Mucous Membrane Pemphigoid
9
AT-A-GLANCE
■ A chronic autoimmune subepithelial blistering disease characterized by erosive lesions of mucous membranes and skin that typically results in scarring in at least some sites of involvement.
■ Lesions commonly involve the oral and ocular mucosae; other sites that may be involved include the nasopharyngeal, laryngeal, esophageal, and anogenital mucosae.
■ A rare disorder with a predominance in the elderly and an estimated incidence of 1 to 2 cases per million annually; females are affected 1.5 to 2 times as often as males.
■ A progressive disorder that may result in serious complications (eg, blindness, loss of the airway, esophageal stricture formation).
■ Severity of involvement in one organ does not necessarily correlate with the presence or severity of disease at other sites.
■ Immunopathologic studies of perilesional mucosa and skin demonstrate in situ deposits of immunoreactants in epithelial basement membranes; circulating anti–basement membrane autoantibodies are detected in the sera of some but not all patients.
■ A variety of different autoantigens are recognized by autoantibodies from patients, suggesting that mucous membrane pemphigoid is not a single nosologic entity but a disease phenotype.
DEFINITION
Mucous membrane pemphigoid (MMP) is a rare chronic autoimmune subepithelial blistering disease characterized by erosive lesions of mucous membranes and skin that typically results in scarring of at least some sites of involvement.1-4
HISTORICAL PERSPECTIVE
In 1794, Wichmann described a chronic blistering disorder characterized by ocular involvement.5
What are thought to be related cases were reported by Thost in 1911.6 In 1949, Civatte separated this disorder from pemphigus on the basis of histopathologic findings.5 Lever affirmed this distinction and suggested that this disorder be named “benign mucous membrane pemphigoid,” a designation now regarded a misnomer given the potential
devastating consequences of MMP.4 Over the years, a variety of alternate designations, such as cicatricial pemphigoid, oral pemphigoid, desquamative gingivitis, ocular pemphigoid, ocular cicatricial pemphigoid, essential shrinkage of the conjunctivae, ocular pemphigus, have been applied to MMP. In 2002, an international consensus conference agreed upon the designation of this disorder as MMP in that this nomenclature is inclusive of patients with disease affecting any mucosal surface and that it emphasizes the mucosal predominant character of this disorder.1 In 2015, an international panel of experts issued a consensus statement providing accurate and reproducible definitions for disease extent, activity, outcome measures, end points, and therapeutic response.7
EPIDEMIOLOGY
MMP has been estimated to have an incidence of 1 to 2 cases per million annually; females are affected 1.5 to 2.0 times as often as males.8-10 MMP has a mean age of onset in the early to middle 60s.10 Although there is no known racial or geographic predilection, the HLADQB1∗0301 allele is significantly increased in frequency in patients with oral, ocular, and generalized bullous pemphigoid; amino acid residues at positions 57 and 71 to 77 of the DQB1 protein may represent a disease-susceptibility marker.11-15
CLINICAL FEATURES
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
Patients typically describe the onset of painful, erosive, and/or blistering lesions on 1 or more mucosal surfaces.16 A few skin lesions on the upper body are also sometimes noted. Associated symptoms are typically site specific. The mouth is the most frequent site of involvement in patients with MMP; it is often the first (and only) site affected.1,5,16 In the mouth, lesions often involve the gingiva, buccal mucosa, and palate (Fig. 55-1); other sites, such as the alveolar ridge, tongue, and lips, are also susceptible. Erosive lesions of the gingiva resulting in tissue retraction and loss are a frequent manifestation of MMP. Other sites of intraoral involvement may show tense blisters that rupture easily or mucosal erosions that form as a consequence of epithelial fragility. Lesions in the mouth may result in a delicate white pattern of reticulated scarring; oral lesions can heal without scarring in some instances. In severe disease, adhesions may develop between the buccal mucosa and the alveolar process, around the
uvula and tonsillar fossae, and between the tongue and the floor of the mouth. Gingival involvement can result in tissue loss and dental complications (eg, caries, periodontal ligament damage, and loss of bone mass and teeth). Ocular involvement in patients with MMP is common and may become sight threatening (Figs. 55-2 and 55-3).17,18 Ocular lesions typically manifest as conjunctivitis that progresses insidiously to scarring. Early ocular disease can be quite subtle and nonspecific. Although disease is usually bilateral, it often begins unilaterally and progresses to both eyes within several years. Patients may complain of burning, dryness, or a foreign-body sensation in one or both eyes; frank blisters on conjunctival surfaces are rarely seen. Early disease is best appreciated by slitlamp examination. Because disease may be localized to the upper tarsal conjunctiva, it may escape detection without eversion of the eyelids. Chronic ocular involvement can result in scarring characterized by shortened fornices, symblepharons (ie, fibrous tracts between bulbar and
9
palpebral conjunctival surfaces), and, in severe disease, ankyloblepharons (ie, fibrous tracts fusing the superior and inferior palpebral conjunctivae with obliteration of the conjunctival sac). Conjunctival scarring also can cause entropion and trichiasis (ie, in-turning of the eyelashes) that result in corneal irritation, superficial punctate keratopathy, corneal neovascularization, corneal ulceration, and/or blindness. Additional ocular complications include scarring of the lacrimal ducts, decreased tear secretion, and loss of mucosal goblet cells leading to decreased tear mucus content and unstable tear films. It is very important for patients with suspected ocular involvement to be examined by an ophthalmologist because early disease may be subtle, only identified by slitlamp examination, and hold potential for severe complications. MMP may be limited to the eyes. Other sites that may be affected by MMP include the nasopharyngeal, laryngeal, esophageal, and anogenital mucosae. Nasopharyngeal lesions can result in discharge, epistaxis, excessive crust formation, impaired airflow, chronic sinusitis, scarring, and tissue loss. Laryngeal involvement may present as hoarseness, sore throat, and/or loss of phonation. Chronic laryngeal erosions, edema, and scarring may result in supraglottic stenosis and airway compromise that eventually necessitates tracheostomy. Esophageal involvement may result in stricture formation, dysphagia, odynophagia, weight loss, and/or aspiration. It has been suggested that esophageal dysfunction and gastroesophageal reflux may elicit or exacerbate laryngeal disease and/or bronchospasm in such patients. Although involvement of the genital and/or rectal mucosae in patients with MMP is relatively rare, it can be a source of substantial pain and morbidity (Fig. 55-4). Cases of urethral stricture, vaginal stenosis, and anal narrowing have developed as a consequence of this disease.
