Vascular Tumors
20
Vascular anomalies commonly present as birthmarks.1
Their classification has historically been problematic, with contradictory and confusing terminology and nomenclature. Although several classification schemes exist, the most widely accepted and comprehensive is the International Society for the Study of Vascular Anomalies (ISSVA) classification, updated in 2014, to incorporate clinical, imaging, histologic and (where known) genetic characteristics.1,2 Vascular anomalies are broadly divided into vascular malformations and vascular tumors.3 Vascular malformations (see Chap. 147) are errors of vascular morphogenesis whereas infantile hemangiomas and other vascular tumors are proliferative. Vascular tumors in the current ISSVA classification are subdivided into (a) benign, (b) locally aggressive or borderline, and (c) malignant; each has unique features based upon histology, biology, clinical appearance, behavior, prognosis, and treatment (Table 118-1). There are several types of vascular tumors, many of which occur in childhood. These tumors also have confusing nosology,4 with descriptive but imprecise terminology such as strawberry, capillary, and cavernous. The nonspecific term hemangioma should be avoided as a standalone term without a qualifying descriptor (eg, infantile, rapidly involuting congenital, lobular capillary, spindle cell).
INFANTILE HEMANGIOMAS
AT-A-GLANCE
■ Infantile hemangiomas are the most common tumor of infancy.
■ Infantile hemangiomas in high-risk anatomic sites are likely to require further workup and treatment.
■ Segmental infantile hemangiomas are associated with greater morbidity than localized infantile hemangiomas.
■ Kasabach-Merritt syndrome does not occur with infantile hemangiomas.
EPIDEMIOLOGY
EPIDEMIOLOGY
Infantile hemangiomas (IHs) are the most common benign tumors of childhood, occurring in approximately 4% to 5% of children.4-7 IHs are distinguished from other vascular tumors and malformations by their unique growth pattern (rapid proliferative phase
with subsequent slower involution.) They are more common in girls (2 to 3:1 ratio), in white, non-Hispanic infants,6,8,9 and infants with low birth weight, and affect up to 30% of premature infants,5,10 especially those weighing less than 2500 g.8,11 Low birth weight appears to be the most significant risk factor for IH development, conferring greater risk than prematurity alone, with a 40% risk increase for every 500 g decrease in birth weight,11 which complements earlier data showing that nearly 1 in 4 infants whose birth weight is less than 1000 g develops IH. Prenatal risk factors include advanced maternal age (older than 30 years of age), preeclampsia, placenta previa, and other placental anomalies.8,12 Preterm infants are more likely to have multiple IHs. IH in the setting of prematurity is less-strongly associated with female predominance.8,13
Complicated IHs are more common in girls; the cause is unknown.14 Chorionic villus sampling is no longer accepted as a significant risk factor.8,11,13
CLINICAL FEATURES
CLINICAL FEATURES
CUTANEOUS FINDINGS
A clinical history of a life cycle marked by characteristic proliferation and involution is critical in the diagnosis of IH (Table 118-2).15
Growth Characteristics—Proliferation: Absence at birth or presence of a nascent IH, often an area of pallor, telangiectasias, or duskiness is characteristic. A fully formed soft-tissue mass at birth is almost certainly another vascular anomaly or other disease process. Most IHs occur on the head and neck, but His can be found at any cutaneous or mucosal site and can, less commonly, involve internal organs (see section “Noncutaneous Findings and Complications”). Almost all IHs with a superficial component become apparent in the first month of life, and most will at least double in size within the first 2 months of life. The early proliferative phase is associated with rapid nonlinear growth with the peak IH growth period occurring between 5.5 weeks and 7.5 weeks of age.16
While 80% of growth has occurred by 3 months of age, 80% of IHs have completed all growth by 5 months of age (Fig. 118-1).17 The late proliferative stage of ongoing slower growth that occurs after peak rapid growth typically ends by 9 months of age; only 3% of IHs have clinically documented growth beyond this age.17
Deep IHs are more likely to have a longer proliferative phase. Large, segmental and parotid gland IHs may also continue to enlarge slowly for months to, rarely, years, which is longer than other IHs.17-19
20
VASCULAR ANOMALIES
VASCULAR TUMORS VASCULAR MALFORMATIONS
BENIGN LOCALLY AGGRESSIVE MALIGNANT SIMPLE COMBINED
Infantile hemangioma Kaposiform hemangioendothelioma Angiosarcoma Capillary malformation (C) CVM, CLM
Congenital hemangioma Retiform hemangioendothelioma Epithelioid hemangioendothelioma Lymphatic malformation (LM) LVM, CLVM
Tufted hemangioma PILA, Dabska tumor Venous malformation (VM) CAVM
Spindle-cell hemangioma Composite hemangioendothelioma Arteriovenous malformation (AVM) CLAVM
Epithelioid hemangioma Kaposi sarcoma Arteriovenous fistula
Pyogenic granuloma
Pyogenic granuloma
CAVM, capillary arteriovenous malformation; CLAVM, capillary lymphatic arteriovenous malformation; CLM, capillary lymphatic malformation; CLVM, capillary lymphatic venous malformation; CVM, capillary venous malformation; LVM, venous malformation; PILA, papillary intralymphatic angioendothelioma. Abbreviated ISSVA classification for Vascular Anomalies by International Society for the Study of Vascular Anomalies is licensed under a Creative Commons Attribution 4.0 International License.
Growth Characteristics—Involution: The rapid and late proliferative growth phases are followed by a slower involution phase that is more variable in length, lasting for months to years (Fig. 118-2). Evidence of involution, often referred to as graying, involves change to a dull red, then gray or milkywhite color, followed by flattening and softening. The change is usually apparent by 1 year of age.17 Smaller hemangiomas typically involute sooner than very large ones.17 More than 90% of IHs have completed involution by 3.5 to 4 years of age.20,21 It is important to recognize that complete involution does not equate with complete resolution of the IH. More than half of children with hemangiomas will be left with residual telangiectasias, fibrofatty tissue, or anetodermic skin. Superficial IHs are more likely to develop residual skin
■Birth history
■Birth history
■Was the child of low birth weight and/or premature?
■Was the child of low birth weight and/or premature?
■Part of a multiple gestational birth?
■Part of a multiple gestational birth?
■Placental abnormalities?
■Placental abnormalities?
■Overall health and past medical history
■Overall health and past medical history
■Is the child feeding well and gaining weight appropriately?
■Is the child feeding well and gaining weight appropriately?
■Have there been any hospitalizations or major illnesses?
■Have there been any hospitalizations or major illnesses?
■History of “birthmark”
■History of “birthmark”
■Was it visible at the time of birth?
■Was it visible at the time of birth?
■Has it changed since birth?
■Has it changed since birth?
■Growth: Is the birthmark growing proportionately or disproportionately with child’s somatic growth? Is it still growing, stable, or shrinking in size? (Serial photographs are helpful.)
■Growth: Is the birthmark growing proportionately or dispropor-
tionately with child’s somatic growth? Is it still growing, stable, or shrinking in size? (Serial photographs are helpful.)
■Any complications such as ulceration, pain or bleeding?
■Any complications such as ulceration, pain or bleeding?
■Any prior treatments?
■Any prior treatments?
■Family history
■Family history
■Is there a family history of infantile hemangiomas or other vascular birthmarks?
■Is there a family history of infantile hemangiomas or other
vascular birthmarks?
changes following involution compared with deep IHs (odds ratio: 8.4).22 Combined IH and those with a steep border or a cobblestoned surface have a significantly higher risk of permanent scarring.21
Classification8: Cutaneous IH morphology can be classified based on tumor depth (superficial, deep, combined/mixed) and distribution (localized, segmental, indeterminate, multifocal).23,24 This morphologic distinction can guide expectations as natural history, prognosis, and possible complications vary based on these classification features. Superficial IHs are the most common of the morphologic subtypes, presenting as bright fuchsia pink to “strawberry” red vascular plaques or nodules; they involve the papillary dermis. In contrast, deep IHs present as a partially compressible, localized subcutaneous tumors with variable prominence depending upon depth in the skin, and either appear with a slight blue hue or the same color as the surrounding skin; they may be noted, on average, 1 month later than superficial IHs. Less frequently, deep IHs are not appreciated until the infant is a few months of age. Deep IHs are more likely to have a longer proliferative phase, rarely growing for months to years beyond the growth phase of other IHs, and later onset of involution compared with their superficial counterparts.17,18 Combined (also called mixed) IHs have clinical features of both deep and superficial IHs. A less-common subtype of IH is distinguished by its minimal-to-absent proliferation and telangiectatic surface, referred to as IH with minimal or abortive growth,25 has been described (Fig. 118-3). Found most commonly on the lower body, especially at acral sites, these IHs express the same histologic markers (eg, GLUT1) as typical proliferative IHs, but for unclear reasons fail to undergo the usual IH trajectory of growth.25,26
In addition to their individual morphology, IHs can be classified based on their distribution on the
2043
20
Infantile hemangioma growth pattern
120
100
IH relative volume (%)
80
60
40
20
0 –2 8 13 18 23 28 Patient age (months)
3
1 day old 5 weeks old 15 months old
33 38 43 48
A B C
2044
body as localized (focal), segmental, indeterminate, or multifocal, which provides valuable prognostic information.23,24 Approximately two-thirds of IHs are localized (focal) and exhibit clear spatial containment as if arising from one central focus (Fig. 118-4).27 On the face, localized IHs tend to present near lines of mesenchymal and ectodermal embryonic fusion.23 Segmental hemangiomas—similar to other segmental dermatologic disease such as vitiligo and neurofibromatosis— are often plaque-like and correspond to a portion of a developmental segment or broad geographic anatomic territory (Fig. 118-5).28 In a large study, 13.1%
of IHs were segmental.29 The patterns of facial segmental IH have been suggested to correspond to neural crest–derived facial prominences and their presence is associated with risk of PHACE (posterior fossa brain malformations, hemangiomas of the face, arterial anomalies, cardiac anomalies, and eye abnormalities).29
Segmental IHs on the lower body confer risk of myelopathy and genitourinary anomalies in LUMBAR (lower body hemangioma and other cutaneous defects, urogenital anomalies, ulceration, myelopathy, bony deformities, anorectal malformations, arterial anomalies, and renal anomalies) syndrome (see section “Lumbar Syndrome”). Indeterminate IHs are those that are not clearly identifiable as localized or segmental; some authors consider them to be “subsegmental” (Fig. 118-6). Multifocal (multiple localized) IHs are less common, with fewer than 3% of infants presenting with 6 or more IHs.27
20
Classification of hemangiomas by subtype not only facilitates communication, but also helps predict risk of complications and need for treatment. A prospective study of more than 1000 children with IHs showed that segmental hemangiomas are 11 times more likely to experience complications and 8 times more likely to receive treatment than localized hemangiomas, even when controlled for size.27
NONCUTANEOUS FINDINGS AND COMPLICATIONS
Certain IHs have a known risk of associated complications and congenital anomalies (Table 118-3). The presence of multifocal or segmental IH is associated with a greater risk of extracutaneous disease.15
Anatomic location is one of the most important factors affecting risk. Involvement of the central face (especially the nose and perioral skin), periocular area, neck, mandibular region, and perineum should alert clinicians to possible increased risk of complications. Even though the vast majority of IHs remain asymptomatic and uncomplicated with no intervention required, complications can include ulceration, severe bleeding (less than 1% of cases), scarring, pain, and infection, with additional complications found in certain sites of involvement (eg, airway obstruction, congestive heart failure, and visual compromise). Approximately 12% of all IHs are complex, requiring specialist referral. Outcomes are greatly dependent upon timing of referral and intervention.
2045
20
ANATOMIC LOCATION/ MORPHOLOGY ASSOCIATED RISK
Facial, large segmental PHACE (posterior fossa malformations, hemangiomas, arterial anomalies, cardiac defects, eye abnormalities, sternal clefting) syndrome
Nasal tip, ear, large facial (especially with prominent deep component)
Permanent scarring, cartilage destruction, disfigurement
Periorbital and retrobulbar Ocular axis occlusion, astigmatism, amblyopia, tear-duct obstruction
Segmental (S3)/”beard area,” central neck Airway infantile hemangioma
Perioral Ulceration, disfigurement, feeding difficulties
Segmental overlying lumbosacral spine Myelopathy, genitourinary anomalies
Perineal, axilla, neck, perioral Ulceration
Multifocal Visceral involvement (especially liver,
Multifocal Visceral involvement (especially liver, GI tract)
GI tract)
SYNDROMES AND ASSOCIATIONS WITH SEGMENTAL INFANTILE HEMANGIOMAS
SYNDROMES AND
ASSOCIATIONS WITH
SEGMENTAL INFANTILE
HEMANGIOMAS
As described above, facial segmental IH patterns correspond to developmental units and have been labeled as 4 segments (S1 to S4): frontotemporal (S1), maxillary (S2), mandibular (S3), and frontonasal (S4).27 The frontotemporal segment (S1) is associated with cerebral, cerebrovascular, and ocular anomalies; the maxillary segment (S2) is less-frequently associated with extracutaneous involvement; the mandibular segment (S3) is correlated with airway IH, ventral developmental defects, and cardiovascular abnormalities; and the frontonasal segment (S4) encompasses the high-risk territory of the nose, including the nasal tip,30 and confers risk of cerebral and cerebrovascular involvement (Fig. 118-7).
Segmental developmental units with corresponding disease manifestation sites
Denotes segmental-specific complications.
2046
PHACE
Facial segmental IHs confer a risk of PHACE,31 which is a neurocutaneous association. It is sometimes referred to as PHACES to denote the additional association of Sternal defects (ie, sternal clefting or supraumbilical raphe).32 For unclear reasons, up to 90% of PHACE cases are in girls.—In addition, up to one-third of patients with facial segmental IHs with a surface area equal to or greater than 22 cm2 have been reported to meet criteria for PHACE syndrome.33-35 The frontotemporal (S1) and mandibular (S3) segments are the segments most highly associated with PHACE. There are infants with extracutaneous features of PHACE who have segmental IHs on the back of the head, upper trunk, or arm, as well as infants who have small IHs or
20
who lack cutaneous IHs entirely.36-39 Diagnostic criteria have been revised (Table 118-4).40
Congenital vascular anomalies of are the most common extracutaneous features in PHACE. Cerebral vascular anomalies occur in greater than 90% of cases, and cardiac abnormalities occur in up to 67% of cases.30,32,33,35,41 Dysplasia, stenosis, hypoplasia, and agenesis of major cerebral and cervical vessels can occur. Coarctation of the aorta has been reported in 19% to 30% of PHACE cases, and aortic arch anomalies are uniquely complex when associated with PHACE, frequently involving long segment of the transversa and/ or descending aorta.40,42 Notably, aortic coarctation in PHACE is frequently associated with aberrant origin of both subclavian arteries arising distal to the coarctation, rendering the blood pressures of the 4 extremities
ORGAN SYSTEM MAJOR CRITERIA MINOR CRITERIA
Arterial anomalies Anomaly of major cerebral or cervical arteriesa
Aneurysm of any of the cerebral arteries
Dysplasiab of the large cerebral arteries Arterial stenosis or occlusion with or without moyamoya collaterals Absence or moderate-severe hypoplasia of the large cerebral and cervical arteries Aberrant origin or course of the large cerebral or cervical arteries except common arch variants such as bovine arch Persistent carotid-vertebrobasilar anastomosis (proatlantal segmental, hypoglossal, otic, and/or trigeminal arteries)
Structural brain Posterior fossa brain anomalies Dandy-Walker complex Other hypoplasia/dysplasia of the mid and/or hind brain
Cardiovascular Aortic arch anomalies Coarctation of the aorta Dysplasiaa
Midline brain anomalies Malformation of cortical development
Ventricular septal defect Right aortic arch/double aortic arch Systemic venous anomalies
Aneurysm Aberrant origin of the subclavian artery with or without a vascular ring
Ocular Posterior segment abnormalities Persistent hyperplastic primary vitreous Persistent fetal vasculature Retinal vascular anomalies Morning glory disc anomaly Optic nerve hypoplasia Peripapillary staphyloma
Anterior segment abnormalities Microphthalmia Sclerocornea Coloboma Cataracts
Ventral/midline Anomaly of the midline chest and abdomen Sternal defect Sternal pit Sternal cleft Supraumbilical raphe
Definite PHACE
IH >5 cm diameter of the head (including scalp) plus 1 major criterion or 2 minor criteria
Ectopic thyroid hypopituitarism Midline sternal papule/hamartoma
IH of the neck, upper trunk, or trunk, and proximal upper extremity plus 2 major criteria
Possible PHACE
IH >5 cm diameter of the head (includ-
No IH plus 2 major criteria
IH of the neck, upper trunk, or trunk, and proximal upper extremity
IH >5 cm diameter of the head (including scalp) plus 1 minor criterion IH of the neck, upper trunk, or trunk, and proximal upper extremity plus 1 major criterion or 2 minor criteria No IH plus 2 major criteria
ing scalp) plus 1 minor criterion
plus 1 major criterion or 2 minor criteria
aInternal carotid artery, middle cerebral artery, anterior cerebral artery, posterior cerebral artery, or vertebrobasilar system.
bIncludes kinking, looping, tortuosity, and/or dolichoectasia. From Garzon MC, Epstein LG, Heyer GL, et al. PHACE syndrome: consensus-derived diagnosis and care recommendations. J Pediatr. 2016;178:24-33.e2. With permission. Copyright © Elsevier.
