Adamantiades–Behçet Disease
22
AT-A-GLANCE
■ Rare disease with worldwide distribution but strongly varying prevalence; certain ethnic groups are mainly affected.
■ A genetically determined disorder with a probable environmental triggering factor.
■ Multisystem occurrence, with oral aphthous ulcers, genital ulcers, papulopustules, erythema nodosum–like lesions, uveitis, and arthropathy as the most common signs.
■ Inflammatory disease representing a neutrophilic vascular reaction or vasculitis.
■ Chronic, relapsing, and progressive course with a potentially poor prognosis (especially in males with systemic presenting signs; mortality, 0%-6%).
Adamantiades–Behçet disease is a multisystem inflammatory disease of unknown etiology. It is classified as a systemic vasculitis involving all types and sizes of blood vessels and characterized clinically by recurrent oral aphthous and genital ulcers, skin lesions, iridocyclitis/posterior uveitis, and arthritis. These findings are occasionally accompanied by vascular, GI, neurologic, or other manifestations.1,2
HISTORICAL ASPECTS
Hippocrates of Kos (460-377 bc) used the designation “στοµατα αϕθωδεα, ελκωδεα” (oral aphthous ulcers) in a probable first description of a patient with the disease (Epidemion Book III, Case 7). The disease is named after Benediktos Adamantiades, a Greek ophthalmologist and Hulûsi Behçet, a Turkish dermatologist, who, in 1931 and 1937, respectively, described patients with the characteristic clinical complex.3 The first international multidisciplinary conference was organized by 2 dermatologists, Drs M. Monacelli and P. Nazarro, in 1964 in Rome, Italy.
EPIDEMIOLOGY
Adamantiades–Behçet disease occurs worldwide with varying prevalence, being endemic in the Eastern and Central Asian and the Eastern Mediterranean countries (along the so-called Silk Road) and rare in Northern European countries, Central and Southern Africa, the Americas, and Australia.4 A prevalence of 80 to 420 patients per 100,000 inhabitants has been reported in Turkey,5 7 to 30 patients per 100,000 inhabitants in
the rest of the Asian continent (Japan, 14-31:100,000; Korea, 35:100,000; Northern China, 14:100,000; Saudi Arabia, 20:100,000; Iran, 17:100,000), and 1.5 to 7.5 per 100,000 in Southern Europe.4 In Northern Europe (0.27-1.18:100,000) and the United States (0.75:100,000), the disease is rare.6,7 The increasing prevalence of the disease is due to its chronic nature. Its annual incidence is low; 0.75 to 1.0 new cases per 100,000 inhabitants were assessed in Japan (1990) and Germany (2005).6 Adamantiades–Behçet disease most often affects patients in their 20s and 30s; however, early and late onsets (first year of life to 72 years) have been reported. Juvenile disease rates are 2% to 21% in different ethnic groups. Its prevalence was estimated to be 0.17:100,000 in France.8,9 In contrast to old Japanese and Turkish reports of male predominance, the male-to-female ratio drastically decreased in the last 20 years.10 Currently, both genders are equally affected overall; however, male predominance is still observed in Arab populations, and female predominance is evident in Korea, China, some Northern European countries, and the United States.
ETIOLOGY AND PATHOGENESIS
The etiology of the disease remains unknown, although genetic factors, infectious agents, environmental pollution, immunologic mechanisms, and endothelial and clotting factors have been implicated and studied intensively.1,2,11 The endemic occurrence along the historical Silk Road, the major involvement of certain ethnic groups (mostly of Turkmenic and Mongol descent), and associated immunogenetic data support the hypothesis that the disease followed the migration of these old nomadic tribes.2,11 On the other hand, the wide variation of the disease prevalence in the same ethnic group in association with different geographic areas of residence indicates an additional environmental triggering factor. Therefore, transfer of genetic material and/or of an unknown exogenous agent may have been responsible for the expansion of the disease.2,11
GENETICS AND IMMUNOGENETICS
GENETICS AND
IMMUNOGENETICS
There is no specific mode of Mendelian transmission in Adamantiades–Behçet disease.2,11-15 Familial occurrence with regional differences has been reported, being more frequent in Korea (15%) than in Japan
22
or China (2%-3%), and in Arab countries, Israel, and Turkey (2%-18%) than in Europe (0%-5%).4,13,14 An earlier disease onset in children compared with their parents, and a higher frequency of familial cases in juveniles than in adults, has been observed.8,13,14
A significant association exists between the disease and human leukocyte antigen-B51 (HLA-B51) in Japan, the Middle East, and the Mediterranean countries; however, this relationship is not as strong in Western countries.12,15,16 The allele also seems to be associated with a more severe prognosis and ocular involvement.4,13,17 Its exact role in the disease mechanism is still unknown; however, it may be involved in the disease development through specific antigen presentation, molecular mimicry with microbial antigens, or participation in linkage disequilibrium with a presently unknown susceptibility gene.14
In an effort to explain the disease process, it has been suggested that Adamantiades–Behçet disease constitutes one of a newly termed group of diseases, the “MHC-I-opathies.”18 Recent work analyzing the peptidome of HLA-B51 suggests that altered peptide presentation by HLA-B51 is vital to the disease process. It is likely that (1) natural killer or other cell interactions, perhaps mediated by leucocyte immunoglobulin-like receptor or killer immunoglobulin-like receptor, are culpable in pathogenesis, or (2) HLA misfolding may lead directly to inflammation. Among the 24 currently described alleles, HLA-B5101 and -B5108 have most frequently been associated with Adamantiades–Behçet disease.19 Shared amino acid residues (defining the Bw4 epitope) are crucial for antigen binding and natural killer cell interactions,20
and Bw4 was also reported to contribute to the severity of the disease.21 Genes possibly associated with the disease have been localized on chromosome 6 in the region between the tumor necrosis factor gene and HLA-B or HLA-C genes, including the major histocompatibility complex class I chain A gene (A6 allele) and genes for heat shock proteins.14-17,19,22 In addition, a susceptibility locus mapped to 6p22-23 was detected.19 Lately, associations on chromosomes 1p31.3 [interleukin (IL) 23R-IL12RB2] and 1q32.1 (IL10) were found by genomewide association studies.20,23 A haplotype association of IL-8 gene with Adamantiades–Behçet disease was also detected.24 Polymorphisms in genes encoding for host effector molecules may contribute to the disease susceptibility and/or severity of the disease, such as in IL-23R reported in a Chinese Han population.25 New gene associations with ERAP-1, CCR1-CCR3, KLRC4, and STAT4 genes have been reported.26
INFECTIOUS PRECIPITANTS
INFECTIOUS PRECIPITANTS
Adamantiades–Behçet disease is not considered contagious as no horizontal transmission has ever been reported. However, viral and bacterial infections have been implicated in initiating immunopathologic pathways, leading to the onset of the disease.2,11,27
2568
VIRAL AGENTS
Early theories of the pathogenesis of Adamantiades– Behçet disease proposed a viral or other infectious etiology.11 Partial transcription of herpes simplex virus Type 1 (HSV-1) DNA in patients’ peripheral blood lymphocytes was reported. HSV-1 DNA was detected in patients’ saliva and oral and genital ulcers, and HSV-1 antibodies were found in patients’ serum.