961
9
The skin is involved in 25% to 35% of patients with MMP. The most frequently affected areas are the scalp, head, neck, and upper trunk (Fig. 55-5). Lesions typically consist of small vesicles or bullae situated on erythematous and/or urticarial bases. Lesions rupture easily and are often seen as small, crusted papules or plaques. The extent and number of cutaneous lesions are generally small; lesions sometimes recur in the same areas. In 1957, Brunsting and Perry described 7 patients with recurrent scarring subepidermal blistering lesions on the head or neck that for many years was considered a variant form of MMP.19 Similar patients were subsequently identified by others.20-25 Although such patients are typically elderly and demonstrate deposits of immunoreactants in epidermal basement membranes like other patients with MMP, Brunsting- Perry pemphigoid predominates in men, lacks mucous membrane involvement, and only in selected cases is associated with immunoglobulin (Ig) G autoantibodies against BP230 (also termed bullous pemphigoid antigen 1 [BPAG1] or dystonin) or BP180 (also termed
962
bullous pemphigoid antigen 2 [BPAG2] or type XVII collagen).21-24 Because some patients with the same clinical, histologic, and immunopathologic features have been reported to have autoantibodies directed against type VII collagen and blister planes beneath the lamina densa, it has been suggested that a subset of such patients may have a localized forms of epidermolysis bullosa acquisita.21,22 Rare patients with this phenotype and autoantibodies directed against laminin-332 and/or other skin autoantigens also have been described.24,25
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
In 2001, a cohort of 35 patients with MMP and IgG autoantibodies directed against laminin-332 (ie, anti– laminin 332 MMP [formerly called antiepiligrin cicatricial pemphigoid]) was shown to have an increased relative risk for cancer.26 Ten patients in this cohort had solitary solid cancers (3 lung, 3 gastric, 2 colon, 2 endometrial); 8 patients developed cancer after the onset of MMP (6 within a year, 7 within 14 months). The time between blister onset and cancer diagnosis was approximately 14 months in 9 of the 10 patients. Eight patients in this cohort died as a consequence of their cancer; all deaths occurred within 21 months. This study showed that this form of MMP has an increased relative risk for malignancy that approximates that for adults with dermatomyositis; as is true for dermatomyositis, the risk for cancer appears to be particularly high in the first year of disease. Other patients with this form of MMP and cancer have been described by numerous sources.26-36 Interestingly, subsequent studies suggest that the relative risk for cancer among patients with ocular or oral MMP and autoantibodies versus integrin subunit β4 or integrin subunit α6, respectively, may be reduced.37,38
COMPLICATIONS
COMPLICATIONS
Site-specific complications of MMP were outlined earlier and are summarized in Table 55-1.
ETIOLOGY AND PATHOGENESIS
RISK FACTORS FOR DISEASE
RISK FACTORS FOR DISEASE
Risk factors for disease include advanced age, female gender, and possession of the HLADQB1∗0301 allele.1,5,16,39 In rare cases, disease may be provoked by prescription drugs, trauma (eg, eye surgery), or hypersensitivity reactions. Autoantibodies directed against autoantigens in epidermal basement membrane are held responsible
SITE POTENTIAL COMPLICATIONS
Mouth
Mucosa Painful, erosive scarring lesions; adhesions between the buccal mucosa and the alveolar process, the uvula and the tonsillar fossae, and the tongue and the floor of the mouth
Gingiva Painful, erosive scarring lesions; loss of gingival tissue; caries; periodontal ligament damage; loss of alveolar bone; loss of teeth
Eyes
Conjunctivae Painful, erosive conjunctivitis; foreign-body sensations; photophobia; scarring; shortened fornices; loss of goblet cells; decrease in tear mucus content and unstable tear films; symblepharons; ankyloblepharons
Eyelids Ectropion; trichiasis; ankyloblepharons
Cornea Corneal irritation; superficial punctate keratopathy; corneal neovascularization; corneal ulcers, blindness
Tear ducts Scarring; occlusion; secondary infection
Nose Discharge; epistaxis; excessive crust formation; impaired airflow; recurrent and chronic sinusitis; scarring and tissue loss
Larynx Hoarseness; impaired phonation; loss of voice; scarring, supraglottic stenosis, airway compromise and loss
Esophagus Dysphagia, odynophagia, impaired swallowing; stricture formation; weight loss; aspiration
Anogenital
Painful erosions; stenosis and strictures;
Anogenital region Painful erosions; stenosis and strictures; secondary infections
region
secondary infections
9
for the pathogenesis of MMP (Fig. 55-6).1,5,16,39 Because different autoantigens are recognized by circulating autoantibodies from different patients with MMP, this disorder is considered a disease phenotype rather than a single nosologic entity. Table 55-2 summarizes the autoantigens recognized by autoantibodies from patients with MMP. Even though autoantibodies directed against some of these autoantigens are pathogenic in vivo (Table 55-2), it is conceivable that other mechanisms may contribute to disease pathogenesis. As scarring is a major pathologic manifestation of MMP, profibrotic processes have been studied in biopsy samples and cultured fibroblasts from lesional tissue. Examples of profibrotic factors that have been identified in such studies include serpin h1; transforming growth factor beta; interleukins 4, 5, and 13; and connective tissue growth factor.40,41 What accounts for the loss of immunologic tolerance to skin in patients with MMP is unknown.
