Actinic Prurigo
17
AT-A-GLANCE
■ A rare chronic pruritic and excoriated papular or nodular eruption of sun-exposed and, to a lesser extent, nonexposed skin.
■ Beginning in childhood, it may remit at puberty, exacerbate most often in summer, and fade in winter.
■ Indigenous peoples of North and South America are disproportionately affected.
■ Association with HLA-DR4, specifically the HLA- DRB1∗0407 and DRB1∗0401 variants
■ Prevention through avoidance of sunlight is the first-line therapy; thalidomide or other immunosuppressive agents may be required.
INTRODUCTION
DEFINITION
DEFINITION
Actinic prurigo (AP) is a chronic sunlight-induced pruritic eruption characterized by papules or nodules, many of which are excoriated. Lesions are distributed mostly on sun-exposed skin but may also occur in sun-protected sites. A family history of the disease may be reported by patients. Indigenous peoples of the Americas are most often affected, and strong associations with specific human leukocyte antigen (HLA) types have been identified.
HISTORICAL PERSPECTIVE
HISTORICAL PERSPECTIVE
AP was first described by Hutchinson in 1878 and was designated by him as “summer prurigo.” Other historical terms for the disease include solar eczema, prurigo solar, hereditary polymorphic light eruption, and hydroa aestivale.1
EPIDEMIOLOGY
AP occurs throughout much of the world. The indigenous populations of North and South America are particularly affected. The disease is estimated to occur in 2% of the Canadian Inuit population.2 In Mexico, AP is seen most commonly in the indigenous and Mestizo (mixed native and European descent) populations living at altitudes greater than 1000 m.3 Less commonly, inhabitants of Europe, United States, Australia, and
Asia are reported to develop AP. In Scotland, less than 1% of all patients evaluated in a specialized photodiagnostic unit were diagnosed with AP.4
In most populations, females are affected more frequently than males, in a ratio ranging from 2:1 to 4:1.5
In Asians, there is a male predominance.6 The eruption has its onset in childhood, usually present by age 10 years.7 The onset of disease appears to occur earliest in Native American populations, at 4 to 5 years of age.5
A positive family history of either AP or polymorphous light eruption is present in about a fifth of patients.8
CLINICAL FEATURES
HISTORY
HISTORY
The eruption is often present all year round, but it is commonly worse in summer. About one third of patients experience symptoms in wintertime.9 Very rarely it is worse in winter, with immunologic tolerance presumably developing during the summer. Exacerbations tend to begin gradually during sunny weather in general rather than after specific sun exposure, although PMLE-like outbreaks may also occur. Patients may not be aware that lesions are provoked by sun exposure. Pruritus is universally present and is typically severe.5,10 Pain or tingling sensations also may be reported, especially in patients with cheilitis.10,11 When conjunctivitis is present, patients report pruritus, photophobia, and lacrimation.5,12
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
The primary lesion of AP is a pruritic papule or nodule that occurs singly or in clusters (Fig. 93-1). Papules and nodules are often excoriated and crusted, and plaques may assume a lichenified or eczematous appearance (Fig. 93-2). Vesicles are not seen unless superinfection is present.5 Sun-exposed areas are most often affected, particularly the forehead, chin, cheeks, ears, and forearms. There is a gradual fading toward habitually covered skin, and the sacral area and buttocks may be mildly affected. Although primary lesions of AP do not lead to scarring, healed facial lesions may leave dyspigmentation, and scarring can occur secondary to excoriations (Fig. 93-3).
