血管腫瘤 (Vascular Tumors)
血管異常 (vascular anomalies) 常以胎記 (birthmark) 的形式表現。¹
其分類在歷史上一直是個難題,充斥著相互矛盾且令人困惑的術語與命名。儘管存在數種分類系統,最被廣泛接受且最全面者為國際血管異常研究學會 (International Society for the Study of Vascular Anomalies, ISSVA) 的分類,並於 2014 年更新,以納入臨床、影像、組織學以及(在已知的情況下)基因特徵。¹,² 血管異常大致分為血管畸形 (vascular malformations) 與血管腫瘤 (vascular tumors)。³ 血管畸形(見第 147 章)是血管形態發生過程中的錯誤,而嬰兒型血管瘤 (infantile hemangiomas) 與其他血管腫瘤則屬於增生性病變。在目前的 ISSVA 分類中,血管腫瘤又再細分為 (a) 良性、(b) 局部侵襲性或交界性,以及 (c) 惡性;每一類根據組織學、生物學、臨床外觀、行為、預後與治療而各具獨特特徵 (Table 118-1)。血管腫瘤有數種類型,其中許多發生於兒童期。這些腫瘤同樣有著令人混淆的病名學 (nosology),⁴ 充斥著描述性但不精確的術語,例如草莓狀 (strawberry)、毛細血管狀 (capillary) 與海綿狀 (cavernous)。應避免單獨使用非特異性的「血管瘤 (hemangioma)」一詞而不加上具區辨性的修飾語(例如:infantile 嬰兒型、rapidly involuting congenital 快速退化型先天性、lobular capillary 葉狀毛細血管型、spindle cell 梭形細胞型)。
國際血管異常研究學會 (ISSVA) 分類
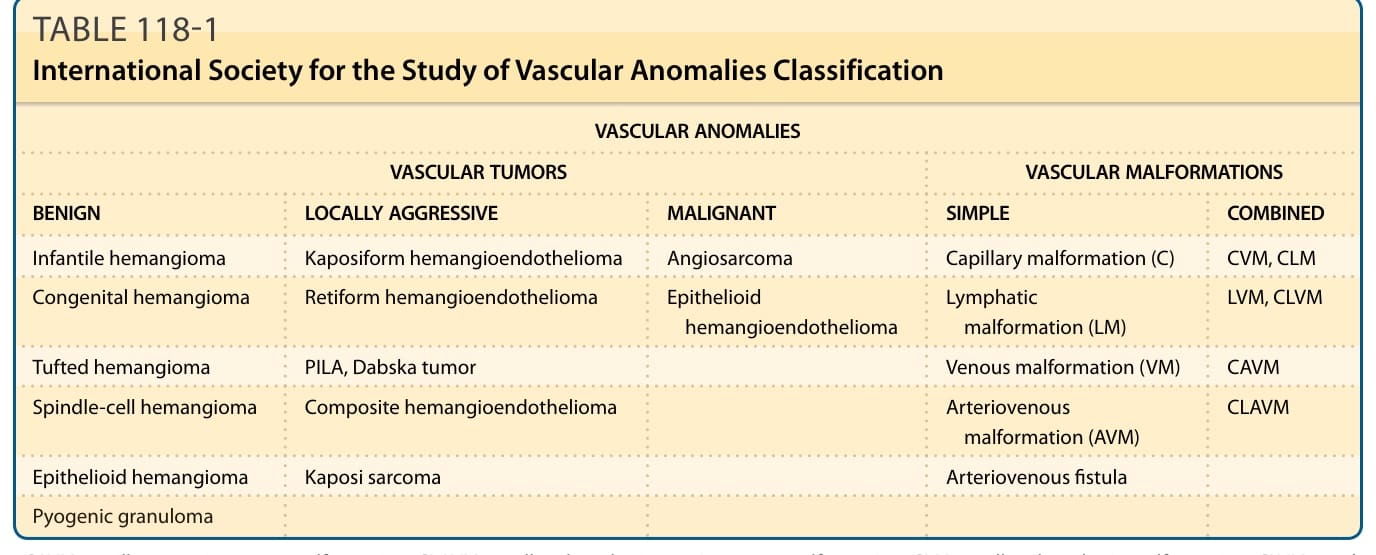
表 118-1:國際血管異常研究學會 (International Society for the Study of Vascular Anomalies) 分類
下表為原書被壓平之分類表的重排版(縮寫定義見表後說明):
| 血管腫瘤 (Vascular Tumors) | 血管畸形 (Vascular Malformations):單純型 (Simple) | 血管畸形:複合型 (Combined) |
|---|---|---|
| 良性 (Benign) | ||
| Infantile hemangioma(嬰兒型血管瘤) | Capillary malformation (C)(毛細血管畸形) | CVM, CLM |
| Congenital hemangioma(先天性血管瘤) | Lymphatic malformation (LM)(淋巴管畸形) | LVM, CLVM |
| Tufted hemangioma(叢狀血管瘤) | Venous malformation (VM)(靜脈畸形) | CAVM |
| Spindle-cell hemangioma(梭形細胞血管瘤) | Arteriovenous malformation (AVM)(動靜脈畸形) | CLAVM |
| Epithelioid hemangioma(上皮樣血管瘤) | Arteriovenous fistula(動靜脈瘻管) | |
| Pyogenic granuloma(化膿性肉芽腫) | ||
| 局部侵襲性 (Locally Aggressive) | ||
| Kaposiform hemangioendothelioma(卡波西樣血管內皮瘤) | ||
| Retiform hemangioendothelioma(網狀血管內皮瘤) | ||
| PILA, Dabska tumor(乳頭狀淋巴管內血管內皮瘤/Dabska 腫瘤) | ||
| Composite hemangioendothelioma(複合性血管內皮瘤) | ||
| Kaposi sarcoma(卡波西肉瘤) | ||
| 惡性 (Malignant) | ||
| Angiosarcoma(血管肉瘤) | ||
| Epithelioid hemangioendothelioma(上皮樣血管內皮瘤) |
縮寫說明:CAVM, capillary arteriovenous malformation(毛細血管動靜脈畸形);CLAVM, capillary lymphatic arteriovenous malformation(毛細血管淋巴管動靜脈畸形);CLM, capillary lymphatic malformation(毛細血管淋巴管畸形);CLVM, capillary lymphatic venous malformation(毛細血管淋巴管靜脈畸形);CVM, capillary venous malformation(毛細血管靜脈畸形);LVM, venous malformation(靜脈畸形);PILA, papillary intralymphatic angioendothelioma(乳頭狀淋巴管內血管內皮瘤)。國際血管異常研究學會 (ISSVA) 之血管異常簡明分類採用 Creative Commons Attribution 4.0 International License 授權。
嬰兒型血管瘤 (Infantile Hemangiomas)
重點一覽 (AT-A-GLANCE)
■ 嬰兒型血管瘤 (infantile hemangiomas) 是嬰兒期最常見的腫瘤。
■ 位於高風險解剖部位的嬰兒型血管瘤可能需要進一步檢查與治療。
■ 節段型 (segmental) 嬰兒型血管瘤的併發症風險高於局限型 (localized) 嬰兒型血管瘤。
■ Kasabach-Merritt 症候群不會發生於嬰兒型血管瘤。
流行病學 (EPIDEMIOLOGY)
嬰兒型血管瘤(infantile hemangiomas, IHs)是兒童期最常見的良性腫瘤,發生於約 4% 至 5% 的兒童。⁴⁻⁷ IH 與其他血管腫瘤及血管畸形不同之處在於其獨特的生長模式(快速增生期 proliferative phase,隨後是較緩慢的退化期 involution)。本病在女嬰較常見(比例為 2 至 3:1),在白人、非西班牙裔嬰兒,⁶,⁸,⁹ 以及低出生體重的嬰兒中較常見,並影響高達 30% 的早產兒,⁵,¹⁰ 尤其是體重低於 2500 g 者。⁸,¹¹ 低出生體重似乎是 IH 發生最重要的危險因子,其風險高於單獨的早產,出生體重每減少 500 g,風險就增加 40%,¹¹ 這與較早期的資料相呼應——出生體重低於 1000 g 的嬰兒中,近 4 分之 1 會發生 IH。產前危險因子包括高齡產婦(大於 30 歲)、子癇前症 (preeclampsia)、前置胎盤 (placenta previa) 及其他胎盤異常。⁸,¹² 早產兒較可能有多發性 IH。在早產情況下的 IH,與女性優勢的關聯較弱。⁸,¹³
複雜性 IH 在女嬰較常見;原因不明。¹⁴ 絨毛膜絨毛取樣 (chorionic villus sampling) 已不再被視為顯著的危險因子。⁸,¹¹,¹³
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (Cutaneous Findings)
以特徵性的增生與退化所構成的生命週期病史,對於 IH 的診斷至關重要 (Table 118-2)。¹⁵
生長特徵——增生期 (Proliferation):出生時不存在,或存在一個初生的 (nascent) IH,常表現為一塊蒼白區、毛細血管擴張 (telangiectasias) 或暗沉 (duskiness) 的區域,是其特徵。出生時即為一個完全成形的軟組織腫塊,幾乎可確定是另一種血管異常或其他疾病過程。大多數 IH 發生於頭頸部,但 IH 可見於任何皮膚或黏膜部位,較少見的情況下也可侵犯內臟器官(見「非皮膚表現與併發症」一節)。幾乎所有具淺層成分的 IH 都在出生後第 1 個月內變得明顯,且大多數會在出生後前 2 個月內至少增大一倍。早期增生期伴隨著快速的非線性生長,IH 生長的高峰期發生在 5.5 週至 7.5 週齡之間。¹⁶
雖然 80% 的生長在 3 個月齡時已完成,但 80% 的 IH 在 5 個月齡時已完成所有生長 (Fig. 118-1)。¹⁷ 在快速生長高峰之後出現的持續較緩慢生長之晚期增生階段,通常在 9 個月齡時結束;只有 3% 的 IH 有臨床記錄到超過此年齡的生長。¹⁷
深部 IH 較可能有較長的增生期。大型、節段型及腮腺 (parotid gland) IH 也可能持續緩慢增大數月,罕見情況下達數年,比其他 IH 更久。¹⁷⁻¹⁹
生長特徵——退化期 (Involution):快速與晚期增生期之後,接續的是較緩慢且長度變異較大的退化期,持續數月至數年 (Fig. 118-2)。退化的徵象常被稱為「灰化 (graying)」,包括顏色變為暗紅,接著轉為灰色或乳白色,隨後是扁平化與軟化。此變化通常在 1 歲時變得明顯。¹⁷ 較小的血管瘤通常比非常大的血管瘤更早退化。¹⁷ 超過 90% 的 IH 在 3.5 至 4 歲時已完成退化。²⁰,²¹ 重要的是要認知到,完全退化並不等同於 IH 的完全消退。超過半數有血管瘤的兒童會留下殘餘的毛細血管擴張、纖維脂肪組織 (fibrofatty tissue) 或鬆弛皮膚 (anetodermic skin)。與深部 IH 相比,淺層 IH 在退化後較可能出現殘餘的皮膚變化(勝算比 odds ratio:8.4)。²² 複合型 (combined) IH 以及具陡峭邊緣或鵝卵石狀 (cobblestoned) 表面者,永久性疤痕的風險顯著較高。²¹
分類⁸:皮膚 IH 的型態可根據腫瘤深度(淺層 superficial、深部 deep、複合/混合型 combined/mixed)與分布(局限型 localized、節段型 segmental、不確定型 indeterminate、多灶型 multifocal)來分類。²³,²⁴ 由於自然病程、預後與可能的併發症會依這些分類特徵而異,此型態學區分有助於指引預期。淺層 IH 是最常見的型態學亞型,表現為鮮亮紫紅至「草莓」紅色的血管性斑塊或結節;侵犯乳頭層真皮 (papillary dermis)。相對地,深部 IH 表現為部分可壓縮、局限性的皮下腫瘤,依其在皮膚中的深度而有不同程度的突出,外觀呈現輕微藍色調或與周圍皮膚同色;平均可能比淺層 IH 晚 1 個月被注意到。較少見的情況下,深部 IH 直到嬰兒數月大時才被察覺。深部 IH 較可能有較長的增生期,罕見地在其他 IH 的生長期之外持續生長數月至數年,且退化發生較其淺層對應者晚。¹⁷,¹⁸ 複合型(也稱混合型 mixed)IH 兼具深部與淺層 IH 的臨床特徵。一種較不常見的 IH 亞型以其極少至無的增生及毛細血管擴張性表面為特徵,稱為「極小或流產型生長之 IH (IH with minimal or abortive growth)」,²⁵ 已有相關描述 (Fig. 118-3)。此型最常見於下半身,尤其是肢端 (acral) 部位,這些 IH 表現出與典型增生性 IH 相同的組織學標記(例如 GLUT1),但因不明原因而未經歷一般 IH 的生長軌跡。²⁵,²⁶
除了個別型態外,IH 也可根據其在身體上的分布分類為局限型(focal)、節段型、不確定型或多灶型,這提供了寶貴的預後資訊。²³,²⁴ 約三分之二的 IH 為局限型(focal),表現出清楚的空間侷限,彷彿源自單一中央焦點 (Fig. 118-4)。²⁷ 在臉部,局限型 IH 傾向出現在間質與外胚層胚胎融合線 (lines of mesenchymal and ectodermal embryonic fusion) 附近。²³ 節段型血管瘤——類似於其他節段型皮膚疾病,如白斑 (vitiligo) 與神經纖維瘤病 (neurofibromatosis)——常呈斑塊狀,並對應於發育節段或廣闊地理性解剖區域的一部分 (Fig. 118-5)。²⁸ 在一項大型研究中,13.1% 的 IH 為節段型。²⁹ 有人提出臉部節段型 IH 的模式對應於神經嵴 (neural crest) 衍生的臉部隆起,而其存在與 PHACE 風險相關(posterior fossa brain malformations 後顱窩腦部畸形、hemangiomas of the face 臉部血管瘤、arterial anomalies 動脈異常、cardiac anomalies 心臟異常與 eye abnormalities 眼部異常)。²⁹