AUTOANTIGEN MW (KDA) LOCATION SSS/ULTRA PASSIVE TRANSFER STUDIES
BP230 230 Epid/HD
BP180 180 Epid/HD-af IgG vs. the NC16A domain of BP180 creates subepidermal blisters in newborn mice that resemble those seen in patients with BP94,95
Integrin β4 ∼205 Epid/HD-af
Integrin α6 ∼120 Epid/HD-af
Laminin 332 400–440 Derm/LL-LD interface Experimental IgG (intact IgG and Fab fragments alone) vs. laminin 332 create subepidermal blisters in newborn and adult mice that resemble those seen in patients with AECP96
Patient IgG creates subepidermal blisters in human skin grafts on immunodeficient adult mice that resemble those seen in patients with AECP97
Type VII collagen 290 Derm/AF Experimental and patient IgG vs. the NC1 domain of type VII collagen create subepidermal blisters in adult mice that resemble those seen in patients with EBA98-100
Type VII
290 Derm/AF Experimental and patient IgG vs. the NC1 domain of type VII collagen create subepidermal
collagen
blisters in adult mice that resemble those seen in patients with EBA98-100
AECP, antiepiligrin cicatricial pemphigoid; AF, anchoring fibril; BP, bullous pemphigoid; BP230, bullous pemphigoid antigen 1; BP180, bullous pemphigoid antigen 2; Derm, dermal; EBA, epidermolysis bullosa acquisita; Epid, epidermal; HD, hemidesmosome; HD-af, hemidesmosome–anchoring filament complexes; IgG, immunoglobulin G; LL-LD interface, lamina lucida–lamina densa interface; Location SSS/Ultra, localization in 1 M NaCl–split-skin/ultrastructural localization in epidermal basement membrane; MW (kDa), molecular weight in kilodaltons.
963
9
BP180 appears to represent a major MMP autoantigen16,42-44; other autoantigens of particular interest include laminin 332, integrin subunit β4, integrin subunit α6, type VII collagen, and BP230.16
Most patients with MMP have IgG anti–basement membrane autoantibodies; some patients have IgA anti–basement membrane autoantibodies alone or in conjunction with IgG anti–basement membrane autoantibodies. The most common IgA autoantigen linked to the MMP phenotype is BP180.45-47
DIAGNOSIS
LIGHT MICROSCOPY
LIGHT MICROSCOPY
Although the findings of light microscopy studies of lesional skin or mucosa from patients with MMP are often nonspecific, they characteristically show a subepidermal blister and a dermal leukocytic infiltrate composed of lymphocytes and histiocytes as well as variable numbers of neutrophils and eosinophils.1,20,48,49 Plasma cells can be seen in mucosal lesions, whereas eosinophils and neutrophils are more commonly seen in skin lesions. Biopsy specimens of older lesions may be relatively “cell poor” and show features that correlate with the noninflammatory character of such sites clinically. Light microscopy studies of older lesions often show fibroblast proliferation and lamellar fibrosis (ie, fibrosis characterized by collagen bundles ordered parallel to the surface epithelium). It is not always possible (or in the case of ocular disease, appropriate) to biopsy blistered mucosa for light microscopy studies.
ELECTRON MICROSCOPY
ELECTRON MICROSCOPY
Ultrastructural studies of lesional skin or mucosa from patients with MMP show that blisters typically develop within the lamina lucida and eventuate in partial or complete destruction of the basal lamina in older lesions.48-52 A generally held idea is that blisters form below those of bullous pemphigoid, because scarring is more common in patients with this disease. Reports of patients with blisters in the sublamina densa region are thought to represent mucosa-predominant forms of epidermolysis bullosa acquisita.
IMMUNOFLUORESCENCE MICROSCOPY
IMMUNOFLUORESCENCE
MICROSCOPY
Direct immunofluorescence microscopy of normalappearing perilesional tissue from patients with MMP shows continuous deposits of immunoreactants in
964
epithelial basement membranes.1,3,5,16,52 The most commonly detected immunoreactants are IgG and C3 (see Fig. 55-6); the predominant subclass of these autoantibodies is IgG4.53 In situ deposits of IgA, IgM, and/ or fibrin are found in some patients.54 Multiple biopsies may be required to demonstrate in situ deposits of immunoreactants in some patients. One study comparing direct immunofluorescence microscopy findings on skin and mucosal samples from 10 patients found immunoreactants more commonly in perilesional mucosal biopsy specimens, suggesting that mucous membranes are the preferred biopsy site for direct immunofluorescence microscopy studies in these patients.52 Subsequent studies showed that splitting tissue samples with 1 M NaCl increases the sensitivity of direct immunofluorescence microscopy and facilitates identification of immunoreactants as well as their relative distribution within epithelial basement membranes.55,56
Indirect immunofluorescence microscopy studies using intact skin or mucosa often find low-titer IgG (and/or IgA) anti–basement membrane autoantibodies in the blood of patients with MMP.1,5,16,52,57
The use of 1 M NaCl split-skin as a test substrate in these studies substantially increases the detection of such autoantibodies.58-61 In such studies, IgG (and/or IgA) binding is usually directed against the epidermal side of 1 M NaCl split-skin, although combined epidermal and dermal or exclusively dermal binding can occur. In fact, this heterogeneity in autoantibody binding patterns was one of the first clues that MMP is a disease phenotype that is associated with different autoantigens (see Table 55-2). Although some studies suggest that the use of human mucosal tissue substrates increases the likelihood of detecting autoantibodies in patients with MMP by indirect immunofluorescence microscopy, other studies have not obtained such results.4,52 Patients with both IgG and IgA anti–basement membrane autoantibodies appear to have a worse prognosis as defined by requirements for medications to control disease as well as overall clinical severity score.45,47
OTHER IMMUNOPATHOLOGY STUDIES
OTHER
IMMUNOPATHOLOGY
STUDIES
Selected cases may require specialized immunochemical studies (eg, immunoblot studies of keratinocyte or skin extracts, immunoprecipitation studies of biosynthetically radiolabeled keratinocytes) to identify the autoantigen targeted by circulating anti–basement membrane autoantibodies from patients with MMP. Perilesional tissue from seronegative patients may be further characterized by immunoelectron microscopy to determine if in situ deposits of immunoreactants reside above or below the lamina densa of epidermal basement membrane.
