Mastocytosis
6
AT-A-GLANCE
MASTOCYTOSIS
■ The hallmark of mastocytosis is a pathologic accumulation of mast cells in tissues.
■ Mastocytosis occurs at any age but children are more commonly affected.
■ Cutaneous findings consist of hyperpigmented macules, papules, or nodules, or a diffuse infiltration of the dermis.
■ Most children only have skin involvement, whereas adults are more likely to have systemic disease.
■ Mastocytosis is usually associated with somatic activating mutations of c-kit with the 816 codon mutation being most common.
■ Many patients have few, if any, symptoms, but some experience varying degrees of flushing, pruritus, hypotension, nausea, dyspepsia, and diarrhea.
■ Most common extracutaneous tissues involved are the bone marrow, liver, spleen, and lymph nodes.
■ Some patients may develop an associated myeloproliferative or myelodysplastic disorder.
■ No effective cure currently exists; treatment is focused on controlling symptoms
MAST CELLS
■ Mast cells are derived from pluripotent stem cells.
■ Stem cell factor is the ligand for KIT and is required for mast cell proliferation and survival.
■ Mast cells release both preformed and newly generated mediators.
EPIDEMIOLOGY
Mastocytosis represents a group of disorders characterized by an abnormal accumulation of mast cells in one or more organs. Although the true incidence of this disease is unknown, most cases arise in children, with approximately 70% of cases occurring by 6 months of age and more than 90% occurring within the first 2 years of life.1-5 Congenital mastocytosis is less common, representing approximately 18% to 31% of childhood cases.2-4 The prevalence of childhoodonset mastocytosis is reported to range from 2.0 to 5.4 cases/1000 population.1,2 The prevalence of adultonset systemic mastocytosis (SM) in Denmark has been reported to be approximately 1/10,000 population over a 14-year period.6 Mastocytosis has no gender preference, and it has been reported in all races.5
While most mastocytosis patients have no family history, there are reports of more than 70 familial cases, including monozygotic twins, some of which were discordant for this disease.7-9
PATHOGENESIS
Mast cells arise from the bone marrow as agranular, undifferentiated, CD34+, KIT+ (CD117) pluripotent progenitor cells. After migrating into tissues, immature mast cells assume their typical granular morphology.10
KIT, a Type III tyrosine kinase is the product of the protooncogene c-kit located on chromosome 4q12. This enzyme is expressed on mast cells, as well as melanocytes, primitive hematopoietic stem cells, primordial germ cells, and interstitial cells of Cajal and serves as the receptor for its ligand, stem cell factor (SCF). Crosslinking of KIT by SCF is essential for mast cell maturation. The gene for SCF is located on chromosome 12, and encodes a protein that localizes to the cell membrane.10,11 Membrane-bound and a soluble forms of SCF exist, both of which are capable of inducing KIT activation. Soluble KIT is thought to arise from chymase-induced cleavage of the membrane bound form.12 SCF is produced by bone marrow stromal cells, fibroblasts, keratinocytes, endothelial cells, as well as reproductive Sertoli and granulosa cells. Mature mast cells require SCF for survival.11,12 Other cytokines that appear important in regulating mast cell growth and differentiation include interleukin-3 (IL-3), IL-4, IL-6, and IL-9 and interferon (IFN)-γ. IL-3 shares a number of signal transduction pathways with SCF, but has minimal direct effects on human mast cell proliferation except in early cultures. IL-4 enhances mast cell function when added to mature cultures. IL-6 increases mast cell mediator concentration13 and IL-9 appears to increase the number of mast cells in culture.14 In developing mast cells, IFN-γ inhibits mast cell proliferation and influences mast cell phenotype and function.13
Mutations in c-kit appear to play a central role in mastocytosis. Somatic mutations in codon 816 of c-kit, lead to amino acid substitutions (D816V, D816Y, D816F, D816I, and D816H), and cause constitutive activation of KIT, resulting in continued mast cell growth and development.15,16 Mutations in this codon are most common in adults with mastocytosis, but also occur in childhood onset disease.2,15,17 Among 50 children with this disorder, 42% had detectable mutations at codon 816 (exon 17), while another 42% had activating mutations in the extracellular and transmembrane regions of KIT (exons 8-11). The remaining 8 children reported in this study had no detectable c-kit mutations. There was no clear correlation between the extent of disease
Activating Mutations D816V, D816Y, D816F, D816I, D816H, S849I, D835Y, N822I, D820G, V560G, V559I, A53D, D572A, F522C, K509I, S476I, C443Y, D419Y, del417-418, ITD 501-502, ITD 502-503, ITD 505-508 Inactivating Mutations E839K, del exon 3-13 Increased Sensitivity to Stem Cell Factor M541L
Activating Mutations D816V, D816Y, D816F, D816I, D816H, S849I, D835Y, N822I, D820G,
V560G, V559I, A53D, D572A, F522C, K509I, S476I, C443Y, D419Y, del417-418, ITD 501-502, ITD 502-503, ITD 505-508 Inactivating Mutations E839K, del exon 3-13 Increased Sensitivity to Stem Cell Factor M541L
and the presence or absence of c-kit mutations among these 50 patients.17 In another report of 22 children and adults with mastocytosis, 11 adult patients and 4 children had 816 activating mutations. Four other pediatric patients with typical lesions of urticaria pigmentosa (UP) lacked changes in this codon, and in 3 other children, an inactivating c-kit mutation (codon E839K) was detected.15 Less common activating c-kit mutations, such asV560G, also have been characterized in some adult patients with mastocytosis,15,16 as have severely truncated, inactive KITs (Table 42-1).18
A transgenic mouse model of mastocytosis has been reported using the human activating D816V c-kit mutation.19 The clinical expression of this disorder ranged from indolent mast cell hyperplasia to invasive mast cell tumors, even though genetically identical animals expressed the same D816V mutation. These clinical and investigative observations indicate that c-kit activating mutations play an important role in the development of mastocytosis. However, the finding of a varied clinical expression of mastocytosis in an animal model along with the fact that some children and adults with mastocytosis have no c-kit mutations
6
or have inactivating c-kit mutations, strongly suggests that other, yet to be defined, factors influence the full clinical expression of this disease. Indeed, mutations in addition to c-kit have been identified in some adults with SM. These include the tumor-suppressor gene TET2, was well as ASXL1, JAK2, SRSF2, DNMT3A, RUNX1, and CBL, all of which may play a role in persistent and more aggressive disease.20,21
CLINICAL FINDINGS
CLASSIFICATION
CLASSIFICATION
Mastocytosis represents a disease spectrum characterized by a diverse phenotypic expression, along with pathologic and genetic findings that affect prognosis. Table 42-2 represents the World Health Organization (WHO) classification of mastocytosis.5 Cutaneous mastocytosis (CM) and indolent SM (ISM) represent the majority of patients with this disease. Most children have CM, which is manifested as UP (Fig. 42-1) or, less commonly, as mastocytomas (Fig. 42-2), or as diffuse CM (Fig. 42-3). SM occurs mostly in adults, and ISM is the most common form. Table 42-3 lists the criteria for SM. SM with an associated clonal hematologic non–mast-cell-lineage disease (SM-AHNMD) occurs almost uniquely in adults, and can be demonstrated, by examining the peripheral blood and bone marrow. Hematologic disorders associated with SM-AHNMD include myeloproliferative and myelodysplastic disorders, such as polycythemia rubra vera, chronic myeloid leukemia, chronic myelomonocytic leukemia, idiopathic myelofibrosis, chronic eosinophilic leukemia and the hypereosinophilic syndrome, acute and chronic lymphocytic leukemia, and non-Hodgkin
VARIANT TERM ABBREVIATION SUBVARIANTS EXAMPLES
Cutaneous mastocytosis
CM Urticaria pigmentosa Diffuse CM Mastocytoma of skin Telangiectasia macularis eruptiva perstans (TMEP)
Indolent systemic mastocytosis
ISM Smoldering SM Isolated bone marrow mastocytosis
Systemic mastocytosis with an associated clonal hematologic non–mast-celllineage disease
SM-AHNMD Acute myelogenous leukemia Myelodysplastic syndrome Chronic myelomonocytic leukemia Chronic eosinophilic leukemia Non-Hodgkin lymphoma
Aggressive systemic mastocytosis ASM Acute vs chronic
Mast cell leukemia MCL
Mast cell sarcoma MCS
Extracutaneous mastocytoma ECM
Extracutaneous mastocytoma ECM
aFor details of the WHO classification of mastocytosis, see Valent P, et al.5
711
6
712
Major
Major
■Multifocal dense infiltrates of mast cells in bone marrow and/or other extracutaneous organs Minor
■Multifocal dense infiltrates of mast cells in bone marrow and/or
other extracutaneous organs Minor
■More than 25% of the mast cells in bone marrow aspirate smears or tissue biopsy sections are spindle shaped or display atypical morphology.
■More than 25% of the mast cells in bone marrow aspirate smears
or tissue biopsy sections are spindle shaped or display atypical morphology.
■Detection of a codon 816 c-kit point mutation in blood, bone marrow, or lesional tissue.
