Antifungals
28
AT-A-GLANCE
■ Topical agents should be used for superficial fungal infections of limited extent.
■ Topical agents have the advantage of low cost, nonprescription availability, ease of use, and high patient compliance.
■ Topical antifungals include imidazoles, allylamines, benzylamines, polyenes, and ciclopirox.
■ A number of new carrier vehicles have been investigated to improve the bioavaliabilty of topical antifungals.
■ Systemic agents should be used to treat onychomycosis and tinea capitis, or when superficial fungal infection affects a large surface area.
■ Attention should be paid to drug–drug interactions when prescribing systemic antifungals.
■ Combination therapy with topical and systemic drugs presents several advantages.
INTRODUCTION/ BACKGROUND
Superficial mycoses, caused mainly by dermatophytes and yeast, are among the most frequent infections worldwide, affecting approximately 20% to 25% of the world’s population.1 Topical antifungals are the preferred option for the treatment of most of these infections, achieving high patient compliance. Moreover, many topical drugs have additional antibacterial action, which is beneficial in cases of superimposed bacterial infections. Further, many also possess antiinflammatory properties, which are advantageous in minimizing the effects of the host local inflammatory reactions to mycotic infection.2 Systemic antifungals are the mainstay treatment of tinea capitis and onychomycosis; also, dermatologists tend to reserve them for extensive, recurrent, or recalcitrant dermatomycosis.3,4
Despite the diversity in the structure of the fungal cell membrane and the unique existence of the mycotic cell wall compared to mammalian cells, the similarities in the metabolic profile between both kingdoms allow for limited numbers of organism-specific targets. Overall, the main target of both systemic and topical antifungals is ergosterol (Fig. 188-1), the fundamental mycotic cell membrane sterol. As of this writing,
the 3 main antifungal categories targeting ergosterol are (1) allylamines and benzylamines, (2) azoles (imidazole and triazoles), and (3) polyenes. Whereas allylamines, benzylamines, and azoles block the biosynthesis of ergosterol, polyenes bind the molecule with high affinity, creating cell membrane pores. Other systemic antifungal agents (griseofulvin and flucytosine) act on intracellular structures via mechanisms similar to cancer chemotherapeutic agents.5,6 Although both are clinically effective, azoles exhibit superior activity against yeasts compared to allylamines, but less activity against dermatophytes.7 Although the most common adverse effects associated with topical therapy are mild, transient, and localized hypersensitivity skin reactions, systemic antifungals demonstrate various degrees of organ toxicity and possible serious drug interactions.3 This chapter aims to discuss the main topical and systemic antifungal agents currently in clinical use (Tables 188-1 through 188-4).
ALLYLAMINES
Two agents represent this family: terbinafine, available in both topical and oral forms, and naftifine, available only for topical application.8
MECHANISM OF ACTION
MECHANISM OF ACTION
Allylamines exhibit differential antifungal activity against Candida species (fungistatic), and dermatophytes (fungicidal). They act through the suppression of ergosterol synthesis by inhibiting the action of squalene epoxidase enzyme, the enzyme that catalyzes the conversion of squalene precursors into ergosterol. The resultant deficiency of ergosterol is responsible for the fungistatic effect, while the buildup of squalene accounts for fungicidal activity.8
NAFTIFINE
NAFTIFINE
Naftifine is a broad-spectrum topical allylamine.
MECHANISM OF ACTION
In addition to its antifungal activity, naftifine has antibacterial activity (gram-positive and gram-negative) as well as antiinflammatory properties secondary to its ability to suppress the synthesis of leukotrienes and prostaglandins.9
28
Mechanism of action of antifungal agents
Acetyl CoA
Squalene precursors
ALLYLAMINES BENZYLAMINES
Squalene epoxidase
Lanosterol
14 alpha demethylase
AZOLES
POLYENE Amorolfine
Ergosterol
Miconazole*
Clotrimazole* Sertaconazole*
Synthesis
Fungal cell wall
Microtubule formation Fungal protein synthesis
Tavorabole
Griseofulvin
Peroxisome
DNA
Peroxide
RNA
Protein precursors
Peroxidase
Ciclopirox Membrane Transporter
*In addition to the classic mechanisms of action as azole antifungals.
PHARMACOKINETICS
The lipophilic properties of naftifine account for its efficient penetration through the stratum corneum. Therapeutic drug levels can persist in stratum corneum up to 5 days following a single application. Only 3% to 6% of the applied drug can be systemically absorbed.9-11
CLINICAL INDICATIONS
Naftifine is indicated for the treatment of interdigital tinea pedis, tinea cruris, tinea corporis, tinea versicolor, and Candida infections.8
DOSAGE AND FORMULATIONS
Naftifine is commercially available as 1% gel and cream. A single daily application of naftifine is usually recommended for 2 to 4 weeks according to the indication (Table 188-1).12
SIDE EFFECTS
The side effects of naftifine are generally minor and include dryness, pruritus, local irritation, and erythema.8
TERBINAFINE
TERBINAFINE
MECHANISM OF ACTION
Terbinafine works through the suppression of squalene epoxidase enzyme, blocking the formation of ergosterol.13
PHARMACOKINETICS
The systemic absorption of topical terbinafine is inconsequential (3%-5%).8 The oral absorption of the drug is not influenced by food intake and, owing to its lipophilic properties, tends to rapidly distribute and accumulate in hair follicles, nails, and skin with minimal concentrations in plasma. It has a long half-life of 17 hours. The oxidative biotransformation of the drug takes place in the liver by the means of CYP2D6 enzyme. Most of the drug is eliminated in urine. A dose adjustment is necessary in patients with advanced renal or liver diseases.8,13-15
INDICATIONS
Topical terbinafine is efficacious in the treatment of all superficial mycoses, including tinea versicolor.8,13 Unless contraindicated, terbinafine is the first line
3437
28
DRUG/CLASS TRADE NAMES USUAL FORMULATION INDICATIONS DOSING REGIME PREGNANCY CATEGORY
Polyenes
Nystatin Mycostatin Nystop Pedi-Dri (Rx)
Cream, powder, ointment Mucosal and cutaneous candidiasis Twice daily B
Azoles
Imidazoles
Clotrimazole Clotrim, Cruex Lotrimin, Mycelex (OTC) Cream, solution, lotion Tinea corporis/cruris/ pedis Twice daily for 2-4 wk B
Econazole Spectazole, Ecoza (Rx) Cream, powder, foam, solution Dermatophytoses, tinea versicolor, cutaneous candidiasis
Once daily (twice for candidiasis) for 2 wk C
Oxiconazole Oxistat (Rx) Cream, lotion Dermatophytoses, tinea versicolor Once-twice daily for 2 wk B
Miconazole Miconazole OTC Cream, ointment, powder or spray Dermatophytoses Twice daily for 2-4 wk C
Sertaconazole Ertaczo (Rx) Cream, lotion, gel, powder, solution Interdigital tinea pedis Twice daily for 4 wk C
Ketoconazole Ketoconazole (Rx) Cream, shampoo Dermatophytoses, tinea versicolor, cutaneous candidiasis
Triazoles
Once daily for 2-6 wk C
Efinaconazole Jublia (Rx) Solution Onychomycosis Once daily for 48 wk C
Allylamines
Terbinafine Lamisil (OTC) Cream, powder, solution, spray, gel Dermatophytoses, tinea versicolor Once or twice daily for 2-4 wk B
Naftifine Naftin (Rx) Cream, gel Dermatophytoses Once-twice daily for 2-6 wk B
Benzylamines
Butenafine Lotrimin Ultra (OTC), Mentax (Rx) Cream, spray, powder Dermatophytoses, tinea versicolor Once-twice daily for 2 wk C
Hydroxypyridones
Ciclopirox olamine Loprox (Rx) Cream, suspension, gel, nail lacquer solution
Morolphines
Amorolfine Loceryl (Rx) Not available in the USA
Dermatophytoses, onychomycosis Twice daily for 2-4 wk; nail lacquer—apply once daily for 48 wk
B
Nail lacquer solution Onychomycosis Apply once daily for 48 wk D
Oxaborole
Tavaborole Kerydin (Rx) Nail lacquer solution Onychomycosis Apply once daily
C
Tavaborole Kerydin (Rx) Nail lacquer solution Onychomycosis Apply once daily for 48 wk C
of treatment of dermatophytosis owing to its fungicidal activity, high efficacy, tolerability, single daily dose, better pharmacokinetic profile, and high affinity to stratum corneum.16 Oral terbinafine exhibits a potent fungicidal action, superior to griseofulvin and itraconazole, against Trichophyton rubrum and T. mentagrophytes. Although considered the first treatment option in dermatophyte onychomycosis, terbinafine does not demonstrate efficacy against nondermatophyte and Candida onychomycosis.15
3438
for 48 wk
