Erythema Elevatum Diutinum
22
AT-A-GLANCE
■ Rare disorder, with around 250 case reports in literature.
■ A chronic leukocytoclastic vasculitis typified by a distinctive clinical pattern of symmetric, erythematous, violaceous, or yellow–brown papules, nodules, or plaques.
■ Sites of involvement often include the skin overlying the joints of the hands and fingers, or extensor surfaces like the elbows, knees, legs, and Achilles tendon. The trunk is usually spared.
■ Coexisting diseases include monoclonal paraproteinemia, lymphoproliferative disorders, chronic infections, autoimmune conditions, and connective tissue disease.
■ Histopathology reveals a leukocytoclastic vasculitis in early-stage disease, and mixed inflammation and fibrosis in late-stage disease.
Erythema elevatum diutinum (EED) is a rare, chronic leukocytoclastic vasculitis (LCV) that presents as largely symmetric papules and plaques, chiefly over joints and upon extensor surfaces, with a pink, erythematous, brown, violaceous, or yellow hue. Cases now recognized as EED were first reported by Hutchinson1,2 in 1878, and Bury3 in 1889. The current nosology, EED, was proposed by Radcliffe-Crocker and Williams in 1894,4 with the last part of the name, diutinum, meaning “long-lasting.” Radcliffe-Crocker and Williams proposed a division of cases into “Bury-type” (more often seen in young women with a personal and/or family history rheumatism), and “Hutchinson-type” (more often seen in older men with gout). In 1932, so-called extracellular cholesterosis of Urbach5 was proposed as a third form. In 1929, Weidman and Besancon classified EED as a vasculitis,6 and this conclusion is now widely held. EED is no longer subdivided, and all forms are considered a spectrum of the disorder.
EPIDEMIOLOGY
The incidence of EED is unknown, with only about 250 cases reported in the literature.7 A case series, culled from a tertiary care center, and spanning 60 years, contained only 13 cases.8 EED may present at any age but is most common in the fourth through sixth decades of life. One of the original cases reported occurred in a 6-year-old.2 Men were thought to be affected more often, but with so few cases reported, it is difficult to ascertain any definitive sexual or ethnic predilection.
CLINICAL FEATURES
EED typically first presents as edematous, erythematous to violaceous papules, nodules, and plaques, distributed relatively symmetrically upon the skin overlying the joints of the fingers, toes, and hands (Fig. 140-1), or upon the extensor surfaces, such as the elbows (Fig. 140-2), wrists, knees (Fig. 140-3), ankles, legs, and Achilles tendon. Purpura, petechiae, vegetative lesions,9 ulcerations, and bullous appearance may occur in association with EED.10
Individual lesions of EED are round to oval, usually smooth and without scale (Fig. 140-4). The lesions are not fixed to the underlying structures. EED is uncommon on the face, ears, buttocks, and genitals. The torso and mucosal membranes are also typically spared. Involvement of the retroauricular area or the palmoplantar skin is uncommon.11-13
Lesions of EED may be asymptomatic, or the condition may be associated with pruritus, pain, burning, stinging, paresthesia, or neuropathy.14 Systemic symptoms, such as arthralgia and fever, may be experienced with the eruption. Over time, EED can coalesce into irregular patterns. Although early lesions of EED are often soft and possibly tender, older lesions may be firm or doughy. Lesions of EED may become more firm, raised, and erythematous in the evening hours.8,15,16
Over time, lesions of EED can become darker brown or violaceous in color. Fibrosis may ensue. Yellow hues may be observed, with the latter resembling xanthomata. The varied appearance of chronic EED may be even more pronounced in HIV-infected persons, and the condition can resemble Kaposi sarcoma or bacillary angiomatosis. The natural course of EED varies. Spontaneous resolution can transpire, usually after 5 to 10 years of disease activity, while in other cases, fixed lesions last decades.17,18 The condition may wax and wane slowly, or flares or crops of disease can erupt rapidly, over weeks or months. Streptococcal infections can precipitate outbreaks. Cold weather can exacerbate symptoms or cause new lesions.
