Cutaneous Paraneoplastic Syndromes
21
CUTANEOUS PARANEOPLASTIC SYNDROMES
Paraneoplastic syndromes refer to the remote effects of underlying neoplastic diseases. The clinical syndromes can occur in various organ systems, including endocrine, neuromuscular, cardiovascular, cutaneous, hematologic, GI, and renal. The symptoms are not the direct effect of metastases or tumor invasion, but may result from substances produced by the tumor (eg. hormones, peptides, or cytokines) or from immunologic or inflammatory reactions between malignant and normal tissues. Cutaneous paraneoplastic syndromes (Table 134-1) are diverse dermatologic entities that suggest the presence of a remote malignancy. In some instances, they may appear before a cancer diagnosis and contribute to the discovery of an occult cancer, or they may also indicate recurrence in a patient with prior remission of cancer. Consequently, prompt recognition of the cutaneous paraneoplastic syndromes may lead to the detection of an underlying malignancy at an early and highly-treatable stage. However, the paraneoplastic manifestations may predate the diagnosis of the related neoplasm by many months or years. If an underlying cancer is not detectible, careful followup should continue for several years. In addition to classic paraneoplastic dermatoses, this chapter also discusses direct tumor involvement of the skin and genodermatoses with malignant potential (Table 134-2). Although paraneoplastic dermatoses are not frequently seen in the clinical setting, recognition of these cancer-associated skin lesions is very important. The dermatologist may play a significant role in diagnosing an occult malignancy at earlier stages of disease.
HYPERKERATOTIC DERMATOSES
ACANTHOSIS NIGRICANS
ACANTHOSIS NIGRICANS
Chap. 137, “Diabetes and Other Endocrine Diseases,” provides additional information on benign acanthosis nigricans.
AT-A-GLANCE
■ Acanthosis nigricans is a cutaneous marker, most commonly of insulin resistance and less frequently of genetic disorders and malignancy.
■ Characterized by symmetric hyperpigmented, hyperkeratotic, verrucous plaques with a velvety texture on intertriginous skin and occasionally mucocutaneous areas.
■ Darker skin pigmentation, insulin resistance, and obesity are more commonly associated with benign acanthosis nigricans.
■ Malignant acanthosis nigricans often appears rapidly in older individuals and can involve atypical areas such as mucosal surfaces.
The term acanthosis nigricans was originally proposed by Unna, although the first cases were malignancy associated and described independently by Pollitzer and by Janovsky in 1890. Curth clinically classified acanthosis nigricans into malignant, benign, or syndromic acanthosis nigricans, or pseudoacanthosis nigricans (obesity related). Today acanthosis nigricans is classified into 2 broad categories: benign (familial, obesity related, hyperinsulinemic states, autoimmune disease associated) or malignant (malignancy associated). Malignant acanthosis nigricans typically occurs in older patients and frequently coexists with other paraneoplastic dermatoses such as tripe palms and the sign of Leser-Trélat.1
EPIDEMIOLOGY
The majority (80%) of acanthosis nigricans occurs idiopathically or in benign conditions such as endocrinopathies, heritable diseases, or drug use. Malignancy-associated acanthosis nigricans is rare, with approximately 1000 cases reported in the literature. It usually occurs in individuals older than 40 years of age, without gender, racial, or genetic predilection.1
CLINICAL FEATURES
The skin lesions first appear as a symmetric hyperpigmentation or a dirty appearance of the skin, followed by thickening and increased skin markings, resulting in hyperpigmented velvety plaques. The colors may vary
21
PARANEOPLASTIC DERMATOSIS MAJOR INTERNAL MALIGNANCY PERCENT WITH CANCER UNIQUE FEATURES
Hyperkeratotic Diseases
Acanthosis nigricans Adenocarcinomas: intraabdominal; gastric cancer (CA) (60%)
Unknown Older age, rapid course, extensive, and oral involvement indicate malignancy associations; more common in benign condition with insulin resistance and metabolic syndromes
Acquired ichthyosis Hodgkin lymphoma most common Unknown Spares flexures, palms, soles; can occur in several benign conditions
Pityriasis rotunda GI and hematologic malignancies; hepatocellular CA most common
30% of Type I Type I: African/Asian; <30 lesions, high rate of malignancy Type II: whites, familial; >30 lesions, cancer rare
Tripe palms Lung CA most common; gastric CA second >90% Coexists with acanthosis nigricans in 70%
Leser-Trélat sign Adenocarcinomas: GI (32%); lymphoproliferative disorders (20%)
Unknown Seborrheic keratoses (SKs) with early onset, eruptive nature, pruritus; coexistence with acanthosis nigricans and/or tripe palms suggests paraneoplastic type
Bazex syndrome Squamous cell carcinoma (SCC) of upper aerodigestive tract
Nearly 100% Acral papulosquamous lesions (ears, nose), paronychia, onychodystrophy
Collagen–Vascular Diseases
Dermatomyositis Women: ovarian and breast CA Men: GI, respiratory tract CA Asians: nasopharyngeal CA
18%-25% Older age, males predominate; refractory or flaring of previously well-controlled disease may signal malignancy
Progressive systemic sclerosis Lung CA 3%-11% Older age of onset, diffuse cutaneous disease, and RNA polymerase III autoantibodies
Reactive Erythemas
Erythema gyratum repens Lung CA 70% Migratory “wood-grain” pattern; erythema
Necrolytic migratory erythema Glucagonoma (pancreatic α cell tumor) Nearly 100% Can be first sign of glucagonoma syndrome; early recognition is important; most have metastatic tumors at the time of diagnosis
Neutrophilic Dermatoses
Sweet syndrome Acute myeloid leukemia (AML) most common 20% Bullous and ulcerated skin lesions, subcutaneous nodules, oral mucosal involvement, anemia, and abnormal platelet count are more common with malignancy
Pyoderma gangrenosum AML most common 4%-20% Atypical presentation more common with malignancy; may be on a continuum with Sweet syndrome; immunoglobulin (Ig) A paraproteinemia is the most frequent associated gammopathy
Dermal Proliferative Diseases
Multicentric reticulohistiocytosis No specific cancer type or location 25% Not parallel to malignancy
80% (gammopathy) Either IgG κ or λ light-chain monoclonal gammopathy; periorbital involvement, especially with necrosis; may develop multiple myeloma, recommend lifelong followup
Necrobiotic xanthogranuloma Monoclonal gammopathy of undetermined significance (MGUS) most common
Disorders of Dermal Deposition
Scleromyxedema MGUS most common; multiple myeloma rare 80% (gammopathy) Most have IgG λ light-chain monoclonal gammopathy; only rarely convert to multiple myeloma, but poor prognosis if conversion occurs
Systemic amyloidosis Multiple myeloma 20% Macroglossia, pinch purpura, carpal tunnel clues to systemic amyloidosis; amyloid light chain (AL) amyloidosis is the most common form associated with malignancy; 1-year mean survival time
2442
(Continued)
(Continued)
21
PARANEOPLASTIC DERMATOSIS MAJOR INTERNAL MALIGNANCY PERCENT WITH CANCER UNIQUE FEATURES
Bullous Disorders
Paraneoplastic pemphigus Lymphoproliferative disorders; non-Hodgkin lymphoma most common; chronic lymphocytic leukemia (CLL) second most common
Nearly 100% Can occur in Castleman disease and thymoma; parallels benign, but not malignant, tumors
Dermatitis herpetiformis Non-Hodgkin lymphoma most common 4% Occurs significantly more often in patients who do not follow a gluten-free diet
Other Changes
Hypertrichosis lanuginosa acquisita Men: lung, colorectal CA Women: colorectal, lung, or breast CA
Nearly 100% Excessive downy hair initially concentrated on face, spreading caudally; usually appears in advanced or metastatic carcinomas
Trousseau syndrome Pancreatic, brain, lung CA 20%-30% Deep vein thrombosis (DVT) of unusual sites (eg. upper limbs),
Trousseau syndrome Pancreatic, brain, lung CA 20%-30% Deep vein thrombosis (DVT) of unusual sites (eg. upper limbs), thrombosis of visceral organs and brain, and arterial thromboembolism suggest cancer association
and include yellow, brown, gray, and black. Although the skin lesions of acanthosis nigricans can occur almost anywhere on the body, they tend to affect the skinfold, flexural, and intertriginous areas. The most commonly involved locations are the axillae, neck, external genitalia, groin, face, inner thighs, antecubital and popliteal fossae, umbilicus, and perianal area. Acrochordons may develop, superimposed on the acanthosis nigricans or on other locations (Fig. 134-1). Hyperkeratosis of the nipple and areola also has been noted. Mucosal involvement tends to occur in the malignant form, but can also be seen in acanthosis nigricans without malignancy and is characterized by papillomatosis and thickening of mucosa with or without hyperpigmentation involving the oral cavity (lips, tongue, buccal mucosa, and palate) (Fig. 134-2), and, rarely, the esophagus, eyes, larynx, and anal and genital mucosae. Typically, patients with acanthosis nigricans are asymptomatic, but pruritus can be a problem in some patients. Oral and esophageal papillomatosis can cause sore mouth and dysphagia.1
The lesions of malignant and benign acanthosis nigricans are indistinguishable. However, malignancyassociated acanthosis nigricans usually appears abruptly and extensively. Mucosal involvement and generalized pruritus are more common. The co-occurrence with florid cutaneous papillomatosis and other paraneoplastic skin lesions, such as tripe palms (see “Tripe Palms” section) and Leser-Trélat sign (see “Leser-Trélat Sign” section), suggests the malignancy-associated form of acanthosis nigricans. Many types of malignancies have been reported in association with malignant acanthosis nigricans. A literature review shows that up to 90% of patients have an associated intraabdominal adenocarcinoma, of which approximately 60% are gastric cancer. Cancers of the uterus, liver, intestinal, ovary, kidney, breast, lung, pancreas, bladder, thyroid, and gallbladder have been reported, as have lymphoma and mycosis fungoides.1
thrombosis of visceral organs and brain, and arterial thromboembolism suggest cancer association
ETIOLOGY AND PATHOGENESIS
The pathogenesis of acanthosis nigricans is poorly understood. In benign acanthosis nigricans, there is evidence that insulin plays a significant role through insulin-like growth factor-1 receptor signaling pathway (see Chap. 137). In acanthosis nigricans associated with inherited syndromes, insulin-like growth factor-1 receptor and fibroblast growth factor receptors may be implicated. In addition, genetic studies also demonstrate activation mutations of fibroblast growth factor receptors.2
In malignancy-associated acanthosis nigricans, it is proposed that the tumor produces transforming growth factor-α that is similar in structure to epidermal growth factor and binds to epidermal growth factor receptors and stimulates keratinocyte proliferation, which leads to development of acanthosis nigricans. This theory was based on observations in which the skin disorders improved or resolved following treatment of the underlying malignancy. For example, elevated urinary transforming growth factor-α and increased expression of epidermal growth factor receptor in lesional skin were noted in a patient with acanthosis nigricans, acrochordons, Leser-Trélat sign, and melanoma. The enhanced expression of this cytokine and its receptor normalized after removal of the melanoma, and the accompanying skin lesions improved postoperatively. In addition, some authors suggest that fibroblast growth factor and insulin-like growth factor-1 also may play a role in the pathogenesis of malignancyassociated acanthosis nigricans.2,3
DIAGNOSIS
Diagnosis of acanthosis nigricans is based on clinical features and distribution of the lesions. Histopathologic study is needed only to confirm diagnosis
2443
21
Carney complex42,43 PRKAR1A Mucocutaneous myxomas, nevi, lentigines, blue nevi Adrenocortical disease Myxomas (cardiac, skin), Sertoli cell testicular cancer Other testicular cancers, breast cancer, thyroid neoplasms, pituitary adenoma, psammomatous melanotic schwannomas, ovarian tumors, osteochondromyxoma
BCC, medulloblastoma Meningiomas, ovarian fibromas and fibrosarcomas, ovarian desmoids, cardiac fibromas, rhabdomyosarcomas, ameloblastoma
Colorectal cancer, kidney cancer, liposarcoma, malignant melanoma
Carcinoid tumors, adrenal cortical tumors, thyroid adenomas
Gardner disease41,45 APC Epidermoid cysts, fibromas, pilomatricomas Congenital hypertrophy of retinal pigment epithelium (CHRPE), supernumerary teeth, osteomas, desmoid tumors
Autosomal Dominant
Conjunctival neuromas, intestinal ganglioneuromas
MEN IIb/III syndrome43 RET Mucosal neuromas, thickened eyelids and lips, circumoral lentigines, diffuse pigmentation of the hands and feet, café-au-lait macules (CALMs)
Optic gliomas, neurofibrosarcoma, juvenile myelomonocytic leukemia Astrocytomas, ependymoma, meningioma, peripheral nerve sheath tumor, pheochromocytoma, hematologic malignancies
Gliomas, astrocytomas, intramedullary ependymomas
Intracranial cortical/subcortical tubers (near 100%), cardiac rhabdomyomas, renal angiomyolipomas, lung lymphangiomyomatosis
Lymphoma-like accelerated phase (85%)
Tonic–clonic seizures, paraventricular calcification, subependymal nodules, retinal hamartomas, learning disabilities, autism
Immunodeficiency, pyogenic infections (Staphylococcus), photophobia, neurodegeneration, mild coagulation defect
Ataxia, nystagmus, choreoathetosis, immunodeficiency, CNS degeneration, growth deficiency, radiosensitivity
Tuberous sclerosis42,43 TSC1, TSC2 Ash-leaf macules, shagreen patches, CALMs, facial angiofibromas, periungual fibromas, dental enamel pits
Reed syndrome41 (hereditary leiomyomatosis and renal cell carcinoma)
Autosomal Recessive
21
(Continued)
Lymphoma-like accelerated phase
Short stature, multiple birth anomalies, microcephaly, bone marrow failure, infection, radial anomalies
21
Hodgkin lymphoma, GI carcinoma
Cutaneous and mucosal SCC, acute myeloid leukemia/myelodysplastic syndrome
Testicular cancer
Pancytopenia, various ocular complications, mental retardation, pulmonary complications, esophageal stricture, microcephaly
Failure of maternal labor, commashaped corneal opacities, cryptorchidism
Dyskeratosis congenita43,44 DKC1 (XLR); TERT/TERD (AD); NOP10 (AR) Leukoplakia, nail dystrophy, reticulate pigmentation, poikilodermatous skin changes, alopecia, premature graying of the hair, palmoplantar hyperkeratosis, adermatoglyphia, hyperhidrosis
Xeroderma pigmentosum (XP)43,46 XP (multiple proteins that form DNA repair complex), XPA to XPG, and XPV
X-Linked Recessive
Sporadic
fibrosarcoma, osteosarcoma
Omphalocele, mental retardation, hemihypertrophy, neonatal hypoglycemia
Familial atypical multiple mole melanoma syndrome (dysplastic nevus syndrome)43,44
in problematic cases. Histopathology of acanthosis nigricans classically shows hyperkeratosis and epidermal papillomatosis. There is only slight acanthosis and usually no hyperpigmentation. Horn pseudocyst formation and increased melanin pigmentation can be observed in some cases. The brown color of the lesion is caused by hyperkeratosis rather than melanin. The
A
B
21
histopathologic findings cannot be distinguished from confluent and reticulated papillomatosis of Gougerot and Carteaud, seborrheic keratosis, or epidermal nevus.
