Pityriasis Rubra Pilaris
4
AT-A-GLANCE
■ A rare inflammatory papulosquamous dermatosis, often self-limiting within a few years.
■ The disease is subclassified into 6 types, including both hereditary and acquired forms.
■ The hereditary form of pityriasis rubra pilaris is linked to CARD14 gain-of-function mutations, the cause of the acquired forms is unknown.
■ Typical features are follicular hyperkeratotic papules that coalesce into scaly, reddishorange–colored plaques, which may progress to erythroderma with well-demarcated islands of normal skin.
■ Distinguishing pityriasis rubra pilaris from psoriasis poses a major diagnostic challenge, particularly in the early phases of the disease.
■ Histopathologic examination reveals hyperkeratosis, alternating parakeratosis and orthokeratosis in a checkerboard pattern, with focal acantholytic dyskeratosis.
■ The most successful treatment options are systemic retinoids, methotrexate, and photochemotherapy (psoralen and ultraviolet A). In recent years, biologics blocking tumor necrosis factor-α and interleukins-12 and -12p40 were shown to be effective.
Pityriasis rubra pilaris (PRP) is a rare, inflammatory skin disease of juvenile or adult onset with distinctive clinical features and a self-limiting or chronic evolution.
EPIDEMIOLOGY
The exact prevalence of PRP is not known. It accounts for approximately 1 in 5000 new patients presenting with skin disease in Great Britain.1 In India this is approximately 1 in 50,000 patients (pityriasis rubra pilaris in Indians.) The disease occurs in all races with an equal male-to-female ratio.2
CLINICAL FEATURES
PRP comprises different entities and most cases are sporadic and have an unknown etiology. Approximately 6.5% of cases of PRP are familial3-5 and linked to CARD14 mutations.6 In 1980, Griffiths proposed classifying PRP into 5 types based on age of onset, clinical course, and prognosis. A sixth, HIV-associated, category was added in later years (Table 29-1).2
Type I (classic adult) is the most common subtype and occurs in more than 50% of all cases. Characteristically, type I PRP starts with erythematous macules forming patches and with follicular hyperkeratotic papules on the upper half of the body (Figs. 29-1A and 29-2). As the disease evolves, a yellow-orange, scaling dermatitis often spreads to a generalized erythroderma over a period of 2 to 3 months (Fig. 29-3). A diagnostic hallmark of PRP is the presence of sharply demarcated islands of unaffected skin (“nappes claires”; Fig. 29-1B). Frequently patients develop a waxy, diffuse, yellowish keratoderma of the palms and soles (Fig. 29-4).7 Nail changes are common in these patients and include nail plate thickening, splinter hemorrhages, and subungual hyperkeratosis. In patients with uniform facial involvement, ectropion frequently develops (Fig. 29-2B). Type II (atypical adult) PRP describes an variant, which affects 5% of PRP patients. It is characterized by its atypical morphologic picture and a long duration of more than 20 years. The clinical picture resembles ichthyosiform scaling, areas of follicular hyperkeratosis, and sparseness of the scalp hair. The cephalocaudal progression observed in type I is missing, and there is less tendency for the patients to develop erythroderma. Type III (classic juvenile) is the clinical counterpart of type I PRP in children. It affects 10% of PRP patients, with the onset usually between the ages of 5 and 10 years.1 The clinical course may be shorter with clearing after 1 year (Fig. 29-5). Type IV (circumscribed juvenile) affects approximately 25% of patients. This type usually manifests in prepubertal children and young adults. It is characterized by well-demarcated hyperkeratotic erythematous plaques on the elbows and knees, resembling localized psoriasis (Fig. 29-6). These lesions do not progress to the more widespread classical type. Palmoplantar keratoderma is characteristic for type IV PRP, but may also be absent.8 The 3-year remission rate is 32%. Type V (atypical juvenile) PRP occurs in 5% of patients and is characterized by an early age of onset and a chronic course. Most patients of the familial PRP belong to this category. These patients were recently shown to harbor a gain-of-function mutation in the CARD14 gene encoding the Caspase Recruitment Domain family member 14 (CARD14).6 It manifests as hyperkeratotic follicular lesions. Some patients present with scleroderma-like features affecting hands and feet. The type VI (HIV-associated) variant was proposed by Miralles and colleagues.9 These HIV-associated cases manifest with follicular papules and prominent follicular plugging. The symmetrically distributed lesions of the extensor surfaces frequently progress to erythroderma. Additional manifestations include acne conglobata, hidradenitis suppurativa, and lichen spinulosus.