PATHOGENESIS
The pathogenesis of EED is not fully understood. A hypothesis is the formation of antigen-antibody complexes leads to deposition of immune complexes in blood vessel walls.19 This leads to complement activation, with chemotaxis of neutrophils, the latter perhaps due to overexpression of IL-8.20 A leukocytoclastic vasculitis ensues, and repeated episodes of this vasculitis lead to fibrosis and other sequelae of late disease.
Evidence in support of this proposed mechanism includes (1) the inducing of lesions with injection of streptokinase, leading to streptokinase-streptodornase complexes21; (2) deposition of IgG, IgM, IgA, and fibrin in vascular and perivascular locations on direct immunofluorescence examination19; and (3) increased C1qbinding activity and enhanced IL-8 responses in the sera of affected patients.20,22
ASSOCIATED CONDITIONS
EED has been associated with a variety of medical conditions: infections (streptococcal, hepatitis B, syphilis, tuberculosis, HIV), monoclonal and polyclonal
22
gammopathies, inflammatory bowel disease and celiac disease, connective tissue disease (rheumatoid arthritis, ankylosing spondylitis, lupus erythematosus, dermatomyositis, relapsing polychondritis, granulomatous polyangiitis), and lymphoproliferative and myelodysplastic conditions (myelodysplasias, lymphoma, leukemia, myeloma) (see Table 140-1). In some series, the most frequent association with EED was an
2563
22
DISEASE CATEGORY SPECIFIC CONDITIONS
Autoimmune/ Connective Tissue Disorders
Rheumatoid arthritis Ankylosing spondylitis Relapsing polychondritis Cutaneous or systemic lupus erythematosus Granulomatosis with polyangiitis Myasthenia gravis Celiac disease Dermatitis herpetiformis Sjogren syndrome Autoimmune keratolysis of the eye
Infectious diseases Chronic bacterial infections HIV-1, HIV-2 Streptococcal infection/rheumatic fever Measles virus Human herpesvirus 6 Syphilis Tuberculosis Hepatitis B
Inflammatory disorders Crohn disease Ulcerative colitis Mixed cryoglobulinemia Pyoderma gangrenosum
Malignancy IgA/IgG paraproteinemia (monoclonal gammopathy/monoclonal gammopathy of undetermined significance/myeloma) Myelodysplasia Polycythemia vera Hairy-cell leukemia Chronic lymphocytic leukemia B-cell lymphoma/non-Hodgkin lymphoma Lymphadenopathy-associated virus/human T-cell lymphotrophic virus Type 3 Lung cancer Prostate cancer
Medications Rifampin Pyrazinamide Isoniazid Streptomycin
Miscellaneous Pulmonary infiltrates/pleural effusions Hypothyroidism Mosquito bites Insulin-dependent diabetes mellitus Hyper-IgD D syndrome Hypereosinophilic syndrome Acro-osteolysis Hemophilia Peripheral ulcerative keratitis
Miscellaneous Pulmonary infiltrates/pleural effusions Hypothyroidism Mosquito bites Insulin-dependent diabetes mellitus Hyper-IgD D syndrome Hypereosinophilic syndrome Acro-osteolysis Hemophilia Peripheral ulcerative keratitis
IgA paraproteinemia,8 although the severity of EED did not appear dependent on total paraprotein levels.
DIAGNOSIS
The diagnosis of EED is suspected when appropriate clinical findings are present, but it is established via skin biopsy. A biopsy to establish EED should contain the full thickness of the dermis and extend into the
2564
upper subcutis. Most often, a deep punch technique is utilized. The histologic findings by light microscopy depend on the temporal course. Leukocytoclastic vasculitis is a central feature of disease process. Early EED often demonstrates neutrophilic infiltrates and neutrophilic pyknotic debris, as well as fibrin, surrounding the upper and middermal vascular plexii (Fig. 140-5). Lymphocytes, histiocytes, and even a few eosinophils, may accompany this neutrophilic infiltrate. Late EED becomes progressively more fibrotic, and perhaps even sclerotic, on occasion.23 In some late-stage cases, it can be difficult to appreciate a vasculitis. In addition, intracellular lipid deposition and rare cholesterol clefts may be seen, especially in admixed histiocytes, may be seen in late-stage disease.24
Direct immunofluorescence studies of EED often reveal the non-specific deposition of IgG, IgM, IgA, C3, and fibrin in perivascular locations.25
There are no laboratory studies to confirm a diagnosis of EED. Because of the range of conditions associated with EED, it is common to consider the following lab tests: complete blood count, comprehensive metabolic panel, HIV screening, immunofixation electrophoresis, streptozyme test, hepatitis B and C screening, antinuclear antibody (ANA), antineutrophil cytoplasmic antibodies, antiphospholipid antibodies, and urinalysis (Table 140-2). Immunofixation electrophoresis has been advocated, over serum protein electrophoresis.8 A chest radiograph should also be considered.