DIFFERENTIAL DIAGNOSIS
The clinical differential diagnoses of malignant acanthosis nigricans include the conditions that present hyperpigmentation and patches or plaques in either flexural areas or a generalized distribution. Psoriasiform dermatitis, atopic dermatitis, pellagra, resolving pemphigus vegetans, parapsoriasis, cutaneous T-cell lymphoma, infections (eg. candidiasis, erythrasma, tinea corporis/cruris), pigmentary disorders (eg. hemochromatosis, Addison disease, Dowling-Degos disease), and others (eg, confluent and reticulated papillomatosis of Gougerot and Carteaud, Becker melanosis, epidermal nevus, and terra firma-forme dermatosis) are included in the differential diagnoses.
CLINICAL COURSE AND PROGNOSIS
Malignant acanthosis nigricans can occur before, during, or after the detection of cancer. It tends to parallel the course of the underlying malignancy. Skin lesions may regress after treatment of the underlying cancers, and recur following relapse or metastasis of the malignancies. Unfortunately, most associated malignancies present at an advanced stage, and thus the prognosis is usually poor.2,4
MANAGEMENT
Evaluation and studies are recommended in suspicious cases to identify the underlying endocrinopathy or malignancy. Older patients presenting with unintentional weight loss, rapid and extensive acanthosis nigricans with mucosal involvement, and have concomitant paraneoplastic skin lesions, should be investigated for underlying malignancy, especially of the GI tract. Cosmetic appearance is often the patient’s primary concern. Management of any co-occurring disease or malignancy often improves and may even resolve the acanthosis nigricans. Topical keratolytics, including the retinoids, and oral retinoids can reduce the appearance of acanthosis nigricans. Other oral medications reported to show improvement include dietary fish oil, metformin, and cyproheptadine, possibly by inhibition of tumor-secreted growth factors in the case of cypropheptadine. Other therapies found beneficial in case reports include calcipotriol, trichloroacetic acid peeling, and longpulsed alexandrite laser.3
ACQUIRED ICHTHYOSIS
ACQUIRED ICHTHYOSIS
Chapter 47, “The Ichthyoses,” provides additional information on acquired ichthyosis.
2447
21
Acquired ichthyosis occurs in adulthood and may be associated with malignancies, endocrine and metabolic diseases, HIV and other infections, autoimmune diseases, nutritional deficiency, and drug reactions. Clinical manifestations include diffuse, symmetrical, plate-like scaling on trunk and extensor extremities, which usually spares flexures, palms, and soles. Malignancies associated with acquired ichthyosis include CD30+ lymphoproliferative disorders, mycosis fungoides, leiomyosarcoma, Kaposi sarcoma, multiple myeloma, and carcinomas of the ovary, breast, lung, and cervix. Hodgkin disease is the most common neoplasm; the ichthyosis usually occurs simultaneously with or after the diagnosis of lymphoma. As a sudden onset of ichthyosis in an adult may be a presenting sign of underlying malignancy, a careful malignancy screening, especially for lymphoma, is important. In lymphomas, acquired ichthyosis can be a cutaneous sign of the disease or a paraneoplastic phenomenon. However, this may be very difficult to identify in some cases, such as in mycosis fungoides, in which clinical and histopathologic findings can be indistinguishable between ichthyosiform mycosis fungoides and acquired ichthyosis related to mycosis fungoides.5
PITYRIASIS ROTUNDA
PITYRIASIS ROTUNDA
Pityriasis rotunda, a rare cutaneous disorder, is considered by some as a variant of acquired ichthyosis. It has been observed in association with chronic diseases, infections, and malignancies. GI (most commonly hepatocellular carcinoma) and hematologic malignancies are frequently reported among the various types of associated malignancies. However, pityriasis rotunda can occur in healthy people as well. Skin manifestations are characteristic, consisting of perfectly round or oval, asymptomatic, well-defined, hypopigmented or hyperpigmented ichthyosiform scaly patches that appear on the trunk and proximal extremities.5 Type 1 pityriasis rotunda is associated with underlying malignant or systemic disease and presents with <30 skin lesions. It is more common in black and East Asian patients. Type 2 or familial pityriasis rotunda occurs in white patients and presents with more than 30 lesions (usually hypopigmented), and is not associated with any underlying disease.5 Treatment of underlying disease may lead to resolution of skin lesions in Type 1 patients. In Type 2 patients, spontaneous improvement may be seen by adulthood. Topical treatments, such as retinoids, salicyclic acid, and lactic acid are useful treatments for the dryness and scaling associated with this condition.5
TRIPE PALMS
TRIPE PALMS
The term tripe refers to a rugose surface of the edible lining of a bovine foregut. It was first implicated in a clinical diagnosis of the keratoderma with
2448
ridging pattern of the palms and fingers in 1963, when a patient reported to his doctor that his hands looked similar to tripe. Tripe palms was first described in the literature by Clarke in 1977. He reported a patient with squamous cell carcinoma of the lung accompanied by malignant acanthosis nigricans and thickening of the palms in tripe pattern. Tripe palms has been recognized as a cutaneous sign of internal malignancy since then.6
EPIDEMIOLOGY
Tripe palms is a rare paraneoplastic dermatosis with approximately 100 cases reported in the literature. The association with malignancy is high, with a greater than 90% occurrence. It is found almost exclusively in adults, and is more common in men than in women.6
CLINICAL FEATURES
The palms are rough, thickened, and velvety with exaggerated dermatoglyphics. The texture of the ventral surface of hand and fingers may be moss-like, cobbled, or honeycombed (Fig. 134-3). Tripe palms usually coexists with malignant acanthosis nigricans, observed in approximately 70% of the patients. Several authors suggest that tripe palms may be a form of palmar acanthosis nigricans. The co-occurrence with other paraneoplastic skin lesions, such as florid cutaneous papillomatosis, pruritus, clubbing of the digits, and the sign of Leser-Trélat, can be also seen.6
Pulmonary and gastric carcinoma account for more than 50% of neoplasms associated with tripe palms. In patients with only tripe palms, pulmonary carcinoma
is the most frequently associated malignancy, especially the squamous cell type. In patients with both tripe palms and acanthosis nigricans, gastric carcinoma is the most common carcinoma, followed by lung carcinoma. Other malignancies less often associated with tripe palms include those of the genitourinary tract, breast, head, and neck.6,7
ETIOLOGY AND PATHOGENESIS
Specific mechanisms of development have not been fully elucidated. Similar to acanthosis nigricans (see “Acanthosis Nigricans” section), there is limited evidence for the role of transforming growth factor-α released by tumor cells, inducing cellular proliferation in the pathogenesis of tripe palms. It is believed that tripe palms can be a variant of malignant acanthosis nigricans.7
DIAGNOSIS
Histopathology of tripe palms includes hyperkeratosis, acanthosis, and papillomatosis. These features closely resemble those pathologic findings observed in acanthosis nigricans and seborrheic keratoses. Additional findings can include dermal mucin and mast cells in approximately 20% of specimens.
DIFFERENTIAL DIAGNOSIS
The clinical differential diagnosis of tripe palms includes pachydermoperiostosis, hypertrophic pulmonary osteoarthropathy, acromegaly, thyroid acropachy, palmoplantar keratoderma, and acanthosis nigricans.
CLINICAL COURSE AND PROGNOSIS
Paraneoplastic tripe palms can occur before (48%), concurrently (21%), or after (31%) the diagnosis of malignancy. In one-third of patients, tripe palms parallels the course of the associated malignancy. The appearance of tripe palms in a known case of malignancy may be a sign of recurrence or metastasis of the tumor.6
MANAGEMENT
A complete malignancy workup, especially to rule out lung and stomach carcinoma, should be performed in all patients with tripe palms because of the high percentage of associated malignancy. A minimum workup should include a medical history, a complete physical examination, routine laboratory studies, a chest roentgenogram, and either a radiographic or an endoscopic evaluation of the upper GI tract. Additional investigations should be indicated by the findings of the initial workup. Treatment of tripe palms is difficult and primarily aimed at the underlying malignancy. There is no specific therapy for tripe palms. Similar to acanthosis nigricans, there are anecdotal reports of improvement
21
with oral retinoids alone and in combination with metformin. Approximately 30% of cases of tripe palms will resolve with treatment of the underlying tumor. However, there are many cases where remission of the cancer had no effect.
LESER-TRÉLAT SIGN
LESER-TRÉLAT SIGN
AT-A-GLANCE
■ Rapid and eruptive increase in number and size of seborrheic keratoses.
■ Pruritus is common.
■ Often occurs with malignant acanthosis nigricans.
■ Adenocarcinomas of the GI tract are the most common associated cancer, followed by lymphoproliferative malignancies.
■ Rarely seen in benign conditions, such as pregnancy, HIV, heart transplantation, acromegaly, erythroderma, and drug reaction.
The sign of Leser-Trélat is characterized by the sudden increase in size and number of seborrheic keratoses (SKs) related to internal malignancy. This phenomenon is attributed to Edmund Leser and Ulysse Trélat, two European surgeons, who observed cherry hemangiomas (but not SKs) in patients with cancer in 1890. In 1900, Hollander was the first person who associated the appearance of numerous SKs with internal malignancy.7,8
EPIDEMIOLOGY
The sign of Leser-Trélat is a rare paraneoplastic dermatosis that occurs with equal frequency between men and women and among different races. Similar to the occurrence of malignancy, this sign is more common in older individuals. This entity has remained controversial because the prevalence of both SKs and cancer is increased in the older population. Moreover, the identification of eruptive SKs is often based on the patient’s self-reporting which is subjective. In a large population-based study of 1752 consecutive cases of SKs, there was no statistical evidence of an increased incidence of internal malignancy compared to the general population. Subanalysis of those presenting with eruptive SKs also failed to demonstrate an increased risk of internal malignancy. In other large studies of patients with SKs and a recent solid tumor, a comparison with age- and sex-matched controls has not demonstrated a difference in either the clinical features or numbers of SKs.9
As such, large epidemiologic studies have not provided the evidence needed to conclusively define this sign as a true paraneoplastic dermatosis.