DIAGNOSTIC ALGORITHM
DIAGNOSTIC ALGORITHM
The diagnosis of MMP should be based on alignment of all clinical, histopathologic, and/or immunopathologic findings.
DIFFERENTIAL DIAGNOSIS
The diagnosis of MMP is suggested when patients present with erosive lesions of mucous membranes, subepidermal bullae, and continuous deposits of immunoreactants in epithelial basement membranes of perilesional tissue. Participants in a 2002 international consensus conference concluded that clinical features and direct immunofluorescence microscopy features are essential findings that should be demonstrated before the diagnosis of MMP is assigned.1
Distinguishing MMP from other autoimmune bullous diseases can be difficult and may require specialized immunopathologic studies. The differential diagnosis of MMP includes other immunobullous diseases (eg, pemphigus vulgaris, paraneoplastic pemphigus, bullous pemphigoid, linear IgA dermatosis, epidermolysis bullosa acquisita), erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug reactions (ie, hypersensitivity reactions or chemotherapy-induced mucositis), lichen planus, and lupus erythematosus (Table 55-3). In respect to early disease in the mouth, MMP may resemble pemphigus vulgaris, paraneoplastic pemphigus, erosive lichen planus, erythema multiforme, Stevens-Johnson syndrome, or early toxic epidermal necrolysis; in
Most Likely
■Pemphigus
■Pemphigus vulgaris
■Paraneoplastic pemphigus
■Other subepidermal immunobullous diseases
■Epidermolysis bullosa acquisita
■Bullous pemphigoid
■Linear immunoglobulin A dermatosis
■Erythema multiforme
■Lupus erythematosus
■Lichen planus
Consider
■Drug-induced hypersensitivity reactions
■Lichen sclerosus (especially in the anogenital area)
Always Rule Out
■Pemphigus (specifically, pemphigus vulgaris, paraneoplastic pemphigus)
■Pemphigus (specifically, pemphigus vulgaris, paraneoplastic
pemphigus)
■Other subepidermal immunobullous diseases
■Other subepidermal immunobullous diseases
■Erythema multiforme
■Erythema multiforme
■Lupus erythematosus
■Lupus erythematosus
■Lichen planus
■Lichen planus
9
respect to early disease in the genital region, MMP may resemble pemphigus vulgaris, bullous pemphigoid, erosive lichen planus, or lichen sclerosus. In the case of ocular disease, cicatrizing or inflammatory conjunctivitis resembling MMP can result from longterm use of certain ophthalmologic preparations (eg, pilocarpine, guanethidine, or ephedrine used in the treatment of glaucoma or idoxuridine used as an antiviral) or exposure to selected biologics (eg, epidermal growth factor receptor tyrosine kinase inhibitors).5,18,39
It has also been demonstrated that some cases of ocular MMP can develop after severe ocular inflammatory injury caused by Stevens-Johnson syndrome or toxic epidermal necrolysis.5,62 Interestingly, the time between the development of Stevens-Johnson syndrome and the onset of ocular MMP in these patients ranges from a few months to more than 30 years. Finally, rare patients with cicatrizing conjunctivitis who lack in situ deposits of immunoreactants in conjunctival or epidermal basement membranes are sometimes encountered. Although immunopathology studies in such patients are negative, because their disease is inflammatory and sight-threatening, they are often treated with immunosuppressive agents as if they have MMP.
CLINICAL COURSE AND PROGNOSIS
MMP is typically a chronic disease. Involvement may be limited to a given anatomic site (eg, the mouth, the eyes) for years; some cases show progressive involvement of other mucosae. Even localized involvement can have a major negative impact on quality of life. In many ways, sites of involvement significantly influence the prognosis of MMP. Uncontrolled or aggressive disease in the eyes, hypopharynx, esophagus, or genital regions can compromise vision, respiration, swallowing, and genitourinary function. MMP rarely goes into spontaneous remission; its treatment is largely determined by its severity and sites of involvement. Scarring can only be prevented in these patients; it cannot be reversed. As noted above, some reports have suggested that patients with high titers and/or dual isotypes of anti–basement membrane autoantibodies (ie, both IgG and IgA anti–basement membrane autoantibodies) tend to have more severe and/or persistent disease.
MANAGEMENT
The primary goal of treatment of MMP is to halt progression of disease and prevent tissue inflammation, destruction, and scarring. Treatment of MMP is largely determined by its site(s) of involvement, its relative severity, and its rate of progression. Treatment regimens are largely derived from clinical experience rather than clinical trials; virtually all treatment interventions are “off-label.”63-65 All patients with MMP require long-term followup because of the possibility for this chronic disease to relapse.
