Sjögren Syndrome
10
AT-A-GLANCE
■ A chronic, multisystem autoimmune disease, characterized by chronic inflammation involving the exocrine glands.
■ Salivary and lachrymal glands are affected predominantly, leading to dry mouth and dry eyes.
■ May occur alone (primary Sjögren syndrome, pSS), or may coexist with other systemic connective tissue disorders (secondary Sjögren syndrome).
■ Extraglandular manifestations of primary Sjögren syndrome include fatigue, Raynaud phenomenon, purpura, arthritis, vasculitis, interstitial pulmonary disease, peripheral or central neuropathy, and autonomic nervous dysfunction.
■ Patients with systemic manifestations are at higher risk of developing lymphoma.
■ Treatment of sicca symptoms is mainly symptomatic, whereas management of extraglandular manifestations is similar to that of other autoimmune diseases.
■ Both innate and adaptive immunity contribute to pSS pathogenesis and pSS is associated with upregulation of the Type 1 interferon signaling pathway.
INTRODUCTION
Sjögren syndrome (SS) is a chronic systemic autoimmune disorder characterized by lymphocytic infiltration and destruction of exocrine glands and epithelia leading to dry mouth, dry eyes, and B lymphocyte hyperreactivity. SS can occur as an isolated disorder; primary Sjögren syndrome (pSS); it also may occur in association with other autoimmune condition such as rheumatoid arthritis, systemic lupus erythematosus (SLE) and other collagen disease; secondary SS. Disease may be localized to the salivary and lachrymal glands, but more than one-third of patients develop systemic manifestations. The clinical presentation can vary from mild sicca symptoms, fatigue, and arthralgias to severe systemic symptoms involving multiple organ systems. A small but definitely number of patients of the pSS develop lymphoma with higher incidence than that of the general population or patients with other autoimmune diseases (Fig. 68-1).
EPIDEMIOLOGY
SS is one of the most common rheumatic autoimmune diseases. SS predominantly affects women, with a
female to male ratio of 9:1. Patients are most commonly diagnosed in their fourth and fifth decades of life, but it can affect any age, including the elderly and children. SS has a worldwide distribution. The estimated annual incidence rate of pSS ranged from 3.9 to 5.3 per 100,000 in several studies.1 The prevalence rates can vary considerably, depending on the classification criteria used. The estimated prevalence rate in the adult general population from previous studies, mainly in whites, was between 0.2% and 2.7%, whereas the estimated prevalence rate from Asian studies was 0.03% to 0.77%.2,3
ETIOLOGY AND PATHOGENESIS
Although the precise pathogenesis of SS remains largely unknown, genetic and epigenetic disposition, various environmental factors, including viral and other pathogenic infections, and hormones have been implicated in the pathogenesis of the disease. Recent studies have shown that both innate and adaptive immunity contribute to the development of pSS; the Type 1 interferon (IFN) signaling pathway plays a central role in the pathogenesis of the disease. Increasing apoptosis in epithelial cells of damaged salivary glands, plasmacytoid dendritic cells (pDCs), autoreactive lymphocytes, and autoantibodies are considered to play significant roles in perpetuating the chronic auto-feedback inflammatory process.4
IMMUNOGENETIC FACTORS
IMMUNOGENETIC FACTORS
The role of genetic factors in SS was recognized in family studies where first-degree relatives of patients had an increased prevalence of SS.5,6 Such family clustering was further observed among first-degree relatives of individuals with anti-Ro/SSA antibodies, regardless of their clinical diagnoses (SS, systemic lupus erythematosus, or even healthy controls).7
There is a well-established association between SS or anti-Ro/SSA and anti-La/SSB antibodies with HLA class genes.8 The genetic loci significantly associated with pSS are HLA DQA1∗0501 and HLA DQB1∗0201 in white patients, but the pattern differs among ethnic groups, studies, and populations.9,10 In 2013, the first genomewide association study (GWAS) identified several susceptibility genes, such as IRF5 (interferon regulatory factor), STAT4 (signal transducer and activator of transcription 4), IL12A (gene regulating innate and adaptive immunity), BLK (B lymphoid tyrosine kinase), and CXCR5 (encoding a protein related to
10
A systemic autoimmune disorder
Recurrent erythema
Peripheral neuropathy
Malignant lymphoma
Chronic thyroidism
Sarcoidosis
lntersitial peumonia
Autoimmune hepatitis
Primary biliary cirrhosis
Renal tubular acidosis
Atrophic gastritis
Myositis
Polyarthritis Multipe joint pain
B-cell activation and localization), as risk variant genes related to SLE and SS.11 IRF5 is an important transcription factor involved in upregulating the Type I IFN pathway, which is activated by toll-like receptor (TLR) signals or viral infection and the production of inflammatory cytokines, whereas STAT 4 is a protein involved in the Type II IFN pathway and activating the adaptive immune system.11-13 TNFAIP3 and TNIP genes were also identified in GWAS, which regulate nuclear factor kappa-light-chain enhancer of activated B-cell (NF-κB) signaling. A TNFAIP3 mutation found in the coding region of TNFAIP3 has been correlated with an increased risk of lymphoma.14
A recent epigenome-wide DNA methylation association study (EWAS) highlighted new links between epigenetic changes and disease-related inflammatory processes in the salivary glands, which is a major target organ in pSS. It also suggests the possibility that epigenetic factors contribute to the pathogenesis of pSS. MicroRNA expression patterns in the salivary glands have been demonstrated linked with the disease.15 Recent studies have shown that the Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression,16 but knowledge in this area is still limited.4
INNATE AND ADAPTIVE IMMUNITY
INNATE AND ADAPTIVE
IMMUNITY
Type I IFN (IFN) is a key cytokine in the innate immune system, activating antigen- presenting cells and influencing autoimmune responses. Increased Type I IFN activity has been shown in the sera of pSS patients,
CNS symptoms Dry eyes
Dry mouth
Pericarditis
Pleurisy
Glomerular nephritis
Protein-losing gastrointestinal disease
Anemia Leukopenia, Thrombocytopenia Aplastic anemia Hyperviscosity syndrome Cryoglobulinemia Primay macroglobulinemia
Raynaud phenomenon
Hypergammaglobulinemic purpura
and IFN-α also has been detected in the minor salivary glands of patients. Many IFN-stimulated genes and proteins are overexpressed, which has been called the “IFN signature.” This has been found in about 50% of pSS patients and confirmed in the peripheral blood and/or salivary glands of patients.17 Previous studies have shown that when the Type I IFN receptor or IFN itself is eliminated, many features of the disease are also eliminated. This supports the theory, based on previous SS research, that Type I IFN has a central role in the pathogenesis of SS.18-22 Discovery of risk variants in IRF5, STAT4, and other susceptibility loci suggests that genetic polymorphism is a major factor in the onset of SS, activating the IFN pathway that regulates T-cell and B-cell immune responses. BAFF (B-cell activating factor), a member of the TNF family and a cytokine produced by the activated IFN signaling pathway, is related to the adaptive immune system. BAFF promotes B-cell maturation, proliferation, and survival. BAFF has been shown to contribute to the pathogenesis of SS. First, levels of BAFF are increased in the salivary glands of SS patients and upregulated by IFN-α.23 Second, transgenic mice that overexpress BAFF develop clinical manifestations of SS and SLE and have an increased risk of developing lymphoma versus wildtype mice.24 Third, BAFF is usually produced in innate immune responses, but after damage to salivary gland cells by RNA viruses or TLR3 ligands, large amounts of BAFF can be produced by epithelial cells. Furthermore, the serum level of BAFF is known to correlate with the levels of anti-Ro/SSA antibodies, anti-La/SSB antibodies, and rheumatoid factor.25 BAFF is also related to follicular structure (also called germinal center [GC]-like structure) formation. GC-like structures are known to be associated with an increased risk of lymphoma and may occur in one-fifth of patients with pSS.26
1171
10
ENVIRONMENTAL FACTORS
ENVIRONMENTAL FACTORS