Terbinafine and griseofulvin are the only FDAapproved drugs for the treatment of tinea capitis in children. However, terbinafine is less effective than griseofulvin in eradicating ectothrix tinea capitis in the pediatric population, particularly, that caused by Microsporum canis and Trichophyton tonsurans, because of its high tendency to accumulate in sebum. Immature preadolescent sebaceous glands produce less sebum, thus resulting in lower drug concentrations.17-19 Many clinical studies have shown efficacy of terbinafine in the treatment
28
DOSING REGIME PREGNANCY CATEGORY DRUG/CLASS TRADE NAMES INDICATIONS ADULT PEDIATRIC
USUAL FORMULATION
Allylamines
Terbinafine Lamisil, Terbinex
- Tablets 250 mg
- Oral granules
Tinea unguiuma Continuous 250 mg/d >20 kg Fingernail for 6 wk Toenail for 9-12 wk Pulse 500 mg/d for 1 wk/m for same duration
Continuous >20 kg 62.5 mg/d 20-40 kg 125 mg/d for same duration
B
T. capitisa 250 mg/d for 2-8 wk 5 mg/kg/dc for 2-4 wk
T. pedis, cruris, corporisb 250 mg/d 2-4 wk
Seborrheic dermatitisb 250 mg/d 4-6 wk
Azoles
Triazoles
- Esophageal candidiasisa 150 mg/d for 2-3 wk after clinical improvement
Fluconazole Diflucan
- Capsule 150 mg
- Tablets 150 mg
- Solution for IV infusion
6 mg/kg/d until clinical improvement, then 3 mg/kg/d for 2 wk
C
-
Vaginal candidiasisa 150 mg once Recurrence 150 mg/wk for 6 mo
-
Cutaneous, mucocutaneous candidiasisa 300 mg/wk for 2 wk
-
T. capitisb 6 mg/kg/d Trichophyton tonsurans for 20 d Microsporum canis 2 wk
-
Onychomycosisb 150-300 mg/wk Fingernail for 6-9 mo Toenail for 9-15 mo
3-6 mg/kg/d Fingernail 12-16 wk Toenail 18-26 wk
-
Tinea pedis, cruris, corporis, barbaeb 150 mg/wk for 2-6 wk
-
Tinea versicolorb 300 mg/wk for 2 wk
Itraconazole Sporanox
- Onychomycosisa Continuous C
Sporanox pluspak Onmel
- Capsules 100 mg
- Cyclodextrin oral solution
200 mg/d Fingernail for 6 wk Toenail for 12 wk Pulse 400 mg/d for 1 wk/mo Fingernail 2 pulses Toenail 3 pulses
Pulse 5 mg/kg/d for 1 wk/mo Fingernail 2 pulses Toenail 3 pulses
- Oropharyngeal candidiasisa Oral solution 100-200 mg/d for 1-2 wk after clinical improvement
15 kg 100 mg/d 15-30 kg 100 mg/d alternating with 200 mg/d 30-45 kg 200 mg/d same duration as adults
(Continued)
3439
28
(Continued)
DOSING REGIME PREGNANCY CATEGORY DRUG/CLASS TRADE NAMES INDICATIONS ADULT PEDIATRIC
USUAL FORMULATION
-
T. capitisb 200 mg/d for 2-8 wk 5 mg/kg/d T. tonsurans for 2-4 wk M. canis for 4-8 wk
-
T. pedis, cruris, corporisb 200 mg/d for 1 wk
-
Pityriasis versicolorb TTT 200 mg/d for 1 wk Prophylaxis 400 mg once every month
Other
- T. capitisa Microsize 500 mg/d or ultramicrosize 300- 375 mg/d for 4-8 wk
Griseofulvin Griseofulicin Griseofulvic Gris-PEG Grifulvin V
-
Tablets
-
T. capitisa Microsize 500 mg/d or
Griseofulvin Griseofulicin Griseofulvic Gris-PEG Grifulvin V
- Tablets Microsize 250, 500 mg Ultramicrosize 125, 165, 250 mg
- Oral suspension 125 mg/ 5 mL
Microsize 250, 500 mg Ultramicrosize 125, 165, 250 mg
2. Oral suspen-
Microsize
C
Microsize 15-2 0 mg/kg/d or ultramicrosize 5-10 mg/kg/d for 6-12 wk
C
ultramicrosize 300- 375 mg/d for 4-8 wk
15-2 0 mg/kg/d or ultramicrosize 5-10 mg/kg/d for 6-12 wk
-
T. cruris,a corporis Same doses as above for 2-4 wk Same doses as above for 2-4 wk
-
T. cruris,a corporis Same doses as above
sion 125 mg/ 5 mL
Same doses as above
for 2-4 wk
for 2-4 wk
-
T. pedisa Microsize 750-1000 mg/d or ultramicrosize 660- 750 mg/d for 4-8 wk
-
T. pedisa Microsize
aFDA-approved indications.
bOff-label indications.
cIndicated for children older than 4 years.
of cutaneous and lymphocutaneous sporotrichosis at a dose of 500 mg twice daily for 2 to 4 weeks beyond the clinical resolution. Terbinafine has also shown efficacy in the treatment of phaeohyphomycosis, chromoblastomycosis, mucormycosis, and maduromycosis.17
DOSAGE AND FORMULATIONS
The following terbinafine formulations are available for topical use—cream, powder, solution, spray, and gel 1% (Table 188-1). Oral terbinafine formulations include 250-mg tablets and oral granules. Because terbinafine can persist in keratinized tissue up to 6 months after the last dose,19 a pulse regimen could represent an alternate treatment strategy for onychomycosis. A daily dose of 250 mg for 1 week monthly continued for 1 year has shown a mycological cure rate of 90% compared to 75% achieved with itraconazole pulse dosage.17 Terbinafine at a daily dose of 250 mg for a period ranging from 2 to 4 weeks is effective in the management of tinea barbae, tinea corporis, tinea cruris, tinea faciei, and moccasin-type tinea pedis (Table 188-2).12
SIDE EFFECTS
The side effects of the topical application are not common (6%) and are typically limited to mild localized cutaneous reaction. In general, the oral route is well
3440
Microsize 10-20 mg/
Microsize 10-20 mg/ kg/d or ultramicrosize 5-10 mg/kg/d for 4-8 wk
750-1000 mg/d or ultramicrosize 660- 750 mg/d for 4-8 wk
kg/d or ultramicrosize 5-10 mg/kg/d for 4-8 wk
tolerated, with fewer side events reported with the pulse regimen compared to continuous dosing. Minimal GI tract upset (eg, abdominal pain, nausea, vomiting, diarrhea), appetite changes, and weight gain are the most common terbinafine adverse effects. A unique side effect is altered taste, which can last up to 6 weeks with or without loss of smell and tongue discoloration. Severe but rare adverse events are hepatotoxicity leading to possible organ failure and hematologic disorders including pancytopenia; these are usually reversible after drug stoppage. Cutaneous hypersensitivity eruptions as severe as Stevens– Johnson syndrome and toxic epidermal necrolysis have been reported, mainly within 4 to 5 weeks of treatment. Drug discontinuation may result in GI and cutaneous adverse events, and headaches, vertigo, and transient optic neuropathy may also occur (Table 188-3).20
DRUG–DRUG INTERACTIONS
Terbinafine exhibits a potent inhibitory effect on CYP2D6, and therefore increases the potential toxicity of drugs metabolized by this enzyme, including nortriptyline, amitriptyline, venlafaxine, and desipramine. In addition, terbinafine reduces the elimination of caffeine and cyclosporine. The concomitant administration of terbinafine with rifampicin or cimetidine
28
DRUG SIDE EFFECTS PRECAUTIONS/CONTRAINDICATIONS MONITORING
Terbinafine
-
Ageusia (altered taste), loss of smell, and tongue discoloration+
-
Hepatotoxicity, hematologic disorders ++
-
GIT upset, aggravate psoriasis, lupus erythematosus
-
Hepatic disease (chronic or active)
-
Renal impairment (creatinine clearance <50 mL/min)
Fluconazole
-
Cardiac abnormalities (torsade de pointes), exfoliative skin reactions, anaphylactic reactions++
-
Headache, myalgia, dizziness, GIT upset+++
-
Hepatic, renal functions abnormalities
-
Baseline LFTs
-
Full CBC
-
BUN, creatinine
-
Plasma level of CYP2D6-metabolized drugs
-
Hepatic and renal impairment
-
In patients with risk for arrhythmias
-
Coadministration with astemizole, terfenadine, cisapride (increased risk of developing torsade de pointes)
-
Coadministration with statins (increased myopathy)
Itraconazole
-
Triad of hypertension, hypokalemia, &edema in elderly+
-
Negative inotropic fulminant hepatitis, Stevens–Johnson syndrome; Anaphylaxis++
-
GIT upset, esp. odious taste with cyclodextrin solution+++
-
Headache, rhinitis, sinusitis, hepatic, renal impairment
-
Baseline LFTs
-
Full CBC
-
Regular LFTs
-
Close monitor of oral hypoglycemic, and blood glucose
-
Heart failure
-
Liver disease
-
Patients with hypersensitivity to other azoles (use with caution)
Griseofulvin
- Hetaotoxicity, pancytopenia++
- Hypersensitivity skin eruptions,
Griseofulvin
-
Hetaotoxicity, pancytopenia++
-
Hypersensitivity skin eruptions, Photosensitivity,
-
GIT upset+++
-
Neurologic problems
-
Patients with risk factors for CHF for developing signs or symptoms of CHF
-
Baseline LFTs
-
Regular LFTs
-
Blood glucose in patients using oral hypoglycemic
-
Plasma level of drugs metabolized with by CYP3A4
-
Porphyria
-
LCF
-
Patients with penicillin sensitivity
-
Liver enzymes after 8 wk of
-
Porphyria
-
LCF
-
Patients with penicillin sensitivity (use with cautious)
-
Females using OCP should change the contraceptive method or add another form
Photosensitivity,
3. GIT upset+++
4. Neurologic problems
- Liver enzymes after 8 wk of continuous use
- BUN, creatinine after 8 wk of continuous use
continuous use
2. BUN, creatinine after 8 wk of
(use with cautious)
4. Females using OCP should change
continuous use
the contraceptive method or add another form
+, Most unique; ++, most serious; +++, most common; LFTs, liver function tests; BUN, blood urea nitrogen; CBC, complete blood picture; CHF, chronic heart failure; CYP, cytochrome P; GIT, gastrointestinal tract; LCF, liver cell failure; OCP, oral contraceptive pills.