BACTERIAL AGENTS
The disease activity has been known to correlate with bacterial infection, particularly Streptococci.11,27 Streptococcus sanguinis dominates the flora of the oral mucosa in patients with the disease and appears to be the most relevant Streptococcus strain as a provoking factor for initiation of the disease.27 Streptococcus antigens and antistreptococcal antibodies are frequently found in the oral mucosa and serum of patients. The involvement of immunoglobulin A protease-producing S. sanguinis is proposed as an explanation for chronic infection leading to initiation of Adamantiades–Behçet disease. High titers of the immunogenic S. sanguinis antigen KTH-1 have been detected in patients. In addition, exposure of the patients to Streptococcus antigens may be a major provoking factor for disease activity. The lipoprotein of Mycoplasma fermentans MALP-404 was found in the serum of patients with Adamantiades– Behçet disease but not in healthy controls.28 Interestingly, MALP-404 contains a peptide motif, which can be presented by HLA-B51. A possible role for bacterial stimulation of monocytes via Toll-like receptor-2 producing neutrophil-stimulating proinflammatory factors in Adamantiades–Behçet disease was detected.29
IMMUNOLOGIC MECHANISMS
IMMUNOLOGIC
MECHANISMS
Immunologic mechanisms are considered to play a major role in the pathogenesis of Adamantiades– Behçet disease.2,11,27 The disease has currently been classified among the autoinflammatory disorders,30
which are caused by primary dysfunction of the innate immune system.
AUTOIMMUNE MECHANISMS
The major microscopic finding at most sites of active disease is an immune-mediated occlusive vasculitis. The pathergy reaction (see section “Clinical Findings”) is induced by the rapid accumulation of neutrophils (hyperchemotaxis) and later by T lymphocytes and monocytes/macrophages at needle prick sites. The disease has been considered to be a typical Th1- mediated inflammatory disease, characterized by elevated levels of Th1 cytokines such as IFN-γ, IL-2, and TNF-α. Recently, some studies reported that
Th17-associated cytokines were increased; thus, Th17 cells and the IL17/IL23 pathway may play important roles in the pathogenesis of Adamantiades–Behçet disease.31 T cells in the peripheral blood and in the involved tissues are increased, and a predominant T helper 1 immune response induced by IL-12 has been demonstrated.32 Patients’ lymphocytes also express CD29 molecules and bind to endothelial cells in active disease. In addition to defective T-cell immunity, B-cell activation is impaired. Circulating immune complexes, together with enhanced neutrophil migration, may be involved; diversity of T cells indicates that specific T-cell responses to several antigens may lead to the variety of symptoms.33 Tropomyosins and the 160-kDa polypeptide kinectin have been detected as autoantigens in Adamantiades–Behçet disease.34,35
HEAT SHOCK PROTEINS
Increased levels of heat shock protein (HSP)–specific antibodies in serum have been found in Adamantiades– Behçet disease.36,37 T cells respond to 60-kDa HSP, and 4 different peptide determinants within 60-kDa HSP identified by T-cell epitope mapping have been suggested to be involved in the pathogenesis of the disease.
CYTOKINE MEDIATORS
Various proinflammatory cytokines, such as IL-1, -8, -12, -17, -23, and tumor necrosis factor-α, are elevated in the sera of patients with Adamantiades– Behçet disease.38-42 In particular, IL-8 seems to play an important role, can also be released by endothelial cells, has a potent effect on the inflammatory response, and is a sensitive marker of disease activity.38-40 Cytokine release may be dependent on the involved organ.38,39,41
ENDOTHELIAL CELLS
The endothelium seems to be the primary target; however, it may just be subject to the bizarre behavior of the immune system.43 An immunoglobulin M-type, 47-kDa cell surface HSP against endothelial α-enolase was identified in the serum of patients with Adamantiades–Behçet disease.44 Plasma endothelin-1 concentrations were found significantly increased, perhaps indicating vasoconstriction and being the direct result of elevated synthesis by injured vascular endothelial cells. Thrombomodulin, a cell surface glycoprotein of vascular endothelium, which is also increased in the plasma of patients with active disease, potentially damages the endothelial cells.
EPIGENETICS
EPIGENETICS
Epigenetic studies on Adamantiades–Behçet disease have shown the role of alterations in the methylation
22
level of interspersed repetitive sequence elements, histone modifications such as H3K4me27 and H3K4me3, upregulation of miR-182 and miR-3591-3p as well as downregulation of miR-155, miR-638, and miR-4488 in the pathogenesis of the disease.45
CLINICAL FINDINGS
Adamantiades–Behçet disease is a chronic, recurrent, multisystem, and, occasionally, life-threatening disorder.1,2,4 Recurrent oral aphthous ulcers, recurrent genital ulcers, skin manifestations, ocular lesions, and arthritis/arthropathy are the most frequent clinical manifestations.1,6 Vascular, GI, neurologic, psychiatric, pulmonary, renal, and cardiac manifestations; epididymitis; and other findings can also occur (Fig. 141-1). The clinical picture usually develops within a few months after the presenting sign; both an acute multisystem presentation and long-term development of the disease over the years are possible. The fulfillment of the International Diagnostic Criteria46 supports the diagnosis.
MUCOCUTANEOUS LESIONS
MUCOCUTANEOUS
LESIONS
Recurrent oral aphthous and genital ulcers are the most frequently observed mucosal manifestations. Oral aphthous ulcers are the presenting sign in more than 80% of the cases.1,4,6 Although recurrent aphthous stomatitis is a common disorder, only a few patients progress to Adamantiades–Behçet disease, and it is not possible to determine in whom or when the transition may occur. Typically, lesions are multiple, painful, 1 to 3 cm in diameter, and sharply margined with a fibrin-coated base and surrounding erythema (Fig. 141-2). Oral aphthous ulcers usually heal without scarring (92%). Genital ulcers may not recur as often and usually heal with a characteristic scar (64%-88%; Fig. 141-3). Spontaneous healing of aphthae occurs within 4 days to 1 month; genital ulcers may persist longer. Large oral ulcerations can also be associated with problems such as pharyngeal involvement, dysphagia, and dyspnea or fistulae involving the pharynx, larynx, trachea, or esophagus. Genital ulcers can occur on the penis, scrotum, vagina, labia, and urethra, and also in the anal, perineal, and inguinal regions. Skin lesions that should be accepted as diagnostically relevant in Adamantiades–Behçet disease should be confined to pustular vasculitic lesions (including pathergy lesions, see Fig. 141-4A), erythema nodosum– like lesions (Fig. 141-4B), Sweet-like lesions, pyoderma gangrenosum–like lesions, and palpable purpuric lesions of necrotizing venulitis (Fig. 141-5).47-49 All of these lesions are characterized in their early stages by a neutrophilic vascular reaction.48 Single acneiform lesions or follicle-based pustules should not be considered relevant.50
2569
22
Adamantiades-Behçet disease: a multisystem disorder
Uveitis
Oral aphthous ulcers
Pulmonary invlovement (embolic lesions, aneurysms, hemorrhage)
Kidney invlovement
Genital ulcers
Prostatitis/epididymitis
Arthritis
Cerebral manifestations (sterile meningoencephalitis, vasa neurorum)
Cardiac involvement (pericarditis, endocarditis)
Arterial aneurysms (vasa vasorum)
Gastrointestinal involvement (gasatritis, ulcers, pseudo- Crohn signs)
Phlebothromosis
To standardize the evaluation of mucocutaneous severity, a Mucocutaneous Activity Index has currently been established.51 This is a specific score that can help with therapeutic decisions and reduce morbidity; however, it still lacks validation.
A B
SYSTEMIC LESIONS
SYSTEMIC LESIONS
Ocular involvement is the major cause of morbidity in patients with Adamantiades–Behçet disease. The most diagnostically relevant lesion is posterior uveitis (also
2570
A
B
called retinal vasculitis), which can lead to blindness (Fig. 141-6A). Other ocular lesions include anterior uveitis, hypopyon (pus in the anterior chamber of the eye, which is now—due to early treatment—uncommon; see Fig. 141-6B), and secondary complications such as cataract, glaucoma, and neovascular lesions.52 Retinal inflammation can lead to vascular occlusion and, ultimately, tractional retinal detachment. Severe vitreous involvement, chronic cystoid macular edema, and possible—presumably also vasculitic—involvement of the optic nerve can result in vision loss.53 Recurrent vasculitic changes can ultimately lead to ischemic optic nerve atrophy.
