Cryoglobulinemia and Cryofibrinogenemia
22
Cryoglobulins are circulating immunoglobulins found in both serum and plasma that reversibly precipitate or gel upon cold exposure. Cryofibrinogens result from precipitation of fibrinogens on cold exposure and are detectable only in plasma samples, not serum. This chapter focuses on conditions that lead to either microvascular occlusion (Type I cryoglobulinemia and cryofibrinogenemia) or small- and medium-sized vessel vasculitis (Type II and III cryoglobulinemia) with emphasis on lesion morphology and cutaneous manifestations.
CRYOGLOBULINEMIA
AT-A-GLANCE
■ Cryoglobulins are circulating immunoglobulin complexes found in plasma or serum that reversibly precipitate in cold temperatures.
■ Cryoglobulinemia may be asymptomatic or cause a clinical syndrome involving the skin: purpura at distal sites is the hallmark. Livedo reticularis, acrocyanosis, ulceration, or gangrene also may be seen.
■ Cryoglobulins are classified based on molecular properties into Types I, II, III (Brouet classification).
■ Type I cryoglobulins consist of a single monoclonal immunoglobulin (typically IgG) or light chain. They occur with hematologic malignancies such as multiple myeloma. If symptomatic, they present with a noninflammatory occlusive vasculopathy.
■ Type II cryoglobulins consist of a monoclonal immunoglobulin (typically IgM) complexed with a polyclonal immunoglobulin (typically IgG).
■ Type III cryoglobulins consist of purely polyclonal immunoglobulin complexes; Type II-III refers to an intermediate state with oligoclonal immunoglobulin.
■ Types II and III are also referred to as mixed cryoglobulinemias. They are caused by chronic hepatitis C virus (HCV) infection in >90% of cases, whereas in a minority of cases no etiology can be identified, designating them as “essential mixed” cryoglobulinemias. They present as the cryoglobulinemic vasculitis syndrome, an immune complex vasculitis involving the skin, neural, and renal tissues.
■ Therapy primarily consists of controlling the underlying disease. In addition, immunosuppressants, plasma exchange, or targeting of cryoglobulin-producing B-cell clones may alleviate the cryoglobulin burden.
INTRODUCTION
INTRODUCTION
DEFINITIONS AND HISTORICAL PERSPECTIVES
In 1933, Wintrobe and Buell first noted the in vitro phenomenon of cryoprecipitation, the cold-induced precipitation of plasma or serum proteins that is reversible upon rewarming at 37°C (98.6°F).1 In 1947, Lerner and Watson described immunoglobulins and mixtures of immunoglobulins with other proteins that precipitate in the cold and called them cryoglobulins.2 In 1974, Brouet introduced the classification of cryoglobulins based on molecular properties that is widely used.2 Cryoglobulinemia describes the presence of cryoglobulins in a patient’s serum (Fig. 144-1). It can be asymptomatic, meaning that its presence usually goes clinically undetected, or it can cause occlusive vasculopathy or the so-called cryoglobulinemic syndrome, which is characterized by immune-complex deposition causing vasculitis that involves the skin and other tissues. The classification scheme of Brouet, estimated frequency, and composition of cryoglobulins is depicted in Table 144-1. Type I cryoglobulins consist of a single monoclonal immunoglobulin (Ig), typically IgM, less commonly IgG, IgA, or free Ig light chains (Bence Jones proteins). Complement components are not routinely found in Type I cryoprecipitates. Type I cryoglobulins are often present in substantial amounts, ranging from 1 to 30 mg/mL. The large molecular size of monoclonal IgM and other molecular characteristics, such as absence of sialic acid moieties, deficient carbohydrate side chains, and weak noncovalent factors may predispose these immunoglobulins to precipitation.3
Type II and Type III cryoglobulins are immune complexes composed of polyclonal IgGs and mono- or polyclonal IgMs classically referred to as “mixed cryoglobulinemias.” Type II and Type III cryoglobulins are typically present in relatively small amounts
22
and generally result from chronic inflammatory states. Type II and Type III cryoglobulins may fix complement. Type II cryoglobulins represent a mixture of 2 Ig components: polyclonal immunoglobulins are associated with a monoclonal Ig that exhibits rheumatoid factor (RF) activity. Typically, a monoclonal IgM RF is complexed with a polyclonal IgG. HCV infection is the classical underlying disease. A mixture of polyclonal immunoglobulins or polyclonal Ig-nonimmunoglobulin cryoprecipitates results in detection of Type III cryoglobulins. Polyclonal IgM– IgG cryoglobulins with complement as an integral component are the most frequent scenario in Type III cryoglobulinemia that is commonly associated with HCV infection or connective tissue diseases. A new type of cryoglobulins, not included in the original Brouet classification, called Type II-III cryoglobulin, containing polyclonal IgG associated with a mixture of polyclonal and monoclonal (oligoclonal) IgM, has been described.4 This type of cryoprecipitate describes an intermediate, developing state between Types III and II, suggesting a continuous transition from a purely polyclonal to a partially monoclonal composition by a process of successive clonal selection.
EPIDEMIOLOGY
EPIDEMIOLOGY
Because of the heterogeneity of clinical presentations, including nonclinically apparent disease, the overall prevalence of this syndrome is probably
2600
underreported but has been estimated at 1 to 7 in 100,000.5 Geographic differences in the distribution of hepatitis C infection are responsible for the predominance of mixed cryoglobulinemia in Southern Europe as compared to Northern Europe or the United States.6
“Essential mixed cryoglobulinemia,” or mixed cryoglobulinemia without an identifiable cause, is more common in females than males with a 3:1 ratio. The average age at onset is 54 years. Many cases formerly known as “essential” have been found to be associated with HCV and other infectious etiologies.7
An association with infectious diseases has been reported in 75% of patients, autoimmune disease in 24%, and hematologic diseases in 7%.8 The vast majority (>90%) of mixed cryoglobulinemias occurs in association with chronic HCV infection. On average, 30% of chronically HCV-infected subjects develop cryoglobulinemia, and this proportion can go up to 90% when patients with very longstanding disease are examined.9-11 On average, symptomatic cryoglobulinemic vasculitis syndrome is present in only 2% to 5% of chronically HCV-infected patients,12 though figures as high as 15% have been proposed.13,14 More recently, non–HCV-related infectious cryoglobulinemic vasculitis has been reported in association with hepatitis B virus (HBV), cytomegalovirus, Epstein–Barr virus, parvovirus B19, HIV, and bacterial, fungal, and mycobacterial infections.15
CLINICAL FEATURES
CLINICAL FEATURES
CUTANEOUS FINDINGS
Cutaneous lesions are the most frequent manifestation, occurring in up to 90% of patients. Type I cryoglobulinemia per se is often asymptomatic and cutaneous signs are related to hyperviscosity and/or thrombosis that induce ischemic vasculopathy presenting as Raynaud phenomenon, digital ischemia (Fig. 144-2) that may progress to gangrene (Fig. 144-3), livedo reticularis, acrocyanosis (Fig. 144-4) or retiform purpura. These vascular occlusive lesions are typically cold induced. Infarction, hemorrhagic crusts, ulcers, and lesions on the head and oral or nasal mucosa are relatively more common in Type I than in mixed cryoglobulinemia.16
Type II and III cryoglobulinemic vasculitis is mediated by deposition of antigen–antibody complexes in small and medium-sized arteries, leading to inflammation of, preferentially, small blood vessel walls (capillaries, venules, or arterioles). Intermittent orthostatic palpable purpura, frequently observed late in the afternoon when highest cryoglobulin concentrations are present, is the most common presentation and particularly affects the lower extremities (Fig. 144-5). The face and the trunk, with the exception of the lower abdomen, are usually spared.2 The clinical signs can vary from sporadic isolated petechiae or erythematous macules to severe vasculitic lesions with ulcerations. Leukocytoclastic vasculitis is the
22
TYPE OF CRYOGLOBULINEMIA (ESTIMATED FREQUENCY) COMPOSITION OF CRYOPRECIPITATES DISEASE ASSOCIATIONS
Type I (25%) Monoclonal IgM (sometimes IgG, IgA), immunoglobulin light chain, complexed to other proteins
Type II (25%) Combination of monoclonal (usually IgM with rheumatoid factor activity) and polyclonal (usually IgG) immunoglobulins
Hematologic disorders
■Multiple myeloma
■Waldenström macroglobulinemia
■Plasma cell dyscrasias, monoclonal gammopathy of undetermined significance (MGUS)
■Other lymphoproliferative diseases with M components
Chronic infection
■Viral (HCV) Autoimmune diseases
■Sjögren syndrome
■Cold agglutinin disease Hematologic disorders
■Waldenström macroglobulinemia
■Chronic lymphocytic leukemia
■B-cell non-Hodgkin lymphoma
Type III (50%) Polyclonal immunoglobulins Chronic infection
Type II and III (frequency
Oligoclonal IgM, intermediate state
Type II and III (frequency unknown) Oligoclonal IgM, intermediate state between the entirely polyclonal Type III and the monoclonal, polyclonal Type II
unknown)
between the entirely polyclonal Type III and the monoclonal, polyclonal Type II
■Viral (HCV, HIV, HBV)
■Bacterial (subacute bacterial endocarditis, leprosy, spirochetal)
■Fungal, parasitic Autoimmune diseases
■Systemic lupus erythematosus
■Rheumatoid arthritis
■Dermatomyositis/polymyositis
■Inflammatory bowel diseases
■Biliary cirrhosis
Chronic infection (HCV) Autoimmune diseases Lymphoproliferative diseases Chronic liver disease Proliferative glomerulonephritis
Chronic infection (HCV) Autoimmune diseases Lymphoproliferative diseases Chronic liver disease Proliferative glomerulonephritis
Data from Crowson AN et al. Cutaneous vasculitis: A review. J Cutan Pathol. 2003;30:161-173; and Kallemuchikkal U, Gorevic PD. Evaluation of cryoglobulins. Arch Pathol Lab Med. 1999;123:119-25.