4
TYPE DESCRIPTION % OF CASES CLINICAL CHARACTERISTICS DISTRIBUTION COURSE
I Classic adult >50 Erythroderma with islands of normal skin (“nappes claires”), follicular hyperkeratosis, waxy diffuse palmoplantar keratoderma
Generalized, beginning on the head and neck, then spreading caudally
Often resolves within an average period of 3 years
II Atypical adult 5 Combination of follicular hyperkeratosis and ichthyosiform lesions on the legs, sparse scalp hair
Generalized Long duration (>20 years)
III Classic juvenile 10 Similar to type I but appears in years 1 or 2 of life Generalized Often resolves within an average period of 1–2 years
IV Circumscribed juvenile 25 Prepubertal children; well-demarcated scaly, erythematous plaques on the elbows and knees, resembling localized psoriasis
V Atypical juvenile, associated with CARD14 mutations
Localized Uncertain, some cases clear in the late teens
5 Begins in first few years, accounts for most familial cases; follicular hyperkeratosis, scleroderma-like appearance of the hands and feet
Generalized Chronic course, improvement with retinoids but relapses when stopped
VI HIV-associated NAb Similar to type I with variable beginning; associated with acne conglobata, hidradenitis suppurativa and lichen spinulosus
VI HIV-associated NAb Similar to type I with variable beginning;
Generalized May respond to
Generalized May respond to antiretroviral triple therapy
associated with acne conglobata, hidradenitis suppurativa and lichen spinulosus
aTypes I to V are according to Griffiths.2
bData not available.
A
antiretroviral triple therapy
B
499
4
A
B
ASSOCIATIONS OF PITYRIASIS RUBRA PILARIS
ASSOCIATIONS OF
PITYRIASIS RUBRA PILARIS
There is an association of PRP with HIV infections, proposed as type VI PRP. In some cases, manifestation of PRP was the first sign of HIV infection,10 and clinical responses occurred with antiretroviral therapy.11
Multiple associations with infections, malignancies, or autoimmune diseases have been noticed, but may occur rather fortuitously.
500
A
B
A B
4
ETIOLOGY AND PATHOGENESIS
The etiology and pathogenesis of sporadic PRP remain elusive. Pathogenic mechanisms associated with infection, such as upper respiratory tract infection and HIV infection, were proposed. In type V PRP, gainof-function mutations in the CARD14 gene linked to autosomal dominant inheritance have been identified.
A B
CARD14 is expressed in the skin and encodes the caspase recruitment domain family member 14, a known activator of nuclear factor-κB signaling. Accordingly, CARD14 levels were increased in affected individuals with PRP, and p65 was found to be activated in the skin. CARD14 mutations also have been described in familial psoriasis.12 A study of 48 patients with PRP type I identified 12.5% of patients as having CARD14 putative mutations.13 A second study of 61 patients with sporadic PRP did not observe CARD14
501
4
mutations.14 Hence, CARD14 mutations may be rare in sporadic cases, and alternate mechanisms may be responsible for activation of the nuclear factor-κB signaling observed in these patients.13
In lesional PRP skin samples from a single patient successfully treated with the anti-IL-12/IL-23 antibody ustekinumab, upregulated expression levels were found for most proinflammatory innate cytokines, including tumor necrosis factor (TNF), IL-6, IL-12, IL-23, and IL-1β. Among adaptive T-cell cytokines, an increase of TH1 cytokines and, in particular, TH17 cytokines IL-17A, IL-17F, and IL-22 was seen.15 Based on these findings, PRP may now be classified as a papulosquamous disease that results from abnormal activation of different inflammatory pathways. Even though PRP and psoriasis have overlapping clinical symptoms, the clinical and histopathologic features of the 2 diseases are distinct. PRP often affects the face, has the typical salmon-colored appearance, presents with classical islands of healthy skin over the trunk, distinct areas of follicular hyperkeratosis, and a waxy palmoplantar keratoderma (see Fig. 29-4). Although psoriasisassociated nail changes are missing, nail plates may be hypertrophic. The histologic picture of psoriasis with hypogranulosis, elongation of the rete ridges, vascular dilation, a high frequency of neutrophils manifesting as intraepidermal Munro microabscesses is not shared with PRP, which presents with histologic features such as alternating horizontal and vertical parakeratosis and orthokeratosis, hypergranulosis, thickening of the rete ridges, follicular hyperkeratosis, lack of neutrophilic infiltration, and limited vascular dilatation (Fig. 29-7). Nevertheless, differentiation between PRP and psoriasis can be difficult in individual cases.
DIAGNOSIS
PRP is typically a clinical diagnosis based on the characteristic skin lesions and the age of onset. Psoriasis is an important differential diagnosis. Initial manifestations of the scalp may mimic seborrheic
502
dermatitis. A biopsy should be taken and helps differentiating PRP from psoriasis.