965
9
LOCAL CARE MEASURES
LOCAL CARE MEASURES
Mild Involvement
SITES LOCAL CARE MEASURES
patient compliance with oral hygiene measures. Some sources recommend use of topical anesthetics for painful intraoral lesions in patients with MMP. Such agents should be applied carefully and locally (ie, not as a “rinse”) before meals, brushing teeth, dental procedures, or other occasions when pain control is required. Care should be taken to avoid introduction of topical anesthetics into pharyngeal, hypopharyngeal, or laryngeal areas to diminish the likelihood of aspiration. Medicated mouthwashes (eg, dexamethasone 100 mcg/mL, 5 mL per rinse) used in a “swishand-spit” regimen for 5 minutes 2 to 3 times each day represents another approach for topical therapy. For oral disease resistant to topical glucocorticoids, these agents can (in some instances) be administered intralesionally. Comanagement of such patients with a dentist familiar with MMP is key. Because of potentially severe complications, ocular, laryngeal, esophageal, and/or anogenital involvement requires aggressive management by teams of physicians familiar with specialized care of these organ systems.1,5,16,64,65 Early and regular evaluation by an ophthalmologist experienced in the management of patients with MMP is also important. Local eye care measures include prompt identification of ocular inflammation and management of eyelid hygiene, ocular lubrication, and secondary ocular infections (infections are more common in patients undergoing treatment with immunosuppressive drugs than in other patients). Patients whose ocular disease is complicated by trichiasis (ie, misdirected eyelashes that grow inward toward the eye and damage the cornea, conjunctivae, or under surface of the eyelid) may benefit from epilation, although this decision is best made by an ophthalmologist. Patients with
Mouth Topical corticosteroid (gels or ointments) twice daily/4 times daily; topical corticosteroids under occlusion (eg, dental trays); topical calcineurin inhibitors; intralesional corticosteroids
Nose Irrigation with isotonic saline twice daily/thrice daily; nasal lubricants; topical corticosteroids (eg, via sprays, inhalers)
Anogenital region Topical corticosteroids; topical calcineurin inhibitors
Skin Topical corticosteroids; topical calcineurin inhibitors
Moderate Involvement
SITES THERAPEUTIC OPTIONS
Mouth, eyes, nose, larynx, esophagus, anogenital region
Local care measures outlined above plus dapsone 50-200 mg daily, prednisone 20-60 mg each morning, or both of these agents simultaneously
Severe Involvement
SITES THERAPEUTIC OPTIONS
Mouth, eyes, nose, larynx,
Local care measures outlined above plus prednisone (1 mg/kg each morning), intravenous immunoglobulin (2 g/kg
Mouth, eyes, nose, larynx, esophagus, anogenital region
Local care measures outlined above plus prednisone (1 mg/kg each morning), intravenous immunoglobulin (2 g/kg body weight over 2-3 days every 2-6 weeks for 4-6 months), or both of these agents simultaneously in conjunction with azathioprine (2-2.5 mg/kg/day), mycophenolate mofetil (1-2.5 g/day), cyclophosphamide (1-2 mg/kg/day), or rituximab (375 mg/m2 weekly × 4 then every 4-6 months as needed, or 1000 mg on days 1 and 15 and then 500 mg at month 6)
esophagus, anogenital region
body weight over 2-3 days every 2-6 weeks for 4-6 months), or both of these agents simultaneously in conjunction with azathioprine (2-2.5 mg/kg/day), mycophenolate mofetil (1-2.5 g/day), cyclophosphamide (1-2 mg/kg/day), or rituximab (375 mg/m2 weekly × 4 then every 4-6 months as needed, or 1000 mg on days 1 and 15 and then 500 mg at month 6)
966
nasal involvement benefit from irrigations, humidified environments, emollients, and topical corticosteroids (sprays or drops). Twice daily irrigation of nasal passages with saline or tap water delivered via bulb syringe, piston syringe, or other nasal irrigation devices removes crusts over mucosal erosions and diminishes the likelihood of secondary complications such as infection and/or bleeding. Irrigations also increase the relative efficacy of topical corticosteroids delivered to nasal passages by spray or drops. Use of intranasal emollients after irrigation of nasal passages limits reaccumulation of crusts. Esophageal involvement requires early diagnosis and aggressive medical management to avert esophageal dysfunction, gastroesophageal reflux, stricture formation, aspiration, laryngeal irritation, and/or bronchospastic pulmonary disease. Soft diets, agents to diminish gastroesophageal reflux and/or gastric acidity, and/or esophageal dilation may be required. Urologists and colorectal surgeons provide important care for the rare patients who develop anogenital lesions. Such patients may require local topical therapy, dilations, stool softeners, and/or evaluation of adjacent internal mucosal surfaces.
