Tuberous Sclerosis Complex
21
AT-A-GLANCE
■ Autosomal dominant syndrome with variable expressivity.
■ Manifested by hamartomatous tumors in multiple organs, including brain (causing seizures), eyes, heart, kidneys, lungs, and skin.
■ Skin lesions occur in nearly all individuals and are important for diagnosis.
■ Skin lesions include hypomelanotic macules, “confetti” lesions, facial angiofibromas, fibrous cephalic plaques, shagreen patches, and ungual fibromas.
■ Hypomelanotic macules appear at birth or shortly thereafter and are most useful in early diagnosis.
■ Although the skin lesions are benign, they may require treatment because of symptoms or disfigurement.
■ Skin lesions can be treated with surgery or topical sirolimus.
Tuberous sclerosis complex (TSC) is a genetic disease caused by mutations in a tumor-suppressor gene, either TSC1 or TSC2, which affects multiple organs, typically the brain, heart, kidneys, lungs, and skin.1
TSC commonly presents with seizures during infancy but some individuals remain undiagnosed until adulthood.2 Most individuals will experience difficulties related to a wide range of cognitive, behavioral, and psychiatric disorders encompassed by the umbrella term TSC-associated neuropsychiatric disorders (TAND).3 Skin lesions adversely affect quality of life4 and dermatologists are frequently called upon to diagnose or treat the skin manifestations of TSC.
EPIDEMIOLOGY
The incidence of TSC is as high as 1 in 6000 live births with a prevalence of approximately 1 in 25,000.5,6 It occurs with equal frequency in males and females and in different races and ethnicities. Hereditary transmission is evident in approximately one-third of patients. Sporadic disease occurs in about two-thirds of patients, caused by de novo mutations.7
CLINICAL FEATURES
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
Individuals with TSC may develop multiple different types of skin findings, including hypomelanotic macules, facial angiofibromas, fibrous cephalic plaques, shagreen patches, and ungual fibromas.8 Some skin findings, such as hypomelanotic macules, may appear at birth, whereas others, such as ungual fibromas, may not appear until adulthood. Most individuals eventually manifest at least 1 skin finding, but there is variability in severity between individuals.9 The ability to recognize TSC-related skin findings is important for diagnosis as skin lesions constitute several of the major and minor features for clinical diagnosis (see section “Diagnosis”).10,11
HYPOMELANOTIC MACULES
Hypomelanotic macules (Fig. 136-1) are observed in more than 90% of children with TSC.12,13 The macules are often present at birth or appear within the first few years of life and may fade or disappear in adulthood.9
The ultraviolet light of a Wood lamp may improve detection, especially in lightly pigmented individuals (Fig. 136-2).8
Hypomelanotic macules typically measure 0.5 to 3.0 cm in diameter. They are off-white and incompletely depigmented as in vitiligo. Some are oval at one end and taper to a point at the other. Such lesions are called ash-leaf spots because of their resemblance to the leaf of the European mountain ash.14 They number from 1 to more than 20. They can be located anywhere on the body, but tend to occur most often on the trunk and buttocks. When located on the scalp, they cause poliosis.12
Three or more hypopigmented macules measuring 5 mm or greater in longest dimension constitutes a major feature for the diagnosis of tuberous sclerosis.10 One or 2, and in rare individuals up to 3, hypomelanotic macules occur in 4.7% of the general population.15 A less-common type of hypopigmentation is the “confetti” skin lesion (Fig. 136-3), which is considered a minor feature for diagnosis.10 It typically occurs on the legs below the knees or on the forearms, and consists of multiple hypopigmented macules 2 to 3 mm in diameter.12,14
FACIAL ANGIOFIBROMAS
Angiofibromas generally appear between 2 and 5 years of age and eventually affect 75% to 90% of patients.9,12,13
These 1 to 3 mm in diameter, pink to red papules have
A
B
21
a smooth surface (Fig. 136-4). They may be hyperpigmented, especially in individuals with darker pigmentation. They occur on the central face and are often concentrated in the alar grooves (Fig. 136-5), extending symmetrically onto the cheeks and to the nose, nasal opening, and chin, with relative sparing of the upper lip and lateral face. They may number from 1 to more
2481
21
than 100. Lesions may coalesce to form large nodules, especially in the alar grooves.12,13 Sometimes lesions occur on the forehead, scalp, or eyelids. Adults may develop angiofibromas on the nipples.16 Angiofibromas may be unilateral in rare cases. Unilateral angiofibromas may indicate a segmental or mosaic defects.17
The development of papules may be preceded by mild erythema that is intensified by emotion or heat. During puberty, angiofibromas may grow in size and number. During adulthood they tend to be stable in size, but redness may gradually diminish.8
Three or more angiofibromas comprise a major feature.10 A solitary angiofibroma is clinically and histologically indistinguishable from the fibrous papule that occurs sporadically as a single lesion in the general population. The development of multiple angiofibromas with onset later in adolescence or early adulthood is not specific for TSC and may, instead, indicate multiple endocrine neoplasia Type 1 or Birt-Hogg-Dubé syndrome.11
FIBROUS CEPHALIC PLAQUE
The fibrous cephalic plaque, previously called forehead plaque, may be congenital or show gradual development over years.12 It is present in approximately 40% of patients.9,12,13 It is an irregular, soft to firm, connective tissue nevus with the color of the normal surrounding skin, red, or hyperpigmented in darkly pigmented individuals (Fig. 136-6). The plaque classically develops on the forehead, but can occur on the scalp, cheeks, and elsewhere on the face. The fibrous cephalic plaque and/or multiple angiofibromas is a major feature for diagnosis.10
SHAGREEN PATCH
The shagreen patch is observed in approximately 50% of patients.9,12,13 It may be present in infancy, but usually becomes apparent later. It is a firm or rubbery irregular plaque ranging in size from 1 to 10 cm (Fig. 136-7).