2449
21
Although large studies have not shown a statistical difference, anecdotal evidence exists demonstrating that this sign may signify an internal malignancy. First, there are reports of eruptive SKs in patients in their 20s with internal malignancies. Clinically, Leser-Trélat sign often coexists with malignant acanthosis nigricans, a more established paraneoplastic phenomenon. Additionally, alterations in growth factor expression differ from control patients. Together, this suggests that the Leser-Trélat sign is a legitimate, but extremely uncommon, paraneoplastic dermatosis.7,8
CLINICAL FEATURES
Clinical features signifying a paraneoplastic phenomenon include numerous, widespread, eruptive SKs, predominantly on trunk and extremities. However, quantification of eruptive SKs in terms of length of the eruptive period and number and size of SKs required for the diagnosis is lacking. Individual SKs found in the sign of Leser-Trélat are similar to normal SKs, both clinically and histologically. Pruritus is a significant feature occurring in approximately half of patients. Cooccurrence with other hyperkeratotic paraneoplastic dermatoses is common, with approximately one-third having acanthosis nigricans. There are some case reports of Leser-Trélat sign coexisting with tripe palms as well.8 The rapid occurrence of pruritic, eruptive SKs, especially in the setting of acanthosis nigricans should alert the clinician to a potential internal malignancy (Fig. 134-4). Adenocarcinomas account for the majority of malignancies described with the sign of Leser- Trélat, in 32% of which the Leser-Trélat sign is associated with GI tract malignancies, particularly gastric
2450
cancer. Lymphoproliferative disorders, including Sézary syndrome, mycosis fungoides, other lymphomas, and leukemia, are the second most common associated malignancies, occurring in approximately 20% of patients. Other tumors, such as lung, bladder, kidney, and ovarian cancers, and melanoma, also have been reported.7
A Leser-Trélat–like eruption or eruptive SKs have been rarely reported in the setting of benign conditions, such as pregnancy, HIV, heart transplantation, acromegaly, and erythroderma, and in association with drugs, such as cytarabine and tumor necrosis factor-α inhibitors.7,10 The term pseudosign of Leser-Trélat has been used to designate nonmalignancy-associated eruptive SKs. Unfortunately, these examples do not help to further define an already confusing and controversial clinical entity.
ETIOLOGY AND PATHOGENESIS
An exact pathogenesis has yet to be elucidated. However, similar to acanthosis nigricans and tripe palms, evidence also exists with the Leser-Trélat sign that an alteration in growth factor homeostasis is contributory. In several instances, a state of increased growth factor expression has been observed, with increased urinary levels of epidermal growth factors and transforming growth factor-α detected in patients with an underlying malignancy and eruptive SKs. Subsequently, growth factor levels decreased following primary tumor resection.7
In addition to increased levels of growth factor expression, lesional skin in paraneoplastic acanthosis nigricans and SKs has alterations in the extracellular matrix. It is unknown what the direct impact of growth factor signaling has on the skin. Proposed ideas include either inducing a hyperproliferative state primarily or by altering the surrounding environment such as via the extracellular matrix. These similar mechanisms further support the idea of a continuum between acanthosis nigricans and the Leser-Trélat sign.
DIAGNOSIS
A skin biopsy may be done to confirm the diagnosis. Histopathologic findings of SK in this syndrome are the same as the common SK and include epidermal hyperkeratosis, papillomatosis, and acanthosis with cystic inclusion of keratinous material (horn pseudocysts).
DIFFERENTIAL DIAGNOSIS
Differential diagnosis of the individual skin lesion in this syndrome is the same as for SKs, consisting of fibroepithelial polyp, epidermal nevus, melanocytic nevus, pigmented basal cell carcinoma, squamous cell carcinoma, malignant melanoma, warty dyskeratoma, verruca vulgaris, and condyloma acuminatum. In extensive and generalized SKs, which can appear in older healthy individuals, it is important to
distinguish the benign presentation from the signs of Leser-Trélat.
CLINICAL COURSE AND PROGNOSIS
Leser-Trélat sign can develop from approximately 5 months prior to 10 months after the diagnosis of malignancy. More than half of the patients have metastatic tumors at the time of diagnosis. The prognosis is poor, with an estimated survival time of 1 year after diagnosis. This eruption parallels the course of the underlying malignancy in some cases, but not as a general rule.
MANAGEMENT
Treatment should be directed toward the underlying tumor. If the lesions are symptomatic or cosmetically concerning to the patient, local treatment of SK, such as α-hydroxy acids, retinoids, trichloroacetic acid, cryosurgery with or without curettage, dermabrasion, laser, and shave removal, can be performed.
ACROKERATOSIS PARANEOPLASTICA OF BAZEX
ACROKERATOSIS
PARANEOPLASTICA OF
BAZEX
AT-A-GLANCE
■ Characteristic cutaneous lesions and distribution symmetrically involving the helices, nose, cheeks, digits, and nails.
■ Lesions evolve from nonspecific dermatitis and paronychia to inflammatory plaques, acral keratoderma, and nail plate changes.
■ Skin lesions often predate detection of internal malignancy.
■ Upper aerodigestive tract malignancy is the most common.
Acrokeratosis paraneoplastica of Bazex was first presented in 1965 at the French Dermatological Society. Bazex and his colleagues described a patient with scaly erythematous lesions of the extremities who had a carcinoma of the pyriform fossa, whose skin lesions cleared after treatment of the carcinoma. The lesions were proposed as a cutaneous marker of internal malignancy.11
This condition is also known as Bazex syndrome, which is an eponym used to describe 2 different clinical entities. The first refers to acrokeratosis paraneoplastica of Bazex, which is discussed in this chapter, and the second is Bazex-Dupré-Christol syndrome, a very rare
21
genodermatosis, characterized by congenital hypotrichosis, follicular atrophoderma, and basal cell carcinomas arising at an early age.
EPIDEMIOLOGY
More than 100 cases of Bazex syndrome have been reported in the medical literature. This condition is highly associated with underlying malignancy. It occurs almost exclusively in males older than age 40 years. Approximately 60% of the associated malignancies are squamous cell carcinomas of the upper aerodigestive tract. The second most common is lung cancer. Other less-common tumor locations are genitourinary and lower GI tract.12
CLINICAL FEATURES
The characteristic cutaneous findings are symmetrical erythematous to violaceous scaly patches or plaques over the acral extremities, ears, and bridge of the nose. Hyperpigmentation tends to appear in dark-skinned individuals. Vesicles and bullae are occasionally observed on the hands and feet. A bulbous enlargement of the distal phalanges has been described. The most common sites of involvement are the nails (77%), ears (76%), fingers (65%), nose (62%), palms (56%)/ hands (51%), and soles (49%)/feet (44%). Several patients have an additional cutaneous paraneoplastic syndrome, which includes acquired ichthyosis, Leser-Trélat sign, and clubbing.12 Bazex and Griffiths described 3 stages of skin lesions that parallel the growth and dissemination of the underlying tumor. In the first stage, the neoplasm is frequently undetected. The helices, nose, fingers, and toes are usually affected in a symmetrical fashion. Early lesions are ill-defined scaly papules, simulating a nonspecific dermatitis. The eruption is classically asymptomatic, but pruritus may be a problem. Paronychia is the first sign of nail involvement (Fig. 134-5) and may be tender. Nail dystrophy, subungual hyperkeratosis, and onycholysis progressing to complete destruction of the nail plate have been reported. During the second stage, the tumor exhibits symptoms, resulting from local extension or metastatic
2451
21
spread, and the skin eruption becomes more extensive. The typical red-to-purple scaly plaques may involve the whole pinna, cheeks, or upper lip. The palms and soles develop a keratoderma that often spares the central volar surfaces, but may lead to painful fissures (Fig. 134-6). Nail plate changes include yellowing, thickening, onycholysis, and ridging, both horizontal and vertical. The final stage is observed when the tumor goes untreated or fails to respond to treatment. All of the above signs and symptoms persist as the papulosquamous lesions begin to appear on other sites, such as trunk, elbows, knees, scalp, and dorsal hands and feet. Rarely, vesicles and bullae may be present, most commonly on the fingers, hands, and feet. Nail changes can be quite variable, ranging from the typical thickening to nail atrophy and loss of the nail plate.
ETIOLOGY AND PATHOGENESIS
The pathophysiology of acrokeratosis paraneoplastica remains unclear. Many authors propose an immunologic mechanism in which the antibodies against the tumor crossreact with keratinocyte and basement membrane antigen, or a T cell-mediated immune response to tumor-like antigens in the epidermis. The findings of immunoglobulins (IgG, IgA, IgM) and complement (C3) along the basement membrane zone in some patients have been observed. Some authors also suggest that epidermal growth factor secreted by the tumor may play a role in epidermal hyperplasia and hyperkeratosis, as mentioned in other hyperkeratotic dermatoses in this chapter. Other hypotheses include low serum levels of vitamin A and zinc.12
2452
DIAGNOSIS
Histopathology is somewhat nonspecific. Psoriasiform dermatitis is the most common pattern. Hyperkeratosis, parakeratosis, and a superficial lymphohistiocytic infiltrate are the main histopathologic features. The interface changes, including vacuolar degeneration, dyskeratotic keratinocytes, lichenoid infiltrate, and melanin-containing macrophages in the dermis, are reported less frequently.12
In the context of histopathologic and characteristic cutaneous findings, acrokeratosis paraneoplastica of Bazex could be a possible diagnosis. If the patients have no history of malignancy, appropriate investigations should be promptly performed.
DIFFERENTIAL DIAGNOSIS
The differential diagnoses include psoriasis, pityriasis rubra pilaris, seborrheic or contact dermatitis, eczematous drug eruption, infections (eg. dermatophytosis, onychomycosis), lupus erythematosus, palmoplantar keratodermas, and mycosis fungoides. The distinguishing clinical feature, which is nearly always present in acrokeratosis paraneoplastica, is involvement of the helices of the ears (Fig. 134-7) and the tip of the nose. Typically, Bazex syndrome is resistant to conventional treatment.
CLINICAL COURSE AND PROGNOSIS
The skin eruption can occur before (67%), simultaneously (18%), or after (15%) the diagnosis of the neoplasm. The cutaneous lesions precede the diagnosis of
the tumor by an average of 12 months. Acrokeratosis paraneoplastica is expected to resolve after successful treatment of the underlying malignancy and can reappear with tumor recurrence. However, nail dystrophy and pigmentary skin changes may persist despite significant improvement in the underlying malignancy and other skin lesions.12
MANAGEMENT
Treatment of the primary neoplasm is the most effective therapy. The skin lesions are typically resistant to standard therapies for hyperkeratotic conditions and few treatments specific for the cutaneous component of acrokeratosis paraneoplastica have been reported. Systemic retinoids have been used with variable success. Isolated reports suggest benefit from oral psoralen and ultraviolet A phototherapy, topical salicylic acid, and corticosteroids.