■Detection of a codon 816 c-kit point mutation in blood, bone t marrow, or lesional tissue.
■Mast cells in bone marrow, blood, or other lesional tissue expressing CD25 or CD2.
■Mast cells in bone marrow, blood, or other lesional tissue express-
ing CD25 or CD2.
■Baseline total tryptase level greater than 20 ng/mL.
■Baseline total tryptase level greater than 20 ng/mL.
aFor details of the WHO classification of mastocytosis, see Valent P, et al.5
and Hodgkin lymphoma. Patients with aggressive SM (ASM) frequently have evidence of impaired liver function, hypersplenism, and/or malabsorption, but they do not have a distinctive hematologic disorder or mast cell leukemia (MCL). Patients with ASM have rapidly increasing mast cell numbers and are difficult to manage medically.5,22 MCL is rare and characterized by multiorgan failure, bone marrow smears demonstrating a greater than 20% mast cell population among the nucleated cell population. In the peripheral blood, mast cells are at least 10% of nucleated cells.5,23 Both acute and chronic forms of MCL have been described; they are differentiated based on symptoms and organ involvement.23 Mast cell sarcomas are rare; they have localized destructive growth, but distant spread is possible. Mast cells in these tumors are highly atypical and immature. Extracutaneous mastocytomas are extremely rare, usually localized to the lung, and consist of mature mast cells.5,22,24,25
CUTANEOUS LESIONS
CUTANEOUS LESIONS
UP was originally described in children and is the most common skin manifestation of childhood-onset CM. UP lesions in children appear as tan to brown papules and, less commonly, as macules, ranging in size from 1.0 to 2.5 cm in diameter (see Fig. 42-1). These lesions may be present at birth or arise during infancy. They frequently appear on the trunk and often spare the central face, scalp, palms, and soles. Cutaneous lesions in adults, on the other hand, are much smaller (approximately 5 mm or less in diameter) reddish-brown macules and papules (Fig. 42-4). On close lesion inspection, variable hyperpigmentation and fine telangiectasias are detectable. In patients with Type I skin, these lesions may lack pigment and appear pink to red. Adult skin lesions are most numerous on the trunk and proximal extremities and appear less frequently on the face, distal extremities, or palms and soles. While adult lesions may appear and resolve
A B
6
over months in individual patients, their number usually increases over years. Most adults with ISM have cutaneous lesions, but skin involvement is less common in patients with more advanced forms of SM (SM-AHNMD, ASM, or MCL).5,22,24,25
Solitary mastocytomas are tan-brown nodules that occur in approximately 10% to 35% of children and frequently appear on the distal extremities (see Fig. 42-2). Their onset is generally before 6 months of age.1-4 Scratching or rubbing skin lesions leads to urtication and erythema known as Darier sign. This reaction is readily demonstrated in childhood UP and mastocytomas, but often is less pronounced in adult skin lesions.24-27 The difference in lesional skin reactivity between children and adults with CM is best explained by the observation that mast cell concentrations in mastocytomas and childhood UP are 150-fold and 40-fold greater than in normal skin, respectively, whereas the mast cell content in lesions of adult mastocytosis is only 8 times greater than normal controls.27
Because trauma to mastocytomas is associated with systemic symptoms such as flushing and hypotension, vigorous scratching of UP lesions or mastocytomas is not advised.1-4,24,26
Diffuse CM (DCM) is seen almost exclusively in infants (see Fig. 42-3), although it may persist into adult life. The skin often has a peau d’orange appearance and yellowish-brown discoloration. Rarely, individual yellow-tan papules have been described; the individual yellow-tan papules are called xanthelasmoidal mastocytosis.26,28 Dermographism with formation of hemorrhagic blisters can occur in the first months to years of life in children with DCM, but these blisters often resolve within several years, even though the DCM may persist (Fig. 42-5).1-4,26 Telangiectasia macularis eruptiva perstans is rare, and is seen almost exclusively in adults. It appears as telangiectatic macules and patches without hyperpigmentation (Fig. 42-6). The Darier sign and pruritus are variable in patients with telangiectasia macularis eruptiva perstans.24-26 Tumor-like growths also have been described in
an adult mastocytosis patient in whose mast cells expressed the V560G c-kit mutation.29
RELATED CLINICAL FINDINGS
RELATED CLINICAL FINDINGS
Most children with CM and adults with CM or ISM have few, if any, symptoms, but children with DCM are more likely to have skin and GI symptoms.1,2 When symptoms occur, they are caused by the release of mast cell mediators, such as histamine, eicosanoids, and cytokines, and often involve several different organs (ie, skin and GI tract or skin and cardiovascular) (Table 42-4). Symptoms and signs of mastocytosis may include: pruritus, flushing, diarrhea, abdominal pain, nausea, bloating, vomiting, gastric reflux, palpitations, dizziness, and/or syncope. Of interest is the relative absence of pulmonary symptoms in mastocytosis patients. Complaints of fever, night sweats, malaise, weight loss, bone pain, epigastric distress, and problems with mentation (cognitive disorganization) often
713
6
signal the presence of SM. Symptoms of mastocytosis can be exacerbated by exercise, heat, or local trauma to skin lesions (childhood UP and mastocytomas). In addition, alcohol, narcotics, salicylates, nonsteroidal antiinflammatory drugs (NSAIDs), polymyxin B, anticholinergics, and some systemic anesthetic agents may induce mast cell mediator release.5,23-26 It has been
suggested that anaphylaxis from hymenoptera stings may be more common in mastocytosis patients, but deaths associated with extensive mast cell mediator release are rare.2,5,30
Extracutaneous disease is extremely uncommon in children, but when present GI symptoms are the most frequent.1,2,4 GI symptoms in adults ranges from 25% to 70%.5,31,32 Diarrhea is the most common symptom and may result from gastric hypersecretion, increased motility, and/or malabsorption. Increased gastric acid secretion is likely a result of elevated histamine release, which may cause gastritis and peptic ulcer disease.33
Malabsorption with associated diarrhea is limited to a subset of patients with usually more advanced disease. While hepatic disease in adults and children with ISM is rare, liver and spleen involvement, including portal hypertension and ascites associated with liver fibrosis, may occur in patients who have SM-AHNMD or ASM.5,24,25,34 Splenomegaly, detected either clinically or by CT scan, has been reported in 50% to 60% of adult SM patients.26,35 However, in 2 other studies, each with more than 140 adult mastocytosis patients, splenomegaly was observed in only 8% to 9% of cases.36,37 Increased numbers of mast cells and eosinophils are frequently observed in the spleen, as are
MEDIATORS BIOLOGIC EFFECTS POSSIBLE CONSEQUENCES
Preformed
Histamine Vasodilation, increased vascular permeability, gastric hypersecretion, bronchoconstriction Hypotension, flushing, urticaria, abdominal pain (peptic, colic), nausea, vomiting, diarrhea, malabsorption CNS symptoms
Heparin Anticoagulant, inhibition of platelet aggregation Prolonged bleeding time Osteoporosis/osteopenia
Tryptase Endothelial cell activation, fibrinogen cleavage, mitogenic for smooth muscle cells Osteoporosis/osteopenia, disruption of cascade systems (clotting, etc)
Chymase
Converts angiotensin I to II, lipoprotein degradation
Cleaves membrane-bound stem cell factor
Newly Synthesized
Hypertension Hyperpigmentation Skin mast cell accumulation (?)