下半身的節段型 IH 在 LUMBAR 症候群中帶來脊髓病變 (myelopathy) 與泌尿生殖系統異常的風險(lower body hemangioma 下半身血管瘤與其他皮膚缺損、urogenital anomalies 泌尿生殖異常、ulceration 潰瘍、myelopathy 脊髓病變、bony deformities 骨骼畸形、anorectal malformations 肛門直腸畸形、arterial anomalies 動脈異常與 renal anomalies 腎臟異常)(見「Lumbar 症候群」一節)。不確定型 IH 是指無法明確辨識為局限型或節段型者;有些作者認為它們是「次節段型 (subsegmental)」(Fig. 118-6)。多灶型(多發性局限型)IH 較不常見,少於 3% 的嬰兒會出現 6 個或以上的 IH。²⁷
依亞型對血管瘤進行分類不僅有助於溝通,也有助於預測併發症風險與治療需求。一項超過 1000 名 IH 兒童的前瞻性研究顯示,即使在控制大小的情況下,節段型血管瘤發生併發症的可能性是局限型血管瘤的 11 倍,接受治療的可能性是 8 倍。²⁷
非皮膚表現與併發症 (Noncutaneous Findings and Complications)
某些 IH 有已知的相關併發症與先天異常風險 (Table 118-3)。多灶型或節段型 IH 的存在與較高的皮膚外疾病風險相關。¹⁵
解剖位置是影響風險最重要的因素之一。侵犯臉部中央(尤其是鼻部與口周皮膚)、眼周區域、頸部、下頷區及會陰,應提醒臨床醫師注意可能增加的併發症風險。儘管絕大多數 IH 在無介入下仍維持無症狀且無併發症,無需治療,但併發症可包括潰瘍、嚴重出血(少於 1% 的病例)、疤痕、疼痛與感染,並在某些侵犯部位有額外的併發症(例如氣道阻塞、充血性心衰竭與視力損害)。約 12% 的所有 IH 是複雜性的,需要專科轉介。結果在很大程度上取決於轉介與介入的時機。
與節段型嬰兒型血管瘤相關的症候群與關聯 (Syndromes and Associations with Segmental Infantile Hemangiomas)
如上所述,臉部節段型 IH 的模式對應於發育單位,並被標記為 4 個節段(S1 至 S4):額顳段 (frontotemporal, S1)、上頷段 (maxillary, S2)、下頷段 (mandibular, S3) 與額鼻段 (frontonasal, S4)。²⁷ 額顳段 (S1) 與腦部、腦血管及眼部異常相關;上頷段 (S2) 較少與皮膚外侵犯相關;下頷段 (S3) 與氣道 IH、腹側發育缺損及心血管異常相關;額鼻段 (S4) 涵蓋了鼻部(包括鼻尖)此一高風險區域,³⁰ 並帶來腦部與腦血管侵犯的風險 (Fig. 118-7)。
PHACE
臉部節段型 IH 帶來 PHACE 的風險,³¹ 這是一種神經皮膚關聯 (neurocutaneous association)。它有時被稱為 PHACES,以表示額外關聯的胸骨缺損 (Sternal defects)(即胸骨裂 sternal clefting 或臍上縫 supraumbilical raphe)。³² 因不明原因,高達 90% 的 PHACE 病例為女性。此外,據報告,臉部節段型 IH 且表面積等於或大於 22 cm² 的病人中,高達三分之一符合 PHACE 症候群的標準。³³⁻³⁵ 額顳段 (S1) 與下頷段 (S3) 是與 PHACE 關聯最高的節段。有些嬰兒具有 PHACE 的皮膚外特徵,其節段型 IH 位於後腦勺、上軀幹或手臂,也有些嬰兒只有小型 IH,或完全沒有皮膚 IH。³⁶⁻³⁹ 診斷標準已經過修訂 (Table 118-4)。⁴⁰
先天性血管異常是 PHACE 中最常見的皮膚外特徵。腦血管異常發生於超過 90% 的病例,心臟異常發生於高達 67% 的病例。³⁰,³²,³³,³⁵,⁴¹ 主要腦部與頸部血管可發生發育不良 (dysplasia)、狹窄 (stenosis)、發育不全 (hypoplasia) 與發育缺失 (agenesis)。據報告,主動脈窄縮 (coarctation of the aorta) 發生於 19% 至 30% 的 PHACE 病例,且與 PHACE 相關的主動脈弓異常異常複雜,常侵犯橫向及/或降主動脈的長節段。⁴⁰,⁴² 值得注意的是,PHACE 中的主動脈窄縮常與兩側鎖骨下動脈異常起源於窄縮處遠端相關,使得四肢血壓在理學檢查時無法呈現反映窄縮的壓力梯度。結構性腦部異常發生於約 40% 的 PHACE 病人,⁴³ 估計範圍從 30% 到大於 80% 不等。⁴⁰ 已知 PHACE 會發生動脈缺血性中風 (arterial ischemic stroke),常以癲癇發作或偏癱 (hemiparesis) 表現。⁴⁴ 雖然大多數 PHACE 嬰兒的風險程度尚不明確,但風險似乎在 Willis 環內或之上的主要腦動脈發生阻塞或顯著狹窄時最高,尤其是合併主動脈窄縮時。⁴⁰
眼部異常較不常見,通常表現為後段異常(例如牽牛花視盤異常 morning glory disc anomaly、視神經發育不全 optic nerve hypoplasia、永存性胎兒血管 persistent fetal vasculature,或視網膜血管異常)或小眼症 (microphthalmia)。內分泌異常,包括甲狀腺功能低下 (hypothyroidism) 與腦垂體功能低下 (hypopituitarism) 及隨後的生長激素缺乏,以及頭痛、言語與語言發展遲緩,也可能發生。⁴⁰,⁴⁵
LUMBAR 症候群
類似於 PHACE,下半身侵犯會陰或腰薦部 (lumbosacral area) 的節段型 IH 是相關脊髓、骨骼與泌尿生殖系統異常的危險因子 (Fig. 118-8)。LUMBAR 症候群描述的是一組合併症狀:下半身血管瘤與其他皮膚缺損 (lower body hemangioma and other cutaneous defects)、泌尿生殖異常 (urogenital anomalies)、潰瘍 (ulceration)、脊髓病變 (myelopathy)、骨骼畸形 (bony deformities)、肛門直腸畸形 (anorectal malformations)、動脈異常 (arterial anomalies) 與腎臟異常 (renal anomalies)。⁴⁶
類似的病人群組也曾以縮寫 PELVIS(perineal hemangioma 會陰血管瘤、external genitalia malformations 外生殖器畸形、lipomyelomeningocele 脂肪脊髓脊膜膨出、vesicorenal abnormalities 膀胱腎臟異常、imperforate anus 肛門閉鎖、skin tag 皮膚贅瘤)與 SACRAL(spinal dysraphism 脊柱裂、anogenital anomalies 肛門生殖器異常、cutaneous anomalies 皮膚異常、renal and urologic anomalies 腎臟與泌尿系統異常,合併腰薦部局部血管瘤)來描述。⁴⁷,⁴⁸ 脊髓病變是最常見的相關皮膚外異常,表現為脊髓栓系 (tethered cord) 或脂肪脊髓(脊膜)膨出。⁴⁶ 在此情況下強烈建議進行 MRI,因為據報告腰薦部超音波因敏感度不佳而會產生偽陰性結果。⁴⁹,⁵⁰ 在 LUMBAR 情況下的 IH 較可能是具有極小或流產型生長型態的 IH,並帶有顯著的潰瘍風險。底層動脈異常與肢體發育不全 (limb hypotrophy) 可能發生。⁴⁶
眼周血管瘤 (Periocular Hemangiomas)
患有眼周血管瘤的嬰兒有不等視 (anisometropia) 與弱視 (amblyopia) 的風險,若未治療,可導致永久性視力喪失 (Fig. 118-9)。⁵¹,⁵² 弱視是最常見的眼部併發症,發生於 40% 至 60% 未治療的眼周 IH 嬰兒。⁵³,⁵⁴ 直徑大於 1 cm 的 IH 帶來較大的弱視風險,顯著降低了啟動治療的閾值。⁵² 對角膜的直接壓迫可產生散光 (astigmatism) 或近視 (myopia),而腫瘤本身的占位效應 (mass effect) 可導致眼瞼下垂 (ptosis)、眼球突出 (proptosis)、視軸阻塞或斜視 (strabismus)。任何眼周區域有血管瘤的病人都應立即接受正式的眼科評估,並在快速與晚期增生期回診。在出現眼科併發症的情況下,應緊急啟動治療以預防長期影響,包括永久性失明。由受過嬰兒照護訓練的眼科醫師進行的標準評估,應包括在睫狀肌麻痺 (cycloplegia) 後以視網膜鏡 (retinoscopy) 進行屈光檢查。當侵犯上眼瞼或眶上區域時,連續性檢查尤其重要。長期併發症可包括視神經萎縮 (optic atrophy)、眼瞼下垂 (blepharoptosis) 與眼球突出。⁵⁵
40% 至 80% 的眼周 IH 在退化後有永久性殘餘變化。⁵⁶
預示退化後視力正常最有利的預後徵象是沒有不對稱的屈光不正。⁵⁴ 在某些病例中,可能需要 MRI 來確定是否有眼球後 (retrobulbar) 侵犯。
「鬍鬚區 (Beard Area)」血管瘤
侵犯耳前、下頷、下巴與頸部皮膚(即所謂的鬍鬚區)的第 3 節段 IH,造成症狀性氣道疾病的風險為 60%。⁵⁷
氣道血管瘤常以在出生後第 4 至 12 週間隱匿發作的雙相喘鳴 (biphasic stridor) 表現,並常被誤診為氣管軟化症 (tracheomalacia)、上呼吸道感染或哮吼 (croup)。若無介入,呼吸窘迫可隨之而來並危及生命。由小兒耳鼻喉科醫師及時評估與治療至關重要。⁵⁸
腮腺 (parotid gland) IH 可能有異常延長的生長期,並可能因 IH 的大量生長與鄰近結構的變形而需要治療。雖然罕見,但已有高輸出量充血性心衰竭 (high-output congestive heart failure) 的報告。⁵⁹,⁶⁰
多灶型血管瘤 (Multifocal Hemangiomas)
約 15% 的嬰兒會有超過 1 個血管瘤。患有多灶型 IH 的嬰兒有較高的非皮膚 IH 風險。對於出現 5 個或以上皮膚 IH 的嬰兒,建議進行篩檢性肝臟與腹部超音波 (Fig. 118-10)。⁶¹,⁶² 罕見情況下,嬰兒可能有數百個病灶。有人建議以「伴或不伴皮膚外疾病之多灶型 IH (multifocal IH with or without extracutaneous disease)」一詞取代較舊的「瀰漫性新生兒血管瘤病 (diffuse neonatal hemangiomatosis)」,因為現在已了解到舊文獻報告的病例將 IH 與伴血小板低下之多灶型淋巴管內皮瘤病 (multifocal lymphangioendotheliomatosis with thrombocytopenia, MLT) 及其他非 IH 多灶型腫瘤混淆。在真正的 IH 中,其他內臟侵犯部位非常罕見。內臟血管瘤,包括侵犯肝臟、消化道與腦部者,也曾在孤立性節段型血管瘤中被報告。⁶³
肝臟血管瘤 (Hepatic Hemangiomas)
肝臟是 IH 最常見的皮膚外部位。即使存在,肝臟 IH 也常無症狀。少數肝臟 IH 會造成發病,且在罕見情況下可危及生命,造成高輸出量充血性心衰竭、甲狀腺功能低下或肝衰竭。肝臟血管瘤可為局灶型、多灶型或瀰漫型。⁶⁴ 局灶型肝臟血管瘤並非真正的 IH,而是類似於發生於肝臟的快速退化型先天性血管瘤 (rapidly involuting congenital hemangioma, RICH)。它們在出生時即完全成形,出現時無皮膚 IH,並可造成中度嚴重的血小板低下與凝血功能障礙,但不如 Kasabach-Merritt 現象的消耗性凝血功能障礙嚴重。多灶型與瀰漫型肝臟血管瘤是真正的 IH。多灶型嬰兒肝臟血管瘤常無症狀,但若發生動靜脈或門靜脈分流,可造成高輸出量充血性心衰竭。瀰漫型嬰兒肝臟血管瘤較不常見,並與高發病率和死亡率相關。在此情況下,大部分肝實質被腫瘤取代,可造成腹腔室隔症候群 (abdominal compartment syndrome) 與嚴重甲狀腺功能低下,後者是腫瘤相關之第 III 型碘甲狀腺胺酸脫碘酶 (Type III iodothyronine deiodinase) 過度產生的結果。可能需要全身性 β-阻斷劑 (β-blocker) 治療,嚴重病例則需合併全身性皮質類固醇或栓塞治療。⁶⁵,⁶⁶ 合併甲狀腺功能低下的病例需要積極的甲狀腺素補充。⁶⁷ 在危及生命的病例中,可考慮肝臟移植。