2482
The surface may appear bumpy with coalescing papules and nodules, or the patch may have the surface appearance of an orange peel. The color may be that of the surrounding skin, or it may be slightly pink or brown. There may be scattered smaller oval papules with or without a larger plaque (Fig. 136-8). The most common location is on the lower back to sacral region; less commonly, the patch is on the mid or upper back, buttocks, or thighs.8
UNGUAL FIBROMAS
Ungual fibromas, also known as Koenen tumors, usually appear after the first decade and eventually affect
more than 85% of adults with TSC.9,13 They are more common on the toes than on the fingers.18
Ungual fibromas measure 1 mm to 1 cm in diameter. They arise from under the proximal nailfold (periungual fibromas) and under the nail plate (subungual fibromas). Periungual fibromas are red papules and nodules that are firm, pointed, and hyperkeratotic, or soft and rounded (Fig. 136-9). They press on the nail matrix and cause a longitudinal groove, and sometimes a groove forms without an evident papule (Fig. 136-10).18 Subungual fibromas are visible through the nail plate as red or white oval lesions or as red papules emerging from the distal nail plate, causing distal subungual onycholysis (Fig. 136-11). In addition to ungual fibromas, individuals with TSC may develop “red comets” (subungual red streaks), splinter hemorrhages, and longitudinal leukonychia.18
The presence of 2 or more ungual fibromas is a major feature for diagnosis of TSC.10 Solitary lesions (also termed acral or acquired digital fibrokeratomas) are also observed in the general population, especially after nail trauma.19
OTHER SKIN LESIONS
Folliculocystic and collagen hamartomas are large plaques with follicular comedo-like openings and cysts observed in association with TSC.20 Molluscum fibrosum pendulum (skin tags) is the name given to
21
multiple fibroepithelial polyps in TSC (Fig. 136-12).12
Skin tags are common in the general population, so these are not useful for diagnosis. Miliary fibromas are patches of multiple minute papules, usually on the neck or trunk that appear like “gooseflesh.”8 Pachydermodactyly is a benign thickening of the proximal fingers that has been observed in a few patients with TSC.18
DENTAL PITTING
Multiple pits of the dental enamel are observed in up to 100% of TSC patients.21 These pits can be tiny pinpoint lesions or larger crater-like lesions (Fig. 136-13). They occur on both deciduous and permanent teeth. The identification of these lesions is enhanced by using a dental plaque stain. Dental pits can also be seen in the general population, albeit with lower prevalence and at lower numbers than in TSC.21 The presence of more than 3 dental pits is a minor feature for diagnosis.10
INTRAORAL FIBROMAS
Approximately 50% of TSC patients have intraoral fibromas (Fig. 136-14).13 These sometimes occur in the
2483
21
first decade but are more common in adulthood. They are most common on the gingivae, but also occur on the buccal and labial mucosa, hard palate, and tongue. Some patients have diffuse gingival overgrowth. Gingival overgrowth is a common side effect of anticonvulsants, especially phenytoin and cyclosporine, but gingival overgrowth can be observed in TSC even in patients not treated with anticonvulsants or immunosuppressive agents.21 Two or more intraoral fibromas are a minor feature for diagnosis.10 Oral fibromas in the general population are typically single and form at sites of trauma, usually on the tongue or buccal mucosa.21
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
BRAIN
Cerebral lesions include cortical dysplasia (tubers and white matter migration lines), subependymal nodules, and subependymal giant cell astrocytomas. Seizures occur in approximately 80% of patients, with onset in most patients in the first 3 years of life. Epilepsy is most likely infantile spasms or focal seizures, but may be tonic, atonic, or tonic–clonic seizures.22 The first-line
2484
treatment of infantile spasms is vigabatrin.23 Seizures are treated with anticonvulsant drugs as used in other forms of epilepsy. Epilepsy surgery may be required for seizures intractable to anticonvulsant therapy.23
Other approaches used to reduce seizures include ketogenic diet, vagus nerve stimulation, and possibly mammalian target of rapamycin (mTOR) inhibitors.22
TAND encompasses a range of behavioral, intellectual, and psychosocial difficulties.3 Approximately half of individuals with TSC will have intellectual disability, ranging from mild to profound. Even those with normal intellectual abilities may have deficits in recall memory, attention, or executive function. Nearly half of individuals with TSC have autism spectrum disorder or attention deficit hyperactivity disorder. Aggressive behavior, impulsivity, and sleep disorders are also common, and adolescents and adults may have anxiety and depressive disorders. TSC affects self-esteem and family and peer relationships.22
Subependymal nodules occur in approximately 80% of individuals with TSC, and subependymal giant cell astrocytomas develop in approximately 5% to 15% of individuals with TSC. Subependymal giant cell astrocytomas may be treated using an mTOR inhibitor or acute obstructive hydrocephalus may require surgical resection and possibly shunting.22,23
HEART
Cardiac rhabdomyomas occur in approximately 80% of infants with TSC and nearly 100% of fetuses with multiple cardiac rhabdomyomas have TSC.24 These neoplasms are often asymptomatic and spontaneously regress, but they may cause fetal hydrops and stillbirth or heart failure shortly after birth. Treatment of cardiac rhabdomyomas with mTOR inhibitors may speed regression. Cardiac arrhythmias are common in TSC, including slow, fast, and irregular rhythms.24
KIDNEYS
Angiomyolipomas are observed in up to 80% of TSC patients and are usually renal but may be hepatic.10,25
Additional renal lesions include renal cysts in approximately 30% and renal cell carcinoma in 2% to 3% of TSC patients. Polycystic kidney disease from contiguous TSC2-PKD1 deletion is present in approximately 1 in 20 patients.25 Renal lesions can cause renal insufficiency, hypertension, and potentially fatal retroperitoneal hemorrhage. Patients may require selective embolization followed by corticosteroids as first-line therapy for angiomyolipomas with acute hemorrhage. Treatment with an mTOR inhibitor is first-line therapy for asymptomatic growing angiomyolipomas.23,25
LUNGS
Lung involvement in TSC includes lymphangioleiomyomatosis (LAM), multifocal micronodular pneumocyte hyperplasia, and clear cell tumors of the lung.10
LAM develops in females with TSC during the third or fourth decade of life and the risk of disease increases with age. Radiographic evidence of LAM was observed in up to 80% of adult females with TSC older than age 40 years.26 It may cause spontaneous pneumothorax, chylothorax, dyspnea, cough, and hemoptysis.27 mTOR inhibitors may be used to treat LAM patients with moderate to severe lung disease or rapid progression.23
EYES
Retinal hamartomas are observed in 30% to 50% of TSC patients.10 The most common type is flat and translucent. Also common are elevated, opaque, and sometimes calcified multinodular “mulberry” lesions.28
Retinal hamartomas are usually stable in size but enucleation has been required for enlarging retinal astrocytomas. Retinal hamartomas can be similar to retinal lesions in neurofibromatosis Type 1 and may appear similar to retinoblastomas.28 TSC patients may also have retinal achromic patches that appear as hypopigmented patches in the retina.10
OTHER ORGANS
Hamartomatous polyps have been observed in the colon and rectum of TSC patients.29 Benign tumors sometimes occur in the spleen, thymus, and thyroid. TSC also may be associated with pituitary, parathyroid, and islet cell tumors.30 Arterial stenotic-occlusive disease and arterial aneurysms including aortic and intracranial aneurysms have been observed.31 Bone lesions in TSC are usually asymptomatic and may be sclerotic (calvaria, pelvis, vertebrae, ribs, and long bones) or cystic (phalanges).32,33
COMPLICATIONS
None of the skin lesions in TSC is prone to malignant degeneration, but the lesions can be a major cosmetic concern for the patient, causing social isolation and difficulties with self-esteem.34 Angiofibromas, facial plaques, and ungual fibromas can be painful or bleed
21
spontaneously or in response to minor trauma. Ungual fibromas can cause nail dystrophy and eventual loss of the nail. Large facial lesions may obstruct vision or occlude the nasal passages.35
ETIOLOGY AND PATHOGENESIS
