Antiviral Drugs
28
AT-A-GLANCE
■ Antivirals are approved for treatment of a variety of viral infections.
■ Antiviral resistance is a growing concern, especially in the treatment of HIV infection.
■ Antivirals work in a number of different ways, and their spectra of activity can be very specific (amantadine) or quite broad (ribavirin).
■ The use of prodrugs of acyclovir and ganciclovir has greatly increased the oral bioavailability of these agents, which allows outpatient treatment of many herpesvirus infections.
The pace of development of new antiviral drugs has been accelerated by the HIV (HIV) epidemic. Progress in our understanding of the molecular biology and pathogenesis of viral diseases has been remarkable. This chapter focuses on the antiviral drugs most likely to be used by dermatologists, as well as those that cause cutaneous side effects. The age of effective antiviral therapy is here, and they are used throughout all disciplines of medicine. We need to be prepared to evaluate patients on a wide variety of antiviral drugs, especially those currently used to treat HIV.
DRUGS FOR THE TREATMENT OF HERPESVIRUS INFECTIONS
See Chaps. 164 and 165.
ACYCLOVIR
ACYCLOVIR
MECHANISM OF ACTION
See Fig. 191-1. Acyclovir, 9-[(2-hydroxyethoxy)methyl] guanine, was the first orally available drug to be widely used for the treatment of herpes simplex virus (HSV) and varicella-zoster virus (VZV) infections. The triphosphate form of the drug is the active form, which has a potent inhibitory effect on herpesvirus-induced DNA polymerases but relatively little effect on host cell DNA polymerase. As such, it has a tremendous margin of safety when used to treat herpetic infections.
Acyclovir triphosphate causes premature termination of the nascent viral DNA chain. HSV- and VZV-induced thymidine kinases result in efficient phosphorylation of acyclovir to acyclovir monophosphate, the first step in drug metabolism. This step is not accomplished efficiently by normal cellular kinases, resulting in greater concentrations of active drug in infected cells.
PHARMACOKINETICS
Although acyclovir is available in oral, IV, and topical formulations, the oral bioavailability is only in the range of 15% to 30%, with the topical even less. Excretion is almost entirely renal, with approximately 62% of the drug remaining unmetabolized. Because of its reliance on renal excretion, the dose must be reduced for patients with a creatinine clearance of less than 50 mL/min. Acyclovir is watersoluble and distributes widely throughout the body, including into the contents of vesicles, cerebrospinal fluid (1:2 ratio from serum concentration), and vaginal secretions. Its half-life is 4 hours in neonates, 2 to 3 hours in children 1 to 12 years old, 2 to 3.5 hours in adults, and approximately 5 hours in hemodialysis patients.
INDICATIONS
■ Symptomatic primary or recurrent HSV1/2 infections
■ Chronic suppression of HSV1/2 infections
■ HSV encephalitis
■ Primary VZV infection, including HIV associated acute retinal necrosis
■ Herpes zoster (shingles)
■ Prevention of perinatal and treatment of neonatal HSV infection
■ HSV gingivostomatitis and orolabial cold sores (off-label)
■ Prevention of HSV, CMV, or VZV reactivation in hematopoietic stem cell transplant and HIV patients (off-label)
■ New onset Bell palsy (off-label)
Acyclovir is indicated for a variety of infections, including that of primary HSV1/2 as well as VZV. In general, treatments for herpes viral infections are most effective when initiated as early as possible, within 24 hours for varicella and 72 hours for herpes zoster onset. A buccal tablet formulation of acyclovir was approved by the FDA in 2013 for treatment of recurrent herpes labialis in immunocompetent adults. Acyclovir given in late pregnancy to women with
28
Mechanisms of action
Prodrug
Prodrug Valacyclovir
Cytoplasm
Acyclovir
Viral thymidine kinase*
Phosphorylation
Acyclovir Monophosphate
Cellular kinases
Phosphorylation
Acyclovir Triphosphate
Active form
Nucleus
Famciclovir
Penciclovir
Viral thymidine kinase*
Phosphorylation
Penciclovir Monophosphate
Cellular kinases
Phosphorylation
Penciclovir Triphosphate Active form
Competitive inhibition Competitive inhibition
Viral DNA polymerase
Acyclovir inserted into nascent DNA chain
Penciclovir inserted into nascent DNA chain
Premature termination of viral DNA synthesis
recurrent genital herpes has been shown to decrease the frequency of genital lesions, though there is some evidence that suggests standard oral dosing of acyclovir results in insufficient levels to prevent viral shedding on delivery.1 As a result, acyclovir treatment is still recommended to neonates of mothers with recent genital HSV infection. In addition, acyclovir has been shown to select for HIV-1 V85I reverse transcriptase variant in vitro,2
which suggests a possible role in reducing HIV viral load. However, it was shown that acyclovir did not significantly prevent transmission of HIV despite a reduction of 0.25log(10) copies per milliliter in HIV RNA in those treated for genital HSV.3 In fact, herpes virus infections in HIV patients is not yet well understood, and the only indicated treatment for HSV that is
3494
independent of HIV status is that of HSV encephalitis. All other indications, while useful on many occasions, remain off-label. Despite its high efficacy, resistance to acyclovir remains an issue with herpes infections. Data from corneal HSV-1 isolates suggests that infections commonly represent mixtures of acyclovir-sensitive and resistant viruses with different thymidine kinase gene sequences. The acyclovir-resistant HSV-1 can establish latency and reactivate intermittently to cause acyclovir-refractory disease.4
DOSING REGIMENS
Treatment regimens with acyclovir vary depending on the indication and route of administration. An
28
CONDITION DOSAGE ROUTE DURATION
Primary HSV infection, immunocompetent host 5 mg/kg every 8 h or 400 mg 3×/d
IV
5-7 d
Oral
7-10 d
Recurrent genital HSV infection, immunocompetent host 400 mg 3×/d or 800 mg 3×/d
Oral
5 d
Oral
2 d4
Recurrent oral labial HSV infection, immunocompetent host 400 mg 5×/d or 5% cream 6×/d
Oral
5 d
Topical
7 d
Mucocutaneous HSV infection, immunocompromised host6 5 mg/kg every 8 h or 400 mg 5×/d
IV
7 d
Oral
14-21 d
Perinatal HSV infection 10-20 mg/kg every 8 h IV 10-21 d
Chronic suppression of HSV infection 400 mg 2×/d or 800 mg every day
Oral <12 mo
HSV encephalitis 10 mg/kg every 8 h IV 14-21 d
Varicella: adolescent >40 kg/adult 800 mg 4×/d Oral 5 d
Varicella: pneumonia or third-trimester pregnancy 800 mg 5×/d or 10 mg/kg every 8 h
Oral
5 d
IV
5 d
Herpes zoster: normal host or not severe disease in immunocompromised host 800 mg 5×/d Oral 7-10 d
Herpes zoster: severe disease or immunocompromised host 10-12 mg/kg every 8 h IV 7-14 d
Herpes zoster: severe disease or immunocompromised host 10-12 mg/kg every 8 h IV 7-14 d
IV dose of 5 mg per kg every 8 hours or oral dose of 400 mg 3 times daily is generally recommended and more successful for patient compliance than 200 mg 5 times daily.5 Chronic suppression requires an oral dose of 400 mg 3 times daily for up to 12 months (Table 191-1A).6 The dose should be adjusted for
patients with a creatinine clearance level of less than 50 mL/min, but drug level assays are not routinely performed. Data for pediatric dosing for <12 years of age is limited but available, particularly recommended for HSV-related neonatal and encephalitis infections (Table 191-1B).7
CONDITION DOSAGE ROUTE DURATION
Primary HSV infection, immunocompetent host 40-80 mg/kg divided into 3-4 doses Oral 5-10 d
Recurrent genital HSV infection, immunocompetent host7 20-25 mg/kg 2×/d Oral —
Primary HSV, immunocompromised host 20 mg/kg 3×/d or 5-10 mg/kg every 8 h
Oral
5-10 d
IV
Mucocutaneous HSV infection, immunocompromised host6 5 mg/kg every 8 h or 400 mg 5×/d
IV
7 d
Oral
14-21 d
Neonatal HSV infection 20 mg/kg every 8 h IV 14-21 d
Chronic suppression of HSV infection 20 mg/kg 2×/d Oral 5-14 d
HSV encephalitis 10-15 mg/kg every 8 h IV 14-21 d
Varicella: >1 y old and <41 kg 20 mg/kg 4x/d Oral 5 d
Herpes zoster:
Herpes zoster: <1 y old ≥1 y old
<1 y old ≥1 y old
HSV, herpes simplex virus.
10 mg/kg every 8 h or 500 mg every 8 h
Oral 7-10 d
10 mg/kg every 8 h or 500 mg every 8 h
Oral 7-10 d
3495
28
SIDE EFFECTS AND PRECAUTIONS
Acyclovir is generally very well tolerated with uncommon reactions such as renal impairment (5% incidence). The major risk for impairment is renal tubular crystallization with rapid IV administration, though interstitial nephritis also has been reported. CNS toxicity is uncommon, but may manifest as lethargy, tremors, or seizures. Patients with these side effects commonly have underlying diseases involving the CNS. Thrombophlebitis is a known complication of infusion, and appears to be related to the high pH of the reconstituted solution (pH 11). It should be warned that acyclovir crosses the human placenta and is excreted in breastmilk. However, it has been determined safe for use (pregnancy category B; see Table 191-2) and recommended for herpes treatment during pregnancy. The drug is also safe for administration during active breast-feeding, so long as the nursing mother does not have active lesions near or on the breast.8 It has been shown to increase the risk of toxicity of foscarnet, mycophenolate, tenofovir, and zidovudine and decrease the therapeutic effects of talimogene laherparepvec, varicella vaccine, and zoster vaccine.
VALACYCLOVIR
VALACYCLOVIR
MECHANISM OF ACTION AND PHARMACOKINETICS
See Fig. 191-1. Valacyclovir, the l-valine ester of acyclovir, was developed to provide increased oral bioavailability of the active drug acyclovir.9 Valacyclovir is readily absorbed from the GI tract and almost entirely converted to acyclovir by intestinal and hepatic esterases. The mechanism of action and spectrum of activity of valacyclovir are identical to those of acyclovir, though improved from its predecessor. Valacyclovir has an oral bioavailability of 55%. Following a 1-g oral dose of valacyclovir, peak plasma concentrations of acyclovir in the range of 5.7 µg/mL are achieved in 1.75 hours, and area-underthe-curve (AUC) concentrations are similar to those achieved with 5 mg/kg of acyclovir given IV. Its
AGENT PREGNANCY CATEGORY
Acyclovir B
Valacyclovir B
Famciclovir B
Ganciclovir and valganciclovir C
Foscarnet C
3496
Cidofovir C
Cidofovir C
half-life ranges from 1.3 to 2.5 hours in children to approximately 30 minutes in adults. Excretion is primarily renal, with 89% metabolized to acyclovir. Like acyclovir, the dose must be adjusted for those with underlying renal impairment.
INDICATIONS
■ Initial and recurrent HSV genital infections
■ Suppression and reduction of transmission of genital HSV infections
■ Herpes zoster (shingles)
■ Herpes labialis (cold sores)
■ Varicella (chickenpox)
Valacyclovir therapy should be initiated at the earliest symptom of an HSV or VZV infection, within 72 hours of first diagnosis or 24 hours of onset of recurrent episode. It is preferred by patients due to its oral efficacy, compared to acyclovir, and ease of use in the outpatient setting. Treatment with valacyclovir has been suggested to decrease the frequency of clinical outbreaks as well as the incidence of viral shedding. Early studies showed a 30% reduction in viral shedding from valacyclovirtreated genital herpes compared to placebo-treated (P <.001).10,11 Recent data showed even more promising results, where valacyclovir given 1 g daily resulted in a 78% reduction in viral shedding compared to placebo, and 79% of subjects had no clinical recurrences while receiving valacyclovir compared with 52% of subjects receiving placebo (P <.01).12 Though less commonly, the drug has further been used to suppress recurrence of ocular herpes disease.13
In the setting of oral herpes simplex, valacyclovir has been used alone or combined with oral corticosteroids. In one study, there were more aborted lesions in the valacyclovir-clobetasol arm than in the placebo group (50% vs 15.8%, P = .04),14 but further study is needed to assess the independent contribution of the corticosteroid. Valacyclovir demonstrates in vitro activity against Epstein-Barr virus (EBV). It is being studied in the settings of infectious mononucleosis, hepatitis, encephalopathy, and posttransplant lymphoproliferative disease. Unfortunately, in a recent trial, valacyclovir treatment was not effective in decreasing peripheral blood EBV viral loads in pediatric liver transplantation patients.15 In healthy volunteers, long-term administration of valacyclovir effectively reduces the number of EBV-infected B cells, although it does not reduce the number of EBV DNA copies per B cell.16
DOSING REGIMENS
Standard doses for adults with herpetic infections range from 0.5 to 1 g 2 to 3 times daily, as listed in Table 191-3.17 The drug also has been evaluated in children with malignancy.18 In this setting, valacyclovir (15 mg/kg) was well tolerated and demonstrated excellent bioavailability.19 Those less than 3 months of age demonstrate decreased clearance of the drug. Among children 3 months through 11 years of age, a 20-mg/kg dose of an extemporaneously compounded
CONDITION DOSAGE ROUTE DURATION
Primary HSV infection, immunocompetent host
1000 mg 2×/d Oral 10 d
Recurrent genital HSV infection, immunocompetent host
500 mg 2×/d Oral 3 d
Recurrent oral labial HSV infection, immunocompetent host
2000 mg 2×/d Oral 1 d
Oral labial HSV infection, immunocompromised host
500 mg 2×/d Oral 5-10 d
Chronic suppression of HSV infection 1000 mg every day Oral Undefined/ indefinite
Varicella: adolescent/ adult 1000 mg 3×/d Oral 5 d
Herpes zoster: normal
1000 mg 3×/d Oral 7 d
Herpes zoster: normal host or not severe disease in immunocompromised host
1000 mg 3×/d Oral 7 d
host or not severe disease in immunocompromised host
HSV, herpes simplex virus.
valacyclovir oral suspension produced favorable acyclovir blood levels and was well tolerated. The authors noted that among children 2 through 5 years of age, a dose increase from 20 to 25 mg/kg resulted in near doubling of the peak concentration [C(max)] and the area under the curve (AUC).20
SIDE EFFECTS AND PRECAUTIONS
Most adverse effects are similar to those with acyclovir, including a potential for acute renal failure as well as CNS effects of agitation, hallucinations, seizures, and encephalopathy.21 Additionally, the drug has been reported to produce both immediate hypersensitivity and a symmetrical drug-related intertriginous and flexural exanthem.22,23 Severe effects of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS) have been reported in patients with AIDS as well as in transplant recipients receiving dosages of 8 g/d. TTP/HUS has not been reported in patients in patients taking conventional dosages (up to 3 g/d) of valacyclovir. It is labeled as pregnancy category B risk. Valacyclovir has been shown to precipitate in renal tubules and should be used with caution in those with renal impairment. Its most severe effects have been observed in elderly and immunocompromised patients, for which it is recommended to give the lowest dose possible. Given its metabolism to acyclovir, which can be detected in breastmilk, breast-feeding mothers should be aware that their nursing infants may receive small, though clinically insignificant, amounts of acyclovir. Common drug interactions
28
include increased toxicity risk of foscarnet, mycophenolate, tenofovir, zidovudine, and decreased therapeutic effects of talimogene laherparepvec, varicella vaccine, and zoster vaccine.