潰瘍 (Ulceration)
潰瘍是 IH 最常見的併發症,在轉診環境中發生於超過 20% 的嬰兒,通常在增生期出現,發作中位年齡為 4 個月。²⁷,⁶⁸ 它最常發生於大型或節段型 IH,以及暴露於潮濕與摩擦的部位,例如口周、肛周與間擦 (intertriginous) 區域 (Fig. 118-11)。⁶⁸,⁶⁹
在小於 3 個月的病人中,早期的灰白色變色是即將發生潰瘍的敏感標記。⁷⁰ 可能發生續發性感染,但多菌種定植 (polymicrobial colonization) 較為典型。⁷¹ 潰瘍的治療 (Table 118-5) 在無抗生素治療的情況下常能成功,不過局部定植與感染可透過局部 mupirocin 或 metronidazole 改善。若懷疑有深部或持續性感染,應開立全身性抗生素。局部傷口照護、屏障保護與疼痛控制對治療至關重要。生物封閉性敷料 (bio-occlusive dressings) 可能有幫助,但其用途常因位置而受限,因為它們在嘴部附近或尿布區域附著不佳。⁷²,⁷³ 在這些區域,厚塗凡士林為基礎的軟膏可能有幫助。疼痛可能是處置上的一大問題。可透過封閉性敷料、口服 acetaminophen 加或不加 codeine,以及每天不超過數次使用極少量的局部 lidocaine 軟膏來將其最小化。⁶⁹ 經過 2 至 3 週適當傷口照護後仍嚴重或未癒合的潰瘍,需要更積極的治療。選項包括全身性 β-阻斷劑治療、局部 β-阻斷劑(例如局部 timolol maleate)與脈衝染料雷射 (pulsed-dye laser, PDL)。在 β-阻斷劑時代較少使用的其他選項包括 becaplermin 0.01% gel、⁷⁴,⁷⁵ 病灶內與全身性皮質類固醇,以及早期切除。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
IH 僅發生於人類,主要由內皮細胞 (endothelial cells) 組成。IH 也含有纖維母細胞 (fibroblasts)、周細胞 (pericytes)、間質細胞 (interstitial cells) 與肥大細胞 (mast cells)。節段型 IH 中所見的模式提示,至少有些 IH 是早在妊娠第 4 至 6 週的發育錯誤的結果。²⁹ 其發生與演變的致病機轉仍然難以捉摸,部分原因是缺乏強健的體外系統或活體動物模型 (Fig. 118-12)。研究持續闡明這些腫瘤增生與退化的複雜病理生理機制。⁵⁶,⁷⁶⁻⁷⁹ 目前的理解認為,血管新生 (angiogenesis,即從現有血管發展出新血管) 與血管生成 (vasculogenesis,即血管的從頭發育) 在 IH 的發展中都很重要。研究顯示 IH 源自胎兒而非母體,⁸⁰ 並表現出與基本靜脈 (cardinal vein,一種早期胚胎血管結構) 相似的未成熟間質特徵。⁸¹
GLUT1 是一種紅血球葡萄糖轉運蛋白,也是 IH 最可靠的組織學標記,在 IH 的所有階段都有表現,對其的認識促成了關於 IH 致病機轉的新假說。⁸²,⁸³ GLUT1 表現在正常皮膚血管中缺乏,但見於胎盤血管以及其他所謂的屏障組織,例如血腦障壁 (blood–brain barrier)。這一點,加上 IH 與人類胎盤共有的其他免疫組織化學標記(Lewis Y antigen、merosin、Fc-γ receptor-IIb、indoleamine 2,3-deoxygenase 與 Type III iodothyronine deiodinase),以及在 DNA 微陣列上發現的相似基因表現特徵,引發了這些腫瘤源自胎盤——來自侵入的血管母細胞 (angioblasts) 並向胎盤表型分化——的推測。⁸³,⁸⁴ 較早期關於栓塞性胎盤組織作為 IH 觸發因子的理論已被推翻,因為 IH 中既無胎盤滋養層標記表現,也無絨毛結構。⁸⁵
增生由原始細胞標記 CD133+ 的表現來區分,⁸⁶,⁸⁷ 此標記見於稱為 HemSCs 的多能幹細胞中;並可在增生性 IH 中證明有內皮前驅細胞(CD31+、CD34+、CD133+ 幹細胞)(HemEPCs) 的存在,但在退化期則缺乏。⁸⁸,⁸⁹ 將從人類 IH 分離出的 HemSCs 植入免疫缺陷小鼠的轉譯研究,結果模擬了 IH 的生命週期,發展出 GLUT1+ 血管並隨後被脂肪細胞取代。⁸⁸ HemSCs 是脂肪細胞的前驅細胞,而退化後 IH 中脂肪細胞的存在是眾所周知的。IH 中的血管生成被理論化為源自循環內皮前驅細胞的純系擴增,且循環內皮前驅細胞可能是襯覆 IH 血管之內皮細胞的前驅細胞。³⁵ 其他研究證明增生中的 IH 有增加的血管新生因子,包括 basic fibroblast growth factor、vascular endothelial growth factor A、insulin-like growth factor 與 matrix metalloprotease 9。⁹⁰ 這些血管新生因子在退化期間被下調。越來越多的證據支持缺氧 (hypoxia) 在 IH 發展中的角色,並假設缺氧會在嬰兒中觸發新血管生成。低出生體重——IH 最重要的危險因子——已知與缺氧相關。⁸,¹² GLUT1 在缺氧情況下被上調。³⁵,⁹¹ 在增生性 IH 上進行的轉譯研究已辨識出除 GLUT1 外的多種缺氧標記,包括 insulin-like growth factor-2 與 vascular endothelial growth factor A。⁹² 組織學特徵可區分 IH 的增生期與退化期,並同樣闡明了致病機轉。在增生期間,內皮細胞增生的純系性與組織學特徵⁹³,⁹⁴ 提示此過程繼發於血管生成,⁹⁵ 而退化則以細胞凋亡 (apoptosis) 與毛細血管腔纖維化 (capillary lumina fibrosis) 為特徵。⁹⁶ 然而,解釋 IH 起始、增生與退化軌跡的確切致病機轉仍有爭議。目前最受支持的解釋認為,缺氧(子宮內或局部組織)是刺激循環內皮前驅細胞(CD34+、CD133+ 幹細胞)的關鍵,⁹⁷ 隨後血管生成⁹⁸ 並伴隨增生以維持組織氧合恆定,⁹⁷ 最終細胞凋亡增加(始於第 1 年末)以啟動退化。⁹⁶
診斷 (DIAGNOSIS)
除了異常困難的病例外,IH 的診斷幾乎完全是臨床性的,基於理學檢查與臨床病史。
輔助檢查 (Supportive Studies)
在某些臨床情境下可能需要輔助檢查 (Fig. 118-13)。必要時,超音波影像具有不需鎮靜、無游離輻射且成本相對低的優點。在杜卜勒超音波 (Doppler ultrasound) 上,IH 是高流量病灶,呈現為界限分明的實質腫塊,腫塊內血流增加。可看到動脈供應血管 (arterial feeder) 與靜脈引流 (venous drainage)。MRI 的使用提供額外解剖細節的好處,但其使用必須權衡幼嬰暴露於吸入性麻醉的風險與增加的成本。目前的證據確實支持在生命早期使用 MRI 與磁振造影血管攝影 (magnetic resonance angiography, MRA) 來檢查 PHACE 症候群。⁴⁰ 一些對嬰兒影像有經驗的中心,可能能在新生兒期於餵食與包裹後成功完成 MRI(「餵食與包裹 feed-and-wrap」技術);影像品質因動作偽影而受損的可能性會隨嬰兒年齡與檢查持續時間而增加。在 MRI 上,IH 呈現為 T2 高訊號、T1 等訊號的腫塊,伴有均勻、明顯的對比強化。在 T2 加權影像上可見內部蜿蜒的流空 (serpiginous flow voids),表示有動脈供應血管。MRA 顯示早期動脈強化。⁹⁹
實驗室檢查 (Laboratory Tests)
甲狀腺功能檢查:甲狀腺功能低下是患有瀰漫型嬰兒肝臟血管瘤之嬰兒的罕見併發症。¹⁰⁰,¹⁰¹ 肝臟腫瘤表現出高量的第 III 型碘甲狀腺胺酸脫碘酶活性,加速甲狀腺素的降解。⁶⁷,¹⁰² 有顯著肝臟血管瘤的嬰兒應評估甲狀腺功能,包括三碘甲狀腺胺酸 (triiodothyronine, T3)(被消耗的荷爾蒙)與甲狀腺刺激素 (thyroid-stimulating hormone),因為甲狀腺素 (thyroxine, T4) 濃度初期可能維持正常。反之,對於不明病因的甲狀腺功能低下嬰兒,即使沒有皮膚血管瘤,也應進行篩檢性肝臟超音波。甲狀腺功能低下與其他內分泌異常也曾在 PHACE 症候群中被報告。
病理 (Pathology)
IH 的組織病理學表現在其生命週期過程中變化顯著。除了最早期的生長階段與退化的最末期外,增生與退化的特徵是混合存在的。作為放射影像的補充,快速增生期的 IH 由無包膜但界限分明的毛細血管腫塊組成,排列成由細隔膜 (fine septae) 或正常周圍軟組織所分隔的小葉 (lobules),由內皮細胞、周細胞、肥大細胞與間質樹突細胞 (interstitial dendritic cells) 構成。這些血管襯覆著飽滿的內皮細胞,具有增大的細胞核與豐富的細胞質,周圍環繞著一圈同樣飽滿的周細胞,且沒有平滑肌細胞。³⁵ 其外觀有些類似於年輕的化膿性肉芽腫(葉狀毛細血管血管瘤 lobular capillary hemangiomas)。在早期 IH 中,細胞增生的標記如 Ki-67 表示內皮與周細胞的增生,且外觀正常的有絲分裂象 (mitotic figures) 顯得豐富。可能可見厚壁的引流靜脈。在退化中的 IH 檢體中,毛細血管開始退化並被脂肪細胞取代;肥大細胞增加,細胞凋亡明顯。IH 內血管的基底膜增厚並玻璃樣變 (hyalinize)。在退化末期,纖維脂肪基質顯著,殘餘的「鬼影 (ghost)」毛細血管中仍留有凋亡碎屑。⁶⁰,⁶⁴ 必要時為確認組織病理檢體的診斷,GLUT1 免疫染色在襯覆 IH 毛細血管的內皮細胞中於 IH 生長的所有階段皆為陽性,可將 IH 與其他血管腫瘤及所有血管畸形區分開來。⁶⁵,⁶⁶,⁷⁶,⁸² 如上所述,IH 也是 CD31+ 與 CD34+。
影像 (Imaging)
頭部、頸部及/或頭皮存在節段型 IH 時,應評估可能的 PHACE,包括頭/頸/胸的 MRI 與 MRA、⁴⁰ 正式的眼科評估與心臟超音波。同樣地,可能的 LUMBAR 症候群之檢查包括腰薦椎、骨盆及/或下肢的 MRI/MRA。在多灶型 IH 情況下對潛在肝臟 IH 的初步評估,需要進行帶杜卜勒血流與頻譜分析的肝臟/腹部超音波(基線超音波 ± 連續影像)。
鑑別診斷 (Differential Diagnosis)
(IH 的鑑別診斷詳見 Table 118-6。)
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
大多數 IH 的預後極佳,會自發性退化且幾乎沒有後遺症,但有顯著少數的 IH 會導致永久性毀容或醫療後遺症。退化後,約半數所有接受治療的病人會獲得正常皮膚,而另一半可能有殘餘的萎縮、疤痕、毛細血管擴張或纖維脂肪軟組織殘留。一項針對 97 名未治療病人的研究顯示,超過 68 名(70%)病人有殘餘皮膚變化。²² 特定的 IH 特徵(如先前各亞分類所概述)與較高的併發症率相關。¹⁷,¹⁰⁴ 影響複雜性 IH 預後最重要的因素是專科轉介處置的時機,包括啟動治療與必要的檢查。某些特徵與增加的併發症風險及治療需求相關(見 Table 118-3)。¹⁷,¹⁰⁴ 對於具有這些特徵的 IH,應依特定臨床情境考慮早期治療。
處置 (MANAGEMENT)
介入 (Interventions)
啟動治療的決定基於許多因素,包括大小與位置、心理社會影響,以及所提議治療的風險與益處 (Fig. 118-14)。對於大多數小型 IH,密切觀察與追蹤的積極不介入 (active nonintervention) 是最適當的方法。尤其是在生命的最初幾個月以及快速增生期,回診應更頻繁,在最年幼的嬰兒中可每 1 至 2 週一次。¹⁰⁵
與父母及照顧者的會診應強調關於 IH 自然病程與預後的衛教,同時處理父母的焦慮¹⁰⁶ 以及對病人與家庭的心理社會影響。¹⁰⁷⁻¹⁰⁹ 類似病灶可能結果的照片常有幫助。許多父母會經歷焦慮,並可能發現自己受到完全陌生人對其孩子血管瘤的評論。¹⁰⁶ 大多數幼兒的父母不認為他們的孩子深受這些反應影響,但臉部 IH 尤其在孩子到達學齡時可造成心理痛苦。¹⁰⁷,¹⁰⁸ 潛在的治療選項應在進入小學前充分討論。對於需要介入的 IH,選項包括藥物、雷射與手術介入。
藥物 (Medications)