TSC is caused by mutations in a tumor-suppressor gene, either TSC1 or TSC2. The TSC1 gene maps to chromosome band 9q34 and consists of 23 exons that encodes a 130-kDa protein called hamartin or TSC1. The TSC2 gene maps to chromosome band 16p13.3 and includes 42 exons that encodes a 200-kDa protein called tuberin or TSC2.7 The mutations observed in patients with TSC are inactivating mutations located anywhere along the sequence of TSC1 or TSC2.7 Consistent with Knudson’s 2-hit hypothesis, most TSC tumors, including skin tumors, show a second somatic mutation that inactivates the wild-type allele.36 TSC1 and TSC2 form a complex that inhibits signaling through the mechanistic target of rapamycin complex 1 (mTORC1) pathway (Fig. 136-15). Loss of TSC1/TSC2 function leads to increased mTORC1 signaling and increased cell growth.37 Sirolimus (rapamycin) inhibits mTORC1 activity and treats TSC tumors. A xenograft mouse model of TSC demonstrated that TSC2-null fibroblast-like cells are the inciting cells in angiofibromas, inducing angiogenesis and hair follicle neogenesis. Treatment with rapamycin normalized the pathologic changes but failed to eradicate the TSC2-null cells.38
RISK FACTORS
RISK FACTORS
Patients with mutations in TSC2 tend to exhibit a more-severe phenotype than those with mutations in TSC1. Immediately adjacent to TSC2 on chromosome 16 is PKD1, the gene mutated in polycystic kidney disease. Some patients with TSC have severe, early-onset renal cystic disease, and most of these patients have a contiguous deletion of TSC2 and PKD1.7 Tuber count and early onset epilepsy is associated with impairment of intellectual abilities.39 Skin manifestations have been reported in association with epilepsy, including higher likelihood of epilepsy in those with angiofibromas or shagreen patch,40 and fibrous forehead plaque with ipsilateral cerebral abnormalities and contralateral seizures.41 Analysis of somatic second-hit mutations in angiofibromas has shown the presence of ultravioletsignature mutations, suggesting that sun exposure may worsen TSC skin findings.36
DIAGNOSIS
The diagnosis of TSC is based on either genetic or clinical criteria (Table 136-1).10 The genetic criterion requires the identification of a mutation in TSC1 or TSC2 that
2485
21
Molecular pathogenesis of tuberous sclerosis complex
A Autosomal dominant Genetics/tumorigenesis Cell signaling C D
Normal cell
B
Fibrous cephalic plaque
Angiofibromas
Dental pitting and oral fibromas
Hypomelanotic macules
Patient cell
Shagreen patch
“Confetti” lesions
Tumor cell
Ungual fibromas
TSC1/TSC2
Rheb-GTP (active)
Rheb-GDP (inactive)
mTORC1
Germline mutation
Cell growth
TSC1/TSC2
Somatic mutation
Rheb-GTP (active)
Rheb-GDP (inactive)
mTORC1
Cell growth
clearly inactivates the function of the TSC1 or TSC2 proteins in DNA from normal tissue. The demonstration of a pathogenic mutation is sufficient to make a definite diagnosis of TSC. Using standard approaches for DNA analysis, approximately 10% to 25% of individuals with TSC will have no mutation identified. Therefore, a normal result does not exclude the possibility of TSC and clinical criteria are still important. The clinical criteria are categorized as major or minor features, with cutaneous and oral lesions comprising 4 of 11 major features and 3 of 6 minor features. Using clinical criteria, a definite diagnosis of TSC requires the presence of 2 or more major features or 1 major feature with 2 or more minor features. A possible diagnosis of TSC is made based on the presence of either 1 major feature or 2 or more minor features.10
2486
SUPPORTIVE STUDIES
SUPPORTIVE STUDIES
PATHOLOGY
A skin biopsy may be useful when internal manifestations of TSC are lacking or if the diagnosis of TSC hinges on skin lesions for satisfying clinical criteria.11
Hypomelanotic macules have normal numbers of melanocytes, in contrast to the lesions of vitiligo, in which melanocytes are absent. The melanocytes in hypomelanotic macules have poorly developed dendritic processes, and melanosomes are decreased in numbers, size, and melanization.42 Angiofibromas contain plump, spindle-shaped, or stellate fibroblastic cells in the dermis among increased numbers of dilated
Genetic Criterion Pathogenic mutation in TSC1 or TSC2 in DNA from normal tissue
Major Features
-
Hypomelanotic macules (≥3, at least 5 mm diameter)
-
Angiofibromas (≥3) or fibrous cephalic plaque
-
Ungual fibromas (≥2)
-
Shagreen patch
-
Multiple retinal hamartomas
-
Cortical dysplasiasa
-
Subependymal nodules
-
Subependymal giant cell astrocytoma
-
Cardiac rhabdomyoma
-
Lymphangioleiomyomatosisb
-
Angiomyolipomas (≥2)b
Minor Features
Minor Features
-
“Confetti” skin lesions
-
Dental enamel pits (≥3)
-
Intraoral fibromas (≥2)
-
Retinal achromic patch
-
Multiple renal cysts
-
Nonrenal hamartomas
-
“Confetti” skin lesions
-
Dental enamel pits (≥3)
-
Intraoral fibromas (≥2)
-
Retinal achromic patch
-
Multiple renal cysts
-
Nonrenal hamartomas
aIncludes tubers and cerebral white matter radial migration lines.
bA combination of the 2 major clinical features (lymphangioleiomyomatosis and angiomyolipomas) without other features does not meet criteria for a definite diagnosis. Definite diagnosis: Pathogenic mutation or 2 major features or 1 major feature with ≥2 minor features. Possible diagnosis: One major feature or ≥2 minor features.
vessels (Fig. 136-16). Collagen fibers are oriented in an onionskin pattern around follicles and vessels. The epidermis shows melanocytic hyperplasia and flattening of rete ridges. Periungual fibromas are similar, but with more extensive hyperkeratosis and a variable increase in vascularity (Fig. 136-17). The shagreen patch has sclerotic bundles of collagen in the reticular dermis, often with reduced elastic fibers (Fig. 136-18).8
21
IMAGING AND LABORATORY TESTING
Besides dermatologic and ophthalmologic examination, the initial evaluation of a patient suspected to have TSC includes MRI of the brain and abdomen.23 MRI of the brain, with and without gadolinium, evaluates for tubers, subependymal nodules, migrational defects, and subependymal giant cell astrocytoma. MRI of the abdomen assesses for renal angiomyolipomas and cysts, as well as extrarenal hamartomas and aortic aneurysms. These imaging studies and additional studies described next are used to confirm the diagnosis of TSC and to determine the extent of disease in those known to have TSC. Additional studies include (a) a baseline routine electroencephalogram even without reported seizures; (b) evaluation for TAND; (c) blood pressure and renal glomerular filtration rate; (d) high-resolution chest computed tomography (HRCT) and pulmonary function testing if
2487
21
symptomatic or as baseline in asymptomatic females 18 years of age or older; (e) an echocardiogram and electrocardiogram to evaluate for rhabdomyomas and arrhythmia in pediatric patients, especially if younger than 3 years of age; and (f) a baseline electrocardiogram at any age.23
MOLECULAR DIAGNOSIS
The identification of a pathogenic mutation in TSC1 or TSC2 is now sufficient for the diagnosis of TSC even without diagnostic clinical findings.10 Genetic testing may yield false-negative or inconclusive results in approximately 10% to 25% of TSC patients.1 Most of these cases are caused by intronic mutations or somatic mosaicism.43 Molecular genetic testing can provide additional information for genetic counseling, and it can be used in prenatal diagnosis. When a pathogenic mutation has been identified, it may be used to screen at-risk family members.1
DIAGNOSTIC ALGORITHM
DIAGNOSTIC ALGORITHM
Individuals with skin findings suggestive of 1 cutaneous major feature of TSC should be queried for TSCassociated symptoms and family history. A thorough skin and oral examination should be performed. Those manifesting at least 1 additional major feature or 2 minor features should undergo TSC diagnostic evaluation to determine baseline disease extent.23 In absence of additional skin or oral lesions, skin biopsy may be performed to confirm the clinical diagnosis (Fig. 136-19). Individuals with skin lesions characteristic of TSC should proceed to complete diagnostic evaluation. Those with skin lesions nonspecific or atypical of TSC should be first considered for other conditions to avoid unnecessary TSC workup.44 Some patients will not meet criteria for a definitive diagnosis. These patients should receive a diagnosis of possible TSC and consideration given to performing a future reevaluation.
Diagnostic and management algorithm for tuberous sclerosis complex
Skin findings suggest 1 major feature of TSC
Lesions not consistent with TSC Lesions nonspecific or atypical of TSC Lesions characteristic of TSC
Consider skin biopsy of:
Facial Papules Localized Hypopigmentation Ungual Papules Connective Tissue Plaque
Melanin & normal melanocyte number
Additional or alternate findings Collagen fibers Additional or alternate findings
Othera
Otherb Otherc Otherd
Other Other
Adult-onset angiofibromase Hypopigmented lesionsf Collagenomasg
Polygonal or ‘ash leaf’ shaped
On scalp or face On back & large
? ? ?