FAMCICLOVIR AND PENCICLOVIR
FAMCICLOVIR AND
PENCICLOVIR
MECHANISM OF ACTION
See Fig. 191-1. Famciclovir, [9-(4-hydroxy-3-hydroxymethylbut-1-yl)guanine], is biotransformed to the active compound penciclovir, [9-(4-hydroxy-3-hydroxy- 3-hydroxymethylbut-1-yl)guanine], which is phosphorylated to penciclovir triphosphate to eventually inhibit HSV-2 polymerase. The initial phosphorylation of penciclovir to penciclovir monophosphate is efficiently carried out by HSV- or VZV-induced thymidine kinases with subsequent phosphorylation to diphosphate and triphosphate forms occurring via cellular kinases. The mechanism by which penciclovir triphosphate competitively inhibits viral DNA polymerases is similar to that of acyclovir, though more DNA chain extension may occur with penciclovir because of its hydroxyl group on the acyclic side chain.
PHARMACOKINETICS
Famciclovir is marketed as an oral formulation, which is converted to penciclovir by deacetylation and oxidation in the liver and intestine. The bioavailability of oral famciclovir is 77%, with a peak plasma concentration of 3.3 µg/mL reached 1 hour after oral administration of 500 mg of famciclovir. The plasma half-life of the converted penciclovir is 2 to 4 hours, with 60% to 70% of the drug excreted in urine unchanged. As with acyclovir, the dose of penciclovir should be reduced in patients with advanced renal dysfunction. Compared with the intracellular half-life of acyclovir triphosphate, that of penciclovir triphosphate is markedly prolonged in both HSV-infected cells (10 to 20 hours) and VZV-infected cells (7 hours), allowing the drug to be administered 2 to 3 times daily. Penciclovir is available as a 1% ointment for the treatment of recurrent oral HSV. At least in some vehicles, topical penetration of penciclovir is superior to that of acyclovir.24 Microemulsion and nanoparticle formulations investigations concluded that these could be a promising vehicle for topical delivery of penciclovir.25-27
INDICATIONS
■ Initial and recurrent HSV genital infections
■ Suppression of frequently recurring genital HSV infections
■ Initial and recurrent HSV labialis (cold sores) infections
■ Herpes zoster (shingles)
■ Varicella infection (chickenpox) in HIV patients (off-label)
3497
28
As with acyclovir and valacyclovir, famciclovir and penciclovir should be initiated as soon as possible after diagnosis, ideally within 72 hours of rash onset. Famciclovir also has been shown to be an effective, welltolerated option for the suppression of genital herpes among individuals with multiple recurrences.28 Famciclovir is also equivalent to acyclovir for treatment of ophthalmic zoster.29
DOSING REGIMENS
Famciclovir is available as an oral agent and penciclovir as a topical cream. Standard adult doses are given in Table 191-4.30 Pediatric doses are generally of increased treatment duration though with similar amount per dose, for example, 500 mg twice daily. In one study of recurrent genital HSV, a 2-day course of 500 mg initially, then 250 mg twice daily, was noninferior to the standard 5-day course of 125 mg twice daily.31 In another study, single-day famciclovir (1000 mg administered twice daily) was similar in efficacy to 3-day valacyclovir (500 mg administered twice daily) for the treatment of recurrent genital herpes.32 Single-day treatment is associated with high patient satisfaction.33
Some data suggest that suppressive treatment may be superior to episodic treatment.34 Studies in children are ongoing, and currently the drug is used less commonly in this population.35,36 Although famciclovir has shown efficacy in general populations, one study of patientinitiated episodic treatment of recurrent genital herpes
CONDITION DOSAGE ROUTE DURATION
Primary HSV infection, immunocompetent host
250 mg 3×/d Oral 7-10 d
Recurrent genital HSV infection, immunocompetent host
125 mg 2×/d or 1000 mg 2×/d
Oral
5 d
Oral
1 d
Recurrent oral labial HSV infection, immunocompetent host
1500 mg every day or 1% penciclovir cream every 2 h
Oral
1 d
Topical
5 d or until lesions healed
Oral labial HSV infection, immunocompromised host
500 mg 2×/d Oral 7 d
Chronic suppression of HSV infection 250 mg 2×/d Oral Up to 1 y
Varicella: adolescent/ adult 500 mg 3×/d Oral 5-7 d
Herpes zoster: normal
500 mg 3×/d Oral 7 d
Herpes zoster: normal host or not severe disease in immunocompromised host
500 mg 3×/d Oral 7 d
host or not severe disease in immunocompromised host
3498
HSV, herpes simplex virus.
in immunocompetent black patients showed efficacy similar to that of placebo.37
SIDE EFFECTS AND PRECAUTIONS
Like acyclovir and valacyclovir, famciclovir is generally well tolerated.38 The dose should be reduced for patients with a creatinine clearance less than 60 mL/min. The drug has been used safely along with hydration in patients with prior renal toxicity related to acyclovir.39 Leukocytoclastic vasculitis has been reported with the drug.40 Common side effects include:
■ headache,
■ nausea,
■ diarrhea, and
■ dizziness.
Serious toxicity is uncommon and the drug is labeled a pregnancy risk factor B. Famciclovir has been shown to diminish the therapeutic effect of talimogene laherparepvec, varicella virus vaccine, and zoster vaccine.
TRIFLURIDINE
TRIFLURIDINE
MECHANISM OF ACTION AND PHARMACOKINETICS
Trifluridine, 5-trifluoromethyl-2′-deoxyuridine, is a pyrimidine nucleoside analog. Trifluridine monophosphate acts as an irreversible competitive inhibitor of thymidylate synthetase, and trifluridine triphosphate inhibits HSV DNA polymerase. Trifluridine triphosphate also inhibits cellular DNA polymerases, although it does so to a lesser extent than viral DNA polymerases. Because of its mechanism, it is considered a potent drug with expected response to treatment within 2 to 7 days. Because of systemic toxicity, trifluridine is approved only for topical application in the form of a 1% ophthalmic aqueous solution. The elimination half-life for the ophthalmic solution is 12 minutes.
INDICATIONS
■ Primary and recurrent HSV keratoconjunctivitis, keratitis
■ Acyclovir-resistant mucocutaneous HSV infections in patients with AIDS (off-label)
Trifluridine is indicated for the treatment of primary keratoconjunctivitis and recurrent epithelial keratitis caused by HSV types 1 and 2. It also has been suggested to benefit acyclovir-resistant HSV infections in AIDS patients. The drug offers a unique and effective delivery route that has similar efficacy to others of its class. A meta-analysis data from 99 randomized trials of 5363 total participants showed no significant difference between topical trifluridine, vidarabine, acyclovir, or ganciclovir in treatment of dendritic epithelial
28
CONDITION DOSAGE ROUTE DURATION
HSV keratoconjunctivitis/ keratitis Induction: 1 drop every 2 h (maximum, 9 drops/24 h) Maintenance: 1 drop every 4 h (maximum, 6 drops/24 h) Topical Maximum, 21 d
Acyclovir-resistant HSV infection in AIDS 1 drop every 8 h Topical 10-14 d or until lesions resolve
Acyclovir-resistant HSV infection in
1 drop every 8 h Topical 10-14 d or until lesions resolve
AIDS
HSV, herpes simplex virus.
keratitis within 1 week of therapy.41 A 2010 systematic review of 106 trials found that use of any of 4 topical antiviral agents (trifluridine, acyclovir ganciclovir, or brivudine) were equally effective and resulted in healing of 90% of the eyes within 2 weeks.42 However, other authors have suggested that although the drug is effective, it may result in delays in reepithelialization of corneal ulcers.43 As a result, there may be additional benefit from topical interferon in those treated with this agent.44
DOSING REGIMENS
Standard dosing regimens consist of 1 drop of 1% solution every 2 hours until reepithelialization of ulcer occurs, followed by 1 drop every 4 hours for another 7 days. Regimens are provided in Table 191-5. It is recommended to consider another form of therapy if no clinical improvement is seen at 7 to 14 days.
SIDE EFFECTS AND PRECAUTIONS
Adverse reactions to trifluridine are generally minor, mainly manifesting as transient local burning or stinging (5%), palpebral edema (3%), epithelial and punctate keratopathy, and stromal edema. It is considered a pregnancy category C risk with systemic application, but the amount available systemically following topical application of the ophthalmic drops has been considered negligible.
DRUGS FOR THE TREATMENT OF CYTOMEGALOVIRUS INFECTIONS
GANCICLOVIR AND VALGANCICLOVIR
GANCICLOVIR AND
VALGANCICLOVIR
MECHANISM OF ACTION AND PHARMACOKINETICS
Ganciclovir, 9-(1,3-dihydroxy-2-propoxymethyl)guanine, is phosphorylated by viral kinases to a substrate that
competitively inhibits the binding of deoxyguanosine triphosphate to DNA polymerase, thereby inhibiting viral DNA synthesis. It is effective in treating cytomegalovirus infections intravenously, though oral bioavailability is only 5%. Valganciclovir, l-valine 2-[(2-amino-1,6-dehydro-6-oxo-9H-purin-9-yl) methoxy]-3-hydroxypropyl ester, is the l-valyl ester of ganciclovir that is more bioavailable. After oral administration, valganciclovir is converted to ganciclovir by intestinal and hepatic esterases with an oral availability of approximately 60% that is increased by 30% when administered with a high-fat meal. After a 900-mg dose of valganciclovir, the maximum plasma concentration of ganciclovir is 5.61 µg/mL, and the plasma concentration–time curve is similar to that achieved with ganciclovir given intravenously at a dose of 5 mg/kg. The elimination half-life is approximately 4 hours, though delayed in those with renal impairment.
INDICATIONS
■ CMV retinitis in immunocompromised patients
■ Suppression and prevention of CMV disease in transplant recipients
■ CMV esophagitis, colitis, or neurologic disease in HIV patients (off-label)
Intravenous ganciclovir and oral valganciclovir are both approved for use in the treatment of cytomegalovirus in immunocompromised patients as well as in prevention of CMV disease in transplant patients.45-47
DOSING REGIMENS
Standard dosing regimens include 5 mg/kg/dose of ganciclovir every 12 hours for induction followed by every 24 hours for maintenance therapy. The equivalent dosing of valganciclovir is 900 mg twice daily induction followed by once-daily maintenance. Dosing summary is provided in Table 191-6. Pediatric dosing regimens also have been published and are similar to adult dosing.48
SIDE EFFECTS AND PRECAUTIONS
Potential side effects include a variety of GI (ie, diarrhea, nausea, and loss of appetite) as well as hematologic side effects (ie, anemia, thrombocytopenia, neutropenia, bone marrow aplasia, and aplastic anemia). Acute
3499
28
CONDITION DOSAGE ROUTE DURATION
CMV retinitis Induction: 900 mg 2×/d 5 mg/kg every 12 h Oral IV 21 d 14-21 d
Maintenance:
Maintenance: 900 mg every day
900 mg every day
Oral
Until CD4 count
Oral
Until CD4 count >100 after 6 mo of HAART
100 after 6 mo of HAART
5 mg/kg every day
IV
5 mg/kg every day
IV
CMV, cytomegalovirus; HAART, highly active antiretroviral therapy.
renal failure may occur, for which doses should be immediately adjusted to half the regular dose. Nonspecific adverse reactions include headache, dizziness, confusion, nervousness, vivid dreams, tremor, weakness, peripheral edema, and pain at the injection site. Known drug interactions include potentiation of imipenem, mycophenolate, probenecid, reverse transcriptase inhibitors, and tenofovir. It is labeled as pregnancy category C and recommended to use contraception during and 30 to 90 days post treatment. Breast-feeding is not recommended, since it is not known if ganciclovir or valganciclovir are excreted into breastmilk.
FOSCARNET
FOSCARNET
MECHANISM OF ACTION
Foscarnet, trisodium phosphonoformate, is a pyrophosphate-containing antiviral drug that noncompetitively inhibits viral DNA polymerases at the pyrophosphate binding site. In contrast to the nucleoside analogs, foscarnet does not require phosphorylation and is therefore active against many strains of virus that are resistant to acyclovir, famciclovir, or ganciclovir as a result of absent or reduced kinase activity.49-51 Salvage therapy with foscarnet plus a thymidine analog has been shown to be effective in patients with advanced-stage HIV disease and viruses harboring multiple drug-resistance mutations including thymidine-associated mutations.52 Unfortunately, resistance to combined ganciclovir and foscarnet therapy has been reported in the setting of dual-strain cytomegalovirus coinfection.53 Valproic acid has been reported to impair the antiviral activity of ganciclovir, cidofovir, and foscarnet.54
PHARMACOKINETICS
Foscarnet is available as an IV preparation that has poor solubility and must be administered by an infusion pump in a dilute solution over 1 to 2 hours. The drug has a half-life of 3 to 4 hours initially with a
3500
terminal component of 88 hours or longer, for which up to 20% of the cumulative dose may be deposited in bone. Twenty-eight percent of the dose is excreted unaltered by the kidney.
INDICATIONS
■ CMV retinitis
■ Acyclovir-resistant HSV infections
■ CMV esophagitis or colitis (off-label)
■ Ganciclovir-resistant CMV infections (off-label)
DOSING REGIMENS
Standard dosing schedules consist of induction therapy of 40 to 90 mg/kg/dose every 8 to 12 hours followed by maintenance therapy of 90 to 120 mg/kg/d. Specific regimens are provided in Table 191-7. The dose must be adjusted accordingly in patients with renal dysfunction.
SIDE EFFECTS AND PRECAUTIONS
Renal toxicity (30%) is the major risk with foscarnet, for which close monitoring of renal function is required. Impairment of renal function typically occurs during the second week of induction therapy and is reversible within 1 week following dose adjustment or discontinuation of therapy. Saline hydration before administration and slow infusion of the drug may reduce nephrotoxicity risk. In addition, the drug has been associated with electrolyte and metabolic abnormalities including hypocalcemia, hyper/ hypophosphatemia, hypomagnesaemia, and hypokalemia, described with up to 48% incidence. Other common adverse reactions include nausea/vomiting with diarrhea (<47%), anemia (33%), granulocytopenia (17%), fever (65%), and headache (26%).