Several factors are considered major environmental triggers that interact with genetic and epigenetic factors to cause disease onset. The strong predominance of females with the disease suggests gender-specific predisposing factors. Although sex hormones are obvious targets, there is no proof that the difference in the pathogenesis between males and females is due to sex hormones alone.27,28
Viruses, especially Epstein–Barr virus (EBV), are known to replicate in oropharyngeal and lachrymal glands, resulting in the hypothesis that these viruses may be involved in the pathogenesis of SS. In fact, genetic material from EBV was detected by DNA hybridization in SS salivary tissue, but it was also found in normal individuals, so the result is controversial.29,30 In a Japanese cohort, defective human T-lymphotropic virus-I genome was isolated from salivary gland tissue.31 Hepatitis C virus32-34 and HIV are also thought of as initiators of the disease, with the presence of chronic inflammation in salivary glands, and presenting clinical symptoms similar to SS.35 Other viruses, such as cytomegalovirus,36 coxsackie A virus,37 and endogenous retroviruses, also have been proposed as causative agents.38 Recent studies further showed that EBV infections promoted the release of Ro/SSA and La/SSB ribonucleoprotein complexes through epithelial apoptosis.39 The EBV-encoded small RNA and La/SSB complex is known to activate Type I IFN expression through activating TLR 3.40 However, no single virus has been clearly implicated in the pathogenesis of SS. Animal model studies support the hypothesis that there can be a delay, for years, between viral infections (such as cytomegalovirus) and the development of SS.36 Stress,41 occupational exposure,42 and personality features also have been proposed as triggers of the disease.43,44
GLANDULAR EPITHELIUM
GLANDULAR EPITHELIUM
In histopathology, sections of salivary and lachrymal glands in pSS are characterized by periductal mononuclear infiltrate. Most of the infiltrating cells are CD4+
T lymphocytes, although CD8+ cytotoxic T cells and plasmacytoid dendritic cells (pDCs) are also detected. Activated B lymphocytes, including immunoglobulinsecreting cells, are also present. The salivary glands are thought to be the first and main target in pSS. The role of the salivary gland epithelial cells in pSS has been established. Studies have indicated that epithelial cells play an active role in disease pathogenesis and this is referred to as “autoimmune epithelitis.”45 First, glandular cells, including duct and acinar epithelial cells, express HLA class II major histocompatibility complex (MHC) molecules and costimulator CD86; these interact with CD28 on T cells and contribute to recruiting inflammatory cells.46 Second, damage to the salivary gland by environmental factors, such as viruses, causes dysfunction in the gland,
1172
leading to activation of epithelial cells, increasing apoptosis, and elevated Type I IFN production by pDCs. Apoptotic keratinocytes are known to relocalize Ro/ SSA and La/SSB from the nucleus to the cell surfaces in a complex, presenting autoantigens to autoantibodies. Thus, epithelial cells may not only be the target of the disease but may also function as antigen-presenting cells, inducing further autoimmune responses through activating TLR and IFN signaling pathways.47-49 Previous studies have shown that epithelial cells express TLR 3, 7, and 9 when in an activated condition.39,50 They can also produce Type I IFN by themselves, inappropriately, through activated TLR 3 within the gland50 and become involved in perpetuating this chronic autofeedback inflammation. Third, such epithelial cells also produce large amounts of BAFF.17 Finally, some epithelial cells express Fas and Fas ligand51 and, as a consequence, undergo apoptosis; others may be destroyed by perforin, granzymes, and other cytotoxins produced by lymphocytes.52 Extraglandular manifestations occur as a result of similar lymphocytic infiltration in other organs. “Autoimmune epithelitis” well describes the systemic nature of SS.
AUTOANTIBODIES
AUTOANTIBODIES
Autoantibodies are hallmarks of systemic autoimmune diseases, including SS. The best-defined autoantibodies in SS are the anti-Ro/SSA and anti-La/ SSB antibodies.53 Although the pathogenic role of any particular autoantibody remains undefined, both are targeted against ribonucleoprotein antigens. Anti-Ro/ SSA recognizes 2 RNA-binding proteins (the 52-kDa or the 60-kDa protein), whereas anti-La/SSB antibodies recognize RNA polymerase III. Anti-Ro/SSA antibodies are found in more than 70% of patients with SS but are not specific for SS and are frequently found in SLE and other autoimmune diseases, even when there is no symptom or sign of oral or ocular dryness. Anti-La/ SSB antibodies are more specific; it is present in 50% of patients with pSS or SS/SLE but is rarely seen in other diseases.54 Although the pathogenic role of these antibodies is undefined, previous studies showed that Ro and La are expressed on the surface of apoptotic epithelial cells48; it is possible that an immune response against these antigens contributes to inflammation in the gland and further immunoreactive pathways. The most compelling in vivo evidence for a pathogenic role for these autoantibodies comes from newborns with fetal heart block, born to women with anti-Ro/SSA and/or anti- La/SSB antibodies. These antibodies can cross the placenta and bind to Ro and La antigens located on the cell surface of fetal myocardial tissue, leading to fetal heart block.55 Other autoantibodies, such as antinuclear antibodies (ANA) and rheumatoid factor (RF), are frequently present in patients with both primary and secondary SS. Although they lack specificity, they are markers of a systemic autoimmune response and thus can help distinguish SS from other causes of salivary or lachrymal gland dysfunction. A large clinical study
performed in Spain suggested the group of patients with anti-Ro/La antibodies had the highest prevalence among most systemic, hematologic, and immunologic alternations (higher frequency of Raynaud phenomenon, altered parotid scintigraphy, positive salivary gland biopsies, peripheral neuropathy, thrombocytopenia, and rheumatoid factor). Hypocomplementemia was associated with a higher frequency of vasculitis, lymphoma, and cryoglobulins, and higher frequencies of parotid enlargement, vasculitis, and leucopenia.56 SSA positivity could also be related to extraglandular manifestations, vasculitis, hematologic abnormalities, and serological hyperreactivity.57,58
Further research has focused on identifying antibodies more specific for SS, such as anti-α-fodrin and antimuscarinic acetylcholine receptor antibodies,59 but the results have been controversial. The major stimulus for saliva production is the binding of acetylcholine to muscarinic acetylcholine receptors. The hypothesis that oral and ocular dryness could result from antibodies antagonizing the muscarinic acetylcholine receptor-3 is intriguing.59 These antibodies have been demonstrated to play an essential role in glandular dysfunction in the non-obese diabetic (NOD) mouse model of SS, possibly through an inhibitory effect on the receptor.60 In humans, however, results are still contradictory because multiple attempts to detect these antibodies with conventional immunologic methods have failed.61,62
A small group of patients positive for anticentromere autoantibody (ACA) present a clinical picture similar to that of limited scleroderma. About 1% to 17% of SS patients have been reported to be positive for ACA.55,63 There is no difference between ACA-positive and ACA-negative patients in terms of female dominance or salivary gland damage and dysfunction. The ACA-positive group usually has a higher prevalence of Raynaud phenomenon and thyroid dysfunction than ACA-negative patients. This group also shows a higher frequency of vasculitis, peripheral neuropathy, and primary biliary cirrhosis, but lymphoma is the same as in the ACA-negative group.64-68 Positivity for antimitochondrial antibody (AMA) also has been demonstrated to be related to primary biliary cirrhosis.69
CLINICAL FINDINGS
Sjögren syndrome (SS) is a multifocal autoimmune disease, with systemic involvement in one-third of patients. The clinical presentation is very wide-ranging, from dry eyes, dry mouth, general fatigue, subfever, myalgia, and arthralgia, to multiple organ dysfunction.70
EXOCRINE GLAND INVOLVEMENT
EXOCRINE GLAND
INVOLVEMENT
The characteristic feature of SS is exocrine gland dysfunction, leading to the classic sicca symptoms of xerostomia (dry mouth) and xerophthalmia or keratoconjunctivitis sicca (dry eyes).
