Cutaneous Necrotizing Venulitis
PART22
Vascular Diseases
AT-A-GLANCE
■ The signature lesions of cutaneous necrotizing venulitis (CNV) are palpable purpura, erythematous to violaceous papules that do not blanch when the skin is pressed.
■ Palpable purpura persist for 1 to 4 weeks and resolve at times with transient hyperpigmentation and/or atrophic scars.
■ CNV may be associated with episodes of recurrent and chronic urticaria and angioedema.
■ Lesions may occur anywhere on the skin but are most common on the lower extremities or over dependent areas such as the back and gluteal regions.
■ CNV may be associated with connective tissue diseases, malignant conditions, cryoglobulinemia, antineutrophil cytoplasmic or antiphospholipid antibody syndromes.
■ CNV has many precipitating causes, but infections and drugs are most common.
■ The most widely recognized subgroup of idiopathic cutaneous necrotizing vasculitis in children is immunoglobulin A vasculitis.
■ Histopathologic criteria include necrosis of the blood vessels with the deposition of fibrinoid material and dermal cellular infiltrates that consist of neutrophils with nuclear debris, mononuclear cells, and extravasated erythrocytes.
■ Therapeutic approaches are divisible into removal of antigen, treatment of any underlying disorder, and treatment of CNV.
Necrotizing angiitis or vasculitis comprises a diverse group of disorders that combine inflammation with necrosis of the blood vessels. The vascular damage may result from immunologic and/or inflammatory
mechanisms. Clinical syndromes are based on criteria that include the gross appearance and the histopathologic alterations of the vascular lesions, the caliber of the affected blood vessels, the frequency of involvement of specific organs, and laboratory abnormalities. Necrotizing vasculitis may be a primary disease, may develop as a feature of a systemic disorder, or may be idiopathic. Although there is no standard classification of the vasculitides, the American College of Rheumatology classification and the International Chapel Hill Consensus criteria are widely used.1,2
Necrotizing vasculitis in the skin predominantly involves venules and is known as cutaneous necrotizing venulitis/vasculitis (CNV), cutaneous small-vessel vasculitis, and leukocytoclastic vasculitis. The occurrence of CNV in association with systemic involvement of the small blood vessels has been termed hypersensitivity angiitis/vasculitis, systemic polyangiitis, and microscopic polyangiitis (see Chap. 139). CNV may be restricted to the skin, may occur in association with an underlying chronic disease, may be precipitated by infections or drugs, or may develop for unknown reasons (Table 138-1). Systemic forms of necrotizing vasculitis that affect larger blood vessels are considered in Chap. 139, “Systemic Necrotizing Arteritis.”
HISTORICAL PERSPECTIVE
Various syndromes of necrotizing vasculitis were described in the 19th century, including necrotizing vasculitis of small blood vessels, described by Schönlein in 1837 and by Henoch in 1847. In 1952, Zeek differentiated systemic hypersensitivity angiitis that involved small blood vessels from polyarteritis nodosa. In various countries, different diagnostic terms were used to describe necrotizing vasculitis of the small blood vessels in the skin, which were perceived to be arterioles in the 1960s but were shown to be venules in the 1970s by multiple investigators.
22
Associated Chronic Disorders
■Rheumatoid arthritis
■Sjögren syndrome
■Systemic lupus erythematosus
■Hypergammaglobulinemic purpura
■Paraneoplastic vasculitis
■Cryoglobulinemia
■Ulcerative colitis
■Cystic fibrosis
■Antineutrophil cytoplasmic or antiphospholipid antibody syndromes
Precipitating Events
■Bacterial, viral, mycobacterial, and rickettsial infections
■Therapeutic and diagnostic agents
Idiopathic Disorders
Idiopathic Disorders
■Immunoglobulin A vasculitis (Henoch-Schönlein purpura)
■Immunoglobulin A vasculitis (Henoch-Schönlein purpura)
■Acute hemorrhagic edema of infancy
■Acute hemorrhagic edema of infancy
■Urticarial venulitis
■Urticarial venulitis
■Erythema elevatum diutinum
■Erythema elevatum diutinum
■Nodular vasculitis
■Nodular vasculitis
■Livedoid vasculopathy
■Livedoid vasculopathy
■Genetic complement deficiencies
■Genetic complement deficiencies
■Eosinophilic vasculitis
■Eosinophilic vasculitis
■Idiopathic
■Idiopathic
EPIDEMIOLOGY
In a population-based study in Minnesota, the overall incidence of CNV was 4.5 per 100,000 person-years. The most common subtypes were cutaneous small-vessel vasculitis in 45% and immunoglobulin (Ig) A vasculitis in 30% of persons with CNV. The most common etiology was idiopathic. CNV was accompanied by
systemic involvement in 4%.3 In a study in France, the 5-year survival rate was 75.6%. Age older than 65 years at the outset of vasculitis was the only factor that was significantly associated with a shorter survival.4 In this study, 18% of patients experienced relapses, especially when vascular thrombosis was identified in the skin biopsy specimen, when an antineutrophil cytoplasmic antibody (ANCA) was present, when hepatic liver enzymes were elevated, and when peripheral neuropathy was present. When the skin was the single organ involved, there were no relapses. IgA vasculitis is the most common form of CNV in children. Geographic variations in vasculitis may reflect an environmental influence, and seasonal variations in the incidence of vasculitis may suggest an infectious etiology.
CLINICAL FEATURES
The skin lesions of CNV are polymorphous. Erythematous to violaceous papules that do not blanch when the skin is pressed, known as palpable purpura, are the signature lesions (Fig. 138-1A). Macules, papules, urticaria/ angioedema, pustules, vesicles, hemorrhagic blisters (Fig. 138-1B), necrosis and ulcers (Fig. 138-1C), and livedo reticularis may be present. Occasionally, there is subcutaneous edema below the area of the dermal lesions. The eruption most often appears on the lower extremities or over dependent areas, such as the back and gluteal regions. The lesions may occur anywhere on the skin but are uncommon on the face, palms, soles, and mucous membranes. The clinical lesions are episodic and may recur over weeks to years. Palpable purpura persist for 1 to 4 weeks and resolve at times with transient hyperpigmentation and/or atrophic scars. Lesional symptoms include pruritus or burning and, less commonly, pain.
A B C
2528
Fever, malaise, arthralgias, or myalgias may accompany an episode of cutaneous vascular lesions, irrespective of a defined underlying cause or associated disease. Systemic involvement of the small blood vessels most commonly occurs in the synovia, GI tract, voluntary muscles, peripheral nerves, and kidneys.