CONDITION DOSAGE ROUTE DURATION
CMV retinitis Induction: 90 mg/kg every 12 h Maintenance: 90 mg/kg every 24 h
IV
14-21 d
IV
Until CD4 count >100 on 6 mo of HAART
Acyclovir-resistant HSV infection 40 mg/kg every 8 h or 60 mg/kg every 12 h
IV 14-21 d
2-3 wk or until clinical resolution
CMV esophagitis/
90 mg/kg
IV 21-42 d or until
CMV esophagitis/ colitis 90 mg/kg every 12 h IV 21-42 d or until clinical resolution
colitis
every 12 h
clinical resolution
CMV, cytomegalovirus; HAART, highly active antiretroviral therapy; HSV, herpes simplex virus.
Careful monitoring for renal impairment and seizures is recommended because of foscarnet’s effects on renal function and electrolytes, respectively. Because of its sodium content, foscarnet use is cautioned in patients with underlying heart failure. It has been determined as pregnancy category C risk, for which ultrasonographic monitoring of amniotic fluid volumes is recommended after 20 weeks to detect oligohydramnios. It not known whether foscarnet is excreted in breastmilk. Known drug interactions include potentiation of adverse side effects of acyclovir/valacyclovir, aminoglycosides, amphotericin B, cyclosporine, QTc-prolonging agents, methotrexate, and tacrolimus. Conversely, loop diuretics and pentamidine have been shown to enhance the toxic effect of foscarnet.
CIDOFOVIR
CIDOFOVIR
MECHANISM OF ACTION AND PHARMACOKINETICS
Cidofovir, (S)-1-[3-hydroxy-2(phosphonylmethoxy)- propyl]cytosine, is a phosphonate nucleotide analog.55 Unlike other nucleoside analogs, it does not require initial phosphorylation by virus-induced kinases and can be converted by host cell enzymes to cidofovir diphosphate, a competitive inhibitor of viral DNA polymerases. Cidofovir is officially approved for IV administration, though a topical formulation can be compounded for off-label usage. It has a volume of distribution of 0.31 L per kg with low CSF penetration; therefore, an IV dose of 5 mg/kg results in a peak plasma concentration of 11.5 µg/mL. Plasma half-life is approximately 2.6 hours with markedly longer intracellular half-life of 24 to 87 hours. Excretion is mainly renal, which can be facilitated by the addition of concomitant probenecid.
INDICATIONS AND CONTRAINDICATIONS
■ CMV retinitis
■ Acyclovir-resistant HSV infections (off-label)
Cidofovir is approved for IV treatment of CMV retinitis particularly in those with AIDS, though it has been shown in multiple incidences to be effective in acyclovir-resistant HSV infections,5,56 both via IV and topical routes, as well as in topical treatments for human papillomavirus infections, Kaposi sarcoma, and molluscum contagiosum.57-65 Given its significant renal metabolism, cidofovir is contraindicated in those with preexisting renal impairment, as evidenced by serum creatinine >1.5 or >2+ proteinuria.
DOSING REGIMENS
For adults, cidofovir may be given at an induction dose of 5 mg/kg once weekly for the first 2 weeks, followed by a 5-mg/kg dose once every 2 weeks (Table 191-8).
28
CONDITION DOSAGE ROUTE DURATION
CMV retinitis Induction: 5 mg/kg every week IV 2 wk
Maintenance: 5 mg/kg every 2 wk
Maintenance:
IV Until CD4 count
IV Until CD4 count >00 after 6 mo on HAART
5 mg/kg every 2 wk
00 after 6 mo on HAART
CMV, cytomegalovirus; HAART, highly active antiretroviral therapy. Note: Cidofovir IV administration must be accompanied by prehydration and probenecid.
Data regarding the use in pediatric patients are accumulating.66
SIDE EFFECTS AND PRECAUTIONS
Common adverse effects consist of renal impairment (59% incidence), which consists of:
■ renal tubular damage,
■ dose-dependent proximal tubular injury (Fanconi-like),
■ proteinuria, and
■ elevated serum creatinine levels.
Other common effects include nausea (48%), alopecia (16%), rash, fever, myalgias, asthenia, headache, abdominal pain, diarrhea, nausea, dyspepsia, flatulence, increased creatinine, pancreatitis, and hypophosphatemia. The incidence of nephrotoxicity can be reduced by vigorous hydration before and after infusion, in addition to coadministration of probenecid. In addition to close monitoring of renal function, the drug should not be used concurrently with tenofovir disoproxil fumarate (TDF) because of a risk of increased tenofovir levels and toxicity. The following rare but possible effects can occur: squamous cell carcinoma associated with intralesional injection of cidofovir to treat human papillomavirus (HPV)–related recurrent respiratory papillomatosis,67-69 and herpes simplex stomatitis associated with prophylactic cidofovir therapy.70
Cidofovir has been labeled pregnancy category C, suggesting possible teratogenic risk. It is thus recommended that women of childbearing potential use effective contraception during and for 1 month following treatment and that men use barrier contraception during and for 3 months following treatment. The indications for treating CMV retinitis during pregnancy are the same as in nonpregnant HIV-infected women; however, therapy with cidofovir should be avoided during the first trimester when possible. It is not known whether cidofovir is excreted in breastmilk, but generally breast-feeding on cidofovir is not recommended because of the potential for serious adverse reactions in the nursing infant.
3501
28
OTHER INFECTIONS
MECHANISM OF ACTION
Interferons are cytokines derived from a variety of cells that interact through high-affinity cell surface receptors. They have broad antiviral and immunomodulating effects that include induction of gene transcription, inhibition of cellular growth, interference with oncogene expression, alteration of cell surface antigen expression, increased phagocytic activity of macrophages, and cytotoxicity of lymphocytes.
PHARMACOKINETICS
Interferon is commonly given subcutaneously, but also may be administered intravenously, intralesionally, or intramuscularly. It has a plasma half-life of 2 to 3 hours after IV administration, and 4 to 6 hours after subcutaneous or intramuscular administration. A prolonged half-life can be obtained with pegylated interferon.
INDICATIONS
Interferon therapy has been approved for a variety of viral infections (IFN-alpha), for multiple sclerosis (IFN-beta), for chronic granulomatous disease (IFNgamma), as well as for a number of neoplastic diseases (IFN-alpha), including melanoma. It also has been useful as off-label therapies for chronic myelogenous leukemia, cutaneous T-cell lymphoma, Behçet disease, desmoid tumor, multiple myeloma, neuroendocrine tumors, renal cell carcinoma, and West Nile virus. In the setting of viral infections, interferon-α has been studied most extensively and includes alphacon-1, alpha-2a/b, and alpha-n3.
VIRAL DISEASES TREATED WITH INTERFERON
■ Condylomata acuminata (intralesional)
■ Chronic hepatitis C infection +/− in combination with ribavirin (IM, subcutaneous [SC])
■ Chronic hepatitis B infection (IM, SC)
■ AIDS-related Kaposi sarcoma (IM, SC)
DOSING REGIMENS
Common dosing regimens are provided in Table 191-9. Pediatric dosing is adjusted by one-third to one-half of adult dosing with recommendation to consider acetaminophen premedication prior to interferon administration to reduce the incidence of some adverse reactions.
3502
CONDITION DOSAGE ROUTE DURATION
Kaposi sarcoma IFNα-2b 30 million units 3×/wk
IM, Subcutaneous Dependent on response
Condyloma acuminata IFNα-2b 1 million units 3×/wk or IFNα-n3 250k units 2×/wk
Intralesional 3 wk
Up to 8 wk
PEG-IFNα-2a 180 µg once a week or PEG-IFNα-2b 1.5 µg/kg once a week or IFN-alfacon-1 15 µg/d or IFNα-2b 3 million units 3×/wk
Chronic hepatitis C (with ribavirin)
Subcutaneous
Dependent on response and genotype
Up to 48 wk
IM, Subcutaneous
18-24 mo
PEG-IFNα-2a
Chronic hepatitis B PEG-IFNα-2a 180 µg once a week or IFNα-2b 10 million units 3×/wk
Chronic
Subcutaneous
48 wk
48 wk
Subcutaneous
hepatitis B
180 µg once a week or IFNα-2b
IM,
16 wk
IM, Subcutaneous
16 wk
Subcutaneous
10 million units 3×/wk
IFN, interferon; PEG-IFN, peginterferon.
SIDE EFFECTS AND PRECAUTIONS
Side effects with interferon are common and often result in discontinuation of the drug. In a majority of patients, flulike symptoms of fever, chills, tachycardia, malaise, myalgia, and headache may occur within 1 to 2 hours of administration. The most common cutaneous side effects include injection site reactions, alopecia, psoriasis, fixed drug eruptions, eczematous drug reactions, sarcoidosis,71 lupus, pigmentary changes, and lichenoid eruptions.72,73 Other adverse effects include bone marrow suppression as well as GI- and hepatotoxicity for which blood counts and metabolic panels should be monitored. Interferon-α has further been shown to cause or aggravate neuropsychiatric events for which patients should be monitored for depression psychosis, cognitive changes, and suicidal and homicidal ideation. Interferon-α should not be used to treat hepatitis B–related cirrhosis or decompensated liver disease because hepatitis flares could lead to further decompensation. Precaution should be taken in prescribing interferon to patients with underlying eye, autoimmune, cardiovascular, ischemic, infectious, or lung disease. Alone, interferon is labeled as pregnancy risk factor C.
Combination therapy of interferon-α with ribavirin is associated with birth defects (category X) and is contraindicated in pregnant women as well as males of pregnant partners. Interferon may enhance the activity of aldesleukin, clozapine, deferiprone, dypnone, methadone, ribavirin, theophylline, tizanidine, zidovudine, and telbivudine.
DRUGS FOR THE TREATMENT OF HIV INFECTION
Drug treatment of HIV infection requires specialized training and experience. The US Department of Health and Human Services (HHS) and the World Health Organization (WHO) publish guidelines for the treatment of HIV. Both sets of guidelines are updated regularly as evidence from ongoing clinical trials emerges and therefore should be consulted routinely. The DHHS guidelines are available at http:// aidsinfo.nih.gov/guidelines, and the WHO guidelines are available at http://www.who.int/hiv/pub/ guidelines.
INTRODUCTION
INTRODUCTION
HIV infection is treated with antiretroviral therapy (ART) which involves the use of multidrug regimens to reduce HIV-associated morbidity, prolong the duration and quality of survival, and prevent HIV transmission. As of June 2016, there are more than 25 available antiretroviral (ARV) drugs in 6 major classes (Table 191-10). Multiple comparative clinical trials have shown that combination therapy involving multiple drugs from different classes is most effective. However, eradication of HIV infection cannot be achieved with available ARV regimens, primarily because the pool of latently infected CD4 T cells is established during the earliest stages of acute HIV infection and persists with a long half-life.74
ARV drugs work by blocking the virus at various points in its 7-stage life cycle. The stages of the HIV life cycle and the classes of ARV drugs that block them are as follows74,75:
- Binding (also called Attachment): The viral envelope glycoprotein binds to the host receptors (CD4) and coreceptors (CC-chemokine receptor 5 (CCR5) or CXC-chemokine receptor 4 (CXCR4)). Entry inhibitors target this stage. The only FDA-approved drug that targets this stage is a CCR5 inhibitor.
- Fusion: The HIV envelope and the host cell membrane fuse enabling the entry of the viral capsid in the host cell. Fusion inhibitors target this stage.
- Reverse Transcription: The viral reverse transcriptase converts its genetic material (HIV RNA) into HIV double-stranded DNA. Nucleoside/ nucleotide analog reverse transcriptase inhibitors
28
Entry Inhibitor Maraviroc (MVC)
Fusion Inhibitor Enfuvirtide (T-20)
Nucleoside and Nucleotide Reverse Transcriptase Inhibitors (NRTIs) Abacavir (ABC) Didanosine (ddI) Emtricitabine (FTC) Lamivudine (3TC) Stavudine (d4T) Tenofovir disoproxil fumarate (TDF) Zidovudine (ZDV, AZT)
Nonnucleoside Reverse Transcriptase Inhibitors (NNRTIs) Delavirdine (DLV) Efavirenz (EFV) Etravirine (ETR) Nevirapine (NVP) Rilpivirine (RPV)
Integrase Strand Transfer Inhibitors (INSTIs) Dolutegravir (DTG) Elvitegravir (EVG) Raltegravir (RAL)
Protease Inhibitors (PIs) Atazanavir (ATV) Atazanavir/ritonavir (ATV/r) Atazanavir/cobicistat (ATV/c) Darunavir (DRV) Darunavir/cobicistat (DRV/c) Fosamprenavir (FPV) Indinavir (IDV) Lopinavir/ritonavir (LPV/r) Nelfinavir (NFV) Saquinavir (SQV) Tipranavir (TPV)
Pharmacokinetic Enhancers/Boosters
Pharmacokinetic Enhancers/Boosters Ritonavir (RTV) Cobicistat (COBI)
Ritonavir (RTV) Cobicistat (COBI)
(NRTIs) and nonnucleoside reverse transcriptase inhibitors (NNRTIs) target this stage.
4. Integration: The viral integrase integrates the viral DNA into the host DNA. Integrase strand transfer inhibitors (INSTIs) target this stage.
5. Replication: HIV protein chains and RNA are produced using the host cell machinery.
6. Assembly: The newly replicated proteins and RNA move to the surface of the host cell and assembly into immature virions.
7. Budding and Maturity: The newly formed virion buds out of the host cell and releases protease that breaks up the viral protein chains, ultimately resulting in mature virions. Protease inhibitors (PIs) target this stage, which results in inhibition of reverse transcription and possibly other downstream stages in the life cycle, including integration.