histopathologic hallmark of mixed cryoglobulinemia and is easily detectable by skin biopsy. Approximately 15% of individuals with circulating cryoglobulins have symptomatology consistent with cryoglobulinemic vasculitis. Erythematous papules, ecchymosis,
and dermal nodules also can be seen.14 Postinflammatory hyperpigmentation is also common (40% of patients).7 Nail-fold capillary abnormalities are common and include dilation, altered orientation, capillary shortening, and neoangiogenesis.17
2601
22
NONCUTANEOUS FINDINGS
The most common extracutaneous manifestations in patients with mixed cryoglobulinemic vasculitis are weakness and arthralgias or arthritis14 but serious internal organ involvement also occur. The association of purpura, arthralgias, and weakness, known as
2602
the “Meltzer triad,” is seen in about 25% to 30% of patients.18,19
Renal involvement is a serious complication, typically manifests early in the course of the disease and can present as a broad range of clinical findings, including hematuria with or without renal insufficiency (41%), isolated proteinuria, nephrotic syndrome (21%) or acute nephritic syndrome.20 Chronic renal insufficiency without significant renal abnormalities and acute renal failure are less common. The incidence of renal disease in cryoglobulinemia varies from 5% to 60% and is typically immune complex mediated (Type II and III), but may also occur secondary to thrombosis (Type I). Membranoproliferative glomerulonephritis (MPGN) is more common in mixed cryoglobulinemia. Neurologic manifestations, typically affecting the sensory peripheral nervous system secondary to epineural vasculitis, frequently complicate the clinical course of patients with mixed cryoglobulinemia. Patients typically describe paresthesias with burning symptoms in the lower limbs, often with nocturnal exacerbation, leading to severely compromised quality of life. Electromyographic and nerve conduction studies demonstrated peripheral neuropathy in up to 80% of patients with mixed cryoglobulinemia,21,22 but many symptombased demographic studies report prevalence of only 5% to 45%. Isolated polyneuropathy is more common with Type II than Type III cryoglobulinemia, but multifocal neuropathy and polyneuropathy are the most common form in Type III cryoglobulinemia.23 Restless leg syndrome may be the major manifestation of peripheral neuropathy in middle-aged women with mixed cryoglobulinemia.24 Peripheral neurologic disease manifests as a progressive, chronic distal mild sensory neuritis and only rarely as acute mononeuritis. Clinically apparent CNS dysfunction is rare. Hyperviscosity due to high levels of monoclonal cryoglobulins in Type I cryoglobulinemia, typically seen in Waldenström macroglobulinemia and less frequently in multiple myeloma, can induce microcirculation impairment and neurologic symptoms: blurring or loss of vision, headache, vertigo, nystagmus, dizziness, sudden deafness, diplopia, ataxia, confusion, dementia, disturbances of consciousness, stroke, seizures, somnolence, or coma. A characteristic retinal venous engorgement (“sausaging”) on funduscopic inspection can serve as a diagnostic clue. Musculoskeletal complaints, typically arthralgias and myalgias, are described in more than 70% of persons with cryoglobulinemia, predominantly in Type II and III disease. Arthralgias classically affect the proximal interphalangeal and metacarpophalangeal joints of the hands, the knees, and ankles. Clear clinical signs of myositis or arthritis are rare.25
Pulmonary involvement. Approximately 40% of patients are symptomatic with dyspnea, cough, or pleuritic pain. Pulmonary function tests often reveal evidence of small airways disease and chest radiographs sometimes show interstitial infiltrates or signs of subclinical alveolitis.26,27 Severe pulmonary disease, for example, bronchiolitis obliterans organizing pneumonia (BOOP), or pulmonary vasculitis are very
uncommon.28 Pulmonary-renal syndrome also has been described.29
Other. Thyroid disorders and diabetes mellitus seem to be associated with cryoglobulinemia.30 Nonspecific abdominal pain can affect 2% to 22%. Intestinal vasculitis of the small mesenteric vessels leading to acute abdomen has been reported.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Type I cryoglobulinemia is associated with hematologic disorders, such as Waldenström macroglobulinemia, multiple myeloma, or lymphoproliferative diseases. Non-Hodgkin B-cell lymphoma is the most frequently encountered hematologic malignancy. Circulating mixed (Types II and III) cryoglobulins are commonly detected in a great number of infectious or systemic disorders (see Table 144-1). The term essential is reserved for instances of mixed cryoglobulinemia in the absence of a well-defined underlying disease and accounts for only a minority of cryoglobulinemic patients. Prevalence of serum anti-HCV antibodies and/or HCV RNA in cryoglobulinemic patients ranges from 70% to 100%. Type II cryoglobulinemia is more strongly associated with HCV than Type III (90% vs 70%, respectively). Presence of cryoglobulins increases with duration of HCV infection: cryoglobulins are found in 55% to 90% of patients with longstanding infection. However, overt cryoglobulinemic syndrome develops in only 2% to 5% of these cases.12 The precise role of viral or host factors contributing to this discrepancy remains largely unknown. Specific HCV genotypes or distinct HLA subtypes, like HLA-DR11 or HLA-DR6 may predispose to extrahepatic systemic manifestations of cryoglobulinemia.31,32 Pathogenesis of mixed cryoglobulinemia is probably a multifactorial and multistep process. Viral HCV antigens exert a chronic stimulus on the host immune system, resulting in specific immune dysregulatory mechanisms with B-cell proliferation and autoantibody production. HCV-related B-cell lymphoproliferative disorders comprise a spectrum of disease, ranging from asymptomatic clonal B-cell expansions to pathogenic cryoglobulinemia and lymphoma. From that perspective, HCV-associated mixed cryoglobulinemia is a benign lymphoproliferative B-cell disease with a potential for subsequent development of B-cell lymphoma.19,33
Development of HCV-associated cryoglobulinemic vasculitis syndrome is associated with longstanding infection, old age, Type II cryoglobulins, higher cryoglobulin levels, and clonal B-cell expansion.34 The mechanism for B-cell activation and proliferation may be explained in part due to the presence of a t(14;18) translocation involving a rearrangement of bcl-2 and inhibition of apoptosis.6
In cases of HCV-associated mixed cryoglobulinemia, there is complex interaction between several
22
inflammatory cytokines that amplifies and perpetuates the autoimmune process. Elevated serum levels of interleukin (IL)-1beta, IL-6, and tumor necrosis factor (TNF)-alpha have been found in these patients35
in addition to T-helper 1 chemokines, CXCL11 and CXCL10.36 High levels of CXCL10 are found in patients with clinically active vasculitis and autoimmune thyroiditis.37