COLLAGEN–VASCULAR DISEASE
DERMATOMYOSITIS
DERMATOMYOSITIS
Chapter 62, “Dermatomyositis,” provides additional information on dermatomyositis. Adult-onset dermatomyositis (DM) is significantly associated with malignancy. The risk of cancer is higher in DM than in polymyositis alone. Approximately 18% to 25% of patients with adultonset DM may have malignancy, which is associated with decreased survival. The malignancy may occur before, simultaneously, or after the diagnosis of DM. The course of DM does not always follow the course of the malignancy. In contrast, juvenileonset DM does not have a strongly increased risk of malignancy. The clinical manifestations of DM with or without associated malignancy are similar. Previous studies reported predictive signs of accompanying malignancy, such as cutaneous necrosis, cutaneous vasculitis, lack of interstitial lung disease, dysphagia, male gender, elevated erythrocyte sedimentation rate, and an older age at onset. However, some of these findings are inconsistent and remain controversial. Recently, anti-p155 antibodies have been reported as a promising cancer marker in adult patients with DM. Types of associated malignancies vary among studies and may reflect differences in malignancy risk across different populations. Lung, breast, ovarian, cervical, pancreatic, gastric, colorectal, and prostate malignancies are frequently reported; hematologic malignancies may also occur in association with DM. It has been noted that nasopharyngeal carcinoma is more common in the Asian population.13
In adult patients with DM, the risk of malignancy is highest in the first year after diagnosis, then steadily declines and remains slightly elevated even after
21
5 years. Therefore, all adult patients with DM should be evaluated for malignancy at the time of diagnosis, followed by long-term surveillance. There is no definitive guidelines for cancer screening in patients with DM. Most authors recommend an initial screening that includes a comprehensive history and physical examination (including a pelvic examination in women), standard laboratory tests (complete blood count, complete metabolic panel, urinalysis, stool occult blood testing), and chest radiography. Further investigation is directed by any abnormal findings. Some clinicians also recommend a colonoscopy and CT scans of the chest, abdomen, and pelvis. Additional age-, gender-, and ethnicity-appropriate malignancy screening, including a transvaginal pelvic sonography, Papanicolaou smear, and mammography for female patients, or an evaluation by an ear-nose-throat physician to exclude nasopharyngeal carcinoma for patients of Asian descent, should be considered.13
PROGRESSIVE SYSTEMIC SCLEROSIS
PROGRESSIVE SYSTEMIC
SCLEROSIS
Chapter 63, “Systemic Sclerosis,” provides additional information on progressive systemic sclerosis. The majority of epidemiologic studies reveal an increased risk of cancer in patients with progressive systemic sclerosis (PSS) compared with general population, and demonstrated that men are at higher risk than women. The frequency of malignancy is approximately 3% to 11%.14 The risk of cancer is higher within the first 12 months after a PSS diagnosis, and may be a paraneoplastic phenomenon. However, other studies included in a newer metaanalysis did not demonstrate an increase in the risk of cancer.15
Lung, bladder, breast, liver, esophageal, and oropharyngeal carcinoma, nonmelanoma skin cancer, and hematologic malignancies have been frequently reported in association with PSS. Lung cancer is the most frequent type of cancer in most studies, and may be associated with the presence of interstitial lung disease, as well as with history of smoking. Interestingly, there is a 25-fold increased incidence of tongue cancer in one U.S. study.14,16
This increased risk of cancer could be the result of damage from chronic inflammation and fibrosis, immunosuppressive therapies for PSS, an environmental exposure, and/or a genetic susceptibility to the development of both cancer and autoimmunity. Although the data are controversial, some studies suggest that the risk of cancer may be greater in patients with diffuse cutaneous disease. Several newer studies confirm an increased risk of cancerassociated PSS in patients with RNA polymerase III autoantibodies or an older age of PSS onset. These groups of patients may benefit from aggressive malignancy screening at diagnosis and long-term cancer surveillance.16
2453
21
REACTIVE ERYTHEMAS
ERYTHEMA GYRATUM REPENS
ERYTHEMA GYRATUM
REPENS
Chapter 46, “Erythema Annulare Centrifugum and Other Figurate Erythemas,” provides additional information on erythema gyratum repens. This particular annular (gyratum is Latin for circle) erythema was thought to be one of the most specific dermatoses associated with underlying neoplasia. However, newer data demonstrate a higher proportion of associated benign conditions than previously reported. Erythema gyratum repens is a rare disease that presents with dramatic appearance and evolution. Nearly 100 cases are reported in the literature. It mostly affects adults in their 60s, men more than women (ratio: 2:1). Numerous serpiginous bands are arranged in a parallel configuration of concentric red swirls over most of the body. This presentation is occasionally referred to as a “wood-grain” appearance (Fig. 134-8). Even more striking is the relatively rapid rate at which lesions migrate (repens is Latin for creeping), estimated at 1 cm per day. A fine scale may be found along the trailing edge of erythema (Fig. 134-9). The hands, feet, and face are commonly spared, except for occasional volar hyperkeratosis. Ichthyosis is present in many cases. Pruritus is universal and may be severe.17
An underlying malignancy is associated with erythema gyratum repens approximately 70% of the time. In more than 80% of cases, the cutaneous eruption appears before the diagnosis of malignancy with a mean period of 7 months (range: 1 to 72 months),
2454
but it can occur simultaneously or after the diagnosis of malignancy. Among patients with associated carcinoma, almost half of cases have lung cancer, whereas 8% have stomach cancer, 7% have esophageal cancer, and 5% have breast cancer. Individual case reports of many other types of associated tumors are published, as well as 5% of cases with an unknown primary. In those individuals with erythema gyratum repens who did not have a detectible underlying malignancy; some cases were idiopathic; some presented with concurrent conditions including concomitant skin disease (pityriasis rubra pilaris, psoriasis, ichthyosis), connective tissue disease (CREST [calcinosis cutis, Raynaud phenomenon, esophageal motility disorder, sclerodactyly, and telangiectasia] syndrome, rheumatoid arthritis), infection (tuberculosis), hypereosinophilic syndrome, or drug reaction (azathioprine).18
The exact etiology of erythema gyratum repens is unknown, but it has been suggested that the tumor may induce a chemical alteration of the normal components of the surrounding tissue. Molecular mimicry ensues as the inflammatory response directed against the tumor crossreacts with benign cutaneous proteins. This theory is supported by documentation of IgG and C3 deposition at the basement membrane of affected skin and bronchial basement membrane in one case associated with lung cancer. Migration characterizes all of the figurate erythemas, but is notably rapid in erythema gyratum repens. The mechanism for this is not clear, although some studies have focused on fibroblast activity. Inflammatory cells and/or fibroblasts may mediate ground substance alterations, which may localize the inflammation that orchestrates the movement of the infiltrate in a patterned mode.17
Treatment of erythema gyratum repens involves locating and treating the primary malignancy. With adequate control of the cancer the rash usually abates, but this may not be possible in cases that are widely metastatic at the time of diagnosis. Otherwise, the eruption is often treatment resistant, although variable results occur with systemic steroids. Topical steroids,
vitamin A, and azathioprine have not been beneficial. The eruption has been known to resolve immediately before death, possibly because of generalized ante mortem immunosuppression.17
NECROLYTIC MIGRATORY ERYTHEMA
NECROLYTIC MIGRATORY
ERYTHEMA
AT-A-GLANCE
■ Painful, eroded, crusted intertriginous, and facial skin eruption.
■ Highly suggestive of pancreatic malignancy (glucagonoma).
■ Half of tumors are metastatic at the time of diagnosis.
■ Pseudoglucagonoma syndrome occurs in absence of glucagonoma.
■ Skin improves with treatment of underlying nutritional aberrations.
In 1942, Becker first described a patient with cutaneous eruptions in association with α-cell pancreatic tumor. In 1966, McGavran found another patient with similar clinical presentation and had hyperglucagonemia. Later, necrolytic migratory erythema (NME) was coined by Wilkinson in 1973 to describe the distinctive rash in patients with pancreatic cancer.19
EPIDEMIOLOGY
NME is a very rare paraneoplastic skin disorder that is considered a hallmark of glucagonoma and is present in more than two-thirds of patients at the time of tumor diagnosis.19 Glucagonoma is an extremely rare, slow-growing neuroendocrine tumor of the α cells of the pancreas. The global incidence is approximately 1 in every 20 million people. It typically presents with glucagonoma syndrome.2 There is no gender or race predilection, and it most commonly affects people in their sixth decade.
CLINICAL FEATURES
The skin lesions of NME are polymorphous, but erosions and crusts are usually apparent. Primary lesions are erythematous patches that evolve into plaques and develop central bullae. The blisters erode rapidly, form crusts, and eventually resolve. The erythematous and eroded annular patches and plaques coalesce into large geographic areas. They are typically painful and pruritic. The eruption disappears and reappears spontaneously over the course of weeks. The distribution of
21
NME is characteristic and includes intertriginous areas (eg. groin, perineum, buttocks, and lower abdomen), the central face (especially perioral), and distal extremities (Fig. 134-10). Mucosal involvement manifests as angular cheilitis, atrophic glossitis, and stomatitis. Dystrophic nails may accompany the syndrome.19,20
Glucagonoma syndrome is characterized by NME, weight loss, sore mouth, diarrhea, diabetes mellitus, deep vein thrombosis, normochromic normocytic anemia, and neuropsychiatric disorders. Weight loss is the most common presenting sign. The glucagonoma tends to grow slowly. Metastasis to the liver, regional lymph nodes, and bone is common, but appears in late stage of the disease. Pseudoglucagonoma syndrome presents identically, but the α cell pancreatic tumor is not present, which may explain why the serum glucagon levels are not elevated. Underlying diseases identified in patients with the pseudosyndrome include liver disease, pancreatitis, celiac sprue, inflammatory bowel disease, acrodermatitis enteropathica, pellagra, and nonpancreatic malignancies.19,20
A
B
2455
21
Because NME can be the first manifestation of the syndromes, early recognition is important and may provide a better outcome.
ETIOLOGY AND PATHOGENESIS
The exact cause of NME remains unclear. The skin rash can be attributed to the metabolic effects of excess glucagon, as resolution of the rash can occur after surgical removal of the glucagonoma or normalizing the glucagon level with medication. Hyperglucagonemia stimulates hepatic gluconeogenesis leading to an increase in blood glucose level. The amino acid consumption in gluconeogenesis pathway contributes to a decrease in serum amino acid. Hypoaminoacidemia resulting in epidermal protein deficiency (histidine and tryptophan) may provoke epidermal necrosis. Glucagon also increases cutaneous levels of arachidonic acid, prostaglandins, and leukotrienes, which may induce a cutaneous inflammatory reaction. However, NME cannot be completely attributed to hyperglucagonemia, because it may appear without elevated levels of glucagon or a pancreatic tumor, as reported in pseudoglucagonoma syndrome. Deficiency of zinc, protein, essential fatty acid, and vitamin B may also play a role in NME development.20
DIAGNOSIS
The histopathology of NME is characteristic, but not pathognomonic. Acute lesions exhibit a striking degree of epidermal necrosis in the upper layers of the stratum spinosum and may detach from the viable epidermis underneath. Neutrophiic infiltrates in the necrotic layer resulting in subcorneal pustules can be observed. Chronic lesions show a psoriasiform dermatitis. Parakeratosis, loss of granular layers, basal vacuolization, and scattered necrotic keratinocytes may be demonstrated. These findings can be also seen in other nutritional deficiencies, graft-versus-host disease, connective tissue disorders, and phototoxic drug eruptions.19,20
Laboratory abnormalities include a dramatically elevated serum glucagon, usually greater than 1000 pg/mL (reference range: 50 to 150 pg/mL). Most patients have hyperglycemia and a normochromic normocytic anemia. Abnormal liver function is present and serum levels of amino acids, total protein, albumin, and cholesterol are low. Occasionally, zinc levels are also decreased. Imaging studies should be performed to detect the pancreatic tumor. Various methods, such as angiography, ultrasonography, CT, MRI, positron emission tomography, octreotide scintigraphy, and somatostatin receptor scintigraphy, have been used to identify the pancreatic tumor.20
DIFFERENTIAL DIAGNOSIS
Other disorders with cutaneous manifestations similar to NME include acrodermatitis enteropathica, nutritional deficiencies, psoriasis, eczema, seborrheic
2456
dermatitis, candidiasis, superficial pemphigus, and side effects of certain chemotherapies.
CLINICAL COURSE AND PROGNOSIS
In NME associated with glucagonoma syndrome, the course of the disease varies according to the stage at which the tumor is diagnosed. If the glucagonoma is not metastatic and can be completely resected, symptoms of the syndrome will resolve. Unfortunately, by the time of diagnosis, tumors are frequently large and metastatic in most cases. Fortunately, the tumors are slow growing and patients may experience symptom improvement by surgically reducing the tumor burden, although survival may not be affected by the procedure.