Leukotrienes Increased vascular permeability, bronchoconstriction, vasoconstriction Bronchospasm, hypotension
Prostaglandins Vasodilation, bronchoconstriction Flushing, urticaria, hypotension CNS symptoms
Cytokines
Stem cell factor Growth and survival of mast cells, chemotaxis of KIT+ cells, melanogenesis Mast cell hyperplasia, focal aggregates
Tumor necrosis factor-α Activation of vascular endothelial cells, cachexia, fatigue Weight loss, fever, fatigue
Transforming growth factor-β Enhanced production of connective tissue components Fibrosis
Interleukin (IL)-1 Activates fibroblasts, lymphocytes and increases neutrophils Fibrosis, fever
IL-5 Eosinophil growth factor Eosinophilia
IL-6 Growth and survival of mast cells Fever, bone pain, osteoporosis/osteopenia
714
Granulocyte-macrophage
Granulocyte and monocyte growth and activation Eosinophilia
Granulocyte-macrophage colony-stimulating factor Granulocyte and monocyte growth and activation Eosinophilia
colony-stimulating factor
varying degrees of fibrosis and hematopoiesis. Lymph node enlargement is uncommon in most patients with ISM, but occurs in patients with more advanced disease. Among 58 SM patients, 15 (26%) had peripheral lymphadenopathy, whereas 11 (19%) had central nodal disease.38 Anemia, leukopenia, thrombocytopenia, and eosinophilia also may occur in association with SM and suggest SM-AHNMD or ASM.5,22,24,25
Musculoskeletal pain affects 19% to 28% of mastocytosis patients. Skeletal lesions occur in patients with SM, but are rare in children with this disorder.1,2,36,39,40 In one large study of 142 adults with mastocytosis, 40% had skeletal involvement.36 Bony lesions may appear as radiopacities, radiolucencies, or a mixture of the two. The skull, spine, and pelvis are most commonly involved. In another study of 58 adult SM patients, 34 (57%) had diffuse bone involvement, whereas only 1(2%) had focal lesions.39 In 199 ISM patients, disease-related lumbar spine osteoporosis was more frequently observed in men. The prevalence of vertebral fractures was 20% in men compared to 14% in women with mastocytosis. This observation has led to the recommendation to consider the diagnosis of mastocytosis in men with unexplained osteroporosis.40
Neuropsychiatric abnormalities have been reported, and include decreased attention span, memory impairment, headache, and irritability. Depression as a consequence of chronic disease or possibly mediated by mast cell mediators may occur.41,42
Information pertaining to pregnancy in mastocytosis is limited. However, there are numerous reports of successful, full-term deliveries in women with mast cell disease.43,44 In a report of 45 pregnancies in 30 mastocytosis patients, 10 (22%) had worsening mast cell–related symptoms, whereas 15 (33%) experienced improved or resolved symptoms. Among the 45 newborns, only 7 were premature, had a low birthweight and/or suffered from respiratory distress. No fatal maternal or fetal outcomes were reported.45
LABORATORY TESTS
The diagnosis of mastocytosis is established by demonstrating increased numbers of mast cells in 1 or more organs. For patients with CM, mast cell infiltrates can be detected in a biopsy of lesional skin using special stains, such as toluidine (Fig. 42-7), Giemsa, or monoclonal antibodies that recognize mast cell tryptase or CD117 (KIT).26,27,46 Biopsy specimens of normalappearing skin from patients with mastocytosis have normal concentrations of mast cells as determined by morphometrics, and thus are not helpful in establishing the diagnosis.27
Detection of circulating mast cell mediators and/or their metabolites can offer indirect evidence of mastocytosis, and their measurement may be useful in patients without cutaneous lesions. Two forms (α and β) of mast cell tryptase have been identified. α-Tryptase is elevated in patients with SM, with or without acute symptoms, whereas increased levels of β-tryptase can be detected both in mastocytosis patients and in allergic patients experiencing anaphylactic reactions.
6
Total (α + β) serum tryptase levels are most commonly measured and correlate with the extent of mast cell disease.47,48 In children, elevated serum tryptase levels correlate with more-extensive skin involvement and a greater number and severity of symptoms.48 Of adult mastocytosis patients with total serum tryptase levels between 20 and 75 ng/mL, 50% had evidence of SM, whereas all patients with levels >75 ng/mL had proven systemic involvement.47 Total serum tryptase levels of >20 ng/mL are currently considered abnormal and represent one of the minor criteria for SM (see Table 42-3).5 Total serum tryptase levels also can provide an estimate of a patient’s overall mast cell burden, and thus serial measurements in adults every 6 to 12 months may prove useful in following disease progression. Determinations of urinary histamine metabolite levels may be worthwhile as a diagnostic test in patients without cutaneous lesions and in whom the diagnosis of mastocytosis is unclear. The major urinary metabolite of histamine, 1,4-methylimidazole acetic acid is often persistently elevated in SM patients and correlates with the extent of mast cell disease. Methylhistamine is next most common urinary histamine metabolite, and can be measured if 1,4-methylimidazole acetic acid levels are not available in commercial laboratories. Certain foods high in histamine content, such as spinach, eggplant, cheeses (eg, Parmesan, Roquefort, and blue), and red wines, can artificially elevate the levels of urinary histamine and its metabolites, and thus should be avoided during the collection process.49
Mast cells can induce bone changes that cause radiographically detectable lesions. Skeletal lesions occur more frequently in adult patients with SM, and are rare in children with this disorder.1,26,36,40 In one large study of 142 adults with mastocytosis, 57 (40%) had skeletal involvement.36 The proximal long bones, skull, spine, ribs, and pelvis are most commonly affected. Skeletal scintigraphy (bone scan) is more sensitive, but less specific, than routine radiographs for detecting and locating active lesions. Thus, radiographs of the skull, spine, and pelvis serve as a reasonable preliminary
715
6
test for detecting bone involvement in mastocytosis patients.36,40
The bone marrow is often involved in patients with SM, and bone marrow biopsies are indicated for patients suspected of having more advanced disease (SM-AHNMD, ASM, MCL).5,22 In a report of 71 adults with mastocytosis, 90% had increased numbers of spindle-shaped, bone marrow mast cells with focal perivascular, peritrabecular, and/or intertrabecular accumulations.26 WHO criteria for SM have been defined and include the bone marrow findings of multifocal dense mast cell aggregates, atypical mast cell morphology, and the expression of CD2 and/or CD25 by bone marrow mast cells (see Table 42-3). A bone marrow biopsy in childhood-onset disease is not recommended unless there is evidence of systemic involvement as demonstrated by hepatosplenomegaly, lymphadenopathy, and/or unexplained peripheral blood abnormalities.1,2,50 Mast cells in bone marrow biopsies are best identified by immunostaining with antitryptase (Fig. 42-8) or CD117 monoclonal antibodies as decalcification interferes with the effectiveness of metachromatic stains.5,22
Roentgenographic abnormalities of the GI tract fall into 3 major categories: (a) peptic ulcers; (b) abnormal mucosal patterns such as mucosal edema, multiple nodular lesions, coarsened mucosal folds, or multiple polyps; and (c) motility disturbances. A biopsy of the GI tract may be indicated for patients in whom the diagnosis of SM is suspected, but who lack skin lesions. Histologic sections of jejunal biopsies show moderate blunting of the villi and may show increased mast cell numbers in association with variable numbers of eosinophils.22,26,31,51 Increased mast cells in bowel biopsies should always be correlated with other signs and symptoms of mastocytosis as these findings also have been reported in patients with irritable bowel syndrome and mast cell activation syndrome.51
Despite the infrequency of significant hepatic disease, liver function tests may be abnormal in up to 50% of SM patients.26,39 Elevated serum IL-6 levels, soluble
716
SCF receptor (CD117), and IL-2 receptor (CD25) levels are associated with SM, but their specificity is not established.52
The initial evaluation of a prepubertal child with mastocytosis generally does not require extensive testing if the history and physical examination do not indicate the presence of SM. In contrast, the presence of noncutaneous signs and symptoms or disease onset in adolescents or adults necessitates a complete blood count (CBC) with differential, liver function test, total serum tryptase level, and skeletal radiographs (Fig. 42-9 and see Table 42-4). While bone marrow biopsies have been recommended by some clinicians for all adult mastocytosis patients, it is the opinion of this author that this procedure is unnecessary for those with a normal CBC. Should the CBC become abnormal in an adult patient, however, then the cause of this abnormality should be investigated by examining the bone marrow.