DIFFERENTIAL DIAGNOSIS
The clinical differential diagnosis of EED depends on the chronicity and symptomatology of the lesions. The main conditions to consider and their differentiating clinical and histologic features are reviewed in Table 140-3. In the setting of HIV-positive persons, the clinical appearance EED can mimic various stages of Kaposi
Complete blood count Comprehensive metabolic panel HIV screening Immunofixation electrophoresis Streptozyme test Hepatitis B and C panel Antinuclear antibodies (ANA) Antineutrophil cytoplasmic antibodies Antiphospholipid antibodies Urinalysis Chest radiograph
Complete blood count Comprehensive metabolic panel HIV screening Immunofixation electrophoresis Streptozyme test Hepatitis B and C panel Antinuclear antibodies (ANA) Antineutrophil cytoplasmic antibodies Antiphospholipid antibodies Urinalysis Chest radiograph
sarcoma (patch, plaque, or nodular disease) and bacillary angiomatosis. Of special mention in the histologic differential diagnosis of EED is granuloma faciale (GF). GF is a rare disorder, characterized by red-brown to violaceous papules and plaques, often with a peau d’orange appearance. The face is the most common location for GF. Histologic similarities between GF and EED include a leukocytoclastic vasculitis with concentric perivascular fibrosis in later lesions. GF is more likely to demonstrate admixed plasma cells and eosinophils,
22
whereas EED is more likely to demonstrate histiocytes and granulomatous areas. Indeed, some dermatopathologists have advocated for the term “localized chronic fibrosing vasculitis” to represent a late-stage common endpoint of GF and EED.26,27
MANAGEMENT
Treatment of early EED can facilitate a dramatic response, and continued therapy is advocated to prevent recrudescence. The goals of therapy include symptomatic relief, clearing of lesions, and appropriate management of associated conditions. Sulfone-based therapies, including dapsone and sulfapyridine, represent first-line agents.10,15,19,21 Rapid improvement may transpire in the first 48 hours, with complete resolution of all lesions over weeks or months. However, lesions often recur rapidly with discontinuation. Although not completely elucidated, it is thought the mechanism of action involves the inhibition of chemotaxis and function of neutrophils. Additional anecdotal therapies, culled from case reports and with varied success and reproducibility, include niacinamide and tetracyclines,28 colchicine,29
addition of nonsteroidal antiinflammatory drugs (diclofenac),30 chloroquine,31 phenformin,32 clofazimine,15 and cyclophosphamide33 as a single agent or combined with plasma exchange.34
DISEASE STAGE DIAGNOSIS DIFFERENTIATING CLINICAL FEATURES DIFFERENTIATING HISTOLOGIC FEATURES
Early
Granuloma Annulare Expanding, annular lesions overlying joints. Disseminated GA more often associated with diabetes. Palisaded and/or interstitial granulomatous infiltrate without vasculitis.
Acute febrile neutrophilic dermatosis (Sweet syndrome)
Reactive condition characterized by tender, erythematous, edematous papules and plaques on the head/neck, upper trunk, and extremities.
Papillary dermal edema with sheets of neutrophils, but without a frank vasculitis.
Rheumatoid neutrophilic dermatitis Associated with rheumatoid arthritis. Presents with asymptomatic papules, plaques, or nodules, on the hands, forearms, neck and trunk.