MANAGEMENT
The underlying cause for hyperglucagonemia must be addressed to eradicate the painful skin disease. For patients with glucagonoma, resection of the tumor is important for symptom relief. Complete surgical removal can be curative if the tumor is confined to the pancreas. This tumor is often resistant to chemotherapy. Somatostatin analog (octreotide, pasireotide, and lanreotide) improves the cutaneous symptoms and may delay tumor progression. Other therapeutic modalities, such as interferon, everolimus (mammalian target of rapamycin inhibitor), sunitinib (tyrosine kinase inhibitor), and peptide receptor radionuclide therapy, have been reported with favorable results. Supplementation to correct zinc, amino acid, or fatty acid deficiencies improves skin lesions in some cases.19
NEUTROPHILIC DERMATOSES
SWEET SYNDROME
SWEET SYNDROME
Chapter 36, “Sweet Syndrome,” provides additional information on Sweet syndrome. Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is characterized by fever, leukocytosis, and skin papules or plaques caused by dermal neutrophils. Malignancy is reported in approximately 21% of patients diagnosed with Sweet syndrome, either hematologic (15%) or solid (6%) malignancy. The most commonly-associated hematologic malignancy is acute myeloblastic leukemia. Other hematologic neoplasms include myeloproliferative neoplasms, diffuse large B-cell lymphoma, Hodgkin lymphoma, myelodysplastic syndrome, and myelofibrosis. The most common solid malignancies associated with Sweet syndrome are carcinomas of the genitourinary organs, breast, and GI tract, most frequently adenocarcinomas (57%).21
Several authors proposed the features to distinguish malignancy-associated Sweet syndrome from the classical form of the disease. Malignancy-associated Sweet
syndrome is less-often preceded by upper respiratory tract infection, and the onset is temporally associated with the new discovery or relapse of cancer. In addition, there is no sex preponderance, whereas female sex is predominant in the classical form.22
Clinically, bullous and ulcerated skin lesions, subcutaneous nodules, and oral mucosal involvement may be more frequently observed in malignancy-associated than in classical Sweet syndrome. Laboratory abnormalities, including anemia (82% to 83%) and abnormal platelet count (68%), are reported in malignancyassociated Sweet syndrome. Although peripheral leukocytosis is one of the diagnostic criteria of Sweet syndrome, neutropenia can be observed in some patients with malignancy-associated Sweet syndrome. A small number of patients with hematologic malignancy demonstrate leukemia cutis concurrently with Sweet syndrome in lesional skin biopsy.21,22
The onset of Sweet syndrome can precede, follow, or appear concurrently with the diagnosis of a patient’s neoplasm. The duration of Sweet syndrome is variable. Recurrence of lesions can occur not only in malignancy-affected patients, but also in individuals with either idiopathic or disease-associated Sweet syndrome. There are no specific guidelines for the treatment of malignancy-associated Sweet syndrome. Systemic corticosteroid and other standard treatments that are used in classical or idiopathic sweet syndrome are the mainstay of therapy. Successful treatment of underlying malignancy may result in complete resolution of malignancy-associated Sweet syndrome, and the reappearance of dermatologic findings may signify relapse of cancer.21,22
PYODERMA GANGRENOSUM
PYODERMA
GANGRENOSUM
Chapter 37, “Pyoderma Gangrenosum,” provides additional information on pyoderma gangrenosum. Pyoderma gangrenosum (PG) is a rare, painful, ulcerating neutrophilic dermatosis associated with various internal diseases. The incidence of malignancy in patients with PG varies among studies, occurring in approximately 4% to 20% of PG patients.23
Hematologic malignancy is the most common associated neoplasm; other associated malignancies include acute and chronic myelogenous leukemia, chronic lymphocytic leukemia, multiple myeloma, myelodysplastic syndrome, polycythemia vera, Hodgkin lymphoma, and cutaneous T-cell lymphoma. Atypical features of PG, such as abrupt onset, superficial lesions, hemorrhagic bullae, and involvement of the upper extremities have been described in 27% of PG patients with underlying hematologic disease or malignancy.24 Improvement of bullous PG with treatment of the underlying malignancy has been documented. Monoclonal gammopathy of undetermined significance has been described by several authors as an associated condition with PG, with IgA as the most
21
frequently associated paraproteinemia.23 These gammopathies are clinically benign, but they occasionally progress to myeloma. Therefore, long-term monitoring is recommended. Underlying solid tumors can also occur in patients with PG. Cancers of various organs, such as breast, prostate, lung, bladder, colon, liver, ovary, larynx, glioblastoma multiforme, and melanoma, have been occasionally reported.23
DERMAL PROLIFERATIVE DISORDERS
MULTICENTRIC RETICULOHISTIOCYTOSIS
MULTICENTRIC
RETICULOHISTIOCYTOSIS
Chapter 117, “Histiocytosis,” provides additional information on multicentric reticulohistiocytosis. Multicentric reticulohistiocytosis (MRH) is a rare, systemic disease presenting with destructive polyarthritis and typical cutaneous lesions. The isolated or grouped reddish brown to skin-colored papules and nodules are seen predominantly on the face and hands with a characteristic “coral bead” appearance of periungual papules. Up to 25% of patients have associated malignancies, and in some cases the diagnosis of multicentric reticulohistiocytosis precedes the diagnosis of cancer.25
There is no predominant type of associated cancer in this disease. Various neoplasms, including breast, lung, thyroid, GI, urogenital, sarcoma, lymphoma, leukemia, and melanoma, have been reported. The course of the disease may be self limtied without joint deformity; waxing and waning; or aggressive with mutilating arthritis. Spontaneous remission usually occurs after many years. It does not run a parallel course with the associated malignancy in the majority of patients. Treatment of the accompanying cancer will resolve joint and skin disease only in some reported cases. It is still debated among several authors whether to label multicentric reticulohistiocytosis as a paraneoplastic disorder. Regardless, a complete workup for underlying malignancy should be performed in every patient diagnosed with multicentric reticulohistiocytosis.26
NECROBIOTIC XANTHOGRANULOMA
NECROBIOTIC
XANTHOGRANULOMA
Chapter 57, “Intercellular IgA Dermatosis (IgA Pemphigus),” provides additional information on necrobiotic xanthogranuloma. Necrobiotic xanthogranuloma is a rare, non– Langerhans cell histiocytosis with a strong association with hematologic disorders. The skin is the most common site of involvement and more than 80% of patients have periorbital lesions. Extracutaneous involvement is rare. The lesions appear as yellowish to red-orange or violaceous papules, plaques, or nodules, with areas
2457
21
of ulceration, telangiectasia, or atrophy. Most of the cases are asymptomatic with an indolent course. Up to 80% of patients have monoclonal gammopathy of IgG type with either a κ or λ light chain.27 The most common associated plasma cell dyscrasias are monoclonal gammopathies of undetermined significance, smoldering multiple myeloma, and multiple myeloma. Other hematologic abnormalities, including non-Hodgkin lymphoma, chronic lymphocytic leukemia, Hodgkin lymphoma, and lymphoplasmacytic lymphoma, may occur in association with necrobiotic xanthogranuloma.27
Patients with necrobiotic xanthogranuloma associated with monoclonal gammopathy are at risk of conversion to multiple myeloma. Consequently, careful monitoring should be continued for early detection of disease progression.27
DERMAL DEPOSITION
SCLEROMYXEDEMA
SCLEROMYXEDEMA
Chapter 67, “Scleredema and Scleromyxedema,” provides additional information on scleromyxedema. Scleromyxedema, the generalized form of papular mucinosis (lichen myxedematosus), is an uncommon disease associated with monoclonal gammopathy and systemic organ involvement. The typical cutaneous manifestations have a predilection for the face, arms, and hands. Cardiopulmonary, rheumatologic, GI, and neurologic involvement is common. The majority (80%) of patients with scleromyxedema has an IgGλ light chain monoclonal gammopathy of undetermined significance. Serum protein electrophoresis should be performed in conjunction with thyroid studies to rule out thyroid dysfunction and myxedema. Fortunately, the paraproteinemia only rarely converts to multiple myeloma, but when it occurs, it portends a poor prognosis. There are several case reports of other hematologic and nonhematologic malignancies among patients with scleromyxedema, including leukemia, Hodgkin or non-Hodgkin lymphoma, and some solid tumors. However, it is uncertain if some of the associated malignancies are incidental or associated with the treatment of scleromyxedema with agents, such as melphalan as a secondary malignancy.28 The course of the disease is usually progressive and major systemic organ involvement may contribute to poor outcome.
SYSTEMIC AMYLOIDOSIS
SYSTEMIC AMYLOIDOSIS
Chapter 124, “The Porphyrias,” provides additional information on systemic amyloidosis. Primary systemic amyloidosis is referred to as AL amyloidosis, in which there is extracellular deposition of fibrils of monoclonal immunoglobulin light chain typically produced by a small plasma cell clone. Secondary (AA) amyloidosis occurs with autoimmune or inflammatory diseases, malignancies, and chronic
2458
infections. Skin manifestations of systemic amyloidosis include “pinch purpura” (periorbital bruising), macroglossia, and, less commonly, waxy skin thickening and subcutaneous nodules. AL amyloidosis is the most common form associated with malignancy. The prognosis is generally poor, with median survival time if left untreated of 12 months. In AL amyloidosis, multiple organs and tissues (kidney, heart, liver, peripheral/autonomic nerve, soft tissue) are generally involved. It is important to distinguish the skin lesions in systemic AL amyloidosis from the less-common, localized, cutaneous variant, which does not progress to multisystem involvement. Multiple myeloma is the most common associated malignancy, and is seen in approximately 20% of patients with AL amyloidosis.29 Other neoplasms, such as non-Hodgkin lymphoma, mucosa-associated lymphoid tissue lymphoma, lymphoplasmacytic lymphoma, and other single case reports of solid-organ tumors, have been rarely reported. For all patients with suspected AL amyloidosis, serum and urine protein electrophoresis/immunofixation electrophoresis, serum free light chain assay, bone marrow biopsy, and skeletal imaging should be performed at baseline to rule out the presence of multiple myeloma.30
Some investigators have described an increased risk of non-Hodgkin lymphoma in transthyretin amyloidosis (ATTR).31 In addition, certain neoplasms, such as hepatocellular carcinoma, renal cell carcinoma, Castleman disease, Hodgkin disease, and adult hairy cell leukemia, can cause reactive or secondary (AA) amyloidosis.30
BULLOUS DISORDERS
PARANEOPLASTIC PEMPHIGUS
PARANEOPLASTIC
PEMPHIGUS
Chapter 53, “Paraneoplastic Pemphigus,” provides additional information on paraneoplastic pemphigus. Paraneoplastic pemphigus is a life-threatening condition with a high mortality rate. Painful, hemorrhagic oral erosions are the earliest characteristic clinical finding. The polymorphous skin eruption, composed of pemphigus-like, bullous pemphigoid–like, erythema multiforme–like, graft-versus-host disease–like and lichen planus–like skin lesions, may involve any site. Multiorgan involvement, such as of the lung, thyroid, kidney, smooth muscle, and GI tract, is documented. The pathogenesis of paraneoplastic pemphigus involves both cellular and humoral immunity. Nearly all patients have an underlying neoplasm, most frequently a lymphoproliferative disease. The most commonly reported associated neoplasms are non- Hodgkin lymphoma, chronic lymphocytic leukemia, Castleman disease, and thymoma. The prognosis is poor in patients with malignancy-associated paraneoplastic pemphigus but may be better in patients with benign tumors.32
DERMATITIS HERPETIFORMIS
DERMATITIS
HERPETIFORMIS
Chapter 59, “Dermatitis Herpetiformis,” provides additional information on dermatitis herpetiformis. Dermatitis herpetiformis is an autoimmune skin condition that presents as a severely pruritic skin eruption with polymorphous lesions and is associated with gluten sensitivity. The majority of studies have demonstrated a significantly increased risk of non-Hodgkin lymphoma in patients with dermatitis herpetiformis, even though the risk of malignancy overall is similar to general population.33,34 It has been noticed that patients with longstanding dermatitis herpetiformis are at particular risk. The reported frequency of associated malignancy in patients with dermatitis herpetiformis is up to 4.3%, predominantly in males.33 However, patients with dermatitis herpetiformis do not appear to have an increased mortality rate despite this association with non-Hodgkin lymphoma.33,34
The lymphomas associated with dermatitis herpetiformis can be either B-cell or T-cell lymphomas, and may occur both in and outside the GI tract as nodal or extranodal disease. In addition, the enteropathyassociated T-cell lymphoma, typically associated with celiac disease, has been reported in dermatitis herpetiformis patients.34 A gluten-free diet may protect against the development of cancer, but more studies are needed to confirm this finding.33,34
BULLOUS PEMPHIGOID
BULLOUS PEMPHIGOID
Chapter 54, “Bullous Pemphigoid,” provides additional information on bullous pemphigoid. Bullous pemphigoid (BP) is an autoimmune blistering disorder that presents in elderly patients with intact skin blisters and pruritus. It remains debated whether there is an increased risk of malignancy in bullous pemphigoid, other than the risk related to the age of the patient. Several studies, including newer large epidemiologic studies, have not found an association between bullous pemphigoid and cancer. Other studies, however, especially from Japan, demonstrated a higher risk of malignancy in bullous pemphigoid patients, compared to the normal population. Many authors discourage a routine cancer screening in bullous pemphigoid patients, except in early-onset pemphigoid, history of malignancy, or standard treatment failure.35
MISCELLANEOUS
HYPERTRICHOSIS LANUGINOSA ACQUISITA
HYPERTRICHOSIS
LANUGINOSA ACQUISITA
Acquired hypertrichosis lanuginosa (“malignant down”) is a rare condition characterized by the
21
relatively-sudden appearance of long, fine, nonpigmented lanugo (Latin for down) hairs. The lanugo hairs most frequently appear on the face and ears early in the course (Fig. 134-11). The hairs may grow to an impressive length; eyebrows and eyelashes may grow to inches long. The long fine hairs also may be seen on the trunk and limbs, including the axillae, but the palms, soles, suprapubic, and genital areas are usually spared. Acquired hypertrichosis lanuginosa often develops in a cephalocaudal direction and may be accompanied by glossitis, hypertrophy of tongue papillae, oral hyperpigmentation, disturbances of taste and smell, acanthosis nigricans, diarrhea, adenopathy, and weight loss.36
This condition is frequently paraneoplastic and can be a marker of an underlying malignancy. It usually appears in advanced or metastasized carcinomas, and thus has a poor prognosis. The most commonly associated malignancies are lung and colorectal cancer in men, and colorectal cancer followed by lung and breast cancer in women. Ovary, uterus, and urinary bladder carcinoma, lymphoma, and leukemia also have been found in patients with acquired hypertrichosis lanuginosa, as have other malignancies.37
Acquired hypertrichosis lanuginosa must be distinguished from hirsutism and hypertrichosis associated with nonmalignant causes, including metabolic and endocrine diseases, such as anorexia nervosa, thyrotoxicosis, and porphyria cutanea tarda (see Chap. 124), or medications such as cyclosporine, phenytoin, penicillamine, spironolactone, psoralens, corticosteroids, interferon, diazoxide, or minoxidil.36,37
Patients presenting with acquired hypertrichosis lanuginosa without history of drug administration or underlying metabolic and endocrine or malignant diseases, should be suspected of an underlying
2459
21
cancer. Appropriate diagnostic evaluations for the occult malignancy, especially of the lung, colon, rectum areas, and breast, should be considered.