22
A
B
The characteristic arthritis is a nonerosive, asymmetric, sterile, seronegative oligoarthritis; however, symmetric polyarticular involvement is common. Joint manifestations frequently occur first in one knee or ankle and then the other as migratory monoarthritis, then in both joints simultaneously, and finally affecting nearly all joints.1 An HLA-B27-positive, erosive sacroiliitis has to be excluded. Systemic vascular involvement can be significant and includes venous occlusions and varices, arterial occlusions, and aneurysms, often being migratory. Cases of large-vein thrombosis (inferior vena cava, cranial venous sinuses) or large-artery aneurysms are potentially fatal.1,2,4,6 Arterial involvement is rather
2571
22
A
B
rare and usually presents in the form of thromboses and, less often, of aneurysms, resulting from multicentric arteritis. Aneurysms may develop in large arteries as a result of vasculitis of the vasa vasorum with penetration of the lamina elastica. Pulmonary artery aneurysms are the principal feature of pulmonary
2572
A
B
involvement in Adamantiades–Behçet disease, occasionally resulting in coughing and hemoptysis. Cardiac involvement can include myocarditis, coronary arteritis, endocarditis, and valvular disease. A wide spectrum of renal manifestations can occur, varying from minimal-change disease to proliferative glomerulonephritis and rapidly progressive crescentic glomerulonephritis. Immune complex deposition is thought to be responsible for the underlying pathogenesis of glomerulonephritis in some cases. Carotid plaques, pulse-wave velocity, and flow-mediated dilation represent clinical biomarkers of prognosis.54
GI complaints can be a symptom for aphthae throughout the GI tract and can rarely result in perforation and peritonitis (0.5%). On the other hand, GI involvement has an acute exacerbating course with ulcers, most commonly in the ileocolonic area.55 These ulcers can be large and deep, causing perforation and
massive bleeding. Inflammatory bowel disease has to be excluded. Sterile prostatitis and epididymitis can be present in male patients without genital ulcers.56 Significant neurologic manifestations occur in approximately 10% of patients and may be delayed in onset.2,4,6
Meningoencephalitis, cerebral venous sinus thrombosis, benign intracranial hypertension, cranial nerve palsies, brainstem lesions, and pyramidal or extrapyramidal lesions have been described. Poor prognosis is associated with a progressive course, relapses after treatment, repeated attacks, and cerebellar symptoms or parenchymal disease. Neurologic manifestations usually present with severe headache. Further symptoms include gait disturbance, dysarthria, vertigo, and diplopia as well as hyperreflexia, epileptic seizures, hemiplegia, ataxia, or a positive Babinski reflex. Psychiatric symptoms, such as depression, insomnia, or memory impairment, are also signs of neurologic involvement.
DIAGNOSIS
The diagnosis of Adamantiades–Behçet disease is based on clinical signs, as pathognomonic laboratory tests or histologic characteristics are absent. There are several sets of diagnostic criteria with the current International Criteria46 being the most accurate among them (Table 141-1).
HISTOPATHOLOGY
HISTOPATHOLOGY
Characteristic histopathologic features of Adamantiades– Behçet disease are vasculitis and thrombosis (Fig. 141-5). Biopsies from early mucocutaneous lesions show a neutrophilic vascular reaction with endothelial swelling, extravasation of erythrocytes, and leukocytoclasia or a fully developed leukocytoclastic vasculitis with fibrinoid necrosis of blood vessel walls.1,6 Although there are reports of lesions that consist primarily of
SYMPTOM POINTS
Ocular lesions (recurrent) 2
Oral aphthosis (recurrent) 2
Genital aphthosis (recurrent) 2
Skin lesions (recurrent) 1
CNS 1
Vascular manifestations 1
Positive pathergy testb 1
Positive pathergy testb 1
aBCD scoring: score ≥4 indicates Adamantiades–Behçet disease.
bThough the main scoring system does not require the pathergy test, if it is conducted, a positive result may be included for 1 extra point.
22
a lymphocytic perivasculitis, most of these lesions are likely older. The neutrophilic vascular reaction should be considered the predominant histopathologic finding.7,49,57
SPECIAL TESTS
SPECIAL TESTS
PATHERGY TEST
A positive pathergy test (hyperreactivity reaction) manifests within 48 hours as an erythematous papule (>2 mm) or pustule at the site of a skin needle prick or after intracutaneous injection of 0.1-mL isotonic saline using a 20-gauge needle without prior disinfection of the injection site (Fig. 141-4A). The skin prick is generally placed at an angle of 45°, 3 to 5 mm intracutaneously on the volar forearm. Erythema without infiltration is considered a negative finding. Provoked oral aphthae and genital ulcers after injection or injury (such as chorioretinitis in the corneal region of the eye after photocoagulation of the ocular fundus region) can also be considered as positive pathergy phenomenon. Broader pathergy phenomena also include the occurrence of aneurysms around vascular anastomoses as well as local recurrence of ulcers after resection of affected bowel segments. Although a positive pathergy reaction is a sign of Adamantiades–Behçet disease, it is not pathognomic, as it can also occur in patients with pyoderma gangrenosum, rheumatoid arthritis, Crohn disease, and genital herpes infection.
RADIOLOGIC FINDINGS
Scintigraphic evidence of arthritis is found in 50% of the patients.6 Cranial MRI allows documentation of hypodense or atrophic changes in the brain. Electroencephalographic detection of diffuse α-waves is considered a positive finding. Vascular lesions can be detected by angiography. Because of the rarity of disease, documentation of characteristic imaging can be supportive for the diagnosis.58
DIFFERENTIAL DIAGNOSIS
Refer to Table 141-2.6,59
CLINICAL COURSE AND PROGNOSIS
The clinical course of Adamantiades–Behçet disease is variable. There can be a delay of up to several years before the diagnosis is made, and this may influence the prognosis. Mucocutaneous and joint manifestations usually occur first. Recurrent erythema nodosum and HLAB51 positivity are risk factors for the
2573
22
■Oculocutaneous/mucocutaneous syndromes
■Oculocutaneous/mucocutaneous syndromes
■Erythema multiforme exudativum and variants, including Stevens–Johnson syndrome
■Erythema multiforme exudativum and variants, including
Stevens–Johnson syndrome
■Vogt–Koyanagi–Harada syndrome
■Vogt–Koyanagi–Harada syndrome
■Reiter disease
■Reiter disease
■Bullous autoimmune diseases: Pemphigus vulgaris, cicatricial mucous membrane pemphigoid, epidermolysis bullosa acquisita
■Bullous autoimmune diseases: Pemphigus vulgaris, cicatricial
mucous membrane pemphigoid, epidermolysis bullosa acquisita
■Viral infections (herpes, coxsackie, echo)
■Viral infections (herpes, coxsackie, echo)
■Syphilis
■Syphilis
■Articulomucocutaneous syndromes
■Articulomucocutaneous syndromes
■Systemic lupus erythematosus
■Systemic lupus erythematosus
■MAGIC syndrome (mouth and genital ulcers with inflamed cartilage)
■MAGIC syndrome (mouth and genital ulcers with inflamed
cartilage)
■Yersiniosis
■Yersiniosis
■Arthropathic psoriasis
■Arthropathic psoriasis
■GI/mucocutaneous syndromes
■GI/mucocutaneous syndromes
■Ulcerative colitis, Crohn disease
■Ulcerative colitis, Crohn disease
■Tuberculosis
■Tuberculosis
■Bowel-associated dermatitis-arthritis syndrome
■Bowel-associated dermatitis-arthritis syndrome
■Aphthae
■Aphthae
■Recurrent aphthous stomatitis (RAS)
■Recurrent aphthous stomatitis (RAS)
■Cyclic neutropenia
■Cyclic neutropenia
■Herpes simplex infection