Using environmental scanning electron microscopy and energy dispersive X-ray spectroscopy microanalysis, higher concentration of microparticles and nanoparticles were found in HCV-associated mixed cryoglobulinemia than in controls, further supporting a role for environmental factors in the pathogenesis of this condition.38
The group of non-HCV-induced mixed cryoglobulinemias is small. An association of cryoglobulinemia with other infectious viral agents, for example, hepatitis B virus,39 and HIV, with or without HCV coinfection, has been demonstrated.40 HIV infection does seem to play a role in the generation of circulating cryoglobulins.41 Parvovirus B19 has been implicated in a mild form of cryoglobulinemic syndrome.42
Another important and well-known association is the one with Sjögren syndrome. According to one study, 16% of patients with primary Sjögren syndrome had cryoglobulins, and of those, 56% had cryoglobulinemic syndrome.43,44 Importantly, Sjögren syndrome patients who meet diagnostic criteria for cryoglobulinemic vasculitis at the time of diagnosis have increased mortality.45 A study by the same group found cryoglobulinemia in 25% of patients with systemic lupus erythematosus.46 A rare association between antiphospholipid syndrome and cryoglobulinemia has recently been reported.47
DIAGNOSIS
DIAGNOSIS
In almost all cases, the possibility of a diagnosis of cryoglobulinemia or cryoglobulinemic syndrome is primarily suggested by the presence of purpura of the distal extremities or by acral cyanosis and necrosis, depending on the potential presence of Type I or Types II and III cryoglobulinemia. Alternatively, the potential diagnosis may be suggested by the presence of a candidate underlying disease. A targeted review of systems and subsequent clinical examination will guide the physician toward the entire picture of organ involvement or differential diagnoses. The laboratory panel must include parameters relevant to both cryoglobulinemia and important differential diagnoses. Table 144-2 presents an algorithm for the diagnostic approach to the patient, including laboratory testing.
LABORATORY TESTING
In clinical practice, cryoglobulin testing is underutilized, most probably due to the expenditure of time and stringent temperature requirements. They can be
2603
22
History and Examination (Including Optional Technical Methods) Ask for
■Cold sensitivity
■Weakness, musculoskeletal complaints, arthralgia
■Alcohol consumption, hereditary liver disease, history of hepatitis
■Known infections (HCV, HBV, HIV)
■Urine abnormalities (hematuria, foamy urine, edema)
■Paresthesias
■Difficulties in concentrating (suggesting CNS involvement) Look for
■Acrocyanosis, Raynaud phenomenon (suggesting Type I cryoglobulinemia)
■Purpura of distal extremities (suggesting Type II and III cryoglobulinemia)
■Peripheral neuropathy (consider electromyographic testing)
■Joint swelling (suggestive for rheumatoid arthritis, SLE)
■Lymph node enlargement (suggesting lymphoma) Technical examination (optional)
■Ultrasonography: Liver abnormalities, splenomegaly (suggesting lymphoma)
■Electrocardiography, echocardiography (suggesting SLE)
■Ophthalmology: Eye involvement, chorioretinitis (suggesting Behçet disease or sarcoidosis)
Mandatory Laboratory Testing in Cryoglobulinemia
■Cryoglobulins (establishing the diagnosis when positive; stringent laboratory workup at 37°C is needed)
■Complete blood count, including differential
■Creatinine, BUN
■Complement levels
■IgA level (suggestive for liver disease or Henoch–Schönlein Purpura)
■ANA, SS-B, SS-A (positive in SLE- and Sjögren syndrome–associated cryoglobulinemia)
■ANCA (possibly positive, or suggesting granulomatosis with polyangiitis [Wegener] or microscopic polyangiitis)
■Rheumatoid factor RF (positive in Type II cryoglobulinemia with a very high titer)
■Urinalysis. If positive, add microscopic examination of urinary sediment
■Hepatitis A, B, C serology
■Look for other viruses, such as parvovirus B19, HIV, if appropriate
■Further workup for hepatic failure if appropriate
Histology
Histology
■Consider skin biopsy
■Consider skin biopsy
■To confirm leukocytoclastic vasculitis (stain for complement and immune complexes)
■To confirm leukocytoclastic vasculitis (stain for complement and
immune complexes)
■To rule out other forms of vasculitis
■To rule out other forms of vasculitis
■Consider renal biopsy (discern between MPGN I and other forms)
■Consider renal biopsy (discern between MPGN I and other forms)
■Consider hepatic biopsy
■Consider hepatic biopsy
falsely negative and should be obtained during active flares and repeated several weeks apart. By current standard laboratory techniques, most healthy individuals will have undetectable circulating cryoglobulins. Serum must be obtained in warm tubes at 37°C in the absence of anticoagulants. Temperature should be kept at 37°C up until laboratory processing. After clotting and centrifugation at 37°C, the separated serum is stored at 4°C and inspected daily for a precipitate. Type I cryoglobulins tend to precipitate within the
2604
first 24 hours (at concentrations >5 mg/mL), whereas Type III cryoglobulins may require 7 days (Fig. 144-1). For calculation of cryocrit (volume of packed cryoglobulins as percentage of original serum volume), the cryoprecipitate has to be spun in a graded (Wintrobe) tube. A cryocrit ≥2% is considered to be positive. Cryocrit levels usually do not correlate with severity and prognosis of disease. Some authors recommend proof of reversibility of the cryoprecipitate by rewarming an aliquot at 37°C for 24 hours.7 For phenotyping and identification of cryoglobulin components, specific immunologic assays are performed at 37°C. Clinical diagnosis of hyperviscosity can be established by measuring serum viscosity with an Oswaldtype viscometer. Reference serum viscosity, measured as flow time through the viscometer of the patient’s serum divided by that of water or saline, is between 1.4 and 1.8, whereas most symptomatic patients have values between 5 and 8. Again, clinical manifestations are often not proportional to serum viscosity. As hypocomplementemia (with the typical pattern of low or undetectable C4 and normal or relatively normal C3 levels) occurs in up to 90% of patients with mixed cryoglobulinemia, C3 and C4 levels, usually measured in nephelometric immunoassays, should be routinely determined. A sudden increase in complement C4, raised to abnormally high levels, can be observed in some patients developing a B-cell lymphoma.48
RF is often positive in Type II and III cryoglobulinemia. Testing for antinuclear antibodies (ANAs, SS-A, SS-B) and other autoantibodies is indicated when there is a clinical suspicion of underlying systemic connective tissue disease, such as SLE or Sjögren syndrome. Although the immunofluorescence ANA assay is the current diagnostic gold standard, titer and specificity of ANAs may vary considerably. Serological studies for hepatitis C and HIV should be conducted. Confirmation of other viral (hepatitis B, Epstein–Barr virus, and cytomegalovirus) or bacterial agents should be considered and may be warranted in the appropriate clinical scenario.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
See Table 144-3 for an overview on differential diagnosis of cryoglobulinemia (and cryofibrinogenemia).