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
Mucosal involvement is not typically seen in Asian patients.6 Cheilitis may be the sole manifestation in
more than half of Native American patients and is evident by edema, scale, crust, and fissures of lip mucosa. Conjunctivitis leads to hyperemia, pterygium, pinguecula, and Trantas dots along the limbus.12
17
COMPLICATIONS
COMPLICATIONS
Mild scarring, especially on the face, and hypopigmentation may result from excoriations associated with AP. Additionally, 2 cases of primary cutaneous B-cell lymphoma arising on the face in patients with AP have been reported.14
ETIOLOGY AND PATHOGENESIS
AP appears to be induced by ultraviolet radiation in that it is more severe in spring and summer and predominates on sun-exposed skin. Additionally, patients with AP often demonstrate abnormal skin phototest responses to UVB and/or UVA radiation.7 UVA is implicated more often than UVB, with most patients demonstrating reduced minimal erythema doses in the UVA or combined UVA/ UVB ranges.5,15 The cytokine TNF-α is overexpressed by keratinocytes in lesions of AP, creating a proinflammatory environment.3 There is a concomitant increase in the number of dermal dendrocytes, and lymphocytes with a TH1 phenotype are recruited to lesional skin.3
Sunlight exposure and solar simulating irradiation may sometimes induce an eruption resembling polymorphous light eruption (PMLE) in patients with AP, and many patients have close relatives with PMLE.8
Therefore, AP may be a slowly evolving, chronic, and excoriated variant of PMLE, and thus also a delayed hypersensitivity reaction. A distinct genetic predisposition to AP has been identified for patients with specific human leukocyte antigen (HLA) subtypes. HLA-DR4 is found most commonly.16 The frequency of the HLA-DR4 allele exceeds 90% in patients from Mexico.17 Specific
1629
17
variants of HLA-DR4 are closely linked to AP, namely, DRB1∗0407 and DRB1∗0401. DRB1∗0407 (DR4) is found in 60% to 70% of patients with AP but in only 4% to 8% of normal DR4-positive controls.5 Additionally, HLA DRB1∗0401 is found in approximately 20% of affected individuals.5 Less frequently identified risk alleles include HLA-A24,18 HLA-A28,17 HLA-B39,17 HLA-B40,19 HLA-Cw3,19 HLA-Cw4,18,20 and HLA-DRB1∗14.2 These immunogenetic features may well be responsible for modulating conventional PMLE into AP. In addition, in some patients, AP appears to transform into PMLE and, in others, PMLE appears to transform into AP,21 all of which suggests a relationship between the 2 disorders. The cutaneous UVR chromophores responsible for the eruption are not known, but they are likely to be diverse.
RISK FACTORS
RISK FACTORS
Genetic risk factors for the development of AP include the aforementioned specific HLA haplotypes. Patients with these genotypes may report a family history of AP or other photosensitive disorder. Environmental risk factors have been explored, and in the Mexican population those risk factors include burning firewood, living with farm animals, and living with animals in the house.10
DIAGNOSIS
SUPPORTIVE STUDIES
SUPPORTIVE STUDIES
LABORATORY TESTING
Assessment of ANA and ENA should be undertaken to exclude subacute cutaneous or other forms of cutaneous LE. The finding of HLA DR4, type DRB1∗0401 or DRB1∗0407 (especially the latter), supports the diagnosis of AP. Patients with moderate or severe presentations of AP may also have elevated serum immunoglobulin E (IgE) levels.22
PHOTOTESTING
Cutaneous phototesting with a monochromator confirms light sensitivity in up to half of cases,7 but, as in PMLE, does not differentiate other photodermatoses. Most patients with positive monochromator testing have reduced minimal erythema doses (MEDs) in the UVA spectrum or in the combined UVA/UVB spectra.4,5,15 Provocation testing with a solar simulator or other broadband sources induces typical lesions of AP in about two-thirds of patients.4,5
PATHOLOGY
Early papular lesions show changes similar to those of PMLE, namely, mild acanthosis, exocytosis of lymphocytes, spongiosis, and mild basilar vacuolar degeneration.23 In the dermis, there is a moderate
1630
lymphohistiocytic superficial and middermal perivascular infiltrate, along with papillary dermal edema.23 Chronic lesions of AP show more acanthosis and hyperkeratosis. Infiltrates of eosinophils may be seen (Fig. 93-4). Histopathologic findings in AP cheilitis tend to be more distinct and specific. Namely, there is a dense nodular lymphoid infiltrate in the lamina propria with formation of lymphoid follicles and expanded germinal centers11 (Fig. 93-5). Eosinophilic spongiosis is also common.11 In persistent lesions, however, excoriations, more acanthosis, variable lichenification, and a dense mononuclear cell infiltrate produce a nonspecific appearance.
DIAGNOSTIC ALGORITHM
See Fig. 93-6.
DIFFERENTIAL DIAGNOSIS
The most challenging diagnosis to distinguish from AP is PMLE, and some consider the 2 diagnoses to be chronicity variants. Some of the clinical features that
suggest a diagnosis of AP rather than PMLE include disease onset in childhood, presence of lesions on both exposed and sun-protected skin, involvement of lip mucosa and conjunctiva, persistence of lesions beyond 4 weeks, occurrence in wintertime, excoriations, and scarring.21 Previously mentioned HLA restrictions are seen in AP but not in PMLE. In a subgroup of patients, overlapping features are seen and a distinction is not possible. Diagnoses to distinguish from AP appear in Table 93-1.