β-阻斷劑 (Beta Blockers):propranolol 在 IH 治療中具有廣泛確立的療效,且與先前的全身性治療選項相比有較有利的風險特性,是美國食品藥物管理局 (FDA) 唯一核准用於治療複雜性 IH 的藥物。對於大多數有顯著發病風險的中度至重度 IH,propranolol 是明確的第一線全身性藥物治療。¹¹⁰⁻¹¹² 與皮質類固醇不同——後者穩定 IH 生長但不導致退化——propranolol 在絕大多數病例中造成消退。停止治療後可發生反彈性生長,但通常對再次治療反應良好。¹¹¹,¹¹² 考慮啟動 β-阻斷劑治療 IH 的關鍵標準包括臉部變形、現存或即將發生的功能損害、預防預期的永久性後遺症,以及減少手術需求。¹¹³ 在其他情境下也曾報告正面結果,包括完全增生的 IH 以及氣道、肝臟與眼眶血管瘤。¹¹¹,¹¹⁴⁻¹¹⁶ 儘管早期有爭議,大多數 PHACE 病人可安全耐受 propranolol,不過建議使用最低有效劑量、考慮較慢的劑量調升,並將藥物分成 3 次給予而非標準的每日兩次給藥,以避免血中峰值濃度的顯著改變。⁴⁰ propranolol 有效治療 IH 的分子機制包括病灶毛細血管收縮(使用後前 48 小時內可見 IH 顏色變化)、血管新生抑制(阻止生長),以及誘導細胞凋亡(促使 IH 消退)。¹¹⁷ propranolol 常見的副作用包括睡眠障礙、肢端發紺 (acrocyanosis),以及無症狀的暫時性血壓或心率下降。罕見觀察到的潛在重要不良反應包括低血糖、症狀性低血壓或心搏過緩、喘鳴、支氣管痙攣與腹瀉。propranolol 的療效已在一項大型、國際、安慰劑對照、隨機試驗,以及眾多較小型的前瞻性與回溯性病例系列和病例報告中被記載,包括一項比較口服皮質類固醇與 propranolol 的小型多中心隨機試驗,以及數項比較性回溯研究。¹¹¹,¹¹²,¹¹⁸ 一項由 456 名病人組成的多中心、隨機、雙盲第 II-III 期試驗,調查口服 propranolol 在 IH 中的療效,涉及 5 個治療組(1 mg/kg/day 持續 3 個月 vs 6 個月、3 mg/kg/day 持續 3 個月 vs 6 個月,以及安慰劑)。3 mg/kg/day 持續 6 個月被發現具有最高的效益風險比,此每日劑量組中 88% 的病人在治療第五週時顯示改善。在此研究中,副作用包括心率下降(平均下降 7 beats per minute,給藥後 1 小時內觀察到心率變化,之後變化極小)與平均收縮壓下降(下降 3 mm Hg)。嚴重不良事件(n = 33)包括:支氣管痙攣、腹瀉(3 mg/kg/day > 1 mg/kg/day)與睡眠障礙(73 propranolol vs 7 安慰劑)。¹¹⁹ 一項涉及 39 個歐洲中心、近 1100 名總病人的世代研究,報告了 propranolol 在 IH 中的使用模式,無論每日維持劑量為何(少於 2 mg/kg/day、2 mg/kg/day、大於 2 mg/kg/day),91.4% 的病人顯示臨床上顯著的改善,不良事件風險則基於每日維持劑量。¹²⁰ 在 IH 中報告的整體反應率為 98%、抗藥率為 0.9%,propranolol 已降低了鼻部特定部位 IH 的手術需求。¹²¹⁻¹²³ 鑑於 propranolol 的親脂性與易穿越血腦障壁的特性,睡眠障礙可以多種方式表現(疲倦、躁動不安、惡夢、失眠),影響 11.4% 的病人,據報告 3.7% 的病人經歷惡夢。從心理學文獻中成人志願者研究外推的 propranolol 理論風險,包括短期與長期記憶、睡眠品質、情緒與精神運動功能的損害。與小兒高血壓相關的疲倦會隨時間改善。¹²⁴,¹²⁵
propranolol 的啟動可在住院或門診環境進行 (Fig. 118-15)。在決定病人是否適合使用 propranolol 時,必須取得家族史以確保無先天性心臟病、心律不整與母親結締組織疾病。對於使用 propranolol 治療 IH 的病人,尚未建立一致接受的標準監測程序。然而,目前提議偏向住院啟動的因素包括年幼(FDA 核准 propranolol 所依據的研究納入了年僅 5 週的嬰兒並採門診啟動)、共病的醫療狀況(例如呼吸或心血管)、社會支持不足,或任何先前存在的血糖疑慮。在門診環境中,可透過預先指導、與餵食一起給予 propranolol、頻繁餵食,以及在口服攝取量減少時減量或暫停劑量來預防低血糖。¹²⁶,¹²⁷ 門診啟動適合大多數新生兒期之後其他方面健康的兒童。目前已發表的指引以及許多(但非全部)醫師主張在門診環境中於給予第一劑時進行 2 小時的心率與血壓監測。劑量通常從 1 mg/kg/day 分 2 次給予開始,每 3 至 7 天以 0.5 mg/kg/day 的增量增加,至目標劑量 2 至 3 mg/kg/day 之間。大多數臨床醫師使用 2 mg/kg/day。在 PHACE 症候群、潰瘍或其他共病情況下,可能需要較慢的劑量調升或較低的目標劑量。Atenolol(β1 選擇性拮抗劑)與 nadolol(非選擇性 β 拮抗劑)已用於嬰兒的 IH 治療,但兩者在美國皆無液體劑型。Atenolol 的主要好處是缺乏支氣管反應性;nadolol 的優點是無法穿越血腦障壁,因此與 propranolol 相比減少了對任何潛在神經認知副作用的疑慮。截至撰寫本文時,僅有少數國際出版品報告 atenolol 與 nadolol 在 IH 中的使用;需要進一步研究。同樣地,調查 propranolol 任何神經認知影響的進一步研究正在進行中。早期證據未顯示使用 propranolol 對神經發育有不良影響,但尚未有此族群的全面神經認知測試報告。¹²⁸
鑑於全身性 β-阻斷劑已證實的療效,為了進一步減少副作用,評估了局部 β-阻斷劑在 IH 治療中的療效。局部 timolol 是一種非選擇性 β-阻斷劑,有核准用於治療小兒青光眼的眼用製劑。¹²⁹
局部 timolol 的效力比 propranolol 強 8 至 10 倍,於 2010 年首次被報告為 IH 治療的有效選項,¹³⁰ 並持續獲得越來越多的接受與應用。1 滴 timolol 0.5% 溶液眼內製劑的全身生體可用率範圍為 39% 至 98%,平均值為 78.3±24.5%。¹³¹
關於局部 timolol 的額外研究顯示,全身吸收會因賦形劑製劑而有差異。與平均血漿濃度為 0.46 至 1.72 ng/mL 的水性 timolol 溶液(0.25% 至 0.5%)相比,timolol 凝膠形成溶液(0.1% 至 0.5%)的開發是為了減少全身吸收,其平均血漿濃度範圍為 0.13 至 0.71 ng/mL。關於穿越 IH 受影響皮膚的吸收所知甚少。數個促成因素可影響獨立於賦形劑之外的全身吸收,包括施用部位(黏膜、潰瘍與封閉區域會增加吸收)、施用大小,以及病人大小(新生兒、尤其是早產兒,因表面積對體積比增加而有較大的吸收)。Timolol 由細胞色素 P450 CYP2D6 代謝;不良代謝者可能有較大的不良反應風險。心率變化與 timolol 的血漿濃度相關,因此在水凝膠製劑中發現較不顯著的心率變化。兩種賦形劑的局部 timolol 對肺部或血壓的影響均可忽略。¹³² 規模最大、檢力最足的多中心回溯性局部 (0.05%) timolol(85% gel foaming solution)研究(n = 731),平均治療持續期間為 9.5 個月(83.5% 的參與者劑量為每日兩次每次 1 滴),報告 3.4% 的不良事件(其中半數為施用部位的局部刺激,並有 3 例支氣管痙攣與 4 例潰瘍發展)。此回溯性研究結論為 timolol 在較薄的 IH 及較長的治療持續期間最有效。使用局部 timolol 時,顏色改善比整體體積、範圍與大小的改善更明顯。¹³³
皮質類固醇 (Corticosteroids):全身性皮質類固醇先前被視為造成變形、危及健康或危及生命之 IH 的第一線治療,鑑於 β-阻斷劑的療效、安全性與改善的耐受性,現在已很少使用。¹³⁴ 皮質類固醇在增生期最有效,在高達 90% 的病例中造成生長減緩或停止,約三分之一有實際縮小。雖然其作用機制尚未充分理解,先前研究提示粒線體細胞色素 b、clusterin/ApoJ(可能的細胞凋亡標記)及/或介白素-6 (interleukin-6) 的上調是皮質類固醇誘導 IH 生長停止的標記。¹³⁵⁻¹³⁷ 使用時,prednisone 或 prednisolone 以 2 至 3 mg/kg/day 的劑量給予,通常持續 4-8 週,隨後依兒童年齡與治療適應症進行不同長度的逐漸減量。一項統合分析顯示,以平均劑量 2.9 mg/kg 治療平均 1.8 個月後才減量,有 84% 的反應率。雖然 3 mg/kg/day(94% 反應)比 2 mg/kg/day(75% 反應)更有效,但較高劑量會發生較多的不良事件。¹³⁸
全身性皮質類固醇的短期併發症包括庫欣樣面容 (cushingoid faces) (71%)、人格改變(易怒/煩躁)(29%)、胃部刺激 (21%)、黴菌感染(口腔或會陰,6%),以及治療期間身高 (35%) 與體重 (42%) 增加的減少。超過 90% 身高增加減少的兒童在 24 個月齡時回到其治療前的生長曲線。¹³⁹,¹⁴⁰ 其他併發症包括高血壓、類固醇誘發的肌病變 (steroid-induced myopathy)、免疫抑制與暫時性腎上腺功能不全。¹⁴⁰,¹⁴¹ 治療醫師應密切監測血壓。¹⁴² 服用超過 2 mg/kg/day 的 prednisone 超過 14 天的兒童被視為有細胞媒介免疫的缺陷。接受高劑量皮質類固醇的嬰兒應延後接種活病毒疫苗。在此情況下曾報告罕見的肺囊蟲肺炎 (Pneumocystis carinii pneumonia) 病例,導致一些醫師在治療期間使用 trimethoprim-sulfamethoxazole 預防。¹⁴³,¹⁴⁴
病灶內皮質類固醇對於位於高風險部位(如嘴唇、鼻尖、臉頰與耳朵)之經選擇、相對較小、局限性的 IH 可為有效的治療。目前此方式用於 IH 是由少數具其使用專長的醫師所維持。不建議對眼周血管瘤注射。若使用,鑑於有視網膜動脈栓塞與失明的報告,應僅由有經驗的眼科醫師執行。¹⁴⁵,¹⁴⁶ 已發表最大型的病灶內類固醇病例系列發現,大多數顯示體積減少超過 50%,最佳結果發生於相對淺層的 IH。不良事件發生於 6.4% 的病人,包括庫欣樣外觀、皮膚萎縮與過敏性休克。¹⁴⁷
僅有少數病例系列報告了第 1 類局部皮質類固醇的療效,主要用於小型、淺層的血管瘤。¹¹⁰,¹⁴⁸,¹⁴⁹
具歷史重要性的藥物(Interferon-α、Vincristine、Imiquimod):Interferon-α、vincristine 與 imiquimod 先前被視為 IH 的第二線或第三線治療選項。鑑於 β-阻斷劑已確立的療效與較有利的副作用特性,已不再提倡使用它們。Interferon-α 帶有潛在神經毒性的顯著風險,特別是痙攣性雙側麻痺 (spastic diplegia)。一項 441 名病人的統合分析顯示,11 名發生不可逆的痙攣性雙側麻痺,16 名發生在停藥後可逆的運動障礙。¹⁵⁰ 所有受影響的病人在開始治療時都小於 1 歲。Vincristine 在血管腫瘤治療中的使用現在保留給複雜性卡波西樣血管內皮瘤 (kaposiform hemangioendothelioma, KHE) 與叢狀血管瘤 (tufted angioma, TA);其用於此適應症的地位未來幾年可能被 sirolimus 取代,但需要進一步研究。¹⁵¹⁻¹⁵³
治療程序 (Procedures)
雷射治療 (Laser Therapy):PDL 原本設計用於治療葡萄酒色斑(即毛細血管畸形),已被用於治療 IH,結果不一。¹⁵⁴ 在 propranolol 被發現之前,PDL 較常用於 IH 的治療。其用途主要受限於其極淺的穿透深度(少於 2 mm)。³⁵ 數篇報告顯示在治療相關潰瘍方面有改善;¹⁵⁵,¹⁵⁶ 其用於減少退化後殘餘的毛細血管擴張與紅斑是廣為接受的。其用於治療增生中的 IH 則有爭議。¹⁵⁷ 在一項前瞻性、隨機、對照研究中,22 名嬰兒接受觀察或帶表皮冷卻的 PDL 治療,PDL 組顯示顯著的顏色改善,但 12 個月追蹤時總表面積與回音深度不變。¹⁵⁸ 另一項前瞻性研究將兒童隨機分配至不帶表皮冷卻的 PDL 組(n = 60)與觀察組(n = 61),兩組在 1 歲時(近乎)完全消退的結果相當,PDL 組有色素減退與質地改變增加的趨勢。在 117 名兒童的 5 年追蹤中,兩組之間的結果相似。¹⁵⁵
其他研究顯示使用 585-nm PDL 或 595-nm PDL 配合不同的能量密度 (fluencies,每單位面積的總能量) 有良好結果,但數篇強調治療對較淺層的血管瘤效果最好,且無法阻止較深成分的生長。¹⁵⁹⁻¹⁶² 已有報告嚴重潰瘍與疤痕,特別是在增生期治療節段型血管瘤時。¹⁶³