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
Prognosis is defined by the severity of the cryoglobulinemic syndrome and by the treatability of the underlying disease causing cryoglobulinemia. In Type I cryoglobulinemia, this will depend on the nature and stage of the hematologic disease.
22
CONDITION DIFFERENTIAL DIAGNOSIS COMMENTS
Type I cryoglobulinemia Cryofibrinogenemia
Thromboembolic conditions:
Conditions that present with Raynaud phenomenon, noninflammatory retiform purpura, digital ischemia, gangrene and/or acrocyanosis due to vascular occlusion
■Inheritable or acquired hypercoagulable states (protein C or S deficiency, antiphospholipid syndrome, lupus anticoagulant)
■Thrombocytopenic disorders (thrombotic thrombocytopenic purpura, idiopathic thrombocytopenic purpura)
■Paroxysmal nocturnal hemoglobinuria
■Disseminated intravascular coagulation
■Atherosclerotic peripheral vascular disease
■Thromboangiitis obliterans
■Livedoid vasculopathy
■Drug-induced (ie, heparin, warfarin)
■Atheroemboli, cholesterol emboli, septic emboli, atrial myxoma Hyperviscosity due to:
■Thrombocytosis (essential or secondary)
■Polyclonal or monoclonal gammopathies that are not cryoglobulins (ie, multiple myeloma, Waldenstrom macroglobulinemia) Primary cutaneous disorders:
■Urticaria
■Livedo or livedoid vasculitis
■Neutrophilic dermatoses
■Lipodermatosclerosis
■Panniculitis
■Idiopathic perniosis (chilblains) Oxalosis Calciphylaxis Hemoglobinopathies Infections (malaria, babesiosis)
Types II and III
Small- and medium-vessel vasculitis:
Types II and III cryoglobulinemia Small- and medium-vessel vasculitis:
■IgA vasculitis
■IgA vasculitis
cryoglobulinemia
Conditions that primarily present with palpable purpura due to small- and medium-vessel vasculitis
Conditions that primarily present
with palpable purpura due to small- and medium-vessel vasculitis
■ANCA-associated (granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, polyarteritis nodosa)
■ANCA-associated (granulomatosis with polyangiitis, eosinophilic granulomatosis with
polyangiitis, microscopic polyangiitis, polyarteritis nodosa)
■Rheumatic vasculitides (lupus erythematosus, rheumatoid arthritis, dermatomyositis, Sjögren syndrome)
■Rheumatic vasculitides (lupus erythematosus, rheumatoid arthritis, dermatomyositis,
Sjögren syndrome)
■Drug-induced
■Drug-induced
■Infection-related (bacterial endocarditis, Rickettsia rickettsia, Neisseria meningitidis)
■Infection-related (bacterial endocarditis, Rickettsia rickettsia, Neisseria meningitidis)
A favorable prognosis in a patient with Type I cryoglobulinemia can only be expected when control of the underlying disease can be achieved. In contrast, patients with cryoglobulinemic vasculitis (Type II or III), have increased associated morbidity and mortality. Consequently, 10-year-survival rates are significantly lower than in the normal population because of renal involvement, intestinal vasculitis and widespread vasculitis, liver and cardiac disease, cardiovascular complications, and myeloproliferative disorders.49-51
These data integrate a heterogeneous population that included hepatitis C–associated cases that were treated by differing approaches ranging from symptomatic steroid therapy to antiviral treatment. Recent studies better elucidate the differences in prognosis and long-term complications in patients with mixed cryoglobulinemia based on HCV status. It seems that patients with non-HCV mixed cryoglobulinemia have lower gammaglobulin levels, increased frequency of renal involvement, 4-fold increased risk of developing B-cell lymphoma, and overall higher mortality rate than their HCV-positive counterparts.33
These observations may be explained in part by the fact that HCV-associated cryoglobulinemic vasculitis can be treated with antivirals. Indeed, in this group of patients, response to antiviral therapy significantly reduces the mortality rate and changes the spectrum of complications. Infection is the most common cause of death in HCV patients with mixed cryoglobulinemia, followed by complications related to liver and cardiac involvement.52
COMPLICATIONS
Acute severe complications directly caused by cryoglobulins are rare with any type of cryoglobulinemia. The organs most vulnerable to complications are the nervous system and the kidneys. In Type I cryoglobulinemia, they are mostly related to an acute vasoocclusive crisis caused by increased serum viscosity leading to acute acral ischemia or to cerebral and renal ischemia, causing stroke or acute renal failure, respectively. In addition, the underlying hematologic disease of Type I cryoglobulinemia can directly cause renal
2605
22
failure (myeloma kidney). At advanced stages, complications are defined by the underlying hematologic disease itself: immunosuppression leading to infection and sepsis, coagulation disorders with severe bleeding, and side effects of hematologic disease–specific treatments. Renal involvement remains a poor prognostic sign in patients with cryoglobulinemic vasculitis, with 15% progressing to end-stage renal disease (ESRD).49
MPGN I from any cause including noncryoglobulinemic ones has a poor prognosis: 50% progress to ESRD within 10 years. Small studies report successful treatment of MPGN I by antiviral strategies.53,54 The prognosis of cryoglobulinemia with no identified underlying disease (essential mixed cryoglobulinemia) is not predictable, and, again, renal involvement is associated with poor prognosis (renal failure in 10% of patients).55
In cases of noninfectious mixed cryoglobulinemic glomerulonephritis, severe infectious and new-onset lymphoma severely impacted long-term outcome.56
Mixed cryoglobulinemic vasculitis may involve the peripheral nerves, leading to life-threatening situations or cause acute-on-chronic renal failure. Advanced stages of the underlying disease (mostly hepatitis C or other types of liver disease) can result in acute liver failure or problems associated with hepatic insufficiency, such as coagulation disorders, malnutrition, ascites, and the hepatorenal syndrome. Skin necrosis and vasculitic lesions can be entry sites for infection. Catastrophic individual patient courses have been described.57
MANAGEMENT
MANAGEMENT
Three levels of treatment can be considered when treating cryoglobulinemia: (1) etiologic treatment, which is aimed at the cure or safe containment of the underlying disease; (2) pathophysiologic treatment, which is aimed at reducing the production of cryoglobulins although the underlying etiology remains untreated; and (3) symptomatic treatment that is aimed at reducing the cryoglobulin burden by removal from plasma or at mitigating the tissue’s vasculitic reaction. Sometimes a combination of these approaches is used. The decision for initiation of a therapy should be informed by the severity of the symptoms, with benefits outweighing risks. Cryoglobulinemia without symptoms does not justify treatment, unless there is an underlying condition that merits therapy.
TYPE I CRYOGLOBULINEMIA
Etiologic treatment is the treatment of choice. The underlying hematologic disease, typically multiple myeloma, Waldenström macroglobulinemia, or lymphoma must be treated with chemotherapy directed by a hematologist according to current standards of care. In addition to this, severely symptomatic hyperviscosity syndrome of Type I cryoglobulinemia can be reduced by repeated plasma exchange or by cryofiltration. This may be necessary as a bridging therapy until therapy of the underlying disease shows an effect. In
2606
plasma exchange, the patient’s plasma is removed by an apheresis membrane and substituted using approximately 3.5 L of donor plasma per session. Cryofiltration operates by passing the patient’s cooled plasma through a specialized membrane unit designed to remove cryoprecipitates without necessitating plasma substitution.58 However, this latter approach may be hampered by rapid clogging of the membrane in the setting of high cryoglobulin concentrations. In severe cases of Type I cryoglobulinemia, it may be necessary that these removal procedures be performed daily over a period of 2 weeks or more.