CLINICAL COURSE AND PROGNOSIS
AP commonly arises in childhood and often improves or resolves in adolescence, although persistence into
17
Most Likely
■Polymorphic light eruption
■Atopic eczema
■Photoexacerbated atopic or seborrheic eczema
■Insect bites
■Prurigo nodularis
Always Rule Out
Always Rule Out
■Scabies
■Scabies
adult life is possible. When the disease begins in childhood (less than age 20 years), eruptions tend to be more severe and acute but remission in adulthood is more likely. When disease begins in adulthood, a milder but
Polymorphic light eruption
Are lesions present despite rigorous avoidance of sunlight? OR Are lesions present in photoprotected sites?
NO
Consider patient’s age
Young adult
Consider photoexacerbated primary dermatosis
YES
Child Elderly
Are pock scars present?
NO
Do excoriations, swallow scars, or dyspigmentation predominate?
Hydroa vacciniforme
Does the patient have a history of atopic or contact dermatitis? OR Does patient pursue outdoor hobbies?
NO YES NO YES
Actinic prurigo
Phototoxic or photoallergic drug eruption
Does the patient take a photosensitizing medication?
Chronic actinic dermatitis
NO YES
Consider onset and duration of lesions
Onset within hours to days, resolution within days to weeks
Onset within minutes, resolution within hours
Polymorphic light eruption
Solar urticaria
1631
17
Treatment algorithm for the management of actinic prurigo
Patient diagnosed with actinic prurigo
Counsel patient regarding strict sun protection and sun-avoidance strategies
High-potency topical corticosteroids or calcineurin inhibitors
Consider hardening
Narrowband UVB or PUVA phototherapy
Add oral antihistamines If pruritus persists
Oral corticosteroids for episodic exacerbations
Thalidomide *
*Starting dose of thalidomide is 50 to 100 mg daily, counsel patient and monitor for side effects including teratogenicity, thromboembolism, and peripheral neuropathy.
more persistent course is seen.9 Persistent cases may assume features of PMLE in adulthood.
MANAGEMENT
INTERVENTIONS
INTERVENTIONS
MEDICATIONS
Controlled clinical trials evaluating medical management of AP are lacking. Higher-potency topical corticosteroids or topical calcineurin inhibitors may be used to relieve the inflammation and pruritus associated with the disease. Oral antihistamines also may be used for treatment of pruritus. Unlike some of the other photoexacerbated dermatoses, antimalarials seem to be ineffective for AP.5
The treatment of choice in more severe or recalcitrant cases is thalidomide, with initial doses of 50 to 100 mg daily for children and 100 to 200 mg daily for adults, preferably given intermittently.5,24
Responses to thalidomide are evident in most patients within several weeks. The most serious complication associated with thalidomide is teratogenicity, so pregnancy must be rigorously avoided. Other potential adverse effects are typically mild, including drowsiness, headache, constipation, and weight gain. An increased risk of thromboembolism and dose-related peripheral (mostly sensory) neuropathy are other potential adverse effects of thalidomide. In cases where thalidomide is unavailable or otherwise not appropriate, oral immunosuppressive
1632
therapy with azathioprine or cyclosporine also may be considered.
PROCEDURES
Phototherapy with narrowband UVB or PUVA may occasionally help, but the treatment response is limited to exposed areas, indicating that phototherapy does not alter the underlying photosensitive process.25
COUNSELING
Sun protection and avoidance strategies represent the cornerstone of management for AP. In less severe cases of AP, sufficient relief may be achieved by restricting sun exposure and by using broad-spectrum, highprotection-factor sunscreens alone.15
TREATMENT ALGORITHM
TREATMENT ALGORITHM
See Fig. 93-7.
PREVENTION
PREVENTION
Prevention of AP flares begins by restricting midday sunlight exposure, wearing sun-protective clothing, and using of broad-spectrum sunscreens. There is no known way to prevent its initial onset.