2047
20
incapable of demonstrating a gradient reflective of the coarctation on physical examination. Structural brain anomalies occur in approximately 40% of PHACE patients,43 with estimates ranging widely from 30% to greater than 80%.40 Arterial ischemic stroke is known to occur in PHACE, and often presents with seizure or hemiparesis.44 Although the degree of risk for most infants with PHACE is unclear, risk appears highest in the setting of occlusion or significant narrowing of a major cerebral artery within or above the circle of Willis, especially with concomitant aortic coarctation.40
Ocular anomalies are less common and typically manifest as posterior segment anomalies (eg, morning glory disc anomaly, optic nerve hypoplasia, persistent fetal vasculature, or retinal vascular anomalies) or microphthalmia. Endocrine abnormalities, including hypothyroidism and hypopituitarism, with subsequent growth hormone deficiency, as well as headaches, and speech and language delays can also occur.40,45
LUMBAR SYNDROME
Similar to PHACE, segmental IHs on the lower body involving the perineum or lumbosacral area are risk factors for associated spinal, bony, and genitourinary anomalies (Fig. 118-8). LUMBAR syndrome describes the constellation of lower body hemangioma and other cutaneous defects, urogenital anomalies, ulceration, myelopathy, bony deformities, anorectal malformations, arterial anomalies, and renal anomalies.46
Similar groups of patients have been described with the acronyms PELVIS (perineal hemangioma, external genitalia malformations, lipomyelomeningocele, vesicorenal abnormalities, imperforate anus, skin
2048
tag) and SACRAL (spinal dysraphism, anogenital anomalies, cutaneous anomalies, renal and urologic anomalies, associated with angioma of lumbosacral localization).47,48 Myelopathy is the most commonly associated extracutaneous abnormality, presenting as tethered cord or lipomyelo(meningo)cele.46 MRI is strongly recommended in this setting as lumbosacral ultrasound is reported to result in false-negative findings given its poor sensitivity.49,50 IHs in the setting of LUMBAR are more likely to be IHs with minimal or abortive growth morphology and carry a notable risk of ulceration. Underlying arterial anomalies and limb hypotrophy can occur.46
PERIOCULAR HEMANGIOMAS
Infants with periocular hemangiomas are at risk for anisometropia and amblyopia, which, if untreated, can lead to permanent visual loss (Fig. 118-9).51,52 Amblyopia is the most common ocular complication, occurring in 40% to 60% of infants with untreated periocular IH.53,54 IHs larger than 1 cm in diameter confer greater risk of amblyopia, significantly lowering the threshold to initiate treatment.52 Direct pressure on the cornea can produce astigmatism or myopia, and the mass effect of the tumor itself can cause ptosis, proptosis, visual axis occlusion, or strabismus. Any patient with a hemangioma in the periocular area should have a prompt formal ophthalmologic evaluation with repeat visits during the rapid and late proliferative phases. In the setting of ophthalmologic complications, treatment should be initiated urgently to prevent longterm effects, including permanent blindness. Standard evaluation with an ophthalmologist trained in the care of infants should include refraction via retinoscopy following cycloplegia. Serial examinations are especially important with involvement of upper eyelid or supraorbital region. Long-term morbidity can include optic atrophy, blepharoptosis, and ocular proptosis.55
Between 40% and 80% of periorbital IHs have permanent residual changes following involution.56
The most favorable prognostic sign to herald normal vision following involution is the absence of asymmetrical refractive error.54 In certain cases, MRI may be necessary to determine the presence of retrobulbar involvement.
“BEARD AREA” HEMANGIOMAS
Segment 3 IHs involving the preauricular, mandibular, chin, and neck skin (or so-called beard area) carry a 60% risk of causing symptomatic airway disease.57
Airway hemangiomas often present with the insidious onset of biphasic stridor between weeks 4 and 12 of life and are often mistakenly diagnosed as tracheomalacia, upper respiratory tract infection, or croup. Without intervention, respiratory distress can ensue and become life-threatening. Prompt evaluation by a pediatric otolaryngologist and treatment is essential.58
Parotid gland IH may have an unusually prolonged growth phase and may require treatment because of
A
C
20
B
D
massive growth of the IH and deformity of adjacent structures. High-output congestive heart failure has been reported, although rarely.59,60
MULTIFOCAL HEMANGIOMAS
Approximately 15% of infants will have more than 1 hemangioma. Infants with multifocal IH have an increased risk of noncutaneous IH. Screening hepatic and abdominal ultrasound is recommended
for infants presenting with 5 or more cutaneous IHs (Fig. 118-10).61,62 In rare cases, infants can have hundreds of lesions. The term multifocal IH with or without extracutaneous disease has been suggested to replace the older term diffuse neonatal hemangiomatosis, given that it is now understood that cases reported in the older literature confused IH with multifocal lymphangioendotheliomatosis with thrombocytopenia (MLT) and other non-IH multifocal tumors. Other sites of visceral involvement are very rare in true IH. Visceral hemangiomas, including those affecting the liver,
2049
20
GI tract, and brain, also have been reported with solitary segmental hemangiomas.63
HEPATIC HEMANGIOMAS
The liver is the most common extracutaneous site for IH. Even when present, hepatic IHs are frequently asymptomatic. A minority of hepatic IHs cause morbidity, and, in rare cases, can be life threatening, causing high-output congestive heart failure, hypothyroidism, or hepatic failure. Hepatic hemangiomas can be focal, multifocal, or diffuse.64 Focal hepatic hemangiomas are not true IHs, but are analogous to rapidly involuting congenital hemangioma (RICH) occurring in the liver. They are fully formed at birth, present without cutaneous IHs, and can cause moderately severe thrombocytopenia and coagulopathy, although not as severe as the consumptive coagulopathy of Kasabach-Merritt phenomenon. Multifocal and diffuse hepatic hemangiomas are true IHs. Multifocal infantile hepatic hemangiomas are often asymptomatic but can cause high-output congestive heart failure if arteriovenous or portovenous shunting occurs. Diffuse infantile hepatic hemangiomas are less common and associated with high morbidity and mortality. In this setting, the majority of the liver parenchyma is replaced by tumor and can cause abdominal compartment syndrome and severe hypothyroidism as a consequence of tumor-related overproduction of Type III iodothyronine deiodinase. Systemic β-blocker therapy and, in severe cases, concomitant systemic corticosteroids or embolization may be required.65,66 Aggressive thyroid hormone replacement is needed in cases with hypothyroidism.67 In life-threatening cases, liver transplantation may be considered.
2050
ULCERATION
ULCERATION
Ulceration is the most common complication of IH, occurring in greater than 20% of infants in a referral setting, typically during the proliferative phase, with a median age of onset of 4 months.27,68 It occurs most commonly in large or segmental IHs and at sites that are exposed to moisture and friction, such as the perioral, perianal, and intertriginous areas (Fig. 118-11).68,69
Early gray-white discoloration in patients younger than 3 months is a sensitive marker of impending ulceration.70 Secondary infection can occur, but polymicrobial colonization is more typical.71 Treatment of the ulceration (Table 118-5) is often successful in the absence of antibiotic therapy, although localized colonization versus infection can improve with the topical mupirocin or metronidazole. If deep or persistent
■Local wound care and barrier protection
■Local wound care and barrier protection
■Compresses (wet to dry saline compresses, Burow solution) for exudative ulcers
■Compresses (wet to dry saline compresses, Burow solution) for
exudative ulcers
■Occlusive dressings
■Occlusive dressings
■Absorbent nonstick dressings, foam dressings
■Absorbent nonstick dressings, foam dressings
■Alginate dressings (exudative ulcers)
■Alginate dressings (exudative ulcers)
■Petrolatum-based ointments; petroleum jelly-impregnated gauze
■Petrolatum-based ointments; petroleum jelly-impregnated gauze
■Zinc oxide paste, hydrophilic petrolatum
■Zinc oxide paste, hydrophilic petrolatum
■Pain control
■Pain control
■Topical lidocaine (very small amount)
■Topical lidocaine (very small amount)
■Acetaminophen with or without codeine
■Acetaminophen with or without codeine
■Topical antibiotics (if secondary infection is suspected)
■Topical antibiotics (if secondary infection is suspected)
■Mupirocin ointment
■Mupirocin ointment
■Metronidazole gel
■Metronidazole gel
■Aggressive therapy in ulcers that are unhealed after 2 to 3 weeks of wound care
■Aggressive therapy in ulcers that are unhealed after 2 to 3 weeks of
wound care
■Systemic β-blocker therapy
■Systemic β-blocker therapy
■Topical β-blocker (timolol maleate)
■Topical β-blocker (timolol maleate)
■Pulsed-dye laser
■Pulsed-dye laser
■Becaplermin 0.01% gel
■Becaplermin 0.01% gel
■Intralesional and systemic corticosteroids
■Intralesional and systemic corticosteroids
■Early excision
■Early excision
infection is suspected, systemic antibiotics should be prescribed. Local wound care, barrier protection, and pain control are essential for treatment. Bio-occlusive dressings may be helpful but their usefulness is often limited by location because they do not adhere well near the mouth or in diaper area.72,73 In these areas, thick applications of petrolatum-based ointments can be helpful. Pain can be a major issue in management. It can be minimized with an occlusive dressing, oral acetaminophen with or without codeine, and the use of very small amounts of topical lidocaine ointment no more than a few times a day.69 Severe ulcers or those that remain unhealed after 2 to 3 weeks of appropriate wound care warrant more aggressive therapy. Options include systemic β-blocker therapy, topical β-blocker (eg, topical timolol maleate), and pulsed-dye laser (PDL). Other options less frequently employed in the β-blocker era include becaplermin 0.01% gel,74,75 intralesional and systemic corticosteroids, and early excision.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
IHs occur exclusively in humans and are primarily composed of endothelial cells. IHs also contain fibroblasts, pericytes, interstitial cells, and mast cells. The patterns found in segmental IHs suggest at least some IHs occur as the result of a developmental error as early as 4 to 6 weeks of gestation.29 The pathogenesis of their development and evolution remains elusive, in
20
part hampered by the lack of a strong in vitro system or in vivo animal model (Fig. 118-12). Research continues to highlight the complex pathophysiology56,76-79 by which these tumors proliferate and involute. Current understanding suggests that both angiogenesis, the development of new blood vessels from existing blood vessels, and vasculogenesis, the de novo development of blood vessels, are important in IH development. Studies show IHs to be of fetal, rather than maternal, origin,80 and demonstrate immature mesenchymal features with similarities to the cardinal vein, an early embryologic vascular structure.81
The recognition that GLUT1, a red blood cell glucose transporter protein and the most reliable histologic marker of IH, which is expressed in all stages of IH, prompted new hypotheses on the pathogenesis of IH.82,83 GLUT1 expression is absent in the normal cutaneous vasculature but is found in placental blood vessels as well as in other so-called barrier tissues, such as the blood–brain barrier. This, together with other immunohistochemical markers shared by IH and human placenta (Lewis Y antigen, merosin, Fc-γ receptor-IIb, indoleamine 2,3-deoxygenase, and Type III iodothyronine deiodinase), and the similar gene expression profiles found on DNA-based microarrays led to speculation that these tumors are of placental origin from invading angioblasts that have differentiated toward a placental phenotype.83,84 Earlier theories of embolic placental tissue as the trigger for IH have been discredited given that both placental trophoblastic marker expression and villous architecture are absent in IH.85
Proliferation is differentiated by the expression of the primitive cell marker, CD133+,86,87 found in pluripotent
Infantile hemangioma pathogenesis
Birth 3 months 5 years Involution Proliferation
Hypoxia
VEGFR2
HIF1α VEGFA
VEGFA ANG1 TIE2/ANG2
PDGFB/IGF2 TBX2/PPARγ
Progenitor cell recruitment Vasculogenesis and angiogenesis Adipogenesis
2051
20
stem cells known as HemSCs, with demonstration of the presence of endothelial progenitor cells (CD31+, CD34+, CD133+ stem cells) (HemEPCs) in proliferative IH but absent in the involution phase.88,89 Translational studies involving implantation of HemSCs isolated from human IH into immunodeficient mice resulted in mimicry of the IH life cycle, with development of GLUT1+ vessels and subsequent adipocyte replacement.88 HemSCs are precursors for adipocytes, and the presence of adipocytes in involuted IH is well known. Vasculogenesis in IH is theorized to occur from the clonal expansion of circulating endothelial progenitor cells and circulating endothelial progenitor cells may be the progenitor of the endothelial cells lining IH vessels.35 Other studies demonstrate increased amounts of angiogenic factors in proliferating IH, including basic fibroblast growth factor, vascular endothelial growth factor A, insulin-like growth factor, and matrix metalloprotease 9.90 These angiogenic factors are downregulated during involution. Increasing evidence supports the role of hypoxia in IH development and hypoxia is postulated to trigger neovasculogenesis in infants. Low birth weight, the most significant risk factor for IH, is known to be associated with hypoxia.8,12 GLUT1 is upregulated in the setting of hypoxia.35,91 Translational studies performed on proliferating IH have identified presence of multiple markers of hypoxia in addition to GLUT1, including insulin-like growth factor-2 and vascular endothelial growth factor A.92 Histologic features differentiate between the proliferative and involuting phases of IH and shed light on the pathogenesis as well. During proliferation, the clonal nature and histologic features of endothelial cell proliferation93,94 suggest the process to be secondary to vasculogenesis,95 whereas involution is marked by
apoptosis and capillary lumina fibrosis.96 However, the definitive pathogenesis to explain trajectory of IH initiation, proliferation, and involution remain controversial. At present, the most supported explanation suggests that hypoxia (in utero or in local tissue) is key97 to stimulation of circulating endothelial progenitor cells (CD34+, CD133+ stem cells), subsequent vasculogenesis98 with ensuing proliferation to maintain tissue oxygenation homeostasis,97 and eventual increased apoptosis (beginning by end of first year of life) to initiate involution.96
DIAGNOSIS
DIAGNOSIS
With the exception of unusually challenging cases, the diagnosis of IH is almost exclusively clinical, based on the physical examination and clinical history.
SUPPORTIVE STUDIES
Supportive studies may be warranted in certain clinical contexts (Fig. 118-13). When necessary, ultrasound imaging offers the advantages of lack of need for sedation, lack of ionizing radiation and relatively low cost. On Doppler ultrasound, IH is a high-flow lesion and appears as a well-defined solid mass with increased vascular flow within the mass. Arterial feeder and venous drainage can be visualized. The use of MRI offers the benefit of additional anatomic detail, but its use must be weighed against both the risk of exposure to inhaled anesthesia in young infants and increased cost. The current weight of evidence does support the use of MRI and magnetic
Infantile hemangioma diagnostic algorithm
Laboratory workup
Consultant referral
Imaging workup
TSH screening Coagulation panel
Cardiology Ophthalmology
Ultrasound/Doppler Echocardiography
• Multifocal IH
• Segmental IH
• Hepatic IH
• Midline lumbosacral IH
• Multifocal IH
• Large IH
• Multifocal intrahepatic IH
MRI/MRA
• Segmental IH
• Periorbital IH
• PHACE syndrome
• Echocardiography and follow up of abnormal findings
• Multifocal IH
• Very large IH
• Lumbosacral IH
• PHACE syndrome workup
Neurology
• In setting of abnormal MRI/MRA of brain
2052
resonance angiography (MRA) in the workup of PHACE syndrome in early life.40 Some centers experienced in the imaging of infants may be able to successfully complete an MRI in the neonatal period after feeding and swaddling (“feed-and-wrap” technique); the likelihood of compromising image quality by motion artifact increases with the age of the infant and the duration of the study. On MRI, IH appears as a T2 bright, T1 isointense mass with homogeneous, avid contrast enhancement. Internal serpiginous flow voids, indicative of arterial feeding vessels, are visible on T2-weighted imaging. MRA demonstrates early arterial enhancement.99
LABORATORY TESTS Thyroid Function Tests: Hypothyroidism is a rare complication in infants with diffuse infantile hepatic hemangiomas.100,101 The liver tumor demonstrates high levels of Type III iodothyronine deiodinase activity, which accelerates the degradation of thyroid hormone.67,102 Infants with significant hepatic hemangiomas should have thyroid function evaluated, including triiodothyronine (T3) (the hormone consumed) and thyroid-stimulating hormone, because thyroxine (T4) levels may initially remain normal. Conversely, screening hepatic ultrasound should be performed in infants with hypothyroidism of unknown etiology even in the absence of cutaneous hemangiomas. Hypothyroidism and other endocrine abnormalities also have been reported with PHACE syndrome.