HCV-ASSOCIATED TYPE II AND III CRYOGLOBULINEMIA IN PATIENTS WITH MILD TO MODERATE DISEASE
The primary therapeutic approach depends on disease severity. Patients with mild to moderate cryoglobulinemic vasculitis without major organ failure are best treated using antiviral agents. Although the effects of the treatment exhibit some delay, the approach has the advantage of offering a potentially complete and sustained remission of the vasculitic syndrome. This has been shown by a series of pioneering studies employing regimens of interferon alone or in combination with ribavirin in patients with HCV and cryoglobulinemia.59-62 Among those therapeutic regimens, the highest response rates were found using the current gold standard of therapy, PEGylated interferon α-2b and ribavirin.59,63 Recovery rates for symptoms are as follows: purpura 87.5%, arthralgia 82%, peripheral neuropathy 74%, and nephropathy 50%. The rate of sustained viral response was similar to large study populations without cryoglobulinemia: 62.5%. The regimen is detailed in the current statements on the management of hepatitis C of the American Gastroenterological Association.64 There are several limitations of antiviral combination therapy. Applicability in renal insufficiency is reduced because of drug accumulation. Recommendations for dosage adjustment have been published in the current Kidney Disease: Improving Global Outcomes (KDIGO) guidelines.65 Although ribavirin is contraindicated at an estimated GFR <50 mL/min, it is a desirable drug rendering interferon therapy markedly more powerful.63,66 Therefore, a dosage regimen guided by plasma levels has been proposed that could allow for off-label administration in settings of a GFR <50 mL/min.67 Of note, ribavirin can cause hemolytic anemia requiring pausing or dosage adjustments. Interdisciplinary collaboration with expert hepatologists and nephrologists is recommended.
HCV-ASSOCIATED TYPE II AND III CRYOGLOBULINEMIA IN PATIENTS WITH SEVERE DISEASE
In the case of organ failure, such as overt renal failure, possibly accompanied by the nephrotic syndrome
or neuropathy, initial pathophysiologic and symptomatic therapy are required to achieve a reasonably rapid response. Traditionally, an immunosuppressive combination therapy using steroids possibly followed by cyclophosphamide, azathioprine, or chlorambucil has been used.68 The use of the chimeric anti-CD20 antibody, rituximab, has recently emerged as a powerful alternative to classic immunosuppression.69
The antibody targets B-lymphocytes, leading to a depletion of 95% of the B-lymphocytic population. Thereby, the cryoglobulin-producing B lymphocytic clone is markedly reduced, leading to marked cryoglobulin level reduction and consecutive remission. Taken together, this represents a pathophysiologic approach to treatment. One recent clinical trial compared the efficacy and safety profile of a combination of pegylated interferon-α (Peg-IFN-α) and ribavirin, with or without rituximab. More patients in the rituximab group had complete response and greater than 80% had sustained clearance of HCV RNA for up to 3 years compared to only 40% of patients in the Peg- IFN-α and ribavirin only group.70 Another similar study found that patients in the Peg-IFN-α/ribavirin/ rituximab arm had a shorter time to clinical remission, better renal response rates, and higher rates of cryoglobulin clearance.71 In light of these findings, some authors recommend rituximab combined with Peg- IFN-α/ribavirin as the preferred therapeutic regimen in patients with severe or refractory/relapsing disease, renal involvement, and in those in whom rapid clinical response is needed.72,73 Though reported to be generally well tolerated in patients with cryoglobulinemic syndrome, rituximab has several limitations. It should not be used in patients with overt skin ulceration because of interference with wound healing. A recent report has pointed to a severe complication with worsening of cryoglobulinemic vasculitis syndrome in patients with high baseline levels of mixed cryoglobulins receiving rituximab.74
TYPE II AND III CRYOGLOBULINEMIC VASCULITIS NOT ASSOCIATED WITH HCV
In Type II and III cryoglobulinemic vasculitis not associated with HCV, therapeutic regimens have not been extensively studied. In mild to moderate disease, conservative measures including the avoidance of cold temperatures, resting, and the use of nonsteroidal antiinflammatory drugs such as colchicine may be sufficient. For more severe cases, a combination of rituximab and oral prednisone showed greater efficacy at achieving complete clinical, renal, and immunologic responses compared to the combination of alkylating agents and prednisone at the expense of more severe infections in the rituximab group. Mortality rates, however, were similar in both groups.75 Therefore, rituximab-based regimens are more effective but should be used judiciously because of the high prevalence of infections.
22
CRYOFIBRINOGENEMIA
AT-A-GLANCE
■ Cryofibrinogenemia results from cryoprecipitation of patients’ native fibrinogen or fibrin by-products in plasma but not serum.
■ Cryofibrinogenemia is classified as essential or secondary (associated with malignancies, collagen vascular diseases, and thrombotic disorders).
■ Cryofibrinogenemia is rare, but probably underestimated in clinical practice.
■ Typical clinical features result from thrombosis (thrombotic phenomena of skin and viscera) and are often life-threatening.
EPIDEMIOLOGY
EPIDEMIOLOGY
Cryofibrinogenemia is often clinically asymptomatic. Of note, 2% to 9% of healthy persons may have demonstrable amounts of cryofibrinogen, usually in concentrations less than 50 mg/L. In the past, cryofibrinogenemia was considered rare, but recent single-center studies indicate that this disorder is possibly underrecognized because of the infrequency with which it causes symptoms, and inconsistencies in laboratory investigations producing falsely negative results. Patients are usually diagnosed between the fifth and seventh decades of life. Initial studies noted the prevalence of cryofibrinogenemia among hospitalized patients between 3.4% and 13%, with a female predilection (female–male ratio 4:1) without age or racial differences. In a study by Saadoun and coworkers, 2312 hospitalized patients were tested for cryofibrinogenemia between 1996 and 2006.76 A total of 515 (22.2%) patients had positive test results, of whom 88% had secondary and 12% had essential cryofibrinogenemia. Another retrospective, single-hospital, 10-year report identified 61 patients having cryofibrinogenemia, which was essential in 18 (29.5%) and secondary in 43 (70.5%) patients.77
CLINICAL FEATURES
CLINICAL FEATURES
Some people with cryofibrinogenemia are asymptomatic, whereas others have clinical features resulting from thrombosis that can be life-threatening when untreated.
CUTANEOUS FINDINGS
Patients with cryofibrinogenemia typically report a temporal association between cold exposure and onset of symptoms. Clinical signs in cryofibrinogenemia are mostly cutaneous and are typically located on cold-exposed areas (hands, feet, buttocks, ear, nose). These cold-sensitive lesions often reflect cold-induced
2607
22
thromboses, increased viscosity, and/or vascular reactivity. Palpable purpura with underlying leucocytoclastic vasculitis is the most frequent clinical presentation. Other cutaneous features can include painful ulcerations, livedo racemosa, Raynaud phenomenon, segmental swelling, lower extremity nodules, painful or pruritic erythema (perniosis) of the extremities, and cold urticaria. Cryofibrinogenemia may clinically simulate calciphylaxis, which is usually seen in patients with ESRD.