Juvenile-Onset Angiofibromas (≥3) Hypomelanotic Macules (≥3) Ungual Fibromas (≥2) Fibrous Cephalic Plaque Shagreen Patch
TSC Diagnostic Evaluation
TSC1/TSC2 Mutation or 2 Major Features of 1 Major + ≥2 Minor Features
No Additional Findings or 1 Minor Feature
Definite TSC Possible TSC
Counseling Counseling
Routine Surveillance Evaluation as Needed
2488
B in Fig. 136-19
■Piebaldism
■Vitiligo
■Waardenburg syndrome
■Postinflammatory hypopigmentation
■Sarcoidosis
■Hypopigmented mycosis fungoides
■Pityriasis alba
■Idiopathic guttate hypomelanosis
■Leprosy
■Nevus anemicus
F in Fig. 136-19
F in Fig. 136-19
■Nevus depigmentosus (congenital isolated or segmental lesion with serrated border)
■Nevus depigmentosus (congenital isolated or segmental lesion
with serrated border)
■Hypomelanosis of Ito (congenital macules arranged in whorls or streaks following Blaschko lines)
■Hypomelanosis of Ito (congenital macules arranged in whorls or
streaks following Blaschko lines)
■Progressive macular hypomelanosis (adolescent or adult onset of nummular, confluent macules concentrated on the trunk)
■Progressive macular hypomelanosis (adolescent or adult onset of
nummular, confluent macules concentrated on the trunk)
■Tuberous sclerosis complex (may also present with hypopigmented macules of Blaschkoid or segmental morphology)
■Tuberous sclerosis complex (may also present with hypopigmented
macules of Blaschkoid or segmental morphology)
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
Individuals with tuberous sclerosis exhibit decreased overall survival compared with the general population. The causes of premature death include renal failure, intractable seizures, obstructive hydrocephalus, cardiac outflow obstruction, arrhythmia, respiratory failure, pneumothorax, and hemorrhage from an aneurysm or a tumor, especially angiomyolipomas.45,46
A in Fig. 136-19
■Acne vulgaris
■Rosacea
■Verruca
■Dermal melanocytic nevi
■Syringomas
■Trichoepitheliomas (multiple familial trichoepithelioma)
■Fibrofolliculomas/trichodiscomas (Birt-Hogg-Dubé syndrome)
■Tricholemmomas (Cowden syndrome)
■Sebaceous tumors (Muir-Torre syndrome)
E in Fig. 136-19
E in Fig. 136-19
■Fibrous papules (single or few)
■Fibrous papules (single or few)
■Multiple endocrine neoplasia Type 1
■Multiple endocrine neoplasia Type 1
■Birt-Hogg-Dubé syndrome
■Birt-Hogg-Dubé syndrome
■Tuberous sclerosis complex adult-onset angiofibromas
■Tuberous sclerosis complex adult-onset angiofibromas
21
C in Fig. 136-19
C in Fig. 136-19
■Digital mucous cyst
■Digital mucous cyst
■Subungual exostosis
■Subungual exostosis
■Subungual corn (heloma)
■Subungual corn (heloma)
■Wart
■Wart
■Pyogenic granuloma
■Pyogenic granuloma
■Squamous cell carcinoma
■Squamous cell carcinoma
■Onychomycosis
■Onychomycosis
■Psoriasis
■Psoriasis
Fatal events may occur at any age. Brain and heart tumors may cause death in infancy, whereas lung and kidney tumors are more likely to cause premature death in adulthood. The prognosis for the individual patient depends on disease expressivity. Some individuals have a normal life span with few medical complications.
MANAGEMENT
MEDICATIONS
MEDICATIONS
The management of TSC skin tumors has changed with the use of mTOR inhibitors. The U.S. Food and Drug Administration has approved oral mTOR inhibitors for treating several TSC-associated tumors, such as everolimus for subependymal giant cell astrocytomas and renal angiomyolipomas, and sirolimus for LAM.47
Most patients treated with an oral mTOR inhibitor for internal tumors will show improvement in their skin lesions.48,49 Therefore, appropriate management may involve monitoring for skin improvement in those on systemic therapy. Oral mTOR inhibitors may delay wound healing, but surgical intervention may be indicated for lesions failing to respond to oral therapy or for urgent clinical situations such as bleeding or functional impairment.11
D in Fig. 136-19
■Nonfamilial elastoma or familial elastoma
■Dermatofibrosis lenticularis disseminata (in Buschke-Ollendorf syndrome)
■Dermal nevi
■Xanthomas
■Granulomatous disease
G in Fig. 136-19
G in Fig. 136-19
■Familial cutaneous collagenomas or eruptive collagenoma
■Familial cutaneous collagenomas or eruptive collagenoma
■Multiple endocrine neoplasia Type 1 collagenomas
■Multiple endocrine neoplasia Type 1 collagenomas
■Sclerotic fibroma (Cowden syndrome)
■Sclerotic fibroma (Cowden syndrome)
■Isolated connective tissue nevus
■Isolated connective tissue nevus
2489
■Tuberous sclerosis complex collagenomas
■Tuberous sclerosis complex collagenomas
21
Systemic treatment with mTOR inhibitors is associated with a variety of adverse effects, such as stomatitis, mouth ulceration, acne-like skin lesions, infections, hypertriglyceridemia, hypercholesterolemia, bone marrow suppression, proteinuria, joint pain, and noninfectious pneumonitis. The potential for these adverse effects limits the use of systemic mTOR inhibitors in the treatment of skin lesions alone.11 To reduce the potential for adverse effects, several investigators have used mTOR inhibitors applied topically. Angiofibromas in most patients became flatter and decreased in redness.50 Hypomelanotic macules increased in pigmentation.51 Adverse effects were minimal and systemic levels were mostly undetectable.50 A doubleblind, placebo-controlled, phase 2 randomized clinical trial showed that topical sirolimus 0.2% gel was safe and effective for treating TSC angiofibromas.52 It is expected that long-term treatment will be required and skin lesions may worsen after discontinuing therapy.
PROCEDURES
PROCEDURES
Indications for treatment of TSC-related skin lesions may include pain, bleeding, functional interference, or disruption of social interactions.11 Angiofibromas have been treated by excision, curettage, chemical peel, cryosurgery, dermabrasion, electrosurgery, and different types of laser procedures.8,11,35,53 Multiple sessions using several approaches may be required for optimal results. Potential complications of surgical treatments include infection, hypertrophic scarring, postinflammatory hyperpigmentation, and hypopigmentation. Treated angiofibromas tend to recur over a couple of years, and new lesions may form.35 Ungual fibromas are usually treated by excision, but these lesions also have a high recurrence rate. Hypomelanotic macules may be temporarily concealed using self-tanning lotions or makeup matched to the person’s skin color. The shagreen patch is usually left untreated, but it can be excised.8,35
COUNSELING
COUNSELING
Upon diagnosing an infant with TSC, a major aspect of care is answering questions and addressing the concerns of the family. Unfortunately, many parents report negative experiences because of physician insensitivity and the provision of inaccurate information and inadequate support.54 Parents should be educated about possible evolution or development of skin lesions and available treatments. Parents should be advised to maintain good sun protection for their children as this may lessen the future severity of angiofibromas.11 Other important educational aspects include informing parents to recognize infantile spasms, discussing the potential need for an individual educational program, and counseling adolescent and adult females about the risks of smoking or estrogen use in oral contraceptives
2490
because of their potential to worsen LAM.23 Patients should be promptly referred to other specialists, social services, and genetic counseling as indicated. Inform individuals and families about organizations dedicated to TSC, such as the Tuberous Sclerosis Alliance (http://www.tsalliance.org).