should be avoided as well, as they are known to increase its toxicity and decrease its efficacy, respectively (Table 188-4).8,13,20
BENZYLAMINES
Benzylamines are structurally and functionally related to allylamines.8
BUTENAFINE
BUTENAFINE
Butenafine is the only available antifungal derivative of benzylamine, and has a chemical formula N-(4-tertbutylbenzyl)-N-methyl-1-naphthalenemethylamine hydrochloride.8
MECHANISM OF ACTION
Similar to allylamines, butenafine inhibits the synthesis of ergosterol, with subsequent enhancement in membrane permeability and leakage of important cellular components, resulting in mycotic cell death.8
INDICATIONS
Butenafine is indicated for the treatment of dermatophyte infections, with efficacy superior to that of the allylamines; additionally, it can be used in cases of pityriasis versicolor (PV) and candidiasis (Table 188-1).8,13
DOSAGE AND FORMULATIONS
The only available commercial formulation is a 1% cream,9 applied once daily for 2 to 4 weeks.8
SIDE EFFECTS
Side effects of butenafine are minimal and include itching, burning, erythema, and contact dermatitis.
AZOLES
The azoles are group of antifungals characterized by 1 or more azole rings. They can be further subclassified into imidazoles, which include ketoconazole, butoconazole, clotrimazole, econazole, luliconazole, miconazole, and sertaconazole, and triazoles, which
3441
28
ANTIFUNGAL AGENT INTERACTING DRUG/CLASS MECHANISM OF INTERACTION RESULTING EFFECTS RECOMMENDATION
Terbinafine
- CYP2D6 metabolizing agents (TCAs, SSRI haloperidol, some BB)
Terbinafine is a CYP2D6 isoenzyme inhibitor ++ Inhibition of their breakdown, and increase their toxicity
Cautious coadministration
- Warfarin Terbinafine is a potential CYP-450 2C9 inducer Decrease the anticoagulant activity and increase the risk of thrombosis
Close monitoring of anticoagulation parameters (ie, INR, PT)
- Rifampicin, phenytoin CYP-450 enzyme inducer Induce the metabolism of terbinafine, decrease its level
Avoid combination, or increase the dose of terbinafine
- Cimetidine CYP2D6 enzyme inhibitor Inhibit the metabolism of terbinafine, decrease its clearance
Avoid combination, or decrease the dose of terbinafine
Fluconazole
-
Terfenadine, astemizole, cisapride Synergism (prolongation of QT interval) Serious arrhythmias ( torsades de pointes) Coadministration is contraindicated
-
CYP3A4-metabolizing agents (eg, CCBs, benzodiazepines, SSRIs, TCA, phenytoin, carbamazepine, cyclosporine, PDIs, warfarin)
Fluconazole is CYP3A4 inhibitor + Increase the plasma level and decrease the clearance of coadministered drug
- CYP2C9 metabolizing agents, phenytoin, oral hypoglycemic agents, (eg, sulfonylurea, rosiglitazone, and nateglinide)
Monitor the level of coadministrated drug or their effects, or consider terbinafine as alternative
Fluconazole is a CYP2C9 isoenzyme inhibitor Clinically evident phenytoin toxicity, hypoglycemia respectively
- 4.CYP3A4 inducers (eg, phenytoin, carbamazepine, phenobarbital, rifampin, rifabutin, INH, nevirapine)
Consider other alternatives (terbinafine, itraconazole, griseofulvin)
Induce the metabolism of fluconazole Decrease its therapeutic effects Avoid combination, or increase the dose of fluconazole
- CYP3A4 inhibitors (clarithromycin, indinavir, ritonavir)
Decrease the metabolism of fluconazole Increase its therapeutic and side effects Avoid combination, or decrease the dose of fluconazole
Itraconazole
- H2 blocker antihistamines (eg, cimetidine, ranitidine) antacids, proton pump inhibitors (eg, omeprazole pantoprazole), oral didanosine
Decrease gastric acidity Decrease itraconazole absorption and bioavailability
- Drug metabolized by cytochrome 3A4 (CYP3A4) isoenzyme (eg, statins, darunavir, CCBs, benzodiazepines, warfarin)
Itraconazole capsules can be taken 2 h after the concurrent drug, or consider alternatives not affected by gastric acidity(eg, itraconazole solution, terbinafine or fluconazole)
Itraconazole is a CYP3A4 inhibitor ++ Decrease the metabolism of these drugs; hence increase their therapeutic and side effects
Monitor the level of coadministrated drug or their effects (eg, close. monitoring of the level of myopathy and myoglobinuria in case of statins), or consider terbinafine as alternative
-
Digoxin, quinidine Itraconazole is a P gp and CYP3A4 inhibitor Digoxin toxicity Terbinafine, fluconazole, or griseofulvin can be safely administered
-
CYP3A4 inducers Increases the hepatic metabolism of itraconazole
-
CYP3A4 inhibitors ( clarithromycin, indinavir, ritonavir)
Decreases the hepatic metabolism of itraconazole
3442
Accelerate the clearance of itraconazole Avoid combination, or increase the dose of itraconazole
Decrease the clearance of itraconazole Avoid combination, or decrease the dose of itraconazole
(Continued)
28
(Continued)
ANTIFUNGAL AGENT INTERACTING DRUG/CLASS MECHANISM OF INTERACTION RESULTING EFFECTS RECOMMENDATION
Griseofulvin
-
Warfarin, phenobarbital, and estrogens Griseofulvin is an enzymatic inducer Increase the metabolism of these drugs Dose adjustment of the concurrent drugs
-
Phenobarbitone Decrease the oral
Failure in griseofulvin
Avoid coadministration
- Phenobarbitone Decrease the oral absorption and induces metabolism of Griseofulvin.