■Herpes simplex infection
■Genital ulcers
■Genital ulcers
■Ulcus vulvae acutum (Lipschütz ulcer)
■Ulcus vulvae acutum (Lipschütz ulcer)
■Herpes simplex infection
■Herpes simplex infection
■Sexually transmitted infections
■Sexually transmitted infections
■Uveitis
■Uveitis
■Other forms of uveitis
■Other forms of uveitis
■Arthritis
■Arthritis
■Ankylosing spondylitis
■Ankylosing spondylitis
■Juvenile rheumatoid arthritis
■Juvenile rheumatoid arthritis
■CNS manifestation
■CNS manifestation
■Multiple sclerosis
■Multiple sclerosis
■Neuro-Sweet disease
■Neuro-Sweet disease
■Lung manifestation
■Lung manifestation
■Sarcoidosis
■Sarcoidosis
development of superficial thrombophlebitis and vision loss,4,13,53 and superficial thrombophlebitis, ocular lesions, and male gender are risk factors for the development of systemic vessel involvement.4,13,56,60 A severe course, including blindness, meningoencephalitis, hemoptysis, intestinal perforation, and severe arthritis, occurs in approximately 10% of patients. Blindness often can be prevented with early aggressive therapy of posterior uveitis. Lethal outcome has been seen in 0 to 6% of affected patients in different ethnic groups. CNS and pulmonary and large vessel involvement, as well as bowel perforation, are the major lifethreatening complications; death may also result as a complication of immunosuppressive therapy. Markers of severe prognosis include HLA-B51 positivity, male gender, and early development of systemic signs.4
Indeed, there is an association of male gender with ocular involvement, vascular involvement, superficial and deep venous thrombosis, cardiac involvement, folliculitis, and papulopustular lesions, whereas females
2574
present more often genital ulcers, erythema nodosum, and joint involvement.56 Onset in childhood does not necessarily predict a poor prognosis.8 Spontaneous remissions of certain or all manifestations of the disease have been observed. Ophthalmic and neurologic sequelae are leading causes of morbidity, followed by severe vascular and GI manifestations, and their effects on morbidity may be cumulative.
TREATMENT
The choice of treatment for patients with Adamantiades–Behçet disease depends on the site and severity of the clinical manifestations of the disease. Recurrent aphthae are most often treated with palliative agents, such as mild diet, avoidance of irritating agents, and potent topical glucocorticoids and local anesthetics61-63; recently, topical hyaluronic acid 0.2% gel 2 times a day over 30 days was found effective64
(Table 141-3). For the topical treatment of genital ulcers and skin lesions, corticosteroid and antiseptic creams can be applied for up to 7 days. Painful genital ulcerations can be managed by topical anesthetics in cream. Corticosteroid injections (triamcinolone acetonide, 0.1-0.5 mL/lesion) can be helpful in recalcitrant
■Mild diet
■Mild diet
■Avoidance of hard, spicy, or salty nutrients and irritating chemicals, such as toasted bread, nuts, oranges, lemons, tomatoes, spices (pepper, paprika, curry), alcohol- or CO2-containing drinks, mouthwashes, toothpastes containing sodium lauryl sulfatea
■Avoidance of hard, spicy, or salty nutrients and irritating chemicals,
such as toasted bread, nuts, oranges, lemons, tomatoes, spices (pepper, paprika, curry), alcohol- or CO2-containing drinks, mouthwashes, toothpastes containing sodium lauryl sulfatea
■Topical treatment of the aphthous oral ulcers includes:
■Topical treatment of the aphthous oral ulcers includes:
■Caustic solutions (silver nitrate, 1%-2%; tinctura myrrha, 5%-10% weight/volume; H2O2, 0.5%; methyl violet, 0.5%) 1-2 times a day
■Caustic solutions (silver nitrate, 1%-2%; tinctura myrrha, 5%-10%
weight/volume; H2O2, 0.5%; methyl violet, 0.5%) 1-2 times a day
■Antiseptic and anti-inflammatory preparations (amlexanox, 5% in oral pastea; triclosan, 0.1% mouthwash solution and in toothpastesa; amyloglucosidase- and glucoseoxidase-containing toothpastesa; hexetidine, 1%, chlorhexidine, 1%-2% mouthwash solutions; benzydamine; camomile extracts); 3% diclofenac in 2.5% hyaluronic acida; hyaluronic acid 0.2% gel; tetracycline mouthwash (as glycerine solution 250 mg/5 mL glycerine) 2 min 4-6 times a daya (caveat: pregnancy); doxycycline in isobutylcyanoacrylatea
■Antiseptic and anti-inflammatory preparations (amlexanox,
5% in oral pastea; triclosan, 0.1% mouthwash solution and in toothpastesa; amyloglucosidase- and glucoseoxidase-containing toothpastesa; hexetidine, 1%, chlorhexidine, 1%-2% mouthwash solutions; benzydamine; camomile extracts); 3% diclofenac in 2.5% hyaluronic acida; hyaluronic acid 0.2% gel; tetracycline mouthwash (as glycerine solution 250 mg/5 mL glycerine) 2 min 4-6 times a daya (caveat: pregnancy); doxycycline in isobutylcyanoacrylatea
■Corticosteroids (triamcinolone mucosal ointment, dexamethasone mucosal paste, betamethasone pastilles) 4 times a day or during the night (ointment/paste) or intrafocal infiltrations with triamcinolone suspension 0.1-0.5 mL per lesion
■Corticosteroids (triamcinolone mucosal ointment, dexametha-
sone mucosal paste, betamethasone pastilles) 4 times a day or during the night (ointment/paste) or intrafocal infiltrations with triamcinolone suspension 0.1-0.5 mL per lesion
■Anesthetics (lidocaine, 2%-5%; mepivacaine, 1.5%; tetracaine, 0.5%-1% gels or mucosal ointments) 2-3 times a day (caveat: allergy)
■Anesthetics (lidocaine, 2%-5%; mepivacaine, 1.5%; tetracaine,
0.5%-1% gels or mucosal ointments) 2-3 times a day (caveat: allergy)
■5-Aminosalicylic acid (5% cream) 3 times a day (reduces the duration of lesions and the pain intensity)
■5-Aminosalicylic acid (5% cream) 3 times a day (reduces the
duration of lesions and the pain intensity)
■Cyclosporine A, 500 mg solution for mouthwash 3 times a day (effective as topical immunosuppressive drug)
■Cyclosporine A, 500 mg solution for mouthwash 3 times a day
(effective as topical immunosuppressive drug)
■Sucralfate suspension, 5 mL × 4 times a daya (for oral aphthous and genital ulcers)
■Sucralfate suspension, 5 mL × 4 times a daya (for oral aphthous
and genital ulcers)
■A close association of smoking with a decrease of recurrences of oral aphthous ulcers has been described.
■A close association of smoking with a decrease of recurrences of
oral aphthous ulcers has been described.
aSmall, randomized, double-blind, placebo-controlled trial against placebo.
ulcerations. They can also be beneficial on panuveitis and cystoid macular edema as a single intravitreal injection (triamcinolone acetonide 4 mg).65,66
Patients with mucocutaneous lesions resistant to topical treatment, those with systemic involvement, and patients with markers of poor prognosis are candidates for systemic treatment.61-63,67-69 Several compounds have been found effective in randomized, double-blind, placebo-controlled trials70-90 (Table 141-4). Additional treatments have been successful in studies with a lower grade of evidence (Table 141-5).2,16,48,71-83 Oral and intravenous prednisolone can be combined with other immunosuppressants, colchicine, dapsone, sulfasalazine, or interferon-α. A synergistic effect with cyclosporine A has been described in patients with ocular involvement. Prednisolone is one of the few medications that can be used during pregnancy. Colchicine can be combined with immunosuppressants and interferon-α. A rapid relapse often occurs after
22
discontinuing cyclosporine A, interferon-α, dapsone, or infliximab.73,76,78
PREVENTION
■ Patients with severe or progressive recurrent aphthous stomatitis should be followed up for years as potential candidates for Adamantiades–Behçet disease, particularly those patients with familial occurrence of the disease.