DIFFERENTIAL DIAGNOSIS
Cutaneous lesions of childhood and adult mastocytosis are so characteristic, that they are rarely confused with other skin disorders. Because UP lesions may urticate, they could initially be mistaken for urticaria. However, individual urticaria lesions last only a few hours and lack the associated hyperpigmentation seen in UP. Rarely, nodular scabies lesions have been confused with UP. Some childhood mastocytosis patients, especially those with DCM, may develop bullae, and, thus, the differential diagnosis for blistering diseases in infants such as bullous impetigo, bullous arthropod bites, linear immunoglobulin A bullous dermatosis, bullous pemphigoid, epidermolysis bullosa, toxic epidermal necrolysis, and incontinentia pigmenti should be considered (Table 42-5). The differential diagnosis of mastocytomas in children includes juvenile xanthogranulomas, Spitz nevi, pseudolymphomas, or, rarely, in resolving lesions, a café-au-lait macule. Adult UP lesions might initially appear as lentigines or atypical melanocytic nevi; however, they usually have an associated erythema (telangiectasia) not seen in these melanocytic lesions. Mastocytosis should be suspected in patients without skin lesions if they have symptoms suggesting mast cell mediator release, and one or more of the following: hepatomegaly, splenomegaly, lymphadenopathy, and/or peripheral blood abnormalities as well as peptic ulcer disease, malabsorption, or unexplained osteoporosis or radiographic/bone scan abnormalities (Table 42-6). In the last few years, patients with nonclonal mast cell activation syndrome have been described. This condition is characterized by symptoms of mast cell mediator release, such as flushing, abdominal cramping, diarrhea, headache, and/or memory/concentration difficulities. However, these patients lack evidence of unregulated mast cell proliferation. Nonclonal mast cell activation syndrome has been reported more frequently in women who experience urticaria/angioedema, respiratory symptoms,
6
Algorithm for a diagnostic evaluation of new-onset mastocytosis
Mast cell mediator symptoms
Hyperpigmented papules, macules, or solitary nodule
Skin biopsy of lesion, CBC with diff LFTs, Serum tryptase level Skeletal radiographs
Elevated serum tryptase plus hepatosplenomegaly or hematologic, LFTs, bone abnormalities
Normal tryptase, CBC, no hepatosplenomegaly
Bone marrow biopsy
No skin findings plus unexplained ulcer or malabsorption hepatosplenomegaly, and/or skeletal or hematologic abnormalities
CBC with diff, LFTs, serum tryptase, 24-h urinary histamine metablolite levels, vasoactiveamines
Normal tryptase urinary histamine metabolite levels
Elevated tryptase and/or urinary histamine metabolites
CBC with differential, LFTs, skeletal radiographs, possible GI or bone marrow biopsy
Explore other diagnoses Repeat serum tryptase, CBC, LFTs annually
Abnormal bone marrow 1 major + 1 minor or 3 minor criteria
Normal Bone marrow follow clinically
Systemic mastocytosis WHO criteria for classification
Normal consider nonclonal mast cell activation syndrome
have drugs as a trigger for their symptoms, and total serum tryptase levels <15 ng/mL.53 In addition, these patients often respond to H1 and H2 antihistamines and leukotriene inhibitors.53,54 Nonclonal mast cell activation syndrome must be differentiated from the broader term mast cell activation disorder, which also includes various forms of mastocytosis.54 In patients with flushing, the diagnosis of a carcinoid tumor or pheochromocytoma should be considered. Mastocytosis patients, however, do not excrete increased amounts of 5-hydroxyindoleacetic acid, and patients with carcinoid tumor or pheochromocytoma do not have histologic evidence of mast cell proliferation or elevated serum tryptase levels.24-26
PROGNOSIS
Pediatric-onset cutaneous disease has a favorable prognosis with 45% to 68% of patients experiencing complete disease regression with a median followup
of 15 to 20 years.1,2 Children born of mothers with ISM reportedly are free of disease.42,44 It has been postulated that children with activating c-kit mutations may represent those patients whose disease persists into adulthood; however, activating c-kit mutations, including D816V, have been identified in up to 84% of children with mastocytosis.17 To date, there has been no correlation of disease severity or outcome in children who have c-kit mutations.2,17 Most adults with skin lesions of mastocytosis have only CM or ISM, and rarely develop more advanced disease having a life expectancy commensurate with the general population. In a study of 145 adults with ISM, the cumulative probabilities of developing more advanced mast cell disease at 10 and 25 years were 1.7% and 8.4%, respectively.37 Patients with ISM who have evidence of hepatosplenomegaly, lymphadenopathy and/or serum tryptase levels of >200 ng/mL have been referred to as smoldering SM. The prognosis of this group is not well defined but is believed to be less favorable than the ISM group.5,22,37 Older patients who experience
717
6
Most Likely
Most Likely
■Diffuse or localized red-brown papules/macules
■Diffuse or localized red-brown papules/macules
■Urticaria
■Urticaria
■Multiple nevi
■Multiple nevi
■Langerhans cell histiocytosis
■Langerhans cell histiocytosis
■Juvenile xanthogranulomas
■Juvenile xanthogranulomas
■Nodular scabies
■Nodular scabies
■Café-au-lait spots
■Café-au-lait spots
■Multiple cutaneous lentiginosis
■Multiple cutaneous lentiginosis
■Bullous lesions
■Bullous lesions
■Linear immunoglobulin A dermatosis
■Linear immunoglobulin A dermatosis
■Bullous impetigo
■Bullous impetigo
■Epidermolysis bullosa
■Epidermolysis bullosa
■Arthropod bite reaction
■Arthropod bite reaction
■Toxic epidermal necrolysis
■Toxic epidermal necrolysis
■Incontinentia pigmenti
■Incontinentia pigmenti
■Solitary papule or nodule
■Solitary papule or nodule
■Congenital nevus
■Congenital nevus
■Juvenile xanthogranuloma
■Juvenile xanthogranuloma
■Pseudolymphoma
■Pseudolymphoma
fading of their lesions continue to exhibit bone marrow lesions typical of their diagnosis, whether ISM or SM-AHNMD.55 Patients with SM-AHNMD have a variable course, which is dependent on the prognosis of their hematologic disorder. One study suggests that
Gastrointestinal
Gastrointestinal
■Peptic ulcer disease
■Peptic ulcer disease
■Ulcerative colitis
■Ulcerative colitis
■Gluten-sensitive enteropathy
■Gluten-sensitive enteropathy
■Hepatitis
■Hepatitis
■Parasitic disease Nonclonal Mast Cell Activation Syndrome
■Parasitic disease Nonclonal Mast Cell Activation Syndrome
■Cardiovascular
■Cardiovascular
■Idiopathic anaphylaxis
■Idiopathic anaphylaxis
■Intrinsic cardiac disease
■Intrinsic cardiac disease
■Endocrine
■Endocrine
■Adrenal tumor
■Adrenal tumor
■Vasoactive intestinal polypeptide tumor
■Vasoactive intestinal polypeptide tumor
■Carcinoid syndrome
■Carcinoid syndrome
■Medullary thyroid carcinoma
■Medullary thyroid carcinoma
■Gastrinoma Neoplastic/Oncologic
■Gastrinoma Neoplastic/Oncologic
■Hypereosinophilic syndrome
■Hypereosinophilic syndrome
■Lymphoma
■Lymphoma
■Myeloma
■Myeloma
■Histiocytosis
■Histiocytosis
■Bone tumor metastases
■Bone tumor metastases
■Paget disease of the bone
■Paget disease of the bone
aSome forms of mast cell disease may not show cutaneous manifestations. These include aggressive systemic mastocytosis, mast cell leukemia, systemic mastocytosis with an associated clonal hematologic non–mast-celllineage disease, and isolated bone marrow mastocytosis.
718
SM-AHNMD patients who express a mutation in the ASXL1 gene have a poorer overall survival than those who express wild-type ASXL1.20 In patients with ASM, the mean survival is 2 to 4 years, but the prognosis may improve with aggressive symptomatic management. The prognosis for MCL is poor, with a mean survival of less than 1 year.23,37 Additional poor prognostic findings in adult mastocytosis patients include detectable D816V mutations in non–mast cell populations, as well as the expression of one or more non–c-kit gene mutations (TETS, SRSF2, ASXLI, ASCL1, RUNXI, and CBL).20,21
TREATMENT
The management of patients with mastocytosis includes counseling patients about the pathophysiology of their disease, avoidance of factors that provoke mast cell mediator release, and management of symptoms associated with these released mediators (Tables 42-7 and 42-8). Mastocytosis patients should be cautioned to avoid potential mast cell degranulating agents such as ingested alcohol, anticholinergic preparations, aspirin, NSAIDs, narcotics, and polymyxin B sulfate. In addition, heat and friction can induce local or systemic symptoms and should be avoided whenever possible. A number of systemic anesthetic agents, including systemic lidocaine, D-tubocurarine, metocurine, etomidate, thiopental, succinylcholine hydrochloride (suxamethonium chloride), enflurane, and isoflurane, have been directly or indirectly implicated in precipitating symptoms of mastocytosis. Recent reports indicate that fentanyl, sufentanil, remifentanil, paracetamol, midazolam, propofol, ketamine, desflurane, sevoflurane, cis-atracurium, pancuronium, and vecuronium bromide, are safe alternative systemic anesthetics for mastocytosis patients. It is recommended that mastocytosis patients undergoing general anesthesia be monitored postoperatively for
TOPICAL SYSTEMIC
First line Topical glucocorticoids Avoidance of triggering factors such as heat, friction, or drugs H1 ± H2 antihistamines
Second line
Psoralen and ultraviolet A light (adults only)
Leukotriene antagonists or 5-lipoxygenase inhibitor
Pulsed-dye laser for adult-type skin lesions and telangiectasia macularis eruptiva perstans
Oral cromolyn sodium Ketotifen
Third
Intralesional
Omalizumab
Third line Intralesional corticosteroids Surgical excision (mastocytoma)
Omalizumab
line
corticosteroids Surgical excision
(mastocytoma)
6
GASTROINTESTINAL CARDIOVASCULAR MUSCULOSKELETAL HEMATOLOGIC
First line
H2 antihistamines, oral cromolyn (children) H1 and H2 antihistamines Subcutaneous epinephrine (anaphylaxis)
Second line
Calcium supplement ± vitamin D supplement 2-Chlorodeoxyadenosine
Proton pump inhibitors Oral glucocorticoids (prophylaxis) Bisphosphonates Systemic chemotherapy appropriate for hematologic disorder
Leukotriene antagonist Nonsteroidal antiinflammatory drugs with caution
Anticholinergics
Third line Omalizumab Oral glucocorticoids
Omalizumab Omalizumab Oral glucocorticoids Local radiation to bony lesions
Allogeneic stem cell
Third line Omalizumab Oral glucocorticoids Omalizumab Omalizumab Oral glucocorticoids Local radiation to bony lesions
Allogeneic stem cell transplantation
transplantation
Note: Cytoreductive therapy is restricted to patients with aggressive variants of mastocytosis (systemic mastocytosis with an associated clonal hematologic non–mast-cell-lineage disease, aggressive systemic mastocytosis, and mast cell leukemia).