TROUSSEAU SYNDROME
TROUSSEAU SYNDROME
The definition of Trousseau syndrome has evolved over time. It originally described the occurrence of migratory thrombophlebitis associated with underlying gastric cancer. Later, it was expanded by several authors to include cancer-associated hypercoagulability with a wide range of clinical manifestations, including abnormal coagulation tests without clinical symptoms, superficial migratory thrombophlebitis, deep vein thrombosis, marantic endocarditis, pulmonary embolism, and massive thromboembolic phenomenon associated with disseminated intravascular coagulation. Venous thromboembolism (VTE) seems to be the most frequent clinical manifestation.38
Clinically, VTE in patients with malignancy can be more severe and more extensive than in patients without malignancy. Deep vein thrombosis of unusual sites, such as upper limbs, thrombosis of visceral organs and brain, and arterial thromboembolism have been reported to occur in cancer-associated cases. Trousseau syndrome composes approximately 20% to 30% of all VTE patients, and occurs in 1% to 8% of patients with underlying malignancy. The incidence has increased, possibly as a result of the more frequent use of newer thrombogenic chemotherapeutic and immunomodulatory agents (eg, thalidomide or lenalidomide) and the longer survival period in cancer patients. Unfortunately, VTE is the second most common cause of death in patients with cancer. Patients with active cancer have a 4 to 7 times higher risk of thrombosis than the normal population, and the risk is highest in certain cancers, such as pancreatic, brain, lung, stomach, and ovarian cancers.38-40
Although the pathogenesis of Trousseau syndrome in cancer patients is unclear, multiple factors are likely to be involved in the thrombogenic process. The combination of abnormal blood flow or stasis, vessel wall injury, and blood hypercoagulability, referred to as the Virchow triad, contributes to thrombus formation. Several studies have described the overexpression of tissue factor by tumor cells and the increase of plasma microparticles, which are involved in the coagulation pathway. Chemotherapy and tumor necrosis may induce inflammatory cytokines resulting in endothelial cell injury. Neutrophil extracellular traps (NETs), released as a result of neutrophil programmed cell death (NETosis), have effects on the intrinsic coagulation pathway. Moreover, several investigators have documented specific signaling pathways that contribute to the procoagulant phenotype of cancer cells, resulting from mutations in oncogenes (eg, K-ras, EGFR, PML/RAR-α, MET) and tumor suppressor genes (eg, p53, PTEN).38,40
As Trousseau syndrome can be a presenting manifestation of an occult cancer, several authors suggest
2460
an age-specific and sex-specific malignancy screening test in patients with idiopathic VTE. Treatment of the underlying malignancy along with anticoagulant therapy is recommended. Low-molecular-weight heparin is the first-line treatment.
GENETIC SYNDROMES INVOLVING SKIN AND/OR MUCOUS MEMBRANES AND ASSOCIATED WITH MALIGNANCY
It is important for clinicians to recognize cutaneous features of genetic conditions that predispose to internal malignancy. In several cases, cutaneous features predate the development of malignancy and increased surveillance may improve overall survival in these patients. For malignancies associated with a genodermatosis, there may be an increased risk for family members to develop cancer and appropriate genetic counseling and testing should be considered.
DIRECT TUMOR INVOLVEMENT OF THE SKIN
CUTANEOUS METASTASES
CUTANEOUS METASTASES
Cutaneous metastases represent involvement of the skin by metastatic spread of a distant primary tumor. The overall incidence of cutaneous involvement is approximately 5%. The most common malignancies to metastasize to the skin in women are breast cancer, followed by colon cancer and melanoma. In men, the most common are lung cancer, followed by colon cancer and melanoma.47
Cutaneous metastases most commonly appear as a rapid onset of solitary or multiple, asymptomatic, skin-colored, mobile, firm, round or oval nodule(s). Metastatic breast cancer can present with various forms of skin lesions. For example, carcinoma erysipelatoides, presents with warm, tender erythematous patches or plaques resembling erysipelas or cellulitis (Fig. 134-12A), but without fever or leukocytosis. Another clinical variant is the leather-like skin changes of sclerosing metastatic breast cancer, known as carcinoma en cuirasse, which may later present as nodules and ulceration (Fig. 134-12B, C). Although carcinoma en cuirasse has been reported as the presenting sign of breast cancer, it more commonly occurs as a local recurrence after treatment of an underlying breast cancer. Both carcinoma erysipelatoides and carcinoma
A
B
C
en cuirasse are not restricted to breast cancer; they also can be seen in lung, kidney, GI tract, and other metastasizing malignancies.48 Metastases from malignant melanoma are usually pigmented (Fig. 134-13). Often there is a bluish tint to the lesion, even if it is deep in the skin. However, even if the primary tumor was pigmented, the metastases may be amelanotic and vice versa. The thorax is the most common site for cutaneous metastasis, as a result of the high frequency of metastatic breast and lung cancers. The scalp is another common site, for metastases from lung (Fig. 134-14), kidney, and breast tumors. On the scalp, the metastatic tumors typically present as single or multiple firm nodules, but scarring alopecia, known as alopecia neoplastica, can be occasionally seen, especially from metastatic breast cancer. The face and neck may be involved by metastases from oropharyngeal carcinomas. Metastases from renal and thyroid carcinoma may be pulsatile and may have a bruit.48
Several histopathologic features may help identify the source of the primary tumor. Microscopically, the collection of neoplastic cells, which usually resemble
21
their malignancy of origin, is generally seen in the dermis and/or subcutaneous tissue. Additional features, such as tumor cells in an “Indian filing” pattern, lymphovascular invasion, necrosis, and a tumor-free “grenz zone,” are also helpful for the diagnosis of metastatic skin lesions. In some types of cancers or poorly differentiated tumors, immunohistochemical staining is often required to achieve the correct diagnosis. For example, the immunohistochemical panel of CK7, CK20, and S-100 is a helpful tool in the diagnoses of breast, lung, and GI cancers and melanoma. Additional
2461
21
recommended markers include TTF-1 (thyroid transcription factor-1; lung), HMB-45 (homatropine methylbromide; melanoma), PSA (prostate-specific antigen; prostate), estrogen/progesterone receptors (breast), and chromogranin (neuroendocrine tumors).47,48
Cutaneous metastases may indicate advanced disease and a poor prognosis. The estimated mean survival after the diagnosis of cutaneous metastases is 50% at 6 months. However, according to recent studies, patients with breast cancer may have a better prognosis than those with other cancers.48
LEUKEMIA AND LYMPHOMA CUTIS
LEUKEMIA AND
LYMPHOMA CUTIS
Chapter 119, “Cutaneous Lymphoma,” provides additional information on leukemia and lymphoma cutis. Cutaneous metastases from leukemia and lymphoma are referred to as leukemia cutis and lymphoma cutis, respectively. Skin manifestations may have similar appearances in both diseases. The lesions generally appear as pink, red, and/or brown-to-purple macules, papules, plaques, and nodules, and are usually firm and painless. Leukemia cutis is an extramedullary manifestation of leukemia, characterized by an invasion of leukemic cells into the epidermis, dermis, or subcutis. The skin eruptions can appear on the trunk, extremities, and head without sites of predilection. Occasionally, erythema, macules, blisters, ulcers, and erythroderma may be seen. Mucosal involvement, especially gum hypertrophy, is common. Nodular or plaque-like infiltration of the face can lead to the development of leonine facies. Rapid and generalized eruption of cutaneous lesions may suggest an acute form of leukemia. Leukemia cutis most frequently develops in patients with acute myeloid leukemia, particularly monocytic (M5) and myelomonocytic (M4) subtypes, but it can also occur in patients with other leukemia subtypes, such as chronic lymphocytic leukemia, chronic myeloid leukemia, chronic myelomonocytic leukemia, and acute lymphocytic leukemia. Most patients with leukemia cutis develop it after a known diagnosis of leukemia, but in rare cases, “aleukemic” leukemia cutis or leukemia cutis without systemic involvement can appear as a presenting manifestation months or years before developing systemic involvement. A skin biopsy is essential to confirm the diagnosis. Immunohistochemical and molecular genetic methods are essential for determining the subtype of leukemia. Treatment is targeted to the specific type of underlying leukemia. Some authors suggest that the appearance of leukemia cutis may indicate aggressive disease and a poor prognosis.49
Lymphoma cutis occurs when systemic lymphoma spreads secondarily to the skin. At times, it may be difficult to distinguish the primary cutaneous lymphoma from lymphoma cutis. Further staging evaluation is needed to document the origins of cutaneous
2462
lymphomas. Cutaneous dissemination can occur in both T-cell and B-cell lymphomas. Systemic anaplastic large-cell lymphoma is a T-cell lymphoma in which skin involvement occurs more commonly in the anaplastic lymphoma kinase–negative subtype. In adult T-cell leukemia-lymphoma, more than half of patients present with skin lesions at the time of diagnosis with chronic and smoldering subtypes resembling mycosis fungoides skin lesions. Cutaneous dissemination in Hodgkin lymphoma is much less frequent than in non- Hodgkin lymphoma. Many types of systemic B-cell lymphomas can also have extranodal spread to the skin (systemic follicular lymphoma, marginal zone lymphoma, or diffuse large B-cell lymphoma), although primary cutaneous B-cell lymphomas may be more common than secondary skin involvement by systemic B-cell lymphomas. In lymphomatoid granulomatosis, skin is the most common extrapulmonary site, with lymphomatoid granulomatosis occurring in a progressive pattern in 50% of cases. Other rare types of B-cell lymphomas with cutaneous involvement include intravascular B-cell lymphoma and precursor B-cell lymphoblastic lymphoma.50
PAGET AND EXTRAMAMMARY PAGET DISEASES
PAGET AND
EXTRAMAMMARY PAGET
DISEASES
Chapter 114, “Paget Disease,” provides additional information on Paget and extramammary Paget diseases. Paget disease of the nipple is a chronic erythematous scaling eruption of the nipple and areola indicating ductal carcinoma of the underlying breast. Extramammary Paget disease most commonly occurs in the anogenital skin, followed by the axillae and penis. It is classified into 3 types. The most common type appears as a primary intraepidermal adenocarcinoma. The second type (25%) is associated with visceral malignancy, most commonly of GI or genitourinary systems. The third type, the least common, is associated with an adjacent sweat gland carcinoma.
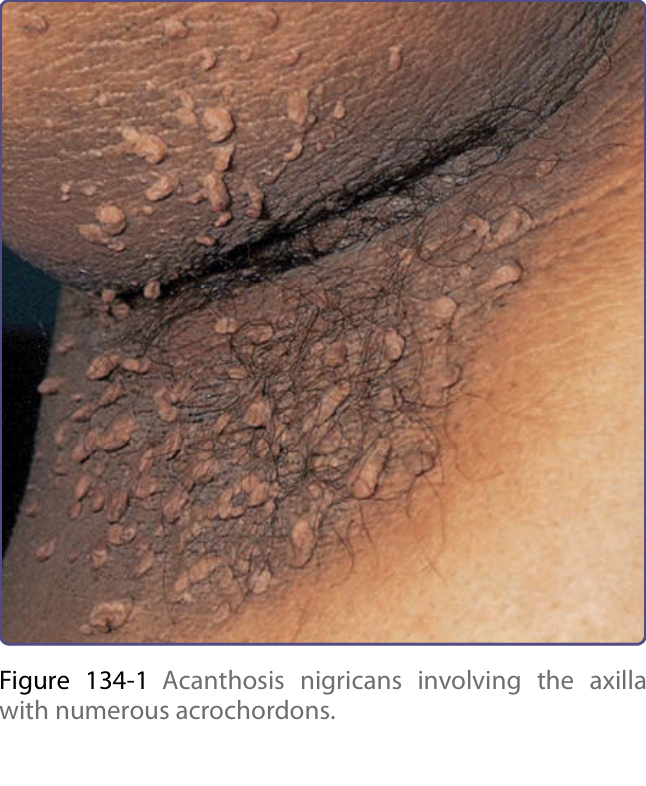
Figure 134-1 Acanthosis nigricans involving the axilla with numerous acrochordons.