ASSOCIATED CHRONIC DISORDERS
ASSOCIATED CHRONIC
DISORDERS
CNV is associated with connective tissue diseases, notably rheumatoid arthritis, Sjögren syndrome, systemic lupus erythematosus (SLE), and hypergammaglobulinemic purpura. It rarely occurs in mixed connective tissue disease, relapsing polychondritis, polymyositis/dermatomyositis, ankylosing spondylitis,5 and scleroderma. In patients with rheumatoid arthritis and CNV, the development of vascular lesions is related to the severity of the disease, which is generally, but not always, seropositive. Patients with rheumatoid arthritis often have involvement of larger vessels with associated peripheral neuropathy, nailfold infarcts, and digital gangrene. In patients with SLE, CNV is associated with exacerbations of the underlying disease. Patients with anti- Ro antibody have a greater risk for the development of CNV. Vasculitis, however, is rare in patients with subacute cutaneous lupus erythematosus. Some women with necrotizing vasculitis without connective tissue disease have anti-Ro antibodies, and their infants may be born with neonatal lupus erythematosus. In patients with Sjögren syndrome, the vascular lesions are located predominantly on the lower extremities and appear after exercise. Both hyperpigmentation and cutaneous ulcers are common features. Patients with Sjögren syndrome and CNV have a higher prevalence of articular involvement, peripheral neuropathy, Raynaud phenomenon, and renal involvement, as well as the presence of anti-Ro antibody. Hypergammaglobulinemic purpura occurs in older women and may be associated with Sjögren syndrome, SLE, or a lymphoproliferative disorder. Dermatomyositis in children, but not in adults, may be associated with vasculitis of the GI tract. Paraneoplastic vasculitis6 describes CNV with associated malignant conditions. Hematologic disorders constitute the most common group of malignant disorders, especially chronic myelomonocytic leukemia, non-Hodgkin lymphoma, Hodgkin disease, chronic lymphocytic leukemia, myelodysplastic syndrome, and acute myelogenous leukemia. Less commonly, CNV is related to solid tumors, especially renal, lung, colon, breast, and prostate. However, the association between CNV and neoplasia is rare. It is not necessary to evaluate all patients with CNV for associated malignant conditions. Cryoglobulins (see Chap. 144), especially mixed Types II and III, may be found in patients with idiopathic CNV and in patients with CNV that is associated
22
with connective tissue diseases, lymphoproliferative disorders, propylthiouracil administration, and hepatitides A, B, and C virus infections. Hepatitis C virus is the most common infection, especially when it is associated with cryoglobulinemia. CNV occurs in patients with cystic fibrosis, inflammatory bowel diseases of the colon, and Behçet disease. ANCAs are associated with various forms of necrotizing vasculitis. ANCAs are present in patients with microscopic polyangiitis and cutaneous vasculitis with hepatitis C virus infection (see Chap. 139). The most common cutaneous feature in patients with ANCAs is palpable purpura. Microscopic polyangiitis is associated with small-vessel systemic vasculitis that involves the cutaneous venules and arterioles, the kidneys with necrotizing and crescentic glomerulonephritis, the lungs with pulmonary hemorrhage or interstitial pneumonia, and perinuclear ANCAs (pANCAs). Erythematous macules, purpura, and livedo reticularis are cutaneous manifestations in microscopic polyangiitis. IgA ANCAs are present in acute IgA vasculitis in children. Various subtypes of antiphospholipid antibodies occur in patients with necrotizing vasculitis. IgA anticardiolipin antibodies were detected in some patients with idiopathic CNV. IgA anticardiolipin, IgA and IgM anti–phosphatidyl serine–prothrombin complex, and IgA antibody-β2 glycoprotein antibodies were detected in adults with IgA vasculitis. IgM anti–phosphatidyl serine–prothrombin complex antibodies were detected in adults with CNV. IgG and IgM anticardiolipin antibodies were detected in some individuals with livedoid vasculitis.
PRECIPITATING INFECTIONS AND DRUGS
PRECIPITATING INFECTIONS
AND DRUGS
Infections7 and drugs8-10 may precipitate episodes of CNV. The most commonly recognized infectious agents are β-hemolytic Streptococcus, Staphylococcus aureus, Mycobacterium leprae, and hepatitides B and C viruses. Transient episodes of urticaria may occur early in the course of hepatitis B virus infection and represent immune complex–induced vasculitis. Cutaneous vasculitis has been recognized in a limited number of individuals with HIV infection; the skin lesions consisted of palpable purpura, which at times had a follicular localization, and cutaneous ulcers. Erythema nodosum leprosum, which appears as cutaneous nodules in lepromatous leprosy, is a form of necrotizing vasculitis that involves capillaries, venules, arterioles, small-to-medium-size arteries, and veins. The vascular lesions occur spontaneously or are precipitated by the administration of chemotherapeutic agents. They may be accompanied by fever, malaise, arthralgias, lymphadenopathy, and polyneuritis. Necrotizing vasculitis that is caused by the direct invasion of the blood vessel wall occurs in septicemia caused by Neisseria meningitidis, Neisseria gonorrhoeae,
2529
22
Pseudomonas, Haemophilus influenzae, Rickettsia, Candida, and infectious endocarditis; in Rocky Mountain spotted fever; and in infections localized at the site of a catheter. Palpable purpura is one of the less-common forms of drug reactions. The most commonly incriminated therapeutic agents were antibiotics, especially β-lactam agents, penicillin, and cephalosporins; nonsteroidal antiinflammatory agents; acetaminophen (paracetamol); allopurinol; anticonvulsants; propylthiouracil; hydralazine; granulocyte colonystimulating factor/granulocyte-macrophage colony-stimulating factor; penicillamine; phenytoin; isotretinoin; methotrexate; and levamisole in cocaine.11 Propylthiouracil12 and hydralazine may cause vasculitis in association with ANCAs. Cutaneous vasculitis also has occurred after the administration of streptokinase, radiocontrast media, and staphylococcal protein A column immunoadsorption therapy. CNV is one of the most frequent autoimmune disorders triggered by anti–tumor necrosis factor biologic agents.13
IDIOPATHIC DISORDERS
IDIOPATHIC DISORDERS
IMMUNOGLOBULIN A VASCULITIS
The most widely recognized subgroup of idiopathic CNV is IgA vasculitis,14-18 which formerly was known as anaphylactoid purpura and Henoch-Schönlein purpura. It comprises 75% of CNV in children and 25% of CNV in adults. Most cases occur in the autumn and winter, and often are preceded by a history of a recent upper respiratory tract infection, especially in children. Sites of involvement include the skin, synovia, GI tract, and kidneys. Symptoms may include abdominal pain, melena, arthralgia, and hematuria. Long-term morbidity from progressive renal disease depends on the degree of initial renal damage. The distribution of skin lesions on the upper and lower extremities is a predictive factor for long-term renal
EPIDEMIOLOGY MANIFESTATIONS
IgA vasculitis (formerly Henoch- Schönlein purpura) 75% of CNV in children 25% of CNV in adults History of recent upper respiratory tract infection; involvement of skin, synovia, GI tract, kidneys; associated with abdominal pain, melena, arthralgia and hematuria; risk for progressive renal disease
Acute hemorrhagic edema of infancy Infants and children <2 years of age Painful, edematous petechiae and ecchymoses on head and distal extremities; may have associated facial edema; lesions may appear targetoid with bullae and necrosis; resolves within 1 to 3 weeks without sequelae
Urticarial venulitis (urticarial vasculitis) Women most often affected; may be associated with serum sickness, connective tissue disorders, hematologic and malignant conditions, infections, or may be idiopathic
Erythematous, indurated wheals with a foci of purpura; may be associated with macular erythema, angioedema, livedo reticularis, nodules and bullae; persists for as long as 5 days; episodes may be chronic and recurrent
Erythema elevatum diutinum Rare; most common in 4th to 6th decades Symmetric, persistent, red-purple or red-brown plaques over joints or extensors and over gluteal area
Nodular vasculitis (erythema induratum) More common in women between 30 and 40 years of age; may be associated with Mycobacterium tuberculosis infection (Erythema induratum) and hepatitis C virus infection
Livedoid vasculopathy (livedoid vasculitis, livedo vasculitis, segmental hyalinizing vasculitis, atrophie blanche)
Tender, red, subcutaneous nodules over calves without systemic manifestations; may be recurrent
More common in women; may occur in association with connective tissue disorders, malignancies, hypercoagulable states, thrombophilia
Chronic recurrent episodic exacerbations; arteriosclerosis or stasis of the lower extremities
Sneddon syndrome May be associated with SLE Livedo racemosa and livedoid vasculopathy are associated with ischemic cerebrovascular lesions, hypotension, and extracerebral arterial and venous thromboses
Eosinophilic vasculitis Idiopathic Recurrent, pruritic, purpuric papular skin lesions,
Eosinophilic vasculitis Idiopathic Recurrent, pruritic, purpuric papular skin lesions, urticarial plaques, and angioedema
2530
urticarial plaques, and angioedema
CNV, cutaneous necrotizing venulitis; IgA, immunoglobulin A; SLE, systemic lupus erythematosus.
involvement. Adults with onset after 60 years of age have a worse prognosis, owing to an increased risk of renal disease. Approximately 23% of IgA vasculitis patients experience a relapse. Longer duration of the initial episode, abdominal pain, joint manifestations, and the lack of an infectious cause were associated with relapses. Complete recovery occurred in 82.7% of those with relapses.