24 hours because delayed anaphylaxis can occur hours after surgery. In contrast to systemic anesthetics, local injections of lidocaine normally can be used safely in patients with mastocytosis unless there is a history of reaction.56,57
There is currently no generally recognized cure for mastocytosis, nor are there effective mast cell stabilizing drugs. Thus, treatment of milder forms of this disorder is focused, in great part, on symptomatic relief. In children and adults who are asymptomatic, no therapy is needed. Chronic administration of H1 antihistamines is often helpful in reducing cutaneous and GI symptoms.22,45 The second-generation H1 antihistamines, cetirizine, loratadine, and fexofenadine, have distinct advantages over first-generation antihistamines because they have longer half-lives and have more specific activity on the H1 receptor.58
Higher-than-recommended doses of combined H1 antihistamines may be required for symptom control. For example, fexofenadine 360 mg in the morning and up to 40 mg of cetirizine at night may be necessary for alleviating histamine-related symptoms. Doxepin, a tricyclic antidepressant, has potent H1 activity and can be used when H1 antihistamines have been ineffective. This agent, however, can cause cardiac QT interval prolongation and should be used with caution in older patients as well as those with a history of cardiac arrhythmias, renal insufficiency, or hepatic disease.57 Both ketotifen and azelastine, antihistamines with potential mast cell–stabilizing properties, appear to be most beneficial in reliving GI symptoms associated with mastocytosis, but neither drug offers a significant advantage over a standard antihistamines.58,59 H2 antihistamines (cimetidine, ranitidine, famotidine, or nizatidine) may be beneficial in patients with symptoms of gastric acid hypersecretion and malabsorption, but also may assist in controlling pruritus, flushing, and wheal formation when administered with an H1 agent.22,45,58 If GI symptoms persist with the use of H2
antihistamines, proton pump inhibitors may be effective secondary treatments.45,58
Disodium cromoglycate (cromolyn sodium) may have some efficacy in the treatment of mastocytosis, particularly in relieving GI complaints in children, but may require higher-than-recommended doses. Cromolyn sodium, however, does not lower plasma or urinary histamine levels in patients with mastocytosis.60
Low-dose aspirin (500 mg twice daily) has been used successfully in some patients to reduce flushing, tachycardia, and syncope. However, aspirin must be used with extreme caution in patients with a history of NSAID intolerance, as it may cause vascular collapse in some patients with mastocytosis and exacerbate peptic ulcer disease.61 Leukotriene inhibitors antagonize cysteinyl leukotriene receptors, and have been reported effective in controlling symptoms of flushing, diarrhea, abdominal cramping, and wheezing in mastocytosis patients.62,63
Omalizumab is a monoclonal antibody against immunoglobulin E that has proven effective in reducing or eliminating symptoms of mastocytosis in adults who are recalcitrant to antihistamines and leukotriene inhibitors. Effective doses range from 150 to 450 mg/month. Although this monthly therapy has proven effective in managing symptoms from mast cell mediator release, it requires continued administration because it does not reduce mast cell proliferation.64-66
Potent topical glucocorticoids applied daily under occlusion for 8 to 12 weeks reduces the number of cutaneous lesions. However, this therapy is impractical for patients with diffuse skin involvement, and lesions of mastocytosis recur with discontinuation of therapy.67 Oral glucocorticoids may have some efficacy in patients with malabsorption, bone disease, abdominal pain, recurrent anaphylaxis, and ascites; this medication, however, should be reserved for patients with advanced disease, and tapered to the
719
6
lowest effective dose.45,57,68 Osteoporosis should be treated with calcium and vitamin D supplementation along with bisphosphonates as needed to maintain normal bone density.57,69
Self-injectable epinephrine syringes should be prescribed for patients with a history of anaphylaxis. However, some experts recommend that all mastocytosis patients should carry preloaded epinephrine syringes with them regardless of their anaphylaxis history.45,57 Patients prescribed epinephrine should be prepared to self-administer this drug, and have a plan for emergency management. If subcutaneous epinephrine is insufficient, intensive therapy for vascular collapse should be instituted in a hospital setting. Patients with recurrent episodes of anaphylaxis also should receive continuous H1 and H2 antihistamines to lessen the severity of attacks. Episodes of vascular collapse in mastocytosis patients may be spontaneous, but have also occurred after insect stings or after administration of iodinated contrast media. In the latter case, premedication with corticosteroids and antihistamines is recommended before such procedures. Methoxypsoralen with ultraviolet A (PUVA) light can relieve pruritus and whealing after 1 to 2 months of treatment.70,71 However, pruritus usually recurs within 3 to 6 months after PUVA discontinuation. Pigmentation induced by PUVA also may camouflage lesions of UP in some adult patients; however, this benefit must be weighed against the increased risk of skin cancers associated with long-term treatment. Cytoreductive therapy should be considered in patients with SM-AHNMD, ASM, MCL, or mast cell sarcoma. The risk-to-benefit ratio must be carefully considered because of the dose-limiting toxicities of the various drugs. IFN-α may be considered for advanced SM. In 2 studies, IFN-α was most efficacious in ameliorating the signs and symptoms of mast cell disease; however, improvement was only transitory.72,73 2-Chlorodeoxyadenosine is efficacious in more advanced SM patients with an overall response rate of approximately 55%.73,74 Even though this drug appears to be the treatment of choice in patients with advanced mastocytosis, myelosuppression may limit its use.74
Splenectomy may improve survival in patients with ASM that have a poor prognosis.75 Radiotherapy has been used to treat refractory bone pain in advanced disease.76
More recently, patients with advanced SM (SM- AHNMD, ASM, and MCL) have been treated with allogeneic hematopoietic stem cell transplantation. In a retrospective review of 57 mastocytosis patients treated with allogeneic hematopoietic stem cell transplantation, 16 (28%) had a complete remission, whereas 12 (21%) had stable disease. All 38 patients with SM-AHNMD achieved complete remission for a time; however, 10 experienced a relapse and 5 died of their associated clonal hematologic non–mast-celllineage disease. Patients with stable disease and SM- AHNMD had a better overall survival than those with progressing mastocytosis or MCL. The overall survival at 1 and 3 years for the entire treatment group was 62% and 55%, respectively.77
720
Targeting the tyrosine kinase KIT has been another important strategy for treating patients with mastocytosis. The first-generation tyrosine kinase inhibitor, imatinib, however, has proven ineffective in patients expressing KIT D816 because it is unable to inhibit this mutated receptor.78,79 Imatinib, however, has been reported to cure a mastocytosis patient who expressed only normal (wild-type) c-kit.80 Second-generation tyrosine kinase inhibitors, dasatinib and nilotinib, inhibit the in vitro growth of D816V-expressing mast cell lines. However, these agents also have been disappointing in their ability to reduce the signs or symptoms in SM patients with the D816 mutation.81-83 The multikinase inhibitor midostaurin, which inhibits both wildtype and mutated c-kit, has been used in patients with advanced SM. Among 116 advanced SM patients receiving this agent, there were no complete remissions, but 53 patients had some response to therapy. Patients with SM- AHNMD had the best response rate (75%) in contrast to MCL patients who responded the least (50%). Unfortunately, these responses were limited in duration.84 Taken together, these observations along with the clinical heterogeneity of mastocytosis, strongly suggest that other molecular abnormalities, in addition to c-kit mutations, play an important role in this disease. Thus it appears that more effective future therapies will be based on the identification of additional altered genes.
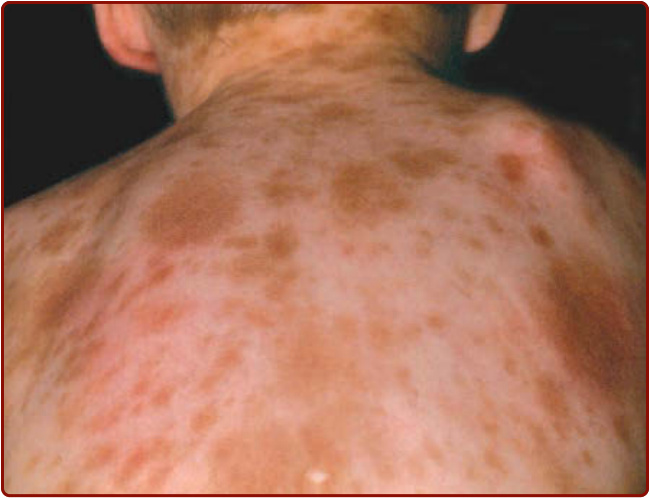
Figure 42-1 Urticaria pigmentosa in a child.
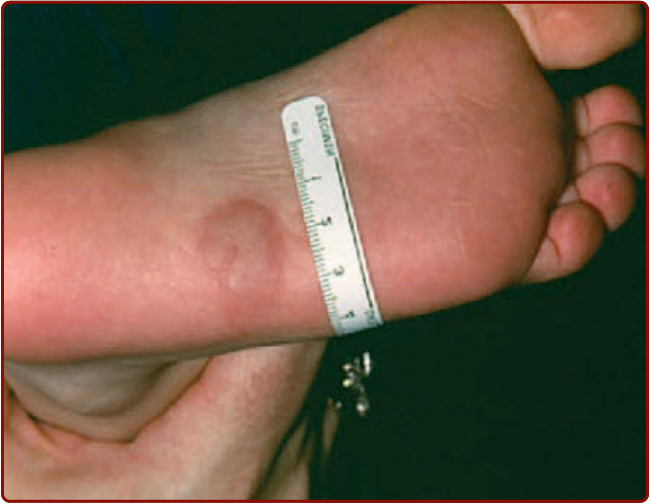
Figure 42-2 Mastocytoma on the sole of a child.