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE, PROGNOSIS MANAGEMENT
Classic adult type I PRP frequently resolves within 3 years. Reoccurrence was observed after periods of subclinical disease in up to 20% of cases. Classic juvenile type III commonly resolves within 1 to
Most Likely Localized
■Psoriasis
■Ichthyosis
■Seborrheic dermatitis of the scalp Generalized
■Psoriasis
■Erythrokeratoderma
Consider Localized
■Follicular eczema
■Lichen planus acuminatus
■Keratosis pilaris Generalized
■Pityriasis lichenoides chronica
■Ichthyosiform erythroderma
Always Rule Out
Always Rule Out
■Generalized (therapy-resistant cases)
■Generalized (therapy-resistant cases)
■HIV infection
■HIV infection
■Cutaneous T-cell lymphoma
■Cutaneous T-cell lymphoma
2 years. A less-favorable prognosis for remission is reported for the atypical variants types II and IV, although some cases of type IV improve in the patient’s late teens. Type V PRP, which is associated with CARD14 mutations, has little or no tendency to resolve spontaneously. PRP responds hesitantly to topical and classic systemic treatment regiments, which makes finding an effective therapy for PRP challenging. As PRP is an infrequent disease of unpredictable duration, conducting doubleblind, placebo-controlled studies addressing the value of individual systemic treatment strategies are difficult. Published treatment recommendations rely on results from small case series and individual cases. From the patient’s point of view, corticosteroids, and salicylic acid in combination with oral retinoids, methotrexate, and tumor necrosis factor (TNF) inhibitors were regarded as most helpful.16 A growing number of reports demonstrate the role of biologics targeting TNF-α, interleukin (IL)-12/IL-23p40 or anti-IL-17 as a valuable second-line treatment option.
TOPICAL TREATMENT
TOPICAL TREATMENT