In addition, 2 drugs are pharmacokinetic (PK) enhancers/boosters used solely to improve the
3503
28
pharmacokinetic profiles of some ARV. These are listed in Table 191-10.74
ART was shown to reduce HIV-related morbidity and mortality, and reduce perinatal and behaviorassociated transmission of HIV. HIV suppression with ART may also decrease inflammation and immune activation thought to contribute to higher rates of cardiovascular and other end-organ damage reported in HIV-infected cohorts. Maximal and durable suppression of plasma viremia delays or prevents the selection of drug-resistance mutations, preserves CD4 T-cell numbers, and confers substantial clinical benefits, all of which are important treatment goals.74
THERAPY INITIATION RECOMMENDATIONS
THERAPY INITIATION
RECOMMENDATIONS
Antiretroviral therapy (ART) is recommended for all HIV-infected individuals to reduce the risk of disease progression, regardless of CD4 count, age group or pregnancy/lactation. Both the HHS and WHO expanded the recommendation of ART to all HIVinfected individuals in 2015. In January 2016, based on new findings, the HHS gave the highest strength and evidence rating for this recommendation.74,76,77
For most patients, ART is initiated soon after the initial diagnosis. However, several conditions increase the urgency for therapy, these include pregnancy, presence of HIV-related complications, opportunistic infections, chronic hepatitis B and C infection, acute symptomatic HIV infection, and CD4 ≤200 cells/µL, rapidly declining CD4 counts, and higher viral loads (eg, >100,000 copies/mL).74
THERAPY REGIMEN RECOMMENDATIONS
THERAPY REGIMEN
RECOMMENDATIONS
The initial ART regimen for a treatment-naive patient generally consists of 2 NRTIs, combined with an INSTI, an NNRTI, or a pharmacologically boosted PI. This strategy has resulted in decreased HIV RNA and increase in CD4 T lymphocyte (CD4) cell in most patients. However, on the basis of individual patient characteristics and needs, an alternative regimen may in some instances be the optimal regimen.74,76
3504
POPULATION PREFERRED FIRST- LINE REGIMEN ALTERNATIVE FIRST-LINE REGIMENS
Adults TDF + 3TC (or FTC) + EFV AZT + 3TC + EFV (or NVP) TDF + 3TC (or FTC) + DTG TDF + 3TC (or FTC) + EFV TDF + 3TC (or FTC) + NVP
Pregnant/ breastfeeding women
TDF + 3TC (or FTC) + EFV AZT + 3TC + EFV (or NVP) TDF + 3TC (or FTC) + NVP
Adolescents TDF + 3TC (or FTC) + EFV AZT + 3TC + EFV (or NVP) TDF (or ABC) + 3TC (or FTC) + DTG TDF (or ABC) + 3TC (or FTC) + EFV TDF (or ABC) + 3TC (or FTC) + NVP
Children 3 y to less than 10 y ABC + 3TC + EFV ABC + 3TC + NVP AZT + 3TC + EFV (or NVP) TDF + 3TC (or FTC) + EFV (or NVP)
Children less
Children less than 3 y ABC (or AZT) + 3TC + LPV/r ABC (or AZT) + 3TC + NVP
ABC (or AZT) +
ABC (or AZT) + 3TC + NVP
than 3 y
3TC + LPV/r
Note: For expansions of the abbreviations used, please refer to Table 191-10.
drugs should be started simultaneously rather than sequentially.74
When initial suppression is not achieved or is lost, rapidly changing to a new regimen with at least 2 active drugs is required. Thus, when prescribing a first-line regimen, second- and third-line strategies should be formulated, keeping in mind the high-level cross-resistance seen among the same class of drugs. Indiscriminate use of the drugs will lead to minimal options for the future. The increasing number of drugs and drug classes makes viral suppression below detection limits an appropriate goal in all patients.74
Viral load reduction to below limits of assay detection in an ART-naive patient usually occurs within the first 12 to 24 weeks of therapy.74
PREVENTION
PREVENTION
PREEXPOSURE PROPHYLAXIS (PrEP)
Oral PrEP of HIV is the use of ARV drugs by HIVnegative people before potential exposure to prevent infection.78 The WHO recommends oral PrEP containing TDF for all populations with substantial risk of HIV infection (defined by an incidence of HIV infection in the absence of PrEP greater than 3%). For such populations, oral PrEP should be offered as an additional prevention choice as part of combination of prevention approaches, including regular HIV testing and counseling, provision of condoms, screening and treatment
28
PROPHYLAXIS TYPE POPULATION REGIMEN
Preexposure prophylaxis (PrEP) All populations at substantial risk TDF (alone or in combination with FTC)
Recommended 2-Drug Regimen Alternative 2-Drug Regimen Preferred Additional 3rd Drug Preferred Additional 3rd Drug Alternative
Postexposure prophylaxis (PEP) Adults and adolescents TDF + 3TC (or FTC) N/A LPV/r or ATV/r RAL, DRV/r or EFV
Children ZDV + 3TC ABC + 3TC TDF + 3TC (or FTC) LPV/r Age-appropriate selection from ATV/r, RAL, DRV, EFV, and NVP
Children ZDV + 3TC ABC + 3TC TDF + 3TC (or FTC)
Note: For expansions of the abbreviations used, please refer to Table 191-10.
for sexually transmitted infections, and adherence counseling.74,78 Furthermore, HIV testing is required before PrEP is offered and regularly while PrEP is taken. Renal function testing using serum creatinine testing is preferred before starting PrEP and quarterly during PrEP use for the first 12 months then annually thereafter. These populations may include people engaging in high-risk sexual behaviors, people who inject drugs, people in prisons and closed settings, and sex workers.78,79 Table 191-12 shows the WHO’s recommend regimen for PrEP.
POSTEXPOSURE PROPHYLAXIS (PEP)
PEP of HIV is the use of ARV drugs by HIV-negative people after exposure to a known or suspected (high-risk) source of HIV infection. PEP should be prescribed following occupational exposure to HIV by health workers and following non-occupational exposures, including unprotected sexual exposure, injecting drug use and exposure following sexual assault. The WHO recommends at least a 2-drug PEP regimen, but prefers 3 drugs. A full 28-day prescription of antiretrovirals should be provided for HIV PEP following initial risk assessment, and enhanced adherence counseling is suggested for all individuals initiating HIV PEP. Data from animal studies suggest that the efficacy of PEP in preventing transmission is time dependent, and every effort should be made to
LPV/r Age-appropriate selection
from ATV/r, RAL, DRV, EFV, and NVP
provide PEP as soon as possible following exposure.79
ADVERSE EFFECTS
ADVERSE EFFECTS
Adverse effects have been reported with the use of all antiretroviral (ARV) drugs and are among the most common reasons cited for switching or discontinuing therapy and for medication nonadherence. Fortunately, newer ARV regimens are less toxic than regimens used in the past. Generally less than 10% of antiretroviral therapy (ART)-naive patients enrolled in randomized trials have treatment-limiting adverse events.74
Several factors may predispose individuals to adverse effects of ARV medications, these include74
■ concomitant use of medications with overlapping and additive toxicities,
■ comorbid conditions that increase the risk of or exacerbate adverse effects,
■ drug–drug interactions that may lead to an increase in drug toxicities, and
■ genetic factors that predispose patients to abacavir (ABC) hypersensitivity reaction.
In general, the overall benefits of ART outweigh its risks and that some non–AIDS-related conditions (eg, anemia, cardiovascular disease, renal impairment) may be more likely in the absence of ART.74
POPULATION RECOMMENDED 2-DRUG REGIMEN ALTERNATIVE 2-DRUG REGIMEN PREFERRED ADDITIONAL THIRD DRUG PREFERRED ADDITIONAL THIRD DRUG ALTERNATIVE
Adults and adolescents TDF + 3TC (or FTC) N/A LPV/r or ATV/r RAL, DRV/r, or EFV
Children ZDV + 3TC ABC + 3TC TDF + 3TC (or FTC) LPV/r Age-appropriate selection from ATV/r, RAL, DRV, EFV, and NVP
Children ZDV + 3TC ABC + 3TC TDF + 3TC (or FTC)
Note: For expansions of the abbreviations used, please refer to Table 191-10.
LPV/r Age-appropriate selection
from ATV/r, RAL, DRV, EFV, and NVP
3505
28
DRUG INTERACTIONS
DRUG INTERACTIONS
Pharmacokinetic (PK) drug–drug interactions between antiretroviral (ARV) drugs and concomitant medications are common, and may lead to increased or decreased drug exposure. In some instances, changes in drug exposure may increase toxicities or affect therapeutic responses. When prescribing or switching one or more drugs in an ARV regimen, clinicians must consider the potential for drug–drug interactions—both those that affect ARVs and those that ARVs affect on other drugs a patient is taking. The magnitude and significance of interactions are difficult to predict when several drugs with competing metabolic pathways are prescribed concomitantly. When prescribing interacting drugs is necessary, clinicians should be vigilant in monitoring for therapeutic efficacy and/or concentrationrelated toxicities.74
The following gives an overview of the pharmacology, mechanism of action, and side effects, particularly mucocutaneous adverse effects, of each class of drugs.
ENTRY INHIBITOR
ENTRY INHIBITOR
The only FDA-approved fusion inhibitor is maraviroc.
PHARMACOLOGY AND MECHANISM OF ACTION
Maraviroc selectively binds to the human chemokine receptor CCR5 present on the cell membrane, preventing the interaction of HIV-1 gp120 and CCR5 necessary for CCR5-tropic HIV-1 to enter cells. Maraviroc is a substrate of CYP3A and P-glycoprotein (P-gp) and its pharmacokinetics are likely to be modulated by inhibitors and inducers of these enzymes/transporters.
SIDE EFFECTS AND PRECAUTIONS Mucocutaneous Side Effects: Severe and potentially life-threatening skin and hypersensitivity reactions have been reported in patients taking maraviroc. This includes cases of Stevens–Johnson syndrome, hypersensitivity reaction, and toxic epidermal necrolysis.
Other Side Effects:
■ Abdominal pain
■ Cough
■ Dizziness
■ Musculoskeletal symptoms
■ Pyrexia
3506
■ Upper respiratory tract infections
■ Hepatotoxicity, which may be preceded by severe rash or other signs of systemic allergic reactions
■ Orthostatic hypotension, especially in patients with severe renal insufficiency
FUSION INHIBITOR
FUSION INHIBITOR
The only FDA-approved fusion inhibitor is enfuvirtide.
PHARMACOLOGY AND MECHANISM OF ACTION
Enfuvirtide is a synthetic peptide that mimics a portion of glycoprotein 41 (gp41), HR1, an HIV envelope glycoprotein required for fusion of the viral envelope with the host cell membrane.80 The drug blocks the formation of a 6-helix bundle structure that is critical for the fusion process. Enfuvirtide is administered via subcutaneous injection. Pharmacokinetics are linear up to a dose of 180 mg. Enfuvirtide does not influence concentrations of drugs metabolized by CYP 3A4, CYP 2D6, or N-acetyltransferase, and has only minimal effects on those metabolized by CYP 1A2, CYP 2E1, or CYP 2C19.
SIDE EFFECTS AND PRECAUTIONS Mucocutaneous Side Effects:
■ Local injection site reactions
■ Induration
■ Erythema
■ Nodules
■ Cysts
Other Side Effects:
■ GI upset
■ Increased risk of bacterial pneumonias
Injection site reactions occur in nearly all patients. Patients should be alert for signs or symptoms of pneumonia.
NUCLEOSIDE AND NUCLEOTIDE REVERSE TRANSCRIPTASE INHIBITORS (NRTIs)
NUCLEOSIDE AND
NUCLEOTIDE REVERSE
TRANSCRIPTASE
INHIBITORS (NRTI
s
)
FDA-approved NRTIs are abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil fumarate, and zidovudine.
PHARMACOLOGY AND MECHANISM OF ACTION
NRTIs compete with the naturally occurring deoxynucleoside substrates for binding to reverse transcriptase (RT). After diffusing into the cytoplasm, they are phosphorylated by intracellular kinases to their active triphosphate forms. The triphosphate form is incorporated into DNA, resulting in chain termination. This group of agents demonstrates activity against HIV-1 and HIV-2, as well as other retroviruses. NRTIs are well absorbed from the GI tract, although there are differences in bioavailability when they are administered with and without food as well as differences in both serum and intracellular half-lives. The volume of distribution, metabolism, and excretion also vary considerably among the different agents.
SIDE EFFECTS AND PRECAUTIONS Mucocutaneous Side Effects: Serious and sometimes fatal hypersensitivity reactions can occur with abacavir. Patients who carry the HLA-B∗5701 allele are at a higher risk of experiencing a hypersensitivity reaction to abacavir. Fat redistribution has been seen in some patients taking emtricitabine. These changes may include an increased amount of fat in the upper back and neck (“buffalo hump”), breast, and around the trunk. Loss of fat from the legs, arms, and face may also happen. Hyperpigmentation primarily of palms and/or soles, but also possibly the tongue, arms, lip, and nails, also have been seen with this medication. Rash is common and includes hypersensitivity reaction, maculopapular rash, pustular rash, and vesiculobullous rash. Rash, pruritus, xerostomia, alopecia, lipodystrophy, Stevens–Johnson syndrome, and vasculitis can occur with didanosine. Alopecia, rash, pruritus, and fat redistribution can be seen with lamivudine. Fat redistribution can be seen with stavudine. Rash, including macular, papular, pustular, vesiculobullous, or urticarial, as well as pruritus and diaphoresis, can be seen with tenofovir. Lipodystrophy, skin/nail pigmentation changes (blue), Stevens–Johnson syndrome, toxic epidermal necrolysis, urticarial, and morbilliform eruption can be seen with zidovudine.
Other Side Effects: The hallmark toxicity of the NRTI class is mitochondrial toxicity, which may manifest as hepatic steatosis, peripheral neuropathy, pancreatitis, dyslipidemia, fat maldistribution, and lipoatrophy. All NRTIs have “black box” warnings in their product labeling regarding the possibility of lactic acidosis syndrome, which is potentially fatal. Common side effects of zidovudine include bone marrow suppression and GI intolerance. Stavudine
28
is associated with lipoatrophy and lactic acidosis. Didanosine can cause pancreatitis and peripheral neuropathy, whereas abacavir is associated with life-threatening hypersensitivity reactions. Tenofovir is associated with headache and nausea. It can elevate didanosine levels and lower those of atazanavir. Lamivudine is generally well tolerated but may produce neutropenia. Onset of bullous pemphigoid has been reported with the combination of lamivudine + didanosine + nelfinavir.81
NONNUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS (NNRTIs)
NONNUCLEOSIDE
REVERSE TRANSCRIPTASE
INHIBITORS (NNRTI
s
)
FDA-approved NNRTIs are delavirdine, efavirenz, etravirine, nevirapine, and rilpivirine.