10
XEROSTOMIA
Oral dryness is a principal symptom of SS, caused by decreased saliva secretion, which is persistent and continuous throughout the day and night and can significantly compromise quality of life. Reduced salivation causes difficulty in chewing and swallowing dry foods. Dryness of the tongue and oral mucosa leads to an altered sense of taste and, at times, produces burning discomfort, especially when eating acidic or spicy foods. Physical examination may reveal a red and fissured tongue with characteristic atrophy of the filiform papillae (Fig. 68-2) or angular cheilitis. Ulcerations may be found, particularly in SS patients with dentures, usually in proximity to the mucosal surface that makes contact with the denture. Normal saliva has antimicrobial properties, so a lack of saliva can predispose to infections. Oral thrush is common and can be manifested as pseudomembranous or erythematous mucosal lesions. Furthermore, patients with SS have an increased incidence of caries. A characteristic feature of caries in SS is its primary location, at the cervical and incisal regions of the teeth. Bilateral salivary gland enlargement typically occurs in the parotid glands of SS patients. It is frequently nontender, and it can be recurrent or chronic. Painful, unilateral parotid enlargement should raise the suspicion of an infection or a salivary gland stone. In cases of persistent unilateral parotid gland enlargement, the presence of lymphoma should be excluded (Table 68-1). Medical causes of oral dryness, such as dehydration, diabetes, viral infections, and drug treatment, should be considered when evaluating a patient for Sjögren syndrome.
1173
10
Bilateral
Viral infections
Mumps Epstein–Barr, cytomegalovirus, coxsackie, influenza HIV, Human T-lymphotropic virus-I (HTLV-I) Hepatitis C
Immune mediated
Sjögren syndrome Sarcoidosis IgG4-related disease Amyloidosis
Metabolic
Diabetes mellitus Hyperlipoproteinemia Hepatic cirrhosis Chronic pancreatitis
Endocrine Acromegaly Gonadal hypofunction
Unilateral Other Alcohol Recurrent parotitis of the
Unilateral
Other
Alcohol Recurrent parotitis of the childhood
childhood
Bacterial infections
Bacterial infections
Neoplasms Mainly lymphomas, salivary
Neoplasms Mainly lymphomas, salivary gland tumor
gland tumor
Sialolithiasis
Sialolithiasis
aMedical causes of oral dryness, such as dehydration, diabetes, viral infections, or drug treatment, should be considered when evaluating a patient for Sjögren syndrome.
KERATOCONJUNCTIVITIS SICCA
Ocular dryness is the other dominant feature of SS. A burning and itching sensation in the eyes, commonly exacerbated by smoke, is caused by lack of tear production. Patients frequently complain of intolerance to contact lenses. Paradoxically, the quantity of tears produced during crying may not be affected. Physical evaluation shows corneal injection and mucous discharge in the lower fornix. Enlarged lachrymal glands have been described in Sjögren patients, but occur less commonly than enlarged salivary glands. The constellation of symptoms and signs indicating dry eyes constitutes keratoconjunctivitis sicca.
OTHER SICCA MANIFESTATIONS
Cutaneous xerosis, a term used to describe dryness of the skin, is very common in SS, with a frequency varying between 23% and 68%. The most common symptoms of xerosis are nonspecific pruritus, burning sensations, and a pin prick–like feeling. Physical examination reveals roughness, fine scaling, and loss of elasticity in the skin. The pathogenesis of xerosis is unknown. Impairment of the sweat glands is considered an important factor because decreased sweating has been reported in SS patients. A recent study indicated that xerosis may be related to increased
1174
epidermal proliferation with disturbed epidermal differentiation.71
Xerostomia predisposes to angular cheilitis, which presents as recurrent, symmetric, itching fissures. Eyelid dermatitis is defined by the presence of erythematous, infiltrated, and lichenified lesions of the eyelids associated with itching and foreign body sensations. Anhidrosis/hypohidrosis also has been reported as an exocrine manifestation, but is rare.72,73
Dryness of the upper respiratory tract can cause epistaxis, hoarseness, and bronchial hyperresponsiveness. Another common complaint in women with the disease is vaginal dryness, which may lead to an increased incidence of vaginal infections and dyspareunia.
EXTRAGLANDULAR INVOLVEMENT
EXTRAGLANDULAR
INVOLVEMENT
SKIN INVOLVEMENT
Cutaneous manifestations are common extraglandular features of SS (Table 68-2).74
Hypergammaglobulinemic Purpura: Purpuric macules are very common in SS (Fig. 68-3). Flat, nonpalpable, blanching purpura has been associated with an entity called benign hyperglobulinemic purpura, characterized by polyclonal hypergammaglobulinemia and rheumatoid factor positivity. Skin biopsies reveal ruptured blood vessels with complement deposition.75
Cutaneous Vasculitis: Cutaneous vasculitis (CV) can present as palpable purpura or urticarial vasculitis and occurs in about 9% of patients.76
■Hypergammaglobulinemic purpura
■Cutaneous vasculitis
■Nonpalpable purpura
■Palpable purpura
■Urticarial vasculitis
■Necrotizing vasculitis
■Annular erythema associated with primary Sjögren syndrome
■Erythema multiforme
■Erythema perstans
■Erythema nodosum
■Oral
■Oral
■Dry mucous membranes
■Dry mucous membranes
■Papillary atrophy of the tongue
■Papillary atrophy of the tongue
■Candidiasis
■Candidiasis
■Angular cheilosis
■Angular cheilosis
■Eyelid dermatitis
■Eyelid dermatitis
■Alopecia
■Alopecia
■Vitiligo
■Vitiligo
Palpable purpura, which does not blanch when pressure is applied to the skin, is due to dermal vasculitis with extravasation of red blood cells, and typically involves the lower extremities and buttocks (Chap. 138). It represents an important marker of more severe disease and is associated with an increased risk of lymphoma development and mortality.55 Histopathologically, palpable purpura can be divided into 2 groups. Neutrophilic inflammatory vascular disease is characterized by a predominantly neutrophilic infiltrate, fibrinoid necrosis, occlusion of the lumen, and extravasation of red blood cells, and is indistinguishable from classical leukocytoclastic vasculitis (Chap. 138). However, mononuclear inflammatory vascular disease is characterized by a mononuclear inflammatory infiltrate, with invasion of the blood vessel walls. Fibrinoid necrosis is present but less prominent. The clinical presentation of these 2 forms are indistinguishable, but neutrophilic inflammatory vascular disease is associated more strongly with markers of systemic autoimmunity, such as antinuclear antibodies and anti-Ro/SSA and anti-La/SSB antibodies, hypergammaglobulinemia, rheumatoid factor, and hypocomplementemia. Cryoglobulinemic vasculitis also can be seen in Sjögren patients and has the same cutaneous manifestations (Chap. 138). Urticarial vasculitis is the second most frequent form of CV in SS and presents as pruritic wheals with erythema (Chap. 138). In contrast to true urticaria, individual lesions last for more than 24 hours and often resolve with hyperpigmentation. Biopsy of the skin lesions demonstrates a perivascular neutrophilic infiltrate, accompanied by leukocytoclasia.
10
Necrotizing vasculitis is not commonly seen in Sjögren syndrome.77 It can present as palpable purpuric lesions of the lower extremities, which may ulcerate, finally resolving within 1 to 4 weeks. They heal with atrophy or scar tissue formation. This form of vasculitis has been observed more frequently in patients with more active disease; it has been associated with arthritis, Raynaud phenomenon, peripheral neuropathy, fever, and pulmonary or glomerular involvement. Antineutrophil cytoplasmic antibodies with perinuclear fluorescence can be found, but are uncommon in SS.