SYSTEMIC THERAPIES
SYSTEMIC THERAPIES
For patients with disease resistant to local therapy or of greater severity, treatment with systemic agents in combination with local care measures is indicated. A number of reports suggest that dapsone (50 to 200 mg by mouth daily) may be effective.70-73 Others have found that patients with MMP do not respond to this agent. Other systemic agents reported to be of benefit in patients with mild disease include sulfapyridine, minocycline, or a combination of tetracycline and niacinamide. For mild or moderate ocular involvement, systemic glucocorticoids (eg, 20 to 60 mg of prednisone by mouth each morning) alone or in conjunction with daily dapsone may be effective. For nonresponsive or severe disease affecting the ocular, pharyngeal, or urogenital epithelia, a combination of systemic glucocorticoids and an additional immunosuppressive is indicated.74-79 In such cases, azathioprine (2.0 to 2.5 mg/kg/day), mycophenolate mofetil (1.0 to 2.5 g/day), or cyclophosphamide (1 to 2 mg/kg/day) are often used in conjunction with daily prednisone (1 mg/kg/day). In this regimen, daily prednisone is tapered gradually over approximately 6 months, and the patient is maintained on the alternate agent alone for an additional 6 to 12 months before further reductions in therapy are pursued. Such combined regimens have had success in halting the progression of severe disease, limiting scarring, and producing longterm remissions. In an effort to avoid adverse effects and complications produced by prolonged treatment with immunosuppressive agents, some groups treat patients with intravenous Ig (ie, intravenous Ig 2 g/kg of body weight administered over 2 to 3 days every 2 to 6 weeks for 4 to 6 months).80-85 Biologic agents that antagonize tumor necrosis factor-α (eg, etanercept,
9
infliximab) have shown efficacy in selected patients with MMP.86 Increasing numbers of patients with MMP are being treated with daily corticosteroids (alone or in combination with the other immunosuppressive agents listed above) plus rituximab (1000 mg on days 1 and 15, 500 mg at month 6; or 375 mg/m2 weekly × 4 then every 4-6 months as needed).87-93
When MMP is stabilized, treatment should be tapered slowly (eg, over 6 to 12 months). Flares of disease justify reinstitution of systemic therapies. Some patients require long-term maintenance therapy. In selected cases, immunopathology studies (eg, indirect immunofluorescence microscopy studies of 1 M NaCl split-skin, autoantibody-specific enzyme-linked immunosorbent assays) can be used to determine if seropositive patients convert to a seronegative status following treatment—a laboratory observation that may identify patients who are better candidates for a reduction (or cessation) of therapy.
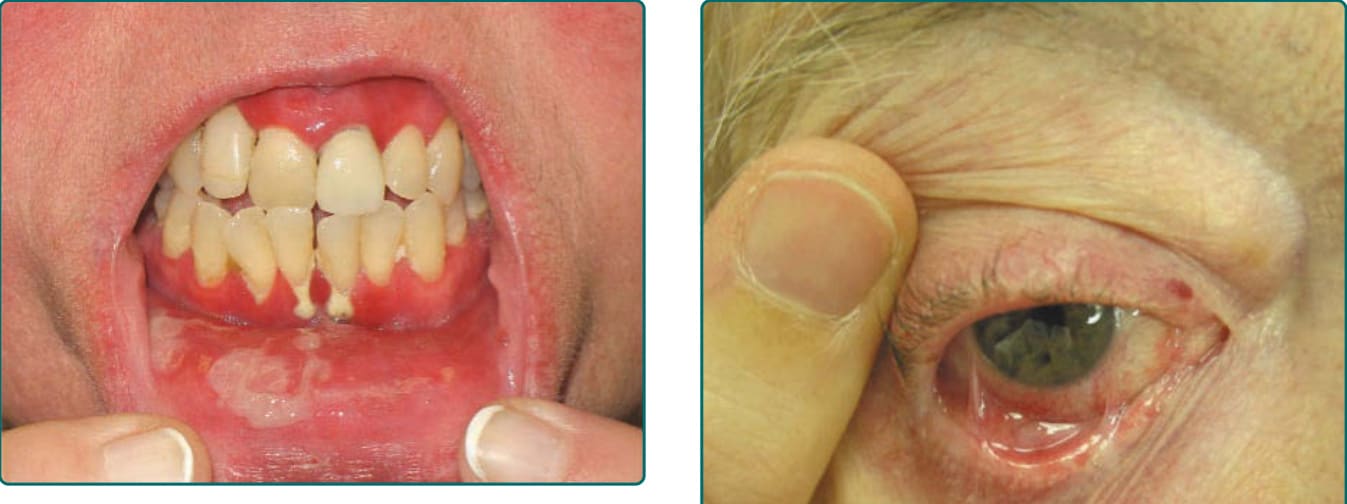
Figure 55-1 Denuded and inflamed sites on the oral mucosa are seen in association with sites of gingiva recession and loss.
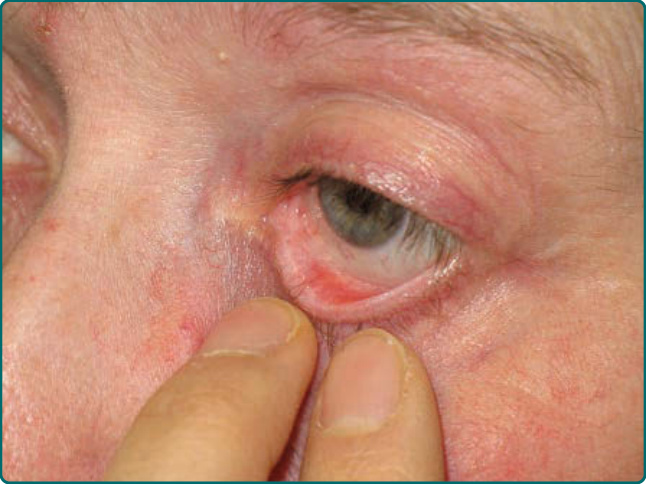
Figure 55-2 The medial aspects of the lower conjunctival fornix and eyelid show shortening, fibrosis, and malaligned eyelashes.