PATHOLOGY
The histopathologic appearance of IH varies markedly over the course of its life cycle. In all but the earliest phase of growth and the very end of involution, features of proliferation and involution are comingled. As a complement to radiologic imaging, IH in the rapid proliferative phase are made up of unencapsulated but well-defined mass of capillaries arranged in lobules divided by fine septae or normal surrounding soft tissue, comprised of endothelial cells, pericytes, mast cells and interstitial dendritic cells. The vessels are lined with plump endothelial cells with enlarged nuclei and abundant cytoplasm with a surrounding rim of similarly plump pericytes in the absence of smooth muscle cells.35 The appearance is somewhat similar to young pyogenic granulomas (lobular capillary hemangiomas). In early IH, markers of cellular proliferation such as Ki-67 denotes proliferation of endothelium and pericytes and normal appearing mitotic figures appear abundant. Thick-walled draining veins may be visible. In involuting IH specimens, capillaries begin to be regress and are replaced with adipocytes; mast cells are increased and apoptosis is notable. The basement membranes vessels within the IH thicken and hyalinize. At the end of involution, fibrofatty stroma is prominent and apoptotic debris in the remaining “ghost” capillaries remains.60,64 When necessary
20
to confirm the diagnosis on histopathologic specimen, GLUT1 immunostaining is positive in the endothelial cells lining IH capillaries at all stages of IH growth, distinguishing IH from other vascular tumors and all vascular malformations.65,66,76,82 IHs are also CD31+ and CD34+, as noted above.
IMAGING
Presence of a segmental IH of the head, neck, and/or scalp warrants evaluation for possible PHACE to include MRI and MRA of the head/neck/chest,40 formal ophthalmologic evaluation, and echocardiogram. Likewise, workup of possible LUMBAR syndrome includes MRI/MRA of the lumbosacral spine, pelvis, and/or lower extremity. Initial evaluation for potential hepatic IHs in the setting of multifocal IHs requires hepatic/ abdominal ultrasound with Doppler flow and spectral analysis (baseline ultrasound ± serial imaging.)
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
The prognosis of most IHs is excellent, with spontaneous involution and little to no sequelae, but a significant minority of IH result in permanent disfigurement or medical sequelae. Following involution, approximately half of all treated patients will attain normal skin, while the other half can have residual atrophy, scarring, telangiectasias, or fibrofatty softtissue remnant. One study of 97 untreated patients demonstrated residual skin changes in more than 68 (70%) of patients.22 Specific IH characteristics (as previously outlined by each subclassification) are associated with increased rates of complications.17,104 The most important factor to affect prognosis in complicated IH is timing of specialist referral for management, including initiation of therapy and workup as necessary. Certain characteristics are associated with an increased risk of complications and need for treatment (see Table 118-3).17,104 Consideration of early treatment should be given to IH with these characteristics, depending on the specific clinical setting.
MANAGEMENT
MANAGEMENT
INTERVENTIONS
The decision to initiate treatment is based on many factors, including size and location, psychosocial implications, and risks and benefits of the proposed therapy (Fig. 118-14). For the majority of small IH,
2053
20
DISORDER AGE OF ONSET CLINICAL EXAMINATION DISTINGUISHING FEATURES
Vascular Anomalies
Kaposiform hemangioendothelioma (KHE) Congenital to infancy or later Firm, reddish-brown to violaceous, plaque or nodule May be associated with Kasabach- Merritt phenomenon: increased
D-dimer, low fibrinogen, severe thrombocytopenia
Tufted angioma As for KHE Similar to KHE ± associated hair Similar to KHE, although considered less severe on spectrum
Rapidly involuting congenital hemangiomas Congenital; full size at birth Often symmetric blue nodule with coarse telangiectasia; rim of pallor High flow
Noninvoluting congenital hemangiomas Congenital; full size at birth or minimal postnatal growth
Similar to rapidly involuting congenital hemangioma (RICH) High flow
Pyogenic granuloma Infancy or childhood (more commonly) Friable vascular papule with brisk intermittent bleeding Rarely may be multiple and congenital
Multifocal lymphangioendotheliomatosis Congenital Varied appearance of papules, plaques, or nodules Thrombocytopenia (waxing and waning), potentially severe GI bleeding
Blue rubber bleb nevus syndrome Congenital, infancy or later Small, compressible, blue to purple papulonodules May be confused for multifocal infantile hemangioma (IH); associated with GI bleeding ± larger venous malformation, often in thigh or pelvis
Capillary malformation (port-wine stain) Congenital Confluent pink to dusky pink; possibly reticulated Reticulated may appear similar to minimal growth IH
Venous malformation Congenital; may not be noted until later Blue-purple compressible; growth gradual and progressive Low flow; may be confused with deep IH
Glomuvenous malformation Congenital in segmental plaque presentation; multifocal present later
Segmental thin plaque pink to violaceous; papules/nodules firm ± tender
Lymphatic malformation Macrocystic cervicofacial present at birth; microcystic often noted later
Autosomal dominant; limited to skin and soft tissue
Deep, skin-colored nodule ± vascular appearing pink to purple blebs Low flow; may be confused with deep IH
Verrucous hemangioma Congenital Plaque, papule, or nodule with keratotic change over time A subtype of lymphatic malformation
Cutis marmorata telangiectatica congenita
Congenital Reticulated, marbled pink to dusky purple ± ulceration ± limb undergrowth May be confused with minimal growth IH or reticulated port-wine stain
Nonvascular Lesions
Myofibroma Congenital to infancy Small to large plaques and nodules May ulcerate and mimick ulcerated IH
Spitz nevus Infancy rarely; childhood commonly Pink to red-brown papule Typically solitary although may have agminated presentation
Juvenile xanthogranuloma Congenital to infancy Pink to red-brown papules or nodules Become yellow with time; central ulceration possible in larger lesions
Dermoid cyst Congenital Skin colored to slightly bluish; firm; typically mobile nodule face and scalp at sites of suture closure/embryonic fusion
May be confused with deep IH
Encephalocele Congenital Bluish nodule, may transilluminate in midline frontal or occipital scalp Consider with midline deep blue nodule
Rhabdomyosarcoma Congenital to first year in most Firm tumoral nodule with overlying telangiectasias; embryonal rhabdomyosarcoma is multifocal and congenital
2054
May be confused with deep or mixed IH
(Continued)
20
(Continued)
DISORDER AGE OF ONSET CLINICAL EXAMINATION DISTINGUISHING FEATURES
Infantile fibrosarcoma Congenital to first 2 years Red to blue; firm and fixed to underlying tissue May be confused with mixed IH, but more often KHE/tufted angioma or RICH/ noninvoluting congenital hemangioma
Langerhans cell histiocytosis Congenital, infancy or later Seborrheic distribution in scalp and diaper area especially; red to brown papules, nodules, petechial papules
Congenital presentation has a morefavorable prognosis than presentation in infancy or later; presentation with larger nodule, often on scalp, possible
Neuroblastoma Congenital to early infancy Blue to violaceous nodule Increased urine homovanillic ± vanillylmandelic acid
Congenital leukemia
Congenital to neonatal Pink, blue, or violaceous firm plaques
May be confused with multifocal IH
Congenital leukemia cutis Congenital to neonatal Pink, blue, or violaceous firm plaques and nodules May be confused with multifocal IH
cutis
and nodules
Adapted from Püttgen KB. Diagnosis and management of infantile hemangiomas. Pediatr Clin North Am. 2014;61(2):383-402.
active nonintervention with close observation and followup is the most appropriate approach. Especially during the first few months of life and during the rapid proliferative phase, visits should be more frequent, as often as every 1 to 2 weeks in the youngest infants.105
Visits with parents and caregivers should emphasize education about the natural course and prognosis of IH, addressing both parental anxiety106 and psychosocial impact on the patient and family.107-109 Photographs of the likely outcome for a similar lesion are often helpful. Many parents experience anxiety and may find themselves subject to comments from complete strangers about their child’s hemangioma.106 Most parents of young children do not think their child is deeply affected by these reactions, but facial IHs, in particular, can cause psychological suffering once the child reaches school-age.107,108 Potential treatment
options should be discussed well ahead of entrance to elementary school. For those IHs warranting intervention, options include pharmacologic, laser, and surgical interventions.
MEDICATIONS Beta Blockers: With widely established efficacy in IH treatment and a more favorable risk profile compared with prior systemic treatment options, propranolol is the only medication approved by the United States Food and Drug Administration (FDA) for the treatment of complicated IH. Propranolol is the clear first-line systemic pharmacologic therapy for most moderate to severe IHs at risk for significant morbidity.110-112 Unlike corticosteroids, which stabilize IH growth but do not cause involution, propranolol
Infantile hemangioma therapeutic algorithm
Identification of IH to benefit from intervention
IH with ulceration
Topical Antimicrobials
Wound Care Other
Indications Factors to Consider
• Nonadherent dressings
• White petrolatum- impregnanted gauze
• Hydrocolloid dressings
• Carboxymethylated high-purity cellulose fibers containing ionic silver
• White petrolatum topical
• Zinc oxide paste
• Emergent basis: potentially fatal complications
• Urgent need: existing or imminent pain, bleeding, functional impairment
• Potential structural anomaly in association with IH
• Patient age
• IH location
• IH size
• Lesional growth phase
• Urgency of treatment
• Complication severity
• Psychosocial potential adverse effects
• Parental and physician preference
• Ointment: mupirocin, bacitracin, gentamicin
• Metronidazole gel
• Oral propranolol
• Topical timolol
• Pulsed-dye laser
• Early excision
• Becaplermin gel
2055
20
causes regression in the vast majority of cases. Rebound growth after cessation of therapy can occur, but usually responds well to retreatment.111,112 Key criteria to warrant consideration of beta-blocker treatment initiation for IH include facial deformity, active or impending functional impairment, early prevention of anticipated permanent sequelae, and mitigation of the need for surgery.113 Positive results also have been reported in other settings, including fully proliferated IHs and airway, liver, and orbital hemangiomas.111,114-116 Despite early controversy, most patients with PHACE can safely tolerate propranolol, although it is recommended to use the lowest effective dose, consider a slower dose titration, and to give the medication in 3 divided doses, rather than the standard twice-daily dosing, to avoid significant alterations in peak blood levels.40 Molecular mechanisms of propranolol in effectively treating IH include lesional capillary vasoconstriction (visible IH color change within first 48 hours of use), angiogenesis inhibition (arresting growth), and induction of apoptosis (bringing about IH regression).117 Common side effects of propranolol include sleep disturbance, acrocyanosis, and asymptomatic transient decrease in blood pressure or heart rate. Rarely observed potential important adverse effects include hypoglycemia, symptomatic hypotension or bradycardia, wheezing, bronchospasm, and diarrhea. Propranolol’s efficacy has been documented in a large, international, placebo-controlled, randomized trial and numerous smaller prospective and retrospective case series and case reports, including a small multicenter randomized trial comparing oral corticosteroids to propranolol and several comparative retrospective studies.111,112,118 A multicenter, randomized, double-blind, Phase II-III trial comprised of 456 patients investigating the efficacy of oral propranolol in IH involved 5 treatment arms (1 mg/kg/day for 3 months vs 6 months’ duration, 3 mg/kg/day for 3 months vs 6 months’ duration, and placebo). Dosing of 3 mg/kg/day for 6 months was found to have the highest benefit-to-risk ratio with 88% of patients on this daily dose demonstrating improvement by the fifth week of treatment. In this study, side effects included a decrease in heart rate (by a mean of 7 beats per minute with observed change of heart rate within 1 hour of dose with minimal change thereafter) and a decrease in mean systolic blood pressure (by 3 mm Hg). Serious adverse events (n =
33) included: bronchospasm, diarrhea (3 mg/kg/day > 1 mg/kg/day), and sleep disorder (73 propranolol vs 7 placebo).119 A study cohort involving 39 European centers with nearly 1100 total patients, reported the use patterns of propranolol in IH, with 91.4% of patients demonstrating clinically significant improvement regardless of daily maintenance dose (less than 2 mg/kg/day, 2 mg/kg/day, greater than 2 mg/kg/day) with risk of adverse events to be based upon daily maintenance dosing.120 With report of overall IH response rate of 98% and 0.9% resistance rate in IH, propranolol has reduced the need for surgery in nasalsite-specific IH.121-123 Given propranolol’s lipophilic nature with ease of crossing the blood–brain barrier, sleep disorder can manifest in several ways (fatigue,
2056
restlessness, nightmares, insomnia), affecting 11.4% of patients, with a reported 3.7% of patients experiencing nightmares. Theoretical risks of propranolol extrapolated from adult volunteer studies in the psychology literature include impaired short-term and long-term memory, sleep quality, mood, and psychomotor function. Fatigue associated with pediatric hypertension improves over time.124,125
Propranolol initiation can take place in the inpatient or outpatient setting (Fig. 118-15). In determining a patient’s candidacy for propranolol, family history must be obtained to ensure absence of congenital heart disease, arrhythmia, and maternal connective tissue disease. Uniformly accepted standard monitoring procedures for patients on propranolol for IH have not yet been established. However, current proposed factors favoring inpatient initiation include young age (the study on which FDA approval of propranolol was granted included infants as young as 5 weeks of age with outpatient initiation), comorbid medical conditions (eg, respiratory or cardiovascular), inadequate social support, or any preexisting concerns for blood glucose. In the outpatient setting, hypoglycemia can be prevented with anticipatory guidance, dosing propranolol with feedings, frequent feedings, and either decreasing or holding the dose in the setting of decreased oral intake.126,127 Outpatient initiation is suitable for most otherwise healthy children beyond the neonatal period. Current published guidelines and many, but not all, physicians advocate 2-hour monitoring in the ambulatory setting for heart rate and blood pressure with first dose administered. Dosing is typically started at 1 mg/kg/day in 2 divided doses and increased by 0.5 mg/kg/day increments every 3 to 7 days to a target dose between 2 and 3 mg/kg/day. Most practitioners use 2 mg/kg/day. Slower dose escalation or lower target dose may be warranted in the setting of PHACE syndrome, ulceration or other comorbid conditions. Atenolol, a β1-selective antagonist, and nadolol, a nonselective β-antagonist, have been used in infants for IH treatment but neither is available in liquid formulation in the United States. The main benefit of atenolol is the lack of bronchial reactivity; nadolol’s advantage is its inability to cross the blood–brain barrier, thereby decreasing concerns for any potential neurocognitive side effects compared with propranolol. Only few international publications reporting the use of atenolol and nadolol in IH are available as of this writing; further studies are warranted. Likewise, further studies investigating any neurocognitive effects of propranolol are ongoing. Early evidence does not demonstrate adverse effects on neurodevelopment with use of propranolol, but comprehensive neurocognitive testing in this population has not been reported.128
In light of the proved efficacy of systemic beta blockers, the efficacy of topical beta blockers in IH treatment were evaluated in an effort to even further decrease side effects. Topical timolol is a nonselective beta blocker available in an ophthalmic preparation approved for the treatment of pediatric glaucoma.129
Eight to 10 times more potent than propranolol, topical
20
Propranolol treatment of infantile hemangioma
Check baseline vitals (VS) and EKG Perform cardiorespiratory examination
Indications for inpatient initiation
• Corrected gestational age <5-8 weeks*
• Comorbid conditions
Normal findings
Propranolol side effects
Common (mild and reversible)
• Changes in sleep
• GI symptoms
• Acrocyanosis Uncommon (severe)
• Hypoglycemia
• Symptomatic hypotension
• Bradycardia
Initiate oral propranolol at 1 mg/kg/day divided into 2-3 daily doses
Reduce initiation dose; gradual increase to 1 mg/kg/day
Abnormal findings
Repeat VS and examination at 1 hour and 2 hours after first dose
Normal findings
Up-titrate dosage regimen as tolerated (counsel parents to check HR 1 hour after each dose increase): 1 mg/kg/day (2-3 daily doses) × 5 days, 1.5 mg/kg/day (2-3 daily doses) × 5 days, 2 mg/kg/day (2-3 daily doses) thereafter
timolol was first reported as an effective option for IH treatment in 2010,130 and continues to gain increasing acceptance and application. The systemic bioavailability of 1 intraocular drop of timolol 0.5% solution ranged from 39% to 98%, with a mean value of 78.3±24.5%.131
Additional research on topical timolol demonstrated differences in systemic absorption based upon vehicle preparation. Compared to aqueous timolol solution (0.25% to 0.5%) with average plasma concentration of 0.46 to 1.72 ng/mL, timolol gel-forming solution (0.1% to 0.5%) was developed to decrease systemic absorption with average plasma concentration rage of 0.13 to 0.71 ng/mL. Little is known about the absorption across IH-affected skin. Several contributing factors can impact systemic absorption independent of vehicle, including application site (mucosa, ulceration, and areas under occlusion increase absorption), application size, and size of patients (neonates and especially premature infants with greater absorption as a result of increased surface-area-to-volume ratios). Timolol is metabolized by cytochrome P450 CYP2D6; poor metabolizers are likely at greater risk for adverse effects. Changes in heart rate correlate with plasma levels in timolol, thus less-significant heart rate changes were found in hydrogel formulation. Topical timolol in either vehicle had negligible pulmonary or blood pressure effects.132 The largest powered multicenter retrospective topical (0.05%) timolol (85% gel foaming solution) study (n = 731) with a 9.5-month