ACUTE HEMORRHAGIC EDEMA OF INFANCY
This uncommon disorder affects infants and children younger than 2 years of age, with a slight predominance in boys.19 It has been classified by some as a variant of IgA vasculitis, while others consider it is a distinct clinical entity. The lesions appear as painful, edematous petechiae and ecchymoses that affect the head and distal portions of the extremities. Facial edema may be the initial sign. The skin lesions may be associated with a target-like appearance and may develop bullae and necrosis. Infections, drugs, or immunizations may be triggering factors. Acute hemorrhagic edema of infancy is distinguished from IgA vasculitis by its age distribution, lack of systemic features, and resolution within 1 to 3 weeks without sequelae.
URTICARIAL VENULITIS
Episodes of recurrent and chronic urticaria and angioedema may be a clinical manifestation of CNV.20
Known as urticarial vasculitis/venulitis, this edematous form of necrotizing venulitis occurs in patients with serum sickness, connective tissue disorders, hematologic and other malignant conditions, an IgMK M component, infections, and, rarely, physical urticarias; after the administration of a variety of therapeutic agents; and as an idiopathic disorder.21 The term hypocomplementemic urticarial vasculitis syndrome (HUVS) has been used to describe patients with more severe systemic manifestations, hypocomplementemia with low Clq levels, and an autoantibody to the collagen-like region of Clq.22
The skin lesions appear as erythematous, occasionally indurated, wheals that may contain foci of purpura (Fig. 138-2). Other skin manifestations include angioedema, macular erythema, livedo reticularis, nodules, and bullae. Although the individual urticarial lesions may last for fewer than 24 hours, they often persist for up to 3 to 5 days. The lesions are pruritic or possess a burning or painful quality; they usually resolve without residua although some individuals may develop contusions or hyperpigmentation. The episodes of urticaria are chronic, range in duration from months to years, and vary in frequency. Approximately 70% of affected individuals are women. The prevalence of this disorder remains unknown. General features include fever, malaise, and myalgia; the lymph nodes, liver, and spleen may be enlarged. Extracutaneous features involve the synovium, kidneys, GI tract, lungs,
22
eyes, heart, CNS, peripheral nerves, and blood vessels. These extracutaneous features are more extensive in patients with HUVS. The natural history of urticarial vasculitis is unknown, although individuals have been described with historic episodes of cutaneous lesions for up to 25 years. In one series of patients followed for 1 year, 40% experienced complete resolution of skin lesions; in another series of individuals followed for as long as 14 years, resolution occurred in only 1 patient. Sjögren syndrome and SLE have developed. Deaths have been reported from pulmonary disease, sepsis, and myocardial infarction. The prevalence of urticarial vasculitis in individuals with chronic idiopathic urticaria is unknown, although series have reported rates that range from 1% to 50%. Most of these data have been reported from histopathologic studies in tertiary referral centers. The prevalence of urticarial vasculitis in a prospective clinical study in a university hospital in India was 11.4%. Schnitzler syndrome23,24 consists of episodes of urticarial vasculitis that is characterized by an infiltrate of neutrophils in the dermis and, less commonly, CNV that occur in association with a monoclonal IgMK M component. Associated features include fever, lymphadenopathy, hepatosplenomegaly, bone pain, a sensorimotor neuropathy, and renal failure. It is a recurrent disorder with periods of spontaneous remission. Evolution into hematologic malignant conditions has been reported in 15% of Schnitzler syndrome patients.
ERYTHEMA ELEVATUM DIUTINUM
Erythema elevatum diutinum occurs as symmetric, persistent, red-purple or red-brown plaques that are predominantly distributed over the joints of extensor surfaces and over the gluteal area (see Chap. 140).
2531
22
NODULAR VASCULITIS
Nodular vasculitis appears as tender, red, subcutaneous nodules over the lower extremities, especially the calves, without systemic manifestations (Fig. 138-3). At times, lesions develop on the thighs, buttocks, trunk, and arms, and ulcerated nodules may be present. Recurrent episodes are common. It is more common in women and has a peak incidence in individuals between 30 and 40 years of age. Erythema induratum is a form of nodular vasculitis, which has been associated with Mycobacterium tuberculosis infection, as demonstrated by polymerase chain reaction amplification for M. tuberculosis DNA in skin biopsy specimens. Various sizes of blood vessels, including venules, are affected. Erythema induratum also is associated with hepatitis C virus infection.
LIVEDOID VASCULOPATHY AND SNEDDON SYNDROME
Livedoid vasculopathy (Fig. 138-4), also known as livedoid vasculitis, livedo vasculitis, segmental hyalinizing vasculitis, and atrophie blanche, occurs as recurrent, painful ulcers of the lower extremities in association with persistent livedo reticularis (livedo racemosa) that often is deep purple in color. Healing results in sclerotic pale areas that are surrounded by telangiectasias termed atrophie blanche (Fig. 138-5). The clinical course is chronic with recurrent episodic exacerbations. Systemic involvement is not a feature of idiopathic livedoid vasculopathy. Many patients have arteriosclerosis or stasis of the lower extremities. Livedoid vasculopathy is more common in women and may occur in patients with connective tissue disorders, in malignant conditions, hypercoagulable states, in thrombophilia, and as an idiopathic
2532
disorder.25 Protein C and protein S deficiencies, factor V Leiden gene mutation, activated protein C resistance, prothrombin gene mutation, hyperhomocysteinemia, and antithrombin deficiency have been reported. Atrophie blanche, however, probably represents the end stage of a variety of forms of vascular damage in the skin. Elevated levels of fibrinopeptide A, homocysteine, and plasminogen activator inhibitor may occur. Pathogenesis has focused on a hypercoagulable state with fibrin thrombi in the lumina of the superficial blood vessels. Some consider this condition to be a thrombogenic vasculopathy rather than a small-vessel vasculitis. Antiphospholipid antibodies have been detected in a few individuals.
Sneddon syndrome is a condition in which livedo racemosa and livedoid vasculopathy are associated with ischemic cerebrovascular lesions, hypotension, and extracerebral arterial and venous thromboses.26,27
This condition may be associated with SLE. Antiendothelial cell antibodies, antiphospholipid antibodies, anti–β2-glycoprotein antibodies, and antiprothrombin antibodies were detected in some patients.
GENETIC COMPLEMENT DEFICIENCIES
Genetic C2 deficiency has been recognized in association with CNV in 3 children and in 2 siblings with human leukocyte antigen (HLA)-A25, HLA-B18, and HLA-DR2 (w15) haplotype. C4 deficiency was present in 1 child with CNV and in 1 adult with CNV, and augmented C4 messenger RNA expression.28
Deficiencies of C4A and C4B isotypes were found in some children and adults with IgA vasculitis. A partial C4B deficiency with C4A1, C4A3, and C4B1, and a null allele B∗QO was reported in a 51-year-old woman with CNV.29 A complete deficiency of complement factor 1 with a homozygous missense mutation in exon 2 of the CF1 gene was reported in a 25-yearold woman with CNV.29
EOSINOPHILIC VASCULITIS
Eosinophilic vasculitis has been described as an idiopathic syndrome in individuals with recurrent, pruritic, and purpuric papular skin lesions, urticarial plaques, and angioedema. Skin biopsy specimens showed an infiltrate that is composed of eosinophils that express CD40 and vascular cell adhesion molecule (VCAM)-1 on endothelial cells of involved vessels. Eosinophilic vasculitis also has been described in some individuals with the hypereosinophilic syndrome (see Chap. 40) and in others with connective tissue disorders. There may be depressed complement levels, peripheral eosinophilia, elevated major basic protein levels, and a prolonged eosinophil survival time.
IDIOPATHIC
CNV that does not meet the criteria for a recognized syndrome is classified as idiopathic. Most cases are idiopathic.