Failure in griseofulvin therapy Avoid coadministration
absorption and induces metabolism of Griseofulvin.
therapy
+, mild potency or needs high doses; ++, high potency; CYP, cytochrome P; BB, beta blockers; CCBs, calcium channel blockers; INH, isoniazid; INR, International Normalized Ratio; PDIs, phosphodiesterase inhibitors; PT, prothrombin time; SSRI, selective serotonin-reuptake inhibitors; TCAs, tricyclic antidepressants.
include fluconazole, itraconazole, efinaconazole, and isavuconazole.5
MECHANISM OF ACTION
MECHANISM OF ACTION
Azoles act through the inhibition of lanosterol demethylase enzyme (14α-demethylase), a fungal cytochrome P450 (CYP)–dependent enzyme that catalyzes the conversion step of lanosterol into ergosterol, thereby blocking the biosynthesis of ergosterol. Subsequently, the reduced level of ergosterol disrupts the membrane permeability. Additionally, the buildup of the 14α-methylated sterols precursors will interfere with cell growth, leading eventually to cell death. Azoles in normal concentrations are typically fungistatic agents; however, high concentrations of azoles can be fungicidal. The heme-containing pocket is the binding site of azoles on the lanosterol demethylase enzyme and variations in its conformation, together with variation in the drug structure, are the determinants of the azole binding affinity and cross resistance.21
IMIDAZOLES
IMIDAZOLES
CLOTRIMAZOLE Mechanism of Action: Clotrimazole is the first developed topical imidazole that can disrupt mycotic phospholipids, resulting in leakage of intracellular iron, degradation of nucleic acids, and suppression of cell respiration.8
Pharmacokinetics: Application of clotrimazole is associated with negligible systemic absorption (0.5%).13
Indications: Clotrimazole can be used for all superficial mycoses, achieving mycological cure rates ranging from 60% to 100% in dermatophytosis, and 80% to 100% in cutaneous candidiasis.13
Dosage and Formulations: Clotrimazole is available as cream, lotion, spray, powder, lozenge,
suppository, and 1% solution (Table 188-1). As with most azoles, clotrimazole is applied twice daily for 2 to 4 weeks according to the indication.8
Side Effects: Similar to other topical antifungals, clotrimazole can result in local skin reaction in the form of itching, erythema, and rash.8,13
Pregnancy Category: B.
KETOCONAZOLE
Ketoconazole was the first marketed oral broad-spectrum antifungal agent.13
Mechanism of Action: Ketoconazole inhibits 14α-demethylase, blocking the conversion of lanosterol into ergosterol. It has fungicidal activity at high concentrations.3 Possible antiinflammatory activity of topical ketoconazole has been postulated.22
Pharmacokinetics: Systemic absorption of topical ketoconazole is negligible, whereas 5% of the drug reaches the hair keratin 12 hours from the first shampoo application.8 The absorption of oral ketoconazole is enhanced by acidic beverages, and decreases with the increase in gastric pH.13
Indications: Ketoconazole is used topically for the treatment of all dermatomycoses and its shampoo formulation is effective in the treatment of seborrheic dermatitis (Table 188-1). However, the prescription of oral preparations has now been markedly restricted by the FDA and other health organizations due to drug interaction and serious side effects on the liver and adrenal glands.23-25
Dosage and Formulations: The commercial formulations in the United States include 5% shampoo, 2% cream, and 200-mg tablets. Ketoconazole shampoo is applied once daily (left for 5-10 minutes) for 1 to 4 weeks in the treatment of PV, and once weekly for prophylaxis (Table 188-1).26 In cases of scalp seborrheic dermatitis, the shampoo is used twice a week for 4 weeks. Oral ketoconazole is used once daily for 10 to 20 days in cases of superficial mycosis.13
3443
28
Side Effects: The adverse effects of the topical drug are generally minimal and uncommon, including itching, allergic reactions, and contact dermatitis. Serious side effects have been recorded with the oral formulation, encompassing liver toxicity, anaphylactic reactions, and marked depression. More common but less serious side effects include nausea, vomiting, diarrhea, abdominal pain, headache, sleeping disturbances, dizziness, and pancytopenia, as well as impotence, gynecomastia, and decreased libido owing to its androgen-opposing properties at both the adrenal and testicular levels.8
Drug–Drug Interactions: Ketoconazole is a strong inhibitor of CYP3A4 and can increase the serum concentrations of coadministered drugs foremost metabolized by this enzyme, leading to a decrease in therapeutic efficacy and increase in adverse effects.8
MICONAZOLE Mechanism of Action: In addition to the mechanism inherent to azoles, miconazole is capable of blocking the mycotic peroxidase enzyme, leading to the accumulation of toxic peroxide and subsequent cell death.8
Pharmacokinetics: Miconazole efficiently penetrates the stratum corneum, with <1% absorbed to systemic circulation.8
Indications: Miconazole cream is an efficacious treatment for tinea cruris, tinea corporis, and tinea pedis, in addition to its efficacy in treating PV and cutaneous candidiasis (Table 188-1).
Dosage and Formulations: The commercial preparations of miconazole include vaginal suppositories (100 mg, 200 mg), gel (2%), cream (2%), ointment, lotion, and powder.8,13
Side Effects: Miconazole side effects include those inherent to topical azoles, with cross-sensitivity recorded with the majority.8
LULICONAZOLE
Luliconazole and sertaconazole are the most recently developed imidazoles.
Mechanism of Action: Luliconazole mechanism of action is the same as azoles, though the modification of its structure renders it less liable to keratin binding and more available for penetration into deeper nail layers. Therefore, many studies are now investigating the use of luliconazole for treating onychomycosis.27
Pharmacokinetics: Luliconazole is characterized by second meta-replaced chlorine on its benzene ring, which is responsible for its superior potency over lanoconazole.28
3444
Indications: Luliconazole is FDA approved for the treatment tinea cruris, tinea pedis, and tinea corporis (Table 188-1).12
Dosage and Formulations: Luliconazole 1% cream is the only available formulation in the United States.29
Side Effects: Mild cutaneous irritation is uncommonly reported with luliconazole.29
SERTACONAZOLE
Sertaconazole is a benzothiophene imidazole derivative.28
Mechanism of Action: Sertaconazole, in addition to the typical action of azoles, is capable of exhibiting fungicidal action at high concentration through binding to nonsterol lipids in the mycotic cell wall, increasing its permeability and resulting in cell lysis. In addition, sertaconazole has antiinflammatory, antibacterial, and antipruritic properties.28
Pharmacokinetics: Insignificant amounts of sertaconazole may reach the systemic circulation after topical application; additionally, sertaconazole forms a drug depot that can persist in the stratum corneum up to 48 hours after the last dose.28
Indications: In the EU, sertaconazole is approved for all dermatomycoses (PV, seborrheic dermatitis, cutaneous candidiasis, and dermatophytosis), whereas in the United States, its license is limited to tinea pedis.28
Dosage and Formulations: Sertaconazole is available as 2% cream and solution (Table 188-1).28
Side Effects: Topical application can result in mild local dryness, burning sensation, and dermatitis.28
Pregnancy Category: C.
EFINACONAZOLE Mechanism of Action: Efinaconazole reduces the ergosterol synthesis in a dose-dependent manner.30
It inhibits lanosterol 14-alphademethylase, which prevents transformation of lanosterol into ergosterol.
Pharmacokinetics: Less efinaconazole is bound to keratin compared to amorolfine and ciclopirox, which allows more drug to penetrate the nail plate. In addition, its lipophilic ester component reduces the surface tension, allowing more nail penetration.28
Indications: Efinaconazole is FDA licensed for the topical treatment of onychomycosis.28
Dosage and Formulations: Efinaconazole 10% solution is applied as a single daily application for 48 weeks (Table 188-1).27,28
Side Effects: Local eczema and ingrown nails have been reported with efinaconazole use.27,28
Pregnancy Category: C.
TRIAZOLES
TRIAZOLES
Both triazoles and imidazoles are 5-membered ring heterocycles. Imidazoles have 2-ring nitrogen atoms, whereas triazoles contain 3 and are less prone to metabolic degradation.