SURVEILLANCE
SURVEILLANCE
After diagnosis, the patient should have periodic surveillance for the development of new lesions or changes in existing lesions.23 MRI of the brain is recommended every 1 to 3 years in asymptomatic TSC patients younger than age 25 years to monitor for development of subependymal giant cell astrocytoma and, more frequently, for positive findings. MRI of the abdomen should be performed every 1 to 3 years to evaluate for progression of angiomyolipomas and renal cystic disease. Additional recommendations include: (a) annual screening for TAND throughout childhood and as needed; (b) annual tests of blood pressure and renal glomerular filtration rate; (c) clinical screening for LAM symptoms and HRCT every 5 to 10 years for asymptomatic at-risk individuals, or, for those with LAM, annual pulmonary function testing and HRCT every 2 to 3 years; (d) electrocardiogram every 3 to 5 years in asymptomatic patients of all ages and, for pediatric patients with asymptomatic cardiac rhabdomyomas, echocardiogram every 1 to 3 years until resolved; (e) annual skin examination; (f) dental examination every 6 months with panoramic radiographs by age 7 years; and (g) annual ophthalmologic evaluation in patients with previously identified ophthalmologic lesions or vision symptoms.23
SCREENING
SCREENING
A child with TSC may be born to parents who do not carry the diagnosis of TSC. This may represent a de novo mutation, parental mosaicism, or it may indicate that 1 parent has a very mild form of TSC that has escaped detection. Both parents should be carefully screened for clinical manifestations or for the pathogenic mutation, if known. Screening begins with examination of the skin, oral mucosa, teeth, and retina. Most affected family members will show TSC lesions with this approach, but additional imaging studies are recommended in their absence.1 If 1 parent has TSC, there is a 50% chance that subsequent children will inherit the mutation. To identify additional family members at risk of TSC, obtain a 3-generation family history.23
If neither parent has TSC, it is possible that there is alternate paternity or undisclosed adoption. Another possibility is that 1 parent is mosaic for a mutation in TSC1 or TSC2.43 This has profound implications for genetic counseling. Whereas it would be extremely unlikely for parents of a child with a de novo mutation to have another affected child, germline mosaicism in
1 parent increases the risk of having another child with TSC. An estimate of the overall risk that apparently unaffected parents will have a second child with TSC is approximately 1% to 2%.1
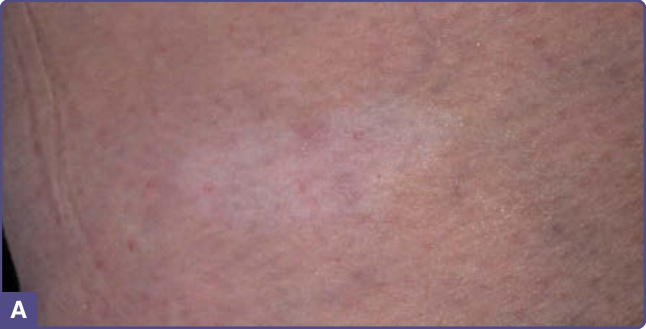
Figure 136-1 Hypomelanotic ash-leaf macules on the lower leg of a child with tuberous sclerosis complex.

Figure 136-2 A, Hypomelanotic macules on the lateral chest of an adult with tuberous sclerosis complex. The macules may be easily overlooked. B, Wood lamp accentuates the macules.
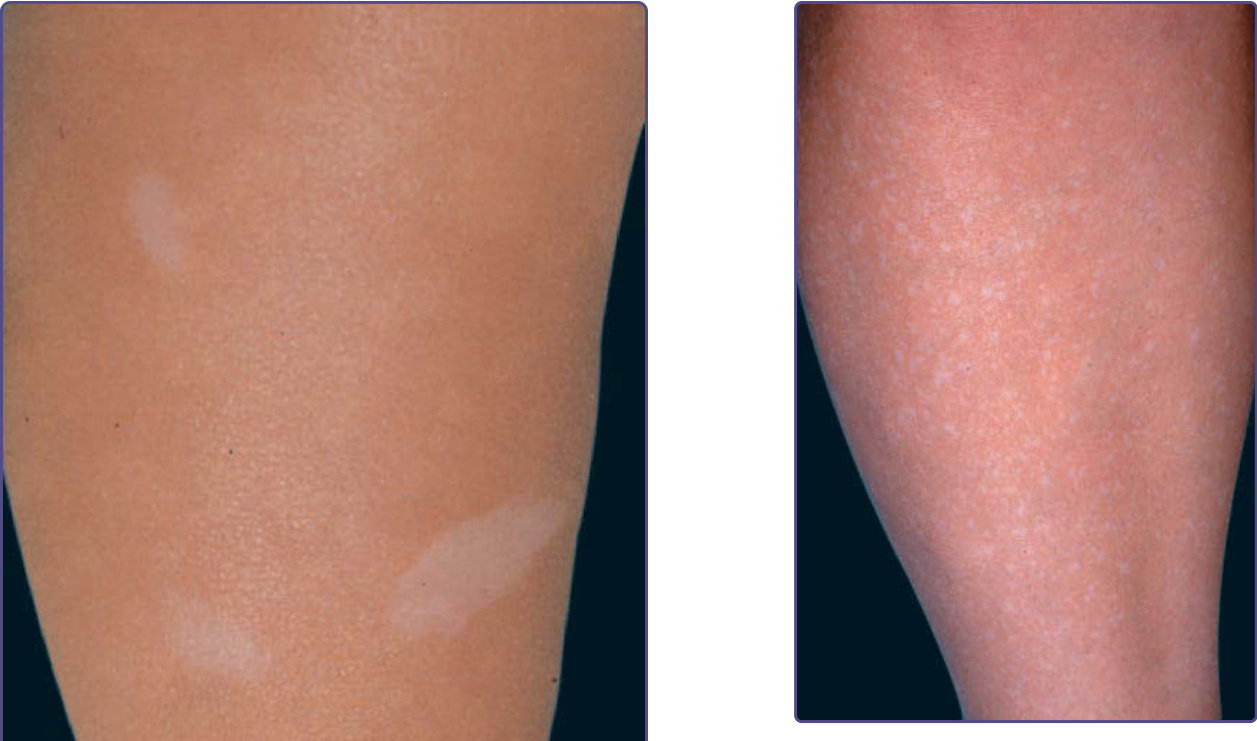
Figure 136-3 Confetti-like hypopigmented macules on the lower leg of an adult with tuberous sclerosis complex.
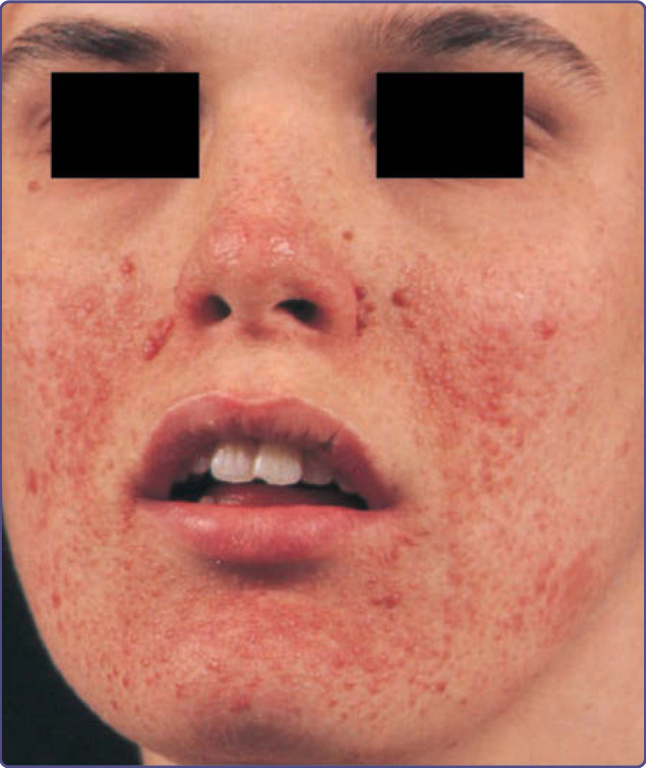
Figure 136-4 Multiple facial angiofibromas on the nose, cheeks, and chin, with relative sparing of the upper lip is commonly observed in tuberous sclerosis complex.