Palisaded and granulomatous neutrophilic dermatosis
Dense dermal neutrophilic infiltrate without vasculitis.
Rare. Associated with rheumatoid arthritis, connective tissue disease, and lymphoproliferative disorders. Presents as erythematous papules on the extensor surfaces of the upper extremities.
Sparse mixed perivascular infiltrate, to interstitial and/or palisaded granulomas with fibrosis, but without vasculitis.
Neutrophilic dermatosis of the hands A localized variant of acute febrile neutrophilic dermatosis and/or pyoderma gangrenosum. Papules, pustules, and ulcerations, typically of the dorsal hands.
Chronic
Sheets of neutrophils. Although vessels may be affected, not considered a primary vasculitis.
Dermatofibroma Limited lesions, typically on lower extremities. Sudden eruptive dermatofibromata may be associated with lupus erythematosus.
Fibrohistiocytic cells intercalated between and among collagen bundles with overlying epidermal induction.
Xanthoma (particularly tuberous xanthomas) Occur on extensor surfaces and overlying tendons (like the Achilles tendon). Foamy, lipid laden histiocytes with minimal fibrosis.
Rheumatoid nodules Associated with rheumatoid arthritis. Found in the deeper soft tissue over the elbow Deeply situated palisaded granulomas with fibrinoid degeneration, but without frank vasculitis.
Multicentric
Associated with destructive arthropathy and telescoping
Histiocytic cells with characteristic “muddy
Multicentric reticulohistiocytoma Associated with destructive arthropathy and telescoping digits. Histiocytic cells with characteristic “muddy rose” or “ground glass” cytoplasm.
reticulohistiocytoma
digits.
2565
rose” or “ground glass” cytoplasm.
22
Systemic corticosteroids are of variable efficacy. The addition of prednisone may be helpful in recalcitrant cases or when faced with dapsone-associated anemia.35
However, in the setting of certain underlying associated illnesses, systemic steroids might be best avoided. Intralesional corticosteroids or high-potency topical corticosteroids can be effective in limited disease. When EED is associated with an underlying condition or infection, appropriate management of that illness can hasten resolution. For example, in HIVpositive patients, antiretroviral therapy should be added to sulfone-based therapy. The treatment of underlying paraproteinemias also improves the symptoms of EED.15
Lastly, even with appropriate treatment, EED may remain nonresponsive or it may recur at various points across a lifetime. Moreover, chronic, fibrotic lesions are unlikely to respond to any therapy.8
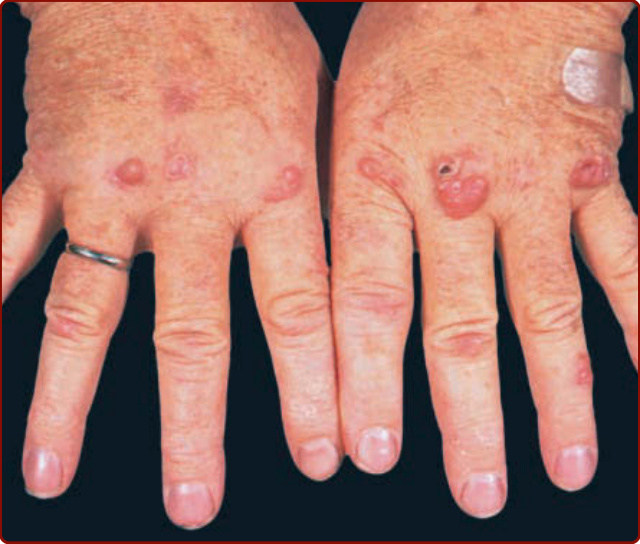
Figure 140-1 Erythema elevatum diutinum. Nodular lesions on dorsal hand present for several years.