NONCUTANEOUS FINDINGS
Nonspecific constitutional complaints of fever and malaise are common. Cryofibrinogenemia has a broad spectrum of clinical manifestations including vessel, kidney, musculoskeletal, and/or neuronal involvement. Various thrombotic events, nephritic or nephrotic syndrome, arthralgia, myalgia, multineuritis, and fever have been reported.78 In a recent study, the main clinical manifestations included purpura (46.6%), skin necrosis (36.6%), and arthralgia (31.6%), with cold sensitivity in 40% and overall thrombotic events occurring in up to 40% of cases. A high cryofibrinogen plasma concentration was a significant predisposing factor for thrombotic events.76 Thrombotic events include cerebrovascular accidents (stroke, ocular thrombi, including retinal arterial and/or venous occlusions), myocardial infarction, limb and bowel ischemia or infarction, and pulmonary emboli, although a causal relationship has not been proven for all described cases.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Precipitation of patients’ native fibrinogen or fibrin byproducts in plasma, but not serum, was first described by Korst and Kratochvil in 1955.79 Cryofibrinogenemia can be classified as primary (also named essential or idiopathic) or secondary. Diagnosis of primary cryofibrinogenemia requires the presence of cryofibrinogens in plasma, absence of cryoglobulins, and one or more compatible clinical features, such as cold-induced thromboses, increases in blood viscosity and/or vascular reactivity, in an otherwise healthy individual. Some authors hypothesize that essential cryofibrinogenemia might be a prerequisite for a secondary disease. Secondary cryofibrinogenemia is diagnosed when an associated disease or drug is present. The most frequently associated disorders include malignancy, infections, and connective tissue and autoimmune diseases (see Table 144-4). Cryofibrinogen is characteristically composed of fibrinogen, fibrin, fibronectin, and/or fibrin degradation products. Other components include albumin, coldinsoluble globulin, factor VIII, and plasma proteins, as well as the plasmin activity inhibitors α1-antitrypsin and α2-macroglobulin. Pathology in cryofibrinogenemia is attributed to “in situ” thrombosis, leading to thrombotic occlusion of small and medium-sized
2608
Endocrine Disorders Diabetes mellitus Hypothyroidism (Hashimoto disease) Graves disease
Infections Viral (VZV, EBV, HC) Bacterial (Klebsiella pneumoniae, Mycoplasma pneumoniae) Severe sepsis
Malignancy Adenocarcinoma (gastric, lung) Hepatocarcinoma Ovarian cancer Prostate cancer
Hematologic Disorders Multiple myeloma Waldenstrom macroglobulinemia Lymphoma (follicular, B cell, T cell) Chronic myelomonocytic leukemia
Autoimmune Diseases Mixed connective tissue disease Sjögren syndrome Dermatomyositis Polymyositis Rheumatoid arthritis Systemic lupus erythematosus Systemic sclerosis
Vasculitis ANCA-associated vasculitis Giant cell arteritis Behçet disease Polyarteritis nodosa Henoch–Schönlein purpura
Cutaneous Disorders Psoriasis Pyoderma gangrenosum
Drugs Oral contraceptive agents
Miscellaneous Sarcoidosis Spondyloarthropathy Inflammatory bowel disease.
Miscellaneous Sarcoidosis Spondyloarthropathy Inflammatory bowel disease.
EBV, Epstein–Barr virus; HCV, hepatitis C virus; VZV, varicella-zoster virus.
dermal vessels and resultant ischemia. Defects in the fibrinolysis process might lead to further clotting in small and medium arteries. Additional immunologic mechanisms may play a significant role in the pathophysiology of cryofibrinogenemia. Secondary forms of cryofibrinogenemia are significantly more frequent in patients with combined cryofibrinogenemia and cryoglobulinemia than in those with isolated cryofibrinogenemia (79 vs 47%).80
Among HCV-infected patients, cryofibrinogenemia is common, and closely correlated with cryoglobulinemia. In a study of 143 patients with HCV infection, 53 (37%) had cryofibrinogen levels >50 mg/L. Fortyseven of these cryofibrinogen-positive patients (89%) had positive tests for cryoglobulins.81
DIAGNOSIS
DIAGNOSIS
LABORATORY TESTING
Laboratory workup is critical for the accuracy of cryofibrinogen detection and ideally includes separation in a temperature-controlled centrifuge. The warm blood specimen should be anticoagulated with citrate, EDTA, or oxalate, but not heparin, which nonspecifically precipitates fibrinogen and may result in a false positive result. The formation of a cryoprecipitate may also lead to a false negative result if the sample was not collected at 37°C. The sample should be processed within an hour. After centrifugation at 37°C, the plasma is placed in a Wintrobe tube, refrigerated at 4°C, and observed for the formation of a precipitate for 72 hours. The cryocrit is quantitated by centrifuging the specimen while it remains cooled to 4°C. Each millimeter of visible precipitate in the Wintrobe tube represents 1% of cryocrit. In parallel, a cryoglobulin test is simultaneously performed in a sample without anticoagulants, to ensure that the plasma precipitate is cryofibrinogen and not cryoglobulin. Affinity-chromatography, immunodiffusion, and/or electrophoresis are used for quantitation and/or identification of the individual cryofibrinogen components using antifibrinogen, anti–heavy-chain and anti–lightchain antibodies.80,82
HISTOLOGIC FEATURES
Irrespective of the anatomic site, cryofibrinogenemia shows an occlusive thrombotic diathesis comprising eosinophilic refractile deposits within vessel lumina with extension into the vessel intima, with or without an accompanying characteristic granulomatous vasculitis.77,83 Cryofibrinogen precipitates have a cylindrical configuration in ultrastructural analysis, which is often displayed within the vessel lumina. Examination of renal deposits showed fibrillary material within glomerular capillary lumina and tubules with unique morphologic features not previously described.78
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE AND
PROGNOSIS
Clinical data regarding prognosis in cryofibrinogenemia are limited. In one study, 3 of 60 patients with
22
essential cryofibrinogenemia died after a mean followup of 85 months.
COMPLICATIONS
COMPLICATIONS
Typical complications due to thrombotic events include gangrene (5%), septicemia (5%), and leg amputation (3.3%).76 In some cases, essential cryofibrinogenemia might suggest a secondary disease, particularly lymphomas.84 In a 2008 report, 27% of patients with primary cryofibrinogenemia developed lymphoma after a 5-year followup period.77
MANAGEMENT
MANAGEMENT
Smoking cessation, avoidance of cold exposure, and eliminating the use of vasoconstricting drugs are nonpharmacologic interventions that are recommended but are only partially effective. In secondary cryofibrinogenemia, specific treatment of the underlying disease can lead to improvement in related symptoms. For essential cryofibrinogenemia, various pharmacologic agents including fibrinolytic approaches and immunosuppressive agents have been proposed. Oral stanozolol (2-4 mg twice daily), a synthetic derivative of testosterone with substantial fibrinolytic properties, has been effective after several days of treatment.85
Streptokinase (sometimes in combination with streptodornase), given intravenously (25,000-200,000 U/d) has a more rapid onset of action than stanozolol. The use of colchicine (0.6 mg twice daily) in combination with high-dose pentoxifylline (800 mg 3 times daily) has been described.86
Combinations of glucocorticoids (prednisone 10-60 mg/d) with low-dose aspirin, for nonsevere cases, or other immunosuppressive agents, for severe disease (azathioprine 150 mg/d or chlorambucil 10 mg/d) also have been used in small studies with some benefit, including successful treatment of acute attacks.87,88
Plasmapheresis might be considered when high levels of cryofibrinogens are present and are associated with monoclonal proteins (as seen in myeloma, Waldenström macroglobulinemia), hyperviscosity, or clinically significant thrombosis. Long-term repeated plasmaphereses and antiimmunoglobulin adsorption improved the symptoms in one patient with secondary cryofibrinogenemia.89
Regular followups are mandatory because of the high risk of symptom recurrence and development of lymphoma years after the initial diagnosis.77,90
ACKNOWLEDGMENTS
The authors acknowledge the significant contributions of Holger Schmid and Gerald S. Braun, the former authors of this chapter.