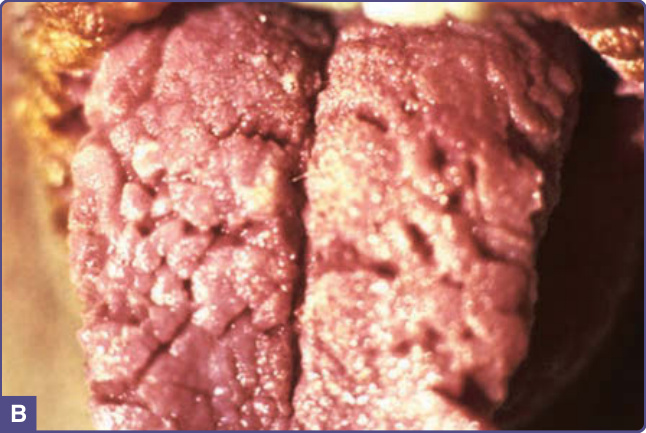
Figure 134-2 Acanthosis nigricans. A, Verrucous and papillomatous growths of the vermilion border of the lip. B, Velvety thickening of the tongue. (A and B are not the same patient.) (A and B, From Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, NY: McGraw-Hill; 2009, with permission.)
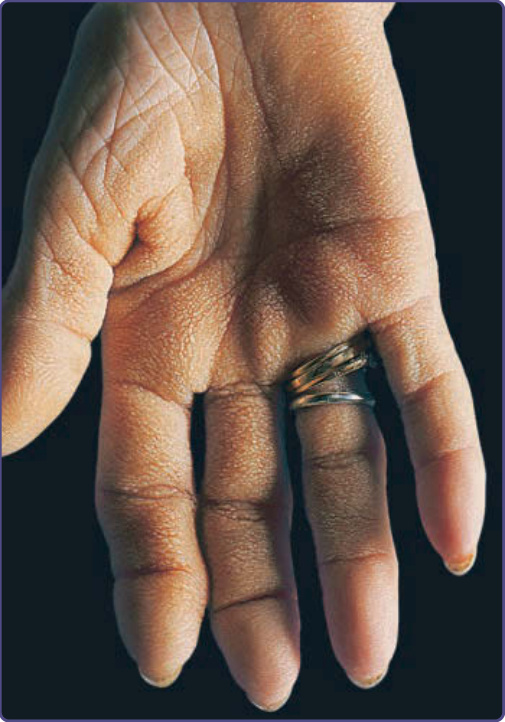
Figure 134-3 Tripe palm. The palmar ridges show maximal accentuation, thus mimicking the mucosa of the stomach of a ruminant.
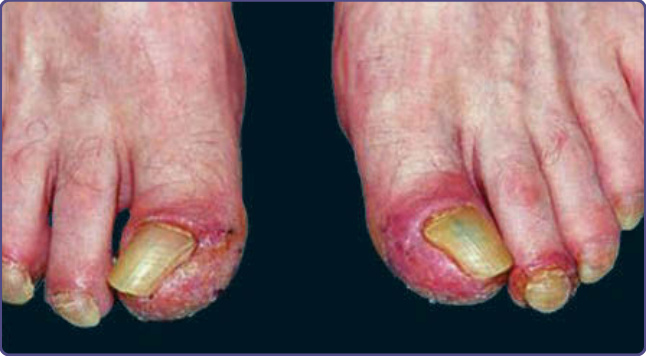
Figure 134-5 Distal edema of toes, painful generalized paronychia, and distal subungual hyperkeratosis in early acrokeratosis paraneoplastica.
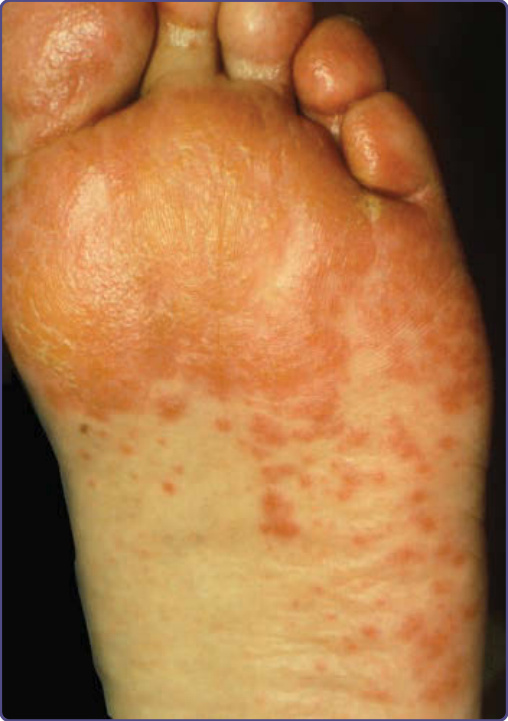
Figure 134-6 Keratoderma characteristically spares central aspects of plantar (and palmar) surfaces in acrokeratosis paraneoplastica.
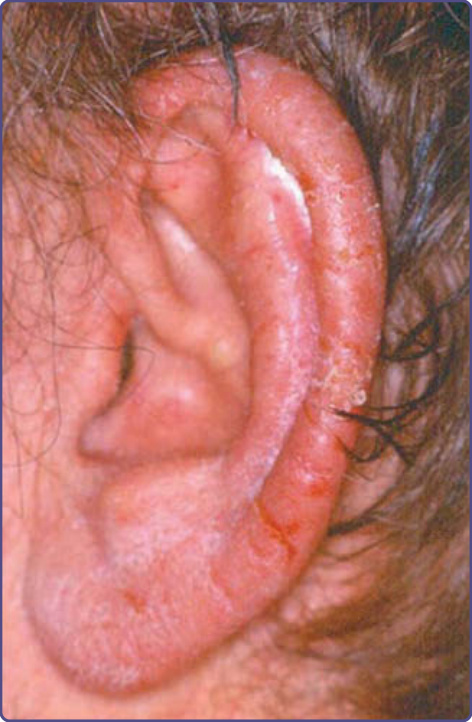
Figure 134-7 Characteristic helical inflammation with scale in acrokeratosis paraneoplastica.
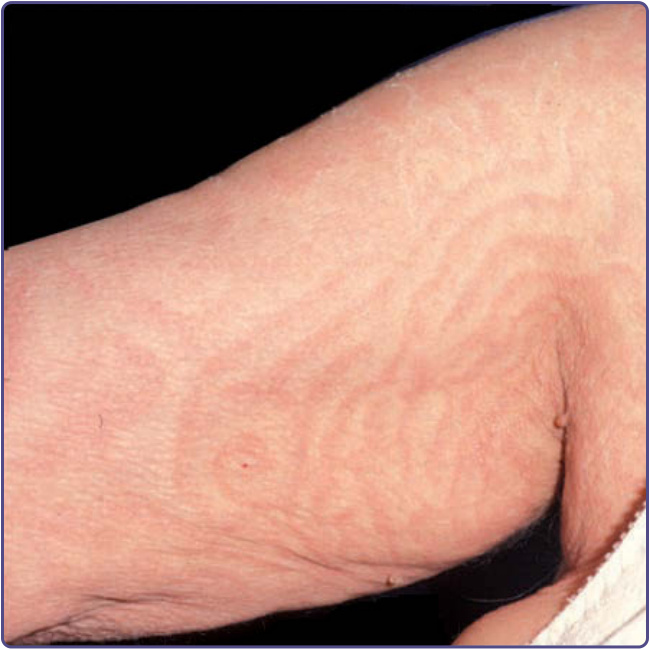
Figure 134-8 Erythema gyratum repens. Serpiginous parallel bands on axilla and arm. (Used with permission from Michael Adler.)
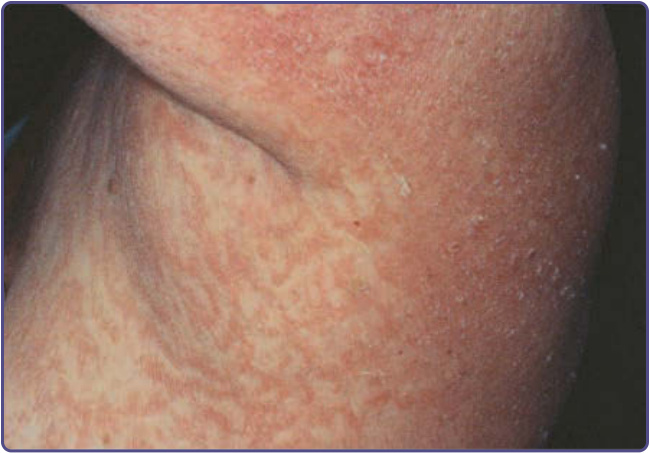
Figure 134-9 Erythema gyratum repens. Characteristic “wood-grain” pattern of erythema and slight scale in a woman with breast cancer. (Used with permission from Jill McKenzie, MD.)
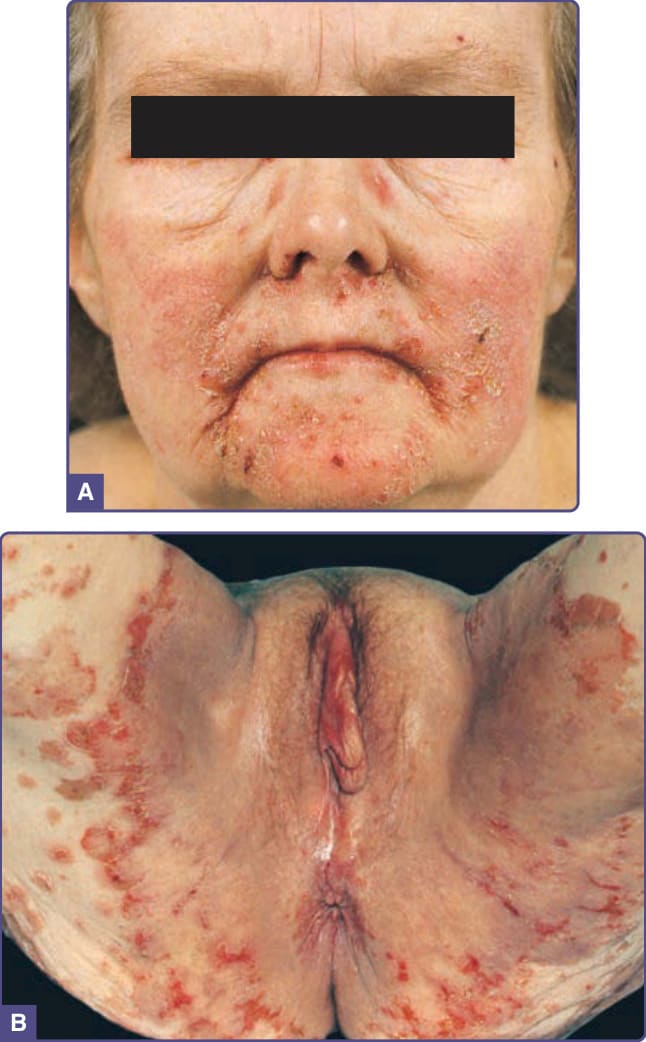
Figure 134-10 The glucagonoma syndrome, necrolytic migratory erythema. Flaccid and papulovesicular lesions (A) with erosions, crusting, and fissures around the orifices, and (B) appearing as geographic, circinate “necrolytic migratory erythema” in the groin. (A and B are not the same patient.)
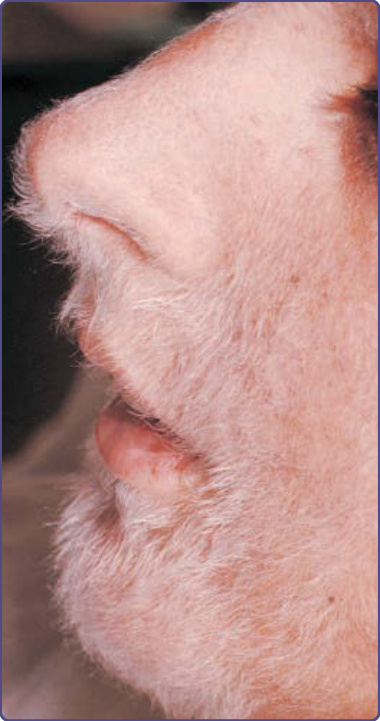
Figure 134-11 Hypertrichosis lanuginosa acquisita in a 19-year-old woman with pancreatic carcinoma.