Local treatment options for PRP regularly do not suffice to control the disease and are used as an adjunct to systemic therapy. As in psoriasis, classical keratolytic treatment is used where hyperkeratosis is the leading sign. To dampen the local inflammatory response, topical corticosteroids are often initially employed and may be followed by topical calcineurin inhibitors. Furthermore, vitamin D3 analogs for the inhibition of various aspects of cutaneous inflammation and epidermal proliferation with enhancement of normal keratinization are successfully used for the treatment of PRP. However, percutaneous resorption of vitamin D analogs restricts treatment to no more than 30% of body surface.
SYSTEMIC TREATMENT
SYSTEMIC TREATMENT
So far, there is no systemic therapy that works for all patients with PRP. Finding a successful therapy for the individual patient can be a challenging task. Oral retinoids generally serve as the first-line therapy in patients with PRP. They block the proliferation of keratinocytes and reduce hyperkeratosis. Acitretin and isotretinoin (1-2 mg/kg/day)17 are effective; good responses to alitretinoin also have been reported. In a substantial proportion of patients, disease manifests at an early age. Although isotretinoin is proven to be effective in prepubertal patients, oral retinoids can induce premature closure of epiphyses or hyperostosis, and should be used with caution. Teratogenicity is another important concern in oral retinoid therapy. Teratogenicity is also a known adverse effect of methotrexate, the second most used drug for the treatment of PRP. In severe cases, methotrexate can be combined with oral retinoids.
4
Psoralen and ultraviolet A (PUVA) alone or in combination with oral retinoids (RE-PUVA) is another efficient treatment option. In addition, narrowband ultraviolet B (UVB) or broadband UVB can be successful. However, because PRP can be aggravated by UV light,18-20 phototesting should be performed before initiating UV treatment. Some authors report about beneficial effects of fumaric acid esters.21,22 The phosphodiesterase 4-inhibitor apremilast also may be a treatment option.23