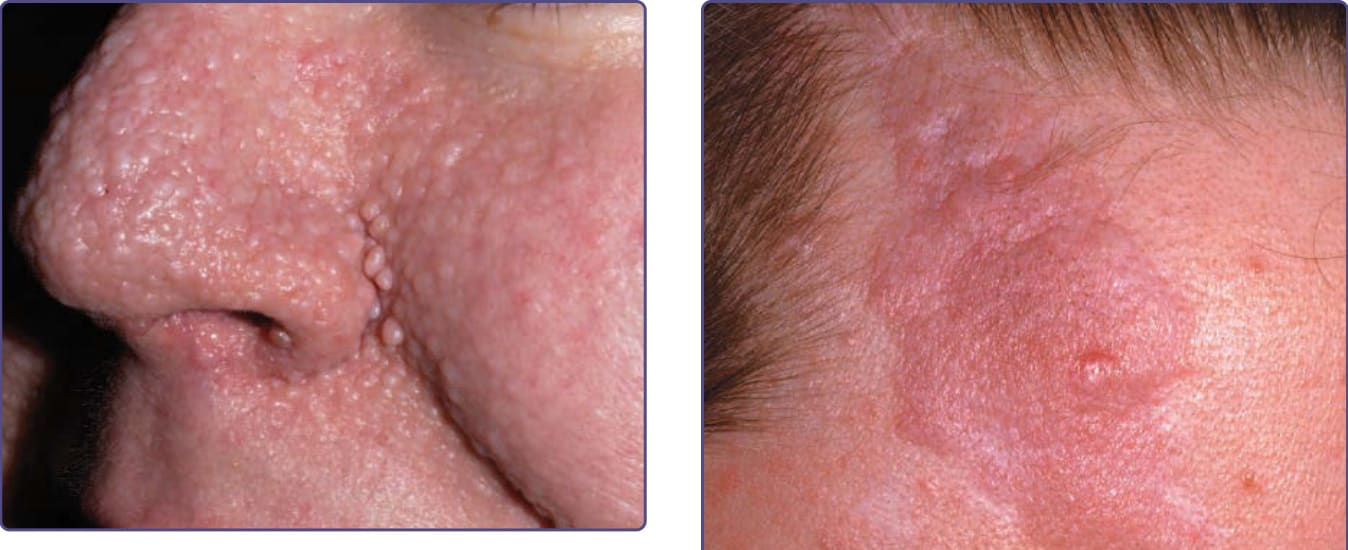
Figure 136-5 Angiofibromas are often larger in the alar grooves and may become less erythematous over time, as in this adult with tuberous sclerosis complex.
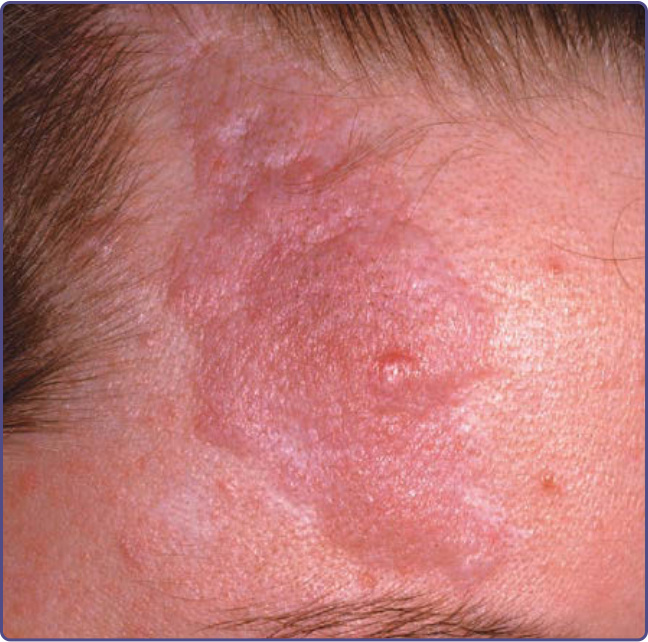
Figure 136-6 The forehead fibrous plaque in tuberous sclerosis complex is often pink-red and has a bumpy surface and irregular outline.
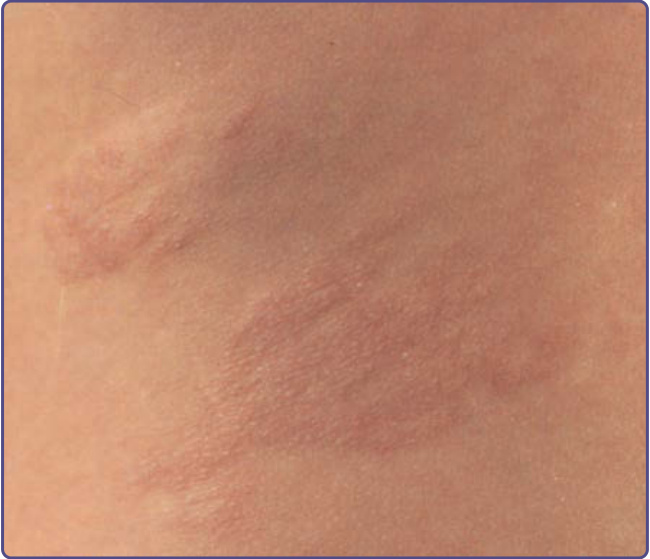
Figure 136-7 The shagreen patch in tuberous sclerosis complex is a firm, bumpy plaque that is usually located on the lower back.
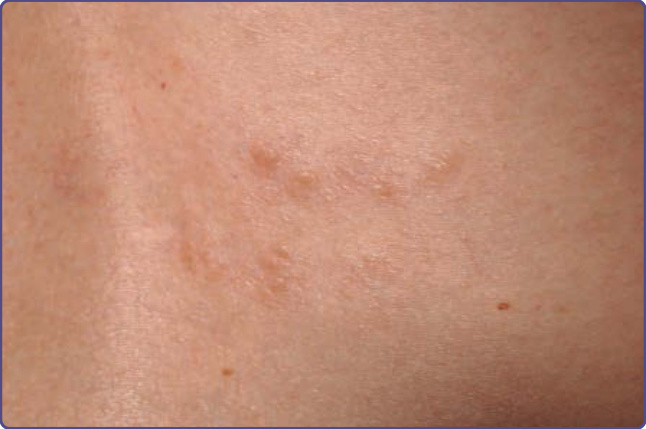
Figure 136-8 The shagreen patch may consist of grouped, dermal papules that are minimally elevated above the skin.
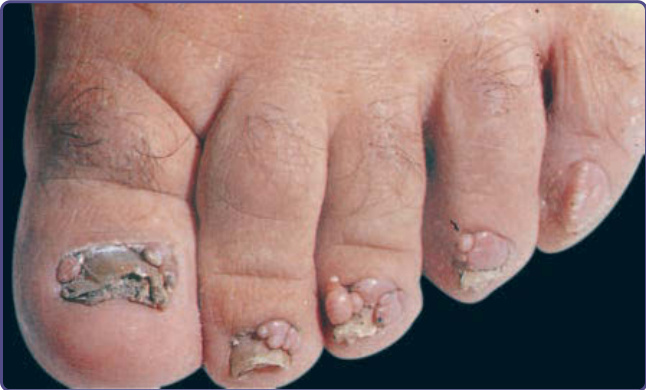
Figure 136-9 Multiple periungual and subungual fibromas on the toes in a patient with tuberous sclerosis complex.
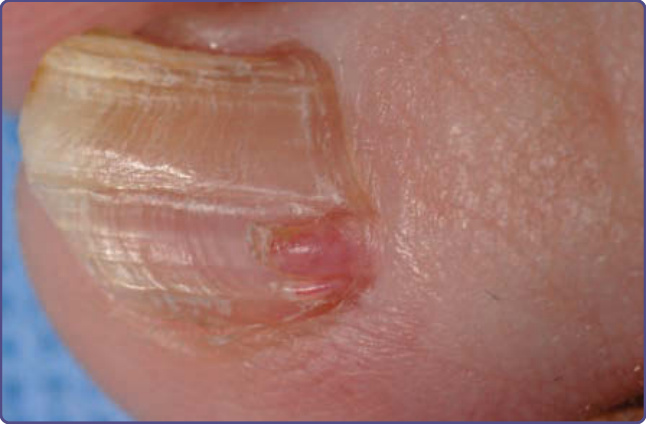
Figure 136-10 Periungual fibromas emanating from the proximal nailfold in tuberous sclerosis complex causes a longitudinal groove in the nail.
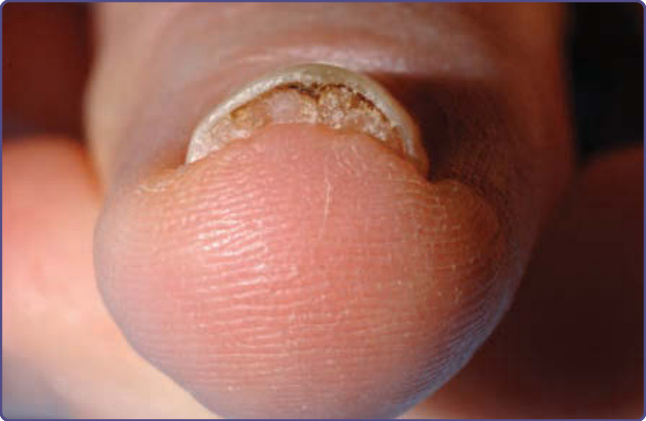
Figure 136-11 Subungual fibromas may disrupt the normal nail anatomy in tuberous sclerosis complex, causing distal onycholysis.