mean duration of treatment (83.5% of participants dosed at 1 drop twice daily) reported 3.4% adverse events (half of these noted to be local irritation at application site with 3 cases of bronchospasm and 4 cases of ulceration development). The retrospective endeavor concluded timolol was most effective in thinner IH and with longer duration of treatment. Color improved more than overall volume, extent, and size with topical timolol use.133
Corticosteroids: Systemic corticosteroids, previously considered first-line treatment of deforming, endangering, or life-threatening IH, are now used rarely given the efficacy, safety and improved tolerability of beta blockers.134 Corticosteroids are most effective during the proliferative phase, causing slowing or cessation of growth in up to 90% of cases, with actual shrinkage in approximately onethird. Although the mechanism of action is not well understood, prior studies suggest the upregulation of mitochondrial cytochrome b, clusterin/ApoJ (possible apoptotic markers), and/or interleukin-6 as markers of corticosteroid-induced cessation of IH growth.135-137 When used, prednisone or prednisolone is given at a dose of 2 to 3 mg/kg/day, typically for 4-8 weeks followed by a tapering of varying length, depending on the age of the child and indication for treatment. A metaanalysis showed an 84% response rate with an average dose of 2.9 mg/kg for a mean of 1.8 months before tapering. Although 3 mg/kg/day
2057
20
is more effective (94% response) than 2 mg/kg/day (75% response), greater adverse events occur at higher doses.138
Short-term complications of systemic corticosteroids include cushingoid faces (71%), personality changes (irritability/fussiness) (29%), gastric irritation (21%), fungal infection (oral or perineal, 6%), and diminished gain of height (35%) and weight (42%) during treatment. More than 90% of children with diminished gain of height return to their pretreatment growth curve by 24 months of age.139,140 Other complications include hypertension, steroid-induced myopathy, immunosuppression, and transient adrenal insufficiency.140,141 Blood pressure should be closely monitored by the treating physician.142 Children taking more than 2 mg/kg/day of prednisone for longer than 14 days are considered to have a deficit in cellmediated immunity. Live viral vaccinations should be deferred in infants receiving high-dose corticosteroids. Rare cases of Pneumocystis carinii pneumonia has been reported in this setting, leading some physicians to use trimethoprim-sulfamethoxazole prophylaxis during treatment.143,144
Intralesional corticosteroids can be an effective treatment for select, relatively small, localized IHs located in high-risk sites such as the lip, nasal tip, cheek, and ear. Current use of this modality for IH is maintained by a minority of physicians with expertise in its use. Injections for periocular hemangiomas are not recommended. If used, it should only be performed by experienced ophthalmologists, given reports of retinal artery embolization and blindness.145,146 The largest published case series of intralesional steroids found that the majority showed a greater than 50% reduction in volume, with the best results occurring in relatively superficial IHs. Adverse events occurred in 6.4% of patients and included cushingoid appearance, cutaneous atrophy and anaphylactic shock.147
Only few case series have reported on the efficacy of Class 1 topical corticosteroids, namely for small, superficial hemangiomas.110,148,149
Medications of Historical Importance (Interferon-α, Vincristine, Imiquimod): Interferon-α, vincristine, and imiquimod were previously regarded as second-line or third-line therapeutic options for IH. Their use is no longer advocated in light of the established efficacy and more favorable side-effect profile of beta blockers. Interferon-α carries the notable risk of potential neurotoxicity, specifically spastic diplegia. A metaanalysis of 441 patients showed 11 developed irreversible spastic diplegia and 16 developed motor disturbances that were reversible on discontinuation of the drug.150 All affected patients were younger than 1 year of age at initiation of therapy. Vincristine’s use in the treatment of vascular tumors is now reserved for complicated kaposiform hemangioendothelioma (KHE) and tufted angioma (TA); its use for this indication may be eclipsed by sirolimus in future years, but further study is warranted.151-153
2058
PROCEDURES Laser Therapy: PDL, originally designed to treat port-wine stains (ie, capillary malformations), has been used to treat IH with varying results.154 PDL was used more frequently in the treatment of IH before the discovery of propranolol. Its usefulness is primarily limited by its minimal depth of penetration (less than 2 mm).35 Several reports have shown improvement in treating associated ulceration155,156; its use in diminishing residual telangiectasias and erythema after involution is well accepted. Its use in the treatment of proliferating IHs is controversial.157 In a prospective, randomized, controlled study, 22 infants treated with either observation or PDL with epidermal cooling demonstrated significant color improvement in the PDL group, but unchanged total surface area and echo depth by 12 months’ followup.158 Another prospective study randomized children to PDL without epidermal cooling (n = 60) and observation (n = 61) groups with comparable results of (near-) complete resolution between groups at age 1 year, with a trend toward increased hypopigmentation and textural change in the PDL group. Outcomes between groups were similar at the 5-year followup of 117 children.155
Other studies have shown good results with either the 585-nm PDL or 595-nm PDL, with varying fluencies (total energy per unit area), but several have emphasized that treatments work best for more superficial hemangiomas and are unable to halt growth of deeper components.159-162 Severe ulceration and scarring, particularly in treating segmental hemangiomas during the proliferative phase, have been reported.163
Although comparative studies are lacking, the same IHs that are reasonable candidates in the proliferative phase for PDL are similarly good candidates for topical timolol treatment. Expert opinion and rudimentary cost analysis support the use of medical therapy with timolol in superficial IHs over the use of PDL in most cases.133 A conservative approach is to reserve PDL primarily for treating ulceration and for hastening resolution of erythema in IH after the proliferative phase is completed.164
Surgical Therapy: Surgical excision may be indicated at any time during the life cycle of an IH, but elective excision in infancy is typically neither necessary nor advocated. Proposed indications for surgical intervention of IH during infancy include an anatomically favorable site amenable to resection, a contraindication or failure of pharmacotherapy, or a high risk of ultimately necessary resection with similar scar regardless of timing of surgery. Timing of surgical intervention depends upon evolution of tumor (whether it continues to regress), anatomic location, degree of deformity, and the age of the child. Certain anatomic locations, such as the nasal tip and lip, often require surgery.165-170 Even earlier excision may be indicated in cases where clinical characteristics, such as pedunculated IH, severe, recalcitrant ulceration, or extremely thick dermal involvement dictate that scarring will inevitably occur. In most instances, it is best
to wait until regression is well under way and a more accurate assessment can be made regarding whether scarring and textural changes have occurred. Surgery during and after involution can allow reconstruction of affected adjacent structures, resection of residual excess skin or fibrofatty tissue, or scar revision. Decisions regarding this can often be made between 3 and 5 years of age, even if involution is not complete.171
Four years serves an appropriate threshold age as most IHs have completed involution and children develop self-awareness, long-term memory, and self-esteem.35
A standard elliptical excision is often performed; however, circular excision followed by a purse-string closure may leave a smaller scar.172 A second stage following purse-string closure may or may not be necessary.
SCREENING
SCREENING
Screening for PHACE is recommended in the setting of segmental IH on the head, in infants without classic segmental IH or with a small IH who have a major anomaly seen in PHACE (eg, coarctation of the aorta or supraumbilical raphe) and in infants with 2 major criteria for PHACE as outlined in consensus guidelines from 2016.40
OTHER VASCULAR TUMORS
AT-A-GLANCE
■ Vascular tumors in the current International Society for the Study of Vascular Anomalies classification are subdivided into (a) benign, (b) locally aggressive or borderline, or (c) malignant.
■ Congenital hemangiomas are fully formed at birth. Rapidly involuting, noninvoluting, and partially involuting congenital hemangiomas are recognized subtypes.
■ Tufted angiomas represent subtle pink or duskyred patches and may evolve into plaques or nodules and have a characteristic histology.
■ Kaposiform hemangioendothelioma is a locally aggressive vascular tumor, morphologically similar to but etiologically distinct from Kaposi sarcoma and can be associated with Kasabach-Merritt phenomenon. Kaposiform hemangioendothelioma exists on a spectrum with tufted angioma.
■ Multifocal lymphangiomatosis with thrombocytopenia consists of cutaneous vascular papules and plaques associated with intermittent thrombocytopenia, often with GI bleeding.
20
■ Spindle-cell hemangioma usually occurs in the extremities most often associated with Maffucci syndrome.
■ Pyogenic granuloma, a rapidly growing papule or nodule with a collarette of scale or eroded surface, is very common. Treatment is excision or electrocautery.
■ Congenital eccrine angiomatous hamartoma is a rare ill-defined plaque associated with increased lanugo hair and sweating.
Many other benign vascular neoplasms occur in both children and adults.173 This section highlights selected important vascular tumors according to the 2014 ISSVA classification, which classifies vascular tumors as (a) benign, (b) locally aggressive or borderline, or (c) malignant. More comprehensive reviews of this subject can be found elsewhere.2,173,174
CONGENITAL HEMANGIOMAS
CONGENITAL
HEMANGIOMAS
Hemangiomas that are fully formed tumors at the time of birth and do not proliferate in postnatal life are referred to as congenital hemangiomas. They are benign and much less common than IH. There are now 3 major subtypes recognized on the basis of their natural history: the rapidly involuting (RICH), noninvoluting (NICH), and partially involuting congenital hemangioma.175,176
Recognition of the partially involuting congenital hemangioma subtype in recent years highlights that these tumors exist on a spectrum. Definitive categorization is often challenging in the immediate newborn period and can often only be made with confidence in retrospect after observation of the clinical behavior. Congenital hemangiomas have similar anatomic sites of predilection, including the head, neck, and extremities, but can occur elsewhere. They are equally common in boys and girls.176
RICH often appears as a raised, violaceous tumor with large, radiating veins or with overlying telangiectasia and a halo of pallor (Fig. 118-16). Central ulceration may be present. Most RICHs involute spontaneously by or before 14 months of age and usually leave residual atrophic anetodermic scar tissue.177 A presumed RICH noted on prenatal ultrasound that involuted before birth has been reported, leaving a scar tissue at the site of the lesion.178
RICHs can be associated with mild to moderate thrombocytopenia and a coagulopathy characterized by elevated fibrin degradation products and low fibrinogen in the neonatal period. In contrast to Kasabach- Merritt phenomenon (discussed in section “Kaposiform Hemangioendothelioma”), the laboratory findings of cutaneous RICHs are often self-limited and uncomplicated with only supportive care generally required.179
2059
20
A
B
C
Hepatic RICHs, which are the focal subtype of hepatic hemangioma, are rare, but at greater risk of complications, with associations with marked thrombocytopenia as well as severe progressive hepatic dysfunction and resultant heart failure. NICHs are less common than RICHs and also present at birth. They are usually flatter than RICHs, presenting as a well-circumscribed round to oval, somewhat indurated or raised soft-tissue mass with overlying telangiectasias and a rim of pallor (Fig. 118-17).
2060
Partially involuting congenital hemangioma was introduced into the vascular anomalies lexicon in 2014 and fewer than 20 cases have been reported. Partial involution occurs over the first 12 to 30 months of life (mean: 18 months) before stabilization occurs (Fig. 118-18).176
Somatic activating missense mutations in the genes GNAQ and GNA11 are reported to cause RICH and NICH. The mutational change, alteration of glutamine at amino acid 209 (Gln209), was the same in both tumors, so does not explain their varied clinical courses.180 The same Gln209 missense mutation is common in uveal melanoma, and postzygotic mosaic mutations in GNAQ and GNA11 codons 183 or 209 have been reported in association with phakomatosis pigmentovascularis and extensive dermal melanocytosis.181
Indications for treatment of congenital hemangiomas are similar to those for IH, including ulceration, impairment of vital function and congestive heart failure. Excision, with or without preoperative embolization, should definitely be considered for ulceration, which can lead to severe hemorrhage, and for postinvolutional skin changes if disfiguring.175 NICH do not go away, but are often asymptomatic; decisions regarding their removal must weigh risks and benefits of the proposed treatment. The distinguishing pathologic features of congenital hemangiomas are lobules of capillaries with plump endothelial cells and pericytes set within densely fibrotic stroma containing hemosiderin deposits, focal lobular thrombosis, and sclerosis.182 They are GLUT1 negative, unlike IH.183 On ultrasound, congenital hemangiomas heterogenous with scattered calcifications possible and are generally confined to the subcutaneous fat. They are high-flow vascular tumors, often showing arteriovenous microfistulas, more common seen in NICH, on Doppler interrogation.184
A
C
20
B
D
TUFTED ANGIOMA
TUFTED ANGIOMA
EPIDEMIOLOGY
TA is a rare benign vascular tumor that also has been called angioblastoma of Nakagawa. Although adult cases have been reported, most cases occur early in childhood and may have a protracted course. Approximately 25% of TA are congenital and 50% appear in the first year of life. There is no sex predilection.
CLINICAL FEATURES
TA display various clinical patterns and are most commonly seen on the neck, trunk, and extremities. They may present as a subtle stain-like area that later thickens, as a large, plaque-like, infiltrated, red or dusky blue-purple lesion, or as an exophytic, firm, rubbery, violaceous, cutaneous nodule (Fig. 118-19). Tenderness and overlying hyperhidrosis may occur.185 TA are usually solitary, but multifocal cases have been reported.186
COMPLICATIONS
Kasabach-Merritt phenomenon (KMP) may develop in TA but this is more common with KHE (see section “Kaposiform Hemangioendothelioma”).
ETIOLOGY AND PATHOGENESIS
Its etiology and pathogenesis are uncertain, although based on its immunostaining pattern, a lymphatic
2061
20
origin is likely. Unlike IH, there are no known gender or gestational age correlates.
DIAGNOSIS
Histologically, both acquired and congenital TAs demonstrate vascular tufts of tightly packed capillaries, randomly dispersed throughout the dermis in a typical “cannonball distribution” with crescentic spaces surrounding the vascular tufts, and lymphatic-like spaces within the tumor stroma.187,188 Unlike IH, TA does not stain with GLUT1.82 It is CD31+; D2-40, LYVE1, and PROX1 lymphatic markers, are only partially positive in surrounding vessels, helping distinguish TA from KHE.189
CLINICAL COURSE AND PROGNOSIS
TAs may persist unchanged or regress completely within a few years.190 TAs that are present at birth or in the first year of life have a greater tendency to spontaneously regress than do those that appear later in life.191 Of those TAs that regress, 95% are reported to do so within 2 years.187
MANAGEMENT
No widely accepted treatment guidelines exist for management of TA. Recommended therapies for TA, with and without KMP, include excision, compression, laser, topical or systemic corticosteroids, propranolol and chemotherapy. See “Kaposiform Hemangioendothelioma” below for a fuller discussion of therapy.
KAPOSIFORM HEMANGIOENDOTHELIOMA
KAPOSIFORM
HEMANGIOENDOTHELIOMA
EPIDEMIOLOGY
KHE is a rare vascular tumor with an estimated prevalence of 0.91 cases per 100,000 children.192 It may be present at birth or develop in early childhood. A series of more than 100 patients reported that 60% present as neonates and 93% in infancy. Rare adult cases have been reported. Three-quarters involve the skin. It is usually reported in association with KMP, which occurs in more than 70% of cases.192
CLINICAL FEATURES
KHE is an infiltrative, aggressive tumor classified as locally aggressive or borderline in the ISSVA classification. It may present as a brown-red stain at birth which begins to thicken and become purpuric, or as plaques or deep-seated nodules and bulky tumors. Most cases involve the skin and musculature. KHE can occur on the extremities, especially overlying joints, trunk, head, neck. Deeper viscera including cervicothoracic, abdominal, and the retroperitoneum can be affected. Notably,
2062
KHE does not occur in the liver. Locoregional lymph node involvement may occur and likely represents multifocal disease. Distant metastases do not occur. Mediastinal or retroperitoneal disease may present with hemothorax and ascites.141 Spontaneous involution is rare. Most reported cases have had associated KMP, but KHE can occur in the absence of a coagulopathy.192-195 KHE may resolve with atrophic or stain-like areas, infiltrated plaques and papules, or nodules. Residual fibrosis is not rare and can result in considerable morbidity.196
COMPLICATIONS Kasabach-Merritt Phenomenon: KMP re- fers to the presence of platelet trapping within a vascular tumor resulting in profoundly severe thrombocytopenia (typically less than 30000/µL) and associated microangiopathic hemolytic anemia and consumption of clotting factors resulting in low fibrinogen, elevated d-dimers, and varying degrees of decreased coagulation factors (Fig. 118-20). It was long considered to be a complication of “hemangioma,” but it is now recognized to be a complication almost exclusively of TA and KHE, not IH.14,197 KMP must be differentiated from the coagulopathy that can arise in association with venous and mixed venous-lymphatic malformations (sometimes erroneously called KMP) as a result of chronic clotting and consumption of clotting factors, but not primarily by platelet trapping,198 which is more likely to be either localized or disseminated intravascular coagulopathy.