As an alternative treatment, anti–TNF-α, anti– IL-12/IL-23p40 (ustekinumab), and anti-IL17 have been used with good results. There are more than 30 published cases of patients with adult PRP type I successfully treated with anti-TNF.24,25 Interestingly, ustekinumab was effective in a number of patients with PRP type I,26-30 in a patient with a CARD14 mutation (type V),30 but not in a patient with type IV PRP.31
An underreporting of negative results when treatment outcomes are published as individual case reports should be considered. In some patients with PRP, extracorporeal photopheresis has been used with success. Evidence for the use of cyclosporine, glucocorticosteroids, or azathioprine is low. PRP associated with HIV infection has responded to triple antiretroviral therapy.32
First Line
Topical
■Emollients (water-in-oil emulsion)
■Keratolytics (salicylic acid, urea)
■Vitamin D3 (calcipotriol)
Systemic
■Retinoids (0.5-0.75 mg/kg acitretin/day)
■Methotrexate (10-25 mg weekly or higher)
■Antiretroviral therapy (HIV-associated variant)
Second Line
Topical
■Glucocorticoids (medium to high potency)
■Vitamin A analogs (tazarotene)
Physical
■Photochemotherapy (topical or systemic PUVA)
■UVA1 phototherapy
■UVB (narrowband) phototherapy
■UVB phototherapy
■Extracorporeal photopheresis
Systemic
Systemic
■TNF-α antagonists
■TNF-α antagonists
■Anti–IL-12/IL-23p40 ustekinumab
■Anti–IL-12/IL-23p40 ustekinumab
■Anti-IL17
■Anti-IL17
■Azathioprine (100-150 mg/day)
■Azathioprine (100-150 mg/day)
■Fumaric acid esters
■Fumaric acid esters
■Cyclosporine (5 mg/kg/day)
■Cyclosporine (5 mg/kg/day)
503
■Apremilast
■Apremilast
4
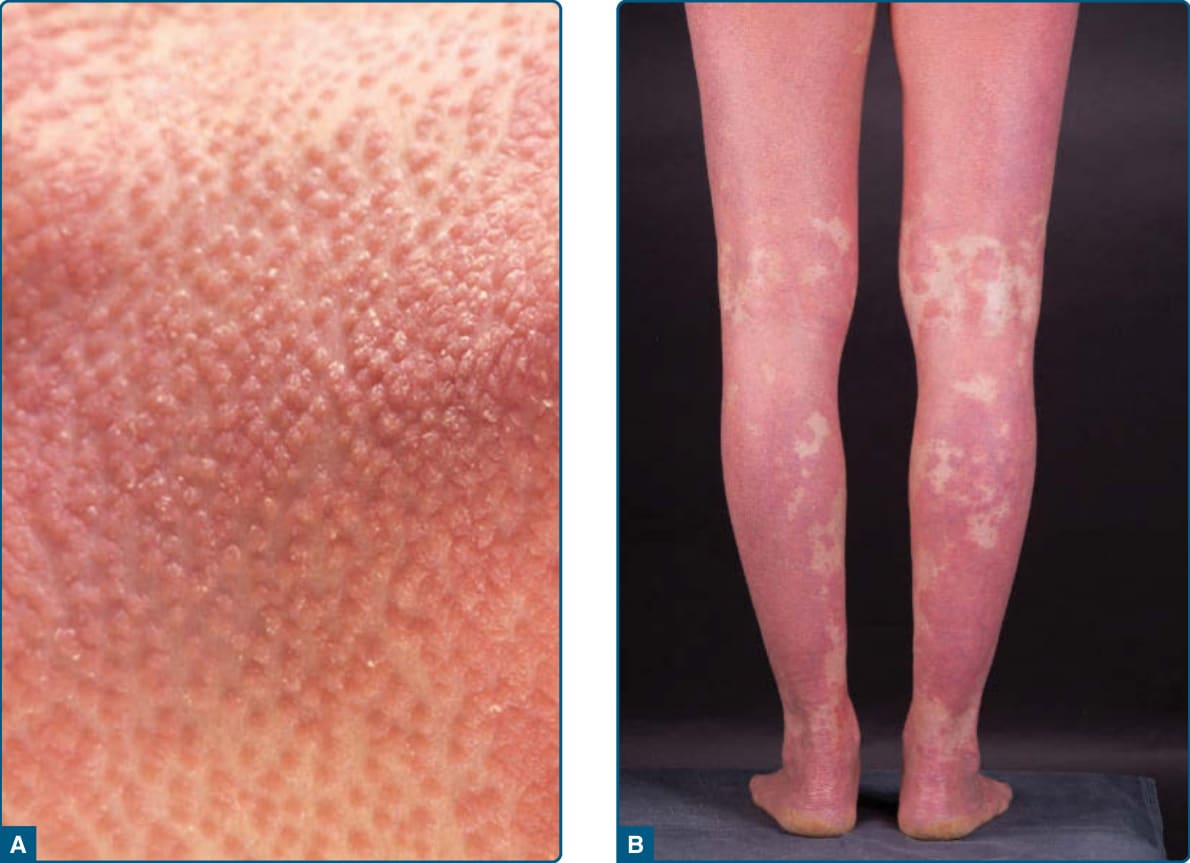
Figure 29-1 A, Follicular papules and B, islands of normal skin “nappes claires” in pityriasis rubra pilaris.
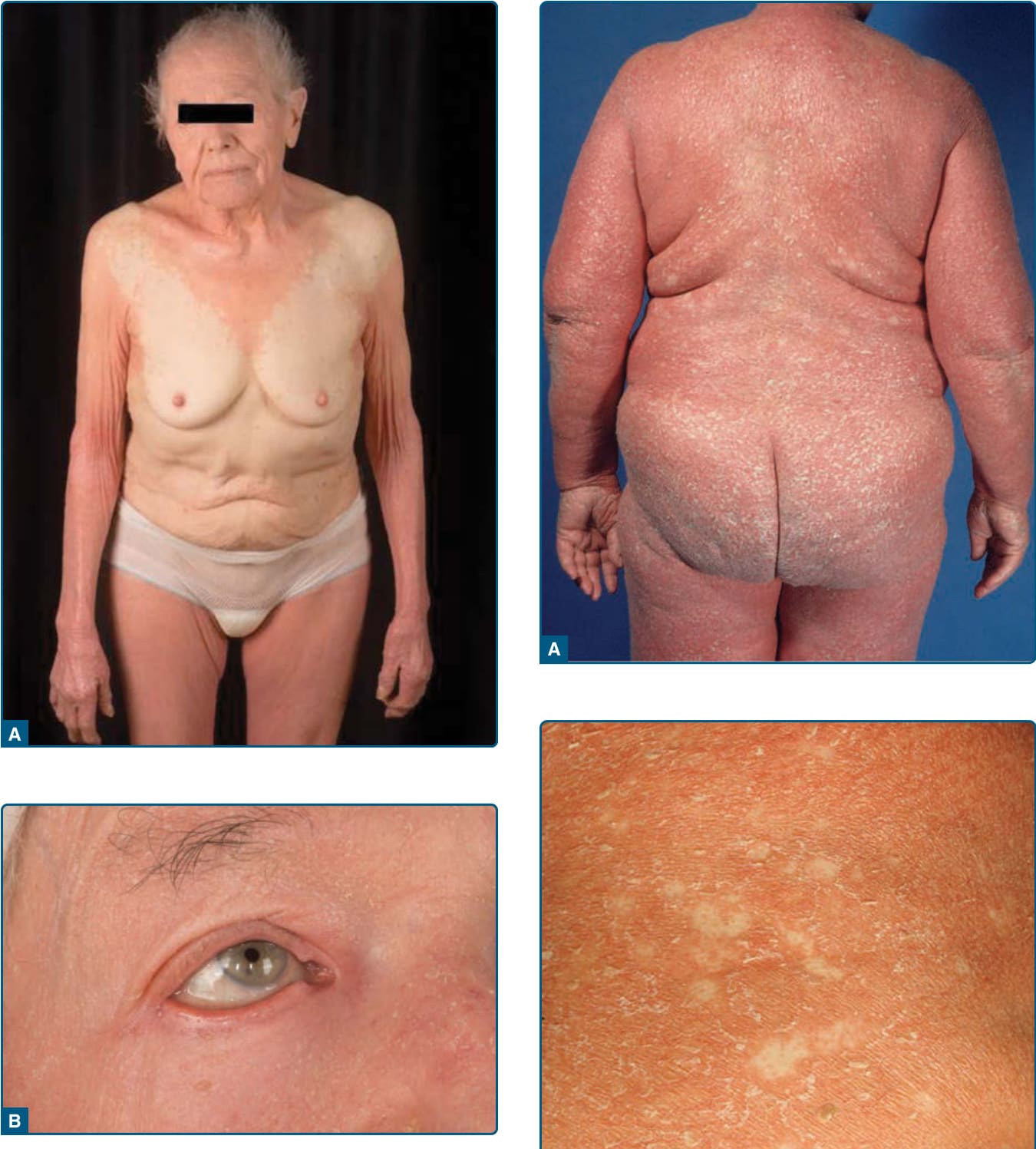
Figure 29-2 Classic adult type I pityriasis rubra pilaris presenting with monomorphic regions of affected skin, sharp margins, diffuse alopecia (A), and ectropion (B).
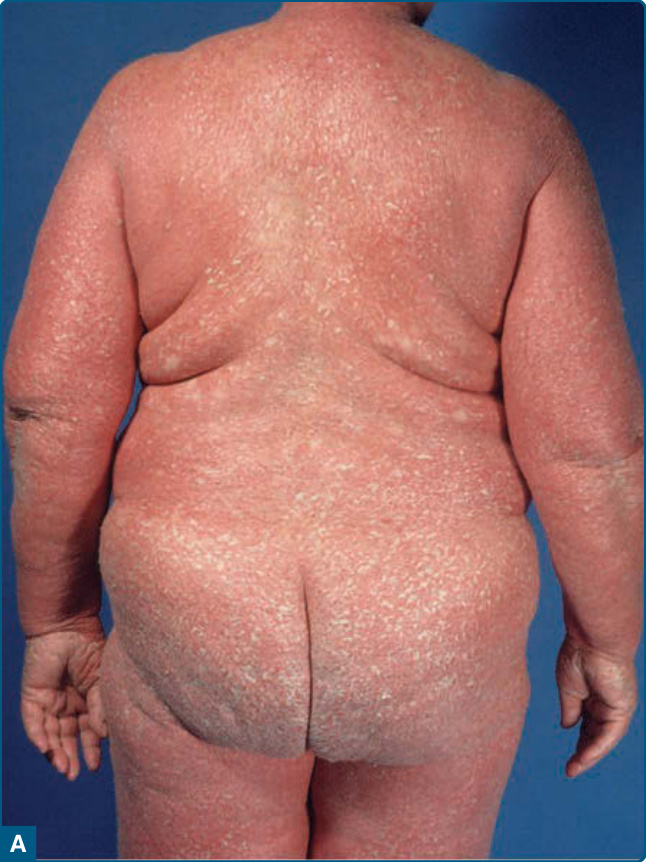
Figure 29-3 Erythrodermic patient (classic adult type I, as in Fig. 29-2) with psoriasiform scaling (A) and island of normal skin (B).