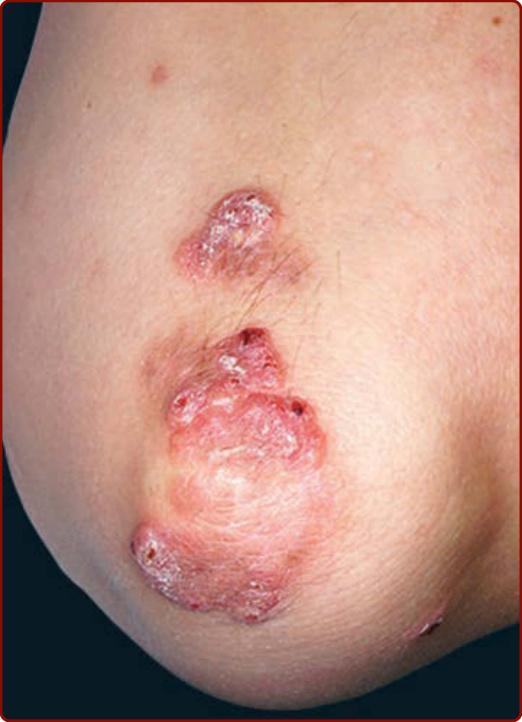
Figure 140-2 Multiple erythematous scaly papules coalescing into plaques on extensor elbow.
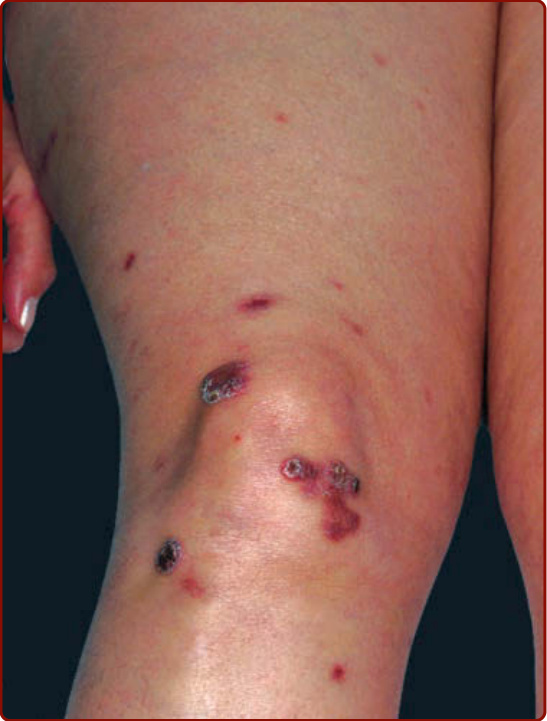
Figure 140-3 Crusted brown and violaceous papules over knee and anterior thigh.
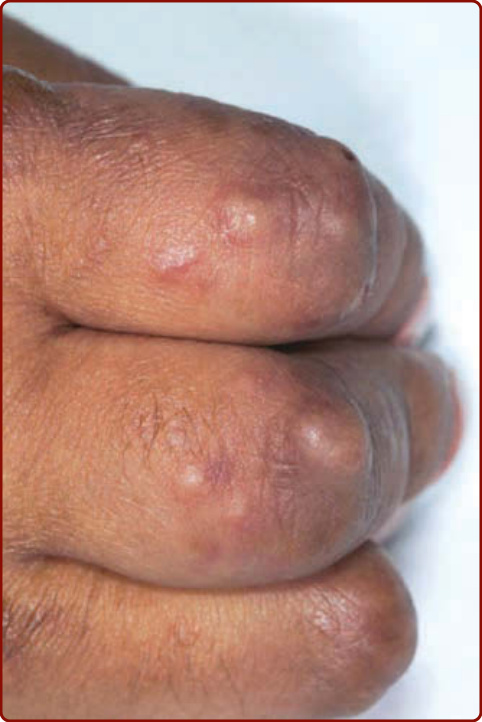
Figure 140-4 Smooth, flesh-colored to slightly erythematous papules on extensor fingers over proximal interphalangeal joints.