ETIOLOGY AND PATHOGENESIS
The pathogenesis of KHE and KMP is unknown. KHE and TA are thought to exist on a spectrum of severity.192
RISK FACTORS
Involvement across more than 1 anatomic region increases the risk of developing KMP, while KHE presenting at a later age, with superficial tumors or tumors involving only bone, appears to have a lower risk of KMP.192
DIAGNOSIS
Diagnosis is based upon clinical and histologic features and laboratory abnormalities, including severe thrombocytopenia, consumption of fibrinogen, elevated d-dimers, decreased coagulation factors to varying degrees, and microangiopathic hemolytic anemia. The histology of KHE is characterized by infiltrative nodules and sheets of spindled endothelial cells with minimal atypia and infrequent mitoses lining slit-like or crescentic vessels containing hemosiderin. Edema, microthrombi, fibrosis and atypical appearing lymphatic vessels can be seen. The tumor is GLUT1 negative. Similar to TA, increased lymphatic spaces reactive with the lymphatic markers including vascular endothelial growth factor receptor-3 and D2-40 suggest a lymphatic origin, similar to that of Kaposi sarcoma.199,200
CLINICAL COURSE AND PROGNOSIS
The prognosis and management of KHE varies with extent and location of the tumor, in addition to the presence of KMP. Although the KHE and TA may persist, the coagulopathy of KMP usually abates by 1 year of age or sooner with treatment.
MANAGEMENT
No one treatment is uniformly effective nor are there widely accepted treatment guidelines for KHE management. KHE complicated by KMP requires more aggressive intervention than KHE in the absence of coagulopathy. Complete surgical excision, when possible, is curative, but is often not feasible given the infiltrative nature of the tumor. Systemic corticosteroids are considered first-line treatment and are often used in combination with vincristine, especially in the setting of KMP. Ticlopidine and aspirin are reported as adjunctive therapies with variable benefit. Arterial embolization may be adjunctive before planned surgery to decrease bleeding risk and may result in temporary improvement of coagulopathy and lesion size but is rarely effective as monotherapy.201 Reports on the use of oral sirolimus, a mammalian target of rapamycin inhibitor, in this condition are promising202,203 showing rapid reversal of the coagulopathy; results from prospective clinical trials are pending. Older literature reports use of radiation therapy, cyclophosphamide, interferon-α, actinomycin-d, and methotrexate, but these are all considered third-line therapies to be used only in the setting of failure of other agents. Platelet transfusions should be avoided unless active bleeding occurs or before surgical procedures.204-208 The use of other blood transfusion products has proved ineffective in correction of the coagulopathy.
20
SPINDLE-CELL HEMANGIOMA
SPINDLE-CELL
HEMANGIOMA
Spindle-cell hemangioma, also called spindle-cell hemangioendothelioma, is a rare benign vascular tumor most often seen with Maffucci syndrome. While the genetic mutation associated with this syndrome has been identified (IDH1 and IDH2, which produce isocitrate dehydrogenases to convert isocitrate to 2-ketoglutarate) and detected in associated cutaneous findings of this condition, the relationship of the mutations and the manifestations of this disorder remains unclear. It can occur at any age and site but the extremities are the most commonly affected. The histology is of a nodular, dense, spindle cell proliferation associated with dilated dysplastic veins. Lesions can be locally aggressive and may recur even after excision.209
PYOGENIC GRANULOMA (LOBULAR CAPILLARY HEMANGIOMA)
PYOGENIC GRANULOMA
(LOBULAR CAPILLARY
HEMANGIOMA)
Pyogenic granuloma (PG) is also known by its correct histopathologic description lobular capillary hemangioma. It is one of the most common vascular tumors of infants and children and can also occur in adults, particularly in pregnant women. PG usually presents as a solitary, red, rapidly growing papule or nodule, often with a subtle collarette of scale (Fig. 118-21). Typical locations include the cheek or forehead, but virtually any body site including the mucous membranes may be affected. The rapid growth of PGs usually presents at a later age than that associated with the proliferative phase of IH, but in very young infants can cause confusion.210
They often develop an eroded surface, with subsequent bleeding which can be profuse, resulting in the
2063
20
moniker the Band-Aid disease. Solitary and agminated PG-like lesions have rarely been reported in association with vascular anomalies including arteriovenous malformations and congenital hemangioma.211,212
PG does not typically involute spontaneously. Even after definitive treatment, recurrence and even satellite lesions surrounding the original PG have been reported.213,214 Simple curettage with electrocautery is usually curative. Other options include excision, laser surgery (carbon dioxide or pulsed-dye laser), and cryotherapy. Imiquimod and timolol have been suggested as effective topical treatment options.215,216
MULTIFOCAL LYMPHANGIOENDO- THELIOMATOSIS WITH THROMBOCYTOPENIA
MULTIFOCAL
LYMPHANGIOENDO
THELIOMATOSIS WITH
THROMBOCYTOPENIA
MLT—also known as cutaneovisceral angiomatosis with thrombocytopenia—is a rare condition that was initially described in 2004, and is characterized by multiple cutaneous vascular papules and plaques that are usually congenital with development of new lesions over time.217 It is currently unclear whether it is a vascular tumor or a vascular malformation. Some patients have prominent exophytic cutaneous vascular tumors, whereas others have subtle blueberry muffin-like papules.218 Most affected infants have intermittent thrombocytopenia and lesions in the GI tract, leading to GI bleeding. Other reported sites of involvement include bones, synovium, lungs, liver, spleen, and brain. It is now understood that many patients previously diagnosed with “diffuse neonatal hemangiomatosis” actually had MLT.219 Skin biopsy specimens demonstrate thin-walled vessels, some hobnailed endothelial cells, and intraluminal papillary projections similar to Dabska tumor.217,220 Endothelial cells stain positively for CD31, CD34, and LYVE1, variably for D2-40, and are negative for GLUT1.186,218 Prognosis varies based on extent of disease and bleeding complications. A metaanalysis reported a 65% mortality rate.219 Effective therapy has been challenging but early experience with sirolimus has been positive, with some authors recommending it as first-line treatment.221,222 A variety of other systemic medications have been reported with variable benefit including corticosteroids, vincristine, thalidomide, interferon- α2a, propranolol, and bevacizumab.218,223
PAPILLARY INTRALYMPHATIC ANGIOENDOTHELIOMA
PAPILLARY
INTRALYMPHATIC
ANGIOENDOTHELIOMA
Papillary intralymphatic angioendothelioma (PILA), also known as Dabska tumor, is a dermal nodule or
2064
a diffuse violaceous swelling on the head, neck, or extremities that is seen almost exclusively in the pediatric-age group. PILA/Dabska tumor is a lowgrade vascular tumor whose rarity has made study of its long-term prognosis and optimal treatment difficult. Histopathology shows enlarged vessels with thick walls surrounded with lymphoid aggregates and smaller lymphatic vessels. The presence of “hobnail” endothelial cells inside tumoral vessels is the sine qua non of PILA/Dabska tumor.185
RETIFORM HEMANGIOENDOTHELIOMA
RETIFORM
HEMANGIOENDOTHELIOMA
Retiform hemangioendothelioma occurs primarily in adults and has significant overlap with PILA/Dabska tumor. There is no noted sex predilection and development occurs in the second to fourth decades of life (mean age: 36 years). Histopathology demonstrates presence of retiform pattern of long, arborizing blood vessels lined by monomorphic hobnail endothelial cells. Most cases noted to have prominent lymphocytic infiltrate with focal presence of hyaline collagenous core comprising papillae, similar to PILA/ Dabska tumor. Recurrence is frequent but metastatic rate is low.224
COMPOSITE HEMANGIOENDOTHELIOMA
COMPOSITE
HEMANGIOENDOTHELIOMA
Composite hemangioendothelioma is a low- to intermediate-grade malignant vascular tumor comprised of a variable combination of benign and malignant vascular components. Identification of at least 2 hemangioendothelioma variants is necessary to render this diagnosis. Epithelioid and retiform variants are most commonly observed, although foci of spindlecell hemangioma and/or angiosarcoma-like foci can also be present.225
TARGETOID HEMOSIDEROTIC HEMANGIOMA
TARGETOID
HEMOSIDEROTIC
HEMANGIOMA
Targetoid hemosiderotic hemangioma is a benign lesion presenting as a violaceous papule, often surrounded by a pale rim and peripheral ecchymotic halo, which fades with time. Lesions usually present on the trunk or extremities and histologically consist of dilated vascular channels within intraluminal papillary projections dissecting into collagen bundles in the subcutis. Extravasated erythrocytes and hemosiderin are present, hence the designation.
CONGENITAL ECCRINE ANGIOMATOUS HAMARTOMA (SUDORIPAROUS ANGIOMA)
CONGENITAL ECCRINE
ANGIOMATOUS
HAMARTOMA
(SUDORIPAROUS ANGIOMA)
Congenital eccrine angiomatous hamartoma is a rare condition characterized by ill-defined plaques with increased lanugo hair and sweating at the site of the lesion. They are usually located on the extremities or abdomen. Diagnosis is established on the basis of characteristic histologic findings: closely packed eccrine sweat glands associated with dilated capillaries, a few dysplastic venous channels, and a dense collagenous matrix.226
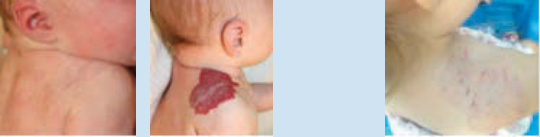
Figure 118-1 Growth characteristics of infantile hemangioma.
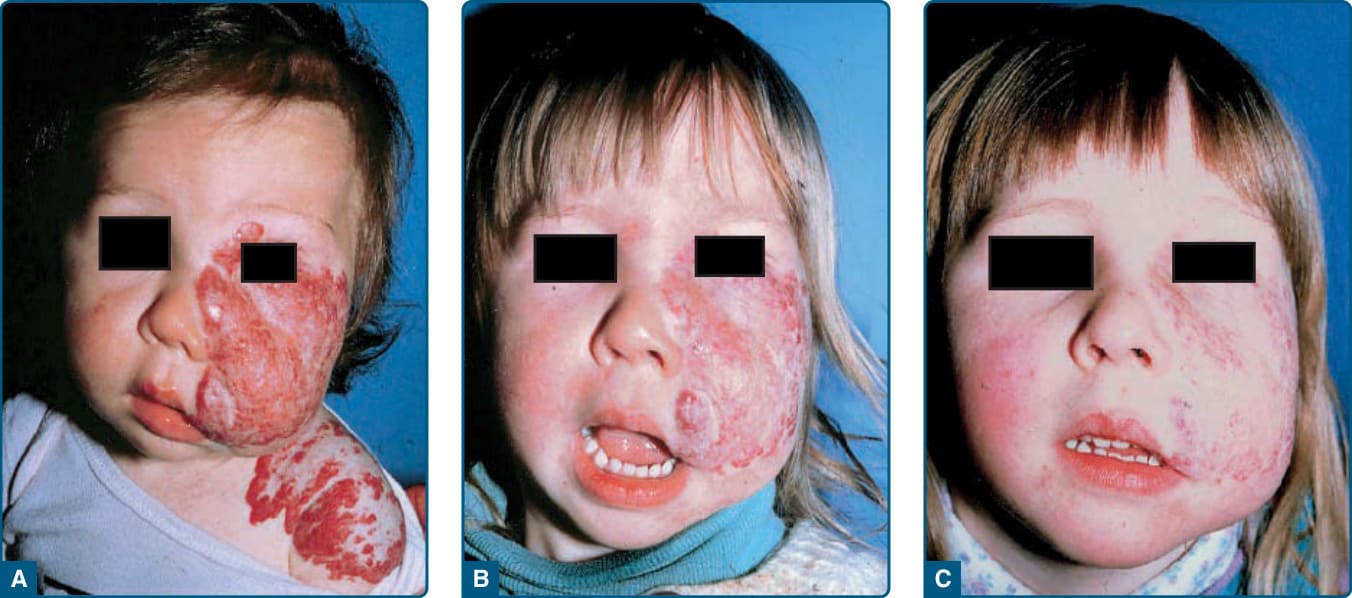
Figure 118-2 Natural history of segmental infantile hemangioma. Note the plaque-like, geographic configuration. A, Age 11 months, peak of proliferative phase. B, Age 2 years, involuting phase. C, Age 4 years, majority of natural involution has occurred.
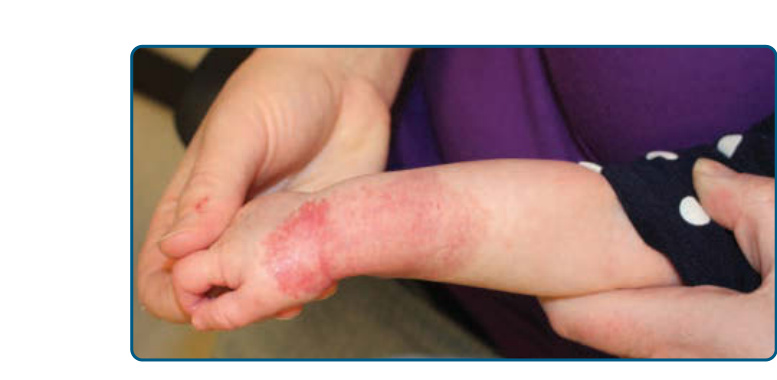
Figure 118-3 An infantile hemangioma with minimal or abortive growth on the right arm and hand of an infant.
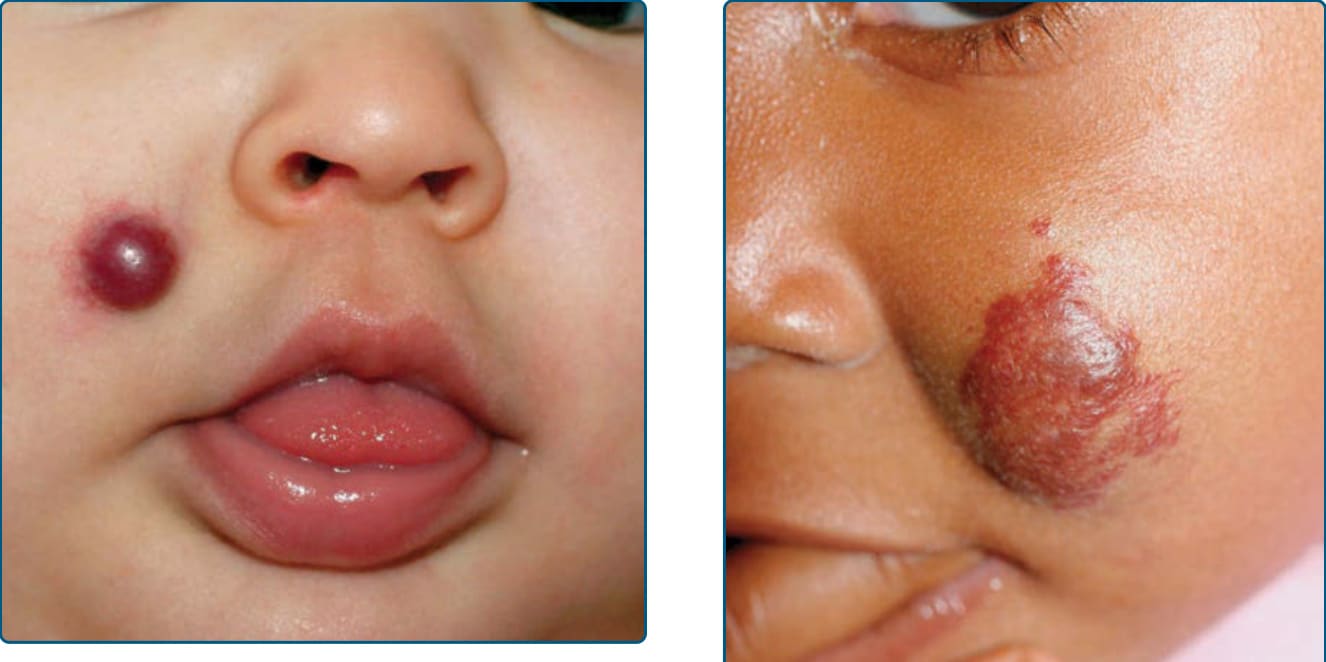
Figure 118-4 Localized infantile hemangioma on the cheek of an infant.