FLUCONAZOLE
Fluconazole is a water-soluble bis-triazole that became FDA approved in the early 1990s.8
Mechanism of Action: Fluconazole inhibits lanosterol 14-demethylase and prevents conversion of lanosterol to ergosterol.27,31
Pharmacokinetics: Fluconazole has an excellent oral bioavailability, attaining nearly 90% (comparable to that achieved by intravenous route). Its pharmacokinetics are linear, predictable, and independent of gastric acidity, food intake, or disease activity. Unlike most other antifungal agents, only a small percentage of fluconazole is bound to plasma protein, resulting in greater amounts of free drug for therapeutic activity.6,32
The drug is also characterized by a long half-life of 25 to 30 hours.4 In addition, the wide distribution in various tissues including skin, nail, vitreous chamber of the eye, and cerebral spinal fluid (CSF), together with a good safety profile especially in immunocompromised patients, have qualified the use of fluconazole for the treatment of invasive fungal infections for almost 3 decades. The drug is poorly metabolized by the liver, and hence the kidney is the main route of drug elimination; therefore, dose adjustment is required with renal insufficiency.6,32
Indications: Fluconazole is one of the most commonly used antifungals in the treatment of esophageal, vaginal, and oropharyngeal candidiasis, and cryptococcal meningitis. It is efficacious against all Candida except C. krusei. Additionally, fluconazole is used as an off-label treatment for onychomycosis, tinea infections, PV, and chronic mucocutaneous candidiasis when itraconazole and terbinafine are contraindicated (Table 188-2).8,13
Dosage and Formulation: Fluconazole is available in the following formulations: 150 mg capsules, tablet, liquid, and solution for intravenous infusion. Fluconazole is used in the treatment of onychomycosis in adults at a weekly dose of 150 to 450 mg for 3 and 6 months for finger and toe nail onychomycosis, respectively. In children, the dose is calculated based on body weight, 3-6 mg/kg/wk for 6 to 12 weeks in cases of finger onychomycosis extended to 9 to 15 weeks in cases of toe nail involvement. The recommended dose in cases of dermatophytosis is 150 mg once a week for
28
2 to 4 weeks in cases of tinea barbae, tinea pedis, and tinea corporis, and 4 to 6 weeks in tinea cruris and tinea capitis. Fluconazole is effective in the treatment of cutaneous and chronic mucocutaneous candidiasis and PV at a dose of 300 mg/wk for 2 weeks and once monthly for prophylaxis (Table 188-2).3,26,33
Side Effects: In general, fluconazole has fewer side effects than other antifungals. The most frequent drug side effects are headache, myalgia, dizziness, nausea, dyspepsia, diarrhea, and abdominal pain. In addition, cardiac abnormalities such as prolonged QT intervals and torsade de pointes are uncommonly reported with fluconazole. Exfoliative cutaneous reactions including Stevens–Johnson syndrome and toxic epidermal necrolysis were rarely described with the drug. Other adverse events include anaphylactic reactions, angioedema, acneform eruption, eye hemorrhage, and neutropenia (Table 188-3).8
Drug–Drug Interactions: Fluconazole is a substrate of the CYP3A hepatic isoenzyme. In addition, it has a moderate suppressing effect on CYP3A and CYP2C9, and a weak inhibitory effect on CYP2C19; therefore, the concurrent administration with drugs metabolized by any of these enzymes could result in deleterious drug interactions. An example would be fatal arrhythmias with the concomitant administration of astemizole, cisapride, pimozide, and terfenadine (Table 188-4).3,8,32
Pregnancy Category: C
ITRACONAZOLE
Itraconazole is a broad-spectrum fungistatic triazole, synthetically derived from ketoconazole through the prolongation of its hydrophobic side chains, promoting the binding of the drug to the apoprotein of the 14α-demethylase enzyme.3
Mechanism of Action: Like other azoles, itraconazole acts through the inhibition of 14-α-demethylase, suppressing the conversion of lanosterol to ergosterol.3
Pharmacokinetics: Itraconazole is a highly lipophilic agent. The capsule formulation requires gastric acidity for optimum absorption, as it delays the gastric emptying and increases the drug dissolution; therefore, it is best administrated after a meal. In contrast, a cyclodextrin oral solution of itraconazole is not influenced by gastric acidity, and is best absorbed when taken on an empty stomach. Like terbinafine, itraconazole has a high affinity to nail and can accumulate there, forming a drug reservoir for 6 to 9 months after discontinuation of the drug. The half-life of itraconazole is typically 21 hours, and it has been postulated that its efficacy as a prophylactic agent is directly proportional to its plasma level, but should not, however, exceed 17 µg/mL by bioassay measurement to avoid drug toxicity. A twice-daily scheduling of doses is required in children because of drug lower plasma concentrations compared to adults.3 Itraconazole undergoes an extensive hepatic metabolism by cytochrome CYP3A4
3445
28
enzyme; as a result, a dose adjustment is required in patients with progressive liver disease. Renal insufficiency and renal dialysis have no influence on the drug plasma concentration. An exception is the intravenous formulation, which should be avoided when the creatinine clearance is less than 30 mL/min.13
Indications: Itraconazole is considered the best choice in treatment of Candida and nondermatophyte onychomycosis; its short course of treatment and good tolerability make it an especially good treatment choice for children.15 Itraconazole is also used as an off-label treatment for many dermatophytoses, including tinea corporis, tinea capitis, tinea pedis, tinea cruris, tinea manuum, and PV (Table 188-2).
Dosage and Formulations: The recommended dose for tinea infections and PV is 100 mg twice daily for 5 days (Table 188-2). For the treatment of onychomycosis, itraconazole is prescribed as a continuous regimen (a daily dose of 200 mg for 6 weeks) or a monthly pulse dose (400 mg/d for 1 week). The dosage of the drug in children is calculated based on body weight (5 mg/kg/d) and both continuous or pulse regimens can be followed (Table 188-2).8,13
Side Effects: The most frequent side effects of itraconazole are GI disorders, especially the characteristic unpleasant taste associated with the hydroxypropyl-b-cyclodextrin vehicle, which can lead to patient noncompliance. Elderly patients on itraconazole often experience a triad of edema, hypertension, and hyperkalemia. In addition, itraconazole has a negative inotropic effect that can predispose to complete heart failure; therefore, it should be avoided in patients with a history of heart disease. Side effects such as hepatitis, jaundice, Stevens–Johnson syndrome, peripheral neuropathy, and adrenal suppression have been infrequently reported (Table 188-3).3
Drug–Drug Interactions: Itraconazole is a hepatic CYP3A4 enzyme substrate and inhibitor, and thus can affect the serum level, efficacy, and toxicity of drugs metabolized by the same enzyme. Similarly, CYP3A4 enzyme inducers and other inhibitors can increase or decrease the metabolism of the drug, respectively, and special considerations should be taken when combining itraconazole with one or more of these drugs. Given the necessity of gastric acidity for the optimum absorption of itraconazole, drugs such as H2 receptor blockers and proton pump blockers will decrease the drug absorption and subsequently its therapeutic effect (Table 188-4).8
Pregnancy Category: C.
POLYENES
NYSTATIN
NYSTATIN
Nystatin is a topical polyene derived from Streptomyces spp.8
3446
MECHANISM OF ACTION
Nystatin has both fungistatic and fungicidal activity. It acts through binding to the mycotic ergosterol, for which its affinity is higher than that of the cholesterol found in mammalian cells. This binding will result in creation of pores in the cell membrane with subsequent leakage of essential fungal intracellular components, resulting in cell death.34
PHARMACOKINETICS
Because of the high toxicity associated with its intravenous administration, nystatin is confined to topical use only. Systemic absorption after cutaneous or mucous membrane application is insignificant.8
INDICATIONS
Nystatin is commonly used in the treatment of cutaneous, oropharyngeal, and vaginal candidiasis. The drug is not effective against dermatophytes or Malassezia spp. (Table 188-1).8,34,35
DOSAGE AND FORMULATIONS
Nystatin is available in cream, ointment, liquid, suspension, powder, and lozenge formulations. A twicedaily application for 4 to 5 days is the recommended dose for cutaneous infections, while the regimen for the treatment of oral candidiasis is a rinse 4 times daily for 14 days (Table 188-1).8,35,36
SIDE EFFECTS
Few side effects were reported with all formulations of nystatin; these include localized hypersensitivity reactions associated with the cutaneous application and manifested as allergic contact dermatitis, erythema, itching, and edema. Nausea, vomiting, and diarrhea have been reported with large doses of oral formulations.8,25
Pregnancy Category: B.