PHARMACOLOGY AND MECHANISM OF ACTION
Drugs in this class are structurally different from the NRTIs. The NNRTIs bind near the catalytic site of reverse transcriptase and alter the enzymes’ ability to change conformation. This increased enzyme rigidity prevents its normal polymerization function and therefore the replication rate of the virus reduces. Nevirapine has greater than 90% bioavailability and a plasma half-life of 24 hours. It is metabolized in the liver and induces its own metabolic pathway. As the metabolism of the drug increases, the dose is increased from once a day during the first 2 weeks to twice per day thereafter. Delavirdine has a bioavailability of 85%. It requires an acidic environment for absorption and should not be given with antacids, H2 blockers, or proton pump inhibitors. Its plasma half-life is 6 hours, and it is metabolized in the liver. The absorption of efavirenz is increased by food. However, it is generally administered on an empty stomach to minimize side effects. The half-life of efavirenz is 40+ hours, and it is metabolized in the liver.82
SIDE EFFECTS AND PRECAUTIONS Mucocutaneous Side Effects: Nevirapine is associated with rash in about 20% of patients and may cause Stevens–Johnson syndrome.83,84 Among HIV-infected patients with CD4 counts <250 cells/µL, higher baseline counts are associated with a higher incidence of rash, requiring discontinuation of the drug.85 The rash appears to be related to the quinone methide formed in the skin by sulfation of the 12-OH metabolite followed by loss of the sulfate.86 Mucosal side effects include whitish plaques, burning, taste disturbance, and xerostomia.87 Treatment with nevirapine
3507
28
should be initiated slowly to minimize the incidence of cutaneous reactions. If the rash is extensive, or if mucous membranes are involved, the drug should be discontinued. Nevirapine is generally not recommended for women with CD4+ T-cell counts higher than 250 cells/mm3 or for men with CD4+ T-cell counts above 400/mm3 because of an increased risk of hepatitis in these patients. Etravirine is associated with a self-limiting rash in 19% of patients.88 Delavirdine is also commonly associated with drug rash.
Other Side Effects:
■ Hepatitis
■ CNS abnormalities
■ Efavirenz is teratogenic
INTEGRASE STRAND TRANSFER INHIBITORS (INSTIs)
INTEGRASE STRAND
TRANSFER INHIBITORS
(INSTI
s
)
FDA-approved INSTIs are dolutegravir, elvitegravir, and raltegravir.
PHARMACOLOGY AND MECHANISM OF ACTION
INSTIs inhibit HIV integrase and thus prevent the integration of HIV-1 DNA into host genomic DNA, blocking the formation of the HIV-1 provirus and propagation of the viral infection.
SIDE EFFECTS AND PRECAUTIONS Mucocutaneous Side Effects: Severe, potentially life-threatening and fatal skin reactions have been reported with the use of raltegravir. This includes cases of Stevens–Johnson syndrome, hypersensitivity reaction, and toxic epidermal necrolysis. Hypersensitivity reactions characterized by rash, constitutional findings, and sometimes organ dysfunction, including liver injury, have been reported with the use of dolutegravir.
Other Side Effects: The most common adverse reactions include insomnia, headache, dizziness, nausea, diarrhea, and fatigue.
PROTEASE INHIBITORS (PIs)
PROTEASE INHIBITORS (PI
s
)
FDA-approved protease inhibitors are atazanavir, atazanavir-cobicistat, darunavir, darunavir-cobicistat, fosamprenavir, indinavir, lopinavir/ritonavir boosting, nelfinavir, ritonavir (used as a pharmacokinetic boosting agent), saquinavir, and tipranavir.
3508
PHARMACOLOGY AND MECHANISM OF ACTION
Protease inhibitors target an enzyme responsible for the proteolytic cleavage of viral polypeptide precursors. They demonstrate activity against HIV-1 and HIV-2 and may be active against other viruses as well. Oral bioavailability and first-pass hepatic metabolism vary among the protease inhibitors and formulations have been altered to provide greater absorption. Saquinavir and lopinavir have poor oral availability and are administered in combination with low-dose ritonavir to improve drug levels. Ritonavir has good oral bioavailability, but side effects limit the dose in most patients. It is often used at low doses to boost blood levels of other protease inhibitors. All protease inhibitors are metabolized via the hepatic cytochrome P450 (CYP) system. This makes them prone to interactions with drugs that induce, inhibit, or are themselves metabolized by CYP enzymes. Ritonavir is used primarily for its CYP effect to inhibit the metabolism of other agents and thus raise their blood levels.
SIDE EFFECTS AND PRECAUTIONS Mucocutaneous Side Effects: The most common side effect of the protease inhibitors to present to dermatologists is lipodystrophy.89,90 The lipodystrophy may relate to inhibition of ZMPSTE24, an enzyme that removes the farnesylated tail of prelamin A. Buildup of this protein appears to be related to acquired lipodystrophy, possibly through an interaction with a transcription factor called sterol regulatory element– binding protein 1.91 Fat distribution may improve with l-acetylcarnitine therapy, and fillers have been employed to reduce the social stigma associated with therapy.92
Other mucocutaneous side effects:
■ Morbilliform rash, generalized erythema, SJS
■ Striae
■ Paronychia, ingrown toenails
■ Pruritus, xerosis
■ Desquamative cheilitis
■ Icterus (especially with atazanavir as it is associated with unconjugated hyperbilirubinemia)
Other Adverse Effects:
■ GI upset
■ Nausea
■ Vomiting
■ Diarrhea
■ Hepatitis
■ Occasional hepatic failure
■ Dyslipidemias (except atazanavir)
■ Alteration in glucose metabolism
■ Significant drug–drug interactions
INVESTIGATIONAL DRUGS
The guidelines are continuously being adjusted and improved based on the latest available research. The currently available drugs are safer, simpler to use, and more potent than previous drugs. These improvements are expected to continue given the current crop of investigational drugs. Table 191-14 summarizes the current investigational drugs.
DRUGS FOR THE TREATMENT OF HEPATITIS C
DRUGS FOR THE
TREATMENT OF HEPATITIS C
Hepatitis C virus (HCV) infection is a major global health problem with 130 to 150 million
DRUG CLASS DRUG NAME MECHANISM OF ACTION
Entry inhibitor AMD-070 Cenicriviroc Fostemsavir INCB-9471 Ibalizumab
28
people estimated to be chronically infected individuals worldwide.93 Up to 85% of patients with acute HCV infection are unable to clear the infection, and become chronically infected if left untreated. For these patients, the risk of progression to liver cirrhosis is estimated to be 15% to 30% within 20 years. These patients are at higher risk of subsequently developing hepatocellular carcinoma.94
HCV belongs to the Flaviviridae family. It is an enveloped, positive-sense RNA virus.95 The lack of a proofreading mechanism during the replication of the HCV RNA genome leads to significant variation. Thus, HCV has been divided into 7 genotypes with distinct geographic distribution. Genotype 1 is the most common genotype worldwide and in the United States. Genotypes 1, 2, 4, and 5 are endemic in Africa, whereas genotypes 3 and 6 are mostly found
Selective, reversible, small molecule CXCR4 chemokine coreceptor antagonist Small-molecule CCR5 coreceptor antagonist Binds to the viral envelope gp120 and interfering with virus attachment to the host CD4 receptor Small-molecule CCR5 coreceptor antagonist Humanized monoclonal antibody that binds to extracellular domain 2 of the CD4 receptor and causes conformational changes Selective CCR5 coreceptor antagonist Humanized IgG4 monoclonal antibody (mAb) that binds to hydrophilic extracellular domains on CCR5 Reversible, small-molecule CCR5 coreceptor antagonist
Monomeric DAPTA PRO-140 Vicriviroc
Nucleoside reverse transcriptase inhibitors (NRTIs)
Amdoxovir Apricitabine Censavudine Elvucitabine PSI-5004 Tenofovir Alafenamide
Inhibits the activity of HIV-1 reverse transcriptase
A prodrug of the nucleotide analog tenofovir
Nonnucleoside reverse transcriptase inhibitor (NNRTI)
Doravirine Noncompetitive inhibitors of HIV-1 reverse transcriptase
Integrase strand transfer inhibitors (INSTI) Cabotegravir Prevents viral DNA integration into the host genome
Protease Inhibitor (PI) TMC-310v911 PIs bind the HIV-1 protease enzyme to inhibit cleavage of viral Gag and Gag-Pol polyproteins, thereby preventing the formation of mature, infectious virus particles.5-7 TMC-310911 is characterized as having a broader in vitro resistance profile than that of currently approved PIs, including darunavir.
Maturation Inhibitor BMS-955176 Binds to HIV-1 Gag, inhibiting the final protease-mediated cleavage event
Gene Therapy Product SB-728-T May provide HIV-infected individuals with a reproducible pool of CD4 T cells permanently resistant to HIV entry, potentially improving immune restoration and leading to functional control of HIV
Histone deacetylase inhibitors Valproic Acid Vorinostat HDAC inhibitors reactivate latent HIV within resting CD4 T cells. When latent HIV is reactivated, it is once again able to produce new virus and replicate. It is hoped that after latent HIV is reactivated, the CD4 T cells in which the virus was hiding are more likely to die off on their own or be recognized and killed by the body’s immune system.8,9 In addition, any new virus that is produced during reactivation can then be prevented from infecting other cells with the use of ongoing ART.8,9 Recent research has shown that additional therapies, together with HDAC inhibitors, may be needed to fully eliminate latent HIV from the body.
Immune modulator Tucaresol Enhances costimulatory signaling to CD4-positive T cells
Microbicides Astodrimer Dapivirine Tenofovir
Blocks HIV attachment to and entry into host CD4 cells Developed as a preexposure prophylaxis. Topical microbicide that inhibit the infection process at the
Microbicides Astodrimer Dapivirine Tenofovir
Blocks HIV attachment to and entry into host CD4 cells Developed as a preexposure prophylaxis. Topical microbicide that inhibit the infection process at the vaginal or rectal mucosa and directly interfere with the HIV replication cycle
3509
vaginal or rectal mucosa and directly interfere with the HIV replication cycle
28
in Asia. Genotype 7 was only recently identified and is thought to have originated in Africa.96
Knowledge of the HCV life cycle paved the way for the development of direct-acting antivirals (DAAs) that have revolutionized the treatment of HCV infection. Briefly, the HCV life cycle consists of the following steps97:
- Attachment: This involves the binding of HCV particles onto hepatocytes and is mediated by multiple cellular factors and components of the enveloped virions.
- Cell entry: Clathrin-mediated endocytosis allows the HCV particles to enter early endosomes.
- Fusion: pH-dependent membrane fusion is induced by the acidic environment in early endosomes. This releases the HCV genome into the cytosol.
- Translation and Replication: The viral genome in the cytosol becomes available for translation by cellular ribosomes into a single polyprotein that is subsequently cleaved by cellular and viral proteases (NS2, NS3/4A) into the 10 mature viral proteins including structural proteins (core, E1, E2, p7) and nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). The replication complex is subsequently assembled at the cellular ER membrane in association with the viral genomic RNA and nonstructural viral proteins (a process thought to be coordinated by NS4B and NS5A). Viral RNA replication is thought to occur at these complexes where NS5B (the viral RNA–dependent RNA polymerase) uses the viral genomic RNA as a template to synthesize a negative-sense intermediate strand. The negative sense intermediate strand is then used as a template to synthesize nascent genomic RNA strands that can be translated into viral proteins, used as templates for further RNA replication, or assembled in virions.
- Assembly: This step is not fully understood but is thought to be dependent on cellular lipid metabolism as well as viral proteins.
- Release: HCV virions are released as lipoviral particles with associated apoE and/or apoB, which are essential for viral infectivity.
DAAs target specific viral proteins with the aim of interrupting critical stages of the HCV life cycle. Approved DAAs as of this writing can be classified based on their mechanism of action as follows:
- Protease (NS3/4A) inhibitors: boceprevir (discontinued), telaprevir (discontinued), paritaprevir, simeprevir, elbasvir, grazoprevir
- NS5A inhibitors: daclatasvir, ledipasvir, ombitasvir
- Polymerase (NS5B) inhibitor, nucleot(s)ide analog: sofosbuvir
- Polymerase (NS5B) inhibitor, nonnucleoside analog: dasabuvir
The goal of HCV is sustained virologic response (SVR), which is defined as undetectable HCV RNA levels at a specified end point after completion of therapy. This end point has historically been set at 24 weeks
3510
from completion of therapy, but SVR at 12 weeks posttreatment has been shown to correlate closely to that at 24 weeks.98 Thus, SVR at 12 weeks has been adopted as the primary efficacy endpoint in more recent clinical trials and clinical practice.99
The exact treatment chosen depends several factors that can affect responses, which include genotype, coinfection with HIV-1, comorbidities, cirrhosis, and prior treatment. This is an area of active research and as new results from clinical trials become available, guidelines outlining the preferred regimens for different patient populations are being updated.93,100 The following website is a useful resource: http://www.hepatitisc.uw.edu/ page/treatment/drugs.
PEG-IFNα AND RIBAVIRIN COMBINATION THERAPY
HCV virus is able to suppress the innate and adaptive immune and proinflammatory responses of the host by various mechanisms. Exogenous PEG-IFNα is used in an attempt to counteract that suppression. Ribavirin has an unclear mechanism of action against HCV, but there is evidence to suggest that it may have an immunomodulatory effect that mediates increased sensitivity to PEG-IFNα.101
PEG-IFNα is administered subcutaneously and is associated with localized injection site reactions in the majority of treated patients. These include erythema, tenderness, pruritus, and rashes at the site of injection.102 The latter most commonly present as pruritic, erythematous patches or plaques with ill-defined borders. These reactions usually resolve spontaneously without treatment. Less commonly (<4%), cutaneous necrosis can occur at the injection site, which can be managed with local wound care and usually resolve in 1 to 2 months. Alternative injection sites can be used for future doses. Other miscellaneous localized skin reactions have been reported, including blistering reactions, granulomatous and suppurative dermatitis, lupus-like reactions, embolia cutis medicamentosa, and localized alopecia.103
Generalized dermatologic adverse events also have been associated with the use of PEG-IFNα, and the addition of ribavirin further increases their incidence. These adverse events can affect compliance and may lead to premature discontinuation of antiviral therapy. The most commonly encountered generalized dermatologic adverse events seen with the combination of PEG-IFNα and ribavirin include alopecia (28% to 36%) and skin rash (21%).104 The skin rash most commonly seen with PEG-IFNα and ribavirin combination therapy has been characterized as eczematous dermatitis (erythematous papules and microvesicles often with excoriation) that is predominantly located over friction sites on the extremities and trunk. It can be associated with generalized pruritis and xerosis. Symptomatic patients can be treated with topical steroids and emollients without discontinuation of antiviral treatment. Steroids can be tapered off once the rash resolves.