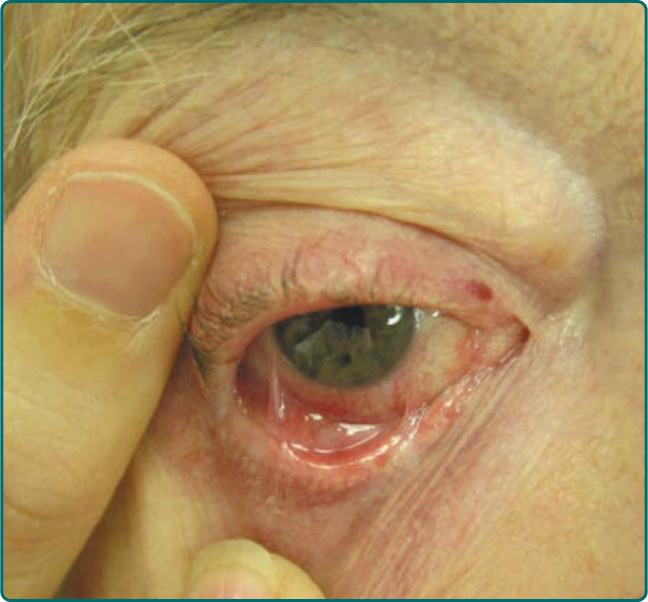
Figure 55-3 Ocular involvement has resulted in conjunctivitis, a shortened conjunctival fornix, and symblepharon formation.
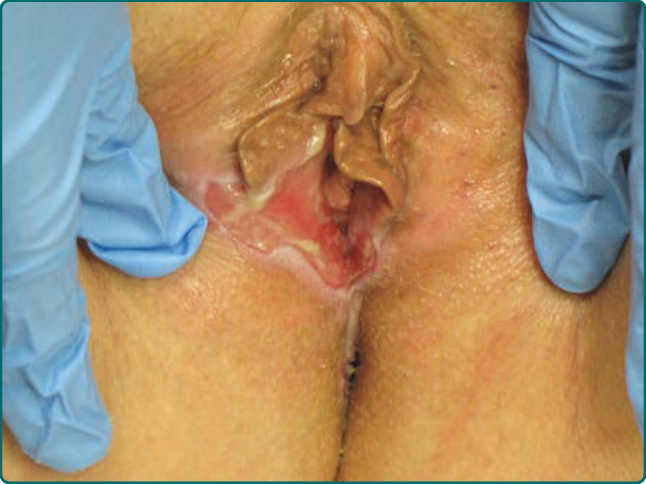
Figure 55-4 Scalloped erosions and sites of denuded vulvar and vaginal mucosae represent painful sites of disease in a patient with mucous membrane pemphigoid.
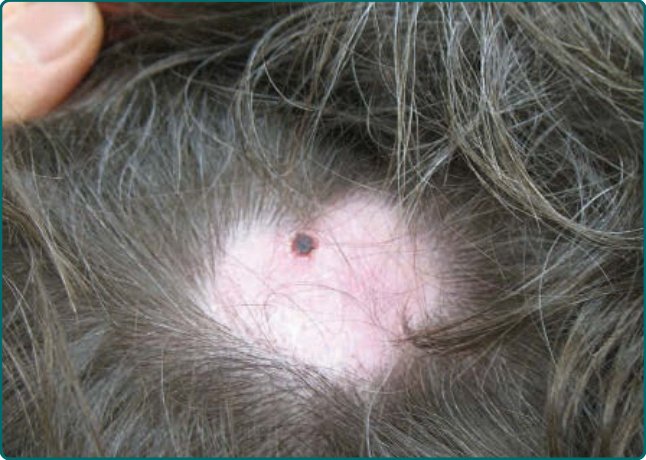
Figure 55-5 The scalp displays scarring alopecia and a focal hemorrhagic crust as a consequence of involvement with mucous membrane pemphigoid.
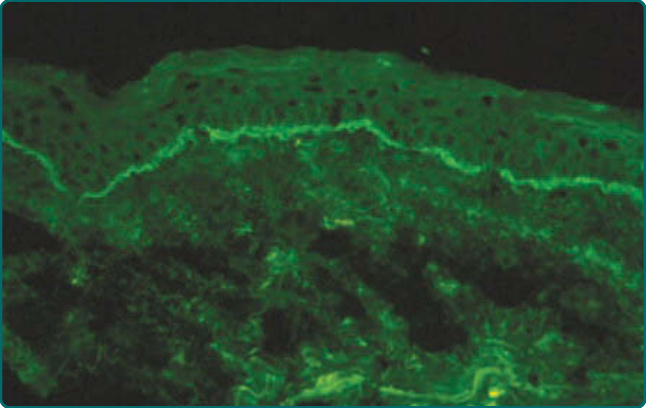
Figure 55-6 Direct immunofluorescence microscopy of normal-appearing perilesional skin from a patient with mucous membrane pemphigoid shows continuous linear deposits of C3 in the epidermal basement membrane.