ETIOLOGY AND PATHOGENESIS
Experimental studies in animal models and observations in humans implicate immune complexes as a major pathobiologic mechanism in the production of CNV. Data obtained in animal models suggest that the localization of immune complexes in venules is related to vasoactive amines and subsequent vasopermeability alterations. Additional factors that are operative in
22
the localization of immune complexes include endothelial cell surface receptors and the defective clearance of immune complexes by the reticuloendothelial system. The most frequently postulated mechanisms in the production of CNV are the local deposition of circulating immune complexes that are formed during antigen excess or the formation of immune complexes in situ in the skin. Immune complexes may activate the complement system and lead to the generation of C5a anaphylatoxin that degranulates mast cells and attracts neutrophils, which release lysosomal enzymes that damage tissue. The neutrophil superoxidegenerating system may produce reactive oxygen products, which also cause tissue injury. The generation of the chemoattractant leukotriene B4 from infiltrating neutrophils enhances the influx of neutrophils. The initial neutrophilic infiltrate contains few CD3, CD4, CD1a, and CD36 cells as these cells are prominent in the later phase, with the adhesion receptors intercellular adhesion molecule-1 (ICAM-1) and lymphocyte function-associated antigen-1. In IgA vasculitis, the fragmentation of neutrophils was attributed to apoptosis on the basis of the detection of inducible nitric oxide synthase and nitrotyrosine in the infiltrates and the detection of interleukin (IL)-8 on vascular endothelial cells. Long penetration PTX3, which inhibits phagocytosis of apoptotic neutrophils by macrophages, was detected in skin biopsy specimens of idiopathic CNV and of IgA vasculitis about blood vessels and at sites of infiltrates with leukocytoclasia. In CNV, circulating immune complexes have been demonstrated in serum as mixed-type cryoglobulins and indirectly by assays that detect C1q precipitins, materials that bind to complement receptors on human lymphocytoid (Raji) cells, materials that bind to monoclonal rheumatoid factor, and substances that function in the antibody-dependent cellular cytotoxicity inhibition assay. The presence of immune complexes is inferred from the occurrence of serum hypocomplementemia with activation of the classic activating pathway and by the detection of increased plasma levels of C4a and C3a anaphylatoxins. In CNV, immune complexes have been detected in lesional tissues by their ultrastructural observation as electron-dense subendothelial deposits; the membraneattack complex, C5b-9, of the complement system has been detected on the surface of endothelial cells and infiltrating neutrophils. Decay-accelerating factor, which is a regulatory complement protein that prevents the assembly of the membrane-attack complex, was not present on the surface of endothelial cells of the superficial dermal microvasculature. Tissue immune complexes also have been detected by direct immunofluorescence techniques as deposited immunoglobulins and complement proteins. In time–course studies of the evolution of cutaneous vascular lesions, immune reactants have been identified in lesions that are less than 24 hours old. Antigens have been identified only in a few instances as bacterial, viral, mycobacterial, or rickettsial proteins by direct immunofluorescence techniques or by the polymerase chain reaction.
2533
22
A role for lymphocytes in the production of CNV is suggested by a perivenular infiltrate in skin lesions that is rich in lymphocytes with large and hyperchromatic nuclei. The lymphocytes express CD3, CD4, CD1a, CD36, ICAM-1, and lymphocyte function-associated antigen-1. Lymphocytes may be activated by immune complexes, by cellular immune mechanisms, and by primary activation in autoimmune disease to produce lymphokines. In CNV, there were increased numbers of factor XIIIa+–derived dendrocytes, which are involved in antigen presentation to T cells. Endothelial cells also can present antigens to and activate T lymphocytes. Activated macrophages secrete chemokines and lysosomal enzymes. γ/δ T cells have been detected in CNV with a neutrophil-rich pattern and with an infectious etiology. In these specimens, a 72-kDa heat shock protein was expressed by endothelial cells and antigen-presenting cells. The participation of mast cells in CNV is suggested by hypogranulated mast cells with shed extracellular granules and by the development of vascular lesions after the intracutaneous injection of histamine in patients with active episodes of CNV. Mast cells can be activated directly by immune complexes through FcγRIII or by C5a. Through the production of histamine, prostaglandin D2, and cysteinyl leukotrienes, the mast cell could alter venular permeability; interendothelial cell gaps have been noted in venules in patients with CNV. Eosinophils and neutrophils may be recruited by mast cell–derived chemotactic factors. The neutral proteases and acid hydrolases of mast cells may facilitate tissue damage, and the release of tumor necrosis factor-α (TNF-α) may increase expression of E-selectin on endothelial cells and facilitate neutrophil recruitment. Evidence for the role of the mast cell also is provided by time–course analyses of the sequential histopathologic changes in individuals with physical urticaria. In a patient with circulating immune complexes and hypocomplementemia, in whom cold and trauma elicited CNV, initial mast cell degranulation was followed by the infiltration of neutrophils, the deposition of fibrin, and venular endothelial cell necrosis. A postulated sequence of events would be the activation of the mast cell by physical stimuli, the release of vasoactive mediators, the deposition of circulating immune complexes with activation of the complement system, the influx of neutrophils, and the development of CNV. Another example of the time–course analysis of CNV in human skin was provided by an individual with exercise-induced vasculitis. At 3 hours, the number of mast cells decreased, and the eosinophil was the first cell to appear around the venules with the deposition of eosinophil peroxidase. TNF-α levels were elevated, E-selectin was expressed on endothelial cells, and an influx of neutrophils appeared with the deposition of neutrophil elastase and the development of CNV. In a study of 10 adult patients with CNV,30 skin biopsy specimens were obtained from early petechial lesions and the later lesions of palpable purpura. The number of mast cells that contained tryptase and chymase were decreased in the early lesions, which
2534
suggested degranulation. The levels of chymase as well as α1-antichymotrypsin continued to decrease with the progression of vasculitis, which suggested inhibition by antichymotrypsin. IgG, IgM, IgA, and fibrin increased in palpable purpura. Early in the course of necrotizing venulitis, endothelial cells show increased expression of ICAM-1 and E-selectin without the expression of VCAM-1in response to TNF-α. Because E-selectin is an adhesion molecule for neutrophils and for skin-homing, memory T lymphocytes, the increase in E-selectin is associated with an infiltrate of neutrophils that express CD11b within the first 24 hours. In the acute phase of IgA vasculitis, endothelin-1, serum insulin-like growth factor-1,31 insulinlike growth factor–binding protein-3, plasma matrix metalloproteinase-9, ICAM-1, VCAM-1, TNF-α, tumor necrosis factor-like inducer of apoptosis weak,32 IL-1β, IL-2 receptor, IL-6, IL-8, IL-33,33 transforming growth factor-β, vascular endothelial cell growth factor, highmobility group box-1,34 visfatin,35 CCL5, CXCLlb, CX3CL,36 and urine leukotriene E4 levels were elevated. However, the pathogenic roles of these mediators remain undefined. IgA antiendothelial cell antibodies bind to endothelial cells and enhance endothelial cell production of IL-8 via the mitogen-activated protein kinase/extracellular signal-regulated kinase pathways. In skin biopsy specimens from patients with idiopathic CNV, hypersensitivity vasculitis, urticarial vasculitis, and IgA vasculitis, E-selectin was detected on endothelial cells of lesions that were less than 48 hours old and was associated with an infiltrate of neutrophils that expressed CD11b. The endothelial cells expressed HLA-DR and very-late-activating antigen-1 but not P-selectin, and the perivascular cells expressed VCAM-1 and HLA-DR. Diminished cutaneous fibrinolytic activity with reduced release of plasminogen activator from venular endothelial cells occurs in patients with CNV; the subsequent reduction in fibrinolytic activity leads to fibrin deposition. Increased levels of plasma thrombomodulin, tissue-type plasminogen activator, and plasminogen activator inhibitor-1 were detected in patients with IgA vasculitis. Cutaneous nerve fibers can release neuropeptides that cause vasodilation, such as substance P, neurokinin A, and calcitonin gene-related peptide. Substance P activates mast cells and macrophages and increases fibrinolytic activity that is mediated by plasminogen activator. Calcitonin gene-related peptide induces expression of E-selectin on endothelial cells and is chemotactic for T lymphocytes. Eosinophils are minor infiltrating cells in CNV except in eosinophilic vasculitis and drug-induced CNV. Eosinophils produce leukotriene C4 and plateletactivating factor, which increase vascular permeability. Eosinophil granule proteins are toxic to endothelial cells and cause the release of mediators from mast cells. Associations have been recognized between smallvessel necrotizing vasculitis and ANCAs, which have specificity for proteins of the cytoplasmic granules of neutrophils and the lysosomes of monocytes.