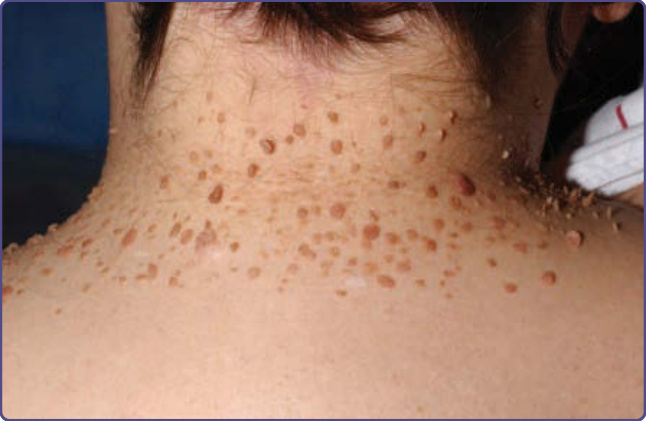
Figure 136-12 Molluscum fibrosum pendulum, another name for multiple skin tags in tuberous sclerosis complex, is observed on the neck in this patient.
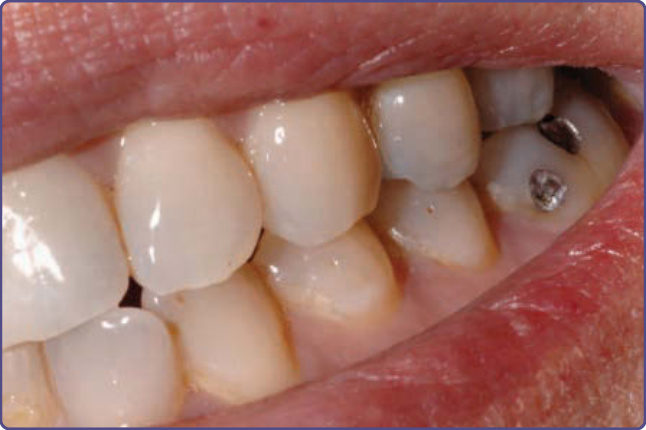
Figure 136-13 Multiple dental enamel pits are seen in nearly all patients with tuberous sclerosis complex.
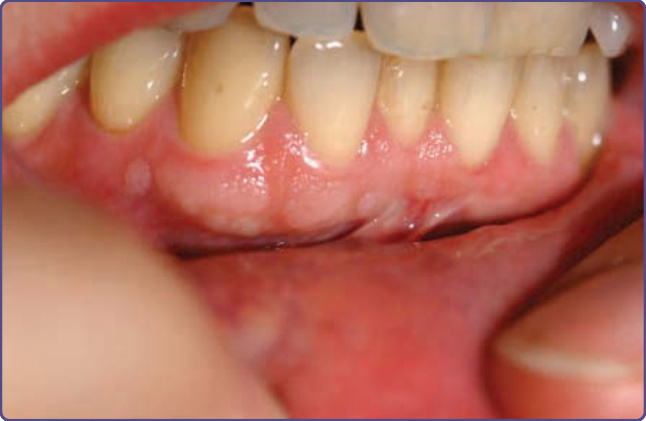
Figure 136-14 Multiple gingival fibromas in an adult with tuberous sclerosis complex.
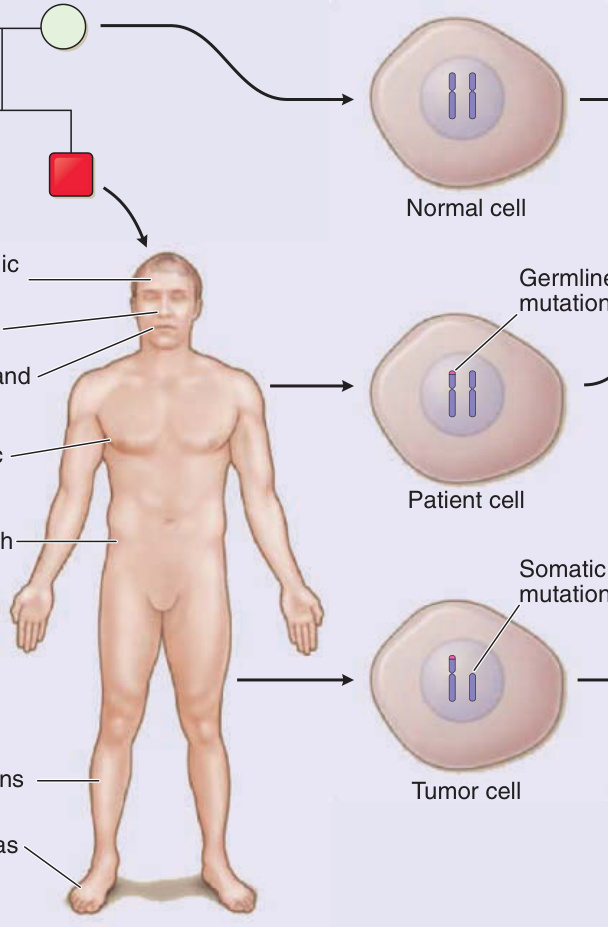
Figure 136-15 Molecular pathogenesis of tuberous sclerosis complex (TSC). A, An example is shown in which a mutation in TSC2 is passed from the father (red square) to the son (red square), whereas the mother (white circle) and another son (white square) are unaffected. B, Skin lesions are observed in the son at the indicated locations. C, A cell from the mother shows 2 normal alleles for TSC2 on chromosome band 16p13.3. A cell from the son shows the TSC2 mutation inherited from his father. A tumor from the son has this germline mutation and a “second-hit” mutation, detectable as loss of heterozygosity at this locus, that inactivates the other allele. D, TSC2, in a complex with TSC1, is a guanosine triphosphatase– activating protein, converting active Ras-homolog enriched in brain (Rheb) guanosine triphosphate (GTP) to inactive Rheb guanosine diphosphate (GDP). Rheb-GTP activates mammalian target of rapamycin complex 1 (mTORC1), a kinase that increases protein translation and cell growth, so TSC2 normally acts to inhibit cell growth. Loss of TSC2 function in tumors results in increased levels of Rheb-GTP, activation of mTORC1, and increased cell growth.
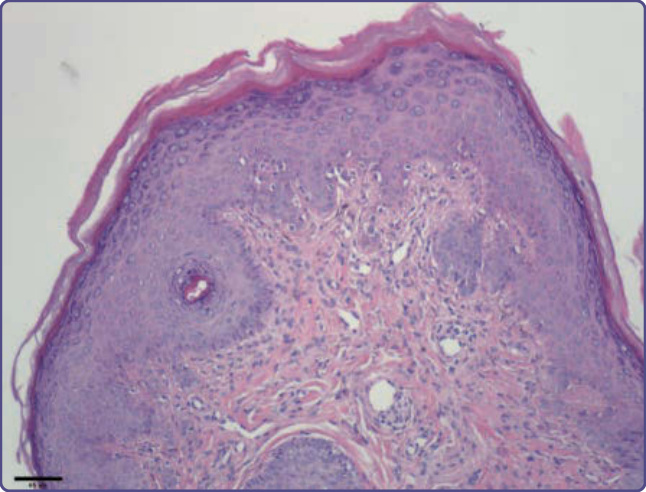
Figure 136-16 Angiofibromas are composed of ectatic vessels and fibroblastic cells that can be large, stellate, and sometimes multinucleated. There is fibrosis that may form concentric rings around vessels and adnexal structures.