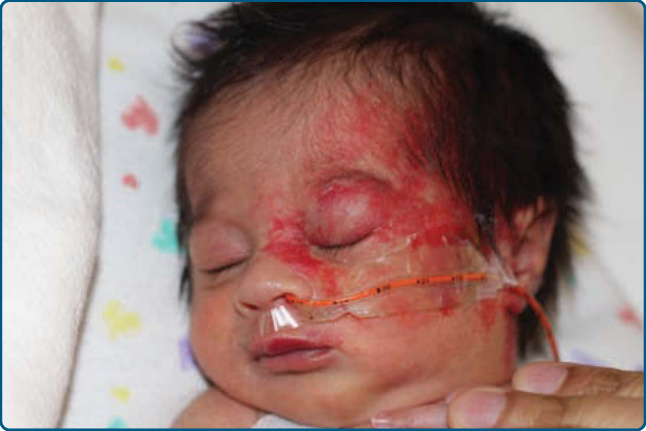
Figure 118-5 Segmental infantile hemangioma on the face, scalp, and neck of a newborn with PHACE.
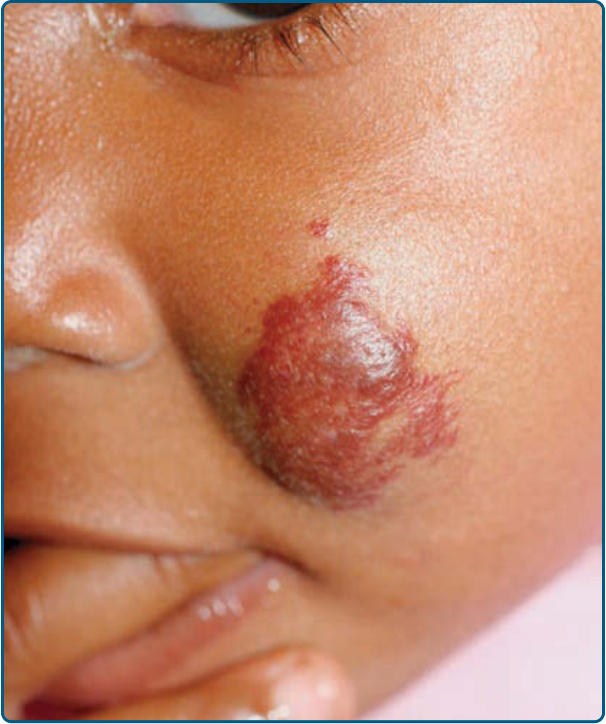
Figure 118-6 Indeterminate infantile hemangioma on the cheek of an infant 14 months of age.
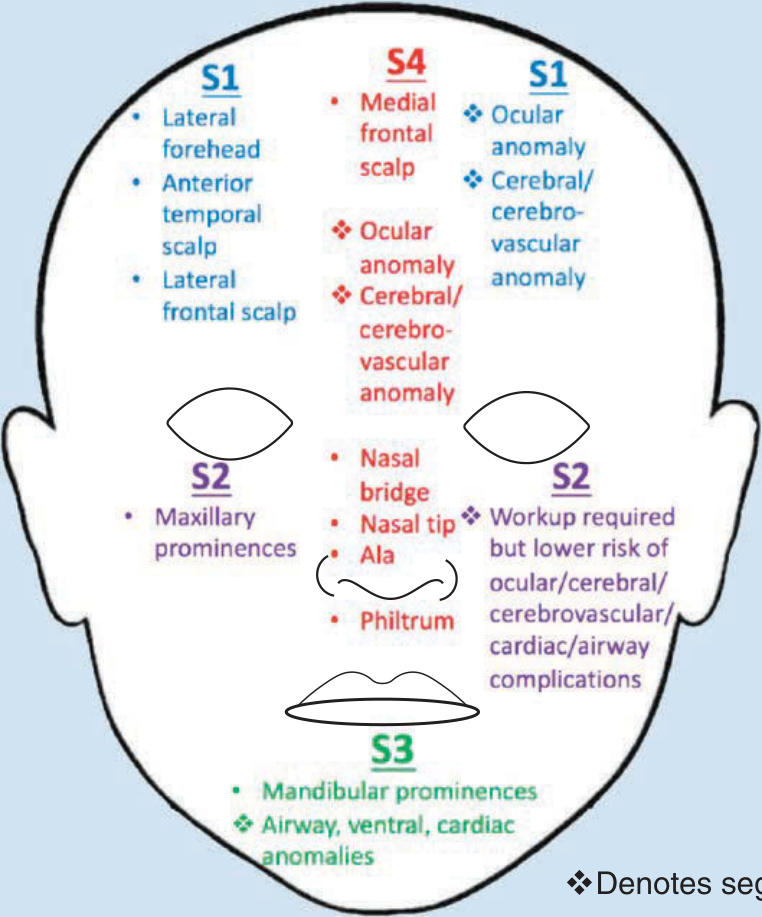
Figure 118-7 Segmental developmental units with corresponding disease manifestation sites. (Data from Haggstrom AN, Lammer EJ, Schneider RA, et al. Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics. 2006;117(3):698-703.)
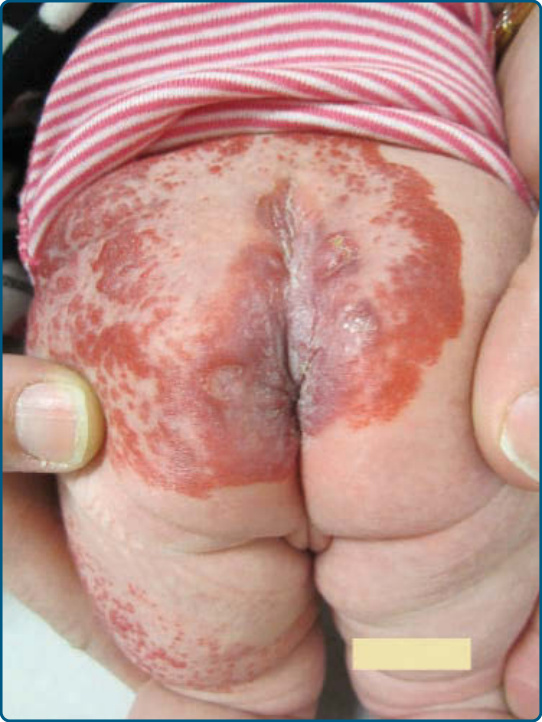
Figure 118-8 A segmental infantile hemangioma of the lower body in an infant with LUMBAR syndrome complicated by painful ulceration.
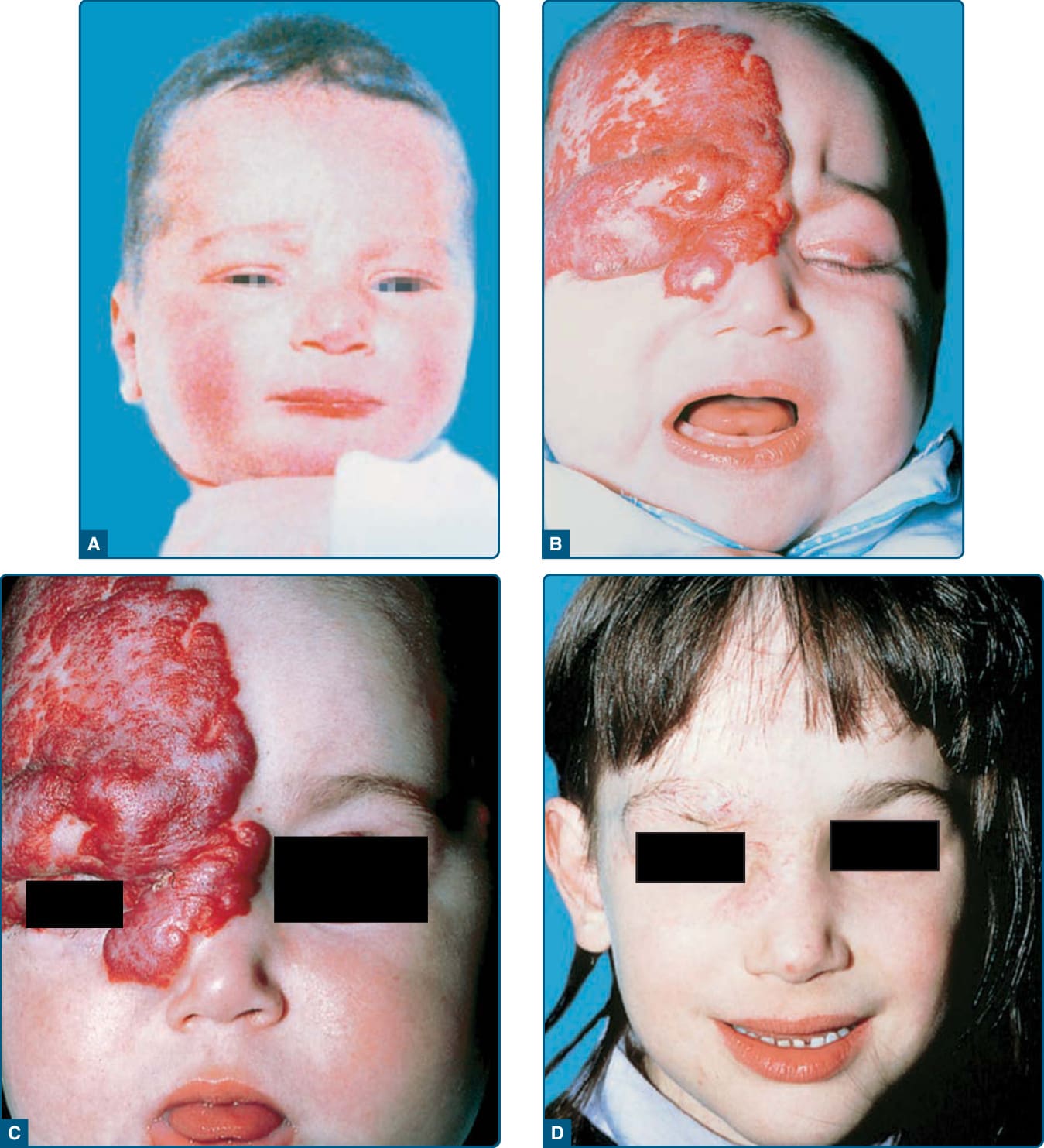
Figure 118-9 Vision-endangering segmental infantile hemangioma treated with systemic glucocorticoids. A, No premonitory signs of tumor seen on infant’s photograph. B, By age 3 months, extensive infantile hemangioma infiltrating the upper lid and surrounding tissue, causing blocked vision. C, Eyelid opened within 2 weeks of glucocorticoid therapy. D, Involuted tumor at age 6 years with residual scarring.
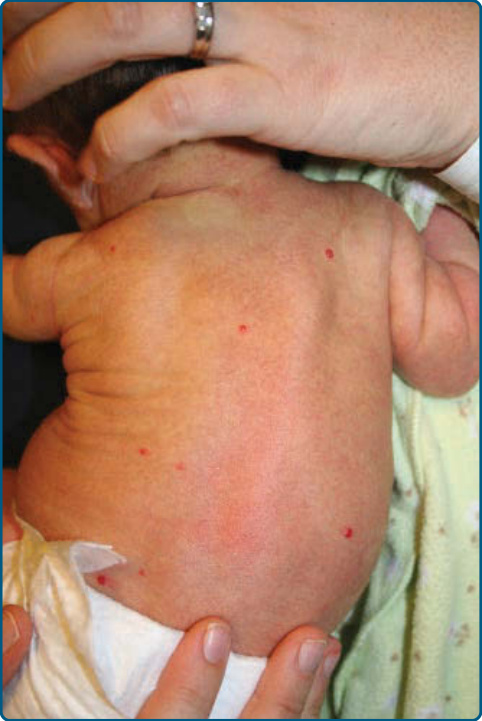
Figure 118-10 Multifocal infantile hemangiomas in an infant. There are multiple, small, superficial, pink papules on the trunk and extremities.
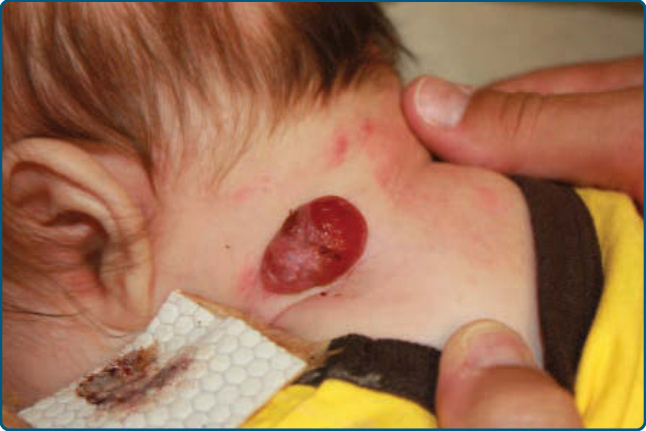
Figure 118-11 A painful ulcerated infantile hemangioma on the neck that required treatment with wound care, pulsed-dye laser, and propranolol.
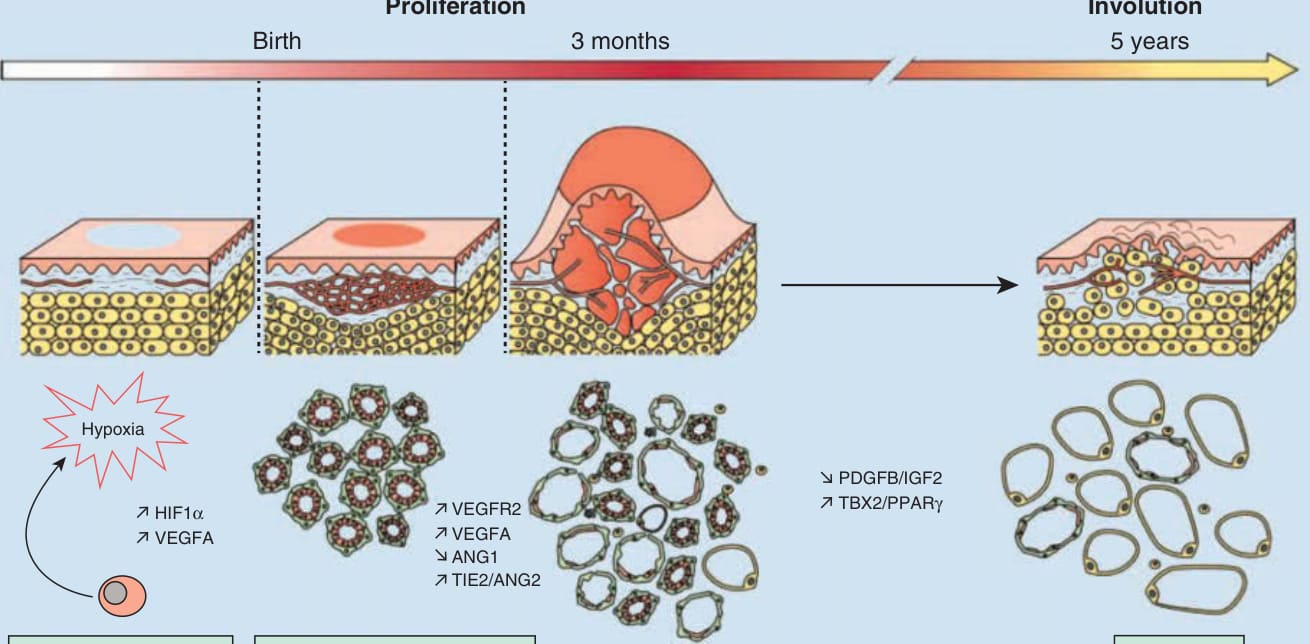
Figure 118-12 Infantile hemangioma pathogenesis. (Figure reprinted from Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. The Lancet. 2017;390(10089):85-94, with permission. Copyright © Elsevier.)