Two forms are recognized: cytoplasmic (cANCAs) that are directed against proteinase 3 and perinuclear (pANCAs) that are directed against myeloperoxidase. TNF-α facilitates neutrophil activation and cell-surface expression of proteinase 3 and myeloperoxidase, which bind to ANCAs and increase the adherence of neutrophils to endothelial cells resulting in endothelial cell injury. Antiendothelial cell antibodies have been detected in the sera of patients with systemic vasculitis, rheumatoid arthritis with vasculitis, microscopic polyangiitis, and Sneddon syndrome. An increased prevalence of the HLA haplotype HLA-A11, Bw35 in patients with CNV and associated connective tissue disorders suggests that genetic factors may be operative. HLA-DRB1 genotype associations were detected in patients with CNV and IgA vasculitis in Spain.
DIAGNOSIS
LABORATORY TESTING
LABORATORY TESTING
The laboratory evaluation of patients with CNV depends on information obtained from the history and physical examination (Table 138-3). An elevated erythrocyte sedimentation rate is the most consistent abnormal laboratory finding. The platelet count usually is normal. Other abnormalities reflect either a coexistent chronic disorder or the involvement of additional organ systems. Occasionally, leukocytosis, anemia, thrombocytosis, abnormal urine sediment, circulating immune complexes, rheumatoid factor, and antinuclear antibodies have been reported in idiopathic disease. Serum complement levels are usually normal. Hypocomplementemia may develop in patients with concomitant connective tissue diseases or
■Erythrocyte sedimentation rate
■Erythrocyte sedimentation rate
■Complete blood count with differential analysis
■Complete blood count with differential analysis
■Platelet count
■Platelet count
■Urinalysis
■Urinalysis
■24-Hour urine protein and creatinine clearance
■24-Hour urine protein and creatinine clearance
■Blood chemistry profile
■Blood chemistry profile
■Serum protein electrophoresis
■Serum protein electrophoresis
■Immunoelectrophoresis
■Immunoelectrophoresis
■Hepatitis B antigen and hepatitis A and C antibodies
■Hepatitis B antigen and hepatitis A and C antibodies
■Cryoglobulins
■Cryoglobulins
■CH50 (50% hemolytic complement)
■CH50 (50% hemolytic complement)
■Antinuclear antibody
■Antinuclear antibody
■Rheumatoid factor
■Rheumatoid factor
■Antineutrophil cytoplasmic antibodies
■Antineutrophil cytoplasmic antibodies
■Antiphospholipid antibodies
■Antiphospholipid antibodies
■Circulating immune complexes
■Circulating immune complexes
■Stool guaiac test
■Stool guaiac test
■Skin biopsy
■Skin biopsy
22
cryoglobulinemia and reflects the associated disease or the composition of the cryoglobulin. Hypocomplementemia also occurs in some individuals with idiopathic CNV and in 40% of individuals with urticarial venulitis. In patients with the HUVS, a low-molecular-weight 7s C1q precipitin, which has been identified as an IgG autoantibody against the collagen-like region of C1q, was detected. Autoantibodies to vascular endothelial cells were detected in patients with HUVS, in patients with SLE and urticarial vasculitis, and in patients with urticarial vasculitis alone. In IgA purpura, serum IgA1 levels are elevated, IgA ANCAs may be present, and urinary endothelin-1 levels may be elevated. In patients with CNV, various types of circulating immune complexes and IgA ANCAs have been described. IgG autoantibodies to IgE and to FcεRlα also have been identified in some patients with urticarial venulitis.
HISTOPATHOLOGY
HISTOPATHOLOGY
In skin biopsy specimens of palpable purpura and urticarial vasculitis stained with hematoxylin and eosin, the histopathologic criteria requisite for the diagnosis of CNV include necrosis of the blood vessels with deposition of fibrinoid material and dermal cellular infiltrates that consist of neutrophils with nuclear debris, mononuclear cells, and extravasated erythrocytes (Fig. 138-6A). The dermal inflammatory infiltrates vary in intensity and are usually perivenular in location, but may be dispersed widely. In patients with CNV associated with bacterial infections, the biopsy specimens showed a greater number of neutrophils.37
Some patients with connective tissue disorders and cutaneous vasculitis have an infiltrate of inflammatory cells composed of eosinophils with deposited major basic protein and decreased numbers of mast cells. Eosinophils may be present in increased numbers in drug-induced CNV. The fibrinoid material consists of fibrin, necrotic endothelial cells, immunoreactants, and antigens. Studies with 1-µm–thick sections38 show 2 distinct cellular patterns in CNV: one rich in neutrophils and the other in lymphocytes. The infiltrate of neutrophils regresses with the persistence of an infiltrate of mononuclear cells; repeat biopsy specimens in some patients consistently demonstrated an infiltrate of mononuclear cells. In a time–course study over 6 days of the evolution of experimentally induced CNV in a patient with physical urticaria, the number of infiltrating neutrophils decreased without a concomitant increase in lymphocytes although the number of monocytes/macrophages was increased at 48 hours and 72 hours. Other features in both cell patterns of CNV include hypogranulated mast cells, macrophages containing debris, and the perivenular and interstitial deposition of fibrin. Venular alterations in both cell patterns consist of endothelial-cell swelling, activation of nuclei, wrinkling of nuclear membranes, necrosis
2535
22
A B
(see Fig. 138-6B), and basement membrane reduplication and thickening. The arterioles are not affected. By direct immunofluorescence techniques, fibrin deposition in venules is routinely identified in biopsy specimens, whereas the deposition of immunoglobulins and complement proteins varies widely.39,40 IgG, IgM, and IgA have been detected. In one study,41 IgA deposits were related to absence of autoimmune and inflammatory disorders, IgM deposits to the presence of autoimmune and inflammatory disorders, and IgG deposits to a positive ANCA. IgA is deposited about blood vessels in the skin, intestines, and kidneys in IgA vasculitis and has become an immunopathologic marker of this condition (Fig. 138-7). In the skin in IgA vasculitis, IgA1 is the dominant subclass that is deposited. C3 is the most frequently detected immunoreactant, but it is the only complement protein that has been sought with frequency in CNV.