OTHER ANTIFUNGALS
CICLOPIROX
CICLOPIROX
Ciclopirox olamine is a synthetic ethanolamine salt derivative of hydroxypyridone.37
MECHANISM OF ACTION
Ciclopirox exhibits broad-spectrum fungicidal and fungistatic, antibacterial, and antiinflammatory activities. It acts through the inhibition of the transmembrane uptake of RNA, DNA, and protein precursors. High drug concentrations (>50 µg/mL) alter the integrity of the cell membrane, leading to leakage of K ions, peptides, and amino acids. Ciclopirox also suppresses
aerobic glycolysis in the yeast cell, and subinhibitory concentrations decrease the adherence of Candida albicans to both vaginal and buccal epithelial cells. The drug is capable of chelating trivalent molecules as Al3+ and Fe3+, cofactors for the enzymes involved in detoxification of hazardous metabolites, mitochondrial electron transport chain, and energy production. The antiinflammatory effect of ciclopirox is attributed to its ability to reduce the synthesis and release of the proinflammatory prostaglandin E2.38
PHARMACOKINETICS
The vaginal application of ciclopirox was associated with 15% to 20% systemic absorption, whereas the systemic absorption from other routes was insignificant.38 Compared to efinaconazole and luliconazole, ciclopirox exhibits the least nail permeation. Ciclopirox is metabolized by glucuronidation and its metabolites are eliminated mainly by the kidney.39
INDICATIONS
Ciclopirox is used in the treatment of various dermatophytoses.11 A ciclopirox monotherapy of onychomycosis has been associated with a cure rate of 5.5% to 8.5% after 48 weeks,40 whereas a combination with oral terbinafine increased the cure rate to 88.2% compared to 64.7% achieved by terbinafine alone.41 Ciclopirox also can be used in the treatment of cutaneous candidiasis, tinea versicolor, and scalp seborrheic dermatitis (Table 188-1).38
DOSAGE AND FORMULATIONS
Ciclopirox can be incorporated in one of the following formulations with either isopropyl or benzyl alcohol as a vehicle: 0.77% cream, gel, 1% shampoo, and 8% nail lacquer solution. For most indications, twicedaily application is required and should be continued for 4 weeks, regardless of clinical improvement. The ciclopirox shampoo should be left for 3 minutes before rinsing. The nail lacquer, on the other hand, should be applied on a daily basis and removed with alcohol once a week before reapplication (Table 188-1).38,42
SIDE EFFECTS
Side effects include mild erythema and itching of the skin adjacent to the application site and infrequent nail alterations.42
AMOROLFINE
AMOROLFINE
MECHANISM OF ACTION
Amorolfine interrupts the ergosterol synthesis at the 14-reduction and the 7-8 isomerization in the ergosterol pathway, resulting in the depletion of
28
ergosterol as well as the accumulation of 24-methylene ignosterol.43
PHARMACOKINETICS
The nail lacquer tends to build a reservoir that continues to release the drug to the nail plate in an exponential pattern. Negligible systemic absorption is associated with topical application. The drug is generally eliminated in urine and stool at a very slow rate.43
INDICATIONS
Amorolfine has both fungistatic and concentration- and time-dependent fungicidal action against yeasts, and exhibits fungicidal activity against most dermatophytes and other filamentous fungi. Amorolfine nail lacquer is approved for the treatment of onychomycosis in the absence of matrix involvement in Europe but not in the United States. The use of amorolfine vaginal tablets has been associated with high clinical cure rates of vulvovaginal candidiasis (90%-95%), though with a high recurrence rate (Table 188-1).
DOSAGE AND FORMULATIONS
Amorolfine is available as a 0.25% cream and 5% nail lacquer. For superficial dermatomycosis, cream is applied once daily for 2 to 4 weeks, whereas the regimen for onychomycosis is a weekly application of nail lacquer until the regeneration of nail (approximately 6 or 12 months in finger- and toenail onychomycosis, respectively) (Table 188-1).44
SIDE EFFECTS
Side effects include mild allergic reaction of adjacent skin.43
Pregnancy Category: D.
TAVABOROLE
TAVABOROLE
Tavaborole is a novel oxaborole antifungal drug that encloses a fluorine atom, decreasing the pH of the boronic center, which inhibits fungal peptide synthesis. Tavaborole exhibits broad-spectrum activity against a variety of fungi, including the dermatophytes (Trichophyton rubrum, Trichophyton mentagrophytes, T. tonsurans, and Epidermophyton floccosum), some Microsporum species, and C. albicans.45
MECHANISM OF ACTION
Tavaborole acts through inhibiting a new target, aminoacyl transfer RNA synthetase (AARS), for which its affinity is a thousand times that of the mammalian cell and thereby selectively inhibits the mycotic protein synthesis.27
3447
28
PHARMACOKINETICS
Tavaborole is incorporated in an alcohol-based solution and is therefore not suitable for ophthalmic, oral, or intravaginal use. It is characterized by low molecular weight, which accounts for enhanced nail penetration. Tavaborole usually undergoes extensive metabolism before finally being eliminated in the urine.27
INDICATIONS
Tavaborole is approved for the topical treatment of onychomycosis (Table 188-1).27
DOSAGE AND FORMULATIONS
Tavaborole is available as a 5% topical solution, applied once daily for 48 weeks (Table 188-1).46
SIDE EFFECTS
The most common side effects are exfoliation, erythema, and dermatitis.47
Pregnancy Category: C.
GRISEOFULVIN
GRISEOFULVIN
Griseofulvin is a metabolic derivative of Penicillium griseofulvum, classically used for the treatment of dermatophytes. It has no activity against yeast and molds.8
MECHANISM OF ACTION
Griseofulvin is a fungistatic drug, blocking the growth and proliferation of the fungal cell. Griseofulvin binds tubulin- and microtubule-associated proteins (MAPs) along the polymerized microtubules, suppressing the formation of the mitotic spindle at the G2/M phase of the cell cycle. This inhibits cell division and forces the cell to undergo apoptosis.48
PHARMACOKINETICS
Griseofulvin is a water-insoluble drug, known for its low bioavailability profile. Most of the drug absorption occurs in the duodenum, and better absorption of griseofulvin can occur either when coated with polyethylene glycol or on coadministration with fatty meals.49,50 Griseofulvin is characterized by its accumulation in the keratin-producing tissues, where it is adherently bound to newly formed keratin, rendering it resistant to fungal penetration. The 6-desmethyl enzyme is responsible for the metabolism of griseofulvin in the liver. The cutaneous elimination of the drug is slower than its elimination from plasma, allowing for extended drug activity even after its discontinuation. Griseofulvin is eliminated from the body mainly through the kidney in the form of metabolites.3
3448
INDICATIONS
Griseofulvin remains the first line of treatment for tinea capitis caused by Microsporum species owing to its higher efficacy compared to terbinafine, and similar efficiency yet lower cost compared to itraconazole and fluconazole.19 It is also the only FDA-approved treatment for pediatric onychomycosis, though its efficacy is limited to dermatophytes only (Table 188-2).15
DOSAGE AND FORMULATIONS
Griseofulvin is available in the following formulations: 250- and 500-mg microsize and 125-, 165-, and 250-mg ultramicrosize tablets, and 125-mg/5-mL oral suspensions.8 Griseofulvin is recommended at a daily dose of 1-g microsize, or 660- to 750-mg ultramicrosize for 4 to 8 weeks in the treatment of tinea pedis and half of these doses for the treatment of tinea cruris and corporis. The recommended daily dosing of griseofulvin for the treatment of tinea capitis in children is dependent on their body weight and is as follows: ultramicrosize 10 to 15 mg/kg, oral suspension 15 to 25 mg/kg, microsize 20 to 25 mg/kg, continued for 1 to 2 months (Table 188-2).3
SIDE EFFECTS
The main side effects associated with griseofulvin are related to hypersensitivity, which ranges from mild urticaria, serum sickness, acute generalized exanthematous pustulosis, subacute cutaneous lupus erythematous, Stevens–Johnson syndrome, and toxic epidermal necrolysis. Photosensitivity, including photo-toxic and photo-allergic reactions, also has been commonly reported with griseofulvin.51 Other cutaneous eruptions include lichenoid eruption, porphyria, and pityriasis rosea.12,13 Rare but serious side effects include hepatotoxicity, leukopenia, thrombocytopenia, and anemia.51 Neurologic problems such as peripheral neuritis, memory loss, confusion, and insomnia were uncommonly recorded in some patients (Table 188-3).52,53
DRUG–DRUG INTERACTIONS
Griseofulvin should not be prescribed concurrently with phenobarbitone because it decreases the absorption and increases the metabolism of griseofulvin.44
Interactions with alcohol, cyclosporine, oral contraceptives, aspirin, and warfarin also have been reported (Table 188-4).54-56
Pregnancy Category: C.