Annular Erythema Associated with Primary Sjögren Syndrome: Annular erythema associated with pSS is found primarily among Asian pSS patients who have anti-Ro/SSA (> 90%) and anti- La/SSB (∼70%) antibodies. The characteristic and typical rash is an erythematous lesion with a wide elevated border and central clearing; fine scale, erosion, or a crust can be seen in some cases.78,79 The lesion usually starts from a small indurate erythema that expands to an annular form; it can sometimes be polycyclic, and may heal without atrophy, scar, or pigmentation (Fig. 68-4). They are localized mainly on the faces of Asian patients (81%), but can also occur on the arms, trunk, and proximal thighs. Sunlight and cold exposure, mental stress, and pregnancy may be triggers of the lesions. Annular erythema associated with pSS occur in Asian patients with an occurrence of up to 47%, but is less common in whites, with an
1175
10
occurrence of 9%.80 Because annular erythema associated with pSS shares a clinical presentation with subacute cutaneous lupus (SCLE), it is still debatable whether annular erythema associated with pSS is a variant of SCLE (annular SCLE), as proposed by Sontheimer in 1979,81 but it seems to represent a distinct clinical and histopathologic entity. The 2 conditions can hardly be distinguished from clinical presentations or laboratory investigations, such as positivity for anti-SSA/Ro and anti-SSB/La antibodies; they even show similar Ro/SS-A autoantibody epitopes and titer responses, but there are some distinct differences between the 2 conditions. First, annular erythema associated with pSS are located mainly on the face (>80%) in Oriental patients, but mainly on the trunk in white patients; second, the percentage of anti-SSA/Ro antibody–positive patients is different between annular erythema associated with pSS in Asians and SCLE in non-Asian patients; third, in histopathologic findings and finally, in HLA DR3 and 8 expression.80,82,83 In histopathology, SCLE presents with liquefaction degeneration in the basal membrane zone, positivity for IgM/IgG in the dermal–epidermal junction with direct immunofluorescence (DIF), and cell infiltration being, basically, in the upper dermis, whereas in annular erythema associated with pSS, the dense cell infiltration is localized mainly around sweat glands, and nuclear dust also can be seen when they have vasculitis. Usually, no vacuolar changes are found in the dermal–epidermal junction, and no other findings suggest lupus erythematosus, such as epidermal atrophy or follicular plugging, and are usually negative for DIF. SS and SLE share common genetic polymorphisms, such as IRF5, STAT4, and BLK11; thus, it is possible patients present similar manifestations in both conditions.84 However, other studies have demonstrated notable differences in populations with risk haplotypes and risk alleles, including HLA.85 Previous studies have also shown that Oriental patients have higher anti-SSA/Ro antibody titers and are more commonly negative for HLA DR3 and DR8, which show a high incidence in American patients.82 It is well known that annular erythema associated with pSS are common in Asians but rare in whites, whereas SCLE are more common in whites but rare in Asians. The similarity between annular erythema associated with pSS and SCLE may be explained as a phenotype variation, from sharing the same genetic polymorphisms but having different risk haplotypes/alleles in different ethnic groups. At present, annular erythema associated with pSS is included in the EULAR-SS Disease Activity Index (ESSDAI), cutaneous domain, and classified as a moderate activity level manifestation in SS.86
Erythema Nodosum: Erythema nodosum, a painful nodular eruption of the anterior surface of the lower extremities, may occur but is rare in SS patients. Its presence should raise suspicion for sarcoidosis. Erythema multiforme–like lesions and superficial illdefined patches (erythema perstans) also have been described in SS.
1176
Other Skin Manifestations Related to Peripheral Circulation and Temperature: Raynaud phenomenon is probably the most common abnormality. It can be seen in 15% to 35% of patients and can precede sicca symptoms by many years. Raynaud phenomenon in SS is not accompanied by telangiectasias, as seen in systemic sclerosis. Calcifications have been described, but are uncommon. Although Raynaud phenomenon is usually mild in pSS, it is a marker of a subgroup with increased risk of extraglandular manifestations.87 Acrocyanosis, pernio-like eruptions (Fig. 68-5), livedo, and periungual hemorrhage also may be observed.
Other Cutaneous Manifestations: Other cutaneous manifestations including photosensitivity, indurated erythema, alopecia, vitiligo vulgaris, lichen planus, sarcoidosis, granuloma annulare, and psoriasis also may be seen in SS, albeit infrequently.
NONCUTANEOUS EXTRAGLANDULAR MANIFESTATIONS Musculoskeletal Manifestations: A symmetric nonerosive polyarthritis is frequently seen in pSS. The distinction between pSS and rheumatoid arthritis may be difficult in this regard; the absence of rheumatoid factor and anti-CCP antibodies, and the absence of erosions on radiographs would favor SS over rheumatoid arthritis. Arthralgias and myalgias are common complaints but true myositis is rare in pSS.
Fatigue and Fibromyalgia: Fatigue is a common complaint in SS, but the pathogenesis remains unknown. Fibromyalgia is a distinct condition, rather similar to SS, and should always be considered in the differential diagnosis.
Thyroid Dysfunction: Thyroid dysfunction is a very common symptom, occurring in 10% to 70% of SS patients. It often presents as fatigue and patients easily become tired.
Visceral Manifestations: Lungs. Dry cough due to dryness of the tracheal mucosa is common (tracheobronchitis sicca). Rarely, patients may develop interstitial pneumonitis. Patients may also develop mucosa-associated lymphoid tissue (MALT) lymphoma in the lungs. Renal and genitourinary manifestations include interstitial cystitis (50%), renal tubular acidosis, interstitial nephritis and, rarely, glomerulonephritis. Gastrointestinal. Dysphagia due to xerostomia and esophageal dysmotility is common. Helicobacter pylori is associated with an increased risk of MALT lymphomas; thus, patients with gastritis should be checked for H. pylori and treated if found positive. An asymptomatic, chemical pancreatitis with high-serum amylase concentrations has been reported in 25% of patients. Other conditions, such as celiac disease, primary biliary cirrhosis, and hypothyroidism may also occur with pSS. Because of these associations, a high index of diagnostic suspicion is warranted to identify and treat these conditions. Neurologic manifestations can be divided into those that involve the CNS, and those that involve the peripheral nervous system and autonomic dysfunction. The peripheral nervous system is involved in ∼20% of patients with pSS.88 However, this can increase to 70% when the patient has cutaneous vasculitis. The most common manifestations are peripheral axonal polyneuropathies, which are typically sensory. Another entity that has been described is a ganglionopathy involving the sensory ganglia of the posterior column. This starts with unilateral peripheral paresthesias, evolving over months to years to deep sensory impairment, positive Romberg sign, generalized areflexia, and ataxia.89 Cranial neuropathies are also frequent. The most common form is unilateral trigeminal neuropathy; it usually spares the ophthalmic division, preserving the corneal reflex. Other cranial neuropathies may lead to Bell palsy (facial nerve), neural deafness, vestibular dysfunction (vestibule-cochlear), and diplopia (oculomotor, trochlear, abducent nerve). The CNS also may be involved, although the prevalence and the spectrum of manifestations remain controversial. Manifestations similar to multiple sclerosis as well as transverse myelitis have been described. The latter is frequently associated with antiaquaporin-4 autoantibodies.
Lymphoma/Lymphoproliferative Disease: The incidence of lymphoma in SS patients is increased 15- to 44-fold, according to various studies90; 4% of patients during the first 5 years, 10% at 15 years, and 18% after 20 years, postdiagnosis, develop lymphomas.91 Most of these are indolent, extranodal, marginal-zone B-cell lymphomas of MALT, but highergrade lymphomas are also seen.90 It has been proposed that at least a proportion of DLBCLs arise from MALT transformation, and lifelong follow-up of patients with pSS and MALT lymphoma is recommended.92
Various clinical and laboratory features have been correlated with an increased risk of lymphoma development. Lymphadenopathy, parotid enlargement,
10
palpable purpura, low C4 serum levels, and cryoglobulins are the most consistent non-Hodgkin lymphoma/ lymphoproliferative disease predictors. Some studies have also identified splenomegaly, low C3 serum levels, lymphopenia, and neutropenia as significant prognostic factors. Histopathologically, the presence of germinal center–like (GC-like) lesions in salivary biopsies of pSS are associated with an enhanced possibility of developing lymphoma. TNFAIP3 and TNIP1 gene variants were identified together with the IRF5, STAT4, and CXCR5 genes in GWAS; polymorphisms in these genes also have been suggested to contribute to GC-like structure formation, and further studies have demonstrated associations between the gene variants and pSS and non-Hodgkin lymphoma.93-95 In contrast, anti-Ro, anti-La, and antinuclear antibodies, rheumatoid factor, male gender, hypergammaglobulinemia, and anemia were not associated with lymphoma/lymphoproliferative diseases.91,96-98
Pregnancy: Recent studies have suggested that women with pSS may experience more complications in pregnancy than healthy controls. Significant increases in the rates of spontaneous abortions, preterm deliveries, and low-body-weight infants were reported. The mechanism of the lower body weight was suggested to be due to underlying placental insufficiency, related to the autoimmune background.99 Moreover, carriers of the Ro/SSA and La/SSB antibody can transmit it through the placental circulation to the fetus. These antibodies (anti-Ro/SSA, anti-La/SSB) can cause congenital heart block (CHB) or neonatal lupus (NLE), characterized by an annular rash with central regression or mild atrophy in the scalp and around the eyes, as well as hepatic and hematologic abnormalities. The skin lesions can be variable and appears in infants at a few weeks of age. Although the rash typically resolves spontaneously at 6-8 months of age, the heart block is permanent and requires the placement of a pacemaker in ∼60% of patients. Expectant mothers with Ro/SSA and La/SSB antibodies should be counseled about this risk, and their fetuses should be followed closely for the development of fetal heart block.