■ Patients with suspected Adamantiades–Behçet disease should be referred early for specialist advice.
■ Male patients with systemic involvement as a presenting sign and/or an early age of onset should be treated systemically because of the poor prognosis.
DRUG DOSE INDICATION REF.
Methylprednisolone 40 mg every 3 wk IM Erythema nodosum (but not orogenital ulcers) 70
Rebamipide 300 mg/d orally (caveat: pregnancy, lactation) Oral ulcers 71
Colchicine
1-2 mg/d orally (caveat: pregnancy, lactation—induces oligozoospermia) Oral aphthous ulcers, genital ulcers, folliculitis, erythema nodosum 72
1.5 mg/d
Erythema nodosum, arthritis, genital ulcers, (oral ulcers in females) Ineffective
73
74
Dapsone 100 mg/d orally (caveat: pregnancy, lactation— methemoglobin increase: ascorbic acid, 500 mg/d) Oral ulcers, genital ulcers, skin lesions, pathergy test 75
Azathioprine 2.5 mg/kg/d (caveat: pregnancy, lactation, severe liver disease, bone marrow depression, severe infection, children)
Recent-onset ocular disease 76
Interferon-α 2a 6 × 106 IU 3 times a week SC (caveat: pregnancy, lactation— induces psychotic signs, psoriasis, myopathy) Oral ulcers, genital ulcers, papulopustular lesions 77
Interferon-α 1000 and 2000 IU/d orally Ineffective 78
Thalidomide 100 or 300 mg/d (caveat: pregnancy, lactation—induces polyneuropathy: minimized at 25 mg/d) Oral ulcers, genital ulcers, papulopustular lesions 79
Cyclosporine A 10 mg/kg/d orally (against colchicine, 1 mg/d orally) (caveat: lactation, renal insufficiency—induces pathologic CNS findings)
Ocular manifestations, oral ulcers, skin lesions, genital ulcers 80
5 mg/kg/d orally (against cyclophosphamide pulses) Visual acuity 81
5 mg/d orally (against conventional treatment) Ocular attacks 82
Adalimumab 80 mg SC (initial dose), 40 mg SC every other week starting 1 wk after the initial dose Intermediate uveitis, posterior uveitis and panuveitis 83,84
Daclizumab 1 mg/kg BW IV every 2 wk Ineffective 85
Etanercept 25 mg 2 times a week orally (caveat: pregnancy, lactation) Oral ulcers, nodular lesions, papulopustular lesions (not pathergy test) 86
Secukinumab 300 mg SC every 2 wk Ineffective 87
Apremilast 30 mg 2 times a day orally over 12 wk Oral ulcers 88
Aciclovir 5 × 800 mg for 1 wk + 2 × 400 mg/d for 11 wk Ineffective 89
Azapropazone 900 mg/d over 3 wk orally Arthritis, ineffective 90
Azapropazone 900 mg/d over 3 wk orally Arthritis, ineffective 90
2575
aEvidence grade A—randomized, double-blind, placebo-controlled trials except otherwise mentioned.
22
DRUG DOSE INDICATION
Corticosteroids 5-60 mg/d prednisolone equivalent orally 100-1000 mg/d IV over 1-3 d (alone or in combination) (caveat: diabetes —induce psychosis)
Active disease Acute exacerbation (particularly uveitis, neurologic manifestations)
Indomethacin 100 mg/d PO mucocutaneous lesions, arthritis
Pentoxifylline Oxpentifylline 300 mg × 1-3/d PO 400 mg × 3/d PO oral ulcers (particularly in children)
Irsoglandine 2-4 mg/d PO Recurrent oral ulcers
Cyclosporin A 3-6 mg/kg/d PO (serum levels: 100-150 ng/mL) (caveat: lactation, renal insufficiency—induces pathologic CNS findings) Uveitis, mucocutaneous signs, thrombophlebitis, acute hearing loss
Tacrolimus 0.05-0.2 mg/kg/d PO (serum levels: 15-25 ng/mL) Refractory uveitis
Interferon-α 9 × 106 IU 3 times a week / 3-9 × 106 IU 5 times a week SC (3 × 106 IU 3 times a week maintenance dose) (caveat: pregnancy, lactation— induces psychotic signs, psoriasis, myopathy) 1.5-3 × 106 IU 3 times a week according to body weight
Ocular lesions, long-term visual prognosis, arthritis, vascular lesions
Corticodependent uveitis in children
Cyclophosphamide 1 g/mo IV bolus (caveat: hemorrhagic cystitis: mesna 200 mg) Uveitis, neurologic manifestations
Chlorambucil 0.1 mg/d orally (2 mg/d maintenance dose) (caveat: cumulative toxicity) Neurologic manifestations, uveitis, thrombosis, mucocutaneous lesions
Methotrexate 7.5-20 mg once a week orally (caveat: pregnancy, lactation, severe bone marrow depression, liver dysfunction, acute infections, GI ulcers, kidney insufficiency)
Severe mucocutaneous lesions, arthritis, progressive psychosis or dementia
Infliximab 5 mg/kg IV days 1, 7, 14, 28 or days 1, 14 / 1, 30 / 1, 45 (caveat: pregnancy, lactation) Acute uveitis, refractory posterior uveitis, neurologic manifestations, intestinal involvement, vascular involvement
Adalimumab
Refractory ocular lesions, GI lesions
Anakinra 100 mg/d SC; escalation to 200 mg/d after 1 mo and 300 mg/d after 6 mo in partial responders (caveat: mild infections) Oral ulcers, ocular lesions
Canakinumab
Ocular lesions
Gevokizumab 30 or 60 mg once in 4 wk IV or SC (on top of a stable immunosuppressive regimen). Ocular lesions
Ustekinumab 90 mg SC at weeks 0, 4, and every 12 wk (caveat: headache) oral ulcers
Alemtuzumab 12 mg/d IV (caveat: infusion reactions, onset of symptomatic thyroid disease) Ocular lesions
Allicin 20 mg 3 times a day orally over 12 wk (caveat: unpleasant smell, nausea, vomiting, abdominal pain, allergic reaction) Oral ulcers, skin lesions
Sulfasalazine 1.5-3 g/d orally GI ulcers
Thalidomide 2 mg/kg/d orally; increased to 3 mg/kg/d if necessary or decreased to
Intestinal involvement (in children)
Thalidomide 2 mg/kg/d orally; increased to 3 mg/kg/d if necessary or decreased to 1-0.5 mg/kg/d according to the response (caveat: neurotoxicity)
1-0.5 mg/kg/d according to the response (caveat: neurotoxicity)
aEvidence grade B—well-conducted open clinical trial.