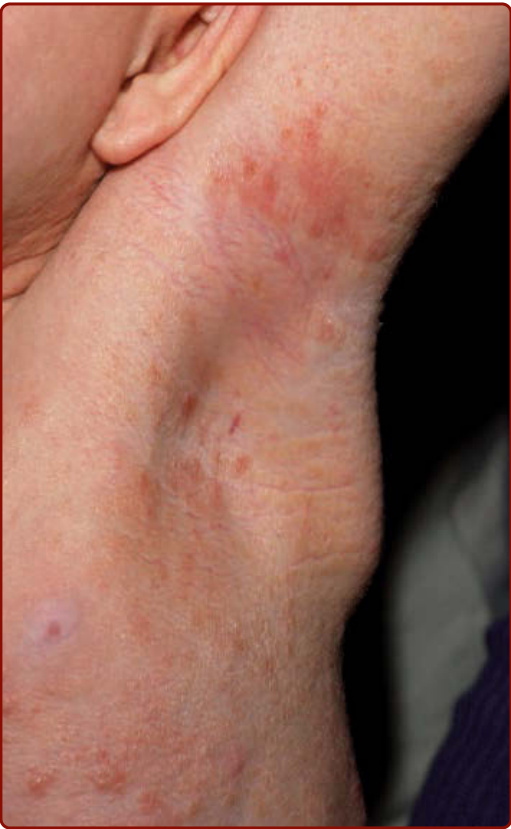
Figure 42-3 Diffuse cutaneous mastocytosis in a child.
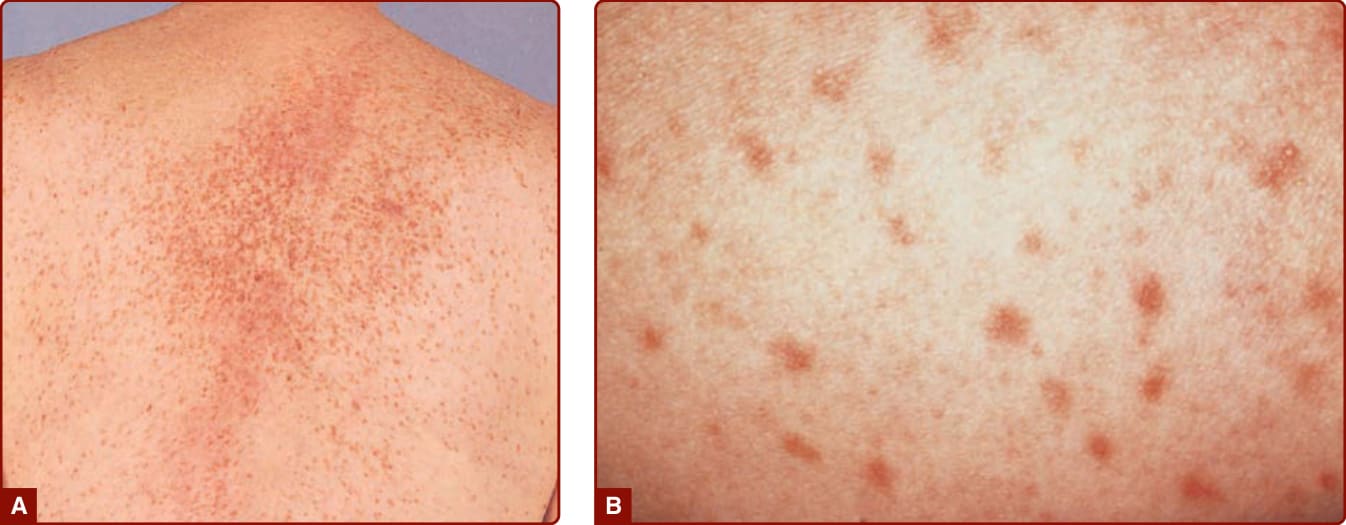
Figure 42-4 Typical red brown papules in an adult with indolent systemic mastocytosis. A, Hundreds of lentigo-like macules are seen on the back of this adult. If vigorously rubbed, these lesions will show urtication and become erythematous, raised, and pruritic. B, Close-up view of the macules.
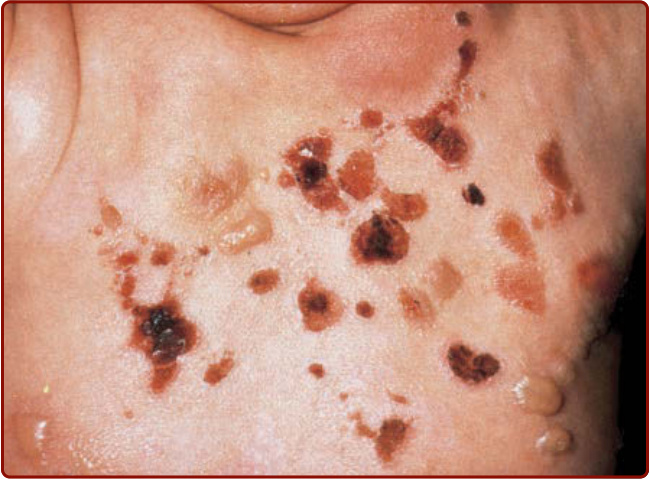
Figure 42-5 Bullous eruption on the back of a child with diffuse cutaneous mastocytosis.
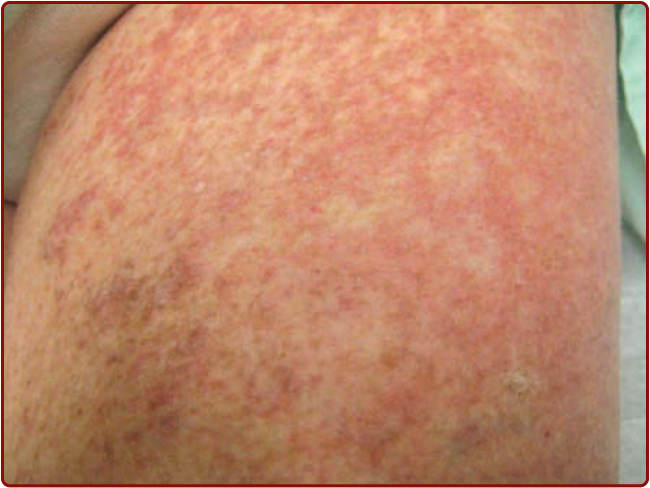
Figure 42-6 Telangiectasia macularis eruptive perstans.
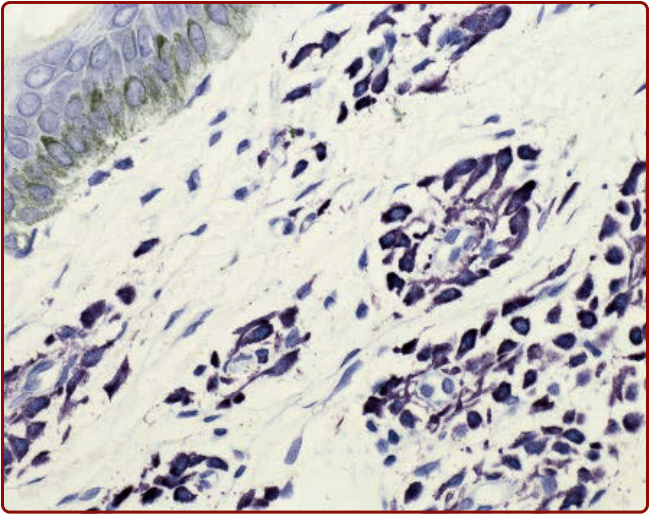
Figure 42-7 Histopathology of urticaria pigmentosa, toluidine blue stain.