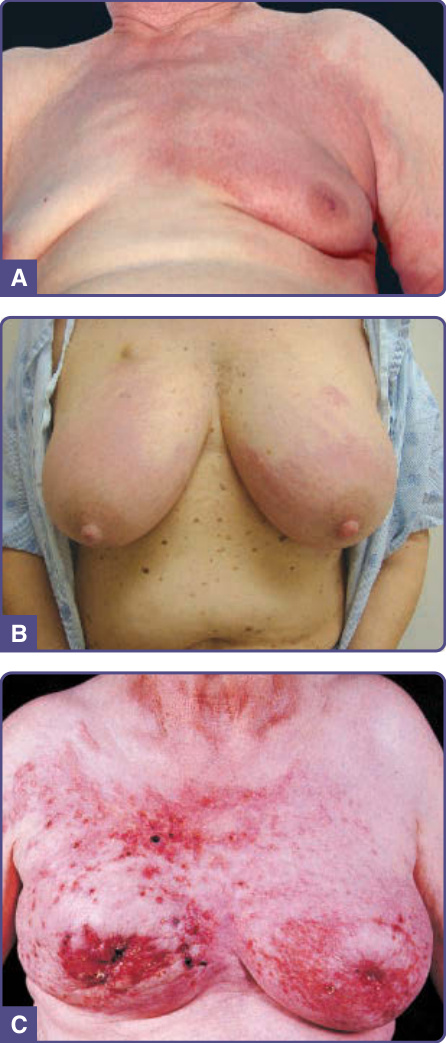
Figure 134-12 A, Carcinoma erysipelatoides. Intralymphatic spread of mammary carcinoma that manifests as erysipelas-like erythema. B, Bilateral cutaneous metastases from underlying breast carcinoma. C, Carcinoma en cuirasse involving both breasts and thoracic wall.
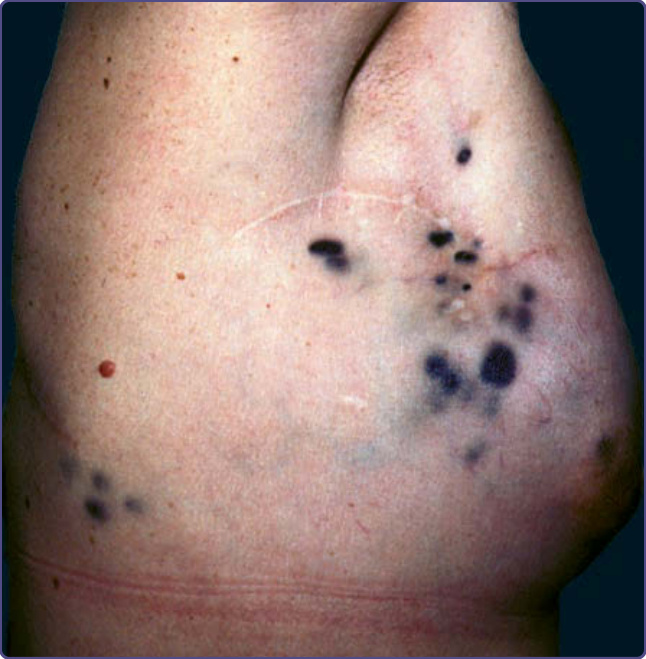
Figure 134-13 Deeply pigmented cutaneous metastases from melanoma.
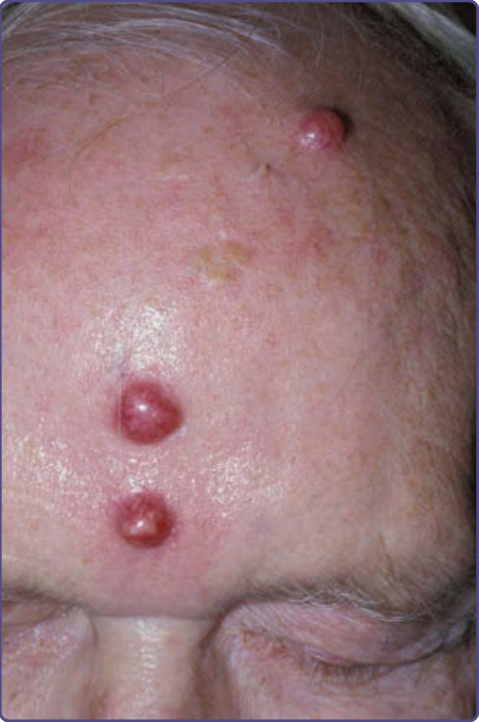
Figure 134-14 Metastatic lung cancer presenting as forehead nodules.
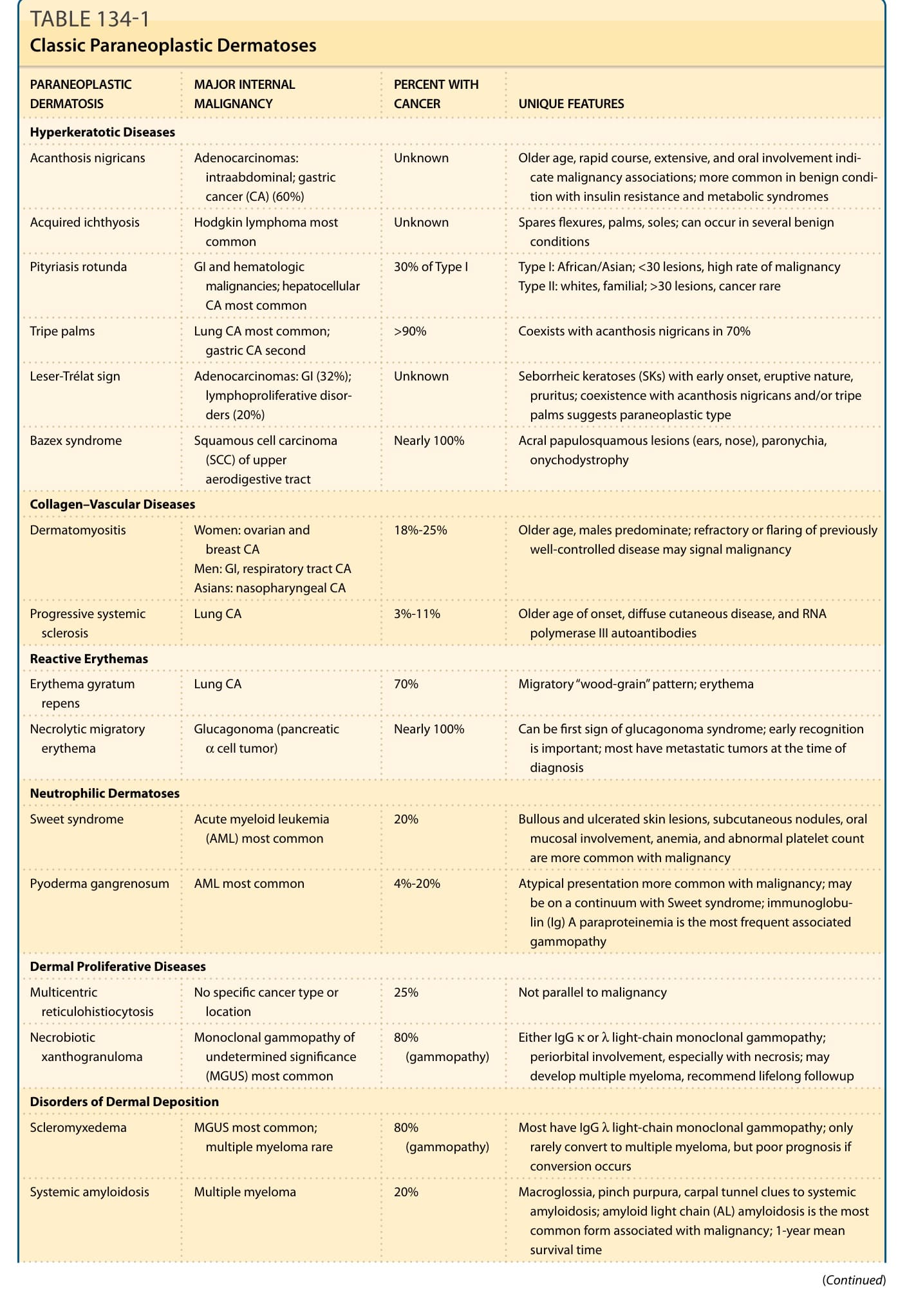
TABLE 134-1 Classic Paraneoplastic Dermatoses
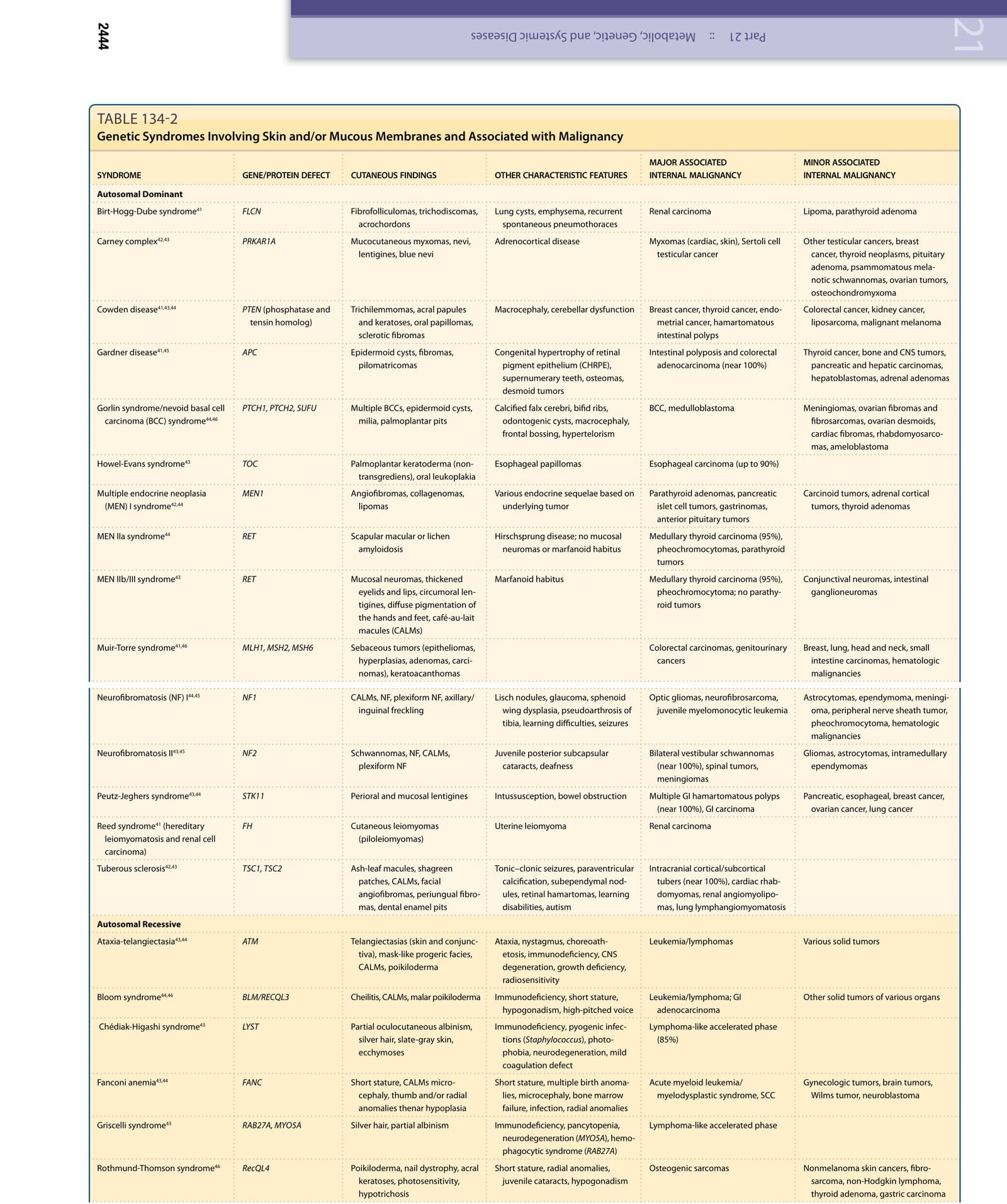
TABLE 134-2