DIAGNOSIS AND DIAGNOSIS TESTS
The diagnosis of SS in a patient with complaints of oral and ocular dryness requires evaluation by a dentist or salivary gland specialist, such as an otorhinolaryngologist, and an ophthalmologist, for specialized tests to quantify mucosal dryness, as well as laboratory evidence of autoantibodies and a salivary gland biopsy for evidence of inflammation within the gland itself. Unlike other autoimmune disorders, SS still lacks universally accepted classification criteria. Several sets of diagnostic criteria have been developed and used by various groups over time, creating some confusion with regard to how to define SS. Recently, an international consensus group agreed on a set of criteria, and this revised American–European classification system
1177
10
I. Ocular Symptoms: A positive response to at least one of the following questions:
- Have you had daily, persistent, troublesome dry eyes for more than 3 mo?
- Do you have a recurrent sensation of sand or gravel in the eyes?
- Do you use tear substitutes more than 3 times a day?
II. Oral symptoms: A positive response to at least one of the following questions:
- Have you had a daily feeling of dry mouth for more than 3 mo?
- Have you had recurrently or persistently swollen salivary glands as an adult?
- Do you frequently drink liquids to aid in swallowing dry food?
III. Ocular signs: Objective evidence of ocular involvement defined as a positive result for at least one of the following 2 tests:
- Schirmer test, performed without anesthesia (≤5 mm in 5 min)
- Rose Bengal score or other ocular dye score (≥4 according to the van Bijsterveld scoring system)
IV. Histopathology: In minor salivary glands (obtained through normal-appearing mucosa) focal lymphocytic sialoadenitis, evaluated by an expert histopathologist, with a focus score ≥1, defined as a number of lymphocytic foci (which are adjacent to normal-appearing mucous acini and contain more than 50 lymphocytes) per 4 mm3 per glandular tissue
V. Salivary gland involvement: Objective evidence of salivary gland involvement defined as a positive result for at least one of the following diagnostic tests:
- Unstimulated whole salivary flow (<1.5 mL/15 min)
- Parotid sialography showing the presence of diffuse sialectasias (punctuate, cavitary, or destructive pattern), without evidence of obstruction in the major ducts
- Salivary scintigraphy showing delayed uptake, reduced concentration, and/or reduced excretion of tracer
VI. Autoantibodies: Presence in the serum of the following autoanti-
VI. Autoantibodies: Presence in the serum of the following autoantibodies: antibodies to Ro/SSA, La/SSB or both
bodies: antibodies to Ro/SSA, La/SSB or both
Reproduced with permission from BMJ Publishing Group Ltd: Vitali C et al. Classification criteria for Sjögren’s syndrome: A revised version of the European criteria proposed by the American- European Consensus Group. Ann Rheum Dis. 2002;61:554.
has been widely accepted (Table 68-3).100 Six features are taken into consideration: subjective complaints and objective evidence of dry eyes or dry mouth, objective evidence of salivary gland inflammation, and the presence of specific autoantibodies in the serum. For the diagnosis of pSS, at least 4 criteria should be present, and at least one of them should be evidence of lymphocytic infiltration or the presence of autoantibodies. Patients with a coexisting connective tissue disease are labeled secondary SS. Exclusions include other diseases and medications that may cause sicca symptoms. In 2012, the American College of Rheumatology provided new criteria (Table 68-4).101 The new criteria require at least 2 of the following 3:
■ Positive serum anti-SSA/anti-SSB antibodies or positive rheumatoid factor (RF) and antinuclear antibody titer ≥1:320,
■ Keratoconjunctivitis sicca with ocular staining score ≥3,
1178
The classification of SS, which applies to individuals with signs/ symptoms that may be suggestive of SS, will be met in patients who have at least 2 of the following 3 objective features:
- Positive serum anti-SSA/Ro and/or anti-SSB/La or (positive rheumatoid factor and ANA titer ≥1:320)
- Keratoconjunctivitis sicca with ocular staining score ≥3 (assuming that individual is not currently using daily eye drops for glaucoma and has not had corneal surgery or cosmetic eyelid surgery in the last 5 y)
- Labial salivary gland biopsy exhibiting focal lymphocytic sialadenitis with a focus score of ≥1 focus/4 mm2
Exclusion Criteria History of head and neck radiation treatment Acquired immunodeficiency syndrome Hepatitis C infection Sarcoidosis Amyloidosis Graft-vs-host disease IgG4-related disease
Exclusion Criteria
History of head and neck radiation treatment Acquired immunodeficiency syndrome Hepatitis C infection Sarcoidosis Amyloidosis Graft-vs-host disease IgG4-related disease
Modified from Shiboski SC, et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: A data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken). 2012;64(4):475-87, with permission.
■ Labial salivary gland biopsy showing focal lymphocytic sialadenitis with a focus score ≥1 focus/4 mm2.
These criteria are thought to be stricter than those previously applied, and so the prevalence may decline.102
ORAL DRYNESS
ORAL DRYNESS
SIALOMETRY
Sialometry involves measurement of the total saliva produced from all salivary glands in a time period of 15 min. The whole unstimulated salivary flow is considered suggestive of SS if it is <1.5 mL in 15 minutes. A “stimulated” salivary flow can be measured after administration of lemon juice or citric acid, and low values are also suggestive of SS.
SALIVARY GLAND SCINTIGRAPHY
Salivary gland scintigraphy (SGS) is a functional study to assess saliva production by measuring the secretion of a radioisotope (99mtechnetium sodium pertechnetate) into the oral cavity. SGS provides functional information about individual salivary glands and can potentially distinguish between decreased production and/ or decreased excretion of saliva. The past decade has seen a shift toward the quantitative evaluation of SGS
based on various parameters generated from time-activity curves.103 Given the high cost and the exposure to radiation, routine use may not yet be justified but it is a useful diagnostic tool in selected cases. Sialography is an imaging method based on retrograde injection of contrast media into the parotid duct. Dilation of salivary ducts and sialolithiases are the most common findings.
OCULAR DRYNESS
OCULAR DRYNESS
The diagnosis of keratoconjuctivitis sicca is based on the demonstration of decreased tear production, corneal damage, or both. Tear production is usually measured using Schirmer test. This is performed by placing a standardized paper strip in the inferior fornix of each eye and measuring the length of filter paper that becomes wet after 5 minutes. The American–European classification system uses a cut-off value of 5 mm in 5 minutes, below which a diagnosis of dry eye is made. An alternative method is staining with a dye (rose Bengal or lissamine green) that preferentially stains the devitalized cornea and conjunctiva. This staining is evaluated through the van Bijsterveld scoring system; a score of 4 or more indicates keratoconjunctivitis sicca. These tests have high sensitivity but low specificity; they identify ocular dryness, but cannot attribute abnormal findings to SS.
LABORATORY TESTING
LABORATORY TESTING
High levels of inflammatory markers, such as high sedimentation rates and signs of chronic inflammation (anemia, hypoalbuminemia), are common in SS patients. Serologic tests may demonstrate hyperglobulinemia in as many as 80% of pSS patients. Autoantibodies commonly include Ro/SSA and La/SSB antibodies, as well as rheumatoid factor and ANA. Hypocomplement has been associated with a higher frequency of vasculitis and lymphoma, and cryoglobulins with higher frequencies of parotid enlargement, vasculitis, and leukoplakia.
HISTOPATHOLOGY
HISTOPATHOLOGY
None of the above diagnostic procedures is specific for SS. The most reliable objective diagnostic feature is seen on a biopsy of the minor salivary gland. A small incision is made on the inner surface of the lip, and a minor salivary gland tissue sample is collected. Evidence of a focal, periductal infiltrate, composed of T and B lymphocytes and few plasma cells, is the histologic hallmark of SS (Fig. 68-6). The degree of lymphocytic infiltration is evaluated semiquantitatively by means of a focus scoring system. Evidence of one or more foci is considered indicative of SS, and a focus is considered a conglomeration of at least 50 lymphocytes per 4 mm2 of glandular tissue. GC-like
10
F
F
A
B
C
structures are thought to be a risk factor for developing lymphoma.104 Atrophy and fat tissue are other findings that may be present in SS patients, but also can be present in healthy, elderly individuals. Although a salivary gland biopsy provides important clues that may lead to a definitive diagnosis, some controversy still exists
1179
10
regarding its sensitivity and specificity. Some patients with pSS, diagnosed on the basis of reduced salivary flow and evidence of anti-SSA and anti-SSB antibodies, have no lymphocytic infiltration in biopsy specimens from the minor salivary glands. Additionally, some healthy individuals may demonstrate lymphocytic foci in their salivary glands.