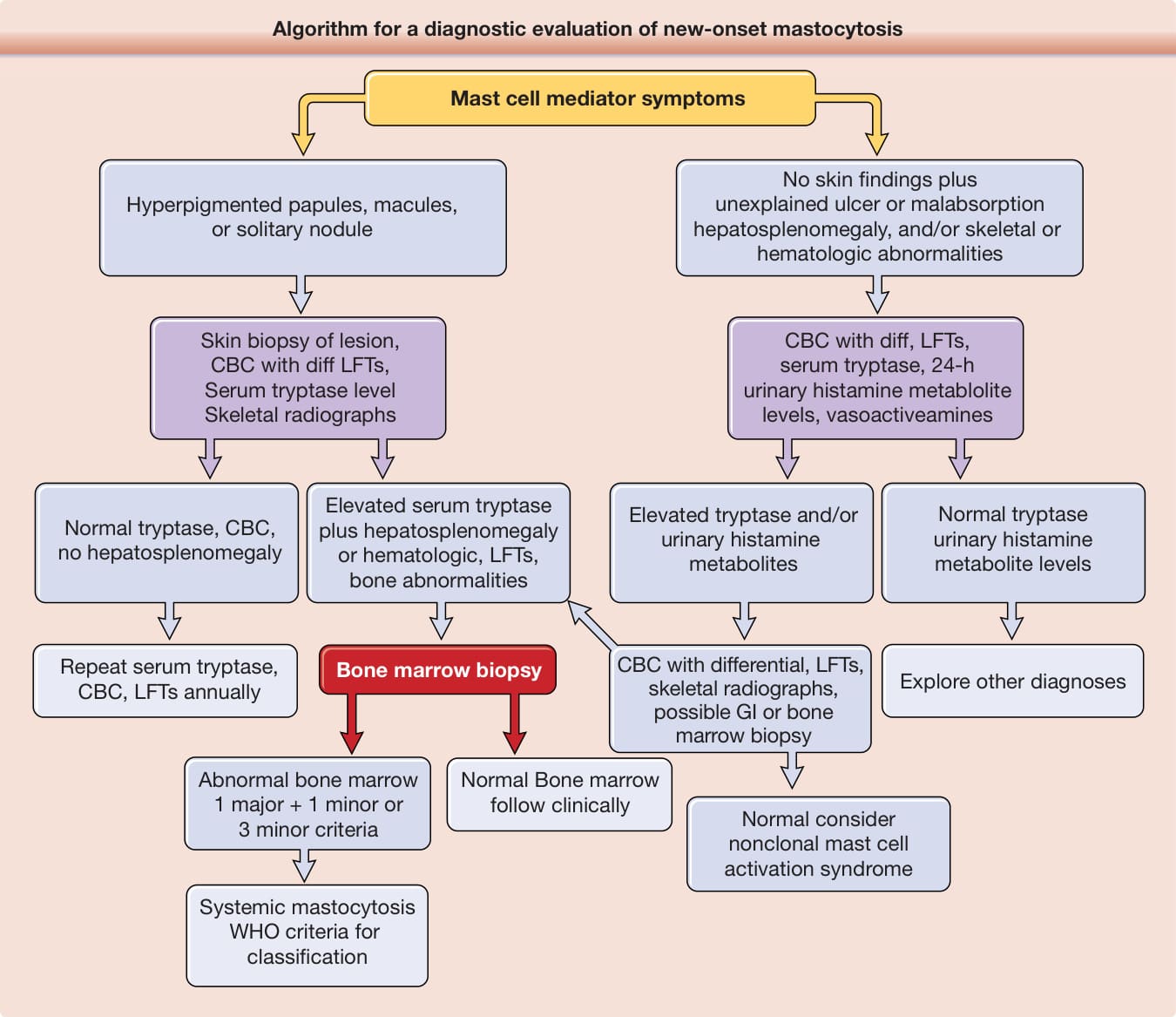
Figure 42-9 Algorithm for a diagnostic evaluation of new-onset mastocytosis (especially in adolescents and adults). CBC, complete blood cell count; LFT, liver function test; Tc, technetium; WHO, World Health Organization. For details of the WHO classification of mastocytosis, see Valent P, et al.5

TABLE 42-1 c-Kit Mutations in Mastocytosis
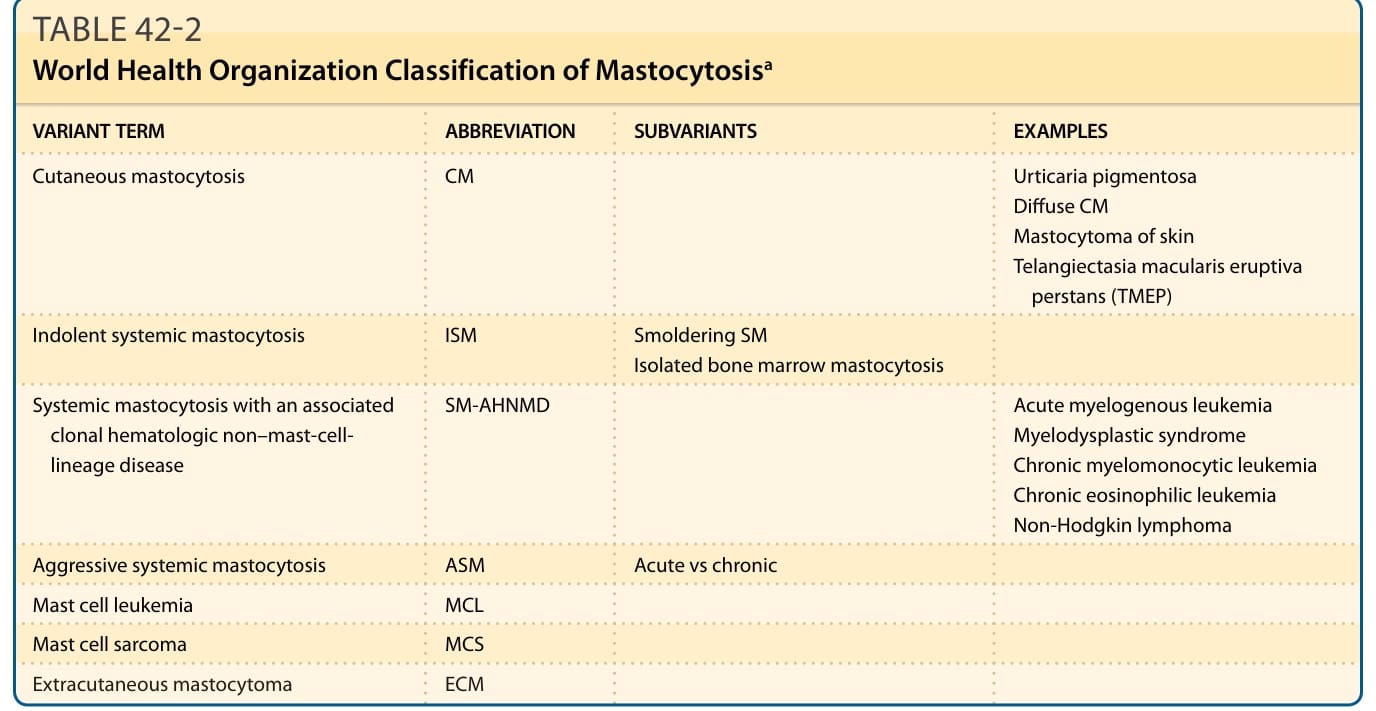
TABLE 42-2 World Health Organization Classification of Mastocytosisa
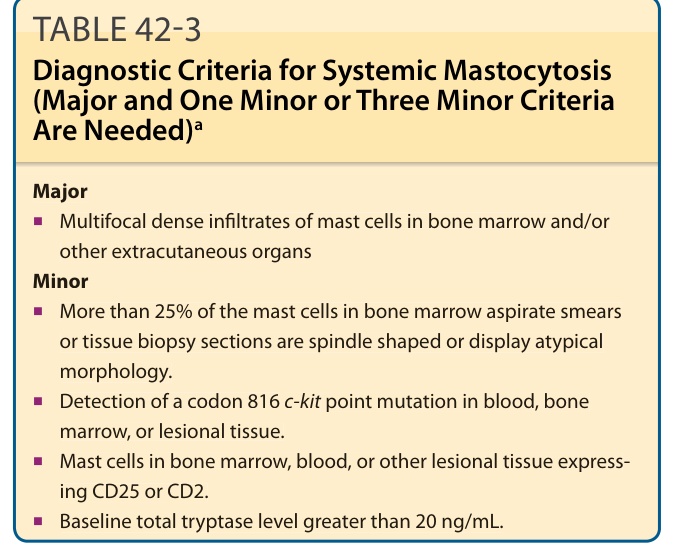