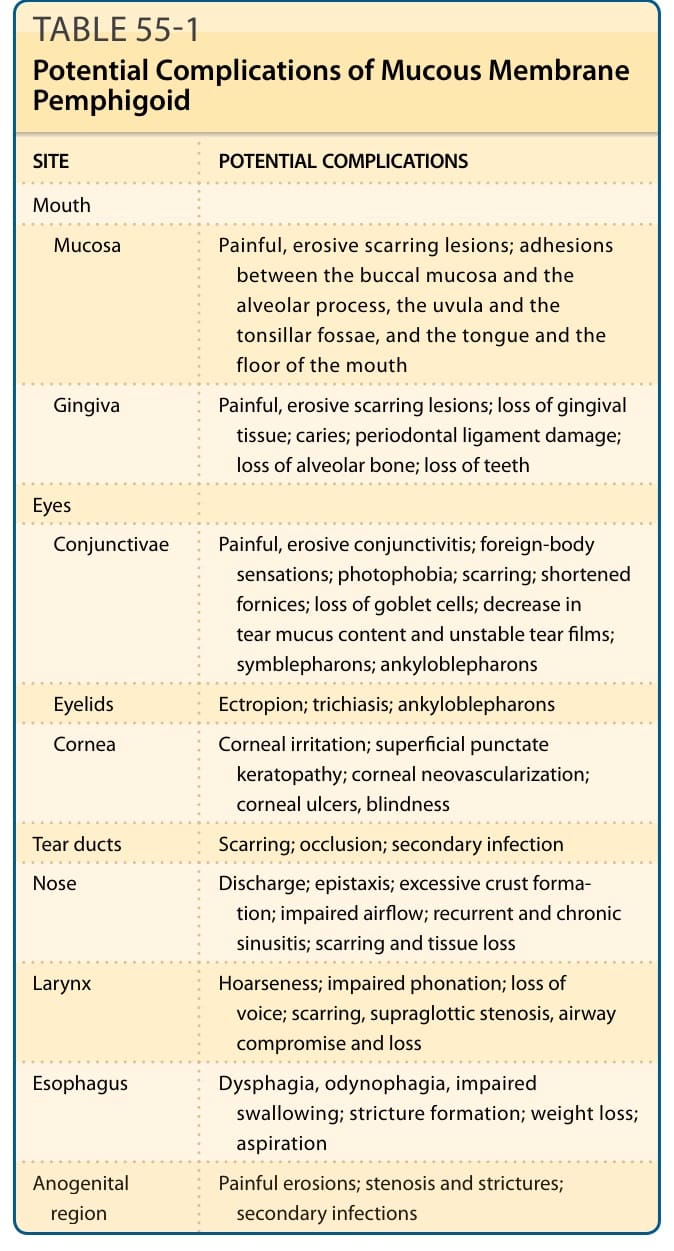
TABLE 55-1 Potential Complications of Mucous Membrane Pemphigoid

TABLE 55-2 Major Mucous Membrane Pemphigoid Autoantigens
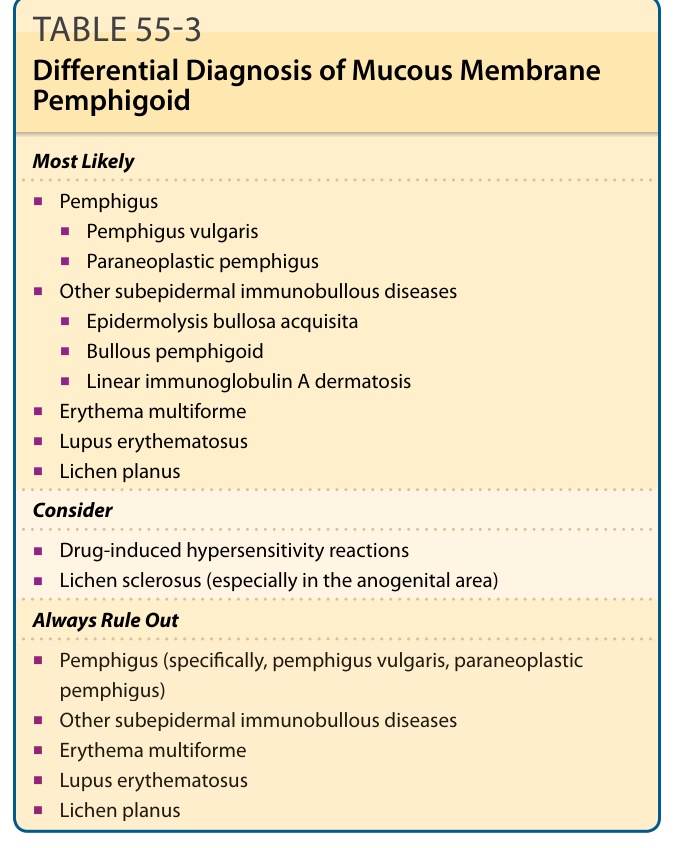
TABLE 55-3 Differential Diagnosis of Mucous Membrane Pemphigoid

Table 55-4 summarizes the treatment options for MMP. Mild lesions of the oral mucosa and skin can often be treated effectively with moderate- to high-potency topical glucocorticoids (or calcineurin inhibitors such as tacrolimus) in a gel or ointment base applied 2 to 4 times each day.64-67 Blotting lesional sites dry with a soft disposable tissue can enhance the adherence and effectiveness of topical agents applied to lesional sites in the mouth. These agents are particularly effective before bed, because oral secretions tend to diminish during sleep. These agents are easily applied, can be used widely on mucosal surfaces, and lack the grittiness encountered with agents formulated in dental pastes. Because it is difficult to maintain contact of topical corticosteroids with mucous membranes (and because lesions often are localized to the gingiva), customized delivery trays to occlude topical glucocorticoids over lesional sites in the mouth are also useful.64,65,68,69 Such delivery trays should be fashioned in soft vinyl and fitted to cover the patient’s gingiva (ie, not the patient’s teeth). Topical agents can be applied under occlusion in such trays for 10 to 20 minutes 1 to 4 times daily for 1 to 2 weeks then tapered (or interrupted for a break in therapy). Patients with intraoral involvement should follow a strict regimen of oral hygiene that includes brushing of the teeth 2 to 3 times each day along with daily flossing and regular dental cleanings 2 to 3 times each year. These interventions are essential to avert the accumulation of plaque, the loss of gingiva, and acceleration of tooth decay. Use of a pediatric toothbrush with soft bristles, toothpaste that lacks sodium lauryl sulfate, and mouthwashes free of alcohol facilitate