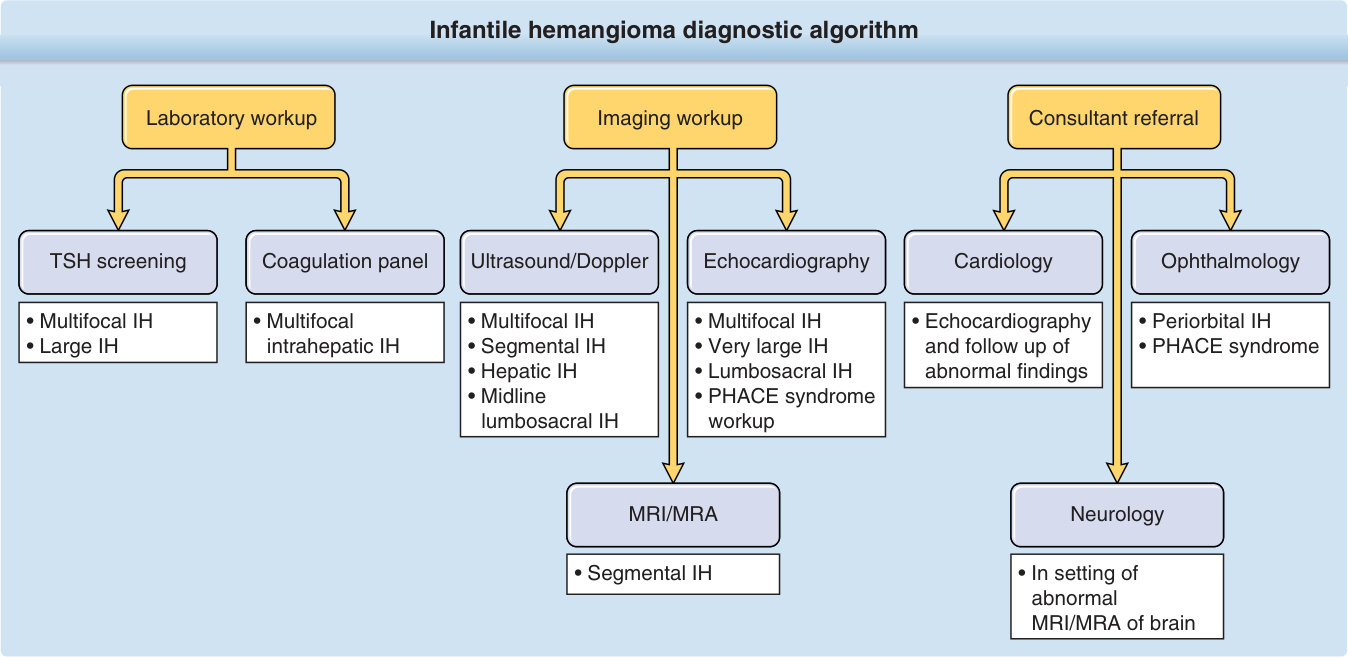
Figure 118-13 Infantile hemangioma (IH) diagnostic algorithm. PHACE, posterior fossa brain malformations, hemangiomas of the face, arterial anomalies, cardiac anomalies, and eye abnormalities; TSH, thyroid-stimulating hormone. (Adapted from Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390(10089):85-94.)
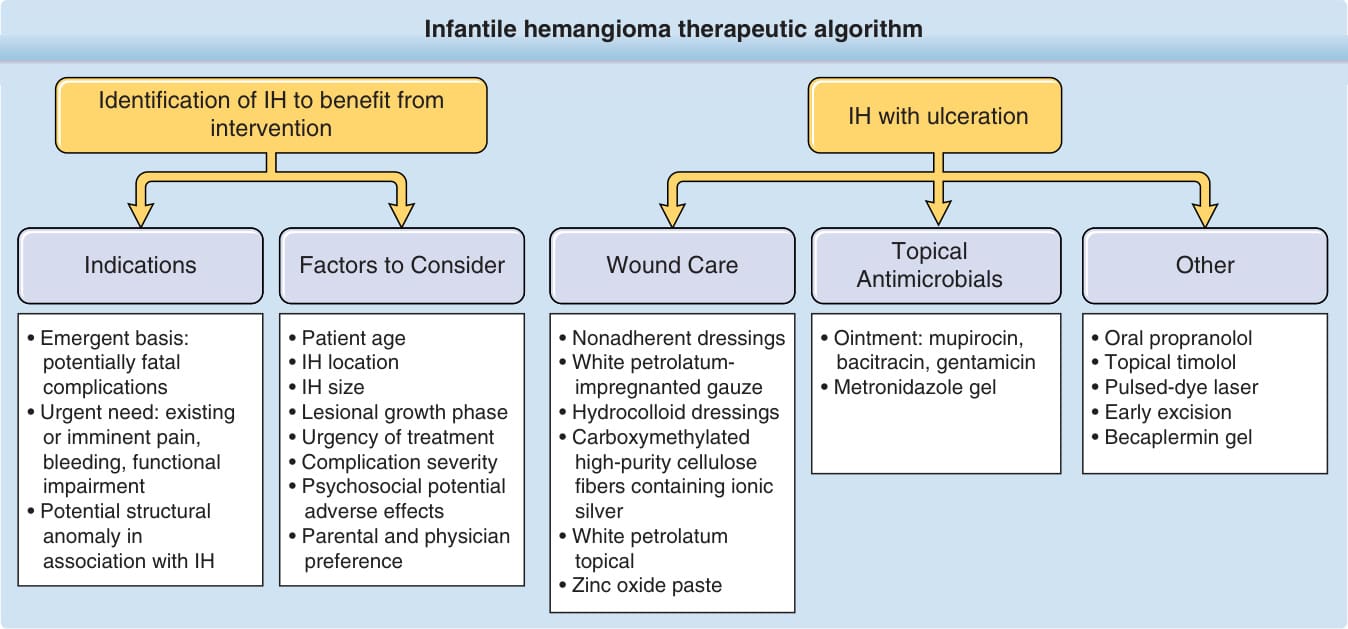
Figure 118-14 Infantile hemangioma (IH) therapeutic algorithm. (Data from Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136(4):e1060-e1104.)
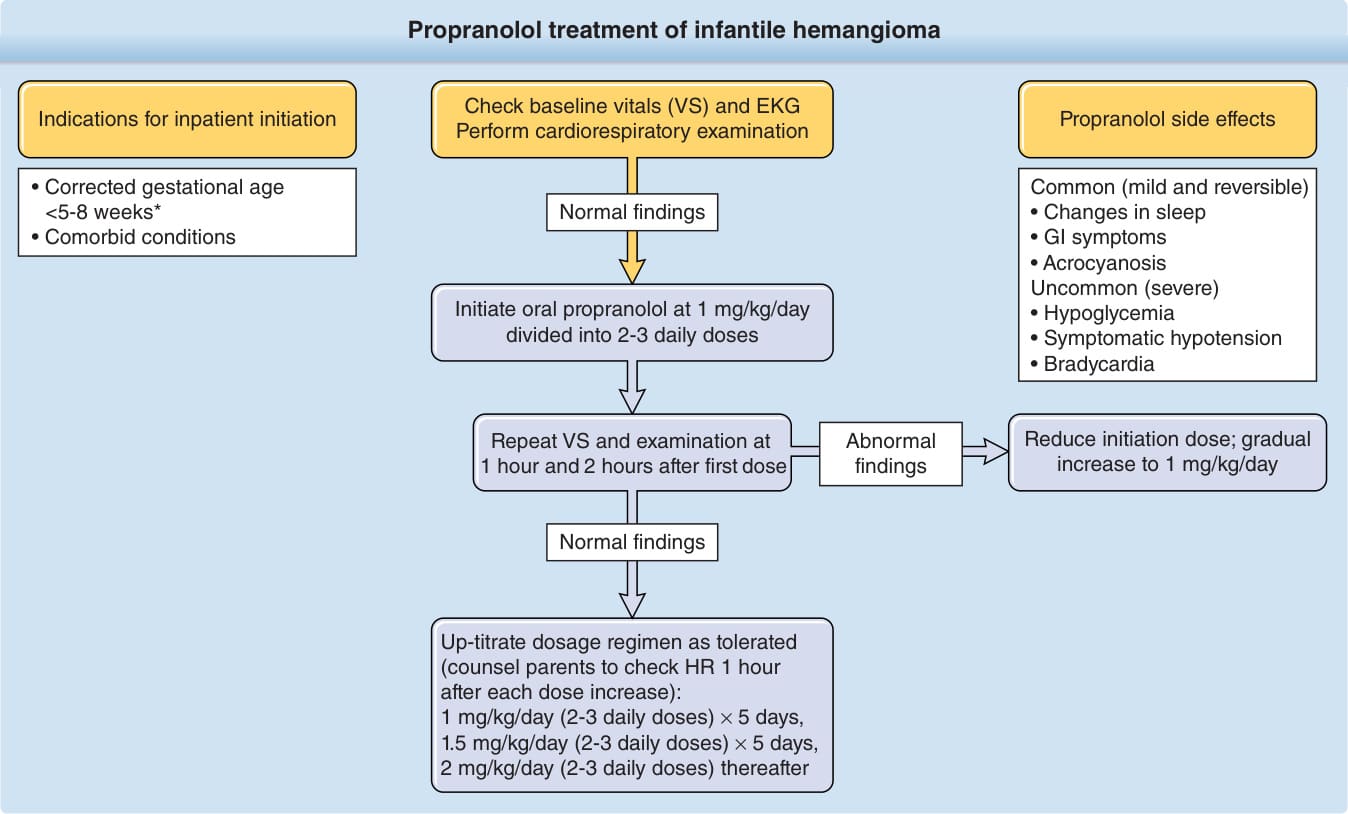
Figure 118-15 Propranolol treatment of infantile hemangioma. ∗Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131(1):128-140; Léauté- Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735-746. (Data from Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131(1):128-140.)
Figure 118-16 Rapidly involuting congenital hemangioma on left scalp. A, Infant with overlying coarse telangiectasias at 1 month of age. B, Early regression at age 5 months. C, Continued rapid involution at age 8 months.
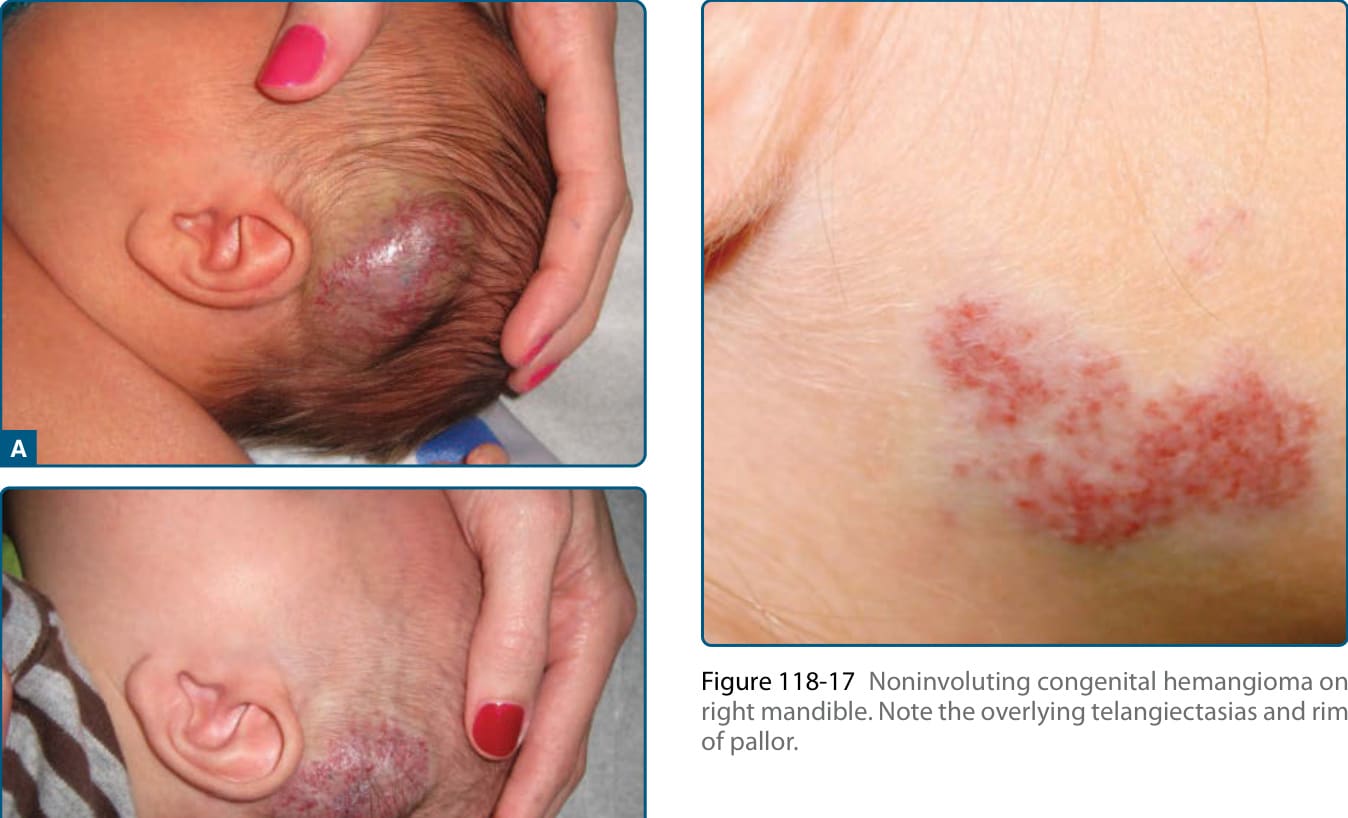
Figure 118-17 Noninvoluting congenital hemangioma on right mandible. Note the overlying telangiectasias and rim of pallor.
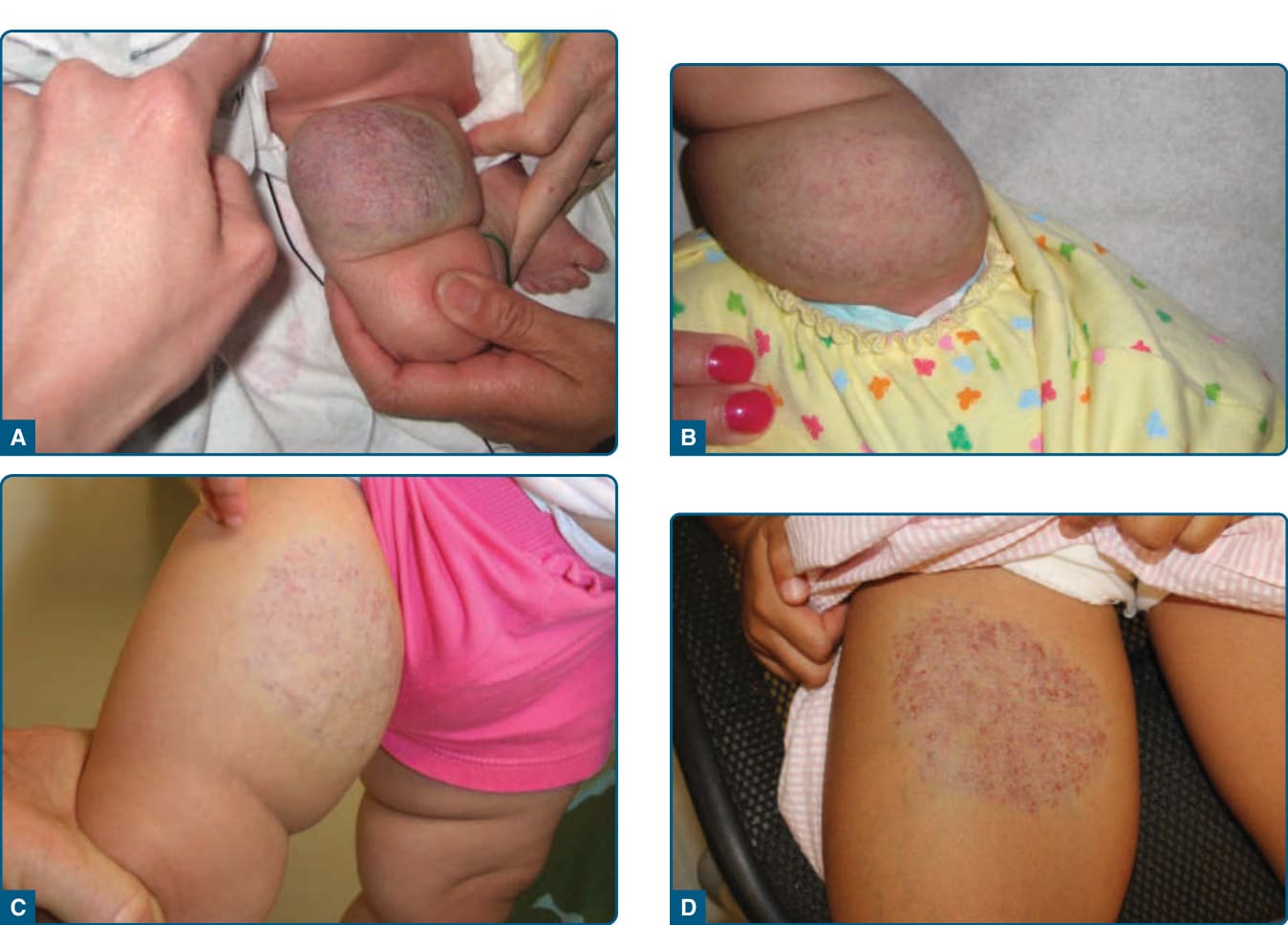
Figure 118-18 Partially involuting congenital hemangioma on the right thigh. Note the overlying telangiectasias and rim of pallor. A, First day of life. B, Early decrease in size at 6 weeks of age. C, At 13 months of age it is broad but flat. D, At 5 years of age no further involution has occurred and there is renewed prominence of superficial vessels.