TABLE 42-3 Diagnostic Criteria for Systemic Mastocytosis (Major and One Minor or Three Minor Criteria Are Needed)a
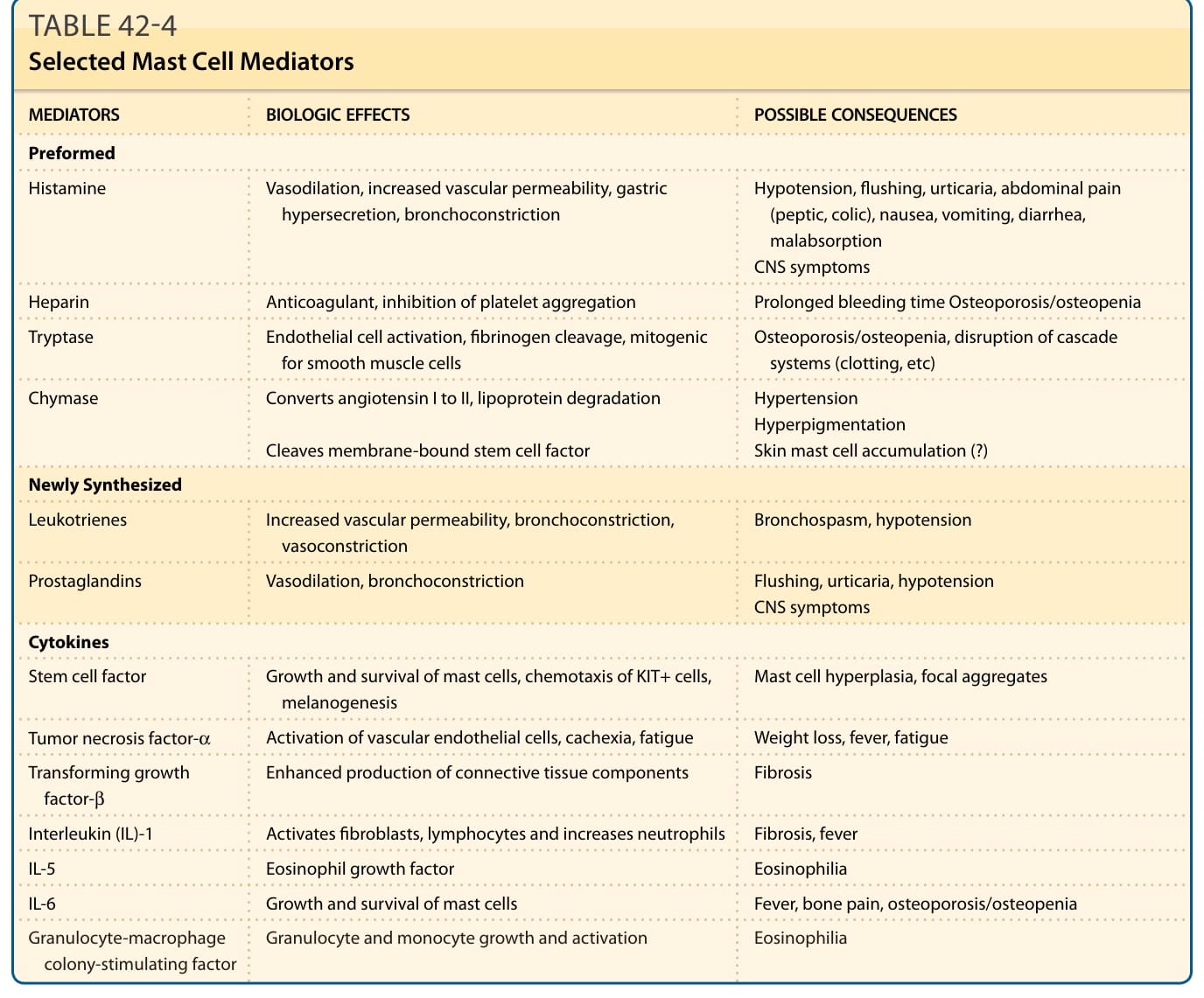
TABLE 42-4 Selected Mast Cell Mediators

TABLE 42-5 Differential Diagnosis of Cutaneous Mast Cell Disease
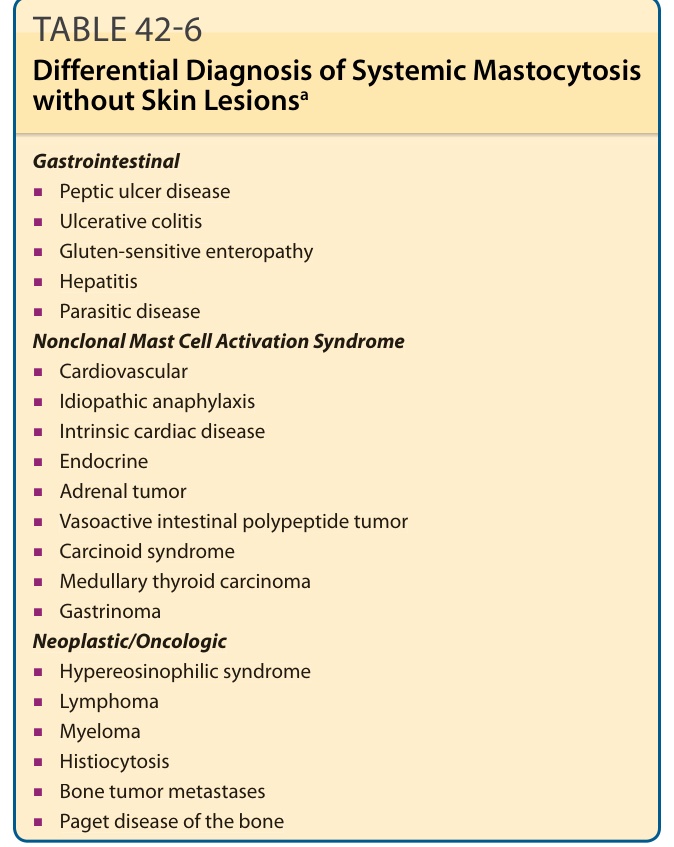
TABLE 42-6 Differential Diagnosis of Systemic Mastocytosis without Skin Lesionsa
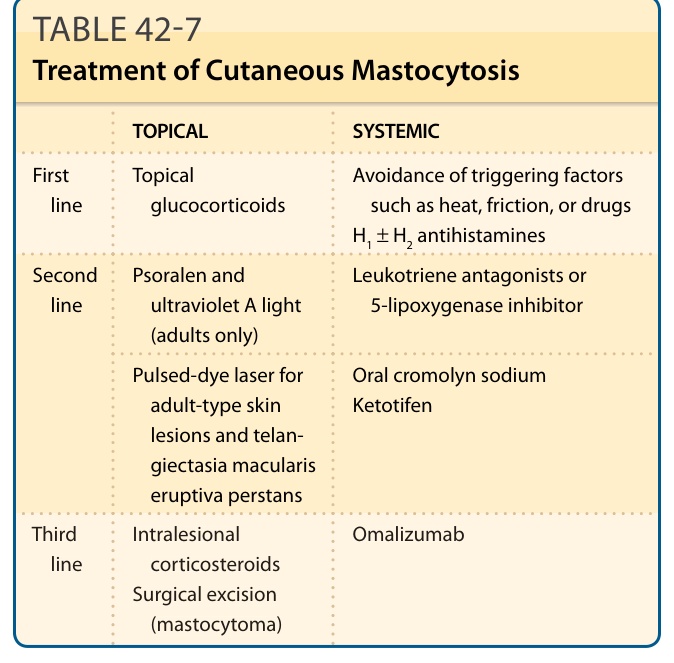
TABLE 42-7 Treatment of Cutaneous Mastocytosis
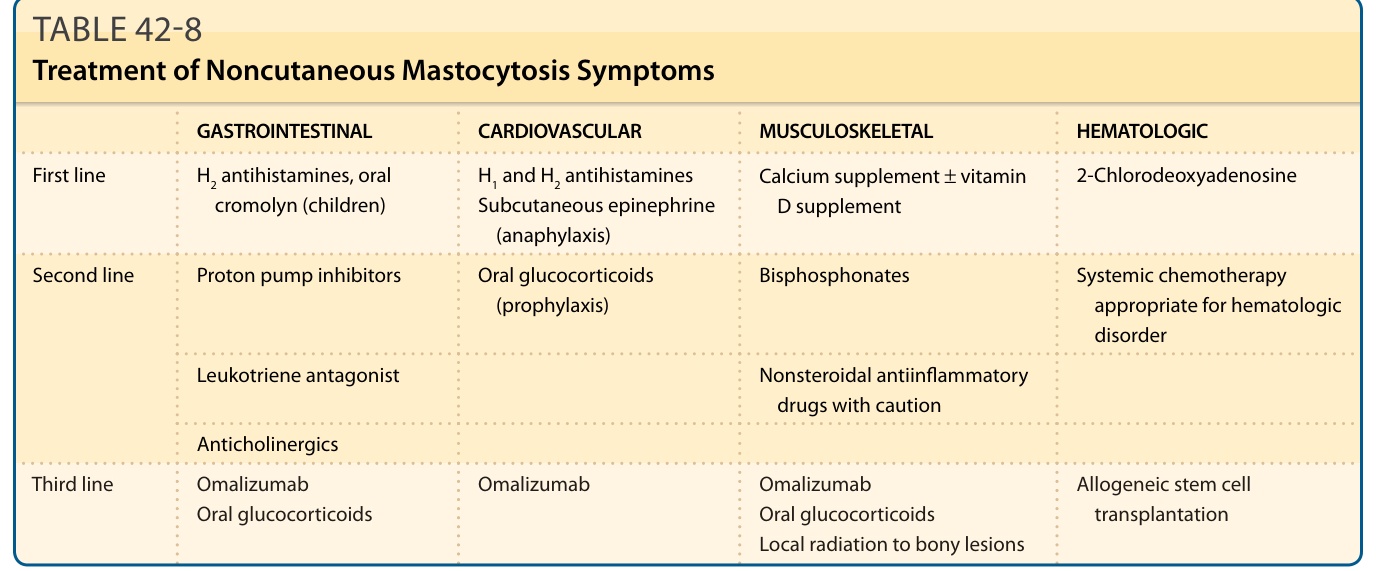
TABLE 42-8 Treatment of Noncutaneous Mastocytosis Symptoms