2609
22

Figure 144-1 Whitish cryoprecipitates after keeping tube at 4°C (39.2°F) for 48 hours and centrifugation.
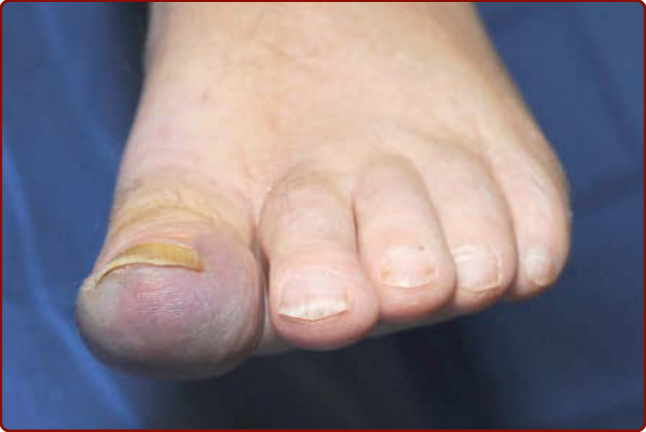
Figure 144-2 Blue discoloration of the distal aspect of the left first toe as the earliest finding of impending digital ischemia in a patient with cryoglobulinemia.
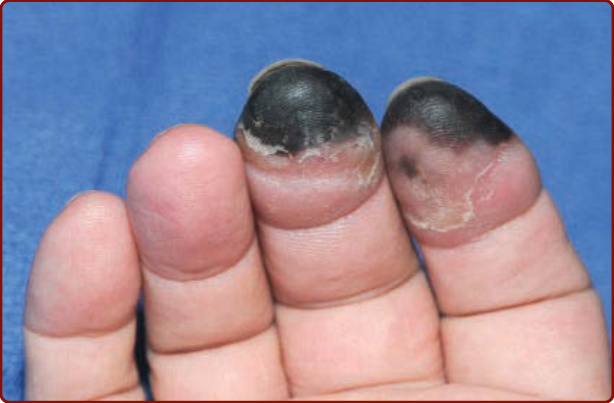
Figure 144-3 Digital gangrene of the distal right second and third fingers in a patient with cryoglobulinemia Type I. Note the well-demarcated color changes and absence of inflammation (erythema).