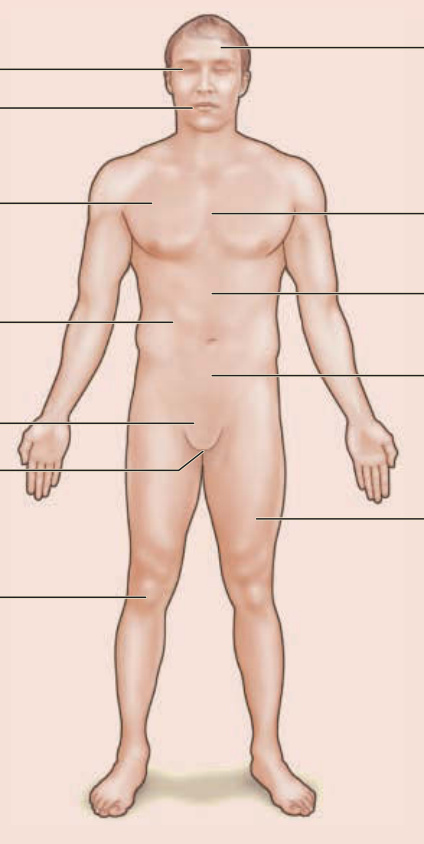
Figure 141-1 Adamantiades–Behçet disease: a multisystem disorder.
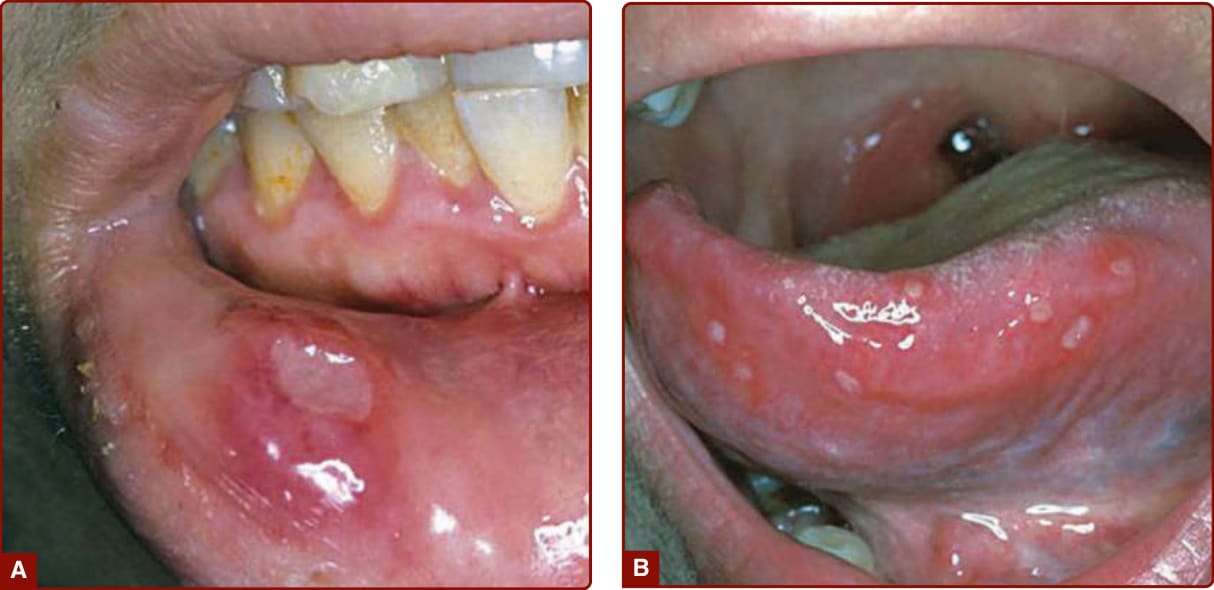
Figure 141-2 (A) Single and (B) multiple oral aphthous ulcers. (A from Altenburg A et al. Epidemiology and clinical manifestations of Adamantiades-Behçet disease in Germany—current pathogenetic concepts and therapeutic possibilities. J Dtsch Dermatol Ges. 2006;4:49, with permission. Copyright © 2006 John Wiley & Sons.)
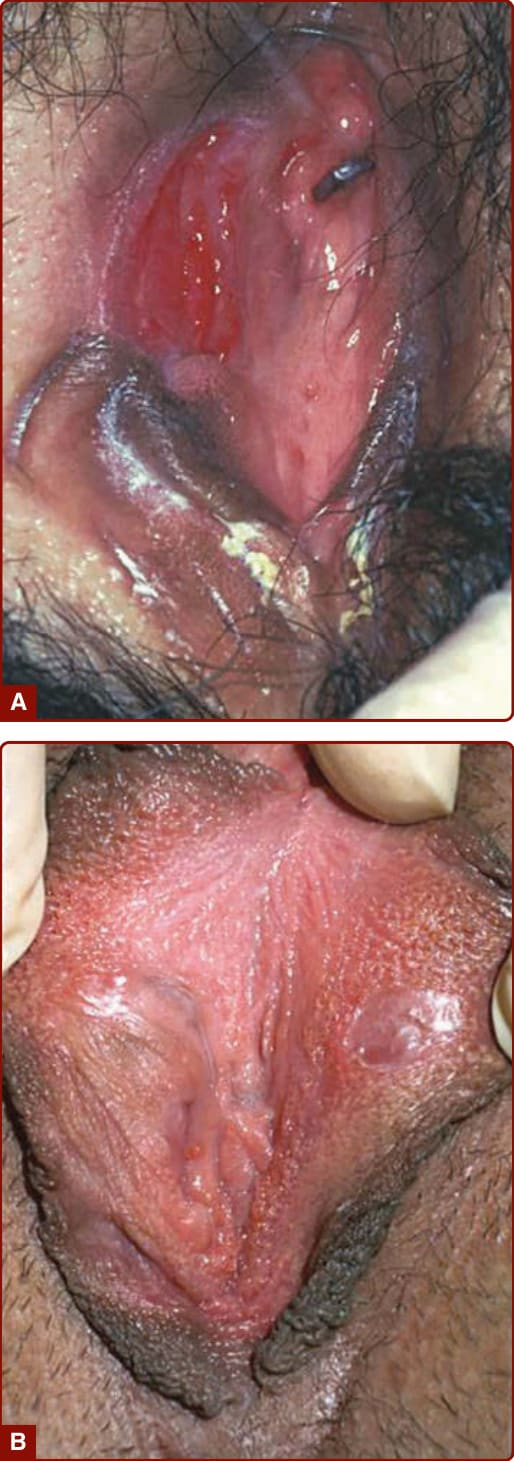
Figure 141-3 (A) Genital ulcer healing with (B) a demarcated flat scar.
