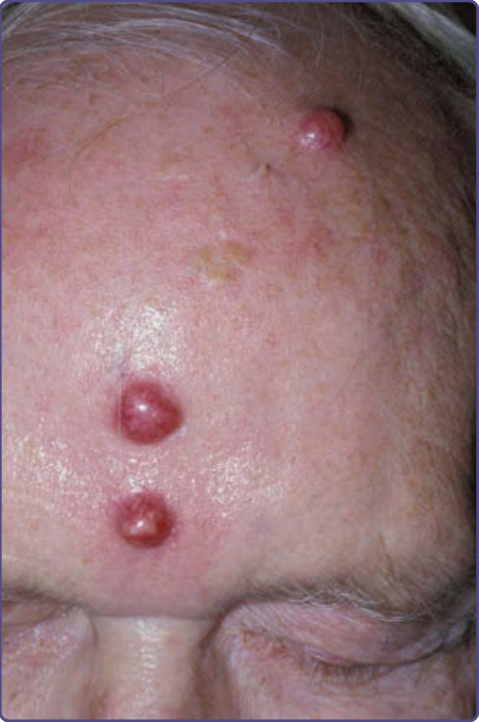
圖 134-14:轉移性肺癌表現為前額結節。
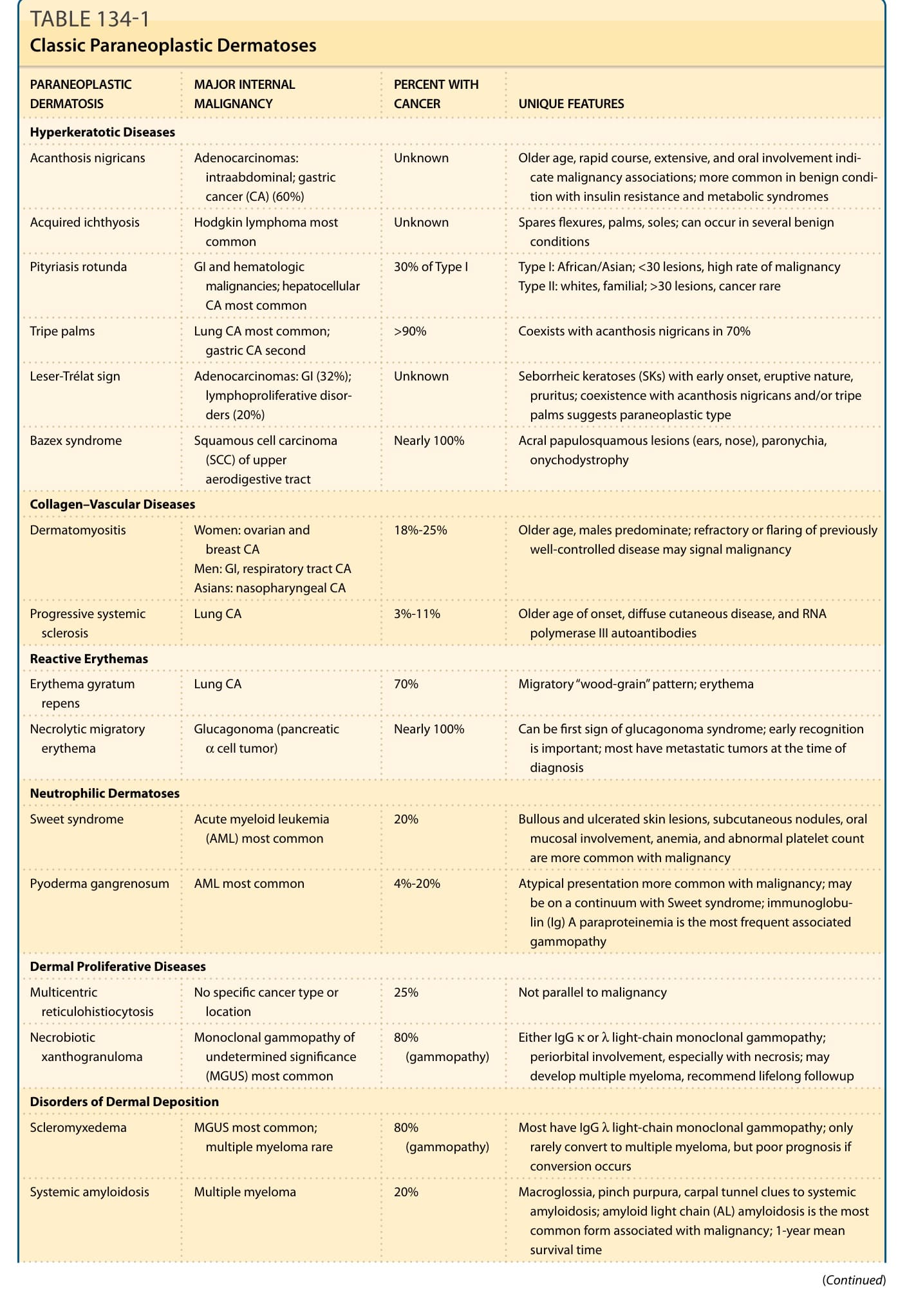
表 134-1:典型副腫瘤皮膚病 (Classic