雖然缺乏比較性研究,但在增生期對 PDL 為合理候選的同一批 IH,對局部 timolol 治療也同樣是良好的候選。專家意見與初步成本分析支持在大多數病例中對淺層 IH 使用 timolol 藥物治療,而非使用 PDL。¹³³ 一種保守的方法是將 PDL 主要保留用於治療潰瘍,以及在增生期完成後加速 IH 紅斑的消退。¹⁶⁴
手術治療 (Surgical Therapy):手術切除可在 IH 生命週期的任何時間有適應症,但嬰兒期的選擇性切除通常既不必要也不被提倡。嬰兒期 IH 手術介入的提議適應症包括:解剖上適合切除的有利部位、藥物治療的禁忌症或失敗,或最終必然需要切除且無論手術時機為何疤痕都相似的高風險。手術介入的時機取決於腫瘤的演變(是否持續退化)、解剖位置、變形程度與兒童年齡。某些解剖位置,如鼻尖與嘴唇,常需要手術。¹⁶⁵⁻¹⁷⁰ 在臨床特徵(如帶蒂的 IH、嚴重頑固性潰瘍,或極厚的真皮侵犯)決定疤痕將無可避免發生的病例中,可能有甚至更早切除的適應症。在大多數情況下,最好等到退化已充分進行,並可對是否已發生疤痕與質地變化做出更準確的評估。退化期間與之後的手術可允許重建受影響的鄰近結構、切除殘餘的多餘皮膚或纖維脂肪組織,或疤痕修整。關於此的決定常可在 3 至 5 歲之間做出,即使退化尚未完成。¹⁷¹
4 歲是適當的閾值年齡,因為大多數 IH 已完成退化,且兒童發展出自我意識、長期記憶與自尊。³⁵
常進行標準的橢圓形切除;然而,圓形切除後接續荷包式縫合 (purse-string closure) 可能留下較小的疤痕。¹⁷² 荷包式縫合後的第二階段手術可能需要也可能不需要。
篩檢 (Screening)
在頭部節段型 IH、沒有典型節段型 IH 或具小型 IH 但具有 PHACE 中所見之主要異常(例如主動脈窄縮或臍上縫)的嬰兒,以及具有 2016 年共識指引所概述之 2 項 PHACE 主要標準的嬰兒中,建議進行 PHACE 篩檢。⁴⁰
其他血管腫瘤 (Other Vascular Tumors)
重點一覽 (AT-A-GLANCE)
■ 在目前的國際血管異常研究學會分類中,血管腫瘤又再細分為 (a) 良性、(b) 局部侵襲性或交界性,或 (c) 惡性。
■ 先天性血管瘤 (congenital hemangiomas) 在出生時即完全成形。已辨識出快速退化型、非退化型與部分退化型先天性血管瘤等亞型。
■ 叢狀血管瘤 (tufted angiomas) 表現為不明顯的粉紅或暗紅色斑塊,可能演變為斑塊或結節,並有特徵性的組織學。
■ 卡波西樣血管內皮瘤 (kaposiform hemangioendothelioma) 是一種局部侵襲性血管腫瘤,型態上類似於但病因上不同於卡波西肉瘤 (Kaposi sarcoma),並可與 Kasabach-Merritt 現象相關。卡波西樣血管內皮瘤與叢狀血管瘤位於同一疾病光譜上。
■ 伴血小板低下之多灶型淋巴管瘤病 (multifocal lymphangiomatosis with thrombocytopenia) 由皮膚血管性丘疹與斑塊組成,並與間歇性血小板低下相關,常伴有消化道出血。
■ 梭形細胞血管瘤 (spindle-cell hemangioma) 通常發生於四肢,最常與 Maffucci 症候群相關。
■ 化膿性肉芽腫 (pyogenic granuloma) 是一種快速生長的丘疹或結節,帶有鱗屑領圈 (collarette of scale) 或糜爛表面,非常常見。治療為切除或電燒灼。
■ 先天性外分泌汗腺血管瘤性錯構瘤 (congenital eccrine angiomatous hamartoma) 是一種罕見、界限不清的斑塊,與胎毛增加及出汗相關。
許多其他良性血管腫瘤同時發生於兒童與成人。¹⁷³ 本節依據 2014 年 ISSVA 分類重點介紹選定的重要血管腫瘤,該分類將血管腫瘤分類為 (a) 良性、(b) 局部侵襲性或交界性,或 (c) 惡性。關於此主題更全面的回顧可見於他處。²,¹⁷³,¹⁷⁴
先天性血管瘤 (Congenital Hemangiomas)
出生時即為完全成形的腫瘤、且在出生後不增生的血管瘤稱為先天性血管瘤。它們是良性的,且遠比 IH 少見。現在根據其自然病程辨識出 3 個主要亞型:快速退化型 (rapidly involuting, RICH)、非退化型 (noninvoluting, NICH) 與部分退化型先天性血管瘤。¹⁷⁵,¹⁷⁶
近年來對部分退化型先天性血管瘤亞型的認識,凸顯了這些腫瘤存在於一個光譜上。確切的分類在新生兒期之初常具挑戰性,且常只能在觀察臨床行為後回溯性地有信心地做出。先天性血管瘤有相似的好發解剖部位,包括頭部、頸部與四肢,但也可發生於他處。它們在男孩與女孩中同樣常見。¹⁷⁶
RICH 常表現為一個隆起的紫色腫瘤,帶有大型放射狀靜脈,或帶有覆蓋其上的毛細血管擴張與一圈蒼白暈 (halo of pallor)(Fig. 118-16)。可能存在中央潰瘍。大多數 RICH 在 14 個月齡時或之前自發性退化,通常留下殘餘的萎縮性鬆弛疤痕組織。¹⁷⁷ 曾報告一個在產前超音波上發現、推測為 RICH 並在出生前退化的病例,在病灶部位留下疤痕組織。¹⁷⁸
RICH 可與輕至中度血小板低下及以纖維蛋白降解產物升高與纖維蛋白原低下為特徵的凝血功能障礙相關,於新生兒期出現。與 Kasabach-Merritt 現象(於「卡波西樣血管內皮瘤」一節討論)相反,皮膚 RICH 的實驗室檢查結果常為自限性且無併發症,一般只需支持性照護。¹⁷⁹
肝臟 RICH 是肝臟血管瘤的局灶型亞型,罕見,但併發症風險較大,與顯著的血小板低下以及嚴重進行性肝功能障礙及繼發的心衰竭相關。NICH 比 RICH 少見,也在出生時出現。它們通常比 RICH 更扁平,表現為界限分明、圓形至橢圓形、有些硬化或隆起的軟組織腫塊,帶有覆蓋其上的毛細血管擴張與一圈蒼白暈 (Fig. 118-17)。
部分退化型先天性血管瘤於 2014 年被引入血管異常的詞彙中,已報告的病例少於 20 例。部分退化發生於生命的前 12 至 30 個月(平均:18 個月),之後才趨於穩定 (Fig. 118-18)。¹⁷⁶
據報告,GNAQ 與 GNA11 基因的體細胞活化錯義突變 (somatic activating missense mutations) 會導致 RICH 與 NICH。其突變改變——胺基酸第 209 位麩醯胺酸 (Gln209) 的改變——在兩種腫瘤中相同,因此無法解釋其不同的臨床病程。¹⁸⁰ 同樣的 Gln209 錯義突變在葡萄膜黑色素瘤 (uveal melanoma) 中常見,且 GNAQ 與 GNA11 密碼子 183 或 209 的合子後鑲嵌突變 (postzygotic mosaic mutations) 曾被報告與斑痣性錯構瘤病色素血管型 (phakomatosis pigmentovascularis) 及廣泛性真皮黑色素細胞增多症 (extensive dermal melanocytosis) 相關。¹⁸¹
先天性血管瘤的治療適應症與 IH 相似,包括潰瘍、重要功能損害與充血性心衰竭。對於可導致嚴重出血的潰瘍,以及若有毀容的退化後皮膚變化,絕對應考慮切除,加或不加術前栓塞。¹⁷⁵ NICH 不會消失,但常無症狀;關於其移除的決定必須權衡所提議治療的風險與益處。先天性血管瘤的區辨性病理特徵是位於緻密纖維化基質內的毛細血管小葉,含有飽滿的內皮細胞與周細胞,基質中含有含鐵血黃素 (hemosiderin) 沉積、局灶性小葉血栓 (lobular thrombosis) 與硬化。¹⁸² 與 IH 不同,它們 GLUT1 陰性。¹⁸³ 在超音波上,先天性血管瘤呈現異質性,可能有散在的鈣化,且一般侷限於皮下脂肪。它們是高流量血管腫瘤,在杜卜勒檢查時常顯示動靜脈微瘻管 (arteriovenous microfistulas),在 NICH 中較常見。¹⁸⁴
叢狀血管瘤 (Tufted Angioma)
流行病學 (Epidemiology)
TA 是一種罕見的良性血管腫瘤,也曾被稱為 Nakagawa 血管母細胞瘤 (angioblastoma of Nakagawa)。雖然曾報告成人病例,但大多數病例發生於兒童早期,並可能有遷延的病程。約 25% 的 TA 是先天性的,50% 出現於出生後第一年。無性別好發傾向。
臨床特徵 (Clinical Features)
TA 表現出各種臨床模式,最常見於頸部、軀幹與四肢。它們可能表現為一塊不明顯的染色狀區域,後來增厚;或表現為一個大型、斑塊狀、浸潤性、紅色或暗藍紫色的病灶;或表現為一個外突性、堅實、橡膠狀、紫色的皮膚結節 (Fig. 118-19)。可能發生壓痛與覆蓋其上的多汗 (hyperhidrosis)。¹⁸⁵ TA 通常為單發,但已有多灶型病例的報告。¹⁸⁶
併發症 (Complications)
Kasabach-Merritt 現象 (KMP) 可能在 TA 中發生,但這在 KHE 中更常見(見「卡波西樣血管內皮瘤」一節)。
病因與致病機轉 (Etiology and Pathogenesis)
其病因與致病機轉不確定,不過根據其免疫染色模式,淋巴起源 (lymphatic origin) 是可能的。與 IH 不同,沒有已知的性別或妊娠年齡相關性。
診斷 (Diagnosis)
組織學上,後天性與先天性 TA 皆顯示緊密堆積的毛細血管所構成的血管叢 (vascular tufts),隨機散布於整個真皮中,呈典型的「砲彈狀分布 (cannonball distribution)」,血管叢周圍有新月形腔隙,腫瘤基質內有淋巴管樣腔隙。¹⁸⁷,¹⁸⁸ 與 IH 不同,TA 不與 GLUT1 染色。⁸² 它是 CD31+;淋巴標記 D2-40、LYVE1 與 PROX1 在周圍血管中僅部分陽性,有助於將 TA 與 KHE 區分。¹⁸⁹
臨床病程與預後 (Clinical Course and Prognosis)
TA 可能維持不變,或在幾年內完全退化。¹⁹⁰ 出生時或出生後第一年即存在的 TA,比生命較晚出現者有較大的自發退化傾向。¹⁹¹ 在會退化的 TA 中,據報告 95% 在 2 年內退化。¹⁸⁷
處置 (Management)
目前沒有廣為接受的 TA 處置治療指引。對於 TA(有或無 KMP)的建議療法包括切除、加壓、雷射、局部或全身性皮質類固醇、propranolol 與化療。關於治療的更完整討論見下文「卡波西樣血管內皮瘤」。
卡波西樣血管內皮瘤 (Kaposiform Hemangioendothelioma)
流行病學 (Epidemiology)
KHE 是一種罕見的血管腫瘤,估計盛行率為每 100,000 名兒童 0.91 例。¹⁹² 它可能在出生時即存在或在兒童早期發展。一個超過 100 名病人的系列報告,60% 以新生兒表現,93% 在嬰兒期表現。已有罕見的成人病例報告。四分之三侵犯皮膚。它通常被報告與 KMP 相關,後者發生於超過 70% 的病例。¹⁹²
臨床特徵 (Clinical Features)
KHE 是一種浸潤性、侵襲性腫瘤,在 ISSVA 分類中歸類為局部侵襲性或交界性。它可能表現為出生時的棕紅色染色,開始增厚並變得紫斑狀 (purpuric),或表現為斑塊、深部結節與龐大腫瘤。大多數病例侵犯皮膚與肌肉組織。KHE 可發生於四肢(尤其是覆蓋關節處)、軀幹、頭部、頸部。較深的內臟,包括頸胸部、腹部與後腹腔,可受侵犯。值得注意的是,KHE 不發生於肝臟。局部區域淋巴結侵犯可能發生,且可能代表多灶型疾病。遠端轉移不會發生。縱膈或後腹腔疾病可能以血胸 (hemothorax) 與腹水 (ascites) 表現。¹⁴¹ 自發性退化罕見。大多數報告的病例有相關的 KMP,但 KHE 可在無凝血功能障礙的情況下發生。¹⁹²⁻¹⁹⁵ KHE 可能以萎縮性或染色狀區域、浸潤性斑塊與丘疹,或結節消退。殘餘纖維化不罕見,並可造成相當大的發病。¹⁹⁶
併發症 (Complications)
Kasabach-Merritt 現象:KMP 指的是血小板被捕陷於血管腫瘤內,導致極度嚴重的血小板低下(通常少於 30000/µL),並伴隨微血管病性溶血性貧血 (microangiopathic hemolytic anemia) 與凝血因子消耗,造成纖維蛋白原低下、d-dimer 升高與不同程度的凝血因子下降 (Fig. 118-20)。它長期以來被認為是「血管瘤」的併發症,但現在認知到它幾乎專屬於 TA 與 KHE 的併發症,而非 IH。¹⁴,¹⁹⁷ KMP 必須與可在靜脈及混合靜脈-淋巴管畸形中發生的凝血功能障礙(有時被錯誤地稱為 KMP)區分,後者是慢性凝血與凝血因子消耗的結果,但並非主要由血小板捕陷所致,¹⁹⁸ 更可能是局部或瀰漫性血管內凝血。