CLINICAL COURSE AND PROGNOSIS
There are 2 patterns of pSS that define 2 distinct disease categories with very different clinical risks. Patients with low complement C4 levels and/or palpable purpura early in their disease course may be classified as a high-risk disease syndrome (Type I). This group comprises ∼20% of pSS diagnoses and carries a significantly increased risk of lymphoproliferative disease; it also has an increased mortality rate. Most severe extraglandular manifestations also occur in this group. Patients without these 2 predictors (80% of all pSS diagnoses) may be reassured that they have a lowrisk (Type II) form of pSS that carries no increased risk of death and, in general, has a more benign course, dominated by sicca symptom.97,105,106
TREATMENT
TREATMENT OF DRY MOUTH (TABLE 68-5)
TREATMENT OF DRY MOUTH
(
)
The treatment of oral dryness is largely symptomatic.107
Nonpharmacologic strategies remain the mainstay of treatment. Adequate hydration, and reducing irritants like coffee, alcohol, and nicotine, as well as the substitution of drugs that can lead to dry mouth (diuretics, tricyclics, antihistamines, β-blockers) when possible are important. Frequent sips of water are recommended initially to maintain moisture. Patients should also be encouraged to use sugar-free candies and chewing gum to increase saliva production. Saliva substitutes are available in the form of gels, oils, and sprays, but the need for frequent application is inconvenient for many patients. Sugar-containing foods should be avoided because they contribute to an increased risk of dental caries and oral candidiasis. Meticulous dental hygiene is essential for SS patients to prevent or treat dental caries. Caries prevention with fluoride application, including prescription-strength toothpaste, fluoride gels, and oral rinses, is further recommended. Oral candidiasis is treated with oral and systemic antifungal therapy. To prevent candidiasis, patients should not wear dentures at night and dentures should be soaked in 2% chlorhexidine. Nystatin or clotrimazole cream can be used to treat angular cheilitis. If dry mouth is not adequately controlled with replacement methods, pharmacologic therapy with
1180
Dry mouth
■Frequent water
■Mechanical stimulation of saliva secretion (sugarfree gum and candies)
■Artificial saliva
■Meticulous oral hygiene
■Caries prevention with fluoride-containing toothpaste and rinses
■Aggressive treatment of candidiasis with topical and systemic antifungals
■Sialogogues: pilocarpine (5 mg orally 3 times/d) or cevimeline (30 mg orally 3 times/d)
Dry eyes
■Frequent use of preservative-free artificial tears
Dry eyes ■Frequent use of preservative-free artificial tears
■Nighttime use of moisturizing ointments
■Nighttime use of moisturizing ointments
■Topical cyclosporine ophthalmic solution (0.05%, one drop every 12 h)
■Topical cyclosporine ophthalmic solution (0.05%,
one drop every 12 h)
■Surgical: punctal plug placement
■Surgical: punctal plug placement
■Sialogogues
■Sialogogues
secretagogue drugs is an option. Two drugs are approved for this indication: (1) pilocarpine (5 mg 4 times/d)108 and (2) cevimeline (30 mg 3 times/d).109
Both act on muscarinic receptors and increase exocrine gland secretion. They are contraindicated in narrowangle glaucoma and uncontrolled asthma. Cholinergic side effects, such as excessive sweating, urinary frequency, flushing, and headaches, are common with both. Tolerability may be increased if treatment is started at a lower dose, which is then increased gradually (Table 68-5).
TREATMENT OF DRY EYES (TABLE 68-5)
TREATMENT OF DRY EYES
(
)
Nonpharmacologic measures are important therapeutic interventions; avoidance of dry, smoky, and windy environments; avoiding contact lenses or opting for those higher in water content; and minimizing medications that inhibit tear production (diuretics, β-blockers, tricyclic antidepressants, antihistamines) are first-line measures. Many tear substitutes are available and are commonly used by patients to alleviate ocular dryness. Preservative-free preparations are preferred, especially if used more than 4 times per day. Methylcellulosecontaining ointments may provide longer relief, but their use is limited to nighttime use because of the risk of blurred vision. A frequently used surgical option is occlusion of the puncta to block tear drainage and, consequently, increase moisture. The occlusion can be transient, with the insertion of collagen or silicone plugs, or permanent, with electrocautery. Cyclosporine 0.05% ophthalmic solution has been approved by the FDA for the treatment of
keratoconjunctivitis sicca.110 Topical corticosteroids are rarely needed and should probably only be prescribed after an ophthalmic examination. Infections may present with aggravation of symptoms or increased mucus secretion and should be promptly treated with topical antibiotics. Pilocarpine and cevimeline can be effective for dry eyes, too, especially in patients with the most severe dryness.109
TREATMENT OF EXTRAGLANDULAR MANIFESTATIONS
TREATMENT OF
EXTRAGLANDULAR
MANIFESTATIONS
The treatment of the musculoskeletal manifestations of SS is similar to that of other systemic rheumatologic diseases.111 Because of reduced saliva production and esophageal dysmotility, these patients have reduced tolerance to nonsteroidal antiinflammatory drugs. Antimalarial drugs, such as hydroxychloroquine, are effective for arthralgia/arthritis, myalgia, fatigue, and annular erythema.112 Dryness of the eyes and mouth may also improve slightly in some cases.113 Visceral manifestations, such as vasculitis, pneumonitis, and glomerulonephritis, and neurologic manifestations are treated with corticosteroids and immunosuppressive drugs, it doses similar to those used in systemic lupus erythematosus.114 Recently, a large-scale cohort study of pSS patients from Spain focused on the adequacy of therapies for the level of systemic activity, measured by the ESSDAI score.115
Treatment of lymphoma in SS is the same as in the population generally. Most SS-associated lymphomas are low-grade B-cell lymphomas, localized to the exocrine glands. For these cases, watchful waiting may be the most appropriate approach. Higher-grade lymphomas require more aggressive treatment with a rituximab, cytotoxic regimen, and/or radiation therapy.
MOLECULAR TARGET THERAPY
MOLECULAR TARGET
THERAPY
Several randomized case–control studies have focused on therapy for SS using biologic agents, such as the TNF-blocking agents, infliximab116 and etanercept,117
but none of the studies showed clinical benefit. There is increasing interest in therapies targeting B cells, such as monoclonal antibodies against CD20 (rituximab) or CD22 (epratuzumab). A recent randomized placebo-controlled study demonstrated the efficacy of rituximab over placebo in patients with active pSS.118
Rituximab improved stimulated salivary flows, laboratory parameters of inflammation, and subjective symptoms. This clinical efficacy was supported by reduced glandular infiltration and morphologic improvements in epithelial cells on salivary gland biopsies.119,120
Other potential targets for biologic therapy include
10
cytokines, such as IL-6 and BlyS (BAFF), interferons, adhesion molecules, and chemokines. However, even if effective, systemic immunomodulatory therapy may be associated with unwanted side effects and may not be justified in patients with pSS whose disease is limited to exocrine glands. An alternative approach is to develop a localized form of immunotherapy by using gene therapy restricted to the salivary and lachrymal glands. This approach would most likely alter the abnormal immune response locally, but possibly avoid the systemic side effects.114,121,122
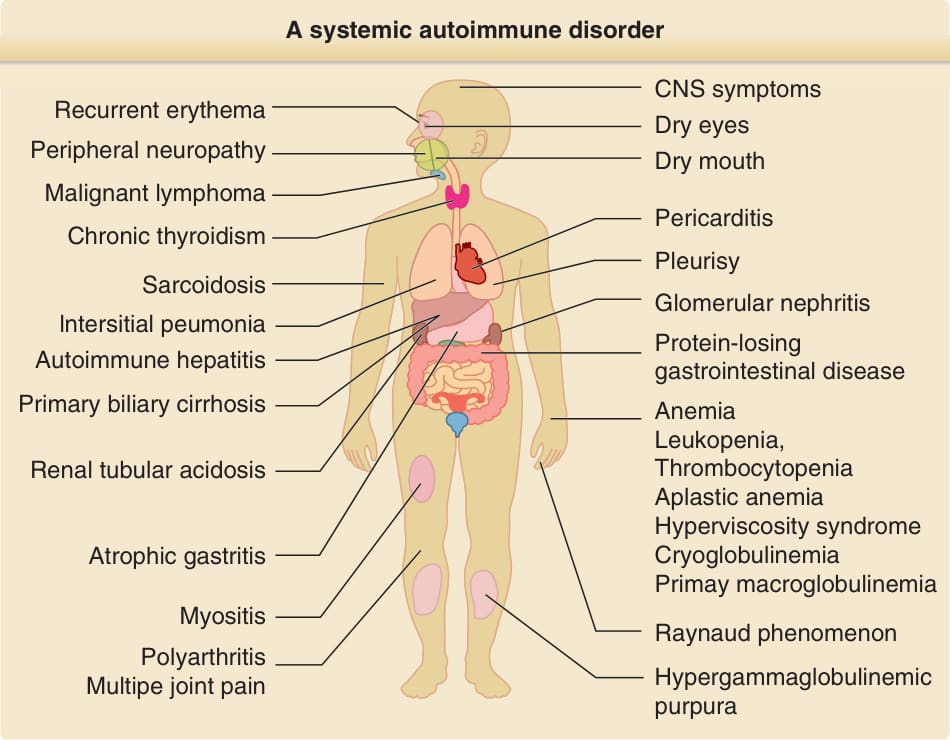
Figure 68-1 Sjögren syndrome. A systemic autoimmune disorder.