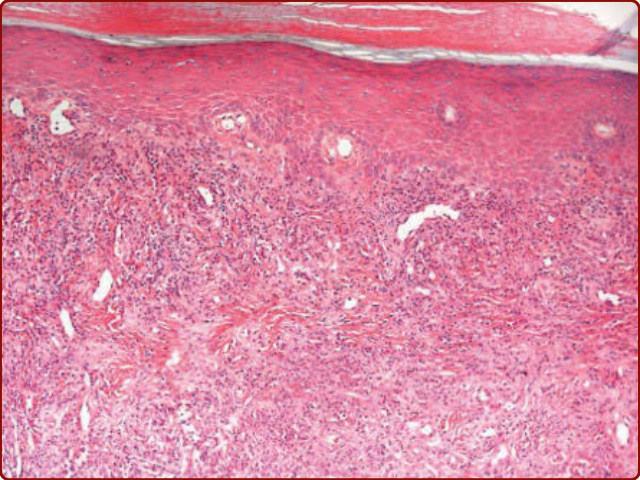
Figure 140-5 Early-stage lesion of erythema elevatum diutinum: Focal leukocytoclastic vasculitis with a fairly dense perivascular and interstitial infiltrate composed of neutrophils, lymphocytes, histiocytes, and leukocytoclastic debris. Mild capillary proliferation is also evident.