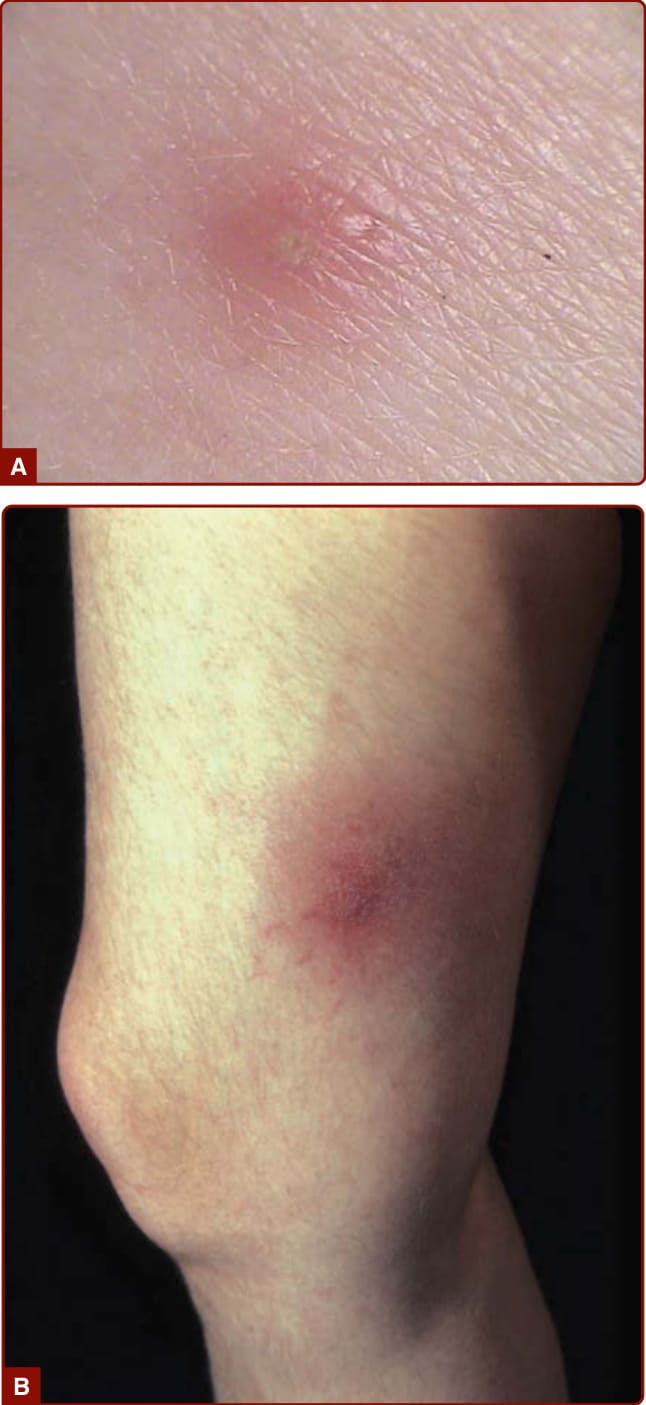
Figure 141-4 (A) Positive pathergy test and (B) erythema nodosum–like lesion in Adamantiades–Behçet disease.
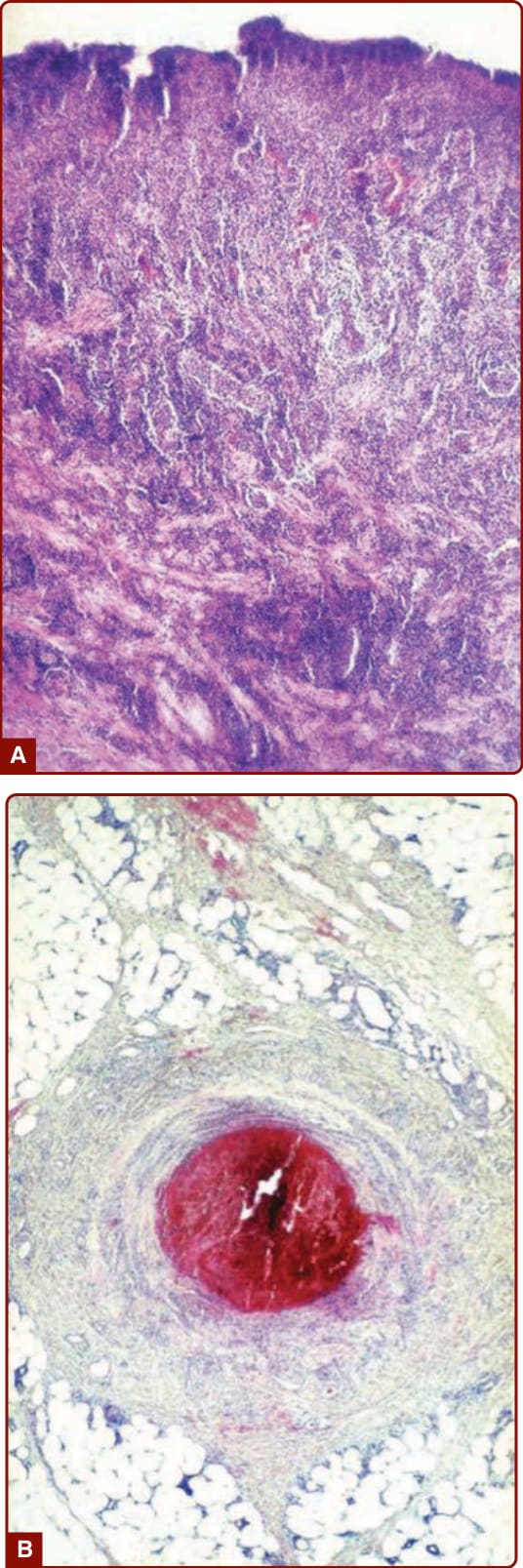
Figure 141-5 A, Abundant mixed inflammatory infiltrate dominated by neutrophils in an oral ulcer of Adamantiades– Behçet disease. B, Vessel thrombosis in an erythema nodosum–like lesion.
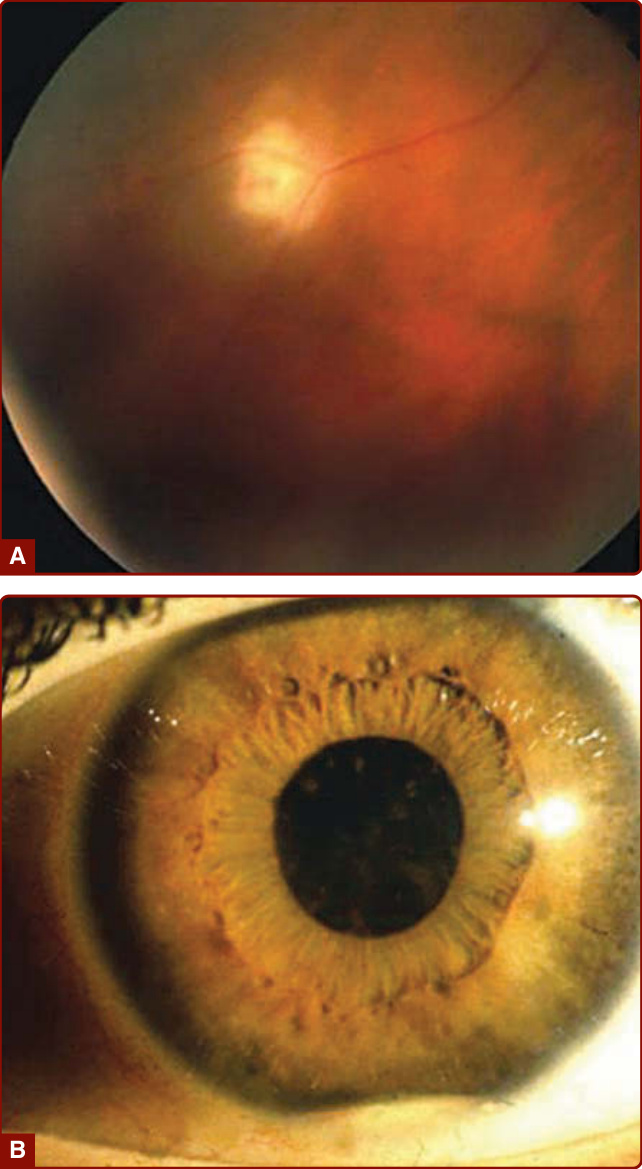
Figure 141-6 A, Posterior uveitis. B, Hypopyon iritis. (From Altenburg A et al. Epidemiology and clinical manifestations of Adamantiades-Behçet disease in Germany—current pathogenetic concepts and therapeutic possibilities. J Dtsch Dermatol Ges. 2006;4:49, with permission. Copyright © 2006 John Wiley & Sons.)