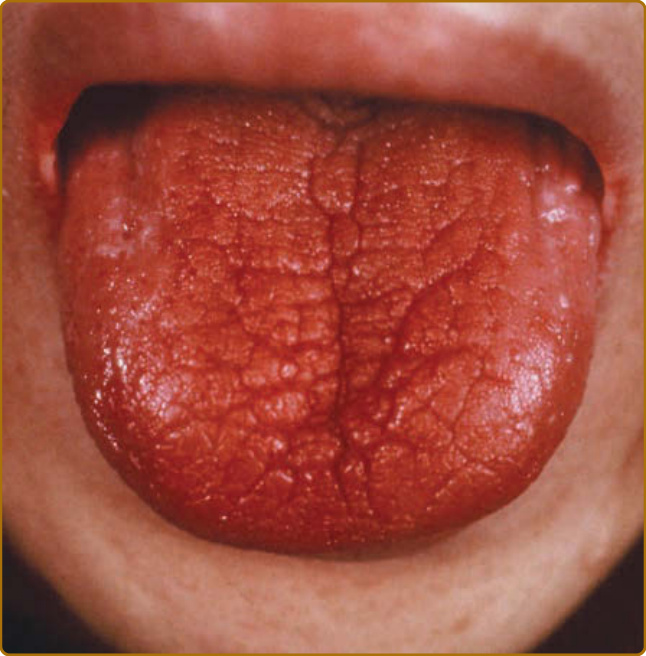
Figure 68-2 The characteristic tongue of patients with Sjögren syndrome. A red and fissured tongue with characteristic atrophy of the filiform papillae.
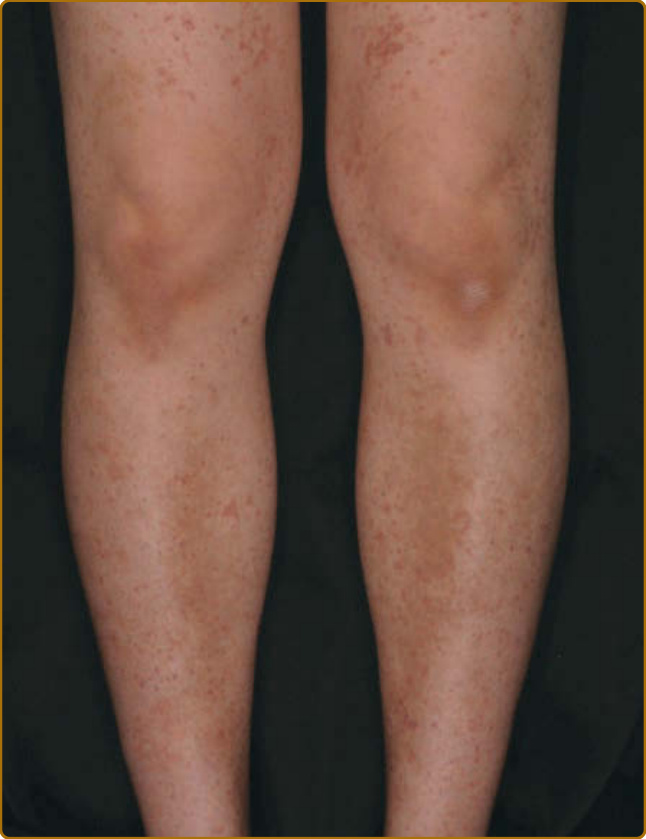
Figure 68-3 Non-palpable purpura with pigmentation in a patient with primary Sjögren syndrome.
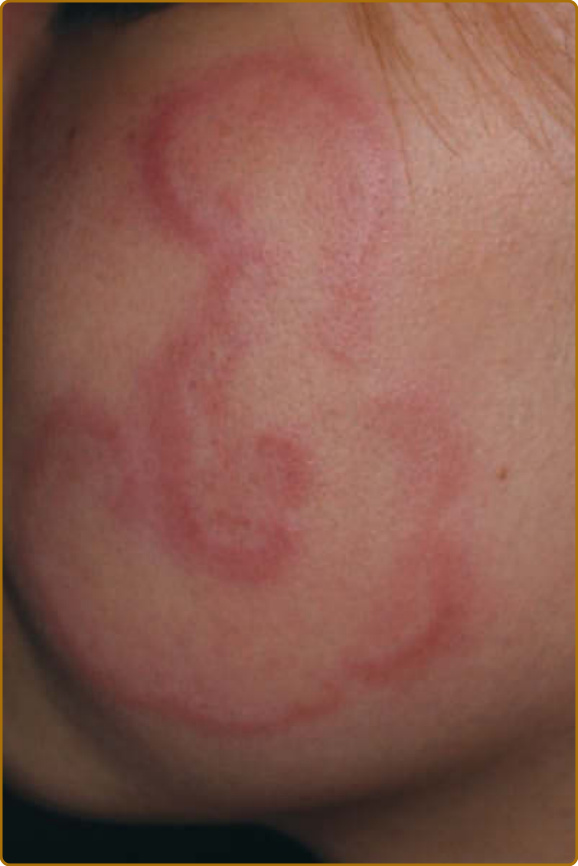
Figure 68-4 Annular erythema associated with primary Sjögren syndrome in a young female patient presented with figurate annular erythema with raised border.
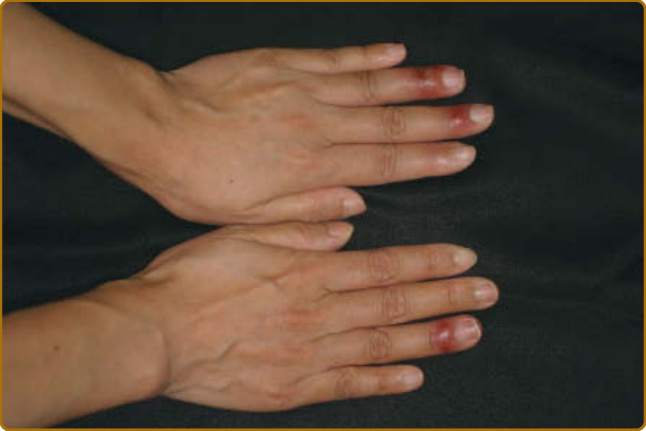
Figure 68-5 Pernio-like eruption in a patient with primary Sjögren syndrome.