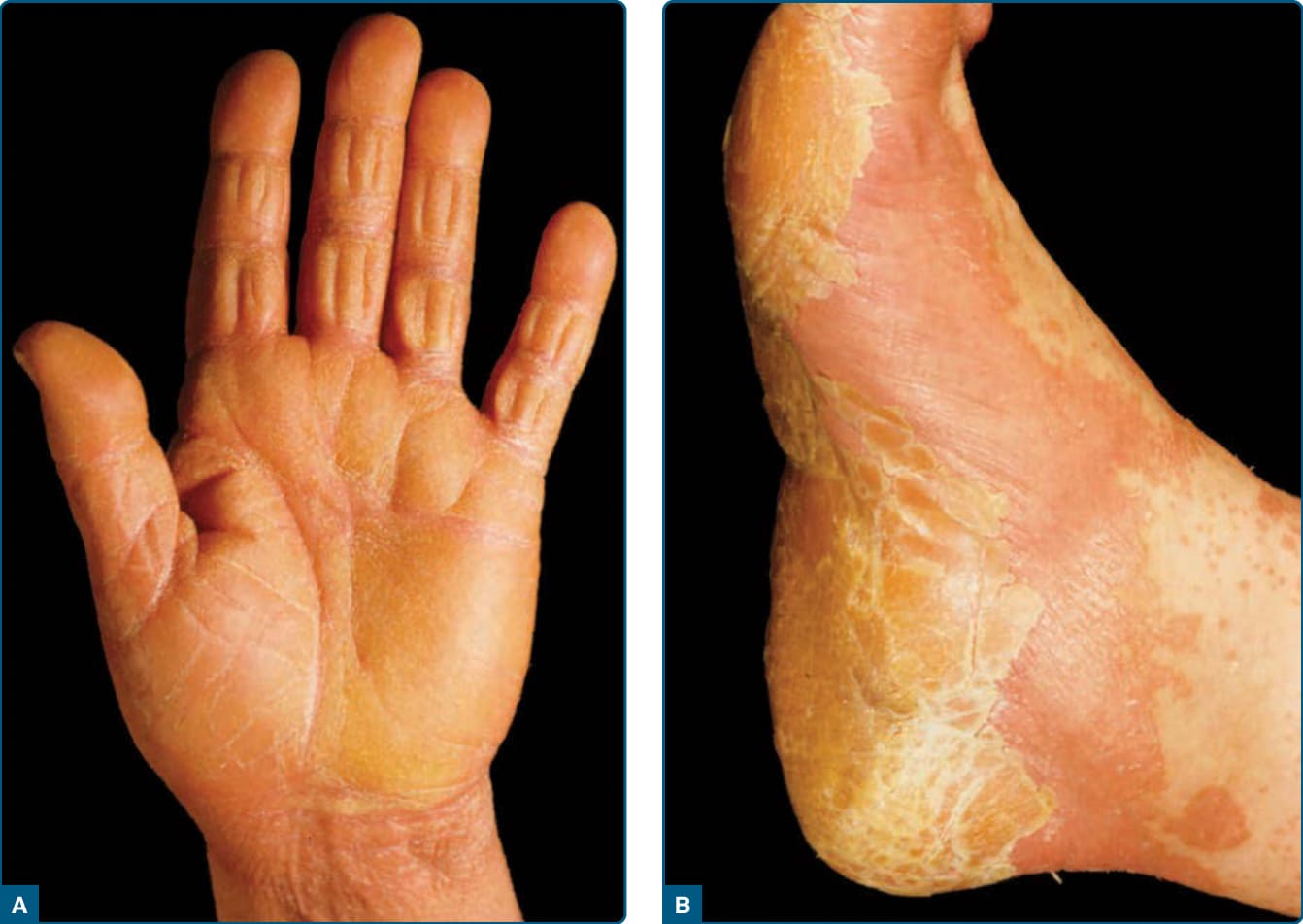
Figure 29-4 Palmar (A) and plantar (B) “waxy” keratoderma in pityriasis rubra pilaris.
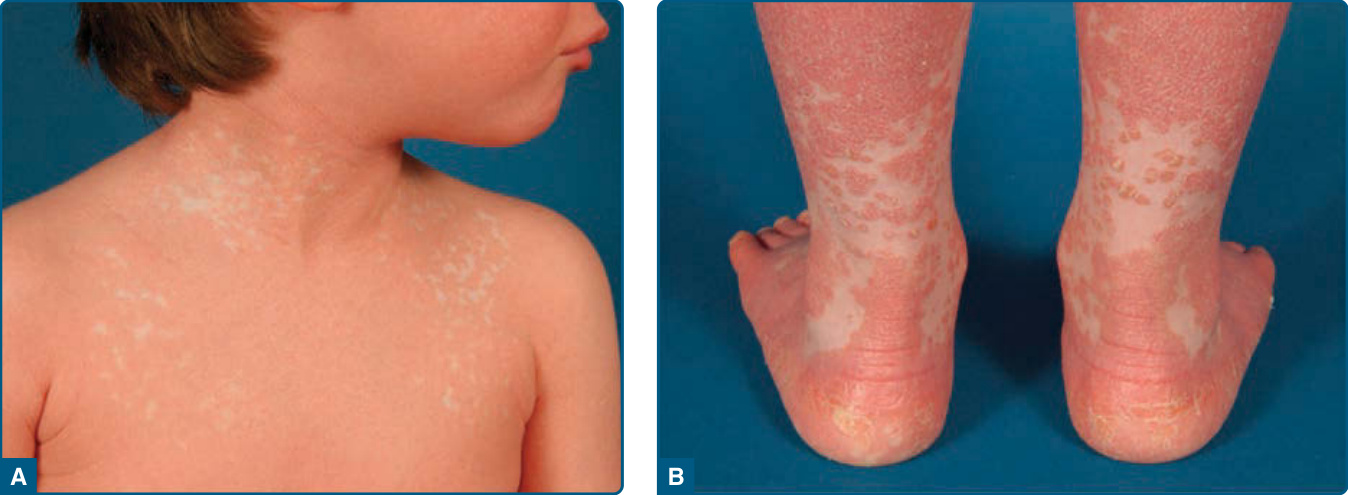
Figure 29-5 Classic juvenile pityriasis rubra pilaris type III. A, Confluence of lesions leads to erythroderma. B, Characteristic scattered islands of unaffected skin are evident.