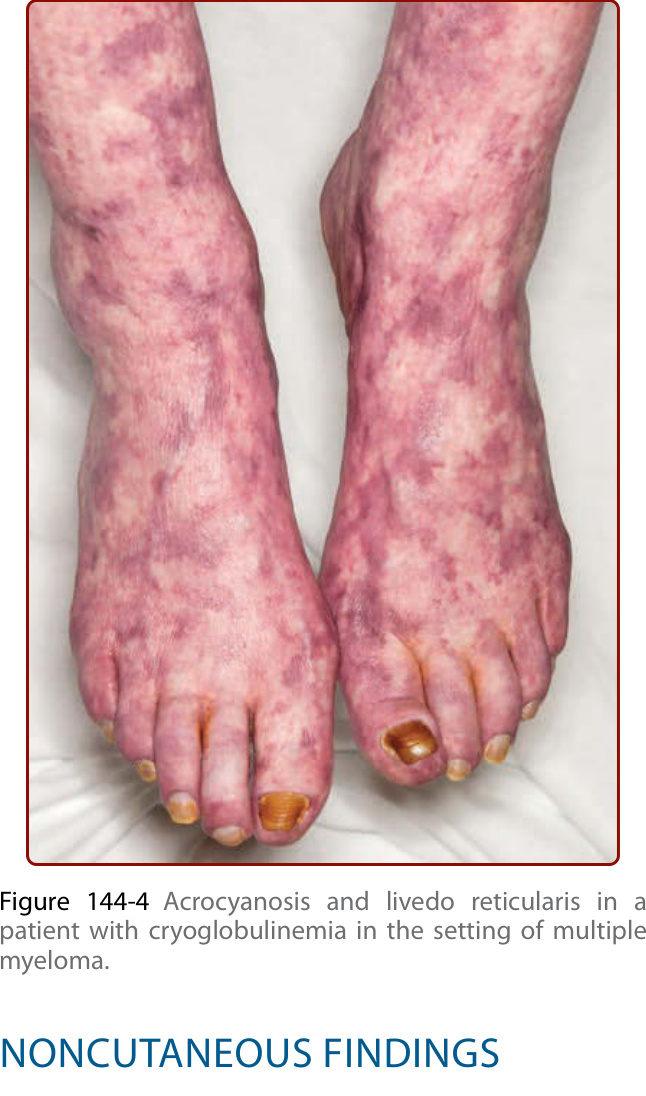
Figure 144-4 Acrocyanosis and livedo reticularis in a patient with cryoglobulinemia in the setting of multiple myeloma.
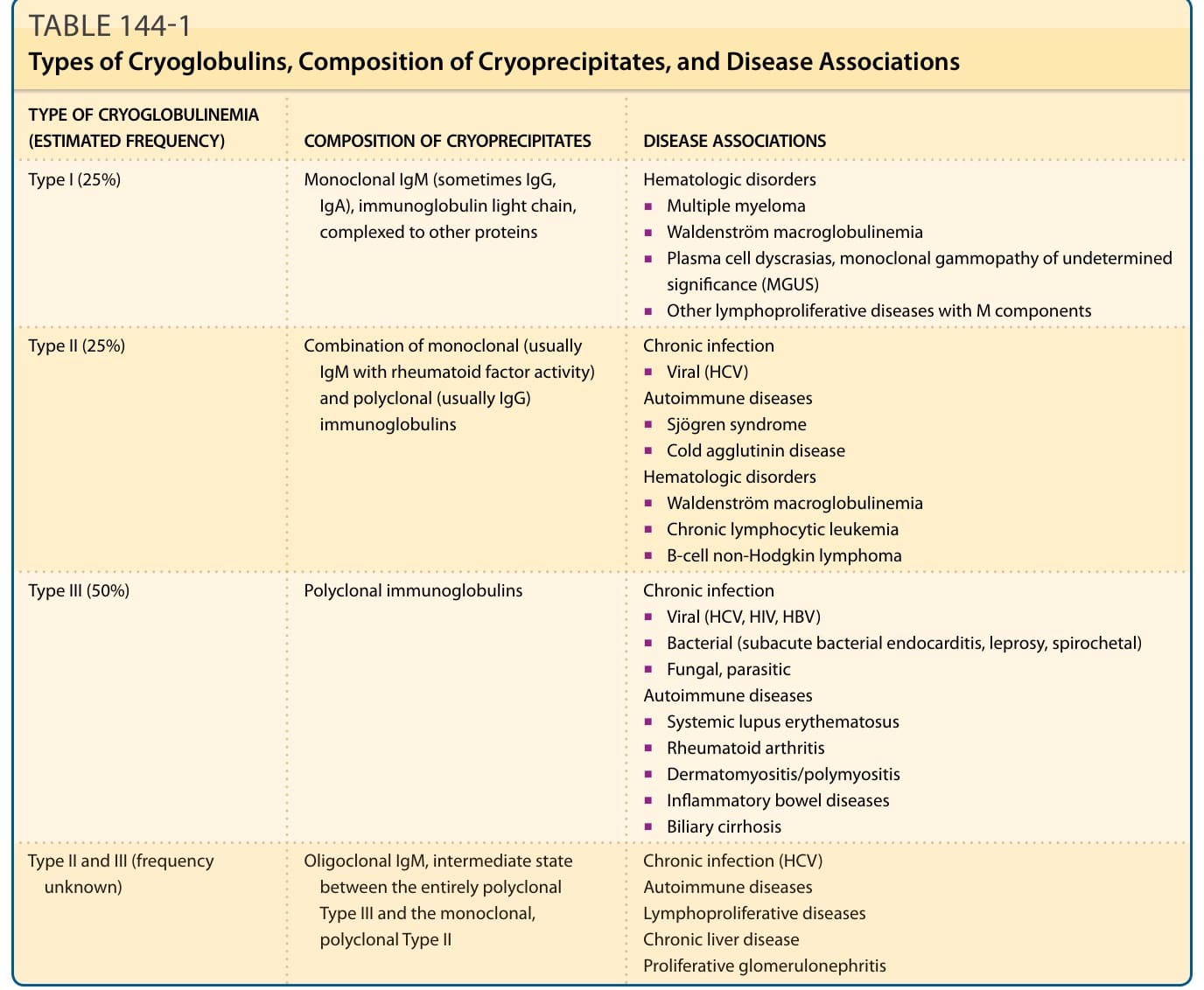
TABLE 144-1 Types of Cryoglobulins, Composition of Cryoprecipitates, and Disease Associations
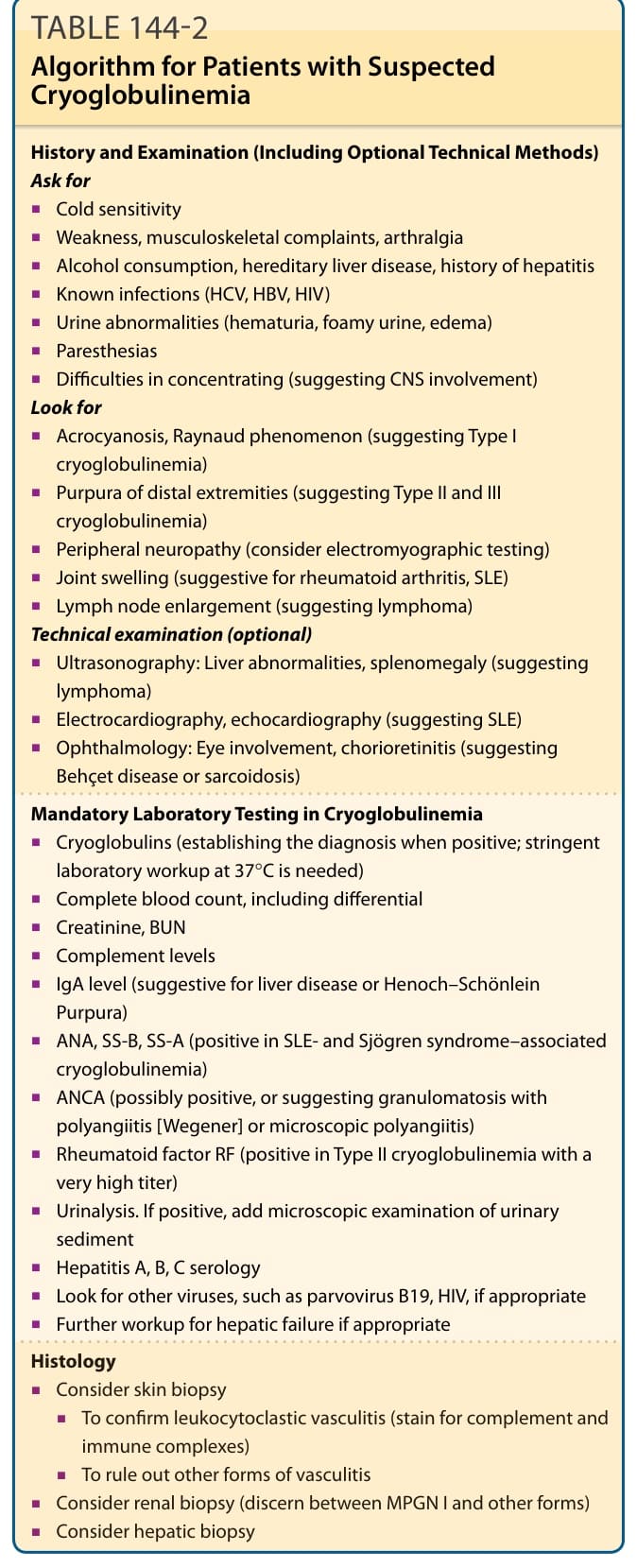
TABLE 144-2 Algorithm for Patients with Suspected Cryoglobulinemia

TABLE 144-3 Differential Diagnosis of Cryoglobulinemia and Cryofibrinogenemia
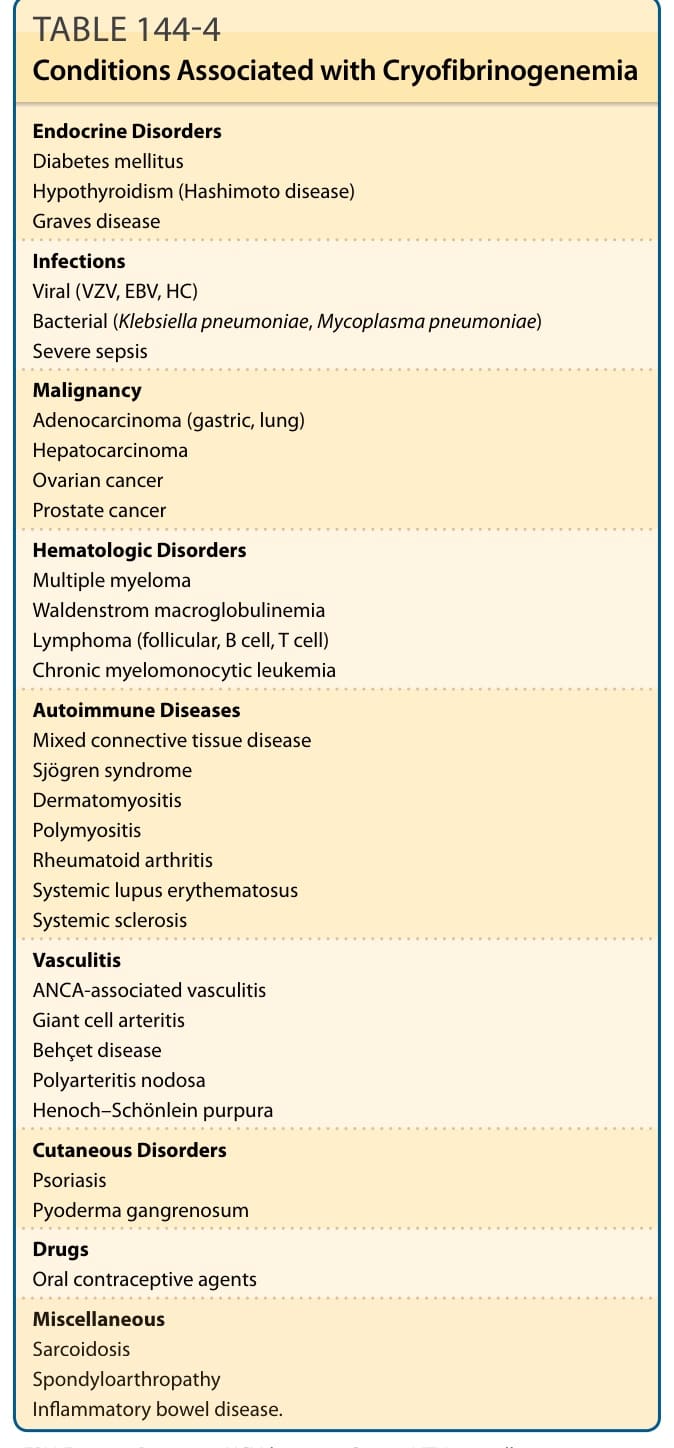
TABLE 144-4 Conditions Associated with Cryofibrinogenemia