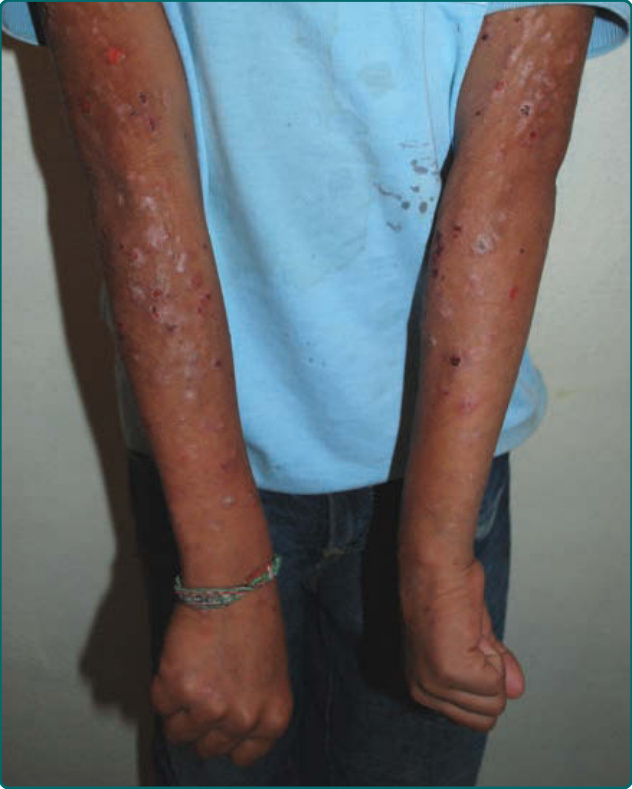
Figure 93-2 Excoriated and lichenified papules and nodules on the forearms of a child with Amerindian ancestry. (Photograph used with permission from Arturo R. Dominguez, MD.)
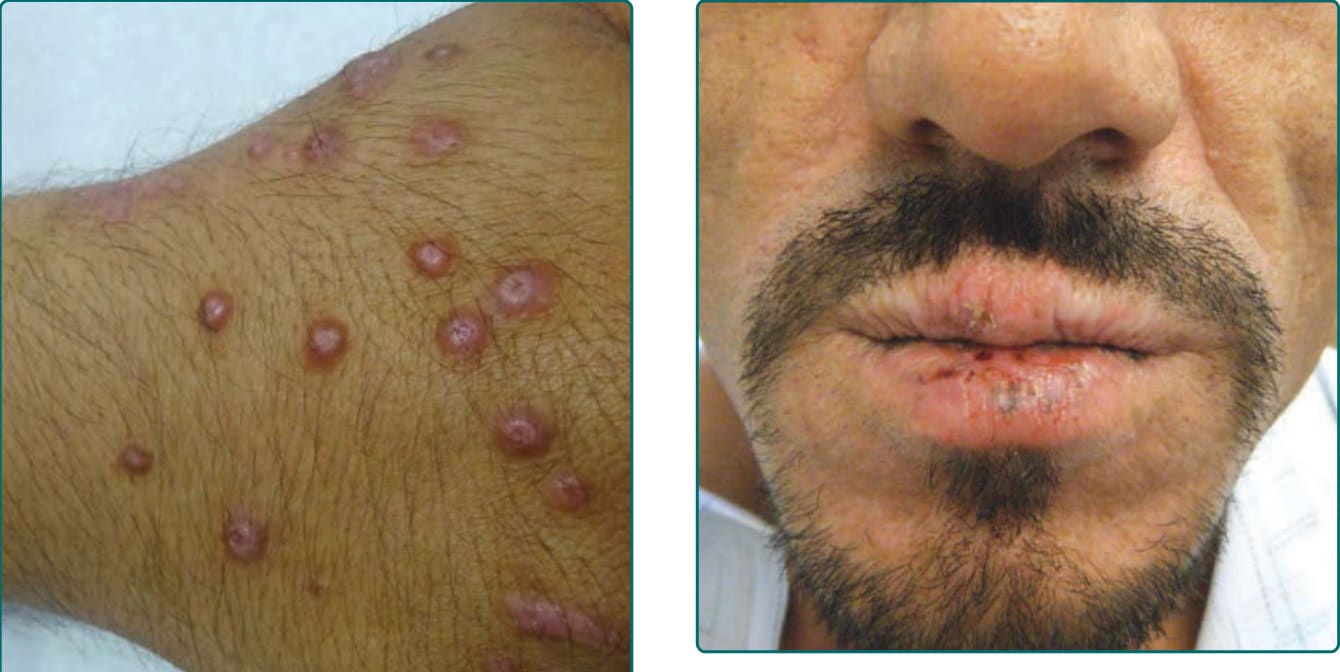
Figure 93-3 Cheilitis of actinic prurigo seen in a Mexican landscape gardener.