2536
DIFFERENTIAL DIAGNOSIS
MANAGEMENT
Therapeutic approaches may be divided into removal of the antigen, treatment of an underlying disorder, and treatment of CNV. Therapeutic approaches in the
DISORDER CLINICAL CONSIDERATIONS
Petechiae or purpura caused by thrombocytopenia The lesions are flat
Trauma to skin of elderly individuals with vascular fragility, dermatoheliosis, chronic aspirin ingestion, and endogenous or iatrogenic hypercorticism
The lesions are flat
Scurvy Hemorrhagic follicular papules with corkscrew hairs over the lower extremities
Disseminated intravascular coagulation (purpura fulminans)
Extensive areas of purpura with a slate-gray color
Septic emboli Finite numbers of hemorrhagic papules, pustules, and vesicles that are distributed over the acral areas
Atheromatous embolism Purpura, livedo reticularis, nodules, and ulcers distributed over acral areas with blue or discolored toes
Calcinosis cutis Indurated purpura, livedo reticularis,
Calcinosis cutis Indurated purpura, livedo reticularis, nodules and ulcers
nodules and ulcers
treatment of necrotizing vasculitis consist of prevention of the deposition of immune complexes, suppression of the inflammatory response, modulation of underlying immunopathologic mechanisms, and local therapy. When the eruption is associated with a precipitating event, withdrawal of the medication or treatment of the infection results in resolution of the cutaneous lesions. If a coexistent chronic disease is present, treatment of the underlying disease may be associated with improvement in the cutaneous vascular lesions. In many cases, CNV is a self-limited condition, and double-blind, placebo-controlled, prospective trials of therapeutic agents are usually lacking. The treatment of CNV (Table 138-5) depends on an analysis of the cutaneous disability as well as on the toxicity and side effects of the therapeutic agents. H1 antihistamines are used in patients with palpable purpura to alleviate lesional symptoms and perhaps to reduce tissue deposition of circulating immune complexes. Nonsteroidal antiinflammatory agents are combined with the H1 antihistamine. Depending on the therapeutic response, colchicine can be added to or substituted for these agents. However, colchicine was shown to have no significant therapeutic effect in a prospective, randomized, controlled trial. If there is no benefit, dapsone or hydroxychloroquine sulfate can be used. If there still is no therapeutic response, a major decision must be made because systemic glucocorticoids, azathioprine, methotrexate, cyclosporine, mycophenolate mofetil, cyclophosphamide, plasmapheresis, intravenous immunoglobulin, infliximab, adalimumab, etanercept, and rituximab are associated with serious side effects. Although all of these agents are reported to benefit some patients, controlled clinical trials are not available. The administration of interferon-α is associated with clearing of cutaneous vasculitis in patients with hepatitis C virus infection. The treatment of patients with urticarial vasculitis is similar to those with palpable purpura. Additional case reports exist on the treatment of patients with intramuscular gold therapy, cyclophosphamide-dexamethasone pulse therapy, mycophenolate mofetil,
■H1 antihistamines
■H1 antihistamines
■Nonsteroidal antiinflammatory agents
■Nonsteroidal antiinflammatory agents
■Colchicine
■Colchicine
■Hydroxychloroquine sulfate
■Hydroxychloroquine sulfate
■Systemic glucocorticoids
■Systemic glucocorticoids
■Methotrexate
■Methotrexate
■Cyclosporine
■Cyclosporine
■Mycophenolate mofetil
■Mycophenolate mofetil
■Intravenous gammaglobulin
■Intravenous gammaglobulin
■Plasmapheresis
■Plasmapheresis
■Infliximab
■Infliximab
■Etanercept
■Etanercept
■Adalimumab
■Adalimumab
■Rituximab
■Rituximab
22
thalidomide, anakinra, cinnarizine, and psoralen plus ultraviolet A photochemotherapy. In an open-label pilot study in 10 patients with urticarial venulitis, improvement was noted after a single, subcutaneous dose of canakinumab.42 In patients with Schnitzler syndrome, IL-1 inhibition is the only completely efficient treatment with the use of anakinra, canakinumab, or rilonacept. In the treatment of livedoid vasculitis, support stockings are useful. Empiric trials of aspirin and dipyridamole, colchicine, dapsone, danazol, low-dose heparin, systemic glucocorticoids, nicotinic acid, lowmolecular-weight dextran, phenformin, ethylestrenol, nifedipine, pentoxifylline, and rivaroxaban43 have been used. Infusions of prostacyclin, prostaglandin E1, intravenous immunoglobulin, rituximab, tissue plasminogen activator, hyperbaric oxygen therapy, and psoralen plus ultraviolet A photochemotherapy have been used successfully in a few patients.
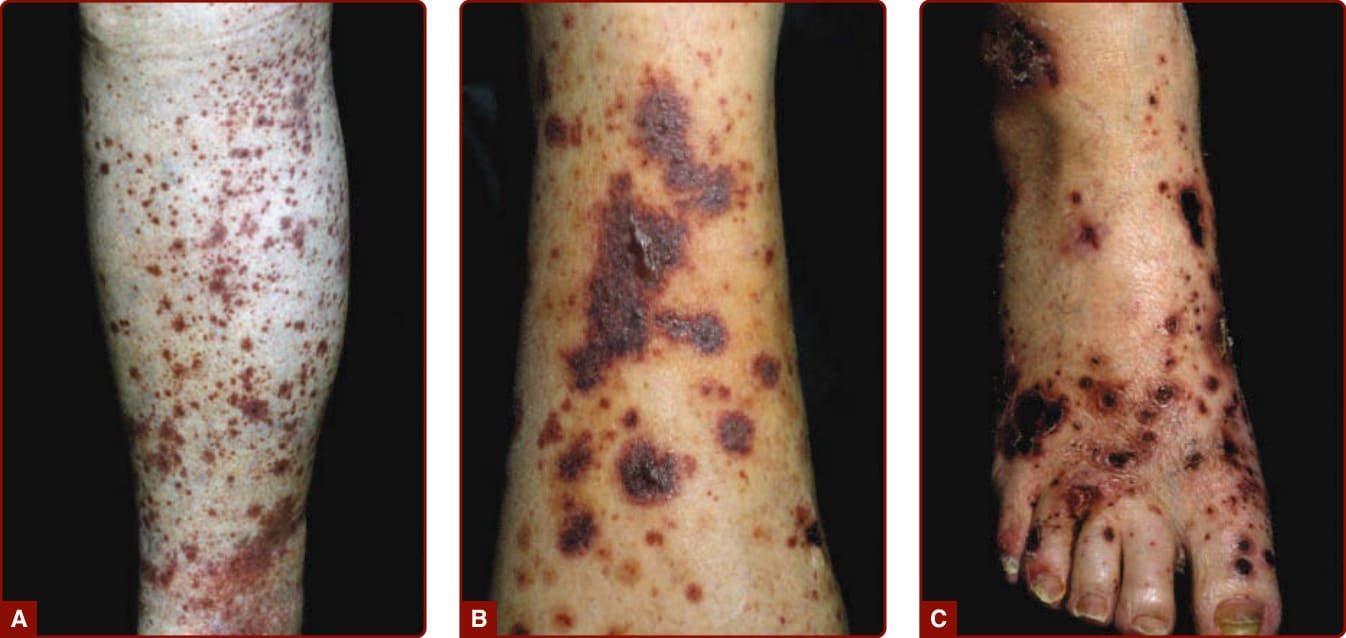
Figure 138-1 A, Palpable purpura on the lower leg in a patient with Henoch-Schönlein purpura. B, Palpable purpura with more-severe tissue damage with hemorrhagic vesicles and bullae. C, With even more tissue damage, multiple necrosis, and ulcers.
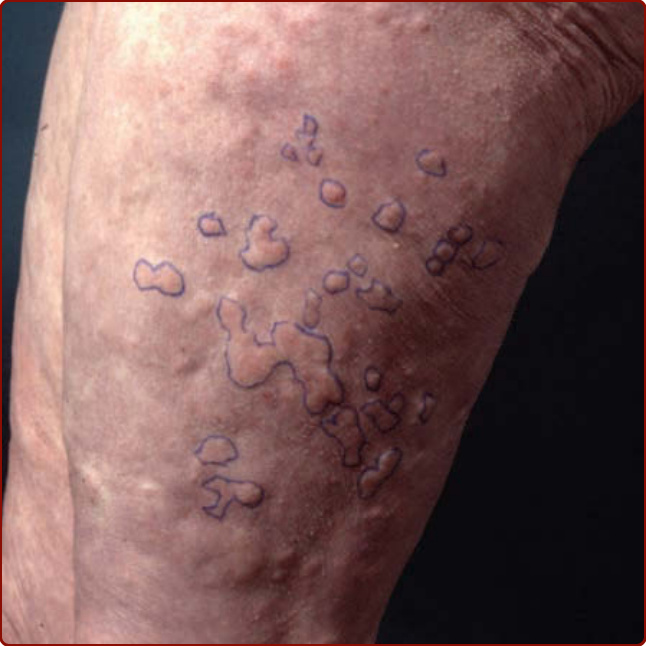
Figure 138-2 Urticarial venulitis. Some of the wheals have been marked 24 hours previously to demonstrate persistent character of the urticaria.