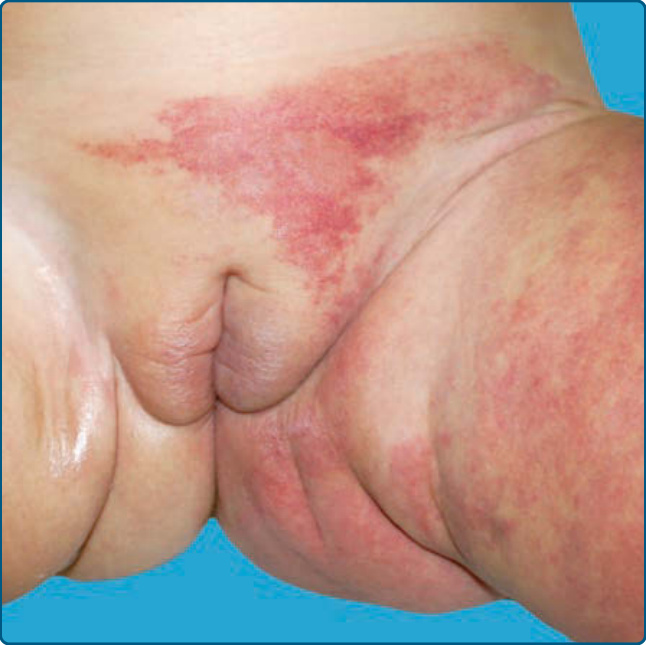
Figure 118-19 Tufted angioma (confirmed by biopsy) of the left leg, buttocks, lower abdomen in an 18-month-old child. Laboratory evaluation revealed elevated D-dimers.
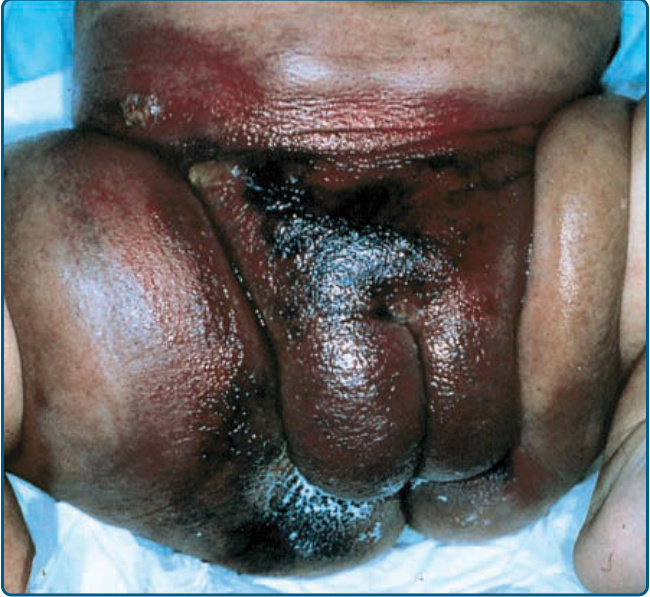
Figure 118-20 Kasabach-Merritt phenomenon secondary to kaposiform hemangioendothelioma (KHE). An indurated, purpuric tumor appeared at age 3 months and is seen here at age 8 months. KHE confirmed by MRI and biopsy. Platelet count <5000/mm3.
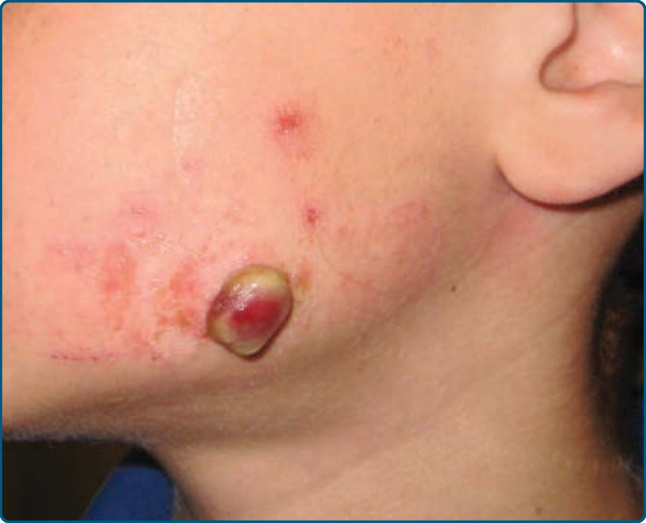
Figure 118-21 Large pyogenic granuloma on left cheek.
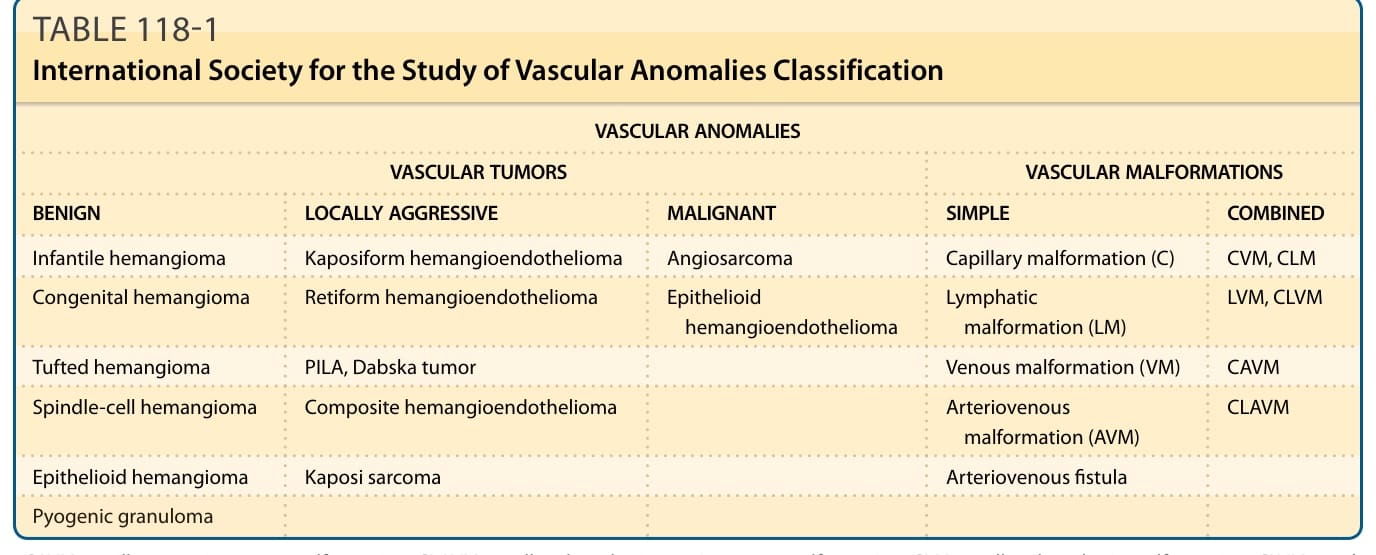
TABLE 118-1 International Society for the Study of Vascular Anomalies Classification

TABLE 118-2 History
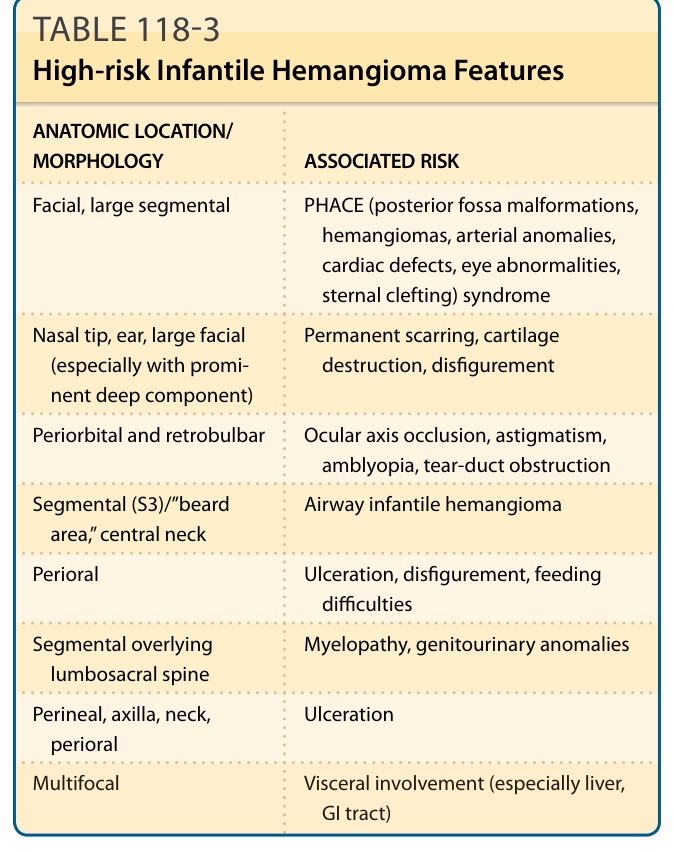
TABLE 118-3 High-risk Infantile Hemangioma Features
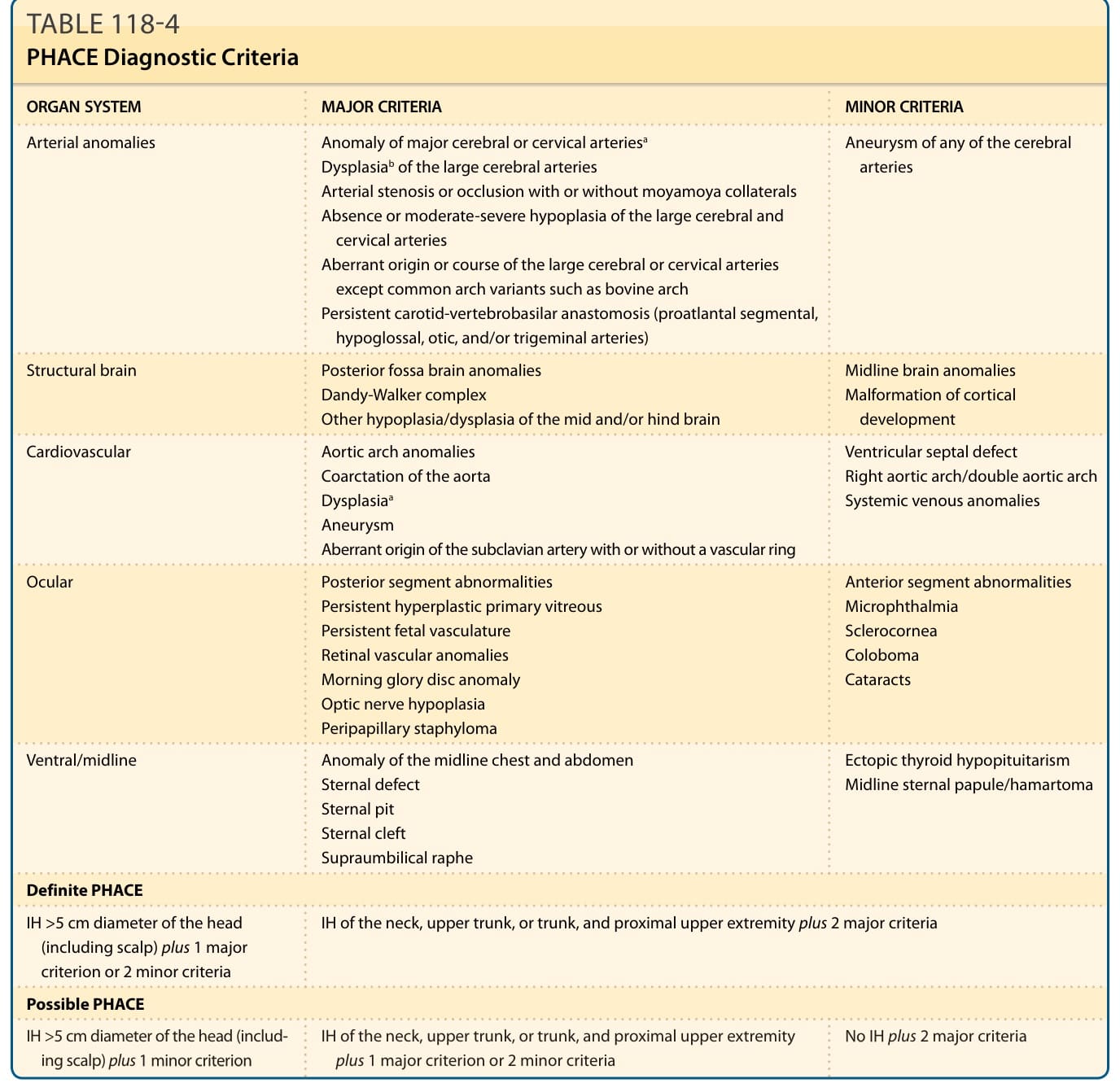
TABLE 118-4 PHACE Diagnostic Criteria

TABLE 118-5 Management of Ulcerations
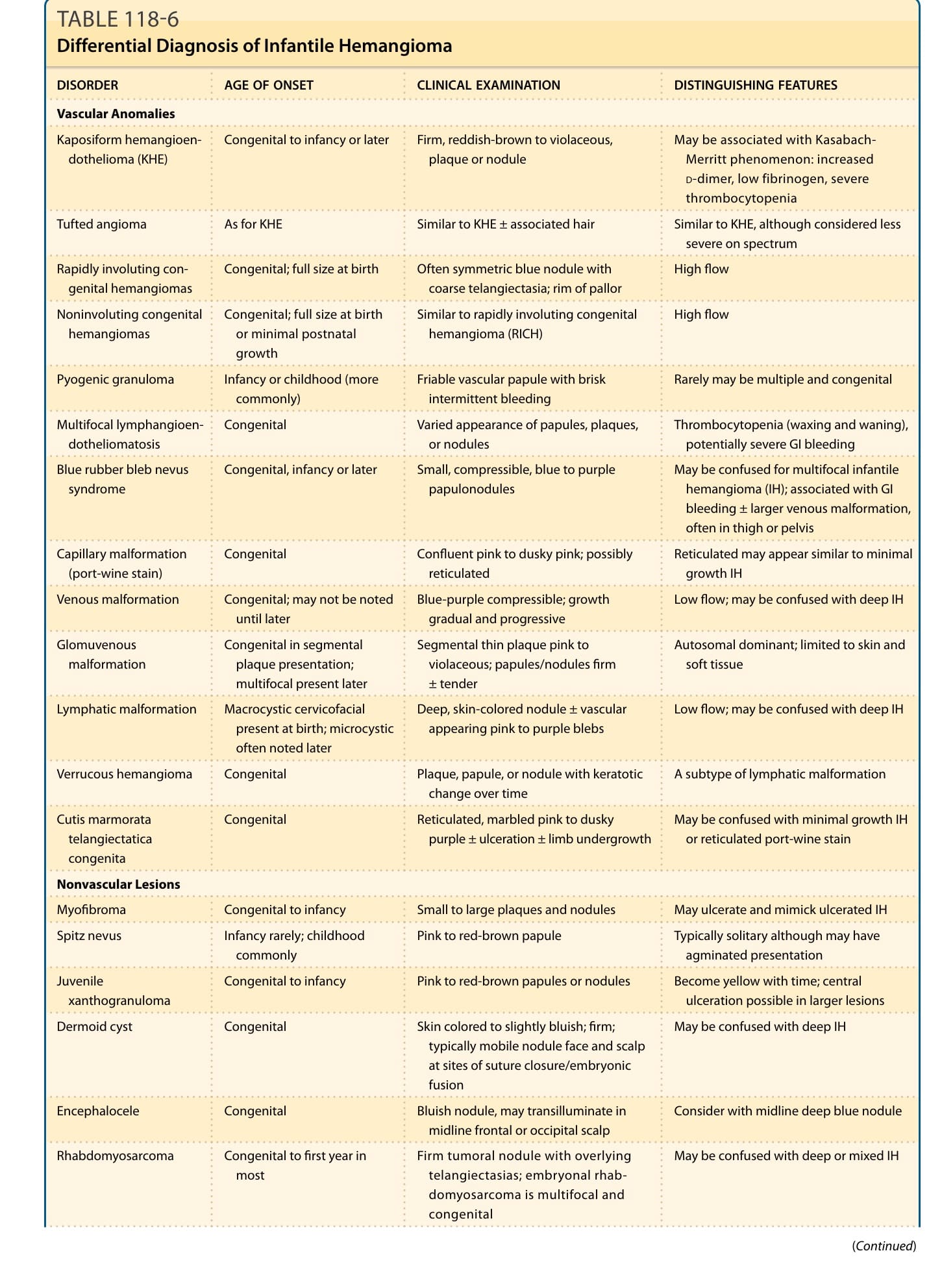
Table 118-6 discusses the differential diagnosis of IH.103