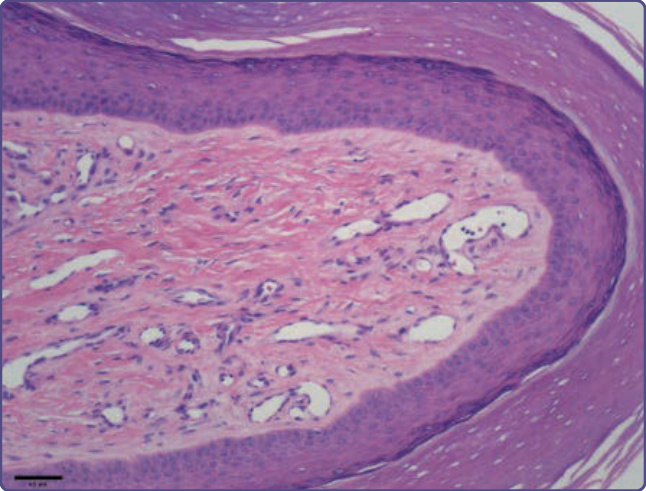
Figure 136-17 Periungual fibromas may exhibit dense collagen bundles and ectatic vessels, sparse to increased fibroblastic cells, and acanthosis and hyperkeratosis.
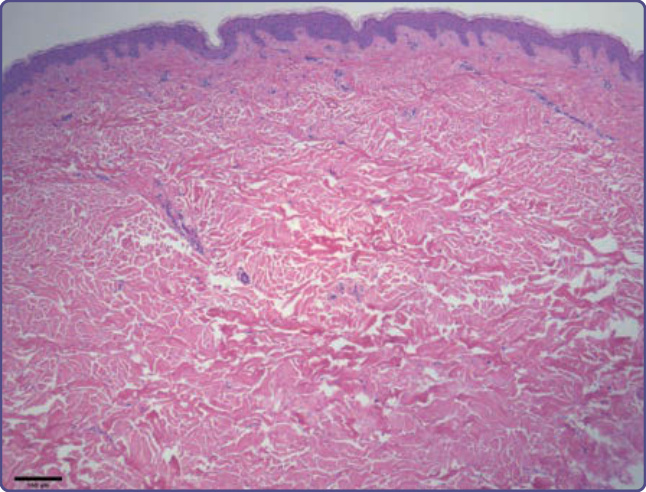
Figure 136-18 The shagreen patch shows thickened collagen bundles that appear disordered. Elastic fibers are decreased in amount compared to normal skin.
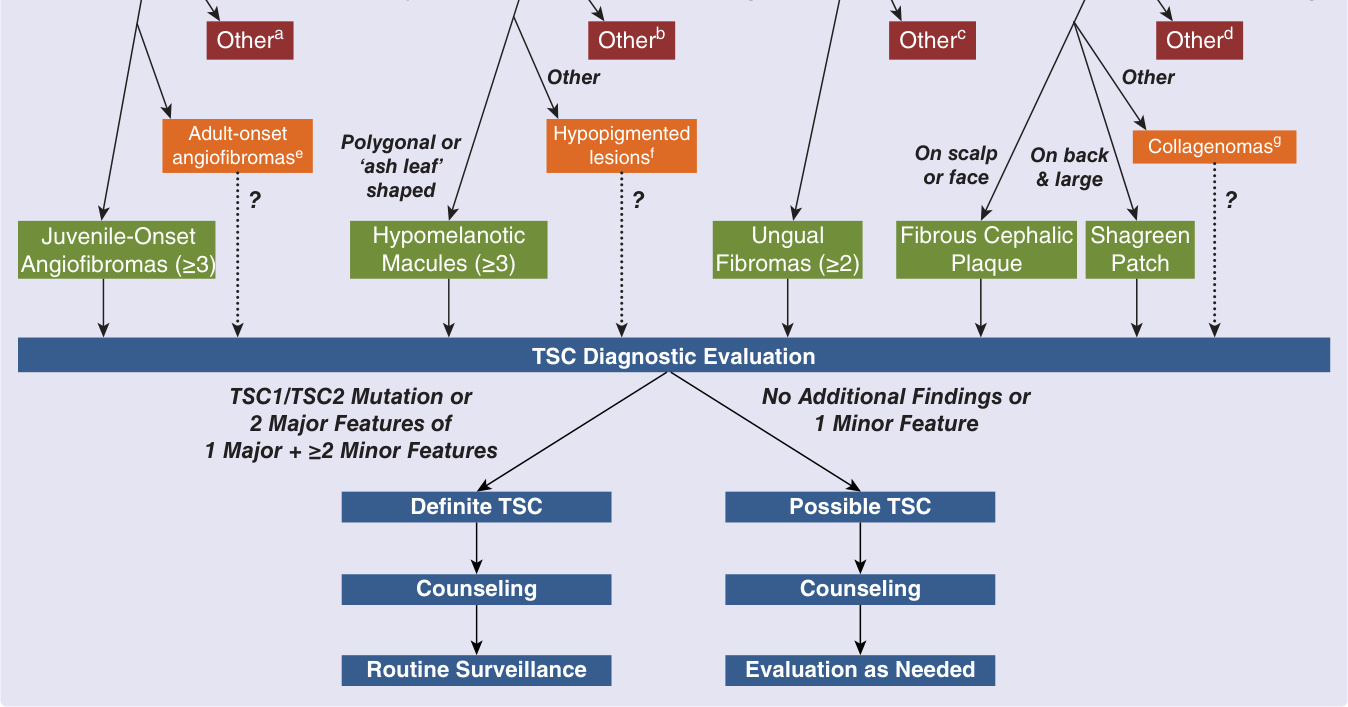
Figure 136-19 Diagnostic and management algorithm for tuberous sclerosis complex (TSC). Skin lesions may be categorized as not consistent (red), nonspecific or atypical (orange), or characteristic (green) of TSC. A and E, see Table 136-3; B and F, see Table 136-2; C, see Table 136-4; D and G, see Table 136-5. (Adapted from Nathan N, Burke K, Moss J, et al. A diagnostic and management algorithm for individuals with an isolated skin finding suggestive of tuberous sclerosis complex. Br J Dermatol. 2017;176(1):220-223.)
TABLE 136-1 Diagnostic Criteria for Tuberous Sclerosis Complex (Updated at 2012 Conference)
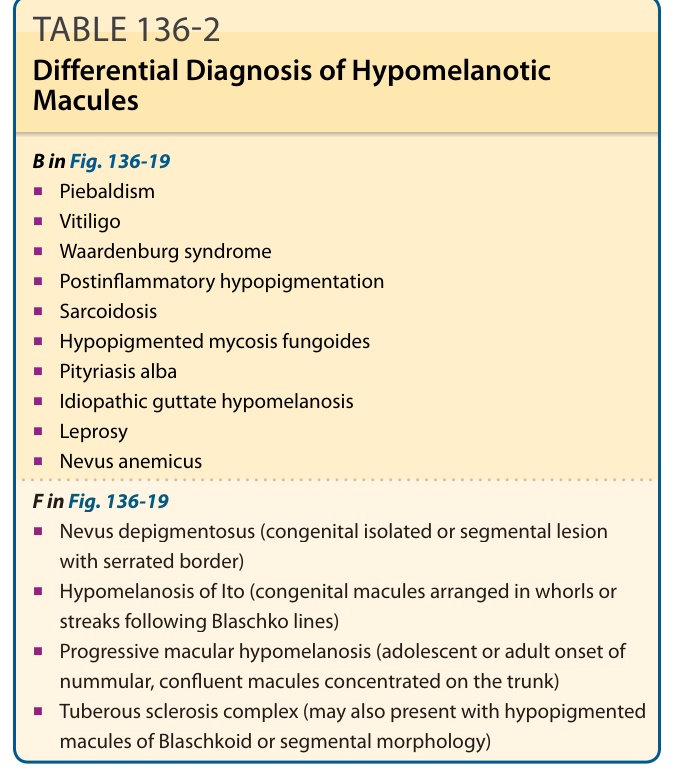
TABLE 136-2 Differential Diagnosis of Hypomelanotic Macules

TABLE 136-3 Differential Diagnosis of Angiofibromas

TABLE 136-4 Differential Diagnosis of Ungual Fibroma

TABLE 136-5 Differential Diagnosis of Shagreen Patch