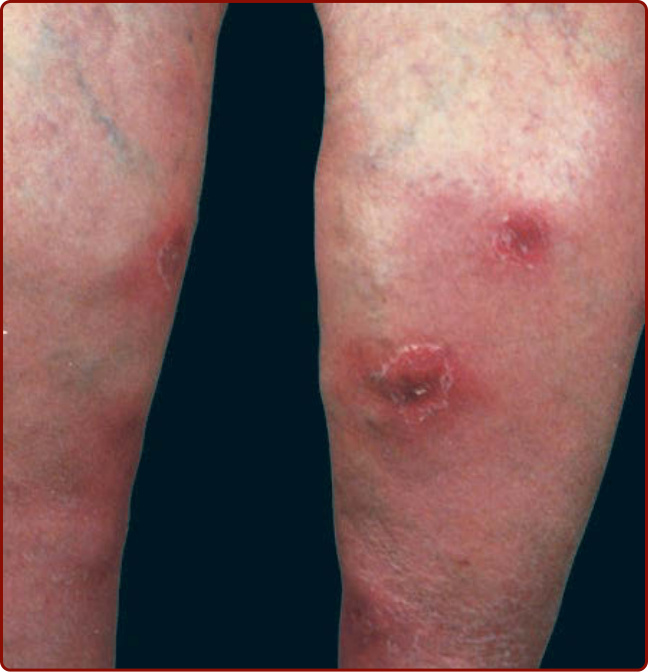
Figure 138-3 Nodular vasculitis with ulcers on lower legs.
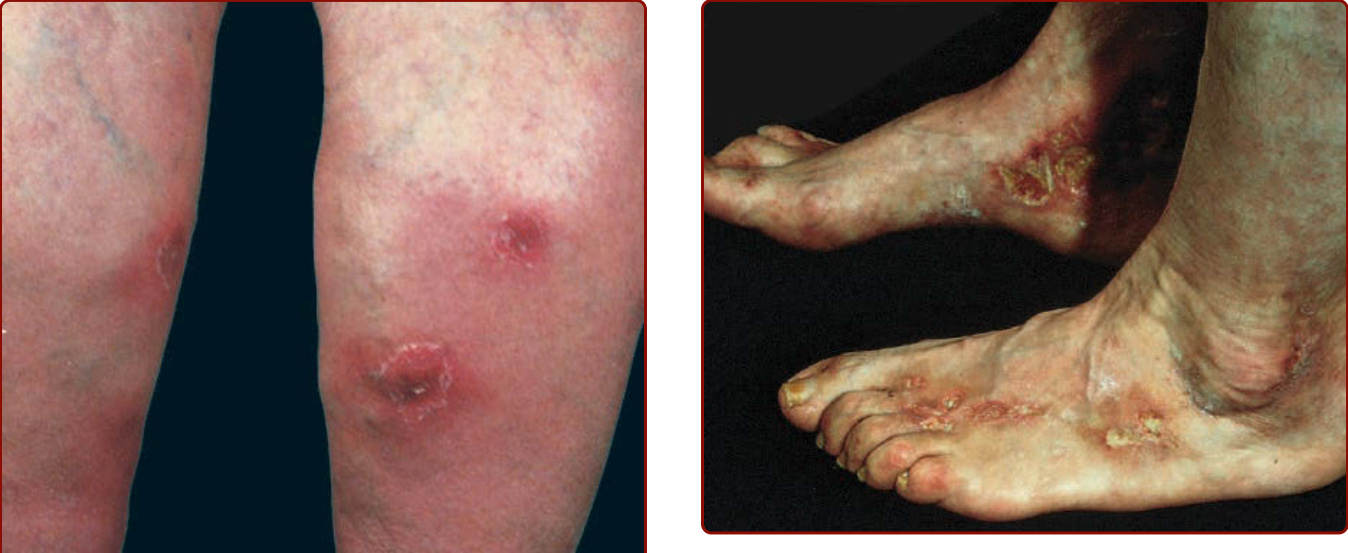
Figure 138-4 Livedoid vasculitis with recurrent, painful ulcers and livedo reticularis.
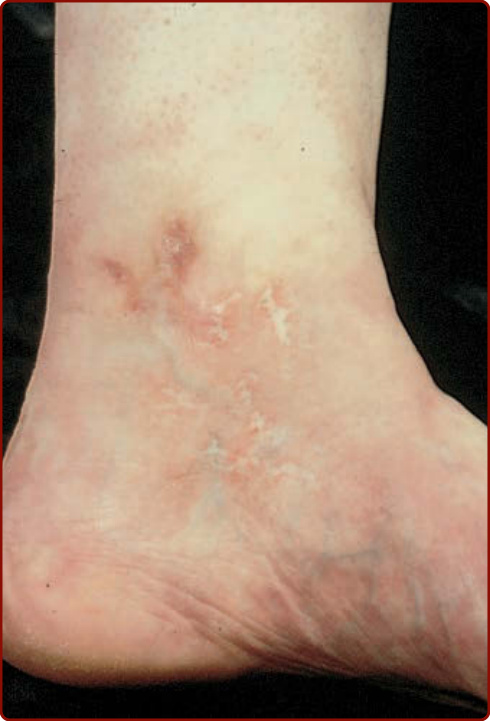
Figure 138-5 Atrophie blanche over the medial malleolus with porcelain-white atrophy, telangiectases, and ulcers.
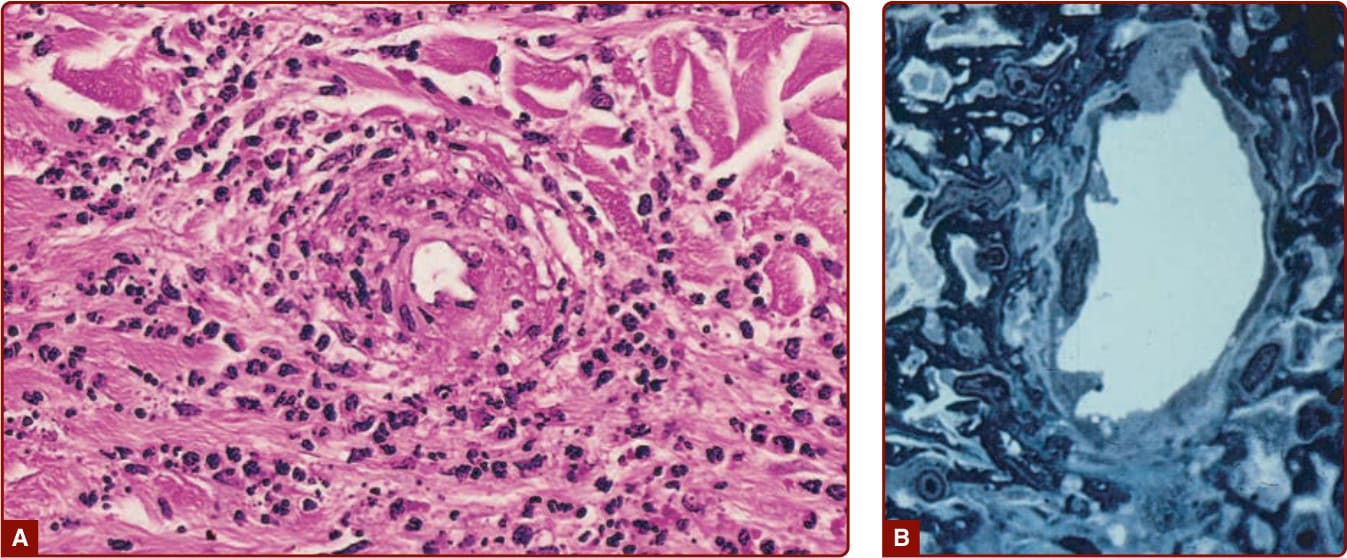
Figure 138-6 A, Perivenular infiltrate of neutrophils with fibrin deposition (hematoxylin and eosin stain, ×50 in the original magnification). B, Endothelial cell necrosis of a venule with perivenular fibrin and neutrophils (1-µm section, Giemsa stain, ×1000 in the original magnification).