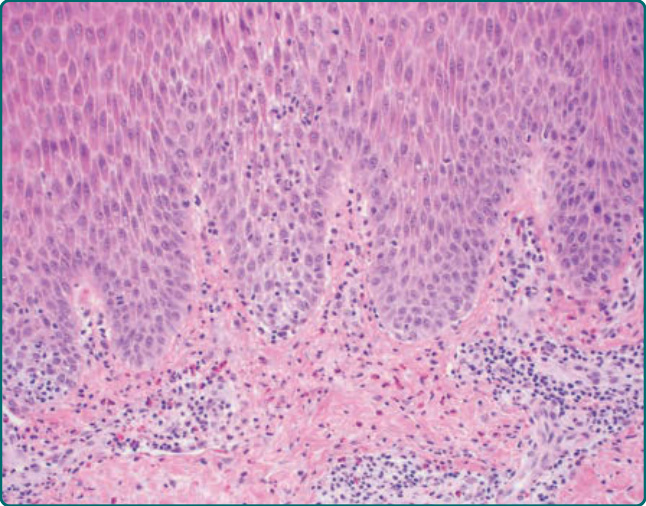
Figure 93-4 Histopathologic features are relatively nonspecific but may show acanthosis, spongiosis, exocytosis of lymphocytes, and an infiltrate of eosinophils.
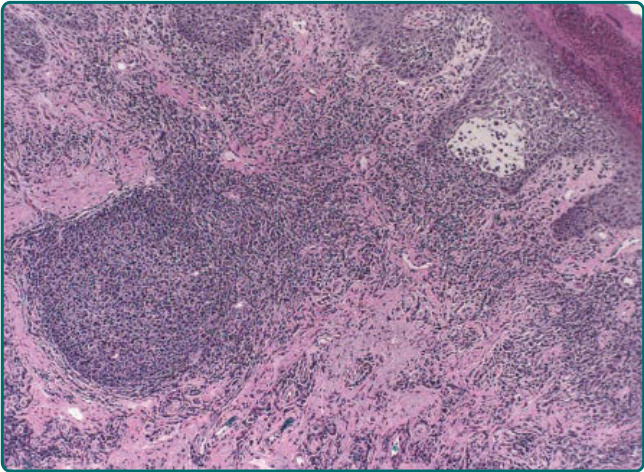
Figure 93-5 Lip biopsy from a patient with actinic prurigo shows a dense lymphohistiocytic infiltrate