Paraneoplastic Dermatoses)
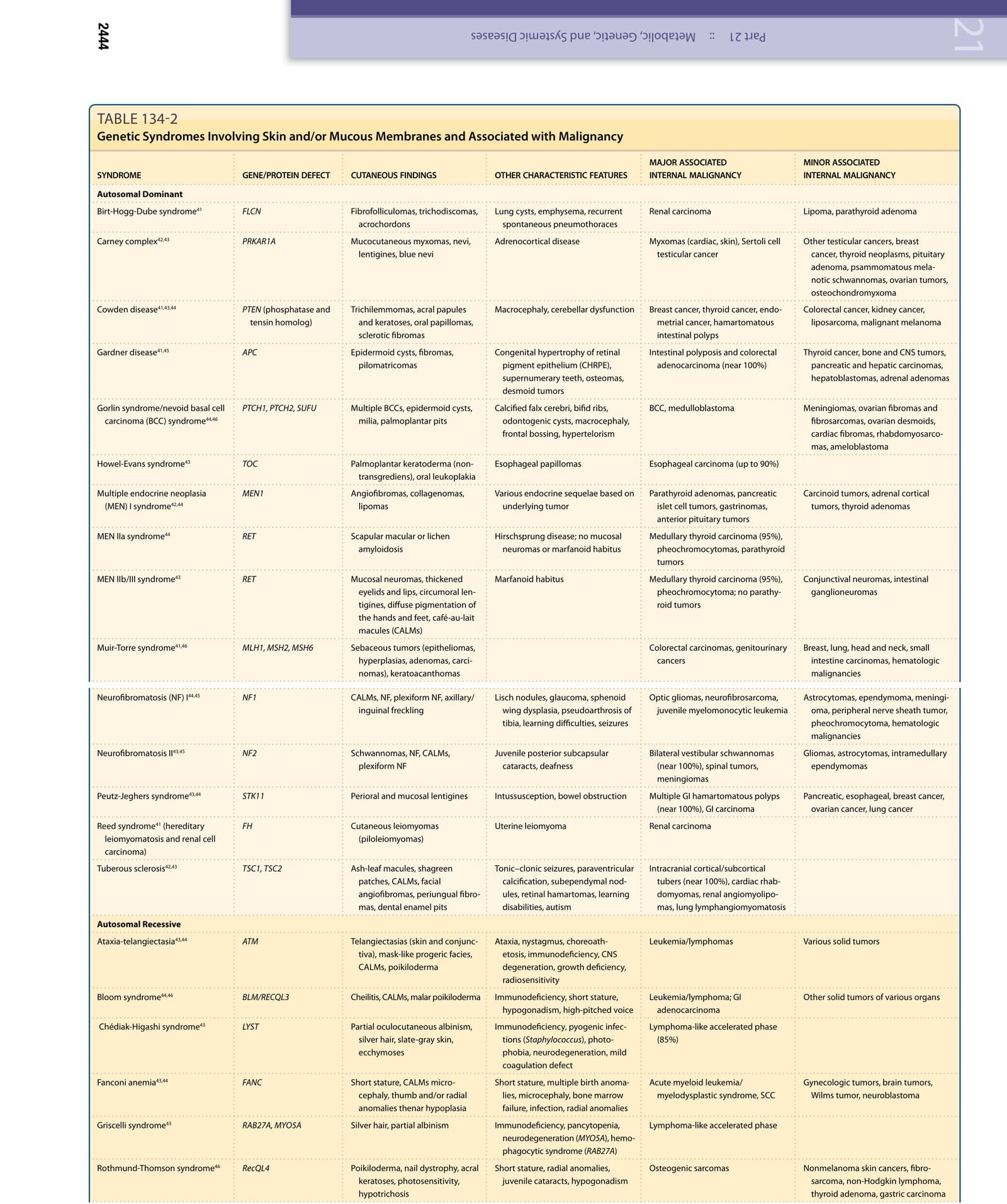
表 134-2:具惡性潛能的遺傳性皮膚病 (Genodermatoses with Malignant Potential)