TABLE 141-1 International Criteria for Behçet Disease46

TABLE 141-2 Differential Diagnosis of Adamantiades– Behçet Disease6,50

TABLE 141-3 Topical Treatment of Oral Aphthous Ulcers63
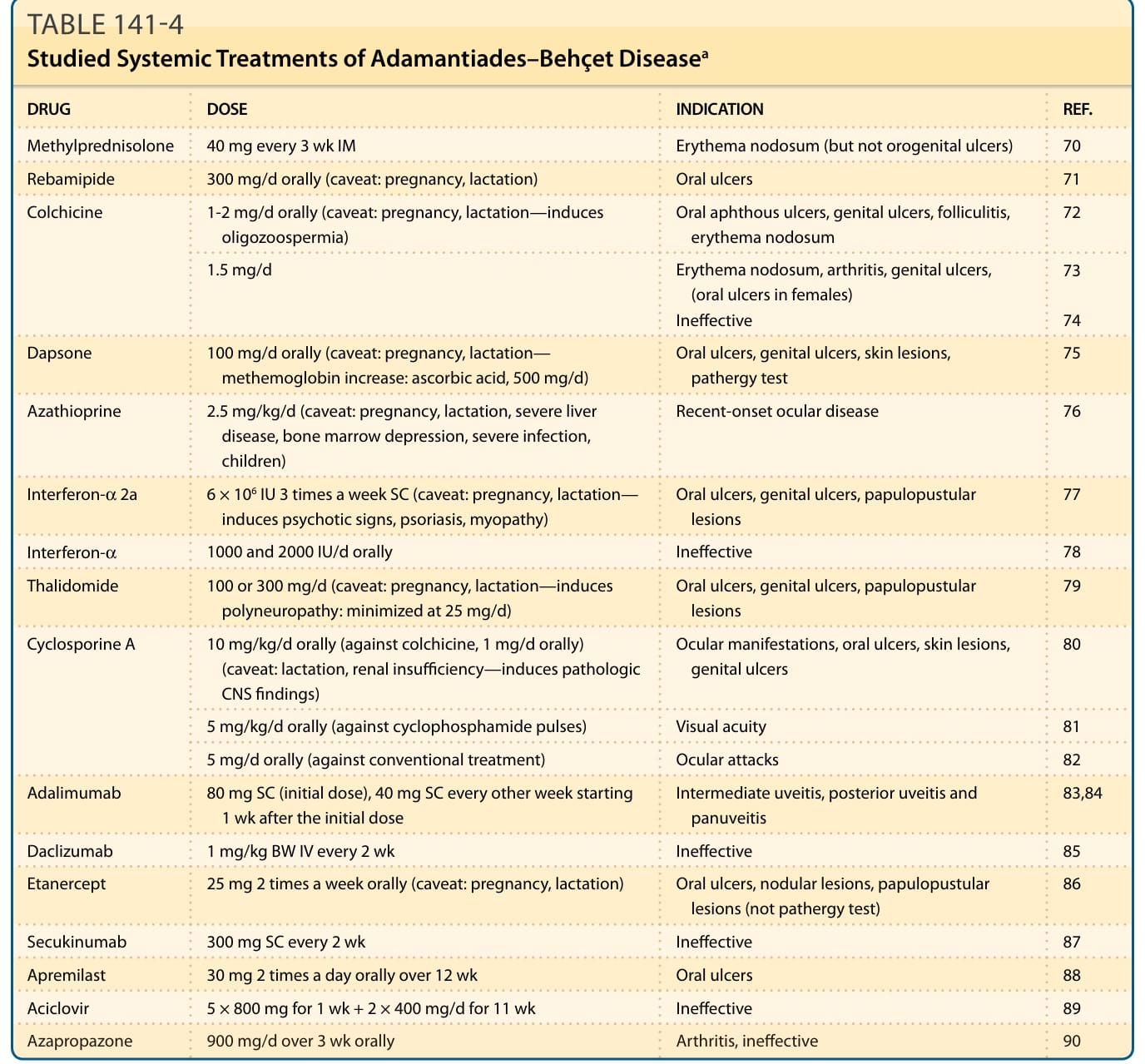
TABLE 141-4 Studied Systemic Treatments of Adamantiades–Behçet Diseasea
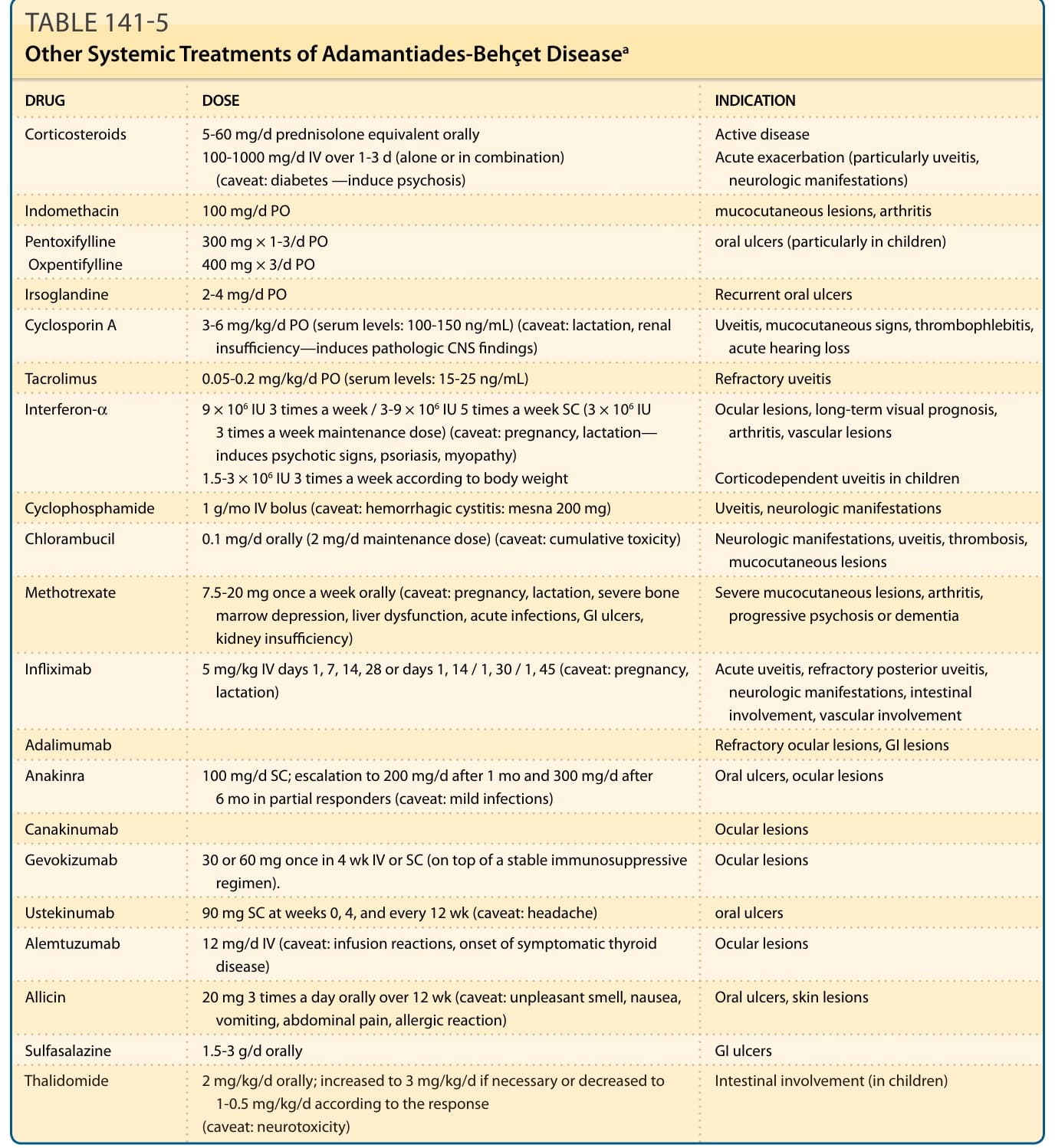
TABLE 141-5 Other Systemic Treatments of Adamantiades-Behçet Diseasea