Systemic anti-histamines and hydroxyzine also can be used for pruritis.102,103
Other forms of dermatitis also have been reported including nummular dermatitis and Meyerson phenomenon, an eczematous eruption centered around preexisting melanocytic nevi. Fixed drug eruptions and lichenoid eruptions (most commonly lichen planus) also have been described in association with PEG- IFNα and ribavirin therapy in HCV-infected patients. PEG-IFNα and ribavirin also have been associated with the exacerbation or unmasking of various autoimmune disorders, many of which have skin manifestations that are important to recognize. These disorders include psoriasis, alopecia aerata, vitiligo, sarcoidosis, systemic lupus erythematosus, systemic sclerosis, polyarteritis nodosa. Some of these conditions were successfully treated with systemic corticosteroids but they often necessitate the discontinuation of antiviral therapy. Topical options for the treatment of psoriasis include steroids, calcipotriol, and PUVA.103
Other side effects of PEG-IFNα include flulike symptoms, fatigue, bone marrow suppression/cytopenias, and mood disturbances. Interferon is contraindicated for patients with liver failure and should be used with caution in patients with compensated cirrhosis. Other contraindications for PEG-IFNα include cytopenias, severe cardiac disease, uncontrolled seizures, uncontrolled psychiatric disease, and pregnancy. Ribavirin also has “black box” warnings for hemolytic anemia and teratogenicity. The former can be severe and most often occurs within 1 to 2 weeks after starting therapy. Therefore, hemoglobin/hematocrit level should be checked at baseline and then 2 and 4 weeks after starting therapy. Given its teratogenicity, ribavirin is contraindicated in pregnancy and in male partners of pregnant women. Pregnancy should also be avoided (by using 2 forms of contraception) during therapy with ribavirin and for at least 6 months after completion of therapy (WHO).
FIRST-GENERATION DAAs
The first-generation protease inhibitors include boceprevir and telaprevir. They were initially used for the treatment of genotype 1 in combination with PEG-IFNα and ribavirin, since they were associated with higher SVR rates. However, compared to PEG- IFNα plus ribavirin alone, these agents were more expensive and were associated with a higher incidence of clinically significant adverse events leading to poor adherence and higher rates of premature discontinuation of therapy. This can subsequently lead to higher rates of resistance and treatment failure.99 Therefore, these regimens are no longer recommended by the most recent guidelines as they have been replaced by safer and more efficacious secondgeneration DAAs.100
The most notable adverse events with boceprevir were anemia, neutropenia, and dysgeusia. However, the latter was not a common cause of discontinuation of therapy. When compared to PEG-IFNα and ribavirin
28
alone, the use of boceprevir was not associated with a significantly increased incidence of serious skin rash. On the other hand, the most notable adverse events associated with telaprevir were rash, pruritis, anorectal discomfort, diarrhea, and anemia. Around 6% to 7% of patients receiving telaprevir discontinued therapy due to skin rash. The rash can occur at any time during treatment with telaprevir, but 50% of the cases occurred within the first 4 weeks of initiation of therapy. Around 90% were classified as Grade 1 or 2 and did not progress. The rash most commonly associated with telaprevir was described as eczematous dermatitis that usually resolved with the discontinuation of therapy. However, telaprevir has been associated with more serious dermatologic adverse reactions, including drug rash with eosinophilia and systemic symptoms (DRESS) and Stevens–Johnson syndrome (SJS). Therefore, patients receiving telaprevir should be instructed to report any skin or mucosal lesions, and any patient with such lesions should be questioned about systemic symptoms. In patients with systemic symptoms or involvement of mucous membranes or a significant portion of body surface area then discontinuation of therapy should be considered. Although the efficacy has not been fully established, mild to moderate rash can be managed with topical steroids and/or oral antihistamines. Systemic corticosteroids should be avoided.105,106
SECOND-GENERATION DAAs
Different combinations of the second-generation DAAs have been approved for HCV treatment.93 There is a trend toward using interferon-free and, whenever possible, ribavirin-free regimens for the shortest effective treatment duration. Indicated and preferred treatment regimens are being actively updated as results of recent and ongoing trials get reported (useful resource: http://www.hepatitisc.uw.edu/page/treatment/ drugs). This section attempts to provide an account of the dermatologic adverse events (exclusive of infectious complications like cellulitis) as well as the most common adverse events reported in recent clinical trials using FDA-approved DAA-based regimens without ribavirin when used over a duration of 12 weeks. It should be noted that the dermatologic side effects previously associated with ribavirin (including rash and pruritis) had higher incidence in patients who received DAA-based regimens that included ribavirin (mostly for treatment-experienced patients with cirrhosis).
■ Simeprevir (Olysio): It is only indicated in combination with sofosbuvir or PEG-IFNα plus ribavirin. The most common adverse events associated with simeprevir were rash, photosensitivity, pruritis, nausea, and transient hyperbilirubinemia. However, the rates of discontinuation of therapy were comparable to those with PEG-IFNα plus ribavirin alone. The skin rash can develop at any time during treatment but most commonly begins within 4 weeks from the initiation of therapy.
3511
28
A photosensitivity reaction with skin rash can also occur and patients should be instructed to minimize exposure to sunlight.107,108
■ Sofosbuvir (Sovaldi): When used in combination with PEG-IFNα plus ribavirin, the most common adverse events include fatigue, nausea, anemia, and neutropenia. There was no significant increase in rate of discontinuation of therapy when compared with PEG-IFNα plus ribavirin alone. When used in combination with ribavirin alone, the most common adverse effects reported were fatigue, headache, nausea and pruritis. However, the rate of discontinuation of this regimen was relatively low (≤1%).107
■ Ledipasvir and sofosbuvir (Harvoni): The most common adverse events were fatigue, headache, nausea, and diarrhea. In the ION-1 Study, up to 7% of patients had rash.109 Mild rash and pruritis were reported in 1% to 2% of patients in the ION-2 and 3 studies.110,111 However, none of the dermatologic adverse events required discontinuation of treatment. No rash was reported in ION-4 study wherein Harvoni was used to treat patients with HCV/HIV-1 coinfection.112
■ Daclatasvir (Daklinza) and sofosbuvir: The most common adverse events were fatigue, headache, nausea, and diarrhea. This combination can also cause bradycardia in patients taking amiodarone (especially if they also take a beta-blocker). Rash was reported in 2% and pruritis in 5% of patients in the 12-week treatment groups.113
■ Elbasvir and grazoprevir: The most common adverse observed in patients were fatigue, headache, and nausea. Skin rash (that did not require discontinuation of therapy) was reported in 2 of 33 patients (6%) in the group receiving elbasvir and grazoprevir without ribavirin.114,115
■ Ombitasvir-paritaprevir-ritonavir and dasabuvir (Viekira Pak): The most common adverse events were fatigue and nausea. Pruritis was reported in 5% to 6% of patients, and skin rash was reported in up to 4% to 5% of patients. The latter did not warrant discontinuation of therapy in any of the treated patients.116,117
■ Sofosbuvir and velpatasvir (Epclusa): The most common adverse effects were headache, fatigue, and nausea. Pruritis was reported in 3% to 4% of patients. However, there were no reports of serious skin rash after 12 weeks of treatment with sofosbuvir and velpatasvir in the clinical trials that have been published as of this writing.118,119
DRUGS FOR THE TREATMENT OF HEPATITIS B
DRUGS FOR THE
TREATMENT OF HEPATITIS B
Hepatitis B virus (HBV) infection is most commonly treated with a nucleoside/tide reverse transcriptase inhibitor (NRTI). Generally, these agents are mostly given orally once daily, with dose adjustment for patients
3512
with impaired renal function (creatinine clearance <50 mL/min). NRTIs act by inhibiting the polymerase activity of HBV. However, their low-level activity against the human mitochondrial DNA polymerase gamma and can lead to mitochondrial toxicity, for which a “Black Box” warning is included in the product labeling of all NRTIs. This may be manifested as myopathy, peripheral neuropathy, hepatic steatosis, pancreatitis, dyslipidemia, lipoatrophy, and lactic acidosis.120
Currently, the preferred agents for HBV treatment are either entecavir or tenofovir based on a favorable side effect profile and lower rates of resistance. The other NRTIs approved for treatment of HBV include lamivudine, adefovir, and telbivudine. Tenofovir and adefovir have been associated with nephrotoxicity and Fanconi syndrome as well as rashes with variable morphology (including maculopapular, urticarial, vesiculobullous, pustular, and lichenoid rashes). Telbivudine has been reported to cause myopathy and peripheral neuropathy. Pancreatitis has been reported with lamivudine. Entecavir is generally well tolerated.121,122
PEG-IFNα-2a also can be used in patients without cirrhosis who are HBeAg positive with good efficacy (27% rate of seroconversion at 1 year) but poor tolerability due to side effects as discussed previously. Of note, PEG-IFNα-2a is contraindicated in patients with decompensated cirrhosis, cytopenias, severe cardiac disease, uncontrolled seizures, and uncontrolled psychiatric disease. It should be used with caution in patients with compensated cirrhosis. For persons with HDV coinfection, the only effective treatment is PEG-IFNα. For persons with HIV coinfection, treatment of HBV and HIV need to be coordinated to utilize drugs active against both viruses (tenofovir, entecavir, lamivudine, and telbivudine).121
Indications for initiation of treatment include the following:
- ALF or decompensated cirrhosis (NRTIs only)
- The presence of significant liver injury or fibrosis, as reflected by elevated ALT levels or biopsyproven inflammation and/or fibrosis plus active HBV viremia (HBV DNA >2000 IU/mL if HBeAg negative, or >20,000 IU/mL if HBeAg positive).
- Immunosuppressive therapy.
For patients treated with PEG-IFNα, the recommended duration of treatment is 48 weeks. For patients treated with NRTIs, the exact duration of therapy varies and depends on multiple factors including HBeAg status, duration of HBV DNA suppression, and presence of cirrhosis/decompensation (AASLD Guidelines for Treatment of Chronic Hepatitis B). If seroconversion of HBeAg or hepatitis B surface antigen (HBsAg) is observed, then treatment can be discontinued. However, the latter is only rarely observed.121,123
DRUGS TO TREAT OTHER VIRAL INFECTIONS
Neuraminidase inhibitors can be used to treat patients presenting with severe influenza infection or those at
high risk for severe infection or complications (such as pregnant women and elderly patients). Neuraminidase inhibitors include oseltamivir, zanamivir, and peramivir. Oseltamivir and peramivir have been associated with serious skin adverse events, including cases of Stevens–Johnson syndrome and erythema multiforme. Other skin side effects have been reported with peramivir including urticaria and maculopapular rash. Ribavirin is used to treat respiratory syncytial virus infection. The dermatologic side effects were discussed in the HCV section.
ACKNOWLEDGMENTS
The authors thank Drs Hay, Reichman, and Elston for their work on this chapter in previous editions. The authors also thank M. Liu for her contribution to this chapter.
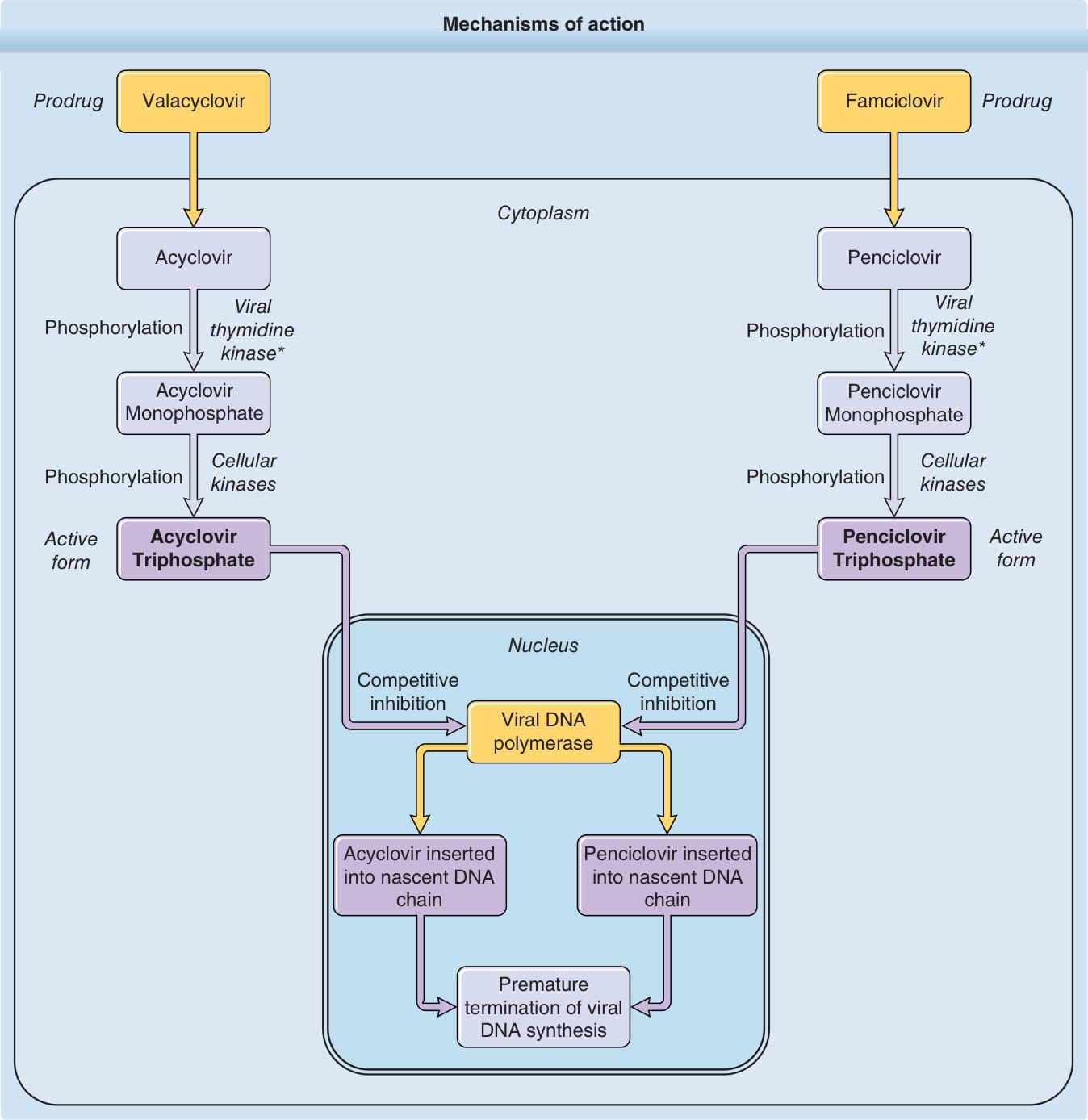
Figure 191-1. Mechanisms of action.