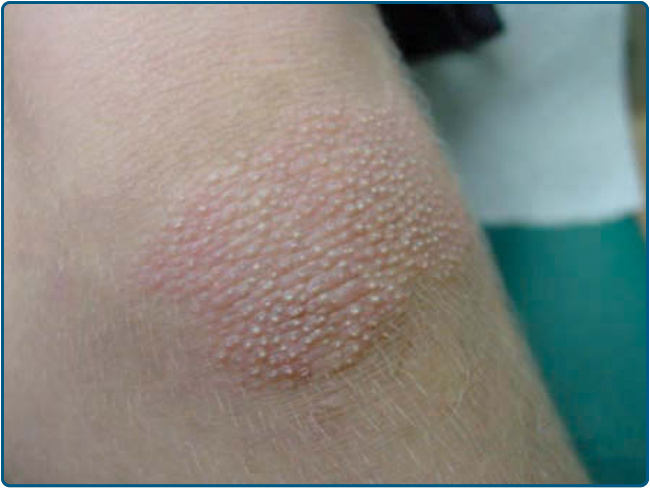
Figure 29-6 Circumscribed juvenile pityriasis rubra pilaris type IV. Well-demarcated hyperkeratotic erythematous plaques are found on the elbows and knees mimicking psoriasis.
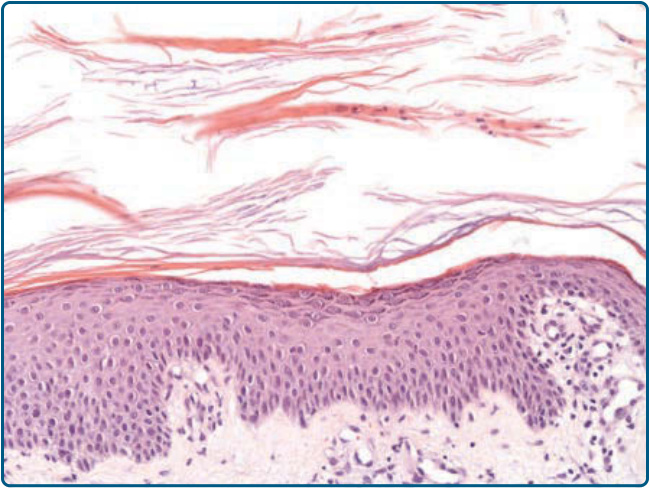
Figure 29-7 Histopathologic features of pityriasis rubra pilaris. Hyperkeratosis, alternating parakeratosis and orthokeratosis in a checkerboard pattern, with focal acantholytic dyskeratosis.