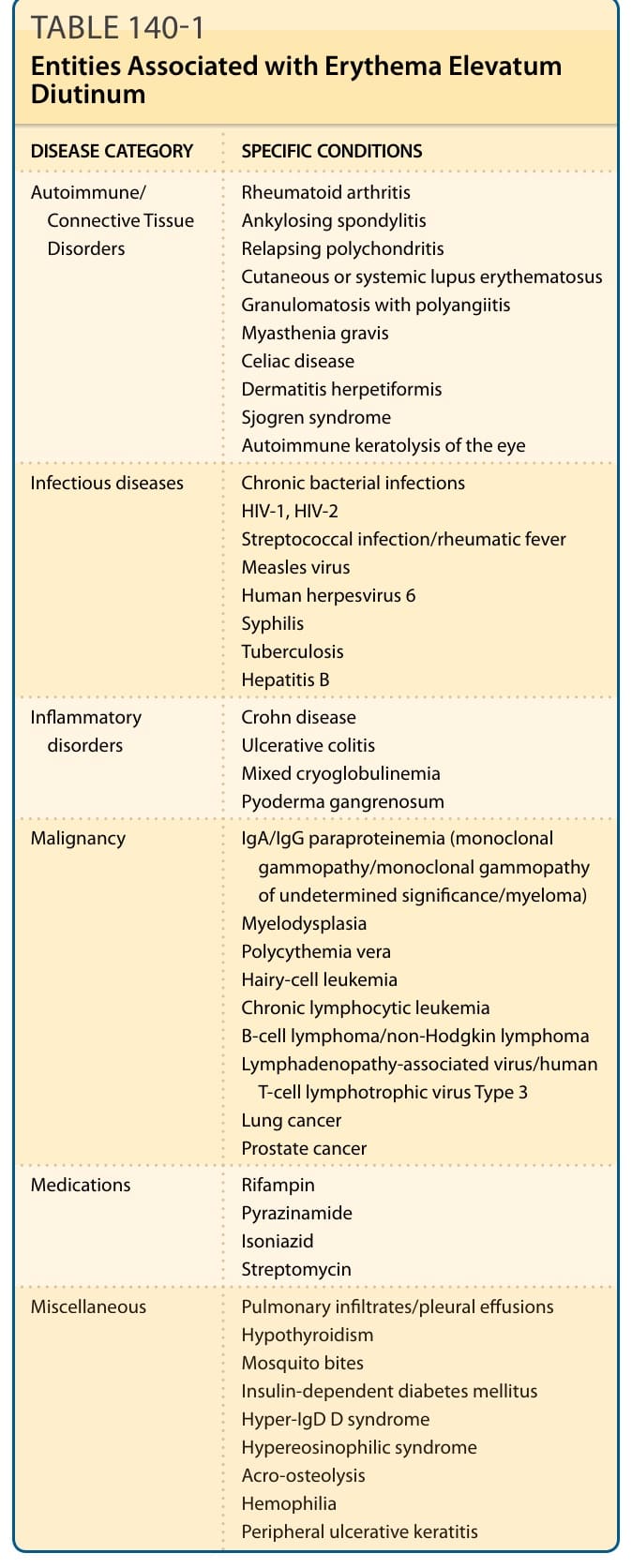
TABLE 140-1 Entities Associated with Erythema Elevatum Diutinum

TABLE 140-2 Laboratory Tests to Consider for a Patient with Erythema Elevatum Diutinum
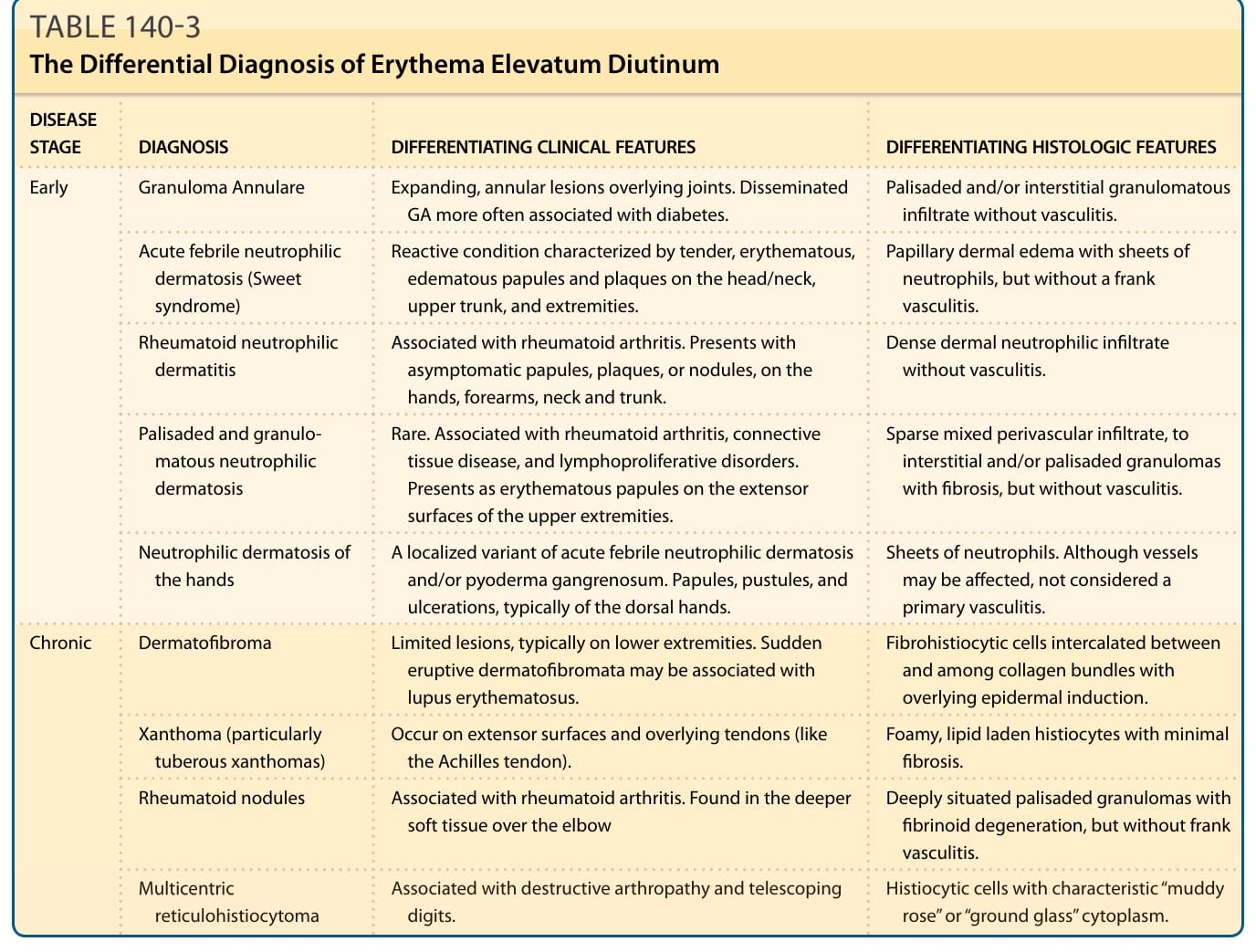
TABLE 140-3 The Differential Diagnosis of Erythema Elevatum Diutinum