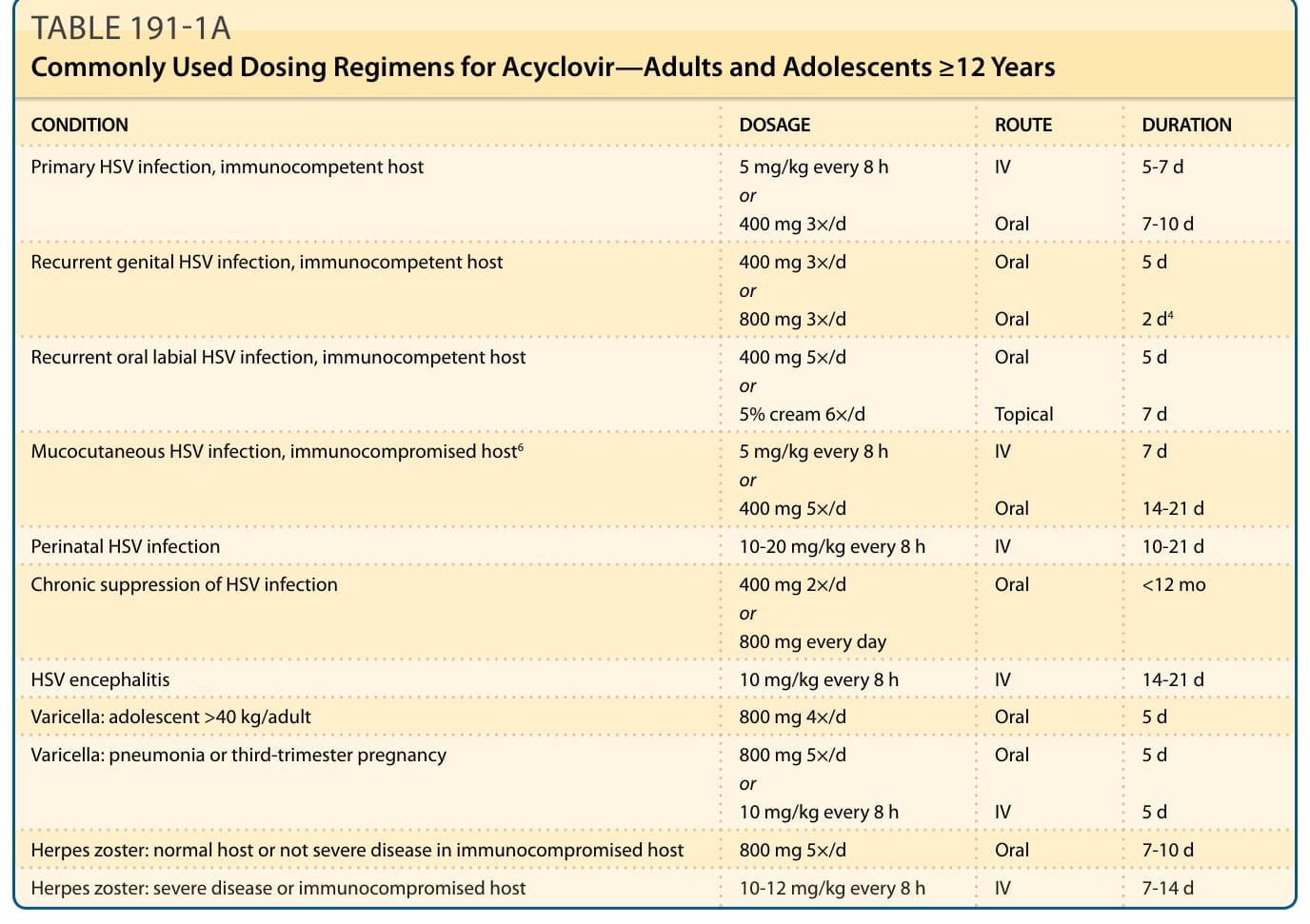
TABLE 191-1A Commonly Used Dosing Regimens for Acyclovir—Adults and Adolescents ≥12 Years
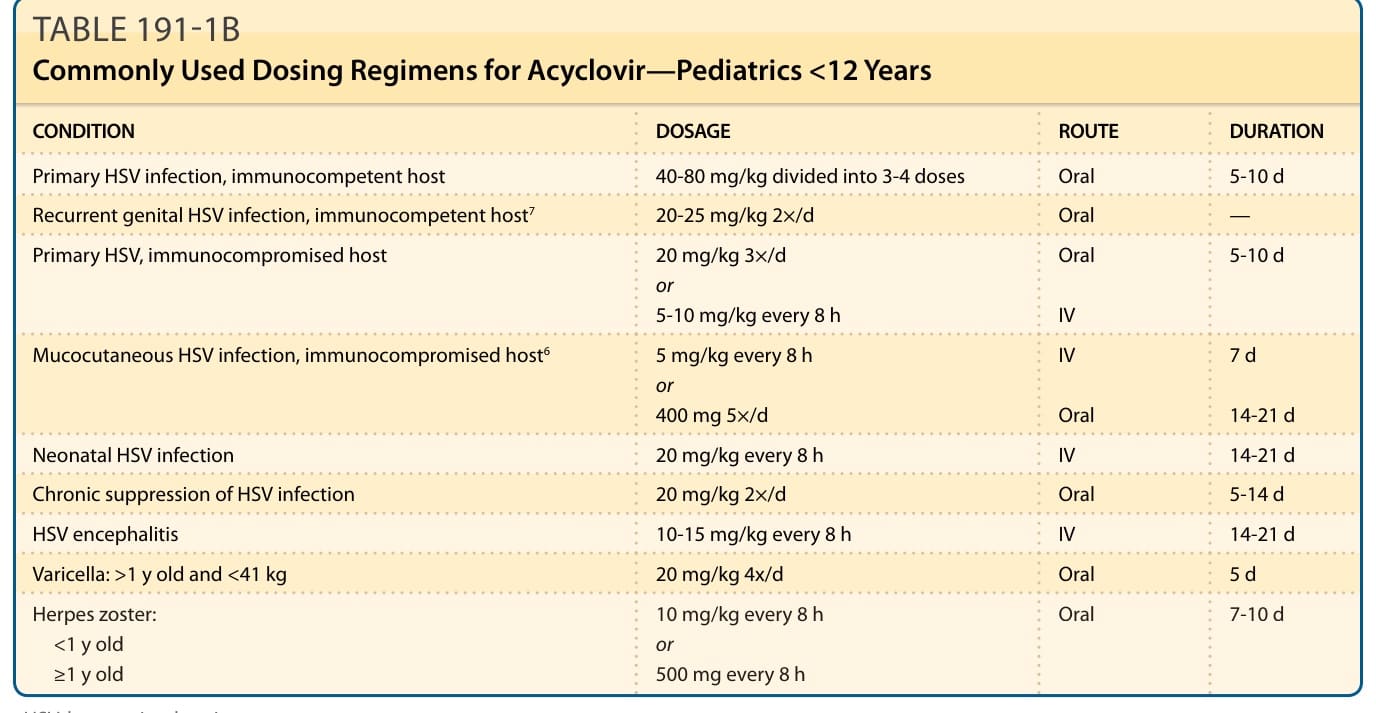
TABLE 191-1A Commonly Used Dosing Regimens for Acyclovir—Adults and Adolescents ≥12 Years

TABLE 191-2 Pregnancy Categories of Systemic Antiviral Agents
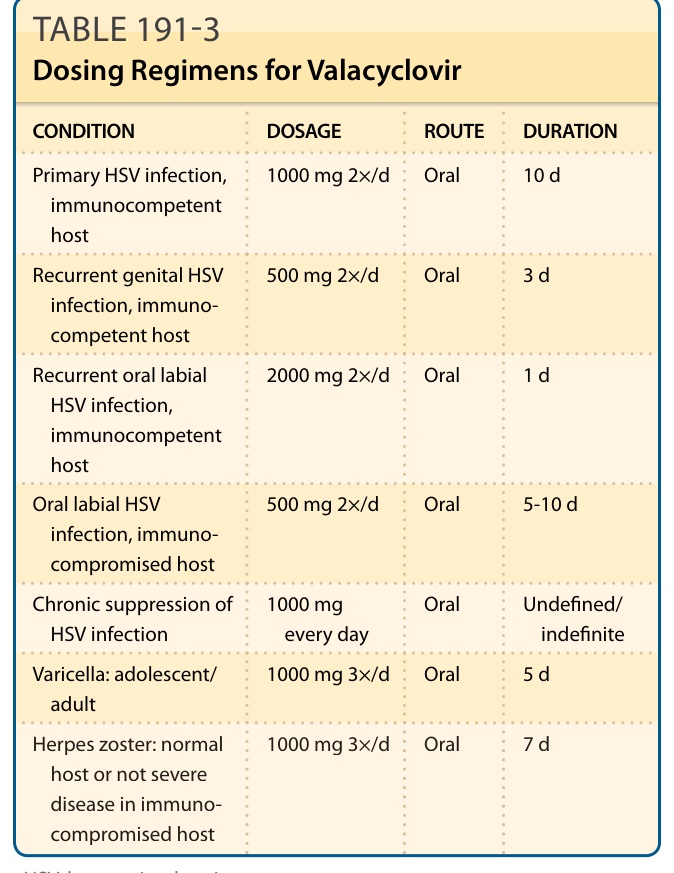
TABLE 191-3 Dosing Regimens for Valacyclovir
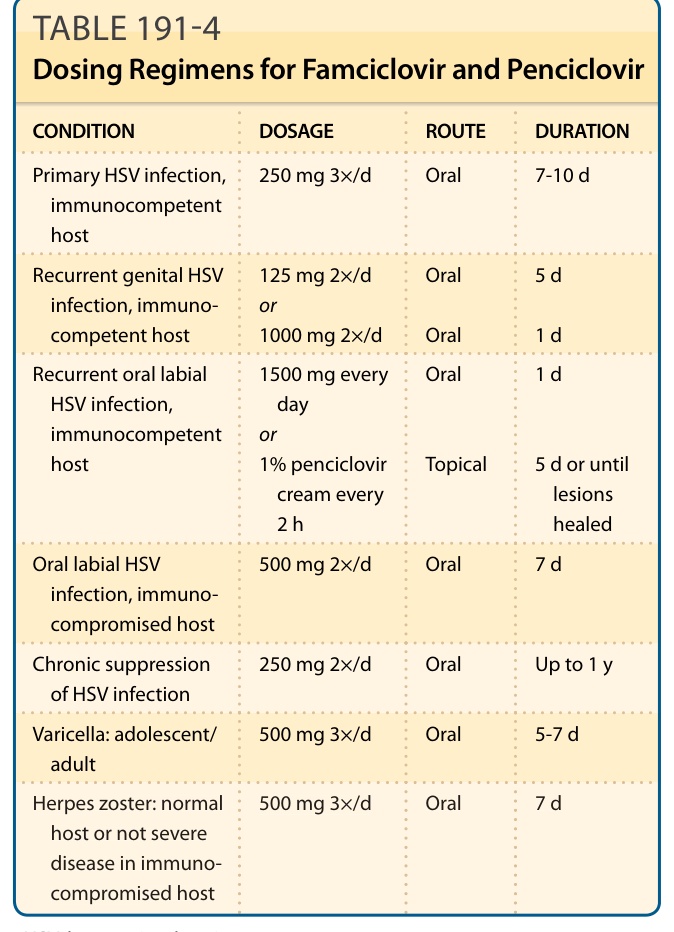
TABLE 191-4 Dosing Regimens for Famciclovir and Penciclovir

TABLE 191-5 Dosing Regimens for Trifluridine
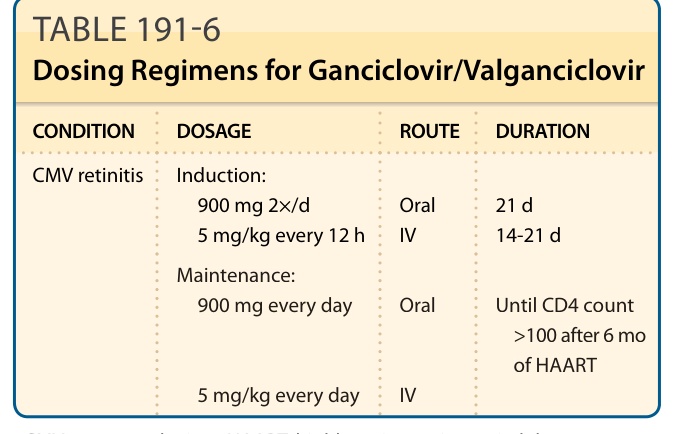
TABLE 191-6 Dosing Regimens for Ganciclovir/Valganciclovir
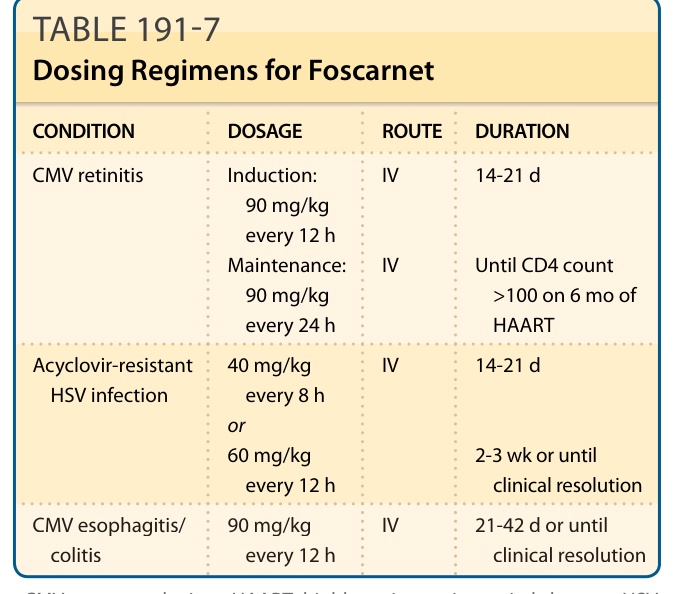
TABLE 191-7 Dosing Regimens for Foscarnet
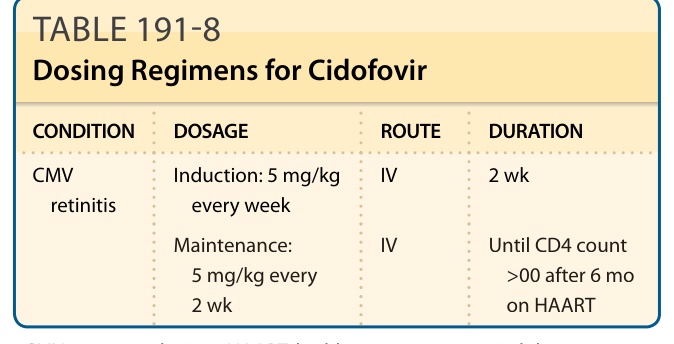
TABLE 191-8 Dosing Regimens for Cidofovir
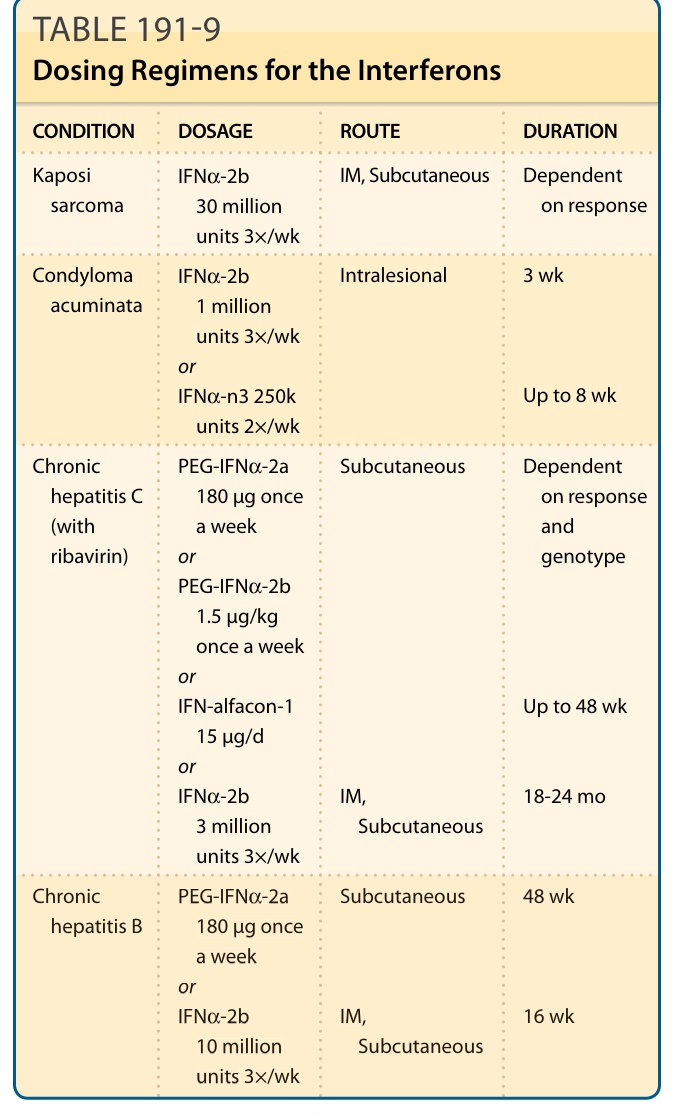
TABLE 191-9 Dosing Regimens for the Interferons