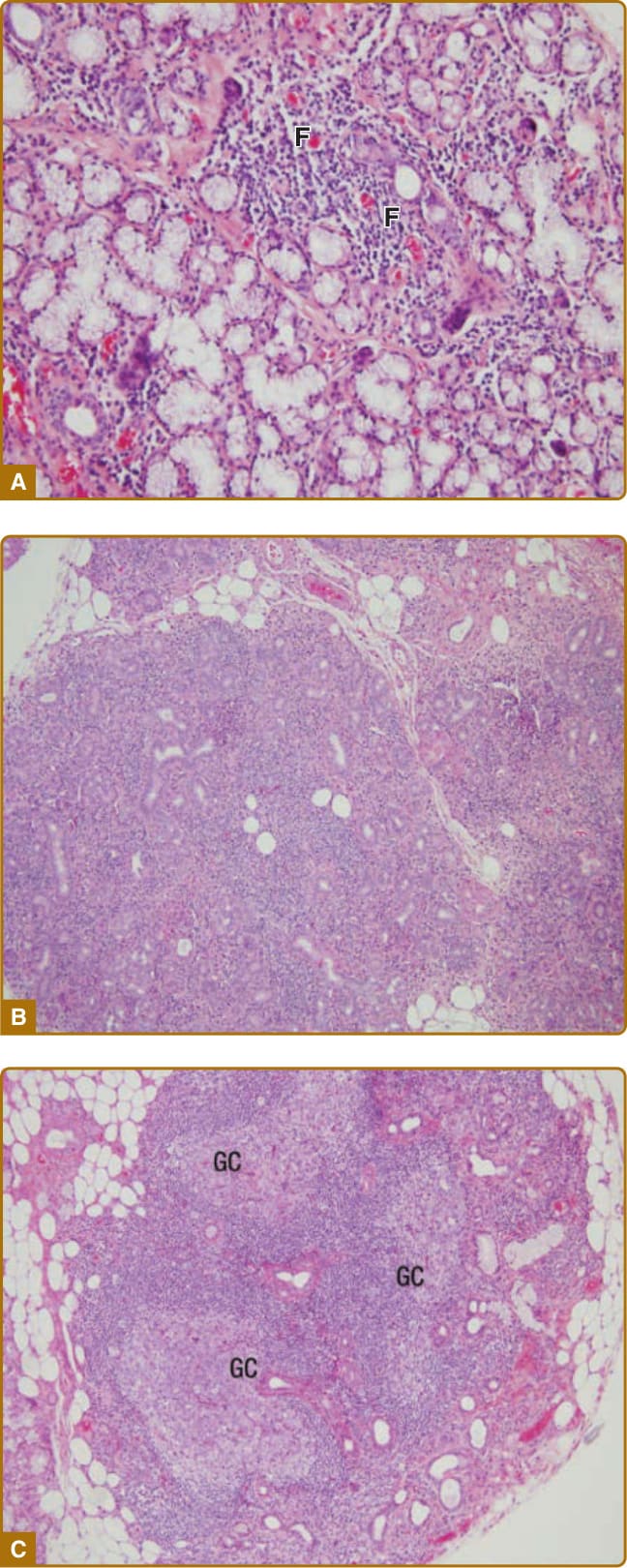
Figure 68-6 Histopathology of the minor salivary gland in Sjögren’s syndrome. The degree of lymphocytic infiltration varies from moderate (A) to diffuse (B). In the most severe forms, germinal center formation can be observed (C). F = lymphocytic focus; GC = germinal center.
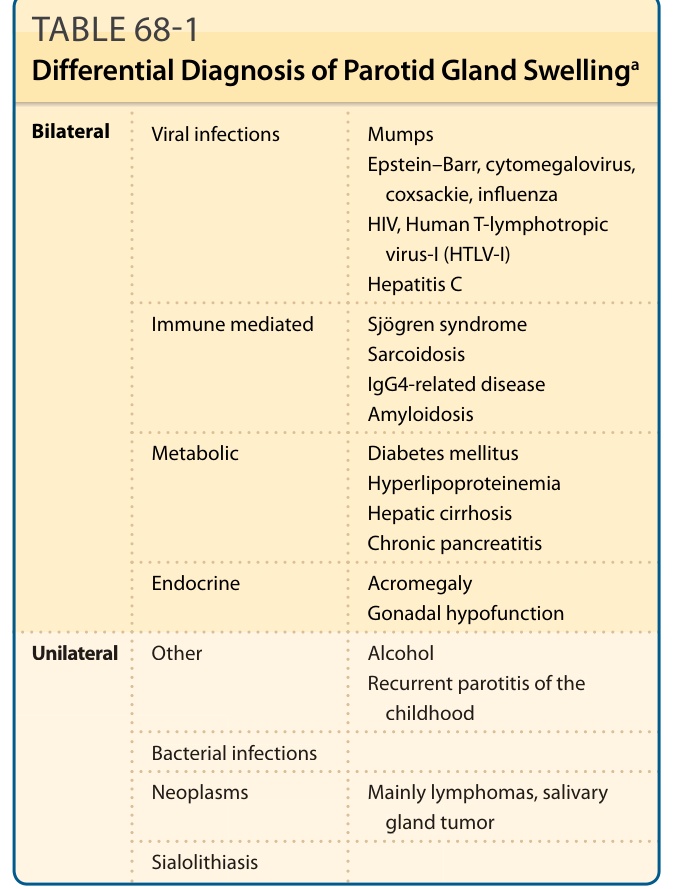
TABLE 68-1 Differential Diagnosis of Parotid Gland Swellinga

TABLE 68-2 Cutaneous Manifestations of Sjögren Syndrome
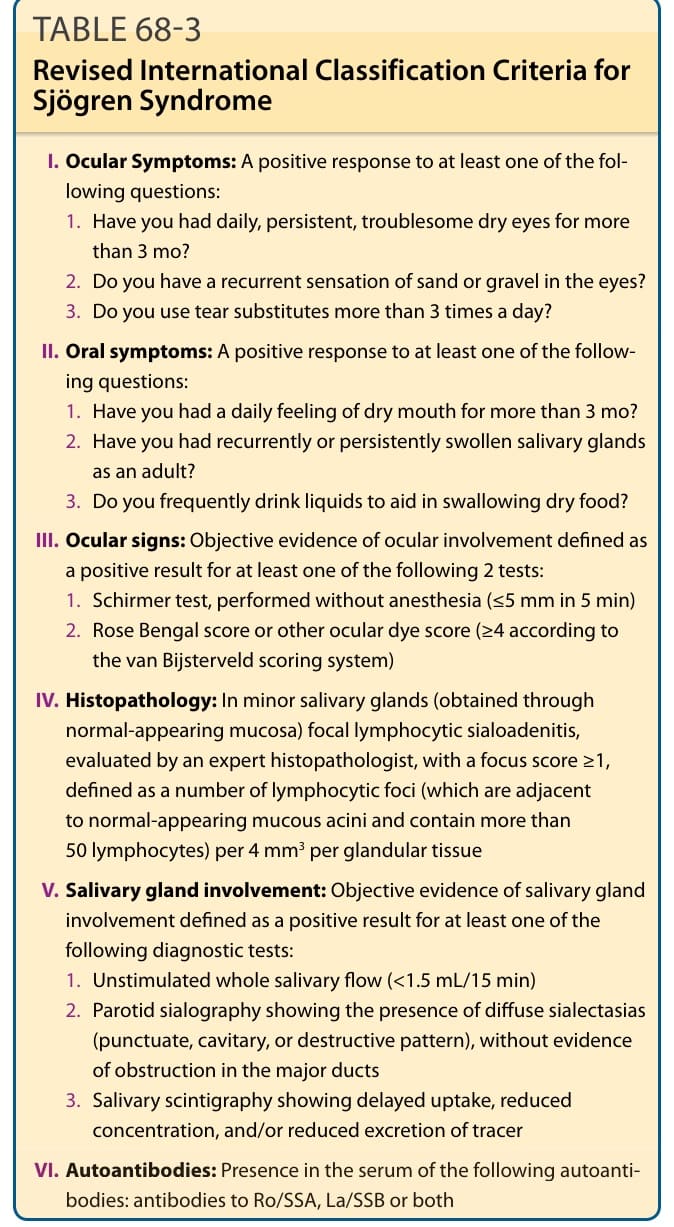
TABLE 68-3 Revised International Classification Criteria for Sjögren Syndrome

TABLE 68-4 Proposed Classification Criteria for Sjögren Syndrome

TABLE 68-5