病因與致病機轉 (Etiology and Pathogenesis)
KHE 與 KMP 的致病機轉不明。KHE 與 TA 被認為存在於一個嚴重度光譜上。¹⁹²
危險因子 (Risk Factors)
侵犯超過 1 個解剖區域會增加發生 KMP 的風險,而較晚年齡表現、淺層腫瘤或僅侵犯骨骼之腫瘤的 KHE,似乎有較低的 KMP 風險。¹⁹²
診斷 (Diagnosis)
診斷基於臨床與組織學特徵及實驗室異常,包括嚴重血小板低下、纖維蛋白原消耗、d-dimer 升高、不同程度的凝血因子下降,以及微血管病性溶血性貧血。KHE 的組織學特徵是浸潤性結節與成片的梭形內皮細胞,具有極少的非典型性與罕見的有絲分裂,襯覆著含有含鐵血黃素的裂隙狀或新月形血管。可見水腫、微血栓、纖維化與外觀非典型的淋巴管。腫瘤 GLUT1 陰性。與 TA 相似,與淋巴標記(包括 vascular endothelial growth factor receptor-3 與 D2-40)反應的淋巴腔隙增加,提示淋巴起源,類似於卡波西肉瘤。¹⁹⁹,²⁰⁰
臨床病程與預後 (Clinical Course and Prognosis)
KHE 的預後與處置隨腫瘤的範圍與位置,以及 KMP 的存在而異。雖然 KHE 與 TA 可能持續存在,但 KMP 的凝血功能障礙通常在 1 歲時或更早(經治療)緩解。
處置 (Management)
沒有任何單一治療能一致有效,也沒有廣為接受的 KHE 處置治療指引。合併 KMP 的 KHE 比無凝血功能障礙的 KHE 需要更積極的介入。在可能的情況下,完全手術切除是治癒性的,但鑑於腫瘤的浸潤性質常不可行。全身性皮質類固醇被視為第一線治療,並常與 vincristine 合併使用,尤其是在 KMP 的情況下。Ticlopidine 與 aspirin 被報告為輔助療法,效益不一。動脈栓塞可在計畫性手術前作為輔助以降低出血風險,並可能造成凝血功能障礙與病灶大小的暫時改善,但作為單一療法則很少有效。²⁰¹ 關於在此病況中使用口服 sirolimus(一種哺乳動物雷帕黴素標靶抑制劑 mammalian target of rapamycin inhibitor)的報告很有前景,²⁰²,²⁰³ 顯示凝血功能障礙的快速逆轉;前瞻性臨床試驗的結果待定。較舊的文獻報告使用放射治療、cyclophosphamide、interferon-α、actinomycin-d 與 methotrexate,但這些都被視為第三線療法,僅在其他藥物失敗的情況下使用。除非發生活動性出血或在手術程序之前,否則應避免輸注血小板。²⁰⁴⁻²⁰⁸ 使用其他輸血血品已證明對矯正凝血功能障礙無效。
梭形細胞血管瘤 (Spindle-Cell Hemangioma)
梭形細胞血管瘤,也稱為梭形細胞血管內皮瘤 (spindle-cell hemangioendothelioma),是一種罕見的良性血管腫瘤,最常見於 Maffucci 症候群。雖然與此症候群相關的基因突變已被辨識(IDH1 與 IDH2,產生將異檸檬酸轉化為 2-酮戊二酸的異檸檬酸脫氫酶 isocitrate dehydrogenases),並在此病況相關的皮膚表現中被檢測到,但突變與此疾病表現之間的關係仍不明確。它可發生於任何年齡與部位,但四肢是最常受影響者。組織學為結節狀、緻密的梭形細胞增生,與擴張的發育不良靜脈相關。病灶可為局部侵襲性,並可能在切除後仍復發。²⁰⁹
化膿性肉芽腫(葉狀毛細血管血管瘤)(Pyogenic Granuloma / Lobular Capillary Hemangioma)
化膿性肉芽腫 (pyogenic granuloma, PG) 也以其正確的組織病理描述——葉狀毛細血管血管瘤 (lobular capillary hemangioma)——而為人所知。它是嬰兒與兒童最常見的血管腫瘤之一,也可發生於成人,特別是孕婦。PG 通常表現為一個單發、紅色、快速生長的丘疹或結節,常帶有不明顯的鱗屑領圈 (collarette of scale)(Fig. 118-21)。典型位置包括臉頰或前額,但實際上任何身體部位包括黏膜都可受影響。PG 的快速生長通常出現於比 IH 增生期相關年齡更晚的年齡,但在非常年幼的嬰兒中可造成混淆。²¹⁰
它們常發展出糜爛表面,隨後出血,出血可能很大量,因而得到「OK 繃疾病 (Band-Aid disease)」的綽號。已有罕見的單發與聚集性 (agminated) PG 樣病灶與血管異常(包括動靜脈畸形與先天性血管瘤)相關的報告。²¹¹,²¹² PG 通常不會自發退化。即使在確定性治療後,仍有原始 PG 周圍復發甚至衛星病灶的報告。²¹³,²¹⁴ 簡單的刮除合併電燒灼通常具治癒性。其他選項包括切除、雷射手術(二氧化碳 carbon dioxide 或脈衝染料雷射)與冷凍治療。Imiquimod 與 timolol 被建議為有效的局部治療選項。²¹⁵,²¹⁶
伴血小板低下之多灶型淋巴管內皮瘤病 (Multifocal Lymphangioendotheliomatosis with Thrombocytopenia)
MLT——也稱為伴血小板低下之皮膚內臟血管瘤病 (cutaneovisceral angiomatosis with thrombocytopenia)——是一種罕見病況,最初於 2004 年被描述,其特徵為多發性皮膚血管性丘疹與斑塊,通常為先天性並隨時間發展出新病灶。²¹⁷ 目前尚不清楚它是血管腫瘤還是血管畸形。有些病人有顯著的外突性皮膚血管腫瘤,而其他病人則有不明顯的藍莓馬芬狀 (blueberry muffin-like) 丘疹。²¹⁸ 大多數受影響的嬰兒有間歇性血小板低下與消化道病灶,導致消化道出血。其他報告的侵犯部位包括骨骼、滑膜、肺、肝、脾與腦。現在已了解到許多先前被診斷為「瀰漫性新生兒血管瘤病」的病人實際上患有 MLT。²¹⁹ 皮膚切片檢體顯示薄壁血管、一些釘頭狀 (hobnailed) 內皮細胞,以及類似於 Dabska 腫瘤的腔內乳頭狀突起。²¹⁷,²²⁰ 內皮細胞對 CD31、CD34 與 LYVE1 染色陽性,對 D2-40 染色不定,且對 GLUT1 陰性。¹⁸⁶,²¹⁸ 預後依疾病範圍與出血併發症而異。一項統合分析報告 65% 的死亡率。²¹⁹ 有效的治療一直具挑戰性,但 sirolimus 的早期經驗是正面的,有些作者建議將其作為第一線治療。²²¹,²²² 已報告多種其他全身性藥物有不等的效益,包括皮質類固醇、vincristine、thalidomide、interferon-α2a、propranolol 與 bevacizumab。²¹⁸,²²³
乳頭狀淋巴管內血管內皮瘤 (Papillary Intralymphatic Angioendothelioma)
乳頭狀淋巴管內血管內皮瘤 (papillary intralymphatic angioendothelioma, PILA),也稱為 Dabska 腫瘤,是一個真皮結節或頭部、頸部或四肢的瀰漫性紫色腫脹,幾乎只見於小兒年齡層。PILA/Dabska 腫瘤是一種低惡性度血管腫瘤,其罕見性使得對其長期預後與最佳治療的研究變得困難。組織病理學顯示厚壁的擴大血管,周圍環繞著淋巴聚集 (lymphoid aggregates) 與較小的淋巴管。腫瘤血管內存在「釘頭狀 (hobnail)」內皮細胞是 PILA/Dabska 腫瘤的必要條件 (sine qua non)。¹⁸⁵
網狀血管內皮瘤 (Retiform Hemangioendothelioma)
網狀血管內皮瘤主要發生於成人,並與 PILA/Dabska 腫瘤有顯著重疊。無顯著的性別好發傾向,發病發生於生命的第二至第四個十年(平均年齡:36 歲)。組織病理學顯示存在由單一型態釘頭狀內皮細胞襯覆的長、分支狀血管所構成的網狀模式。大多數病例可見顯著的淋巴球浸潤,並局灶性存在構成乳頭的玻璃樣膠原核心,類似於 PILA/Dabska 腫瘤。復發頻繁,但轉移率低。²²⁴
複合性血管內皮瘤 (Composite Hemangioendothelioma)
複合性血管內皮瘤是一種低至中惡性度的惡性血管腫瘤,由良性與惡性血管成分的可變組合構成。要做出此診斷,必須辨識出至少 2 種血管內皮瘤變異型。上皮樣與網狀變異型最常被觀察到,不過也可存在梭形細胞血管瘤及/或血管肉瘤樣 (angiosarcoma-like) 病灶的病灶。²²⁵
靶狀含鐵血黃素性血管瘤 (Targetoid Hemosiderotic Hemangioma)
靶狀含鐵血黃素性血管瘤是一種良性病灶,表現為一個紫色丘疹,常被一圈蒼白邊緣與周邊瘀斑暈 (ecchymotic halo) 環繞,並隨時間消退。病灶通常出現於軀幹或四肢,組織學上由擴張的血管腔道組成,腔內乳頭狀突起切入皮下的膠原束中。存在外滲的紅血球與含鐵血黃素,因而有此命名。
先天性外分泌汗腺血管瘤性錯構瘤(汗腺血管瘤)(Congenital Eccrine Angiomatous Hamartoma / Sudoriparous Angioma)
先天性外分泌汗腺血管瘤性錯構瘤是一種罕見病況,特徵為界限不清的斑塊,在病灶部位有胎毛增加與出汗。它們通常位於四肢或腹部。診斷基於特徵性的組織學發現確立:緊密堆積的外分泌汗腺,與擴張的毛細血管、少數發育不良的靜脈腔道,以及緻密的膠原基質相關。²²⁶
圖表 (Figures and Tables)
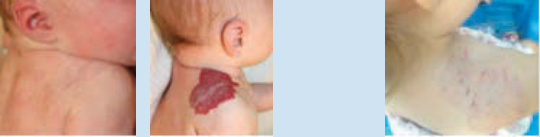
圖 118-1:嬰兒型血管瘤的生長特徵 (Growth characteristics of infantile hemangioma)。
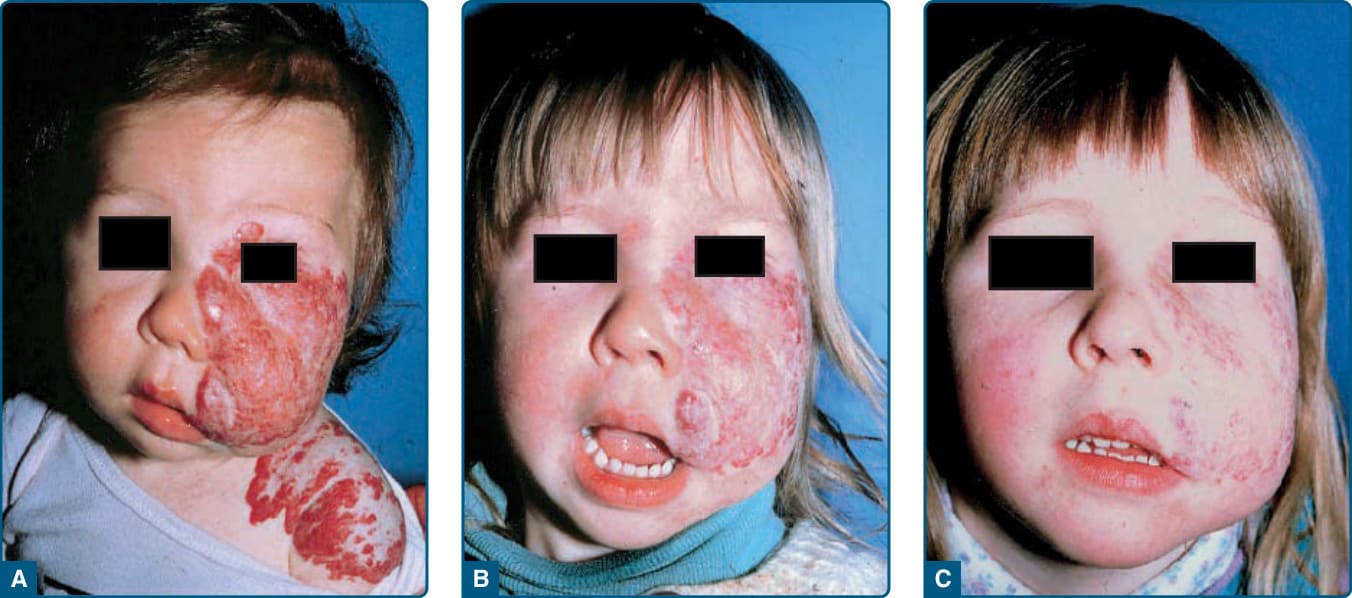
圖 118-2:節段型嬰兒型血管瘤的自然病程。注意其斑塊狀、地理性的構型。A,11 個月齡,增生期高峰。B,2 歲,退化期。C,4 歲,大部分的自然退化已發生。
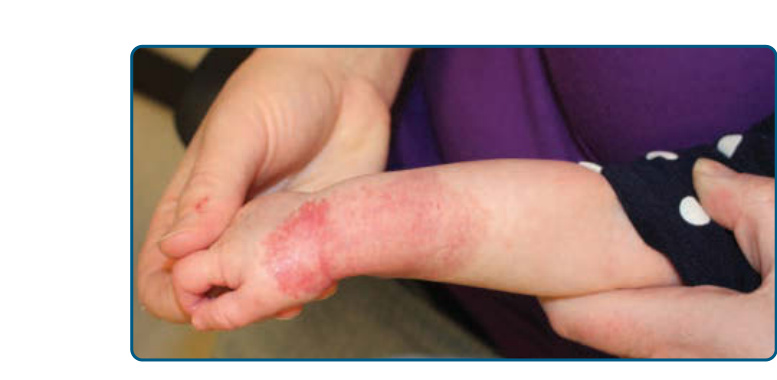
圖 118-3:一名嬰兒右臂與右手上具極小或流產型生長的嬰兒型血管瘤。
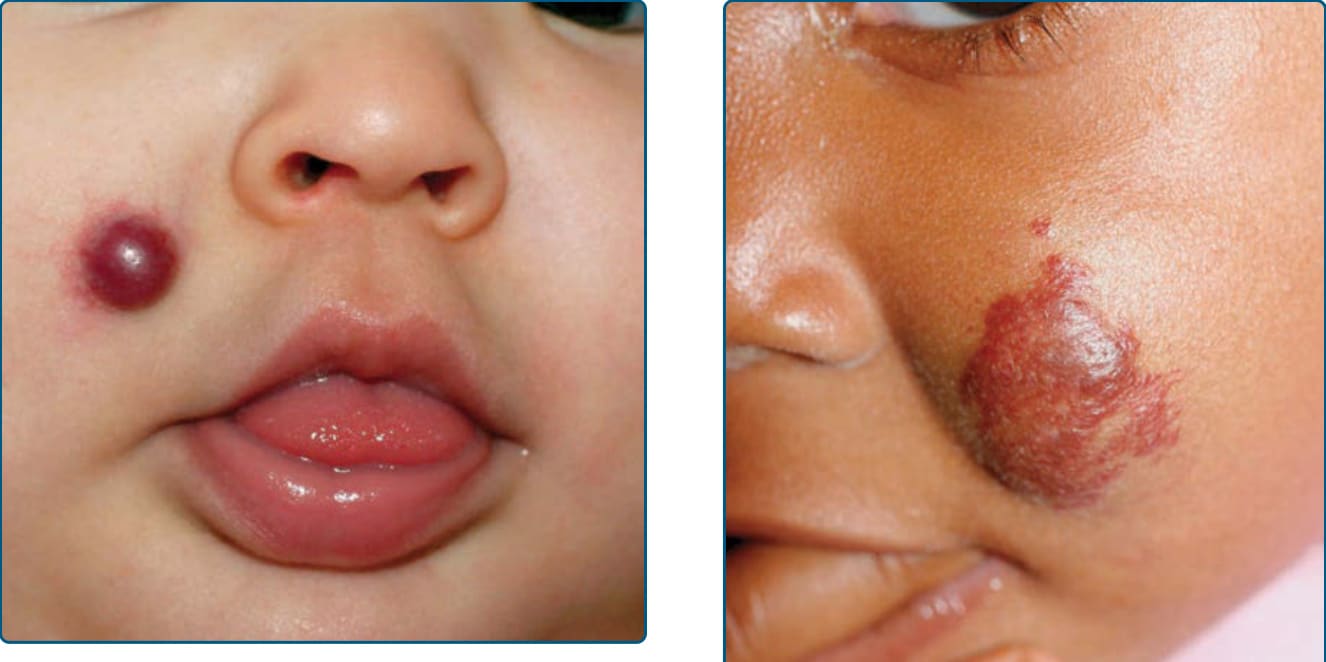
圖 118-4:一名嬰兒臉頰上的局限型嬰兒型血管瘤。
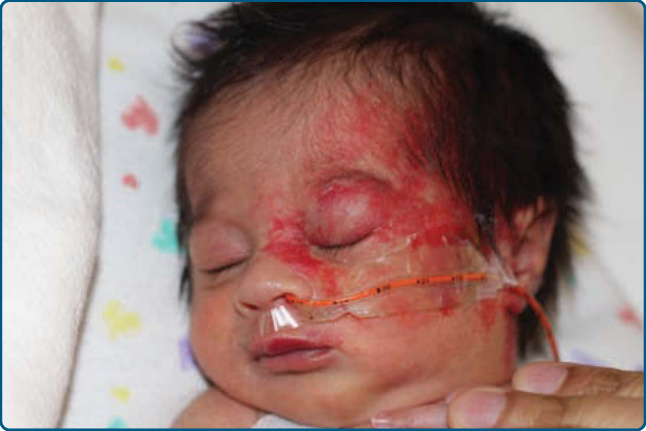
圖 118-5:一名患有 PHACE 之新生兒臉部、頭皮與頸部的節段型嬰兒型血管瘤。
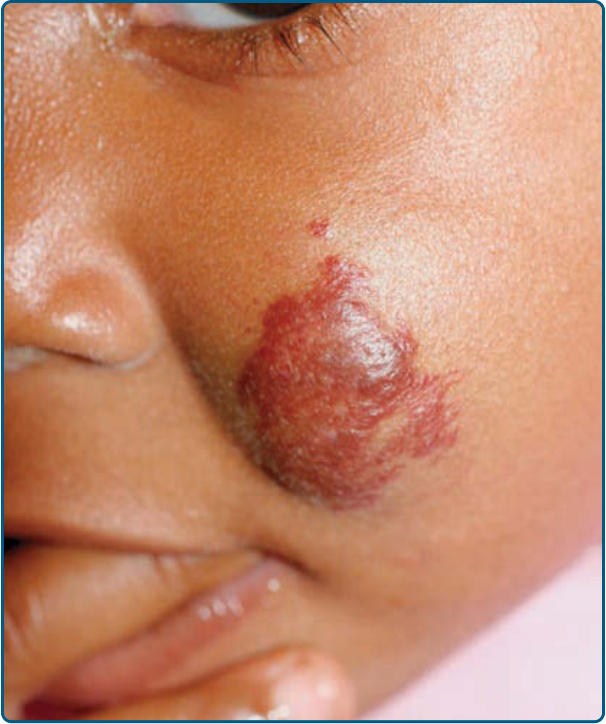
圖 118-6:一名 14 個月齡嬰兒臉頰上的不確定型嬰兒型血管瘤。
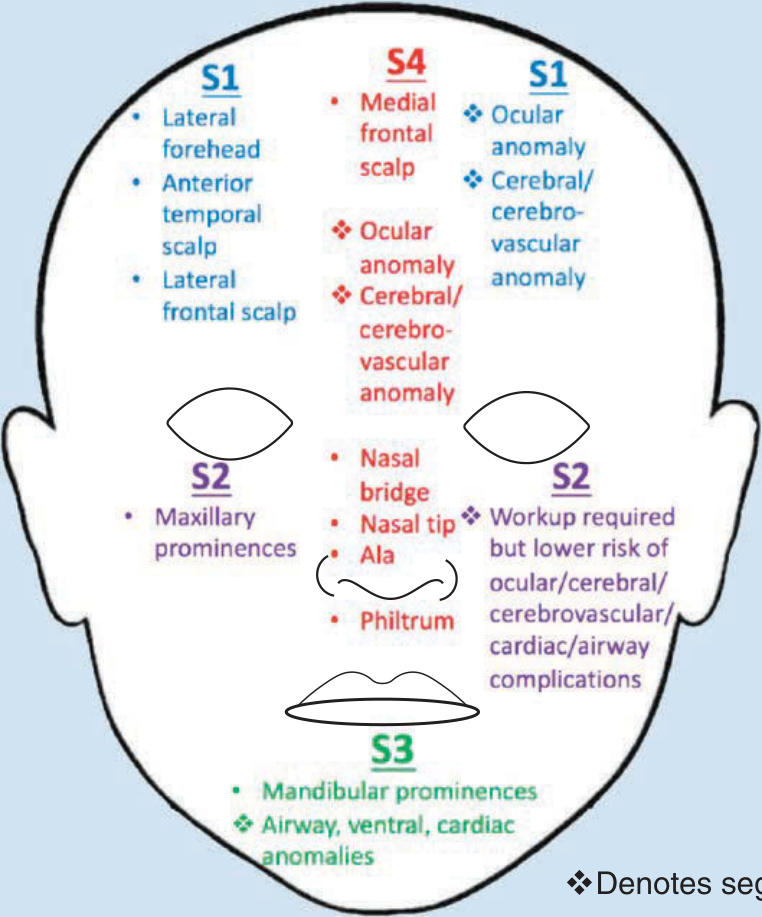
圖 118-7:節段型發育單位與對應的疾病表現部位。(Data from Haggstrom AN, Lammer EJ, Schneider RA, et al. Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics. 2006;117(3):698-703.)
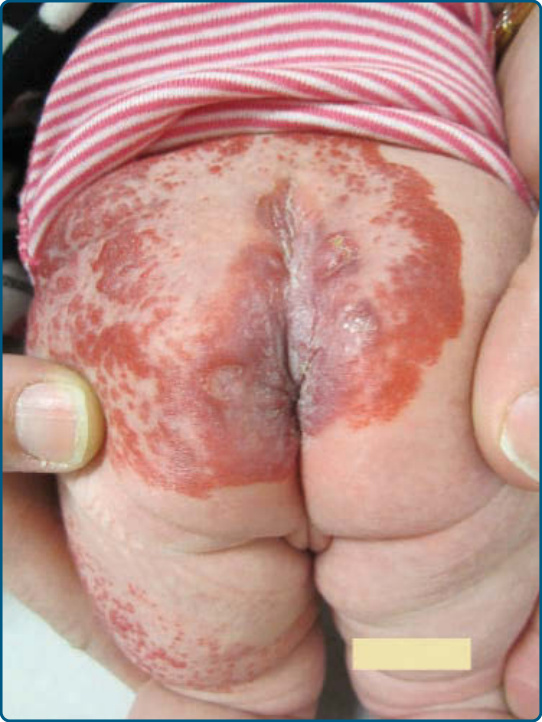
圖 118-8:一名患有 LUMBAR 症候群、併發疼痛性潰瘍之嬰兒下半身的節段型嬰兒型血管瘤。
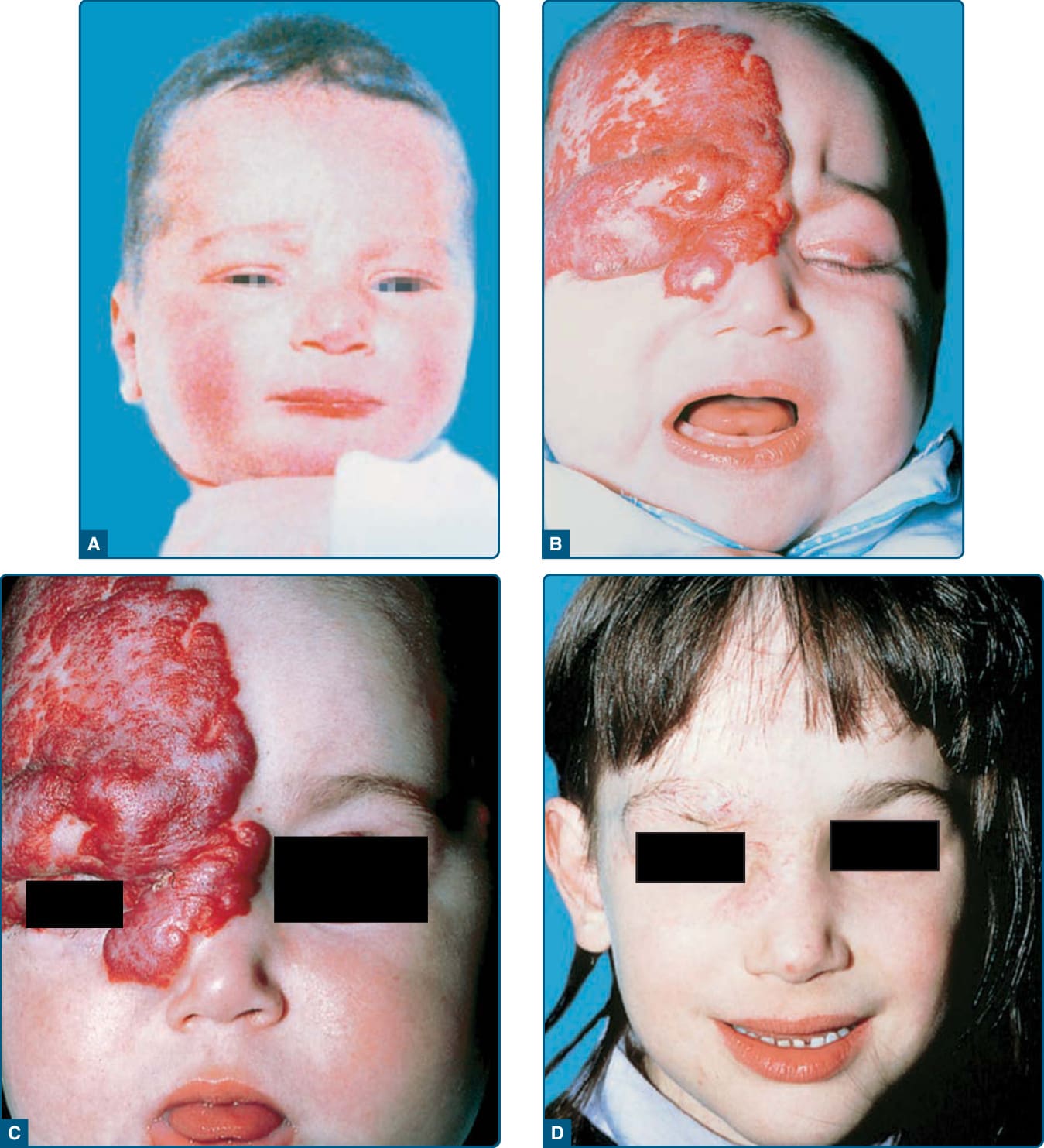
圖 118-9:以全身性糖皮質素治療的危及視力之節段型嬰兒型血管瘤。A,嬰兒照片上未見腫瘤的前兆徵象。B,至 3 個月齡,廣泛的嬰兒型血管瘤浸潤上眼瞼與周圍組織,造成視力阻擋。C,糖皮質素治療後 2 週內眼瞼張開。D,6 歲時退化的腫瘤伴殘餘疤痕。
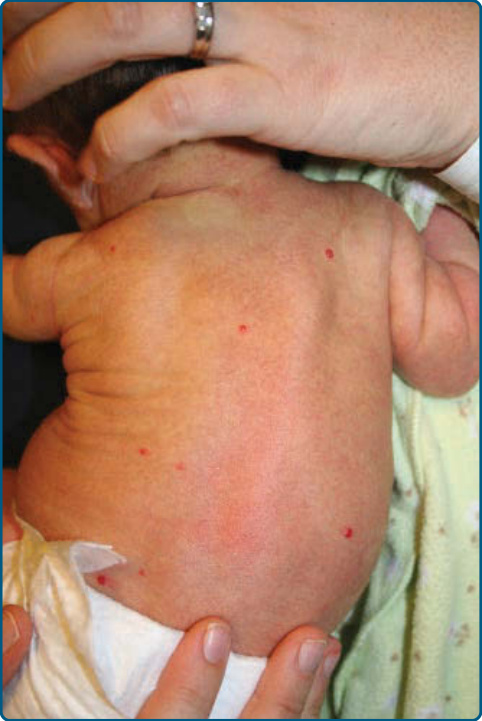
圖 118-10:一名嬰兒的多灶型嬰兒型血管瘤。軀幹與四肢上有多個小型、淺層、粉紅色丘疹。
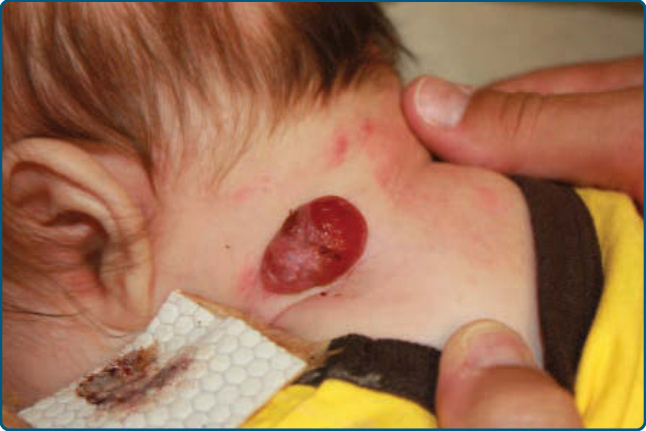
圖 118-11:頸部一個疼痛性潰瘍的嬰兒型血管瘤,需以傷口照護、脈衝染料雷射與 propranolol 治療。
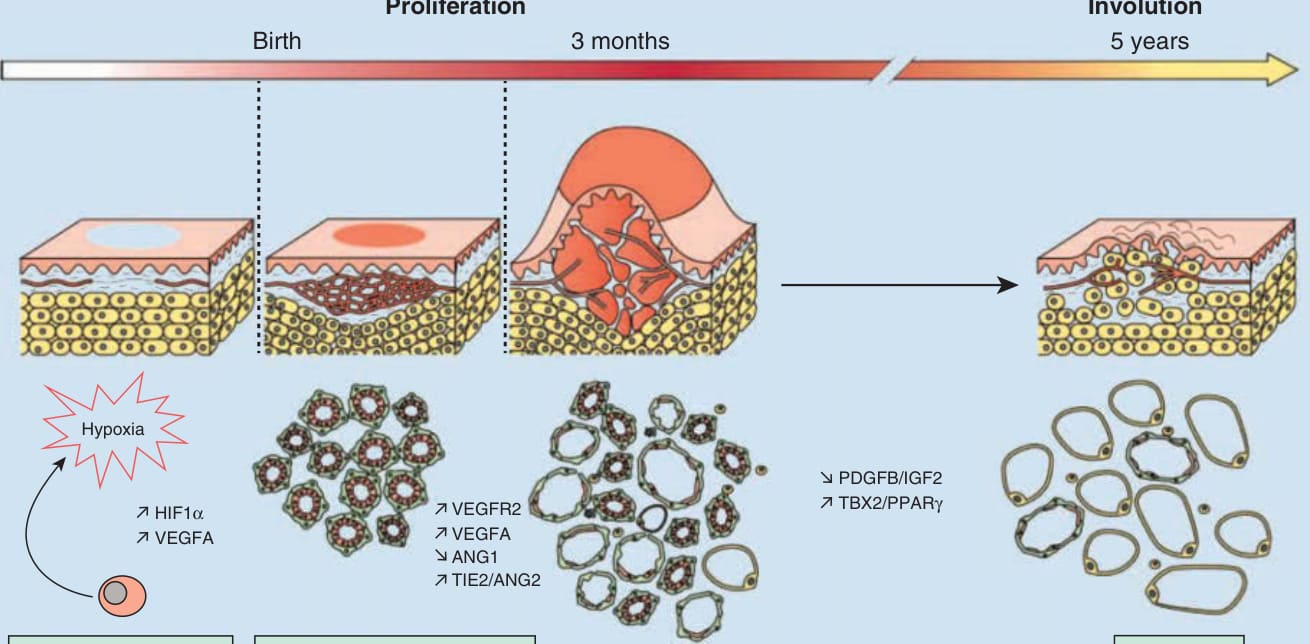
圖 118-12:嬰兒型血管瘤的致病機轉。(Figure reprinted from Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. The Lancet. 2017;390(10089):85-94, with permission. Copyright © Elsevier.)