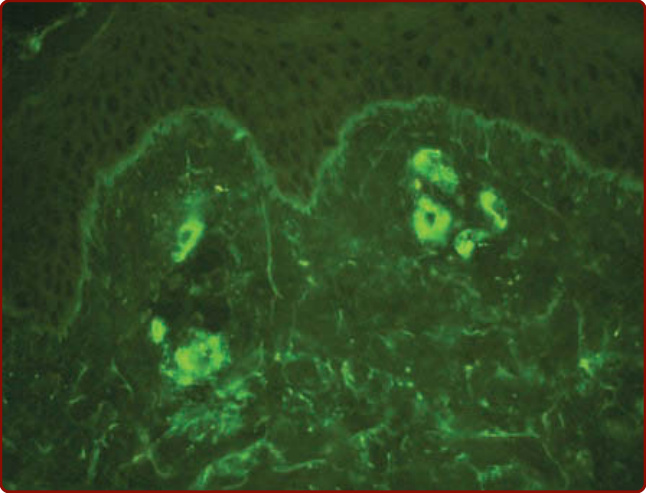
Figure 138-7 Immunofluorescence study of the skin showing deposition of IgA in the venules of the superficial dermis. (Image courtesy of Dr. James E. Fitzpatrick and used with permission.)

TABLE 138-1 Cutaneous Necrotizing Venulitis: Associated Disorders and Events
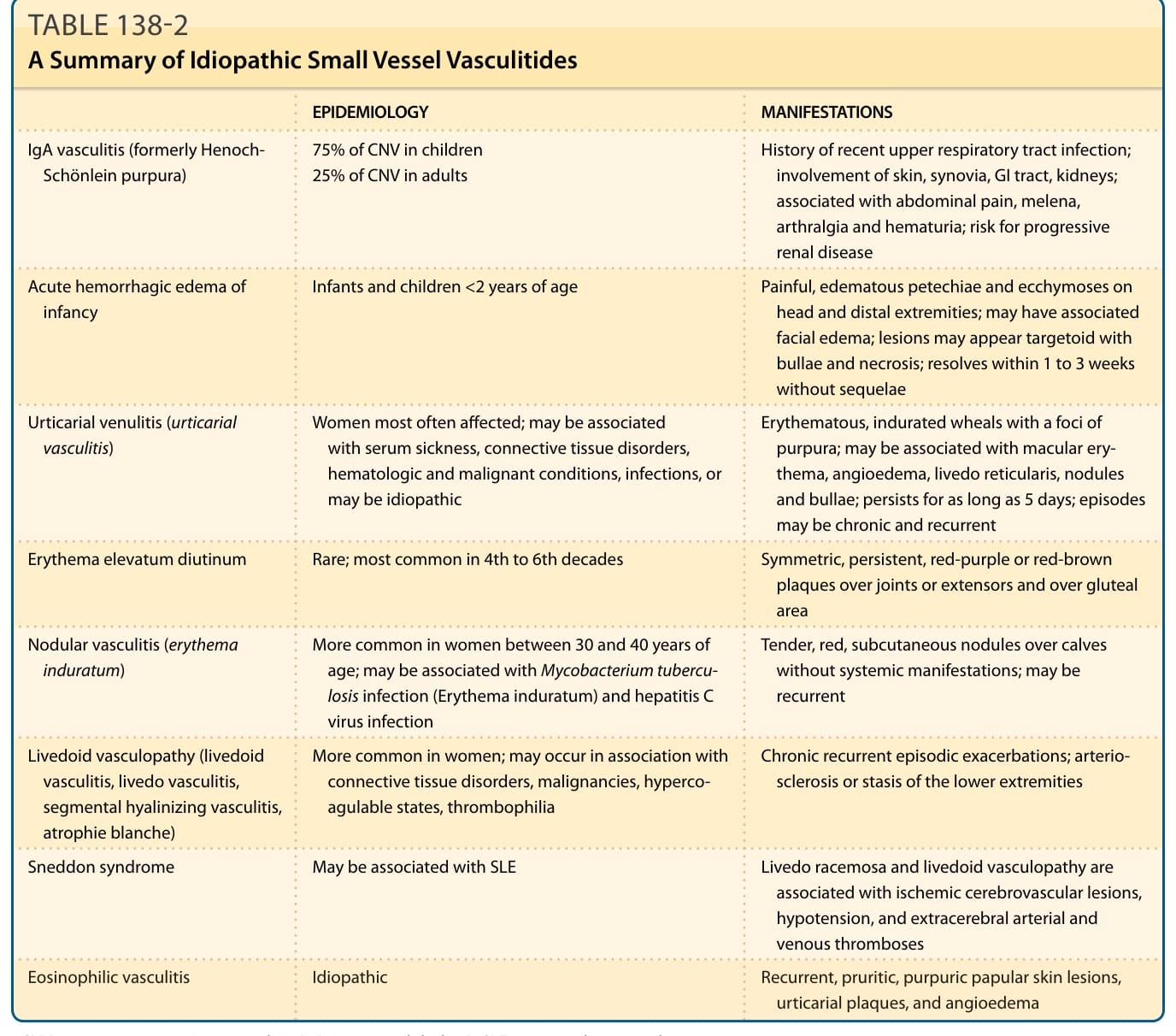
TABLE 138-2 A Summary of Idiopathic Small Vessel Vasculitides

TABLE 138-3 Laboratory Evaluation of Cutaneous Necrotizing Venulitis
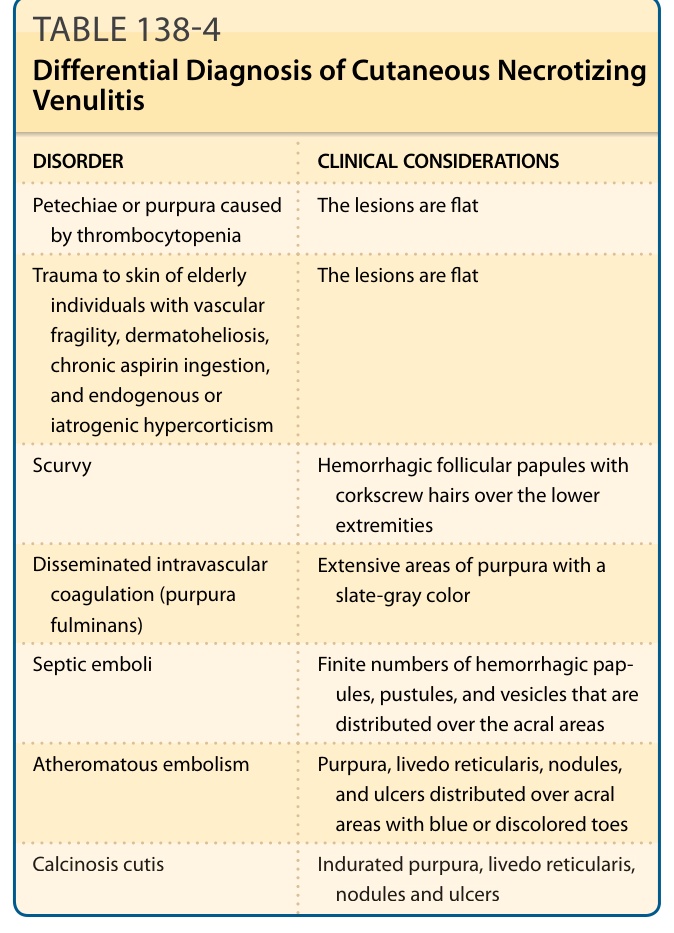
Table 138-4 outlines the differential diagnosis of CNV.

TABLE 138-5 Agents Used in the Treatment of Cutaneous Necrotizing Venulitis