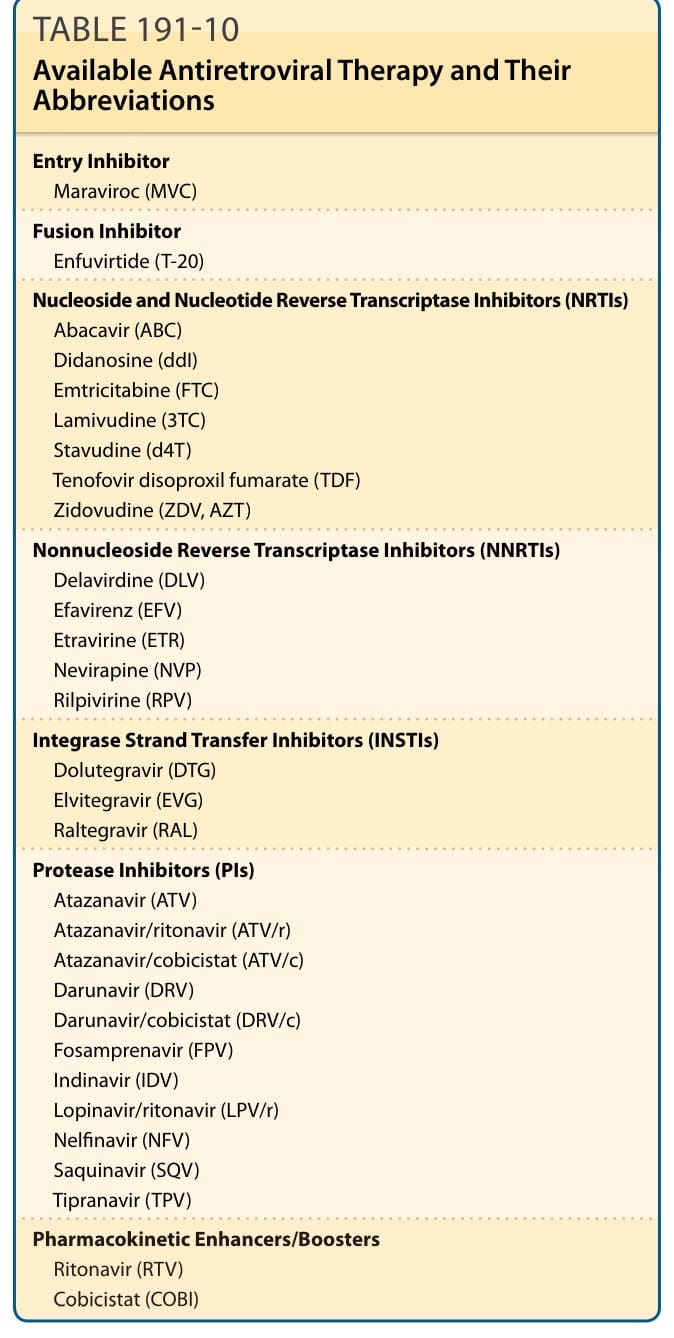
TABLE 191-10 Available Antiretroviral Therapy and Their Abbreviations
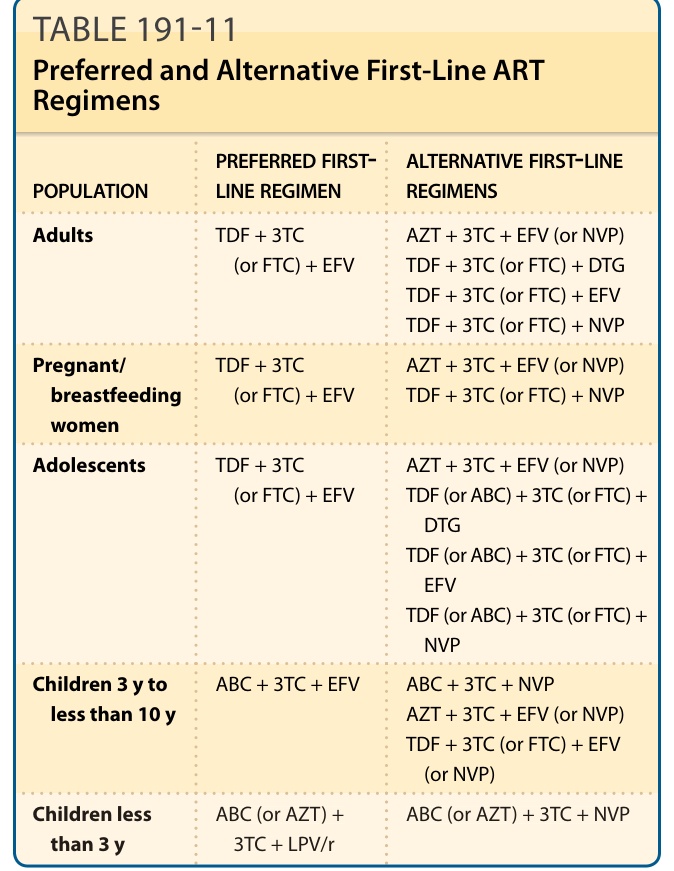
Table 191-11 shows the recommended and alternative first-line regimens based on the latest WHO guidelines. Given the large number of excellent options for initial therapy, selection of a regimen for a particular patient should be guided by factors such as viral load, CD4 count, virologic efficacy, side-effect profile, toxicity, pill burden, dosing frequency, previous exposure to drugs, drug–drug interaction potential, resistance testing results, comorbid conditions, and cost. When initiating a combination ART, all
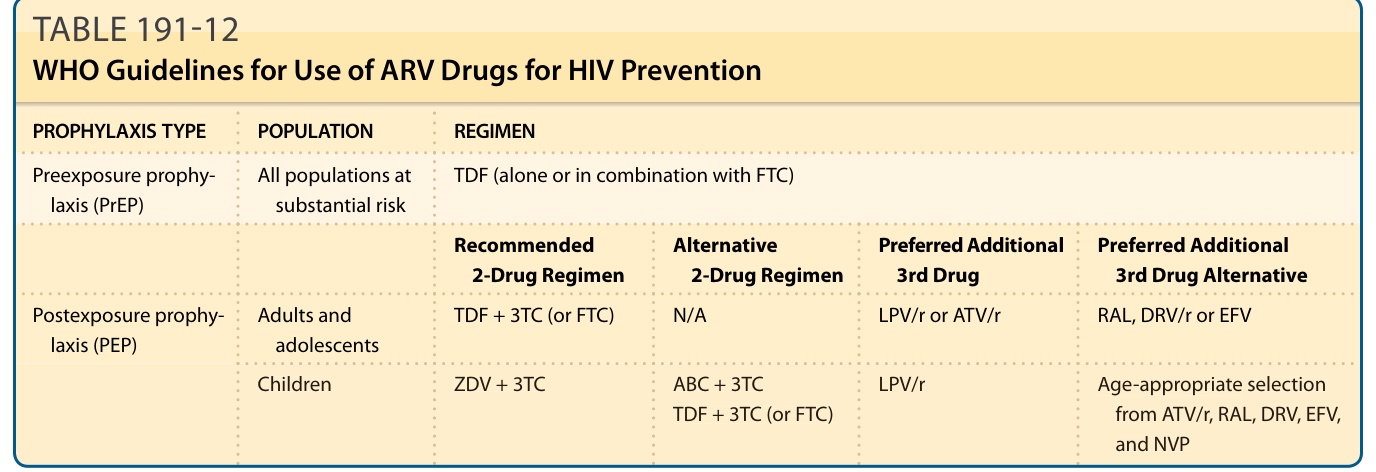
TABLE 191-12 WHO Guidelines for Use of ARV Drugs for HIV Prevention
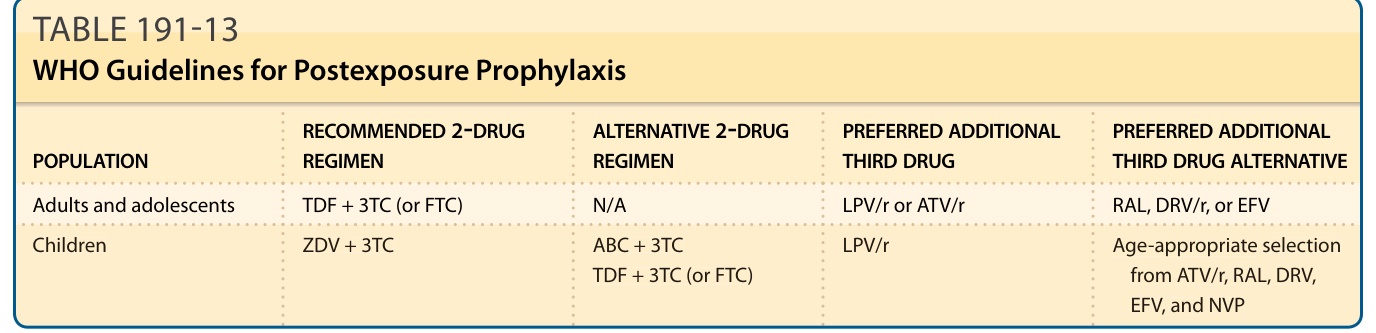
TABLE 191-13 WHO Guidelines for Postexposure Prophylaxis
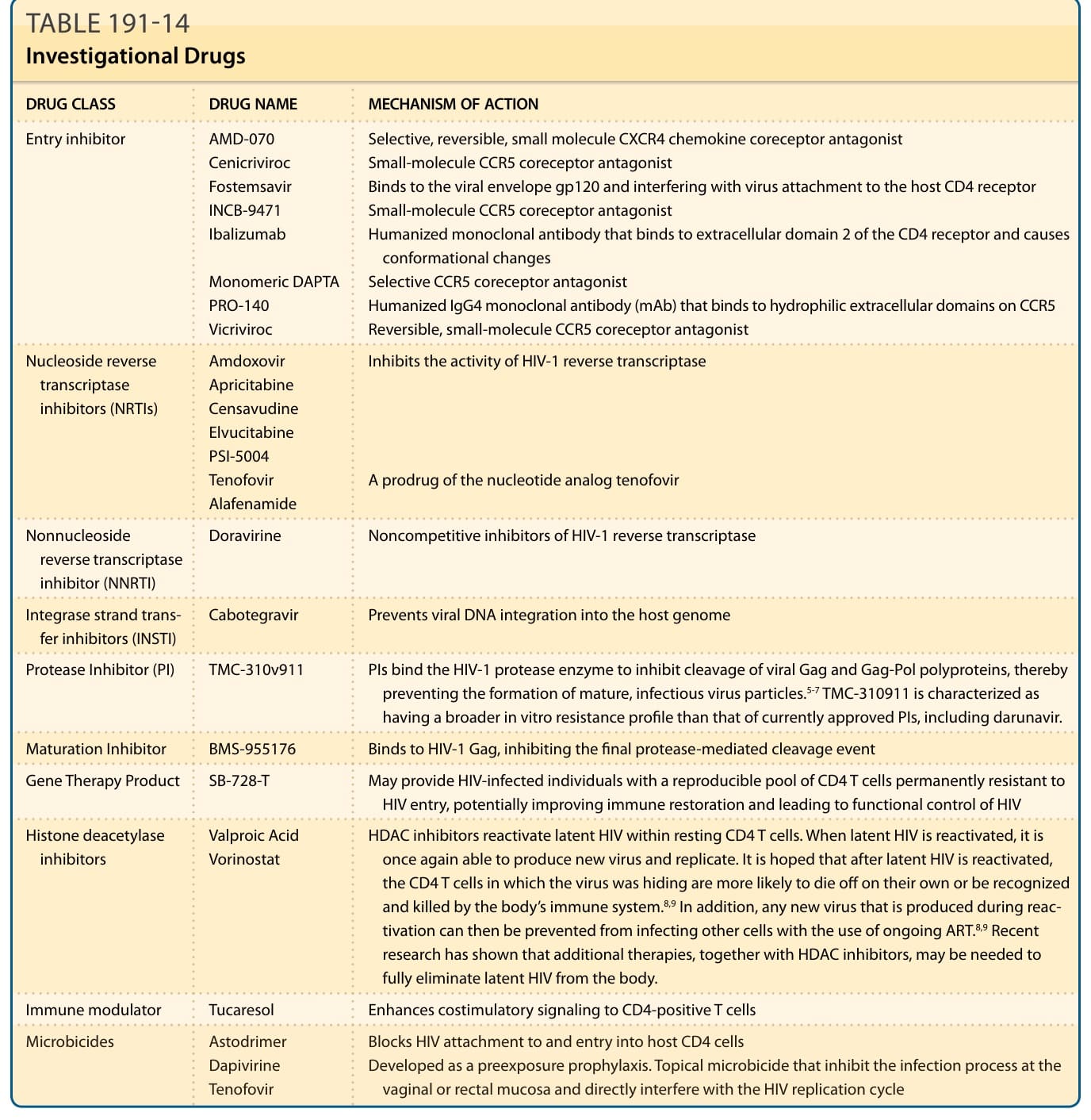
TABLE 191-14 Investigational Drugs