NEW CARRIER VEHICLES AND FORMULATIONS
A number of new carrier vehicles have been investigated to improve the bioavailability of topical antifungals and enhance their therapeutic effects. Different mechanisms include incorporation of the drug in
a lipid core matrix to achieve a maintained drug release up to 24 hours and to prevent light degradation of the drug. New carriers include micelles, solid lipid nanoparticles and nanostructured lipid carriers, and drug microemulsions. Vesicular carrier systems include liposomes, niosomes, transferosomes, ethosomes, and penetration enhancer vesicles.57,58
COMBINATION THERAPY
For the treatment of onychomycosis, combination therapy of a topical agent with a systemic antifungal provides several advantages, including higher rates of fungal killing, prevention of drug resistance, and expansion of spectrum activity.3,15,59 Further, because the duration of therapy is shorter and oral medications can be used at lower doses, combination therapy decreases drug-related toxicity.15,59,60 A case in which combination therapy can be of particular use is in onycholysis secondary to onychomycosis, as the separation of the nail plate from the subungual tissue interrupts the transport of drug from the nail plate to the nail bed and vice-versa. Using topical as well as oral therapy would ensure the nail plate and nail bed both are in contact with the antifungal agent.3,59-61
In the case of superficial skin infections, the addition of topical steroids to antifungal therapy, either in the same formulation or separately, is controversial. Because of the concern about the side effects associated with the abuse of topical steroids, many studies have discouraged this combination and suggested the use of topical antifungals with antiinflammatory properties instead.12
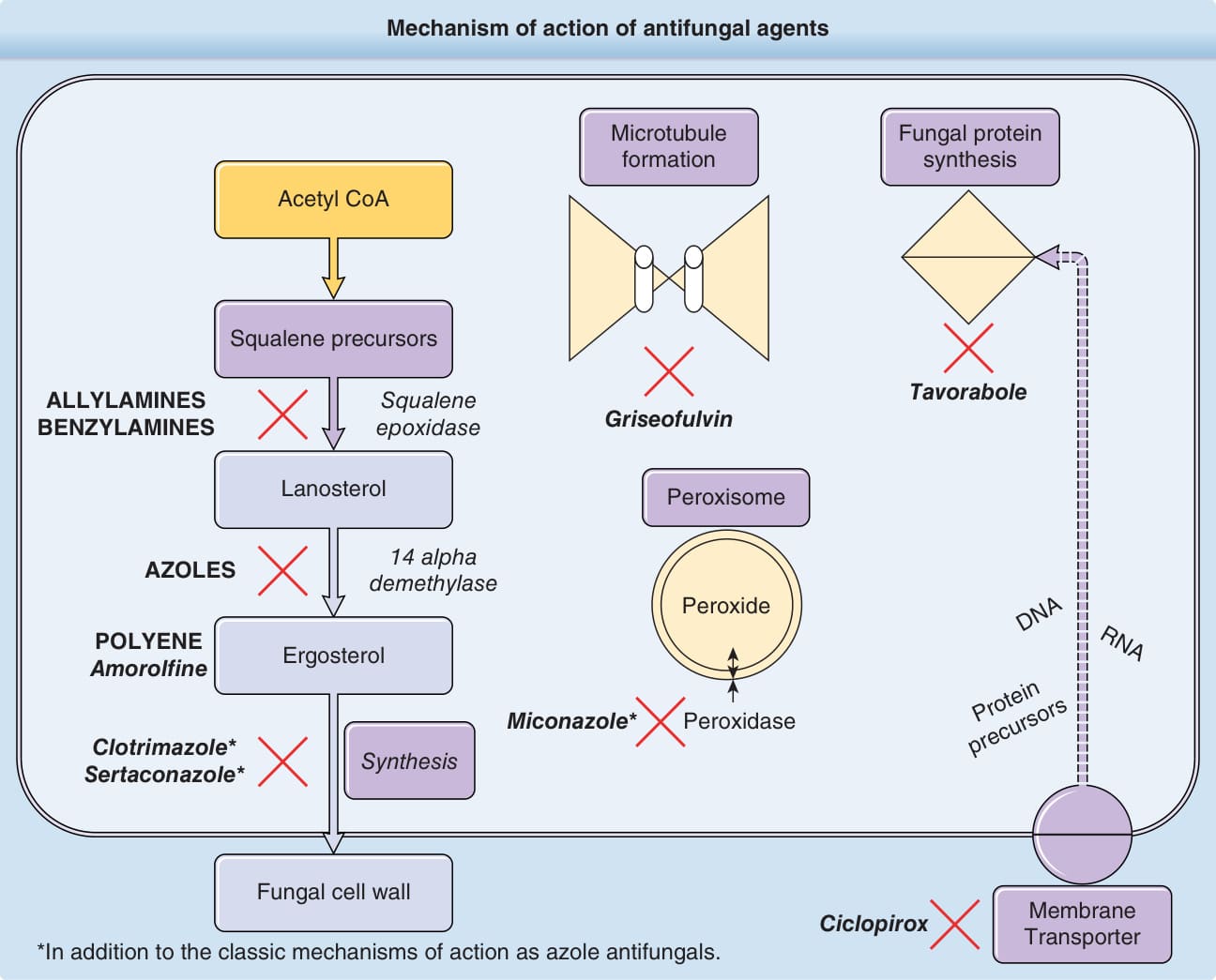
Figure 188-1 Mechanism of action of antifungal agents.
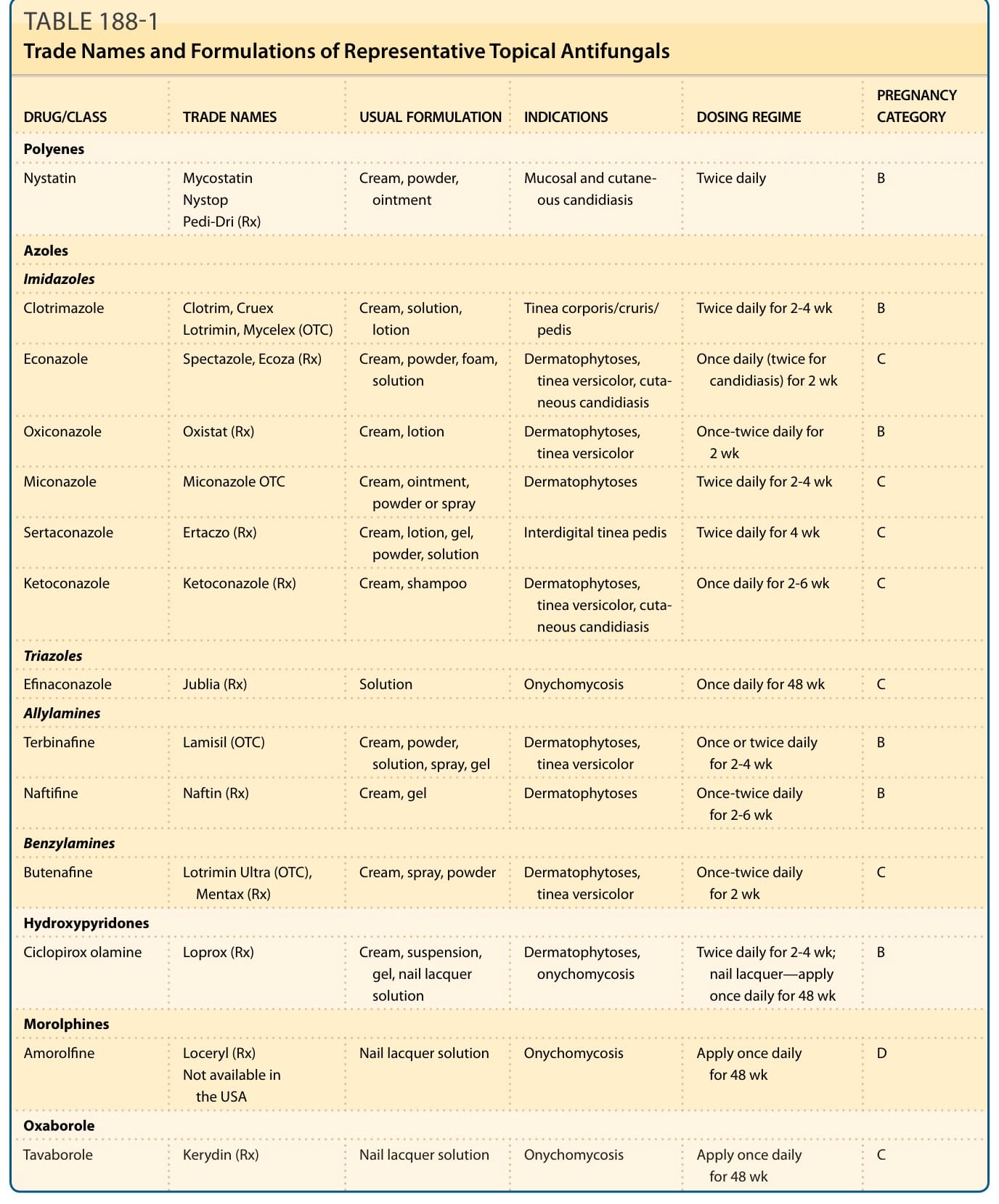
TABLE 188-1 Trade Names and Formulations of Representative Topical Antifungals
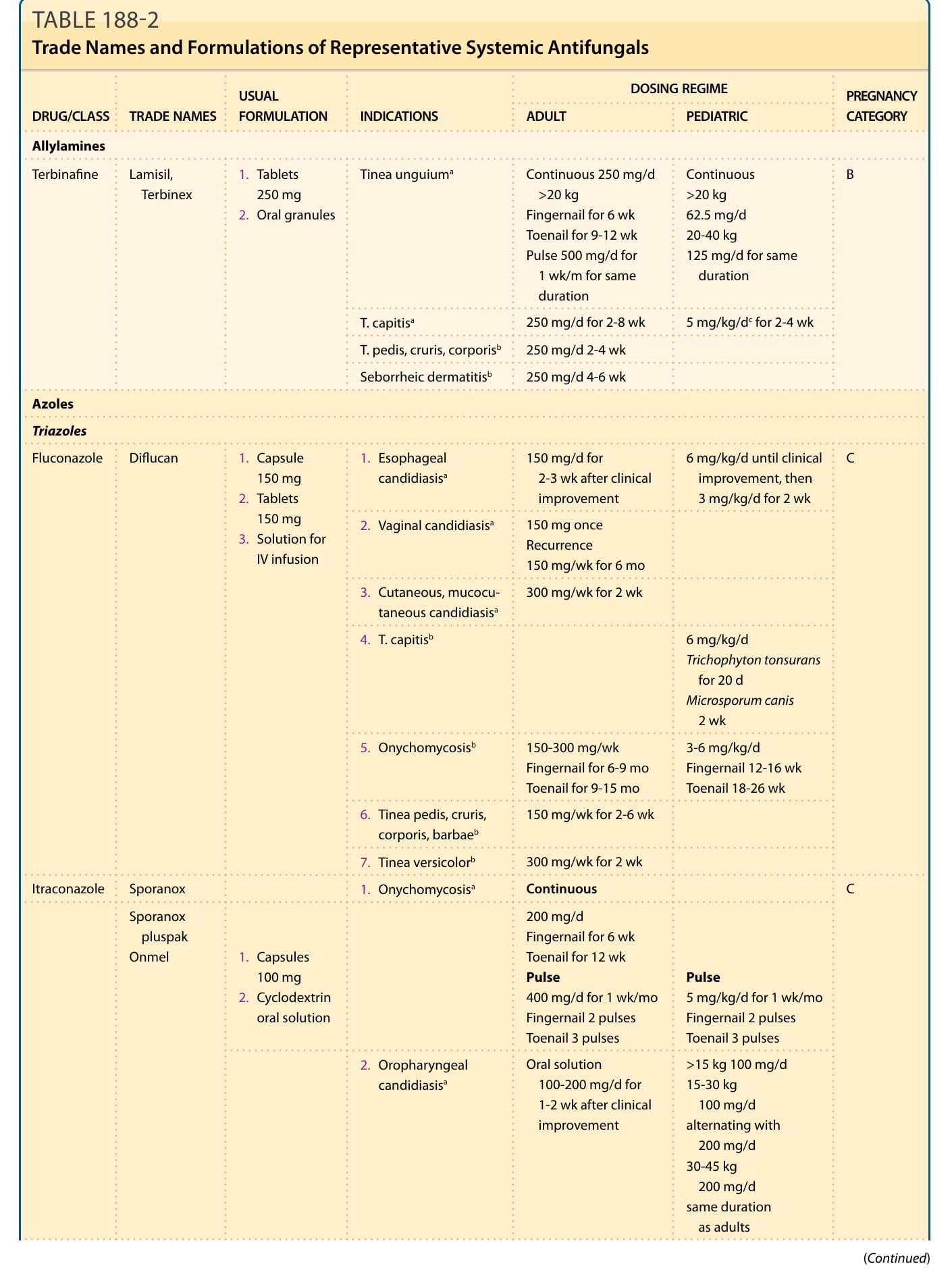
TABLE 188-2 Trade Names and Formulations of Representative Systemic Antifungals

TABLE 188-3 Side Effects, Contraindications, and Monitoring of Representative Systemic Antifungals
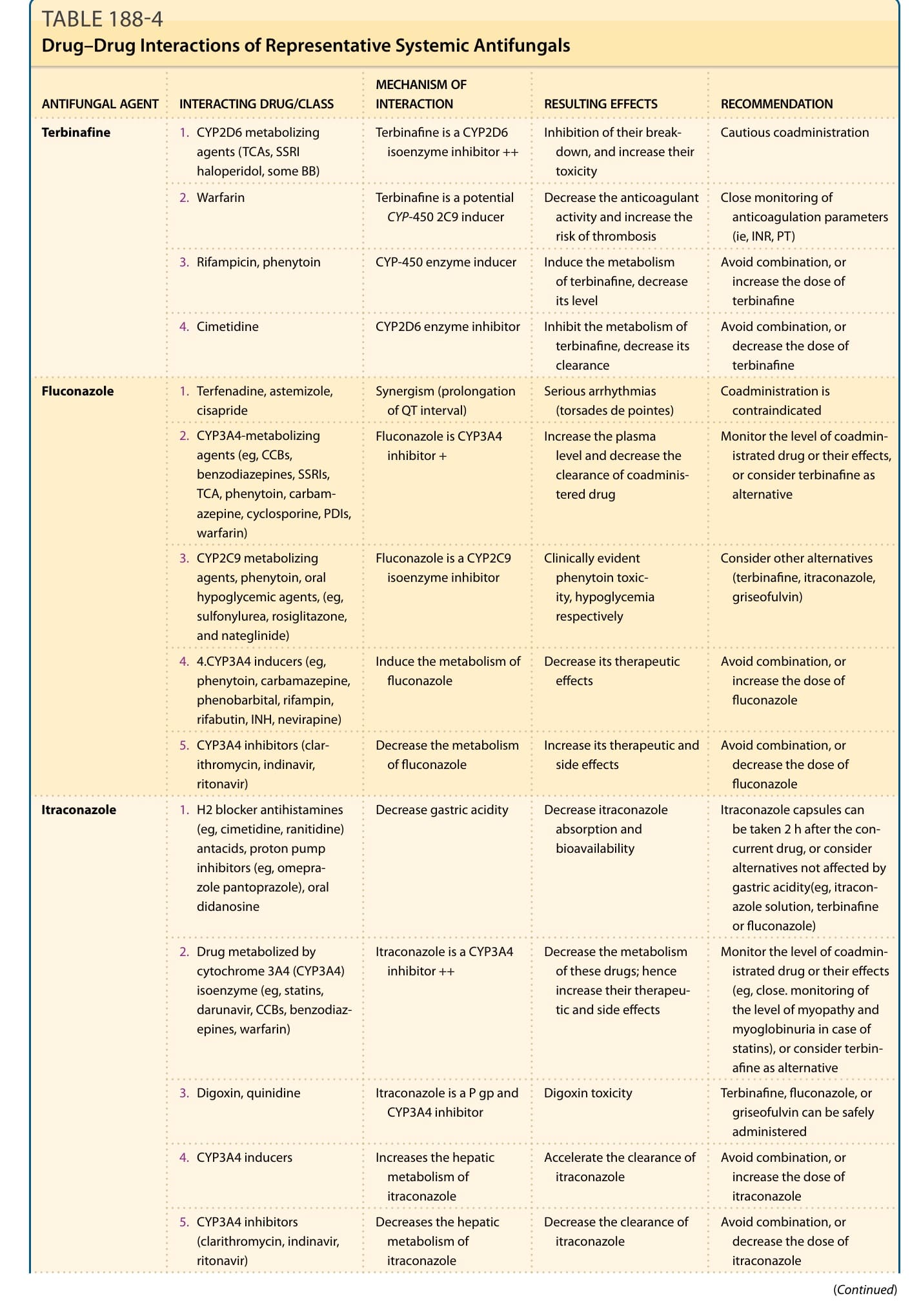
TABLE 188-4 Drug–Drug Interactions of Representative Systemic Antifungals