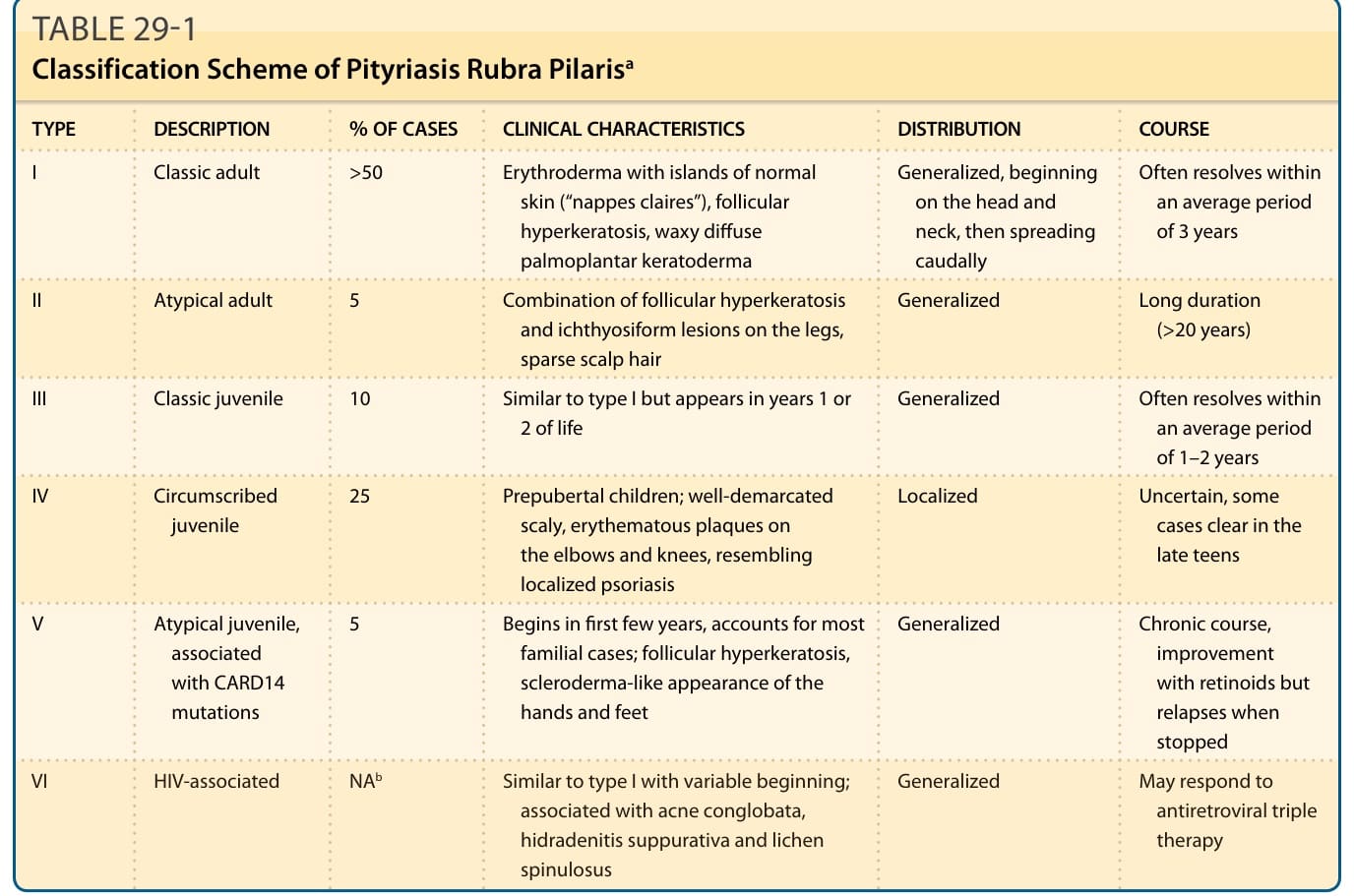
TABLE 29-1 Classification Scheme of Pityriasis Rubra Pilarisa

Table 29-2 outlines the differential diagnosis of pityriasis rubra pilaris.
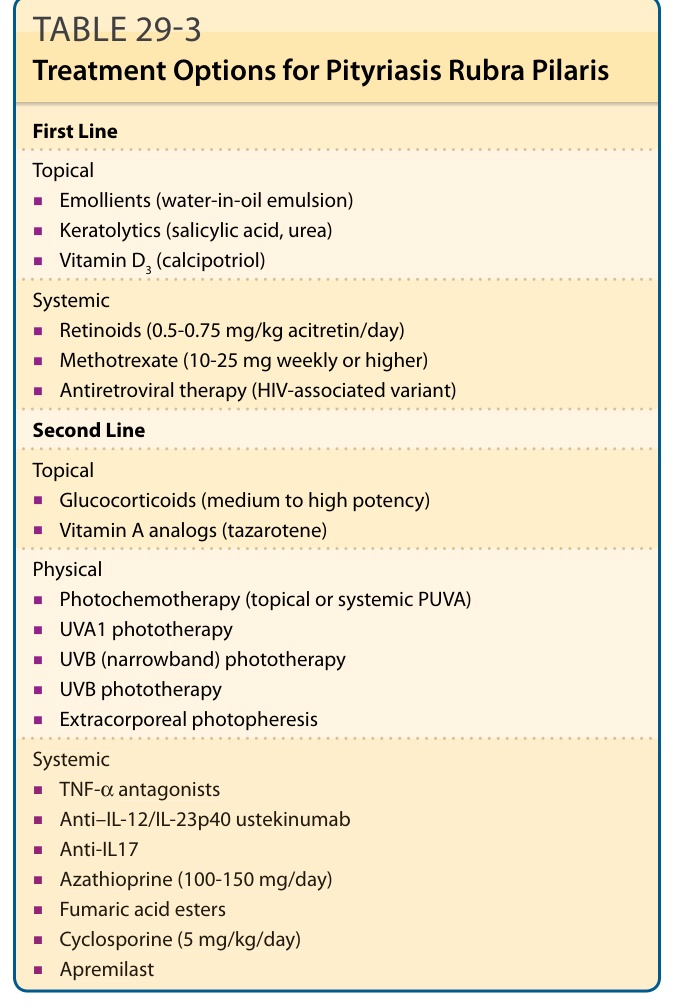
Table 29-3 lists the current therapeutic options (including drug doses) for PRP used in adults and children.