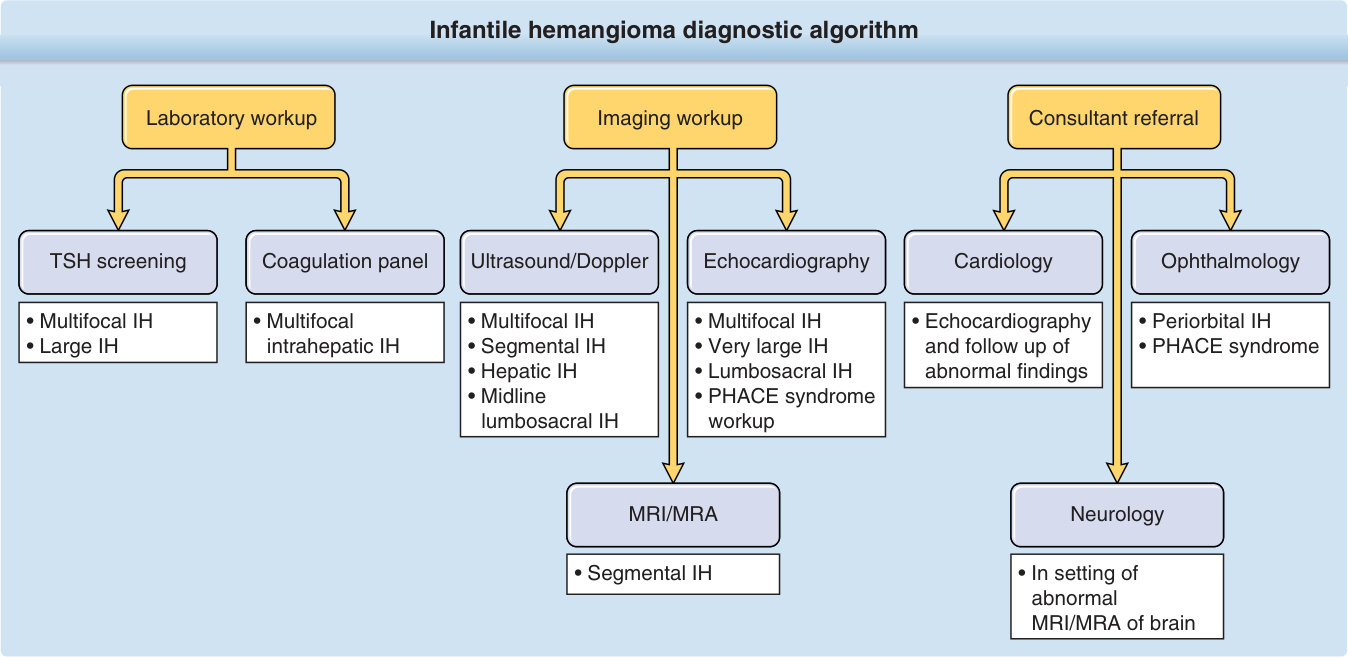
圖 118-13:嬰兒型血管瘤 (IH) 診斷演算法。PHACE,posterior fossa brain malformations, hemangiomas of the face, arterial anomalies, cardiac anomalies, and eye abnormalities;TSH,thyroid-stimulating hormone。(Adapted from Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390(10089):85-94.)
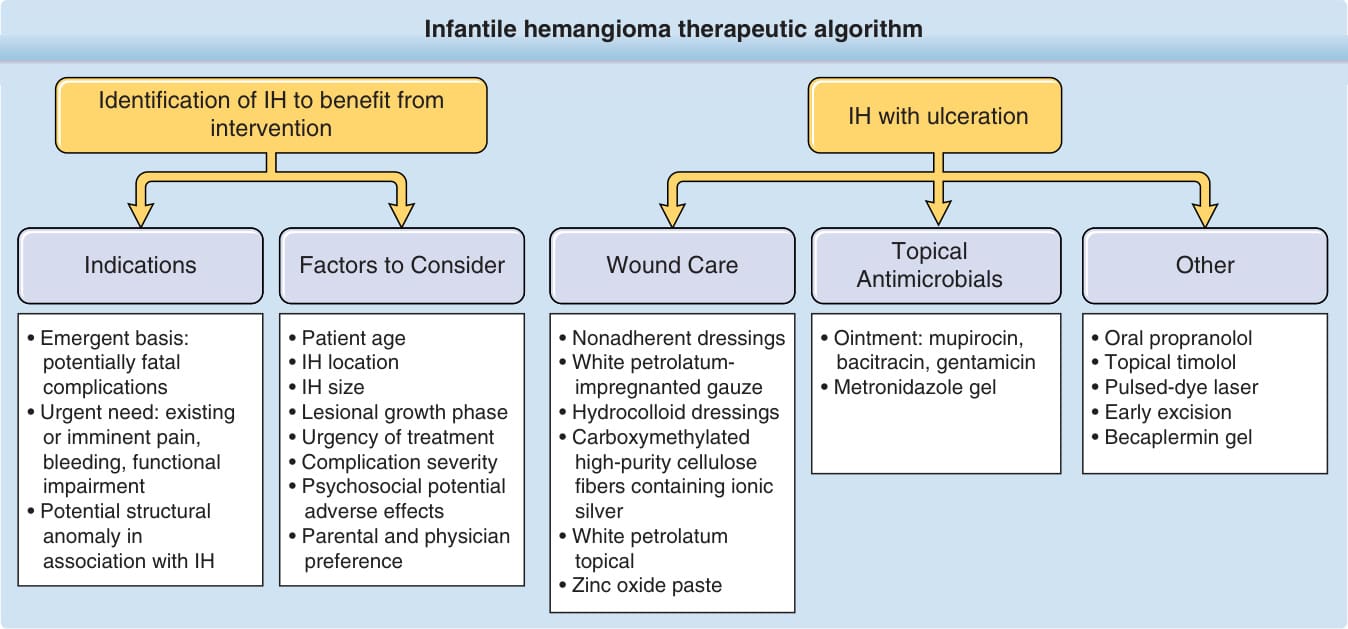
圖 118-14:嬰兒型血管瘤 (IH) 治療演算法。(Data from Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136(4):e1060-e1104.)
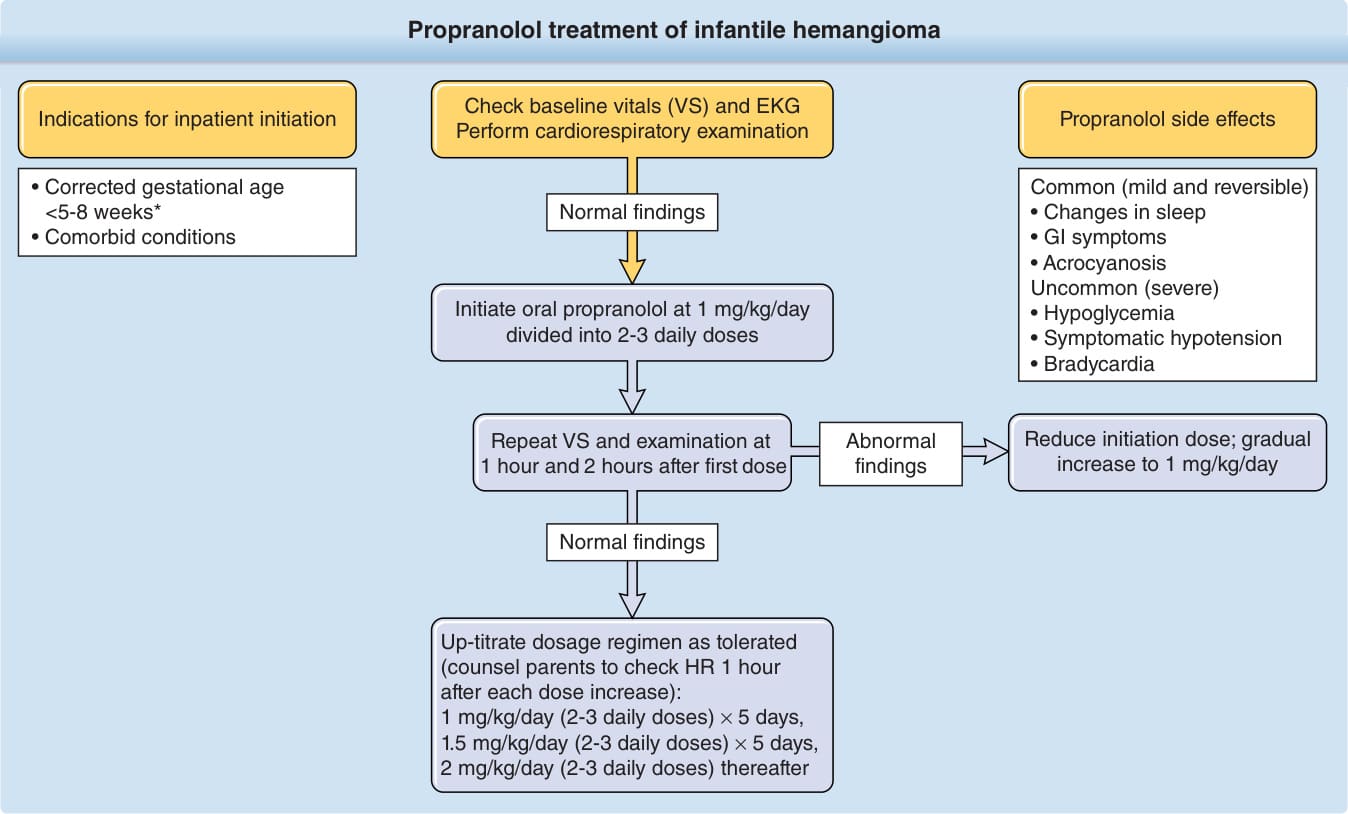
圖 118-15:嬰兒型血管瘤的 propranolol 治療。∗Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131(1):128-140; Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735-746. (Data from Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131(1):128-140.)
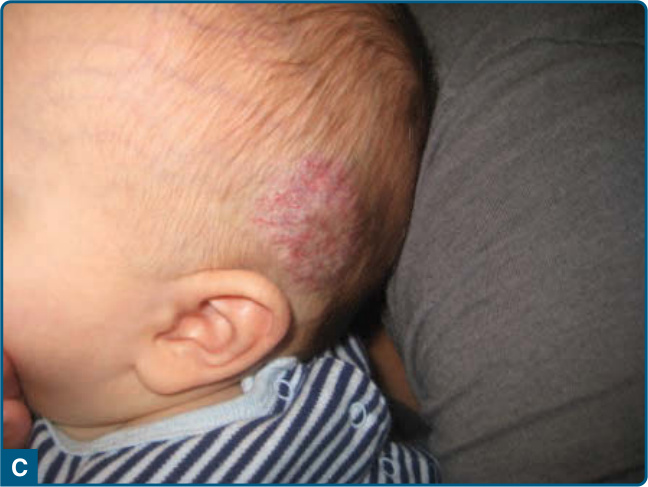
圖 118-16:左側頭皮的快速退化型先天性血管瘤。A,1 個月齡的嬰兒,覆蓋其上有粗糙的毛細血管擴張。B,5 個月齡時的早期退化。C,8 個月齡時持續快速退化。
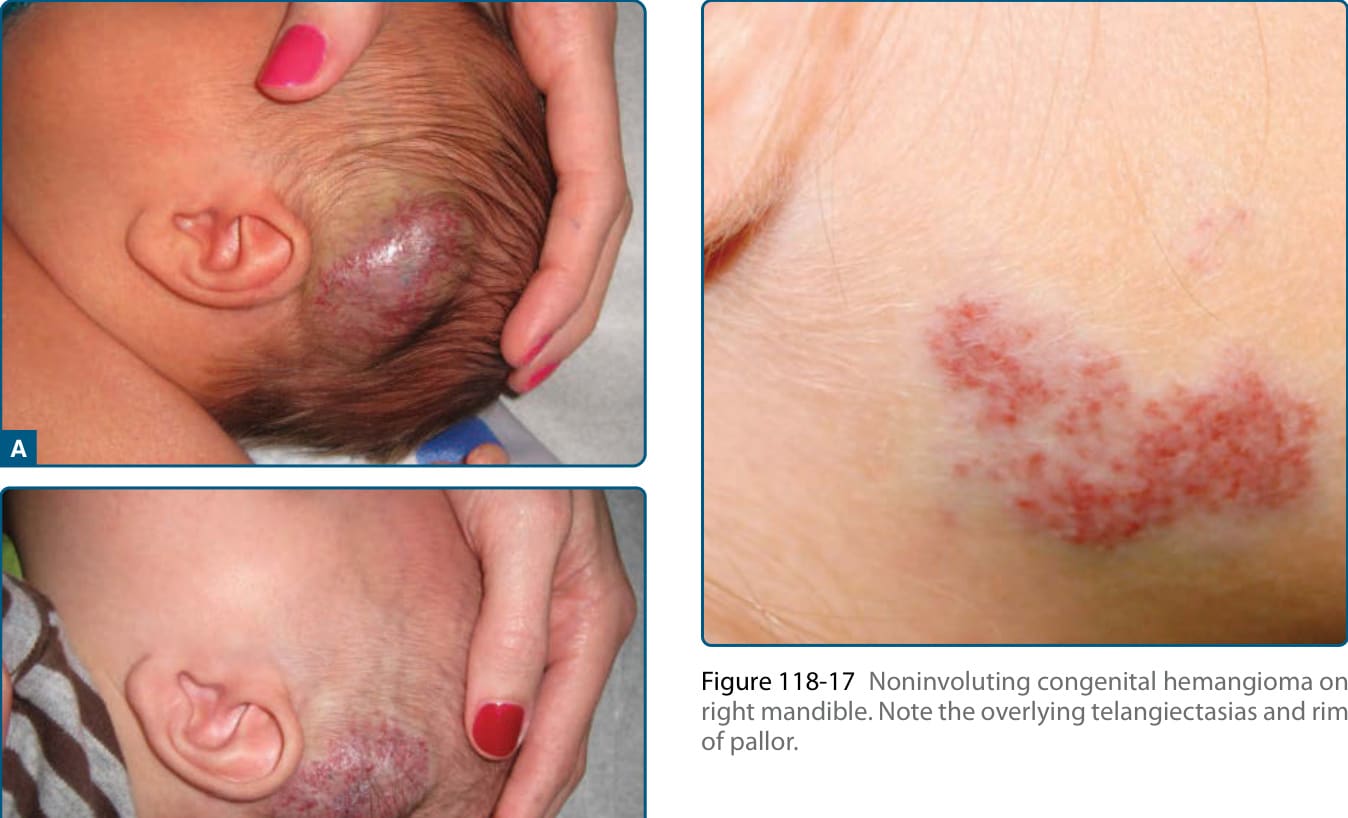