as well as a lymphoid follicle in the lamina propria.
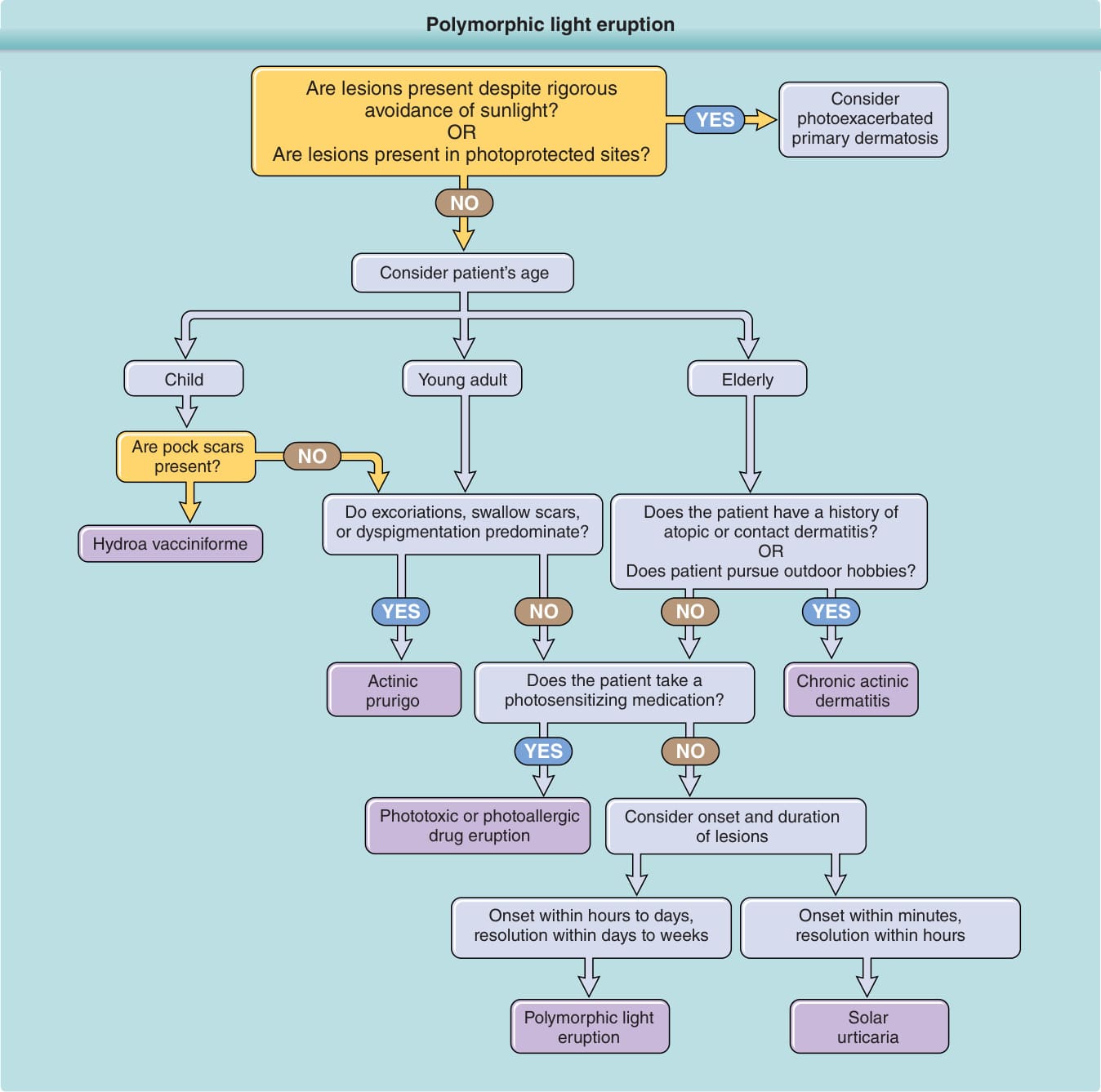
Figure 93-6 Diagnostic algorithm for abnormal responses to ultraviolet radiation.
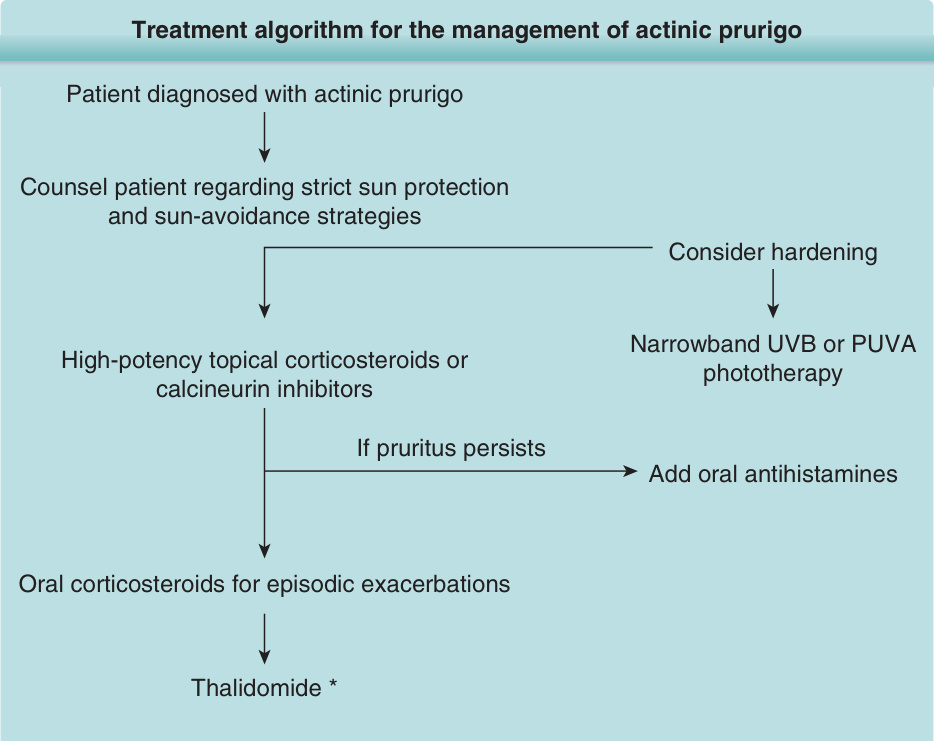
Figure 93-7 Treatment algorithm for the management of actinic prurigo.

TABLE 93-1 Differential Diagnosis of Actinic Prurigo