圖 118-17:右側下頷的非退化型先天性血管瘤。注意覆蓋其上的毛細血管擴張與一圈蒼白暈。
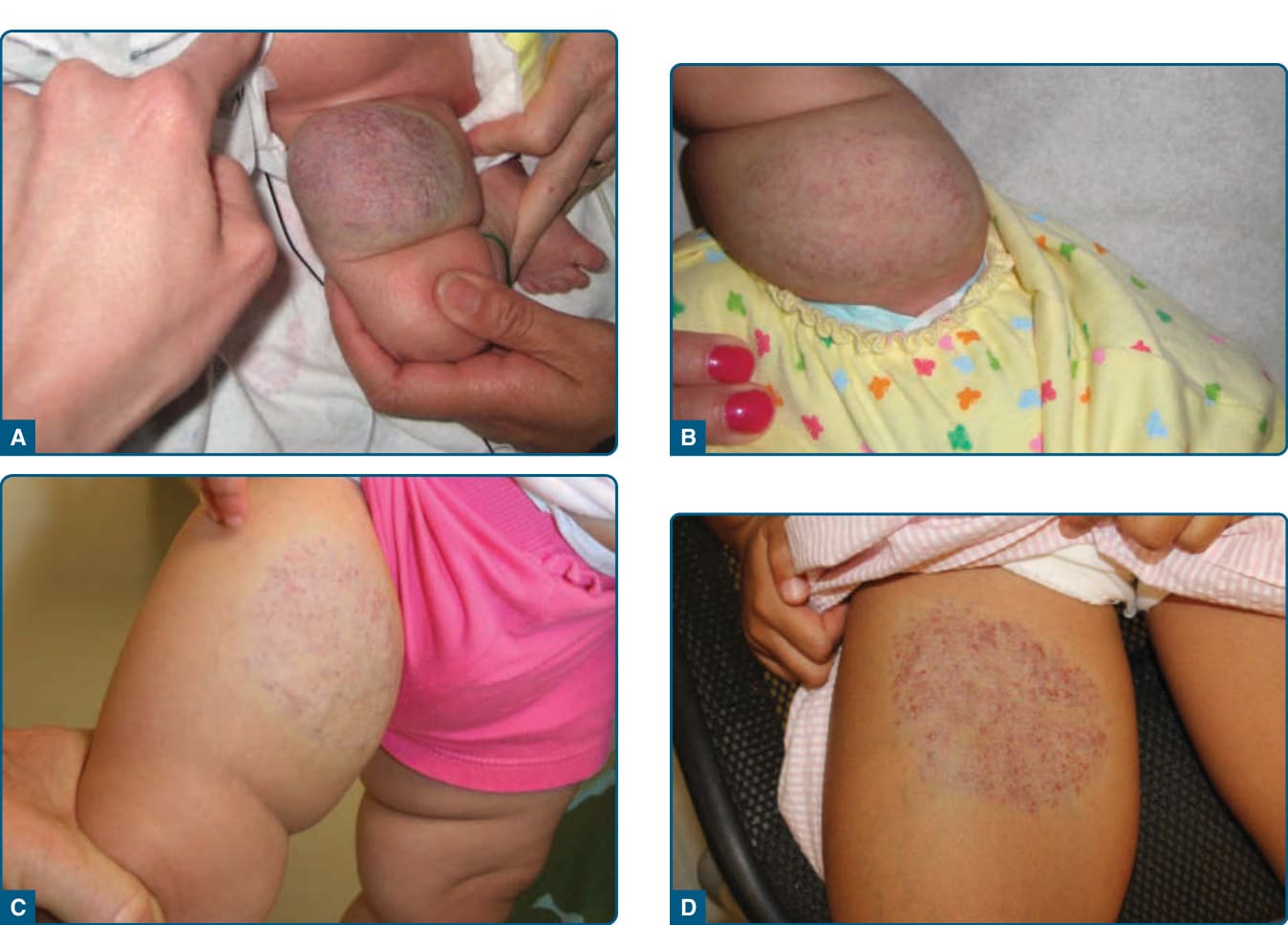
圖 118-18:右側大腿的部分退化型先天性血管瘤。注意覆蓋其上的毛細血管擴張與一圈蒼白暈。A,出生第一天。B,6 週齡時大小早期減少。C,13 個月齡時寬廣但扁平。D,5 歲時未再進一步退化,且淺層血管重新變得明顯。
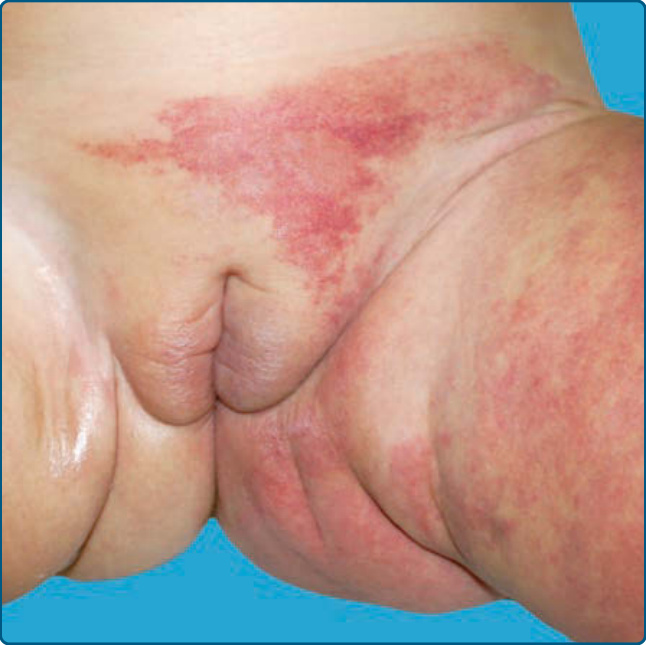
圖 118-19:一名 18 個月大兒童左腿、臀部、下腹部的叢狀血管瘤(經切片證實)。實驗室評估顯示 D-dimer 升高。
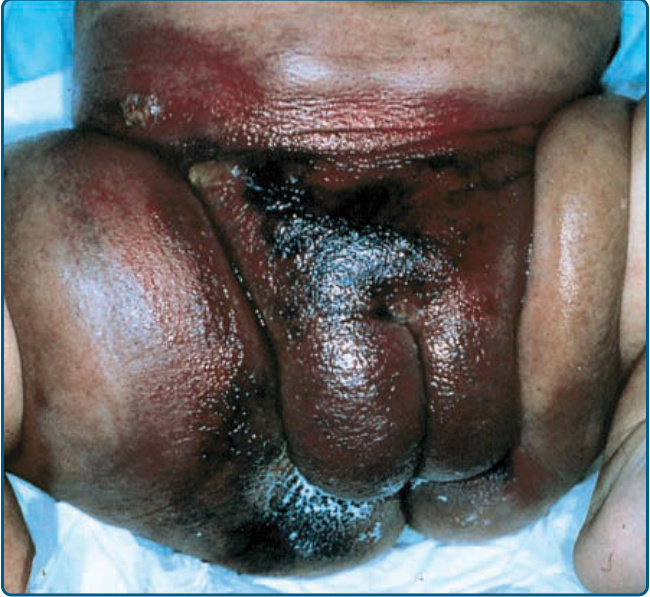
圖 118-20:繼發於卡波西樣血管內皮瘤 (KHE) 的 Kasabach-Merritt 現象。一個硬化、紫斑狀的腫瘤於 3 個月齡時出現,此處所見為 8 個月齡時。KHE 經 MRI 與切片證實。血小板計數 <5000/mm³。
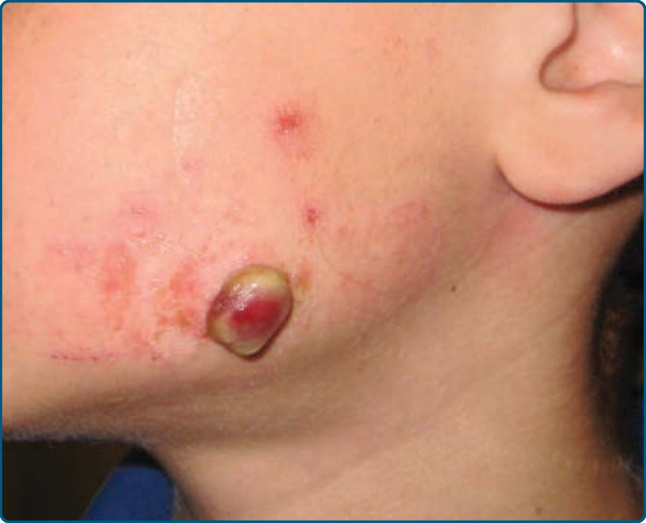
圖 118-21:左臉頰的大型化膿性肉芽腫。
表 118-1:國際血管異常研究學會 (International Society for the Study of Vascular Anomalies) 分類

表 118-2:病史 (History)
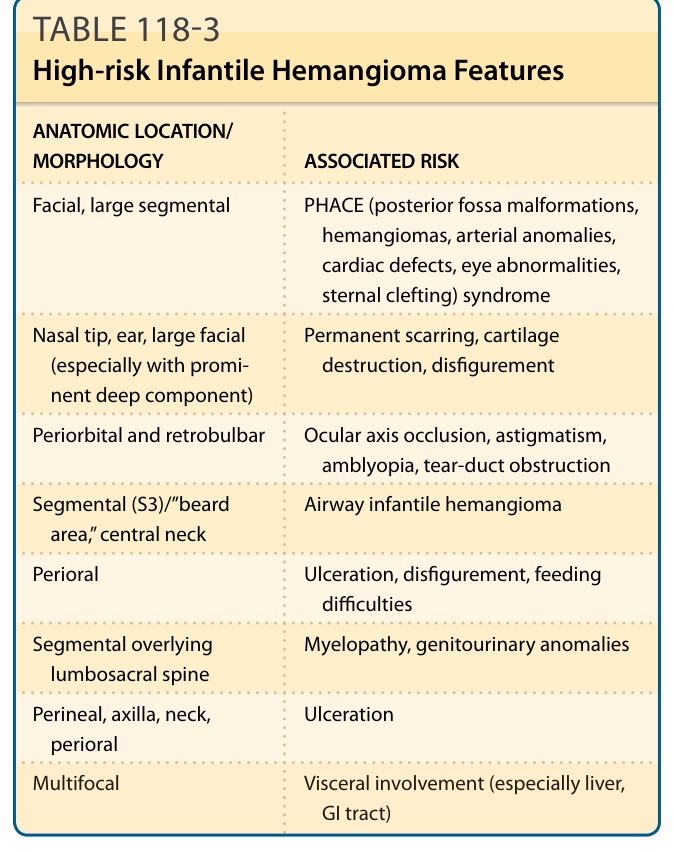
表 118-3:高風險嬰兒型血管瘤特徵 (High-risk Infantile Hemangioma Features)
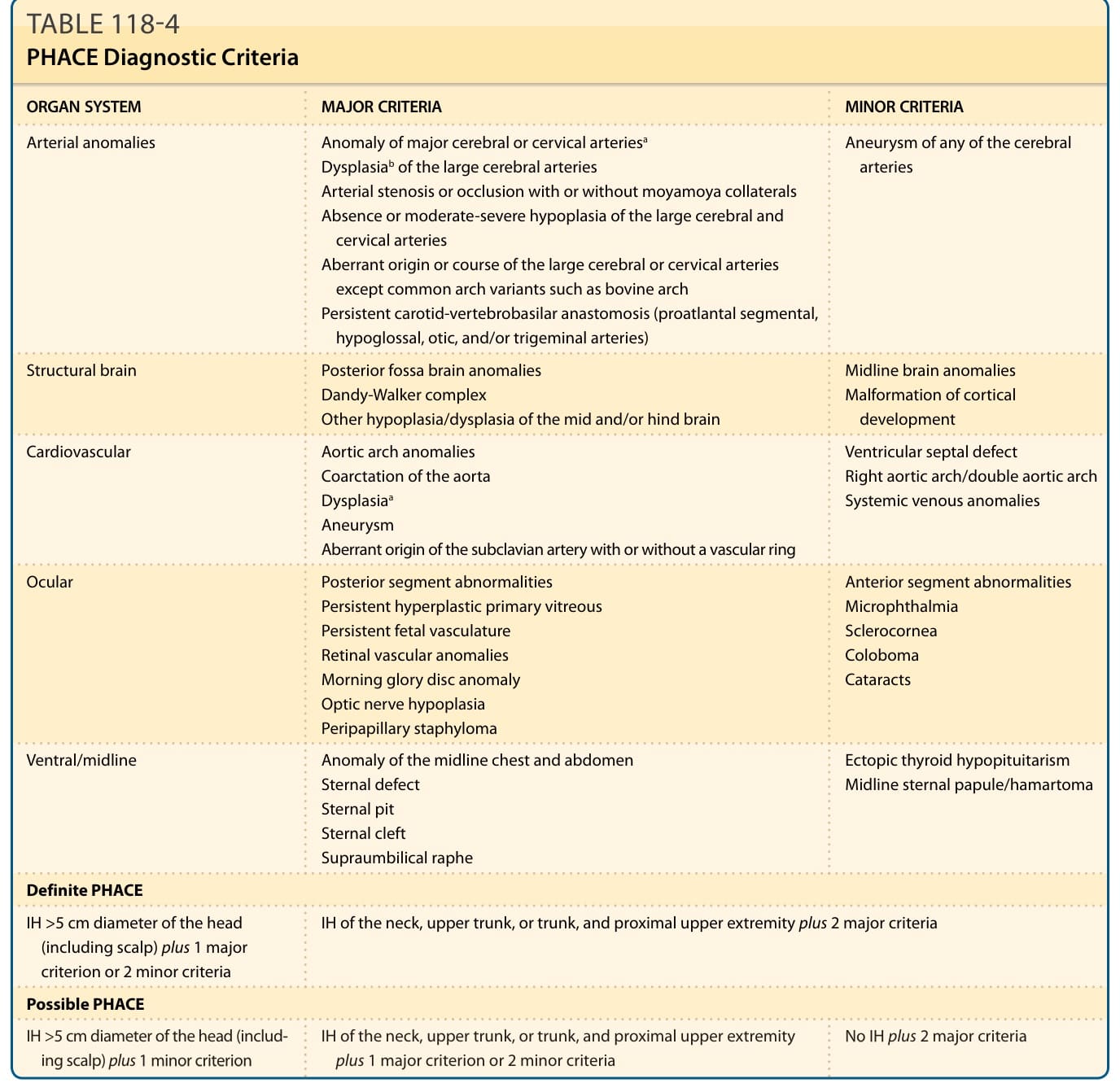
表 118-4:PHACE 診斷標準 (PHACE Diagnostic Criteria)

表 118-5:潰瘍的處置 (Management of Ulcerations)
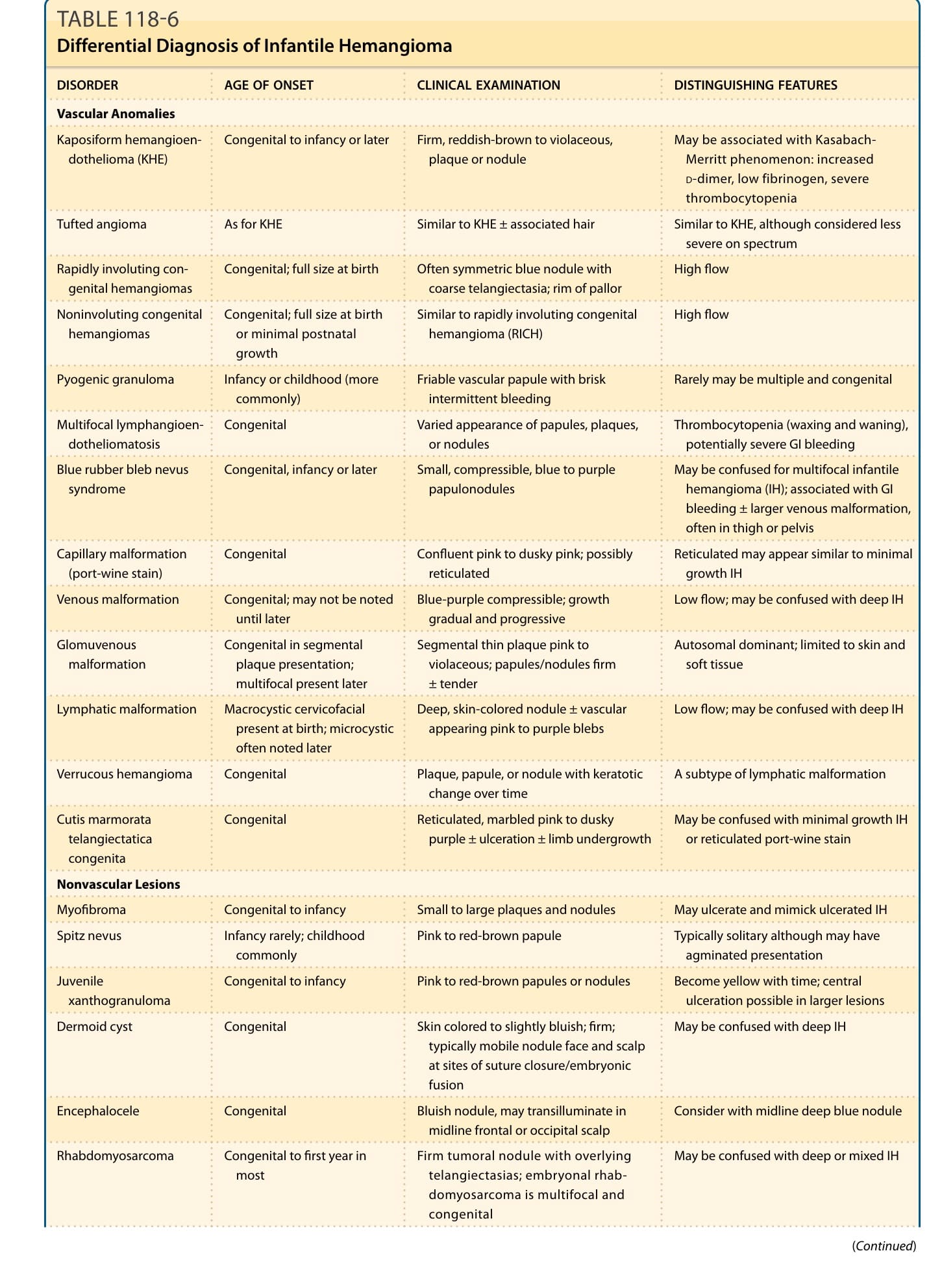
表 118-6:討論 IH 的鑑別診斷。¹⁰³